03.005 anemias hemolíticas corpusculares
TRANSCRIPT

8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 1/9
Concepto y valoración analítica
Las anemias hemolíticas de causa corpuscular constituyen ungrupo heterogéneo de entidades, caracterizadas por unacortamiento en la vida media del hematíe en sangre periféricao por un aumento de la apoptosis intramedular poreritropoyesis ineficaz. La causa es siempre congénita decarácter hereditario y el hematíe presentará característicasestructurales o funcionales alteradas. Estas patologías puedenacompañarse de anemia si la médula ósea no es capaz decompensar mediante hiperplasia eritroide la destrucciónincrementada de los hematíes.
Los eritrocitos son elementos celulares altamenteespecializados en el transporte de oxígeno, por lo cual suestructura está notablemente simplificada para el desarrollode dicha función. Su membrana presenta característicasúnicas en cuanto a deformabilidad para atravesar loscapilares hasta 10 veces más estrechos que su diámetro, y sucitoplasma carente de núcleo consta de un reducidometabolismo enzimático para mantener la integridadfuncional de la hemoglobina (Hb). Las anemiascorpusculares pueden ser debidas a alteraciones en la síntesisde hemoglobina, del sistema enzimático o a alteraciones enla composición de la membrana. Los hallazgos de
laboratorio serán comunes a las anemias hemolíticasadquiridas, episodios de hemólisis aguda o crónica en lashemoglobinopatías cualitativas, en las alteraciones demembrana y en los déficits enzimáticos. Lashemoglobinopatías cuantitativas (talasemias) cursanpredominantemente con microcitosis y eritropoyesis ineficaz(o aborto intramedular). Hemoglobinopatías cuantitativas
Estructura genética de la hemoglobina
La hemoglobina, el componente eritrocitario másabundante del hematíe, es una proteína de 68 kD
Medicine 2004; 9(20): 1276-1285 127761
ACTUALIZACIÓN
Anemias hemolíticascorpusculares J. Sánchez García, A. Torres Gómez,
J. Serrano López y J.M. García CastellanoServicio de Hematología y Hemoterapia. Laboratorio de Eritropatología.
Hospital Universitario Reina Sofía. Córdoba.
PUNTOS CLAVE
Concepto. Las anemias hemolíticas
corpusculares agrupan alteraciones
congénitas del hematíe con alta prevalencia
en nuestro medio, en la mayoría de los
casos en forma de portadores silentes
o formas clínicamente leves. La ausencia
de déficits carenciales y la historia
familiar compatible, orientan el diagnóstico,
cuya confirmación definitiva se realizará
en laboratorios especializados de
Eritropatología.
Hemoglobinopatías cuantitativas. Según las zonas
geográficas, hasta un 5% de la población puede
ser portador de rasgo talasémico β+ o de deleciónde uno o dos genes alfa. Cursan con microcitosis,
ferritina normal y número de hematíes normal o
elevado y no precisan tratamiento. El correcto
diagnóstico es imprescindible en los casos que
precisen consejo genético.
Enzimopatías. El déficit de glucosa-6-fosfato-
deshidrogenasa (G-6PDH) es frecuente en nuestro
medio, caracterizándose por crisis hemolíticas al
contacto con habas o medicación oxidante. El
tratamiento es básicamente preventivo.
M embranopatías. La esferocitosis hereditariaprovoca en la mayoría de los pacientes hemólisis
crónica compensada, sin anemia y con
esplenomegalia y elevación de bilirrubina
indirecta. Algunos casos se benefician de
esplenectomía.
Hemoglobinuria paroxística nocturna. La
hemoglobinuria paroxística nocturna es una
enfermedad poco frecuente, caracterizada por
anemia con hemólisis aguda y hemoglobinuria de
curso insidioso y tendencia a la pancitopenización
y fenómenos trombóticos.
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.

8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 2/9
constituida por 4 subunidades proteicas que forman unaestructura globular con cavidades para alojar 4 gruposhemo y en la región central el 2-3 difosfoglicerato (fig. 1A).Las 4 subunidades proteicas o cadenas globínicas son detipo alfa o de tipo beta e iguales dos a dos. Las cadenas tipoalfa constan de 141 aminoácidos y los genes que controlansu síntesis se localizan en el cromosoma 16 en un grupo queincluye en dirección 5’-3’: los genes embrionarios ξ, trespseudo-genes y los genes α1 y α2 funcionantes en la vidaadulta. Las cadenas tipo beta contienen 146 aminoácidos y los genes que controlan su síntesis se localizan en elcromosoma 11, en otro grupo que incluye en dirección 5’-3’: el gen embrionario ε, los genes fetales γ G y γ A y losgenes δ y β de la vida adulta (fig. 1B). La estructura essimilar para todos los genes globínicos y comprende laregión promotora (CAT box y ATA box), región CAP, genestructural con tres exones y dos intrones, y cola depoliadenilación. En la vida adulta la hemoglobina principal
(95%-98%) es la HbA (α2β2), y en mucha menorproporción (1%-3%) la HbA2 (α2δ2) y la HbF (< 1%) (α2γ 2).
Fisiopatología de las hemoglobinopatíascuantitativas
Las hemoglobinopatías cuantitativas o talasemias son ungrupo de enfermedades genéticas caracterizadas por lareducción o ausencia de las síntesis de uno o más tipos decadenas globínicas. Se clasifican en alfa o beta-talasemias
según el tipo de cadena cuya síntesis esté comprometida. En
condiciones fisiológicas la síntesis de cadenas alfa y beta esequilibrada para configurar una hemoglobina normal, peroen las talasemias habrá un desequilibrio con exceso decadenas codificadas por el gen no defectuoso.
En las beta-talasemias, las cadenas alfa sobrantesprecipitan en el interior de los eritroblastos provocando sudestrucción precoz intramedular (eritropoyesis ineficaz) y por alteración en el metabolismo, mitosis y alteraciones en la
membrana. Así mismo el hierro libre no incorporado a losgrupos hemo produce radicales libres y oxidación de lasproteínas1. También los hematíes en la circulación sufrenprocesos de fagocitosis parcial en el bazo.
En las alfa-talasemias, el exceso de cadenas tipo beta (β oγ ) no ocasiona precipitados porque son más solubles y estables, pero forman agregados en forma de tetrámeros:hemoglobina H (β4) (fig. 2) o hemoglobina Barts (γ 4). Sinembargo, en la circulación periférica estas hemoglobinas noson funcionantes para el transporte de oxígeno, conprecipitación y acortamiento de la vida media del hematíe.
Alfa-talasemia
Las alfa-talasemias se producen generalmente por delecioneso pérdida de material genético en los genes alfa y en unaminoría de pacientes la alfa-talasemia se produce pormecanismos no delecionales. En nuestro medio2, laalteración más frecuente es la deleción de 3,7 Kb de ADN y con menos frecuencia las deleciones 4,2 Kb y 3,5 Kb. Existendeleciones más amplias que engloban ambos geneslocalizados en el mismo cromosoma que se denominansegún el área geográfica donde son prevalentes (MED, SEA,BRIT, SPAN, etc.). Por último existen casos excepcionales
de alfa-talasemia adquiridas que se asocian a retraso mentalpor ruptura de la porción distal del brazo corto delcromosoma 16 (síndrome ATR-16) o a síndromesmielodisplásicos por deleciones secundarias3 o mutacionesen un gen controlador4 (ATRX) situado en el cromosoma X.
La deleción puede afectar a un solo gen (-α /αα), a dosgenes situados en distinto cromosoma (-α /-α), a dos genessituados en el mismo cromosoma (– – /αα), a tres genes oenfermedad de HbH, o pérdida de los 4 genes o hidropesíafetal por HbBarts (fig. 3). El portador silente o deleción enun solo gen presenta características normales o solamenteuna discreta hipocromía o tendencia microcítica. El rasgotalasémico o deleción en dos genes son clínicamente
ENFERMEDADES DE LA SANGRE (I)
1278 Medicine 2004; 9(20): 1276-1285 62
α1 β
1
α
2β2Deoxi-hemoglobina
2 - 3
D P G
α1 β
1
α
2β2Oxi-hemoglobina
O2 O2
O2 O2
Cromosoma 16 Cromosoma 11
5’ 3’ 5’ 3’
ζ α 1 α 2 ε Gγ Aγ δ βεαζ γ δ β
ζ2 ε2 ζ2 γ 2 α2 ε2 α2 γ 2 α2 δ2 α2 β2Hb Gow er s I Hb Por tland Hb Gow er s II Hb F(< 1%) Hb A2(1-3%) Hb A(95-98%)
A
B
Fig. 1. Estructura de la hemoglobina (A). Evolución de la sí ntesis de lascadenas de hemoglobina (B).
Fig. 2. Precipitados de
hemoglobina H (β4) en
alfatalasemia.
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.

8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 3/9
asintomáticos aunque presentan microcitosis e hipocromíaen sangre periférica con cifra de hematíes normal. Laenfermedad por HbH o deleción en tres genes presentaanemia microcítica de carácter variable con datos dehemólisis crónica y hematopoyesis extramedular conesplenomegalia. La hidropesía fetal o HbBarts esincompatible con la vida con muerte intraútero o alnacimiento por hematopoyesis extramedular en hígado y bazo y edemas generalizados.
El diagnóstico se basa inicialmente en la presencia demicrocitosis en grado variable con patrón de hierro normal.El patrón de hemoglobinas en la electroforesis es normal,pudiendo detectarse un 5%-30% HbH (o cuerpos H con
tinción de azul cresil) principalmente en las formas dedeleción en tres genes. Los portadores silentes y rasgostalasémicos precisan para su confirmación estudiosmoleculares.
El tratamiento para la Enfermedad por HbH es elsoporte transfusional, y para las formas de hidropesía fetaldetectadas precozmente, la terapia transfusional intraúteroha conseguido buenos resultados.
Beta-talasemia
Las beta-talasemias constituyen un grupo genéticamentemuy heterogéneo, siendo poco frecuentes las deleciones y habiéndose descrito más de 200 mutaciones que afectan a latranscripción del ADN o a la traducción de ARNm conmarcada distribución geográfica. En nuestro medio, lasmutaciones que afectan la transcripción del ADN incluyenmutaciones en una base del ADN que condicionan laaparición de un codón de terminación (codón 39, C-T) y las inserciones/deleciones de 1-4 bases con desplazamientode lectura (frameshift mutation) surgiendo posteriormenteun codón de terminación (codón 8/9 +G). Las mutacionesque afectan al procesamiento del ARNm son aquellas quese sitúan en las uniones de los intrones y exones de tal
forma que se impide o dificulta la retirada del intrón (IVS-
I -1 G-A; IVS-I-6 T-C) o en el interior de los intrones(IVS-I-110 G-A) creando un ARNm no funcional. Se handescrito también mutaciones en las regiones promotoras y reguladoras (CAT y ATA box), región CAP y en el sitio depoliadenilización.
Las manifestaciones clínicas son muy heterogéneas variando según la lesión genética subyacente (β+ o β0), elestado de homo o heterocigosidad y la interacción con
otras alteraciones de la hemoglobina. Las formas clínicasminor son asintomáticas con microcitosis pero sóloocasionalmente anemia leve. Las formas de beta-talasemiaintermedia provocan la presencia de anemia microcíticaque generalmente no precisa transfusiones con algunas delas manifestaciones clínicas generalmente leves derivadasde la hematopoyesis incrementada (deformidadesesqueléticas, dolores óseos, retraso del crecimiento) y extramedular (hepato-esplenomegalia). La forma de beta-talasemia mayor (anemia de Cooley) constituye la formamás grave con anemia transfusión-dependiente, hepato-esplenomegalia gigante, retraso en el crecimiento y
alteraciones craneofaciales y esqueléticas por hiperplasiade la médula ósea. La alteración en el metabolismo delhierro por aumento de captación y sobrecarga derivada delas transfusiones provoca hemocromatosis secundaria conafectación hepática, pancreática, paratiroides y cardíaca.
Asimismo presentan susceptibilidad incrementada a lasinfecciones (por bloqueo del sistema retículo-endotelial[SRE] y disminución de producción de opsoninastermolábiles) y a los procesos trombóticos.
El diagnóstico se basa en el estudio de sangre peri-férica con microcitosis marcada, presencia de hematíesen diana o codocitos, punteado basófilo y eritroblastoscirculantes (fig. 4). En las formas heterocigotas existe un
aumento de la HbA2 y niveles de HbF normales odiscretamente elevados. En las formas homocigotas, losniveles de HbF están elevados en el 10%-90% con HbA2
variable.El tratamiento de las formas mayores incluye soporte
transfusional para mantener cifras de hemoglobina óptimas
ANEMIAS HEMOLÍTICAS CORPUSCULARES
Medicine 2004; 9(20): 1276-1285 127963
Normal Rasgo alfa + Rasgo alfa +homocigoto
Rasgo alfa O Enfermedad HbH Hb Bart’s (HF)
Fig. 3. Formas cl í nicas de alfa-talasemia según el número de genes afectos.
Fig. 4. M orfologí a de hematí es con rasgo talasémico.
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.

8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 4/9
para evitar retraso del crecimiento. La terapia quelante delhierro (desferroxamina parenteral) es fundamental paraevitar daño cardíaco y hepático. La esplenectomía se indicaen los casos de altas necesidades transfusionales otrombopenia asociada. El trasplante de médula ósea dedonante HLA idéntico familiar no afecto o no-emparentadoconstituye una modalidad curativa5, especialmente si serealiza a edades tempranas. Actualmente se investigan
modalidades de terapia génica6.
Hemoglobinopatías cualitativas
Las hemoglobinopatías estructurales se producen pormutaciones que ocasionan cambios en algunos aminoácidosde las cadenas globínicas. La cantidad de hemoglobinasintetizada es normal pero presentará característicasfisiológicas distintas a la HbA normal, dependiendo del lugardonde el aminoácido erróneo esté situado. Existen más de1.000 hemoglobinopatías estructurales descritas que afectan
a aminoácidos situados en la superficie de la molécula (HbS y hemoglobinopatías asintomáticas), en el interior en lasuniones α1β1 (hemoglobinopatías inestables) o en lasuniones α1β2 (hemoglobinopatías con alteración en laafinidad por el O2), y en las zonas de contacto con el grupohemo que producen la estabilización del hierro en formaférrica (hemoglobinopatías M). Existen también mutacionesque provocan una Hb estructuralmente anómala con síntesisdisminuida (hemoglobinopatías talasémicas) (tabla 1).
Hemoglobinopatía S
La hemoglobina S es el resultado de la sustitución del ácidoglutámico de la posición 6 de la cadena globínica beta por
valina, debido a una mutación GAG-GTG en el codón desíntesis. Los portadores heterocigotos de esta mutacióntendrán menos del 50% de HbS y son clínicamenteasintomáticos. Esta mutación es de alta incidencia endeterminadas zonas geográficas como África (20%-40% de
la población) y raza negra del continente americano (8%-10% de la población). La presencia de la mutación en lapoblación africana confiere una ventaja al ser estos hematíesmenos susceptibles a infecciones por Plasmodium falciparum.Cuando la mutación se encuentra en estado homocigoto,aparece la anemia de células falciformes cuya clínica vienedeterminada por la facilidad que tiene la HbS a agregarse. LaHbS oxigenada es tan soluble como la HbA, pero en estados
de deoxigenación (ectasia vascular, acidosis o infecciones)pierde solubilidad y se agregan 30 tetrámeros que inician laformación de filamentos de doble cadena que se agregan enhaces de 14 hebras (cuerpos tactoides), que producen lafalciformación irreversible con el típico hematíe en hoz. Deforma secundaria se afecta la membrana con alteración enbombas iónicas y deshidratación y se afectan las víasmetabólicas por estrés oxidativo. El hematíe falciformepresenta adhesión incrementada a los endotelios vascularesprovocando crisis vasooclusivas y se destruye finalmentemediante hemólisis intra y extravascular. Los pacientespresentan además mayor susceptibilidad a infecciones y
tendencia trombótica.La clínica viene determinada por la presencia de anemiacrónica y retraso del crecimiento con exacerbaciones enepisodios agudos que pueden afectar a cualquier órgano: sis-tema nervioso (infartos-hemorragias), pulmones (distrésrespiratorio agudo), hígado (colelitiasis, colecistitis, isquemiahepática), riñones (hematuria, alteraciones tubulares,glomerulares, insuficiencia renal crónica), infartosesplénicos, retinopatía, infartos óseos, úlceras cutáneas y miocardiopatía.
El diagnóstico se realiza mediante las pruebas defalciformación y la presencia de HbS en electroforesis dehemoglobinas. El tratamiento incluye medidas de soporte
(como analgesia, transfusiones, antibióticos en procesosinfecciosos), agentes que incrementan la síntesis de HbF(hidroxiurea, butirato, azacitidina, decitabina), y agentes
vasodilatadores (óxido nitroso o arginina). Otros fármacos conpropiedades antiagregantes, antioxidantes, inhibidores de lapolimerización y de la deshidratación están siendo estudiados.La modalidad potencialmente curativa es el trasplante
alogénico de médula ósea dedonante sano HLA idéntico conbuenos resultados si se realizaprecozmente7 y la terapia génica queestá en investigación8.
Enzimopatías
El sistema metabólico del hematíedepende de 4 vías fundamentales:glucólisis anaerobia, metabolismodel glutatión, metabolismonucleotídico y sistema diaforásico.En todas ellas, el sistemaenzimático es el formado durantela etapa de eritroblasto nucleado y reticulocito, sin posibilidad de
regenerarse durante la vida del
ENFERMEDADES DE LA SANGRE (I)
1280 Medicine 2004; 9(20): 1276-1285 64
TABLA 1
Hemoglobinopatí as estructurales
Hemoglobina M utación Alteración Clí nica
HbS β6 Glu-Val Pérdida de solubilidad Anemia falciforme
HbC β6 Glu-Lys Pérdida de solubilidad Dianocitos
Cristalización Leve anemia
HbM His-tyr Fe en estado oxidado Cianosis
Hb alta afinidad por oxígeno Varias Desplazamiento izquierdo curva disoc iac ión Eritroc itosis
Hb baja afinidad por oxígeno Varias Desplazamiento derec ho c urva disoc iac ión Cianosis
Inestables Varias Inestabilidad al calor, oxidantes, etc. Crisis hemolíticaso hemólisis crónica
Cuerpos Heinz
HbE β26 Glu-Lys Hemo globinopatía talasémica Microcitosis
Curso leve
HbLepore Crossing-over Hemoglobinopatía talasémica Talasemia
Genes δ y βminor/ mayor
Hb: hemoglobina.
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.

8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 5/9
hematíe. La fuente principal de energía es la glucosa que escatabolizada en la vía de la glucólisis anaerobia (Embden-
Meyerhoff) hasta piruvato o lactato generando dos moles deadenosina trifosfato (ATP). El 2-3 difosfoglicerato que actúacomo modulador de la oxigenación de la Hb es sintetizadodesde esta vía. Parte de la glucosa se deriva a la vía de lashexosas monofosfato (pentosas fosfatos) mediante la enzimaglucosa-6-fosfato-deshidrogenasa (G-6PDH) quetransforma la glucosa-6-fosfato a 6-fosfo-gluconato (fig. 5).Este paso genera nicotinamida adenosina dinucleótidofosfato [NADPH] que por la acción de la glutatión reductasaconvierte el glutatión oxidado en reducido, imprescindiblepara mantener la integridad eritrocitaria al eliminar radicalesoxidantes.
El metabolismo nucleotídico es necesario para el
mantenimiento del ATPeritrocitario con enzimas comoadenosin-deaminasa (ADA),pirimidin-5-nucleotidasa (P5’N) oadenilato-kinasa (AK). El sistemadiaforásico emplea NADH(sistema principal) y NADPH(sistema secundario) diaforasas
para mantener el hierro hemínicoen forma reducida.
Desde la descripción del déficitde G-6PDH como causa de anemiahemolítica enzimopática, se handescrito déficits enzimáticos entodas las vías metabólicas, algunosde los cuales cursan con hemólisisgeneralmente crónica y en algunoscasos con alteraciones sistémicasneurológicas o musculares (tabla 2).
Déficit de glucosa-6-fosfato-deshidrogenasa
Es el déficit enzimático másfrecuente con una ampliaincidencia en la poblaciónmundial y en especial en las áreasasiáticas, africanas (zonas depaludismo endémico) y mediterráneas. En nuestro país la
prevalencia es variable (0,1%-1,5%) de la población. El
gen que codifica la síntesis de G-6PDH está localizado enel cromosoma X y consta de 13 exones. Se han descritomás de 50 mutaciones puntuales exónicas que condicionan
varios tipos funcionales de G-6PDH9,10. El tipo I(variantes raras o esporádicas) cursa con ausencia total deactividad en eritrocitos y leucocitos, lo cual condicionaanemia hemolítica crónica y tendencia a infecciones. Eltipo II (variante mediterránea, la más frecuente en nuestromedio) cursa con ≤ 5% de actividad eritrocitaria con crisishemolíticas agudas inducidas por habas o medicamentos.El tipo III (variantes africanas o asiáticas) cursan con 5%-15% de actividad con hemólisis agudas. Existenmutaciones que no condicionan pérdida de actividadenzimática y son por tanto asintomáticas. La herencia estáligada al cromosoma X y por tanto la expresividad clínicaes propia de hombres o mujeres homocigotas. Las mujeresheterocigotas son susceptibles a padecer crisis hemolíticassegún el grado de inactivación del gen X afecto.
La clínica se inicia generalmente en las primeras décadasde la vida en forma de síndrome hemolítico agudo de iniciobrusco, con ictericia y hemoglobinuria. El desencadenantesuele ser la ingesta de determinados medicamentos (tabla 3)con actividad potencialmente oxidante. Puede asociarse afavismo, o crisis hemolíticas tras ingesta o contacto conhabas, debido posiblemente a su actividad oxidante. Otros
desencadenantes son las infecciones, el embarazo o
ANEMIAS HEMOLÍTICAS CORPUSCULARES
Medicine 2004; 9(20): 1276-1285 128165
Glucosa
Glucosa-6-P
Fuctosa-6-P
Fructosa-1,6-difosfato
DihidroxiacetonaFosfato
P-
P-
6-PG-lactona
Glucólisis anaerobia
G6PDH
6-fosfogluconato
Vía hexosaMonoP
Ribulosa
Ribosa 5 Xilulosa 5P
Gliceraldehido 3fosfato
+sedoheptulosa
Gliceraldehido 3-fosfato
1,3 P- glicerat o
3 P- glicerato
2 P- glicerato
Fosfoenolpiruvato
Piruvato
Lactato
P+
P+
2,3 P-glicerato
P+
PVK
Shunt de Luebering-Rapaport
Fig. 5. Principale s ví as enzimáticas del eritrocito.
TABLA 2
Principales enzimopatí as eritrocitarias
Ruta enzimática/enzima Hemólisis Clí nica asociada
Metabolismo óxido-reductor
Glucosa-6-P-desh idrogenasa Agudas +/+++++ No
Glutatión-sintetasa Crónica + Oxiprolinuria/neuropatía
Glucólisis anaerobia
Piruvato-kinasa Crónica +/++++ No
Glucosa-P-isomerasa Crónica +/++++ Glucogenosis/retraso mental
Triosa-P-isomerasa Crónica +/+++ Neuropatía/infecciones
Metabolismo nucleotídico
Pirimidin-5-nuc leotidasa Crónica +/++ No
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.

8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 6/9
descompensaciones diabéticas. Los cuadros suelen serautolimitados y de corta duración.
El diagnóstico se realiza mediante la dosificaciónenzimática y comprobación de la mutación en el ADN porbiología molecular. En las crisis hemolíticas es frecuenteencontrar hematíes con disposición excéntrica de lahemoglobina. La incubación de lasangre con azul cresil provoca laaparición de cuerpos de Heinz y laincubación con fenilhidracinaprovoca la formación deprecipitados de hemoglobina.
El tratamiento esfundamentalmente preventivoevitando el contacto con lassustancias oxidantes y soportetransfusional en las crisis intensas.
Déficit de piruvato-cinasa
Es el segundo déficit más frecuentedespués del de G-6PDH. Seproduce por mutaciones en losgenes (PK-RL para eritrocitos y PK-M para leucocitos) que selocalizan en el cromosoma 15. La
herencia es autosómica recesiva
con expresividad clínica principalmente en pacienteshomocigotos o dobles heterocigotos. Produce una hemólisiscrónica sin relación con medicamentos. La sangre periféricamuestra con frecuencia hematíes espiculados.
Membranopatías
La membrana del hematíe consta de una doble bicapalipídica en la que se insertan proteínas integrales. En la carainterna se sitúa una red fibrilar constituida por proteínas delesqueleto. Su configuración especial y el exceso desuperficie con relación al volumen condicionan ladeformabilidad y elasticidad que el hematíe precisa (fig. 6).Las proteínas integrales son glucoproteínas cuyo grupohidrocarbonato se sitúa en el exterior (responsable degrupos sanguíneos) y son principalmente la banda 3 y lasglucoforinas. La banda 3 es la más abundante, actúa comocanal transportador de aniones y porta los antígenos Ii y
ABH. Las glucoforinas (GF) presentan fragmentos
glucídicos en el exterior para los determinantes antigénicos(MNS en GF-A, Ss en GF-B y Gerbich en GF-C).La red fibrilar del esqueleto se compone por dímeros
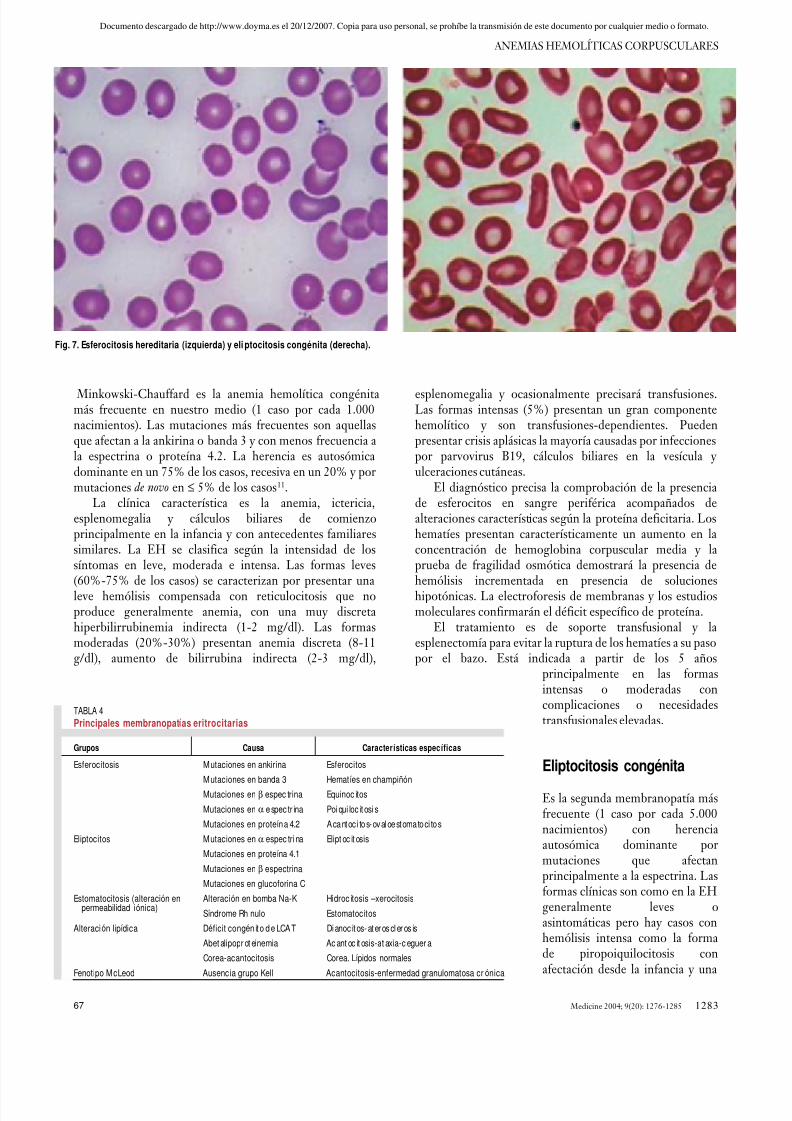
de espectrina α y espectrina β unidas de forma helicoidalque se unen entre sí en dirección caudal. Esta red interiorde espectrina se une a la banda 3 mediante la ankirina y alas glucoforinas mediante la actina-tropomiosina y proteína 4.1. Las alteraciones congénitas de la membranaeritrocitaria más comunes (esferocitosis y eliptocitosis)(fig. 7) afectan a las proteínas citadas, y con menosfrecuencia a la permeabilidad iónica o alteraciones en labicapa lipídica (tabla 4).
Esferocitosis hereditaria
La esferocitosis hereditaria (EH) o enfermedad de
ENFERMEDADES DE LA SANGRE (I)
1282 Medicine 2004; 9(20): 1276-1285 66
TABLA 3
Fármacos con potencial oxidante que pueden provocar crisis hemol í ticasen pacientes afectos de déficit de glucosa-6-fosfato-deshidrogenasa
Grupo farmacológico Acción intensa Acción moderada
Antipiréticos Acetanilida AAS
Analgésicos Acetaminofén
Fenilbutazona
Sulfamidas y sulfonas Sulfametoxazol Sulfadiacina
Sulfapiridina Sulfacitidina
Sulfacetamida Sulfisoxazol
Sulfanilamida
Tiazolsulfona Sulfoxona
Antipalúdicos Primaquina Cloroquina
Pamaquina Quinina
Pentaquina Quinidina
Nitrofuranos Nitrofurantoína
Nitrofurazona
Varios Ácido nalidíxico Anilina
Azul de metileno Vitamina C
Fenilhidrazina Vitamina K
Naftaleno Cloranfenicol
DifenilhidraminaIsoniazida
L-dopa
Probenecid
Pirimetamina
Trimetoprim
Colchicina
Menadiona
AAS: ácido acetilsalic ílico.
FosfolípidosBanda 3
Glicofor ina A
Glicoforina B Glicoforina C
ColesterolActina
Espectrina beta
Proteína 4. 3Proteína 4. 2
Ankirina
Espectrina alf a
Fig. 6. Esquema de l a estructura de la membrana del hematí e.
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.
8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 7/9
Minkowski-Chauffard es la anemia hemolítica congénitamás frecuente en nuestro medio (1 caso por cada 1.000nacimientos). Las mutaciones más frecuentes son aquellasque afectan a la ankirina o banda 3 y con menos frecuencia ala espectrina o proteína 4.2. La herencia es autosómicadominante en un 75% de los casos, recesiva en un 20% y pormutaciones de novo en ≤ 5% de los casos11.
La clínica característica es la anemia, ictericia,esplenomegalia y cálculos biliares de comienzoprincipalmente en la infancia y con antecedentes familiaressimilares. La EH se clasifica según la intensidad de lossíntomas en leve, moderada e intensa. Las formas leves(60%-75% de los casos) se caracterizan por presentar unaleve hemólisis compensada con reticulocitosis que noproduce generalmente anemia, con una muy discretahiperbilirrubinemia indirecta (1-2 mg/dl). Las formasmoderadas (20%-30%) presentan anemia discreta (8-11g/dl), aumento de bilirrubina indirecta (2-3 mg/dl),
esplenomegalia y ocasionalmente precisará transfusiones.Las formas intensas (5%) presentan un gran componentehemolítico y son transfusiones-dependientes. Puedenpresentar crisis aplásicas la mayoría causadas por infeccionespor parvovirus B19, cálculos biliares en la vesícula y ulceraciones cutáneas.
El diagnóstico precisa la comprobación de la presenciade esferocitos en sangre periférica acompañados dealteraciones características según la proteína deficitaria. Loshematíes presentan característicamente un aumento en laconcentración de hemoglobina corpuscular media y laprueba de fragilidad osmótica demostrará la presencia dehemólisis incrementada en presencia de solucioneshipotónicas. La electroforesis de membranas y los estudiosmoleculares confirmarán el déficit específico de proteína.
El tratamiento es de soporte transfusional y laesplenectomía para evitar la ruptura de los hematíes a su pasopor el bazo. Está indicada a partir de los 5 años
principalmente en las formasintensas o moderadas concomplicaciones o necesidadestransfusionales elevadas.
Eliptocitosis congénita
Es la segunda membranopatía másfrecuente (1 caso por cada 5.000nacimientos) con herenciaautosómica dominante pormutaciones que afectanprincipalmente a la espectrina. Lasformas clínicas son como en la EHgeneralmente leves oasintomáticas pero hay casos conhemólisis intensa como la formade piropoiquilocitosis conafectación desde la infancia y una
ANEMIAS HEMOLÍTICAS CORPUSCULARES
Medicine 2004; 9(20): 1276-1285 128367
Fig. 7. Esferocitosis hereditaria (izquierda) y eli ptocitosis congénita (derecha).
TABLA 4
Principales membranopatí as eritrocitarias
Grupos Causa Caracterí sticas especí ficas
Esferocitosis Mutaciones en ankirina Esferocitos
Mutaciones en banda 3 Hematíes en champiñón
Mutaciones en β espec trina Equinoc itos
Mutaciones en α e spec tr ina Poiqui loc itosi s
Mutaciones en proteína 4.2 Acantoci tos-ovaloestomatoci tos
Eliptocitos Mutaciones en α espec tri na Elipt oc it osis
Mutaciones en proteína 4.1
Mutaciones en β espectrina
Mutaciones en glucoforina C
Estomatocitosis (alteración en Alteración en bomba Na-K Hidroc itosis –xerocitosispermeabilidad iónica)
Síndrome Rh nulo Estomatocitos
Alteración lipídica Déficit congén ito d e LCA T Dianoc itos-ateroscl eros is
Abet alipopr ot einemia Ac ant oc it osis-at axia-c eguer a
Corea-acantocitosis Corea. Lípidos normales
Fenotipo McLeod Ausencia grupo Kell Acantocitosis-enfermedad granulomatosa crónica
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.
8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 8/9
marcada inestabilidad del hematíe al calor por mutacioneshomocigotas o doble heterocigoto de la espectrina.
Hemoglobinuria paroxística nocturna
La hemoglobinuria paroxística nocturna (HPN), oenfermedad de Marchiafava-Micheli, es un síndrome
hemolítico debido a un defecto adquirido no-congénito de lamembrana del hematíe. El nombre es debido a los cuadroshemolíticos agudos intravasculares con hemoglobinuria deaparición nocturna. Sin embargo, la HPN cursageneralmente con cuadros hemolíticos crónicos insidiososcon leuco y trombopenia asociada y presencia incrementadade fenómenos trombóticos. Es una enfermedad pocofrecuente con aparición principalmente a partir de la terceradécada de la vida.
Fisiopatogenia
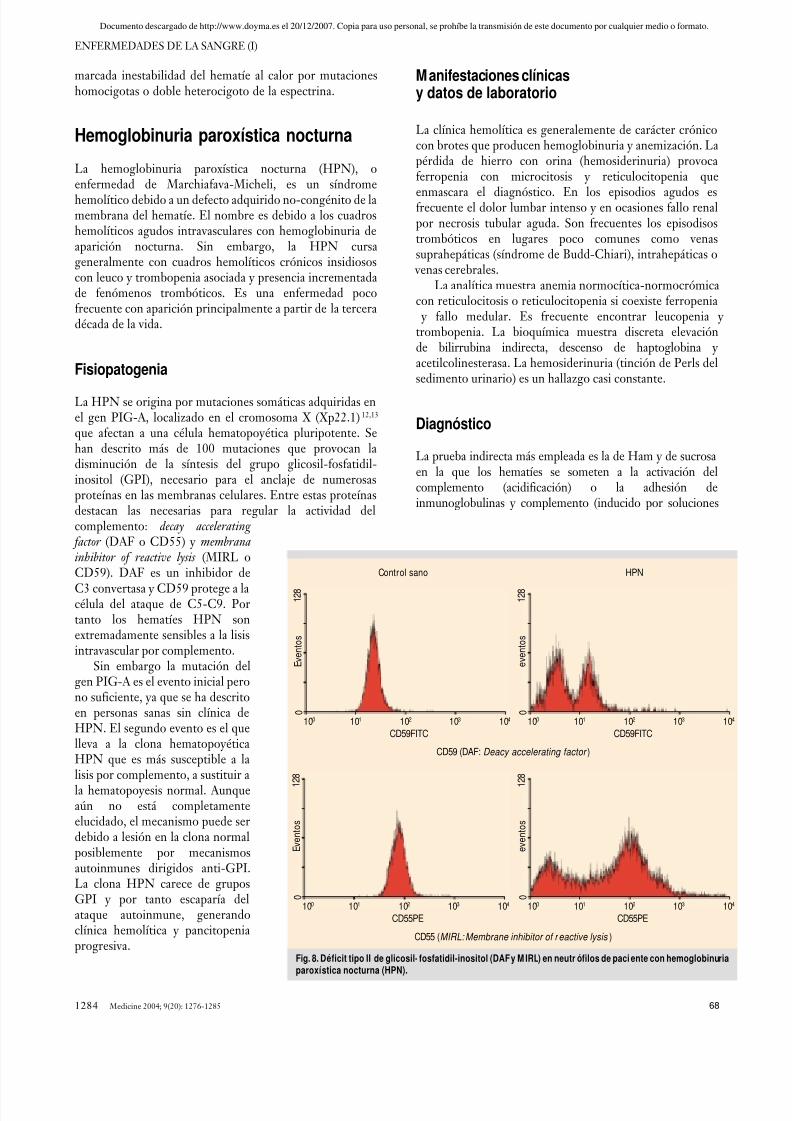
La HPN se origina por mutaciones somáticas adquiridas enel gen PIG-A, localizado en el cromosoma X (Xp22.1)12,13
que afectan a una célula hematopoyética pluripotente. Sehan descrito más de 100 mutaciones que provocan ladisminución de la síntesis del grupo glicosil-fosfatidil-inositol (GPI), necesario para el anclaje de numerosasproteínas en las membranas celulares. Entre estas proteínasdestacan las necesarias para regular la actividad delcomplemento: decay accelerating
factor (DAF o CD55) y membranainhibitor of reactive lysis (MIRL oCD59). DAF es un inhibidor de
C3 convertasa y CD59 protege a lacélula del ataque de C5-C9. Portanto los hematíes HPN sonextremadamente sensibles a la lisisintravascular por complemento.
Sin embargo la mutación delgen PIG-A es el evento inicial perono suficiente, ya que se ha descritoen personas sanas sin clínica deHPN. El segundo evento es el quelleva a la clona hematopoyéticaHPN que es más susceptible a lalisis por complemento, a sustituir ala hematopoyesis normal. Aunqueaún no está completamenteelucidado, el mecanismo puede serdebido a lesión en la clona normalposiblemente por mecanismosautoinmunes dirigidos anti-GPI.La clona HPN carece de gruposGPI y por tanto escaparía delataque autoinmune, generandoclínica hemolítica y pancitopeniaprogresiva.
Manifestaciones clínicasy datos de laboratorio
La clínica hemolítica es generalemente de carácter crónicocon brotes que producen hemoglobinuria y anemización. Lapérdida de hierro con orina (hemosiderinuria) provocaferropenia con microcitosis y reticulocitopenia que
enmascara el diagnóstico. En los episodios agudos esfrecuente el dolor lumbar intenso y en ocasiones fallo renalpor necrosis tubular aguda. Son frecuentes los episodisostrombóticos en lugares poco comunes como venassuprahepáticas (síndrome de Budd-Chiari), intrahepáticas o
venas cerebrales.La analítica muestra anemia normocítica-normocrómica
con reticulocitosis o reticulocitopenia si coexiste ferropenia y fallo medular. Es frecuente encontrar leucopenia y trombopenia. La bioquímica muestra discreta elevaciónde bilirrubina indirecta, descenso de haptoglobina y acetilcolinesterasa. La hemosiderinuria (tinción de Perls delsedimento urinario) es un hallazgo casi constante.
Diagnóstico
La prueba indirecta más empleada es la de Ham y de sucrosaen la que los hematíes se someten a la activación delcomplemento (acidificación) o la adhesión deinmunoglobulinas y complemento (inducido por soluciones
ENFERMEDADES DE LA SANGRE (I)
1284 Medicine 2004; 9(20): 1276-1285 68
Fig. 8. Déficit tipo II de glicosil- fosfatidil-inositol (DAF y M IRL) en neutrófilos de paci ente con hemoglobinuriaparoxí stica nocturna (HPN).
1 2 8
e v e n t o s
0
100 101 102 103 104
CD59FITC
1 2 8
E v e n t o s
0
100 101 102 103 104
CD59FITC
Control sano HPN
CD59 (DAF: Deacy accelerating factor )
CD55 (MIRL: Membrane inhibitor of r eactive lysis )
1
2 8
e v e n t o s
0
100 101 102 103 104
CD55PE
1
2 8
E v e n t o s
0
100 101 102 103 104
CD55PE
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.

8/3/2019 03.005 Anemias hemolíticas corpusculares
http://slidepdf.com/reader/full/03005-anemias-hemoliticas-corpusculares 9/9
isotónicas de sucrosa pero de baja fuerza iónica) en suero delpaciente o de sujetos sanos, produciendo hemólisis en amboscasos. Actualmente la prueba directa diagnóstica es lademostración del déficit parcial (tipo II) o total (tipo III) deCD55 y CD59 en hematíes o leucocitos mediante citometríade flujo (fig. 8). Este déficit se ha observado también aldiagnóstico en algunos pacientes con aplasia medular osíndromes mielodisplásicos.
Tratamiento
El soporte transfusional con concentrado de hematíesdesleucotizados (y mejor aún lavados, además, pero no en
vez de) y la hiperhidratación para evitar el fallo renal, seemplean en las crisis hemolíticas. El empleo de andrógenos,esteroides o gammaglobulina antitimocítica puede producirelevación de las cifras de hemoglobina en algunos pacientes.Si se producen fenómenos trombóticos se emplea heparina(aunque puede inducir crisis hemolíticas) y anticoagulación
oral prolongada. La modalidad curativa es el trasplantealogénico de médula ósea, reservado para pacientes jóveneso formas evolucionadas hacia aplasia medular que dispongande un donante HLA-genéticamente idéntico.
Bibliografía
• Importante •• Muy importante
✔ Metaanálisis
✔ Ensayo clínico controlado
✔ Epidemiología
✔1. Schrier SL, Stanley L. Pathophysiology of thalassemia. Curr OpinHematol 2002;9:123-6.
✔2. • Villegas A, Ropero P , Gon zález FA, Anguita E, Espinos D. T hethalassemia syndromes: molecular characterization in the spanishpopulation. H emoglobin 2001;25:273-83.
✔3. Steensma DP, Viprakasit V, Hendrick A, Goff DK, Leach J, Gibbons RJ,et al. Deletion of the alpha-globin gene cluster as a cause of acquired al-pha-thalassemia in myelodysplastic syndrome. Blood 2004;103:1518-20.
✔4. Gibbons RJ, Pellagatti A, Garrick D, Wood WG, Malik N, Ayyub H, etal. Identification of acquired somatic mutations in the gene encodingchromatin-remodeling factor ATRX in the alpha-thalassemiamyelodysplasia syndrome (ATMDS). Nat Genet 2003;34:446-9.
✔5. Lucarelli G, Andreani M, Angelucci E.The cure of thalassemia by bonemarrow transplantation. Blood Rev 2002;16:81-5.
✔6. •• May C, Rivella S, Chadburn A, Sadelain M. Successfultreatment of murine beta-thalassemia intermedia by transfer of thehuman beta-globin gene. Blood 2002;99:1902-8.
✔7. Atweh GF, DeSimone J, Saunthararajah Y, Fathallah H, Weinberg RS,Nagel RL, et al. Hemoglobinopathies. Hematology (Am Soc Hematol
Educ Program) 2003;14-39.
✔8. Vermylen C. Hematopoietic stem cell transplantation in sickle celldisease. Blood Rev 2003;17:163-6.
✔9. Mehta A, Mason PJ, Vulliamy TJ. Glucose-6-phosphate dehydrogenasedeficiency. Baillieres Best Pract Res Clin Haematol 2000;13:21-38.
10. • Beutler E . G6P D deficiency. Blood 1994;84:3613-36.11. Eber S, Lux SE. Hereditary spherocytosis-defects in proteins that
connect the membrana skeleton to the lipid bilayer. Semin Hematol2004;41:118-41.
12. • Karadimitris A, Luzzatto L. T he cellular patogenesis of paroxysmal nocturnal haemoglobinuria. Leukemia 2001;15:1148-52.
13. Rosti V. The molecular basis of paroxysmal nocturnal hemoglobinuria.Haematologica 2000;85:82-7.
ANEMIAS HEMOLÍTICAS CORPUSCULARES
Medicine 2004; 9(20): 1276-1285 128569
Documento descargado de http://www.doyma.es el 20/12/2007. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato.