“propuesta para validaciÓn de sistemas automÁticos y de...
TRANSCRIPT

INSTITUTO POLITÉCNICO NACIONAL ESCUELA SUPERIOR DE INGENIERÍA MECÁNICA Y ELÉCTRICA
UNIDAD PROFESIONAL “ADOLFO LÓPEZ MATEOS”
TESIS
“PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA”
QUE PARA OBTENER EL TÍTULO DE INGENIERO EN CONTROL Y AUTOMATIZACIÓN
PRESENTA VANESSA JAZMÍN GONZÁLEZ CUÉLLAR
ASESORES: ING. ARTURO ROLANDO ROJÁS SALGADO
ING. JOSÉ ANTONIO MARTÍNEZ HERNÁNDEZ
AGOSTO 2008

AGRADECIMIENTOS
A mis PADRES, María de La Luz Cuéllar y Roberto González por darme la vida, por verme crecer y cuidarme, por haber sembrado en mí esa semillita de seguir estudiando, prepararme y terminar la Carrera, ayudarme con el sustento de mis estudios, por estar siempre ahí cuando los necesito, por la confianza que siempre tuvieron en mí, por todos los valores que me transmitieron, por forjarme la entereza que tengo al día de hoy y por hacer de mi la persona que en estos momentos soy. A mis HERMANOS: IVONNE, ROBERTO y CRISTIAN por apoyarme cuando solicite su ayuda y sus ganas de que siempre saliera adelante. Me siento orgullosa de tenerlos como hermanos y sigamos apoyándonos. A GERARDO, por siempre apoyarme, creer en mí, ayudarme, enseñarme y estar a mi lado en ese sueño de prepararme y tener una Carrera Profesional que ahora es una realidad. Además de haber pasado tantos momentos en Voca 7 y ESIME Zacatenco. A mis AMIGOS con quienes compartí mi época estudiantil y todas esas cosas que la Voca 7 y ESIME fueron testigos. A NELY por apoyarme, por darme su amistad todos estos años, por creer en mí y todos los momentos que juntas pasamos para terminar la Ingeniería. A CECI por todo su apoyo. A mis PROFESORES DE TODAS LAS MATERIAS por transmitirme sus conocimientos, a mis PROFESORES ASESORES: Ing. Arturo Rojas e Ing. José Antonio Martínez por facilitarme la elaboración de esta Tesis. Por último, pero no por eso menos importante al IPN y ESIME por permitirme formar parte de su Comunidad y ser Politécnica de corazón y estar orgullosa de ello y llevar con el mismo orgullo el lema de “La Técnica al Servicio de la Patria”. GRACIAS por todo y por siempre.
Vanessa

Página i
OBJETIVO Analizar los requisitos del Proyecto de Norma Oficial Mexicana PROY-NOM-059-SSA1-2004 para los sistemas automáticos y de cómputo y presentar una propuesta para validar dichos sistemas tomando como referencia la Guía para la validación de sistemas automáticos GAMP4.

Página ii
JUSTIFICACIÓN Un aspecto que está tomando gran importancia en la Industria Farmacéutica son sus sistemas automáticos, los cuales requieren cumplir con ciertos requisitos para su validación. Es importante que los estudiantes en la Carrera de Ingeniería en Control y Automatización conozcan el tema ya que esto les permitirá desempeñarse en ese medio, ya sea como Ingenieros de Validación, Ingenieros de Proyectos, Ingenieros de Soporte o Ingenieros de Servicio. Este conocimiento les permitirá ofrecer sus servicios como Desarrolladores de documentación o Coordinadores / Supervisores de Validación. O en el caso de que formen parte de una empresa de Automatización y Control, que dé servicio y soporte a Laboratorios Farmacéuticos, sean capaces de desarrollar la documentación de diseño y mantenimiento correspondiente a los proyectos que les asignen. Actualmente los Proveedores que dan servicio a la Industria Farmacéutica además de ofrecer un equipo, su instalación y capacitación ofrecen toda la documentación de validación basada en la GAMP4 y es un hecho que no hay Ingenieros con experiencia al respecto. Si lo vemos desde otra perspectiva este documento puede ser la base para crear una Empresa que ofrezca este Servicio. Cabe mencionar que nosotros, como Ingenieros en Control y Automatización conocemos la parte técnica de los Sistemas y Procesos, lo que nos facilita desarrollar este tipo de validaciones. Los cursos formales y literatura acerca de este tema tienen costos un tanto elevados para algunas comunidades estudiantiles y egresados, por lo que mi tesis les permitirá el acceso a este conocimiento.

Página iii
INTRODUCCIÓN La Industria Farmacéutica debe cumplir con ciertos requisitos para la fabricación y la distribución de sus productos con el fin de garantizar la Calidad de los mismos. Cada Agencia emite sus leyes o normas para establecer los requisitos que se deben cumplir para la fabricación de los medicamentos que se comercialicen en su País. El control del cumplimiento de estos requisitos en nuestro País es ejercido por la Secretaría de Salud a través de la Norma Oficial Mexicana NOM-059-SSA1-1993. Dentro de estos requisitos se encuentra la validación de sus Sistemas Computacionales, Equipos Automáticos y la Documentación que esta validación lleva implícita. Los cuáles son los temas que desarrollaré a largo de esta tesis. Esta validación involucra aspectos como: llevar a cabo el ciclo de vida de los sistemas donde entre otras cosas se definen las especificaciones de requerimientos y de diseño, protocolos de calificación, procedimientos y controles de cambios. Durante el desarrollo de los temas mencionare términos internacionales usados en inglés ya que son empleados en este contexto. La lectura de esta tesis le permitirá conocer el proceso de validación de una manera desglosada, práctica, sencilla y con un lenguaje de fácil comprensión. También le dará el punto de partida para profundizar aún más en el tema y adoptar las regulaciones de otros Países. La tesis se encuentra estructurada en cuatro capítulos, en ellos se destacan los conceptos relevantes que permiten el desarrollo de esta tesis. En el capítulo primero se describen los antecedentes de la validación de sistemas automáticos y de cómputo desde los años ochentas hasta nuestros días, además del marco normativo nacional e internacional (EU, Australia, Argentina) acerca de este tema. En el capítulo segundo se presentan los conceptos teóricos acerca de la Validación, el Ciclo de Vida y la GAMP (Good Automated Manufacturing Practice, Buenas Prácticas de Automatización para Manufactura). Se detalla la definición de la Validación de Sistemas desde varios puntos de vista de la normatividad vigente y se introduce de manera general a este proceso. En cuanto al Ciclo de Vida se menciona su definición, los elementos que la conforman (fases y entregables) y sus modelos. Respecto a la GAMP se da una descripción general de su objetivo, alcance, su visión general para la validación y los procesos de validación para los Sistemas de Tecnología de Información y para los Sistemas de Control de Procesos.

Página iv
En el capítulo tercero se encuentra la descripción de la propuesta que hago para la Validación de Sistemas Automáticos y de Cómputo en la Industria Farmacéutica. Desde la elaboración del Plan de Validación, el Plan del Proyecto y Calidad, las Especificaciones de Requerimientos y de Diseño, para después la redacción y ejecución de Pruebas llevando la rastreabilidad de los requerimientos hasta llegar a la elaboración del Reporte de Validación. Además de tomar en cuenta el mantenimiento del estado validado del equipo o sistema y los procedimientos de operación. Y por último la elaboración del Plan de Retiro cuando el sistema salga de operación. El capítulo cuarto describe el Plan de implementación de mi propuesta y los beneficios que aportará. Además en el desarrollo de la tesis se encuentran figuras, tablas y formatos para presentar la propuesta de una manera esquemática, así como también se cuenta con conclusiones, glosario, acrónimos y bibliografía.

Página v
ÍNDICE OBJETIVO ..................................................................................................................................... i
JUSTIFICACIÓN........................................................................................................................ ii
INTRODUCCIÓN....................................................................................................................... iii
ÍNDICE.............................................................................................................................................v
CAPÍTULO 1. ANTECENTES Y MARCO NORMATIVO.........................................................1 1.1 Antecedentes....................................................................................................................2 1.2 Marco Normativo ............................................................................................................3
1.2.1 Normatividad Farmacéutica. ..................................................................... 3 1.2.2 Normatividad Farmacéutica en México. ................................................... 5
CAPÍTULO 2. VALIDACIÓN, CICLO DE VIDA Y GAMP (GOOD AUTOMATED MANUFACTURING PRACTICE, BUENAS PRÁCTICAS DE AUTOMATIZACIÓN PARA MANUFACTURA)......................................................................................................7 2.1 Validación........................................................................................................................8
2.1.1 Definición de Validación de Sistemas. ..................................................... 8 2.1.2 Introducción al proceso de Validación de Sistemas.................................. 9
2.2 Ciclo de Vida. ................................................................................................................10 2.2.1 Definición de Ciclo de Vida.................................................................... 10 2.2.2 Elementos de un Ciclo de Vida. .............................................................. 10 2.2.3 Modelos de Ciclo de Vida....................................................................... 11 2.2.4 Fases del Ciclo de Vida........................................................................... 14
2.3 La GAMP4 como herramienta para la validación de sistemas automáticos..................15 2.3.1 Objetivo de GAMP4 ............................................................................... 15 2.3.2 Alcance.................................................................................................... 15 2.3.3 Visión general de validación en la GAMP4............................................ 15 2.3.4 Actividades y desarrollo de la validación. .............................................. 17 2.3.5 Responsabilidades de usuario. ................................................................ 19 2.3.6 Proceso de validación de Sistemas IT. .................................................... 21 2.3.7 Proceso de validación de Sistemas de Control de Procesos.................... 23
CAPÍTULO 3. PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA. ........................................28 3.1 Análisis. .........................................................................................................................29
3.1.1 Correspondencia entre mi Propuesta con el Proyecto de Norma PROY-NOM-059-SSA1-2004, la GAMP4 y NMX-CC-9001-IMNC-2000. ..... 31

Página vi
3.2 Plan de Validación.........................................................................................................37 3.3 Especificación de requerimientos de usuario (URS) y Especificación funcional (FS)..43 3.4 Plan del Proyecto y Calidad (QPP, Quality and Project Plan).......................................54 3.5 Matriz de Rastreabilidad de Requerimientos.................................................................61 3.6 Especificación de Diseño (DS, Desing Specification)...................................................66 3.7 Pruebas al Equipo o Sistema..........................................................................................74
3.7.1 Principios fundamentales que deben seguir las pruebas ......................... 78 3.7.2 Pruebas de Revisión de Diseño. .............................................................. 79 3.7.3 Pruebas de Calificación de Instalación (Installation Qualification, IQ).. 82 3.7.4 Pruebas de Calificación de Operación (Operational Qualification, OQ).83 3.7.5 Pruebas de Calificación de Desempeño (Performance Qualification,
PQ)………………………...……………………………………………83 3.8 Registro de Capacitación. ..............................................................................................89 3.9 Reporte de Validación. ..................................................................................................89 3.10 Mantenimiento del Estado Validado. ............................................................................92 3.11 Retiro. ............................................................................................................................97 3.12 Buenas Prácticas. .........................................................................................................102
3.12.1Buenas Prácticas de Documentación. ................................................... 103 3.12.2Buenas Prácticas de Prueba. ................................................................. 103 3.12.3Buenas Prácticas de Ingeniería. ............................................................ 105
CAPÍTULO 4. IMPLEMENTACIÓN Y BENEFICIOS. ..........................................................106 4.1 Recursos humanos para la implementación.................................................................107 4.2 Inversión para la implementación................................................................................108
4.2.1 Costos Directos ..................................................................................... 108 4.3 Compromiso de las Gerencias y de la Dirección.........................................................110 4.4 Plan de Implementación. .............................................................................................111 4.5 Beneficios de la validación..........................................................................................113
4.5.1 Beneficios de negocio. .......................................................................... 113 4.5.2 Beneficios sociales. ............................................................................... 117 4.5.3 Beneficios al aplicar mi propuesta ........................................................ 117
CONCLUSIONES.........................................................................................................................119
GLOSARIO Y ACRÓNIMOS .....................................................................................................120
GLOSARIO ...................................................................................................................................120
ACRÓNIMOS ...............................................................................................................................123
BIBLIOGRAFÍA...........................................................................................................................125

CAPÍTULO 1. ANTECENTES Y MARCO NORMATIVO
CAPÍTULO 1.
“ANTECEDENTES Y MARCO NORMATIVO”
Objetivo particular: Describir los antecedentes que dieron lugar al requerimiento de Validación de Sistemas automáticos y de Cómputo y establecer las bases y el contexto legal que envuelve tal requerimiento.

ANTECEDENTES Y MARCO NORMATIVO
Página 2 de 125
Capítulo 1. Antecedentes y Marco Normativo Para iniciar con el tema de la Validación de Sistemas Automáticos y de Cómputo empezaré por describir sus antecedentes y la normatividad que regula la Industria Farmacéutica.
1.1 ANTECEDENTES.
En los años ochentas dentro de la normatividad se mencionaba de manera general requerimientos de validación de equipos y software. Pero fue hasta finales de los años ochentas y principios de los noventas donde la validación de sistemas automáticos y de cómputo tomó gran importancia que la que se había tenido hasta ese momento aunque en las regulaciones ya estaban establecidos dichos requerimientos. Esa importancia fue tomada debido a que a pesar de que las guías concernientes al tema habían estado disponibles ya por algún tiempo, estos sistemas en la Industria Farmacéutica habían sido sometidos a un bajo escrutinio regulatorio en comparación con otras áreas y la interpretación de las guías regulatorias para este ámbito era menos madura que en otras áreas convencionales. El interés por estos sistemas dentro del ramo farmacéutico se incremento gracias al aumento en su uso y su complejidad, por lo tanto fue necesario mejorar el entendimiento y la interpretación de las regulaciones. Además de una mayor comunicación dentro de la Industria Farmacéutica, así como también con los Proveedores y las Agencias Regulatorias. Lo anterior aunado a los cambios macroeconómicos y de globalización de esos años como el Acuerdo General sobre Aranceles y Comercio (GATT, General Agreement on Tariffs and Trade), el Tratado de Libre Comercio (TLC) y la creación de la Organización Mundial del Comercio (OMC) han hecho que esto tenga importancia para la importación y exportación de estos productos. Como dato interesante: entre 1992 y 1998 la FDA (Food and Drug Administration) analizó 3140 retiros del mercado (recalls) de dispositivos médicos, encontrando que 242 de ellos (7.7 %) se atribuían a fallas de software. Dentro de estos 242 casos de software, 192 (79%) fueron causados por defectos de software que se producían por hacer cambios al software después de que la producción y distribución de los productos era iniciada. Estos datos estadísticos refuerzan la importancia en la atención y cumplimiento de los requerimientos en la validación de sistemas automáticos y de cómputo. Ahora la mayoría de las farmacéuticas están trabajando en cumplir con este requerimiento regulatorio de validación de sistemas automáticos y de cómputo, el cual es solo una parte de toda la regulación que involucra la industria de este ramo. En México por ejemplo hasta este momento la norma vigente NOM-059-SSA1-1993 contempla la parte de validación pero no ahonda en la descripción del proceso. Esta

ANTECEDENTES Y MARCO NORMATIVO
Página 3 de 125
importancia se está dando en el proyecto de Norma PROY-NOM-059-SSA1-2004 donde la sección de validación ya expone los requerimientos que se necesitan para la validación de equipos y sistemas críticos e involucra los sistemas computarizados. En Australia y Argentina introducen estos requerimientos en 2002 y 2005, respectivamente. La validación de equipo de cómputo en Estados Unidos es un requerimiento desde 1997 de la regulación de Sistema de Calidad y de CFR 21 (Code of Federal Regulations) Parte 11 correspondiente a Firmas y Registros Electrónicos. Actualmente existe el Fórum GAMP (Good Automated Manufacturing Practice) que es el grupo que se formó para promover el entendimiento de las regulaciones de equipos automáticos y de cómputo. En nuestros días este Fórum es un subcomité técnico de ISPE (International Society of Pharmaceutical Engineering) y una de sus prioridades, cuando se creó, fue el establecimiento de Guías para Proveedores de sistemas automáticos a la Industria Farmacéutica. La versión preliminar o borrador de estas guías estuvo disponible para comentarios de los Proveedores de dichos sistemas y otros interesados en Marzo 1994. Desde entonces dichas guías se han ido mejorando y actualizando de tal manera que son unas de las Guías más reconocidas a nivel mundial para el cumplimiento de los requerimientos a nivel Internacional. El apego a estas guías facilita el cumplimiento con la FDA y otras Agencias Europeas que son calificadas como las más exigentes en este ámbito. Ahora abordemos, en la siguiente sección, las características de los requerimientos de la normatividad farmacéutica para ubicarnos en este marco.
1.2 MARCO NORMATIVO
1.2.1 Normatividad Farmacéutica.
Cada País es responsable de emitir su normatividad en cuestión de fabricación de medicamentos con el fin de que se garantice la calidad de los mismos para su población. En México le corresponde a la Secretaría de Salud establecer los requerimientos para tal fin y lo hace empleando como marco de referencia la Norma Oficial Mexicana NOM-059-SSA1-1993, Buenas Prácticas de fabricación para establecimientos de la industria químico farmacéutica dedicados a la fabricación de medicamentos. Las secciones 5.6.3, 5.7.4 y 9.11 especifican algunos aspectos de validación. Sin embargo se tiene el Proyecto de Norma Oficial PROY-NOM-059-SSA1-2004, donde se detallan un poco más estos requerimientos en las secciones 10.5 y 14. En las cuales se establecen los requerimientos para los Equipos automáticos, mecánicos y electrónicos y para la Validación. En Estados Unidos la ley referente a los Alimentos y Drogas es el CFR-21 (CFR en inglés es Code of Federal Regulations). La Agencia regulatoria de este País es la

ANTECEDENTES Y MARCO NORMATIVO
Página 4 de 125
FDA (Food and Drug Administration). Las secciones que corresponden a las Buenas Prácticas de Manufactura para productos farmacéuticos son Parte 210 y 211, la de firmas y registros electrónicos es la Parte 11 y la de Validación de Software y equipos automáticos se encuentra dentro de la Parte 820. Entre otros países, por ejemplo, podemos citar Australia con la TGA (Therapeutic Goods Administration) y su código Australian Code of Good Manufacturing Practice for Medicinal Products. Donde en el Anexo 11 menciona los requerimientos a cumplir para los sistemas computarizados y en el Anexo 15 los referentes a la Calificación y Validación. Y Argentina con la ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica) por medio de la Disposición 2819/2004, especifica los requerimientos para la Calificación y Validación en la sección 4 y en el Anexo II. La siguiente lista presenta algunos de los requerimientos que son solicitados en las regulaciones de México, EU, Argentina y Australia.
Elaboración de un Plan de Validación. Capacitación de Personal. La validación debe ser considerada como parte del ciclo de vida de un
sistema computarizado. Especificaciones de Diseño. El software es un componente crítico, que debe ser producido conforme
un sistema de Aseguramiento de Calidad. Seguridad para el acceso a los sistemas (control de acceso y rastreo de
auditoría). Control de cambios. Respaldos, resguardo y recuperación. Rastreabilidad. Atención de fallas y problemas. Calificación de Diseño. Calificación de Instalación. Calificación de Operación. Calificación de Desempeño. Mantenimiento del estado validado. Control de cambios. Revalidación. Auditoría de Rastreo. Firmas y registros electrónicos.
Para el contexto de esta tesis tomaremos como base la Normatividad Mexicana, aunque en algunos puntos hagamos referencia a otros aspectos importantes a nivel internacional.

ANTECEDENTES Y MARCO NORMATIVO
Página 5 de 125
1.2.2 Normatividad Farmacéutica en México.
Dentro de la NOM-059-SSA1-1993 indica que se deben llevar a cabo estudios de validación para los sistemas involucrados en los procesos de fabricación. Y con respecto a los equipos automáticos y electrónicos señala que deben ser calibrados, inspeccionados y asegurar y respaldar la información que manejan. En esta versión vigente de la Norma se puede observar que no se desglosan los requerimientos necesarios para cumplir con la validación señalada, pero actualmente existe el Proyecto de Norma Oficial PROY-NOM-059-SSA1-2004 en el cual ya se toman en consideración estos puntos. El PROY-NOM-059-SSA-2004 específica que los equipos, sistemas críticos y computacionales que impacten la calidad del producto deben ser calificados y validados. Para llevar a cabo esta validación nos presenta los requerimientos que la integran, a grandes rasgos son los siguientes.
Hacer un Plan de la validación Documentación Calificación Llevar un control de cambios en este sentido Mantener el Sistema Validado
Es un hecho que si se comparan los requerimientos que marca el Proyecto de Norma Mexicana para los sistemas computacionales con los que solicitan las Normas de Australia, Argentina y/o EU, que estoy tomando como referencia, resultan muy básicos y todavía ambiguos. La sección del proyecto de norma mexicana que hace referencia a los sistemas computacionales es la 14.8 y solo hace referencia a lo siguiente:
14.8 Sistemas computacionales. 14.8.1 Deben validarse los sistemas y aplicaciones computacionales relacionados con: 14.8.1.1 Transferencias de materiales y producto. 14.8.1.2 Disposición de materiales y producto. 14.8.1.3 Control de procesos y análisis. 14.8.1.4 Control de sistemas críticos.
Y si lo comparamos por ejemplo con la de Australia que tiene todo un anexo dedicado al punto de sistemas computarizados donde tiene 22 puntos con los requerimientos respecto al tema vemos que el proyecto de norma sigue un poco deficiente con el cumplimiento a nivel internacional. Si una empresa mexicana quisiera exportar a EU, Australia y/o Argentina, por ejemplo, no le bastaría cumplir con los requisitos de la mexicana sino tendría

ANTECEDENTES Y MARCO NORMATIVO
Página 6 de 125
muchos puntos pendientes por cubrir para alcanzar los requerimientos de otras legislaciones. Por ejemplo el PROY-NOM-059-SSA-2004 no contempla requerimientos acerca de auditoría de rastreo (audit trail) para los sistemas computarizados, los cuales si son mencionados por la TGA y FDA. Además hasta este PROY-NOM-059-SSA-2004 se solicita la existencia de un Plan para definir los requerimientos del producto, procesos, sistemas críticos y servicios y que su diseño esté conforme a los requerimientos definidos. Este punto es muy importante ya que está directamente relacionado con la ISO 9001:2000 en la sección de Diseño.
Después de abordar el esquema regulatorio me resta mencionar que el desarrollo de la tesis está basado para el cumplimiento con el Proyecto de Norma PROY-NOM-059-SSA1-2004 que entrará en vigor en un futuro y que finalmente es donde ya se contemplan los requerimientos para la validación de sistemas de cómputo y sistemas automáticos; además de que ofrecerá la base para el cumplimiento con otras regulaciones a nivel internacional.

CAPÍTULO 2. VALIDACIÓN, CICLO DE VIDA Y GAMP (GOOD AUTOMATED MANUFACTURING PRACTICE, BUENAS PRÁCTICAS DE AUTOMATIZACIÓN PARA
MANUFACTURA)
CAPÍTULO 2.
“VALIDACIÓN, CICLO DE VIDA Y GAMP (GOOD AUTOMATED MANUFACTURING
PRACTICE, BUENAS PRÁCTICAS DE AUTOMATIZACIÓN PARA MANUFACTURA).”
Objetivos particulares:
Introducir al proceso de validación de los sistemas automáticos y de cómputo.
Describir el Ciclo de Vida. Introducir a la guía GAMP4 (en Inglés Good Automated
Manufacturing Practice, en español Buenas Prácticas de Automatización para Manufactura).

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 8 de 125
Capítulo 2. Validación, Ciclo de Vida y GAMP (Good Automated Manufacturing Practice, Buenas Prácticas de Automatización para Manufactura). Después de conocer los antecedentes generales y el marco normativo de la validación de sistemas computarizados, abordemos la definición de la Validación, el tema del Ciclo de Vida y los objetivos de la Guía de las Buenas Prácticas de Automatización para Manufactura.
2.1 VALIDACIÓN.
2.1.1 Definición de Validación de Sistemas.
Hablar de validación comprende varios aspectos del proceso de manufactura farmacéutica desde los procesos, sistemas, equipos y servicios hasta la limpieza de las áreas. Pero empecemos por ver que hay alrededor de la palabra Validación. A continuación se muestran algunas definiciones del Proceso de Validación. La NOM-059-SSA1-1993 nos indica que la validación es “la evidencia documentada que demuestra que a través de un proceso específico se obtiene un producto que cumple consistentemente con las especificaciones y los atributos de calidad establecidos”. Por otro lado la definición de Validación que nos dan en la GAMP4 es “establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes”. En español “establecer evidencia documentada que provea un alto grado de aseguramiento de que un proceso específico producirá consistentemente un producto cumpliendo con especificaciones y atributos de calidad predeterminados”. En el Código Australiano de Buenas Prácticas de Manufactura para Productos Medicinales podemos encontrar que el Proceso de Validación es “the documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its predetermined specifications and quality attributes”. En español “La evidencia documentada de que el proceso opera dentro de parámetros establecidos, y puede desempeñarse efectiva y reproduciblemente para producir un producto medicinal conociendo de manera predeterminada sus especificaciones y atributos de calidad”.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 9 de 125
De las definiciones anteriores se puede rescatar las siguientes ideas:
La validación es una evidencia documentada de un proceso. A través de ese proceso se obtiene un producto, que en este caso es un
producto farmacéutico. Este proceso y producto cumplen con especificaciones y atributos de
calidad predeterminados. El proceso de fabricación de ese producto debe ser eficaz y reproducible.
Por lo tanto, reuniendo las ideas anteriores, la validación es la evidencia documentada de que a través de un proceso eficaz y reproducible, un producto farmacéutico es obtenido de acuerdo a las especificaciones, atributos de Calidad predeterminados y principios de buenas prácticas de manufactura. La definición está de alguna manera apegada al proceso de obtención un producto, pero sin embargo si hablamos de un sistema de manera general podemos decir que la validación de sistemas es la generación de evidencia documentada que nos proporcione un alto grado de aseguramiento de que el sistema funcionará como se requiere cuando se ponga en uso.
2.1.2 Introducción al proceso de Validación de Sistemas.
La importancia de validar un sistema es que nos asegura su funcionamiento y el beneficio que se obtiene es el Cumplimiento Legal. La validación involucra que los sistemas críticos y los equipos de producción deben ser probados tomando en cuenta su diseño, su instalación y su operación. Cuando se tiene un sistema nuevo por instalar, su validación será de una manera gradual y tomando en cuenta algo muy importante: la participación y compromiso del usuario y del proveedor. En otras palabras validar un sistema involucra:
Tener su Plan de validación y aseguramiento de la Calidad. Desarrollo de documentos donde se especifique los requerimientos y el
diseño del sistema. Desarrollo de Planes de Prueba y Protocolos de Calificación de Instalación,
Operación y Desempeño. Sus cambios deben estar controlados. Desarrollo de PNO’s para su administración. Ser un sistema seguro, es decir, que tenga protección que no permita la
modificación o perdida de datos. Tener respaldo de la información. Su documentación debe ser completa, exacta, ordenada y disponible.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 10 de 125
Mantenimiento y seguimiento del estado validado. Plan de Retiro.
2.2 CICLO DE VIDA.
En algunos casos se hace referencia al termino Ciclo de Vida del Sistema y en otros al termino Ciclo de Vida del proyecto, en realidad cualquiera de los dos términos son correctos depende del contexto en donde se emplee sólo habrá que ver si en realidad se tiene un proyecto como tal o si se está validando a un sistema que no pertenece a un proyecto pero en ambos se está utilizando el concepto de Ciclo de Vida como herramienta para su desarrollo.
2.2.1 Definición de Ciclo de Vida.
El Ciclo de Vida se define como un conjunto de fases sucesivas empleadas para generar un producto, un proceso o un servicio. Cada una de estas fases sucesivas se divide a su vez en actividades específicas para el cumplimiento del objetivo de cada fase. El agrupamiento de estas actividades depende, además del objetivo de cada fase, del producto, proceso o servicio final que se pretende obtener y de los requerimientos que haya que cumplir para su obtención.
El dividir los proyectos o los procesos en fases nos asegura la reducción de la complejidad de los mismos. El empleo del concepto de Ciclo de Vida nos permite:
Definir las actividades a ser ejecutadas. Tener un mayor control sobre los tiempos. Subcontratar servicios (para el desarrollo de actividades específicas). Controlar el logro del objetivo principal mediante la obtención de objetivos
específicos. Tomar decisiones al término de cada fase en base a los resultados
obtenidos.
2.2.2 Elementos de un Ciclo de Vida. Los elementos que integran un Ciclo de Vida son:1
Fase (s): Una fase dentro del Ciclo de Vida es un conjunto de actividades
relacionadas entre sí con un objetivo dentro del desarrollo de un proyecto o proceso.
El detalle en la definición de cada fase dependerá del tamaño del proyecto y de su complejidad. El propósito de la división en fases es que el contenido de cada una sea manejable, controlado y con una relación lo más simple posible con las demás fases.
1 Elementos del Ciclo de Vida según artículo de Ciclo de Vida en la página de Internet http://www.getec.etsit.upm.es/docencia/gproyectos/planificacion/cvida.htm

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 11 de 125
Entregables: Son los productos intermedios que se deben generar en cada fase. En inglés el término utilizado para los entregables es “deliverables”.
Estos productos intermedios pueden ser equipos, componentes,
documentos, programas, software, manuales, entre otros. Los entregables al final de cada fase del proyecto nos ayudan a evaluar la marcha del proyecto en cumplimiento con los requisitos previamente establecidos.
2.2.3 Modelos de Ciclo de Vida.
Los modelos de Ciclo de Vida son:1
Ciclo de Vida Lineal.
Este modelo consiste en descomponer el proyecto o el proceso en fases que suceden de manera lineal. Cada fase se realiza una sola vez y siempre tienen una secuencia, cada fase tiene una fase o actividad precedente y una fase o actividad que le procede. Este modelo consta de un solo ciclo.
Una de las características de este modelo es que el proyecto o proceso se pueda dividir de manera que cada fase no necesite realimentación de otra, es decir, que el resultado obtenido de una fase no realimente a una fase anterior. Aunque es valida la realimentación correctiva. Este modelo es el más empleado por su sencillez. Ver figura 2.1.
Figura 2.1. Ejemplo de Modelo de Ciclo de Vida Lineal.
1 Modelos del Ciclo de Vida según artículo de Ciclo de Vida en la página de Internet http://www.getec.etsit.upm.es/docencia/gproyectos/planificacion/cvida.htm

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 12 de 125
Ciclo de Vida con Prototipado.
Este modelo es empleado cuando no se conocen con exactitud las especificaciones de un producto o proyecto y es necesario tener una retroalimentación de los resultados obtenidos, evaluarlos, hacer correcciones y entonces obtener las especificaciones completas. En este modelo regularmente se definen especificaciones iniciales para hacer un prototipo (un producto parcial y provisional), se desarrolla y en base a los resultados se repiten las fases de definición, diseño y construcción; con lo que llevan a cabo dos ciclos. Comúnmente este modelo es empleado en desarrollo avanzado. Ver figura 2.2.
Figura 2.2 Ejemplo de Modelo de Ciclo de Vida Prototipado.
Ciclo de Vida en espiral.
Este modelo es empleado cuando no basta con una sola evaluación del prototipo, como en el modelo anterior, de modo que es necesario hacer más evaluaciones y correcciones para eliminar incertidumbres en las especificaciones y diseño del sistema, producto o proyecto. En este modelo se tienen más de dos ciclos, y en cada uno de ellos se obtiene un prototipo mejorado hasta alcanzar el prototipo deseado, ya que en cada ciclo se van resolviendo las dudas de las especificaciones.
El modelo se representa por un bucle en espiral, donde los cuadrantes son, habitualmente, las fases de desarrollo del prototipo. En cada vuelta el prototipo gana “madurez” hasta que en una vuelta la evaluación es aprobada y se abandona el bucle. Ver figura 2.3.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 13 de 125
Figura 2.3. Ejemplo de Modelo de Ciclo de Vida en Espiral.
El ciclo de vida se puede aplicar en distintos campos, por ejemplo, si se aplica en una investigación básica el resultado esperado son los conocimientos científicos y no necesariamente se tienen diversos entregables. O por ejemplo en el caso de una investigación científica avanzada ya podría hablarse de una división de actividades en ciertas fases con objetivos bien definidos al término de cada una de ellas.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 14 de 125
2.2.4 Fases del Ciclo de Vida.1
El ciclo de vida se clasifica en fases o etapas, a continuación muestro las que nos sugieren en el artículo Ciclo de Vida en la página de Internet citada al pie de esta página.
Fase de definición (¿qué hacer?)
Estudio de viabilidad. Conocer los requisitos que debe satisfacer el sistema (funciones y
limitaciones de contexto). Asegurar que los requisitos son alcanzables. Formalizar el acuerdo con los usuarios. Realizar una planificación detallada.
Fase de diseño (¿cómo hacerlo? Soluciones en costo, tiempo y calidad) Identificar soluciones tecnológicas para cada una de las funciones del
sistema. Asignar recursos materiales para cada una de las funciones. Proponer (identificar y seleccionar) subcontratar. Establecer métodos de validación del diseño. Ajustar las especificaciones del producto.
Fase de construcción Generar el producto o servicio pretendido con el proyecto. Integrar los elementos subcontratados o adquiridos externamente. Validar que el producto obtenido satisface los requisitos de diseño
previamente definidos y realizar, si es necesario, los ajustes necesarios en dicho diseño para corregir posibles lagunas, errores o inconsistencias.
Fase de mantenimiento y operación Operación: asegurar que el uso del proyecto es el pretendido. Mantenimiento (nos referimos a un mantenimiento no habitual, es decir,
aquel que no se limita a reparar averías o desgastes habituales -este es el caso del mantenimiento en productos software, ya que en un programa no cabe hablar de averías o de desgaste):
1 Fases del Ciclo de Vida según artículo de Ciclo de Vida en la página de Internet http://www.getec.etsit.upm.es/docencia/gproyectos/planificacion/cvida.htm

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 15 de 125
2.3 LA GAMP4 COMO HERRAMIENTA PARA LA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS.
Dentro del ámbito farmacéutico se ha desarrollado una herramienta que proporciona una ayuda para la validación y el cumplimiento de las expectativas regulatorias actuales para los sistemas automáticos, es decir, es una guía para validar dichos sistemas. Esta guía es llamada GAMP por sus siglas en Inglés Good Automated Manufacturing Practice, en español podemos decir que son las Buenas Prácticas de Automatización para Manufactura.
Esta guía surgió a partir de la necesidad de la interpretación y entendimiento de las regulaciones, debido a que las reglas estaban establecidas pero se requería una mayor comunicación e uniformidad en los conceptos.
Por otro lado también proporciona una guía para el desarrollo y mantenimiento de las Buenas Prácticas, ya que en su conjunto cubre todos los requerimientos para la validación y cumplimiento con las Buenas Prácticas de Manufactura.
Una de las características de la GAMP4 es que no está dirigida a un solo grupo de personas. Está dirigida a usuarios y proveedores de sistemas automáticos, ambos de la Industria Farmacéutica.
2.3.1 Objetivo de GAMP4
Su objetivo es asistir a las Industrias que se dedican al cuidado de la Salud, incluyendo Farmacéutica, Biotecnológica y de Dispositivos Médicos, a lograr la validación y cumplimiento de los sistemas automáticos.
2.3.2 Alcance
La GAMP4 proporciona un alcance general para todos los sistemas automáticos, aunque reconoce que todos los sistemas no son iguales y tienen diferencias significantes.
Esta guía aplica para desarrollar el Ciclo de Vida de sistemas nuevos. Los sistemas existentes no entran en el alcance de esta guía.
Por otro lado la guía especifica que no todas las actividades descritas aplican a todos los proyectos, ya que el líder de proyecto y/o el área usuaria deberán de especificar los requerimientos de validación necesarios para cada sistema.
2.3.3 Visión general de validación en la GAMP4
La GAMP4 nos dice que tradicionalmente y observándola de manera general, la validación ha consistido en los siguientes puntos:

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 16 de 125
IQ Calificación de la Instalación por sus siglas en Inglés Installation Qualification.
OQ Calificación de Operación por sus siglas en Inglés Operational Qualification.
PQ Calificación de Desempeño por sus siglas en Inglés Performance Qualification.
DQ Calificación de Diseño por sus siglas en Inglés Design Qualification.
Y nos presentan un esquema general de las actividades de validación que se muestra a continuación:
Especificación y acuerdos de lo que se requiere.Elaborar las revisiones de Diseño.
PLANEACIÓN
ESPECIFICACIÓN
PLAN DEPRUEBAS
(IQ, OQ,PQ)
PRUEBAS(IQ, OQ,PQ)
REVISIÓN DERESULTADOS
La Planeación se llevará a cabo mediantela elaboración del Plan de Validación.
Elaboración de un documento donde sedescriba como se probará el sistema.
Ejecución de pruebas y colección deresultados.
Conclusión de resultados para mostrar que elsistema funciona como fue especificado.
Figura 2.4 Actividades Generales de Validación.
También nos muestra mediante la siguiente figura 2.5 la estructura básica de las especificaciones y las calificaciones. Donde podemos observar la relación entre los tipos de calificación para verificar los requerimientos previamente establecidos y diseñados en la etapa correspondiente.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 17 de 125
Verifica
Figura 2.5 Estructura básica y relación de Especificaciones y Pruebas de Calificación.
2.3.4 Actividades y desarrollo de la validación.
La GAMP4 nos presenta mediante la figura 2.6 el concepto de la validación, mostrando las actividades de validación y de desarrollo en cada etapa del ciclo de vida. En dicha figura se puede observar a la par, las actividades que hay que desarrollar en cierta etapa del ciclo de vida del proyecto y a que parte de la validación corresponden dichas actividades.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 18 de 125
REVISIONES DE CÓDIGO
Actividades a desarrollar
Fases en el Ciclo de Vida
Actividades de Validación
Especificación de Requerimientos de UsuarioEspecificaciones Funcionales
Especificación de Diseño de HardwareEspecificación de Diseño de SoftwareEspecificación de Diseño del Modulo de SoftwareEspecificaciones Mecánicas y EléctricasEspecificación de Diseño de RedEspecificación de Configuración del Paquete o Empaque
Manufactura y Ensamble del HardwareMódulos de Código de SoftwareManufactura y Ensamble de EquipoManufactura y Ensamble de Red
Pruebas de hardwarePruebas del Módulo de SoftwarePruebas del Integración de SoftwarePruebas de equipoPruebas de la Configuración del Paquete o Empaque
Instalación de HardwareInstalación de SoftwareInstalación de EquipoInstalación de RedPruebas de aceptación de hardwarePruebas de aceptación de red
Pruebas de aceptación de Sistema
Mantenimiento
Control de Cambios
PLANEACIÓN Y ESPECIFICACIÓN
DISEÑO
CONSTRUCCIÓN
PRUEBA
INSTALACIÓN
PRUEBAS DE INSTALACIÓN
OPERACIÓN
PLAN DE VALIDACIÓN
EVALUACIÓN DEL PROVEEDOR
REVISIONES DE DISEÑO
REVISIONES DE DISEÑO
REVISIONES DE CONSTRUCCIÓN
MONITOREO DEL PROVEEDOR
CALIFICACIÓN DE INSTALACIÓN
CALIFICACIÓN OPERACIONAL
CALIFICACIÓN DE DESEMPEÑO
REPORTE DE VALIDACIÓN
MANTENIMIENTO DEL ESTADO
VALIDADO
Figura 2.6 Actividades de Validación contra el Desarrollo de Ciclo de Vida.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 19 de 125
La figura anterior 2.6 muestra de manera general las actividades de validación, pero por otro lado, la GAMP4 para abordar el proceso de validación hace una diferencia entre los sistemas de Tecnología de la Información (IT) y los Sistemas de Control de Proceso. Para el propósito de dicha guía un Sistema de Tecnología de la Información (IT) incluye Planificación de Recursos de la Empresa (ERP, Enterprise Resource Planning), Sistemas de Administración de Información de Laboratorio (LIMS, Laboratory Information Management System), administración de producción y sistemas de base de datos. Y un sistema de control de proceso incluye grandes sistemas de control como los de Adquisición de Datos y Supervisión de Control (SCADA, Supervisory Control and Data Acquisition), Sistemas de Control Distribuido (DCS, Distributed Control System), sistemas controlados por un Controlador Lógico Programable (PLC, Programmable Logic Controller) y sistemas integrados (embedded systems) instalados en manufactura.
A continuación les presento las responsabilidades del usuario en el proceso de validación, el proceso de validación para los sistemas IT y para los Sistemas de Control de Proceso que nos sugiere la GAMP4.
2.3.5 Responsabilidades de usuario.
La GAMP4 en su capítulo de “Ciclo de Vida de la Validación” (“Validation Life Cycle”) nos explica las responsabilidades que tiene el usuario dentro del proceso de la validación y la importancia de la relación entre usuario y el proveedor. Cada usuario dentro de su empresa debe tener especificadas las políticas que debe seguir para validar, estas políticas deben contemplar las tareas mencionadas en la siguiente Tabla 2.1.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 20 de 125
No. DE PASO TAREA O ACTIVIDAD DESCRIPCIÓN
1 Identificación del Sistema. Cada sistema automático debe ser evaluado e identificado como Sistema regulatorio GMP.
2 Elaboración del URS.El URS debe definir de manera clara y precisa lo que el usuario quiere que haga el sistema, algunas restricciones y definir requerimientos regulatorios y de documentación.
3
Determinar la estrategia de validación* Evaluación del riesgo.
* Estimación de los componentes del sistema.* Evaluación del proveedor.
Una evaluación de riesgo inicial debe ser llevada durante el Plan de Validación.
Los componentes del sistema deben ser estimados y categorizados para determinar el acercamiento a la validación requerida.
Los proveedores deben ser formalmente evaluados como parte del proceso de selección a proveedores y la planeación para la validación. La decisión de si se realizará una auditoría a Proveedores debe ser documentada y basada en la evaluación de riesgo y en la categorización de componentes.
4 Elaboración del Plan de Validación.El Plan de Validación debe definir las actividades, procedimientos y responsabilidades para establecer el sistema. Es típico definir la evaluación de riesgo que será tomada.
5 Revisión y aprobación de especificaciones, incluyendo la descripción del sistema. El usuario debe revisar y aprobar las especificaciones elaboradas
por el proveedor.
6 Monitoreo del desarrollo del Sistema. El usuario debe monitorear el desarrollo y las actividades de configuración contra el Plan acordado.
7 Revisión del código fuente. El usuario debe asegurar que el código fuente sea revisado adecuadamente durante el desarrollo del Sistema.
8 Revisión y aprobación de las especificaciones de prueba.
El usuario debe revisar y aprobar las especificaciones de prueba antes de ejecutarlas formalmente.
9 Desarrollo de las pruebas. El usuario puede ser involucrado en las pruebas como testigo durante la ejecución o como revisor de los resultados.
10 Revisión y aprobación de los reportes de prueba. El usuario debe aprobar los reportes de prueba y los resultados
asociados.
11 Elaboración del Reporte de Validación.El reporte de validación debe resumir todos los entregables y actividades, además de proveer evidencia de que el sistema está validado.
12 Mantenimiento del Sistema.Una vez que el sistema ha sido aceptado, el usuario debe establecer los procedimientos adecuados para la administración y operación del sistema.
13 Retiro del Sistema. El usuario debe administrar el reemplazo o el retiro del uso del sistema automático.
Tabla 2.1. Responsabilidades del usuario del Sistema.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 21 de 125
2.3.6 Proceso de validación de Sistemas IT.
La GAMP4 aborda el proceso de validación de los sistemas IT a través de los proveedores. En su capítulo “Sistema de Administración para los proveedores de Sistemas de Tecnología de la Información” (“Management System for Suppliers of IT Systems”) nos sugiere un modelo de validación basado en el concepto de ciclo de vida.
Con el objetivo de que los proveedores jueguen su rol en la producción de sistemas validados la GAMP4 recomienda que los proveedores sigan un sistema de administración formal y de preferencia basado en estándares tales como las series de la ISO 9000. Esta sección de la Guía sugiere un modelo basado en el concepto de ciclo de vida para el desarrollo y soporte subsecuente de un sistema automático.
El modelo descrito va encaminado a proveedores de Sistemas de Tecnología de la Información, como ERP, LIMS, administración de la producción y sistemas de base de datos.
Muchos sistemas IT para la industria farmacéutica actualmente están basados en productos de software y paquetes, los cuales están configurados cumpliendo los requerimientos del usuario. De tal manera que estos productos vienen con documentación de soporte y donde sea posible esta información puede ser empleada en la validación del ciclo de vida. Adicionalmente módulos de software son a veces requeridos para ofrecer o satisfacer una funcionalidad específica tal como interfases y reportes. El diseño y desarrollo de cada software debe ser documentado.
Esta sección aplica tanto para sistemas configurados específicamente como para sistemas de solución común o para la combinación de ambos.
El modelo de ciclo de vida que nos sugiere la Guía para validar estos sistemas esta mostrado en la figura 2.7. Este modelo cubre las fases de Planeación, Especificación, Diseño, Construcción, Pruebas, Instalación, Pruebas de aceptación y Operación.
No todos los documentos son requeridos para todos los productos, por lo que el proveedor deberá elaborar su Plan de Calidad y Proyectos para identificar los documentos que son requeridos.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 22 de 125
(Mantenimiento del estado validado)
PLANEACIÓN Y ESPECIFICACIÓN
ESPECIFICACIÓN DE REQUERIMIENTOS DE
USUARIOPLAN DE VALIDACIÓNGAMP4
EVALUACIÓN DEL PROVEEDOR
PLAN DEL PROYECTO Y CALIDAD
ESPECIFICACIÓN FUNCIONAL
DISEÑO Y CONSTRUCCIÓN
INSTALACIÓN Y PRUEBAS
PRUEBAS DE ACEPTACIÓN
OPERACIÓN
ESPECIFICACIÓN DE DISEÑO DEL
HARDWARE
ESPECIFICACIÓN DE PRUEBAS DE
ACEPTACIÓN DEL HARDWARE
ESPECIFICACIÓN DE DISEÑO DEL
SOFTWARE
ESPECIFICACIÓN DE PRUEBAS DE
INTEGRACIÓN DEL SOFTWARE
ESPECIFICACIÓN DE
CONFIGURACIÓN DEL PAQUETE
ESPECIFICACIÓN DE PRUEBAS DE
CONFIGURACIÓN DEL PAQUETE
ESPECIFICACIÓN DE DISEÑO DEL MODULO
DE SOFTWARE
ESPECIFICACIÓN DE PRUEBAS DEL MODULO DE
SOFTWARE
PRODUCCIÓN DE HARDWARE
PRODUCCIÓN DE SOFTWARE
PAQUETE DE CONFIGURACIÓN
RESULTADOS Y PRUEBAS DE
ACEPTACIÓN DE HARDWARE
RESULTADOS Y PRUEBAS DEL MODULO DE SOFTWARE
RESULTADOS Y PRUEBAS DE
INTEGRACIÓN DE SOFTWARE
VERIFICACIÓN DE LA CONFIGURACIÓN
RESULTADOS Y PRUEBAS DE
ACEPTACIÓN DEL SISTEMA
MANTENIMIENTO
CONTROL DE CAMBIOS
ACEPTACIÓN DEL SISTEMAESPECIFICACIONES DE PRUEBA
Figura 2.7. Modelo de Ciclo de Vida para validación de Sistemas IT.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 23 de 125
2.3.7 Proceso de validación de Sistemas de Control de Procesos.
La GAMP4 en su capitulo “Validación de Sistemas de Control de Procesos” (“Process Control System Validation”) considera cómo las actividades de ciclo de vida pueden ser aplicadas a un rango de control, monitoreo, o sistemas analíticos diseñados a administrar la producción o procesos de laboratorio.
Los sistemas de control de proceso son usados para automatizar los procesos de manufactura o de laboratorio por medio de colección de datos que provienen de instrumentos de medición o a través de algoritmos de control o bloques lógicos pre-programados o configurados, regulando dispositivos finales de control como válvulas o bombas para controlar equipos y/o procesos.
La función primaria de un sistema de control de proceso es producir un producto o proveer datos para soporte de manufactura o de laboratorio. Algunos sistemas de control de proceso también proveen funcionalidad en la administración de los procesos o están ligados con el procesamiento de datos de aplicaciones como parte de una computadora integrada a manufactura.
Las buenas prácticas de ingeniería deben ser aplicadas al desarrollo e implementación de sistemas de control de proceso. Las buenas prácticas de ingeniería son la aplicación de los métodos o estándares establecidos a través del proyecto de ciclo de vida para entregar apropiadamente soluciones costo-beneficio.
2.3.7.1 Modelos de Ciclo de Vida
La validación del ciclo de vida de un Sistema de Control de Proceso está definida en la figura 2.8 y aplica para un amplio rango de sistemas, desde sistemas de control robusto en industrias grandes hasta sistemas de control pequeños instalados en equipos usados como secundarios o ligados con equipo de laboratorio montado.
La relación entre las fases de diseño y de pruebas es compleja. La verificación de que se ha conocido la intención de los requerimientos y diseño debe hacerse en la parte mas apropiada del ciclo de vida dependiendo del tipo y madurez del sistema que está siendo desarrollado o configurado, del nivel del plan de pruebas y de la complejidad de los requerimientos a probar.
La Guía en Commissioning y Calificación de la Sociedad Internacional para la Ingeniería Farmacéutica (Guide on Commissioning and Qualification by the International Society for Pharmaceutical Engineering, ISPE) da una guía de como soportar o sustentar la calificación con actividades de Commissioning. La rastreabilidad a través del ciclo de vida asegura que todos los requerimientos y el diseño han sido entendidos.
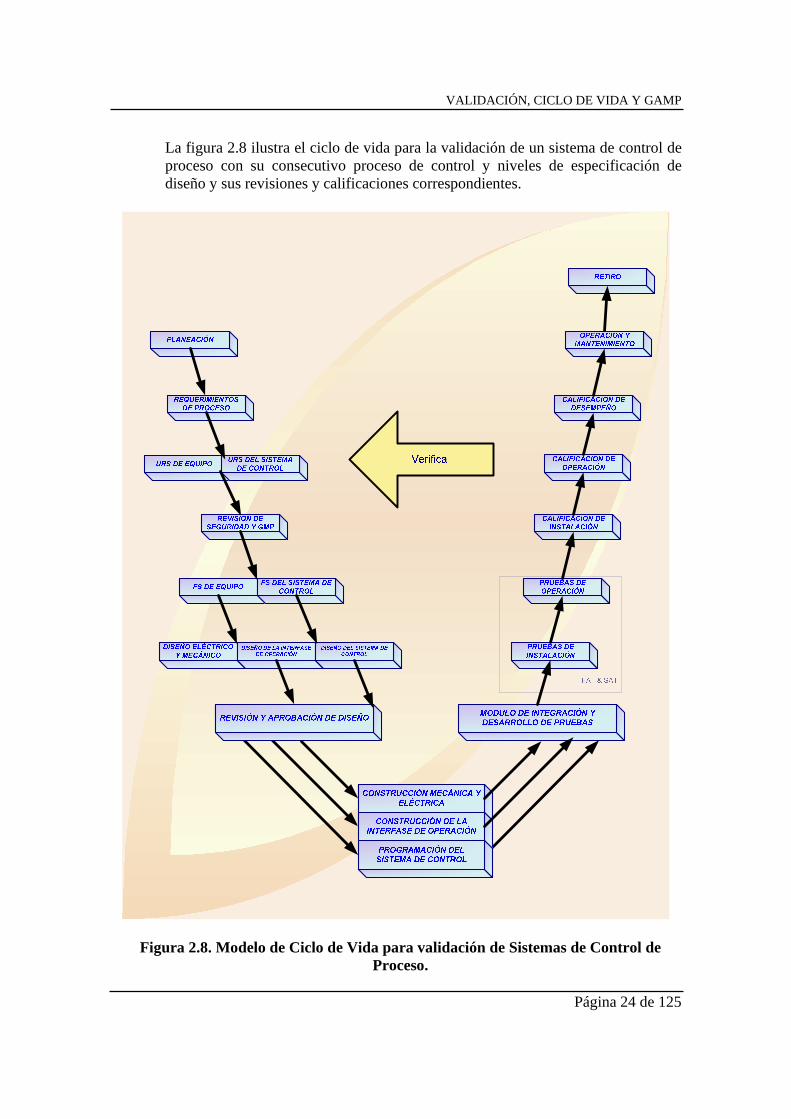
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 24 de 125
La figura 2.8 ilustra el ciclo de vida para la validación de un sistema de control de proceso con su consecutivo proceso de control y niveles de especificación de diseño y sus revisiones y calificaciones correspondientes.
Figura 2.8. Modelo de Ciclo de Vida para validación de Sistemas de Control de Proceso.

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 25 de 125
2.3.7.2 Tipos de Sistema de Control de Proceso
La GAMP4 distingue dos Sistemas de Control de Proceso: Sistemas Integrados (Embedded Systems) y Sistemas Autónomos (Standalone Systems).
Sistemas Integrados (Embedded Systems): Son Sistemas basados en microprocesadores tales como un Circuito Integrado Programable (IC), un Controlador Lógico Programable (PLC) o una Computadora con el solo propósito de controlar o monitorear una pieza o parte de manufactura o equipo analítico, el cual es usualmente entregado como una parte de ese determinado equipo. Ejemplos: máquinas de llenado, máquinas de empaque o Cromatógrafos de Líquidos de Alto Desempeño (HPLC, High performance liquid chromatography).
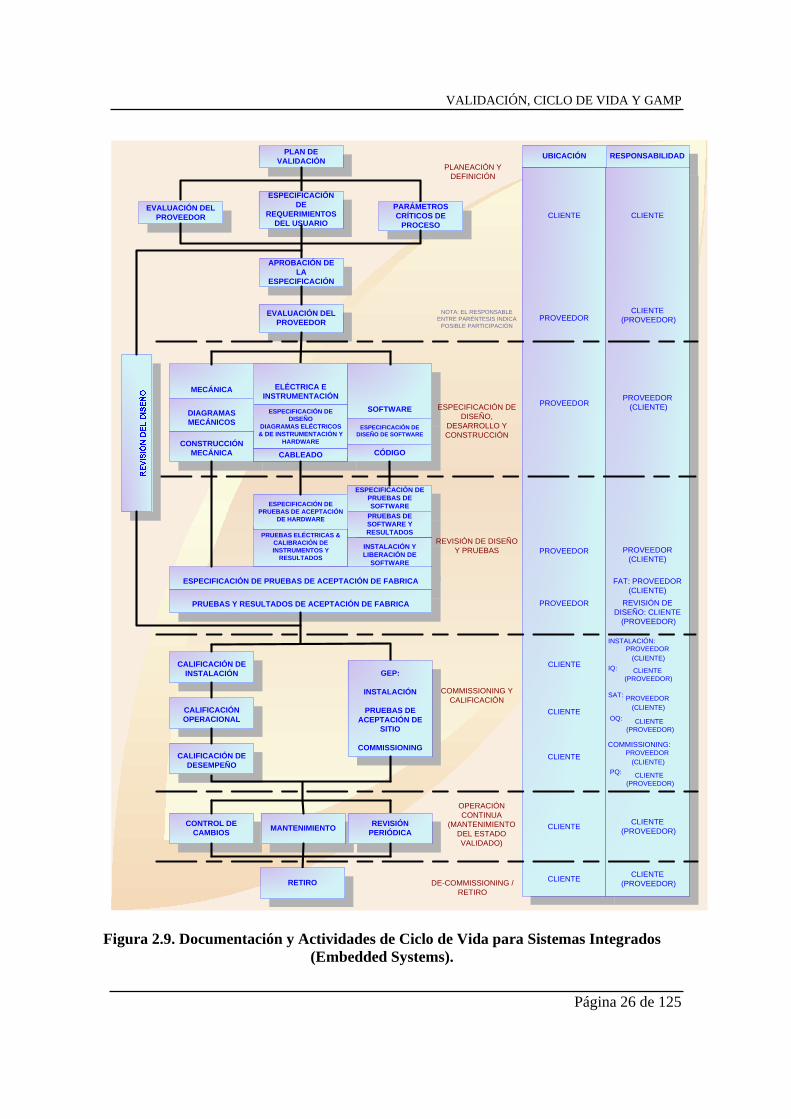
En la figura 2.9 podemos ver el modelo que nos presenta la GAMP4 para este tipo de sistemas.
Sistemas Autónomos (Standalone Systems): Son aquellos sistemas que son configurados o hechos a la medida, sistemas contenidos en si mismos que son componentes de una aplicación de un proceso automático de manufactura. Éstos son integrados como sistemas de cómputo independientes, separados del equipo de Planta, para su conexión a instrumentación de campo o dispositivos reguladores y a cada uno. Estos sistemas pueden ser integrados verticalmente con un nivel alto de administración de datos. Ejemplos: controladores multi-loop o Controlador Lógico Programable (PLC, Programmable Logic Controller) que controlan parte de un proceso, sistemas de Adquisición de Datos y Supervisión de Control (SCADA, Supervisory Control and Data Acquisition), Sistemas de Control Distribuido (DCS) que controlen procesos de Planta y Sistemas de Administración de Edificios (BMS, Building Management Systems).
En la figura 2.10 podemos ver el modelo que nos presenta la GAMP4 para este tipo de sistemas.
VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 26 de 125
RESPONSABILIDAD
CONTROL DE CAMBIOS
ESPECIFICACIÓN DE PRUEBAS DE SOFTWARE
PRUEBAS DE SOFTWARE Y RESULTADOS
INSTALACIÓN Y LIBERACIÓN DE
SOFTWARE
ESPECIFICACIÓN DE PRUEBAS DE ACEPTACIÓN DE FABRICA
CONSTRUCCIÓN MECÁNICA CABLEADO
PLAN DE VALIDACIÓN
ESPECIFICACIÓN DE
REQUERIMIENTOS DEL USUARIO
EVALUACIÓN DEL PROVEEDOR
PARÁMETROS CRÍTICOS DE
PROCESO
APROBACIÓN DE LA
ESPECIFICACIÓN
EVALUACIÓN DEL PROVEEDOR
ELÉCTRICA E INSTRUMENTACIÓN
SOFTWARE
MECÁNICA
ESPECIFICACIÓN DE DISEÑO
DIAGRAMAS ELÉCTRICOS & DE INSTRUMENTACIÓN Y
HARDWARE
ESPECIFICACIÓN DE DISEÑO DE SOFTWARE
CÓDIGO
DIAGRAMAS MECÁNICOS
PRUEBAS Y RESULTADOS DE ACEPTACIÓN DE FABRICA
PRUEBAS ELÉCTRICAS & CALIBRACIÓN DE INSTRUMENTOS Y
RESULTADOS
ESPECIFICACIÓN DE PRUEBAS DE ACEPTACIÓN
DE HARDWARE
CALIFICACIÓN DE INSTALACIÓN
CALIFICACIÓN OPERACIONAL
CALIFICACIÓN DE DESEMPEÑO
GEP:
INSTALACIÓN
PRUEBAS DE ACEPTACIÓN DE
SITIO
COMMISSIONING
MANTENIMIENTO REVISIÓN PERIÓDICA
RETIRO
UBICACIÓN
CLIENTE
PROVEEDOR
PROVEEDOR
PROVEEDOR
PROVEEDOR
CLIENTE
CLIENTE
CLIENTE
CLIENTE
CLIENTE
CLIENTE
CLIENTE (PROVEEDOR)
PROVEEDOR (CLIENTE)
PROVEEDOR (CLIENTE)
CLIENTE (PROVEEDOR)
CLIENTE (PROVEEDOR)
FAT: PROVEEDOR (CLIENTE)
REVISIÓN DE DISEÑO: CLIENTE
(PROVEEDOR)
INSTALACIÓN: PROVEEDOR
(CLIENTE)
CLIENTE (PROVEEDOR)
IQ:
SAT: PROVEEDOR (CLIENTE)
CLIENTE (PROVEEDOR)
OQ:
COMMISSIONING:PROVEEDOR
(CLIENTE)
CLIENTE (PROVEEDOR)
PQ:
PLANEACIÓN Y DEFINICIÓN
ESPECIFICACIÓN DE DISEÑO,
DESARROLLO Y CONSTRUCCIÓN
REVISIÓN DE DISEÑO Y PRUEBAS
COMMISSIONING Y CALIFICACIÓN
OPERACIÓN CONTINUA
(MANTENIMIENTO DEL ESTADO VALIDADO)
DE-COMMISSIONING / RETIRO
NOTA: EL RESPONSABLE ENTRE PARÉNTESIS INDICA
POSIBLE PARTICIPACIÓN
Figura 2.9. Documentación y Actividades de Ciclo de Vida para Sistemas Integrados
(Embedded Systems).

VALIDACIÓN, CICLO DE VIDA Y GAMP
Página 27 de 125
RESPONSABILIDADPLAN DE VALIDACIÓN
ESPECIFICACIÓN DE REQUERIMIENTOS
DEL USUARIO
EVALUACIÓN DEL PROVEEDOR
PARÁMETROS CRÍTICOS DE
PROCESO
APROBACIÓN DE LA ESPECIFICACIÓN
EVALUACIÓN DEL PROVEEDOR
UBICACIÓNCLIENTE
PROVEEDOR
CLIENTE
CLIENTE (PROVEEDOR)
PLANEACIÓN Y DEFINICIÓN
NOTA: EL RESPONSABLE ENTRE PARÉNTESIS INDICA
POSIBLE PARTICIPACIÓN
CLIENTE
CLIENTE
CLIENTE
CLIENTE
PRUEBAS DE ACEPTACIÓN DE FABRICA
(CONTRA PUNTO 1)
CONTROL DE CAMBIOS
DATOS DE DISEÑO DE PROCESO
CALIFICACIÓN DE INSTALACIÓN
CALIFICACIÓN OPERACIONAL
CALIFICACIÓN DE DESEMPEÑO
GEP:
INSTALACIÓN
PRUEBAS DE ACEPTACIÓN DE SITIO
COMMISSIONING
MANTENIMIENTO REVISIÓN PERIÓDICA
RETIRO
REV
ISIÓ
N D
EL D
ISEÑ
O
PROVEEDOR
PROVEEDOR
PROVEEDOR
CLIENTE
CLIENTE
CLIENTE
CLIENTE
PROVEEDOR (CLIENTE)
PROVEEDOR (CLIENTE)
CLIENTE (PROVEEDOR)
CLIENTE (PROVEEDOR)
FAT: PROVEEDOR (CLIENTE)
REVISIÓN DE DISEÑO: CLIENTE
(PROVEEDOR)
INSTALACIÓN: PROVEEDOR
(CLIENTE)
CLIENTE (PROVEEDOR)
IQ:
SAT: PROVEEDOR (CLIENTE)
CLIENTE (PROVEEDOR)
OQ:
COMMISSIONING:PROVEEDOR
(CLIENTE)CLIENTE
(PROVEEDOR)PQ:
ACUERDO DE DISEÑO
COMMISSIONING Y CALIFICACIÓN
OPERACIÓN CONTINUA
(MANTENIMIENTO DEL ESTADO VALIDADO)
DE-COMMISSIONING / RETIRO
PLAN DE CALIDAD CON
CONTRATISTAS
ESPECIFICACIÓN DE DISEÑO FUNCIONAL
ESPECIFICACIÓN DE PRUEBAS DE
ACEPTACIÓN
PLAN DE CALIDAD DEL SISTEMA DE
PROVEEDORES
(5) C&I APLICACIÓN
HOJAS DE DATOS DE DISEÑO
C&I APLICACIÓN INGENIERÍA Y DIAGRAMAS
ESPECIFICACIÓN DE DISEÑO
COMPUTADORA H/W
(2) ESPECIFICACIÓN DE PRUEBAS DE
HARDWAREESPECIFICACIÓN DE DISEÑO S/W
(3) ESPECIFICACIÓN DE PRUEBAS DE
INTEGRACIÓN MODULO DE SOFTWARE
PRODUCCIÓNCOMPUTADORA
H/W
ESPECIFICACIÓN DE DISEÑO
MODULO S/W
(2) ESPECIFICACIÓN DE PRUEBAS MODULO DE SOFTWARE
CONFIGURACIÓN / CÓDIGO MODULO S/W
C&I INSPECCIÓN / CALIBRACIÓN (CONTRA
PUNTO 5)
COMPUTADORA H/W PRUEBAS (CONTRA
PUNTO 2)
MODULO S/W PRUEBAS (CONTRA
PUNTO 4)
MODULO S/W INTEGRACIÓN DE PRUEBAS (CONTRA
PUNTO 4)
SISTEMA DEL PROVEEDOR PRUEBAS DE INTEGRACIÓN (CONTRA PUNTOS 1, 2 Y 3)
DESARROLLO Y DISEÑO
CONSTRUCCIÓN DEL SISTEMA Y
DESARROLLO DE PRUEBAS
REVISIÓN DEL DISEÑO Y PRUEBAS
DE ACEPTACIÓN
PROVEEDORPROVEEDOR
(CLIENTE)
PROVEEDORPROVEEDOR
(CLIENTE)
PROVEEDORPROVEEDOR
(CLIENTE)
PROVEEDORPROVEEDOR
(CLIENTE)
PROVEEDORPROVEEDOR
(CLIENTE)
CLIENTE
Figura 2.10. Documentación y Actividades de Ciclo de Vida para Sistemas Autónomos
(Standalone Systems).

CAPÍTULO 3. PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA
INDUSTRIA FARMACÉUTICA.
CAPÍTULO 3.
“PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO
PARA LA INDUSTRIA FARMACÉUTICA”
Objetivo particular: Describir mi propuesta de validación de Sistemas Automáticos y de Cómputo a través del desarrollo del Ciclo de Vida, para el cumplimiento del Proyecto de Norma Oficial Mexicana PROY-NOM-059-SSA1-2004 utilizando como herramienta base la guía GAMP4.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 29 de 125
Capítulo 3. Propuesta para Validación de Sistemas Automáticos y de Cómputo para la Industria Farmacéutica. En el capítulo 2 abordamos los temas de Validación, el Ciclo de Vida como recurso para la validación de sistemas y la GAMP4 como herramienta para la implementación de la metodología para dicha validación. Ahora en este capítulo presento mi propuesta de validación, tomando en cuenta la propuesta de la GAMP4 y el sistema de validación que he llevado en mi experiencia.
3.1 ANÁLISIS.
Desde mi perspectiva la GAMP4 propone 3 caminos para validar los sistemas, ya que marca la diferencia para sistemas IT, para sistemas integrados y para sistemas autónomos.
Mi propuesta consiste en dar un solo esquema de validación general que se pueda aplicar a cualquier clasificación de sistema y que contemple toda la información necesaria para el cumplimiento del Proyecto de Norma Oficial Mexicana y proporcione la base para el cumplimiento regulatorio de otros Países.
La Figura 3.1 muestra mi propuesta para el proceso de validación, donde se pueden observar las fases que propongo en las que se divida la vida de los sistemas y las actividades y/o entregables que se tienen para cada una de ellas. El alcance de esta propuesta es sólo para los sistemas GMP de Impacto Directo a la calidad del producto.
La inquietud de reunir el proceso de validación en un solo esquema es tener un solo camino que nos muestre los pasos para cumplir con la validación y que cumpla para cualquier clasificación del sistema, ya sea IT o de proceso. Además de que se evita la repetición de información, ya que si analizamos los 3 esquemas que plantea la GAMP4 de manera general tienen los mismos entregables y algunos vistos desde distintas perspectivas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 30 de 125
RESPONSABILIDAD
PROCEDIMIENTOS/ GUIAS/CAPACITACIÓN
ESPECIFICACIÓN DE DISEÑO [a]
PLAN DE VALIDACIÓN
ESPECIFICACIÓN DE REQUERIMIENTOS (PRIMERA
VERSIÓN) [a]
EVALUACIÓN Y SELECCIÓN DEL
PROVEEDOR
CALIFICACIÓN DE INSTALACIÓN
CALIFICACIÓN OPERACIONAL
CALIFICACIÓN DE DESEMPEÑO
MANTENIMIENTO DEL SISTEMA DURANTE SU OPERACIÓN
RETIRO
UBICACIÓN
CLIENTE
PROVEEDOR
PROVEEDOR
CLIENTE
CLIENTE
CLIENTE
CLIENTE PROVEEDOR
PROVEEDOR CLIENTE
PLANEACIÓN Y ESPECIFICACIÓN
DISEÑO Y CONSTRUCCIÓN
INSTALACIÓN Y PRUEBAS
OPERACIÓN DEL SISTEMA
RETIRO
PLAN DEL PROYECTO Y CALIDAD
PRUEBAS DE REVISIÓN DE DISEÑO
REPORTE DE VALIDACIÓN
Resultados satisfactorios
ESPECIFICACIÓN DE REQUERIMIENTOS
[b]
ESPECIFICACIÓN DE DISEÑO [b]
SI
NO
MATRIZ DE RASTREABILIDAD
Resultados satisfactorios
SI
NOCORRECCIÓN
DEL PROBLEMA
ESPECIFICACIÓN DE REQUERIMIENTOS (SEGUNDA
VERSIÓN) [a]
CLIENTE CLIENTE
CLIENTE
CLIENTE
CLIENTE PROVEEDOR
CLIENTE PROVEEDOR
PROVEEDORPROVEEDOR
CLIENTE
CLIENTE PROVEEDOR
CLIENTE PROVEEDOR
CLIENTE CLIENTE
CLIENTE
CLIENTE CLIENTE
CLIENTE PROVEEDOR
CLIENTE PROVEEDOR
CLIENTE PROVEEDOR
Figura 3.1. Ciclo de Vida propuesto.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 31 de 125
3.1.1 Correspondencia entre mi Propuesta con el Proyecto de Norma PROY-NOM-059-SSA1-2004, la GAMP4 y NMX-CC-9001-IMNC-2000. La propuesta que expongo en esta tesis tiene correspondencia con el Proyecto de Norma PROY-NOM-059-SSA1-2004, con la Guía GAMP4 y con la Norma NMX-CC-9001-IMNC-2000, en las tablas 3.1, 3.2 y 3.3 se presentan este análisis. A continuación se presentan dichas tablas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 32 de 125
Tabla 3.1. Correspondencia entre el Proyecto de Norma PROY-NOM-059-SSA1-2004 y mi Propuesta.
PROY-NOM-059-SSA1-2004 PROPUESTA DE VALIDACIÓN Equipo automático, mecánico y electrónico. 10.5 Los sistemas computarizados instalados en los equipos para el control del proceso de fabricación deben estar validados. 10.5.2 1.2 Marco Normativo.
3.0
Propuesta para Validación de Sistemas Automáticos y de Cómputo para la Industria Farmacéutica.
Con el fin de asegurar la exactitud de los datos manejados por estos sistemas, se debe implementar un sistema de protección de los mismos para evitar modificaciones a las fórmulas o registros efectuadas por personal no autorizado. 10.5.3 1.2 Marco Normativo. Se debe mantener un respaldo en copias fieles, cintas o microfilms, de toda la información archivada en las computadoras o los sistemas relacionados, para asegurar que la información emitida por estos sistemas es exacta, completa y que no existen modificaciones inadvertidas. 10.5.4 1.2 Marco Normativo. Validación. 14 1.2 Marco Normativo. 2.1 Validación.
3.0
Propuesta para Validación de Sistemas Automáticos y de Cómputo para la Industria Farmacéutica.
Política. 14.1 1.2 Marco Normativo. 3.2 Plan de Validación. Planeación para la validación. 14.2 3.2 Plan de Validación. Documentación. 14.3 3.2 Plan de Validación. 3.4 Plan del Proyecto y Calidad 3.9 Reporte de Validación Calificación. 14.4 3.7 Pruebas al Equipo o Sistema. Sistemas computacionales. 14.8 1.2 Marco Normativo. Mantenimiento del estado validado. 14.11 3.10 Mantenimiento del Estado Validado
Control de Cambios. 15 3.10 Mantenimiento del Estado Validado

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 33 de 125
Tabla 3.2. Correspondencia entre mi Propuesta y la GAMP4.
PROPUESTA DE VALIDACIÓN GAMP4
Antecedentes y Marco Normativo 1.0 1.0Introducción a la GAMP (Introduction to GAMP)
Antecedentes 1.1 1.1¿Cómo surgió la iniciativa GAMP? (How did the GAMP Initiative start?)
Validación, Ciclo de Vida y GAMP 2.0 6.0Generalidades de la Validación (Validation Overview)
Validación 2.1 Propuesta para Validación de Sistemas Automáticos y de Cómputo para la Industria Farmacéutica. 3.0 7.0
Ciclo de Vida de Validación (Validation Life Cycle)
Análisis 3.1 8.0
Sistema de Administración para Proveedores de Sistemas de TI (Management System for Suppliers of IT Systems)
9.0
Validación de Sistemas de Control de Proceso (Process Control System Validation)
Plan de Validación 3.2 7.5
Validación, Plan del Proyecto y Calidad (Validation, Quality and Project Plan)
8.1.2 Planeación (Planning)
9.1 Planeación (Planning)
Especificación de Requerimientos de usuario (URS) y Especificación Funcional (FS). 3.3 7.3
Especificación de Requirimientos de Usuario (User Requirements Specification)
8.1.3
Especificación, Diseño y Construccción (Specification, Design and Construction)
9.2
Especificación y Diseño (Specification and Design)
Plan del Proyecto y Calidad 3.4 7.5
Validación, Plan del Proyecto y Calidad (Validation, Quality and Project Plan)
8.1.2 Planeación (Planning)
9.1 Planeación (Planning)
9.7 Revisión del Diseño (Design Review)

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 34 de 125
Matriz de Rastreabilidad de Requerimientos
3.5 8.2.1 Revisiones (Reviews)
9.7 Revisión de Diseño (Design Review)
Especificación de Diseño 3.6 8.1.3
Especificación, Diseño y Construccción (Specification, Design and Construction)
9.2
Especificación y Diseño (Specification and Design)
Pruebas al Equipo o Sistema 3.7 7.9
Pruebas (Testing)
8.4
Especificaciones de Prueba (Test Specifications)
9.7 Revisión del Diseño (Design Review)
9.12
Desarrollo de Pruebas (Development Testing)
9.13
Pruebas de Aceptación (Aceptance Testing)
9.15 Calificación (Qualification)
Registro de Capacitación 3.8 7.11.2
Capacitación (Training)
8.2.4
Capacitación del Usuario por el Proveedor (Training of Supplier Staff)
Reporte de Validación 3.9 7.10
Reporte de Validación (Validation Reporting)
9.16
Reporte de Validación (Validation Report)
Mantenimiento del Estado Validado 3.10 7.11
Mantenimiento del Estado Validado (Maintaining the Validated State)
9.17
Mantenimiento del Estado Validado (Maintaining the Validated State)
Retiro 3.11 9.18
Retiro (Retirement)
7.11.14 Retiro del Sistema (System Retirement)
Buenas Prácticas 3.12 11
Definiciones de Buenas Prácticas (Good Practice Definitions)

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 35 de 125
Tabla 3.3. Correspondencia entre mi Propuesta y la Norma NMX-CC-9001-IMNC-2000.
PROPUESTA DE VALIDACIÓN NMX-CC-9001-IMNC-2000
Propuesta para Validación de Sistemas Automáticos y de Cómputo para la Industria Farmacéutica. 3.0 Plan de Validación 3.2 5.4 Planificación Especificación de Requerimientos de usuario (URS) y Especificación Funcional (FS). 3.3 5.2 Enfoque al Cliente
7.2.1Determinación de los requisitos relacionados con el Producto
7.3.2Elementos de Entrada para el Diseño y Desarrollo
Plan del Proyecto y Calidad 3.4 5.4 Planificación
7.1Planificación de la realización del Producto
7.3.1 Planificación del Diseño y Desarrollo 7.3.4 Revisión del Diseño y Desarrollo Matriz de Rastreabilidad de Requerimientos 3.5 5.2 Enfoque al Cliente
7.2.2Revisión de los requisitos relacionados con el Producto
7.5.3 Identificación y Trazabilidad Especificación de Diseño 3.6 7.3 Diseño y Desarrollo 7.3.1 Planificación del Diseño y Desarrollo 7.3.3 Resultados del Diseño y Desarrollo
Pruebas al Equipo o Sistema 3.7 7.2.2Revisión de los requisitos relacionados con el Producto
7.3 Diseño y Desarrollo 7.3.1 Planificación del Diseño y Desarrollo 7.3.4 Revisión del Diseño y Desarrollo 7.3.5 Verificación del Diseño y Desarrollo 7.3.6 Validación del Diseño y Desarrollo
7.4.3Verificación de los Productos comprados
Registro de Capacitación 3.8 6.2.2 Competencia, toma de conciencia y formación
Reporte de Validación 3.9 7.3.6 Validación del Diseño y Desarrollo

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 36 de 125
Mantenimiento del Estado Validado 3.10 4.2.3 Control de los documentos 8.2.2 Auditoría interna
8.2.3Seguimiento y medición de los procesos
8.2.4 Seguimiento y medición del producto 8.4 Análisis de datos 8.5.1 Mejora continua 8.5.2 Acciones correctivas 8.5.3 Acciones preventivas

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 37 de 125
3.2 PLAN DE VALIDACIÓN
El primer paso que nos marca la GAMP4 (para los 3 tipos de sistemas), dentro de la etapa de Planeación y Especificación, para iniciar con el ciclo de vida de un sistema es elaborar un Plan de Validación. De hecho nos habla del desarrollo de un Plan Maestro de Validación y de los Planes de validación para proyectos o sistemas.
Un Plan de Validación es el documento donde se definen las actividades de validación, las responsabilidades y los procedimientos a seguir para llevarla a cabo. En cada Empresa dependerá el nombre que se le quiera dar y el alcance que tenga, los más comunes son Plan Maestro de Validación, Plan de Validación y Guías de Validación.
Las siglas en inglés para los términos que utilizaremos para la descripción de estos documentos de validación son: VMP para el Plan Maestro de Validación (Validation Master Plan) y VP para un Plan de Validación (Validation Plan).
Usualmente en el VMP se definen todas las áreas y elementos que requieren validación dentro de una Compañía y se define el plan general para el cumplimiento de esas validaciones específicas. Este Plan no lo comentaremos a fondo ya que el alcance de ese Plan es toda la Planta (limpieza de áreas de producción y equipos, procesos de producción, edificios, instalaciones, equipos y sistemas automáticos y críticos, etc) y nosotros hablaremos del VP que nos definirá como validar en específico uno de los elementos que comprende el VMP.
La importancia del VP radica en que este Plan junto con la Especificación de Requerimientos son la base para definir el Plan del Proyecto y Calidad, entre Usuario y Proveedor.
La GAMP4 nos dice que para cada sistema GMP (Good Manufacturing Practice, Buenas Prácticas de Manufactura) se debe elaborar un Plan de Validación, pero también aclara que cada Compañía establece la estructura en que desarrollara sus Planes.
En esta parte me parece importante mencionar que para un sistema nuevo es conveniente hacer su Plan de Validación individual, pero en el caso de sistemas pequeños que se puedan agrupar o en el caso de aplicar un Plan general de validación para varios equipos iguales en la Planta lo que conviene es hacer un solo Plan que aplique a esos determinados sistemas o equipos.
El Plan de validación es la clave para todo el proceso de validación, en él se puede describir la estrategia de validación que se seguirá o hacer referencia al documento donde está descrito.
Un aspecto importante dentro de la documentación son las firmas y hablando del VP debe ser firmado por el área de Aseguramiento de Calidad y por los usuarios finales

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 38 de 125
del sistema o equipo. El significado de las firmas debe ser definido. La responsabilidad de elaborar el VP recae en el propietario del sistema o en el líder del proyecto.
El contenido de un Plan de Validación debe definir:
Las actividades requeridas. Cómo esas actividades se llevarán a cabo y quiénes son los responsables de
ello. Cuáles deben ser los resultados. Cuáles serán los requisitos para la aceptación. Cómo se mantendrá la validación del sistema durante su ciclo de vida.
La GAMP4 menciona que puede haber la necesidad de generar una serie de reportes para indicar el avance en el proyecto o la aprobación de etapas parciales del proyecto. Si este fuera el caso, deberá indicarse en el VP que se emitirán dichos reportes.
La GAMP4 contempla los siguientes puntos como contenido de un Plan de Validación:
1. Introducción y alcance. 2. Estructura organizacional. 3. Evaluación de la criticidad. 4. Estrategia de validación. 5. Entregables de la validación. 6. Criterios de aceptación. 7. Control de cambios. 8. Procedimientos. 9. Capacitación. 10. Administración de documentos. 11. Mantenimiento del estado validado. 12. Glosario.
A continuación les presento la Plantilla que propongo para desarrollar un Plan de validación para un proyecto. Estoy tomando como base el contenido y la estructura que propone la GAMP4, pero adiciono algunas secciones que me parece importante incluir. Los datos propios de la empresa como derechos reservados, logos de confidencialidad, deben ser seguidos conforme a las políticas de la empresa.
En mi experiencia, el primer documento que realizo para la validación de sistemas GMP es un Plan de aseguramiento de Calidad, más no un Plan de Validación, como sugiere la GAMP4. Esto es porque los planes para validar los sistemas están contemplados en VMP’s generales. Por mi parte sugiero que se realice el VP y posteriormente en otra etapa el Plan de Calidad, ya que ambos tienen características especificas e importantes. De esta manera el VP se puede realizar desde el punto de vista de usuario y el QPP (Plan del Proyecto y Calidad, Quality and Project Plan) desde el punto de vista del acuerdo de Usuario y Proveedor.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 39 de 125
PLANTILLA 1. PROPUESTA DE CONTENIDO DE UN PLAN DE VALIDACIÓN
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio. En este registro de cambios cuando un documento es elaborado se puede incluir la fecha de emisión del documento, quién lo elaboró y que es su primera emisión. Esta sección es importante porque nos muestra todo el historial de cambios que ha tenido el documento desde su emisión, lo cuál apoya al control de cambios que se debe llevar a lo largo de la validación.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo. Esta sección de firmas tiene relación con la sección de descripción de roles y responsabilidades, ya que el número de firmas debe asegurar el cumplimiento de los requerimientos de todos los Departamentos. Se pueden incluir tantas áreas firmantes como sean necesarias para el cumplimiento del objetivo del proyecto.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 40 de 125
5. Introducción. Esta sección debe dar un bosquejo general del contenido del documento, recomiendo describir los antecedentes del proyecto y de una manera breve en que consistirá el proyecto.
6. Objetivo.
En esta sección se definen el o los objetivos del documento. Es importante que estos sean precisos para no caer en ambigüedades.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo. En esta sección se puede mencionar el periodo en el cual se realizará la siguiente revisión al documento (en el caso de que aplique).
8. Referencias.
Es importante mencionar documentos con los que tenga relación este Plan que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, VMP’s, etcétera.
9. Descripción de los roles y responsabilidades.
La GAMP4 señala que en esta sección se deben describir los roles y responsabilidades como mínimo del Líder o administrador del Proyecto, del departamento de Aseguramiento de Calidad y de los Dueños del sistema.
10. Evaluación de la criticidad del sistema.
Aquí se debe describir la evaluación de la criticidad del sistema, ésta debe incluir un alto nivel de valoración de la información o recurrir a alguna fuente de información relacionada a este tema. Cada empresa debe tener establecidos los criterios de clasificación de sus sistemas. Recomiendo en base a mi experiencia y la GAMP4 cubrir lo siguiente:
1. Clasificación de Software / Sistema
Clasificación de Software / Sistema Descripción
Categoría 1: Sistemas Operativos / Infraestructura
Están disponibles en el mercado como Sistemas Operativos. Aplicaciones que están desarrolladas para ejecutarse bajo la plataforma de estos sistemas operativos. También incluye la infraestructura para Sistemas Operativos y Redes, aplicaciones

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 41 de 125
web, y software de servidor. Ejemplos: Windows XP, Unix, Windows NT, DOS, etc. Los sistemas de esta categoría no están sujetos a una validación específica, ya que su funcionalidad es probada indirectamente durante las pruebas involucradas en la validación de la aplicación. El nombre del sistema operativo y su versión deben ser documentados en la Especificación de Diseño y verificados en la Calificación de Instalación.
Categoría 2: Sistema Firmware
Sistemas que no están personalizados ni en hardware ni software. Es una combinación de hardware con instrucciones que dan como resultado un software de solo lectura en ese dispositivo. Dentro de esta clasificación caen instrumentos y controladores. Ejemplos: Sistemas que trabajan a partir de un PLC u otro controlador, básculas, lectores de código de barras, cronómetros, etc. Para los sistemas firmware, su nombre, versión y configuración deben ser documentados en la Especificación de Diseño y verificados en la Calificación de Instalación. Su operación también debe ser probada en un protocolo.
Clasificación 3: Sistema Estándar
Estos sistemas están disponibles en el mercado como software estándar, (llamados en inglés “off-the-shelf”) y proveen una solución de negocio o de proceso de manufactura. No son configurados para definir procesos de manufactura o de negocio. El sistema puede ser adaptado con cambio de datos para operar dentro de un ambiente operativo específico pero no son adaptados para conformar un usuario específico. Ejemplos: Paquetes de análisis estadístico, software de instrumentos de laboratorio como un HPLC, etc. Su nombre y versión deben ser documentados en la Especificación de Diseño y verificados en la Calificación de Instalación. Su operación también debe ser probada en un protocolo.
Clasificación 4: Sistema Configurable
Software configurable que provee interfases y funciones estándar que habilitan la configuración de un usuario específico de procesos de negocio o manufactura. Envuelve módulos de software configurados de manera predefinida y la posibilidad de personalizar dichos módulos. Ejemplos: ERP, SCADA, etc.
Clasificación 5: Sistema Personalizado
Estos sistemas son desarrollados para cubrir necesidades específicas de una Empresa. Incluyen la construcción y personalización del sistema completo o subsistemas. No es un software o sistema comercial. Ejemplo: Desarrollos en Oracle.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 42 de 125
11. Estrategia de Validación.
Esta sección debe describir la estrategia de validación a ser aplicada, haciendo referencia a los procedimientos de sistemas automáticos a ser seguidos y el modelo de ciclo de vida a ser aplicado. Esta sección se basa en la determinación de la criticidad del sistema. Conforme la GAMP4 también se pueden definir los requerimientos de calificación de hardware y software y si hay algún impacto en el sistema de infraestructura. Si la validación se realizara en etapas, es en este documento donde se pueden definir esas etapas, sus entregables y como se pondrá el sistema o una determinada parte del sistema en funcionamiento. Si la estrategia de validación no incluye la auditoría al proveedor esta sección debe justificar el porque no se contempla. En concreto la GAMP4 recomienda que se describa: El modelo del ciclo de vida. La evaluación del riesgo del proceso. Las categorías de hardware y software. Las salidas y entradas requeridas para cada nivel o etapa del proyecto. Los criterios de aceptación para cada nivel o etapa.
12. Documentos a realizar en la validación (entregables).
Esta sección debe describir los documentos a elaborar, incluyendo las responsabilidades de elaboración, revisión y aprobación de cada uno de ellos. Conviene hacer una tabla donde se indiquen las etapas del proyecto, que documentos se elaborarán y los responsables.
13. Criterios de aceptación. Aquí se debe definir de manera general los criterios de aceptación para el sistema.
14. Control de Cambios.
Esta sección debe mencionar cómo se llevarán a cabo los controles de cambio para la implementación del nuevo equipo o sistema y como se llevará el control de cambios en el desarrollo de los mismos. Se puede hacer referencia a los procedimientos existentes para estos fines.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 43 de 125
15. Capacitación.
Se deben establecer los requerimientos de capacitación tanto del equipo de proyecto como a los usuarios del sistema.
16. Calendario de actividades y recursos.
Esta sección da un esbozo de las actividades de validación, tiempos de finalización y los recursos asignados para cada tarea.
17. Mantenimiento del estado validado.
En esta sección se deben definir las acciones y procedimientos a seguir para mantener el estado del sistema validado.
18. Glosario, Acrónimos y Términos
Esta sección contiene la definición de los términos que pueden no ser familiares para el lector.
19. Anexos En esta sección se puede incluir información necesaria para la completa comprensión del documento.
3.3 ESPECIFICACIÓN DE REQUERIMIENTOS DE USUARIO (URS) Y ESPECIFICACIÓN FUNCIONAL (FS).
El siguiente documento que sugiere la GAMP4 es la Especificación de Requerimientos de Usuario y más adelante la Especificación Funcional. Por otro lado en mi experiencia hasta el momento después del QP (primer entregable) se elabora una Especificación de Requerimientos, en la cual no se hace diferencia a cuales son los requerimientos de usuario y cuales son los requerimientos funcionales, simplemente son requerimientos del sistema. Lo que observo es que si se debe hacer una especificación de requerimientos de usuario ya que a partir de ésta se derivan los requerimientos funcionales.
Por ejemplo, si el requerimiento de usuario es tener un área con una temperatura de 18 °C, el requerimiento funcional será que se requiere tener un sistema de aire acondicionado y un lazo de control que permita tener en esa área especifica una temperatura de 18 °C. Y si por el contrario no sabemos previamente que el usuario necesita 18 °C en esa área, el requerimiento puede ser tan ambiguo como: se requiere un sistema que permita controlar la temperatura en el área. ¿Pero en qué temperatura? ¿En qué rangos? Los sistemas de control de temperatura se seleccionan dependiendo de qué temperatura tengan que mantener o controlar.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 44 de 125
Una especificación de requerimientos de usuario es el documento donde se describe lo que el sistema debe hacer y es recomendable y usual que sea escrita por el usuario. La Especificación de Requerimientos de Usuario puede ser enviada a los proveedores como parte del proceso de la selección de proveedores.
La GAMP4 nos sugiere tomar en cuenta las siguientes indicaciones para desarrollar un URS:
Cada requerimiento debe tener una referencia única y no ser mas largo de 250
palabras, para que exista rastreabilidad.
Ningún requerimiento debe ser duplicado ni contradicho.
Debe expresar requerimientos no diseños.
Cada requerimiento debe poder ser probado y verificado.
Los requerimientos deben ser entendidos por el usuario y por el proveedor, no utilizar ambigüedades ni jerga.
Se puede establecer la diferencia entre requerimientos esenciales y requerimientos deseables.
El contenido que nos recomienda la GAMP4 para un URS es:
1. Introducción. 2. Sección de generalidades o vista rápida. 3. Sección de requerimientos operacionales (funciones, datos, interfases y
ambiente). 4. Sección de restricciones. 5. Sección de Ciclo de Vida. 6. Sección de Glosario.
La Especificación Funcional es normalmente elaborada por el proveedor y describe el detalle de las funciones del equipo o sistema, es decir, describe lo que el sistema hará. La primera versión del FS puede ser la primera respuesta del proveedor a las necesidades de los clientes, después las siguientes versiones pueden ser complementadas con las revisiones del usuario. Al tener el FS se tiene una salida de la etapa anterior que describe las funciones del sistema, al mismo tiempo que se tiene la entrada para el siguiente entregable que es el de Diseño.
Es importante que el FS sea escrito de tal forma que resulte entendible para ambas partes, usuario y proveedor. Se recomienda que sea firmado tanto por el usuario como por el proveedor para establecer el compromiso.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 45 de 125
Si el sistema es pequeño se recomienda que se maneje el FS y el DS en un mismo documento y así evitar la emisión de los dos, reduciendo con ello tiempo y esfuerzos. Pero si por el contrario es un sistema robusto, resulta de ayuda que se realicen diversos FS, de tal manera que se divida en secciones y se vaya abordando el sistema por secciones definidas.
Los protocolos de calificación deben probar todas las funciones especificadas.
El FS debe cubrir todos los aspectos físicos y funcionales definidos en el URS y se debe identificar algún incumplimiento con el URS.
La GAMP4 menciona que dependiendo de las circunstancias del proyecto algunos detalles de la funcionalidad del sistema no pueden ser definidos completamente hasta que ciertas etapas del proyecto sean alcanzadas, y que la documentación de esto puede ser hasta la elaboración de la especificación de diseño. Es importante mencionar que este documento debe ser actualizado bajo un control de cambios y durante toda la vida del sistema. Todos los cambios que se hagan al sistema, por pequeños que sean, deben ser establecidos bajo requerimientos en su URS y FS.
La GAMP4 recomienda seguir lo siguiente al desarrollar un FS:
Todas las limitaciones deben ser definidas explícitamente. Las ambigüedades, duplicaciones y contradicciones deben ser evitadas. Deben usarse convenciones al nombrar. Cada función descrita debe ser probada. La especificación debe ser lo suficientemente clara de tal forma que cada
requerimiento pueda ser rastreable de manera individual.
El contenido que nos recomienda la GAMP4 para un FS es:
1. Introducción. 2. Sección de generalidades o vista rápida. 3. Sección de Funciones. 4. Sección de Datos. 5. Sección de Interfases. 6. Sección de Atributos no funcionales. 7. Sección de Glosario. 8. Apéndices.
Mi propuesta es tener un solo documento para la especificación de requerimientos de usuario y la especificación funcional. De modo que en lugar de tener dos documentos independientes como lo plantea la GAMP4 yo propongo tener solo uno que contemple los 2 contenidos. Donde la primera versión corresponda a establecer los requerimientos de usuario y el mismo documento en una segunda versión indique la especificación funcional para cumplir con los requerimientos del usuario. A

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 46 de 125
continuación les presento la plantilla que propongo para el contenido de un URS y un FS en un mismo documento.
PLANTILLA 2. PROPUESTA DE CONTENIDO DE UNA ESPECIFICACIÓN DE REQUERIMIENTOS (RS, Requirements Specification)
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo. Esta sección de firmas tiene relación con la sección de descripción de roles y responsabilidades, ya que el número de firmas debe asegurar el establecimiento de todos los requerimientos.
5. Introducción. Esta sección debe dar un bosquejo general del contenido del documento, recomiendo describir los antecedentes del proyecto y de una manera breve en que consistirá el proyecto y que características se requieren del sistema.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 47 de 125
6. Objetivo. En esta sección se definen el o los objetivos del documento. Es importante que estos sean precisos para no caer en ambigüedades.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo.
8. Referencias.
Es importante mencionar documentos con los que tenga relación este documento que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, VMP’s, etcétera.
9. Descripción de los roles y responsabilidades.
La GAMP4 señala que en esta sección se deben describir los roles y responsabilidades como mínimo del Líder o administrador del Proyecto, del departamento de Aseguramiento de Calidad y de los Dueños del sistema. Se pueden incluir tantas áreas firmantes como sean necesarias para el cumplimiento del objetivo del proyecto.
10. Descripción general del Sistema.
Esta sección es utilizada para proporcionar una descripción general de la arquitectura y funciones que se pretende tenga el sistema.
11. Requerimientos de usuario.
Esta sección es de suma importancia debido a que es la base para que el proveedor diseñe el sistema y para que las pruebas que se realizaran al sistema sean definidas. Es la esencia del documento. Es importante hacer hincapié en que cada uno de los requerimientos debe ser definido de tal manera que se asegure que pueda ser probado. Esto es porque algunas veces se solicita un requerimiento muy ambiguo. Además de que deben ser identificados por un número único y deben ser numerados de tal manera que puedan ser actualizados ya sea por adición o por eliminación de un requerimiento.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 48 de 125
Al numerar los requerimientos que tiene el usuario, los podemos clasificar como sigue:
Requerimientos funcionales.
Aquí se tienen que definir las funciones del sistema requeridas y el modo de operación que se necesita. Se debe incluir la descripción del proceso incluyendo el desempeño y el manejo de tiempo que se requiera. Si se tiene una filosofía de control como requerimiento debe ser incluida aquí. También es importante mencionar los requerimientos en caso de falla del sistema y la seguridad que debe cumplir. Se recomienda mencionar cuales funciones son críticas.
Requerimientos de software.
En esta sección se deben describir las características y funciones con las que debe contar el software para el sistema.
Requerimientos de hardware.
Si se conocen los requerimientos de hardware se deben incluir en esta sección, de lo contrario al tener establecidos los requerimientos funcionales y los de software, los requerimientos de hardware podrán ser definidos.
Requerimientos de datos.
La descripción de los requerimientos de datos incluye:
Capacidades de almacenamiento. Velocidades de transmisión de datos. Acceso a los datos. Definición de datos críticos, incluyendo rangos, límites. Características de respaldo. Seguridad e integridad de la información, se recomienda cumplir los
requerimientos del CFR 21 parte 11.
Requerimientos de interfases.
Es este punto se deben definir los requerimientos de interfases, como lo son las interfases de usuarios, las interfases con otros sistemas y las interfases con otros equipos, dispositivos, etc. Si se tiene un protocolo especial o específico dentro de la empresa que deba ser utilizado debe mencionarse en este punto.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 49 de 125
Requerimientos ambientales.
Aquí se debe describir las características ambientales que se requieren en el área donde estará el sistema, especificando las variables a controlar y los rangos.
Requerimientos de usuarios.
Los requerimientos de usuarios como son: niveles de usuarios y privilegios de cada nivel deben ser especificados para que sean contemplados por el diseñador del sistema. Se deben tomar en cuenta todos los usuarios que tendrán interacción con el sistema, por ejemplo para su operación, mantenimiento, soporte y administración.
Requerimientos de seguridad.
Se deben especificar los factores de seguridad que se requiere cumpla el Sistema, factores como protección del software y hardware contra accesos accidentales o malintencionados para su uso, modificación, destrucción y divulgación.
Requerimientos de reportes.
Se pueden definir las características que deben cubrir los reportes que se requiere proporcione el Sistema; por ejemplo, formatos, nombres, información, forma de presentar la información como gráficas, tablas, etc.
Requerimientos de Auditoría de Rastreo (Audit Trail), firmas y registros
electrónicos. En el caso de que se requiera cumplir con estos puntos de CFR 21 Parte 11 de la FDA.
Requerimientos generales adicionales.
Esta sección es para incluir los requerimientos que no caigan en ninguna de las clasificaciones anteriores o requerimientos deseables.
12. Restricciones y dependencias.
En esta sección se deben mencionar los factores que afectan los requerimientos solicitados al proveedor. Factores como restricciones y dependencias que existan en la Empresa, algunos ejemplos son los siguientes:
Escalas de tiempos. Si se cuenta con hardware que pueda o deba ser utilizado. Plataformas de Sistemas Operativos que deban ser empleados.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 50 de 125
Políticas, estrategias, reglas. Capacidad de expansión. Tiempo de vida esperado. Disponibilidad del nuevo sistema.
13. Ciclo de Vida.
En esta sección se pueden definir los requerimientos del desarrollo de Ciclo de Vida. Estos requerimientos pueden ser para que los cumpla el proveedor al desarrollar el sistema o para que conozca cómo se lleva el proceso de validación en la Empresa. Se pueden tocar puntos como los siguientes:
Estándares, procedimientos, guías, políticas, que deban cumplir al desarrollar el sistema.
Pruebas mínimas que deben entregar para la aceptación de sus trabajos. Entregables que se les solicite elaborar. Documentos como manuales, planos, programación. Capacitaciones que deben ser impartidas. Si se requiere soporte posterior a la implementación.
A partir de esta sección será la segunda versión.
14. Funciones requeridas del Sistema.
Esta sección debe describir las funciones que serán suministradas por el sistema para que se cumplan los requerimientos del usuario, la descripción debe ser con el más alto nivel de detalle en su descripción. Aquí se deben describir las funciones incluyendo su modo de operación. Para la descripción de estos requerimientos la GAMP4 nos recomienda:
El objetivo de la función, los detalles de su uso, incluyendo su interfase con otras partes del sistema.
Desempeño: como respuesta, exactitud, rendimiento, transferencia de datos. Estas características deben ser cuantificables y no ser ambiguas.
Seguridad y garantía: estos puntos deben incluir acciones en el caso de falla del software o hardware seleccionado, revisión o validación de datos de entrada, redundancia, restricciones de acceso, tiempos de salida y recuperación de datos.
Funciones que son configurables. Los requerimientos funcionales deben tener rastreabilidad con los
requerimientos de usuarios, para asegurar su cumplimiento.
15. Datos requeridos en el sistema.
Esta sección debe definir los datos con los cuales el sistema trabajará e interactuará y los siguientes puntos deben ser considerados:

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 51 de 125
Definición de los datos, de manera jerárquica y explicando cómo los objetos complejos se convierten o construyen objetos simples. Los parámetros críticos deben ser subrayados o resaltados.
Accesos.
Capacidad de datos, tiempo de retención y detalles de archivo de los datos.
Integridad y seguridad de datos.
16. Interfases requeridas.
En esta sección se debe definir si hay alguna interfase del sistema, mencionando como es la interacción del sistema con esa interfase o la interacción entre los sistemas. Es de gran ayuda describir que provee esa interfase y que requiere, así como también tomar en cuenta todas las interfases que puedan existir: interfases con el usuario, entre sub-sistemas, con otros equipos, con otros sistemas. Para el caso de que la interfase sea parte de un sistema GMP la seguridad de la misma es de gran relevancia. Los aspectos que se deben considerar para la definición de las interfases según la GAMP4 son:
Datos de recepción y de envío. Tipo de datos, formato, rangos y significado de las variables. Velocidad de transferencia de datos. Protocolo de comunicación. Compartimiento, creación, duplicación, uso, almacenamiento o destrucción
de datos. Mecanismos de llamado y de interrupción. Comunicación a través de parámetros, datos comunes entre áreas o
mensajes. Acceso directo a datos internos. Posibles errores, recuperación y reporte. Seguridad.
17. Atributos no funcionales requeridos.
En esta sección se debe contemplar y definir los requerimientos no funcionales como disponibilidad, confiabilidad, redundancia, control de errores, mantenimiento, expansión, mejoras, saturación de la capacidad, cambios en el ambiente o entorno, tiempo de vida, etc.
18. Glosario, Siglas y Términos Esta sección contiene la definición de los términos que pueden no ser familiares para el lector.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 52 de 125
19. Anexos
En esta sección se puede incluir información necesaria para la completa comprensión del documento.
Después de la propuesta para Especificación de Requerimientos, vamos a analizar los modelos que nos ofrece la GAMP4 para los diferentes tipos de sistemas para la fase de Planeación / Especificación.
Como podemos ver para los Sistemas IT se tiene el siguiente modelo:
Figura 3.2. Planeación y Especificación – Sistemas IT.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 53 de 125
Para los sistemas integrados y los autónomos es el mismo modelo en esta fase y es el siguiente:
PLAN DE
VALIDACIÓN
ESPECIFICACIÓN DE
REQUERIMIENTOS DEL USUARIO
EVALUACIÓN DEL PROVEEDOR
PARÁMETROS CRÍTICOS DE
PROCESO
APROBACIÓN DE LA
ESPECIFICACIÓN
EVALUACIÓN DEL PROVEEDOR
PLANEACIÓN Y DEFINICIÓN
Figura 3.3. Planeación y Especificación – Sistemas Control de Proceso.
Como se puede observar para ambos modelos se contempla el Plan de Validación, la especificación de requerimientos de usuario, la evaluación del proveedor y una aprobación de la especificación funcional. La diferencia en los modelos es que para los Sistemas IT es contemplado un Plan del Proyecto y Calidad y para los Sistemas de Control de Proceso no.
Y entonces el modelo de mi propuesta para esta etapa de Planeación y Especificación es:
Figura 3.4. Planeación y Especificación – Propuesta.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 54 de 125
3.4 PLAN DEL PROYECTO Y CALIDAD (QPP, QUALITY AND PROJECT PLAN).
Este QPP se elaborará después de la primera versión del RS, donde se especificaron los requerimientos del usuario y después de haber elegido al proveedor o desarrollador del proyecto. Después de que esté aprobado el QPP se debe seguir con la Especificación de Requerimientos segunda versión para aprobar los requerimientos funcionales.
Tanto el Plan de Proyecto como el Plan de Calidad son un acuerdo entre el usuario y el proveedor y son documentos con un carácter contractual.
El Plan de Calidad define las acciones, entregables, responsabilidades y procedimientos que cumplirán con los requerimientos de calidad y de validación del usuario. Mientras que el Plan de Proyecto detalla las fases, actividades y tiempos del mismo. La GAMP4 aborda estos documentos en uno solo, propone una combinación de ambos Planes.
Este Plan dentro del Ciclo de Vida de los Sistemas tiene como objetivos definir:
Cómo será aplicado el Sistema de Administración de Calidad del proveedor
(QMS, Quality Management System). La organización del Proyecto. Actividades a ser realizadas. Responsabilidades y tiempos. Acuerdos para los entregables comprometidos. Ajustes en la configuración. Personalización del sistema. Software personalizado.
Las secciones que la GAMP4 recomienda para este documento son:
1. Sección de Introducción. 2. Sección de generalidades o vista rápida. 3. Sección del Plan de Calidad (Requerimientos de Calidad del Usuario y
Sistema de Calidad del Proveedor). 4. Sección del Plan del Proyecto (Organización del Proyecto, Entregables y
Actividades).
Para la emisión de este Plan, la GAMP4 marca algunas responsabilidades que debe tener el usuario; entre ellas están:
Asegurarse de que todos los requerimientos definidos en el VP estén
cubiertos dentro de este Plan.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 55 de 125
Revisar y aprobar el Plan para asegurar que durante todas las etapas del proyecto se cumplirá con los requerimientos de negocio, calidad y cumplimiento.
Monitorear el cumplimiento del proveedor con las actividades comprometidas a través de revisiones, reportes y auditorías.
Revisar periódicamente el Plan para asegurar su cumplimiento, de lo contrario asegurarse de su actualización para reflejar los cambios en los acuerdos de ambas partes.
Asegurarse que todos los entregables están completamente definidos. Establecer un acuerdo dentro del Plan para las actividades de instalación y
entrega. Revisar, en la entrega, que todos los requerimientos contractuales han sido
cubiertos particularmente los de tipo regulatorio. Asegurarse de que todas las deficiencias y limitaciones han sido
identificadas y documentadas y que un plan de acción haya sido acordado para dar seguimiento a estos puntos.
Acuerdos envueltos con partes como mantenimiento, procedimientos de identificación de fallas y acciones correctivas.
Analizando las figuras 2.7 (Modelo de Ciclo de Vida para la validación de Sistemas IT), 2.9 (Documentación y Actividades de Ciclo de Vida para Sistemas Integrados (Embedded Systems)) y 2.10 (Documentación y Actividades de Ciclo de Vida para Sistemas Autónomos (Standalone Systems)) donde se muestra el modelo del ciclo de vida para los sistemas IT, Integrados y Autónomos, respectivamente, se puede observar que la GAMP4 para los sistemas IT si contempla como tal un Plan de Proyecto y Calidad (después del Plan de Validación, de la Especificación de Requerimientos de Usuario y de la Evaluación del Proveedor), para los sistemas Integrados no menciona nada de Plan de Proyecto y Calidad y para los sistemas Autónomos sólo menciona un Plan de Calidad con Contratistas y de Proveedores (en la etapa de Acuerdos de Diseño).
No veo alguna razón porque la GAMP4 no contempla este Plan en la figura 2.9 de Documentación y Actividades de Ciclo de Vida para Sistemas Integrados, da a entender que se tiene que realizar ese Plan para establecer esa relación contractual entre el Proveedor y el Cliente aunque no lo especifica como tal en el diagrama. Esto porque en la sección de Planeación para Sistemas de Control de Procesos hace referencia a que el Proveedor debe desarrollar dicho Plan de Proyecto y de Calidad, aunque no da mas información y/o descripción del mismo.
Otro punto por el cual pienso que no lo contempla es porque un sistema Integrado está más delimitado y puede no ser tan grande en comparación con un Sistema Autónomo o un IT. Entendiendo por delimitado que es un sistema que consta de un microprocesador, un controlador o un equipo que solo se configura o se programa pero no se personaliza para las necesidades de la empresa y por lo tanto no sea necesario hacer ese Plan del Proyecto y de Calidad. Es decir, esos equipos los tienen en muchas empresas como una pieza específica o un equipo analítico, no como un

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 56 de 125
sistema personalizado y diseñado específicamente, que es único y sólo una empresa lo tiene.
Partiendo de la información del párrafo anterior y de mi experiencia lo que propongo es incluir este documento (Plan de Proyecto y de Calidad) para cualquiera que sea el tipo de sistema debido a la importancia de comprometer al Proveedor con el cumplimiento de los lineamientos marcados en el VP y acordar las fechas de entrega de avances y finalización del proyecto. Mas bien lo extenso del documento y las secciones que contenga dependerán de lo robusto del sistema y del proyecto.
La importancia de este documento es asegurar la Calidad durante todo el proceso del proyecto, llegar a acuerdos escritos con el proveedor permite hacerlo adquirir responsabilidad con las fechas y en cumplir con lo dicho, al mismo tiempo de tener por escrito cual será el proceso de revisión de diseño para cumplimiento del proyecto de Norma PROY-NOM-059-SSA1-2004.
Además agregaré otra sección a este documento y será el Plan de Revisión de Diseño. Por el momento solo explicaré cual es el objetivo de incluirlo aquí y en que consiste, en la sección 3.7 de Pruebas al equipo o sistema abordaré a fondo este tema de Revisión de Diseño.
Por ahora me limito a mencionar que el proyecto de Norma PROY-NOM-059-SSA1-2004 indica que es necesario tener una calificación del Diseño y demostrar que el equipo cumple con las especificaciones de Diseño. Para cumplir con este punto dentro de mi propuesta está elaborar un Plan donde se defina como se realizará este proceso de Revisión de Diseño y propongo incluirlo como otra sección en el mismo Plan del Proyecto y Calidad.
Para proyectos que involucren la configuración de paquetes comerciales los proveedores deberán saber las condiciones o requerimientos de las revisiones. Esto debe ser verificado desde la evaluación del proveedor. Las revisiones de diseño por parte del usuario deben enfocarse en las actividades de configuración y personalización.
Las revisiones de Diseño evalúan los entregables contra los estándares y requerimientos, identificando problemas y proponiendo las acciones correctivas requeridas. Están planeadas y revisan sistemáticamente las especificaciones, el diseño y el desarrollo y deben ser planeadas para ocurrir en etapas apropiadas durante el ciclo de vida. Son una parte importante en el proceso de verificación.
La GAMP4 recomienda tomar en cuenta los siguientes puntos para el Plan de Revisión del Diseño:
El alcance y los objetivos de la revisión. Qué método o proceso se seguirá. Quienes estarán involucrados. Cuáles serán los entregables de estas pruebas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 57 de 125
La profundidad y el rigor del proceso de revisión debe reflejar el impacto GMP del Sistema, su complejidad y la experiencia del proveedor y del equipo del proyecto.
PLANTILLA 3. PROPUESTA DEL PLAN DEL PROYECTO Y CALIDAD
Esta plantilla propuesta debe ser usada para definir los Planes del Proyecto y de Calidad para proyectos desarrollados por proveedores, incluyendo el proceso o plan de revisión del Diseño del Sistema. Este plan es un documento clave contractual y por lo tanto debe ser aprobado por los representantes autorizados adecuados del proveedor y usuario.
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 58 de 125
5. Introducción. Esta sección debe dar un bosquejo general del contenido del documento, recomiendo describir los antecedentes del proyecto y de una manera breve en que consistirá el proyecto.
6. Objetivo.
En esta sección se definen el o los objetivos del documento. Es importante que estos sean precisos para no caer en ambigüedades.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo.
8. Referencias.
Es importante mencionar documentos con los que tenga relación este documento que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, VMP’s, etcétera. También se pueden mencionar normas y guías nacionales o internacionales que se deban cumplir.
9. Descripción de los roles y responsabilidades.
En esta sección se deben describir los roles y responsabilidades como mínimo del Líder o administrador del Proyecto, del departamento de Aseguramiento de Calidad y de los Dueños del sistema. Se pueden incluir tantas áreas firmantes como sean necesarias para el cumplimiento del objetivo del proyecto.
Plan de Calidad Esta sección debe ser elaborada para definir las actividades de verificación de la validación y de la calidad, las responsabilidades y los procedimientos que se seguirán.
10. Requerimientos de Calidad del usuario
Se debe listar todos los requerimientos de calidad del usuario de la compañía. Los requerimientos de calidad del usuario tienen prioridad y están sobre el sistema de calidad del proveedor. En el caso de desarrollo de código fuente describir los requerimientos para el control y administración que se debe llevar conforme el sistema del Cliente.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 59 de 125
Se debe listar todos los requerimientos de calidad del usuario de la compañía. Los requerimientos de calidad del usuario tienen prioridad y están sobre el sistema de calidad del proveedor.
11. Sistema de Calidad del proveedor
Esta sección debe describir cómo el proveedor cumple con los requerimientos de calidad del usuario. Se deben definir las actividades a ser emprendidas, los procedimientos a seguir y las responsabilidades. Las actividades a considerar son:
Especificaciones. Revisiones de diseño. Estándares de programación y revisión de códigos. Pruebas. Instalación. Migración de datos. Pruebas de aceptación. Administración de documentos. Control de cambios. Administración de la configuración. Administración de problemas y riesgos. Capacitación.
Plan de Revisión del Diseño Esta sección debe ser elaborada para definir y acordar con el proveedor el proceso de revisión del Diseño del Sistema.
12. Alcance de la revisión del Diseño
Esta sección debe describir el alcance que tendrán las revisiones del diseño.
13. Método o acuerdos para la revisión del Diseño
Aquí se debe mencionar qué método se seguirá para llevar a cabo las revisiones y cuáles serán los entregables de estas pruebas y/o revisiones. Se puede incluir cual será el medio de comunicación, los períodos de revisión y asistentes a las revisiones.
La frecuencia de las revisiones dependerá de la naturaleza del proyecto, sin perder de vista la importancia de las mismas.
Se recomienda llevar un control de seguimiento de las reuniones, revisiones, pendientes y responsables; por ejemplo por medio de minutas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 60 de 125
Plan del Proyecto Por cada proyecto se debe generar un plan
14. Organización del proyecto
Esta sección debe incluir un organigrama que muestre:
El equipo del proyecto del proveedor y sus puestos. El contacto del proveedor para propósitos de que las quejas del cliente puedan ser incluidas.
La interfase entre el equipo del proyecto del proveedor y su función de aseguramiento de calidad dentro de la organización.
Usuarios quienes serán los contactos de la Compañía.
15. Entregables
En esta sección se deben definir todos los entregables comprometidos para emitir en el proyecto o para el sistema. Se tiene que mencionar como serán identificados y en que formato se harán. Si la empresa tiene procedimientos o guías donde se especifiquen estos puntos solo se tendrá que hacer referencia a ellos.
16. Actividades
Para esta sección se puede utilizar un Diagrama de Gantt. Se debe definir en esta sección los siguientes aspectos:
Puntos clave del proyecto. Actividades del proyecto, incluyendo revisión de diseño y de códigos
fuentes. Responsables de las actividades. Fechas planeadas para el inicio y termino de cada actividad. Dependencia de actividades.
17. Glosario, Siglas y Términos
Esta sección contiene la definición de los términos que pueden no ser familiares para el lector.
18. Anexos En esta sección se puede incluir información necesaria para la completa comprensión del documento.
Conforme el diagrama de mi propuesta (Figura 3.1) al término del Plan del Proyecto y Calidad debe seguir la segunda versión del RS. Para el momento en que se tenga que emitir la segunda versión del RS, el Proveedor ya debe estar seleccionado y los acuerdos con el mismo establecidos en el QPP. Por lo tanto en esta etapa el Proveedor

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 61 de 125
debe hacer formal su propuesta con la especificación funcional del sistema cumpliendo con los acuerdos y responsabilidades indicados en el QPP. Después de que el Proveedor presente su propuesta y ésta sea aprobada por el Laboratorio, se debe iniciar con la Matriz de Rastreabilidad de Requerimientos, documento mostrado en la siguiente sección y posteriormente con la Especificación de Diseño abordada en la sección 3.6.
3.5 MATRIZ DE RASTREABILIDAD DE REQUERIMIENTOS.
En la terminología de la ISO la Rastreabilidad es la habilidad de trazar la historia, aplicación o ubicación de lo que está bajo consideración.
De acuerdo con la GAMP4 la rastreabilidad es el establecimiento de la relación entre 2 o más productos del proceso de desarrollo, por ejemplo, el punto en que encajan los requerimientos y el diseño de un componente de software.
El proceso de la rastreabilidad permite asegurar:
Que todos los requerimientos son cumplidos y que el diseño apropiado de los elementos pueden ser rastreado.
Que todos los requerimientos son verificados y que las actividades de verificación o prueba pueden ser rastreadas para demostrar que los requerimientos han sido cumplidos.
En la terminología de la ISO, la rastreabilidad muestra que las entradas del diseño están ligadas a las salidas del diseño y que han sido verificadas. Así como la Matriz de Rastreabilidad verifica y demuestra que todo el diseño está cubierto, también puede ayudar considerablemente a la evaluación y administración de cambios.
La información de este proceso es regularmente plasmada en forma tabular, de ahí su nombre Matriz de Rastreabilidad. La Matriz puede tomar varias formas, dependiendo del detalle de sus objetivos y del alcance que se tenga que cubrir. Para un sistema o proyecto se pueden elaborar más de un tipo de matriz.
La elaboración de las Matrices de Rastreabilidad debe iniciar en la etapa de especificación en conjunto con la Especificación de los Requerimientos del Usuario y debe ser actualizada y mantenida durante el desarrollo del ciclo de vida a través de la aprobación de las especificaciones finales de prueba (revisión de diseño y protocolos de calificación). Es decir con cada uno de los siguientes documentos otra columna de la matriz deberá ser actualizada: Requerimientos Funcionales, Especificación y revisión de Diseño y Protocolos de Calificación de Instalación, Operación y Diseño.
La matriz se puede actualizar en cada etapa del Ciclo de Vida y aprobarse completa al tener la aprobación para ejecución de los protocolos de calificación. La matriz terminada puede pasarse a firmas de aprobación junto con los protocolos de

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 62 de 125
calificación a los involucrados para que puedan verificar que todos los requerimientos fueron retados exitosamente. La matriz debe resguardarse como evidencia de que los requerimientos han sido implementados y retados. Esta matriz debe ser mantenida como parte del Control de Cambios Operacional de tal manera que continúe reflejando el sistema actual.
La estructura que se le de a los documentos a los cuales se hará referencia en la matriz de rastreabilidad es de importancia ya que puede tener un impacto en el proceso de rastreabilidad.
Al igual que los documentos de requerimientos y de diseño se puede elaborar más de una matriz de rastreabilidad para el proyecto o sistema, dependiendo si se dividió al sistema en sub-sistemas o se maneje todo en uno solo.
PLANTILLA 4. PROPUESTA DE LA MATRIZ DE RASTREABILIDAD DE REQUERIMIENTOS
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 63 de 125
5. Introducción. Esta sección debe dar un bosquejo general del contenido del documento y del proyecto.
6. Objetivo.
En esta sección se definen el o los objetivos del documento. Es importante que estos sean precisos para no caer en ambigüedades.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo.
8. Referencias.
Es importante mencionar documentos con los que tenga relación este documento que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, VMP’s, Especificaciones de Requerimientos, de Diseño, Protocolos, etcétera.
9. Descripción de los roles y responsabilidades.
En esta sección se deben describir los roles y responsabilidades como mínimo del Líder o administrador del Proyecto, del departamento de Aseguramiento de Calidad y de los Dueños del sistema. Se pueden incluir tantas áreas firmantes como sean necesarias para el cumplimiento del objetivo del proyecto.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 64 de 125
10. Matriz de Rastreabilidad.
A continuación se muestra la matriz de rastreabilidad de los requerimientos del Sistema donde se podrá observar que todos los requerimientos del usuario fueron diseñados y retados exitosamente.
1. Requerimiento del Usuario (número del requerimiento y descripción breve)
2. Requerimiento Funcional (número del requerimiento y descripción breve)
3. Especificación de Diseño (número de la sección y descripción breve)
4. Pruebas de Aceptación IQ (número de la prueba y título de la misma)
5. Pruebas de Aceptación OQ (número de la prueba y título de la misma)
6. Pruebas de Aceptación PQ (número de la prueba y título de la misma)
1.0
2.0
2.1.1
2.1.2
1. Requerimiento del Usuario
Todos los requerimientos deben ser listados para dar seguimiento a su rastreabilidad, haciendo referencia a su identificador único.
2. Requerimiento Funcional
Colocar la referencia al requerimiento funcional que cumple con el requerimiento del usuario.
3. Especificación de Diseño Colocar la referencia al elemento o sección de los documentos de diseño que cumple con el requerimiento previamente establecido.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 65 de 125
4. Calificación de Instalación Colocar la referencia a la(s) prueba(s) del protocolo que retan el requerimiento previamente establecido.
5. Calificación de Operación
Colocar la referencia a la(s) prueba(s) del protocolo que retan el requerimiento previamente establecido.
6. Calificación de Desempeño Colocar la referencia a la(s) prueba(s) del protocolo que retan el requerimiento previamente establecido.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 66 de 125
11. Glosario, Siglas y Términos
Esta sección contiene la definición de los términos que pueden no ser familiares para el lector.
12. Anexos En esta sección se puede incluir información necesaria para la completa comprensión del documento.
3.6 ESPECIFICACIÓN DE DISEÑO (DS, DESING SPECIFICATION).
El siguiente documento que sugiere la GAMP4 es la Especificación de Diseño, lo cual coincide con mi experiencia y cumple con una secuencia lógica. Después de tener los requerimientos que debe cumplir el sistema se tienen las bases para elaborar la configuración y el diseño del mismo.
Una Especificación del Diseño es el documento donde se describe cómo el sistema o equipo cumple con los requerimientos establecidos a fin de asegurarse que se cumplen las funciones para lo que fue planeado. Esta Especificación debe contener el suficiente detalle para permitir la construcción y mantenimiento del sistema.
Dentro de esta Especificación se tienen ciertas partes del sistema para describir. Por ejemplo la sección de hardware debe describir el hardware donde operará el software y cómo será conectado a otro sistema existente, interfases o a equipos instalados en Planta.
La sección de Software debe incluir una descripción de los componentes, módulos, aplicaciones o sub-sistemas que conformarán el software que cumplirá con las funciones requeridas del Sistema. Cada módulo de software debe ser descrito de tal manera que se cuente con toda la información para llegar a la elaboración del código.
En el caso de que el sistema incluya el diseño de una red se deben describir los elementos físicos, el protocolo de comunicación, la interacción con el hardware, otros sistemas u otras redes. Esto incluye el tipo de cables, interruptores, servidores, clientes, fuentes de energía, tarjetas, conexiones. Las redes, dentro de lo posible, deben ser construidas con dispositivos estándar y contar con procedimientos de mantenimiento definidos y aceptables.
La GAMP4 para presentar el ejemplo de lo que debe contener una Especificación de Diseño la divide en 2 partes; una plantilla para el software y una plantilla para el hardware.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 67 de 125
Dentro de la Plantilla de Diseño de Hardware (HDS, Hardware Design Specification) recomienda incluir:
1. Introducción. 2. Sección de generalidades o vista rápida. 3. Sección de Sistemas de Cómputo. 4. Sección de Entradas y Salidas. 5. Sección de ambiente. 6. Sección de suministro eléctrico. 7. Glosario. 8. Y para los Sistemas Integrados de Control de Proceso menciona que se debe
incluir: diagramas eléctricos, P&ID, diagramas de los paneles de control y de ubicación de equipos.
Para el ejemplo de la Especificación de Diseño del Software (SDS, Software Design Specification) la GAMP4 hace una división más que es la de hacer una Especificación de Diseño de Software y una Especificación de Diseño del módulo de Software y para ellas recomienda incluir:
Especificación de Diseño de Software:
1. Introducción. 2. Sección de generalidades o vista rápida. 3. Sección de descripción del Sistema. 4. Sección de datos del Sistema. 5. Sección de descripción del módulo. 6. Glosario.
Especificación de Diseño del Módulo del Software:
1. Introducción. 2. Sección de generalidades o vista rápida del módulo. 3. Sección de datos del módulo. 4. Sección de descripción del sub-programa. 5. Sección de datos del sub-programa. 6. Glosario.
La GAMP4 para esta Especificación de Diseño de Software señala que si es un Sistema Integrado de Control de Proceso se debe incluir: diagramas de flujo, una descripción detallada de los algoritmos usados y la descripción del contenido de las pantallas empleadas.
Mi propuesta para elaborar un documento de Especificación de Diseño es tener un solo documento donde se incluyan todos los aspectos de diseño, como son sus elementos de hardware y software, su configuración, sus entradas, salidas, conexiones, interfases, diagramas, planos, etc. De tal manera que no se tenga que emitir 2 o 3 tipos de documentos de diseño para un solo Sistema. Aunque coincido con la flexibilidad de poder dividir un Sistema que sea muy grande en sub-sistemas mas pequeños para facilitar la visualización y modelo de su diseño.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 68 de 125
Algo que observo en la GAMP4 es que marca una diferencia con los Sistemas Integrados al recomendar incluir ciertas secciones solo si se trata de ese tipo de Sistemas, en mi experiencia desarrollando este tipo de documentación lo que yo recomiendo es incluir ese tipo de secciones no solo para Sistemas Integrados, sino también para Sistemas Autónomos e incluso Sistemas IT. Las características del Sistema son las que marcan si es importante o no incluir secciones de diagramas de flujo, arquitectura del sistema, diagramas eléctricos, de control, de tubería e instrumentación, código fuente, planos de distribución de equipos en Planta, dentro de un Sistema o de una Máquina, entre otros.
Lo anterior lo puedo ejemplificar tomando como referencia un Sistema SCADA que cae en la clasificación de Sistemas Autónomos. Para la comprensión de un Sistema de este tipo y poder dar el soporte, mantenimiento, uso, mejoras, etcétera, es importante tener planos eléctricos, de ubicación de sensores en la Planta, diagramas de conexión de los controladores, diagramas de redes de control, sentidos de flujo, diseño de pantallas de monitoreo, que entre otros puntos son básicos para que el operador del sistema pueda interactuar con él y el sistema cumpla con las funciones para las que fue diseñado.
A continuación expongo mi propuesta para la Plantilla de Diseño de un Sistema donde incluyo todas las secciones que considero importantes para este documento. Tomé como base los ejemplos de la GAMP4 y los documentos que desarrollo en mi experiencia profesional. Recomiendo incluir diagramas con la estructura e interfases del sistema.
Conforme el Plan de Revisión de Diseño esta Especificación de Diseño deberá ser actualizada hasta tener que emitir la versión en base a lo que se probó y cumple exitosamente con lo requerido, es decir, la versión final será después de las pruebas de Revisión del Diseño.
PLANTILLA 5. PROPUESTA DE CONTENIDO DE UNA ESPECIFICACIÓN DE DISEÑO
Es importante que el cuerpo del documento de diseño esté de tal manera que se pueda hacer la relación con la especificación de requerimientos, es decir, que la rastreabilidad se pueda identificar de manera fácil y práctica.
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 69 de 125
Numero de identificación del documento. (Usualmente cada empresa tiene establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo. Esta sección de firmas tiene relación con la sección de descripción de roles y responsabilidades, ya que el número de firmas debe asegurar el cumplimiento con todos los requerimientos.
5. Introducción. Esta sección debe dar un bosquejo general del contenido del documento, recomiendo describir los antecedentes del desarrollo del sistema.
6. Objetivo.
En esta sección se definen el o los objetivos del documento. Es importante que estos sean precisos para no caer en ambigüedades.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo. En esta sección se puede mencionar el periodo en el cual se realizará la siguiente revisión al documento (si aplica).
8. Referencias.
Es importante mencionar documentos con los que tenga relación este documento que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, VMP’s, PV, URS, etcétera.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 70 de 125
9. Descripción de los roles y responsabilidades. La GAMP4 señala que en esta sección se deben describir los roles y responsabilidades como mínimo del Líder o administrador del Proyecto, del departamento de Aseguramiento de Calidad y de los Dueños del sistema. Se pueden incluir tantas áreas firmantes como sean necesarias para el cumplimiento del objetivo del proyecto.
10. Descripción del Sistema
En esta sección se deberá incluir la descripción general del Sistema incluyendo diagramas que faciliten el Diseño del mismo. Se puede incluir los módulos que formarán el sistema y la descripción del objetivo de cada uno de ellos. Así como las interfases entre módulos y con sistemas externos.
11. Datos del Sistema
Esta sección debe mostrar un resumen de los datos del sistema y definir los objetos principales de los datos. Se recomienda que los datos sean definidos en un orden jerárquico y de formal tal que se pueda entender el proceso desde que se tiene los objetos simples hasta la construcción de los objetos más complejos y el sistema de datos final. La descripción puede incluir lo siguiente:
Bases de datos y archivos de donde colectan datos. Descripción de la estructura. Archivos. Registros. Tipos de datos.
La estructura de cada archivo y datos debe ser identificada de manera única.
12. Descripción de Módulos de Software o Programación
Para la descripción de cada módulo programado se debe incluir la siguiente información como base:
Nombre y versión. Marca. Operación del módulo (puede incluir el código). Interfases con otros módulos. Factores de tiempo específicos. Verificación o validación de datos y aviso de errores. Que datos son tomados / utilizados por este módulo.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 71 de 125
Se recomienda mencionar los módulos u aplicaciones del software que no serán utilizados o configurados.
13. Datos del módulo del software
En esta sección debe incluir los datos del módulo y definir los objetos principales del mismo.
Si es necesario tener una sub-sección de módulos de software o programas para entrar en el detalle de su diseño se pueden llenar las siguientes secciones.
14. Descripción de sub-módulos o sub-programas
Por cada uno se debe contemplar la descripción de lo siguiente:
Operación del sub-programa (se puede incluir código). Parámetros de salida y entrada. Algún problema secundario que pudiera causar el sub-programa. Lenguaje de programación incluyendo versión. Referencia a estándares de programación.
15. Datos del sub-módulo o sub-programa
Aquí se puede describir los datos que sean propios de este sub-módulo o sub-programa, si aplica.
16. Interfases
Incluir en esta sección las características de las interfases internas y externas del Sistema. Mencionar los datos que convertirá o transferirá, además de referenciar otros sistemas que también utilicen estas interfases. Incluir las interfases con los usuarios.
17. Sistema de Cómputo
Esta sección será incluida para describir las características del hardware que conformará el Sistema y mostrar la configuración del mismo para cumplir con los requerimientos establecidos previamente. Esta descripción debe incluir:
Descripción del CPU (Unidad central de procesamiento): modelo, marca, memoria, velocidad de procesamiento.
Interfases de comunicación entre el CPU y demás componentes, por ejemplo tarjeta de red, puerto serial, puerto paralelo, etc.
Descripción del Controlador: modelo, marca, memoria, velocidad de procesamiento.
Capacidad de almacenamiento: tamaño de discos duros, capacidad de cintas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 72 de 125
Medios de almacenamiento: Disco de escritura, grabador de cintas. Periféricos: mouse, teclado, escáner, monitor, impresoras, plotters, lectores
de códigos de barras, de huella digital, tarjetas magnéticas, etcétera. Conexiones: especificaciones de los cables y de los conectores, diagramas
de conexiones y de red. Configuración detallada como arreglo de interruptores y direcciones de
dispositivos.
18. Seguridad
En esta sección se debe especificar como quedaron configurados los niveles y privilegios de usuario, describiendo los permisos que tiene cada uno de los usuarios dentro de la aplicación. Si el sistema tiene configurados parámetros de seguridad deben mencionarse en esta sección, parámetros como: mínimo o máximo número de caracteres para configurar una clave de acceso, tipo de caracteres que conformaran la clave, etc.
19. Entradas y Salidas.
Especificar en esta sección las entradas y salidas siguientes: DI (Digital Input, Entrada Digital). DO (Digital Output, Salida Digital). AI (Analog Input, Entrada Analógica). AO (Analog Output, Salida Analógica). Pulse counters (Contadores de pulsos).
20. Características de instrumentos.
Especificar las características de los instrumentos del sistema como:
Exactitud. Aislamiento. Rangos de voltaje y corriente. Tipo y número de tarjetas de interfase. Encendido.
21. Ambiente.
Describir el ambiente de operación del hardware, como temperatura, humedad, interferencias externas, seguridad física, interferencia de radio-frecuencia, o electromagnetismo.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 73 de 125
22. Suministro Eléctrico.
Describir las características requeridas de la alimentación eléctrica del hardware por ejemplo: filtros, carga, protección a tierra y alimentación ininterrumpible (UPS, uninterruptible power supply).
Esta sección aplica cuando la Especificación de Diseño es para un Sistema Integrado (Embedded System).
23. Detalle del Sistema u Sub-Sistema Integrado. Aquí se debe incluir lo siguiente:
Diagrama de flujo o alguna representación, donde se muestre cómo es alcanzada cada función individual de las máquinas (incluyendo generación de alarmas o advertencias y las estrategias de restauración de las mismas).
Descripción detallada de los algoritmos utilizados. Guía de mensajes. Descripción gráfica de todas las pantallas de interfase.
24. Diagramas / Planos.
En esta sección se debe incluir la siguiente información:
Diagramas de ubicación de los Equipos, dispositivos y sistemas dentro de la Planta.
Diagramas del panel de control y arreglos interiores y exteriores. Diagramas de localización de sensores e instrumentos en las máquinas. Diagramas de cableado eléctrico. Diagramas mecánicos, eléctricos, neumáticos, etc. Diagramas de Tubería e Instrumentación. Referencias a estándares, normas y/o guías relevantes al equipo o sistema
que se estén cumpliendo. Cumplir con las normas y/o nomenclatura de la ISA.
25. Restricciones y dependencias.
En esta sección se deben mencionar los factores que afectan los requerimientos solicitados al proveedor. Factores como restricciones y dependencias que existan en la Empresa, algunos ejemplos son los siguientes:
Escalas de tiempos, rangos de medición. Si se cuenta con hardware que pueda o deba ser utilizado. Plataformas de Sistemas Operativos que deban ser empleados. Políticas, estrategias, reglas. Capacidad de expansión.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 74 de 125
Tiempo de vida esperado. Disponibilidad del nuevo sistema.
26. Reportes.
Definir en esta sección los reportes que proporcionará el Sistema y sus características.
27. Respaldos Automáticos.
Especificar en esta sección, si aplica, la configuración que tendrá el sistema para la ejecución automática de los respaldos, incluyendo los datos a ser respaldados, la frecuencia del respaldo y el medio en el que se respaldará.
28. Glosario, Siglas y Términos
Esta sección contiene la definición de los términos que pueden no ser familiares para el lector.
29. Anexos En esta sección se puede incluir información necesaria para la completa comprensión del documento.
3.7 PRUEBAS AL EQUIPO O SISTEMA.
Para abordar está sección de pruebas al Equipo o Sistema, empezaré por hacer referencia a los siguientes puntos del Proyecto de Norma Oficial PROY-NOM-059-SSA1-2004 en su sección “Calificación”:
La primera etapa de la validación de los equipos y sistemas nuevos o
modificados es la Calificación del Diseño (CD). La Calificación de Instalación (CI) debe realizarse en sistemas y equipo nuevo o
modificado. La Calificación Operacional (CO) debe seguir a la CI. La Calificación de Desempeño (CE) debe seguir a la terminación satisfactoria
de la CI y la CE.
La Norma hace referencia al término Calificación como la evaluación de las características de los elementos del proceso.
De lo anterior puedo rescatar que para el cumplimiento de estos puntos de la norma es necesario ejecutar Protocolos de Calificación. La primera calificación que pide la norma es la de Diseño, el cual es un nuevo término para mí ya que en mi experiencia este documento de Calificación de Diseño no se contempla, solo se elaboran los de Calificación de Instalación, de Operación y de Desempeño. Antes de seguir con todo

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 75 de 125
el contenido de esta sección me gustaría hacer un paréntesis y analizar esta parte de la Calificación del Diseño ya que en mi propuesta de Validación de los Sistemas Automáticos y de Cómputo la incluiré como Pruebas de Revisión de Diseño pero desde el punto de vista de un proceso de revisión.
Investigando en la GAMP4 acerca de esta cuestión menciona que este término de Calificación de Diseño ha sido introducido recientemente dentro de la documentación y que tiene como propósito la verificación del diseño de sistemas, equipos e instalaciones.
En la GAMP4 el proceso que equivale a la Calificación de Diseño es la Revisión de Diseño, la cual involucra revisiones planeadas de especificaciones, diseño y desarrollo a lo largo del ciclo de vida. Estas revisiones de diseño evalúan los entregables contra los estándares y requerimientos, identificando problemas y proponiendo las acciones correctivas requeridas.
Mi propuesta para cumplir con la Calificación de Diseño que es solicitada en el proyecto de Norma Oficial PROY-NOM-059-SSA1-2004 es establecer un Plan de Revisión de Diseño y documentar estas revisiones a través de Pruebas de Diseño. El Plan de Revisión de Diseño permite revisar los avances en el desarrollo del proyecto, confirmar que se está cumpliendo con los requerimientos del usuario o del cliente y que se está dentro de los tiempos estimados. En la sección 3.4 Plan del Proyecto y Calidad, puntos 12 y 13 de la Plantilla 3, se describe a más detalle este Plan de Revisión y las características que debe cumplir. En esta sección de Pruebas nos enfocaremos en como podemos documentar estas revisiones.
Analizando lo referente a pruebas que nos presenta la GAMP4, se puede observar en la figura 2.7 (Modelo de Ciclo de Vida para la validación de Sistemas IT), que considera la elaboración de una Especificación de Prueba por cada Especificación de Diseño dentro de la etapa de Diseño y Construcción. Después de esa etapa considera otras pruebas en la etapa de Instalación que consisten en unas Pruebas de Aceptación de hardware y software. Y por último considera las Pruebas de Aceptación del Sistema Integrado.
Las Especificaciones de Pruebas para los Sistemas IT deben abarcar los siguientes elementos, conforme la GAMP4:
Especificación de Pruebas de Módulo(s) de Software. Especificación de Pruebas de Configuración de paquetes. Especificación de Pruebas de Integración de Software. Especificación de Pruebas de Aceptación de Red. Especificación de Pruebas de Aceptación de Hardware. Especificación de Pruebas de Aceptación del Sistema.
Las pruebas FAT son las Pruebas de Aceptación de Fábrica (Factory Acceptance Tests), las cuales son ejecutadas en las instalaciones del Proveedor antes de liberar el equipo o sistema para instalarlo y probarlo en el sitio final con el Cliente.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 76 de 125
Las pruebas SAT son las Pruebas de Aceptación en Sitio (Site Acceptance Tests) y son realizadas para verificar que el equipo / sistema opera de la manera correcta en su ambiente de operación real y comunicado con las interfases internas y externas.
Para los Sistemas de Control de Proceso la GAMP4 considera 3 tipos de pruebas, los cuales son: Pruebas de Desarrollo, las pruebas de Aceptación (FAT y SAT) y las Pruebas de Calificación. Además de la calibración e inspección de los instrumentos.
Como se puede observar en la figura 2.9 (Documentación y Actividades de Ciclo de Vida para Sistemas Integrados (Embedded Systems)) considera la elaboración de Especificaciones de Prueba y la ejecución de las Pruebas FAT dentro de la etapa de Revisión de Diseño y Pruebas. Después de esa etapa sigue la de Commissioning y Calificación donde incluye las Pruebas SAT de Aceptación y las Pruebas de Calificación del Sistema Integrado.
Como se puede observar en la figura 2.10 (Documentación y Actividades de Ciclo de Vida para Sistemas Autónomos (Standalone Systems)) considera la elaboración de Especificaciones de Prueba dentro de la etapa de Desarrollo y Diseño. Posteriormente se contemplan pruebas de hardware, software e integración en la etapa de Construcción del Sistema y Desarrollo de Pruebas. Después sigue la etapa de Revisión del Diseño y Pruebas de Aceptación (FAT). Para terminar en la etapa de Commissioning y Calificación con pruebas SAT y Calificaciones (IQ, OQ y PQ). Mi propuesta en esta sección de pruebas es describir el contenido que debe contemplar cada uno de los entregables de pruebas para la metodología, además de definir los tipos de entregables. Como lo expuse en los párrafos anteriores, entre los entregables de prueba que considera la GAMP4 están: pruebas de desarrollo, especificaciones de pruebas por módulos y de integración, pruebas de aceptación en fábrica, pruebas de aceptación en sitio, pruebas de calificación de instalación, operación y desempeño, commissioning y aspectos solo de calibración. Dentro de mi propuesta consideraré las Pruebas de Revisión de Diseño, Protocolos de Calificación de Instalación, de Operación y de Desempeño. Considero que las pruebas FAT pueden entrar en las Pruebas de Revisión de Diseño. Y por otro lado pienso que en lugar de hacer pruebas SAT y probablemente repetirlas en la calificación solo se realicen los protocolos de calificación donde quedarán asentados los resultados ya sean correctos o con fallas y la solución a las mismas.
Con lo anterior no quiero restar importancia a las pruebas FAT y SAT. Cada una de ellas tiene sus objetivos e importancia y para algunas Industrias tienen mucho valor. Incluso para equipos robustos y que no se tiene ninguna experiencia con ellos es conveniente realizar las pruebas FAT para tener el primer contacto con el equipo y no solo ver la operatividad sino aspectos de instalación. Las pruebas de Commissioning no están en el alcance de este documento ya que éstas van dirigidas a los sistemas de Impacto Indirecto.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 77 de 125
ESPECIFICACIÓN DE REQUERIMIENTOS (PRIMERA
VERSIÓN, USUARIO)
ESPECIFICACIÓN DE REQUERIMIENTOS (SEGUNDA
VERSIÓN, FUNCIONAL)
ESPECIFICACIÓN DE DISEÑO
CONSTRUCCIÓN DEL SISTEMA
CALIFICACIÓN OPERACIONAL
CALIFICACIÓN DE INSTALACIÓN
CALIFICACIÓN DE DESEMPEÑOValida
Verifica
Verifica
PRUEBAS DE REVISIÓN DE DISEÑO
Verifica
La relación que existe de cada tipo de protocolo con los documentos de las etapas de especificación y diseño se puede ver en la siguiente figura 3.5 (Relación entre Protocolos de Calificación y Documentos de Especificación), donde podemos observar que todo lo especificado es probado por la misma metodología que se lleva. Si observamos la figura 3.5, vemos como mediante las Pruebas de Diseño se verifica que el Diseño propuesto del sistema es conveniente para el propósito proyectado y se tiene la seguridad de que será construido en tiempo y de manera funcional. Después con la Calificación de Instalación se verifica que se cumple con el Diseño aprobado y en Sitio. Posteriormente con la Calificación de Operación se verifica que el equipo o sistema funciona como se esperaba. Y por último con la Calificación de Desempeño se valida que el equipo o sistema rinde efectiva y reproduciblemente a los requerimientos del usuario.
Figura 3.5. Relación entre Protocolos de Calificación y Documentos de Especificación.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 78 de 125
3.7.1 Principios fundamentales que deben seguir las pruebas
Las pruebas deben cumplir con los siguientes principios.
Las pruebas deben reflejar la arquitectura y la complejidad del sistema que está siendo desarrollado.
Las pruebas deben ser repetibles. Los resultados de las pruebas deben ser reproducibles y verificables. Los resultados de las pruebas deben ser documentados en hojas de pruebas
en el momento de la ejecución y de manera indeleble. Las hojas de pruebas deben incluir los resultados esperados y los criterios de
aceptación previamente definidos. Cada prueba debe contener un estado de pasa o falla que confirme el
resultado de la prueba y una sección para que el ejecutor de la prueba incluya su firma y fecha.
Las pruebas deben cubrir todas las áreas relevantes del sistema. Las pruebas deben ser planeadas y ejecutadas por personal calificado. Los incidentes encontrados durante las pruebas deben ser documentados y la
solución administrada. Probar todos los casos, es decir, contemplar las condiciones de error del
sistema además de los casos ideales. Si se requiere hacer alguna corrección se debe trazar una línea sobre lo que
se escribió erróneamente y poner las iniciales del documentador y la fecha. Las evidencias que se generen durante las pruebas deben ser anexadas,
firmadas por el ejecutor y tener la referencia cruzada al Paso de Prueba correspondiente.
Estos puntos son importantes porque con ellos podemos tener un protocolo de pruebas para referencia y poderlo tomar en un momento determinado para repetir las pruebas si se tiene algún problema con el sistema o para una posterior revisión del mismo. El Proyecto de Norma Oficial PROY-NOM-059-SSA1-2004 en su sección “Mantenimiento al Estado Validado” indica que en los Protocolos correspondientes se debe definir la vigencia de las calificaciones y validaciones. Además de que la vigencia de las Calificaciones de Ejecución o Desempeño y las validaciones no puede ser mayor a 5 años, al término de la cual debe llevarse a cabo la recalificación o revalidación. Por lo que es significativo mencionar que si se hacen cambios al equipo o sistema se debe llevar a cabo una recalificación o revalidación, que demuestre la funcionalidad del sistema.
Para los sistemas IT es usual que se trabaje en un sistema de pruebas y después se ponga en producción o tener incluso esos dos ambientes independientes para que

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 79 de 125
cualquier cambio se realice primero en el ambiente de pruebas y después se pase a producción ya que probó que es funcional y que no afecta. Otro punto de relevancia es que deben elaborarse procedimientos y/o guías donde se especifique como elaborar protocolos de calificación, como recolectar los resultados y corregir errores encontrados. Se puede tener el caso de que se tomen las pruebas del proveedor como pruebas de calificación, por ejemplo la migración de una base de datos y éstas deberán cumplir con lo especificado en los procedimientos y guías del cliente para que se puedan tomar como formales. Es común que se contrate a Proveedores especializados para el desarrollo exclusivo de este tipo de documentación.
A continuación describiré el objetivo, características y el contenido que propongo de los entregables de prueba para el cumplimiento de la validación de los sistemas, conforme mi propuesta.
3.7.2 Pruebas de Revisión de Diseño.
La Revisión del Diseño es la revisión formal principal de las especificaciones de Diseño, lo cual permitirá que el diseño cumpla los requerimientos del usuario. La definición que establece el proyecto de Norma Oficial PROY-NOM-059-SSA1-2004 para la Calificación de Diseño es la verificación documentada de que el diseño propuesto de las instalaciones, sistemas y equipo es conveniente para el propósito proyectado, por lo tanto la documentación de estas Pruebas de Revisión de Diseño debe asegurar:
Que los criterios para la validación final sean claros. Que las correcciones conduzcan a un resultado exitoso, es decir, que
cumplan con los criterios de aceptación del equipo o sistema, además de incluir el seguimiento de los errores encontrados, su solución y su cierre.
Que el diseño final esté disponible en tiempo y que satisfaga los requerimientos del cliente. Esto se logra ya que se va avanzando en el desarrollo de cada módulo o elemento y se revisa periódicamente.
En el caso de equipos, que funcionen correctamente antes de entregarlos al Cliente.
Que se compare el sistema que se está diseñando con planos, isométricos, Diagramas de Tubería e Instrumentación (DTI’s), diagramas de flujo, esquemas de hardware y redes.
Mi propuesta es que estas pruebas sean elaboradas y ejecutadas por el Proveedor o Desarrollador del Sistema durante el diseño del mismo, de manera que formen parte de las mismas revisiones y correcciones. Posteriormente en la fecha de revisión las deberá presentar al equipo del Proyecto del Cliente quién las revisará y hará sus comentarios acerca de si las pruebas y el avance satisfacen lo acordado para ese periodo de tiempo.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 80 de 125
Estas revisiones conducirán a cada nivel de detalle de la especificación. Cada revisión debe involucrar los usuarios finales, diseñadores y Aseguramiento de Calidad para que los resultados de la revisión cumplan con los requerimientos. Por lo tanto la aceptación de los Reportes de Revisión del Diseño por parte de los interesados, preferentemente a través de firmas, es muy importante. Este proceso de revisión no deberá permitir que se siga adelante en el proceso de validación del sistema hasta estar seguros de que los resultados de las revisiones son exitosos. Una vez que el Equipo del proyecto determine que los resultados de las Pruebas de Revisión de Diseño son favorables para cubrir los requerimientos del usuario se deberá firmar la aprobación de la instalación del equipo o sistema en Sitio. Esta aprobación se puede firmar al final de las Pruebas en la última revisión. Estas pruebas deben incluir pruebas al software, al hardware, a la integración de los mismos y a los instrumentos y equipos eléctricos. Pueden ser tan básicas como el encendido y apagado de equipos, hasta probar una secuencia de control o flujo de información. Estas pruebas pueden ser por módulos conforme el desarrollador va avanzando en el diseño. Además de que sirven como base para las pruebas de calificación de instalación, operación y desempeño que llevará a cabo el Cliente en Sitio. El Cliente puede definir previamente en el QPP las características que deben cumplir estos entregables de pruebas de revisión. También se puede permitir que el Proveedor las documente siguiendo su propio sistema de Calidad, para ello se recomienda que el Cliente revise las capacidades del mismo, tomando en cuenta sus métodos, herramientas, políticas, procedimientos, reglas y convenciones de programación, métodos de revisión, pruebas, seguridad y confidencialidad. A estas Pruebas de Revisión de Diseño las clasifico de la siguiente manera:
Pruebas Unitarias: son realizadas para asegurar que cada elemento
(software o hardware) cumple con el Diseño predefinido.
Pruebas de Integración: son desarrolladas para retar la interacción entre sub-sistemas o componentes.
Pruebas del Sistema: son ejecutadas para evaluar que el Sistema integrado funciona como se esperaba.
Pruebas a instrumentos y equipos eléctricos: son realizadas para verificar que el instrumento o equipo opera cumpliendo las características para las que fue diseñado.
Cabe mencionar que para los Sistemas Comerciales estas pruebas son realizadas y resguardas por el Fabricante responsable del software y hardware relacionado,

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 81 de 125
siguiendo su propio Sistema de Administración de la Calidad; por ejemplo un Sistema Operativo Windows XP o un PLC. Para los Sistemas Personalizados (o hechos a la medida) donde el software y hardware es desarrollado únicamente para una empresa es donde se tiene directamente la aplicación de estas pruebas. A continuación describo el contenido que propongo tienen que llevar las Pruebas de Revisión de Diseño.
PLANTILLA 6. PROPUESTA PARA LAS PRUEBAS DE REVISIÓN DE DISEÑO
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Proveedor o Desarrollador y/o logo. Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control, incluso podría ser una identificación que proporcione el mismo Laboratorio).
Fecha de emisión del documento. Versión del documento emitido (puede ser siguiendo el control de
versiones del Laboratorio).
2. Hojas de Pruebas
Se pueden elaborar unas hojas de prueba genéricas que pueden ser llenadas al momento de la prueba. Recomiendo tener una plantilla que contenga espacios delimitados para colocar los siguientes datos:
Título de la Prueba. Pasos de la prueba. Resultados obtenidos en cada paso de prueba. Conclusión de los resultados de la prueba, especificando si se cumplió con el
objetivo del diseño, programación o configuración del sub-sistema / equipo que se está retando o si la prueba tendrá que ser repetida.
En el caso de repetir la prueba describir brevemente cual fue el problema y como se soluciono y si los resultados de la repetición de la prueba fueron exitosos.
Firma(s) de quien(es) ejecuta(n) la prueba.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 82 de 125
Los pasos de prueba podrían ser llenados previamente y tenerlos impresos, los que si tienen que ser llenados en el momento de la prueba son los resultados obtenidos.
3. Aprobación de instalación en Sitio.
En esta sección se debe incluir la conclusión de que el Diseño propuesto es conveniente y aprobado para instalarse en el Sitio.
3.7.3 Pruebas de Calificación de Instalación (Installation Qualification, IQ).
El Protocolo IQ permitirá proveer evidencia documentada de que el Sistema o Equipo fue instalado en su ambiente de producción, de la manera correcta y conforme las especificaciones aprobadas, configuradas y los requerimientos del Sistema.
Este Protocolo debe permitir confirmar lo siguiente:
Que el hardware, software, equipo, dispositivos, instrumentos o cualquier
elemento comprado cumple con los requisitos de compra especificados. Que el software fue instalado correctamente y documentar las licencias si
aplica. Que los elementos de hardware / equipo fueron instalados y/o ensamblados e
identificados en sitio de la manera correcta y conforme a diagramas de instalación en caso de aplicar.
Alimentaciones de energía eléctrica y conexiones a tierra. Conexiones de interfases o en campo. Que los instrumentos de control y monitoreo hayan sido calibrados e
instalados correctamente. Funciones básicas de operación del Sistema como: encendido, apagado,
arranque, paro, introducción de datos, etc. Comunicación. Funciones básicas de los equipos. Pruebas de continuidad. Señales de entradas y salidas de control (analógicas y digitales). Que la instalación de la red cumple con las especificaciones predefinidas. Que las instalaciones de tubería y servicios estén instalados conforme a
planos. En el Protocolo se debe documentar:
Nombre, versión de software y forma de asegurar que se instaló correctamente, para cada una de las aplicaciones.
Nombre, marca, modelo, número de serie y características del hardware. Direcciones y/o protocolos de comunicación del hardware. Planos y diagramas relacionados al sistema.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 83 de 125
Verificación de la alimentación eléctrica de los equipos. Conexión a tierra de los componentes. Las condiciones ambientales donde fue instalado el equipo y que cumplen
con las que recomienda el fabricante. Si alguna prueba no paso, es decir, que no cumplió con el resultado esperado
se deberá registrar y dar seguimiento a su solución. En este caso se puede levantar un incidente, el cual se puede considerar como un evento no esperado, como puede ser un resultado erróneo en la prueba o una falla en el sistema o equipo.
3.7.4 Pruebas de Calificación de Operación (Operational Qualification, OQ).
El Protocolo de Calificación de Operación permite tener evidencia documentada de que el Sistema / Equipo funciona en el ambiente de producción en el que fue instalado y que ejecuta todas las funciones para las que fue diseñado dentro de los rangos especificados.
El OQ puede incluir pruebas como:
Comunicación con Bases de Datos, Interfases, subsistemas. Arranque y Paro del Sistema. Aspectos de seguridad. Secuencias de Control. Reto de los límites inferiores y superiores, condiciones del “peor caso” y
generación de alarmas. Pruebas de cumplimiento con CFR-21 (cuando aplique). Pruebas de “stress” del sistema, es decir, pruebas donde se verifique la
respuesta del sistema ante condiciones como horarios pico de transmisión de datos, conexión remota del número máximo de usuarios, envío de información y comandos.
Respaldo de información y su recuperación.
3.7.5 Pruebas de Calificación de Desempeño (Performance Qualification, PQ).
El PQ puede incluir las siguientes pruebas:
Pruebas que demuestren que el equipo se desempeña de acuerdo a los
parámetros y especificaciones de los productos o procesos. Rendimiento del sistema. Numero de tareas realizadas en un determinado tiempo. Control de variables de proceso dentro de rango. Reto de los límites inferiores y superiores, condiciones del “peor caso” y
generación de alarmas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 84 de 125
La propuesta del contenido de los Protocolos de Calificación la presentaré de manera general, es decir, propondré una plantilla que se puede llenar y adecuar dependiendo del tipo de calificación que se vaya a realizar: IQ, OQ o PQ. PLANTILLA 7. PROPUESTA PARA PROTOCOLOS DE CALIFICACIÓN (IQ, OQ, PQ).
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo. Esta sección de firmas tiene relación con la sección de descripción de roles y responsabilidades, ya que el número de firmas debe asegurar la conformidad de todos los Departamentos involucrados.
5. Introducción. Esta sección debe dar un bosquejo general del contenido del documento, recomiendo también describir el sistema o equipo de manera general.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 85 de 125
6. Objetivo. En esta sección se definen el o los objetivos del documento. Es importante que estos sean precisos para no caer en ambigüedades.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo. Mencionar lo que estará dentro del alcance de las pruebas, así como también lo que no se probará y la justificación de ello.
8. Referencias.
Es importante mencionar documentos con los que tenga relación este documento que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, VMP’s, URS, FS, DS, Normas Oficiales Mexicanas, lineamientos Internacionales, etcétera.
9. Descripción de los roles y responsabilidades.
Se deben describir los roles y responsabilidades como mínimo del Líder o administrador del Proyecto, del departamento de Aseguramiento de Calidad, de los Dueños del sistema, del Área Técnica, del ejecutor de las pruebas y los revisores de las mismas.
10. Descripción general del procedimiento a seguir.
La descripción consiste en decir como o donde (dentro del Protocolo) se documentarán los pasos de la prueba, los requisitos previos, los resultados esperados, los resultados encontrados, si la prueba Paso o Fallo, las firmas de los ejecutores y revisores, los incidentes encontrados y el resumen final. Si se tiene un PNO específico dentro del Sistema de Calidad donde describa todo el proceso de Calificación y sus formatos se puede hacer referencia al mismo. También se puede hacer mención de las pruebas que se realizarán en todo el Protocolo. Por ejemplo, para mi propuesta: Se utilizará un formato para documentar las pruebas donde se especificarán los pasos de prueba, sus requisitos previos, los resultados esperados y encontrados, el estatus de Pasa o Falla. Los incidentes se registrarán y cerrarán en el formato incluido en la sección de Incidentes de las Pruebas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 86 de 125
11. Pruebas de Instalación, Operación o Desempeño.
En esta sección se deben incluir las pruebas que reten las características que estén dentro del alcance, objetivo y tipo de Protocolo. A continuación se muestra un formato práctico para documentar las pruebas. Algunas secciones solo aplican a un tipo de Protocolo y se mencionará donde aplique.
SISTEMA / MÓDULO Nombre del Sistema que se está probando / Nombre del módulo del sistema que se probará. Caso de Prueba _1_.
Nombre del caso de Prueba.
Características del Equipo, Hardware, Dispositivo o Módulo de Prueba:
Nombre del Equipo, Modelo, Marca, número de serie, fecha de calibración, en el caso de software la versión, Fabricante, etc.
Requisitos previos: Elementos necesarios antes de iniciar la prueba por ejemplo: • Pruebas de revisión de Diseño o Protocolos de Calificación previos. • Instalación de algún instrumento. • Certificados de calibración. • Etcétera.
Criterios de Aceptación • La prueba se considerará exitosa si todos los resultados encontrados son iguales a los esperados o en el caso de tener incidentes que sean resueltos.
• El sistema debe controlar la temperatura en el rango de -2 a 6 ºC. (por ejemplo para un protocolo de calificación de operación).
No. Instrucciones del Paso de la Prueba Resultado esperado Resultado encontrado PASA o FALLA
Prueba ejecutada por
Fecha de la ejecución
1 2 3
Se registra el incidente 1 del Paso de Prueba 2. Documentación de Incidentes: Se registra el incidente 2 del Paso de Prueba 3. Anexo 1. Pantalla con la versión del software. Anexo 2. Gráfica de humedad obtenida.
Se han generado los siguientes anexos como evidencia de las pruebas: Anexo 3…….. Firma del Revisor y Fecha
Llenar y/o adecuar este formato las veces que sea necesario para completar cada uno de los protocolos.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 87 de 125
12. Incidentes de las Pruebas.
En este punto se deben documentar los incidentes encontrados en las Pruebas, de tal manera que se especifique la prueba donde el resultado obtenido no sea igual al resultado esperado o donde ocurrió la falla, la explicación del incidente, los cambios requeridos, la repetición de la prueba y los resultados y las firmas del Ejecutor y Revisor de la solución del incidente. En el siguiente formato pueden documentarse los incidentes encontrados en la ejecución de las Pruebas.
SISTEMA: Caso de Prueba y paso de la misma donde se encontró el incidente:
Descripción del Incidente No. _1_:
Ejecutor: Fecha:
La realización de otras pruebas o de todo el protocolo se ven afectadas:
( ) SI ( ) NO Cuáles:
Acciones necesarias para corregir el incidente:
Ejecutor: Fecha: Revisor: Fecha:
Los cambios propuestos fueron hechos:
( ) SI ( ) NO Ejecutor: Fecha: Revisor: Fecha:
Resultados de la re-ejecución de la Prueba:
Ejecutor: Fecha: Revisor: Fecha:
Cierre del incidente: Aprobador del cierre: Fecha:

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 88 de 125
13. Resumen de los resultados de la Calificación.
En este resumen se debe describir de manera general los resultados de las pruebas, indicando que los requerimientos del sistema o equipo fueron retados obteniendo resultados exitosos y que el sistema puede pasar a la siguiente Calificación o salir a producción según aplique. Se puede incluir la siguiente tabla para resumir los resultados de las Pruebas del Protocolo.
TABLA DE RESULTADOS SISTEMA: Número total
de Pruebas:
Caso de Prueba: No. de ejecución
1 2 3
PASO o FALLO Fecha: PASO o FALLO Fecha: PASO o FALLO Fecha: PASO o FALLO Fecha: PASO o FALLO Fecha: PASO o FALLO Fecha:
Ejecutor: Fecha: Revisor: Fecha: Aprobador por parte de Calidad: Fecha:
14. Anexos.
Aquí se pueden incluir los anexos que sean necesarios para dar evidencia de los resultados de las pruebas.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 89 de 125
3.8 REGISTRO DE CAPACITACIÓN.
Se recomienda que durante la fase de Instalación y Pruebas de Calificación se desarrollen los procedimientos de operación, uso y administración del Equipo o Sistema. Ver sección 3.10 Mantenimiento del Estado Validado donde se da una descripción más a fondo de estos procedimientos. Después de terminar con las calificaciones de manera exitosa se debe proceder con el proceso de capacitación al personal involucrado conforme procedimientos implementados en el Laboratorio para tal proceso o como se haya especificado en el VP y/o QPP. La capacitación y su registro es un factor de suma importancia ya que mediante este proceso se prepara a los usuarios para el uso del Equipo o Sistema implementado.
3.9 REPORTE DE VALIDACIÓN.
El Reporte de Validación debe ser elaborado como una conclusión de la validación que se hizo en el Proyecto, resumiendo las actividades de la misma, si hubo desviaciones al VP y el estado del equipo o sistema comparado con los propósitos del proyecto y los requerimientos previamente establecidos. En este documento también se hace una revisión de todos los entregables que se comprometieron en el VP, dando evidencia de su emisión. Las responsabilidades de su emisión deben establecerse en el VP.
La GAMP4 señala que cual sea la escala o alcance de un proyecto siempre debe emitirse un Reporte de Validación. Y menciona que se puede elaborar un reporte por fase del proyecto o un reporte final que abarque todas las fases. El contenido que la GAMP4 recomienda para este documento es:
Introducción y Alcance. Resumen de resultados por fase. Resultados de la fase de ejecución. (Pre-requisitos, Ambiente del proyecto,
Cambios de alcance, Detalles de la fase de ejecución como resultados o interrupciones que pongan en riesgo la operación.
Reporte y resolución de desviaciones. Estatus de la Fase (en el caso de que el Reporte de validación sea por fases del
proyecto). Capacitación. Mantenimiento del estado validado. Glosario. Apéndices.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 90 de 125
A pesar de que la GAMP4 no coloca al Reporte de Validación como un documento en las figuras 2.7 (Modelo de Ciclo de Vida para la validación de Sistemas IT), 2.9 (Documentación y Actividades de Ciclo de Vida para Sistemas Integrados (Embedded Systems)) y 2.10 (Documentación y Actividades de Ciclo de Vida para Sistemas Autónomos (Standalone Systems)), en el esquema de mi propuesta si está contemplado. Mi recomendación para este Reporte es elaborarlo al finalizar el Proyecto y no por fases, es decir, en un solo documento describir los resultados de la validación de todo el proyecto. El contenido que propongo para la elaboración de un Reporte de Validación es el siguiente:
PLANTILLA 8. PROPUESTA DEL REPORTE DE VALIDACIÓN.
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo. Esta sección de firmas tiene relación con la sección de descripción de roles y responsabilidades, ya que el número de firmas debe asegurar el cumplimiento con todos los requerimientos.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 91 de 125
5. Introducción. Esta sección debe dar un bosquejo general del contenido del documento.
6. Objetivo.
En esta sección se definen el o los objetivos del documento. Es importante que estos sean precisos para no caer en ambigüedades.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo.
8. Referencias.
Es importante mencionar documentos con los que tenga relación este documento que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, VMP’s, todos los documentos del ciclo de vida, etcétera. En este documento en particular es importante que se haga referencia al VP del proyecto.
9. Resumen.
Elaborar un breve resumen del resultado de la validación en comparación con lo establecido en el VP.
10. Cambios y Desviaciones
En ocasiones, como resultado de ciertas circunstancias durante la vida del proyecto es necesario modificar el alcance original. Estos cambios y desviaciones deben ser mencionados y justificados.
11. Capacitación.
En esta sección se debe confirmar que el personal involucrado en el uso del Sistema o Equipo ya fue capacitado y que la capacitación está documentada.
12. Entregables
En esta sección se debe incluir una tabla que muestre los entregables comprometidos en el VP y los entregables elaborados dentro del Proyecto para asegurar que fueron emitidos.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 92 de 125
13. Conclusión
Dar la conclusión de que el Sistema está listo para uso en un ambiente de Producción conforme a la información descrita en este Reporte de Validación y su aprobación.
14. Glosario, Siglas y Términos
Esta sección contiene la definición de los términos que pueden no ser familiares para el lector.
15. Anexos En esta sección se puede incluir información necesaria para la completa comprensión del documento.
3.10 MANTENIMIENTO DEL ESTADO VALIDADO.
Una vez que el equipo o sistema es validado y puesto en operación, se debe mantener en dicho estado definiendo algunas medidas para asegurar la integridad de los elementos de configuración: hardware, software, datos y documentos asociados durante si uso. En mi experiencia para cumplir con ello lo que se hace es controlar ese estado por medio del cumplimiento de procedimientos locales. Dentro del proyecto de Norma PROY-NOM-059-SSA1-2004 es contemplado este mantenimiento particularmente en la sección 14.11 Mantenimiento del estado validado, donde se menciona que se debe contar, entre otras cosas, con:
Sistema de control de cambios. Sistema de calibración. Programa de mantenimiento preventivo. Sistema de calificación de personal. Sistema de auditorías técnicas. Sistema de desviaciones. Recalificar o revalidar cuando haya cambios significativos a programas y/o
sistemas.
Si nos referimos a la GAMP4, recomienda tomar en cuenta puntos como:
Procedimientos operacionales. Capacitaciones. Administración y resolución de problemas. Administración del sistema. Respaldos y recuperación.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 93 de 125
Administración de la seguridad. Retiro del sistema.
Y hace hincapié en que para los Sistemas IT se requiere una consideración particular en:
Control de cambios. Administración de la configuración. Administración de las versiones de software producidas. Planeación de la continuidad del negocio.
Y para los Sistemas de Control de Proceso menciona la importancia de la calibración que se debe cumplir de los sensores y dispositivos de medición. Para mi propuesta acerca de las actividades que se deben llevar a cabo para mantener el estado validado una vez que el sistema está en uso, tomaré en cuenta las recomendaciones de la GAMP4 y como he mantenido este estado validado de los sistemas que he tenido bajo mi experiencia.
PROPUESTA DE MEDIDAS PARA MANTENER EL ESTADO VALIDADO DE LOS SISTEMAS / EQUIPOS.
Dentro de mi propuesta está el cubrir todos los puntos enlistados abajo, a través de Procedimientos que formen parte del Sistema de Administración de Calidad de la Planta donde se implemente (es decir, del Sistema de Calidad del Usuario). De tal forma que se lleve el control de las medidas para el mantenimiento del estado validado y lo garantice. Para cumplir con algunas de estas actividades será necesario involucrar al proveedor, principalmente en las actividades de mantenimiento, soporte y nuevas implementaciones. Antes de pasar a los puntos que permitirán el mantenimiento de la validación, tocaré el tema de los Procedimientos Operacionales. Éstos deberán ser elaborados y aprobados previamente a la puesta en uso del equipo o sistema en producción. En un procedimiento se puede abordar solo un punto de los que abajo se enlistan o agrupar varios que estén relacionados de alguna manera. Estos procedimientos deben reflejar el Sistema que está en funcionamiento y deben ser mantenidos durante todo el ciclo de vida del equipo o sistema. La manera que propongo para agruparlos es hacer un Procedimiento de Uso y Operación del Sistema y uno para la Administración del Sistema. A continuación se enlistan los puntos que se deben cubrir y en que procedimiento se puede contemplar.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 94 de 125
1. Procedimiento de Operación y Uso del Sistema o Equipo.
Este procedimiento debe describir las instrucciones para operación y uso del equipo o sistema en el ambiente de producción. En él se debe especificar cómo encenderlo, apagarlo, funcionamiento básico, actividades para llevar a cabo el proceso de manufactura que le corresponda, etc.
2. Procedimiento de Administración del Sistema o Equipo.
En este procedimiento se debe describir como se cumplirán o seguirán los siguientes puntos que formarán parte de la Administración del Sistema / Equipo para mantenerlo en estado validado.
2.1 Control de Cambios.
Una vez que el Sistema está en fase de operación, cualquier cambio que se quiera realizar al hardware, software, datos, documentación o instrumentación tendrá que ser llevado bajo un Proceso formal de Control de Cambios. En este proceso se debe documentar en que consiste el cambio, el impacto o riesgo al implementarlo, la actualización a documentos “vivos”, la justificación del porque se quiere hacer ese cambio y el diseño de lo que se cambiara, para poder analizar si se tienen las suficientes bases para implementar el cambio. En este documento se pueden especificar o anexar especificaciones de diseño del cambio, esquemas, especificaciones de nuevos dispositivos, etc. Es importante tener en cuenta que para un cambio realizado se deben ejecutar ciertas pruebas de recalificación o revalidación.
2.2 Capacitación y calificación del personal.
Se deben implementar planes y procedimientos para la capacitación en el uso y la operación del equipo o sistema, de tal manera que se asegure que todo el personal que los utilice esté capacitado para ello, incluyendo el soporte o mantenimiento que se le pueda dar al mismo.
2.3 Administración y resolución de problemas.
Para los sistemas automáticos y de cómputo es necesario tener el historial de sus fallas o problemas, de tal forma que sean documentados, revisados, priorizados, escalados y cerrados. Esto con el fin de formar parte del monitoreo de desempeño y tener una retroalimentación para las mejoras del sistema.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 95 de 125
2.4 Respaldo, resguardo, retención y recuperación.
Se deben asegurar los respaldos de software y datos, resguardarlos para protegerlos de alguna pérdida accidental o daño malicioso, mantenerlos durante el tiempo de retención y su correcta recuperación.
2.5 Contratos de Servicio.
En algunos casos es usual y práctico que se hagan contratos de mantenimiento preventivo y correctivo de los equipos o sistemas con los proveedores. Esto dependerá de lo crítico que sea el Sistema
2.6 Administración del Sistema.
Dentro de las actividades de administración que se deben definir están:
Actividades de limpieza. El uso de herramientas de software. Refacciones. Manejo de desviaciones. Controles de acceso al equipo / sistema / software.
2.7 Administración de la Configuración.
Las actividades de administración de la configuración que deben precisarse son:
Identificar y definir los componentes del sistema. Control de modificaciones y versiones de los elementos de
configuración, incluyendo parches. Registros y reportes del estatus y de las modificaciones de los
elementos. Asegurar la consistencia y veracidad de los elementos. Control de la entrega, manejo y almacenamiento de los elementos.
2.8 Administración de la Seguridad.
Los Sistemas automáticos y datos deben ser protegidos adecuadamente de pérdidas y daños accidentales o intencionados, además de cambios no autorizados. El procedimiento que cubra este punto deberá contener la forma en que se administrará la seguridad de los accesos, incluyendo la alta y baja de usuarios, la administración de virus, de contraseñas y medidas de seguridad. Este procedimiento deberá ser aprobado antes de que el sistema se ponga en uso. La seguridad aplica para todos los niveles de usuario, ya sea operadores, supervisores o administradores.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 96 de 125
2.9 Monitoreo de Desempeño.
El impacto de una falla del sistema puede variar dependiendo de la criticidad del sistema o equipo automático. Donde sea apropiado el desempeño del sistema debe ser monitoreado para capturar los problemas en una manera pronta. Esto también puede hacer posible que se anticipe a las fallas a través del uso de herramientas y técnicas de monitoreo. La necesidad de monitorear el desempeño debe ser considerada y las actividades requieren ser calendarizadas y documentadas.
2.10 Plan de Continuidad del Negocio.
Los planes o procedimientos que soporten la continuidad del negocio, incluyendo planes de recuperación en caso de desastres y planes de contingencia, deben ser especificados, probados y aprobados antes de que el sistema sea aprobado para uso. Los temas que se deben considerar incluyen: fallas catastróficas de hardware y software, incendios, inundaciones e incumplimientos de la seguridad. Lo que significa que la operación debe estar disponible en caso de alguna falla, por ejemplo si algunos datos son requeridos de manera inmediata como es en los llamados “recall” que son los retiros del mercado de algún determinado producto.
Todo el software, incluyendo código fuente, compiladores, sistema operativo, etc., respaldos y documentación debe estar disponible para inspeccionar y dar continuidad al negocio. Las copias de software deben ser retenidas en áreas seguras y protegidas. En caso necesario de tener acceso al software, se deben establecer acuerdos formales para ello, por ejemplo mediante una bitácora de registro.
2.11 Calibración.
Se debe implementar un plan de calibraciones de los instrumentos que formen parte del Sistema. Este plan no tiene que ser específico para el sistema, pueden adicionarse los instrumentos al sistema de calibración de la Planta.
2.12 Mantenimiento preventivo.
Se deben adicionar los nuevos equipos y sistemas al sistema de mantenimiento que se utilice en la Planta.
2.13 Recalificaciones / Revalidaciones.
Conforme el proyecto de Norma PROY-NOM-059-SSA1-2004 se debe llevar a cabo una recalificación o revalidación del equipo o sistema, por lo que se tendrá que tener un programa que permita controlar estas revalidaciones después de un

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 97 de 125
período de 5 años de vigencia de las mismas. Además de que en cada protocolo que se elabore se deberá especificar su vigencia.
Otro punto que se debe especificar en los procedimientos de calificación o de control de las revalidaciones es que cuando se realicen cambios significativos a los programas, equipos o sistemas también se deben hacer recalificaciones o revalidaciones.
2.14 Revisión y evaluación periódica.
Para verificar que se estén cumpliendo todos los puntos anteriormente mencionados para mantener el estado validado de los sistemas, se deben programar revisiones a los mismos. Estas revisiones se pueden abordar a través de auditorías internas a los equipos y sistemas automáticos de la Planta. Estas auditorías deben contemplar aspectos técnicos para el cumplimiento del proyecto de Norma PROY-NOM-059-SSA1-2004 y también si el sistema está siendo operado de acuerdo con las regulaciones aplicables, políticas y procedimientos de la Compañía. Dentro de estas auditorías se debe asegurar el levantamiento de las observaciones encontradas y su seguimiento hasta ser solucionadas.
3.11 RETIRO.
La fase de Retiro dentro del Ciclo de Vida de los Sistemas es importante debido a que en ella se define la disposición del hardware, software, datos y documentación del Sistema que será retirado y los períodos de retención de los mismos. Además de definir los métodos de migración de datos al nuevo sistema, en caso de que aplique. En mi experiencia para esta fase se elabora un documento llamado “Plan de Retiro”, donde se describen a detalle todas las actividades que mencioné en el párrafo anterior. Por otro lado la GAMP4 no menciona alguna recomendación para hacer un Plan o algún documento específico para llevar a cabo este desmantelamiento, pero enlista los siguientes puntos que deben ser cubiertos para el Retiro de un Sistema.
Establecer procedimientos que cubran el retiro del sistema. Definir qué evidencia documental será retenida de las acciones tomadas
durante el retiro. Definir qué registros GMP serán mantenidos y sus períodos de retención y
cuales registros serán destruidos. La necesidad de migrar registros al nuevo sistema y la posibilidad de
archivarlos en el nuevo sistema, y el método de verificación y documentación de este proceso.
Habilidad de recuperar los registros migrados en el nuevo sistema.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 98 de 125
La retención de hardware y software del sistema a retirar con fines de retención o recuperación no es recomendada como una solución a largo de tiempo.
Procedimientos de administración de archivo que cubran medios, almacenamiento, ubicación, identificación e integridad.
Que documentación del sistema que será retirado se requiere mantener para futuras consultas regulatorias y durante qué periodo de tiempo se retendrán.
Mi propuesta es hacer un Plan de Retiro para esta última etapa del Ciclo de Vida de un Sistema y el contenido que propongo es el siguiente:
PLANTILLA 9. PROPUESTA DE UN PLAN DE RETIRO.
1. Carátula del documento.
La carátula debe proporcionar información como:
Nombre del Laboratorio Farmacéutico y/o logo. Nombre del documento. Numero de identificación del documento. (Usualmente cada empresa tiene
establecido un procedimiento para asignar números o claves a sus documentos de ciclo de vida para su manejo y control).
Fecha de emisión del documento. Versión del documento emitido.
2. Índice. 3. Registro de cambios al documento.
Este registro de cambios debe proporcionar información de los cambios hechos al documento, por ejemplo: fecha de modificación, persona que realizó el cambio y en que consistió el cambio.
4. Sección de firmas.
En toda la documentación es importante esta sección ya que en ella deben ser incluidas las firmas de la persona que elabora el documento, de los revisores y de los aprobadores. Las firmas deben definir bajo qué autoridad se está firmando y con qué objetivo. Esta sección de firmas tiene relación con la sección de descripción de roles y responsabilidades, ya que el número de firmas debe asegurar el cumplimiento con todos los requerimientos. Para este documento se tienen dos secciones de firmas:

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 99 de 125
Firmas de pre-aprobación del Plan de Retiro Estas firmas tendrán la responsabilidad de revisar y aprobar las actividades programas y plasmadas en este documento para retirar el Sistema.
Firmas de aprobación de la ejecución del Plan de Retiro
Estas firmas tendrán la responsabilidad de revisar y aprobar la ejecución de este Plan de Retiro, las firmas indicarán que el Sistema fue retirado exitosamente de acuerdo con las actividades programas y plasmadas en el Plan. En el anexo 1 (de esta plantilla) se especificará un resumen de las actividades realizadas conforme el Plan para que pueda ser evaluada la ejecución del mismo.
5. Introducción.
Esta sección debe dar un bosquejo general del contenido del documento.
6. Objetivo.
En esta sección se definen el o los objetivos del documento.
7. Alcance. La importancia de esta sección es que en ella se define el alcance del documento, señalando los límites del objetivo del mismo.
8. Referencia
Es importante mencionar documentos con los que tenga relación este documento que se está desarrollando o los documentos que sirvieron como base para su elaboración, por ejemplo políticas, procedimientos, guías, etcétera.
9. Razón del Retiro del Sistema
En esta sección se debe especificar brevemente la razón del retiro o desmantelamiento del sistema.
10. Impacto del Retiro
Describir que impacto tiene el retiro del sistema sobre las áreas usuarias, otros sistemas, procedimientos de Planta y otros. Además de definir cual será el proceso de notificación del apagado del sistema a los usuarios.
11. Migración de base de datos.
En el caso de que se haga una migración de base de datos del sistema a retirar al sistema nuevo, se tendrá que describir en esta sección cómo se llevará a cabo ese proceso y como se asegurará la recuperación de la misma.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 100 de 125
12. Disposición y retención de hardware, software, datos y documentación.
En esta sección se debe enlistar todos los elementos de hardware, software, datos y documentación del sistema, así como mencionar la disposición que tendrán o el tiempo de retención de los mismos. El hardware puede que sea utilizado por otra área, someterse a venta o definitivamente confinarse. Para el software, datos y documentación vigente se tiene que evaluar y definir el tiempo de resguardo, conforme a las políticas de retención con las que debe contar la empresa. La documentación se refiere a todos los documentos de validación que se tienen del sistema. Esta retención es importante para la disponibilidad de los elementos ante alguna auditoría u otros propósitos, por lo que también se debe indicar en las tablas siguientes la ubicación de los elementos.
Listado de Hardware del Sistema
Equipo / Hardware
Marca y Modelo
Número de serie
Activo Fijo Ubicación actual
Disposición del equipo
Servidor PC Cliente Impresora Computadora Móvil
Listado del Software del Sistema
Software / Aplicación Versión Ubicación del Archivo de
instalación
Tiempo de
retención
Fecha programada
de destrucción Instalado en:

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 101 de 125
Para los datos se debe hacer un respaldo si es que no se tiene hecho y resguardarlo conforme a las políticas de retención de la empresa.
Tabla: Listado de Datos
Dato / Archivo Ubicación
actual Fecha de Respaldo Ubicación del Respaldo
Tiempo de
retención
Fecha programada de
destrucción Respaldo de la Base de Datos
Reportes
Los documentos de los Sistemas deben estar resguardados en algún archivo bajo controles de resguardo y acceso a la información, se deben listar en la siguiente tabla mencionando las políticas de retención de la empresa.
Tabla: Listado de Documentación
Documento / Última Versión
Formato del Documento
(Electrónico o en Papel)
Localización / Archivo de Resguardo
Tiempo de retención
Fecha programada de
destrucción
Plan de Validación Plan del Proyecto y Calidad
Especificación de Requerimientos
Especificación de Diseño
13. Reemplazo del Sistema
Aquí se debe mencionar de manera breve y general el Sistema que reemplazará el Sistema a desmantelar así como las actividades para ejecutar ese cambio. Se debe describir si ambos sistemas trabajaran en paralelo por un determinado tiempo o si se debe apagar el sistema a retirar para poner en funcionamiento el nuevo, o de cualquier forma hacer referencia al documento donde se describe esto.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 102 de 125
En la siguiente tabla se pueden listar las actividades, duración de las mismas y recursos para la desmantelar el Sistema. Los recursos pueden ser humanos o materiales.
Tabla: Actividades para el reemplazo del Sistema
Actividad Duración Área responsable / Recursos
para su ejecución Elaboración y pre-aprobación del Plan.
Elaboración y resguardo de respaldos
Actividades de Transición al nuevo Sistema
Des-instalación de los equipos Des-instalación del software
14. Glosario, Siglas y Términos Esta sección contiene la definición de los términos que pueden no ser familiares para el lector.
15. Anexos En esta sección se puede incluir información necesaria para la completa comprensión del documento.
Anexo 1. Resumen de la ejecución del Plan de Retiro.
Para la aprobación de la ejecución del Plan en este anexo se deben resumir las actividades realizadas para su cumplimiento. Si hubo desviación a alguna actividad deberá especificarse y documentar lo que se realizo finalmente. Este anexo será revisado por los aprobadores y les permitirá evaluar si el retiro del Sistema fue realizado conforme a lo especificado.
3.12 BUENAS PRÁCTICAS.
Durante el proceso de validación de un equipo o sistema es importante tener presente y seguir los conceptos de Buenas Prácticas, que se describen a continuación. Además de llegar a un acuerdo entre Proveedor y Cliente para su cumplimiento.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 103 de 125
3.12.1 Buenas Prácticas de Documentación.
Las Buenas Prácticas de Documentación aseguran el control en la creación, revisión, aprobación, distribución y archivo de los documentos. Este control permite tener las bases de rastreabilidad y administración para las actividades de validación. Estas Prácticas requieren el acatamiento de los siguientes puntos:
Definición de los formatos de la documentación, incluyendo estilo y número de referencia.
Control de versiones de los documentos, además de especificar si su estatus es borrador o aprobado.
Almacenamiento seguro de la documentación e información de acuerdo a procedimientos definidos de tiempo de retención de los mismos. La seguridad implica protección contra daños accidentales o maliciosos.
Sometimiento de la documentación a revisión para la identificación de problemas o errores y la corrección de los mismos.
Las aprobaciones deben ser documentadas por medio de una firma incluyendo la fecha en que se está firmando y deberá indicar la responsabilidad de la firma.
Los documentos aprobados deben ser resguardados y estar disponibles para su consulta. Se debe establecer un control de copias de documentación aprobada, además de controles para que los documentos usados sean los vigentes.
Los cambios a documentos aprobados deben ser revisados y aprobados. Se debe documentar el cambio dando una descripción del mismo y mencionando si hay documentos afectados para su actualización.
Los documentos que salen de vigencia deben ser resguardados e identificados como fuera de vigencia o cancelados.
3.12.2 Buenas Prácticas de Prueba.
Las Buenas Prácticas de Prueba aseguran que las pruebas cubran todos los aspectos de los equipos y sistemas, además de que sean lo suficientemente bien ejecutadas y documentadas que permitan la rastreabilidad de la prueba, los resultados, el manejo de las desviaciones y las personas responsables de cada actividad. Las pruebas deben hacer referencia a especificaciones y ser reproducibles a lo largo de todo el ciclo de vida. Los requerimientos deben ser rastreables a una prueba específica. Las Buenas Prácticas de Prueba requieren que:
Las pruebas sean ejecutadas de acuerdo a un procedimiento o protocolo de pruebas predefinido y preaprobado.

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 104 de 125
Las pruebas sean basadas en documentos formales que estén regidos bajo controles de versión.
Las pruebas no sean iniciadas antes de la aprobación del procedimiento o protocolo de prueba
Las pruebas sean planeadas y ejecutadas por personal capacitado y calificado. El personal debe contar con habilidades técnicas, conocimiento del equipo o sistema y capacitación en Buenas Prácticas de Prueba.
Los ejecutores y aprobadores de las pruebas sean, tanto como sea posible, independientes y no ser el autor del protocolo o el diseñador del equipo o sistema.
La documentación de la prueba incluya el nombre y puesto de: autor(es), revisores y aprobadores.
Las firmas de autor(es), revisores y aprobadores estén acompañadas de la fecha en que se está firmando.
El procedimiento o protocolo de prueba esté descrito con suficiente detalle que permita la repetición de pruebas.
Todas las pruebas muestren la firma y la fecha del ejecutor y del revisor o verificador.
Cada prueba tenga criterios de aceptación de manera predeterminada. Durante la ejecución de la prueba, los resultados sean registrados
directamente en las hojas de resultados. Cualquier evidencia sea firmada, fechada y referenciada a la prueba
relacionada de manera única. La documentación de las pruebas y resultados incluyan las actividades y
observaciones necesarias para la reconstrucción y evaluación de las pruebas. El registro manual de las pruebas sea legible. Las correcciones sean cruzadas con una línea (de manera que quede legible
el contenido original) y acompañarse de la firma de la persona que está haciendo la corrección, la fecha en que se está haciendo y una breve explicación de la corrección. De ninguna manera se debe dejar ilegible el texto original.
Cada prueba sea terminada con una conclusión de que la prueba cumple con los criterios de aceptación.
Todos los incidentes sean registrados y rastreables a lo largo de la corrección y re-ejecución de la prueba para su cierre. Donde aplique, las correcciones de los incidentes deben incluir pruebas de regresión para verificar que las correcciones no introduzcan nuevos problemas en otros módulos o subsistemas.
Los instrumentos críticos y equipos de prueba sean calibrados. Se debe dar evidencia documental del certificado de calibración.
Después de la ejecución, los resultados sean revisados para hacer alguna corrección y asegurar la finalización de las pruebas de manera completa. La revisión debe incluir que todas las pruebas hayan sido ejecutadas, que todos los documentos relevantes se hayan anexado, que todos los criterios de

PROPUESTA PARA VALIDACIÓN DE SISTEMAS AUTOMÁTICOS Y DE CÓMPUTO PARA LA INDUSTRIA FARMACÉUTICA
Página 105 de 125
aceptación hayan sido cumplidos y que se hayan registrado todos los incidentes.
La ejecución de pruebas sea auditada por un representante del Área Usuaria o de Aseguramiento de Calidad.
3.12.3 Buenas Prácticas de Ingeniería.
Las Buenas Prácticas de Ingeniería son la implementación a lo largo de la vida del proyecto de métodos y estándares de Ingeniería establecidos. Esta implementación debe asegurar la ejecución de actividades de ingeniería de acuerdo a sistemas y procesos de administración de calidad, basados en un ciclo de vida apropiado y bien definido. También debe asegurar un manejo apropiado de los Requerimientos, Diseño, Revisiones y Pruebas. Estas Prácticas deben asegurar que la metodología para el desarrollo de ingeniería y software generan entregables que soportan los requerimientos para la calificación y validación en la Industria Farmacéutica. Éstas deben incluir:
Diseño e instalación que tome en cuenta todos los aspectos de buenas prácticas, seguridad, salud, ambiente, ergonomía, operación, mantenimiento, conocimiento de guías y normas.
Administración del proyecto, diseño de Ingeniería, construcción e instalación competente y profesional.
Documentación apropiada que incluya conceptos de Diseño, diagramas de Diseño y “como instalado” (“as-built”), registros de prueba y manuales de operación y mantenimiento.

CAPÍTULO 4. IMPLEMENTACIÓN Y BENEFICIOS.
CAPÍTULO 4.
“IMPLEMENTACIÓN Y BENEFICIOS”
Objetivo particular: Describir la implementación de mi propuesta para validar Sistemas Automáticos y de Cómputo, para el cumplimiento del proyecto de Norma PROY-NOM-059-SSA1-2004 y los beneficios de la misma.

IMPLEMENTACIÓN Y BENEFICIOS
Página 107 de 125
Capítulo 4. Implementación y Beneficios. Después de describir mi propuesta en el capítulo anterior, donde expuse la metodología para validar los sistemas, sus etapas y los documentos que se tienen que elaborar, presentaré la implementación de la misma y los beneficios que nos ofrece. Para describir la implementación del Sistema de Validación tomaré como base una empresa que no tiene implementado un Sistema de Validación para Sistemas Automáticos y de cómputo de manera formal y que no tiene la cultura del Ciclo de Vida de los Sistemas. La localización de la empresa es México y se tiene el contexto de que el proyecto de Norma PROY-NOM-059-SSA1-2004 entrará en vigor y actualmente si la empresa no implementa y/o mejora su sistema de validación para estos sistemas no cumplirá con todos los requisitos para el cumplimiento de la nueva versión de la norma mexicana. Primero tocaré el tema de los Recursos, después el Plan de Implementación y finalmente los Beneficios. Entre los recursos para la implementación están los recursos humanos, la inversión y el compromiso de las Gerencias y Dirección. A continuación los abordo.
4.1 RECURSOS HUMANOS PARA LA IMPLEMENTACIÓN
Los recursos humanos necesarios para implementar el sistema de validación de sistemas automáticos y de cómputo son:
Un Administrador de este Sistema de Validación (ASV). Esta persona debe
conocer el entorno global de requerimientos de validación de estos sistemas, transmitir el conocimiento a los Líderes de Proyectos y Líderes del Sistema de Validación, coordinar las actividades de validación de estos sistemas en la Planta o Empresa y ser responsable del cumplimiento con las regulaciones en este ámbito.
Líderes del Sistema de Validación (LSV). Su responsabilidad será validar los
sistemas heredados y los sistemas nuevos desde que inicie un proyecto y mantener ese estado validado durante todo su ciclo de vida. Serán líderes dedicados exclusivamente a validación de sistemas, no a todo el proyecto. Es decir cada área debe tener sus proyectos de manera individual con su respectivo Líder de Proyecto, pero será apoyado para el desarrollo de la documentación por un Líder del Sistema de Validación (LSV). El Líder del Proyecto deberá proporcionar toda la información necesaria y apoyo técnico al LSV para la elaboración de los documentos.
Revisores Técnicos: Su responsabilidad será revisar toda la documentación que se genere durante los proyectos de sus áreas, serán revisores desde la perspectiva técnica. Serán responsables de asegurar que el Sistema / Equipo

IMPLEMENTACIÓN Y BENEFICIOS
Página 108 de 125
nuevo cumple con todas las características y funciones necesarias para lo que fue requerido. Esta responsabilidad se les puede asignar a empleados existentes, sumándosela a sus actividades actuales. La persona que revise la documentación desde el punto de vista técnico debe ser el experto en el sistema, experto en su funcionamiento, en lo que se espera del mismo, en el cumplimiento de la Normatividad aplicable y aspectos de Ingeniería.
Usuarios: Su responsabilidad será revisar la documentación desde el punto de vista usuario / cliente interno, la cual consistirá en asegurar que el sistema cumple con todos los requerimientos establecidos para que se tengan las funciones necesarias en el área.
Dueños de los Sistemas: Después de finalizar un determinado proyecto los sistemas son entregados a los usuarios o al responsable que hará uso del mismo. Por lo tanto a este usuario le llamaremos Dueño del Sistema y su responsabilidad será asegurar que su sistema se mantiene validado después de que se lo hayan entregado como nuevo y validado. Para cumplir con esta responsabilidad se podrá apoyar de los líderes dedicados a validación a quienes solicitará la actualización de los documentos “vivos” para mantener su sistema “como construido”.
Calidad: El Departamento de Calidad es el responsable de asegurar el cumplimiento de las regulaciones de la Industria, las políticas, los procedimientos y guías, al momento de revisar la documentación de los proyectos.
Como podemos ver para la implementación del sistema sólo se requiere contratar al ASV y a los LSV, los demás recursos ya existen en la Planta y solo se tendría que considerar su organización y capacitación en la Validación de los Sistemas. Con esto cubrimos el punto del proyecto de Norma PROY-NOM-059-SSA1-2004 14.2.3.2 que pide la estructura organizacional para las actividades de validación.
4.2 INVERSIÓN PARA LA IMPLEMENTACIÓN
Los costos de inversión para la implementación del Sistema los podemos especificar como costos directos.
4.2.1 Costos Directos
Los elementos que permitirán saber un aproximado de la inversión directa que es necesaria hacer para esta implementación se concentran en la siguiente Tabla 4.1.

IMPLEMENTACIÓN Y BENEFICIOS
Página 109 de 125
Tabla 4.1 Recursos y costos de inversión.
PERSONAL DE NUEVO INGRESO NECESARIO PARA LA IMPLEMENTACIÓN
Recursos humanos Número de recursos
Inversión mensual
$ MN
Tiempo considerado
para el cálculo (meses)
Inversión anual por el recurso
$ MN
Administrador del Sistema de Validación (ASV) 1 $18.000,00 12 $216.000,00
Líderes del Sistema de Validación (LSV) 5 $15.000,00 12 $900.000,00
RECURSOS MATERIALES NECESARIOS PARA LA IMPLEMENTACIÓN
Recurso Tipo de Unidad
Número de unidades
Inversión por unidad
Inversión del recurso
Papelería Papelería mensual 12 $2.500,00 $30.000,00
Equipos de cómputo Equipo 6 $14.000,00 $84.000,00
Bibliografía A (GAMP) 1 $5.000,00 $5.000,00
Capacitación del ASV
Bibliografía B (Libro de la ISA de Pruebas a Sistemas
Automáticos) 1 $800,00 $800,00
Total $1.235.800,00
Dentro de los recursos humanos se considera al Administrador del Sistema de Validación (ASV) y los Líderes del Sistema de Validación (LSV) quienes durante todo el año trabajaran en la implementación del Sistema conforme surjan los proyectos y en la alineación de los sistemas existentes. El ASV será responsable de la implementación formal del Sistema, alineando el contenido de mi propuesta en guías y procedimientos locales. Tomará cursos extra que considere necesarios, realizará la investigación que necesite y será responsable de capacitar a los LSV y personal necesario en Planta. En la siguiente tabla se muestra la inversión que se tendría el siguiente año a la implementación, que consiste principalmente en los recursos humanos y la papelería necesaria para la elaboración de los documentos del sistema de validación. Con lo que se concluye que los costos de inversión anuales aproximados para el cumplimiento en la validación de los sistemas automáticos y de cómputo es de $1,200,000.00 MN. Costos que deberá considerar la Dirección dentro de la asignación del Presupuesto para este Departamento de Validación.
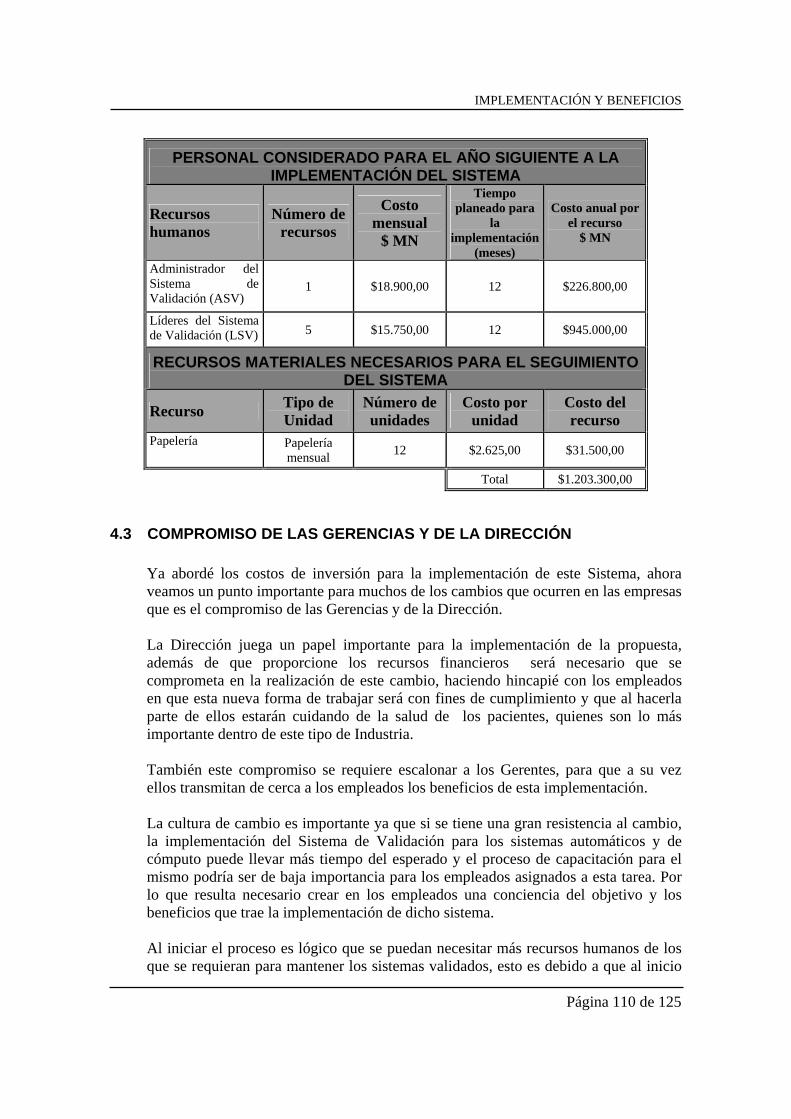
IMPLEMENTACIÓN Y BENEFICIOS
Página 110 de 125
PERSONAL CONSIDERADO PARA EL AÑO SIGUIENTE A LA IMPLEMENTACIÓN DEL SISTEMA
Recursos humanos
Número de recursos
Costo mensual
$ MN
Tiempo planeado para
la implementación
(meses)
Costo anual por el recurso
$ MN
Administrador del Sistema de Validación (ASV)
1 $18.900,00 12 $226.800,00
Líderes del Sistema de Validación (LSV) 5 $15.750,00 12 $945.000,00
RECURSOS MATERIALES NECESARIOS PARA EL SEGUIMIENTO DEL SISTEMA
Recurso Tipo de Unidad
Número de unidades
Costo por unidad
Costo del recurso
Papelería Papelería mensual 12 $2.625,00 $31.500,00
Total $1.203.300,00
4.3 COMPROMISO DE LAS GERENCIAS Y DE LA DIRECCIÓN
Ya abordé los costos de inversión para la implementación de este Sistema, ahora veamos un punto importante para muchos de los cambios que ocurren en las empresas que es el compromiso de las Gerencias y de la Dirección. La Dirección juega un papel importante para la implementación de la propuesta, además de que proporcione los recursos financieros será necesario que se comprometa en la realización de este cambio, haciendo hincapié con los empleados en que esta nueva forma de trabajar será con fines de cumplimiento y que al hacerla parte de ellos estarán cuidando de la salud de los pacientes, quienes son lo más importante dentro de este tipo de Industria. También este compromiso se requiere escalonar a los Gerentes, para que a su vez ellos transmitan de cerca a los empleados los beneficios de esta implementación. La cultura de cambio es importante ya que si se tiene una gran resistencia al cambio, la implementación del Sistema de Validación para los sistemas automáticos y de cómputo puede llevar más tiempo del esperado y el proceso de capacitación para el mismo podría ser de baja importancia para los empleados asignados a esta tarea. Por lo que resulta necesario crear en los empleados una conciencia del objetivo y los beneficios que trae la implementación de dicho sistema. Al iniciar el proceso es lógico que se puedan necesitar más recursos humanos de los que se requieran para mantener los sistemas validados, esto es debido a que al inicio

IMPLEMENTACIÓN Y BENEFICIOS
Página 111 de 125
del proceso la carga de trabajo es mayor porque se abordan los Procedimientos y/o Guías que establecerán los lineamientos para esta validación, se inicia con todos los sistemas nuevos y hay que generar todos los documentos, pero una vez validados sólo es necesario la administración de su configuración, contando ya con la experiencia en Ciclo de Vida y de la Validación.
4.4 PLAN DE IMPLEMENTACIÓN.
Después de analizar los recursos para la implementación es necesario plasmarlo en un Plan, el cual presento a continuación de manera general, indicando las actividades, responsables y tiempos estimados. Como se puede observar el Plan tiene un tiempo de implementación de 1 año.
Después del Plan doy una descripción detallada de las actividades.
Descripción detallada de las actividades:
1. Cursos de inducción y capacitaciones del ASV (Administrador del Sistema de
Validación) y de los LSV’s (Líderes del Sistema de Validación). Como estoy considerando que este personal será de nuevo ingreso debemos considerar unos días para que reciban los cursos de inducción al Laboratorio que prestarán sus servicios.
2. Adquisición de bibliografía recomendada en la Tabla 4.1. El ASV deberá
adquirir la bibliografía recomendada para su lectura y análisis como parte de las bases que debe adquirir para su Puesto.

IMPLEMENTACIÓN Y BENEFICIOS
Página 112 de 125
3. Capacitación del ASV y de los LSV. Esta será una autocapacitación que
incluye la lectura, análisis y comprensión de la propuesta, de la bibliografía recomendada en la Tabla 4.1 u otras que resulten de la investigación de los interesados. Además del proyecto de Norma PROY-NOM-059-SSA1-2004.
4. Elaboración del listado de los Sistemas y Equipos a validar. Incluyendo
sistemas heredados y de los proyectos que estén naciendo.
5. Elaboración del Plan Maestro de Validación de Sistemas Automáticos y de Cómputo. Después de tener el listado de sistemas a validar será necesario elaborar el Plan Maestro de Validación (VMP) para formalizar los sistemas que se encuentran dentro del alcance, las actividades y responsables.
6. Elaboración del Procedimiento que describirá la metodología. Esta tarea
consiste en la elaboración y emisión del Procedimiento que indicará la metodología a seguir para la validación de los Sistemas Automáticos y de Cómputo, con el objetivo de definir los pasos a seguir para que dichos sistemas GMP sean validados, documentados y administrados.
7. Emisión de las plantillas. Esta acción consiste en tomar las plantillas
propuestas en este documento y adecuarlas a los requerimientos de Guías y Procedimientos vigentes del Sistema de Calidad del Laboratorio, incluyendo sus logos, confidencialidad, etc.
8. Capacitación de los Líderes de Proyectos y de las personas que fungirán los
roles de Revisores, Usuarios, Dueños del Sistema y Calidad. Esta tarea radica en que el ASV capacite a los demás roles dentro de la validación una vez que se tiene el procedimiento de la metodología, el listado de sistemas y equipos, el Plan de Validación y las Plantillas.
9. Validación de Sistemas y Equipos Heredados. Consiste en hacer la
documentación de Ciclo de Vida de los sistemas que existan antes de la implementación de la metodología. Se elaborarán todos los documentos como lo marca la Figura 3.1 (Ciclo de Vida propuesto), exceptuando el proceso de Evaluación y Selección del Proveedor, ya que los sistemas serán existentes. Además de sus PNO’s, si estos ya existen solo se tendrá que revisar si es necesaria alguna actualización, de lo contrario será necesaria su elaboración.
10. Validación de los Sistemas y Equipos en Proyecto. Consiste en llevar a la
práctica la metodología de validación en los proyectos que estén etapa de desarrollo.
Es importante notar que los tiempos estimados para las actividades 9 y 10 de validación dependerán mucho del número de equipos y sistemas con los que cuente el

IMPLEMENTACIÓN Y BENEFICIOS
Página 113 de 125
Laboratorio Farmacéutico, cuanto más grande sea el Laboratorio y menor el tiempo para este cumplimiento se requerirá de la contratación de más LSV’s.
Por lo tanto el sistema de Validación quedará implementado en 1 año de manera completa, en forma y oficial.
4.5 BENEFICIOS DE LA VALIDACIÓN.
Después de abordar los recursos, los costos de inversión y el compromiso para la implementación, veamos los beneficios que le aportará a la Dirección y a la Planta Farmacéutica el implementar este Sistema. Los beneficios de la validación los podemos clasificar de primera instancia en dos: beneficios de negocio y sociales. A continuación se mencionan los principales beneficios que tiene una industria farmacéutica y la sociedad al tener sistemas automáticos y de cómputo validados.
4.5.1 Beneficios de negocio.
El principal beneficio de negocio que tiene validar los sistemas automáticos y de cómputo es el Cumplimiento regulatorio. Adicional a este beneficio, se tienen beneficios tecnológicos, operativos y de conocimiento.
4.5.1.1 Beneficios regulatorios y económicos
Mi propuesta está diseñada para responder a los requerimientos regulatorios no sólo de México, sino, de otros Países y con ello obtener un beneficio de negocio. Ya que al poder responder a las auditorías de distintos Países se tiene apertura al comercio y un aumento en las ventas.
Por lo tanto al mejorar nuestra forma de documentar y validar conforme lo solicitan las regulaciones se abre el mercado de distribución y venta, lo cuál es redituable de manera económica obteniendo de entrada una participación en el mercado y una posición importante y competitiva en el mismo. Los beneficios regulatorios de practicar la validación de sistemas automáticos y de cómputo es contar con evidencia escrita de lo que se hizo para poder demostrar que se está trabajando de acuerdo a las regulaciones y que la empresa es capaz de realizar investigaciones en el caso que sean necesarias. También permite demostrar que las actividades se realizaron de manera correcta, que se evitan errores, que se obtienen los resultados esperados, que se cumple con las funciones para las que fueron diseñados los sistemas, existe forma de saber quién hizo una determinada actividad, qué se hizo, cuándo se hizo.

IMPLEMENTACIÓN Y BENEFICIOS
Página 114 de 125
El tener los sistemas validados y aplicar el ciclo de vida permite responder al requisito básico y obligatorio de que todas las actividades y operaciones relacionadas directamente con los medicamentos deben registrarse y documentarse. Mi propuesta permite tener la rastreabilidad requerida para los sistemas, es decir, permite reconstruir la historia del sistema, desde que nace hasta que muere. El validar un sistema permite tener claro el objetivo y el alcance del nuevo sistema desde el inicio del proyecto, al igual que asegurar la calidad. Permite planear la implementación del mismo teniendo en claro lo que el sistema debe hacer y cómo lo va a hacer. Esta planeación junto con una buena organización y dirección permite cumplir los propósitos en tiempo, dinero y satisfacción al cliente.
4.5.1.2 Beneficios operativos
A un sistema que está bien definido, diseñado y especificado es fácil darle soporte, mantenimiento, monitoreo y control; resultando menos perdida de tiempo. El tener un sistema definido, dividido en subsistemas y bien delimitados facilita la pronta atención y respuesta a alguna falla. Esto nos lleva a reducción de tiempos lo cual es un término monetario para la empresa. La información presentada de manera correcta y objetiva ayuda a la toma de decisiones además soportan y justifican dicha decisión. Si en el desarrollo del proyecto participan los operadores del sistema y/o equipo permitirá que ellos desde el diseño estén involucrados en el funcionamiento y conozcan el objetivo, la operación y el proceso.
Tener documentada la configuración de los equipos permite que ante alguna contingencia se pueda consultar el documento en base al cual se realizó la configuración y volver a poner en marcha el equipo y/o sistema, sin tener paros en las líneas de producción esperando que llegue el proveedor experto del sistema y sin poner en riesgo la operación.
4.5.1.3 Beneficios de conocimiento
Adquirir experiencia en un determinado tema o equipo proporciona un soporte muy fuerte para que seamos seleccionados dentro del ambiente competitivo a nivel mundial. La intensa competitividad hace que sean elegidos los proveedores con más experiencia ya que son más confiables. La experiencia hace el conocimiento y dominio de los equipos y procesos, entonces este conocimiento también ayuda a mejorar las técnicas utilizadas y con ello asegurar la calidad y dar resultados esperados. Al presentar la información de manera gráfica es más fácil la comprensión del proceso o del tema que se está tratando y estos gráficos, en los documentos del ciclo de vida, quedan resguardados para su consulta.

IMPLEMENTACIÓN Y BENEFICIOS
Página 115 de 125
Estos conocimientos también se adquieren a través del análisis de los documentos existentes. El analizar, también permite hacer mejoras al sistema, ya que se analizan las funciones y procesos existentes y se puede visualizar la reducción de tiempos de proceso, de tiempos muertos, ahorros de energía, etc. El qué debe hacer el sistema y el cómo lo va a hacer manejado en la Especificación de Requerimientos y en la Especificación de Diseño desde las primeras etapas del ciclo de vida permite también tener el beneficio para el negocio del conocimiento. El validar un sistema permite reunir, desde un principio toda la información necesaria para conocer el sistema. El conocimiento se obtiene de la siguiente manera:
El proceso de validación ayuda a la transferencia del conocimiento del
proveedor al usuario o la organización. En un sistema nuevo es esencial ir adquiriendo el conocimiento desde
los inicios del proyecto, lo que permite identificar puntos de mejora y puntos críticos.
El conocimiento del sistema puede ser mantenido a lo largo de la vida del sistema.
El proceso que involucra el sistema se puede entender de manera más fácil, de forma más digerida y transparente si se lleva a cabo la validación.
La información va a estar plasmada en documentos y / o archivos electrónicos, de manera tal que serán accesibles para todo el personal interesado.
Típicamente cuando un equipo o proceso requiere validación, mucha de la información requerida para este proceso está disponible con el proveedor. El conocimiento necesita ser transferido al usuario de la organización. La validación ayuda en esta transferencia y genera información acerca del equipo o sistema durante la operación. Esto es particularmente práctico para nuevos productos elaborados usando nuevos sistemas y equipo. Por lo tanto es esencial que un mecanismo esté disponible para identificar y capturar información, sistemáticamente, con el fin de que el conocimiento requerido sea generado y mantenido con el usuario de la organización. El conocimiento que puede ser adquirido a través del proceso de validación incluye:
Entendimiento del proceso: la información obtenida en un proceso a
través de la transferencia y validación del proceso, además de la información disponible por parte de los proveedores, ofrece un

IMPLEMENTACIÓN Y BENEFICIOS
Página 116 de 125
entendimiento minucioso o profundo de ese proceso. El conocimiento va a ser tan minucioso y extenso como se requiera.
Eficiencia Operacional Adquirida: un entendimiento profundo del proceso usualmente mejora la eficiencia en el arranque inicial, de manera general, entre más grande sea la eficiencia de inicio, mayor será la eficiencia al termino a través de la aplicación continua de procesos mejorados.
Reducción de fallas y riesgos: La información generada a través de la validación permite que las partes críticas de un proceso sean identificadas. Medidas apropiadas pueden ser tomadas para prevenir que los límites de fallas sean alcanzados.
Mantenimiento de Estándares de Calidad: Los resultados y tendencias en un proceso de control proveen alertas acerca de cambios ligeros en un proceso. Lo anterior aunado a un entendimiento de los más significantes parámetros de proceso, obtenidos del proceso de validación, provee una invaluable herramienta para la toma de decisiones requeridas durante el proceso para el mantenimiento del cumplimiento.
La reducción de costos de operación y la mejora en la rentabilidad se encuentran entre los beneficios del negocio como resultado de los beneficios del conocimiento. Tales beneficios continúan generándose a través de la vida de trabajo del sistema, mientras se tengan procedimientos apropiados de administración operacional. Los beneficios aparecen desde el momento que un sistema inicia su operación, pero se incrementan a lo largo de las mejoras hechas. Tales mejoras pueden ser hechas también por los operadores, quienes tienen el conocimiento del proceso de manera detallada.
4.5.1.4 Beneficios tecnológicos
Los beneficios tecnológicos vienen como resultado de los conocimientos y experiencia que van adquiriendo los empleados en los equipos y procesos. Al ir adquiriendo mejor tecnología se promueve el desarrollo y la industrialización y con ello llegamos al beneficio de ser altamente competitivos. Si esta tecnología aunada al conocimiento y experiencia se deja de manera escrita, es decir, documentada se cuenta con una parte valiosa en la empresa. Esta documentación sirve como base para que el personal de nuevo ingreso adquiera el conocimiento necesario para dar soporte, mantenimiento, o la responsabilidad que vayan a desarrollar, para seguir con el desarrollo de la empresa. Si a las personas se les enseña a transmitir sus conocimientos y experiencia a través de documentos, al paso del tiempo la empresa tendrá un conjunto de

IMPLEMENTACIÓN Y BENEFICIOS
Página 117 de 125
conocimientos del sistema y los equipos lo que le dará un valor invaluable para seguir desarrollando tecnología y desarrollo en el mercado. Por ejemplo, en el caso de sistemas de adquisición de datos podemos ver que si tenemos un sistema que no cumple con la parte de firmas electrónicas, se desarrollará tecnología al adquirir un sistema de adquisición de datos que ya cumpla con ello, pero es muy importante tener la base de las características del sistema anterior y se documentarán las características del siguiente para realizar mejoras al sistema. Este conocimiento plasmado en documentos también permite no depender de terceras personas, como proveedores, ya que con el conocimiento en papel otras personas lo pueden absorber. Una empresa puede ser competitiva cuando se mantiene actualizada y a la vanguardia tecnológica.
4.5.2 Beneficios sociales.
Si hablamos de los beneficios sociales tenemos que el hecho de hacer pruebas a nuestro sistema, el tener controlados sus cambios y tener un monitoreo continuo de su desempeño permite fabricar medicamentos con calidad. Calidad que se verá reflejada en la salud de los pacientes, al ofrecerles medicamentos limpios, fabricados en las condiciones ambientales adecuadas y con los más rigurosos niveles de calidad.
En el caso de Laboratorios Farmacéuticos Mexicanos tener un proceso de validación, confirmar el cumplimiento regulatorio y tener la cultura de mejora continua les permite posicionarse en el ambiente competitivo no sólo nacional sino también en el internacional. Lo que se vería reflejado también en la economía del País. Para validar los sistemas se requiere de personal calificado y dedicado a este proceso. Por lo que, ya sea un Laboratorio Nacional o Extranjero generará empleos donde la sociedad mexicana podrá emplearse. Los profesionistas solicitados para desarrollar este tipo de actividades suelen ser, entre otros, Ingenieros Químicos, Ingenieros Farmacéuticos, Ingenieros Industriales, Ingenieros en Control y Automatización e Ingenieros en Mecatrónica o carreras a fin.
4.5.3 Beneficios al aplicar mi propuesta
Los beneficios que tiene mi propuesta para la validación son:
Presentar a la comunidad de Ingeniería en Control y Automatización esta opción de desarrollo profesional en el ambiente laboral o para la creación de una Empresa que ofrezca estos servicios.

IMPLEMENTACIÓN Y BENEFICIOS
Página 118 de 125
Proporciona de manera general el contexto de la Validación de Sistemas
Automáticos y de Cómputo, lo cuál sirve de introducción al personal que tiene un primer contacto con la Industria Farmacéutica.
Doy una propuesta con la explicación del objetivo de cada documento y los formatos con lo que deben contener, así la Empresa que lo requiera implementar (Laboratorio Farmacéutico, Integradores, Proveedores) sólo debería adecuarlo a los formatos de Guías o Procedimientos que rijan.
Compendio de los puntos que debe incluir un determinado entregable en un
solo formato que en la GAMP4 viene mencionado en más de una sección, por ejemplo la especificación de diseño.
Tener un organigrama para la tarea de validación de sistemas automáticos y
de cómputo que permita tener personal capacitado y adquiriendo conocimientos que posteriormente reduzcan el tiempo de atención y reparación de fallas, el tiempo de los procesos de documentación, el tiempo de solicitudes de cambios y su implementación, además de la mejora de los sistemas y una experiencia muy amplia en los sistemas para poder responder a futuras auditorías.
Con la implementación de mi propuesta ofrezco una estructura de validación
común para todos los equipos y sistemas.
Contempla la parte de recuperación y audit trail que permitiría cumplir con la TGA, FDA y ANMAT y que México aún no lo contempla en el proyecto de norma PROY-NOM-059-SSA1-2004.
Con el ciclo de vida que propongo se están cubriendo varios puntos de Normatividad Internacional, con mi propuesta se tendría la base para un proyecto más ambicioso de exportación a otros Países incluyendo EU.
El ciclo de vida es recomendado por guías de EU, por ejemplo, General
Principles of Software Validation.

Página 119 de 125
CONCLUSIONES La Propuesta para Validación de Sistemas Automáticos y de Cómputo para la Industria farmacéutica que expongo en esta Tesis responde a los requerimientos del Proyecto de Norma Oficial Mexicana PROY-NOM-059-SSA1-2004, además de dar las bases para el cumplimiento de la normatividad de otros Países. La implementación de la misma en un Laboratorio Farmacéutico le permitirá estar a la vanguardia y ser una Industria competitiva a nivel nacional e internacional. El modelo de ciclo de vida propuesto en las 5 fases: Planeación y Especificación, Diseño y Construcción, Instalación y Pruebas, Operación del Sistema y Retiro, es sencillo y está descrito de manera general para cubrir a varios tipos de sistemas. El seguimiento de la metodología propuesta permite que la satisfacción del cliente, esté garantizada durante todo el ciclo de vida a través del cumplimiento de los requerimientos de usuario. Este documento nos permitió conocer la posición de México a nivel mundial en cuanto a los requerimientos de la validación de Sistemas Automáticos y de Cómputo. Con las tablas de correspondencia, mostradas en la sección 3.1 Análisis, se puede ver el cumplimiento de la Propuesta con la normatividad y guías relacionadas, mencionadas a lo largo de la misma. El análisis de toda la documentación necesaria para la implementación del proceso de validación de sistemas automáticos y de cómputo, en mi experiencia me ha permitido hacer mejoras en cuanto la organización de la información, identificar la información valiosa y lo importante que tiene el hecho de plasmar la información de manera directa, exacta y entendible para las personas no importando su especialidad. Por otro lado, este análisis, también permite hacer mejoras al sistema, ya que se analizan los procesos existentes y se puede visualizar la reducción de tiempos de elaboración de documentos y la liberación del sistema o equipo a Producción. Por último cabe mencionar que no hay que perder de vista, como personas involucradas en la validación de sistemas en la Industria Farmacéutica, que estos sistemas y equipos producen medicamentos para uso humano y la validación juega un papel trascendental en la salud de los pacientes.

Página 120 de 125
GLOSARIO Y ACRÓNIMOS
GLOSARIO Auditoría: Proceso sistemático, independiente y documentado para obtener evidencia y evaluar su objetividad para determinar el cumplimiento de los criterios. Automatización: Es el uso de sistemas de control como computadoras para controlar máquinas y procesos industriales, reduciendo la necesidad de la intervención humana. Buenas Prácticas de Fabricación: Conjunto de lineamientos y actividades relacionadas entre sí, destinadas a garantizar que los productos farmacéuticos elaborados tengan y mantengan la identidad, pureza, concentración, potencia e inocuidad, requeridas para su uso. NOTA: En lugar de la palabra Fabricación es común también el uso de la palabra Manufactura. En inglés el término empleado es Good Manufacturing Practice (GMP). Calidad: Grado en que un conjunto de características inherentes cumple con las necesidades o expectativas establecidas, generalmente implícitas u obligatorias. La calidad de un medicamento está determinada por su identidad, pureza, contenido o potencia y cualesquiera otras propiedades químicas, físicas, biológicas o del proceso de fabricación que influyen en su aptitud para producir el efecto para el cual se destina. Calificación: Evaluación de las características de los elementos del proceso. Ciclo de Vida de un Sistema: Período de tiempo que inicia cuando un sistema es creado y termina cuando el sistema sale de operación. Cliente: Organización o persona que recibe un producto o servicio. Commissioning: Término en inglés empleado para referirse a la adecuada administración, planeación y documentación desde el punto de vista de Ingeniería para la puesta en marcha y la entrega de los sistemas. Control: Comparación de los resultados obtenidos en el seguimiento de objetivos previamente definidos. Criterio de aceptación: Especificación del producto y el criterio de aceptar o rechazar con base en niveles de calidad de aceptación o rechazo, asociado a un plan de muestreo. Elementos necesarios que forman parte de la liberación o rechazo de un lote o de unidades fabricadas. Especificación: Documento que describe de manera precisa, completa y verificable, los requerimientos, diseño u otras características de un sistema o componente.

Página 121 de 125
Eficacia: Es la capacidad de lograr un efecto deseado o esperado. Eficiencia: Relación entre la energía obtenida (energía útil) del funcionamiento y la energía suministrada o consumida por la máquina o el proceso. Hardware: Es la parte física (tangible) de una computadora y más ampliamente de cualquier dispositivo electrónico. Ingeniería: Arte de aplicar los conocimientos científicos a la invención, perfeccionamiento o utilización de la técnica en todas sus determinaciones. Esta aplicación se caracteriza por utilizar principalmente el ingenio de una manera más pragmática y ágil que el método científico, puesto que una actividad de ingeniería, por lo general, está limitada a un tiempo y recursos dados por proyecto Medicamento: Toda sustancia o mezcla de sustancias de origen natural o sintético que tenga efecto terapéutico, preventivo o rehabilitatorio, que se presente en forma farmacéutica y se identifique como tal por su actividad farmacológica, características físicas, químicas y biológicas. Parámetro de Proceso: Variable que define o controla un proceso y que puede afectar las salidas. Peor Caso: Condición o conjunto de condiciones que abarcan límites y circunstancias superiores e inferiores de procesamiento, dentro de procedimientos de operación normalizados, que poseen la mayor oportunidad de falla en el producto o en el proceso cuando se compara con condiciones ideales. Tales condiciones no inducen necesariamente a fallas en el producto o proceso. Proceso: Conjunto de actividades que interrelacionadas transforman ciertas entradas en ciertas salidas. Proceso de Validación: Establecimiento de evidencia documentada que provea un alto grado de aseguramiento de que un proceso especifico producirá consistentemente un producto cumpliendo con especificaciones y atributos de calidad predeterminados. Proveedor: Organización o persona que proporciona un producto o servicio. Rastreabilidad: Capacidad de reconstruir la historia, localización de un elemento o de una actividad, por medio de registros de identificación. Rendimiento: Relación entre la energía obtenida (energía útil) del funcionamiento y la energía suministrada o consumida por la máquina o el proceso. Requerimiento: Condición o capacidad solicitada por un usuario para resolver un problema o cumplir un objetivo, especificación o estándar.

Página 122 de 125
Revalidación: Repetición de la validación del proceso para proveer un aseguramiento de que cambios en el proceso / equipo introducidos de acuerdo con los procedimientos de control de cambios no afecten adversamente las características del proceso y la calidad del producto. Revisión: Actividad asumida para determinar lo apropiado, adecuado y efectivo del tema en cuestión para alcanzar los objetivos establecidos. Seguridad: Protección del hardware y software de accesos, uso, modificación, destrucción o divulgación de manera accidental o maliciosa. La seguridad también va dirigida al Personal, datos, comunicaciones e instalaciones. Sistema: Conjunto de elementos cuyas propiedades se interrelacionan e interactúan entre sí para contribuir a un determinado objeto. Sistemas Críticos: Aquellos que tiene impacto directo en los procesos y productos. Sistema de Cómputo: Sistema que contiene una o más computadoras, periféricos y un software asociado. Sistema de Control: Ordenamiento de componentes físicos conectados de tal manera que él mismo pueda comandar, dirigir o regularse a sí mismo o a otro sistema. Sistema Automático: Sistema que utiliza una computadora para controlar equipos y procesos. Tiene un paso más allá de la mecanización y capacidad de repetición, reduce la necesidad de la intervención del hombre. Software: Conjunto de programas, instrucciones y reglas informáticas para ejecutar ciertas tareas en una computadora. Usuario: Persona(s) que opera o interactúa directamente con un equipo o sistema. Verificación: Confirmación, a través de la provisión de evidencia objetiva que requerimientos específicos han sido cumplidos. Validación: Es la evidencia documentada de que a través de un proceso eficaz y reproducible, un producto es obtenido de acuerdo a las especificaciones, atributos de Calidad predeterminados y principios de buenas prácticas de manufactura.

Página 123 de 125
ACRÓNIMOS
ACRÓNIMO DEFINICIÓN AI Analog Input, Entrada Analógica
ANMAT Administración Nacional de Medicamentos, Alimentos y Tecnología Médica.
AO Analog Output, Salida Analógica ASV Administrador del Sistema de Validación
BMS Building Management Systems, Sistemas de Administración de Edificios
CFR Code of Federal Regulations, Código de Regulaciones Federales
BPF, BPM o GMP
Cualquiera de estos 3 términos se utiliza para referirse a las Buenas Prácticas de Manufactura. BPF – Buenas Prácticas de Fabricación BPM – Buenas Prácticas de Manufactura GMP – Good Manufacturing Practice
CD Calificación de Diseño CE Calificación de Desempeño CI Calificación de Instalación CO Calificación de Operación CPU Central Processing Unit, Unidad Central de Procesamiento DCS Distributed Control System, Sistema de Control Distribuido DI Digital Input, Entrada Digital DQ Design Qualification, Calificación de Diseño DO Digital Output, Salida Digital DS Design Specificaction, Especificación de Diseño DTI’s Diagramas de Tubería e Instrumentación
ERP Enterprise Resource Planning, Planificación de Recursos de la Empresa
EU Estados Unidos FAT Factory Acceptance Test, Pruebas de Aceptación de Fábrica
FDA Food and Drug Administration, Administración de Alimentos y Drogas
FS Functional Specification, Especificación Funcional
GAMP Good Automated Manufacturing Practice, Buenas Prácticas de Automatización para Manufactura
GATT General Agreement on Tariffs and Trade GEP Good Engineering Practices, Buenas Prácticas de Ingeniería
HDS Hardware Design Specification, Especificación de Diseño de Hardware
HPLC High Performance Liquid Chromatography, Cromatógrafos de Líquidos de Alto Desempeño
IC Integrated Circuit, Circuito Integrado IQ Installation Qualification, Calificación de Instalación
ISPE International Society of Pharmaceutical Engineering, Sociedad Internacional de Ingeniería Farmacéutica
ISO International Organization for Standardization, Organización Internacional de Normalización

Página 124 de 125
IT Information Technology, Tecnología de la Información
LIMS Laboratory Information Management System, Sistemas de Administración de Información de Laboratorio
LSV Líderes del Sistema de Validación NOM Norma Oficial Mexicana OMC Organización Mundial del Comercio OQ Operational Qualification, Calificación de Operación
P&ID Piping & Instrumentation Diagram, Diagramas de Tubería e Instrumentación
PLC Programmable Logic Controller, Controlador Lógico Programable PNO Procedimiento Normalizado de Operación PQ Performance Qualification, Calificación de Desempeño PROY Proyecto QMS Quality Management System, Sistema de Administración de Calidad QP Quality Plan, Plan de Calidad QPP Quality and Project Plan, Plan del Proyecto y Calidad RS Requirements Specification, Especificación de Requerimientos SAT Site Acceptance Test, Pruebas de Aceptación en Sitio
SCADA Supervisory Control and Data Acquisition, Adquisición de Datos y Supervisión de Control
SDS Software Design Specification, Especificación de Diseño de Software
TGA Therapeutic Goods Administration, Buena Administración Terapéutica
TLC Tratado de Libre Comercio
UPS Uninterruptible Power Supply, Fuente de Alimentación Ininterrumpible
URS User Requirements Specification, Especificación de Requerimientos de Usuario
VMP Validation Master Plan, Plan Maestro de Validación VP Validation Plan, Plan de Validación

Página 125 de 125
BIBLIOGRAFÍA
• NOM-059-SSA1-1993, Buenas Prácticas de Fabricación para Establecimientos de la Industria Químico Farmacéutica dedicados a la Fabricación de Medicamentos.
• PROY-NOM-059-SSA1-2004, Buenas Prácticas de Fabricación para Establecimientos de la Industria Químico Farmacéutica dedicados a la Fabricación de Medicamentos.
• EU CFR-21 (Code of Federal Regulations), Partes 210 y 211 (Buenas Prácticas de Manufactura, Abril 2007), Parte 11 (Firmas y registros electrónicos, Agosto 2001) y Parte 820 (Validación de Software y equipos automáticos, Abril 2007).
• General Principles of Software Validation; Final Guidance for Industry and FDA Staff, January 2002.
• Australian Code of Good Manufacturing Practice for Medicinal Products, Agosto 2002, Anexo 11 (Sistemas computarizados) y Anexo 15 (Calificación y Validación).
• Disposición Argentina 2819/2004, sección 4 (Calificación y Validación) y Anexo II (Calificación y Validación).
• NMX-CC-9001-IMNC-2000 Sistemas de Gestión de la Calidad – Requisitos.
• GAMP4 Guide for Validation of Automated Systems, Diciembre 2001, Publicada
por ISPE
• Matt Seaver, Implementación de la ISO 9000:2000, Año 2002, Editorial Panorama
• Artículo Ciclo de Vida en página electrónica: http://www.getec.etsit.upm.es/docencia/gproyectos/planificacion/cvida.htm
• Página electrónica de la Real Academia Española (RAE):
http://www.rae.es/rae.html