determinacion de la estructura de una proteina mediante metodos computacionales
DESCRIPTION
DETERMINACION DE LA ESTRUCTURA DE UNA PROTEINA MEDIANTE METODOS COMPUTACIONALES. Modelado por homología. Dra. Cristina Marino Buslje Septiembre 2006. Preedición de la estructura de una proteína. Es una de las tareas mas significantes abordadas en la biología estructural computacional. - PowerPoint PPT PresentationTRANSCRIPT

DETERMINACION DE LA ESTRUCTURA DE UNA PROTEINA MEDIANTE
METODOS COMPUTACIONALES
Modelado por homología
Dra. Cristina Marino BusljeSeptiembre 2006

1i9b.pdb
Es una de las tareas mas significantes abordadas en la biología estructural computacional. Tiene por objeto determinar la estructura tridimensional de proteínas a partir de su secuencia de aminoácidos. En términos mas formales, es la predicción de la estructura terciaria por su estructura primaria. Dada la utilidad del conocimiento de la estructura de proteínas en tareas tan valiosas como el diseño de drogas, este es un campo altamente activo de investigación.
Preedición de la estructura de una proteína
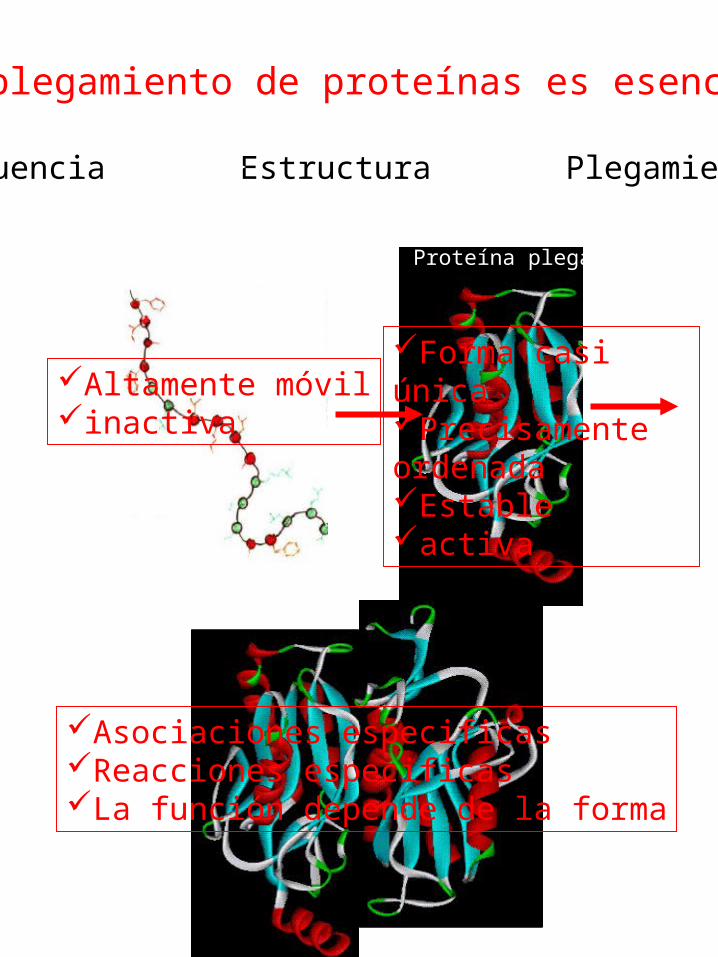
El plegamiento de proteínas es esencial
Secuencia Estructura Plegamiento
Proteína desplegada Proteína plegada
Altamente móvilinactiva
Forma casi únicaPrecisamente ordenadaEstableactiva
Asociaciones especificasReacciones especificasLa función depende de la forma

1tc2.pdb

Dentro de una proteína, un dominio estructural (dominio) es un elemento que se estabiliza por si mismo y generalmente se pliega independientemente del resto de la proteína. Muchos dominios no son únicos de las proteínas producto de un gen o una familia de genes, sino que aparecen en una gran variedad de proteínas. Los dominios son frecuentemente nombrados según la función biológica en la que intervienen en la proteína donde aparecen predominantemente, por ejemplo, el "calcium-binding domain” de la calmodulina
Dominio estructural
LAS PROTEINAS ESTAN FORMADAS POR DOMINIOS

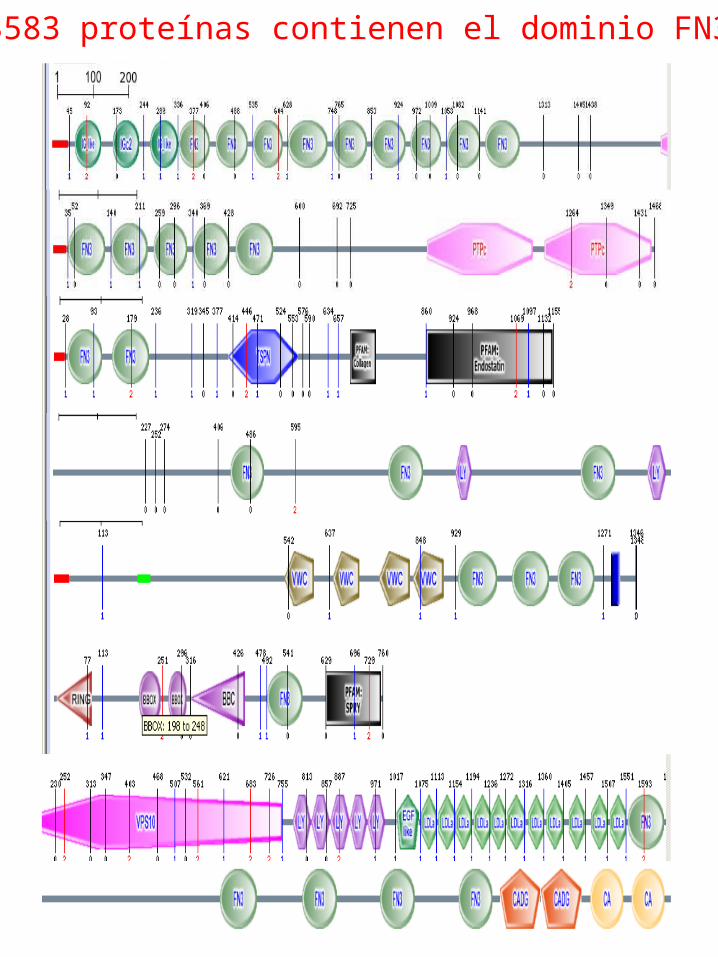
3583 proteínas contienen el dominio FN3
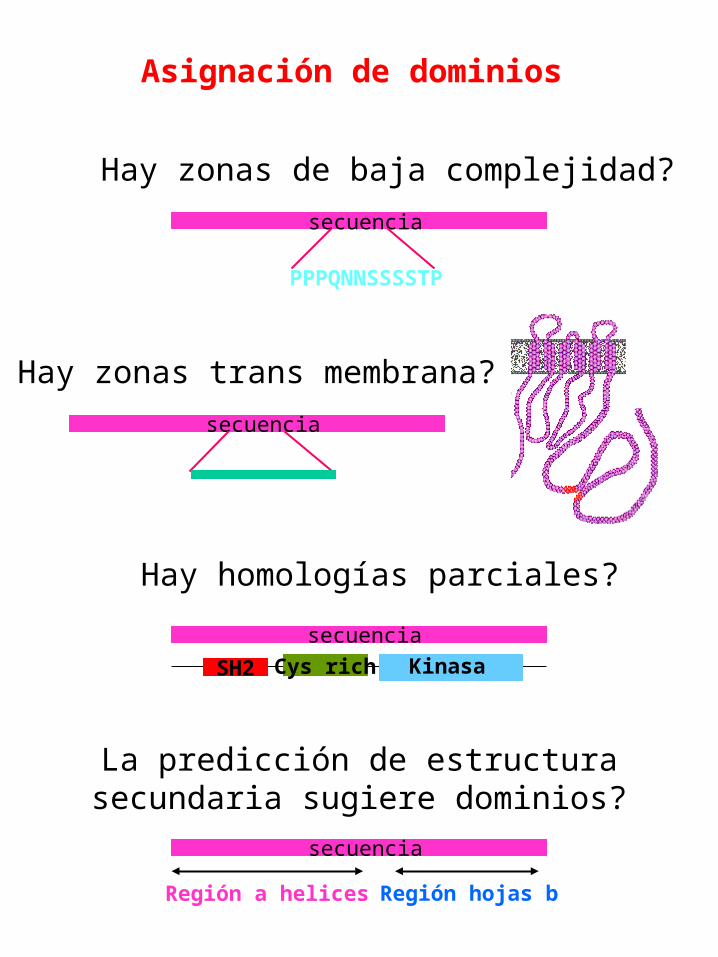
Asignación de dominios
Hay homologías parciales?
secuencia
SH2 Cys rich Kinasa
Hay zonas de baja complejidad?
secuencia
PPPQNNSSSSTP
La predicción de estructura secundaria sugiere dominios?
secuencia
Región a helices Región hojas b
Hay zonas trans membrana?
secuencia

Superfamily: PRTase-like
Lineage:1.Root: scop 2.Class: Alpha and beta proteins (a/b) Mainly parallel beta sheets (beta-alpha-beta units) 3.Fold: PRTase-likecore: 3 layers, a/b/a; mixed beta-sheet of 6 strands, order 321456; strand 3 is antiparallel to the rest 4.Superfamily: PRTase-like
Families:1.Phosphoribosyltransferases (PRTases)2.Phosphoribosylpyrophosphatesynthetase
Jerarquía SCOPE. Ej: 1tc2
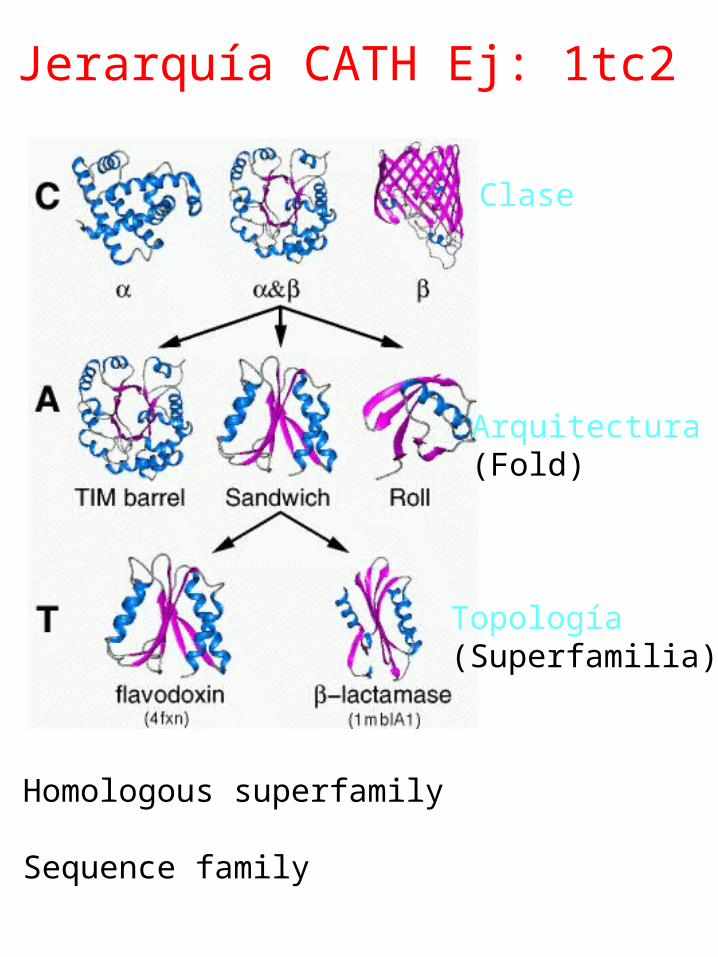
Jerarquía CATH Ej: 1tc2
Clase
Arquitectura(Fold)
Topología (Superfamilia)
Homologous superfamily
Sequence family
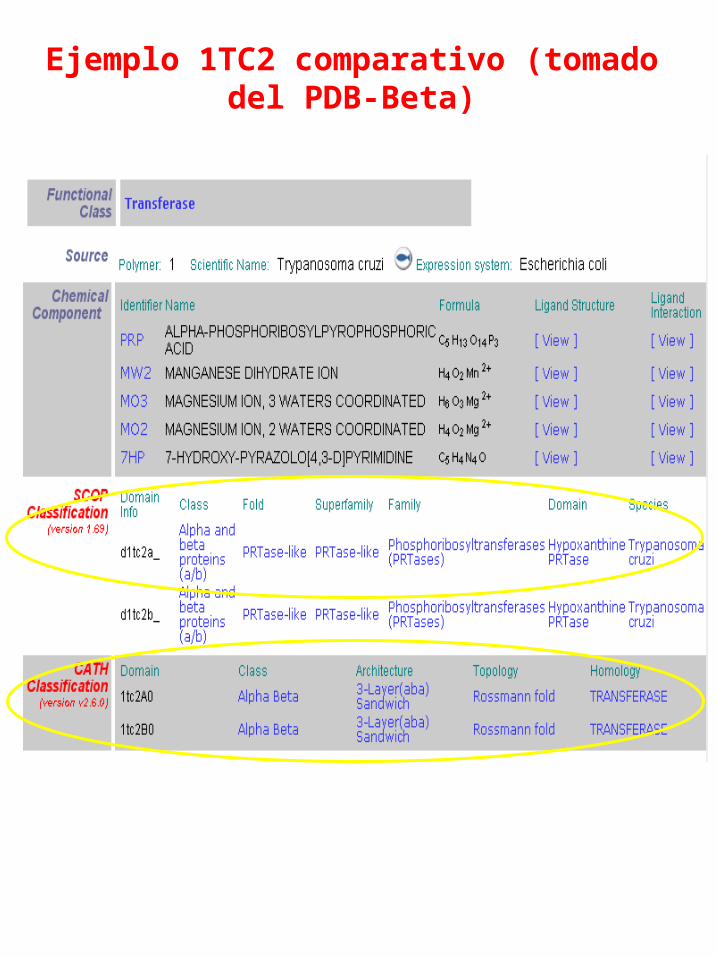
Ejemplo 1TC2 comparativo (tomado del PDB-Beta)

“Homology modelling”
“Fold recognition Modelling”
“Ab initio”
Typos de Modelado

“Comparative protein modelling (Homology Modelling)”
Usa estructuras previamente resueltas como puntos de partida o moldes
se basa en la razonable suposición de que dos proteínas homologas compartirán estructura similar

Predecir la estructura de una proteína basándose solo en su secuencia. Las predicciones “ab initio” recaen en la hipótesis termodinámica del plegamiento, que postula que las estructuras nativas de una secuencia proteica, corresponde a un mínimo global de energía libre. Actualmente hay tres problemas principales a los que debe enfrentarse este campo de investigación que son básicamente:
definir un sistema de “scoring” que diferencie estructuras nativas de las no nativas.
definir un campo de fuerzas para el cual la estructura con mínima energía libre global coincida con la estructura que conocemos como “nativa”.
El tiempo de computación necesario que requiere una cadena polipeptídica para recorrer todas las posibilidades conformacionales.
De novo “Ab initio” protein modelling

Pasos para el modelado por homologia (“homology modelling”)
Busqueda de la proteína homologa(”molde” en general)
Alineamiento
Selección del modelo
Refinamiento del modelo
Validación del modelo
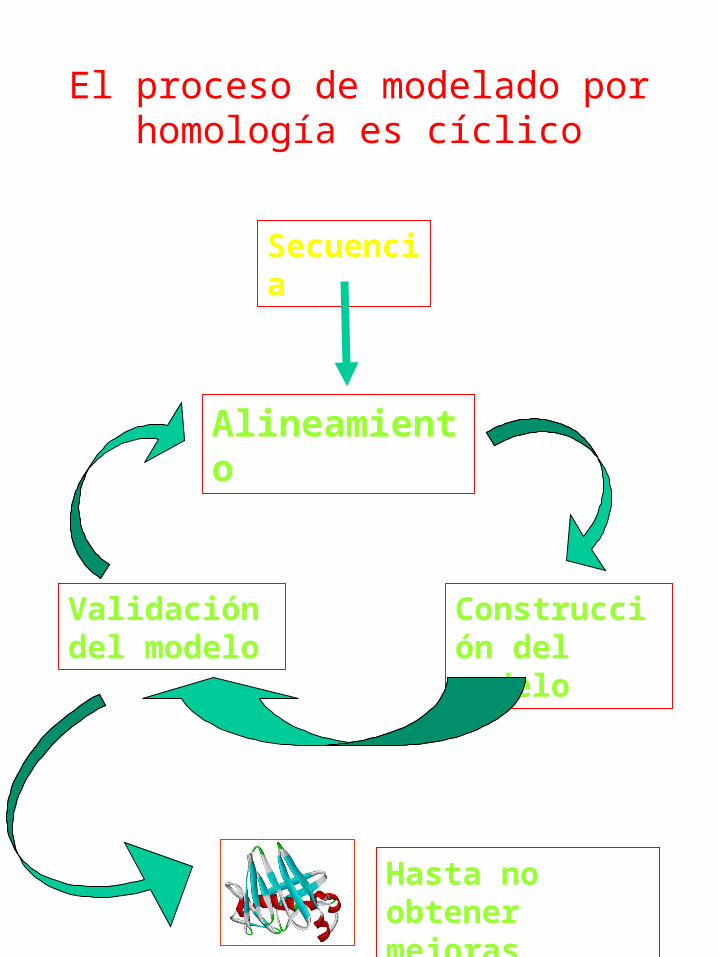
El proceso de modelado por homología es cíclico
Secuencia
Alineamiento
Construcción del modelo
Validación del modelo
Hasta no obtener mejoras
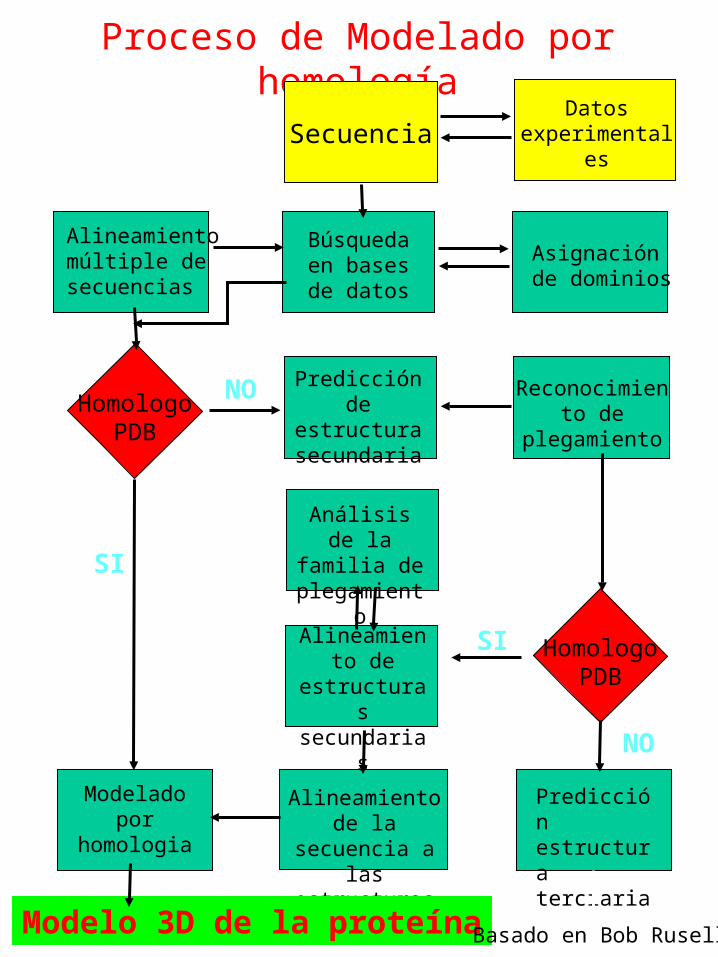
Proceso de Modelado por homología
SecuenciaDatos
experimentales
Alineamiento múltiple de secuencias
Búsqueda en bases de
datos
Asignación de dominios
Modelado por
homologia
Alineamiento de la secuencia
a las estructuras
Predicción estructura terciaria
HomologoPDB
HomologoPDB
Predicción de estructura secundaria
Reconocimiento de
plegamiento
NO
SI
SI
NO
Análisis de la familia de plegamiento
Alineamiento de
estructuras secundarias
Modelo 3D de la proteína Basado en Bob Rusell

Cosas a tener en cuenta para evaluar una predicción de
plegamiento (Fold recognition):
Correr mas de un programa de “Fold recognition”
En lo posible, correrlo sobre mas de un homologo.
Evaluar todas las salidas de un programa (no la primera) la solución puede estar entre las 10.
Función de la proteína de estructura desconocida.
Función de la proteína de estructura conocida.
La familia de plegamiento “FOLD FAMILY”.
Predicción de estructura secundaria.

Datos experimentales
Tener todo dato experimental en mente a la hora de hacer un trabajo predictivo.
Comprobar si la predicción concuerda con los resultados experimentales. En caso negativo, habrá que re plantearse lo hecho.

Puentes disulfuro, restringen las posiciones de las cisteinas en el espacio.
Datos espectroscópicos. Dan información del contenido de estructura secundaria.
Mutagénesis dirigida, da información a cerca de que residuos intervienen en el centro activo o lugares de unión.
Conocimiento de lugares proteolíticos, modificaciones post-transduccionales, glucosilaciones, sugieren residuos accesibles.
Sitios antigénicos.
Etc.
Datos experimentales pueden guiar el proceso de predicción ej:
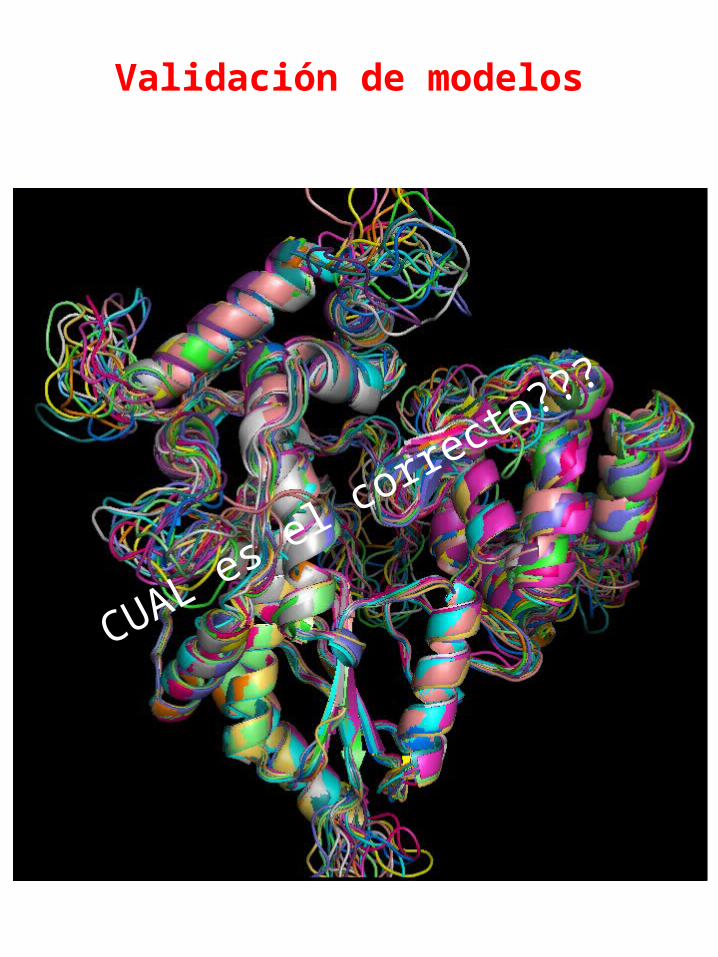
Validación de modelos
CUAL es el corre
cto???

Validación de modelos
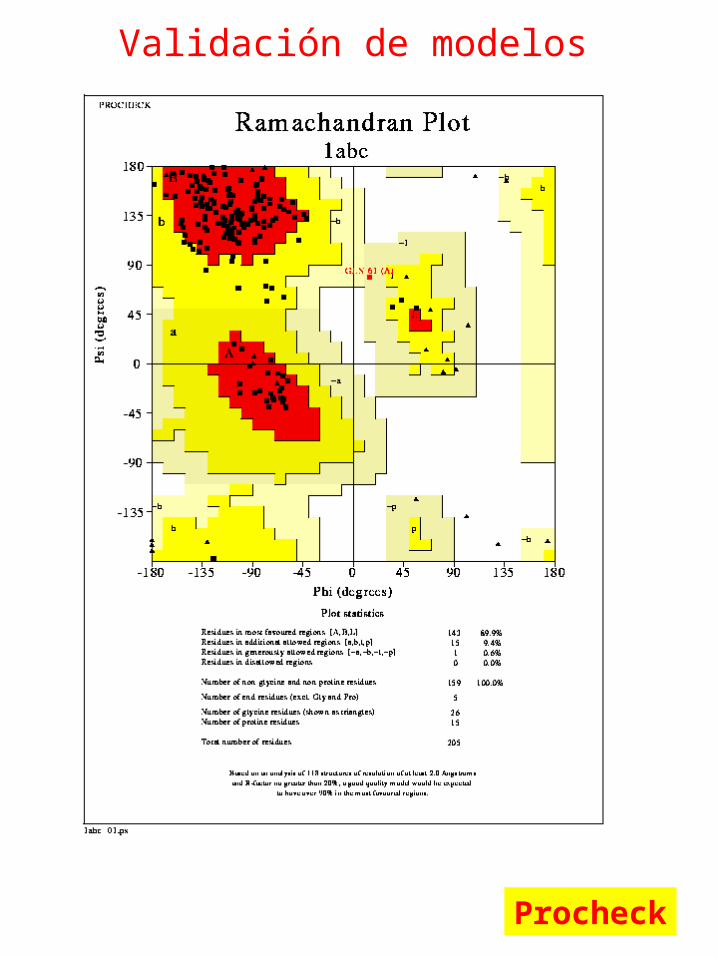
Validación de modelos
Procheck
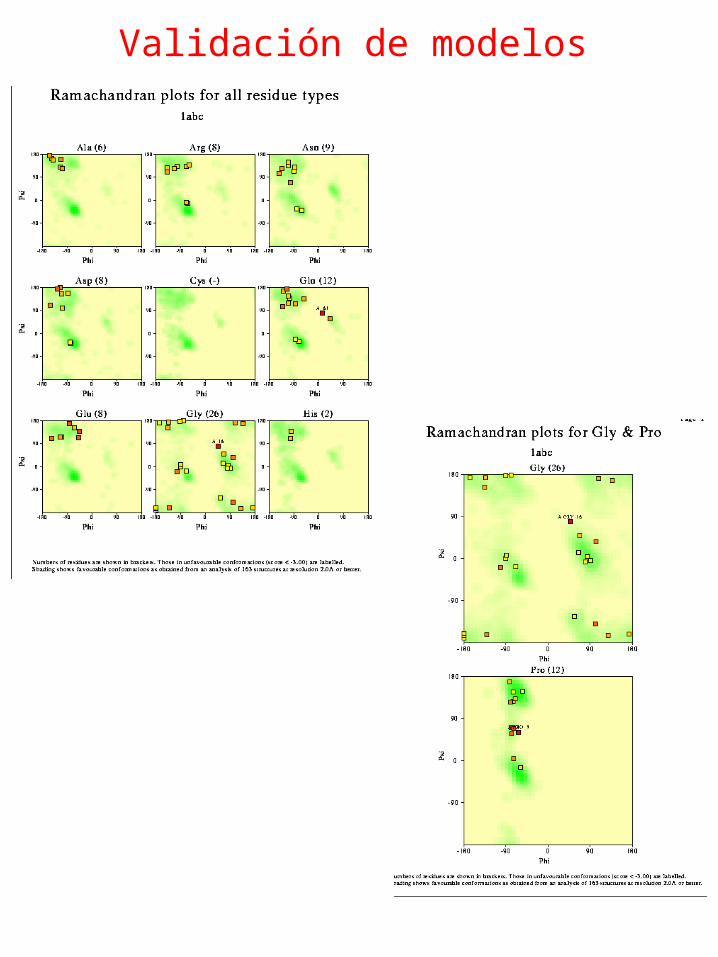
Validación de modelos
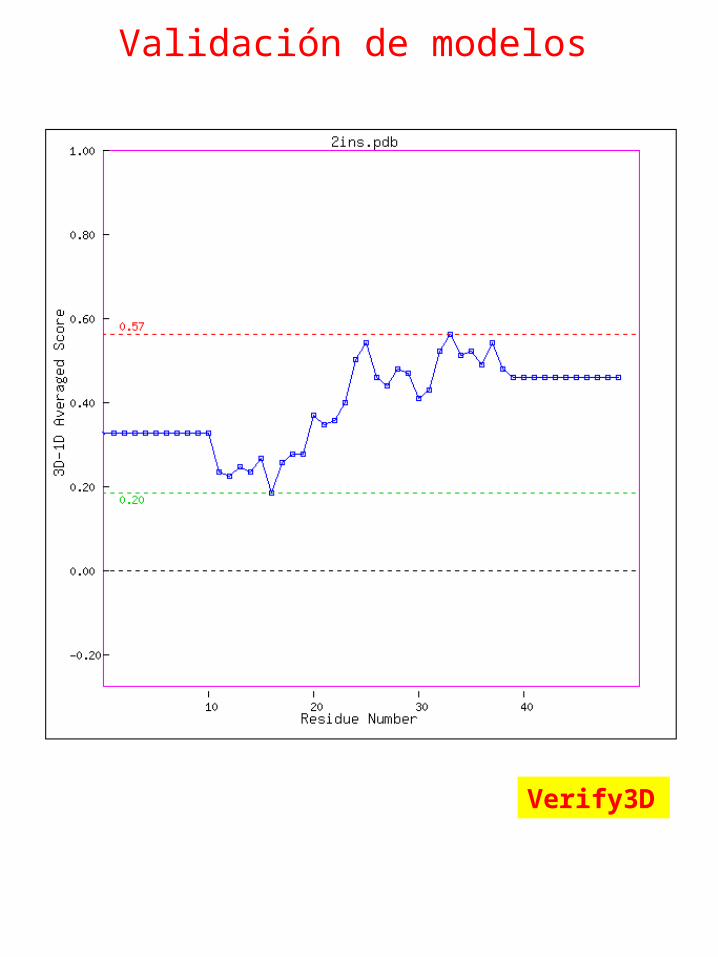
Validación de modelos
Verify3D

Validación de modelos
Datos experimentalesProcheckVerify3DProsa IIErratWhatCheckProQ…

Modelado de Loops
El modelado de Loops constituye un problema de “homology Modelling” dentro de la proteína.
Modelado “Knowledge based” (por homología)
Modelado “ab initio” (loop building)
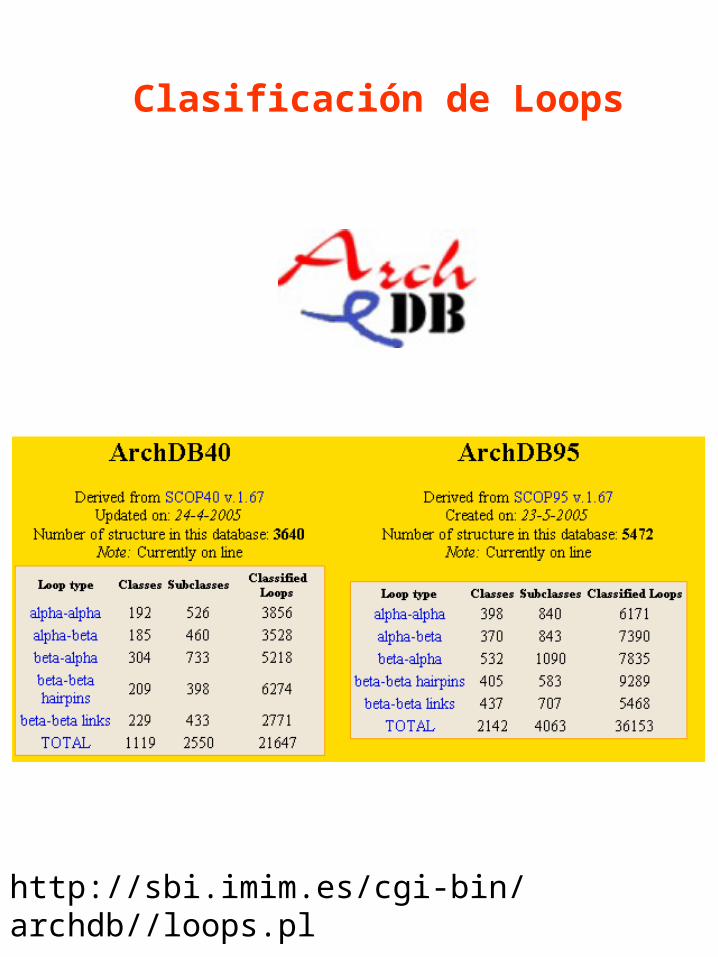
http://sbi.imim.es/cgi-bin/archdb//loops.pl
Clasificación de Loops

Numero de clases Vs. Longitud del loop en ArchDB
Clasificación de Loops
Fernández Fuentes, Narcís. Tesis doctoral UAB 2004
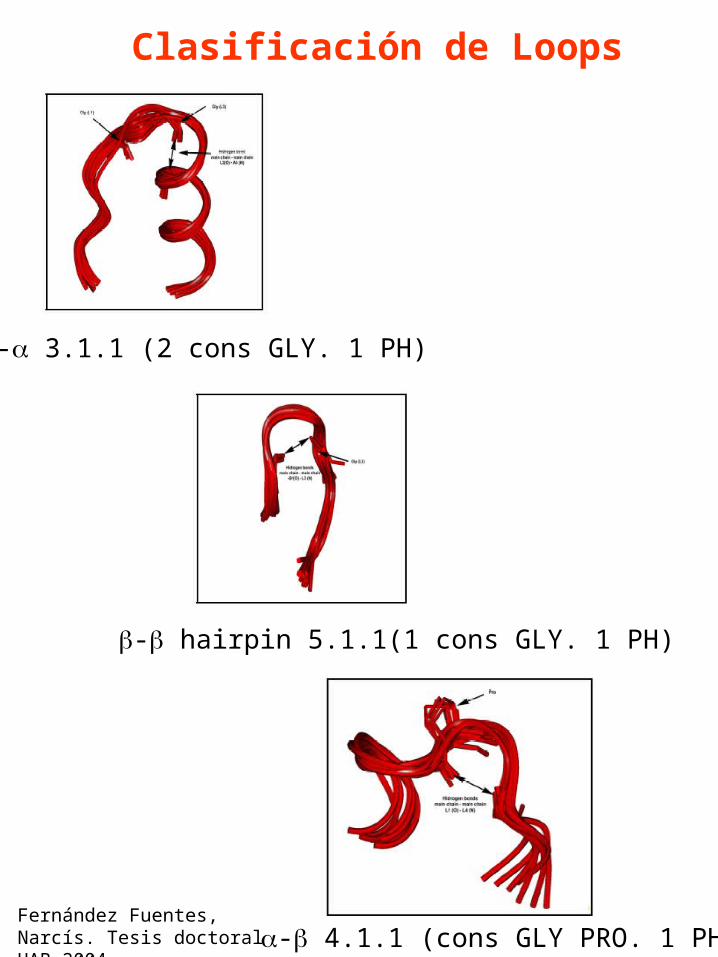
Fernández Fuentes, Narcís. Tesis doctoral UAB 2004
Clasificación de Loops
- 3.1.1 (2 cons GLY. 1 PH)
- hairpin 5.1.1(1 cons GLY. 1 PH)
- 4.1.1 (cons GLY PRO. 1 PH)

Modelado de Loops
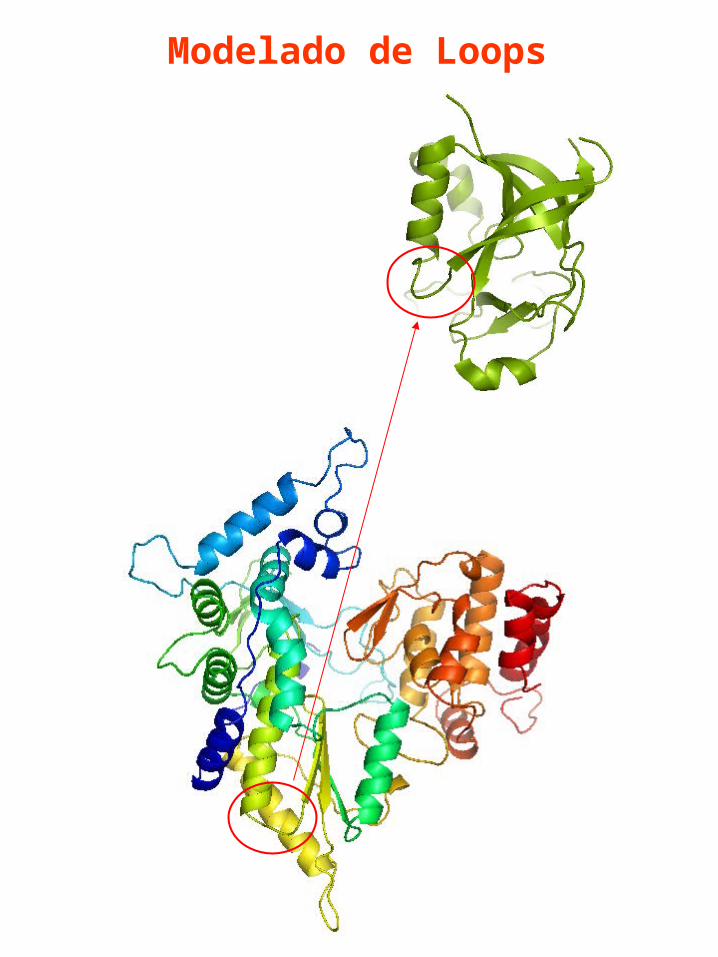
Modelado de Loops

http://manaslu.aecom.yu.edu/loopred/
http://www-cryst.bioc.cam.ac.uk/servers.html
http://alto.compbio.ucsf.edu/modloop/
ej: Knowlege based
Modelado de Loops
ej: By satisfaction of spatial restrains

Errores mas frecuentes en modelos creados por “homology modelling”
Mala elección del “template”
Mal alineamiento entre el “template” y la proteína de estructura desconocida.
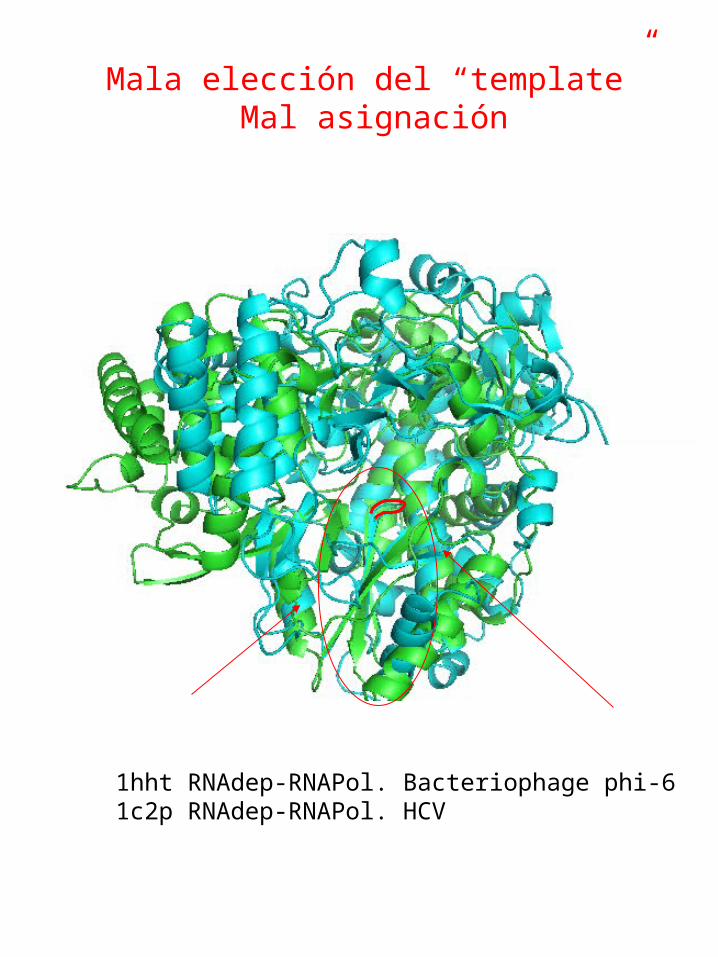
1hht RNAdep-RNAPol. Bacteriophage phi-6 1c2p RNAdep-RNAPol. HCV
Mala elección del “template”Mal asignación

Mala elección del “template”Mal asignación
Ribbon diagrams of RNA-dependent RNA polymerases shown from a similar vantage point
RHDV
PV HCV
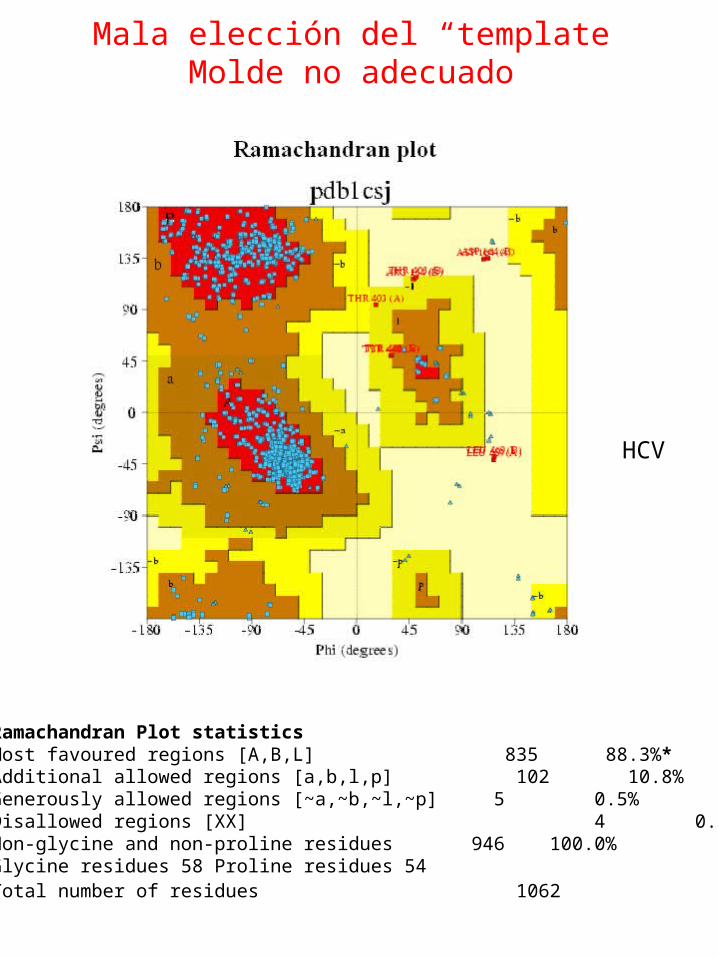
Mala elección del “template”Molde no adecuado
Ramachandran Plot statisticsMost favoured regions [A,B,L] 835 88.3%* Additional allowed regions [a,b,l,p] 102 10.8%Generously allowed regions [~a,~b,~l,~p] 5 0.5% Disallowed regions [XX] 4 0.4%* Non-glycine and non-proline residues 946 100.0% Glycine residues 58 Proline residues 54 Total number of residues 1062
HCV
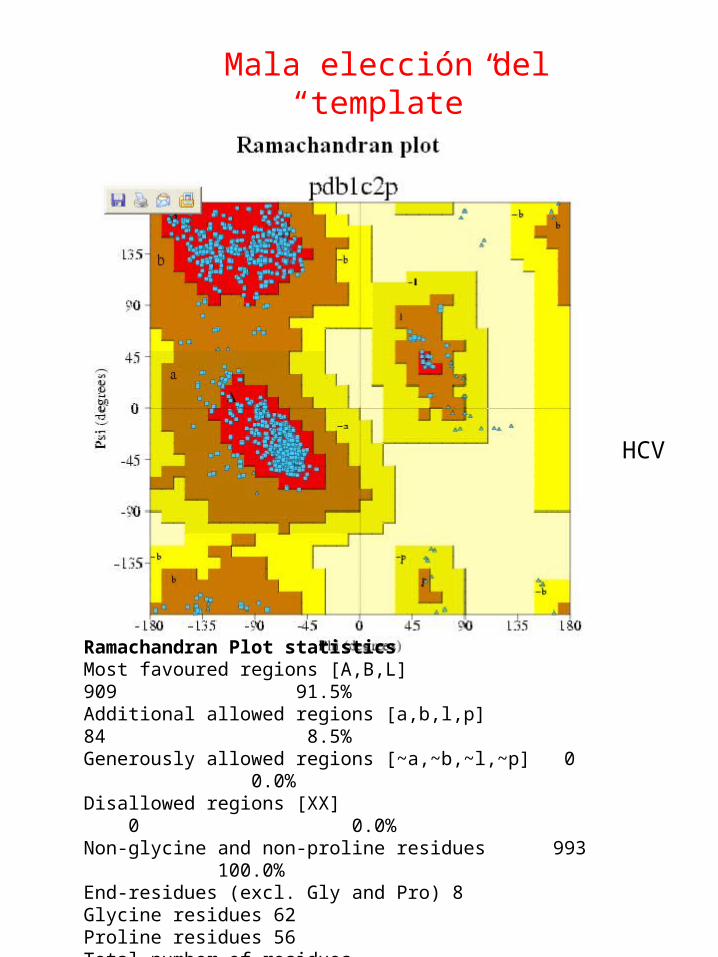
Ramachandran Plot statisticsMost favoured regions [A,B,L] 909 91.5% Additional allowed regions [a,b,l,p] 84 8.5% Generously allowed regions [~a,~b,~l,~p] 0 0.0% Disallowed regions [XX] 0 0.0% Non-glycine and non-proline residues 993 100.0% End-residues (excl. Gly and Pro) 8 Glycine residues 62 Proline residues 56 Total number of residues 1119
Mala elección del “template”
HCV

Alineamiento parcialmente erróneo
82-93

Alineamiento parcialmente erróneoSuperposición molde modelo
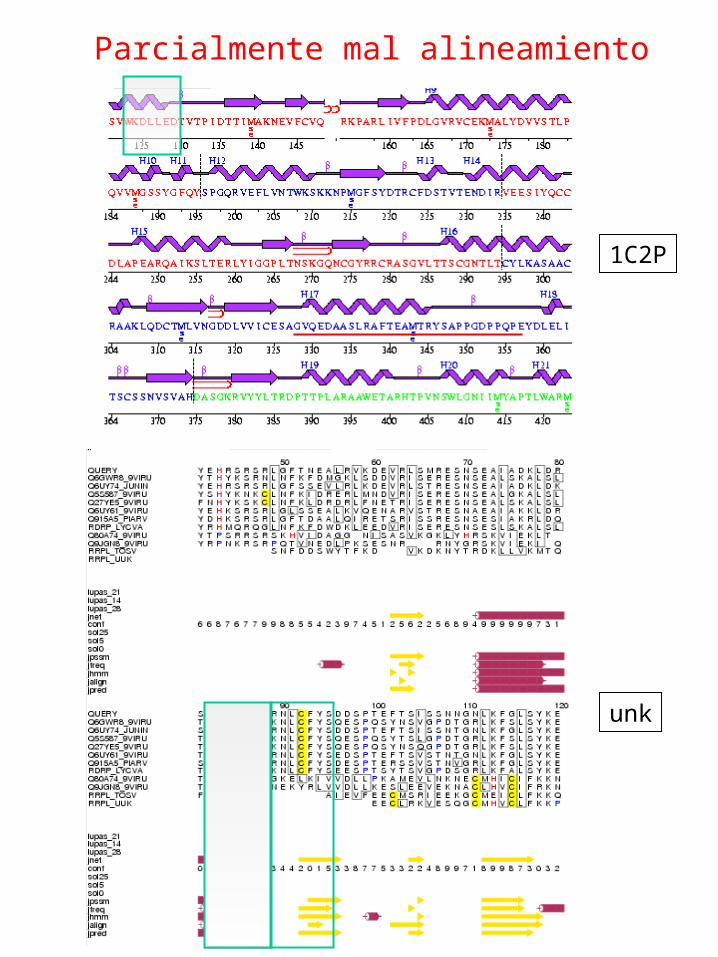
Parcialmente mal alineamiento
1C2P
unk

Modelado de Loops
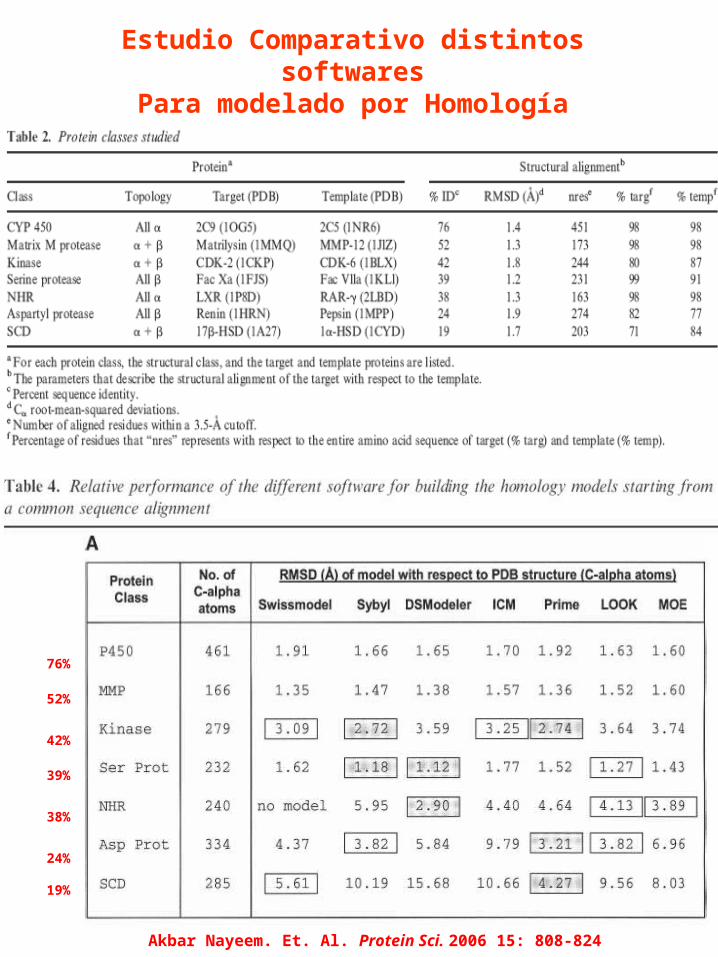
76%
52%
42%
39%
38%
24%
19%
Akbar Nayeem. Et. Al. Protein Sci. 2006 15: 808-824
Estudio Comparativo distintos softwares
Para modelado por Homología

http://predictioncenter.org/casp6/Casp6.html
6th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction Gaeta (Italy) December 2004
Critical Assessment of Techniques for Protein Structure Prediction Asilomar Conference Center, Pacific Grove, CA November 2006 (CASP7), 2006
Estado del arte: CASP

CAPRI: Critical Assessment of PRediction of Interactions
CAPRI community wide experiment on the comparative evaluation of protein-protein
docking for structure predictionHosted By EMBL/EBI-MSD Group
Estado del arte: CAPRI

EJEMPLO DE ESTUDIOS ESTRUCTURALES
Utilidad de un modelo
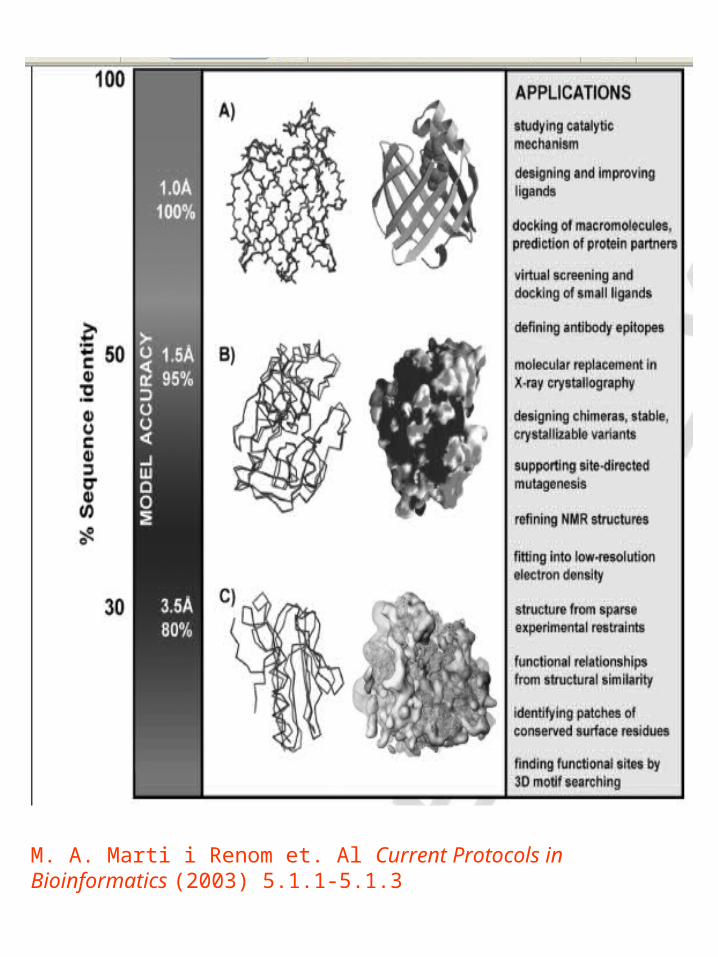
M. A. Marti i Renom et. Al Current Protocols in Bioinformatics (2003) 5.1.1-5.1.3

Construcción de estructuras supramolecularesIR
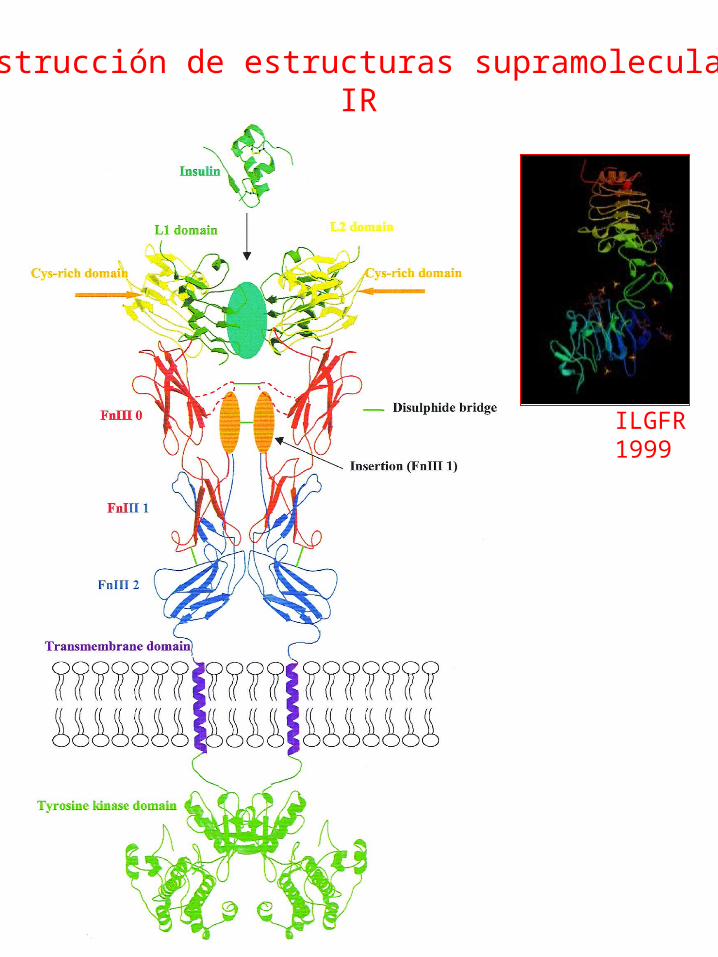
Construcción de estructuras supramolecularesIR
ILGFR1999

Identification of functional residues
• Residues with backbone dihedral angles in strained conformation.
• Clusters of charged residues.• Cavities or clefts in the protein
structure.• Surface properties such as
hydrophobicity, planarity, size or shape.
• Energetics of the protein structure.
• Surface mapping of phylogenetic information.
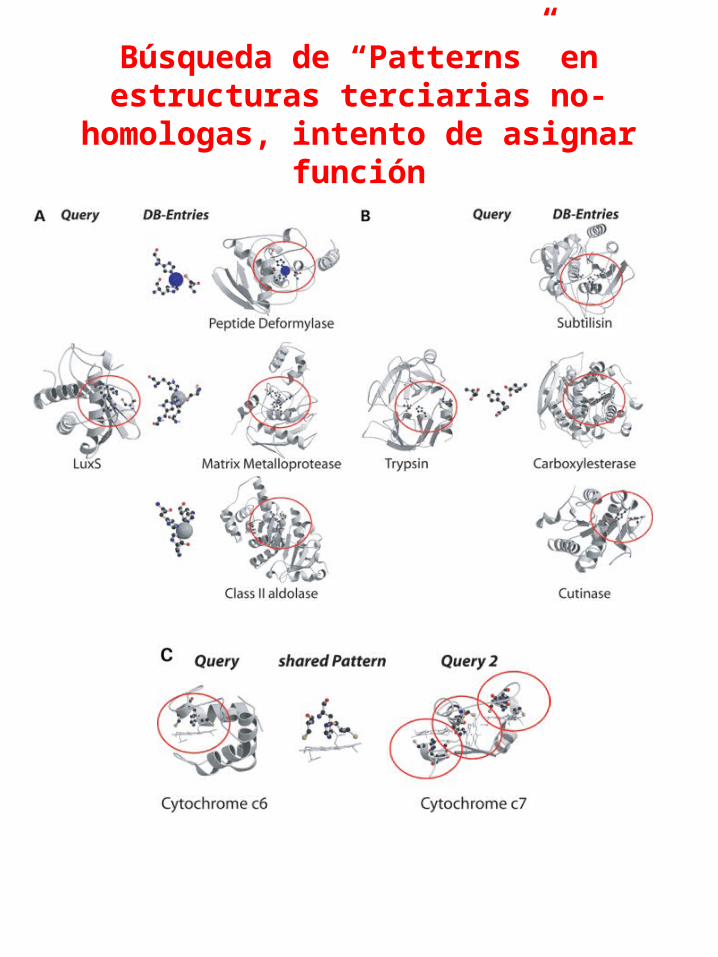
Búsqueda de “Patterns” en estructuras terciarias no-
homologas, intento de asignar función

Trazado evolutivo de residuos
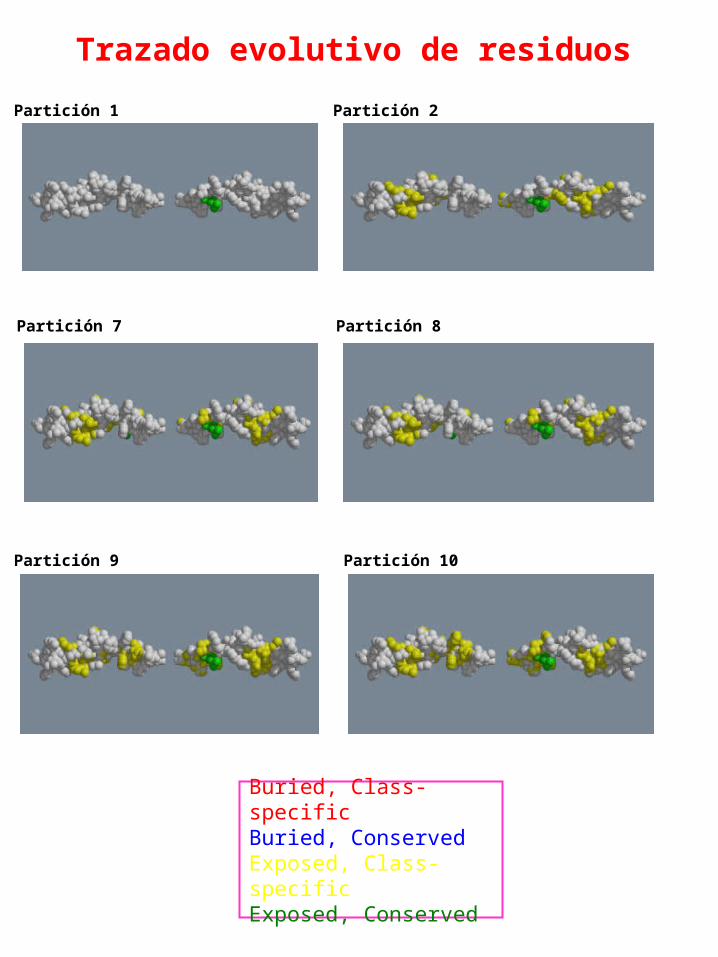
Partición 1 Partición 2
Partición 7 Partición 8
Partición 9 Partición 10
Buried, Class-specificBuried, ConservedExposed, Class-specificExposed, Conserved
Trazado evolutivo de residuos
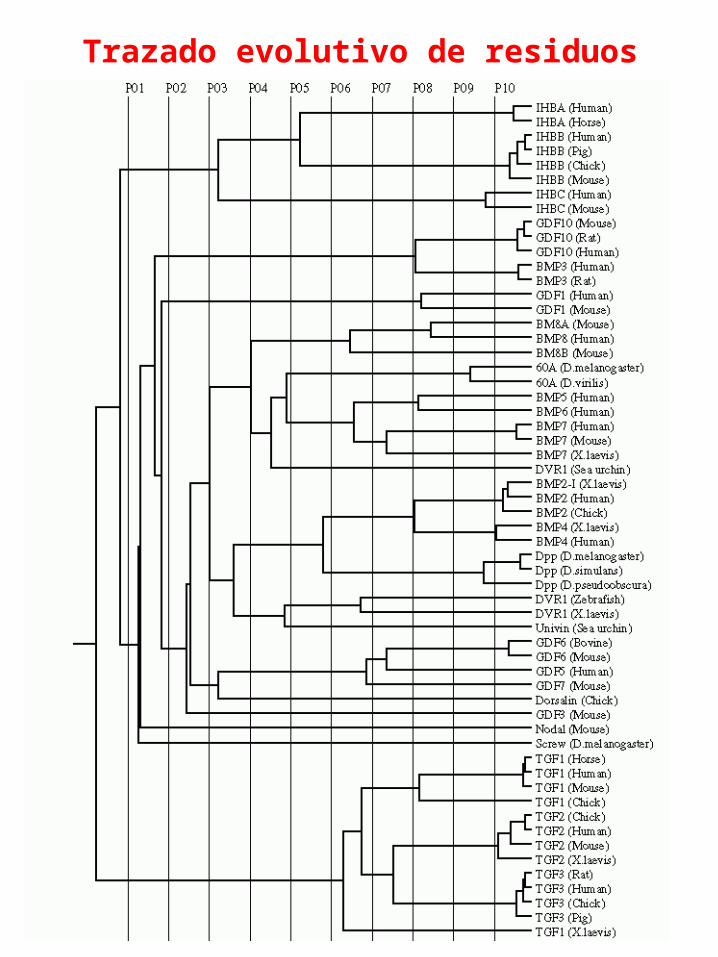
Trazado evolutivo de residuos
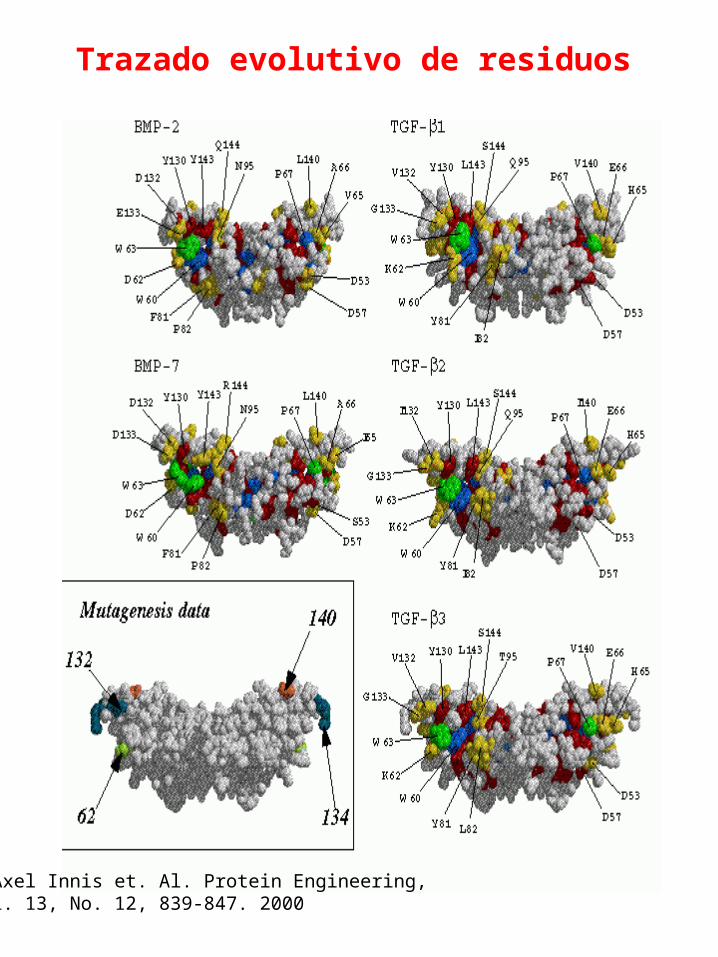
Trazado evolutivo de residuos
C.Axel Innis et. Al. Protein Engineering, Vol. 13, No. 12, 839-847. 2000

Dilucidación del mecanismo de transducción de señal (Interacción
Proteína-Proteína)
II
I
I
II
II
I
I
II
II
I
I
II
I
I
II
II
2tgi.pdb
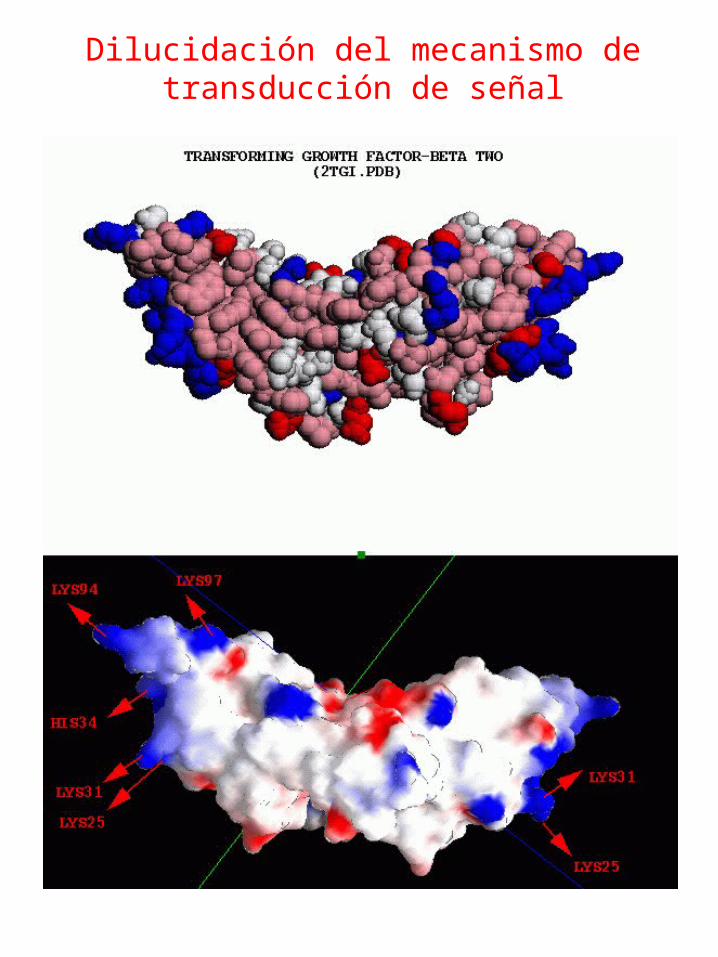
Dilucidación del mecanismo de transducción de señal

Dilucidación del mecanismo de transducción de señal

Dilucidación del mecanismo de transducción de señal

Dilucidación del mecanismo de transducción de señal
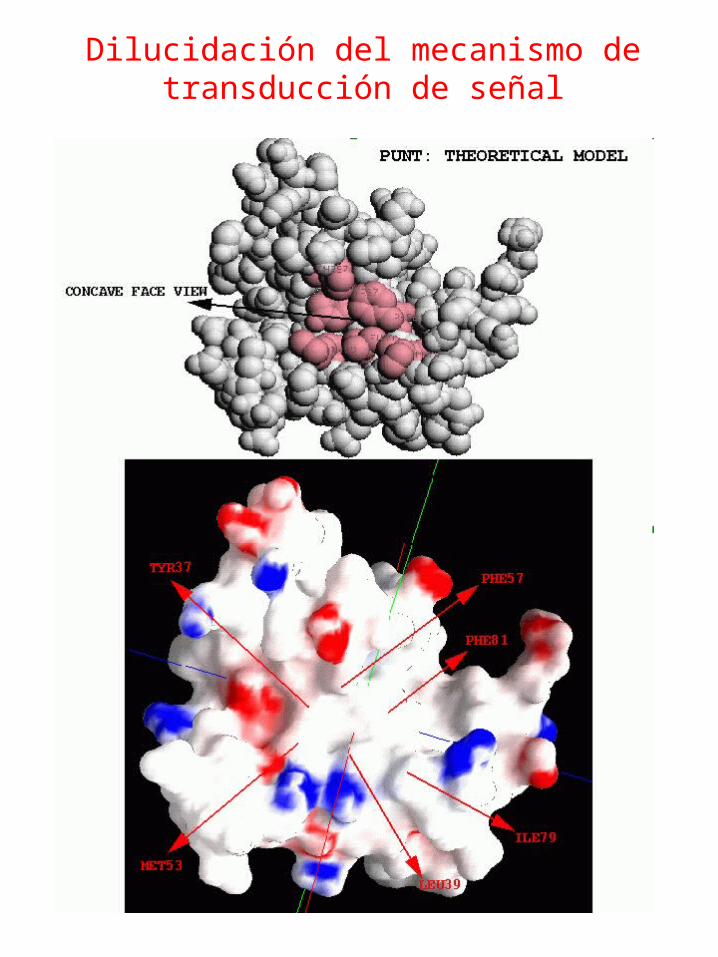
Dilucidación del mecanismo de transducción de señal

Dilucidación del mecanismo de transducción de señal
Parantu K. Shah, Cristina Marino Buslje, R. Sowdhamini Proteins: Structure, Function, and GeneticsVolume 45, Issue 4. 2001
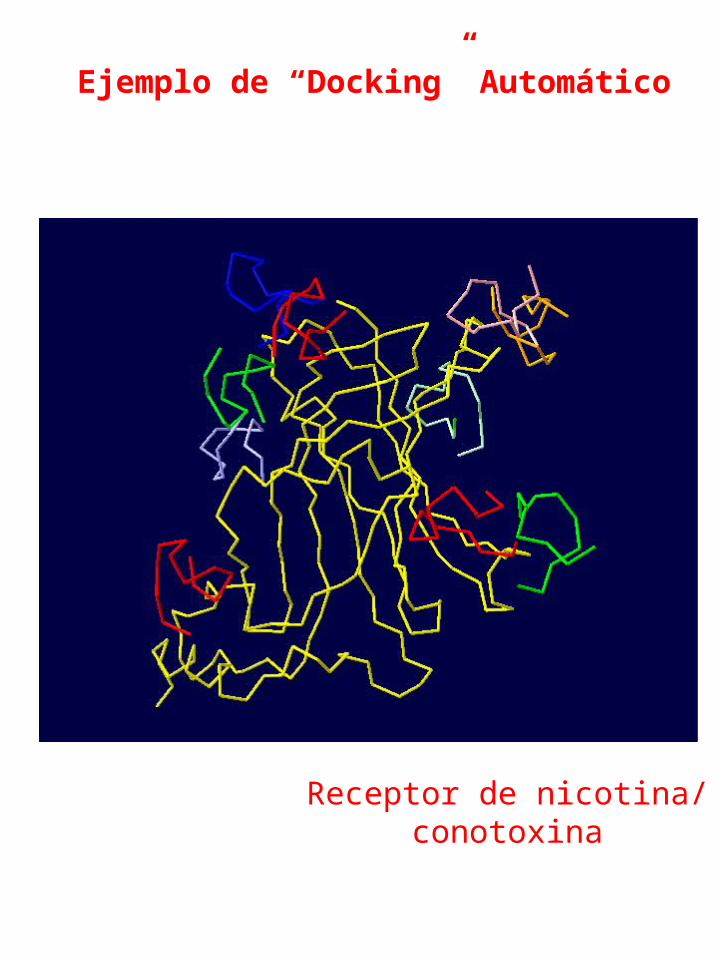
Ejemplo de “Docking” Automático
Receptor de nicotina/conotoxina
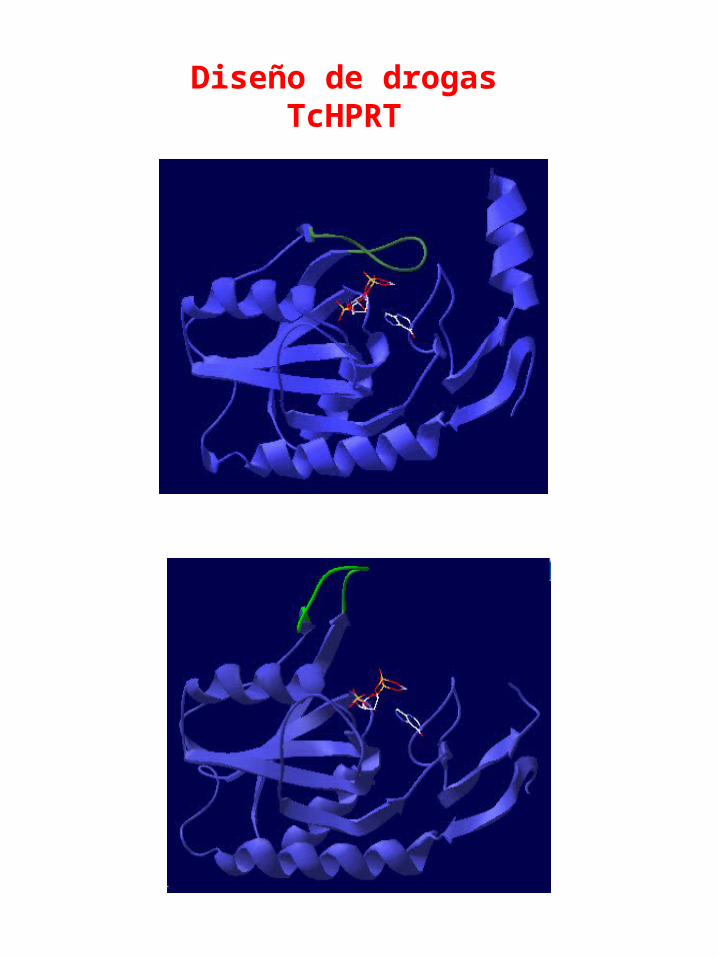
Diseño de drogasTcHPRT
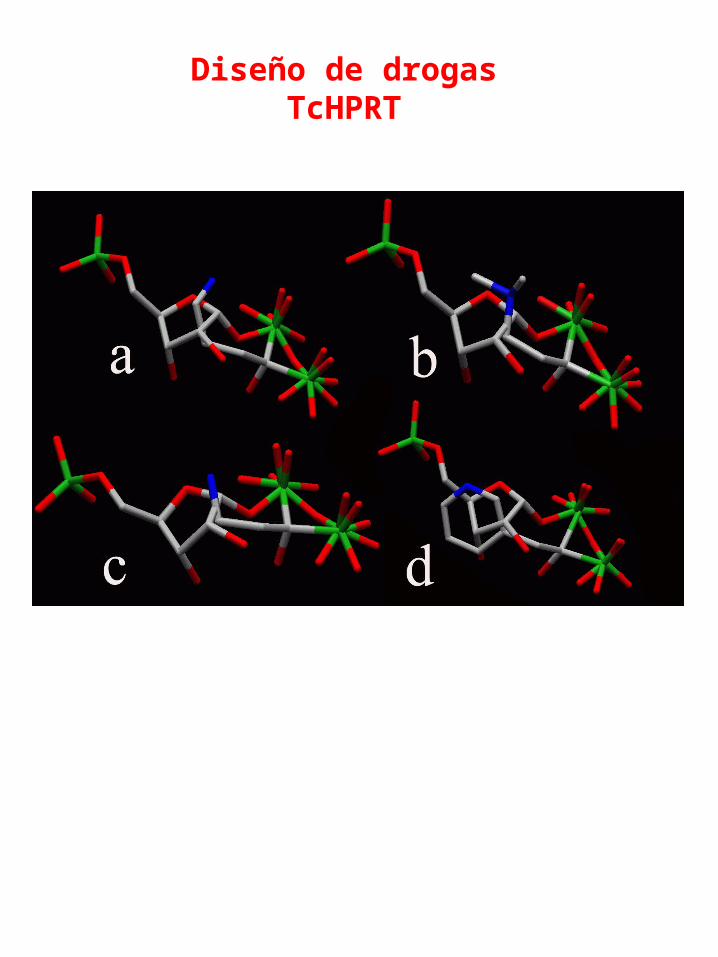
Rigid superimposition on PRPP of: a) ALN ;b) OLP; c) PAM; d) RIS. Blue: N, Green: P, Grey: C, Red: O.
Daniel Fernandez, et. al.
Diseño de drogasTcHPRT
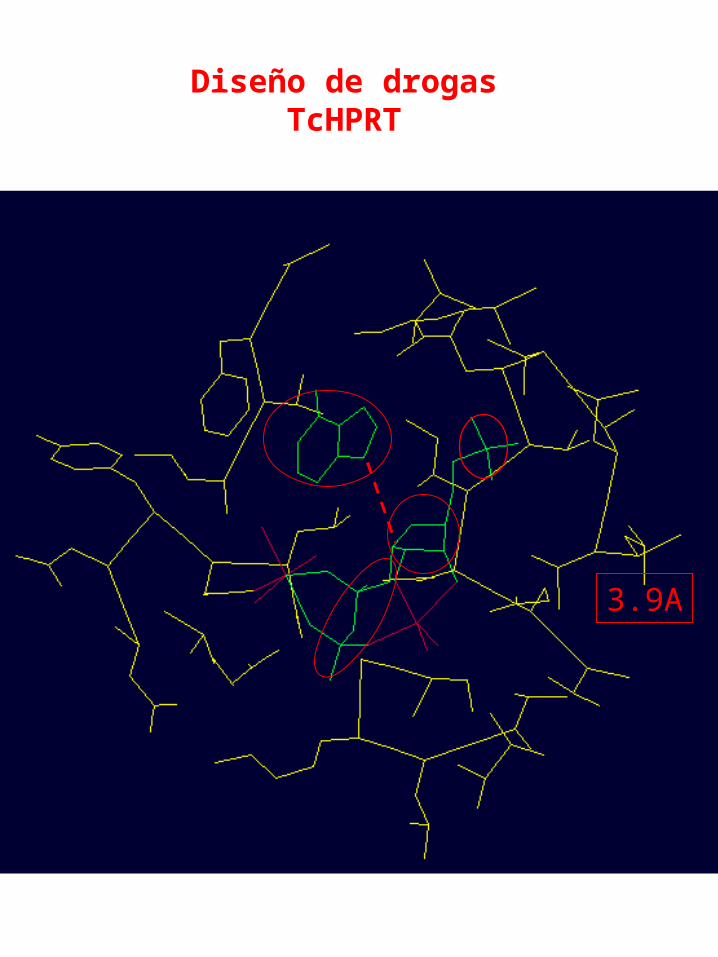
Diseño de drogasTcHPRT
3.9A
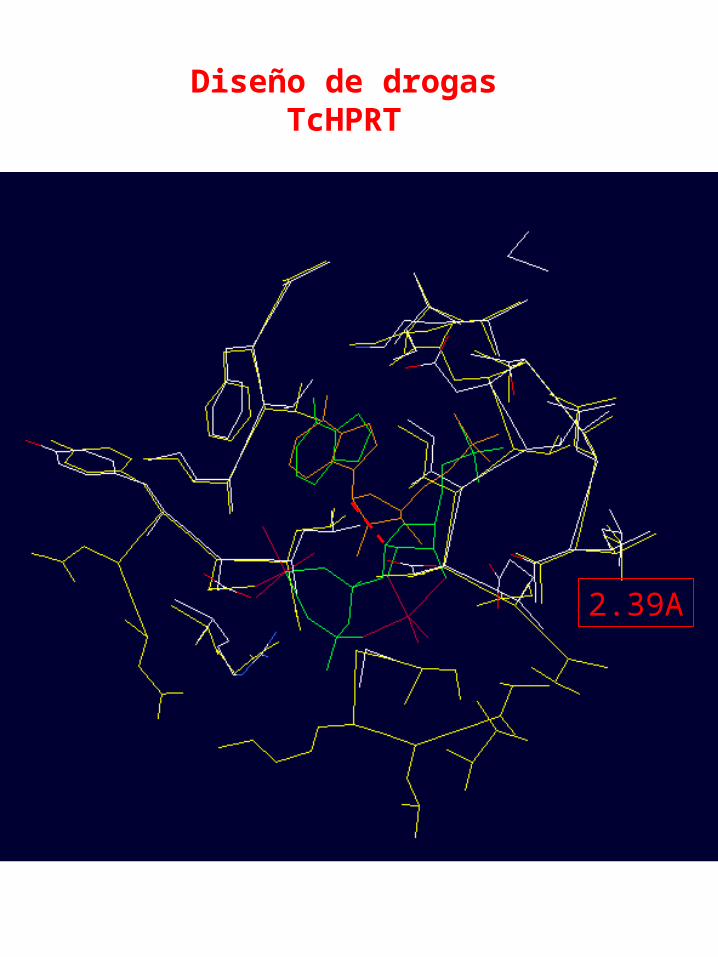
Diseño de drogasTcHPRT
2.39A

Explicación de datos experimentales y diseño de nuevos experimentos.
* RNA-dependent RNA-polimeraseTacaribe virus* 1c2p.pdb
8.8 %identity
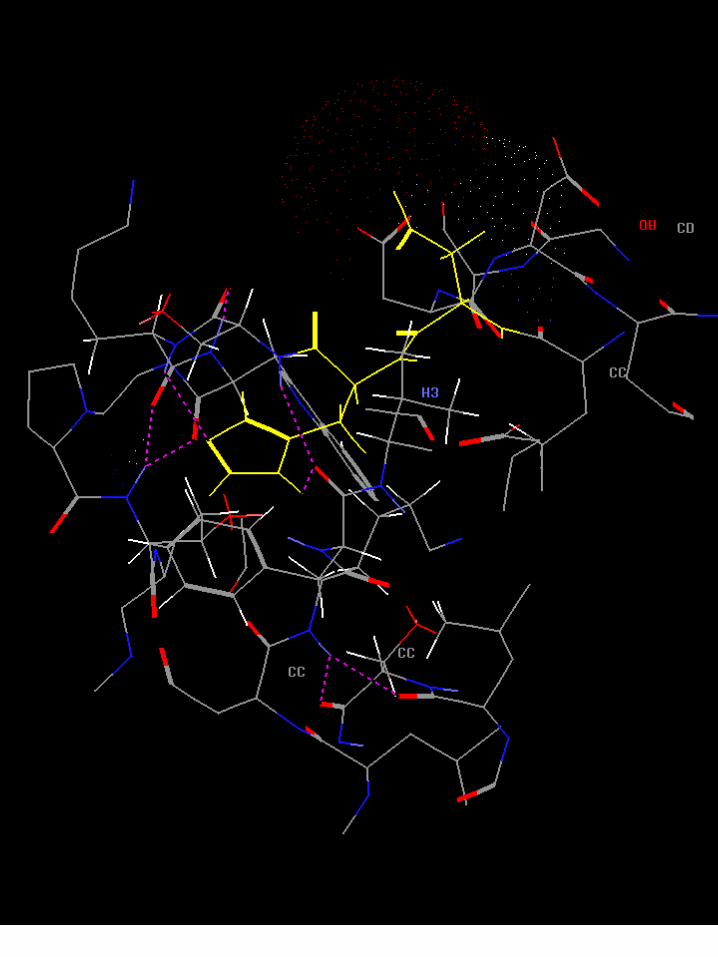
1e3p.pdb