anemias hemolÍticas por alteraciones congÉnitas del
TRANSCRIPT

4
But. Soc. Cat. Pediatr., 41, 315, 1981
ANEMIAS HEMOLÍTICAS POR ALTERACIONESCONGÉNITAS DEL METABOLISMO Y LA MEMBRANA
ERITROCITARIA. ESTADO ACTUAL DE LOSMÉTODOS DIAGNÓSTICOS
Joan Lluis Vives Corrons *, Lluis Berga**,Josep Lluis Aguilar*, Josep M. a Jou * i Anna Ester *
1. Introducción
Los métodos de estudio aplicados al diagnóstico etiológico de lasanemias hemolíticas han experimentado durante los últimos años ungran avance. A ello ha contribuido tanto la fácil accesibilidad de loshematíes a partir de una muestra de sangre como el perfeccionamientode los métodos empleados para su estudio que aglutinan los conocimientosrnás recientes en disciplinas tan diversas como la morfología óptica yultraestructural, la enzimología, el análisis proteico y de permeabilidad ymás recientemente el comportamiento físico o «reológico» de los hematíesen suspensión.
El estudio de la morfología eritrocitaria ha constituido desde siemprkisiabto nel procedimiento más simple y en muchos casos el más eficaz para el diag-
‘‘EiLloTECAuöstico de las anemias hemolíticas. Tal sucede en la esferocitosis heredi-taria (EH) donde su omisión puede conducir a múltiples e innecesariasdeterminaciones analíticas. En muchos casos, la morfología óptica con-vencional del frotis puede complementarse con el empleo de colorantesvitales. Entre ellos destacan el azul de metileno nuevo (NMB) para eIrecuento de reticulocitos y el azul de Cresil brillante (ACB) para el aná-lisis de posibles precipitados hemoglobínicos intraeritrocitarios (hemoglo-binas inestables). La incubación de una muestra de sangre con agentes
(*) Laboratori Central d'Hematologia i Escola Professional d'Hematologia«Farreras Valentí». Hospital Clinic i Provincial. Universitat de Barcelona
(*") Escola Técnica Superior d'Enginyers. Universitat Politécnica de Bar-celona
315

oxidantes, como por ejemplo la acetilfenilhidrazina (APH), constituye unprocedimiento muy sencillo para valorar el poder reductor eritrocitario.Cuando éste se halla disminuido tal como sucede ,por ejemplo, en eldéficit de glucosa-6-fosfato deshidrogenasa (G6PD), la incubación de loshematíes en APH da lugar a abundantes precipitados hemoglobínicosdenominados «cuerpos de Heinz».
El simple examen de la morfología eritrocitaria mediante el micros-copio óptico y la realización de pruebas de laboratorio tan sencillas comoel estudio de la resistencia osmótica eritrocitaria (ROE) o la prueba de lahemólisis en medio ácido (prueba de Ham-Dacie) son muchas veces sufi-cientes para confirmar el diagnóstico.
Cuando ello no es así, se impone el empleo de otros procedimientoscuya naturaleza vendrá determinada por la historia clínica y exploraciónfísica del paciente.
Dentro del grupo de anemias hemolíticas congénitas no hemoglobino'páticas, la más frecuente es la EH y en segundo lugar el déficit de G6PD( 32 33 ). En la mayoría de los casos, no obstante, el comportamiento clínicodel paciente permite establecer el diagnóstico ya que mientras la EHcursa con anemia hemolítica crónica, el déficit de G6PD lo hace con herno
-lisis aguda en general después de la ingesta de algún medicamento (sulfa-midas o aspirinas) o de habas (favismo).
En el presente trabajo se revisan los procedimientos actuales para eldiagnóstico etiológico de las anemias hemolíticas no hemogloninopáticascuya naturaleza y sistemática de aplicación vendrá siempre determinadapor la clínica y los antecedentes familiares del paciente.
2. Aspectos clínicos
Como en cualquier tipo de patología, el diagnóstico de la anemia h e-mol tica debe basarse siempre en la realización de una historia clínica de
-tallada y examen físico minucioso del paciente. En muchos casos esto essuficiente para establecer una orientación etiológica que deberá confir
-marse mediante la realización de las correspondientes pruebas compl e-mentarias ( 1 ' 32 ").
En la orientación etiológica de un proceso hemolítico son de granvalor, determinados datos tales corno la edad del paciente, el inicio Ycaracterísticas del cuadro anémico, el origen étnico y la existencia o no deantecedentes familiares. La edad del paciente es importante por cuanto enlos niños, una anemia hemolítica tiene casi siempre un origen congénitomientras que en los adultos es más probable que sea adquirida. Los ante'•cedentes personales de ictericia neonatal o de frecuentes «hepatitis», asícomo la existencia de antecedentes familiares son también de gran valorpara conocer el inicio y las características del síndrome hemolítico.
316

El origen étnico tiene valor cuando el paciente pertenece a un áreageugráfica donde es frecuente un determinado tipo de anemia hemolíticaCongénita. En nuestro medio mediterráneo, las causas más frecuentes deanemia hemolítica congénita no hemoglobinopática son la esferocitosishereditaria (EH), que cursa con un cuadro clínico de anemia crónica, y eldéficit de glucosa-6-fosfato deshidrogenasa (G6PD) que lo hace en formade hernölisis aguda. Las otras causas de anemia hemolítica congénita sonmucho más raras (Tabla I). La exploración física minuciosa del pacientetiene también un gran valor orientativo. Así, cuando la anemia hemolítica
TABLA I
CAUSAS DE ANEMIA HEMOLITICA CONGENITA
DEFECTOS CONGENITOS DE LA MEMBRANA ERITROCITARIA
Esferocitosis hereditaria (EH)b) Eliptocitosis congénita (EC)c) Piropoiquilocitosis congénita (PPC)d) Estomatocitosis congénita e Hidrocitosise) Dessicitosis (Xerocitosis)
2. HEMOGLOBINOPATIAS
a) Hemoglobinopatías estructurales— Hemoglobinopatía S (anemia falciforme) y otras hemoglobino-
patías estructurales— Hemoglobinas M (Metahemoglobinemia)
b) Talasemias— a-talasemia— 5-talasemia— Oß-talasemia— Hemoglobina Lepore— Hemoglobinas con alargamiento de una cadena globínica.
3. ERITROENZIMOPATIAS
a) Déficit de glucosa-6-fosfato deshidrogenasa (G6PD)b) Déficit de piruvatokinasa (PK)e) Déficit de fosfoglucosaisomerasad) Déficit de Pirimidina 5'-nucleotidasa (P5N)e) Enzimopatías con afectación y/o muscular
— Déficit de Triosa fosfatoisomerasa (TPI)— Déficit de Fosfofructokinasa (PFK)— Déficit de Fosfogliceratokinasa (PGK)— Déficit de Glutation sintetasa (GS)
f) Exceso de Adenosina deaminasa
317

se acompaña de esplenomegalia (palpable o radiológica) y/o signos delitiasis biliar debe considerarse siempre un mecanismo crónico. La litiasisbiliar en un niño o en un sujeto joven es en muchos casos el primer sigiloclínico de la EH, la cual, al igual que otras formas de anemia hemoltficacrónica, suele acompañarse de alteraciones óseas características tales cornOdeformaciones craneofaciales e imágenes radiológicas peculiares («cráneoen cepillo»). Por el contrario, la aparición de una anemia aguda accmp a
-fiada de emisión de orinas oscuras (hemoglobinuria) en ausencia de esple-nomegalia, debe orientar hacia una hemólisis por defecto del poder reduc-tor eritrocitario (déficit de G6PD). En este caso el interrogatorio debedescartar la existencia de ingesta previa de medicamentos o habas.
La asociación de anemia hemolítica crónica no esferocítica (AHCNE)a manifestaciones extrahematológicas tales como transtornos neurológicoso musculares, deben orientar el diagnóstico hacia ciertos defectos cong é
-nitos del metabolismo ,en realidad excepcionales (déficit de triosa iwine"rasa, fosfogliceratokinasa y fosfofructokinasa) ").
3. Exámenes hematológicos básicos
Los exámenes hematológicos básicos son, excepto la morfología ecitaria, de escasa ayuda en la catalogación etiológica de una anemialítica congénita no hemoglobinopática.
El hemograma suele caracterizarse por una anemia de intensidad va-riable, normocroma y normocítica que se acompaña de un aumento, engeneral intenso, del número de reticulocitos. Los índices eritrocitario5 sue"len hallarse dentro de los límites normales excepto el volumen corpuscularmedio (VCM) que puede estar aumentado ( > 100 fl) especialmente cu and°la cifra de reticulocitos es elevada. En un síndrome hemolítico secundarioa un trastorno estructural de la membrana conocida como piropoiquilo-citosis congénita (" 7 ) se ha descrito una gran disminución del VCM ( > 25fl). Las alteraciones de la concentración corpuscular media de la hert ogi°-bina (CCMH) pueden ser orientativas de ciertos trastornos de la n enl-brana eritrocitaria tales como la EH, ciertas formas raras de alteración con-génita de la misma (estomatocitosis y desincocitosis congénitas). En la e°y desicocitosis congénita el valor de CCMH suele hallarse aumentado o en ellímite superior de normalidad ( > 36 g/d1) mientras que en la estomac tosiscongénita, caracterizada por un aumento del contenido acuoso eritroc tari°(hidrocitosis), el valor de la CC1V1H suele hallarse disminuido (26-28 g/d1) (52)'
Al contrario de lo que sucede con los parámetros hematológicos bási"cos, el examen de la morfología eritrocitaria a partir de un frotis sanguíneocorrectamente realizado y teñido puede ser de gran valor diagnóstico. Debetenerse en cuenta, no obstante, que la realización del frotis somete a loshematíes a unos efectos mecánicos capaces de producir deformacion es Y
aplanamientos que en ocasiones pueden enmascarar pequeñas alteraciones
ritro-errio-
318

Morfológicas ( 3 ) • Ello puede evitarse mediante la observación directa en elMicroscopio óptico de una muestra de sangre total fijada inmediatamentedespués de la extracción, en glutaraldehido ( 1 ') o mediante el empleo delMicroscopio electrónico de barrido (MEB). Con todo, un buen frotis sanguí-neo correctamente realizado y teñido permite, en la mayoría de los casos,establecer el diagnóstico. Tal sucede en la EH donde los hematíes se carac-terizan por su pequeño diámetro y su mayor contenido hemoglobínico (es-ferocitos). Otra alteración morfológica fácil de apreciar mediante el simpleexamen morfológico de los hematíes es la eliptocitosis congénita, caracteri-zada por la presencia en sangre periférica de un porcentaje superior al 25 %de eliptocitosis o hematíes ovalo-eliptocíticos.
Las eritroenzimopatías congénitas no suelen, en general, acompañarsede alteraciones morfológicas eritrocitarias características aunque cuandoexisten son de gran valor diagnóstico. Tal sucede en el déficit de G6PDintnediatamente después de una crisis hemolítica aguda, donde se puedeapreciar una distribución anómala de la hemoglobina por la cual esta sehalla concentrada en uno de los extremos del hematíe. Tales hematíes reci-ben el nombre de «eccentrocitos» (Fig. 1) y ponen de manifiesto una faltade poder reductor eritrocitario frente a los agentes oxidantes ( 19 ). Las en-
FIGURA 1: Eccentrocitos en un caso de déficit de G6PD y Favismo (MGGx800).
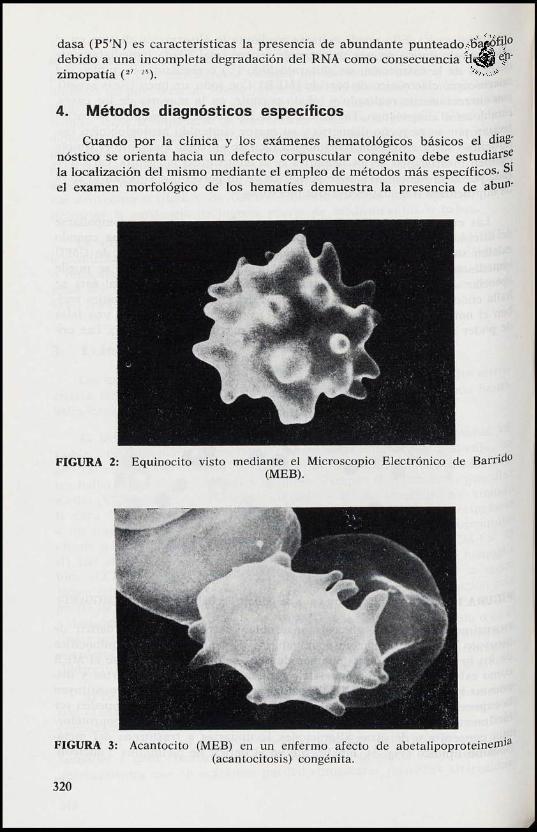
troenzimopatías de las glucosis anaerobia y especialmente el déficit dePiruvato kinasa (PK) suelen acompañarse de transformación equinocíticade los hematíes por la cual algunos de ellos se observan mediante el MEBCorno esferocitos con una superficie repleta de prominencias cortas y dis-Puestas regularmente (Fig. 2). En estos casos los «equinocitos» constituyenla expresión morfológica del descenso del ATP eritrocitario ( 3 ) y pueden sertäeilmente diferenciados de los acantocitos propios de la abetalipoproteine-n'Ida congénita o de otras alteraciones secundarias a trastornos del meta-bolismo lipídico (Fig. 3). Finalmente en el déficit de pirimidina 5'nucleoti-
319
dasa (P5'N) es características la presencia de abundante punteado basófil°debido a una incompleta degradación del RNA como consecuencia de la en-zimopatía (2'
4. Métodos diagnósticos específicos
Cuando por la clínica y los exámenes hematológicos básicos el diag"nöstico se orienta hacia un defecto corpuscular congénito debe estudiarsela localización del mismo mediante el empleo de métodos más específicos . Si
el examen morfológico de los hematíes demuestra la presencia de abuil -
FIGURA 2: Equinocito visto mediante el Microscopio Electrónico de Barrido(MEB).
FIGURA 3: Acantocito (MEB) en un enfermo afecto de abetalipoproteine1Tli3(acantocitosis) congénita.
320

dantes esferocitos, la práctica de una resistencia osmótica eritrocitaria(ROE) permitirá completar el diagnóstico. Por el contrario, si las altera-ciones morfológicas no son orientativas, deberá procederse a descartar unaenzimopatía, una hemoglobinopatía estructural o una alteración de mem-brana diferente a la EH o eliptocitosis congénita.
4. 1. Alteraciones congénitas de la membrana eritrocitaria
Recientemente se ha atribuido gran importancia a la elevada capacidadde deformación (deformabilidad eritrocitaria) como factor decisivo en lasupervivencia normal de los hematíes en la circulación sanguínea. Graciasa su deformabilidad estas células pueden circular a lo largo de capilarescuyo calibre es muy inferior al de su propio diámetro ( hm) y atravesar lainicrocirculación esplénica ( 2 ). Esta propiedad depende de tres factores: a)la forma del hematíe o relación superficie/volumen, b) la viscosidad hemo-globínica y c) las propiedades viscoelásticas de la membrana. La alteraciónde uno o varios de estos factores dificulta el paso de los hematíes a través
TABLA II
ALTERACIONES MORFOLOGICAS ERITROCITARIASMAS DESTACADAS QUE PUEDEN APARECER EN EL CURSO
DE DIVERSAS ENFERMEDADES
ENFERMEDAD
— ANEMIA HEMOLITICA AUTO-INMUNE
— a BETALIPOPROTEINEMIACONGENITA (ACANTOCITO-SIS)
— HEPATOPATIAS CRONICAS— INSUFICIENCIA RENAL
CRONICA— ANEMIA HEMOLITICA MI.
CROANGIOPATICA— MIELOFIBROSIS CON META-
PLASTA MIELOIDE— ANEMIA FERROPENICA
— ANEMIA MEGALOBLASTICAY PERNICIOSA
— ALCOHOLISMO CRONICODISERITROPOYESIS ADQUI-RIDA
,4LTERACION MORFOLOGICA
ESFEROCITOS Y MICROESFE-ROCITOS
ACANTOCITOS («spur-cells»)
ACANTOCITOS y/o DIANOCITOSESQUINOCITOS («burr-cells»)
ESQUISTOCITOS (HEMATIESEN CASCO)
DACRIOCITOS (HEMATIES ENLAGRIMA)
ELIPTOCITOS, DIANOCITOS,MICROCITOS
NIACROOVALOCITOS
MACROCITOSANISOPOIQUILOCITOSIS, ANI-
LLOS DE CABOT, PUNTEADOBASOFILO.
321

de la microcirculación y especialmente del «filtro esplénico» produciendosu rotura en el interior de los capilares (hernölisis extravascular) ( 2 "). Laalteración de la forma puede apreciarse mediante el simple examen morfo-lógico del frotis sanguíneo o el de una suspensión de hematíes fijados englutaraldehido, y en muchos casos es suficiente para orientar etiológiea-mente un determinado proceso patológico. Las alteraciones de la membranase acompañan casi siempre de modificaciones morfólógicas que muchasveces son secundarias a procesos patológicos diversos (Tabla II) pero queen casos más raros obedecen a defectos intríneco o congénitos de la misma(2j.
La anemia hemolítica crónica por defecto congénito de la membrana,es, excepto la EH y EC, de diagnóstico difícil ya que en la mayoría de loscasos su mecanismo íntimo se desconoce. No obstante, y debido a que elnúmero de pacientes con anemia hemolítica «corpuscular» no eritroenzimo-pätica ni hemoglobinopática que quedan sin catalogación etiológica es rela-tivamente elevado (aproximadamente un 15-20 %) el interés por el estudiode la membrana eritrocitaria ha aumentado notablemente en los últimosarios ('"). Las principales alteraciones congénitas de la membrana eritr°-citaria identificadas hasta la actualidad vienen resumidas en la Tabla 1 yentre ellas destaca por su frecuencia la Esferocitosis Hereditaria.
4.1.1. Esferocitosis Hereditaria (EH). En nuestro medio la EH cons-tituye la causa más frecuente de anemia hemolítica constitucional o con"
génita, por lo que los métodos aplicados a su dianóstico son de gran interésclínico. Aunque la historia clínica del paciente o la existencia de anteceden"tes familiares son, en general, muy orientativos, el diagnóstico viene confir"mado siempre por la presencia de un número variable de esferocitos ensangre periférica y un descenso de la resistencia osmótica eritrocitaria(ROE) especialmente después de incubar la sangre 24 horas a 37° C (11'Las características de la curva de ROE varían de acuerdo con el porcentajede esferocitos (Figura 4) el cual a su vez puede variar ampliamente de unenfermo a otro. Ello obedece a las características especiales de penetranciagenética de ésta enfermedad por la cual pueden existir sujetos afectos enlos que prácticamente no se observan esferocitos ni manifestaciones afín-cas de ningún tipo (portadores asintomáticos). En tales casos el estudio dela ROE carece de la sensibilidad suficiente para confirmar el diagnósticoy es necesario recurrir a métodos menos habituales y que no siempre Per-miten establecer el diagnóstico de forma definitiva. Entre ellos destaca n elmicroscopio electrónico de barrido (MEB) y el estudio «in vitro» de ladeformabilidad eritrocitaria (
El Microscopio Electrónico de Barrido (MEB) permite apreciar e°11mayor precisión y detalle la morfología eritrocitaria ya que se parte de unasuspensión sanguínea y se evita el efecto mecánico producido sobre los he-matíes al realizar el frotis. Mediante esta técnica se ha podido apreciar qUeen la mayoría de los casos de EH, los esferocitos propiamente dichos con,'tituyen sólo una pequeña fracción del total de hematíes, siendo la mayo
322
de ellos discocitos o estomatocitos en diferentes etapas de transformacióndiseo-estomatocitica (Figuras 5 y 6). Debido a que la EH suele acompañarsede un importante estímulo eritropoyetico con salida a sangre periférica delunnerosos reticulocitos inmaduros (Reticulocitos tipo I), éstos pueden sertambién fácilmente apreciados mediante el MEB (Fig. 7).
El estudio de la defonnabilidad eritrocitaria puede constituir tambiénun método diagnóstico complementario por cuanto los esferocitos y esto-Inatocitos son menos deformables que los hematíes normales ( 2 '). La defor-//labilidad eritrocitaria puede determinarse mediante diferentes métodos
100
u)
O
50
oae
5 6
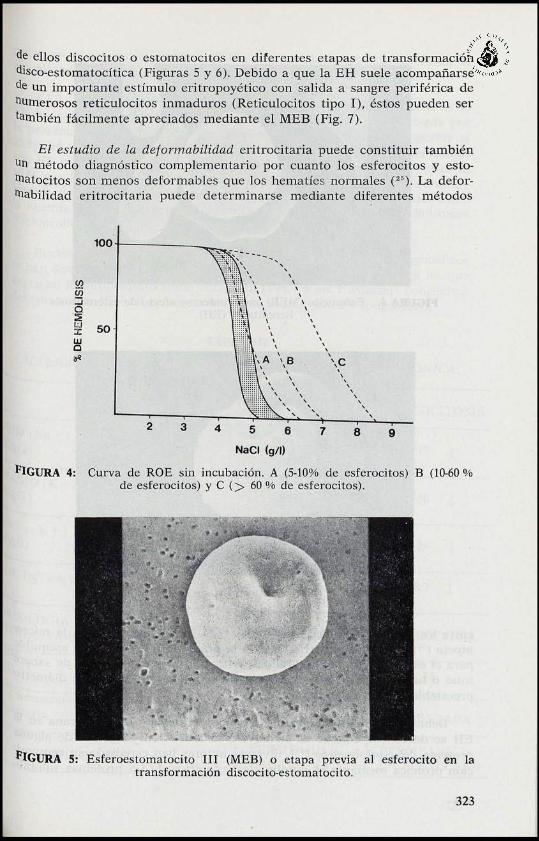
NaCI (g/1)
'FIGURA 4: Curva de ROE sin incubación. A (5-10% de esferocitos) B (10-60 %de esferocitos) y C (> 60 % de esferocitos).
FIGURA 5: Esferoestomatocito III (MEB) o etapa previa al esferocito en latransformación discocito-estomatocito.
323
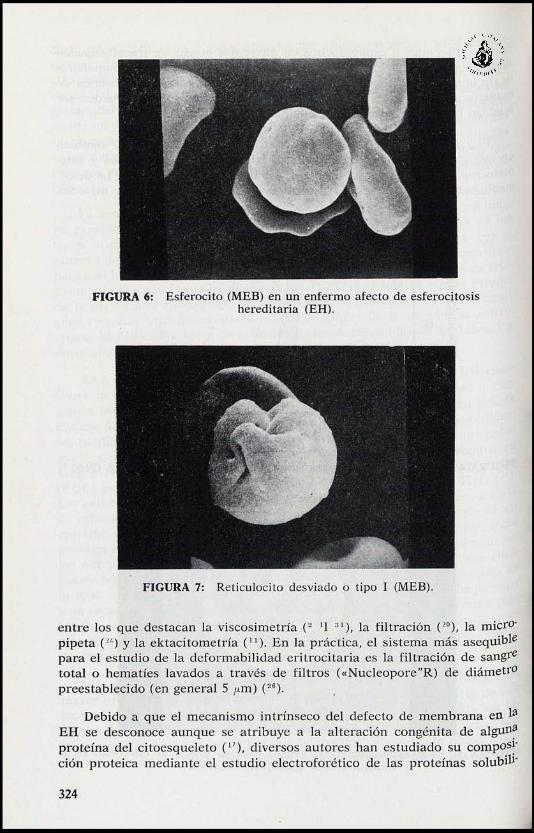
FIGURA 6: Esferocito (MEB) en un enfermo afecto de esferocitosishereditaria (EH).
FIGURA 7: Reticulocito desviado o tipo I (MEB).
entre los que destacan la viscosimetría ( 2 '1 3 '), la filtración ("), la micro'pipeta (") y la ektacitometría ("). En la práctica, el sistema más asequiblepara el estudio de la deformabilidad eritrocitaria es la filtración de sangretotal o hematíes lavados a través de filtros («Nucleopore"R) de diámetropreestablecido (en general 5 /un) (26).
Debido a que el mecanismo intrínseco del defecto de membrana en laEH se desconoce aunque se atribuye a la alteración congénita de algunaproteína del citoesqueleto ( diversos autores han estudiado su composi-ción proteica mediante el estudio electroforético de las proteínas solubili"
324

zadas en dodecilsulfato sódico (SDS). Los resultados obtenidos hasta laactualidad han sido, no obstante, contradictorios ( 6 3 6 ).
4.1.2. Otras alteraciones congénitas de membrana eritrocitaria. A la41 le sigue en frecuencia la eliptocitosis congénita (EC) caracterizada porla Presencia en sangre periférica de eliptocitos en porcentaje superior al25 % del total de hematíes. A diferencia de la EH, el número de enfermoscort EC que presentan clínica de anemia hemolítica crónica es inferior al10 % por lo que su diagnóstico diferencial debe establecerse con otras en-fermedades que pueden acompañarse de eliptocitosis secundaria tales comola anemia ferropénica y megaloblástica, la talasemia y ciertos síndromesInieloproliferativos (32).
Recientemente y gracias al desarrollo de nuevos métodos diagnósticosSe han descrito otras alteraciones congénitas de la membrana entre las quedestacan la piropoiquilocitosis congénita (PPC) y los trastornos congénitosdel transporte Na+1K+ (estomatocitosis y desicocitosis congénitas).
TABLA III
ALTERACIONES CONGENITAS DE LA PERMEABILIDAD IONICA
NORMAL HIDROCITOSIS DESICOCITOSIS
k)bicI (Na+)
11441/1)4-10 98 I, 19
; IO (K+)1) 96-104 40 60
+ IONES<c„...11) TOTALES
K+)100-104 138 I, 79
INTRA CELULAR 68 % 77.6 % 58.7
OLOGIAWCITARIA
DISCOTICOS ESTOMATOCITOS DESICOCITOS
14) 32-36 32 36
' ik-esistencia osmótica:itaria. NORMAL DISMINUIDA AUMENTADA
MECANISMOPERMEABILI-DAD PASIVA Na+
4, PERMEABILI-I DAD PASIVA K+
: 325
Ä

La PPC fue descrita por Zarkowsky y colaboradores ("') y se caracterizapor un aumento de la membrana eritrocitaria a la sensibilidad térmica. Sudiagnóstico puede realizarse incubando los hematíes del paciente a --F . 440 yobservando la aparición de intensa anisocitosis, poiquilocitosis, microesf ero-citosis y abundantes formas bizarras. En los hematíes normales estas alter a
-ciones morfológicas no aparecen hasta que la temperatura alcanza los 500C
(") • La PPC se acompaña de un descenso muy intenso del VCM que Puedellegar a alcanzar valores próximos a 25 fl.
Los trastornos congénitos del transporte jónico obedecen a un defect°de la membrana por el cual existe un aumento de la permeabilidad pasivaal sodio (estomatocitosis congénita o hidrocitosis) o al potasio (desicocitosiso xerocitosis) ( 2 ') (Tabla III).
En la estomatocitosis congénita debido a la mayor permeabilidad Pasi-va al sodio, éste presenta una concentración intraeritrocitaria superior a lanormal. Este defecto no es compensado por una suficiente salida de potasioy el contenido iónico global así como el del agua intraeritrocitaria se hallan
aumentados (hidrocitosis). La alteración morfológica de este síndrome serala estomatocitosis o hematíes con una hendidura en su región central(Figura 8). El mayor contenido acuoso del hematíe tiene como consecuenciauna dilución de la hemoglobina y un descenso de la CC1V1H ( < 30 gic11).
En la desicocitosis (xerocitosis, para algunos autores), el aumento de lapermeabilidad pasiva del potasio produce una disminución de la concentra'ojón eritrocitaria de este ion que no llega a ser compensada por un aument°,del contenido en sodio. Debido a ello, la concentración jónica global asicomo el contenido acuoso del hematíes se hallan disminuidos (deshidratacióneritrocitaria) dando lugar a alteraciones morfológicas eritrocitarias Pecti-liares (desicocitos) fácilmente apreciables mediante el MEB (Fig. 9) . t3
FIGURA 8: Estomatocitos (MGG x 1000)
326

FIGURA 9: Desicocitos (MEB) o hematíes con forma «bizarra» que asemejauna silla de montar.
deshidratación eritrocitaria condiciona una mayor concentración hemo-globínica y por tanto de la CCMH ( < 36 g/dl).
Debido a que las otras formas de anemia hemolítica congénita incluidala EH no suelen cursar con alteraciones de la concentración iónica intra-eritrocitaria, el diagnóstico de la estomatocitosis y decicocitosis congénitasPrecisan de la determinación del contenido en sodio y/o potasio eritrocita-rio lo cual suele realizarse mediante un fotómetro de llama como el quenormalmente se usa en clínica para determinar el sodio y potasio plasma-ticos (").
4.2. Eritroenzimopatías
Desde la descripción del déficit de G6PD por Carson y colaboradoresen el ario 1956 ( ' 2 ) el conocimiento de las eritroenzimopatías como causade hemölisis congénita ha experimentado un gran avance. En la actualidadexisten descritas deficiencias congénitas de prácticamente todas las enzi-mas de la glucOlisis anaerobia algunas de las cuales se acompañan sólode AHCNE pero otras de manifestaciones extrahematológicas tales comotrastornos neurológicos o musculares (Tabla 1). No obstante, las eritroen-zimopatías más frecuentes son el déficit de G6PD, PK, glucosafosfatoiso-merasas (PGI), y probablemente el de P5N, por lo que en presencia de unaelnica compatible el diagnóstico deberá orientarse hacia alguna de ellas(") • En la actualidad diferentes procedimientos informan desde la simpleexistencia de un déficit de actividad enzimática hasta las características yMecanismos moleculares de la enzimopatía (1 3 ).
4.2.1. Estudio de la actividad enzimática. La detección de una eritro-enzimopatía puede hacerse de dos formas: a) mediante métodos cualita-
327

tivos y b) mediante métodos cuantitativos o análisis propiamente dichode la actividad enzimática.
Los métodos cualitativos se han aplicado principalmente a la deteccióno despitaje del déficit de G6PD y con mucho menor éxito al de otras enzi-mopatías de la glucólisis eritrocitaria ('"). Para la detección cualitativa del
déficit de G6PD el Comité Internacional para la Estandarización en H e-
matología (ICSH) recomienda la llamada prueba de la «mancha fluor es-cente» ( 9 ) que debido a la sencillez de su realización (Figura 10) puede ap1i-
100/41 10,.!
O
10' __>10/41
Whatman n°1
G6P
6PG
NADP*
NADPH2
8 340 nm
I G6PD
640 nri2;',.•\
FLUORESCENCIA
FIGURA 10: Prueba de la mancha fluorescente para el diagnóstico del déficitG6PD. R: Mezcla reactiva, Whatman 0'1 papel de filtro. S: Sangre.
carse al escrutinio de numerosos casos simultáneamente o al estudio depoblaciones ( 34 ). Asimismo, la presencia de mosaicismo en las mujeresportadoras o heterozigotas para el déficit de G6PD puede ponerse tambiénfácilmente de manifiesto mediante un método citoqumico basado en inca-pacidad de los hematíes deficientes en G6PD para reducir la metahemo-globina Los métodos cualitativos aplicados a la detección del déficitde PK se han basado también en el principio de la «mancha fluorescente"y son por tanto similares a los empleados para el déficit de G6PD ( 5 ). Noobstante las características moleculares del déficit de PK, diferentes a lasdel de G6PD, dan lugar a frecuentes falsos negativos» que en cierta formainvalidan la fiabilidad del método para detectar esta enzimopatía ( I " ).
Los métodos cuantitativos son los únicos recomendados para la con-firmación diagnóstica de una eritroenzimopatía ( 13 ). En general, aunquecon ligeras diferencias, todos ellos se basan en la elaboración de una reac-ción enzimätica específica consistente en la mezcla de substrato/s, coen
-zima/s, e iones en un medio tamponado donde la enzima a estudiar cons-tituye la etapa limitante. A esta reacción se acopla la transformación deun nucleótido adenílico (NAD o NADP) deSde su forma oxidada a reducida
128

O viceversa. Debido a que el NADH y el NADPH presentan una absorciónespecífica a 340 nm, la medida espectrofotometrica de su velocidad de for-mación o desaparición permitirá determinar la actividad enzimática. (Fi-
gura 11).
E
NADP (P)
NAD (P) H 2
Pv= At
1U = CANTIDAD DE ENZIMA NECESARIA
PARA PRODUCIR 1 p. MOL DE P POR
MINUTO A 37° C.
FIGURA II: Fundamento del método para la determinación de la actividadde las enzimas eritrocitarias. E: Enzima; S: Substrato; P: Producto.
El análisis cuantitativo de las actividades enzimáticas eritrocitarias serealiza a partir de hemolizados totalmente desprovistos de contaminaciónleucocitaria. Esto es especialmente importante en la determinación dela actividad PK ya que el déficit eritrocitario de esta enzima se acompañade una actividad PK leucocitaria normal o incluso elevada, de forma quela contaminación del hemolizado por leucocitos podría enmascarar la de-tección de la enzimopatía ( i "). Al objeto de sistematizar la metodología apli-
cada al análisis de las actividades enzimáticas eritrocitarias, en la prácticaclínica el ICSH ha unificado la preparación del hemolizado y ha estandari-zado los diferentes métodos de análisis correspondientes a cada una de
329

TABLA IV
VARIACIONES FISIOLGICAS DE LAS ACTIVIDADES ENZIMATICAS
1. AUMENTO DE ACTIVIDAD
* Reticulocitos y Hematíes fetales— Hexokinasa— Aldolasa— Triosafosfatoisomerasa— Piruvatokinasa— Glucosa-6-fosfato deshidrogenasa— Fosfogluconato deshidrogenasa
Hematíes fetales exclusivamente— Fosfoglucosaisomerasa— Glicerinaldehido 3 fosfato deshidrogenasa— Fosfogliceratokinasa— Enolasa
2. DISMINUCIONES DE ACTIVIDAD
Hematíes fetales
— Fosfofructokinasa— Adenilatokinasa— Glutationperoxidasa— Acetilcolinesterasa— NADPH-metahemoglobina reductasa (diaforasa)
* Diseritropoyesis adquirida— Piruvatokinasa— Adenilatokinasa
las enzimas a determinar ('). La elaboración de esta metódica ha supuest°una gran mejora en la detección de cualquier enzimopatía pero especial"mente de la del déficit de PK ya que evita por completo la contaminaciónleucocitaria del hemolizado y permite además detectar variantes con cialé"tica anormal en las que el déficit sólo se manifiesta cuando la enzima sehalla en presencia de pequeñas concentraciones de substrato. En talescasos, si se utiliza el método convencional, es decir, una concentraciónsaturante de substrato, la enzimopatía puede pasar desapercibida ( 7 13)*
Asimismo en la interpretación de los resultados de un estudio enzilná-tico eritrocitario debe valorarse siempre la edad del paciente y el númer°de reticulocitos. En los recién nacidos y niños menores de un ario ciertasenzimas presentan una actividad inferior a la de la edad adulta (deficiell"
330

cias enzimáticas fisiológicas) ,por lo que si este hecho no se considera ade-cuadamente, pueden producirse importantes errores de interpretación (")(Tabla IV). El número de reticulocitos es importante por cuanto éstosPoseen actividades enzimáticas en muchos casos varias veces superioresa las del hematíe adulto. Debido a ello, un exceso de reticulocitos puedetambién enmascarar un déficit enzimático produciendo una falsa «activi-dad normal» en enzimas que por la cifra de reticulocitos, deberían presen-tar una actividad varias veces superior a la normal (30).
4.2.2. Estudio de las propiedades moleculares. Una vez identificadala enzimopatía y siempre que sea posible, es aconsejable realizar un aná-lisis de sus propiedades moleculares.
Estos estudios escapan del interés clínico inmediato pero son muyÚtiles para comprender mejor el mecanismo fisiopatológico de la hemólisis.La enzimopata mejor estudiada desde este punto de vista es el déficit deG6PD. Esta enzimopatía junto a su frecuencia, se caracteriza por un ele-vado polimorfismo genético, de forma que en la actualidad se han descritoalrededor de 150 variantes moleculares distintas, muchas de las cualesson asintomáticas pero otras (aproximadamente 64) se acompañan deACHNE ('). Debido al interés genético que suponen los estudios molecula-res del déficit de G6PD, la Organización Mundial de la Salud (OMS) hasistematizado su metódica al objeto de que los resultados obtenidos endiferentes laboratorios sean comparables ('). Los parámetros molecularesrecomendados para identificad una variante así como la clasificación generalde las variantes más frecuentes se resumen en las (Tablas V y VI).
Recientemente el ICSH ha estandarizado también la metódica para elestudio molecular de la PK al objeto de poder identificar sus posibles va-
TABLA V
PARAMETROS IMPRESCINDIBLES PARA LA CARACTERIZACION DE LAG6PD (OMS, 1967)
" ACTIVIDAD ERITROCITARIA (U/g Hb)" MOVILIDAD ELECTROFORETICA EN GEL DE ALMIDON A pH 8.0" AFINIDAD APARENTE POR LOS SUBSTRATOS
a) GLUCOSA-6-Fosfato (Km-G6P)b) NADP (Km-NADP)UTILIZACION DE LOS ANALOGOS DEL SUBSTRATOa) 2 Deoxiglucosa-6-fosfato (2dG6P)b) deamino NADP (D-amino NADP)CURVA DE pH OPTIMO
--- ESTABILIDAD TERMICA A 46° C.
331

riantes ( 23 ). No obstante, tanto ésta como la correspondiente a la G6PD, se
hallan sólo al alcance de laboratorios especializados o de Referencia.
TABLA VI
CLASIFICACION GENERAL DE LAS VARIANTES DE G6PDDE ACUERDO CON SUS CARACTERÍSTICAS CLINICO-MOLEULARES
CLASE ACTIVIDAD PROPIEDADES MOLECULARES CLINICA
0% INESTABILIDAD HEMOLISISCINETICA DESFAVORABLE CRONICA
ASINTOMATICA2 5-10 % INESTABILIDAD
CINETICA FAVORABLEHEMOLISISAGUDA
ASINTOMAT1CA
10-40% VARIABLE HEMOLISISAGUDA
60-100 % VARIABLE ASINTOMATICA
AGRADECIMIENTOS
Queremos expresar nuestro agradecimiento a las srtas. M. José Ferräny Ni. José Insá por su asistencia técnica en la elaboración de la iconografíade MEB presentada en este trabajo y a la srta. Teresa Martínez por stilabor de mecanografiado.
Bibliografía
/. Allard, C.; Mohandas, N., Bessis,M.: Red Celi deformability changes inhemolytic anemias stimated by difrac-tometric me t ho ds (Ektacytometry).Blood Cells 3: 290-21, 1977.
2. Berga, L.; Vives Corrons, J.L.; Fe-liu, E.; Woessner, S.; Rozman, C.: As-pectos de la hemorreología y deforma-bilidad eritrocitarias en el diagnóstico
hematológico. Salvat Ed. (Barcelona)Pendiente de publicación, 1981.
3. Bessis, M.: Reinterpretation desfrotis sanguins. Ed. Masson y SPrirl-ger, 1976.
4. Betke, K.; Beutler, E.; Brevvel.'C.J., Kirkman, H.N., Luzzato, L.;tulsky, A.G.; Ramot, B.; Siniscalco,Standarization of procedures for die
332

study of glucose-6-phosphate dehydro-genase. Report of a WHO scientificgroup. WHO Rech Rep. Ser. num. 366,1967.
5. Beutler, E.: A series of new scree-fing procedures for pyruvate-kinasedeficiency, glucose-6-phosphate dehy-drogenase defiicency and glutathionereductase deficiency. Blood, 28: 553-556,1966.
6. Beutler, E.; Guinto, E.; Johson,C .: Human red celi protein Kinase innormal subpects and Patient with he-reditary spherocytosis, sickle cell di-sease and autoimmune hemolytic ane-mia. Blood. 48: 887-98, 1976.
7. Beutler, E.; Blume, KG.; Kaplan,J.C.; Lohr, G.W.; Ramot, B., Valentine,W.N.: International Committee forStandatization in Haematology. Re-eommended methods for red cell en-zYme analysis. Brit. J. Haematol 35:331-340, 1977.
8. Beutler, E.: Hemolytie Anemia indisorders of Red Cell metabolism. Ple-num Medical Book Co. (New York andLondon), 1978.
9. Beutler, E.; Blume, K.G.; Lohr,G .W.; Ramot, B.; Valentine, W.N.: In-ternational Committee for Standadi-zation in Haematology RecommendedScreening test for glucose-6-phosphatedehydrogenase (G6PD) deficiency. Brit.
Haematol. 43 465-467, 1979.10. Boivin, P.; Galdós, A.; Lestradet,
Enzymopathies. Fas I.' Masson etCle (Paris) 1971
11. Brereton, W.; Murph y, J.R.:kheologic studies of deoxygenated nor-mal and hereditary spherocytosis bloodand separated erythrocytes. J. Lab.Clin. Med. 83: 112-18, 1974.
12. Car so n, P.E.; Flanagan, C.L.;C.E.; Alving, A.S.: Enzymatic
deficiency in primaquine-sensitive ery-throcyte Science 124: 584-490, 1956.
13. Enzymopathies. Clinics Haema-Mlogy . Sauders and Co. (London), 1981.
1
▪
4. Feo, C.j.; Thernia, C.; Subtil, E.;Leblond, P.F.: Observation of echinocy-tosis in eight patients. A phase con-trast and SEM study. Brit. J. Haema-101. 40: 519-526, 1978:315. Gall, J.C., Brewer, G.J., Dem, R.
Studies of glucose-6-phosphate de-
hydrogenase activity of individual ery-throcytes. The methemoglobin elutiontest for identification of female hete-rozygous for G6PD deficiency. Amer.J. Hum. Genet. 171: 359-368, 1965.
16. Glader, BE.; Fortier, N.; Alba-la, MM., Nathan, DG.: Congenital he-molytic anemia associated with dehy-drated erythrocytes and increased no-tassium loss. N. Engl. J. Med. 291:491-496, 1974.
17. Greenquist, A.C.; Shohet, S.B.:ATP dependent phosphorilation of amembrane protein in normal and he-reditary spherocytosis red cells. Blood42 997-1006, 1973.
18. Haemolytic Anaemias. Clinics inHaematology. Saunder and Co. (Lon-don) 1975.
19. Jou, J.M.; Vives Corrons, J.L.;Carreras: Favismo y «Eccent roci tos i s ».Sangre 25: 111-114, 1980.
20. La Celle, PL.; Weed, R.1.: Re-versibility of abnormal deformabilityand permeability of the hereditaryspherocyte (abstr). Blood 34:858, 1967.
21. Leblond, P.F.; De Boisfleury,Bessis, M.: Erythrocyte shape in here-ditary spherocytosis. A scanning dec-tron microscopy study and relations-hip to deformability. Nouv. Rev. F.Hematol. 13: 873-83, 1973.
22. Mentzer, W.C.; Lubin, B.H.; Em-mons, S.: Correction of permeabilitydefect in hereditary stomatocytosis bydimethyl adipimidate. N. Engl. J. Med.294: 1200-1204, 1976.
23. Miwa, S.; Boivin, P.; Blume, K.G.; Arnold, H.; Black, J.A.; Kahn, A.;Staat, G.E.; Nakashima, K., Tanaka,R., Paglia, DE.; Valentine, W.N.; Yos-hida, A.; Beutler, E.: Recommendedmethods for the characterization ofred cell pyruvate kinase variants. Brit.J. Haematol. 43: 275-86, 1979.
24. Murphy, J.R.: The influence ofpH and temperature on some physicalproperties of normal erythrocytes anderythrocytes from patients with here-ditary spherocytosis. J. Lab. Clin. Med.69: 758-75, 1967.
25. Piomelli, S.; Yachnin, S.: Cu-rrent topics in Hematology. Vol. I,Alan Liss (N.Y.) 1978.
333

26. Reid, Hl., A.I. Barnes, P.S. Lock,J.A. Dormandy and J.L. Dormandy: Asimple method for measuring erythro-cyte deformability. J. Clin. Pathol. 29:855, 1976.
27. Vives Corrons, J.L., MontserratCosta, E.; Rozman, C.: Hereditary He-molytic Anemia with Erythrocyte Py-rimidine 5'Nucleotidase Deficiency inSpain. Clinical Biological and FamilialStudies. Human Geneties. 34: 285-292,1976.
28. Vives Corrons, J.L., MontserratCosta, E.; Pujades, A.; Woessner, S.;Rozman, C: Déficit congénito de piri-midina 5'Nucleotidasa eritrocitaria. Unnuevo tipo de eritroenzirnopatía conhemólisis. Sangre 21: 827-835, 1976.
29. Vives Corrons, J.L., Rozman, C.:Anemias bemoltíicas eritroenzimopäti-cas. Aspectos fisiopatológicos y clíni-cos. Med. Clin. 67: 317-321, 1976.
30. Vives Corrons, J.L., Pujades, M.A.; Rozman, S.: Enzimas eritrocitariasL Estudio comparativo entre los valo
-res normales de actividad en adultossanos, en adultos con reticulocitosis yen hematíes de cordón umbilical. Med.Clin. 68: 52-8, 1976.
31. Vives Corrons, J.L.; Berga, L.;Aguilar, .L.; Martínez, M.; Insa,Rozman, C.; Jou, J.M.: Corportam ien-to reológico de los hematíes en la es-
ferocitosis hereditaria. Sangre. 25: 192-201, 1980.
32. Vives Corrons, J.L. en «Manualde Hematología »de Sans Sabrafen Ycolaboradores. Ed. Doyma. 1981.
33. Vives Corrons, J.L., Pujades,M.A.: Application of the ICSFI recorn-mended screening test for G6PD defi
-ciency to Hospital population (en pre-paración), 1981.
34. Wintrobe: Clinical HematologY5th Ed. Lea Febiger, 1974.
35. Wolfe., L.Z.; Lux, S.E: Membra-ne protein phosphorylation of intadnormal and hereditary spherocytic(HS) red cells (Abstr). Blood 48: 963,1963.
36. Zarkowski, H.S.; Mohandas, N.,Speaker, C.B., Sholet, S.A. A conge-nital haemolytic anaemia with th er-mal sensibity of the erythecyte melwbrane. Brit. J. Haematol, 29: 537-542'1975.
334