valorización de diferentes cultivos lignocelulósicos para...
TRANSCRIPT

Valorización de diferentes cultivos lignocelulósicospara la fabricación de pasta de papel:
Valorización de diferentes cultivos lignocelulósicospara la fabricación de pasta de papel:
Caracterización química, modificación estructural de sus constituyentes orgánicos durante los procesos de cocción y blanqueo y aplicaciones biotecnológicas
Caracterización química, modificación estructural de sus constituyentes orgánicos durante los procesos de cocción y blanqueo y aplicaciones biotecnológicas
Gisela Marques SilvaSevilla, 2010

Valorización de diferentes cultivos lignocelulósicos para la fabricación de pasta de papel: Caracterización química, modificación estructural de sus constituyentes orgánicos durante los procesos de cocción y blanqueo y aplicaciones biotecnológicas
Memoria que presenta
Gisela Marques Silva para optar al título de Doctor en Ciencias Químicas por la Universidad de Sevilla. Sevilla, a 13 de Abril de 2010.

Valorización de diferentes cultivos lignocelulósicos para la fabricación de pasta de papel: Caracterización química, modificación estructural de sus constituyentes orgánicos durante los procesos de cocción y blanqueo y aplicaciones biotecnológicas
Visado en Sevilla, a 13 de Abril de 2010
LOS DIRECTORES
Dr. D. José C. del Río Andrade Investigador Científico del CSIC
IRNAS-CSIC
Dra. Dña. Ana Gutiérrez Suárez Investigador Científico del CSIC
IRNAS-CSIC
EL TUTOR
Dr. D. Alfonso Guiraúm Pérez Catedrático de la Universidad de Sevilla
Memoria que presenta Gisela Marques Silva para optar al grado de Doctor en Ciencias Químicas por la Universidad de Sevilla.

DOCTOR D. LUIS CLEMENTE SALAS, DIRECTOR DEL INSTITUTO DE
RECURSOS NATURALES Y AGROBIOLOGÍA DE SEVILLA DEL
CONSEJO SUPERIOR DE INVESTIGACIONES CIENTÍFICAS
CERTIFICA: Que la presente Memoria de Investigación titulada “Valorización
de diferentes cultivos lignocelulósicos para la fabricación de pasta de papel:
Caracterización química, modificación estructural de sus constituyentes
orgánicos durante los procesos de cocción y blanqueo y aplicaciones
biotecnológicas”, presentada por Gisela Marques Silva para optar al grado de
Doctor en Ciencias Químicas, ha sido realizada en el Departamento de
Biotecnología Vegetal, bajo la dirección de los Drs. D. José C. del Río Andrade
y Dña. Ana Gutiérrez Suárez, reuniendo todas las condiciones exigidas a los
trabajos de Tesis Doctorales.
En Sevilla, a 13 de Abril de 2010

AGRADECIMIENTOS
Este trabajo se ha llevado a cabo en el Instituto de Recursos Naturales y Agrobiología de Sevilla (IRNAS-CSIC). Ha sido financiado por una beca I3P de postgrado del CSIC, por una beca FPI del Ministerio de Educación y Ciencia, por los proyectos nacionales de investigación AGL2005-01748 y AGL2008-00709, y por el proyecto europeo NMP2-CT-2006-026456.
Quiero expresar mi sincero agradecimiento a las personas que tanto directa como indirectamente han hecho posible la realización de esta Tesis:
A los Dres. José Carlos del Río y Ana Gutiérrez, directores de esta Tesis, por todo lo que me han aportado tanto a nivel científico, por sus conocimientos y enseñanzas, como a nivel personal, por la confianza en su trato personal y por sus consejos, y por estar siempre que los he necesitado. Por su esfuerzo y dedicación en esta Tesis.
Al Prof. Ángel T. Martínez, del Centro de Investigaciones Biológicas (CIB-CSIC, Madrid), por ofrecerme la posibilidad de realizar en el CIB algunas estancias breves, aportándome numerosos y valiosos conocimientos, y por haber seguido el desarrollo de esta Tesis.
Al Prof. Dmitry Evtuguin, de la Universidad de Aveiro (Aveiro, Portugal), por ofrecerme la posibilidad de realizar dos estancias en su grupo de investigación, por acogerme como un miembro más de su familia y por aportarme valiosos conocimientos. Por su apoyo y consejos, y su excelente trato personal. También quiero agradecer al Dr. José Antonio Gamelas y a la Dra. Paula Pinto por su apoyo durante las estancias en la Universidad de Aveiro.
Al Prof. Alfonso Guiraúm, Catedrático de la Universidad de Sevilla, tutor de esta Tesis, por toda su ayuda en la parte burocrática.
A mi compañero de laboratorio durante los primeros años de la Tesis y amigo, el Dr. Jorge Rencoret, por ayudarme siempre que lo he necesitado tanto en el laboratorio como fuera, por su alegría, dándole siempre una vida muy suya al laboratorio sin dejar de lado la profesionalidad.
A mis compañeras la Dra. Isabel María Rodríguez y Setefilla Molina, con las que he coincidido en el inicio y mediados de esta Tesis, y a mis compañeros PepijnPrinsen, Alejandro Rico y Esteban Babot con los que he coincidido en el final de esta Tesis. Un agradecimiento muy especial a Esteban por su apoyo y los buenos momentos compartidos.
A Dña. Trinidad Verdejo por hacer las pirólisis de mis numerosas muestras.

Al Prof. Jesús Jiménez-Barbero, al Dr. Iñaki Santos y a Lidia Nieto del CIB-CSIC, por sus múltiples análisis de NMR.
A Gerardo Artal (CELESA) por suministrarme las muestras de diversas fibras y sus pastas, al Dr. Javier Romero (ENCE) por las pastas de eucalipto y al Dr. Manuel J. Díaz Blanco (Universidad de Huelva) por las muestras de caña común y tagasaste.
Al Dr. García Hortal (UPC, Terrassa) por las imágenes proporcionadas de las fibras elementales de las muestras de lino, cáñamo, kenaf, yute, sisal y abacá que se muestran en la sección de Material y Métodos de esta Tesis.
A mis compañeros del IRNAS, Rocío, Mari Trini, Agüi, Alegría, María Fernanda, Fátima, José María, Antonio y Jaime, que me acompañaron durante el inicio de esta Tesis, y en particular a Fátima Sopeña que también me acompañó durante casi toda la Tesis brindándome muy buenos momentos y consejos y por estar siempre allí incluso durante su post-doc en el extranjero.
A los compañeros del CIB, Mario, Yuta, María, Ángeles, Miguel, Elvira, Davinia, Vero, Helena, Aitor, Eva y Beatriz, y en particular a Celia Méndez por su apoyo en mi primera estancia en el CIB y por los buenos momentos brindados. Quiero agradecer también al Dr. Javier Ruiz-Dueñas por su apoyo en las estancias realizadas en el CIB.
A María Jesús Ortega, madre de Aitor del CIB, aunque sólo la conozco por Internet, por sus e-mails y por proporcionarme una de las fotos que se muestra en la sección de Introducción de esta Tesis.
Y por último a mi hermana, a mi madre y a Augusto por el apoyo brindado aunque estén lejos. A Dani por su paciencia, por escucharme hablar de experimentos que le son completamente ajenos y por estar ahí cuando más lo necesito, así como a su familia de Sevilla que desde que los conozco me han brindado todo su apoyo.
Gracias a todos y gracias también, tan sólo por su existencia, a una nueva personita subacuática que ahora llevo dentro…

ABREVIATURAS
AQ Antraquinona ABTS 2,2’-azinobis(3-etilbenzotiazolin-6-sulfonato) BSTFA� N,O-bis-(trimetilsilil)-trifluoroacetamida
�C� Desplazamiento químico del carbono
�H Desplazamiento químico del protón
CED Cobre (II)-etilendiamina COSY Espectroscopia de correlación (“Correlation Spectroscopy”) DBO Demanda biológica de oxígeno DCM Diclorometano DMAC N,N-dimetilacetamida DMSO Dimetilsulfóxido DTT Ditiotreitol 2D-NMR Espectroscopía de Resonancia Magnética Nuclear bidimensional 3D-NMR Espectroscopía de Resonancia Magnética Nuclear tridimensional ECF Secuencia de blanqueo libre de cloro elemental (“elemental
chlorine free”) FAO Organización de las Naciones Unidas para la Agricultura y
Alimentación (“The Food and Agriculture Organization of the United Nations”)
FID Detector de ionización de llama (“flame ionization detector”) G Unidad guayacilpropano (o guayacilo) GC Cromatografía de gases (“gas chromatography”) GC/MS Cromatografía de gases/espectrometría de masas (“gas
chromatography/mass spectrometry”) H Unidad 4-hidroxifenilpropano (o 4-hidroxifenilo) HBT 1-Hidroxibenzotriazol HPLC Cromatografía líquida de alta resolución HSQC Espectroscopía 2D de correlación heteronuclear de cuanto simple
(“heteronuclear single-quantum correlation”)

HexA Ácidos hexenurónicos ICP-OES Espectrometría de emisión óptica con plasma acoplado
inductivamente (“inductively coupled plasma-optical emission spectrometry”)
ID Diámetro interno (“internal diameter”) IK Índice Kappa IPTG Isopropil tio-�-D-galactopiranósido ISO Organización Internacional para la Estandarización,
Documentación e Información (“International Organization forStandardization”)
ITD Detector de trampa de iones (“ion trap detector”) Ligninaox Productos de degradación oxidativa de la lignina LiP Lignina peroxidasa Mw Masa molecular (“molecular weight”) MnP Manganeso peroxidasa MWL Lignina de madera molida (“milled wood lignin”) NMR Espectroscopía de Resonancia Magnética Nuclear (“nuclear
magnetic resonance”) O Etapa de deslignificación con oxígeno (en secuencia de blanqueo) PCA Ácido p-cumárico POM polioxometalato POM 1- Primera etapa POM en los ensayos de deslignificación -POM 2- Segunda etapa POM en los ensayos de deslignificación POMox Polioxometalato oxidado POMred Polioxometalato reducido PoP Doble etapa de blanqueo con peróxido de hidrógeno, la primera
bajo oxígeno presurizado ppb Partes por billón ppm Partes por millón Py-GC/MS Pirólisis acoplada a cromatografía de gases/espectrometría de
masas (“pyrolysis-gas chromatography/mass spectrometry”) Q Etapa de quelato (en secuencia de blanqueo) rpm Revoluciones por minuto

S Unidad siringilpropano (o siringilo) SPE Extracción en fase sólida (“solid phase extraction”) TAPPI Technical Association of the Pulp and Paper Industry TCF Secuencia de blanqueo totalmente libre de cloro (“totally chlorine
free”) TMAH Hidróxido de tetrametilamonio TMP Pasta termomecánica (“thermomechanical pulp”) TMSD Trimetilsilildiazometano TMS Trimetilsililo TOCSY Espectroscopia de correlación total (“Total Correlation
Spectroscopy”) U Unidad de actividad enzimática UV/VIS Espectroscopía de ultravioleta/visible VP Peroxidasa versátil � Coeficiente de extinción molar

ÍNDICE
RESUMEN ..........................................................................................................1
1. INTRODUCCIÓN.........................................................................................5
1.1. CULTIVOS LIGNOCELULÓSICOS .....................................................5 1.1.1. Fibras procedentes de cultivos madereros ......................................5 1.1.2. Fibras procedentes de cultivos agrícolas ........................................7
1.2. ESTRUCTURA Y COMPOSICIÓN QUÍMICA DE LOS MATERIALES LIGNOCELULÓSICOS................................................9
1.2.1. Celulosa.........................................................................................10 1.2.2. Hemicelulosas ...............................................................................11 1.2.3. Lignina ..........................................................................................13 1.2.4. Componentes de bajo peso molecular ..........................................20
1.3. UTILIZACIÓN DE CULTIVOS LIGNOCELULÓSICOS: PRODUCCIÓN DE PASTA DE CELULOSA .....................................23
1.3.1. Procesos de pasteado ....................................................................24 Procesos mecánicos ...................................................................24 Procesos químicos .....................................................................24 1.3.2. Procesos de blanqueo....................................................................26
1.4. PROBLEMÁTICA PLANTEADA POR LA PRESENCIA DE LIGNINA Y LÍPIDOS EN LA PRODUCCIÓN DE PASTA DE CELULOSA...........................................................................................28
1.5. BIOTECNOLOGÍA EN EL SECTOR DE LA PASTA DE CELULOSA...........................................................................................29
1.5.1. Degradación enzimática de la lignina...........................................30 1.5.2. Degradación enzimática de lípidos: Control del pitch .................33
2. OBJETIVOS ................................................................................................37
3. MATERIAL Y MÉTODOS .......................................................................41
3.1. MATERIALES ......................................................................................41 3.1.1. Cultivos lignocelulósicos..............................................................41
Lino ...........................................................................................41

Cáñamo .....................................................................................42 Kenaf .........................................................................................43 Yute ...........................................................................................44 Sisal ...........................................................................................45 Abacá ........................................................................................46 Curauá .......................................................................................47 Caña común...............................................................................47 Tagasaste...................................................................................47
3.1.2. Pastas de papel ..............................................................................48 Pastas de fibras no madereras ...................................................48 Pastas de fibras madereras ........................................................48
3.1.3. Enzimas y mediadores ..................................................................49 Lipoxigenasas ...........................................................................49 Peroxidasas................................................................................49 Polioxometalatos.......................................................................50
3.2. MÉTODOS ANALÍTICOS ...................................................................51 3.2.1. Aislamiento y análisis de los compuestos lipofílicos de las
fibras y pastas............................................................................51 Fraccionamiento de los compuestos extraíbles
lipofílicos mediante SPE...........................................................51 Métodos de derivatización de los compuestos extraíbles
lipofílicos ..................................................................................53 Análisis de los extractos lipofílicos mediante GC y GC/MS........53 3.2.2. Aislamiento y análisis de la lignina de las fibras y pastas............54
Determinación del contenido en lignina ...................................54 Aislamiento de la lignina de las fibras......................................55 Análisis de la lignina mediante Py-GC/MS..............................55 Análisis de la lignina mediante DFRC .....................................56 Análisis de la lignina mediante 2D-NMR ................................59
3.2.3. Aislamiento y análisis de las hemicelulosas de las fibras y pastas.........................................................................................62
Preparación de la holocelulosa y aislamiento de los xilanos ......................................................................................62
Análisis de azúcares neutros tras hidrólisis ácida....................62

Análisis de azúcares neutros y ácidos urónicos tras metanolisis ácida ......................................................................63
Determinación del peso molecular de los xilanos mediante SEC............................................................................63
Análisis de la estructura de los xilanos mediante NMR ..........64 Determinación del contenido en ácidos hexenurónicos ...........64 3.2.4. Otros análisis.................................................................................65 Determinación de la fracción hidrosoluble de las fibras ..........65 Determinación del contenido en cenizas de las fibras..............65 Análisis de metales y otros elementos en las fibras .................66 3.2.5. Tratamientos enzimáticos de las pastas ........................................66
Tratamientos con lipoxigenasas................................................66 Tratamientos con POM y peroxidasa versátil...........................67 Determinación de las propiedades de las pastas .......................69 Determinación de la blancura ISO........................................69 Determinación del índice kappa ...........................................69 Determinación de la viscosidad intrínseca ...........................70 Determinación del contenido en ácidos hexenurónicos .......72
4. REFERENCIAS ..........................................................................................75
5. RESULTADOS Y DISCUSIÓN.................................................................91
Publicación I: Marques G., Rencoret J., Gutiérrez A., del Río J.C. (2010) Evaluation of the chemical composition of different non-woody plant fibers used for pulp and paper manufacturing. The Open Agriculture Journal (in press) ......................................................93
Publicación II: del Río J.C., Marques G., Rencoret J., Martínez A.T. and Gutiérrez A. (2007) Occurence of naturally acetylated lignin units. Journal of Agricultural and Food Chemistry, 55, 5461-5468. ...........................................................................................111
Publicación III: del Río J.C., Rencoret J., Marques G., Gutiérrez A., Ibarra D., Santos J.I., Jiménez-Barbero J., Zhang L. and Martínez A.T. (2008) Highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. Journal of Agricultural and Food Chemistry, 56, 9525-9534.............127
Publicación IV: Marques G., Gutiérrez A. and del Río J.C. (2007) Chemical characterization of lignin and lipophilic fractions from

leaf fibers of curaua (Ananas erectifolius). Journal of Agriculture and Food Chemistry, 55, 1327-1336................................151
Publicación V: Coelho D., Marques G., Gutiérrez A., Silvestre A.R.D. and del Río J.C. (2007) Chemical characterization of the lipophilic fraction of Giant reed (Arundo donax) fibers used for pulp and paper manufacturing. Industrial Crops and Products, 26, 229-236.. ........................................................................................173
Publicación VI: Marques G., Gutiérrez A. and del Río J.C. (2008) Chemical composition of lignin and lipids from tagasaste (Chamaecytisus proliferus spp. palmensis). Industrial Crops and Products, 28, 29-36. .....................................................................187
Publicación VII: Marques G., del Río J.C. and Gutiérrez A. (2010) Lipophilic extractives from several nonwoody lignocellulosic crops (flax, hemp, sisal, abaca) and their fate during alkaline pulping and TCF/ECF bleaching. Bioresource Technology, 101, 260-267.................................................................................................203
Publicación VIII: Marques G., Gutiérrez A., del Río J.C. and Evtuguin D.V. (2010) Acetylated heteroxylan from Agavesisalana and its behavior in alkaline pulping and TCF/ECF bleaching. Carbohydrate Polymers, (doi: 10-1016/j.carbpol.2010.02.043)................................................................221
Publicación IX: Marques G., Gamelas J.A., Ruiz-Dueñas F.J., del Río J.C., Evtuguin D.V., Martínez A.T. and Gutiérrez A. (2010) Delignification of eucalypt kraft pulp with manganese-substituted polyoxometalate assisted by fungal versatile peroxidase. Bioresource Technology, 101, 5935-5940. .....................241
Publicación X: Marques G., Molina S., Babot E.D., Lund H., del Río J.C. and Gutiérrez A. Exploring the potential of a fungal manganese lipoxygenase to remove lipophilic extractives from paper pulps. Bioresource Technology (in preparation).......................255
6. CONCLUSIONES .....................................................................................273
7. ANEXOS ....................................................................................................277

RESUMEN
La presente Tesis plantea el estudio de la composición química de los principales constituyentes de diferentes cultivos lignocelulósicos utilizados como materia prima para la fabricación de pastas de celulosa de alta calidad, poniendo especial énfasis en la composición de la fracción lipofílica (responsable de la formación de los denominados depósitos de pitch) y de la lignina (cuya composición y estructura influyen decisivamente en el proceso de deslignificación), así como en la composición de las hemicelulosas. Entre los materiales estudiados se incluyen fibras no madereras del tallo de varias angiospermas dicotiledóneas, tales como lino (Linum usitatissimum), kenaf (Hibiscus cannabinus), cáñamo (Cannabis sativa) y yute (Corchorus capsularis), así como fibras procedentes de hojas de angiospermas monocotiledóneas como sisal (Agave sisalana), abacá (Musa textilis) y curauá (Ananas erectifolius). Otras fibras estudiadas fueron las procedentes de la caña común (Arundo donax) y de podas de árboles de tagasaste (Chamaecytisusproliferus spp. palmensis). Se estudió también la evolución de los constituyentes de los materiales lignocelulósicos durante la producción de pasta de papel. Para ello, se seleccionaron diversas pastas de celulosa a lo largo de los procesos de pasteado (cocción sosa-antraquinona) y de blanqueo (procesos TCF y ECF). Finalmente, se ensayaron dos procedimientos biotecnológicos basados en la utilización de enzimas fúngicas para la eliminación tanto de lignina como de lípidos residuales en pastas de celulosa.
Los resultados obtenidos muestran que las diferentes materias primas estudiadas se caracterizan, en general, por un alto contenido en polisacáridos y un bajo contenido en lípidos y lignina, lo que las hace, en principio, favorables para la producción de pasta de celulosa. Los compuestos lipofílicos presentes en las fibras, analizados por GC y GC/MS, incluyen principalmente ácidos grasos, hidroxiácidos, alcoholes, ceras, alcanos y esteroles libres y conjugados (en forma de ésteres y glicósidos), entre otros. Los análisis indican que el contenido y composición de las diferentes clases de lípidos varía considerablemente entre las distintas fibras. Además, las diferentes clases de lípidos muestran distinto comportamiento durante los procesos de cocción y blanqueo. Así, las ceras se hidrolizan durante la cocción alcalina mientras que los ácidos grasos se disuelven. Por el contrario, los alcanos, alcoholes grasos, esteroles y triterpenoles, hidrocarburos esteroidales, cetonas y glicósidos de esteroles tienen baja solubilidad en agua y son difíciles de eliminar de la pasta, por lo que sobreviven a la cocción. Se observó que entre los compuestos que sobreviven a la cocción, los esteroles libres se eliminan durante el blanqueo ECF pero resisten al blanqueo TCF, mientras que los glicósidos de esteroles se eliminan tanto en el blanqueo TCF como ECF. Finalmente, mientras que los ácidos grasos
1

insaturados se eliminan durante los procesos de blanqueo ECF y TCF, los ácidos grasos saturados, así como los alcanos y alcoholes grasos sobreviven a estas secuencias de blanqueo.
En cuanto a las ligninas, su estructura y composición se estudió tanto por métodos degradativos (pirólisis-GC/MS y DFRC) como espectroscópicos (2D-NMR). Los análisis mostraron un predominio de unidades de tipo siringilo (S) en el caso de las fibras liberianas de kenaf y yute, así como en todas las fibras de hojas (sisal, abacá y curauá). Por el contrario, las fibras de cáñamo, lino y caña común mostraron un predominio de unidades de tipo guayacilo (G). Esto fue especialmente evidente en las ligninas de lino y cáñamo, con una relación S/G de 0,1. La mayor relación S/G de las ligninas de kenaf, yute, sisal y abacá hace que estas fibras sean más fáciles de deslignificar a causa del menor grado de condensación de la lignina, a pesar de tener un mayor contenido en lignina. Los principales enlaces entre las unidades de lignina fueron de tipo aril-éter �-O-4� en todas las fibras estudiadas. También se observaron enlaces condensados �-5�/�-O-4� (fenilcumarano), �-�� (resinol) y �-1�/�-O-�� (espirodienona). La mayor proporción de enlaces no condensados �-O-4� se encontró en las ligninas de kenaf, sisal, abacá y curauá, las cuales al tener también mayor proporción de unidades S son más fácilmente deslignificables. Por otro lado, en las ligninas de kenaf, sisal, abacá y curauá se encontraron unidades aciladas (con acetatos y/o p-cumaratos) en el carbono � de la cadena lateral y predominantemente sobre unidades S. Se demostró que la acilación tiene lugar a nivel de monómero y que el sinapil acetato, y otros monómeros acilados, se comportan como auténticos monómeros de la lignina. Se demostró también, que el nivel de acilación de la lignina estaba relacionado con un alto contenido en unidades S y enlaces �-O-4�, así como con un menor contenido en enlaces �-��.
También se estudió la composición química de las hemicelulosas y las modificaciones de las mismas durante los procesos de pasteado y blanqueo. El estudio de las hemicelulosas tiene importancia debido a que los polisacáridos se disuelven y/o degradan parcialmente durante el pasteado, lo que afecta al rendimiento del proceso y a la calidad de las pastas de celulosa. Los resultados mostraron que las hemicelulosas de las fibras liberianas presentan una mayor variabilidad en cuanto a su composición en azúcares neutros que las fibras procedentes de hojas. Así, mientras que en las fibras de lino y cáñamo predominan la manosa y la galactosa, en el kenaf y yute el monosacárido predominante es la xilosa. Por otro lado, en todas las fibras de hojas estudiadas (sisal, abacá y curauá) se observó un predominio de la xilosa. Un estudio en profundidad de la estructura de las hemicelulosas de sisal mostró que están constituidas fundamentalmente por un glucuronoxilano acetilado cuya cadena principal está formada por unidades de �-D-xilopiranosa parcialmente ramificada con residuos glucuronosilos. Esta hemicelulosa sufre una
2

despolimerización y desacetilación significativa durante el proceso de pasteado. Los grupos acetilo residuales que quedaban en la pasta cruda se eliminaron completamente tras el blanqueo.
Finalmente, se estudiaron dos procedimientos biotecnológicos basados en el uso de enzimas fúngicas para la eliminación de la lignina residual de pastas así como de los compuestos extraíbles lipofílicos responsables de la formación de depósitos de pitch durante el proceso de fabricación de pasta de papel. Estos procedimientos incluyeron la utilización de un sistema compuesto de un polioxometalato y una enzima de tipo peroxidasa producida por el hongo Pleurotus eryngii, y una lipoxigenasa producida por el hongo Gaeumannomycesgraminis. Los resultados obtenidos mostraron la eficacia del sistema polioxometalato-peroxidasa para eliminar la lignina residual de la pasta y de la lipoxigenasa para degradar parte de los compuestos lipofílicos responsables de la formación de los depósitos de pitch.
La presente Tesis incluye los siguientes apartados: i) una introducción general sobre los cultivos lignocelulósicos, su interés industrial, su estructura y composición, y los procesos utilizados para la producción de pasta de celulosa, así como los principales problemas que plantean algunos de sus constituyentes y algunas soluciones biotecnológicas a estos problemas; ii) los objetivos perseguidos en la Tesis; iii) una descripción detallada de los materiales estudiados y los métodos analíticos empleados; iv) las referencias citadas en el primer y tercer apartado; v) los resultados obtenidos y su discusión, que se presentan en forma de publicaciones; vi) las principales conclusiones; y vii) una lista de tablas que se muestran como Anexos.
3

1

1. Introducción
INTRODUCCIÓN 1.1. CULTIVOS LIGNOCELULÓSICOS
Los cultivos lignocelulósicos incluyen especies tanto de origen agrícola como forestal y poseen un gran interés industrial. Entre los principales usos de los cultivos lignocelulósicos se encuentra la producción de pasta de celulosa. Por otro lado, estos cultivos presentan un gran potencial como materia prima en el contexto de las futuras biorrefinerías para la producción de biocombustibles y otros productos de interés, como alternativa al petróleo.
La principal fuente de fibra de celulosa virgen utilizada actualmente en la fabricación de pasta de celulosa la constituyen los cultivos de fibras madereras, mientras que las fibras no madereras se utilizan en menor proporción. La amplia disponibilidad y concentración de madera en zonas de fácil acceso, el elevado contenido en fibras, el coste de manejo, transporte y facilidad de almacenamiento, así como la estabilidad de la materia prima y su comportamiento durante el proceso de obtención de celulosa, han apoyado el uso de la misma en la industria de la pasta de papel. Sin embargo, existe en la actualidad un renovado interés en el uso de plantas de origen no maderero debido a, entre otras razones, la gran disponibilidad de residuos agrícolas. Éstos constituyen una fuente abundante de fibras de bajo coste, siendo a veces la única fuente aprovechable de fibras en determinadas zonas geográficas, principalmente en países en vías de desarrollo. La gran variedad de características, dimensiones fibrosas y composición química de estas fibras les confieren un gran potencial como materias primas (García Hortal 2007). Además, en los países desarrollados, se utilizan para la fabricación de pastas de celulosa para papeles especiales.
1.1.1. Fibras procedentes de cultivos madereros
Las fibras madereras provienen de especies vegetales que desarrollan un tronco donde se acumulan preferentemente las mejores fibras. Las coníferas constituyen el primer cultivo forestal a escala mundial para la obtención de pasta de papel, aunque también existe un importante mercado de pastas de frondosas (Figura 1).
Las coníferas presentan fibras largas (3 a 5 mm), que son óptimas para la fabricación de papeles de elevada resistencia mecánica. Las coníferas, en términos económicos generales, son más valiosas que las frondosas, ya que sus troncos son más largos y rectos, su madera es uniforme, ligera y blanda, por lo que es más fácil de trabajar, y presentan una mayor proporción de elementos
5

1. Introducción
fibrosos que son más adecuados para la mayoría de las calidades papeleras (García Hortal 2007). Las principales coníferas usadas para la fabricación de pasta de papel son la Picea y el pino.
La madera de frondosas, por otro lado, es una madera más dura, de fibras cortas (entre 0,75 y 2 mm) que dan lugar a pastas menos uniformes. El papel fabricado con maderas de frondosas es más débil que los fabricados con maderas de coníferas pero su superficie es más lisa, y por lo tanto, es mejor para papel de escritura. Otra de las ventajas es que el crecimiento de las especies de frondosas utilizadas para la fabricación de pasta de papel es más rápido que el de las coníferas, dando lugar a mayor cantidad de fibra en menos tiempo. Las principales frondosas utilizadas en el sector papelero son el eucalipto, el chopo y el abedul.
Figura 1. Ejemplos de especies madereras usadas para la producción de pasta de celulosa, incluyendo coníferas como la Picea (izquierda) y frondosas como el abedul (derecha).
6

1. Introducción
1.1.2. Fibras procedentes de cultivos agrícolas
Las fibras procedentes de cultivos agrícolas constituyen una excelente materia prima alternativa a las fibras madereras para la producción de pasta de celulosa. Uno de los hechos apremiantes que conducen a la utilización de materia prima no maderera es su conocida abundancia que sobrepasa la utilización actual.
En general, las fibras de plantas no madereras tienen una estructura menos densa y más porosa, lo que implica un menor requerimiento de energía y productos químicos para la separación de las fibras durante la producción de pasta de papel. Además, presentan ciclos de crecimiento más cortos, alcanzando la madurez más rápidamente que las especies madereras y en muchos casos los rendimientos de pasta obtenidos son mayores (Tabla 1). Algunas pastas de fibras largas no madereras, tienen propiedades superiores a las mejores pastas del mercado de coníferas, pues son extremadamente resistentes. El principal inconveniente de este tipo de materias primas es que la mayoría sólo están disponibles en ciertas épocas del año.
Tabla 1. Rendimiento promedio de algunas materias primas (Pierce 1991).
Especies
Rendimiento materia seca (t/ha)
Rendimiento Pasta (t/ha)
Trigo 2,5 1,1 Avena 1,6 0,7 Centeno 2,2 1,1 Arroz 3,0 1,2 Caña de azúcar (bagazo) 9,0 4,2 Bambú 4,0 1,6 Miscanthus sinensis 12,0 5,7 Canary grass 6,0 3,0 Caña común 9,0 4,3 Kenaf 15,0 6,5 Cáñamo 12,0 6,7 Frondosa de zona templada (abedul) 3,4 1,7 Frondosa de crecimiento rápido (Eucalyptus) 15,0 7,4 Conífera escandinava 1,5 0,7 Conífera de crecimiento rápido 8,6 4,0
7

1. Introducción
Las fibras no madereras se pueden clasificar en tres categorías: i) fibras procedentes del tallo de diversas plantas como lino, cáñamo, kenaf y yute, y de hojas como abacá y sisal; ii) residuos agrícolas como la paja de trigo, maíz y arroz o el bagazo de caña; y iii) hierbas silvestres como bambú o hierba elefante. Actualmente, las fibras no madereras representan una alternativa para la producción de pasta de celulosa en países con baja disponibilidad de madera y en los que disponen de abundantes residuos agrícolas fibrosos o cultivos de plantas fibrosas no madereras. Así, el uso de estas fibras para la producción de pasta de celulosa ha ido aumentando, especialmente en los países en vías de desarrollo, como India, China y algunos países latinoamericanos.
En los países desarrollados, las fibras no madereras se usan principalmente para la producción de papeles especiales. En España, existen varias empresas que fabrican pasta de papel a partir de fibras no madereras. Entre ellas destaca la empresa CELESA que utiliza fibras liberianas (del tallo) de lino, cáñamo y yute, y fibras de hojas de sisal y abacá para fabricar pasta de celulosa para papeles especiales de distintas características, tales como papeles para cigarrillos, filtros especiales o papeles dieléctricos (Figura 2). Dicha empresa ha suministrado la mayoría de las fibras y sus respectivas pastas de papel que se han estudiado en esta Tesis.
Papeles para circuitos eléctricos
Bolsas de té
Papel para cigarrillos
Papeles para filtros
Papelesdecorativos
Papel para bolsas de vacío Lino
Cáñamo
Kenaf
Yute
Sisal
Abacá
Figura 2. Papeles especiales (izquierda) obtenidos de las pastas de papel fabricadas por la empresa CELESA (Tortosa, Tarragona) y sus principales materias primas (derecha).
8

1. Introducción
1.2. ESTRUCTURA Y COMPOSICIÓN QUÍMICA DE LOS MATERIALES LIGNOCELULÓSICOS
Los materiales lignocelulósicos, incluyendo los productos de origen agrícola y forestal, representan la mayor fuente de energía y materia orgánica renovables de la biosfera. Son materiales heterogéneos cuya estructura y composición química varían dentro de amplios límites y condicionan su utilización industrial y la posible aplicación de métodos biotecnológicos. Los principales componentes de estos materiales son los polímeros constituyentes de todas las paredes celulares de materiales vegetales: celulosa, hemicelulosas y lignina (Figura 3), y una serie de compuestos de bajo peso molecular solubles en agua o en solventes orgánicos, así como pequeños contenidos en proteína y sales minerales (Fengel y Wegener 1984, Sjöström 1993).
Enlaces dehidrógeno
Celulosa Lignina Hemicelulosas
Lignina
Hemicelulosas
Celulosa
Figura 3. Representación esquemática de los principales constituyentes de la pared vegetal correspondiente a una angiosperma no leñosa (adaptado de Bidlack et al. 1992).
9

1. Introducción
1.2.1. Celulosa
La celulosa es el componente principal de las células vegetales, que comprende aproximadamente del 10 al 20% del peso seco de las hojas, entre un 43 y un 47% de la madera de coníferas, entre un 42 y un 44% de la madera de frondosas y el 90% del peso de las fibras de algodón (Streitwieser y Heathcock 1983, Aitken et al. 1988). Estructuralmente, es un polímero lineal constituido por unidades de �-D-glucopiranosa unidas por enlaces glicosídicos � (1�4), en los que dos moléculas de glucosa se unen con eliminación de una molécula de agua entre dos hidroxilos de los carbonos 1 y 4. La configuración � sólo es posible por la rotación de la unidad de glucosa siguiente del eje C1-C4 del anillo de piranosa, por eso la unidad de cadena de celulosa que se repite es la celobiosa (disacárido), con una longitud de 1,03 nm (Fengel y Wegener 1984, Sjöström 1993, Sjöström y Westermark 1999). Los numerosos grupos hidroxilo favorecen la formación de enlaces de hidrógeno intra e inter-moleculares, formando cada unidad de glucosa dos enlaces intramoleculares y uno intermolecular (Figura 4). Los enlaces de hidrógeno intermoleculares se establecen con otras cadenas que están en el mismo plano, así como con cadenas en planos superiores e inferiores, de este modo, las cadenas de celulosa se unen dando lugar a la formación de microfibrillas, y la unión de éstas entre sí a la fibra de celulosa, cuyos agregados forman la pared celular (Lennholm y Henriksson 2007).
OO
HOOH
HO
OO
HOOH
O
OH
O
HOOH
HO
OO
HOOH
O
OH
OO
HOOH
HO
OO
HOOH
O
OH
O
HO OH
HO
OO
HOOH
O
OH
OO
HOOH
HO
OO
HOOH
O
OH
O
HOOH
HO
OO
HOOH
O
OH
�
�
�
�
1
1
1
1
4
4
4
Unidad celobiosa
Figura 4. Estructura de la celulosa donde se muestra la unidad de celobiosa, la conformación � (1�4) y los enlaces por puentes de hidrógeno intra e inter-moleculares.
10

1. Introducción
En su estructura supramolecular, la celulosa se organiza en zonas cristalinas y zonas amorfas. Son los enlaces de hidrógeno inter-moleculares los que permiten una estructura ordenada, esto es, una alta cristalinidad. En las zonas amorfas, el número de enlaces por puentes de hidrógeno establecidos es menor y bastante más desorganizado que en las zonas cristalinas, siendo por lo tanto la celulosa amorfa más fácil de disolver y más reactiva, pues la accesibilidad a los grupos hidroxilo es mayor (García Hortal 2007, Annergren 1996). Las propiedades de los materiales lignocelulósicos están relacionadas con el grado de polimerización de la molécula de celulosa, que es de al menos 15000 (Brett y Waldron 1996). La resistencia del papel es debida, en parte, a la resistencia individual de las cadenas de celulosa, que diminuye si estas se degradan.
1.2.2. Hemicelulosas
Las hemicelulosas comprenden aproximadamente del 25 al 30% del peso seco de la madera de coníferas, entre un 20 y 43% de la madera de frondosas, entre un 12 y 18% de las fibras liberianas de lino y un 12% de las fibras de hojas de sisal (Aitken et al. 1988, Fengel y Wegener 1984, García Hortal 2007). Actúan como matriz de soporte para las microfibrillas de celulosa y estructuralmente son más complejas que la celulosa.
Las hemicelulosas son polisacáridos heterogéneos constituidos por una cadena lineal de diferentes monosacáridos unidos principalmente por enlaces � (1�4) y en algunos casos � (1�3), de la que parten diversas ramificaciones (Sjöström 1993). Los principales monosacáridos que las constituyen incluyen pentosas (D-xilosa y L-arabinosa), hexosas (D-glucosa, D-galactosa, D-manosa, L-ramnosa y L-fucosa), y ácidos urónicos (ácido D-glucurónico y ácido D-galacturónico) (Figura 5) con un grado de polimerización entre 200 y 300, siendo más fáciles de disolver y de degradar que la celulosa (Sjöström y Westermark 1999). Contrariamente a la celulosa, la naturaleza de las hemicelulosas varía entre las diferencies especies (Tabla 2). En el caso de las maderas de coníferas se suele apreciar una mayor cantidad de hexosanos, como la manosa y galactosa, siendo predominantes los galactoglucomananos y los glucomananos (García Hortal 2007, Sjöström y Westermark 1999) aunque también se observan en las coníferas los arabinoglucuronoxilanos (Sjöström y Alén 1999). La xilosa es más abundante en las frondosas donde predominan pentosanos como los glucuronoxilanos (Fengel 1989, Sjöström 1993, Shimizu 2001) aunque también se observan glucomananos (García Hortal 2007, Sjöström y Alén 1999). En las plantas no madereras, las hemicelulosas presentan una gran variedad en su composición dependiendo de la especie, siendo en algunas, como en el lino, predominantes la manosa y la galactosa (Morrison et al. 1999) y en otras, como en el kenaf, la xilosa (Han 1998, Neto et al. 1996).
11

1. Introducción
O
H
HO
H
HO
H
H
OHH
H
OH
�-D-Xilosa
O
H
HO
H
HO
H
H
OHHOH
OH
�-D-Glucosa
O
H
HO
H
HO
H
H
OHH
COOH
OH
Ácido �-D-Glucurónico
O
OH
H
H
HO
H
OH
OHH
H
H
�-L-Arabinopiranosa
O
H
HO
H
HO
OH
H
HHOH
OH
�-D-Manosa
O
H
H3CO
H
HO
H
OH
OHH
COOH
H
Ácido �-D-4-O-Metilglucurónico
OH
H
HOH2C
H
H OH
HO H
O
�-L-Arabinofuranosa
O
OH
H
H
HO
H
OH
OHHH
OH
�-D-Galactosa
O
OH
H
H
HO
H
OH
OHH
COOH
H
Ácido �-D-Galacturónico
PENTOSAS HEXOSAS ÁCIDOS HEXURÓNICOS
O
OH
H
OH
H
H
OH
OH
H
H
�-L-Ramnosa
O
H
HO
OH
H
H
OH
OH
H
H
�-L-Fucosa
DESOXI-HEXOSAS
Figura 5. Monosacáridos componentes de las hemicelulosas (adaptado de Fengel y Wegener 1984).
12

1. Introducción
Tabla 2. Tipos y estructuras simplificadas de las principales hemicelulosas en diversos materiales lignocelulósicos, X, xilosa; A, arabinosa; G, glucosa; Gal, galactosa; M, manosa; Ac, grupo acetilo; Gl, ácido 4-O-metilglucurónico (adaptado de García Hortal 2007)
Tipo hemicelulosa Estructura simplificada Presencia
Glucuronoxilanos Frondosas, plantas no madereras
Glucomananos Coníferas, frondosas
Galactoglucomananos Coníferas
Arabinoglucuronoxilanos Coníferas, plantas no madereras
Tipo hemicelulosa Estructura simplificada Presencia
Glucuronoxilanos Frondosas, plantas no madereras
Glucomananos Coníferas, frondosas
Galactoglucomananos Coníferas
Arabinoglucuronoxilanos Coníferas, plantas no madereras
X- X - X - X
Ac Gl7
X- X - X - X
Ac Gl7
G - M - G - M - MG - M - G - M - M
X - X - X- X - X5
Gl A2
X - X - X- X - X5
Gl A2
G- M - M - M
Gal Ac
G- M - M - M
Gal Ac
Las hemicelulosas, con estructura ramificada y amorfa, son muy hidrofílicas y desempeñan un papel fundamental en el proceso de fabricación de papel al promover el hinchamiento de la fibra y aumentar su plasticidad, flexibilidad y capacidad de enlace, con la consiguiente mejora de la densidad de la hoja. Sin embargo, durante el secado de la pasta también tienden a mantener dura o rígida la fibra, lo que puede impedir la subsiguiente rehidratación de la pasta (García Hortal 2007).
1.2.3. Lignina
Después de la celulosa, la lignina es el polímero más abundante en el mundo vegetal, representando entre un 25 y un 33% de la madera de coníferas y entre un 18 y un 34% de la madera de frondosas (Aitken et al. 1988). En el caso de las plantas no madereras hay un menor porcentaje de lignina con respecto a las especies madereras, con un 8-9% para fibras de hojas (abacá y sisal), entre un 3-13% para fibras liberianas (lino, cáñamo, yute y kenaf), entre un 12 y un 21% para pajas (paja de arroz, paja de trigo) y entre un 19 y un 22% para cañas
13

1. Introducción
(azúcar, bambúes) (García Hortal 2007). La lignina actúa como aglomerante de las fibras debido a su carácter hidrófobo siendo una de las moléculas orgánicas más recalcitrantes.
Estructuralmente, la lignina es un heteropolímero aromático con una estructura tridimensional irregular, constituida por unidades de fenilpropano con diferentes patrones de substitución y unidas por diferentes tipos de enlaces, que varían considerablemente entre las especies vegetales e incluso dependiendo de la edad (Freudenberg y Lehmann 1963), de la parte del árbol/planta (Bland 1966), tipo de células (Fergus y Goring 1970, Hardell et al. 1980a, 1980b) y del lugar de la pared celular donde se sintetice (Fergus y Goring 1970, Fukushima y Terashima 1991, Christierini et al. 2005), por lo que la lignina no puede ser descrita por una fórmula simple.
Los precursores de la lignina son los alcoholes p-hidroxicinamílicos (Figura 6), que incluyen los alcoholes p-cumarílico (4-hidroxicinamílico, I), coniferílico (4-hidroxi-3-metoxicinamílico, II) y sinapílico (4-hidroxi-3,5-dimetoxicinamílico, III), que difieren entre sí en el número de grupos metoxilo sustituyentes. Estos precursores se sintetizan a su vez a partir de la fenilalanina a través de la ruta de los ácidos cinámicos (Higuchi 1997, Boerjan et al. 2003, Freudenberg y Neish 1968, Adler 1977, Ralph et al. 2004). Recientemente, se ha descrito la existencia de otros precursores de la lignina, tales como derivados acilados (acetatos y/o p-cumaratos) de los correspondientes alcoholes p-hidroxicinamílicos (IV y V) (del Río et al. 2004, 2007, 2008a, 2008b, Martínez et al. 2008) observados en diversas plantas angiospermas, así como alcoholes dihidroxicinamílicos (VI), o aldehídos cinamílicos (VII), observados en la lignina de especies modificadas genéticamente (Ralph et al. 1997, 1998, Sederoff et al. 1999). Su deshidrogenación oxidativa, catalizada por peroxidasas o lacasas en presencia de peróxido de hidrógeno u oxígeno, respectivamente, conlleva a la formación de radicales fenoxilo estabilizados por resonancia que luego se acoplan entre sí y con el polímero creciente de lignina mediante diversos tipos de enlaces (Figura 7).
Aunque la variedad de uniones para formar el polímero de lignina es amplia (Figura 8), se pueden diferenciar dos tipos: uniones de tipo éter y uniones de tipo carbono-carbono. La formación de enlaces éter-alquil-arílico es la más favorable termodinámicamente, como es el caso del enlace �-O-4�, en el que se encuentran involucrados la posición � del monolignol radical y el radical fenoxilo del polímero de lignina creciente. En menor proporción existen uniones de tipo aril-aril éter, como por ejemplo la unión 4-O-5�. Los enlaces de tipo carbono-carbono, conocidos también como enlaces condensados, son más difíciles de romper que los de tipo éter, e incluyen las uniones de dos cadenas alifáticas (�-�� resinol), la unión de un carbono de un anillo bencénico con el de una cadena alifática (�-1� y �-5� fenilcumarano) y las uniones entre carbonos de
14

1. Introducción
dos anillos bencénicos (5-5�). Se ha descrito que el enlace 5-5� no se encuentra tal cual, sino en forma de trímero, ya que incorpora una nueva unidad mediante un enlace ���-O-4� y un enlace ���-O-4�, dando lugar a una estructura de tipo dibenzodioxocina (Karkunen et al. 1995). Igualmente, estudios recientes indican que la mayoría de las uniones �-1� se encuentran en forma de espirodienonas (Zhang y Gellersted 2001, Zhang et al. 2006).
OH
O
O
OH
OH
OH
OMeHO
OH
OH
(OMe)(MeO) (OMe)(MeO)
V VI VII
OH
OH
OH
OH
OH
OH
OH
O
(OMe)
O
(MeO)
I II III IV
��
�
OMe(MeO)OMe
Figura 6. Estructuras de precursores de la lignina: I, alcohol p-cumarílico; II, alcohol coniferílico; III, alcohol sinapílico; IV, derivado acetilado de los alcoholes p-hidroxicinamílicos; V, derivados p-cumaroilados de los alcoholes p-hidroxicinamílicos; VI, alcohol 5-hidroxiconiferílico y VII, aldehídos correspondientes a los alcoholes p-hidroxicinamílicos.
15

1. Introducción
OH
HO
(MeO)
HO
O
HO O
Lignina
O
HO
O
OH
HO
OR
OxidaciónAcoplamiento
radicalar ROH
Lignina Lignina
Lignina
(MeO)OMe (MeO) OMe
(MeO)
OMe (MeO) OMe
OMeOMe
(MeO)
OMe
(MeO)
OMe
(MeO)
b
OH
OH
OMe
O
OH
OMe
O
OH
OMe
O
OH
OMe
O
OH
OMe
O
OH
OMe
- (e- + H+)
PeroxidasaLacasa
a
��
�
Figura 7. Síntesis de la lignina: (a) deshidrogenación del alcohol coniferílico y formas resonantes del radical fenoxilo (adaptado de Adler 1977) y (b) mecanismo de la unión de los monolignoles libres al polímero de lignina (Freudenberg y Neish 1968).
La cantidad de lignina, su distribución a través de las paredes celulares y la estructura básica de la misma, difieren según su origen entre coníferas, frondosas y fibras no madereras. En la lignina de las coníferas la estructura que se repite predominantemente es la unidad guayacilo (G), que contiene un único grupo metoxilo en el anillo de fenilpropano y deriva del alcohol coniferílico (más de 95% de las unidades estructurales). En el caso de la lignina de las maderas de frondosas, hay predominantemente dos unidades que se repiten, la unidad guayacilo (G) y la unidad siringilo (S), conteniendo esta última dos grupos metoxilo por núcleo de fenilpropano y deriva del alcohol sinapílico (Parhan 1983, Sarkanen y Hergert 1971, Lin y Dence 1992). Por otro lado, la lignina de fibras no madereras presenta unidades del tipo p-hidroxifenilo (H), procedentes del alcohol p-cumarílico, y unidades S y G, en proporciones variables dependiendo de la planta. Las unidades G, al contrario que las S, tienen un único grupo metoxilo y la posición C-5 está libre y disponible para la formación de enlaces carbono-carbono, por lo que ligninas con mayor cantidad de unidades G tienen una estructura más condensada y por lo tanto la lignina se degrada con mayor dificultad. En la Figura 9 se muestran modelos estructurales del polímero de lignina de coníferas y frondosas, y en la Figura 10 se muestran modelos estructurales del polímero de lignina de algunas plantas no madereras.
16

1. Introducción
� �
�
12
34
5
6
1’6’
5’
4’3’
2’HO
O
O
HO
OMe
MeO
�-O-4�
MeO
OMe
O
O
OH
OMe OMe
OMe
MeO
HO
12
34
5
6
�-O-4 �
1’6’5’
4’ 3’
2’
�
��
O
O
OMe
OMe
OMe
4-O-5 �
23
45
6
1 2’1’
6’5’
4’3’
�
1´2´
3´4´
5´
6´
O
O
OHHO
�� �´ �´�´
O
1''2''
3''4''
5''
6''
MeO
O
2
1
4 3
56MeO
MeO
�-1 � /�-O-� �espirodienona
O
O O
OH
OMe
OMeMeO
MeO
1’2’
3’4’
5’
6’1
6
54
3
2
1’’
2’’
3’’
5’’
4’’
6’
��� ���
5-5 � /� ��-O-4 � /� ��-O-4 �dibenzodioxocina
OMe
OMe
OMe
���
12
34
5
6
O
1’2’
3’4’
5’
6’
HO
O
MeO
�-5 � /�-O-4 �fenilcumarano
�
�
�
1
23
4
56
�’
�’
�’
1’
2’3’
4’
5’6’
O
O
O
O
OMe
MeO
�-� � /�-O-� � /�-O-� �resinol
O
O
HO
HO
MeO OMeOMe
OMe
�-1 �
12
34
5
6
3’2’
1’6’
5’4’
OMe
OMe
OMe
OMe
��
�
Figura 8. Enlaces tipo éter y carbono-carbono presentes en el polímero de lignina.
17

1. Introducción
Figura 9. Modelos del polímero de lignina en maderas: (a) conífera (Picea) (Brunow 2001) y (b) frondosa (álamo) (Boerjan et al. 2003).
18
A B
(a) (b)

1. Introducción
19
(a)
OHOMe
OMeHO
O O
OOMe
OHO
OH
OMe
OMeHO
OO
O OMeMeO
OO
MeOO
OMe
OH
HO
O
OMe
OH
OOMeMeO
HO OMe
OMeO
OH
OMeMeOOHO
OO
OMe
OMe
OH
OMeO
MeO
OHO
O
OMe
O OH
MeO
O OMe
O
OHHO
MeO
OMe
O
O
O
O
OH
O
OHO
OMe
O
OMe
OH
OH
O
OMe
MeO
O OH
HO OMe
MeO
O
O
O
(b)
O
OMeMeOO
O
HO
OO
OMe
OMe
OH
OMeO
MeO
OHO
O
OMe
MeO
O
O OH
O
MeO
OMe
O
HO
OHHO
O
OMe
OHO
O
MeO
OMe
O
OHO
O
OMe
MeO
O
OH
O
OMe
OMeHO
OO
MeO OMe
OHO
OOMe
OMe
HO
OO
OH
O
O
OMe
OMe
HO
OO
MeO OMe
OHOH
O
O
HO
O
OH
O
O O
O
HO
HO
O
OH
O OMe
MeOHO
OHO
MeO
O
OH
HO
MeO
OMe
O
O OH
OMeMeO
Figura 10. Modelos del polímero de lignina en plantas no madereras: (a) kenaf y (b) abacá (del Río et al. 2008a).

1. Introducción
La lignina aparece también asociada a los polisacáridos en la pared celular y es esta asociación la que determina la rigidez y la resistencia estructural del material. Las hemicelulosas están asociadas a la lignina principalmente a través de las unidades de arabinosa, xilosa y galactosa por enlaces de tipo glicosídico, éter bencílico y éster bencílico formando complejos lignina-polisacáridos (Watanabe 2003).
1.2.4. Componentes de bajo peso molecular
Además de los carbohidratos (celulosa y hemicelulosas) y lignina, existen en los materiales lignocelulósicos pequeñas cantidades de componentes que no influyen en la estructura morfológica de las células pero que tienen gran influencia en el procesamiento de estos materiales. Algunos de estos componentes protegen a la madera de los insectos y son responsables de su color, olor y gusto. Atendiendo a su solubilidad se pueden dividir en compuestos extraíbles en solventes apolares, que incluyen los extraíbles lipofílicos, y compuestos extraíbles en disolventes polares (extraíbles hidrofílicos), así como diversos compuestos insolubles tales como sustancias proteicas, pécticas y de naturaleza inorgánica (García Hortal 2007, Hillis 1962, Fengel y Wegener 1984, Rowe 1989, Sjöström 1993).
Los extraíbles lipofílicos (Figura 11) incluyen típicamente alcanos (a), alcoholes grasos (b), aldehídos (c), ácidos grasos (d), esteroles (e), ácidos resínicos (f), ceras (ésteres de ácidos grasos con alcoholes de cadena larga, g) y glicéridos (ésteres de ácidos grasos con glicerol, h). Los esteroles pueden encontrarse libres o esterificados con ácidos grasos (ésteres de esteroles, i) y también pueden estar formando glicósidos y acilglicósidos (Gutiérrez y del Río 2001), siendo el más abundante el 3�-D-glucopiranósido (j).
Los extraíbles lipofílicos afectan negativamente al proceso de fabricación de pasta de celulosa así como al producto final, formando depósitos insolubles comúnmente denominados depósitos de pitch, que se describen más adelante en el apartado 1.4. Debido a su alto grado de pegajosidad, los esteroles libres y conjugados se encuentran en el origen de muchos depósitos de pitch (Back y Allen 2000, del Río et al. 1998, 2000). El estudio de los compuestos extraíbles lipofílicos de cada una de las materias primas constituye un requisito fundamental para identificar los compuestos que originan los depósitos de pitch y diseñar estrategias adecuadas para su control.
20

1. Introducción
aOHOH
b
cH
O
H
O
H
O
OH
O
OH
O
d
HOHOHO
e
O
O
O
O
i jO
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH
gO
O
O
O
FF
COOHCOOH
FF
COOHCOOH
f
h
CO-O-CH2
CO-O-CH
CO-O-CH2
CO-O-CH2
CO-O-CHCO-O-CH
CO-O-CH2CO-O-CH2
Figura 11. Estructuras de compuestos representativos de las principales familias de extraíbles lipofílicos: (a) pentacosano, (b) docosanol, (c) octacosanal, (d) ácido palmítico, (e) sitosterol, (f) ácido abiético, (g) octacosanil hexadecanoato, (h) trilinoleína, (i) sitosteril linoleato, (j) sitosteril 3�-D-glucopiranósido.
21

1. Introducción
Por otro lado, los extraíbles polares engloban diferentes compuestos fenólicos libres de bajo peso molecular (Figura 12), los cuales incluyen precursores de la lignina (ácidos p-hidroxicinámicos y aldehídos p-hidroxicinamílicos), ácidos bencenocarboxílicos relacionados (ácido p-hidroxibenzoico, vainíllico y siríngico), aldehídos y cetonas aromáticas (p-hidroxibenzaldehído, vainillina, siringaldehído y propioguayacona), e incluyen taninos hidrolizables (ésteres del ácido gálico y sus dímeros), flavonoides (estructuras derivadas del anillo de flavona, 2-fenilbenzopirona) y taninos no hidrolizables (varias unidades de flavonoides condensadas). Además de incrementar el consumo de reactivos durante la cocción, estos compuestos pueden dificultar las reacciones de pasteado impidiendo la difusión de los reactivos en la materia prima, y los taninos, cuando están presentes en cantidades importantes, forman complejos coloreados con cationes metálicos afectando el color de las pastas de papel y su blanqueabilidad (García Hortal 2007).
OH
O
O
H
O
OH
O
OHO
OHO
OH
O
OH
O
O
OHO
OH
OHHO
O
O
a b c d
e f Figura 12. Estructuras de compuestos representativos de los compuestos extraíbles polares: (a) Ácido siríngico, (b) ácido p-hidroxibenzoico, (c) vainillina, (d) acetosiringona, (e) ácido gálico y (f) 2-fenilbenzopirona.
22

1. Introducción
1.3. UTILIZACIÓN DE CULTIVOS LIGNOCELULÓSICOS: PRODUCCIÓN DE PASTA DE CELULOSA
La fabricación de pasta de celulosa consiste básicamente en la separación de las fibras de celulosa de la madera u otros materiales fibrosos a través de procesos mecánicos y/o químicos (Fengel y Wegener 1984, Sjöström 1993). Se cree que la fabricación de papel tuvo su origen en China hacia el año 100 d.C. y para su fabricación se utilizaban trapos, cáñamo, paja y hierba como materias primas, que se golpeaban contra morteros de piedra para separar la fibra original. Aunque con el tiempo ganó terreno la mecanización, hasta el siglo XIX siguieron utilizándose los métodos de producción por lotes y las fuentes de fibra agrícolas. Las primeras máquinas continuas de papel se patentaron a finales del siglo XIX y principios del siglo XX. Entre 1844 y 1884 se desarrollaron los primeros métodos para la obtención de pasta a partir de madera, una fuente de fibra más abundante que los trapos o hierbas; estos métodos implicaban la abrasión mecánica y la aplicación de diversos procedimientos químicos. La fabricación de papel fue una labor artesana e individualizada, pero con los descubrimientos de la ciencia y los avances tecnológicos, así como con el desarrollo y expansión de la cultura, la industria de fabricación del papel se desarrolló a un ritmo acelerado. La industria de pasta celulósica muestra aún una tendencia creciente en su producción, según los datos de la FAO (FAO 2004) de la Tabla 3.
Tabla 3. Estadísticas sobre la capacidad de producción de papel en los principales países productores (2003-2008) (FAO 2004)
2003 2004 2005 2006 2007 2008
Capacidad total , 1000Mt
Países desarrollados 244169 247669 251387 254304 255786 256589
Norteamérica 114550 114325 115062 115405 115405 115405
Europa 91894 95866 98824 101360 102827 103620
Oceanía 4049 4143 4208 4246 4261 4271
Otros 33676 33335 33293 33293 33293 33293
Países en desarrollo 21201 21774 22434 22964 23296 23493
África 2681 2688 2721 2865 2865 2872
América Latina 13181 13469 13841 14052 14219 14261
Asia 5339 5617 5872 6047 6212 6360
23

1. Introducción
El proceso de producción de pasta de celulosa comprende fundamentalmente el proceso de pasteado y el proceso de blanqueo. El proceso de pasteado tiene por objeto separar las fibras de celulosa del resto de los componentes de la madera, fundamentalmente de la lignina ya que las fibras de celulosa se encuentran cementadas por ella. Por otro lado, el blanqueo de la pasta tiene por objeto disolver o modificar la lignina residual que no se elimina durante el pasteado, para mejorar las propiedades de la pasta y consecuentemente del producto final.
1.3.1. Procesos de pasteado
Dependiendo de las características de las fibras, el tratamiento aplicado para destruir o debilitar los enlaces interfibras varía, con la finalidad de obtener una pasta de características adecuadas y el mayor rendimiento posible. Los procesos de obtención de pasta de papel se clasifican básicamente en mecánicos y químicos. Combinaciones de éstos dan lugar a procedimientos intermedios o semiquímicos.
Procesos mecánicos
El pasteado mecánico tiene como objeto la separación física de las fibras, realizándose el desfibrado por fragmentación mecánica, utilizando molinos y refinadores de discos. La fabricación de pastas mecánicas ofrece la ventaja de dar como resultado rendimientos elevados (hasta un 98% del material inicial), obteniéndose pastas ventajosas para algunos tipos de papel por su rigidez, volumen y opacidad (García Hortal 2007). Sin embargo, como en este proceso la lignina sólo se ablanda (no se disuelve), el alto contenido en lignina va en detrimento de la calidad del papel ya que las fibras muy lignificadas son rígidas, poco flexibles, no están bien unidas entre sí, proporcionando papeles con bajas características de resistencia y muy sensibles al envejecimiento óptico.
Procesos químicos
En el pasteado o cocción química, la deslignificación se lleva a cabo con la ayuda de agentes químicos ácidos o básicos, en digestores o reactores a altas temperaturas y presiones. La pasta se produce con disolución de la lignina que se encuentra entre las fibras del material lignocelulósico y los productos de degradación se disuelven en la lejía de la cocción. En el pasteado químico, se eliminan muchos de los componentes no fibrosos de la materia prima y los rendimientos son normalmente del 35 al 65%, sin embargo, la pasta se blanquea mejor y el producto es más resistente y de mejor calidad que en el caso de los procesos mecánicos (Sjöström 1993).
24

1. Introducción
Los procesos de pasteado químico pueden realizarse en condiciones alcalinas, como el pasteado a la sosa y el proceso kraft, o en condiciones ácidas como el pasteado al sulfito. Otro tipo de procesos utiliza solventes orgánicos (pasteado organosolv) (Gilarranz et al. 1999).
El proceso a la sosa es el más antiguo y el más simple de los procesos químicos alcalinos. En este proceso, la fibra se somete a un proceso de cocción con sosa cáustica y vapor a alta presión y temperatura. El hidróxido de sodio es un producto muy útil para la deslignificación de materias primas vegetales, principalmente de maderas, pajas de cereales y plantas fibrosas en general. En este proceso, se puede utilizar antraquinona (AQ) como catalizador ya que presenta dos efectos fundamentales como son la aceleración del proceso de deslignificación alcalino y la estabilización de los carbohidratos, mejorando los rendimientos respecto al proceso convencional en las mismas condiciones de operación (Abarca y Blanco 2008).
El proceso kraft para la obtención de pasta de papel es un proceso químico alcalino que deriva del proceso a la sosa. En este proceso, además de hidróxido de sodio se utiliza sulfuro sódico, siendo estos agentes de cocción conocidos como lejías blancas. El proceso se lleva a cabo en digestores que pueden ser tanto discontinuos como continuos, en los que se introducen las astillas junto a las lejías blancas llevándose a cabo la cocción a elevada temperatura (150-170ºC) y presión. Generalmente el proceso tiene lugar con una concentración de reactivos del 16-20% (expresados como peso de Na2O, en relación al peso de la madera). Este tipo de pasteado permite obtener pastas con una gran resistencia, aunque con menor rendimiento (entre un 40 y 60%), ya que se elimina mucha cantidad de lignina (hasta el 90%) (García Hortal y Colom 1992, Santos et al. 1997). La ventaja de este proceso es que requiere tiempos de cocción relativamente cortos pues el sulfuro acelera la deslignificación reduciendo la degradación del material celulósico y produciendo así pastas de mejor calidad. Para este proceso, se pueden utilizar todo tipo de maderas, aunque los mejores resultados se obtienen con maderas de frondosas.
El proceso al sulfito es un proceso químico ácido donde se utilizan sulfitos y bisulfitos para la deslignificación. Es un proceso más fuerte que el proceso alcalino y permite una mejor separación de la celulosa. Este proceso está limitado en cuanto al tipo de materia prima, pues no se pueden utilizar maderas de coníferas ya que a pH bajos los fenoles y los ácidos resínicos se condensan con la lignina formando complejos insolubles y coloreados que manchan la pasta. El licor de cocción es una disolución de ácido sulfuroso (H2SO3) y bisulfito de calcio (Ca(HSO3)2), que se prepara disolviendo dióxido de azufre en agua y haciéndola reaccionar con CaCO3. Los digestores operan a temperaturas comprendidas entre los 125 y 180ºC según la aplicación que se quiera dar al producto final (papel, cartón, etc.), obteniéndose rendimientos entre el 40 y
25

1. Introducción
60%. En estos procesos también se degradan los hidratos de carbono por rotura de los enlaces glicosídicos, lo que provoca una disminución del grado de polimerización todavía mayor que en los procesos kraft siendo la pasta resultante menos resistente, pero por lo contrario estas pastas son más fáciles de blanquear. El método al sulfito ha sido relegado en parte por el proceso kraft (Bryce 1990).
Alternativamente, se han desarrollado los procesos organosolv que utilizan solventes orgánicos para la deslignificación. Estos procesos presentan una mayor selectividad y por lo tanto, dan lugar a rendimientos mayores. Por otro lado, permiten la utilización de cualquier materia prima fibrosa (coníferas, frondosas y plantas no madereras) dando lugar a la obtención de pastas con bajo contenido en lignina que pueden ser blanqueadas sin el uso de compuestos clorados. Se han empleado multitud de disolventes orgánicos (etanol, metanol, butanol, alcohol bencílico, glicerol, glicol, etilenglicol, trietilenglicol, fenol, acetona, ácido fórmico, ácido acético, dioxano, dimetilsulfóxido, hexametilendiamina, etc.) puros o en disolución acuosa, con la adición o no de catalizadores. Los elevados precios de los reactivos, la dificultad en su recuperación y en muchos casos su elevada toxicidad, ha favorecido el uso de alcoholes alifáticos de bajo peso molecular (etanol y metanol) como solventes para los procesos organosolv (Herrero et al. 2002). Estos solventes combinan su alta velocidad de deslignificación en condiciones de operación favorables y su fácil recuperación. Sin embargo, en general, las propiedades de resistencia de las pastas organosolv son inferiores a las pastas kraft.
1.3.2. Procesos de blanqueo
En el proceso de blanqueo se trata químicamente la pasta de celulosa para eliminar o modificar la lignina residual que queda después del proceso de cocción. Los componentes coloreados de la lignina se degradan, disuelven o se decoloran (Sjöström 1993). El proceso de blanqueo se lleva a cabo hasta el punto de blancura que se pretende, por lo que el número de etapas dependerá de la calidad de la pasta que se desee obtener (Figura 13). Los reactivos comerciales más utilizados para el blanqueo son el cloro gas, el hipoclorito, el peróxido de hidrógeno y el dióxido de cloro; y el álcali utilizado es el hidróxido de sodio que se usa en la operación de extracción alcalina.
26

1. Introducción
Figura 13. Diferentes grados de blancura de una pasta de celulosa.
el blanqueo (blanqueo ECF, elemental ch
todos de blanqueo menos agresivos y que se
fábricas con objeto de adaptarlas a las modernas tecnologías ECF y TCF.
El blanqueo ha sido la etapa de la producción de pasta de celulosa que ha sufrido más cambios durante los últimos años. La decisión de eliminar el cloro molecular y, en algunos casos también el dióxido de cloro, de las secuencias de blanqueo se debe a la necesidad de reducir las emisiones de compuestos clorados orgánicos, de haluros orgánicos absorbibles y dioxinas en los efluentes de las plantas de blanqueo. El desarrollo de leyes más restrictivas con respecto a los procesos contaminantes ha llevado a una parte importante de la industria europea de pasta y papel a introducir secuencias de blanqueo totalmente libres de cloro (blanqueo TCF, totally chlorine free) (Brooks et al. 1994). Estas secuencias incluyen blanqueo con peróxido de hidrógeno, oxígeno y ozono. Otra parte de la industria papelera mundial, ha eliminado el cloro elemental pero continúa utilizando dióxido de cloro en
lorine free).
El desarrollo del blanqueo con oxígeno ha sido bastante lento por la degradación de la celulosa y demás polisacáridos de la madera. Las ventajas del peróxido de hidrógeno se apoyan en su facilidad de manipulación y aplicación, su versatilidad y la naturaleza relativamente inocua de los productos de reacción. La novedad de las secuencias de blanqueo TCF obliga a solucionar nuevos problemas que surgen al introducir mé
describen en el apartado siguiente.
En España, diversas empresas productoras de pasta de papel, incluyendo ENCE y CELESA (que han suministrado pastas de papel para la presente Tesis), han realizado un considerable esfuerzo de inversión y modernización de sus
27

1. Introducción
1.4
clorados producidos en el blanqueo con cloro. Sin embargo, algunos de los extraíbles lipofílicos que son destruidos por el
teres de glicerol se saponifican y los ácidos grasos y resínicos se disuelven. Los ésteres de esteroles, los esteroles libres y las ceras, se saponifican más lentamente que los ésteres de glicerol, no forman jabones solubles como en el caso de los ácidos libres, por lo que tienen tendencia a depositarse (Gutiérrez et al. 2001).
. PROBLEMÁTICA PLANTEADA POR LA PRESENCIA DE LIGNINA Y LÍPIDOS EN LA PRODUCCIÓN DE PASTA DE CELULOSA
Una parte de los problemas originados durante la producción de pasta de celulosa está relacionada con los compuestos extraíbles lipofílicos de los materiales lignocelulósicos. Estos compuestos causan tanto problemas medioambientales como problemas técnicos durante el proceso de producción. Entre los compuestos lipofílicos más problemáticos están los ácidos grasos libres, ácidos resínicos, ceras, alcoholes, esteroles tanto libres como esterificados, glicéridos, cetonas y otros compuestos (Hillis 1962, Fengel y Wegener 1984, Rowe 1989, Gutiérrez et al. 1999). Durante el proceso de producción de pasta de celulosa los compuestos lipofílicos se liberan formando partículas coloidales que pueden unirse y formar gotas que luego se depositan en la pasta o en la maquinaria formando los llamados “depósitos de pitch” (Figura 14). La formación de estos depósitos da lugar a importantes pérdidas económicas como consecuencia de pastas contaminadas, paradas en la producción, así como por el coste de los aditivos químicos utilizados para el control del pitch (Hillis 1989, Allen 2000). Además, algunos compuestos lipofílicos de los materiales lignocelulósicos también tienen un impacto negativo sobre el medio ambiente, considerándose algunos de ellos como primera fuente de toxicidad cuando se liberan en los vertidos (Ali y Sreekrishnan 2001, Rigol etal. 2004). Esto es especialmente importante en los procesos modernos donde el blanqueo con cloro ha sido sustituido por el blanqueo libre de cloro elemental (ECF) o totalmente libre de cloro (TCF). El blanqueo ECF evita los problemas asociados a la formación de compuestos
dióxido de cloro no se eliminan en el blanqueo TCF, ya que estas secuencias que utilizan oxígeno y peróxido de hidrógeno no afectan prácticamente a la fracción lipídica de las materias primas.
La problemática del pitch es muy compleja porque varía con la materia prima así como con el proceso empleado para la fabricación de pasta y papel. En el caso de las pastas mecánicas, los depósitos de pitch muestran una composición similar a los extractos lipofílicos de la materia prima. En el caso de las pastas alcalinas, sólo algunos de los compuestos extraíbles presentes en la materia prima sobreviven al proceso de cocción. En condiciones alcalinas los és
28

1. Introducción
Figura 14. Imagen de una gota de resina en el árbol (izquierda) y de un depósito de pitch en una pasta kraft TCF (cedidas por María Jesús Ortega y Javier Romero, respectivamente).
de los productos derivados e la lignina, responsable del color de las pastas.
1.5
Por otro lado, la lignina está también relacionada con la problemática existente en la producción de pasta de celulosa ya que la variabilidad en su composición y estructura influye decisivamente en el proceso de deslignificación. Por otro lado, la formación de compuestos oxidados de la lignina durante el proceso de pasteado (lignina residual) es responsable del color oscuro de las pastas. La fabricación de pastas de papel mediante tecnologías menos contaminantes ha traído consigo nuevos problemas en el blanqueo de la pasta, que no se daban al utilizar reactivos más agresivos (aunque también más contaminantes) y/o en sistemas con un menor grado de cierre en los circuitos. De momento ni el oxígeno ni la combinación de oxígeno y peróxido pueden igualar la eficacia de la cloración para la eliminaciónd
. BIOTECNOLOGÍA EN EL SECTOR DE LA PASTA DE CELULOSA
La producción de pasta y papel ha sido tradicionalmente un proceso industrial con un fuerte impacto medioambiental. El gran incremento en la demanda de papel ha agravado el impacto negativo sobre el medio ambiente, por lo que se han desarrollado leyes más restrictivas con respecto a los procesos contaminantes. Por consiguiente, las empresas papeleras han tenido que realizar un considerable esfuerzo de inversión y modernización de sus fábricas con objeto de adaptarlas a tecnologías más limpias y además, con un mayor grado de cierre en los circuitos para reducir los efluentes líquidos. La biotecnología aplicada a este sector ofrece nuevas posibilidades de utilizar métodos biológicos
29

1. Introducción
basados en el uso de hongos y enzimas para reducir o remediar el impacto medioambiental, reduciendo el consumo de reactivos químicos, así como el gasto energético durante la fab
oblemas de pitch y la mejora en la reutilización de las aguas del proceso.
o de la pasta de papel y el control del pitch, que se mencionan a continuación.
1.5.1. Degradación enzimática de la lignina
997). En la Figura 15 se pueden observar las estructuras de estas tres enzimas.
ricación de pasta de papel.
Durante los últimos años, el número de aplicaciones enzimáticas en la industria de la pasta de celulosa ha aumentado considerablemente, y varias han alcanzado o se están acercando a su uso comercial. Éstas incluyen el uso de xilanasas para ayudar al blanqueo, la deslignificación directa con enzimas oxidativas, el ahorro de energía de refino con celulasas, así como la reducción de depósitos de pitch con lipasas (Bajpai 1999, 2006). Además de las enzimas, los tratamientos microbianos también tienen una potencial aplicación para aumentar la eficiencia en la fabricación de pasta de celulosa, para la reducción de los pr
En la presente Tesis, se incluye el estudio de enzimas para dos de estas aplicaciones, como son el blanque
El uso de enzimas en la industria papelera ha crecido rápidamente a partir de mediados de los años 80. Las enzimas más utilizadas en el blanqueo de las pastas son enzimas hidrolíticas como las xilanasas, que se utilizan para limitar el uso de cloro en los procesos de blanqueo de la pasta (Viikari et al. 1994). Las xilanasas no actúan directamente sobre la lignina, sino catalizando la hidrólisis de los xilanos que se encuentran entre las microfibrillas de la celulosa y la lignina. Sin embargo, enzimas de tipo oxidoreductasa (lacasas y peroxidasas) tienen mayor potencial que las xilanasas porque actúan directamente sobre la lignina. Durante años se concedió mayor atención a las peroxidasas ligninolíticas que a las lacasas en la degradación de la lignina y en el desarrollo de aplicaciones biotecnológicas (Paice et al. 1995) ya que los bajos potenciales redox de las lacasas (0.3 a 0.8 V) comparados con los de las peroxidasas ligninolíticas (>1 V) sólo permiten a las lacasas la degradación directa de compuestos fenólicos de bajo potencial redox, que constituyen únicamente un 20% del total de la lignina (Kawai et al. 1987a, 1987b). El interés por las lacasas como biocatalizadores industriales en la producción de pasta de papel se ha incrementado enormemente tras el descubrimiento de compuestos mediadores que amplían la acción de la lacasa a sustratos no fenólicos, lo que aumenta el potencial en la degradación de la lignina y de otros compuestos aromáticos (Call y Mücke 1
30

1. Introducción
(b)(a) (c)
Figura 15. Enzimas de interés en la industria de la pasta y papel: (a) xilanasa, (b) lacasa y (c) peroxidasa versátil.
Las peroxidasas catalizan la oxidación de una gran variedad de compuestos tanto orgánicos como inorgánicos en presencia de peróxidos. Hay dos tipos de peroxidasas ligninolíticas: la lignina peroxidasa (LiP) y las manganeso peroxidasas (MnP) (Tien y Kirk 1983, Glenn et al. 1983, Kuwahara et al. 1984). La LiP oxida compuestos aromáticos de alto potencial redox, como el alcohol veratrílico (alcohol 3,4-dimetoxibenzílico) y dímeros modelo de lignina de tipo no fenólico. Esta enzima es una glicoproteína con hierro protoporfirínico IX como grupo prostético, dependiente de H2O2 para su actividad. Inicialmente es oxidada por peróxido de hidrógeno, oxidando núcleos aromáticos de la molécula de lignina (fenólicos y no fenólicos), generando radicales catiónicos. Estos interactúan espontáneamente con nucleófilos (principalmente H2O) y con oxígeno molecular, generando una “combustión enzimática” donde los enlaces C-C e C-O se rompen, despolimerizando la lignina y abriendo los anillos aromáticos. Las MnP son glicoproteínas con hierro protoporfirínico IX como grupo prostético y dependiente de H2O2 para su actividad, pero la oxidación por esta enzima es también dependiente de la disponibilidad de iones manganeso. Un tercer tipo de peroxidasa ligninolítica, la VP (peroxidasa versátil), se ha descrito por primera vez en Pleurotus eryngii (Martínez et al. 1996, Ruiz-Dueñas et al. 1999a, Camarero et al. 1999) y se caracteriza por combinar propiedades catalíticas de las otras dos peroxidasas ligninolíticas (Ruiz-Dueñas et al. 1999b) pudiendo oxidar lignina y compuestos de manganeso. En la Figura 16 se puede observar una vista axial de la región del hemo de la VP donde se muestra los tres sitios de la oxidación y el ciclo catalítico de ésta enzima. La VP se ha utilizado en esta Tesis en ensayos de deslignificación con polioxometalatos (POMs).
31

1. Introducción
Alcohol veratrílico
(VA)
Asp175Grupo Hemo
Glu40
Hélice B
Figura 16. Vista axial de la región del hemo de la VP que muestra los tres sitios de la oxidación: Mn2+ (que incluye tres residuos acídicos de amino-ácidos), ABTS (en el límite del grupo hemo) y VA (a través de Trp164) (a) y ciclo catalítico propuesto para la VP (b) (Ruiz-Dueñas y Martínez 2010, Pérez-Boada et al. 2005).
Los POMs son catalizadores de oxidación conocidos por sus síntesis orgánicas homogéneas y heterogéneas y se han sugerido como alternativas a los reactivos de blanqueo basados en cloro y al blanqueo convencional alcalino con oxígeno (Gamelas et al. 2007). Se utilizan en procesos de deslignificación oxidativa con oxígeno, cuyo objetivo principal es la oxidación selectiva de la lignina residual, pudiendo ser regenerados y reutilizados en el proceso de deslignificación. Se han intentado regenerar POMs por reoxidación con lacasas, pero los resultados no fueron muy esperanzadores para su aplicación industrial debido a los elevados tiempos de reoxidación, que además no era completa (Gamelas et al. 2007, Gaspar et al. 2007, Tavares et al. 2004, Gamelas et al. 2008). En esta Tesis se han utilizado por primera vez peroxidasas para la reoxidación de POMs. En la Figura 17 se puede observar el ciclo catalítico propuesto para la oxidación de la lignina por el POM así como su reoxidación por la VP. Los POMs se caracterizan por ser aniones que se pueden visualizar estructuralmente como conjuntos agregados metal-oxígeno (Pope 1983). La unidad básica y más común, los octaedros, está formada por un metal rodeado de seis oxígenos (MO6) (Weinstock et al. 1997). Además de M y O, otros elementos que usualmente son designados por X, pueden formar parte de la estructura de los POMs. Los POMs con estructura de Keggin son los más importantes y los más estudiados actualmente ya que son más estables y fácilmente disponibles (Gamelas et al. 2003). El anión de Keggin está compuesto por un tetraedro central XO4 rodeado por 12 octaedros MO6, metal-oxígeno, compartiendo aristas y vértices. Los octaedros se encuentran en cuatro grupos M3O13, que comparten los átomos de oxígeno de los vértices formando el POM con la fórmula [XM12O40]m-, en que XM12 es la fórmula abreviada.
Mn2+
Trp164·
ABTS(fenoles)
agua
Glu36
Mn2+
Mn3+
Mn2+ Mn3+
C-IIA[Fe4+=O]
ROOH
ROH
C-IA[Fe4+=O P·]
VP[Fe3+]
C-IB[Fe4+=O Trp·]
C-IIB[Fe3+ Trp·]
VAVA·
ABTS
ABTS·+
ABTS ABTS·+
VA
VA·
(b)(a)
32

1. Introducción
H2O2H2O
Compuesto 1 (enzima activa)
Ligninaox
H2O
Lignina
Ligninaox
Compuesto 2
Lignina
POMred
POMoxPOMred
Mn3+
Mn2+
Mn2+
POMox
Mn3+
H2O2H2O
Compuesto 1 (enzima activa)
Ligninaox
H2O
Lignina
Ligninaox
Compuesto 2
Lignina
POMred
POMoxPOMred
Mn3+Mn3+
Mn2+Mn2+
Mn2+Mn2+
POMox
Mn3+Mn3+
(b)(a)
Figura 17. Representación poliédrica de un polioxometalato con estructura de Keggin (a) y ciclo catalítico propuesto para la oxidación de la lignina y reoxidación del POM por la VP en los ensayos de deslignificación realizados durante esta Tesis (b).
1.5.2. Degradación enzimática de lípidos: Control del pitch
El control enzimático del pitch en pastas mecánicas de coníferas mediante el uso de lipasas, se puso en práctica como una operación rutinaria en la producción industrial de papel a principios de los años 90 (Hata et al. 1996) y fue el primer caso en que una enzima se aplicó con éxito en el proceso de producción papelero. Las lipasas (Figura 18a) son un grupo de hidrolasas producidas por una gran variedad de organismos. Los tratamientos de pasta con lipasas se iniciaron en Japón con una enzima de Candida cylindracea (Irie 1990) y se continuaron en diversas pruebas de fábrica utilizando una lipasa mejorada y comercializada por Novo Nordisk (actualmente Novozymes) bajo el nombre comercial de Resinase® (Matsukura et al. 1990, Fujita et al. 1991, 1992, Gutiérrez et al. 2001), que es capaz de hidrolizar aproximadamente el 95% de los triglicéridos presentes en una pasta mecánica de pino. A lo largo de los años siguientes, varias fábricas en Japón y China han introducido esta tecnología del control del pitch basada en lipasas en pasteados mecánicos. Las lipasas actúan sobre los glicéridos pero no sobre otros extraíbles lipídicos. Teniendo en cuenta que los triglicéridos se hidrolizan con facilidad en el pasteado alcalino (kraft y sosa), las lipasas no son de interés para el control del pitch en estos procesos, así como cuando otros compuestos como esteroles libres y conjugados, alcoholes grasos y alcanos, son los responsables del pitch (del Río et al. 1998, 1999, 2000, Gutiérrez y del Río 2005). Otras enzimas de tipo hidrolasa estudiadas son las esterol esterasas, que también podrían ser efectivas para el control del pitch ya que los ésteres de esteroles son a menudo causa de pitch, debido a su
33

1. Introducción
pegajosidad y resistencia a la cocción alcalina. Sin embargo, estas enzimas liberan esteroles libres que son tan problemáticos como sus ésteres.
Por otro lado, las lacasas constituyen un grupo de enzimas oxidativas que ha sido objeto de gran interés en el desarrollo de tecnologías respetuosas con el medio ambiente (Mayer y Staples 2002). Como se ha comentado anteriormente, la acción directa de las lacasas en principio está restringida a las unidades fenólicas, pero en presencia de mediadores redox (Bourbonnais y Paice 1990, Call 1994) amplían la acción de la lacasa a sustratos no fenólicos, lo que aumenta su potencial en la degradación de lignina y de otros compuestos aromáticos. Recientemente, se ha mostrado la gran eficacia del sistema lacasa-mediador en la eliminación de extraíbles lipofílicos de pastas de coníferas, frondosas así como de fibras no madereras (Gutiérrez et al. 2006a, 2006c, 2006b, 2007, 2009, Molina et al. 2008).
Finalmente, hay que mencionar que se ha sugerido recientemente el uso de una lipoxigenasa (de soja) para el control del pitch en pasta termomecánica de coníferas (Zhang et al. 2007). Las lipoxigenasas (Figura 18b) son una clase de dioxigenasas que contienen hierro (no en forma hemo) y catalizan la oxigenación de ácidos grasos insaturados y sus ésteres. El descubrimiento reciente de una lipoxigenasa fúngica (que contiene manganeso) ha revelado la existencia de otro tipo de lipoxigenasas con una habilidad única para oxidar ácidos grasos (Hamberg et al. 1998; Su and Oliw 1998). Esta lipoxigenasa se ha evaluado en la Tesis para la eliminación de lípidos y lignina.
(b)(a)
Figura 18. Estructura de una lipasa (a) y de una lipoxigenasa (b).
34

1. Introducción
35

2

2. Objetivos
OBJETIVOS
En esta Tesis se aborda el estudio exhaustivo de la composición química de los principales constituyentes de diversos cultivos lignocelulósicos utilizados como materia prima para la fabricación de pasta de papel, así como de la evolución de los mismos durante el proceso de cocción y blanqueo. Estos estudios tienen por objeto obtener un mejor aprovechamiento industrial de estas fibras, que ayudará a optimizar los procesos de cocción y blanqueo utilizando tecnologías menos contaminantes, incluyendo la biotecnología.
Los objetivos específicos de esta Tesis son los siguientes:
- Realizar una caracterización química de los diferentes cultivos lignocelulósicos seleccionados, poniendo especial énfasis en la composición de lípidos, lignina y hemicelulosas.
- Estudiar la modificación estructural de los constituyentes orgánicos de los diferentes cultivos lignocelulósicos durante los procesos de cocción y blanqueo.
- Estudiar y desarrollar diferentes aplicaciones biotecnológicas que permitan degradar tanto la lignina residual como los compuestos extraíbles lipofílicos presentes en las pastas de papel mediante tecnologías menos contaminantes.
37

2. Objetivos
39

3

3. Material y Métodos
MATERIAL Y MÉTODOS
3.1. MATERIALES
En esta Tesis se ha estudiado la composición química de fibras de diversos cultivos agrícolas utilizadas para la fabricación de pastas de celulosa de alta calidad y el comportamiento de sus componentes a lo largo del proceso de fabricación de dichas pastas. Entre los materiales estudiados se encuentran fibras del tallo de varias plantas anuales, tales como lino, kenaf, cáñamo y yute; y fibras procedentes de hojas de sisal, abacá y curauá. También se seleccionaron pastas crudas y pastas blanqueadas (TCF y ECF). Tanto las muestras correspondientes a las materias primas como sus pastas fueron suministradas por la empresa Celulosa de Levante S.A., CELESA (Tortosa, Tarragona). Por otro lado, también se estudiaron otras fibras de potencial aplicación en el sector procedentes de tallos de caña común, así como fibras de residuos de poda de árboles de tagasaste, que fueron suministradas por la Universidad de Huelva. Finalmente, para la realización de los diversos tratamientos biotecnológicos se utilizaron pastas de eucalipto y de lino, suministradas por las empresas ENCE (Pontevedra) y CELESA, respectivamente.
3.1.1. Cultivos lignocelulósicos
Lino
El lino (Linum usitatissimum) (Figura 19) es una planta herbácea dicotiledónea de la familia de las Lináceas, originaria de Asia que se cultiva por el aceite de su semilla y por las fibras de sus tallos. Se cultiva principalmente en regiones frías y templadas como Europa, Asia, Australia, Argentina y Brasil.
Figura 19. Planta de lino (izquierda) y morfología de sus fibras elementales (derecha) (García Hortal 2007).
41

3. Material y Métodos
42
De las 150 especies del género Linum, sólo la especie L. usitatissimum produce fibras útiles comercialmente. Las plantas de lino textil alcanzan una altura de 0,9-1,25 m y un diámetro de 0,25-0,5 cm. Los haces fibrosos se extraen fácilmente por un procedimiento denominado enriado, que consiste en un proceso hidrolítico en el que enzimas de hongos y bacterias disuelven las pectinas que mantienen adheridas las fibras, proporcionando unas fibras muy puras y de alta calidad que tienen una longitud de 30 a 90 cm. La fibra elemental tiene una longitud comprendida entre 10 y 55 mm y un diámetro de 12 a 30 �m, estando entre las más altas de todas las fibras utilizadas en la industria papelera, siendo excedida sólo por la fibra de algodón (García Hortal 2007).
La pasta procedente de fibras de lino es ideal para la producción de papeles delgados a los que se exige una gran resistencia, papeles densos y permanentes, tales como el papel para cigarrillos, papeles muselina, papel biblia y papeles para imprimir de bajo gramaje. Debido a su pureza y resistencia, a su capacidad para soportar un refinado intenso y por consiguiente dar una pasta extremadamente porosa, es ideal para bolsas de té, papeles para registro, papeles de seguridad y papel moneda.
Cáñamo
El cáñamo (Cannabis sativa) (Figura 20) es una planta herbácea dicotiledónea de la familia de las Cannabináceas, originaria de Asia central que se cultiva por el aceite y la proteína de su semilla así como por las fibras de sus tallos. Se cultiva principalmente en los países de la antigua URSS y en China, pero se encuentra también en la India, países de Europa Central, Paquistán, Turquía, Italia y Colombia. Es una planta que se acomoda a todos los climas, muy competitiva con las malas hierbas, por lo que no exige grandes inversiones de herbicidas (Struik et al. 2000).
Figura 20. Planta de cáñamo (izquierda) y morfología de sus fibras elementales (derecha) (cedida por García Hortal).

3. Material y Métodos
Las plantas de cáñamo presentan tallos que pueden alcanzar alturas de 1 a 5 m y diámetros entre 5 y 10 mm. Los haces fibrosos son más largos que los de lino, de 100-300 cm, más rígidos y gruesos. La fibra elemental tiene características morfológicas similares a la del lino, aunque no son tan uniformes, son menos transparentes y con numerosos nudos. Las fibras secundarias, más cortas, son más delgadas y más lignificadas (García Hortal 2007).
La pasta procedente de cáñamo, al igual que la pasta de lino, es ideal para la producción de papeles especiales que requieren una fibra fuerte, tales como el papel para cigarrillos, papel biblia, filtros de café, bolsitas de té, papel aislante y pañales.
Kenaf
El kenaf (Hibiscus cannabinus) (Figura 21) es una planta herbácea dicotiledónea de la familia de las Malváceas, probablemente originaria de África. Se cultiva en regiones tropicales y subtropicales de la India, Sudeste de Asia y América Central. Es una planta de crecimiento rápido, que se está desarrollando como fuente de fibras para la fabricación de papel.
Las plantas de kenaf crecen en delgadas cañas de hasta 6 m de altura y 4 cm de diámetro. En el tallo, al igual que en el lino y cáñamo, se distinguen dos regiones distintas. Las fibras de la corteza exterior (bast), que constituye alrededor de 30-40% del tallo, son moderadamente largas, mientras que las fibras del núcleo (core) son más cortas. La longitud de las fibras liberianas es de 2 a 6 mm, sin embargo, cabe destacar que las fibras liberianas de las plantas tardías son más cortas que las de las plantaciones normales, ya que el crecimiento vegetativo de esta planta está influenciado por la duración del día (García Hortal 2007, Pande y Roy 1998).
Figura 21. Planta de kenaf (izquierda) y morfología de sus fibras elementales (derecha) (García Hortal 2007).
43

3. Material y Métodos
44
Se pueden obtener pastas de kenaf tanto de la corteza exterior como del núcleo, siendo las fibras largas de la corteza exterior especialmente adecuadas para la fabricación de papeles de calidad especial. Las pastas de kenaf son ideales para papel de prensa, impresión y escritura, y se pueden usar en la mayoría de calidades de papel.
Yute
El yute (Corchorus capsularis) (Figura 22) es una planta herbácea dicotiledónea de la familia de las Tiliáceas, que crece principalmente en climas húmedos y cálidos de Paquistán, India, Bangladesh, Brasil y otros países tropicales. Sus haces fibrosos, que tienen una longitud de 20 a 50 cm y contienen de 10 a 30 fibras elementales, se usan principalmente para la producción de sacos, tapices, cuerdas, textiles y materiales para embalaje.
Las plantas de yute presentan tallos que pueden alcanzar alturas de 2,5-3 m y 10-20 mm de diámetro. La fibra elemental tiene una longitud variable de 1,5 a 5 mm, el menos elevado de las otras fibras liberianas mencionadas, y un ancho de 10 a 25 �m. Los tallos son enriados en estanques de agua, donde se liberan los haces de fibras liberianas de la corteza y la porción leñosa de la planta (García Hortal 2007, Han 1998).
Dureza y durabilidad son las principales características aportadas por las fibras de yute en mezclas con pastas kraft. Las pastas mecánicas o poco cocidas de yute, debido a la mayor lignificación de sus haces fibrosos respecto a los del lino y cáñamo, se usan para cartones, papeles de estraza y de embalaje, y las pastas químicas y blanqueadas se usan para papeles finos como bolsas de té y papel de fumar.
Figura 22. Planta de yute (izquierda) y morfología de sus fibras elementales (derecha) (García Hortal 2007).

3. Material y Métodos
Sisal
El sisal (Agave sisalana) (Figura 23) es una planta robusta de climas tropicales originaria de América Central y México, cultivada actualmente en Brasil, Venezuela, Tanzania, Kenia, Mozambique, Angola, Madagascar y otras zonas tropicales. Es una planta monocotiledónea de la familia de las Agaváceas y sus fibras, denominadas fibras duras, más rígidas y bastas que las fibras liberianas, se han utilizado durante mucho tiempo en las industrias textil y de cordelería.
La planta de sisal crece hasta alturas de 2 m, con un tronco corto de 15-23 cm de diámetro. Las fibras se extraen de las hojas, que tienen entre 100 y 150, tras ser cortadas y descortezadas. El descortezado se debe hacer tan pronto como sea posible una vez que se cortan las fibras, para reducir el deterioro de las mismas. El espesor, longitud y resistencia de la fibra depende de la madurez de la hoja y de su posición a lo largo de ésta, ya que las hojas más maduras contienen las fibras más largas y bastas, y las fibras más gruesas se hallan en el extremo terminal de la hoja.
La hoja madura del sisal mide 1-2 m de largo, 10-15 cm de ancho y 6 mm de espesor (en el centro) y las fibras se disponen longitudinalmente en la hoja. Los haces fibrosos tienen longitudes de 60-150 cm, y la fibra elemental tiene una longitud de 1-8 mm y un ancho de 8-40 �m, (García Hortal 2007, McDougall etal. 1993).
Las fibras del sisal se aplican fundamentalmente en la fabricación de cuerdas y papeles especiales como bolsas de té, papeles dieléctricos y papel de filtro, debido, sobre todo, a la alta porosidad de sus pastas. Por otra parte, también se utilizan como refuerzo para los papeles delgados (Moore 1996)
Figura 23. Plantas de sisal (izquierda) y morfología de sus fibras elementales (derecha) (García Hortal 2007).
45

3. Material y Métodos
46
Abacá
El abacá (Musa textilis) (Figura 24) es una planta perenne nativa de Filipinas y que se cultiva actualmente también en Indonesia y América tropical. Es una planta monocotiledónea que pertenece a la familia de las Musáceas y sus fibras también están clasificadas como fibras duras.
La planta se asemeja a la del platanero, pero con hojas más pequeñas y frutos no comestibles. Los tallos alcanzan alturas de 3 a 7,5 m y diámetros de 12 a 30 cm y consisten en un corazón central envuelto por vainas foliares. Las fibras útiles comercialmente se encuentran en las vainas externas del tallo, que se extraen por descortezado puramente mecánico de las vainas, de las que se separan los haces por simple rasgadura.
Los haces fibrosos son muy largos, de hasta 2 m y la longitud de la fibra elemental varía de 2,5 a 12 mm según el espesor y la posición en el tallo de la vaina madre, donde las vainas más externas tienen fibras más cortas, más gruesas y de color más oscuro, por lo que son las de menor calidad (García Hortal 2007, Moore 1996).
Los papeles producidos por las fibras de abacá son altamente porosos, por lo que son ideales para filtros y envolturas de embutidos. Se considera una excelente materia prima para papeles de alta calidad, como papel moneda, pañales, servilletas, papel tisú, accesorios para hospitales, etc. El abacá de primera calidad también se emplea mezclado con algodón o pasta de madera en la fabricación de papeles superfinos, papel para imprimir de bajo gramaje, de registro, moneda y seguridad y, sobre todo, papel para filtro poroso de uso en laboratorio o industrial.
Figura 24. Planta de abacá (izquierda) y morfología de sus fibras elementales (derecha) (cedida por García Hortal).

3. Material y Métodos
Curauá
El curauá (Ananás erectifolius) es una planta herbácea de fruto no comestible (Figura 25a) perteneciente a la familia de las Bromeliáceas, de origen americano y hábitat tropical, muy común en el Amazonas. Tiene un tallo tan corto que parece carecer de él y con hojas rígidas. Normalmente produce entre 20 y 24 hojas, proporcionando aproximadamente 2 kg de fibra. En la última década, ha ganado reconocimiento comercial como material para composites en la industria del automóvil (Leao et al. 1998, Silva et al. 2001). La fibra del curauá también se ha propuesto como una materia prima alternativa para la producción de pastas químicas en Brasil.
Caña común
La caña común (Arundo donax) es una planta monocotiledónea perenne, perteneciente a la familia de las Poáceas que parece ser originaria de Asia y que ha colonizado el área del Mediterráneo, en países como Portugal y España. Es considerada una de las mayores gramíneas, con una estructura tubular segmentada semejante al bambú (Figura 25b), con alturas entre 2 y 8 m (Seca etal., 2000). Debido a la fácil adaptabilidad de esta gramínea a diferentes condiciones ecológicas, a la elevada productividad de biomasa y capacidad de cultura intensiva, es una de las especies no madereras más atractivas como fuente alternativa de fibras para la industria de pasta de papel (Shatalov y Pereira 2002).
Tagasaste
El tagasaste (Chamaecytisus proliferus spp. palmensis) es un arbusto robusto de crecimiento rápido perteneciente a la familia de las Fabáceas. Es nativo de las Islas Canarias y se cultiva en Australia, Nueva Zelanda y otros países. Debido a su alto contenido proteico, se usa como alimento para el ganado y también como cultivo fijador del nitrógeno para mejorar la fertilidad del suelo. Con el fin de fomentar la formación de tallos múltiples, el arbusto debe ser podado con regularidad, lo que conduce a una alta acumulación de residuos de poda. Estos residuos se han evaluado recientemente como materia prima alternativa para la producción de pasta de celulosa (Díaz et al. 2004, López et al. 2004, Jiménez et al. 2006, 2007, García et al. 2008). En la Figura 25c se muestra una fotografía de un arbusto de tagasaste.
47

3. Material y Métodos
48
a
b
c
Figura 25. Diversas plantas estudiadas como materias primas potenciales para la producción de pasta de celulosa: (a) curauá, (b) caña común y (c) tagasaste.
3.1.2. Pastas de papel
Pastas de fibras no madereras
Se han estudiado pastas de lino, cáñamo, sisal y abacá, tanto crudas (proceso de cocción sosa-AQ) como blanqueadas (procesos de blanqueo TCF y ECF) suministradas por CELESA. Las pastas crudas se obtuvieron por una cocción sosa-AQ que emplea hidróxido de sodio y antraquinona (hasta 0,05%) como agentes de cocción, durante 2-4 h a una temperatura de 160-170 ºC.
Las pastas blanqueadas se obtuvieron tras secuencias de blanqueo TCF y ECF. La secuencia de blanqueo TCF (Q-Po), incluyó una etapa quelato (Q), seguida de una etapa con peróxido de hidrógeno con oxígeno presurizado (Po). La etapa de quelato se lleva a cabo para capturar los iones metálicos y evitar que dichos iones destruyan el peróxido de hidrógeno. La secuencia de blanqueo ECF usada (D-Po) incluyó una etapa de dióxido de cloro (D), seguida por una etapa de peróxido de hidrógeno con oxígeno presurizado (Po).
Pastas de fibras madereras
Para la realización de los diferentes tratamientos biotecnológicos se usó pasta kraft cruda (no blanqueada) de eucalipto (E. globulus) suministrada por ENCE.

3. Material y Métodos
3.1.3. Enzimas y mediadores
En esta Tesis se han estudiado dos aplicaciones biotecnológicas: (i) enzimas de tipo lipoxigenasa para la degradación de lípidos y lignina en pastas kraft de eucalipto y pastas sosa-AQ de lino, y (ii) un sistema compuesto de un polioxometalato y una enzima de tipo peroxidasa para la degradación de lignina en pastas kraft de eucalipto.
Lipoxigenasas
La lipoxigenasa utilizada se obtuvo del hongo ascomiceto Gaeumannomycesgraminis y fue suministrada por la empresa Novozymes (Bagsvaerd, Dinamarca). La unidad de actividad de esta enzima se define como la cantidad de enzima que da lugar a un aumento en la Absorbancia a 234 nm, de 0,001 por minuto a pH 7 y 30ºC, cuando se usa el ácido linoleico como sustrato (volumen de reacción = 1,0 mL y camino óptico = 1 cm). Los tratamientos enzimáticos con lipoxigenasa incluyeron reacciones con varios lípidos modelo, como alcanos (nonacosano), alcoholes (octacosanol), ácidos grasos (ácido linoleico), esteroles libres (sitosterol) y ésteres de esteroles (colesteril linoleato), representativos de los extraíbles lipofílicos de las pastas estudiadas. El sitosterol fue suministrado por Calbiochem y los demás compuestos por Sigma-Aldrich.
Peroxidasas
Se utilizó la peroxidasa versátil del hongo basidiomiceto Pleurotus eryngii (VP), producida en el Centro de Investigaciones Biológicas (CIB, CSIC, Madrid) como se describe a continuación.
La cepa de E. coli se cultivó en matraces de 1 L con 500 mL de medio TB (Terrific Broth) y 100 μg/mL de ampicilina. El medio TB consiste en un medio tamponado para el crecimiento de la cepa de E. coli transformada con el plásmido portador del gen que codifica la VP de P. eryngii. Se incubó a 37ºC y 200 rpm durante 3 horas. Se indujo la expresión añadiendo isopropil tio-�-D-galactopiranósido (IPTG) a una concentración final de 1 mM y se continuó con la incubación durante 4 horas más. El “pellet” bacteriano, que contiene la proteína recombinante en forma de cuerpos de inclusión, se obtuvo por centrifugación, se resuspendió en tampón de lisis (tampón con Tris-HCl pH 8,0 50 mM, EDTA pH 8,0 10 mM y DTT 5 mM), se le añadió lisozima a una concentración final de 2 mg/mL y se incubó durante 1 hora en hielo. Se le añadió 0,1 mg/mL DNAasa, se sonicó en 3 ciclos de 1 minuto a 20.000 Hz de frecuencia y se centrifugó 30 min a 12.500 rpm para favorecer la precipitación de los cuerpos de inclusión.
49

3. Material y Métodos
50
Una vez lavados los cuerpos de inclusión con una solución de Tris-HCl pH 8,0 20 mM, EDTA pH 8,0 1 mM y DTT 5 mM, se resuspendieron en el mínimo volumen posible de la misma solución de lavado, se homogenizaron y centrifugaron. Se resuspendieron de nuevo los cuerpos de inclusión en el mínimo volumen de solución desnaturalizante (solución de Tris-HCl pH 8,0 50 mM, EDTA pH 8,0 1 mM, DTT 5 mM y urea 8 M), se incubaron a temperatura ambiente durante 15 min y se centrifugaron para eliminar los restos no disueltos. El sobrenadante, con la proteína completamente desplegada, se diluyó 5 veces con una solución de Tris-HCl pH 8,0 50 mM, EDTA pH 8,0 1 mM y DTT 1 mM, para llegar a una concentración final de urea de 1,6 M y de 1-2 mg/mL de proteína. Esta solución se añadió a la mezcla de replegado (CaCl2 5 mM, glutatión oxidado 0,5 mM, Tris-HCl pH 9,5 50 mM y hemina 20 μM) en una proporción 1:10 para obtener una concentración final de urea de 0,16 M y se incubó en oscuridad a temperatura ambiente durante 16 horas.
La mezcla de replegado con la enzima activada se concentró por filtración tangencial hasta 80-100 mL, a través de una membrana de 3 KDa de tamaño de poro (Membrane Casette de Filtron) con una bomba peristáltica (Masterflex modelo 7518-02), y posteriormente por ultrafiltración con membrana de 3-10 KDa de tamaño de poro en un sistema Amicon® de Millipore hasta 20-40 mL. Se dializó la mezcla concentrada en una solución tampón acetato (acetato 20 mM, pH 4,3 y CaCl2 1 mM) y el material insoluble precipitado se eliminó por centrifugación. Se dializó de nuevo en solución tampón tartrato (tartrato 10 mM, pH 5,5 y CaCl2 1 mM) y se purificó por cromatografía líquida de alta resolución (HPLC) en un equipo Äkta de Amersham Pharmacia Biotech de intercambio aniónico previamente equilibrada con el mismo tampón. La proteína se eluyó con un gradiente de NaCl de 0 a 0,3 M en tampón tartrato (tartrato 10 mM, pH 5,5, CaCl2 1 mM) con un flujo de 2 mL/min. Las fracciones obtenidas se dializaron con tartrato 10 mM a pH 5,0 y se realizó el espectro UV-VIS (espectrofotómetro Shimazdu UV-160) para verificar la correcta incorporación del hemo durante el proceso de replegado. La concentración de la enzima pura se determinó a partir de su coeficiente de extinción molar (�406= 150.000 M-1cm-1) (Ruiz-Dueñas et al. 1999b). La enzima se congeló en nitrógeno líquido y se conservó a -80ºC hasta su utilización.
Polioxometalatos
El POM utilizado fue [SiW11MnIII(H2O)O39]5- denominado SiW11MnIII en forma simplificada. La solución acuosa del POM se preparó a partir de �-K8[SiW11O39]•13H2O, KMnO4 y Mn(CH3COO)2•4H2O. Se disolvieron 9,6 g de K8[SiW11O39]•13H2O en 13 mL de agua, a 95ºC. En vasos separados se disolvieron, a temperatura ambiente, 0,095 g de KMnO4 en 10 mL de HCl 0,6mM y 10 mL de agua y 0,6 g de Mn(CH3COO)2•4H2O en 10 mL de agua. Al

3. Material y Métodos
vaso conteniendo K8[SiW11O39]•13H2O a 95ºC se le adicionó poco a poco y consecutivamente las disoluciones de los otros dos vasos y se dejó unos 25 minutos a 95ºC. Se guardó la disolución preparada hasta el día siguiente para filtrarla y diluirla para 100 mL de modo que la concentración final fuera de aproximadamente 30 mM.
3.2. MÉTODOS ANALÍTICOS
3.2.1. Aislamiento y análisis de los compuestos lipofílicos de las fibras y pastas
El análisis de los compuestos extraíbles lipofílicos de las fibras y pastas requirió su aislamiento previo. Dichos compuestos se extrajeron con acetona en un extractor de tipo Soxhlet durante 8 horas. A continuación se evaporó el disolvente a sequedad en un rotavapor y la cantidad de extracto se determinó por gravimetría. Los extractos lipofílicos obtenidos se redisolvieron en CHCl3 para su posterior análisis por cromatografía de gases (GC) y cromatografía de gases/espectrometría de masas (GC/MS), descritos más adelante.
Fraccionamiento de los compuestos extraíbles lipofílicos mediante SPE
Para una caracterización más detallada de los compuestos presentes en los extractos lipofílicos, se procedió a su aislamiento y purificación mediante SPE (extacción en fase sólida) según un método previamente descrito (Gutiérrez et al. 1998, 2004), tal como se muestra en la Figura 26.
Los extractos lipídicos se fraccionaron en cartuchos (500 mg) de aminopropilo (Waters, Millipore). Los extractos secos (5-10 mg) se resuspendieron en un volumen mínimo (<0,5 mL) de hexano-CHCl3 (4:1) con el que se cargó el cartucho, previamente acondicionado con hexano (4 mL). La primera fracción se eluyó con 8 mL de hexano, que contiene los compuestos más apolares como ésteres de esteroles, ceras e hidrocarburos; la segunda fracción se eluyó con 6 mL de hexano-CHCl3 (5:1), rica en triglicéridos; la tercera con 10 mL de CHCl3, que contiene principalmente esteroles y alcoholes libres y por último la cuarta fracción se eluyó con 10 mL de una mezcla de éter etílico-ácido acético (98:2), conteniendo compuestos ácidos. Cada fracción aislada se secó con nitrógeno, se pesó y se procedió a su análisis por GC y GC/MS.
51

3. Material y Métodos
52
Esteroles
Figura 26. Esquema del fraccionamiento de un extracto lipídico por SPE (Gutiérrez et al. 2004).
10 20 30 min
Ésteres de esteroles Triglicéridos
Hidrocarburos esteroidales
EscualenoÁcidos grasos
Esteroles
Ésteres de esteroles Triglicéridos
Hidrocarburos esteroidales
EscualenoÁcidos grasos
20 30 min10
Ésteres de esteroles
Escualeno
Hidrocarb.esteroidales
Ceras
10 20 30 min
Hexano 8 mL
A
Ésteres de esteroles
Escualeno
Hidrocarb.esteroidales
Ceras
10 20 30 min
Ésteres de esteroles
Escualeno
Hidrocarb.esteroidales
Ceras
10 20 30 min10 20 30 min
Hexano 8 mL
A
Hexano 8 mL
Hexano 8 mL
A
Triglicéridos
10 20 30 min
Hexano /CHCl3(5:1) 6mL
B
Triglicéridos
10 20 30 min
TriglicéridosTriglicéridos
10 20 30 min10 20 30 min
Hexano /CHCl3(5:1) 6mL
B
Hexano /CHCl3(5:1) 6mL
B
Hexano /CHCl3(5:1) 6mL
BB
CHCl310 mL
10 20 30 min
Esteroles
C
CHCl310 mL
10 20 30 min
Esteroles
CHCl310 mLCHCl310 mL
10 20 30 min
Esteroles
10 20 30 min
Esteroles
10 20 30 min10 20 30 min
Esteroles
C
Éter/AcOH(98:2) 10 mL
Ácidos grasosC16
C18:1
C18:2
C18C22C24
C26C28C30
10 20 30 min
C20
D
Éter/AcOH(98:2) 10 mL
Ácidos grasosC16
C18:1
C18:2
C18C22C24
C26C28C30
10 20 30 min
C20
Ácidos grasosC16
C18:1
C18:2
C18C22C24
C26C28C30
10 20 30 min10 20 30 min
C20
DAcondicionamiento
Hexano 4 mL
Fase aminopropilo
Extracto lipídico total enHexano /CHCl3 (4:1) 0.5 mL
Acondicionamiento
Hexano 4 mL
Fase aminopropilo
Extracto lipídico total enHexano /CHCl3 (4:1) 0.5 mL
Hexano 4 mL
Fase aminopropilo
Extracto lipídico total enHexano /CHCl3 (4:1) 0.5 mLExtracto lipídico total enHexano /CHCl3 (4:1) 0.5 mL
Hexano 4 mL
Fase aminopropilo

3. Material y Métodos
Métodos de derivatización de los compuestos extraíbles lipofílicos
Para el análisis por GC y GC/MS es esencial que los compuestos existentes en la muestra sean suficientemente volátiles, por lo que es necesario recurrir a métodos de derivatización cuando los compuestos a analizar no son volátiles, esto es, métodos de conversión de ciertos compuestos en otros que sean compatibles con el método analítico de GC/MS. Las técnicas empleadas en esta Tesis incluyeron la metilación de grupos carboxilo y la silanización de grupos hidroxilo.
La metilación de los grupos carboxílicos de ácidos grasos libres, hidroxiácidos y grupos fenólicos se realizó con (trimetilsilil)diazometano (TMSD) suministrado por Sigma-Aldrich. Para ello, una vez seca la muestra, se añadió 100 �l de metanol y 50 �l de una solución de TMSD 2,0 M en hexano y se mantuvo 20 min en el baño de ultrasonidos. A continuación se secó con nitrógeno, para resuspenderla en CHCl3 y analizarla por GC/MS.
La silanización de los grupos hidroxilo de alcoholes, esteroles, etc, se realizó con N,O-bis-(trimetilsilil)-trifluoroacetamida (BSTFA) suministrado por Sigma-Aldrich. Para ello, una vez seca la muestra, se añadió 0,2 mL de BSTFA y 0,1 mL de piridina. A continuación se calentó a 70ºC durante 2 h y se secó con nitrógeno. Posteriormente, se redisolvió en CHCl3 para analizarla por GC/MS.
Análisis de los extractos lipofílicos mediante GC y GC/MS
Para el análisis de los compuestos extraíbles lipofílicos por GC y GC/MS, las características de las columnas cromatográficas utilizadas fueron las adecuadas para separar e identificar los compuestos de alto peso molecular como ceras, ésteres de esteroles, triglicéridos, etc. Previamente se habían realizado estudios sobre procedimientos para el análisis de los extractos lipofílicos de maderas (Gutiérrez et al. 1998a, 2004) por GC y GC/MS en donde se usaron diversas columnas de diferente longitud y diferentes programas de temperatura. En estos estudios, las columnas capilares seleccionadas para el análisis de lípidos por GC fueron de longitud corta (5 m) ya que proporcionan una conveniente elución y separación de lípidos de alto peso molecular en un corto período de tiempo (20 min). Columnas menores de 5 m no son convenientes ya que no proporcionan la resolución necesaria para análisis cuantitativos. En el caso de los análisis por GC/MS, los cromatogramas obtenidos tienen que ser reproducibles con los obtenidos por GC usando columnas capilares de 5 m. No obstante, en el sistema GC/MS, debido a las condiciones de alto vacío a las que opera, no se pueden usar columnas tan cortas, por lo que se usaron columnas de 10-15 m. Esta longitud de columna es apropiada para el análisis de lípidos de alto peso
53

3. Material y Métodos
54
molecular por GC/MS proporcionando resultados en un período de tiempo corto (30 min).
Los análisis cromatográficos de los extractos lipofílicos, tanto de las muestras derivatizadas como sin derivatizar, se llevaron a cabo en un cromatógrafo de gases Agilent 6890N equipado con un detector de ionización de llama (FID) y una columna capilar corta de sílice fundida (DB-5HT, J&W; 5 m x 0,25 mm ID y 0,1 �m de espesor de película). El programa de calentamiento del horno comenzó a 100°C (1 min), seguido de un incremento de temperatura hasta 350°C (3 min) a 15ºC/min. Las temperaturas del inyector y del detector se mantuvieron a 300°C y 350°C, respectivamente. El gas portador que se utilizó fue Helio y la inyección se realizó en modo splitless.
El análisis mediante GC/MS se llevó a cabo en un cromatógrafo de gases Varian 3800 acoplado a un detector de trampa de iones (ITD, Varian 4000), usando una columna capilar de sílice fundida (DB-5HT, J&W; 12 m x 0,25 mm ID, con espesor de película de 0,1 �m). El horno se calentó de 120°C (1 min) a 380°C (5 min) a 10ºC/min. La línea de transferencia se mantuvo a 300ºC. La temperatura del inyector se programó de 120ºC (0,1 min) a 380°C con una rampa de 200ºC/min y manteniéndose hasta el final del análisis. El gas portador utilizado fue Helio. La identidad de cada componente se determinó por comparación de sus espectros de masas con los espectros existentes en las librerías (Wiley y NIST) y con espectros publicados anteriormente, por sus fragmentaciones y, cuando fue posible, por comparación con patrones suministrados por Sigma-Aldrich (octadecano, ácido palmítico, sitosterol, colesteril oleato y sitosteril 3�-D-glucopiranósido). Los picos cromatográficos se cuantificaron a partir de sus áreas en los cromatogramas. Se utilizó una recta de calibrado, realizada con los patrones anteriormente descritos. En todos los casos se obtuvo un coeficiente de correlación mayor de 0,99.
3.2.2. Aislamiento y análisis de la lignina de las fibras y pastas
Determinación del contenido en lignina
El contenido en lignina de las muestras se determinó por el método Klason según la norma Tappi T222 om-88 (Tappi 2004), con algunas modificaciones. En este método, las muestras molidas de las distintas fibras seleccionadas, libres de compuestos extraíbles, se sometieron a una hidrólisis con H2SO4 al 72% (p/p), a 30ºC durante 1 h. Posteriormente, la solución se diluyó hasta alcanzar una concentración del 4% en H2SO4 y se autoclavaron (1h a 110ºC). A continuación, las muestras se filtraron, guardándose los primeros 100 mL para el posterior análisis de los azúcares libres y el residuo insoluble (lignina Klason) se

3. Material y Métodos
lavó con agua destilada hasta pH neutro y se secó para su cuantificación gravimétrica.
Aislamiento de la lignina de las fibras
La lignina se extrajo de las muestras, previa eliminación de los compuestos lipofílicos e hidrosolubles de las fibras, según el protocolo desarrollado por Björkman (1956) que consiste en extraer la lignina de la muestra finamente molida. El grado de molienda deseado se alcanzó utilizando un molino de bolas centrífugo modelo Retsch S100 durante 100 h. La lignina de las muestras finamente molidas se extrajo con dioxano-agua (9:1, utilizando 250 mL por cada 10 g de muestra) durante 12 h. Posteriormente, se centrifugó (25 min, 4ºC, 11000 rpm) y se recogió el sobrenadante (que contiene la lignina) en un matraz, repitiéndose este proceso dos veces de forma consecutiva, y se secó en rotavapor a 40ºC. A continuación, el residuo seco se disolvió en una mezcla de ácido acético:agua (9:1), añadiéndose 20 mL por cada gramo de residuo seco. La lignina se precipitó en agua destilada (225 mL por cada gramo de lignina) en constante agitación y se recogió tras centrifugación. Una vez seco el residuo, se trituró en un mortero de ágata para facilitar su disolución en una mezcla 1,2-dicloroetano-etanol (2:1) y de nuevo se volvió a centrifugar a baja velocidad (5000 rpm durante 5 min) recuperando el sobrenadante, que contiene la lignina. Éste se dispersó gota a gota, sin agitar, en éter dietílico (225 mL es suficiente para 0,5-1 g de lignina), precipitando de nuevo la lignina. Se volvió a centrifugar (5000 rpm durante 5 min) y el residuo se resuspendió en éter de petróleo en el que se dejó durante 12 horas. Finalmente, se centrifugó y se secó mediante corriente de nitrógeno y se conservó a 4ºC, preservándola de la luz y el aire para evitar su oxidación hasta el momento del análisis. Con este método se obtuvo una lignina poco degradada y representativa de la lignina nativa de las fibras.
Análisis de la lignina mediante Py-GC/MS
La pirólisis es un método degradativo que transforma compuestos complejos no volátiles en una mezcla de fragmentos volátiles por descomposición térmica en ausencia de oxígeno (Meier y Faix 1992; Fullerton y Franich 1983) que se lleva a cabo habitualmente a temperaturas de 400-800ºC. En la pirólisis se producen roturas de los enlaces por acción del calor, ya que cuando la energía aplicada a la molécula es mayor que la energía de enlaces específicos ocurre la disociación de éstos de una forma predecible y reproducible, pudiéndose obtener información sobre la molécula original a través del análisis de los productos de degradación. Los fragmentos resultantes de la pirólisis se pueden separar por GC e identificar por MS. La Py-GC/MS es un método poderoso para el análisis de materiales lignocelulósicos, especialmente de la lignina. La lignina se piroliza
55

3. Material y Métodos
56
produciendo una mezcla de compuestos fenólicos que resultan de la rotura no sólo de enlaces éter, sino también de ciertos enlaces C-C, reteniendo estos fenoles las características de sustitución del polímero de lignina y siendo posible por lo tanto identificar los diferentes componentes de ligninas provenientes de unidades H, G y S. La Py-GC/MS presenta diversas ventajas frente a otros métodos degradativos, pues es una técnica analítica rápida que proporciona resultados en apenas un paso, que necesita poca cantidad de muestra y una simple preparación de la misma. También presenta ventajas frente a los métodos clásicos de análisis de la lignina, pues no es necesario aislar la lignina de la muestra, permitiendo su análisis in situ.
En la presente Tesis, la pirólisis de las muestras se llevó a cabo en un pirolizador Frontier Laboratories Ltd., a 500ºC durante 10 s. El pirolizador estaba conectado a un cromatógrafo de gases Agilent 6890 con una columna capilar HP 5MS (30 m x 0,25 mm ID, y un espesor de película de 0,25 �m) acoplado a un espectrómetro de masas Agilent 5973 N. El cromatógrafo se programó de 50ºC (1 min) a 100ºC con un incremento de 30ºC/min y de 100 a 300ºC con un incremento de 10ºC/min. La temperatura final se mantuvo durante 10 min. El gas portador utilizado fue Helio con un flujo controlado de 1 mL/min. Para las pirólisis en presencia de hidróxido de tetrametilamonio (TMAH), se adicionó 0,5 μL de TMAH 25% a 100 μg de muestra. Los compuestos obtenidos mediante Py-GC/MS se identificaron por comparación con la literatura (Faix et al. 1990, Ralph y Hatfield 1991) y con los incluidos en las librerías de espectros de masas Wiley y NIST. Se calcularon las áreas molares para los productos de la pirólisis (lignina y carbohidratos), se normalizó al 100% y se hizo una media entre las repeticiones de las pirólisis. La desviación estándar fue inferior al 5% de la media.
Análisis de la lignina mediante DFRC
El método de degradación química de la lignina conocido como DFRC (derivatization followed by reductive cleavage) es un método simple y poderoso que rompe de manera selectiva y eficiente los enlaces éter �-O-4 y �-O-4 presentes en la lignina. Permite el análisis cuantitativo de las unidades estructurales de la lignina eterificadas y también ofrece información sobre los enlaces carbono-carbono a través del análisis de las estructuras diméricas que se forman.
El método DFRC incluye dos pasos fundamentales: (i) la solubilización de la lignina por bromación y acetilación con bromuro de acetilo y (ii) la fragmentación reductora de los enlaces aril éter en la lignina con polvo de zinc. La identificación de los productos resultantes de la degradación (monómeros y dímeros) por GC/MS proporciona información valiosa sobre la estructura de la

3. Material y Métodos
lignina (Lu y Ralph 1997a, 1997b, 1998). Una de las ventajas de este método es que deja intacto el carbono � de la cadena lateral de la lignina, por lo que lo hace muy adecuado para estudiar la presencia de unidades aciladas (con acetatos, p-cumaratos o p-hidroxibenzoatos) en el carbono �. Sin embargo, el método de DFRC usa reactivos acetilantes que interfieren en el análisis de los grupos acetatos nativos en la lignina, pero con las modificaciones apropiadas, mediante la sustitución de la acetilación por la propionilación (DFRC’), es posible obtener una información significativa sobre la presencia de unidades acetiladas en la lignina nativa (Ralph y Lu 1998). En la Figura 27a se puede observar la rotura selectiva de los enlaces éter en el método DFRC y en la Figura 27b la modificación de este método (DFRC’) para el análisis de ligninas naturalmente acetiladas.
La degradación DFRC se llevó a cabo con 10 mg de lignina aislada que se trató con bromuro de acetilo en ácido acético (8:92) durante 2 h a 50ºC. Después de proceder a la eliminación del disolvente en rotavapor, se disolvió en una mezcla de dioxano/ácido acético/agua (5:4:1) con polvo de zinc y se dejó en agitación durante 30-40 min a temperatura ambiente. A continuación se ajustó el pH hasta un valor inferior a 3 por adición de HCl 3% y se pasó la disolución a un embudo de extracción para separar las fases orgánica y acuosa por extracción líquido-líquido con diclorometano. A la fase orgánica extraída se le añadió sulfato de sodio anhidro y se filtró, recogiéndose el filtrado para eliminación del disolvente en rotavapor. El residuo que quedó tras la evaporación, se acetiló durante 1 h con anhídrido acético, diclorometano y piridina. Se evaporó el disolvente en rotavapor con etanol, se resuspendió en diclorometano y se procedió a su análisis por CG/MS. En el caso de la DFRC’ el protocolo aplicado fue el mismo pero sustituyendo todos los reactivos acetilantes por reactivos con propionilo.
Los análisis mediante GC/MS se llevaron a cabo en un cromatógrafo de gases Varian Star 3400 acoplado a un detector de trampa de iones (ITD, Varian Saturn 2000), usando una columna capilar de sílice fundida (DB-5HT, J&W; 12 m x 0,25 mm ID, con espesor de película de 0,1 �m). El horno se calentó de 50°C (0,2 min) a 100°C a 30ºC/min y se elevó a 300ºC a 5ºC/min. El inyector y la línea de transferencia se mantuvieron a 300ºC. El gas portador utilizado fue Helio. La cuantificación de los monómeros se realizó usando tetracosano como patrón externo asumiendo factores de respuesta similares a los obtenidos por Lu y Ralph (1997).
57

3. Material y Métodos
58
OCH3CH3OO
HO
HOO
OCH3CH3OO
HO
HOO
OCH3CH3OOAc
OAc
Zn
Ac2O/Py OCH3CH3OOAc
OAc
OCH3CH3OOAc
OAc
Zn
Ac2O/Py
OCH3CH3OO
Figura 27. Rotura selectiva de los enlaces éter: (a) Método DFRC (Lu y Ralph 1997a) y (b) Método DFRC modificado (DFRC’) para el análisis de ligninas naturalmente acetiladas (Ralph y Lu 1998).
RO
HOO
OCH3CH3OO
RO
HOO
OCH3CH3OO
AcO
BrO
AcBr
OCH3CH3OO
AcO
BrO
OCH3CH3OO
AcO
BrO
AcBr
OR
OCH3CH3OOAc
OR
Zn
Ac2O/Py OCH3CH3OOAc
OR
OCH3CH3OOAc
Zn
Ac2O/PyOCH3CH3O
O
RO
BrO
AcBr
OCH3CH3OO
RO
BrO
OCH3CH3OO
RO
BrO
AcBr
OCH3CH3OO
HO
HOO
OCH3CH3OOProp
OProp
OCH3CH3OO
PropO
BrO
PropBr
Prop2O/Py
Zn
OCH3CH3OO
HO
HOO
OCH3CH3OO
HO
HOO
OCH3CH3OOProp
OProp
OCH3CH3OOProp
OProp
OCH3CH3OO
PropO
BrO
PropBr
Prop2O/Py
Zn
OCH3CH3OO
AcO
HOO
OCH3CH3OOProp
OAc
OCH3CH3OO
AcO
BrO
PropBr
Prop2O/Py
Zn
OCH3CH3OO
AcO
HOO
OCH3CH3OO
AcO OAc
HOO
OCH3CH3OOProp
OAc
OCH3CH3OOProp
OCH3CH3OO
AcO
BrO
OCH3CH3OO
AcO
BrO
PropBr
Prop2O/Py
Zn
(b)
(a)

3. Material y Métodos
Análisis de la lignina mediante 2D-NMR
La NMR, tanto de 1H como de 13C, ofrece una información detallada de la estructura de la lignina, incluyendo los diferentes tipos de unidades y los enlaces que se establecen entre ellas (Robert 1992). En los últimos años, se ha desarrollado la NMR bidimensional (2D-NMR) y tridimensional (3D-NMR), en las que se establecen correlaciones 1H-13C, entre otras, que resuelve señales que aparecían solapadas en los espectros unidimensionales (Capanema et al. 2001, Ralph et al. 2001, Liitiä et al. 2003). La 2D-NMR se considera en la actualidad la técnica más potente para el análisis de la estructura de la lignina y a través de ella se han podido identificar nuevas sub-estructuras tales como las dibenzodioxocinas (Karhunen et al. 1995) o las espirodienonas (Zhang y Gellerstedt 2001, Zhang et al. 2006).
El HSQC (Heteronuclear Single Quantum Correlation) proporciona correlaciones a través de acoplamiento escalar a un enlace entre un protón y el heteronúcleo al que está directamente unido. Los espectros HSQC de la lignina presentan tres regiones bien diferenciadas: región alifática, región alifática oxigenada y región aromática (Figura 28). La región alifática oxigenada (Figura 29) es la más importante para el estudio de la estructura de la lignina ya que en esta zona se encuentran las correlaciones de la mayoría de los enlaces que tienen lugar entre las distintas unidades estructurales. La región aromática (Figura 30) es la más importante desde el punto de vista de la composición de la lignina, ya que en esta zona aparecen las correlaciones de las distintas unidades H, G y S.
ppm (t2)0.05.010.0
0
50
100
150ppm (t1
Región aromática
Región alifática oxigenada
Región alifática
0
50
100
150
C (ppm)
H (ppm)10 5 0
Figura 28. Espectro HSQC de la lignina aislada de sisal donde se pueden observan sus tres regiones características.
59

3. Material y Métodos
60
�-OAc
-OMe
�-OH
�
� (�-Oac)
� (�-OH)
�
�
�
�
�’
Figura 29. Región alifática oxigenada del espectro HSQC de la lignina aislada de sisal y sus correspondientes estructuras.
��� 1’
6’5’
123
456
4’3’ 2’HO
O
O
OMe
MeOO
O
OMeMeO
R
���
123
456
1’6’
5’4’
3’2’HO
O
O
HO MeO
OMe
OMeMeO
��
�
1
23
45
6
� ’� ’
� ’1’2’
3’4’
5’6’
O
O
O
O
OMe
OMe
MeO
OMe
�
1´2´3´4
´
5´
6´
O
O
OHHO
�� �´ �´
�´
O
OMeO 1
23
456
OMeMeO2
1
4 3
56MeO
OMeMeO
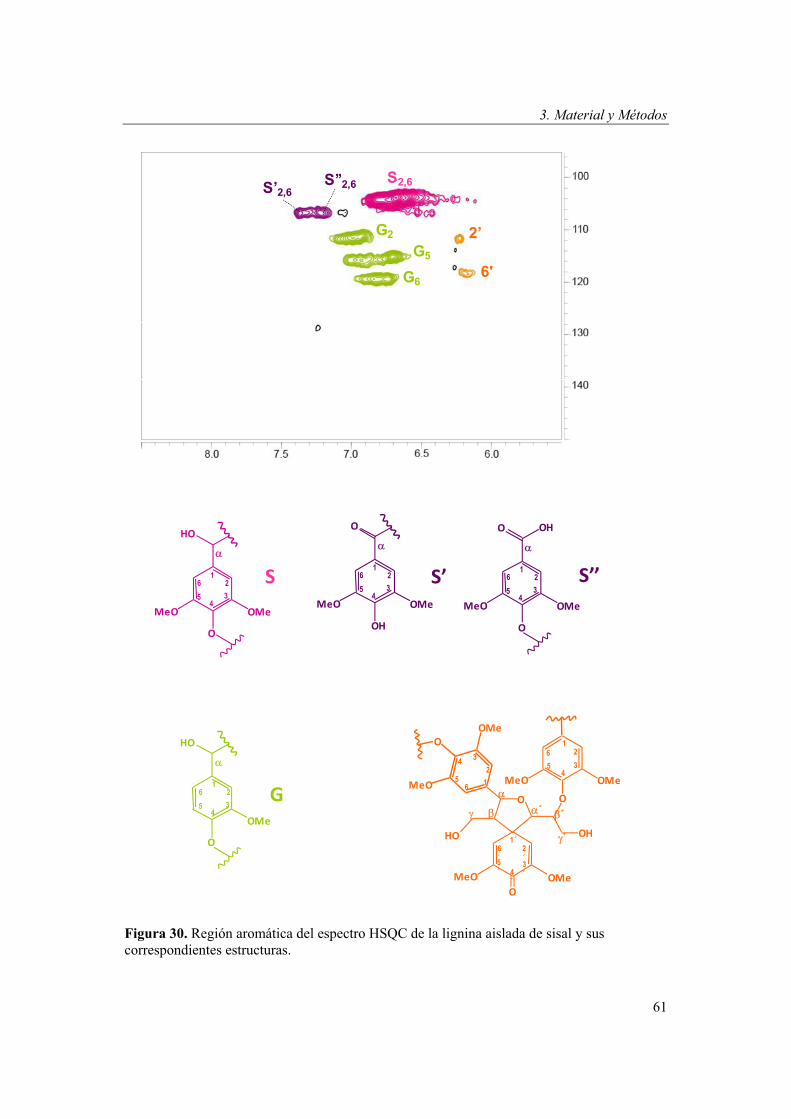
3. Material y Métodos
2’
6'
S2,6S’2,6S’’2,6
G2G5
G6
�
123
456
HO
O
OMeMeO
S
�
123
456
O
OH
OMeMeO
S’
�
123
456
HO
O
OMe
G
S’’�
123
456
O
O
OMeMeO
OH
�
1´2´3´4
´
5´
6´
O
O
OHHO
�� �´ �´
�´
O
123
456
OMeMeO
OMeO
21
4 3
56MeO
OMeMeO
Figura 30. Región aromática del espectro HSQC de la lignina aislada de sisal y sus correspondientes estructuras.
61

3. Material y Métodos
62
Los espectros de NMR de ligninas se registraron en un espectrómetro Bruker AVANCE 500 MHz, equipado con una sonda triple con gradientes en el eje z, a una temperatura de 298ºK. Se disolvieron 40 mg en 0,75 mL de dimetilsulfóxido deuterado (DMSO-d6). Los experimentos HSQC se realizaron empleando una matriz de datos de 256 incrementos en la dimensión indirecta, y adquiriendo 1024 puntos en la dimensión de adquisición. La constante de acoplamiento 1JCH utilizada fue de 140 Hz. La intensidad de las señales en los espectros HSQC dependen del valor de esta constante así como del tiempo de relajación T2(Zhang y Gellerstedt 2007). Por ello, la integración de las señales se realizó por separado en cada una de las regiones del espectro, utilizando las señales correspondientes a correlaciones C-H químicamente análogas, con constantes de acoplamiento 1JCH similares. En la región alifática oxigenada, las abundancias relativas de las diferentes subestructuras de la lignina se estimaron mediante la integración de las correlaciones C�-H�. En la región aromática, las correlaciones 1H-13C de las unidades S y G se usaron para estimar las relaciones S/G.
3.2.3. Aislamiento y análisis de las hemicelulosas de las fibras y pastas
Preparación de la holocelulosa y aislamiento de los xilanos
Las holocelulosas se obtuvieron por deslignificación de las fibras y pastas crudas (5,0 g) con ácido peracético 10% (pH 3,5) durante 20 minutos a 85ºC. Una vez ocurrida la deslignificación, la holocelulosa se filtró, se lavó con acetona y agua destilada caliente y se secó para su determinación gravimétrica.
Los xilanos se aislaron de las correspondientes holocelulosas (molidas previamente) con DMSO a 50ºC durante 24 h y se precipitaron con un exceso de 7:2:1 etanol:metanol:agua acidificado con ácido acético a 4ºC durante 3-4 días. El precipitado se recogió tras centrifugación, se lavó con metanol y se secó sobre vacío. En el caso de las pastas blanqueadas, los xilanos se aislaron directamente de dichas pastas sin previo aislamiento de las holocelulosas correspondientes.
Análisis de azúcares neutros tras hidrólisis ácida
Los xilanos aislados se sometieron a una hidrólisis con H2SO4 72% durante 3 horas a 20ºC, seguido de otra hidrólisis con H2SO4 4% durante 2,5 horas a 100ºC. Los monosacáridos neutros se determinaron como acetatos de alditol por cromatografía de gases (Selvendran et al. 1979).

3. Material y Métodos
El análisis por cromatografía de gases se realizó en un equipo Varian 3350 con un detector FID y columna DB-225 J&W (30 m × 0,25 mm ID y un espesor de película de 0,15 μm). El programa de temperatura se programó de 220ºC (5 min) a 230ºC con un incremento de 2ºC/min. La temperatura final se mantuvo durante 5 min. Las temperaturas del inyector y detector fueron de 230ºC, respectivamente. La cuantificación se realizó con ayuda de curvas de calibración realizadas con patrones.
Análisis de azúcares neutros y ácidos urónicos tras metanolisis ácida
La fibra o pasta seca (4,5 mg) se sometió a metanolisis ácida añadiendo 2 mL de una disolución de HCl 2 M en metanol anhidro a 100ºC durante 4 horas (Sundberg et al. 1996). Una vez frío, se adicionó 80 μL de piridina para neutralizar la disolución ácida y se adicionó 1 mL de una disolución 0,1 mg/mL de sorbitol como patrón interno. Se extrajo 2 mL del sobrenadante, se secó en un rotavapor a 40-50ºC, se resuspendió en 70 μL de piridina y se le añadió 150 μL de hexametildisiloxano y 80 μL de trimetilclorosilano para promover la sililación durante 12 horas a temperatura ambiente.
Los análisis de las muestras sililadas se llevaron a cabo en un cromatógrafo de gases Hewlet-Packard 5890 equipado con un detector de masas MSD series II, usando Helio como gas portador (35 cm/s) y una columna capilar (DB-1 J&W; 30 m x 0,32 mm ID y 0,25 �m de espesor de película). El horno se calentó de 100°C a 175°C a 4ºC/min y de 175ºC a 290ºC a 12ºC/min. La línea de transferencia se mantuvo a 300ºC. La temperatura del detector (FID) fue de 290ºC. La identidad de cada componente se determinó por comparación de sus espectros de masas con los espectros existentes en las librerías (Wiley y NIST) y con espectros publicados anteriormente (Sundberg et al. 1996, Bertaud et al. 2002, Bleton et al. 1996).
Determinación del peso molecular de los xilanos mediante SEC
El peso molecular de los xilanos solubles en Me2SO se valoró mediante cromatografía de exclusión molecular. Los xilanos aislados con Me2SO se disolvieron en una disolución al 10% de LiCl en N,N-dimetilacetamida (DMAC) y luego se diluyeron con DMAC hasta una concentración de aproximadamente 0,5% (5 mg/mL). Los análisis por SEC se realizaron en columnas de PLgel 10μm (Mixed B 300 × 7,5 mm) con una pre-columna PLgel 10μm (Polymer Laboratories, UK) usando un sistema PL-GPC 110 (Polymer Laboratories). Las columnas, el inyector y el detector se mantuvieron a 70ºC durante el análisis. El eluyente fue LiCl en DMAC 0,1 M con un flujo controlado de 0,9 mL/min. Las columnas se calibraron con patrones (Polymer
63

3. Material y Métodos
64
Laboratories) en un rango de 0,8-100 kDa. El volumen de muestra que se inyectó fue de 100 μL.
Análisis de la estructura de los xilanos mediante NMR
Los xilanos aislados se analizaron tanto por 1H-NMR como por 2D-NMR. Los espectros 1H-NMR se obtuvieron en un espectrómetro Bruker AVANCE 300. Se utilizó como patrón interno ( 0,00) 3-(trimetilsilil) propionato de sodio-d4. Los experimentos 1H-NMR se realizaron a 30ºC con un pulso de 90º, un tiempo de relajación de 16 s y adquiriendo 400 puntos.
Los espectros 2D-NMR se obtuvieron en un espectrómetro Bruker AVANCE 300. Los experimentos 2D 1H-1H COSY se realizaron a 50ºC usando una secuencia COSY patrón (pulso de 90º y un tiempo de relajación de 2 s). Los experimentos 2D 1H-1H TOCSY (mix= 0.050 s) se realizaron empleando una anchura espectral de 2185 Hz en ambas dimensiones a 60ºC. El tiempo de relajación fue de 2 s. Se emplearon 128 acumulaciones para cada FID, adquiriendo 1024 puntos en t1 × 512 en t2. Finalmente, los experimentos HSQC se obtuvieron a 50ºC empleando una anchura espectral de 12,000 Hz (F1) y de 2000 Hz (F2), una matriz de datos de 2048 × 1024 incrementos y 128 acumulaciones. El tiempo de relajación fue de 2 s y la constante de acoplamiento 1JCH utilizada fue de 148 Hz.
Determinación del contenido en ácidos hexenurónicos
Durante el proceso kraft, los ácidos 4-O-metil-D-glucurónicos, presentes en la cadena lateral de los xilanos, se convierten en los ácidos insaturados correspondientes (ácidos hexenurónicos, HexA) a través de la pérdida del grupo metoxilo. Los HexA interaccionan con los agentes químicos de blanqueo y otros reactivos disminuyendo la blancura de las pastas. Los HexA interfieren en el método de determinación del índice kappa, ya que reaccionan con el permanganato de potasio, haciendo que se obtenga un valor más elevado del contenido de lignina residual que el real (Göran y Liebing 1996).
El contenido en HexA se determinó a través del método de la hidrólisis ácida, por el que los ácidos hexenurónicos se convierten selectivamente en derivados del furano por hidrólisis en tampón formato de sodio a pH 3,0 (Vuorinen et al. 1999). La determinación de la cantidad de HexA, se basa en la cuantificación de los derivados del furano que se forman a través del análisis del espectro de UV/VIS.
Se dejaron impregnar aproximadamente 0,75 g de pasta en 0,75 mL de tampón formato de sodio 10 mM sobre agitación durante una noche. Esta

3. Material y Métodos
mezcla se transfirió al reactor, se substituyó el aire por nitrógeno, se encendió la agitación mecánica y se llevó la temperatura hasta 110ºC durante 1h. Una vez acabada la reacción se filtró la pasta y se lavó, recogiendo siempre el líquido de filtrado, diluyéndose todo hasta 500 mL. Se leyó entonces el valor de la absorbancia a 245 nm y 480 nm. Se realizó también un ensayo en blanco.
El contenido en HexA existentes en las pastas se determinó mediante la fórmula:
CmAA
�
7,8)( 480245
Siendo,
C = cantidad de HexA en la pasta (meq/Kg)
A245 = Absorbancia a 245 nm
A480 = Absorbancia a 480 nm
M = peso de la pasta seca (Kg)
8,7 mM-1cm-1 = coeficiente de extinción molar a 245 nm con relación a los HexA.
3.2.4. Otros análisis
Determinación de la fracción hidrosoluble de las fibras
El porcentaje de compuestos hidrosolubles en las fibras se determinó según la norma Tappi T 207 om-88 (Tappi 2004). Para ello, los cartuchos de las muestras extraídas con acetona, una vez secos, se colocaron en matraces con 100 mL de agua destilada y se tuvieron en un baño a 100ºC durante 3 h, al cabo de las cuales el extracto se concentró en rotavapor y se secó a 100ºC para su determinación gravimétrica.
Determinación del contenido en cenizas de las fibras
El contenido en cenizas se determinó mediante la norma Tappi 211 om-85 (Tappi 2004). Para ello se depositaron 200 mg de cada una de muestras en crisoles de porcelana previamente tarados y se introdujeron en la mufla a 575 ºC durante 6 h. Para tararlos se limpiaron con HCl y se introdujeron en la mufla a 575ºC durante 1 h. Posteriormente se sacaron los crisoles de la mufla y se
65

3. Material y Métodos
66
pesaron una vez que alcanzaron la temperatura ambiente. Los contenidos en cenizas se expresaron como porcentajes de la materia prima inicial.
Análisis de metales y otros elementos en las fibras
Las fibras seleccionadas, una vez lavadas y secas, se molieron en un molino de cuchillas y se les realizó una digestión con 4 mL de HNO3 concentrado por 0,5 mg de muestra, dejándolas 15 min en un horno microondas (Jones y Case 1990). Posteriormente se filtraron con filtro Whatman del número 2, y se recogieron en un matraz que se enrasó hasta 50 mL. La concentración de metales en la disolución obtenida se determinó por espectrometría de emisión por plasma (ICP-OES) en un espectrómetro Termo Jarrel Ash, modelo IRIS Advantages.
3.2.5. Tratamientos enzimáticos de las pastas
Tratamientos con lipoxigenasas
Los tratamientos de pastas de eucalipto y lino con lipoxigenasa se realizaron con 5 g de pasta seca, al 1% de consistencia (peso/peso) en tampón dihidrógeno fosfato sódico 100 mM (pH 7). La dosis de enzima fue de 10 mg lipoxigenasa/g de pasta de eucalipto y 20 mg de lipoxigenasa/g de pasta de lino, la temperatura de 30ºC, y el tiempo de reacción de 4 horas. Los tratamientos se realizaron en matraces de 1L, con burbujeo de oxígeno en un baño térmico con agitación (170 rpm). En una etapa posterior, las pastas al 5% de consistencia, se sometieron a una etapa de blanqueo con peróxido, usando H2O2 al 3% (peso/peso) y NaOH 1,5% (peso/peso), ambos referidos al peso de la pasta seca, a 90ºC durante 2 horas, en unas bolsitas selladas de plástico termorresistente. Los controles para la evaluación de la acción de la lipoxigenasa se trataron bajo las mismas condiciones pero sin enzima.
Una vez tratadas las pastas, se extrajeron con acetona en un extractor de tipo Soxhlet durante 6 horas. A continuación se evaporó el disolvente a sequedad en un rotavapor y los extractos lipofílicos obtenidos se redisolvieron en CHCl3 para su posterior análisis mediante GC y GC/MS, en las condiciones descritas anteriormente para el análisis de los compuestos lipofílicos. Se realizaron análisis posteriores de las propiedades de las pastas (blancura ISO, índice kappa, viscosidad intrínseca y ácidos hexenurónicos).
También se llevaron a cabo reacciones enzimáticas con compuestos modelo. Para las reacciones con compuestos modelo, se usó 1 mg de cada compuesto y se añadió 0,1 mg de lipoxigenasa, y Tween 20 como dispersante (1% v/v). El

3. Material y Métodos
tratamiento se realizó a pH 7, usando tampón dihidrógeno fosfato sódico 100 mM, durante 2 horas. Se burbujeó oxígeno dentro de los matraces de reacción, y la reacción se llevó a cabo en un baño con agitación a una velocidad de 100 rpm. En los controles de los experimentos, los lípidos se someten a las mismas condiciones de reacción, sin lipoxigenasa. En una etapa posterior se realizó una fase de peróxido, en la que se añadía a cada matraz de reacción 50 �L de H2O2 al 30% (p/v) y 37,5 �L de NaOH 5N, se taparon los matraces y se colocaron en el baño térmico de agitación, a 90ºC y 100 rpm, durante 2 horas. Las dispersiones de los lípidos se secaron en rotavapor, se recogieron con cloroformo-metanol (1:1), que se secó luego con nitrógeno, y se redisolvieron en cloroformo para su análisis mediante GC y GC/MS.
Tratamientos con POM y peroxidasa versátil
Los ensayos de blanqueo de pasta kraft de eucalipto con POM se llevaron a cabo en un reactor PARR modelo 4842 (0,25 L) equipado con un sistema de control de temperatura y mecanismo de agitación (Figura 31).
Figura 31. Sistema usado para los ensayos de deslignificación.
67

3. Material y Métodos
68
Previamente a la realización de los ensayos de deslignificación, se optimizó la cinética de oxidación del POM reducido (SiW11MnII) con peroxidasa versátil (VP) para conocer las cantidades a utilizar de peróxido y enzima en los diferentes ensayos de deslignificación. La reoxidación del POM por la VP fue monitorizada por espectroscopía de UV/VIS. El color amarillo del POMred, SiW11MnII, cambia gradualmente durante la oxidación con la VP a un color rosa característico del POMox, SiW11MnIII, (Figura 32a), mostrando una banda de transición d-d* con un máximo de Absorbancia a 490-495 nm (Figura 32b). Se realizaron varias pruebas en tampón acetato 0,1 M, pH 4,5 con objeto de maximizar la oxidación del POM con VP/H2O2, variando para esto la relación H2O2/POM y POM/VP. Las lecturas se realizaron a un valor de longitud de onda fijo (490 nm) para intervalos de tiempo de 1 minuto, considerando � de (SiW11MnIII)= 327 y de (SiW11MnII)= 19 en cm-1mol-1L. Todas las medidas espectrofotométricas se realizaron en un espectrofotómetro Jasco V-560 UV/Vis a temperatura ambiente.
Una vez conocidas las condiciones óptimas para la reoxidación del POM, se procedió a la realización de los diferentes ensayos de deslignificación. Para cada uno de los ensayos, se dejó la pasta (8 g) en agua destilada (600 mL) durante una noche en agitación constante. Se filtró, se pasó al reactor y se adicionó 67 mL de tampón acetato de sodio 0,2 M pH 4,5 (concentración final de 0,1 M), disolución de POM concentrado (entre 12,5 a 13,5 mL para que la concentración final de POM en la mezcla fuera de aproximadamente 3 mM) y agua destilada hasta 134 mL de volumen total (considerando todavía la contribución del agua retenida en la pasta filtrada), para obtener una consistencia final del 6%. Se presurizó el reactor con pO2=5 bar, se encendió la agitación mecánica y se llevó hasta la temperatura de 110ºC. Una vez acabado el ensayo, se filtró la pasta y se lavó con agua destilada (POM 1-). En los casos en que los líquidos de filtrado conteniendo el POM fueron reoxidados (-VP-), se adicionó a estos la enzima y el peróxido de hidrógeno en las cantidades necesarias (obtenidas a través de los ensayos de optimización descritos anteriormente), manteniendo agitación constante y a temperatura ambiente (para no desnaturalizar la enzima), durante aproximadamente 20 min (aunque a los seis minutos la reoxidación ya se estimaba como completa). El filtrado conteniendo el POM reoxidado se añadió nuevamente a la pasta filtrada reiniciando una nueva etapa de deslignificación (-POM 2-).
Finalmente, todas las pastas deslignificadas se sometieron a una extracción alcalina con NaOH (-E) 2% durante 1 hora a 70 ºC. Después de las extracciones alcalinas, se lavaron las pastas con agua destilada, hasta alcanzar pH neutro en el líquido de filtrado y se dejaron secar a temperatura ambiente durante 4 días. Se realizaron análisis posteriores de las propiedades de las pastas (blancura ISO, índice kappa, viscosidad intrínseca y ácidos hexenurónicos).

3. Material y Métodos
(a) (b) Figura 32. (a) Colores característicos de SiW11MnIII (izquierda) y de SiW11MnII (derecha) y (b) Espectros de UV/VIS de disoluciones acuosas de a: SiW11MnII, b-d: diferentes porcentajes de oxidación hasta e: SiW11MnIII (la oxidación máxima).
Determinación de las propiedades de las pastas
Determinación de la blancura ISO
Las medidas de blancura en porcentaje ISO de las pastas fueron realizadas en ENCE y UPC (Terrassa) en el caso de los tratamientos con lipoxigenasas y en el Instituto Raíz de Aveiro (Portugal) en el caso de las pastas tratadas con POM y peroxidasa versátil.
El método para la determinación de la blancura ISO mide el factor de reflectancia difusa en azul (grado de blancura ISO) de pastas de papel y cartones. El alcance de esta norma está restringido a pastas de papel y cartones blancos o casi blancos.
Determinación del índice kappa
El procedimiento estándar utilizado en la industria para determinar el grado de deslignificación en una pasta química es la determinación del índice kappa por la norma TAPPI T 236cm-85 (Tappi 2006) y consiste en el volumen en mL de una disolución de KMnO4 0,1 N consumido por 1 g de pasta. La lignina de la pasta reacciona con el permanganato y la cuantificación del permanganato consumido se determina por valoración con tiosulfato de sodio.
Con ayuda de una batidora de mano, se desintegraron los gramos de pasta necesarios para el ensayo (ver Anexo 1) en 140 mL de agua destilada y se lavó el pie de la batidora con 50 mL de agua. Con agitación constante, se adicionó
69

3. Material y Métodos
70
una mezcla de 25 mL de KMnO4 0,1 N y 25 mL de H2SO4 0,2 N. Al cabo de 5 minutos se midió la temperatura y después de 10 minutos se paró la reacción con 5 mL de KI 1,0 N, se colocaron unas gotas de solución indicadora de almidón al 0,2% y se valoró el I2 liberado con una disolución de Na2S2O3 0,2 N. Previamente, se realizó siempre un ensayo en blanco que no contenía pasta.
El índice kappa se calculó por la expresión:
� )25(013,01 tw
fpIK ��
��
���
� �
1,0)( Nabp
Con:
IK = índice kappa.
p = Volumen de permanganato de potasio 0,1 N consumido en el ensayo (mL).
f = factor de corrección para un consumo de 50% de permanganato de potasio y que depende de p (Anexo 2).
w = peso de pasta seca (g).
b = volumen consumido de tiosulfato de sodio para determinación del blanco (mL).
a = volumen consumido de tiosulfato de sodio para determinación de la muestra (mL).
N = normalidad de la disolución de tiosulfato de sodio.
t = Temperatura del medio de reacción (ºC).
Determinación de la viscosidad intrínseca
La viscosidad de las pastas está directamente relacionada con el grado de polimerización de las moléculas de celulosa y por lo tanto con la resistencia de

3. Material y Métodos
las fibras. Se determinó la viscosidad intrínseca de las pastas a través de la norma SCAN-CM 15:85 (SCAN 1994). Este método permite determinar la viscosidad de las pastas celulósicas solubles en una disolución de cobre (II)-etilendiamina (CED) en un viscosímetro capilar.
Se pesó la cantidad de pasta necesaria para el ensayo (ver Anexo 3) y se transfirió a frascos de 61 mL, se adicionó 25 mL de agua destilada y 5 hilos de cobre. Se colocaron los frascos a agitar durante aproximadamente 30 minutos en un agitador de brazos. Seguidamente, se adicionó 25 mL de CED 1,0 M para dar lugar a la disolución de los polisacáridos. Se completó el volumen del frasco con disolución de CED 0,5 M usando una bureta, se cerraron los frascos para no dejar burbujas de aire y se colocaron de nuevo sobre agitación durante otros 30 minutos. Finalmente, se anotó el tiempo de escurrido de 1 mL de pasta disuelta en CED, repitiendo la lectura 3 veces para cada frasco, usando un viscosímetro capilar a una temperatura controlada de 25ºC.
La viscosidad relativa de las pastas fue determinada por la expresión:
�rel = h×tn
Con:
h = constante del viscosímetro, obtenida por calibración (0,0928 s-1).
tn = tiempo de escurrido (s).
A partir de la tabla que se encuentra en anexo (Anexo 4), se lee el valor del producto [�]×C, que corresponde al valor de la viscosidad relativa obtenido:
�rel = [�]×C
Con:
[�] = viscosidad intrínseca (mL/g).
C = concentración de la pasta seca en CED (g/mL).
71

3. Material y Métodos
72
Determinación del contenido en ácidos hexenurónicos
La determinación del contenido de ácidos hexenurónicos (HexA) en las pastas tratadas con POM se realizó según el procedimiento explicado anteriormente en el apartado 3.2.3.

3. Material y Métodos
73

4

4. Referencias
REFERENCIAS
Abarca, R. y Blanco, M. L. (2008) Soda- Antraquinone Pulp of Tectona grandis in Costa Rica. V Congreso Iberoamericano de investigación en celulosa y papel, CIADICYP, Octubre 2008, Guadalajara, Jalisco, México.
Adler, E. (1977) Lignin chemistry - Past, present and future. Wood Science and Technology, 11, 169-218.
Aitken, I., Cadel, F. y Voillot, C. (1988) Constituants fibreux des pates papiers et cartons pratique de l'analyse, 1st edition.
Ali, M. y Sreekrishnan, T. R. (2001) Aquatic toxicity from pulp and paper mill effluents: A review. Advances in Environmental Research, 5, 175-196.
Allen, L. H. (2000) Pitch control in paper mills. En: Pitch control, wood resin and deresination (Eds.: Back, E.L. y Allen, L.H.), TAPPI Press, Atlanta, pp. 307-328.
Annergren G. E. (1996) Pulp bleaching. Principles and practice. Capítulo VII-3: Strength properties and characteristics of bleached chemical and (chemi)mechanical pulps. Ed. C. W. Dence y D. W. Reeve. Tappi press, pp: 717-748
Back, E. L. y Allen, L. H. (2000) Pitch control, wood resin and deresination. TAPPI Press, Atlanta.
Bajpai, P., Anand, A., Sharma, N., Mishra, S. P., Pramod K. Bajpai P. K. y Lachenal, D. (2006) Enzymes improve ECF Bleaching of pulp. BioResources, 1(1), 34-44.
Bajpai, P. (1999) Application of enzymes in the pulp and paper industry. Biotechnology Progress, 15, 147-157. ISSN 8756-7938
Bajpai, P. (2004) Biological bleaching of chemical pulps. Critical Reviews in Biotechnology, 24, 1-58.
Bidlack, J., Malonge, M. y Benson, R. (1992) Molecular structure and component integration of secondary cell walls in plants. Proceedings of the Oklahoma. Academy of Science, 72, 51-56.
Björkman, A. (1956) Studies on finely divided wood. Part I. Extraction of lignin with neutral solvents. Svensk Papperstindning, 13, 477-485.
Bertaud, F., Sundberg, A. y Holmbom, B. (2002). Evaluation of acid methanolysis for analysis of wood hemicelluloses and pectins. Carbohydrate Polymers, 48, 319-324.
75

4. Referencias
Bland, D. E. (1966) Colorimetric and chemical identification of lignins in different parts of Eucalyptus botryoides and their relation to lignification. Holzforschung, 20, 12-16.
Bleton, J., Mejanelle, P., Sansoulet, J., Goursaud, S. y Tchapla, A. (1996) Characterization of neutral sugars and uronic acids after methanolysis and trimethylsilylation for recognition of plant gums. Journal of Chromatography A, 720, 27-49.
Boerjan, W., Ralph, J. y Baucher, M. (2003) Lignin biosynthesis. Annual Review of Plant Biology, 54, 519-546.
Bourbonnais, R. y Paice, M. G. (1990) Oxidation of non-phenolic substrates. An expanded role for laccase in lignin biodegradation. FEBS Letters, 267, 99-102.
Brett, C. y Waldron, K. (1996) Physiology and Biochemistry of Plant Cell Walls.
Brooks, R. T., Edwards, L. L., Nepote, J. C. y Caldwell, M. R. (1994) Bleach plant closeup and conversion to TCF: A case study using mill data and computer simulation. Tappi Journal, 77, 83-92.
Brunow, G. (2001) Methods to reveal the structure of lignin. Biopolymers.Vol.1 - Lignin, Humic substances and Coal, 1, 89-116.
Bryce J.R. (1990) Producción de pulpa al sulfito. En Pulpa y Papel: Química y Tecnología Química. Vol. 1. Casey, J.P. Ed Limusa, México, 29-64.
Call, H. P (1994) Verfahren zur Veränderung, Abbau oder Bleichen von Lignin, ligninhaltigen Materialien oder ähnlichen Stoffen. Patent (International) WO 94/29510.
Call, H. P. y Mücke, I. (1997) History, overview and applications of mediated lignolytic systems, especially laccase-mediator-systems (Lignozym(R)-process). Journal of Biotechnology, 53, 163-202.
Camarero, S., Sarkar, S., Ruiz-Dueñas, F. J., Martínez, M. J. y Martínez, A. T. (1999) Description of a versatile peroxidase envolved in natural degradation of lignin that has both Mn-peroxidase and lignin-peroxidase substrate binding sites. The Journal of Biological Chemistry, 274, 10324-10330
Capanema, E. A., Balakshin, M. Y., Chen, C.-L., Gratzl, J. S. y Gracz, H. (2001) Structural analysis of residual and technical lignins by 1H-13C correlation 2D NMR-spectroscopy. Holzforschung, 55, 302-308.
Christierini, M., Ohlsson, A. B., Berglund, T. y Henriksson, G. (2005) Lignin isolated from primary walls of hybrid aspen cell cultures indicates significant differences in lignin structure between primary and secondary cell wall. PlantPhysiology and Biochemistry, 43, 777-785.
76

4. Referencias
del Río, J. C., Gutiérrez, A. y González-Vila, F. J. (1999) Analysis of impurities occurring in a totally-chlorine free-bleached Kraft pulp. Journal of Chromatography A, 830, 227-232.
del Río, J. C, Gutiérrez, A., González-Vila, F. J., Martín, F. y Romero, J. (1998) Characterization of organic deposits produced in the Kraft pulping of Eucalyptusglobulus wood. Journal of Chromatography A, 823,457-465
del Río, J. C., Gutiérrez, A. y Martínez, A. T. (2004) Identifying acetylated lignin units in non-wood fibers using pyrolysis-gas chromatography/mass spectrometry. Rapid Communications in Mass Spectrometry, 18, 1181-1185.
del Río, J. C., Marques, G., Rencoret, J., Martínez, A. T. y Gutiérrez, A. (2007) Occurrence of naturally acetylated lignin units. Journal of Agriculture and Food Chemistry, 55, 5461-5468.
del Río, J. C., Rencoret, J., Marques, G., Gutiérrez, A., Ibarra, D., Santos, J. I., Jiménez-Barbero, J., Zhang, L. y Martínez, A.T. (2008a) Highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. Journal of Agriculture and Food Chemistry, 56, 9525-9534.
del Río, J. C., Rencoret, J., Marques, G., Gutiérrez, A., Ibarra, D., Santos, J. I., Jiménez-Barbero, J. y Martínez, A.T. (2008b) Occurrence and structural characteristics of highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. 10th European Workshop on lignocelluloses and Pulp. Stockholm Sweden, 26-29 August.
del Río J. C., Romero J. y Gutiérrez A. (2000) Analysis of pitch deposits produced in Kraft pulp mills using a totally chlorine free bleaching sequence. Journal of Chromatography A, 874, 235-245
Díaz, M. J., Alfaro, A., García, M. M., Eugenio, F. M. E., Ariza, J. y López, F. (2004) Ethanol pulping from tagasaste (Chamaecytisus proliferus L.F. ssp. Palmensis). A new promising source for cellulose pulp. Industrial & Engineering Chemistry Research, 43, 1875-1881.
Evtuguin, D. V., Neto, C. P., Silva, A. M. S., Domingues, P. M., Amado, F. M. L., Robert, D. y Faix, O. (2001) Comprehensive study on the chemical structure of dioxane lignin from plantation Eucalyptus globulus wood. Journal of Agricultural and Food Chemistry, 49, 4252-4261.
Faix, O., Meier, D. y Fortmann, I. (1990) Thermal degradation products of wood. A collection of electron-impact (EI) mass spectra of monomeric lignin derived products. Holz Roh-Werkstoff, 48, 351-354.
FAO (2004) Base de datos de la FAO "FAOSTAT".
77

4. Referencias
Fengel, D. (1989) Chemistry and morphology of quebracho colorado wood. International Symposium on Wood and Pulping Chemistry, Raleigh, 1-3.
Fengel, D. y Wegener, G. (1984) Wood: Chemistry, ultrastructure, reactions. De Gruyter, Berlin.
Fergus, B. J. y Goring, D. A. I. (1970) The location of guaiacyl and syringyl lignins in birch xylem tissue. Holzforschung, 24, 113-117.
Freudenberg, K. y Lehmann, B. (1963) Radioactive isotopes and lignin. X. Investigation of a lignin preparation labeled with 14C. Chemische Berichte, 96, 1850-1854.
Freudenberg, K. y Neish, A. C. (1968) Constitution and biosynthesis of lignin. Springer-Verlag, New York.
Fuji, K., Ichikawa, K., Node, M. y Fujita, E. (1979) Hard acid and soft nucleophile system. New efficient method for removal of benzyl protecting group. Journal of Organic Chemistry, 44, 1661-1664.
Fujita, Y., Awaji, H., Matsukura, M. y Hata, K. (1991) Enzymic pitch control in papermaking process. Kami Pa Gikyoshi, 45, 905-921
Fujita, Y., Awaji, H., Taneda, H., Matsukura, M., Hata, K., Shimoto, H., Sharyo, M., Sakaguchi, H. y Gibson, K. (1992) Recent advances in enzymic pitch control. Tappi Journal, 75 (4), 117-122
Fukushima, K. y Terashima, N. (1991) Heterogeneity in formation of lignin. XIV. Formation and structure of lignin in differentiating xylem of Ginkgo biloba. Holzforschung, 45, 87-94.
Fullerton, T. J. y Franich, R. A. (1983) Lignin analysis by pyrolysis-GC-MS. Holzforschung, 37, 267-269.
Gamelas J. A. F., Cavaleiro A., Santos I., Balula M. 2003. Os polioxometalatos. Do anião de Keggin às nanocápsulas. Boletim da Sociedade Portuguesa de Química. 90, 45-51.
Gamelas, J. A. F., Evtuguin, D. V. y Gaspar, A. R. (2008) Transition metal complexes in the delignification catalysis. In: Varga, B., Kis, L. (Eds.), Transition metal chemistry: New research. Nova Science Publishers, Inc., New York, pp. 15-57.
Gamelas J. A. F., Pontes A. S. N., Evtuguin D. V., Xavier A. M. R. B. y Esculcas A. P. (2007) New polyoxometalete-laccase integrated sistem for kraft pulp delignification. Biochemical Engineering Journal, 33, 141-147.
78

4. Referencias
Gaspar, A. R., Gamelas, J. A. F., Evtuguin, D. V. y Neto, C. P. (2007) Alternatives for lignocellulosic pulp delignification using polyoxometalates and oxygen: a review. Green Chemistry, 9, 717-730.
García Hortal. (2007) Fibras Papeleras. Ediciones UPC (Universitat Politècnica de Catalunya), Terrassa (Spain).
García Hortal, J. A. y Colom, J. F. (1992) El proceso al sulfato. Vol. I. Universitat Politècnica de Catalunya, Terrassa (Spain).
García, M. M., López, F., Alfaro, A., Ariza, J., Tapias, R. (2008) The use of tagasaste (Chamaecytisus proliferus) from different origins for biomass and paper production. Bioresource technology, 99(9), 3451-7.
Gellerstedt, G. y Li, J. (1996) An HPLC method for the quantitative determination of hexenuronic acid groups in chemical pulps. Carbohydrate Research, 294, 41-51.
Gellerstedt, G. Pranda J. y Lindfors E-L. (1994) Structural and molecular properties of residual birch kraft lignins. Journal of Wood Chemistry and Technology, 14(4), 467-482.
Gilarranz, M. A., Oliet, M., Rodríguez, F. and Tijero, J. (1999) Methanol-based pulping of Eucalyptus globulus. Canadian Journal of Chemical Engineering, 77, 515-521.
Glenn, J. K., Morgan, M. A., Mayfield, M. B., Kuwahara, M. y Gold, M. H. (1983) An extracellular H2O2-requiring enzyme preparation involved in lignin biodegradation by the write rot basidiomycete Phanerochaete chrysosporium. Biochemical and Biophysical Research Communications, 114, 1077-1083.
Grabber, J. H., Quideau, S. y Ralph, J. (1996) p-Coumaroylated syringyl units in maize lignin: Implications for �-ether cleavage by thioacidolysis. Phytochemistry, 43, 1189-1194.
Gutiérrez, A. y del Río, J. C. (2001) Gas chromatography-mass spectrometry demonstration of steryl glycosides in eucalypt wood, kraft pulp and process liquids. Rapid Communications in Mass Spectrometry, 15, 2515-2520.
Gutiérrez, A. y del Río, J. C. (2005) Pitch episodes produced during the manufacturing of high-quality paper pulps from hemp fibers. BioresourseTechnology, 96, 1445-1450.
Gutiérrez, A., del Río, J. C., González-Vila, F. J. y Martín, F. (1998) Analysis of lipophilic extractives from wood and pitch deposits by solid-phase extraction and gas chromatography. Journal of Chromatography A, 823, 449-455.
79

4. Referencias
Gutiérrez, A., del Río, J. C., González-Vila, F. J. y Martín, F. (1999) Chemical composition of lipophilic extractives from Eucalyptus globulus Labill. wood. Holzforschung, 53, 481-486.
Gutiérrez, A., del Río, J. C., Ibarra, D., Rencoret, J., Romero, J., Speranza, M., Camarero, S., Martínez, M. J. y Martínez, A. T. (2006a) Enzymatic removal of free and conjugated sterols forming pitch deposits in environmentally sound bleaching of eucalypt paper pulp. Environmental Science and Technology, 40, 3416-3422.
Gutiérrez, A., del Río, J. C. y Martínez, A. T. (2004) Chemical analysis and biological removal of wood lipids forming pitch deposits in paper pulp manufacturing. In Spencer, J. F. T. (ed), Protocols in Environmental Microbiology. Humana Press, Totowa, USA.
Gutiérrez, A., del Río, J. C. y Martínez, A. T. (2009) Microbial and enzymatic control of pitch in the pulp and paper industry. Applied Microbiology and Biotechnology, 82, 1005-1018.
Gutiérrez, A., del Río, J. C., Martínez, M. J. y Martínez, A. T. (2001) The biotechnological control of pitch in paper pulp manufacturing. Trends in Biotechnology, 19, 340-348.
Gutiérrez, A., del Río, J. C., Rencoret, J., Ibarra, D. y Martínez, A. T. (2006b) Main lipophilic extractives in different paper pulp types can be removed using the laccase-mediator system. Applied Microbiology and Biotechnology, 72, 845-851.
Gutiérrez, A., del Río, J. C., Rencoret, J., Ibarra, D., Speranza, A. M., Camarero, S., Martínez, M. J. y Martínez, A. T. (2006c) Sistema enzima-mediador para el control de los depósitos de pitch en la fabricación de pasta y papel. Patent (International) ES2282020B1.
Gutiérrez, A., Rencoret, J., Ibarra, D., Molina, S., Camarero, S., Romero, J., del Río, J. C. y Martínez, A. T. (2007) Removal of lipophilic extractives from paper pulp by laccase and lignin-derived phenols as natural mediators. EnvironmentalScience & Technology, 41, 4124-4129.
Hamberg, M., Su, C. y Oliw, E. (1998) Manganese lipoxygenase - Discovery of a bis-allylic hydroperoxide as product and intermediate in a lipoxygenase reaction. The Journal of Biological Chemistry, 273, 13080-13088.
Han, J. S. (1998) Properties of Nonwood Fibers. Proceedings of the Korean Society of Wood Science and Technology Annual Meeting.
80

4. Referencias
Hardell, H. L., Leary, G. J., Stoll, M. y Westermark, U. (1980a) Variations in lignin structure in defined morphological parts of birch. Svensk Papperstidn, 83, 71-74.
Hardell, H. L., Leary, G. J., Stoll, M. y Westermark, U. (1980b) Variations in lignin structure in defined morphological parts of spruce. Svensk Papperstidn, 83, 44-49.
Hata, K., Matsukura, M., Taneda, H. y Fujita, Y. (1996) Mill-scale application of enzymatic pitch control during paper production. In: Viikari, L. and Jeffries, T. W. (eds) Enzymes for pulp and paper processing. ACS, Washington, pp 280-296.
Higuchi, T. (1997) Biochemistry and molecular biology of wood. Springer Verlag, London.
Hillis, W. E. (1962) Wood extractives. Academic Press, London.
Hillis, W. E. y Sumimoto, M. (1989) Effect of extractives on pulping. In Rowe, J. W. (ed), Natural products of woody plants. II. Springer-Verlag, Berlin, pp. 880-920.
Hoareau, W., Trindade, W. G., Siegmund B., Castellan, A. y Frollini E. (2004) Sugar cane bagasse and curaua lignins oxidized by chlorine dioxide and reacted with furfuryl alcohol: characterization and stability. Polymer Degradation and Stability, 86, 567-576.
Irie, Y. (1990) Enzymatic pitch control in papermaking system. Abs 1990 Papermakers Conference, 23-25 April, Atlanta.
Jiménez, L., Pérez, A., Rodríguez, A. y de La Torre, M.J. (2006) New raw materials and pulping processes for production of pulp and paper. Afinidad 63 (525), 362-369.
Jiménez, L., Pérez, A., de la Torre, M.J., Moral A. y Serrano, L. (2007) Characterization of vine shoots, cotton stalks, Leucaena leucocephala and Chamaecytisus proliferus, and of their ethyleneglycol pulps. BioresourceTechnology, 98, 3487-3490.
Jones, J. B. y Case, V. W. (1990) Sampling, handling, and analyzing plant tissue samples. Soil Testing and Plant Analysis, 389-427.
Karhunen, P., Rummakko, P., Sipila, J., Brunow, G. y Kilpeläinen, I. (1995) Dibenzodioxocins - A novel type of linkage in softwood lignins. TetrahedronLetters, 36, 169-170.
81

4. Referencias
Kawai, S., Umezawa, T. y Higuchi, T. (1987a) p-Benzoquinone monoketals, novel degradation products of �-O-4 lignin model compounds by Coriolus versicolor and lignin peroxidase of Phanerochaete chrysosporium. FEBS Letters, 210, 61-65.
Kawai, S., Umezawa, T., Shimada, M., Higuchi, T., Koide, K., Nishida, T., Morohoshi, N. y Haraguchi, T. (1987b) C�-C� cleavage of phenolic �-1 lignin substructure model compound by laccase of Coriolus versicolor. MokuzaiGakkaishi, 33, 792-797.
Kuwahara, M., Glenn, J. K., Morgan, M. A. Y Gold, M. H. (1984) Separation and characterization of two cellular H2O2-dependent oxidases from ligninolytic cultures of Phanerochaete chrysosporium. FEBS Letters, 1969, 247-250.
Lapierre, C., Monties, B. y Rolando, C. (1985) Thioacidolysis of lignin: Comparison with acidolysis. Journal of Wood Chemistry and Technology, 5, 277-292.
Lapierre, C., Monties, B. y Rolando, C. (1986) Preparative thioacidolysis of spruce lignin: Isolation and identification of main monomeric products. Holzforschung, 40, 47-50.
Lapierre, C., Pollet, B. y Monties, B. (1991) Thioacidolysis of spruce lignin: GC-MS analysis of the main dimers recovered after Raney nickel desulphuration. Holzforschung, 45, 61-68.
Leao, A. L., Rowell, R. y Tavares, N. (1998) Applications of natural fibres in automotive industry in Brazil-Thermoforming process. In: Science and Technology of Polymers Advanced Materials. Emerging Technologies and Business Opportunities; Prasad, P. N., Mark, J. E., Kandil, S. H., Kafafi, Z. H., Eds., Plenum Press: New York, 755-761.
Lennholm, H. y Henriksson, G. (2007) Chapter 4. Cellulose. In Fibre and Polymer Technology, K. I. I. (ed), Ljungberg Textbook Pulp and Paper Chemistry and Technology Book 1. Wood Chemistry and Technology.
Liitiä, T. M., Maunu, S. L., Hortling, B., Toikka, M. y Kilpeläinen, I. (2003) Analysis of technical lignins by two- and three-dimensional NMR spectroscopy. Journal of Agricultural and Food Chemistry, 51, 2136-2143.
Lin, S. Y. y Dence, C. W. (1992) Methods in lignin chemistry. Springer-Verlag, Berlin.
López, F., Alfaro, A., García, M.M., Diaz, M.J., Calero, A.M. y Ariza, J. (2004) Pulp and paper from tagasaste (Chamaecytisus proliferus ssp palmensis). Chemical Engineering Research and Design, 82, 1029-1036.
82

4. Referencias
Lu, F. y Ralph, J. (1997a) Derivatization followed by reductive cleavage (DFRC method), a new method for lignin analysis: protocol for analysis of DFRC monomers. Journal of Agricultural and Food Chemistry, 45, 2590-2592.
Lu, F. y Ralph, J. (1997b) The DFRC method for lignin analysis. Part 1. A new method for ��aryl ether cleavage: lignin model studies. Journal of Agricultural and Food Chemistry, 45, 4655-4660.
Lu, F. y Ralph, J. (1998) The DFRC method for lignin analysis. 2. Monomers from isolated lignin. Journal of Agricultural and Food Chemistry, 46, 547-552.
Martínez A. T., Rencoret J., Marques G., Gutiérrez A., Ibarra D., Jiménez-Barbero J., and del Río J. C. (2008) Monolignol acylation and lignin structure in some nonwoody plants: A 2D-NMR study. Phytochemistry, 69, 2831-2843.
Martínez, M. J., Ruiz-Dueñas, F. J., Guillén, F. y Martínez, A. T. (1996) Purification and catalytic properties of two manganese-peroxidase isoenzymes from Pleurotus eryngii. European Journal of Biochemistry, 237, 424-432.
Matsukura, M., Fujita, Y. y Sakaguchi, H. (1990) On the use of Resinase™ for pitch control. Novo Publication. A, 6122, 1-7.
Mayer, A. M. y Staples, R. C. (2002) Laccase: new functions for an old enzyme. Phytochemistry, 60, 551-565.
McDougall, G. J., Morrison, I. M., Stewart, D., Weyers, J. D. B. y Hillman, J. R. (1993) Plant fibres: Botany, chemistry and processing for industrial use. Journal of the Science of Food and Agriculture, 62, 1-20.
Meier, D. y Faix, O. (1992) Pyrolysis-gas chromatography-mass spectrometry. In Lin, S. Y. and Dence, C. W. (eds), Methods in lignin chemistry. Springer-Verlag, Berlin, pp. 177-199.
Molina, S., Rencoret, J., del Río, J. C., Lomascolo, A., Record, E., Martínez, A. T. y Gutiérrez, A. (2008) Oxidative degradation of model lipids representative for main paper pulp lipophilic extractives by the laccase-mediator system. AppliedMicrobiology and Biotechnology, 80, 211-222.
Morrison, W. H. III., Akin, D. E., Himmelsbach, D. S. y Gamble, G. R. 1999. Chemical, microscopic, and instrumental analysis of graded flax fibre and yarn. Journal of the Science of Food and Agriculture, 79, 3-10.
Neto, C. P., Seca, A. M. L., Fradinho, D., Coimbra, M. A., Domingues, F. M. J., Evtuguin, D., Silvestre, A. J. D. y Cavaleiro, J. A. S. (1996) Chemical composition and structural features of the macromolecular components of Hibiscus cannabinus grown in Portugal. Industrial Crops and Products, 5, 189-196.
83

4. Referencias
Node, M., Hori, H. y Fujita, E. (1976) Demethylation of aliphatic methyl ethers with a thiol and boron trifluoride. Journal of the Chemical Society, Perkin Transactions, 1, 2237-2240.
Norma Finlandesa TYÖOHJE C/6 15/07/98.
Paice, M. G., Bourbonnais, R., Reid, I. D., Archibald, F. S. y Jurasek, L. (1995) Oxidative bleaching enzymes: A review. Journal of Pulp and Paper Science, 21, J280-J284.
Pande, H. y Roy, D. N (1998) Influence of fibre morphology and chemical composition on the papermaking potential of kenaf fibres. Pulp & Paper Canada, 99(11), 31-34.
Parhan A., Gray R. L. (1990) The practical identification of wood pulp fibers. Tappi Press. USA.
Pérez-Boada, M., Ruiz-Dueñas, F. J., Pogni, R., Basosi, R., Choinowski, T., Martínez, M. J., Piontek, K. y Martínez, A. T. (2005) Versatile peroxidise oxidation of high redox potential aromatic compounds: site-directed mutagenesis, spectroscopic and crystallographic investigations of three long-range electrón transfer pathways. Journal of Molecular Biology, 354, 385-402.
Pope M. T. Springer Verlag. (1983) Heteropoly and Isopoly Oxometalates. Inorganic Chemistry Concepts, V 8. Berlin.
Ralph, J. y Hatfield, R. D. (1991) Pyrolysis-GC-MS characterization of forage materials. Journal of Agricultural and Food Chemistry, 39, 1426-1437.
Ralph, J., Hatfield, R. D., Piquemal, J., Yahiaoui, N., Pean, M., Lapierre, C. y Boudet, A. M. (1998) NMR characterization of altered lignins extracted from tobacco plants down-regulated for lignification enzymes cinnamyl-alcohol dehydrogenase and cinnamoyl-CoA reductase. Proceedings of the National Academy of Sciences, 95, 12803-12808.
Ralph, J., Lapierre, C., Lu, F. C., Marita, J. M., Pilate, G., van Doorsselaere, J., Boerjan, W. y Jouanin, L. (2001) NMR evidence for benzodioxane structures resulting from incorporation of 5-hydroxyconiferyl alcohol into lignins of O-methyltransferase-deficient poplars. Journal of Agricultural and Food Chemistry, 49, 86-91.
Ralph, J. y Lu, F. (1998) The DFRC method for lignin analysis. 6. A simple modification for identifying natural acetates in lignin. Journal of Agricultural and Food Chemistry, 46, 4616-4619.
84

4. Referencias
Ralph, J., MacKay, J. J., Hatfield, R. D., Omalley, D. M., Whetten, R. W. y Sederoff, R. R. (1997) Abnormal lignin in a loblolly pine mutant. Science, 277, 235-239.
Rigol, A., Latorre, A., Lacorte, S. y Barceló, D. (2004) Bioluminescence inhibition assays for toxicity screening of wood extractives and biocides in paper mill process waters. Environmental Toxicology and Chemistry, 23, 339-347.
Robert, D. (1992) Carbon-13 nuclear magnetic resonance. In Lin, S. Y. and Dence, C. W. (eds), Methods in lignin chemistry. Springer-Verlag, Berlin, pp. 250-273.
Rolando, C., Monties, B. y Lapierre, C. (1992) Thioacidolysis. In Lin, S. Y. and Dence, C. W. (eds), Methods in lignin chemistry. Springer-Verlag, Berlin, pp. 334-349.
Rowe, J. W. (1989) Natural products of woody plants. I and II. Chemicals extraneous to the lignocellulosic cell wall. Springer-Verlag, Berlin.
Ruiz-Dueñas, F. J. y Martínez, A. T. (2010) Structural and functional features of peroxidases with a potential as industrial biocatalysts. In: Torres, E. y Ayala, M. (eds.). Biocatalysis Based on Heme Peroxidases (ISBN: 978-3-642-12626-0). Springer.
Ruiz-Dueñas, F. J., Martínez, M. J. y Martínez, A. T. (1999a) Heterologous expression of Pleurotus eryngii peroxidase confirms its ability to oxidize Mn2+
and different aromatic substrates. Applied and Environmental Microbiology, 65, 4705-4707.
Ruiz-Dueñas, F. J., Martínez, M. J. y Martínez, A. T. (1999b) Molecular characterization of a novel peroxidase isolated from the ligninolytic fungus Pleurotus eryngii. Molecular Microbiology, 31, 223-236.
Santos, A., Rodríguez, F., Gilarranz, M. A., Moreno, D. y García-Ochoa, F. (1997) Kinetic modeling of kraft delignification of Eucalyptus globulus. Industrial & Engineering Chemistry Research, 36, 4114-4125.
Saito, K. y Fukushima, K. (2005) Distribution of lignin interunit bonds in the differentiating xylem of compression and normal woods of Pinus thunbergii. Journal of Wood Science, 51, 246-251.
Sarkanen, K. V. y Hergert, H. L. (1971) Classification and distribution. In Sarkanen, K. V. and Ludwig, C. H. (eds), Lignins - Occurrence, Formation, Structure and Reactions. Wiley-Interscience, New York, pp. 43-94.
Scandinavian Pulp Paper and Board Committee, 1994. SCAN Test Methods. Sweden.
85

4. Referencias
Seca, A. M., Cavaleiro, J. A. S., Domingues, F. M. J., Silvestre, A. J. D, Evtuguin, D. y Neto, C. P. (2000) Structural characterization of the lignin from the nodes and Internodes of Arundo donax Reed. Journal of Agricultural and Food Chemistry, 48, 817-824.
Sederoff, R. R., MacKay, J. J., Ralph, J. y Hatfield, R. D. (1999) Unexpected variation in lignin. Current Opinion in Plant Biology, 2, 145-152.
Selvendran, R. R., March, J. F. y Ring, S. G., (1979). Determination of aldoses and uronic acids content of vegetable fiber. Analytical Biochemistry, 96, 282-292.
Shatalov, A. A. y Pereira, H. (2002) Influence of stem morphology on pulp and paper properties of Arundo donax L. reed. Industrial Crops and Products, 15, 77-83.
Shimizu, K. (2001) Wood and Cellulosic Chemistry, 2nd edition, Eds. D.N.-S. Hon, N. Shiraishi, Marcel Dekker Inc., New York, USA, 177-214.
Sigoillot, C., Camarero, S., Vidal, T., Record, E., Asther, M., Pérez-Boada, M., Martínez, M.J., Sigoillot, J.C., Asther, M., Colom, J. y Martínez, A.T. (2005) Comparison of different fungal enzymes for bleaching high-quality paper pulps. Journal of Biotechnology, 115, 333–34.
Silva, G. S., Assis, M. B. y Barbosa, W. L. R. (2001) Investigaçao fitoquímica e microbiologica da espécie Ananas erectifolius (curauá). ReV. Virtual Iniciaçao Acad. UFPA, 1.
Sjöström, E. y Westermark, U. (1999) Chemical Composition of Wood and Pulps: Basic constituents and Their Distribution. In: Analytical Methods in Wood Chemistry, Pulping and Papermaking. E. Sjöström and R. Alén (Eds.), Springer-Verlag, Berlin, Germany, Chap. 1, pp. 1-19.
Sjöström, E. (1981) Wood Chemistry: Fundamentals and Applications. AcademicPress, Orlando, pp. 68–82.
Sjöström, E. (1993) Wood chemistry. Fundamentals and Applications. Academic Press, San Diego.
Streitwieser, A. Jr. y Heathcock, C. H. (1983). Química Orgánica. 3ª edición. Importecnica, S.A. 726-730.
Struik, P. C., Amaducci, S., Bullard, M. J., Stutterheim, N. C., Venturi G. y Cromack, H. T. H. (2000) Agronomy of fibre hemp (Cannabis sativa L.) in Europe. Industrial Crops and Products, 11, 107-118.
Su, C. y Oliw, E. (1998) Manganese lipoxygenase - Purification and characterization. The Journal of Biological Chemistry, 273, 13072-13079.
86

4. Referencias
Sundberg, A., Sundberg, K., Lillandt, C. y Holmbom, B. (1996). Determination of hemicelluloses and pectins in wood and pulp fibers by acid methanolysis and gas chromatography. Nordic Pulp and Paper Research Journal, 4, 216-226.
Tappi (2004) 2004-2005 TAPPI Test Methods. TAPPI Press, Norcoss, GA 30092, USA.
Tappi (2006) 2006-2007 TAPPI Test Methods. TAPPI Press, Norcoss, GA 30092, USA.
Tavares, A. P. M., Gamelas, J. A. F., Gaspar, A. R., Evtuguin, D. V. y Xavier, A. M. R. B. (2004) A novel approach for the oxidative catalysis employing polyoxometalate-laccase system: application to the oxygen bleaching of kraft pulp. Catalysis Communications, 5, 485-489.
Tien, M. y Kirk, T. K. (1983) Lignin-degrading enzyme from the hymenomycete Phanerochaete chrysosporium Burds. Science, 221, 661-663.
Viikari, L., Kantelinen, A., Sundquist, J. y Linko, M. (1994) Xylanases in bleaching - From an idea to the industry. FEMS Microbiology Reviews, 13, 335-350.
Vuorinen, T., Fagerström, P., Buchert, J., Tenkanen, M., y Teleman, A. (1999). Selective hydrolysis of hexenuronic acid groups and its application in ECF and TCF bleaching in kraft pulps. Journal of Pulp and Paper Science, 25(5), 155-162.
Watanabe, T. (2003) Analysis of native bonds between lignin and carbohydrate by specific chemical reactions. In Springer-Verlag, B. H. (ed), Timell, T.E. (Ed.). Springer series in wood science, Association between lignin and carbohydrates in wood and other plant tissues. pp. 91-130.
Weinstock, I. A., Atalla, R.H., Reiner, R. S., Moen, M.A. Hammel, K. E., Houtman, C. J., Hill, C. L. y Harrup, M. K. (1997) A new environmentally bening technology for transforming wood pulp into paper. Engineering polyoxometalates as catalysts for multiple processes. Journal of Molecular Catalysis A: Chemical, 116, 59-84.
Zhang, L. y Gellerstedt, G. (2001) NMR observation of a new lignin structure, a spiro-dienone. Chemical Communications, 2744-2745.
Zhang, L. M. y Gellerstedt, G. (2007) Quantitative 2D HSQC NMR determination of polymer structures by selecting suitable internal standard references. Magnetic Resonance in Chemistry, 45, 37-45.
87

4. Referencias
Zhang, L. M., Gellerstedt, G., Ralph, J. y Lu, F. C. (2006) NMR studies on the occurrence of spirodienone structures in lignins. Journal of Wood Chemistry and Technology, 26, 65-79.
Zhang, X., Nguyen, D., Paice, M. G., Tsang, A. y Renaud, S. (2007) Degradation of wood extractives in thermo-mechanical pulp by soybean lipoxygenase. Enzyme and Microbial Technology, 40, 866-873.
88

4. Referencias
89

5

5. Resultados y discusión
RESULTADOS Y DISCUSIÓN
Los resultados obtenidos durante la Tesis y la discusión de los mismos se muestran a continuación en forma de publicaciones. Durante la realización de esta Tesis se han publicado los resultados principales de los estudios realizados sobre: i) la composición química de la lignina, lípidos y hemicelulosas de diversos cultivos lignocelulósicos (Publicaciones I-VI); ii) el comportamiento de los constituyentes orgánicos de diversos cultivos lignocelulósicos durante la fabricación de pastas de papel de alta calidad, que incluyen procesos de cocción sosa-AQ y blanqueo TCF y ECF (Publicaciones VII-VIII); y iii) el desarrollo de dos procedimientos biotecnológicos para la degradación de lignina y lípidos residuales en pastas de papel (Publicaciones IX-X).
91

5. Resultados y discusión
Publicación I: Marques G., Rencoret J., Gutiérrez A., del Río J.C. (2010) Evaluation of the chemical composition of different non-woody plant fibers used for pulp and paper manufacturing. The Open Agriculture Journal (in press).
92

5. Resultados y discusión
Evaluation of the chemical composition of different non-woody plant fibers used for pulp and paper manufacturing
Gisela Marques, Jorge Rencoret, Ana Gutiérrez, José C. del Río
Instituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, P.O. Box 1052, 41080-
Seville, Spain
Abstract The chemical composition of several non-woody plant fibers (bast fibers from flax, hemp, kenaf, jute; leaf fibers from sisal, abaca and curaua; and giant reed), which are used as raw materials for pulp and papermaking, has been evaluated. Particular attention was paid to the composition of the lipophilic compounds and the structure of the lignin polymer since they are important components of the fiber that strongly influence the pulping and bleaching performances. Keywords: non-woody fibers; flax, hemp, kenaf, jute, sisal, abaca, giant reed, paper pulp; lipophilic extractives; lignin 1. Introduction An alternative to woody raw materials for pulp and paper production in developing countries is the use of non-woody fibers from field crops and agricultural residues. In developed countries, non-woody fibers are mainly used for the production of specialty papers, i.e., tea bags, filter papers, bank notes, etc. On the other hand, there is a growing need within Europe to consider alternative agricultural strategies that move an agricultural industry purely focused on food production to one that also supplies the needs of other industrial sectors, such as paper and textiles. Non-wood fibers, therefore, could become important raw materials in this transformation [1-3]. The main sources of non-woody raw materials are agricultural residues from monocotyledons, including cereal straw and bagasse, or plants grown specifically for the fiber, such as bamboo, reeds, and some other grass plants such as flax, hemp, kenaf, jute, sisal, or abaca. Non-woody plants offer several advantages including short growth cycles, moderate irrigation requirements and low lignin content, which in principle would result in reduced energy and chemicals consumption during pulping [4].
Plant fibers are constituted by three structural polymers (the polysaccharides cellulose, and hemicelluloses and the aromatic polymer lignin) as well as by some minor non-structural components (i.e. proteins, extractives, minerals). Pulping and bleaching performances are highly dependent on the relative content, structure and reactivity of the plant components. In particular, the lignin content and its composition in terms of p-hydroxyphenyl (H), guaiacyl (G) and syringyl (S) moieties and the different inter-unit linkages are important factors
93

5. Resultados y discusión
94
in pulp production affecting the delignification rate. It has been shown that higher S/G ratios in woods implied higher delignification rates, less alkali consumption and therefore higher pulp yield [5]. On the other hand, among the non-structural components, lipophilic extractives present special relevance due to their high impact in paper pulp manufacturing [6]. Lipophilic extractives include different classes of compounds (i.e. alkanes, fatty alcohols, fatty acids, free and conjugated sterols, terpenoids, triglycerides and waxes), which have different behavior during pulping and bleaching [6-8]. These lipophilic compounds, even when present in low amounts in the raw material, may play an important role during the industrial wood processing for pulp and paper production since they are at the origin of the so-called pitch deposits. Pitch deposition is a serious problem in the pulp and paper industry being responsible for reduced production levels, higher equipment maintenance costs, higher operating costs, and an increased incidence of defects in the finished products, which reduces quality and benefits [6].
In order to maximize the exploitation of non-woody plant fibers for paper pulp production, a more complete understanding of its chemistry is required. Most studies have been devoted to the chemical characterization of woody materials, while studies on non-woody fibers have been comparatively scarce. In this context, the main objective of this work is to revise and evaluate the chemical composition of different non-woody plant fibers used for pulp and papermaking, that will help improving the industrial processes in which they are used as raw materials. 2. Analytical methodologies 2.1. Samples The samples selected for this study were bast fibers from flax (Linum usitatissimum), hemp (Cannabis sativa), kenaf (Hibiscus cannabinus) and jute (Corchorus capsularis); leaf fibers from sisal (Agave sisalana), abaca (Musa textilis) and curaua (Ananas erectifolius); as well as giant reed (Arundo donax). 2.2. Chemical analyses For hemicellulose and Klason lignin content estimation, milled samples were extracted with acetone in a Soxhlet apparatus for 8h and subsequently extracted with hot water (3h at 100 ºC). The acetone extracts were evaporated to dryness and resuspended in chloroform for chromatographic analysis of the lipophilic fraction. Klason lignin was estimated as the residue after sulfuric acid hydrolysis of the pre-extracted material. The acid-soluble lignin was determined, after filtering off the insoluble lignin, by spectrophotometric determination at 205 nm wavelength. Neutral sugars from polysaccharide hydrolysis were analyzed as alditol acetates by GC according to Tappi rules T222 om-88 and T249 om85, respectively [9]. Ash content was estimated as the residue after 6h at 575 �C.

5. Resultados y discusión
2.3. Analysis of lipids The broad range of molecular masses of lipophilic extractives and their structural diversity represent two important difficulties for their chemical analysis. High-temperature, short-length (5m) capillary columns with thin films was used for the rapid identification and quantification of lipophilic wood extractives with no prior derivatization nor fractionation [10], resulting in an optimal analysis of high-molecular-weight lipids such as waxes, sterol esters, and triglycerides. This method enables elution and separation of compounds with a wide range of molecular weights (from fatty and resin acids to sterol esters and triglycerides) in the same chromatographic analysis. For GC-MS, medium-length high-temperature capillary columns (12 m) were used [10]. When a more accurate characterization of some compounds was required, the extracts were fractionated by solid-phase extraction (SPE) procedures [10-11]. 2.4. Analysis of lignin Pyrolysis coupled to gas chromatography/mass spectrometry (Py-GC/MS) was used for the “in situ” analysis of the chemical composition of the lignin, in terms of their H:G:S distribution. Lignin is thermally degraded to produce a mixture of relatively simple phenols, which result from cleavage of ether and certain C-C inter-unit linkages. The released methoxylated phenols retain the substitution patterns of the different lignin monomers, and it is thus possible to identify components from the p-hydroxyphenylpropanoid (H), guaiacylpropanoid (G) and syringylpropanoid (S) lignin units [5, 12, 13]. For a more detailed structural study, the milled wood lignins were isolated according to a known procedure [14] and analyzed by bidimensional nuclear magnetic resonance (2D-NMR). 2D-NMR can provide information of the structure of the whole macromolecule and is a powerful tool for lignin structural elucidation since signals overlapping in the 1H and 13C NMR spectra are resolved revealing both the aromatic units and the different interunit linkages present in lignin [15-18]. 3. Characterization of the selected non-woody plant fibers 3.1. Morphological characteristics of the fibers The morphological characteristics of a fiber, such as fiber length and width, are important parameters in estimating pulp qualities. Fiber length is the most important physical property for pulping as it generally influences the tearing strength of paper. Greater the fiber length, higher will be the tearing resistance of paper. On the other hand, longer fibers tend to give a more open and less uniform sheet structure. Table 1 shows the morphological characteristics of the non-woody fibers used for this study [19]. An important feature of non-wood fibers is the wide variability among the lengths of the fibers of different species. Some of these fibers have short lengths (i.e. giant reed, with only 1180 �m fiber
95

5. Resultados y discusión
96
length), similar to the short fibers of hardwoods, while others, and particularly flax and hemp bast fibers, present remarkably high lengths (up to 28000 �m fiber length). Table 1. Morphological characteristics (length and width) of the selected fibers [19]. Fiber Source Length (�m) Width (�m) L/W ratio Bast fibers flax 28000 21 1350:1 hemp 20000 22 1000:1 kenaf 2740 20 135:1 jute 2000 20 100:1 Leaf fibers sisal 3030 17 180:1 abaca 6000 20 300:1 curaua n.a. n.a. n.a. Reeds giant reed 1180 15 78:1 Woods for comparison softwoods 3000 30 100:1 hardwoods 1250 25 50:1 n.a. not available
Among the studied fibers, flax and hemp pulps have traditionally been used as the primary furnish for cigarette paper (burning tube), where strength, opacity and control of air permeability are required. Banknote paper often incorporates flax or hemp to enhance general strength characteristics. Jute pulp is used for high porosity papers. Its fiber length plus low diameter makes it very suitable for finishing paper purposes. Sisal and abaca pulps have an unusually high tearing resistance and high porosity and are well suited for the production of papers where high strength and high porosity are required. 3.2. Raw chemical composition of the fibers The chemical composition of the main constituents of the selected non-woody fibers is shown in Table 2. In general, they are characterized by a high polysaccharide content and low contents of lignin, lipids and ash [20-22]. Giant reed presents the lowest holocellulose content and the higher content of lignin, which makes it less interesting for pulp and papermaking. The low lignin content of the rest of the fibers, with a lignin content as low as 2.9 % in flax, is in principle advantageous for pulping. Moreover, the acetone extractives content is also low, and usually less than 2%, except for curaua fibers (5.3% of total

5. Resultados y discusión
fiber weight). However, most of the acetone extracts in curaua corresponds to polar compounds, while only 1.3% corresponds to lipophilic compounds, which were estimated by redissolving the acetone extracts in chloroform. Thus, in general, the lipophilic content of the selected non-woody fibers ranges from 0.5 to 1.3%. Finally, the ash content for all the selected fibers was low in comparison to other raw materials used for pulp and papermaking, as the cereal straws, with an ash content generally higher than 15% [1]. Therefore, according to their chemical composition, most of these fibers seem suitable raw materials for pulp and papermaking. Table 2. Composition of the main constituents of the selected fibers (% of dry matter) [20-22].
Ash Acetone extractives
Water-solubles Klason lignin
Acid-soluble lignin
Holocellulose
Bast fibers flax 1.5 0.7 1.3 2.9 1.6 92.0 hemp 2.0 0.5 1.2 4.6 1.5 90.3 kenaf 1.8 1.0 1.1 11.4 3.0 81.9 jute 2.4 0.5 0.4 13.3 2.8 81.6 Leaf fibers sisal 1.0 0.7 2.3 5.9 3.0 85.0 abaca 0.9 0.5 1.7 7.7 1.4 85.6 curaua 1.3 5.3 5.1 4.9 1.6 92.5 Reeds giant reed 4.2 1.6 8.5 24.7 n.d. 49.8
3.3. Carbohydrate composition of the fibers The results of the analyses of neutral sugars of the non-woody fibers selected for this study are reflected in Table 3. The hemicelluloses fraction of the bast fibers presents a higher variability than those of the leaf fibers. Thus, hemicelluloses from flax and hemp are mainly constituted by mannose followed by galactose, while the hemicelluloses from kenaf and jute are predominantly constituted by xylose. On the other hand, all the leaf fibers (sisal, abaca and curaua) show a predominance of xylose. Finally, giant reed presents a strikingly high content of xylose, that amounts up to 39.2% of the total neutral sugars.
97

5. Resultados y discusión
98
Table 3. Composition of neutral monosaccharides (as percentage of total neutral carbohydrates) [20-22].
Rhamnose Arabinose Xylose Mannose Galactose Glucose
Bast fibers flax 0.4 0.9 1.1 8.8 3.5 85.3 hemp 0.4 0.6 1.0 9.9 1.6 86.4 kenaf 0.5 2.1 10.5 4.9 0.5 81.5 jute 0.5 1.5 7.9 4.2 0.5 85.4 Leaf fibers sisal 0.3 1.9 12.0 3.6 0.6 81.7 abaca 0.3 1.6 7.5 3.5 0.3 86.9 curaua 0.0 2.7 8.0 3.5 0.2 85.6 Reeds giant reed 0.0 3.4 39.2 0.3 0.8 56.3 3.4. Lipid composition of the fibers As shown in Table 2, all the studied fibers present low extractives contents. However, due to the wide structural heterogeneity of the compounds that may occur and their different behavior during pulping, the knowledge of the chemical nature of these components, especially the lipophilic compounds, is important in order to predict and control the eventual pitch problems that may occur during pulping and bleaching and to establish appropriate methods and strategies for their control.
The composition of the lipids present in the different fibers was studied by GC and GC-MS and is shown in Table 4. The main lipid classes found in the non-woody fibers are shown in Figure 1 and consists mainly of alkanes (A), fatty alcohols (B), aldehydes (C), fatty acids (D), sterols (G), sterol esters (H), sterol glycosides (I), steroid hydrocarbons (J), steroid ketones (K) and waxes (L). Other compounds found are alkyl ferulates (M), glycerides (N), -hydroxy monoesters (O) and -hydroxy acylesters of glycerol (P). The detailed composition of the lipophilic compounds present in these fibers has been addressed [7, 8, 22-27]. The content and composition of the different lipid classes vary considerable among the fibers. In the case of flax bast fibers the predominant lipophilic compounds are fatty acids and aldehydes, accounting for 34% and 23% of total extract, respectively, followed by ester waxes (18%) and fatty alcohols (13%). Fatty acids are also the predominant compounds (27% of total extracts) in hemp bast fibers, followed by alkanes (15%), free sterols (12%) and steroid hydrocarbons (12%). The predominant lipophilic compounds in

5. Resultados y discusión
kenaf and jute are fatty acids (28 and 35% respectively), followed by ester waxes (26 and 27% respectively). Among the selected leaf fibers, free sterols and fatty acids predominate in both sisal (20 and 24%, respectively) and abaca (45 and 19%, respectively), while in curaua fibers, fatty acids and ester waxes predominate (38 and 34%, respectively), followed by free sterols (10%). Finally, in giant reed, the predominant lipophilic compounds are fatty acids (40%), followed by free sterols (19%) and ester waxes (15%).
Generally, these fibers are pulped by an alkaline process, usually soda/anthraquinone pulping. Therefore, we discuss the behavior and fate of the different fiber components during alkaline cooking. In this context, the lipids present in these fibers can be classified, in general terms, into two principal groups, namely fatty acids (including �- and -hydroxyfatty acids) and neutral components, including wax esters, long-chain n-fatty alcohols, alkanes, and steroids and triterpenoids. The different lipids classes have different behavior during cooking and bleaching [7, 8]. The wax esters, which are abundant lipophilic compounds in some of these fibers (i.e. flax and curaua), are hydrolyzed during alkaline cooking and the fatty acids dissolved. At sufficiently high pH (as in alkaline pulping), the acids dissociate and form fatty acid soaps and can thus dissolve in water to quite a high extent. By contrast, alkanes, fatty alcohols, sterols and triterpenols, steroid hydrocarbons and ketones, and steryl glycosides do not form soluble soaps under the alkaline pulping conditions and therefore survive cooking. These compounds have a very low solubility in water and are difficult to remove, and therefore can be at the origin of pitch deposition. The low amounts of these neutral compounds in most of the fibers, and particularly the low abundances of free and conjugated sterols, which have a high propensity to form pitch deposits [28-30] would point to a low pitch deposition tendency of the lipophilics from these fibers. On the other hand, fatty acid soaps are effective solubilizing agents facilitating the removal from pulp of these sparingly soluble neutral substances. Therefore, the ratio of saponifiables-to-unsaponifiables has been suggested to be a better index for predicting pitch problems than the total amount of lipids (Back and Allen, 2000). In fact, the higher abundances of unsaponifiable compounds (neutrals) with respect to the saponifiable ones is the main cause for pitch problems during pulping of some woods, such as aspen or eucalypt [28-31]. Fatty alcohols, alkanes and sterols are among the compounds responsible for pitch deposits formed during pulping of nonwoody plants [8, 32]. In most of the fibers, as in flax, hemp, kenaf or jute fibers, the content of free fatty acids (including �- and -hydroxyfatty acids) is high, and therefore the fatty acid soaps formed during alkaline pulping may possess sufficient micellar-forming properties to carry the less polar compounds into solution. However, in other fibers, such as sisal and particularly abaca, the fatty acids amounts up to only 20% of total lipophilic compounds, and therefore they would be more prone to produce pitch deposition.
99

5. Resultados y discusión
100
�
O
KJ
AOHB
CH
O
OH
O
D
HOG O
O
H
IO
OH
O
OHHO
CH2OH
LO
O
O
OHO
O
O
OCH3
OH
O
M
O-CH2
HO-O-CH
HO-O-CH2
O
N
O-CH2
HO-O-CH
HO-O-CH2
OHO
P
HOOH
O
E
OH
O
OHF
O
K
OO
KJJ
AAOHB OHOHB
CH
O
CH
O
H
O
H
O
OH
O
OH
O
D
HOG
HOHOHOG O
O
HO
O
O
O
H
IO
OH
O
OHHO
CH2OH
IO
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH
LO
O
LO
O
O
O
O
OHO
OO
OHO
OO
OHO
O
O
OCH3
OH
O
M
O-CH2
HO-O-CH
HO-O-CH2
O
N
O
OCH3
OH
O
M
O
OCH3
OH
OO
OCH3
OH
O
M
O-CH2
HO-O-CH
HO-O-CH2
O
N
O-CH2
HO-O-CH
HO-O-CH2
O
O-CH2
HO-O-CH
HO-O-CH2
OOO
N
O-CH2
HO-O-CH
HO-O-CH2
OHO
P
O-CH2
HO-O-CH
HO-O-CH2
OHO
O-CH2
HO-O-CH
HO-O-CH2
OOHO
P
HOOH
O
E
HOOH
OHO
OH
O
OHOH
O
E
OH
O
OHFOH
O
OHOH
O
OHF
Figure 1. Structures of compounds representing the main classes of lipophilic extractives found in the different fibers selected. : A, pentacosane; B, docosanol; C, octacosanal; D, palmitic acid; E, 26-hydroxyhexacosanoic acid; F, 2-hydroxytetracosanoic acid; G, sitosterol; H, sitosteryl linoleate; I, sitosteryl 3�-D-glucopyranoside; J, stigmasta-3,5-diene; K, stigmasta-3,5-dien-7-one; L, octacosyl hexadecanoate; M, trans-docosanylferulate; N, 1-monodocosanoylglycerol; O, docosanyl, 16-hydroxyhexadecanoate; P, 1-mono(24-hydroxytetracosanoyl)glycerol.

5. Resultados y discusión
Table 4. Composition of lipophilic extractives (mg/100g) in the different fibers [8, 22-26]. Bast fibers Leaf fibers Reeds
flax hemp kenaf jute sisal abaca curaua giant reed
n-alkanes (A) 27 43 27 5 15 - - 8
fatty alcohols (B) 220 2 13 13 8 <1 55 19
n-aldehydes (C) 371 25 1 - 1 - - 8
fatty acids (D) 552 78 33 13 11 9 81 114
-hydroxyfatty acids (E) - - - 3 6 1 142 -
�-hydroxyfatty acids (F) 11 9 - 10 7 1 23 -
free sterols/triterpenols (G) 92 36 5 4 20 25 62 53
sterol/triterpenol esters (H) 6 7 1 - <1 1 9 7
sterol glycosides (I) 5 13 <1 1 2 2 27 15
steroid hydrocarbons (J) 14 30 2 2 14 3 12 13
steroid/triterpenoid ketones (K) 33 27 4 3 3 4 6 4
n-alkyl ferulates (M) - - - - 6 3 - -
ester waxes* (L, N, O, P) 284 17 30 20 8 7 222 42
* including mono- and diglycerides and -hydroxyfatty acid esters. 3.5. Lignin composition of the fibers The lignin content of the non-woody plant fibers selected for this study (estimated as Klason lignin) ranges from 2.9 for flax bast fiber to 24.7 for giant reed (Table 2). The low lignin content of most of the fibers seems to be advantageous for their use in paper pulp manufacturing, as they would require fewer chemicals and less drastic conditions during pulping and bleaching. However, not only the content but the lignin composition also strongly affects delignification rates, chemical consumption and pulp yields [5, 33]. In general, the efficiency of pulping is directly proportional to the amount of syringyl (S) units in lignin [5]. The G units have a free C-5 position available for carbon-carbon inter-unit bonds, which make them fairly resistant to lignin depolymerization in pulping, while the S lignin is relatively unbranched and has a lower condensation degree and therefore is easier to delignify. The higher reactivity of the S lignin with respect to the G lignin in alkaline systems is known [34, 35] and, therefore, the lignin S/G ratio directly affects the delignification behavior. Higher S/G ratios would imply higher delignification rates, less alkali consumption and, therefore, higher pulp yield [5, 33]. The lignin composition of the selected fibers was characterized “in situ” by Py-GC/MS [20-24, 26, 36]. The relative composition of the H, G and S-lignin units
101

5. Resultados y discusión
102
for the non-woody fibers studied here is listed in Table 5. A predominance of S- over G-lignin was found in the bast fibers of kenaf [24] and jute [20, 36] and in all the leaf fibers of sisal, abaca and curaua [20, 22, 23]. By contrast, the bast fibers from flax and hemp, as well as the giant reed showed a predominance of G-lignin [20, 21, 26]. This is especially evident in the lignin of flax, with an extremely low S/G ratio of 0.1. The low S/G ratio of the lignins from flax and hemp, despite having very low lignin contents (less than 5% Klason lignin), makes them fairly resistant to alkaline delignification. Also, the high lignin content of giant reed (24.7 % Klason lignin) together with its low S/G ratio of 0.7 makes it especially hard to delignify. By contrast, the rest of the fibers (kenaf, jute, sisal, abaca, curaua) present both high S/G ratios and low lignin contents, which will make them easily delignifiable under alkaline pulping, requiring lower energy and less drastic conditions. Table 5. Composition of H, G and S moieties for all the raw materials studied by Py-GC/MS [20-25, 35].
Bast fibers Leaf fibers Reeds
flax hemp kenaf jute sisal abaca curaua giant reed
% H 56,4 12.8 1.3 2.1 1.3 20.2 29.8 25.6
% G 40.8 53.0 15.4 32.2 18.7 13.5 29.1 44.2
% S 2.8 34.2 83.3 65.7 80.0 66.2 41.1 30.2
S/G ratio 0.1 0.6 5.4 2.0 4.3 4.9 1.4 0.7
For a more complete structural characterization of the lignins from these non-
woody fibers, the milled-wood lignins (MWL) were isolated and analyzed by 2D-NMR spectroscopy [36-38]. Signals from S and G lignin units were observed in all spectra, whereas signals for p-hydroxyphenyl (H) lignin units could only be detected in the HSQC spectra of flax and hemp lignins, in agreement with the high amounts of these units observed by Py-GC/MS and shown in Table 5. In general, the relative proportions of the different lignin units (S/G ratios in Table 6) are in close agreement with the Py-GC/MS data shown above. In addition, prominent signals corresponding to p-coumarate structures were observed in the lignins of abaca and curaua [37]. In these lignins, p-coumaric acid has been reported to be esterified to the lignin polymer [23, 37, 39]. The side-chain region of the HSQC spectra gave additional information about the different inter-unit linkages (i.e. �-O-4� aryl ether, �-�� resinol, �-5� phenylcoumaran, �-1�/�-O-�� spirodienones, etc) present in the structure of these lignins. The main substructures found in these lignins are depicted in Figure 2. The relative abundances of the main inter-unit linkages present in the MWL of the non-woody fibers selected for this study are shown in Table 6. �-O-4� aryl ether substructures (I) were predominant in all of the lignins. Interestingly, the lignins from kenaf, sisal, abaca and curaua are

5. Resultados y discusión
especially enriched in �-O-4� structures (more than 84% of all side-chains) [37]. Phenylcoumaran (�-5� linkages) substructures (II) were observed in most of the fibers, being especially abundant in flax and hemp, but were completely absent in abaca. The presence of these low amounts of phenylcoumaran substructures was expected due to the low levels of guaiacyl lignin units in these samples. Resinol (�-�� linkages) substructures (III) were also observed in important amounts in flax, hemp, and jute, and in low amounts in kenaf and sisal, but were completely absent in abaca and curaua lignins. Finally, spirodienone structures (IV) were also present, although in lower amounts in most of the fibers, being absent in flax and hemp. The high abundance of non-condensed linkages in the lignins of kenaf, sisal, abaca and curaua makes them particularly easily to delignify, in contrast to the rest of the lignins, with a high content of condensed linkages, particularly in flax and hemp lignins.
Table 6. Structural characteristics (relative abundance of the main interunit linkages as percentages of side-chains involved, percentage of �-acylation and S/G ratio) observed from the HSQC spectra of the MWL of selected fibers (curaua, hemp, kenaf, jute, sisal and abaca) [35-37].
Bast fibers Leaf fibers flax hemp kenaf jute sisal abaca curaua
Linkage relative abundance (% of side-chains involved) �-O-4� alkyl-aryl units (I, I', I'') 71 69 84 72 89 94 94 Phenylcoumarans (II) 16 9 2 4 2 0 2 Resinols (III) 13 22 8 16 4 0 0 Spirodienones (IV) 0 0 6 4 5 6 4 Percentage of �-acylation 0 0 58 4 68 80 69 S/G ratio 0.1 0.8 5.6 2.0 3.9 8.7 4.9
Interestingly, the spectra of some of these lignins (kenaf, sisal, abaca, curaua)
revealed the presence of intense signals corresponding to acylated �-carbon (Figure 2, structures I' and I'') [37]. An estimation of the percentage of �-acylation of the lignin side-chain was calculated by integration of the signals corresponding to the hydroxylated and acylated �-carbon (Table 6) and ranged from 4% in jute lignin to 80% in abaca lignin. The high level of acylation of the �-carbon has been correlated with the high abundances of �-O-4� linkages and the low abundances of the �-�� resinol structures [37, 38]. The nature of the acyl group esterifying the �-carbon was studied by the so-called Derivatization Followed by Reductive Cleavage (DFRC) degradation method [40, 41], which selectively and efficiently cleaves �-ether and �-ether linkages but leaves �-esters intact. This method allowed confirming that p-coumarate groups are
103

5. Resultados y discusión
104
attached at the �-carbon of abaca and curaua lignins, and predominantly on syringyl units [37, 41]. In addition, acetate units were also found esterifying the �-carbon in the lignins of all the studied fibers, although at different extents. In all cases, acetate and p-coumarate groups were found to be preferentially attached to syringyl units [37, 41-43]. It must be noted that, although these ester moieties will, in principle, consume additional alkaline reagents during cooking, it has been shown above that the highly acylated lignins are extremely enriched in easily hydrolysable non-condensed �-O-4� linkages, which will be more amenable to delignification.
��
�
123
456
1’6’
5’4’
3’2’HO
O
O
MeOO
O
OMe
I' I''
��
�
123
456
1'’6'’
5'’4'’
3'’2'’HO
O
O
OMe
O
O
HO
�´�´
�´2´
3´
5´6´
1´
4´
MeO
��
�
123
456
1’6’
5’4’
3’2’HO
O
O
HO MeO
OMeMeO
I
���
123
456
1’2’3’
4’5’6’
OHO
O
OMe
OMe
II
�
1´2´
3´4´
5´6´
O
O
OHHO
�� �´ �´�´
O
1''2''3''
4''5''6''
OMe
OMeO
21
4 3
56
OMe
IV
�
�
�
1
23
45
6
�’
�’
�’
1’2’
3’4’
5’6’
O
O
O
O
OMe
OMe
III
OMe OMe
MeO
OMe
MeO
MeO
MeO
OMe MeO MeO
MeO
Figure 2. Main substructures present in the lignins studied here: I, �-O-4� linked substructures; I�, �-O-4� linked substructures with acetylated �-carbon; I��, �-O-4� linked substructures; with p-coumaroylated �-carbon; II, phenylcoumaran structures formed by �-5� and �-O-4� linkages; III, resinol structures formed by �-� �, �-O-��, and �-O-R� linkages; IV, spirodienone structures formed by �-1�, and �-O-� � linkages. 4. Conclusions The chemical composition of different non-woody plant fibers used as raw materials for pulp and papermaking has been summarized, with especial emphasis in the chemistry of lipids and lignin and their fate during alkaline

5. Resultados y discusión
pulping. This study offers valuable information that will lead to a better industrial utilization of these non-woody plant species of high socioeconomic interest. Acknowledgements This study has been supported by the Spanish Projects AGL2005-01748 and AGL2008-00709 and the EU BIORENEW project (NMP2-CT-2006-26456). We thank CELESA (Tortosa, Spain) and University of Huelva for providing the samples. G.M. thanks the Spanish Ministry of Education for a FPI fellowship. J.R. thanks the Spanish CSIC for a I3P fellowship. References [1] Moore G. Nonwood Fibre Applications in Papermaking, Pira International,
Leatherhead, Surrey, UK, 1996.
[2] Paavilainen L. European prospects for using nonwood fibres. Pulp Pap Int 1998; 61-86.
[3] Saijonkari-Pahkala K. Non-wood plants as raw materials for pulp and paper. PhD Thesis, University of Helsinki, Finland, 2001; pp. 101.
[4] Hurter RW, Riccio FA. Why CEOS don’t want to hear about nonwoods-or should they? In: TAPPI Proceeding, NA Nonwood Fiber Symposium, Atlanta, GA, USA, 1998; 1-11.
[5] del Río JC, Gutiérrez A, Hernando M, Landín P, Romero J, Martínez AT. Determining the influence of eucalypt lignin composition in paper pulp yield using Py-GC/MS. J Anal Appl Pyrol 2005; 74: 110-115.
[6] Back EL, Allen LH. Pitch Control, Wood Resin and Deresination, Tappi press, Atlanta, GA., 2000; pp. 392.
[7] Gutiérrez A, del Río JC. Lipids from flax fibers and their fate after alkaline pulping. J Agric Food Chem 2003; 51: 4965-4971.
[8] Marques G, del Río JC, Gutiérrez A. Lipophilic extractives from several nonwoody lignocellulosic crops (flax, hemp, sisal, abaca) and their fate during alkaline pulping and TCF/ECF bleaching. Biores Technol 2010; 101: 260-267.
[9] Technical Association of the Pulp and Paper Industry. Test methods, 1992-1993. TAPPI, Atlanta, Ga. 1993.
105

5. Resultados y discusión
106
[10] Gutiérrez A, del Río JC, González-Vila FJ, Martín F. Analysis of lipophilic extractives from wood and pitch deposits by solid-phase extraction and gas chromatography. J Chromatogr A 1998; 823: 449-455.
[11] Christie WW. In Christie WW, Ed. Solid-phase extraction columns in the analysis of lipids. Advances in Lipid Methodology—One, The Oily Press, Dundee, Scotland 1992; pp. 1–18.
[12] Faix O, Meier D, Fortmann I. Thermal degradation products of wood. A collection of electron of electron-impact (EI) mass spectra of monomeric lignin derived products. Holz Roh- Werkst 1990; 48 (9): 351-354.
[13] Ralph J, Hatfield RD. Pyrolysis-GC/MS characterization of forage materials. J Agric Food Chem 1991; 39 (8): 1426-1437.
[14] Björkman A. Studies on finely divided wood. Part I. Extraction of lignin with neutral solvents. Sven Papperstidn 1956; 59: 477-485.
[15] Capanema EA, Balakshin MY, Chen CL, Gratzl JS, Gracz H. Structural analysis of residual and technical lignins by 1H-13C correlation 2D NMR-spectroscopy. Holzforschung 2001; 55: 302-308.
[16] Capanema EA, Balakshin MY, Kadla JF. Quantitative characterization of a hardwood milled wood lignin by nuclear magnetic resonance spectroscopy. J Agric Food Chem 2005; 53 (25): 9639–9649.
[17] Liitiä TM, Maunu SL, Hortling B, Toikka M, Kilpeläinen I. Analysis of technical lignins by two- and three-dimensional NMR spectroscopy. J Agric Food Chem 2003; 51 (21): 2136–2143.
[18] Ralph J, Marita JM, Ralph SA, Hatfield RD, Lu F, Ede RM, Peng J, Quideau S, Helm RF, Grabber JH, Kim H, Jimenez-Monteon G, Zhang Y, Jung HJG, Landucci LL, MacKay JJ, Sederoff RR, Chapple C, Boudet AM. Solution-state NMR of lignin. In: Argyropoulos DS, Ed. Advances in Lignocellulosics Characterization, Tappi Press, Atlanta, GA 1999; 55-108.
[19] García Hortal JA. Fibras Papeleras, Edicions UPC, Barcelona, Spain, 2007; pp. 243.
[20] Rodríguez MI. Caracterización química de plantas hebáceas utilizadas para la fabricación de pasta de papel de alta calidad. PhD Thesis, University of Seville, Spain 2006.

5. Resultados y discusión
[21] Coelho DS Estudo sistemático da composiçao quimica das fibras de Arundo donax e a sua evoluçao durante a producto de pasta de papel através do processo organosolv. MSc Thesis, University of Aveiro, Portugal, 2006.
[22] Marques G, Gutiérrez A, del Río JC. Chemical characterization of lignin and lipophilic fractions from leaf fibers of curaua (Ananas erectifolius). J Agric Food Chem 2007; 55: 1327-1336.
[23] del Río JC, Gutiérrez A. Chemical composition of abaca (Musa textilis) leaf fibers used for manufacturing of high quality paper pulps. J Agric Food Chem 2006; 54 (13): 4600–4610.
[24] Gutiérrez A., Rodríguez IM, del Río JC. Chemical characterization of lignin and lipid fractions in kenaf bast fibers used for manufacturing high-quality papers. J Agric Food Chem 2004¸ 52: 4764-4773.
[25] del Río JC, Marques G, Rodríguez IM, Gutiérrez A. Chemical composition of lipophilic extractives from jute (Corchorus capsularis) fibers used for manufacturing of high-quality paper pulps. Ind Crops Prod 2009; 30: 241-249.
[26] Gutiérrez A, Rodríguez IM, del Río JC. Chemical characterization of lignin and lipid fractions in industrial hemp bast fibers used for manufacturing high-quality paper pulps. J Agric Food Chem 2006; 54: 2138-2144.
[27] Coelho DS, Marques G, Gutiérrez A, Silvestre ARD, del Río JC. Chemical characterization of the lipophilic fraction of Giant reed (Arundo donax) fibres used for pulp and paper manufacturing. Ind Crops Prod 2007; 26: 229-236.
[28] del Río JC, Gutiérrez A, González-Vila FC, Martín F, Romero J. Characterization of organic deposits produced in kraft pulping of Eucalyptus globulus wood. J Chromatogr A 1998; 823: 457–465.
[29] del Río JC, Romero J, Gutiérrez A. Analysis of pitch deposits produced in kraft pulp mills using a totally chlorine free bleaching sequence. J Chromatogr A 2000; 874: 235–245.
[30] Gutiérrez A, del Río JC. Gas chromatography/mass spectrometry demonstration of steryl glycosides in eucalypt wood, kraft pulp and process liquids. Rapid Commun Mass Spectrom 2001; 15: 2515–2520.
[31] Chen T, Wang Z, Zhou Y, Breui, C, Aschim OK, Yee E, Nadeau L. Using solid-phase extraction to assess why aspen causes more pitch problems than softwoods in kraft pulping. Tappi J 1995; 78: 143-149.
107

5. Resultados y discusión
108
[32] Gutiérrez A, del Río JC. Chemical characterization of pitch deposits produced in the manufacturing of high-quality paper pulps from hemp fibers. Biores Technol 2005; 96: 1445-1450.
[33] González-Vila FJ, Almendros G, del Río JC, Martín F, Gutiérrez A, Romero J. Ease of delignification assessment of wood from different Eucalyptus species by pyrolysis (TMAH)-GC/MS and CP/MAS 13C NMR spectrometry. J Anal Appl Pyrol 1999; 49: 295-305.
[34] Chang HM, Sarkanen KV. Species variation in lignin. Effect of species on the rate of Kraft delignification. Tappi press 1973; 56: 132–134.
[35] Tsutsumi Y, Kondo R, Sakai K, Imamura H. The difference of reactivity between syringyl lignin and guaiacyl lignin in alkaline systems. Holzforschung 1995; 49 (5): 423-428.
[36] del Río JC, Rencoret J, Marques G, Li J, Gellerstedt G, Jiménez-Barbero J, Martínez AT, Gutiérrez A. Structural characterization of the lignin from jute (Corchorus capsularis) fibers. J Agric Food Chem 2009; 57: 10271-10281.
[37] del Río JC, Rencoret J, Marques G, Gutiérrez A., Ibarra D, Santos JI, Jiménez-Barbero J, Zhang L, Martínez AT. Highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. J Agric Food Chem 2008; 56: 9525-9534.
[38] Martínez AT, Rencoret J, Marques G, Gutiérrez A, Ibarra D, Jiménez-Barbero J, del Río JC. Monolignol acylation and lignin structure in some nonwoody plants: A 2D NMR study. Phytochemistry 2008; 69: 2831-2843.
[39] Sun RC, Fang JM, Goodwin A, Lawther JM, Bolton J. Fractionation and characterization of ball-milled and enzyme lignins from abaca fibre. J Sci Food Agric 1999; 79: 1091-1098.
[40] Lu F, Ralph J. Derivatization followed by reductive cleavage (DFRC method), a new method for lignin analysis: protocol for analysis of DFRC monomers. J Agric Food Chem 1997; 45 (7): 2590-2592.
[41] del Río JC, Marques G, Rencoret J, Martínez AT, Gutiérrez A. Occurrence of naturally acetylated lignin units. J Agric Food Chem 2007; 55:5461-5468.
[42] Ralph J, Lu F. The DFRC method for lignin analysis. 6. A simple modification for identifying natural acetates in lignin. J Agric Food Chem 1998; 46: 4616-4619.

5. Resultados y discusión
[43] Ralph J. An unusual lignin from kenaf. J Nat Prod 1996; 59 (4): 341-342.
109

5. Resultados y discusión
Publicación II: del Río J.C., Marques G., Rencoret J. Martínez A.T. and Gutiérrez A. (2007) Occurence of naturally acetylated lignin units. Journal of Agricultural and Food Chemistry, 55, 5461-5468.
110

5. Resultados y discusión
Occurrence of naturally acetylated lignin units
José C. del Río†, Gisela Marques†, Jorge Rencoret†, Ángel T. Martínez‡, Ana Gutiérrez†
†Instituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, P.O. Box 1052, 41080-
Seville, Spain ‡Centro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid, Spain
Abstract In this work, we have studied the occurrence of native acetylated lignin in a large set of vascular plants, including both angiosperms and gymnosperms, by a modification of the so-called Derivatization Followed by Reductive Cleavage (DFRC) method. Acetylated lignin units were found in all angiosperms selected for this study, including mono- and eudicotyledons, but were absent in the gymnosperms analyzed. In some plants (e.g. abaca, sisal, kenaf or hornbeam), lignin acetylation occurred at a very high extent, exceeding 45% of the uncondensed syringyl lignin units. Acetylation was observed exclusively at the �-carbon of the lignin-side chain and predominantly on syringyl units, although a predominance of acetylated guaiacyl over syringyl units was observed in some plants. In all cases, acetylation appears to occur at the monomer stage and sinapyl and coniferyl acetates seem to behave as real lignin monomers participating in lignification. Keywords: lignin, angiosperms, gymnosperms, eudicotyledons, monocotyledons, coniferyl acetate, sinapyl acetate, abaca, sisal, kenaf, hornbeam, Derivatization Followed by Reductive Cleavage (DFRC).
1. Introduction
Lignin is a principal structural component of cell walls in higher terrestrial plants. In addition to structural support and pathogen defense, lignin functions in water transport as a hydrophobic constituent of vascular phloem and xylem cells. Lignins are complex polymers formed by the dehydrogenative polymerization of three main monolignols, p-coumaryl, coniferyl and sinapyl alcohols. Gymnosperm lignins are mainly formed from coniferyl alcohol, together with small proportions of p-coumaryl alcohol. Angiosperm lignins are mainly formed from coniferyl and sinapyl alcohols with small amounts of p-coumaryl alcohol. A considerable variation in the contribution of these three alcohol precursors is observed in lignins from annual plants (1-4). After their synthesis, the lignin monomers are transported to the cell wall where they are polymerized in a combinatorial fashion by free-radical coupling mechanisms in a reaction mediated by peroxidases, generating a variety of structures within the lignin polymer (5-7).
111

5. Resultados y discusión
112
Some lignins are known to be naturally acetylated at the �-carbon of the side chain. Acetates have been reported to occur in the lignin of a limited number of hardwoods and non-woody plants (1, 8, 9). In particular, kenaf bast lignin has been found to be extensively �-acetylated and predominantly on syringyl units, although the role of such a highly acetylated lignin is not yet known. Recent studies have provided strong evidence that sinapyl acetate is implicated as a monomer in lignification in kenaf bast fibers and that the naturally acetylated polymeric lignin derives not from acetylation of the lignin polymer but from polymerization of pre-acetylated monolignols (9, 10). Other acids are also known to occur naturally acylating lignin; thus, grass lignins are partially p-coumaroylated and some hardwood lignins such as poplar, aspen or willow are p-hydroxybenzoylated (7, 11-18).
Naturally acetylated lignins may also occur in other plants but their occurrence has probably being missed due to the limitations of the analytical procedures used for their isolation and/or structural characterization. Natural acetates present on lignin might have been hydrolyzed and removed when using traditional isolation methods (such as alkaline extraction often applied to non-wood lignins) and degradative procedures for chemical characterization (such as nitrobenzene oxidation, CuO oxidation or thioacidolysis). Indeed, for spectroscopic analysis, e.g. using Nuclear Magnetic Resonance (NMR), lignin is frequently acetylated for improved solubility and spectroscopic properties and, therefore, native acetates cannot be seen. Recently, we reported the occurrence of acetylated lignins in some herbaceous plants, including kenaf, jute, sisal and abaca, characterized by a high syringyl (S) to guaiacyl (G) ratio, by the use of Pyrolysis coupled to Gas Chromatography-Mass Spectrometry (Py-GC/MS) (19-21), although the method used hindered the determination of the extent of acetylation. In this work we have studied the occurrence and abundance of native acetylated lignin units in the milled wood lignins (MWL) isolated from a wide set of vascular plants, including gymnosperms and angiosperms (mono- and eudicotyledons). For this purpose, we have used a modification of the so-called Derivatization Followed by Reductive Cleavage (DFRC) degradation method (22-24). DFRC is a simple and powerful method which selectively and efficiently cleaves �-ether and �-ether linkages and allows quantitative analysis of structural units in etherified lignin, and also provides some information on carbon-carbon linked lignin by analysis of the dimeric structures released. DFRC includes two key steps, (i) solubilization of lignin by bromination and acetylation with acetyl bromide and (ii) reductive cleavage of the aryl ether bonds in lignin with zinc dust. Identification of the resulting monomeric and dimeric degradation products by GC/MS gives valuable information on the lignin structure. However, and most importantly, DFRC leaves �-esters intact allowing the analysis of native �-acylated lignin. Thus, the method has allowed to confirm that p-coumarate groups are attached at the �-carbon of grass lignins, predominantly on syringyl units (17). However, the DFRC method uses

5. Resultados y discusión
acetylating reagents that interfere in the analysis of native acetates in lignin, but with appropriate modification by substituting acetylation by propionylation (25), it is possible to obtain significant information and clues about the occurrence and extent of native lignin acetylation. In this paper, we use this method to investigate the presence of naturally acetylated lignin units in a set of vascular plants, including angiosperms and gymnosperms. 2. Material and methods 2.1. Samples The plant samples selected for this study are listed in Table 1. They consist of both woody and nonwoody angiosperms (mono- and eudicotyledons) and gymnosperms. Among the woody angiosperms, wood of beech (Fagus sylvatica), European hornbeam (Carpinus betulus), aspen (Populus tremula), and eucalyptus (Eucalyptus globulus) were selected. The nonwoody angiosperm samples consisted of bast fibers obtained from the stalk phloem layer of bamboo (Bambusa sp.), hemp (Cannabis sativa), kenaf (Hibiscus cannabinus) and jute (Corchorus capsularis); leaf fibers of sisal (Agave sisalana) and abaca (Musa textilis); and coir, a coarse fiber obtained from the outer shell of coconut from the palm tree (Cocos nucifera). Among gymnosperm woods, Scots pine (Pinus sylvestris) and Norway spruce (Picea abies) were selected for this study. Wood and nonwoody plants were finely ground to sawdust using a knife mill (Analysenmühle A10, Janke and Kunkel GmbH, Staufen, Germany) before analysis. MWL was extracted from finely ball-milled (150 h) plant material, free of extractives and hot water soluble material, using dioxane-water (9:1, v/v), followed by evaporation of the solvent, and purified as described (26). The final yields ranged from 5-15% of the original lignin content. Extension of milling time, which would increase yield, was avoided in order to prevent chemical modifications on the lignin structure. 2.2. DFRC A modification of the standard DFRC method by using propionyl instead of acetyl reagents (DFRC´) was used (25). Lignins (10 mg) were stirred for two hours at 50 ºC with propionyl bromide in propionic acid (8:92, v/v). The solvents and excess of bromide were removed by rotary evaporation. The products were then dissolved in dioxane/propionic acid/water (5:4:1, v/v/v), and 50 mg powered Zn was added. After 40 min stirring at room temperature, the mixture was transferred into a separatory funnel with dichloromethane and saturated ammonium chloride. The aqueous phase was adjusted to pH < 3 by adding 3% HCl, the mixture vigorously mixed and the organic layer separated. The water phase was extracted twice more with dichloromethane. The combined dichloromethane fractions were dried over anhydrous NaSO4 and the filtrate was evaporated in a rotary evaporator. The residue was subsequently propionylated
113

5. Resultados y discusión
114
for 1 h in 1.1 mL of dichloromethane containing 0.2 mL of propionic anhydride and 0.2 mL pyridine. The propionylated lignin degradation compounds were collected after rotary evaporation of the solvents, and subsequently analyzed by GC/MS. 2.3. GC/MS analysis The GC/MS analyses were performed with a Star 3400 GC (Varian) equipped with a Saturn 2000 ion trap detector (Varian) using a 12m x 0.25 mm i.d., 0.1 �m, DB-5HT length capillary column (J&W Scientific, Folsom, CA, USA). The oven was heated from 50 (held 0.2 min) to 100 ºC at 30 ºC/min, then raised to 300 ºC at 5ºC/min, and held for 5 min at the final temperature. The injector and transfer line were kept at 300 ºC. Helium was used as the carrier gas at a rate of 2 mL/min.
Quantification of the released individual monomers was performed using tetracosane as external standard and by assuming similar response factors as those of the acetylated monomers reported in Lu and Ralph (22), although without authentication on our instrument. Molar yields were calculated on the basis of molecular weights of the respective propionylated (or acetylated) compounds. 3. Results and discussion The MWL isolated from the different vascular plants selected for this study, including angiosperms (mono- and eudicotyledons) and gymnosperms, were analyzed by the modified DFRC´ method developed by Lu and Ralph (25) in order to investigate the occurrence of naturally acetylated lignin moieties. However, we have to emphasize here that the monomeric degradation products released by DFRC´ originate from etherified lignin units since this method only cleaves �- and �-aryl ether bonds. The abundance of guaiacyl lignin units, which are predominantly in condensed form (i.e. forming 5-5´ linkages), may also be underestimated.
The chromatograms of the DFRC´ degradation products of selected MWL samples are shown in Figure 1. All the analyzed lignins released the cis and trans isomers of guaiacyl (c-G and t-G) and syringyl (c-S and t-S) lignin monomers (as their propionylated derivatives) in different proportions, arising from normal (�-OH) units in lignin. In addition, the presence of originally �-acetylated guaiacyl (c-Gac and t-Gac) and syringyl (c-Sac and t-Sac) lignin units could also be clearly observed in the chromatograms of most of the analyzed lignins. The structures and mass fragments of these compounds arising from �-OH and from �-acetylated lignin units are depicted in Figure 2. Acetylation occurred exclusively at the �-carbon of the lignin side-chain, as already reported for kenaf lignin (1, 25). In all samples, coniferyl and sinapyl acetates presented a

5. Resultados y discusión
115
Tab
le 1
. Abu
ndan
ce (
Mol
ar Y
ield
s) o
f th
e D
FRC
´ D
egra
datio
n M
onom
ers
of th
e M
WL
Isol
ated
fro
m th
e D
iffer
ent P
lant
s Se
lect
ed f
or
This
Stu
dy, S
/G R
atio
s and
Rel
ativ
e A
bund
ance
s of A
cety
late
d Li
gnin
Moi
etie
s.
M
onom
ers (
�mol
/g li
gnin
)
O
rder
Fa
mily
Sp
ecie
s N
ame
G
Gac
S
S ac
S/G
%
S aca
%
Gac
b A
ngio
sper
ms
Mon
ocot
yled
ons
A
spar
agal
es
Aga
vace
ae
Agav
e si
sala
na
Sisa
l 12
2 12
4 10
8 37
8 2.
0 77
.7
50.4
Are
cale
s A
reca
ceae
C
ocos
nuc
ifera
C
oir
819
5
17
4 1
4 0.
2 7.
4 0
.6
Poa
les
Poa
ceae
Ba
mbu
sa sp
. B
ambo
o 25
6 1
3 28
0
4
1.1c
1.2
c 4
.8
Zi
ngib
eral
es
Mus
acea
e M
usa
text
ilis
Aba
ca
50
3
21
131
3.0c
80.3
c 5
.6
Eudi
coty
ledo
ns
Fa
gale
s Fa
gace
ae
Fagu
s syl
vatic
a B
eech
12
6
2 16
5 2
0 1.
4 10
.8
1.6
Faga
les
Bet
ulac
eae
Car
pinu
s bet
ulus
H
ornb
eam
14
6
4 23
0 18
5 2.
8 44
.6
2.7
Ros
ales
C
anna
bace
ae
Can
nabi
s sat
iva
Hem
p 28
6
2 17
7
2
0.6
1.1
0
.7
M
alva
les
Mal
vace
ae
Hib
iscu
s can
nabi
nus
Ken
af
390
38
543
780
3.1
59.0
8
.9
M
alva
les
Mal
vace
ae
Cor
chor
us c
apsu
lari
s Ju
te
299
1
336
23
1.2
6.4
0
.3
M
alpi
ghia
les
Salic
acea
e Po
pulu
s tre
mul
a A
spen
65
1
5 66
2
8
1.0
1.2
0
.8
M
yrta
les
Myr
tace
ae
Euca
lypt
us g
lobu
lus
Euca
lypt
15
4
8 27
5
3
2.3
1.1
4
.9
Gym
nosp
erm
s
Con
ifera
les
Pina
ceae
Pi
cea
abie
s Sp
ruce
52
0 0
0
0
0.0
-
0
.0
C
onife
rale
s Pi
nace
ae
Pinu
s syl
vest
ris
Pine
40
2 0
0
0 0.
0
-
0.0
a %
S ac i
s the
per
cent
age
of a
cety
late
d S
units
resp
ect t
o th
e to
tal S
uni
ts. b %
Gac
is th
e pe
rcen
tage
of a
cety
late
d G
uni
ts re
spec
t to
the
tota
l G u
nits
. c Som
e am
ount
s of
�-p-
coum
aroy
late
d S
units
wer
e fo
und
(27
an 1
1 �m
ol/g
lign
in fo
r bam
boo
and
abac
a, re
spec
tivel
y) a
nd w
ere
incl
uded
in th
e es
timat
ion
of to
tal S
uni
ts fo
r cal
cula
tion
of S
/G a
nd %
S ac.

5. Resultados y discusión
116
predominance of the trans- over the cis- form, as also occurred with the corresponding non-acetylated alcohols.
The results from the DFRC´ analysis of the MWL selected for this study, namely the molar yields of the released monomers, the S/G ratios and the percentages of naturally acetylated guaiacyl (%Gac) and syringyl (%Sac) lignin moieties, are presented in Table 1. The yields of the released monomers were in the same range as previously observed in the DFRC degradation of other isolated lignins (17, 24). As shown in Table 1, naturally acetylated lignin units were found to occur in all angiosperms analyzed in the present study, including both mono- and eudicotyledons. However, no traces of acetylated lignin units could be found in the MWL of the two gymnosperms (pine and spruce) studied here. The data also indicated that in most lignin samples acetylation occurred predominantly on syringyl units, whereas only traces of acetylated guaiacyl units were detected, although in bamboo and eucalyptus lignins, with a low extent of acetylation, this occurred preferentially on guaiacyl units. We can exclude acetylation as an artifact produced during the lignin isolation protocol since MWL from pine and spruce (where no traces of acetylated units could be detected) were also isolated using the same procedure as the rest of the samples. Indeed, acetates were found predominantly on S lignin units and exclusively at the �-carbon, which suggests that they are naturally present. And finally, direct DFRC´ of some whole fibers (such as sisal and kenaf), without previous lignin isolation, gave also similar results.
The occurrence of naturally acetylated lignin units seems to be widespread among angiosperms and restricted only to this group of vascular plants, being particularly abundant in syringyl-rich lignins. Especially important is the high extent of lignin acetylation observed in the MWL from the herbaceous plants abaca, sisal and kenaf, and in the hardwoods hornbeam and, in a minor extent, beech, all of them characterized by high S/G ratios. However, we also noted the acetylation of S units in coir lignin, which is characterized by a very low S/G ratio (0.2). The high extent of acetylation of kenaf lignin has been previously reported by NMR and DFRC´ (1, 25). The occurrence of naturally acetylated lignin units was also reported in kenaf, jute, sisal and abaca by Py-GC/MS of the whole fibers, although the extent of acetylation could not be determined due to the limitations of the technique (19-21). Interestingly, the high extent of acetylation of sisal MWL included both S units (78%) and G units (50%), whereas in the case of abaca, kenaf or hornbeam lignins, acetylation occurred almost predominantly on S units (80, 59 and 45%, respectively) and only a minor degree of acetylation was observed on G units (6, 9 and 3%, respectively).

5. Resultados y discusión
Abaca
Hornbeam
Kenaf
Sisal
t-S
c-S
t-Sac
t-Gac
t-G
c-G c-Sac
t-S
c-S
t-Sac
c-Sac
t-G
c-Gt-Gac
t-Sac
c-Sac
t-S
c-S
t-G
c-G
t-Gacc-Gac
t-Sac
t-S
c-Sc-Sac
t-G
c-G
t-Gac
c-Gac
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
10 15 20
Retention time (min) Figure 1. Chromatograms of the DFRC´ degradation products of selected MWL from sisal, kenaf, hornbeam and abaca. c-G, t-G, c-S and t-S are the cis- and trans-guaiacyl and syringyl monomers, respectively. c-Gac, t-Gac, c-Sac and t-Sac are the originally acetylated cis- and trans-guaiacyl and syringyl monomers, respectively.
On the other hand, it has been recently and elegantly proved that acetylated lignin in kenaf derives from polymerization of pre-acetylated monolignols and not from acetylation of the lignin polymer (9, 10). Acetylation of lignin at monomer stage only partially affects the course of the coupling reactions, and
117

5. Resultados y discusión
118
these acetylated monolignols can still undergo the ��O–4-coupling reactions that incorporate them into the lignin polymer. Obviously, the resulting acetylated lignin polymer is produced and has the mechanical properties required by the plant. However, the presence of �-acetylated monolignols alters to some extent the structure of the lignins because the �-OH group of a monolignol participates in some post-coupling reactions, such as ��� coupling, internally trapping the quinone methide. With the �-OH group acetylated, such internal reactions are no longer possible and the quinone methide must be rearomatized by trapping an external nucleophile, usually water, and forming as a result new ��� products in the lignin different to the expected resinol structures formed from non-acetylated monolignols (9). Figure 3 shows the different tetrahydrofuran structures arising from the ��� homo- and cross-coupling of the two sinapyl (acetylated and non-acetylated) monolignols. It is clear that structures I and II can only be formed if sinapyl alcohol is pre-acetylated and then undergoes ��� coupling. These ��� homo- (I) and cross-coupled (II) structures arising from pre-acetylated sinapyl alcohol were found in kenaf lignin by DFRC´ and 2D-NMR (9, 10), which unequivocally demonstrates that sinapyl alcohol is pre-acetylated prior to lignification and that sinapyl acetate behaves as a real monolignol in kenaf lignin.
OCH3
OProp
OProp
GM+= 292; [M+-56]= 236
OCH3
OProp
O-C-CH3
=
O
Gac
M+= 278; [M+-56]= 222
OCH3CH3OOProp
OProp
SM+= 322; [M+-56]= 266
OCH3CH3OOProp
O-C-CH3
=
O
Sac
M+= 308; [M+-56]= 252 Figure 2. Structures and mass fragments of the �-OH (G and S) and naturally �-acetylated (Gac and Sac) lignin monomers released after DFRC´ of MWL.

5. Resultados y discusión
O
CH3O
OCH3
OH
OCH3CH3O
OH
CH3-C-O
=
O
HO� �
�
���
OCH3CH3OOH
OH
��
�
��
�
OCH3CH3OOH
O-C-CH3
=O
+���
cross-coupling
II
OCH3
CH3O
HO
OCH3
OCH3
OH
O
O-C-CH3
=
O
CH3-C-O
=
O
� �
��
��
��
�
OCH3CH3OOH
O-C-CH3
=
O
���
homo-coupling
I
��
�
OCH3CH3OOH
O-C-CH3
=
O
+
OCH3CH3OOH
OH
��
�
���
homo-coupling
OCH3
CH3O
HO
OH
OCH3
OCH3
O
O
IIIOCH3CH3O
OH
OH
��
�
+�
�
� �
�
�
Figure 3. Structures of the tetrahydrofuran dimers arising from the ��� coupling of sinapyl alcohol and sinapyl acetate. I: ����coupling product of two sinapyl acetates; II: ����coupling product of a sinapyl alcohol and a sinapyl acetate; III: ����coupling product (syringaresinol) of two sinapyl alcohols.
We also investigated the presence of the tetrahydrofuran structures arising from ��� coupling of sinapyl acetates in the MWL selected for this study, by using the DFRC´ method. The DFRC´ degradation products of the tetrahydrofuran structures depicted in Figure 3 are presented in Figure 4, and their mass spectra have already been published (9). Compounds I´, II´a and II´b, arising from originally acetylated sinapyl alcohol, together with compound III´ arising from the resinol structure, were found in most of the samples analyzed. Figure 5 shows the chromatograms (sum of the single ion chromatograms of the respective base peaks) of the DFRC´ degradation products of the tetrahydrofuran
119

5. Resultados y discusión
120
structures arising from ����coupling of sinapyl alcohol (and its acetylated counterpart) in selected samples. It is interesting to note the relatively high amounts of compounds I´, II´a and II´b, arising from native acetylated lignin units in kenaf and especially in sisal lignin and the complete absence of ��� coupling structures in abaca lignin. However, we also observed that the respective tetrahydrofuran structures arising from the ����coupling of the acetylated guaiacyl counterparts could not be found, even in the case of sisal lignin (with 50% acetylated ether-linked G units). The presence of compound I´(arising from structure I in lignin) and compounds II´a and II´b (arising from structure II) in the DFRC´ degradation products of sisal, kenaf, jute, hornbeam, and other lignins clearly indicates that in these samples sinapyl alcohol is pre-acetylated and behave as a real monolignols participating in post cross-coupling reactions. Therefore, it is possible that in all angiosperms, in which we have shown the occurrence of acetylated lignin units, sinapyl and possibly coniferyl acetates participate in lignification as true lignin precursors. This implies that the naturally acetylated polymeric lignins in all these plants derive not from acetylation of the lignin polymer but from polymerization of pre-acetylated monolignols, as already suggested (9). Thus, the traditional concepts of both lignin biosynthesis and structure must be reconsidered. This indicates, in agreement with other authors (7, 27) that the lignification process is very flexible, and that the definition of lignin must not be restricted to a polymer of the three traditional hydroxycinnamyl alcohols.
The relative abundance of the compounds released in Figure 5 gives some additional information. In sisal, the relative molar abundance of the acetylated versus the non-acetylated sinapyl alcohols forming ��� linkages is 44:56, with a predominance of the non-acetylated sinapyl alcohol, whereas their relative molar abundances in ether-linked structures is 78:22, with a strong predominance of sinapyl acetate. This indicates that sinapyl acetate has a lower affinity to form ��� linkages than the normal sinapyl alcohol and therefore those lignins having a high extent of acetylation would produce lower amounts of ��� linkages. This is in agreement with the high proportions of �-O-4 substructures present in sisal lignin, as indicated by 2D-NMR (data not shown). Interestingly, abaca MWL, with a very high extent of acetylation (80% of S units), lacks ��� linkages, including those arising from normal non-acetylated lignin units (Figure 5), and produces almost exclusively ��O-4 substructures. Similar results were observed after normal DFRC and 2D-NMR (data not shown). Therefore, it seems that the high extent of �-acetylation would favor the formation of a predominantly ������lignin structure. The difficulty in finding ��� substructures arising from naturally acetylated guaiacyl lignin in sisal, despite the high abundance of this type of units, would be perhaps due to the low amounts of ��� linkages produced.

5. Resultados y discusión
CH3O
PrO
OPr
OPr
CH3O
OPr
OCH3CH3O
III´; M+= 644; [M-56]+= 588
IIIDFRC´
CH3O
PrO
CH3O
OPr
OCH3CH3O
I´; M+=616; [M-56]+= 560
IDFRC´
O-C-CH3
=
O
O-C-CH3=
O
CH3O
PrO
OPr
CH3O
OPr
OCH3CH3O
II´a; M+= 630; [M-56]+= 574
CH3O
PrOOPr
CH3O
OPr
OCH3CH3O
II´b; M+= 630: [M-56]+= 574
IIDFRC´
+
O-C-CH3=O
O-C-CH3
=
O
Figure 4. Aryltetralin products resulting from the DFRC´ degradation of the di- (I´), mono- (II´a and II´b) and none-acetylated (III´) ��� coupling structures. The molecular mass and base peaks are indicated under the structures.
Although it is now evident that native acetylated lignin units are widespread, and probably ubiquitous, in angiosperms, the role of such lignin acetylation in the plant is not yet known. Some studies indicated that �-acylation with p-coumarates may function as radical transfer carriers to help sinapyl alcohol incorporate into lignin when the wall peroxidases have a low reactivity with sinapyl alcohol directly (28). However, this is not the case for acetylated lignin monomers and the function of such acetylated lignin remains unknown.
121

5. Resultados y discusión
122
35 36 37 38 39 40Retention time (min)
I´
II´a,II´b
III´Sisal
I´
II´a,II´b
III´
Kenaf
II´a,II´b
III´
Hornbeam
Abaca
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
Figure 5. Detail of reconstructed (sum of the ions at m/z 560, 574 and 588) chromatograms of the DFRC´ degradation products of selected MWL from sisal, kenaf, hornbeam and abaca, showing the presence of aryltetralin ��� products containing two (I´), one (IIá and II´b) and none (III´) native acetates.
The high extent of lignin acetylation of sisal and other Agaves (preliminary results on another species, Agave americana, by DFRC´ of the whole fiber, without previous lignin isolation, also indicate more than 75% of acetylated S units) can provide some answers about its role in this plant. Since the resultant

5. Resultados y discusión
acetylated lignin polymer is more hydrophobic than normal lignin, the role of lignin acetylation could be associated with drought tolerance, as already advanced by Ralph (29). Sisal, as other Agaves, is a drought tolerant desert perennial plant successfully copying with high temperatures and desiccation. Agaves are characterized by tissue succulence and Crassulacean Acid Metabolism to minimize water loss. The presence of a highly acetylated lignin in Agaves will increase hydrophobicity of the vascular tissues thus helping to reduce water loss in the plant. However, abaca, despite being highly acetylated, has not succulent leaves and is not drought tolerant, and therefore the highly acetylated lignin might have a different role for this plant. In this case, the role of the high extent of lignin acetylation could be more related with the lower affinity of acetylated monolignols to form ��� linkages producing a more non-condensed lignin enriched in �-O-4 linkages. Whatever the reason for the occurrence of acetylated lignin in plants, it seems that the mechanism for lignin acetylation confers the plant a high flexibility to produce different types of lignins with different degree of acetylation to adapt to different environmental conditions.
In conclusion, the presence of naturally acetylated lignin units, which in some plants make up to 80% of the uncondensed S lignin, has been largely underestimated. This has mainly been due to the analytical methodologies used for their isolation and structural characterization, which are not appropriate for the analysis of native acetylated lignin. Therefore, all subsequent lignin structural studies should take into account the occurrence of these moieties and accordingly adapt the methodological protocols used. Acknowledgements This study has been supported by the Spanish MEC (project AGL2005-01748) and EU contract NMP2-CT-2006-26456. JR thanks the Spanish CSIC for an I3P fellowship; GM thanks the Spanish Ministry of Education for a FPI fellowship. Literature cited [1] Ralph, J. An unusual lignin from kenaf. J. Nat. Prod. 1996, 59, 341-342.
[2] Sun, R.C.; Fang, J.M.; Goodwin, A.; Lawther, J.M., Bolton, A.J. Fractionation and characterization of ball-milled and enzyme lignin from abaca fibre. J. Sci. Food Agric. 1999, 79, 1091-1098.
[3] Sun, R.C.; Sun, X.F.; Wang, S.Q.; Zhu, W.; Wang, X.Y. Ester and ether linkages between hydroxycinnamic acids and lignin from wheat, rice, rye, and barley straws, maize stems, and fast-growing poplar wood. Ind. Crops & Prod. 2002, 15, 179-188.
123

5. Resultados y discusión
124
[4] del Río, J.C.; Gutiérrez, A.; Rodríguez, I.M., Ibarra, D., Martínez, A.T. Composition of non-woody plant lignins and cinnamic acids by Py-GC/MS, Py/TMAH and FT-IR. J. Anal. Appl. Pyrol. 2007, 79, 39-46.
[5] Ros Barceló, A. Lignification in plant cell walls. Int. Rev. Cytol. 1997, 176, 87-132.
[6] Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546
[7] Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P. F.; Marita, J. M.; Hatfield, R. D.; Ralph, S. A.; Christensen, J. H. et al. Lignins: natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 2004, 3, 29-60.
[8] Sarkanen, K.V.; Chang, H. -M.; Allan, G.C. Species variation in lignins. III. Hardwood lignins. Tappi 1967, 50, 587-590.
[9] Lu, F.; Ralph, J. Preliminary evidence for sinapyl acetate as a lignin monomer in kenaf. Chem. Commun. 2002, 90–91.
[10] Lu, F.; Ralph, J. Novel ����structures in lignins incorporating acylated monolignols. In Proceedings of 13th International Symposium on Wood, Fiber, and Pulping Chemistry, Auckland, New Zealand, May 16-19, 2005; Volume 3, pp. 233-237.
[11] Smith, D. C. C. p-Hydroxybenzoate groups in the lignin of aspen (Populus tremula). J. Chem. Soc. 1955, 2347–2351.
[12] Nakano, J.; Ishizu, A.; Migata, N. Studies on lignin. XXXII. Ester groups of lignin. Tappi 1961, 44, 30–32.
[13] Monties, B.; Lapierre, C. Donnés récentes sur l’hétérogénéite de la lignine (Recent data on the heterogeneity of lignin). Physiol. Veg. 1981, 19, 327-348.
[14] Landucci, L. L.; Deka, G. C.; Roy, D. N. A. 13C NMR study of milled wood lignins from hybrid Salix clones. Holzforschung 1992, 46, 505-511.
[15] Ralph, J.; Hatfield, R. D.; Quideau, S.; Helm, R. F.; Grabber, J. H.; Jung, H. -J. G. Pathway of p-coumaric acid incorporation into maize lignin as revealed by NMR. J. Am. Chem. Soc. 1994, 116, 9448-9456.
[16] Sun, R. C.; Fang, J. M.; Tomkinson, J. Fractional isolation and structural characterization of lignins from oil palm trunk and empty fruit bunch fibres. J. Wood Chem. Technol. 1999, 19, 335–356.
[17] Lu, F. and Ralph, J. Detection and determination of p-coumaraloylated units in lignin. J. Agric. Food Chem. 1999, 47, 1985-1992.

5. Resultados y discusión
[18] Meyermans, H.; Morreel, K.; Lapierre, C.; Pollet, B.; De Bruyn, A.; Busson, R.; Herdewijn, P:; Devreese, B.; Van Beeumen, J.; Marita, J. M.; et al. Modification in lignin and accumulation of phenolic glucosides in poplar xylem upon down-regulation of caffeoyl-coenzyme A O-methyltransferase, an enzyme involved in lignin biosynthesis. J. Biol. Chem. 2000, 275, 36899–36909.
[19] del Río, J. C.; Gutiérrez, A.; Martínez A. T. Identifying acetylated lignin units in non-wood fibers using pyrolysis-gas chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 1181-1185.
[20] Gutiérrez, A.; Rodríguez, I. M.; del Río J. C. Chemical characterization of lignin and lipid fractions in kenaf bast fibers used for manufacturing high-quality papers. J. Agric. Food Chem. 2004, 52, 4764-4773.
[21] del Río, J. C.; Gutiérrez, A. Chemical composition of abaca (Musa textilis) leaf fibers used for manufacturing of high quality paper pulps. J. Agric. Food Chem. 2006, 54, 4600-4610.
[22] Lu, F.; Ralph, J. Derivatization followed by reductive cleavage (DFRC method), a new method for lignin analysis: protocol for analysis of DFRC monomers. J. Agric. Food Chem. 1997, 45, 2590-2592.
[23] Lu, F.; Ralph, J. The DFRC method for lignin analysis. Part 1. A new method for ��aryl ether cleavage: lignin model studies. J. Agric. Food Chem. 1997, 45, 4655-4660.
[24] Lu, F.; Ralph, J. The DFRC method for lignin analysis. 2. Monomers from isolated lignin. J. Agric. Food Chem. 1998, 46, 547-552.
[25] Ralph, J.; Lu, F. The DFRC method for lignin analysis. 6. A simple modification for identifying natural acetates in lignin. J. Agric. Food Chem. 1998, 46, 4616-4619.
[26] Björkman, A. Studies on finely divided wood. Part I. Extraction of lignin with neutral solvents. Sven. Papperstidn. 1956, 59, 477-485.
[27] Sederoff, R. D.; MacKay, J.; Ralph, J.; Hatfield, R. Unexpected variation in lignin. Curr. Opin. Plant Biol. 1999; 2: 145.
[28] Takahama, U.; Oniki, T.; Shimokawa, H. A possible mechanism for the oxidation of sinapyl alcohol by peroxidase-dependent reactions in the apoplast: enhancement of the oxidation by hydroxycinnamic acids and components of the apoplast. Plant Cell Physiol. 1996, 37, 499-504.
[29] Ralph, J. Elucidation of new pathways in normal and perturbed lignification. Appita 2005, 3-13.
125

5. Resultados y discusión
Publicación III: del Río J.C., Rencoret J., Marques G., Gutiérrez A., Ibarra D., Santos J.I., Jiménez-Barbero J., Zhang L. and Martínez A.T. (2008) Highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. Journal of Agricultural and Food Chemistry, 56, 9525-9534.
126

5. Resultados y discusión
Highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants
José C. del Río†, Jorge Rencoret†, Gisela Marques†, Ana Gutiérrez†, David Ibarra‡, J. Ignacio
Santos‡, Jesús Jiménez-Barbero‡, Liming Zhang¥, Ángel T. Martínez‡
†Instituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, P.O. Box 1052, 41080-
Seville, Spain ‡Centro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid, Spain
¥Royal Institute of Technology (KTH), Fiber and Polymer Technology, SE-100 44 Stockholm, Sweden
Abstract The structure of lignins isolated from the herbaceous plants sisal (Agave sisalana), kenaf (Hibiscus cannabinus), abaca (Musa textilis) and curaua (Ananas erectifolius) has been studied upon spectroscopic (2D-NMR) and chemical degradative (Derivatization Followed by Reductive Cleavage) methods. The analyses demonstrate that the structure of the lignins from these plants is highly remarkable, being extensively acylated at the �-carbon of the lignin side-chain (up to 80% acylation) with acetate and/or p-coumarate groups, and preferentially over syringyl units. While the lignins from sisal and kenaf are �-acylated exclusively with acetate groups, the lignins from abaca and curaua are esterified with acetate and p-coumarate groups. The structures of all these highly-acylated lignins are characterized by a very high syringyl/guaiacyl ratio, a large predominance of �-O-4´ linkages (up to 94% of all linkages) and a strikingly low proportion of traditional �-�´�linkages, which indeed are completely absent in the lignins from abaca and curaua. The occurrence of �-�´ homo-coupling and cross-coupling products of sinapyl acetate in the lignins from sisal and kenaf indicates that sinapyl alcohol is acetylated at monomer stage and that, therefore, sinapyl acetate should be considered as a real monolignol involved in the lignification reactions. Keywords: lignin, herbaceous plants, sinapyl acetate, sinapyl p-coumarate, 2D-NMR, HSQC, DFRC, sisal, kenaf, abaca, curaua 1. Introduction Lignins are complex natural biomacromolecules characteristics of vascular plants, where they provide mechanical support. In addition, lignin waterproofs the cell wall, enabling transport of water and solutes through the vascular system, and plays a role in protecting plants against pathogens (1). The lignin polymer results from the random oxidative coupling of p-hydroxycinnamyl monolignols mediated by laccases and/or peroxidases (2, 3). The three primary
127

5. Resultados y discusión
128
monolignols are p-coumaryl, coniferyl and sinapyl alcohols, which produce, respectively, p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) phenylpropanoid units when incorporated into the lignin polymer.
However, it is now widely accepted that other monomers also participate in coupling reactions giving rise to the lignin macromolecule. This is the case of �-acylated (with acetate, p-hydroxybenzoate and/or p-coumarate groups) lignins which have been discovered in many plants. Different grass lignins are partially p-coumaroylated and some hardwood lignins such as poplar, aspen or willow lignins are p-hydroxybenzoylated (3-11). Acetylated lignin units have also been reported to occur in many plants (12-16). Characteristic products from sinapyl and coniferyl acetate coupling have been detected upon degradative techniques (Py-GC/MS and derivatization followed by reductive cleavage, DFRC) in the lignin of several plants characterized by having a high S/G ratio such as sisal, kenaf, abaca and jute (15, 16). Previous studies have shown that lignin from these plants is acetylated exclusively at the �-position of the side-chain and that this acetylation occurred predominantly on S-units (12-16). Moreover, these studies have provided strong evidence that sinapyl acetate is implicated as a monomer in lignification in several plants and that the naturally acetylated lignin derives not from acetylation of the lignin polymer but from polymerization of pre-acetylated monolignols (14, 16, 17). The same seems also to occur with sinapyl p-coumarate and sinapyl p-hydroxybenzoate (17-19).
We have recently shown, using a previously developed modification of the DFRC method to allow detection of natural acetate groups (13) (the so-called DFCR´ methodology) that lignin �-acetylation is widespread, and probably ubiquitous, among angiosperms, although at different extents, but is absent from conifers (16). Moreover, the lignins of many plants (e.g. the non-woody sisal, kenaf, abaca or the hardwood hornbeam) are particularly extensively acetylated (up to 80% of the S-lignin moieties) (16). However, the DFRC degradation method only cleaves �- and �-aryl ether bonds allowing only the analysis of the monomeric degradation products released from non-condensed etherified lignin units. Therefore, the DFRC method, although extremely useful, does not give information of the whole macromolecule, and the extent of lignin acylation may be actually different from that estimated.
Spectroscopic techniques, and particularly 1D- and 2D-NMR, can provide information of the structure of the whole macromolecule and are powerful tools for lignin structural elucidation (7, 20-24). Therefore, they can be very useful to estimate the actual extent of lignin acylation. The main advantage of spectroscopic techniques over degradation methods is the analysis of the whole lignin structure and direct determination of the different lignin moieties and inter-unit linkages. In this paper, we study the structure of some naturally extensively acylated lignins occurring in several herbaceous plants (namely sisal, kenaf, abaca and curaua) by a combination of chemical degradative (DFRC) and spectroscopic (2D-NMR) techniques.

5. Resultados y discusión
2. Material and methods 2.1. Samples The plant samples selected for this study consist of bast fibers obtained from the stalk phloem layer of kenaf (Hibiscus cannabinus) and leaf fibers of sisal (Agave sisalana), abaca (Musa textilis) and curaua (Ananas erectifolius). The fibers were finely ground to sawdust using a knife mill (Analysenmühle A10, Janke and Kunkel GmbH, Staufen, Germany) before analysis. Milled-wood lignin (MWL) was extracted from finely ball-milled (150 h) plant material, free of extractives and hot water soluble material, using dioxane-water (9:1, v/v), followed by evaporation of the solvent, and purified as described (25). The final yields ranged from 5-15% of the original lignin content. Extension of milling time, which would increase yield, was avoided to prevent chemical modifications on the lignin structure. 2.2. DFRC (derivatization followed by reductive cleavage) The DFRC degradation was performed according to the developed protocol (26-28). Lignins (10 mg) were stirred for two hours at 50ºC with acetyl bromide in acetic acid (8:92). The solvents and excess of bromide were removed by rotary evaporation. The products were then dissolved in dioxane/acetic acid/water (5:4:1, v/v/v), and 50 mg powered Zn was added. After 40 min stirring at room temperature, the mixture was transferred into a separatory funnel with dichloromethane and saturated ammonium chloride. The pH of the aqueous phase was adjusted to less than 3 by adding 3% HCl, the mixture vigorously mixed and the organic layer separated. The water phase was extracted twice more with dichloromethane. The combined dichloromethane fractions were dried over anhydrous NaSO4 and the filtrate was evaporated in a rotary evaporator. The residue was acetylated for 1 h in 1.1 mL of dichloromethane containing 0.2 mL of acetic anhydride and 0.2 mL pyridine. The acetylated lignin degradation products were collected after rotary evaporation of the solvents, and subsequently analyzed by GC/MS. To asses the presence of naturally acetylated lignin units, a modification of the standard DFRC method by using propionylating instead of acetylating reagents (DFRC´) was used in the present study (13, 16).
The GC/MS analyses were performed with a Varian model Star 3800 GC equipped with an ion trap detector (Varian model Saturn 4000) using a medium-length (15 m) capillary column (DB-5HT, 5 m × 0.25 mm I.D., 0.1 �m film thickness) from J&W Scientific. The oven was heated from 120 (1 min) to 330 ºC at 6 ºC/min, and held for 4 min at the final temperature. The injector was programmed from 60ºC to 350ºC at a rate of 200ºC/min and held until the end of the analysis. The transfer line was kept at 300 ºC. Helium was used as the carrier gas at a rate of 2 mL/min. Quantification of the released individual monomers was performed using tetracosane as external standard, and by assuming similar
129

5. Resultados y discusión
130
response factors as those of the acetylated monomers previously reported (26), although without authentication on our instrument. Molar yields were calculated on the basis of molecular weights of the respective acetylated and/or propionylated compounds.
2.3. NMR spectroscopy NMR spectra were recorded at 25 ºC on a Bruker AVANCE 500 MHz equipped with a z-gradient triple resonance probe. Around 40 mg of lignin were dissolved in 0.75 mL of deuterated dimethylsulfoxide (DMSO-d6) and 2D-NMR spectra were recorded in HSQC (heteronuclear single quantum correlation) experiments. The spectral widths were 5000 Hz and 13200 Hz for the 1H- and 13C-dimensions, respectively. The number of collected complex points was 2048 for 1H-dimension with a recycle delay of 5 s. The number of transients was 64, and 256 time increments were always recorded in 13C-dimension. The 1JCH used was 140 Hz. The J-coupling evolution delay was set to 3.2 ms. Squared cosine-bell apodization function was applied in both dimensions. Prior to Fourier transform the data matrixes were zero filled up to 1024 points in the 13C-dimension. The central solvent peak was used as an internal reference (C 40.1; H 2.50 ppm). HSQC cross-signals were assigned by combining the results of the different experiments, and comparing with the literature (20-23, 29-32). A semiquantitative analysis of the intensities of the HSQC cross-signal intensities was performed (29, 33). Since the cross-signal intensity depends on the particular 1JCH value, as well on the T2 relaxation time, a direct analysis of the intensities is impossible. A more accurate quantification of the 2D HSQC NMR data analysis was achieved by using the recently published method in which the errors caused by T2 relaxation, off-resonance effect and coupling constant deviation could mostly be eliminated (34). Thus, the integration on the cross-signals was performed separately for the different regions of the HSQC spectra, which contain signals that correspond to chemically analogous carbon-proton pairs. For these signals, the 1JCH coupling value is relatively similar, their chemical shifts are also similar to each other hence the error of off-resonance effect is small, and therefore can be used semiquantitatively to estimate the relative abundance of the different species. In the aliphatic oxygenated region, inter-unit linkages were estimated from C�-H� correlations to avoid possible interference from homonuclear 1H-1H couplings, and the relative abundance of side-chains involved in inter-unit linkages were calculated. In the aromatic region, C2,6-H2,6 correlations from S units and C2-H2 plus C6-H6 correlations from G units were used to estimate the S/G ratio of lignin, and p-coumaric acid content was estimated from its C2,6-H2,6 correlation signal. Lignin acylation was estimated from the intensities of C�-H� correlations in acylated and non-acylated side-chains.

5. Resultados y discusión
3. Results and discussion MWL is a lignin preparation considered as the most representative of the whole native lignin in the plant (25), in spite of its low yield and the possibility of some modifications during milling (35). Therefore, in this work, the MWL isolated from the selected herbaceous plants (sisal, kenaf, abaca and curaua) were analyzed by 2D-NMR and DFRC to get insight into their structural characteristics. However, we must still keep in mind that the results obtained here reflect only the structure of isolated MWL, which only represents a part of the whole lignin in the plant. 3.1. HSQC-NMR spectra of highly acetylated lignins The HSQC NMR spectra of the different MWL showed three regions corresponding to aliphatic, side-chain and aromatic 13C-1H correlations. The aliphatic (non-oxygenated) region showed signals with no structural information (except for the presence of acetate signals at �C/�H 20.7/1.74 ppm) and therefore is not discussed in detail. The side-chain regions (�C/�H 50-90/2.5-5.5 ppm) and the aromatic regions (�C/�H 95-150/5.5-8.5 ppm) of the MWL selected for this study are shown in Figures 1 and 2, respectively, and the main substructures found in these lignins are depicted in Figure 3. The main lignin cross-signals assigned in the HSQC spectra are listed in Table 1.
In the side-chain region, cross-signals of methoxyls (�C/�H 56.2/3.73 ppm) and side-chains in ��O-4´ substructures were the most prominent in all lignins. Interestingly, the spectra clearly show the presence of intense signals corresponding to acylated �-carbon in the range from �C/�H 63.5/3.83 and 4.30 ppm in all lignin samples, together with the presence of signals from normal hydroxylated �-carbon (at �C/�H 60.2/3.30 and 3.70 ppm). The HSQC spectra indicate that these lignins are extensively acylated and that acylation occurs exclusively at the �-position of the lignin side-chain. HSQC can not identify the nature of the ester, although it was shown in previous studies that acetates (and p-coumarates) occurred in these lignins (12, 13, 15, 16, 36). Traces (less than 0.5% of acylated �-carbon) of a signal at �C/�H 5.87/74.66 ppm corresponding to acylated �-carbon were found in the HSQC of sisal and kenaf MWL although it was absent in the rest of the lignins studied here. This signal could be due to a migration of the acetyl group from the �-carbon to the �-carbon in the lignin side-chain, as already advanced by Ralph (12).
An estimation of the percentage of �-acylation of the lignin side-chain was calculated from the HSQC spectra by integration of the signals corresponding to the hydroxylated and acylated �-carbon (Table 2), and ranged from 58% in kenaf bast lignin up to 80% in abaca lignin. Although kenaf lignin has already been known for long to be highly acetylated (12), up to our knowledge, this is the first time that this type of highly acylated lignin has been described in other
131

5. Resultados y discusión
132
plants. The high extent of lignin acylation observed in different herbaceous plants (including both mono- and eudicotyledoneous) indicates that this type of lignin might be more frequent than previously thought. Naturally acetylated lignins may also occur in many other plants but their occurrence has probably being biased due to the limitations of the analytical procedures used for their isolation and/or structural characterization. Natural acetates present on lignin might have been hydrolyzed and removed when using traditional isolation methods (such as alkaline extraction often applied to non-wood lignins) and degradative procedures for chemical characterization (such as nitrobenzene oxidation, CuO oxidation or thioacidolysis). Indeed, for spectroscopic analysis, e.g. using NMR, lignin is frequently in vitro acetylated for improved solubility and spectroscopic properties, which prevented the detection of natural lignin acetylation.
�C
65
�H
60
70
75
80
85
55
3.03.54.04.55.0
(a) MeO
A�A'�
A'�D�
D�´
D�
carbohydrates
C�
C�
C�
A�
�C
�H
60
65
70
75
80
85
55
3.03.54.04.55.0
(b) MeO
carbohydrates
D�
C�
A�A'�
C�
A'�A�
D�
D�´ C�
�C
60
65
70
75
80
85
55
3.03.54.04.55.0�H
(d) MeO
carbohydrates
A�A'�/A''�
A'�/A''�A�
D�
D�´
�C
60
65
70
75
80
85
55
3.03.54.04.55.0�H
(c) MeO
carbohydrates
A�A'�/A''�
A��/A´��/A´´�
A'�/A''�D�
D�´ A�
D�
A��/A´� A��/A´�
A��/A´��/A´´�
Figure 1. Expanded side-chain region, �C/�H 50-90/2.5-5.5 ppm, of the HSQC spectra of the lignins from (a) sisal, (b) kenaf, (c) abaca and (d) curaua. Carbohydrate signals are presented in grey color. See Table 1 for signal assignment and Figure 3 for the main lignin structures (A-D) identified.

5. Resultados y discusión
110
120
130
140
100�C
6.06.57.07.58.0�H
S2,6 (C�=O)S2,6
G2
G5G6
D2`
D6`
(a)
110
120
130
140
100
6.06.57.07.58.0�H
�C
D2`
D6`
S2,6 (C�=O)S2,6
G2
G5
G6
(b)
110
120
130
140
100
6.06.57.07.58.0
�C
�H
S2,6 (C�=O)S2,6
G2
G6
D2`
D6`
A''2´,6´
A''�´
A''�´
A''3´,5´
G5
(c)
110
120
130
140
100
6.06.57.07.58.0�H
�C
S2,6 (C�=O)S2,6
G2
G6
G5
(d)
A''2´,6´
A''�´
A''3´,5´
A''�´
D2`
D6`
S/G 3.9 S/G 5.6
S/G 8.7 S/G 4.9
Figure 2. Expanded aromatic region, �C/�H 95-150/5.5-8.5 ppm, of the HSQC spectra of the lignins from (a) sisal, (b) kenaf, (c) abaca and (d) curaua. See Table 1 for signal assignment and Figure 3 for the main lignin structures (A-D) identified. G and S are the guaiacyl and syringyl aromatic units, respectively.
The side-chain region of the HSQC spectra gives also additional information about the inter-unit linkages present in the structure of these lignins. All the spectra showed prominent signals corresponding to ��O-4´ aryl ether linkages. The C�-H� correlations in ��O-4´ substructures were observed at �C/�H 72.3/4.86 ppm (structures A, A' and A''), while the C�-H� correlations were observed at �C/�H 86.5/4.10 ppm in normal �-OH ��O-4´ aryl ether substructures (A) but shifted to �C/�H 83.6/4.32 ppm in �-acylated ��O-4´ aryl ether substructures (A', A''). ��O-4´ aryl ether substructures were highly predominant in all the lignins analyzed here although other substructures were also observed. Small signals corresponding to spirodienone (��1´, ��O��´ linkages) substructures (D)� can be observed in the spectra of sisal, kenaf, abaca and curaua lignins. Signals of spirodienone C�-H�, C�´-H�´ and C�-H� correlations were observed at �C/�H 85.4/4.64, 85.4/4.80 and 56.1/3.09 ppm, respectively. Spirodienone substructures were previously reported in the lignin
133

5. Resultados y discusión
134
Table 1. Assignment of Main Lignin 13C-1H Cross-Signals in the MWL HSQC Spectra Shown in Figures 1 and 2.
�C/�H (ppm) Assignment 53.7/3.12 C�-H� in ���' (resinol) substructures (C) 56.1/3.09 C�-H� in ���'�(spirodienone) substructures (D) 60.0/3.38-3.71 C�H� in �-O-4' substructures (A) 63.8/3.83-4.30 C�H� in �-acylated �-O-4' substructures (A' and A'') 71.7/3.81 and 4.17 C�-H� in ���' (resinol) substructures (C) 72.3/4.86 C�-H� in �-O-4' substructures (A, A' and A'') 82.1/5.12 C�-H� in ���'�(spirodienone) substructures (D) 83.6/4.32 C�-H� in �-acylated �-O-4' substructures (A' and A'') 85.4/4.64 C�-H� in ���' (resinol) substructures (C) 85.4/4.80 C�´-H�´ in ���'�(spirodienone) substructures (D) 86.5/4.10 C�-H� in �-OH �-O-4' substructures (A) 87.7/5.45 C�-H� in phenylcoumaran substructures (B) 103.8/6.68 C2-H2 and C6-H6 in syringyl units 106.7/7.36 and 7.21 C2-H2 and C6-H6 in oxidized (C�=O) syringyl units 111.5/6.99 C2-H2 in guaiacyl units 111.6/6.23 C2´-H2´ in ���'�(spirodienone) substructures (D) 114.3/6.24 C�´-H�´ in p-coumaroylated substructures (A'') 115.2/6.71 and 6.94 C5-H5 in guaiacyl units 116.2/6.77 C3´-H3´ and C5´-H5´ in p-coumaroylated substructures (A'') 118.3/6.19 C6´-H6´ in ����(spirodienone) substructures (D) 119.5/6.83 C6-H6 in guaiacyl units 130.5/7.4 C2´-H2´ and C6´-H6´ in p-coumaroylated substructures (A'') 145.1/7.39 C�´-H�´ in p-coumaroylated substructures (A'')
from kenaf bast fibers by Zhang et al. (37). Phenylcoumaran (��5´ linkages) substructures (B) were also found, although in very small proportions. Very weak signals corresponding to C�-H� correlations of phenylcoumaran substructures at �C/�H 87.7/5.45 ppm were observed in the spectra of sisal, kenaf and curaua lignins, but were absent in the spectrum of abaca. The presence of these low amounts of phenylcoumaran substructures was expected due to the very low levels of guaiacyl lignin units in all these samples. Finally, resinol (���´ linkages� substructures (C) were clearly observed in the spectrum of kenaf. Signals for the C�-H�, C�-H� and the double C�-H� correlations of resinol substructures were observed at �C/�H 85.4/4.64, 53.7/3.12 and 71.7/3.81 and 4.17 ppm, respectively. Resinol substructures could also be observed, although in very small traces, in the spectrum of sisal, but were completely absent in the spectra of abaca and curaua lignins. The relative abundances of the main inter-unit linkages present in the MWL selected for this study were calculated from the HSQC spectra and are shown in Table 2. All these highly acetylated lignins share a common characteristic, the strikingly high proportion of ��O-4´ ether linkages (up to 94% of all linkages) and a very low proportion of condensed linkages (i.e. ���´, ���´ and ���´). Some of these condensed linkages (���´ and ���´) are even absent in some lignins (abaca and curaua).

5. Resultados y discusión
Table 2. Structural characteristics (percentage of �-acylation, relative abundance of the main inter-unit linkages, and S/G ratio) observed from the HSQC spectra of the selected MWL. sisal kenaf abaca curaua � Percentage of �-acylation 68 58 80 69 Linkage relative abundance (% of side-chains involved) �-O-4' alkyl-aryl ether 89 84 94 94 �-1' (spirodienone) 5 6 6 4 �-5' (phenylcoumaran) 2 2 0 2 �-�' (syringaresinol) 4 8 0 0
S/G ratio 3.9 5.6 8.7 4.9
The main cross-signals in the aromatic region of the HSQC spectra (Figure 2)
correspond to the aromatic rings of the different lignin units. Signals from syringyl- (S) and guaiacyl- (G) lignin units can be observed in all spectra. The syringyl units show a prominent signal for the C2,6-H2,6 correlation at �C/�H 103.8/6.68 ppm, while guaiacyl units showed different correlations for C2-H2 (�C/�H 111.5/6.99 ppm), C5-H5 (�C/�H 115.2/6.71 and 6.94) and C6-H6 (�C/�H 119.5/6.83 ppm). Signals corresponding to C2,6-H2,6 correlations in C�-oxidized S-lignin units were observed at �C/�H 106.7/7.36 and 7.21 ppm. No signals for p-hydroxyphenyl (H) lignin units could be detected in the HSQC spectra of these lignins. An estimation of the relative proportions of the S and G-lignin units in the HSQC spectra revealed that all the lignins selected for this study present a very high S/G ratio, ranging from 3.9 in sisal to 8.7 in abaca (Table 2). Other signals present in this region of the HSQC spectra are from spirodienone substructures (D) with C2´-H2´ and C6´-H6´ correlations at �C/�H 111.6/6.23 and 118.3/6.19, respectively. Prominent signals corresponding to p-coumarate structures were observed in the lignins of abaca and curaua. Cross-signals corresponding to the correlations C2´,6´-H2´,6´ at �C/�H 130.5/7.40 ppm and C3´,5´-H3´,5´ at �C/�H 116.2/6.77 ppm of the aromatic ring and signals for the correlations of the unsaturated C�´-H�´ at �C/�H 145.1/7.39 and C�´-H�´ at 114.3/6.24 ppm of the p-coumarate unit in structure A'' of Figure 3, were observed in this region of the HSQC spectra of abaca and curaua. In abaca lignin, p-coumaric acid has already been reported to be esterified to the lignin polymer (8, 16, 35, 38). 3.2. Degradation Followed by Reductive Cleavage (DFRC and DFRC´) The HSQC data shown above indicate that these lignins are extensively acylated at the �-position of the side-chain, but cannot provide additional information on
135

5. Resultados y discusión
136
the nature of the acyl group (besides the occurrence of acetate and p-coumarate moieties). A sensitive and selective method is therefore needed to reveal the
��
�
12
34
5
6
1’6’
5’4’
3’2’HO
O
O
OMe
MeOO
O
OMeMeO
A' A''
��
�
12
34
5
6
1'’6'’
5'’4'’
3'’2'’HO
O
O
OMeMeO
O
O
HO
�´�´
�´2´
3´
5´6´
1´
4´
MeO
OMe
��
�
12
34
5
6
1’6’
5’4’
3’2’HO
O
O
HO
OMe
MeO
OMeMeO
A
���
12
34
5
6
1’2’
3’4’
5’
6’
OHO
O
OMe
OMeMeO
B
�
1´2´
3´4´
5´
6´
O
O
OHHO
�� � ´�´
�´
O
1''2''
3''4''
5''
6''
OMeMeO
OMeO
2
1
4 3
56MeO
OMeMeO
D
�
�
�
1
23
4
56
�’
�’
� ’
1’
2’3’
4’
5’6’
O
O
O
O
OMe
OMe
MeO
OMe
C Figure 3. Main structures present in the highly acylated lignins studied here: (A) ���-4´ aryl ether linkages; (A') ���-4´ aryl ether linkages with acetylated �-carbon; (A'') ���-4´ aryl ether linkages with p-coumaroylated �-carbon; (B) phenylcoumaran structures formed by ��5´ and ���-4´ linkages; (C) resinol structures formed by ���´, ��O��´and ��O��´ linkages; and (D) spirodienone structures formed by ��1´,��O-�´ linkages. nature of the acyl group that is esterifying the �-carbon of the lignin side-chain and to know to which lignin moiety it is attached. The DFRC degradation method, which cleaves �- and �-ether linkages in the lignin polymer leaving �-esters intact (26-28), seems to be the most appropriate method for the analysis of native �-acylated lignin.
DFRC analysis of the lignin samples selected for this study allowed confirming that p-coumarate groups are attached to the �-carbon of abaca and curaua lignins, and predominantly on syringyl units (Figure 4). Saturated p-coumarate (dihydro-p-coumarate) esterified to sinapyl alcohol (as its acetate derivative, Sdpc) was expected to be the major DFRC degradative compound,

5. Resultados y discusión
according to Lu and Ralph (9), and this was the only degradation product that was quantified in our previous paper (16). However, a closer look to other major degradation compounds produced upon DFRC of abaca and curaua lignins indicated the release of important amounts of the unsaturated counterpart, that is, intact sinapyl p-coumarate (as its acetate derivative, Spc), that was biased, and therefore not quantified, in our previous paper. Therefore, in this work, we have now taken into account both compounds to quantify the total abundance of sinapyl p-coumarate units present in these lignins (Table 3). Trace amounts of the respective coniferyl p-coumarate could also de detected in abaca and curaua lignins. Moreover, some amounts of free p-coumaric acid (as its acetate derivative) could be observed among the DFRC degradation products of curaua lignin as a broad peak (Figure 4), that could probably correspond to p-coumaric acid moieties linked to lignin through ��O-4´ aryl-ether bonds.
The original DFRC degradation method, however, does not allow the analysis of native acetylated lignin because the degradation products are acetylated during the degradation procedure, but with appropriate modification of the protocol by substituting acetylating reagents with propionylating reagents, DFRC´, it is also possible to obtain information about the occurrence of native lignin acetylation (13). Figure 5 shows the chromatograms of the DFRC´ products released from the lignin samples selected in this study. All the analyzed lignins released the cis and trans isomers of guaiacyl (c-G and t-G) and syringyl (c-S and t-S) lignin monomers (as their propionylated derivatives) arising from normal �-OH units in lignin. In addition, the presence of originally �-acetylated guaiacyl (c-Gac and t-Gac) and syringyl (c-Sac and t-Sac) lignin units could also be clearly observed in the chromatograms of all of the selected lignins indicating that acetylation occurred exclusively at the �-carbon of the lignin side-chain, as already observed in the HSQC spectra. Table 3. Abundance (Molar Yields) of the DFRC and DFRC´ degradation monomers of the MWL isolated from the different plants selected for this study, and relative abundances of the different acylated (acetylated and p-coumaroylated) lignin moieties. Monomers (�mol/g lignin) G Gac Gpc S Sac Spc %Sac
a %Spcb %Gac
c %Gpcd S/G
sisal 122 124 0 108 378 0 78 0 50 0 2.0 kenaf 390 38 0 543 780 0 59 0 9 0 3.1 abaca 50 3 1 21 131 124 48 45 6 2 5.1 curaua 250 252 3 515 595 195 46 15 50 1 2.6
a %Sac is the percentage of acetylated S units respect to the total S units. b %Spc is the percentage of p-coumaroylated S units respect to the total S units. c %Gac is the percentage of acetylated G units respect to the total G units. d %Gpc is the percentage of p-coumaroylated G units respect to the total G units
137

5. Resultados y discusión
138
5 10 15 20 2Retention time
5(min)
(a)
(b)
tS
tS
cS
cS
tG
tG
pC
Spc
SpcSpcSdpc
Spc
SpcSpc
Sdpc
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
Figure 4. Chromatograms of the DFRC degradation products of the MWL from (a) abaca and (b) curaua, showing the presence of sinapyl alcohol esterified to p-coumarate moieties. Sdpc and Spc are the sinapyl alcohol esterified with dihydro-p-coumarate and p-coumarate, respectively (as their acetyl derivative). c-G, t-G, c-S and t-S are the normal cis- and trans-guaiacyl and syringyl monomers, respectively, (as their acetyl derivatives).
The results from the DFRC and DFRC´ analysis of the MWL selected for this study, namely the molar yields of the released monomers, the percentages of naturally acetylated guaiacyl (%Gac) and syringyl (%Sac) and p-coumaroylated guaiacyl (%Gpc) and syringyl (%Spc) lignin moieties, and the S/G ratios, are presented in Table 3. The data indicate that a high extent of �-acetylation occurs

5. Resultados y discusión
in all lignins studied here, and that p-coumaric acid is also found partially esterifying the lignin of abaca and curaua, in agreement with the NMR data. In all cases, acetate and p-coumarate groups are preferentially attached to syringyl units, as previously noted for other lignins (7-9, 12-16, 39). Interestingly, the high extent of acetylation observed in sisal and curaua also included the G-lignin units (around 50% of acetylation in both cases). By contrast, in kenaf and abaca lignins, the �-carbon of G-lignin units is mostly not esterified. On the other hand, the high extent of acylation of the lignin monomers observed by the DFCR (and DFRC´) method, which can only analyze non-condensed lignin moieties, is in accordance with the high extent of lignin acylation observed by HSQC technique, which allows the analysis of the entire MWL structure, including both condensed and non-condensed linkages. This fact indicates that both condensed and non-condensed moieties have similar extent of acylation. However, we must now convey again that the results presented here reflect only the structure of the isolated MWL, which only represent a small part of the entire native lignin. However, similar lignin S/G ratio and acylation degree have been found by “in situ” analysis in HSQC spectra of the whole cell wall material (without lignin isolation) at the gel state (40), indicating that MWL can still be considered as the most representative preparation for the plant native lignin, in spite of its low yield.
Previous papers describing the structure of some of these lignins have failed to detect their high levels of acetylation. A recent paper describing the structure of sisal lignin (41) did not detect the high levels of acetylation, despite of using spectroscopic techniques. Probably, this was due to the method used for isolation (acidolysis) that might have hydrolyzed the acetyl groups, or to a misassignment of the spectral bands. Previous structural studies on abaca lignin (8, 16, 36, 38), using different degradation methods, also suggested the occurrence of p-coumaroylated units attached to the �-carbon of the lignin side-chain. The presence of acetylated �-carbons was also observed in abaca fibers directly by Py-GC/MS (15) although other authors failed to detect their presence (8).
On the other hand, the question to whether acylated lignin derives from polymerization of acylated monolignols or from acylation of the lignin polymer has recently been addressed and sinapyl acetate has been demonstrated to behave as a monomer in lignification participating in coupling reactions (14, 16, 17, 42). Part of the evidence comes from the �–�´ coupling reactions. If the �-carbon of a monolignol is pre-acylated, the formation of the normal �–�´ resinol structures can not occur because the absence of free �-hydroxyls needed to re- aromatize the quinone methide moiety. Instead, new tetrahydrofuran structures are formed from the ���´ homo- and cross-coupling of two sinapyl (acylated and non-acylated) monolignols, as advanced by Lu and Ralph (14) (Figure 6). It is clear that tetrahydrofuran structures I and II can only be formed if sinapyl alcohol is pre-acetylated (at monomer stage) and then undergoes ���´ coupling.
139

5. Resultados y discusión
140
(a)
(b)
(c)
5.0 7.5 10.0 12.5 15.0
Retention time (min)
(d)
t-Sac
t-S
c-Sc-Sac
t-G
c-G
t-Gac
c-Gac
t-Sac
t-S
c-Sc-Sac
t-G
c-Gt-Gacc-Gac
t-Sac
t-S
c-Sc-Sac
t-G
c-Gt-Gac
t-Sac
t-S
c-Sc-Sac
t-G
c-G
t-Gac
c-Gac
100
rela
tive
inte
nsity
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
0
100re
lativ
e in
tens
ity
100
0
0
Figure 5. Chromatograms of the DFRC´ degradation products of MWL from (a) sisal, (b) kenaf, (c) abaca and (d) curaua. c-G, t-G, c-S and t-S are the normal cis- and trans-guaiacyl and syringyl monomers, respectively (as their propionylated derivatives). c-Gac, t-Gac, c-Sac and t-Sac are the originally acetylated cis- and trans-guaiacyl and syringyl monomers, respectively (as their propionylated derivatives).

5. Resultados y discusión
Therefore, the presence of these tetrahydrofuran substructures in the lignin polymer would be indicative of the occurrence of pre-acylated monolignols. In this work, we have investigated the presence of the tetrahydrofuran structures arising from ���´ coupling of sinapyl acetate in the MWL selected for this study by DFRC´. The expected DFRC´ degradation products of the tetrahydrofuran ���´ structures, as suggested by Lu and Ralph (14), are also indicated in Figure 6. Figure 7 shows the reconstructed chromatograms (sum of the single ion chromatograms of the respective base peaks) of the DFRC´ degradation products of the expected tetrahydrofuran dimers arising from the ���´�coupling of the sinapyl monolignols. Interestingly, compounds derived from the DFRC´ of homo-coupling (I´) and cross-coupling (II´a and II´b) of sinapyl acetate were clearly observed in the lignins of sisal and kenaf, clearly indicating that in these lignins sinapyl alcohol is preacetylated and behaves as a real monolignol participating in post-coupling reactions. However, in the case of abaca and curaua lignins, no traces of any type of ���´ linkage (including syringaresinol and the new tetrahydrofuran structures) could be detected after DFRC, in agreement with the absence of these linkages observed in the HSQC spectra.
The presence of cross-coupling structures of sinapyl alcohol and sinapyl acetate indicates that both monolignols are produced simultaneously by the plant. Moreover, the relative abundance of the compounds released in Figure 7 gives some additional information. In sisal, the relative molar abundance of the acetylated versus the non-acetylated sinapyl alcohols forming ���´ linkages (taking into account that dimer I´ consists of two sinapyl acetates, dimers II´ consist of one sinapyl acetate and one sinapyl alcohol, and dimer III´ consists of two sinapyl alcohols) is 44:56, with a slight predominance of the non-acetylated sinapyl alcohol, whereas their relative molar abundances in ether-linked structures is 78:22, with a strong predominance of sinapyl acetate. A similar trend is also observed in kenaf lignin, where the relative molar abundance of the acetylated versus the non-acetylated sinapyl alcohols forming ���´ linkages is23:77, with a predominance of the non-acetylated sinapyl alcohol, whereas their relative molar abundances in ether-linked structures is 59:41, with a strong predominance of sinapyl acetate. This indicates that sinapyl acetate has a lower tendency to form ���´ linkages than the normal sinapyl alcohol and therefore those lignins having a high extent of acetylation would produce lower amounts of ���´ linkages, as already advanced (16). This means that, probably, the high level of lignin acetylation is related in some way with the low presence of ���´ linkages. This is in agreement with the high proportions of �-O-4´ aryl ether substructures and the low proportion of ���´ substructures present in these lignins, as observed in the HSQC spectra shown above. Therefore, it seems that the high extent of �-acetylation would favor the formation of a predominantly �-O-4 lignin structure, which is indeed devoid of �-�´ linkages.
141

5. Resultados y discusión
142
�
Figu
re 4
���´
hom
o-co
uplin
gD
FRC
´�
�
�
OC
H3
CH
3OO
H
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
O
+
O-C
-CH
3
=
O
OC
H3
CH
3O HO
OC
H3
OC
H3
OH
O
��
��
��
I
O-C
-CH
3
=
OC
H3-
C-O=
OC
H3O PrO
CH
3O
OP
r
OC
H3
CH
3O
I´; M
+ =61
6; [M
-56]
+ = 5
60
O-C
-CH
3
=
O
O-C
-CH
3
=
O
���´
hom
o-co
uplin
gO
CH
3C
H3O
OH
OH
��
�
OC
H3
CH
3OO
H
OH
��
�
+
OC
H3
CH
3O HO
OH OC
H3
OC
H3
OO
III
�
�
��
�
�
CH
3O PrO
OP
rO
Pr
CH
3O
OP
r
OC
H3
CH
3O
III´;
M+ =
644
; [M
-56]
+ = 5
88
DFR
C´
OC
H3
CH
3OO
H
OH
��
�
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
O
+��
�´cr
oss-
coup
ling
DFR
C´
O
CH
3O
OC
H3
OH
OC
H3
CH
3OO
H
HO
��
�
��
�
II
CH
3-C
-O=
O
CH
3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´a;
M+ =
630
; [M
-56]
+ = 5
74CH
3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´b;
M+ =
630
: [M
-56]
+ = 5
74
+
O-C
-CH
3
=
O
O-C
-CH
3
=
O
Figu
re 4
���´
hom
o-co
uplin
gD
FRC
´�
�
�
OC
H3
CH
3OO
H
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
O
+
O-C
-CH
3
=
O
OC
H3
CH
3O HO
OC
H3
OC
H3
OH
O
��
��
��
I
O-C
-CH
3
=
OC
H3-
C-O=
OC
H3O PrO
CH
3O
OP
r
OC
H3
CH
3O
I´; M
+ =61
6; [M
-56]
+ = 5
60
O-C
-CH
3
=
O
O-C
-CH
3
=
O��
�´ho
mo-
coup
ling
���´
hom
o-co
uplin
gD
FRC
´�
�
�
OC
H3
CH
3OO
H
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
O
+
O-C
-CH
3
=
O
��
�
OC
H3
CH
3OO
H
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
OO
-C-C
H3
=
O
+
O-C
-CH
3
=
OO
-C-C
H3
=
O
OC
H3
CH
3O HO
OC
H3
OC
H3
OH
O
��
��
��
I
O-C
-CH
3
=
OC
H3-
C-O=
O OC
H3
CH
3O HO
OC
H3
OC
H3
OH
O
��
��
��
I
O-C
-CH
3
=
OO
-C-C
H3
=
OC
H3-
C-O=
OC
H3-
C-O=
OC
H3O PrO
CH
3O
OP
r
OC
H3
CH
3O
I´; M
+ =61
6; [M
-56]
+ = 5
60
O-C
-CH
3
=
O
O-C
-CH
3
=
O
CH
3O PrO
CH
3O
OP
r
OC
H3
CH
3O
I´; M
+ =61
6; [M
-56]
+ = 5
60
O-C
-CH
3
=
OO
-C-C
H3
=
O
O-C
-CH
3
=
OO
-C-C
H3
=
O
���´
hom
o-co
uplin
gO
CH
3C
H3O
OH
OH
��
�
OC
H3
CH
3OO
H
OH
��
�
+
OC
H3
CH
3O HO
OH OC
H3
OC
H3
OO
III
�
�
��
�
�
CH
3O PrO
OP
rO
Pr
CH
3O
OP
r
OC
H3
CH
3O
III´;
M+ =
644
; [M
-56]
+ = 5
88
DFR
C´
���´
hom
o-co
uplin
gO
CH
3C
H3O
OH
OH
��
�
OC
H3
CH
3OO
H
OH
��
�
+
OC
H3
CH
3O HO
OH OC
H3
OC
H3
OO
III
�
�
��
�
�
CH
3O PrO
OP
rO
Pr
CH
3O
OP
r
OC
H3
CH
3O
III´;
M+ =
644
; [M
-56]
+ = 5
88
DFR
C´
���´
hom
o-co
uplin
g��
�´ho
mo-
coup
ling
OC
H3
CH
3OO
H
OH
��
�
OC
H3
CH
3OO
H
OH
��
�
+
OC
H3
CH
3OO
H
OH
��
�
OC
H3
CH
3OO
H
OH
��
�
+
OC
H3
CH
3O HO
OH OC
H3
OC
H3
OO
III
�
�
��
�
�
OC
H3
CH
3O HO
OH OC
H3
OC
H3
OO
III
�
�
��
�
�
CH
3O PrO
OP
rO
Pr
CH
3O
OP
r
OC
H3
CH
3O
III´;
M+ =
644
; [M
-56]
+ = 5
88
CH
3O PrO
OP
rO
Pr
CH
3O
OP
r
OC
H3
CH
3O
III´;
M+ =
644
; [M
-56]
+ = 5
88
DFR
C´
OC
H3
CH
3OO
H
OH
��
�
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
O
+��
�´cr
oss-
coup
ling
DFR
C´
O
CH
3O
OC
H3
OH
OC
H3
CH
3OO
H
HO
��
�
��
�
II
CH
3-C
-O=
O
CH
3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´a;
M+ =
630
; [M
-56]
+ = 5
74CH
3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´b;
M+ =
630
: [M
-56]
+ = 5
74
+
O-C
-CH
3
=
O
O-C
-CH
3
=
O
OC
H3
CH
3OO
H
OH
��
�
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
O
+
OC
H3
CH
3OO
H
OH
��
�
��
�
OC
H3
CH
3OO
H
O-C
-CH
3
=
OO
-C-C
H3
=
O
+��
�´cr
oss-
coup
ling
���´
cros
s-co
uplin
gD
FRC
´OO
CH
3O
OC
H3
OH
OC
H3
CH
3OO
H
HO
��
�
��
�
II
CH
3-C
-O=
OC
H3-
C-O=
O
CH
3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´a;
M+ =
630
; [M
-56]
+ = 5
74CH
3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´b;
M+ =
630
: [M
-56]
+ = 5
74
+
O-C
-CH
3
=
O
O-C
-CH
3
=
OC
H3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´a;
M+ =
630
; [M
-56]
+ = 5
74CH
3O PrO
OP
r
CH
3O
OP
r
OC
H3
CH
3O
II´b;
M+ =
630
: [M
-56]
+ = 5
74
+
O-C
-CH
3
=
OO
-C-C
H3
=
O
O-C
-CH
3
=
OO
-C-C
H3
=
O
�
Fi
gure
6. S
truct
ures
of t
he te
trahy
drof
uran
dim
ers
aris
ing
from
the�
���´
cou
plin
g of
sin
apyl
alc
ohol
and
sin
apyl
ace
tate
. I:��
��´ c
oupl
ing
prod
uct o
f tw
o si
napy
l ace
tate
s; I
I: ��
�´ c
oupl
ing
prod
uct o
f a
sina
pyl a
lcoh
ol a
nd a
sin
apyl
ace
tate
; III
: ���
´�cou
plin
g pr
oduc
t (sy
ringa
resi
nol)
of tw
o si
napy
l al
coho
ls. T
he a
rylte
tralin
pro
duct
s ex
pect
ed f
rom
the
DFR
C´
degr
adat
ion
of th
ese
tetra
hydr
ofur
an m
oiet
ies
are
also
sho
wn,
with
indi
catio
n of
thei
r m
olec
ular
wei
ght a
nd b
ase
peak
in th
eir m
ass s
pect
ra. A
dapt
ed fr
om L
u an
d R
alph
(14)
. OOOOO

5. Resultados y discusión
20.0 22.5 25.0 27.5 30.0 32.5
Retention time (min)
(a)
(b)
(c)
(d)
I´
II´a,II´b
III´
I´
II´a,II´b
III´
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
0
100
rela
tive
inte
nsity
Figure 7. Detail of reconstructed chromatogram (sum of the characteristic ions at m/z 560, 574 and 580) of the DFRC´ degradation products of the MWL from (a) sisal, (b) kenaf, (c) abaca and (d) curaua, showing the presence of aryltetralin ���´ products containing two (I´), one (II´a and II´b) and no (III´) native acetates.
143

5. Resultados y discusión
144
It has been reported that “in vitro” peroxidase-H2O2 oxidation of equimolar amounts of sinapyl alcohol and �-acylated sinapyl alcohol produced equal amounts of the expected ���´ coupled and cross-coupled products (I, II and III) shown in Figure 6, in a ratio 1:2:1, suggesting that the coupling reactions were insensitive to the acylation of the �-carbon (17, 18). However, this is not the case of what we have seen that occur in the plants, where sinapyl acetate seems to have lower tendency to form ���´ linkages and, therefore, a high abundance of acetylated lignin monomers will ultimately produce a lignin with very low levels of ���´ structures during lignification. Therefore, a discrepancy exists between what is observed in vitro and in the plant. Moreover, as indicated by Lu and Ralph (17), from the two possible stereoisomers of the ���´ homo-dimerization product of sinapyl acetate (structure I in Figure 6), the isomer produced during “in vitro” coupling reactions is not the same that is present in plants. Whether there is a protein (or any other mechanism) in the plant controlling the ���´ coupling reaction needs additional investigations. 3.3. Structural features of highly acylated lignins The lignins selected for this study share some common structural features. First, they are characterized for being extensively acylated (with either acetate or p-coumarate groups), exclusively at the ��carbon of the lignin side-chain, and preferentially over syringyl units. Moreover, all these lignins present a high predominance of syringyl over guaiacyl lignin units, a very high predominance of ��O-4´ linkages and a very low proportion of ���´ and ��5´ linkages and other condensed bonds that make these lignins very linear and unbranched. In particular, sisal and kenaf lignins present a high extent of �-acylation, exclusively with acetate groups, and preferentially on S-lignin moieties in the case of kenaf lignin and over both S- and G-lignin moieties in the case of sisal lignin. In both cases, ��O-4´ aryl ether linkages predominate although some ���´ (resinol) and ��1´ (spirodienones) linkages are observed in low proportions. On the other hand, the structure of abaca lignin is assembled mostly with syringyl units with a high extent of acylation of the �-carbon with both acetate and p-coumarate groups. ��O-4´ linkages are also predominant in this lignin. No ���´ linkages are present but some ��1´ (spirodienones) linkages can be observed in abaca lignin. Finally, the lignin of curaua has a predominance of S-lignin units, a predominance of ��O-4´ linkages and a high extent of acylation at the �-carbon with acetate and p-coumarate groups, acetate groups being also esterifying to a high extent the �-carbon of G-lignin units. In general, all these structural features make these lignins very different from the structural models already proposed for softwood (43, 44) and hardwood (45). lignins. All subsequent lignin structural studies, including those in plant genetics or plant breeding projects, should take into account the possible occurrence of lignin

5. Resultados y discusión
acylation, which in many plants takes place at very high levels, as seen above, and which have often been overlooked in the past; otherwise the conclusions drawn may not be representative of the real native lignin structure. 4. Conclusions The structure of the MWL isolated from the herbaceous plants sisal, kenaf, abaca and curaua has been elucidated by 2D-NMR and DFRC techniques. The analyses indicated that the lignins from these plants are extensively acylated at the �-carbon of the lignin side-chain (with either acetate and/or p-coumarate groups) and preferentially on syringyl moieties. The structure of these highly acetylated lignins can be essentially regarded as syringyl units linked mostly through ��O-4´ ether bonds, where the �-carbons of the side-chains are extensively acylated. The lignin polymer is therefore extremely linear and unbranched. The study of highly acylated lignins will significantly contribute to redefine the structure of lignin and to complete the lignin biosynthetic pathway. Acknowledgements This study has been supported by the Spanish MEC (projects AGL2005-01748 and BIO2007-28719-E) and the EU contract NMP2-CT-2006-26456 (BIORENEW). JR thanks the Spanish CSIC for an I3P fellowship; GM thanks the Spanish Ministry of Education for a FPI fellowship. References (1) Sarkanen, K. V.; Ludwig, C. H. Definition and Nomenclature. In Lignins:
Occurrence, Formation, Structure, and Reactions, Sarkanen, K.V. and Ludwig, C.H., Ed. Wiley- Intersci.: New York, 1971: pp 1-16.
(2) Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546.
(3) Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P. F.; Marita, J. M.; Hatfield, R. D.; Ralph, S. A.; Christensen, J. H.; Boerjan, W. Lignins: natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 2004, 3, 29-60.
(4) Smith, D. C. C. p-Hydroxybenzoate groups in the lignin of aspen (Populus tremula). J. Chem. Soc. 1955, 2347-2351.
(5) Nakano, J.; Ishizu, A.; Migata, N. Studies on lignin. XXXII. Ester groups of lignin. Tappi 1961, 44, 30-32.
(6) Landucci, L. L.; Deka, G. C.; Roy, D. N. A. 13C NMR study of milled wood lignins from hybrid Salix clones. Holzforschung 1992, 46, 505-511.
145

5. Resultados y discusión
146
(7) Ralph, J.; Hatfield, R. D.; Quideau, S.; Helm, R. F.; Grabber, J. H.; Jung, H. -J. G. Pathway of p-coumaric acid incorporation into maize lignin as revealed by NMR. J. Am. Chem. Soc. 1994, 116, 9448-9456.
(8) Sun, R. C.; Fang, J. M.; Goodwin, A.; Lawther, J. M.; Bolton, J. Fractionation and characterization of ball-milled and enzyme lignins from abaca fibre. J. Sci. Food Agric. 1999, 79, 1091-1098.
(9) Lu, F.; Ralph, J. Detection and determination of p-coumaraloylated units in lignin. J. Agric. Food Chem. 1999, 47, 1985-1992.
(10) Meyermans, H.; Morreel, K.; Lapierre, C.; Pollet, B.; De Bruyn, A.; Busson, R.; Herdewijn, P.; Devreese, B.; Van Beeumen, J.; Marita, J. M.; Ralph, J.; Chen, C.; Burggraeve, B.; Van Montagu, M.; Messens, E.; Boerjan, W. Modification in lignin and accumulation of phenolic glucosides in poplar xylem upon down-regulation of caffeoyl-coenzyme A O-methyltransferase, an enzyme involved in lignin biosynthesis. J. Biol. Chem. 2000, 275, 36899-36909.
(11) Crestini, C.; Argyropoulos, D. S. Structural analysis of wheat straw lignin by quantitative 31P and 2D NMR spectroscopy. The occurrence of ester bonds and �-O-4 substructures. J. Agric. Food Chem. 1997, 45, 1212-1219.
(12) Ralph, J. An unusual lignin from kenaf. J. Nat. Prod. 1996, 59, 341-342.
(13) Ralph, J.; Lu, F. The DFRC method for lignin analysis. 6. A simple modification for identifying natural acetates in lignin. J. Agric. Food Chem. 1998, 46, 4616-4619.
(14) Lu, F.; Ralph, J. Preliminary evidence for sinapyl acetate as a lignin monomer in kenaf. Chem. Commun. 2002, 90-91.
(15) del Río, J. C.; Gutiérrez, A.; Martínez A. T. Identifying acetylated lignin units in non-wood fibers using pyrolysis-gas chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 1181-1185.
(16) del Río, J. C.; Marques, G.; Rencoret, J.; Martínez A. T.; Gutiérrez, A. Occurrence of naturally acetylated lignin units. J. Agric. Food Chem. 2007, 55, 5461-5468.
(17) Lu, F.; Ralph, J. Novel ����structures in lignins incorporating acylated monolignols. Appita 2005, 233-237.
(18) Lu, F.; Ralph, J.; Morreel, K.; Messens, E.; Boerjan, W. Preparation and relevance of a cross-coupling product between sinapyl alcohol and sinapyl p-hydroxybenzoate. Org. Biomol. Chem. 2004, 2888-2890.

5. Resultados y discusión
(19) Morreel, K.; Ralph, J.; Kim, H.; Lu, F.; Goeminne, G.; Ralph, S. A.; Messens, E.; Boerjan, W. Profiling of oligolignols reveals monolignols coupling conditions in lignifying poplar xylem. Plant Physiol. 2004, 136, 3537-3549.
(20) Ralph, J.; Marita, J. M.; Ralph, S. A.; Hatfield, R. D.; Lu, F.; Ede, R. M.; Peng, J.; Quideau, S.; Helm, R. F.; Grabber, J. H.; Kim, H.; Jimenez-Monteon, G.; Zhang, Y.; Jung, H. -J. G.; Landucci, L. L.; MacKay, J. J.; Sederoff, R. R.; Chapple, C.; Boudet, A. M. Solution-state NMR of lignin. In Advances in lignocellulosics characterization, Argyropoulos, D. S., Ed.; Tappi Press: Atlanta, 1999; pp 55-108.
(21) Ralph, S. A.; Ralph, J.; Landucci, L. NMR database of lignin and cell wall model compounds; US Forest Prod. Lab., One Gifford Pinchot Dr., Madison, WI 53705, 2004 (http://ars.usda.gov/Services/docs.htm?docid=10491) (accessed: July 2006): 2004.
(22) Capanema, E. A.; Balakshin, M. Y.; Kadla, J. F. A comprehensive approach for quantitative lignin characterization by NMR spectroscopy. J. Agric. Food Chem. 2004, 52, 1850-1860.
(23) Capanema, E. A.; Balakshin, M. Y.; Kadla, J. F. Quantitative characterization of a hardwood milled wood lignin by nuclear magnetic resonance spectroscopy. J. Agric. Food Chem. 2005, 53, 9639-9649.
(24) Balakshin, M. Y.; Capanema, E. A.; Chen, C.- L.; Gracz, H. S. Elucidation of the structures of residual and dissolved pine kraft lignins using an HMQC NMR technique. J. Agric. Food Chem. 2003, 51, 6116-6127.
(25) Björkman, A. Studies on finely divided wood. Part I. Extraction of lignin with neutral solvents. Sven. Papperstidn. 1956, 59, 477-485.
(26) Lu, F.; Ralph, J. Derivatization followed by reductive cleavage (DFRC method), a new method for lignin analysis: protocol for analysis of DFRC monomers. J. Agric. Food Chem. 1997, 45, 2590-2592.
(27) Lu, F.; Ralph, J. The DFRC method for lignin analysis. Part 1. A new method for ��aryl ether cleavage: lignin model studies. J. Agric. Food Chem. 1997, 45, 4655-4660.
(28) Lu, F.; Ralph, J. The DFRC method for lignin analysis. 2. Monomers from isolated lignin. J. Agric. Food Chem. 1998, 46, 547-552.
147

5. Resultados y discusión
148
(29) Liitiä, T. M.; Maunu, S. L.; Hortling, B.; Toikka, M.; Kilpeläinen, I. Analysis of technical lignins by two- and three-dimensional NMR spectroscopy. J. Agric. Food Chem. 2003, 51, 2136-2143.
(30) Ämmälahti, E.; Brunow, G.; Bardet, M.; Robert, D.; Kilpeläinen, I. Identification of side-chain structures in a poplar lignin using three-dimensional HMQC-HOHAHA NMR spectroscopy. J. Agric. Food Chem. 1998, 46, 5113-5117.
(31) Ibarra, D.; Chávez, M. I.; Rencoret, J.; del Río, J. C.; Gutiérrez, A.; Romero, J.; Camarero, S.; Martínez, M. J.; Jiménez-Barbero, J.; Martínez, A. T. Lignin modification during Eucalyptus globulus kraft pulping followed by totally chlorine free bleaching: a two dimensional nuclear magnetic resonance, Fourier transform infrared, and pyrolysis-gas chromatography/mass spectrometry study. J. Agric. Food Chem. 2007, 55, 3477-3490.
(32) Ibarra, D.; Chávez, M. I.; Rencoret, J.; del Río, J. C.; Gutiérrez, A.; Romero, J.; Camarero, S.; Martínez, M. J.; Jiménez-Barbero, J.; Martínez A. T. Structural modification of eucalypt pulp lignin in a totally chlorine free bleaching sequence including a laccase-mediator stage. Holzforschung 2007, 61, 634-646.
(33) Heikkinen, S.; Toikka, M. M.; Karhunen, P. T.; Kilpeläinen, I. A. Quantitative 2D HSQC (Q-HSQC) via suppression of J-dependence of polarization transfer in NMR spectroscopy: Application to wood lignin. J. Am. Chem. Soc. 2003, 125, 4362-4367.
(34) Zhang, L.; Gellerstedt, G. Quantitative 2D HSQC NMR determination of polymer structures by selecting the suitable internal standard references. Magn. Reson. Chem., 2007, 45(1), 37-45.
(35) Holtman, K. H.; Chang, H. M.; Jameel, H.; Kaddla, J. F. Quantitative 13C NMR characterization of milled wood lignins isolated by different milling techniques. J. Wood Chem. Technol. 2006, 26, 21-34.
(36) del Río, J. C.; Gutiérrez, A.; Rodríguez, I. M.; Ibarra, D.; Martínez, A. T. Composition of non-woody plant lignins and cinnamic acids by Py-GC/MS, Py/TMAH and FT-IR. J. Anal. Appl. Pyrolysis 2007, 79, 39-46.
(37) Zhang, L; Gellerstedt, G.; Ralph, J.; Lu, F. NMR studies on the occurrence of spirodienone structures in lignins. J. Wood Chem. Technol. 2006, 26, 65-79.

5. Resultados y discusión
(38) del Río, J. C.; Gutiérrez, A. Chemical composition of abaca (Musa textilis) leaf fibers used for manufacturing of high quality paper pulps. J. Agric. Food Chem. 2006, 54, 4600-4610.
(39) Grabber, J. H.; Quideau, S.; Ralph, J., p-Coumaroylated syringyl units in maize lignin; implications for �-ether cleavage by thioacidolysis. Phytochemistry 1996, 43, 1189-1194.
(40) Rencoret, J.; Marques, G.; Gutiérrez, A.; Nieto, L.; Santos, J. I.; Jiménez-Barbero, J.; Martínez, A. T.; del Río, J. C. “In situ” analysis of lignin by 2D-NMR of wood (Eucalyptus globulus and Picea abies) and non-woody (Agave sisalana) plant materials at the gel state. Proc. EWLP-2008, Stockholm, 26-29 August.
(41) Megiatto, J. D.; Hoareau, W.; Gardrat, C.; Frollini, E.; Castellain, A. Sisal fibers: surface chemical modification using reagent obtained from a renewable source; characterization of hemicellulose and lignin as model study. J. Agric. Food Chem. 2007, 55, 8576-8584.
(42) Ralph, J. What makes a good monolignol substitute? In The Science and Lore of the Plant Cell Wall Biosynthesis, Structure and Function, Hayashi, T., Ed. Universal Publishers (BrownWalker Press): Boca Raton, FL, 2006; pp 285-293.
(43) Adler, E. Lignin chemistry – past, present and future. Wood Sci. Technol. 1977, 11, 169-218.
(44) Brunow, G. Methods to Reveal the Structure of Lignin. In: Hofrichter M & Steinbüchel A, (ed), Lignin, Humic Substances and Coal, Vol 1, 2001, pp. 89–116, Wiley-VHC, Weinheim.
(45) Nimz, H. Beech lignin- Proposal of a constitutional scheme. Agnew. Chem. 1974, 13(5), 313-321.
149

5. Resultados y discusión
Publicación IV: Marques G., Gutiérrez A. and del Río J.C (2007) Chemical characterization of lignin and lipophilic fractions from leaf fibers of curaua (Ananas erectifolius). Journal of Agriculture and Food Chemistry, 55, 1327-1336.
150

5. Resultados y discusión
Chemical characterization of lignin and lipophilic fractions from leaf fibers of curaua (Ananas erectifolius)
Gisela Marques, Ana Gutiérrez and José C. del Río
Instituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, P.O. Box 1052, 41080-Seville, Spain
Abstract The chemical composition of leaf fibers of curaua (Ananas erectifolius), an herbaceous plant native of Amazonia, was studied. Special attention was paid to the content and composition of lignin and lipophilic compounds. The analysis of lignin in the curaua fibers was performed “in situ” by pyrolysis-gas chromatography/mass spectrometry (Py-GC/MS) and showed a lignin composition with a p-hydroxyphenyl:guaiacyl:syringyl units (H:G:S) molar proportion of 30:29:41 (S/G molar ratio of 1.4). The presence of p-hydroxycinnamic acids (p-coumaric and ferulic acids) in curaua fibers was revealed upon pyrolysis in the presence of tetramethylammonium hydroxide. On the other hand, the main lipophilic compounds, analysed by GC/MS, were series of long-chain n-fatty acids, n-fatty alcohols, �- and �-hydroxyacids, monoglycerides, sterols and waxes. Other compounds, such as �-hydroxy monoesters and �-hydroxy acylesters of glycerol were also found in this fiber in high amounts. Keywords: Curaua; Ananas erectifolius; lipids; lignin; pyrolysis; hydroxy monoesters; glyceryl esters; paper pulp. 1. Introduction An alternative to wood raw materials for pulp and paper production in developing countries is the use of non-woody fibers from herbaceous field crops. In developed countries non-woody fibers are mainly used for the production of specialty papers, i.e. tea bags, filter papers, bank notes, etc. The main sources of non-woody raw materials are agricultural residues from monocotyledons, including cereal straw and bagasse. Bamboo, reeds and some other grass plants such as flax, hemp, kenaf, jute, sisal or abaca are also grown or collected for the pulp industry but increased attention has been paid in recent years to find new non-wood raw materials for pulp production.
Curaua (Ananas erectifolius), an herbaceous plant native of the Amazonian region and member of the bromeliad family, has been recognized since pre-Columbian days for its valuable fibers. In the last decade, it has gained commercial recognition as material for composites for automotive industry [1-4]. The curaua fiber has also been promoted for paper pulp in Brazil [5] and it is
151

5. Resultados y discusión
152
being now investigated as an alternative lignocellulosic material for the production of chemical pulps.
Studies on the chemical composition of curaua fibers are important to evaluate this fiber as a potential raw material for pulp and papermaking, however only limited studies have been performed so far on this interesting fiber [1-4]. In this work, we have performed a chemical characterization of curaua fibers, paying especial attention to the content and composition of the lipophilic compounds and the structural characterization of lignin, since these two organic fractions are of high importance during pulping and papermaking. It is known that the efficiency of pulping is directly proportional to the amount of syringyl (S) units in lignin [6, 7]. This is because the S-lignin is mainly linked by more labile ether bonds, is relatively unbranched and has lower condensation degree that G-lignin [8, 9]. Indeed, the S-lignin has higher reactivity in alkaline systems than G-lignin [10]. On the other hand, the lipophilic compounds present in raw materials cause significant environmental and technical problems in the manufacturing of paper pulp. During pulping, lipids are released from the fibers forming colloidal pitch, which can deposit in either pulp or machinery causing production troubles [11-13]. Moreover, such extractives might also contribute to the toxicity of paper pulp effluents and products [14, 15].
In the present study, the lignin in curaua fibers was characterized “in situ” by using analytical pyrolysis coupled to gas chromatography/mass spectrometry (Py-GC/MS), which is a powerful analytical tool for the rapid analysis of complex polymer mixtures, including lignocellulosic materials [16, 17]. Pyrolysis in the presence of a methylating reagent, tetramethylammonium hydroxide (TMAH), was used for the analysis of p-hydroxicinnamic acids (p-coumaric and ferulic acids). On the other hand, the lipid composition in curaua fibers was analyzed by gas chromatography (GC) and gas chromatography/mass spectrometry (GC/MS), using short- and medium-length high-temperature capillary columns, respectively [18], which enable the elution and analysis of intact high molecular weight lipids such as waxes, sterol esters, and triglycerides. 2. Material and methods 2.1. Samples Curaua (Ananas erectifolius) fibers were supplied by CELESA pulp mill (Tortosa, Spain). The dried samples were milled using a knife mill (Janke and Kunkel, Analysenmühle). For the isolation of lipids, the milled samples were extracted with acetone in a Soxhlet apparatus for 8 h. The acetone extracts were evaporated to dryness and weighted. Then, the extracts were resuspended in chloroform for chromatographic analysis of the lipophilic fraction. Two replicates were used for each sample, and all samples were subjected to GC and GC/MS analyses. For carbohydrate analysis and estimation of the Klason lignin

5. Resultados y discusión
content, the acetone extracted samples were subsequently extracted with hot water (3 h at 100 ºC) to remove the water-soluble material. Holocellulose was isolated from the pre-extracted fibers by delignification for 4 hours using the acid chlorite method (19). The �-cellulose content was determined by removing the hemicelluloses from the holocellulose by alkali extraction (19). Klason lignin was estimated as the residue after sulfuric acid hydrolysis of the pre-extracted material according to Tappi rule T222 om-88 [20]. The acid-soluble lignin was determined, after filtering off the insoluble lignin, by spectrophotometric determination at 205 nm wavelength. Neutral sugars from polysaccharide hydrolysis were analyzed as alditol acetates by GC according to Tappi rule T249 om85 [20]. Ash content was estimated as the residue after 6 h at 575 ºC. The general composition (as percent of whole fiber) was as follows: holocellulose, 92.5%; �-cellulose, 66.4%; ash, 1.3%; acetone extractives, 5.3%; water-soluble extract, 5.1%; Klason lignin, 4.9%; acid-soluble lignin, 1.6%. The composition of neutral monosaccharides (as percent of total neutral carbohydrates) included arabinose, 2.7%; xylose, 8.0%; mannose, 3.5%; galactose, 0.2%; and glucose, 85.6%. No uronic acid determination was performed in this study. The composition of metals and other elements was analyzed by inductively coupled plasma spectrophotometry (ICP-OES) after oxidation with concentrated HNO3 under pressure in a microwave digestor, with the following results: K, 2770 ppm; Ca, 2025 ppm; Mg, 945 ppm; Mn, 120 ppm; Na, 95 ppm; Al, 86 ppm; Fe, 82 ppm; Sr, 10 ppm; Zn, 4 ppm. 2.2. Solid Phase Extraction (SPE) fractionation For a better characterization of the different homologous series, the lipid extracts were fractionated by a SPE procedure using aminopropyl-phase cartridges (500 mg) from Waters Division of Millipore (Mildford, MA, USA), as already described [18]. Briefly, the dried chloroform extracts were taken up in a minimal volume (< 0.5 mL) of hexane:chloroform (4:1) and loaded into the cartridge column previously conditioned with hexane (4 mL). The cartridge was loaded and eluted by gravity. The column was first eluted with 8 mL of hexane and subsequently with 6 ml of hexane:chloroform (5:1), then with 10 mL of chloroform and finally with 10 mL of diethyl ether:acetic acid (98:2). Each isolated fraction was dried under nitrogen and analyzed by GC and GC/MS. 2.3. GC and GC/MS analyses The GC analyses of the extracts were performed in an Agilent 6890N Network GC system using a 5 m × 0.25 mm i.d., 0.1 �m DB-5HT fused silica capillary column from J&W Scientific (Folsom, CA, USA). The temperature program was started at 100 ºC with a 1-min hold and then raised to a final temperature of 350 ºC at 15 ºC/min, and held for 3 min. The injector and flame-ionization detector temperatures were set at 300 and 350 ºC, respectively. Helium was used
153

5. Resultados y discusión
154
as the carrier gas at a rate of 5 mL/min, and the injection was performed in splitless mode. Peaks were quantified by area, and a mixture of standards (octadecane, palmitic acid, sitosterol and cholesteryl oleate) was used to elaborate calibration curves. The data from the two replicates were averaged. In all cases the standard derivations from replicates were below 10% of the mean values.
The GC/MS analyses were performed with a Varian model Star 3400 GC equipped with a model Saturn 2000 ion trap detector using a medium-length (12 m) capillary column of the same characteristics described above. The oven was heated from 120 ºC (1 min) to 380 ºC at 10 ºC/min and held for 5 min. The transfer line was kept at 300 ºC. The injector was temperature programmed from 120 ºC (0.1 min) to 380 ºC at a rate of 200 ºC/min and held until the end of the analysis. Helium was used as the carrier gas at a rate of 2 mL/min. Methylation with trimethylsilyldiazomethane and silylation with bis(trimethylsilyl)trifluoroacetamide (BSTFA) was used when required. Compounds were identified by comparing their mass spectra with mass spectra in Wiley and NIST libraries, by mass fragmentography, and when possible, by comparison with authentic standards. 2.4. Py-GC/MS The pyrolysis of curaua fibers (approximately 100 �g) was performed in duplicate with a model 2020 micro-furnace pyrolyzer (Frontier Laboratories Ltd., Yoriyama, Japan) directly connected to an Agilent 6890 GC/MS system equipped with a 30 m × 0.25 mm i.d., 0.25 �m HP 5MS fused silica capillary column. The detector consisted of an Agilent 5973 mass selective detector (EI at 70 eV). The pyrolysis was performed at 500 °C. The final temperature was achieved at a rate of 20 °C/min. The GC/MS conditions were as follows: oven temperature was held at 50 °C for 1 min and then increased up to 100 °C at 30 °C/min, from 100 to 300 °C at 10 °C/min and isothermal at 300 °C for 10 min. The carrier gas used was helium with a controlled flow of 1 ml/min. For the pyrolysis in the presence of TMAH, approximately 100 �g of sample was mixed with 0.5 �L of 25% TMAH. The pyrolysis was carried out as described above. The compounds were identified by comparing the mass spectra obtained with those of the Wiley and NIST computer libraries and that reported in the literature [16, 17]. Relative peak molar areas were calculated for carbohydrate and lignin pyrolysis products. The summed molar areas of the relevant peaks were normalized to 100%, and the data for two repetitive pyrolysis experiments were averaged. The relative standard deviation for the pyrolysis data was less than 5%.

5. Resultados y discusión
3. Results and discussion The curaua fiber was characterized by a high holocellulose and �-cellulose contents (92.5 and 66.4, respectively), and a low lignin content (6.5% of the total fiber weight). This lignin content is similar to other non-wood fibers such as flax or hemp and lower than other non-wood fibers such as kenaf or abaca [21-26]. The extractives content (5.3% of total fiber weight) is very high, and much higher than other nonwood fibers, which are usually less than 1% [21-26]. However, most of the acetone extract corresponds to polar compounds, while only 1.3% corresponded to lipophilic compounds, which were estimated by redissolving the acetone extracts in chloroform. On the other hand, the hemicellulose fraction was mainly constituted by xylose. Finally, the ash content (1.3% of total fiber weight) was low in comparison to cereal straw [27], and the composition of the different metals revealed a predominance of Ca and K, and a very low content of other metals. 3.1. Lignin composition The lignin composition of curaua fibers was analyzed “in situ” by Py-GC/MS. The Py-GC/MS chromatogram of curaua fibers is shown in Figure 1 and the identities and relative molar abundances of the released compounds are listed in Table 1. The Py-GC/MS of curaua fibers released predominantly compounds arising from carbohydrates, with only minor amounts of lignin-derived phenols. Carbohydrate pyrolysis products represented 88% on average and phenols from lignin represented only 12% of the total identified compounds, which is in agreement with the low lignin content estimated as Klason lignin. Among the lignin derived compounds, the pyrogram of curaua fibers showed compounds derived from p-hydroxyphenyl (H), guaiacyl (G) and syringyl (S) lignin units, with a slight predominance of the S units. The main lignin-derived compounds identified were 4-methylphenol (13), guaiacol (14), 4-vinylphenol (19), 4-vinylguaiacol (23), syringol (24), 4-ethylsyringol (33), 4-vinylsyringol (34) and trans-4-propenylsyringol (40). The relative molar distribution of the different lignin units (H:G:S) estimated by Py-GC/MS were 30:29:41 with a S/G molar ratio of 1.4. The predominance of S-lignin observed in the curaua fiber is advantageous for delignification during pulping because the S-lignin is relatively unbranched and has lower condensation degree than H- and G-lignins. Moreover, S-lignin is more reactive in alkaline media [10].
It must be noted that the relatively high abundances of 4-vinylphenol (19) observed in the pyrogram of curaua fibers could be due to the presence of p-coumaric acid, which upon pyrolysis will decarboxylate to produce 4-vinylphenol [28]. p-Hydroxycinnamic acids (p-coumaric and ferulic acids) occur widely in the cell walls of herbaceous plants forming cross-linkages between lignin and polysaccharides [29-34]. The presence of p-hydroxycinnamic acids constitutes a complication for lignin analyses by analytical pyrolysis since they
155

5. Resultados y discusión
156
yield pyrolysis products similar to those of corresponding lignin units. However, this problem can be solved by the use of pyrolysis in the presence of TMAH (Py/TMAH), which prevents decarboxylation and releases intact p-hydroxycinnamic acids (as their methyl derivatives), in addition to different lignin degradation products [28, 35-38].
Py/TMAH of curaua fibers released significant amounts of the methyl derivative of p-coumaric acid (25% of the lignin and cinnamic acids released products) as well as minor amounts of the methyl derivative of ferulic acid (5% of the lignin and cinnamic acids released products). p-Hydroxycinnamic acids are present in curaua fiber in relatively high amounts (cinnamic acids/lignin ratio of 0.4, estimated after Py/TMAH) and agrees with the relatively high content of 4-vinylphenol released by Py-GC/MS. Studies on maize [39], wheat [40] and other grasses including bamboo [41] revealed that p-coumaric acid is esterified at the �-position of lignin side-chains, and predominantly to S units [41, 42]. Therefore, probably the major part of the p-coumaric acid in curaua fibers also attaches at the �-position of the lignin side-chain by ester bonds. The relatively high content of p-hydroxycinnamic acids in curaua fibers would also be advantageous for pulping since ester bonds are easily cleaved during cooking.
1
2 3
4
5
6
7
8
9
10
11,12
1413
15
17
16
19
18
20
2122
23
24
2526
27 29
30 31
32,33
34
35 36
37
38
40
39 4441 42 43
2 4 6 8 10 12 14 16 18 20 22Retention time (minutes)
28
Figure 1. Py-GC/MS chromatogram of curaua fibers. The identities and relative molar abundances of the compounds are listed in Table 1.

5. Resultados y discusión
Table 1. Identification and Relative Molar Abundances (%) of the Compounds Released after Py-GC/MS of Curaua Fibers.
No Compound Mass Fragments MW Origin %1 acetic acid 45/60 60 C 35.82 2-hydroxypropanal 43/74 74 C 3.13 (3H)-furan-2-one 55/84 84 C 3.14 1,3-hydroxydihydro-2-(3H)-furanone 58/102 102 C 6.05 (2H)-furan-3-one 55/84 84 C 1.46 2-furaldehyde 67/95/96 96 C 6.47 cyclopent-1-ene-3,4-dione 54/68/96 96 C 0.98 (5H)-furan-2-one 55/84 84 C 4.29 2,3-dihydro-5-methylfuran-2-one 55/69/98 98 C 8.5
10 4-hydroxy-5,6-dihydro-(2H)-piran-2-one 58/85/114 114 C 2.111 3-hydroxy-2-methyl-2-cyclopenten-1-one 55/85/112 112 C 1.112 2-hydroxy-3-methyl-2-cyclopenten-1-one 55/85/112 112 C 4.813 4-methylphenol 77/107/108 108 LH 0.714 guaiacol 81/109/124 124 LG 0.515 2 furoic acid, methyl ester 67/95/126 126 C 1.316 4-methylguaiacol 95/123/138 138 LG 0.117 3,4-dihydroxybenzaldehyde 81/109/137/138 138 L 0.518 catechol 64/81/92/110 110 L/C 1.019 4-vinylphenol 65/91/120 120 LH/pCA 2.220 5-hydroxymethyl-2-furaldehyde 69/97/109/126 126 C 1.921 3-methoxycatechol 60/97/125/140 140 L 0.322 4-ethylguaiacol 122/137/152 152 LG 0.223 4-vinylguaiacol 107/135/150 150 LG 1.124 syringol 111/139/154 154 LS 0.725 eugenol 131/149/164 164 LG 0.226 4-propylguaiacol 122/136/166 166 LG <0.127 vanillin 109/151/152 152 LG 0.328 cis-isoeugenol 131/149/164 164 LG <0.129 4-methylsyringol 125/153/168 168 LS 0.230 trans-isoeugenol 131/149/164 164 LG 0.131 acetoguaiacone 123/151/166 166 LG 0.132 levoglucosane 60/98 162 C 7.733 4-ethylsyringol 167/182 182 LS 0.834 4-vinylsyringol 137/165/180 180 LS 0.935 4-allylsyringol 167/179/194 194 LS 0.136 4-propylsyringol 123/167/196 196 LS 0.137 cis-4-propenylsyringol 167/179/194 194 LS 0.238 syringaldehyde 167/181/182 182 LS 0.239 4-propinylsyringol 106/131/177/192 192 LS 0.140 trans-4-propenylsyringol 167/179/194 194 LS 0.541 trans-coniferaldehyde 107/135/147/178 178 LG 0.142 acetosyringone 153/181/196 196 LS 0.143 syringylacetone 123/167/210 210 LS 0.144 trans-sinapaldehyde 137/165/180/208 208 LS 0.1
%H 29.8 %G 29.1 %S 41.1 S/G 1.4 %L 11.7 %C 88.3 L/C 0.13
C, carbohydrates; L, lignin; LH, p-hydroxyphenyl lignin units, H; LG, guaiacyl lignin units, G; LS, syringyl lignin units, S; pCA, p-coumaric acid.
157

5. Resultados y discusión
158
3.2. Lipid composition The total lipid extract of the curaua fibers accounted for 1.3% of the total fiber weight. The extracts were analyzed by GC and GC/MS according to the method developed by Gutiérrez et al. [18]. The chromatogram of the curaua extracts (as methyl ester and TMSi ether derivatives) is shown in Figure 2 and the detailed list with the identities and abundances of the main compounds present are summarized in Table 2. The main compounds identified were series of n-fatty acids, n-fatty alcohols, �- and �-hydroxyacids, monoglycerides, sterols and waxes. Other series of high molecular weight compounds such as �-hydroxy monoesters and �-hydroxy acylesters of glycerol, as well as sterol esters and sterol glycosides, were also present in important amounts. The structures of the main lipophilic compounds identified in the curaua extract are shown in Figure 3. The �-hydroxy fatty acids both in free or esterified form (forming esters with both fatty alcohols and glycerol), was the main series of compounds present in the extracts.
5 10 15 20 2Retention time (minutes)
5
�OH20
Al22
�OH22
Al24
�OH24
OH22
OH24
M24
M26
OHM24
M28
OHM26
OHM28
W40
W42
CG
SG
W38
FA16:1
FA18:2+
FA18:1
FA16FA18 FA20
FA22FA24
12
3,4
CE SE
5 10 15 20 2Retention time (minutes)
5
�OH20
Al22
�OH22
Al24
�OH24
OH22
OH24
M24
M26
OHM24
M28
OHM26
OHM28
W40
W42
CG
SG
W38
FA16:1
FA18:2+
FA18:1
FA16FA18 FA20
FA22FA24
12
3,4
CE SE
Figure 2. GC/MS chromatogram of the methyl ester and TMSi ether derivative of the lipid extract from curaua fibers. FA(n), n-fatty acid series; Al(n), alcohol series; W(n), wax series; �OH(n) and �OH(n), �� and �-hydroxy fatty acids series; M(n), monoglyceride series; �OHM(n), �-hydroxy acylesters of glycerol series; SG, sitosteryl 3�-D-glucopyranoside; CG, campesteryl 3�-D-glucopyranoside;1, campesterol; 2, ergostanol; 3, sitosterol; 4, stigmastanol; CE, campesterol ester; SE, sitosterol ester; n denotes the total carbon atom number.

5. Resultados y discusión
Table 2. Composition and Abundance (mg/Kg) of Lipophilic Compounds in Curaua Fibers compound mass fragments (m/z) MW abundance fatty acids 813.2n-tetradecanoic acid 73/117/145/285* 285* 4.3n-pentadecanoic acid 73/117/145/299* 299* 3.39-hexadecenoic acid 55/69/236/254 254 7.6n-hexadecanoic acid 60/73/129/256 256 162.59,12-octadecadienoic acid 67/81/280 280 23.09-octadecenoic acid 55/69/264 282 91.6n-octadecanoic acid 60/73/129/284 284 262.0n-eicosanoic acid 60/73/129/312 312 24.2n-docosanoic acid 60/73/129/340 340 138.2n-tetracosanoic acid 60/73/129/368 368 96.5 �-hydroxy fatty acids 1423.316-hydroxyhexadecanoic acid 311/343/359# 374# 20.218-hydroxyoctadecanoic acid 339/371/387# 402# 28.420-hydroxyeicosanoic acid 367/399/415# 430# 30.822-hydroxydocosanoic acid 395/427/443# 458# 235.024-hydroxytetracosanoic acid 423/455/471# 486# 367.026-hydroxyhexacosanoic acid 451/483/499# 514# 532.528-hydroxyoctacosanoic acid 479/511/527# 542# 119.230-hydroxytriacontanoic acid 507/539/555# 570# 90.2 �-hydroxy fatty acids 226.52-hydroxyeicosanoic acid 73/117/355* 472* 62.72-hydroxydocosanoic acid 73/117/149/383* 500* 19.92-hydroxytetracosanoic acid 73/117/411* 528* 79.22-hydroxyhexacosanoic acid 73/117/439* 556* 64.7
fatty alcohols 552.4n-eicosanol 75/103/355* 370* 8.9n-docosanol 75/103/383* 398* 247.2n-tetracosanol 75/103/411* 426* 242.2n-hexacosanol 75/103/439* 454* 44.3n-octacosanol 75/103/467* 482* 9.8 sterols 618.7campesterol 55/145/213/382/400 400 56.9ergostanol 215/402 402 145.7sitosterol 145/213/396/414 414 226.4stigmastanol 215/416 416 189.7 tocopherols 31.4�-tocopherol 165/205/430 430 31.4 steroid hydrocarbons 119.4ergostatriene 135/143/380 380 23.8
159

5. Resultados y discusión
160
ergostadiene 81/147/367/382 382 14.4stigmastadiene 81/147/381/396 396 71.8stigmasta-3,5,22-triene 135/143/394 394 2.6stigmasta-3,5-diene 81/147/381/396 396 6.8
steroid ketones 57.9stigmasta-3,5-dien-7-one 174/269/410 410 7.7stigmast-4-en-3-one 124/229/412 412 24.4stigmastadienone isomer 57/136/174/269/410 410 16.4stigmastane-3,6-dione 245/287/428 428 9.4
sterol esters 89.4campesterol ester 49.3sitosterol ester 40.1 steryl glycosides 264.9campesteryl 3�-D-glucopyranoside
204/217/361/383* 850* 141.3
sitosteryl 3�-D-glucopyranoside 204/217/361/397* 864* 123.6 waxes 173.2C36 201/229/257/285/536 536 3.5C37 243/257/550 550 3.3C38 257/564 564 56.8C39 243/257/271/285/299/578 578 5.9C40 257/285/313/592 592 46.5C40:1 264/283/590 590 2.9C41 257/271/285/299/313/327/341/355/606 606 2.3C42 257/285/313/341/620 620 24.9C42:1 264/283/618 618 1.2C43 257/271/285/299/313/327/355/369/634 634 1.5C44 257/285/313/341/648 648 19.1C46 257/285/313/341/369/397/676 676 5.3 �-hydroxy monoesters 369.5C36 73/129/237/311/609* 624* 29.1C37 73/129/237/311/623* 638* 3.0C38 73/129/237/311/339/637* 652* 229.8C40 73/129/237/311/339/367/395/665* 680* 95.6C42 73/129/237/311/339/367/395/693* 708* 11.7C44 73/129/367/395/721* 736* 0.3 monoglycerides 714.61-monotetradecanoylglycerol 73/103/129/147/343/431* 446* 5.11-monohexadecanoylglycerol 73/103/129/147/371/459* 474* 5.31-monooctadecanoylglycerol 73/103/129/147/399/487* 502* 5.31-monoeicosanoylglycerol 73/103/129/147/427/515* 530* 31.61-monodocosanoylglycerol 73/103/129/147/455/543* 558* 288.21-monotetracosanoylglycerol 73/103/129/147/483/571* 586* 179.31-monohexacosanoylglycerol 73/103/129/147/511/599* 614* 168.2

5. Resultados y discusión
1-monooctacosanoylglycerol 73/103/129/147/539/627* 642* 23.41-monotriacontanoylglycerol 73/103/129/147/567/655* 670* 8.2 �-hydroxy acylesters of glycerol
960.8
1-mono(22-hydroxydocosanoyl)glycerol
73/103/129/147/203/486/543/631* 646* 116.8
1-mono(24-hydroxytetracosanoyl)glycerol
73/103/129/147/203/514/571/659* 674* 482.8
1-mono(26-hydroxyhexacosanoyl)glycerol
73/103/129/147/203/542/599/687* 702* 301.7
1-mono(28-hydroxyoctacosanoyl)glycerol
73/103/129/147/203/570/627/715* 730* 59.5
tr traces,* as TMSi ether derivates, # as methyl ester and TMSi ether derivates.
Waxes (esters of fatty acids to fatty alcohols) were also important components
of the curaua fiber extracts and were found in the range from C36 to C46. Among the waxes, the GC/MS analysis revealed that each chromatographic peak consisted of a complex mixture of different long-chain fatty acids esterified to different long-chain fatty alcohols. The identification and quantification of the individual long-chain esters in each chromatographic peak was resolved based on the mass spectra of the peaks. The mass spectra of long-chain esters are characterized by a base peak produced by a rearrangement process involving the transfer of 2H atoms from the alcohol chain to the acid chain giving a protonated acid ion [24, 43-45]. Therefore, the base peak gives the number of carbon atoms in the acid moiety and the molecular ion the total number of carbon atoms in the ester. It is possible then to determine the individual contribution of the esters to every chromatographic peak by mass spectrometric determination of the molecular ion and the base peak. Quantification of individual esters was accomplished by integrating areas in the chromatographic profiles of ions characteristic for the acidic moiety. The detailed structural composition and abundance of the high molecular weight waxes identified in the curaua fiber is shown in Table 3. The esterified fatty acids ranged from C12 to C25 and the esterified fatty alcohols ranged from C16 to C30. Waxes with unsaturated fatty acids (C40:1 and C42:1) were also found in lower amounts, the unsaturated fatty acid being in all cases oleic acid.
161

5. Resultados y discusión
162
Table 3. Composition of the Different Waxes (mg/kg) Identified in Curaua Fibers. Wax Fatty acid:fatty alcohol Abundance
wax C36 3.5
C12:C24 0.2 C14:C22 1.1 C16:C20 2.0 C18:C18 0.2
wax C37 3.3 C16:C21 0.9 C15:C22 2.4
wax C38 56.8 C16:C22 56.8
wax C39 5.9 C15:C24 0.9 C16:C23 2.6 C17:C22 2.2 C18:C21 0.2 C19:C20 <0.1
wax C40 46.5 C16:C24 25.3 C18:C22 15.5 C20:C20 5.7
wax C40:1 2.9 C18:1:C22 2.9
wax C41 2.3 C15:C26 0.1 C16:C25 0.6 C17:C24 0.6 C18:C23 0.4 C19:C22 0.4 C20:C21 0.2 C21:C20 <0.1 C22:C19 <0.1 C23:C18 <0.1 C24:C16 <0.1
wax C42 32.2 C16:C26 3.4 C18:C24 4.0 C20:C22 22.1 C22:C20 2.7
wax C42:1 1.2

5. Resultados y discusión
C18:1:C24 1.2
wax C43 1.5 C15:C28 <0.1 C16:C27 <0.1 C17:C26 0.1 C18:C25 0.1 C19:C24 0.1 C20:C23 0.2 C21:C22 0.7 C22:C21 0.2 C23:C20 0.1 C24:C19 <0.1 C25:C18 <0.1
wax C44 19.1 C16:C28 0.9 C18:C26 0.5 C20:C24 1.7 C22:C22 16.0
wax C46 5.3 C16:C30 0.2 C18:C28 0.2 C20:C26 0.2 C22:C24 1.8 C24:C22 2.9
tr: traces.
Other waxes, consisting of a complex mixture of different long-chain �-hydroxy fatty acids esterified to different long-chain fatty alcohols, were also found in high amounts. These waxes are similar to those described among the waxes normally secreted by bees [46-48]. The mass spectrum of the TMSi ether derivative of a selected �-hydroxy monoester (C38) is shown in Figure 4. The mass spectrum of this compound is characterized by a base peak at m/z 637 corresponding to the [M-15]+ fragment ion and a fragment formed by the loss of the fatty alcohol at m/z 311. The elimination of trimethylsilanol (TMSOH) from the molecular ions also can be observed at m/z 563 [49, 50]. As occurred with the esters of fatty acids with fatty alcohols, each chromatographic peak is composed of a complex mixture of compounds. Quantification of individual compounds was performed by integrating the chromatographic profiles of the characteristic ions. The detailed structural composition of the �-hydroxy monoesters is shown in Table 4. The esterified �-hydroxy fatty acids ranges from C16 to C22 and the esterified fatty alcohols ranged from C20 to C26.
163

5. Resultados y discusión
164
Table 4. Composition and abundance (mg/kg) of the different �-hydroxy monoesters identified in curaua fibers. �-Hydroxy monoester �-hydroxy fatty acid: fatty alcohol abundance �-hydroxy monoester C36 29.1 �-OHC16:C20 29.1 �-hydroxy monoester C37 3.0 �-OHC16:C21 3.0 �-hydroxy monoester C38 229.8 �-OHC16:C22 223.9 �-OHC18:C20 5.9 �-hydroxy monoester C40 95.6 �-OHC16:C24 59.5 �-OHC18:C22 35.2 �-OHC20:C20 0.9 �-hydroxy monoester C42 11.7 �-OHC16:C26 4.8 �-OHC18:C24 0.7 �-OHC20:C22 5.3 �-OHC22:C20 0.9 �-hydroxy monoester C44 0.3 �-OHC20:C24 0.1 �-OHC12:C22 0.2
�-Hydroxy fatty acids esterified to glycerol were also found in high amounts in the curaua fiber. The mass spectra of the TMSi derivatives of �-hydroxy acylesters of glycerol are characterized by the presence of an abundant fragment arising from the loss of a methyl group at [M-15]+. The cleavage between the C-2 and C-3 carbons in the glyceryl moiety gives rise to the fragments at m/z 103 and [M-103]+. Other diagnostic ions are derived from the glyceryl moiety- i.e. at m/z 205 as a result of the cleavage between the C-2 and C-1 (the esterified carbon), and at m/z 219 due to the loss of the acyloxy moiety. The same loss of the acyloxy group from M+· and M-15+, but with the H rearrangement, gives rise to the ions at m/z 218 and 203, respectively. Other significant ions in the low-mass region occur at m/z 73 (the TMSi group), m/z 129 (the glycerol carbon backbone with a TMSi group [H2C=CH�CH=O+�Si(CH3)3]) and m/z 147 (produced by the rearrangement of two TMSi groups) [51]. The �-hydroxy fatty acids esterified to the glycerol ranges from C22 to C28. The structure and mass spectrum of the TMSi derivative of 1-mono-(22-hydroxydocosanoyl)glycerol is shown in Figure 5.

5. Resultados y discusión
HO HO
O
OH
O
OHHO
CH2OH
OH
O
HOOH
O
OH
O
OH
OH
HO HO
O
OH
O
OHHO
CH2OH
O
OHO
O-CH2
HO-O-CH
HO-O-CH2
O
O-CH2
HO-O-CH
HO-O-CH2
OHO
A
B
C
D
E
F
G
H I J K
L M
HOHO HOHO
O
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH
OH
O
OH
O
OHOH
O
HOOH
OHO
OH
O
OHOH
O
OH
O
OHOH
O
OH
OHOH
HOHO HOHO
O
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH
O
OHO
OO
OHO
O-CH2
HO-O-CH
HO-O-CH2
O
O-CH2
HO-O-CH
HO-O-CH2
OOO
O-CH2
HO-O-CH
HO-O-CH2
OHO
O-CH2
HO-O-CH
HO-O-CH2
OOHO
A
B
C
D
E
F
G
H I J K
L M
Figure 3. Structures of the main lipids present in the curaua fibers. A: stearic acid, B: n-docosanol, C: 26-hydroxyhexacosanoic acid, D: 2-hydroxytetracosanoic acid, E: docosanyl, 16-hydroxyhexadecanoate, F: 1-monodocosanoylglycerol, G: 1-mono(24-hydroxytetracosanoyl)glycerol, H: campesterol, I: ergostanol, J: sitosterol, K: stigmastanol, L: campesteryl 3�-D-glucopyranoside, M: sitosteryl 3�-D-glucopyranoside.
165

5. Resultados y discusión
166
�
100%
55
129
237
311
563
637
73327
343
[C15H31COO-H2O]+
[M-TMSOH]+
[M-15]+
[M-C22H45OH-H]+
[M-15-C22H45OH]+
m/z100 200 300 400 500 600
100%
55
129
237
311
563
637
73327
343
[C15H31COO-H2O]+
[M-TMSOH]+
[M-15]+
[M-C22H45OH-H]+
[M-15-C22H45OH]+
m/z100 200 300 400 500 600
Figure 4. Mass spectrum of trimethylsilylated hydroxy monoester C38.
n-Fatty alcohols ranging from C20 to C28 were present in the curaua extracts with the presence of only the even carbon atom homologs, docosanol (C22) and tetracosanol (C24) being the most abundant. Monoglycerides, accounting for 690.7 mg/Kg of the fibers, were present in important amounts, from C14 to C30, C22 (1-monodocosanoylglycerol) being the most prominent. Di- and triglycerides were only identified in trace amounts.
Sterols were also present among the lipids of curaua fibers in high amounts. Sitosterol was the most abundant among the free sterols with the presence of minor amounts of stigmastanol, ergostanol and campesterol. Lower amounts of sitosterol and campesterol could also be found in ester form. Sterol glycosides, such as sitosteryl and campesteryl 3�-D-glucopyranosides were also identified in high amounts, the former being the most predominant. The identification of steryl glycosides was accomplished, after BSTFA derivatization of the lipid extract, by comparison with the mass spectra and relative retention times of authentic standards [52]. Finally, other compounds identified among the curaua fiber extractives were �-tocopherol, several steroid hydrocarbons and steroid ketones, as reflected in Table 2.
In conclusion, curaua fiber is characterized by a high content of holocellulose and �-cellulose and low lignin content which would make this fiber suitable for papermaking. Moreover, the lignin composition indicates a slight predominance of S-lignin units (S/G molar ratio of 1.4). On the other hand, the high extractive content can be considered as a detrimental aspect, however most of the acetone extracts are due to polar compounds and only 1.3% corresponds to lipophilic compounds. Indeed, most of the lipophilic compounds are easily saponifiable, and therefore can be hydrolyzed and dissolved during alkaline cooking.

5. Resultados y discusión
�
100 200 300 400 500 600 m/z
100%73
129
203
265
321
381 411 486 543
631
147103
[M-145]+ [M-88]+
[M-15]+
O
OTMS
OTMS
TMSO
O
100 200 300 400 500 600 m/z
100%73
129
203
265
321
381 411 486 543
631
147103
[M-145]+ [M-88]+
[M-15]+
O
OTMS
OTMS
TMSO
O
Figure 5. Mass spectrum and structure of the TMSi ether derivative of 1-mono(22-hydroxydocosanoyl)glycerol. Acknowledgements This study has been supported by the Spanish MEC (project AGL2005-01748). We thank CELESA (Tortosa, Spain) for providing the curaua fibers. Literature cited [1] Fujihashi, G. A.; Barbosa, W. L. R. Ananas erectifolius (curauá): padronização dos extractos, frações e do material vegetal. Revista Científica da UFPA. Vol 3, 2002 .
[2] Silva, G. S.; Assis, M. B.; Barbosa, W. L. R. Investigação fitoquímica e microbiologica da espécie Ananas erectifolius (curauá). Revista Virtual de Iniciação Académica da UFPA. Vol 1, 2001.
[3] Hoareau, W.; Trindade, W. G.; Siegmund B.; Castellan, A.; Frollini E. Sugar cane bagasse and curaua lignins oxidized by chlorine dioxide and reacted with furfuryl alcohol: characterization and stability. Polym. Degrad. Stabil. 2004, 86, 567-576.
[4] Kelley, S. S.; Rowell, R. M.; Davis, M.; Jurich, Ch. K.; Ibach R. Rapid analysis of the chemical composition of agricultural fibers using near infrared spectroscopy and pyrolysis molecular beam mass spectrometry. Biomass Bioenergy. 2004, 27, 77-88.
[5] Leao, A.L., Rowell, R., and Tavares, N., 1998. Applications of natural fibres in automotive industry in Brazil - Thermoforming process. In: (P.N. Prasad, J.E.
167

5. Resultados y discusión
168
Mark, S.H. Kandil, and Z.H. Kafafi, eds) Science and Technology of Polymers Advanced Materials. Emerging technologies and business opportunities. Plenum press, New York, pp 755-761.
[6] González-Vila, F. J.; Almendros, G.; del Río, J. C; Martín, F.; Gutiérrez, A.; Romero, J. Ease of delignification assessment of wood from different Eucalyptus species by pyrolysis (TMAH)-GC/MS and CP/MAS 13C NMR spectrometry. J. Anal. Appl. Pyrol. 1999, 49, 295-305.
[7] del Río, J. C.; Gutiérrez, A.; Hernando, M.; Landín, P.; Romero, J.; Martínez, A. T. Determining the influence of eucalypt lignin composition in paper pulp yield using Py-GC/MS. J. Anal. Appl. Pyrol. 2005, 74, 110-115.
[8] Nimz, H. Beech lignin- Proposal of a constitutional scheme. Angew. Chem. Int. Edit. 1974, 13, 313-321.
[9] Adler, E. Lignin chemistry - Past, present and future. Wood Sci. Technol. 1977, 11, 169-218.
[10] Tsutsumi, Y.; Kondo, R.; Sakai, K.; Imamura, H. The difference of reactivity between syringyl lignin and guaiacyl lignin in alkaline systems. Holzforschung 1995, 49, 423-428.
[11] Hillis, W. E.; Sumimoto, M. Effect of extractives on pulping. In Natural Products of Woody Plants II; Rowe, J. W., Ed.; Springer-Verlag, Berlin, Germany, 1989; pp 880-920.
[12] Back, E. L.; Allen, L. H. Pitch Control, Wood Resin and Deresination; Tappi Press: Atlanta, GA, 2000.
[13] Gutiérrez, A.; del Río, J. C.; Martínez, M. J.; Martínez, A. T. The biotechnological control of pitch in paper pulp manufacturing. Trends Biotechnol. 2001, 19, 340-348.
[14] Ali, M.; Sreekrishnan, T. R. Aquatic toxicity from pulp and paper mill effluents: A review. Adv. Environ. Res. 2001, 5, 175-196.
[15] Rigol, A.; La Torre, A.; Lacorte, S.; Barceló, D. Bioluminiscence inhibition assays for toxicity screening of wood extractives and biocides in paper mill process waters. Environ. Toxicol. Chem. 2003, 23, 339-347.
[16] Faix, O.; Meier, D.; Fortmann, I. Thermal degradation products of wood. A collection of electron of electron-impact (EI) mass spectra of monomeric lignin derived products. Holz Roh- Werkst. 1990, 48, 351-354.
[17] Ralph, J.; Hatfield, R. D. Pyrolysis-GC/MS characterization of forage materials. J. Agric. Food Chem. 1991, 39, 1426-1437.

5. Resultados y discusión
[18] Gutiérrez, A.; del Río, J. C.; González-Vila, F. J.; Martín, F. Analysis of lipophilic extractives from wood and pitch deposits by solid-phase extraction and gas chromatography. J. Chromatogr. A. 1998, 823, 449-455.
[19] Browning, B.L. Methods of Wood Chemistry, vol. II; Wiley-Interscience Publishers, New York, USA, 1967.
[20] Technical Association of the Pulp and Paper Industry. Test methods, 1992-1993. TAPPI, Atlanta, GA. 1993.
[21] Gutiérrez, A.; Rodríguez, I. M.; del Río, J. C. Chemical characterization of lignin and lipid fractions in kenaf bast fibers used for manufacturing high-quality papers. J. Agric. Food Chem. 2004, 52, 4764-4773.
[22] van Dam, J. E. G.; van Vilsteren, G. E. T.; Zomers, F. H. A.; Shannon, W. B.; Hamilton, I. T. Increased application of domestically produced plant fibers in textiles, pulp and paper production and composite materials. Directorate-General XII, Science, Research and Development, European Commision: Brussels, 1994.
[23] del Río, J. C.; Gutiérrez, A. Chemical composition of abaca (Musa textilis) leaf fibers used for manufacturing of high quality paper pulps. J. Agric. Food Chem. 2006, 54, 4600-4610.
[24] Gutiérrez. A.; del Río, J. C. Lipids from flax fibers and their fate in alkaline pulping. J. Agric. Food Chem. 2003, 51, 4965-4971.
[25] Gutiérrez. A.; del Río, J. C. Lipids from flax fibers and their fate in alkaline pulping. J. Agric. Food Chem. 2003, 51, 6911-6914.
[26] Gutiérrez, A.; Rodríguez, I. M.; del Río, J. C. Chemical characterization of lignin and lipid fractions in industrial hemp bast fibers used for manufacturing high-quality paper pulps. J. Agric. Food Chem. 2006, 54, 2138-2144.
[27] Moore, G. Nonwood Fibre Applications in Papermaking; Pira International, Leatherhead, Surrey, UK, 1996
[28] del Río, J. C.; Martín, F.; González-Vila, F. J. Thermally assisted hydrolysis and alkylation as a novel pyrolytic approach for the structural characterization of natural biopolymers and geomacromolecules. Trends Anal. Chem. 1996, 15, 70-79.
[29] Lam, T. B. T.; Kadoya, K.; Liyama, K. Bonding of hydroxycinamic acids to lignin: ferulic and p-coumaric acids are predominantly linked at the benzyl position of lignin, not the �-position, in grass cell walls. Phytochemistry 2001, 57, 987-992.
169

5. Resultados y discusión
170
[30] Sun, R. C.; Sun, X. F.; Zhang, S. H. Quantitative determination of hydroxycinnamic acids in wheat, rice, rye, and barley straws, maize stems, oil palm frond fiber, and fast-growing poplar wood. J. Agric. Food Chem. 2001, 49, 5122-5129.
[31] Grabber, J. H.; Ralph, J.; Hatfield, R. D. Cross-linking of maize walls by ferulate dimerization and incorporation into lignin. J. Agric. Food Chem. 2000, 48, 6106-6113.
[32] Scalbert, A.; Monties, B.; Lallemand, J. Y.; Guittet, R.; Rolando, C. Ether linkage between phenolic acids and lignin fractions from wheat straw. Phytochemistry 1985, 24, 1359-1362.
[33] Sun, R. C.; Sun, X. F.; Wang, S. Q.; Zhu, W.; Wang, X. Y. Ester and ether linkages between hydroxycinnamic acids and lignins from wheat, rice, rye, and barley straws, maize stems, and fast-growing poplar wood. Ind. Crop Prod. 2002, 15, 179-188.
[34] Lam, T. B. T.; liyama, K.; Stone, B. A. Cinnamic acids bridges between cell wall polymers in wheat and Phalaris internodes. Phytochemistry 1992, 31, 1179-1183.
[35] del Río, J. C.; McKinney, D. E.; Knicker, H.; Nanny, M. A.; Minard, R. D. Hatcher, P. G. Structural characterization of bio- and geo-macromolecules by off-line thermochemolysis with tetramethylammonium hydroxide. J. Chromatogr. A 1998, 823, 433-448.
[36] Martín, F.; del Río, J. C.; González-Vila, F. J.; Verdejo, T. Thermally assisted hydrolysis and alkylation of lignins in the presence of tetra-alkylammonium hydroxides. J. Anal. Appl. Pyrol. 1995, 35, 1-13.
[37] Challinor, J. M. Characterization of wood by pyrolysis derivatization-gas chromatography/ mass spectrometry. J. Anal. Appl. Pyrol. 1995, 35, 93-107.
[38] Clifford, D. J.; Carson, D. M.; Mackinney. D. E.; Bortiatynski, J. M.; Hatcher, P. G. A new rapid technique for the characterization of lignin in vascular plants: thermochemolysis with tetramethylammonium hydroxide (TMAH). Org. Geochem. 1995, 23, 169-175.
[39] Ralph, J.; Hatfield, R.D.; Quideau, S.; Helm, R.F.; Grabber, J.H.; Jung, H.-J.G. Pathway of p-coumaric acid incorporation into maize lignin as revealed by NMR. J. Chem. Soc. 1994, 34, 1-12.
[40] Crestini, C.; Argyropoulos, D.S. Structural analysis of wheat straw lignin by quantitative 31P and 2D NMR spectroscopy. The occurrence of ester bonds and �-O-4 substructures. J. Agric. Food Chem. 1997, 45, 1212-1219.

5. Resultados y discusión
[41] Lu, F.C.; Ralph, J. Detection and determination of p-coumaroylated units in lignins. J. Agric. Food Chem. 1999, 47, 1988-1992.
[42] Grabber, J.H.; Quideau, S.; Ralph, J. p-Coumaroylated syringyl units in maize lignin: Implications for �-ether cleavage by thioacidolysis. Phytochemistry 1996, 43, 1189-1194.
[43] Sharkey, Jr. A. G.; Shultz, J. L.; Friedel, R. A. Mass spectra of esters. Formation of rearrangement ions. Anal. Chem. 1959, 31, 87-94.
[44] Moldovan, Z.; Jover, E.; Bayona, J. M. Systematic characterization of long-chain aliphatic esters of wool wax by gas chromatography-electron impact ionisation mass spectrometry. J. Chromatogr. A 2002, 952, 193-204.
[45] Reiter, B.; Lechner, M.; Lorbeer, E.; Aichholz, R. Isolation ad characterization of wax esters in fennel and caraway seed oils by SPE-GC. J. High Res. Chromatogr. 1999, 22, 514-520.
[46] Tulloch, A. P. The composition of beeswax and other waxes secreted by insects. Lipids. 1969, 5, 247-258.
[47] Tulloch, A. P. Beeswax: structure of the esters and their component hydroxy acids and diols. Chem. Phys. Lipids. 1971, 6, 235-265.
[48] Aichholz, R.; Lorbeer, E. Investigation of combwax of honeybees with high-temperature gas chromatography and high-temperature gas chromatography-chemical ionization mass spectrometry: I. High-temperature gas chromatography. J. Chromatogr. A. 1999, 855, 601-615.
[49] Aichholz, R.; Lorbeer, E. Investigation of combwax of honeybees with high-temperature gas chromatography and high-temperature gas chromatography-chemical ionization mass spectrometry: II. High-temperature gas chromatography-chemical ionization mass spectrometry. J. Chromatogr. A. 2000, 883, 75-88.
[50] Kimpe, K.; Jacobs, P. A.; Waelkens, M. Mass spectrometric methods prove the use of beeswax and ruminant fat in late Roman cooking pots. J. Chromatogr. A. 2002, 968, 151-160.
[51] Graça, J.; Schreiber, L.; Rodrigues, J.; Pereira, H. Glycerol and glyceryl esters of �-hydroxyacids in cutins. Phytochemistry. 2002, 61, 205-215.
[52] Gutiérrez, A.; del Río, J. C. Gas chromatography/mass spectrometry demonstration of steryl glycosides in eucalypt wood, kraft pulp and process liquids. Rapid Commun. Mass Spectrom. 2001, 15, 2515-2520.
171

5. Resultados y discusión
Publicación V: Coelho D., Marques G., Gutiérrez A., Silvestre A.R.D. and del Río J.C. (2007) Chemical characterization of the lipophilic fraction of Giant reed (Arundo donax) fibers used for pulp and paper manufacturing. Industrial Crops and Products, 26, 229-236.
172

5. Resultados y discusión
Chemical characterization of the lipophilic fraction of Giant reed (Arundodonax) fibres used for pulp and paper manufacturing
Dora Coelho1,2, Gisela Marques1, Ana Gutiérrez1, Armando J.D. Silvestre2, José C. del Río1
1 Instituto de Recursos Naturales y Agrobiología, Consejo Superior de Investigaciones
Científicas, P.O. Box 1052, 41080-Seville, Spain 2 Departamento de Química, Universidade de Aveiro, 3810-193 Aveiro, Portugal
Abstract The chemical composition of lipophilic extractives from Arundo donax stems (including nodes and internodes), used for pulp and papermaking, was studied. The lipid fraction was extracted with acetone and redissolved in chloroform, and then fractionated by solid-phase extraction (SPE) on aminopropyl-phase cartridges into four different fractions of increasing polarity. The total lipid extract and the resulting fractions were analysed by gas chromatography and gas chromatography-mass spectrometry, using short- and medium-length high-temperature capillary columns, respectively. The main compounds identified in the fibres included series of long-chain n-fatty acids, n-alkanes, n-aldehydes, n-alcohols, monoglycerides, free and esterified sterols and triterpenols, steryl glucosides, steroid hydrocarbons and steroid and triterpenoid ketones. Minor amounts of other compounds such as diglycerides, waxes and tocopherols were also identified among the lipids of A. donax. Keywords: Arundo donax, lipophilic extractives, pitch, fatty acids, sterols, steryl glucosides, GC, GC/MS. 1. Introduction In the last decades, fast growing plants have received particular attention as alternative sources of cellulose fibres (van Dam et al. 1994; Moore, 1996). These non-wood plants are the common fibre source for paper pulp production in developing countries where wood fibres are not available. In the developed world, although wood is still by far the main raw material for pulp and paper manufacture, a market exists for high-value-added papers from these fibres. Arundo donax L. (giant reed) is a widely distributed naturally growing perennial rhizomatous grass with a segmented tubular structure like bamboo (Seca et al., 2000), which has been considered as one of the promising non-wood plants for pulp and paper industry (Shatalov and Pereira, 2002). The easy adaptability to different ecological conditions, the annual harvesting period and the high biomass productivity (32-37 t per year-1ha-1 of dry biomass) reached by intensive cultivation (Vecchiet et al., 1996), combined with appropriate chemical composition (Shatalov et al., 2001), make A. donax very attractive as an alternative source of fibres (Shatalov and Pereira, 2005).
173

5. Resultados y discusión
174
To improve the utilisation of A. donax fibres, it is necessary to broaden the knowledge of structural features of its components. Previous chemical research on A. donax includes chemical composition, general features of macromolecular components (Pascoal Neto et al., 1997) and structures of isolated hemicelluloses (Driss et al., 1973, Joseleau and Barnoud, 1974, 1975, 1976). A few studies on the lignin composition (Joseleau and Barnoud, 1976, Joseleau et al., 1976, Faix et al., 1989) showed that it is composed of guaiacyl- and syringyl-propane units with minor amounts of p-hydroxyphenylpropane units (Faix et al., 1989) and associated with phenolic acids (Tai et al., 1987). However, until now no studies about the composition of A. donax lipophilic fraction have been performed.
The amount and composition of lipophilic extractives is an important parameter in wood processing for pulp and paper production and it is dependent on factors such as the plant species, age, and growth location. The different lipid classes have different chemical behaviour during pulping and bleaching (Gutiérrez and del Río, 2003; Freire et al., 2005). The lipophilic extractives are also responsible for the formation of sticky deposits on the machinery, giving rise to dark spots in bleached pulp and paper, the so-called pitch, both with negative economic impact on pulp and paper industry (del Río et al., 1998, 2000; Gutiérrez et al., 2004; Gutiérrez and del Río, 2005; Silvestre et al., 1999). The accumulation of lipophilic compounds leads also to higher chemical consumption during pulping and bleaching and therefore increasing production costs. On the other hand, extractives or their derivatives, might contribute to the toxicity of paper pulp effluents and products (McCubbin and Folke 1995; Rigol et al., 2003). The detailed identification of such lipophilic components is therefore an important step in the study of the behaviour and fate of extractives during pulp and paper production and consequently in the search for new solutions to control pitch deposition as well as to decrease effluent toxicity.
In the present paper, the chemical composition of the lipophilic extractives from A. donax fibres was studied. Gas chromatography (GC) and GC-mass spectrometry (GC/MS) using, respectively, short- and medium-length high-temperature capillary columns with thin films, that enable elution and separation of high-molecular-mass lipids such as waxes, steryl esters and triglycerides, are employed. For a more detailed characterization of the different homologous series and other minor compounds, the extract was fractionated by a simple solid-phase extraction (SPE) method using aminopropyl phase cartridges, as described previously (Gutiérrez et al., 1998, 2004). 2. Experimental 2.1. Samples Samples of A. donax L. reed stems (including nodes and internodes) were supplied by University of Huelva, Spain. The samples were air-dried and milled using a knife mill (Janke and Kunkel, Analysenmühle). For the isolation of

5. Resultados y discusión
lipids, the milled samples were Soxhlet extracted with acetone for 8h. The lipophilic extractives were obtained by redissolving the dried acetone extract in chloroform and evaporated to dryness under nitrogen. 2.2. Solid Phase Extraction (SPE) fractionation The chloroform extracts (5-20 mg) were fractionated by a SPE procedure in aminopropyl phase cartridges (500 mg) from Waters (Dvision of Millipore, Milford, MA, USA), as already described (Gutiérrez et al., 1998, 2004). Briefly, the dried extract was taken up in a minimal volume (<0.5 mL) of hexane:chloroform (4:1) and loaded into the cartridge column previously conditioned with hexane (4 mL). The cartridge was loaded and eluted by gravity. The column was first eluted with 8 mL of hexane and subsequently with 6 mL of hexane:chloroform (5:1), then with 10 mL of chloroform and finally with 10 mL of diethyl ether:acetic acid (98:2). Each isolated fraction was dried under nitrogen. 2.3. GC and GC/MS analyses For identification and quantification, the total extracts and the SPE fractions were analysed by GC and GC/MS. For GC analysis, a Hewlett-Packard HP 5890 gas chromatograph equipped with split-splitless injector and a flame ionization detector (FID) system was used (Hewlett-Packard, Hoofddorp, Netherlands). The injector and detector temperatures were set at 300 and 350ºC, respectively. Duplicate samples (1 �L) were injected in the splitless mode. Helium was used as the carrier gas. The capillary column used was a 5m × 0.25 mm i.d., 0.1 �m film thickness, high-temperature, polyimide-coated fused silica tubing DB-5HT from J&W Scientific (Folsom, CA), especially processed for use at 400ºC. The oven was temperature programmed from 100ºC (1 min) to 350ºC (3 min) at 15ºC/min. Peaks were quantified by area and a mixture of standards (tetracosane, hexadecanoic acid, sitosterol, cholesteryl oleate and triheptadecanoin) was used for quantitation. The data from the two replicates was averaged.
The GC/MS analysis were performed on a Varian Star 3400 gas chromatograph (Varian, Walnut Creek, CA) coupled with an ion-trap detector (Varian Saturn) equipped with a high-temperature capillary column (DB-5HT, 15 m×0.25 mm i.d., 0.1 �m film thickness; J&W). Helium was used as carrier gas at a rate of 2 ml/min. The oven was heated from 120ºC (1 min) to 380ºC (5 min) at 10ºC/min. The temperature of the injector during the injection was 120ºC, and 0.1 min after injection was programmed to 380ºC at a rate of 200ºC/min and held for 10 min. The temperature of the transfer line was set at 300ºC. Bis(trimethylsilyl)trifluoroacetamide (BSTFA) silylation was used when required. Compounds were identified by comparing their mass spectra with
175

5. Resultados y discusión
176
mass spectra in Wiley and NIST libraries, by mass fragmentography, and, when possible, by comparison with authentic standards.
3. Results and discussion The total acetone extract from A. donax fibres accounted for 1.56% of total fibre weight. The lipophilic – chloroform soluble – compounds represented 0.62%, while the remaining 0.94% corresponded to polar compounds non-soluble in chloroform. The lipid extracts were analyzed by GC and GC/MS according to the method previously described (Gutiérrez et al. 1998, 2004). The GC/MS chromatogram of the A. donax fibres extract, as trimethylsilyl (TMS) derivatives, is shown in Figure 1. For a better characterization of the compounds present in the lipid extracts, these were subsequently fractionated by SPE in aminopropyl-phase cartridges into four major fractions of increasing polarity. The chromatograms of the different SPE fractions are shown in Figure 2. The first fraction (A), eluted with hexane, was enriched in steryl esters, waxes and hydrocarbons. The second fraction (B), eluted with hexane:chloroform (5:1), contained steroid ketones. The third fraction (C), eluted with chloroform, contained sterols, fatty alcohols and mono- and diglycerides. A final fraction (D)
5 10 15 20
Retention time (min)
Sitosterol
SG
StG
CG
FA18
FA18:2
FA18:1
FA16
FA28
FA20MG18
FA30
MG16
Campesterol
FA17 MG26FA24
Stigmasterol
FA26
Steryl glycosides
Steryl/triterpenyl esters
30
Figure 1. GC/MS chromatogram of the derivatized (TMS) chloroform extract of Arundo donax fibres. FA: fatty acids; MG: monoglycerides; CG: campesteryl 3�-D-glucopyranoside; StG: stigmasteryl 3�-D-glucopyranoside; SG: sitosteryl 3�-D-glucopyranoside.

5. Resultados y discusión
�-Sitosterol + Stigmastanol + �-amyrin
CampesterolStigmasterol
7-oxositosterol
�-amyrin
Ak 27
Ak 31Ak 25
Ak 29
Steryl esters
5 10 15 20 25
Cycloartenone
�-amyrenone
�-amyrenone
5 10 15 20 25
5 10 15 20 25
FA18
FA26
FA18:1+
FA18:2
FA16
FA28
FA24
5 10 15 20 25
A
B
C
D
Figure 2. GC/MS chromatograms of the different SPE fractions isolated from the A. donax fibres extracts. Fraction A, eluted with 8 mL of hexane; fraction B, eluted with 6mL of hexane:chloroform (5:1); fraction C, eluted with 10 mL of chloroform; and fraction D, eluted with 10 mL diethyl ether:acetic acid (98:2). FA: fatty acids; AK: n-alkanes.
177

5. Resultados y discusión
178
Table 1. Chemical composition of lipophilic extractives in Arundo donax reed (mg/Kg of fibre). Each value is the average of two extractions with variation coefficients within 0.1-4.5% .
Compound Mass Fragments MW Amount n-Alkanes 77.9 n-docosane 57/71/85/310 310 0.5 n-tricosane 57/71/85/324 324 0.2 n-tetracosane 57/71/85/338 338 0.6 n-pentacosane 57/71/85/352 352 6.3 n-hexacosane 57/71/85/366 366 3.9 n-heptacosane 57/71/85/380 380 15.8 n-octacosane 57/71/85/394 394 6.7 n-nonacosane 57/71/85/408 408 37.0 n-triacontane 57/71/85/422 422 0.8 n-hentriacontane 57/71/85/436 436 5.4 n-dotriacontane 57/71/85/450 450 0.3 n-tritriacontane 57/71/85/464 464 0.4 Steroid hydrocarbons 127.4 ergostatriene 135/143/380 380 14.5 ergostadiene 81/147/367/382 382 9.3 estigmastadiene 81/147/381/396 396 8.4 estigmasta-3,5,22-triene 135/143/394 394 49.2 estigmasta-3,5-diene 81/147/381/396 396 46.0 Fatty acids 1137.7 n-tetradecanoic acid 73/117/132/145/285/300 * 300* 3.5 n-pentadecanoic acid 73/117/132/145/299/314 * 314* 1.8 n-hexadecanoic acid 60/73/129/256 256 276.3 n-heptadecanoic acid 73/117/132/145/327/342 * 342 * 10.0 9,12-octadecadienoic acid 67/81/280 280 30.0 9-octadecanoic acid 55/69/264 282 55.7 n-octadecanoic acid 60/73/129/284 284 73.6 n-nonadecanoic acid 73/117/132/145/355/370 370* 3.1 n-eicosanoic acid 60/73/129/312 312 50.0 n-heneicosanoic acid 55/69/129/326 326 3.3 n-docosanoic acid 60/73/129/340 340 35.7 n-tricosanoic acid 60/73/129/354 354 25.3 n-tetracosanoic acid 60/73/129/368 368 55.7 n-pentacosanoic acid 60/73/129/382 382 33.5 n-hexacosanoic acid 73/117/132/145/453/468 * 468 * 144.1 n-heptacosanoic acid 73/117/132/145/467/482 482* 14.3 n-octacosanoic acid 73/117/132/145/482/497 * 497 * 134.9 n-nonacosanoic acid 73/117/132/145/495/510 * 510 * 53.9 n-triacontanoic acid 73/117/132/145/509/525 * 525 * 109.9 n-hentriacontanoic acid 73/117/132/145/523/538 538* 6.2 n-dotriacontanoic acid 73/132/145/117/537/552 * 552 * 16.9 Fatty alcohols 194.3 n-hexacosanol 75/103/439* 454* 33.4

5. Resultados y discusión
n-octacosanol 75/103/467* 482* 54.9 n-triacontanol 75/103/495* 510* 57.7 n-dotriacontanol 75/103/523* 538* 48.3 Aldehydes 81.6 n-hexacosanal 82/96/362 380 10.4 n-octacosanal 82/96/390 408 22.9 n-triacontanal 82/96/418 436 48.3 Sterols/Triterpenols 528.1 campesterol 55/145/213/382/400 400 90.6 stigmasterol 55/81/255/394/412 412 46.4 sitosterol 145/213/396/414 414 281.0 stigmastanol 215/416 416 71.9 7-oxo-sitosterol 135/161/187/396/428 428 6.5 �-amyrin 189/203/218/409/426 426 8.2 �-amyrin 189/203/218/409/426 426 23.5 Tocopherol 17.7 �-tocopherol 151/416 416 6.8 �-tocopherol 165/430 430 10.9 Triterpenoid and steroid ketones 43.9 �-amyrenone 189/203/218/409/424 424 10.2 �-amyrenone 189/203/218/409/424 424 5.9 cycloartenone 189/205/313/409/424 424 14.2 stigmasta-3,5-dien-7-one 174/269/410 410 3.2 stigmast-4-en-3-one 124/229/412 412 4.6 stigmast-4-en-3,6-dione 137/398/408/411/426 426 3.6 stigmastane-3,6-dione 245/287/428 428 2.5 Steryl /triterpenyl esters 68.1 sitosteryl ester 147/381/397 - 16.1 �-amyrinyl ester 189/203/218 - 14.0 �-amyrinyl ester 189/203/218 - 38.0 Steryl glucosides 151.6 campesteryl 3-�-D-glucopyranoside 204/217/361/383 * 850 * 30.6 stigmasteryl 3-�-D-glucopyranoside 204/217/361/395 * 864 * 8.0 sitosteryl 3-�-D-glucopyranoside 204/217/361/397 * 862 * 113.0 Monoglyceride 367.5 2,3-dihydroxypropyl tetradecanoate 73/103/129/147/343/431 * 446 * 5.5 2,3-dihydroxypropyl hexadecanoate 73/103/129/147/371/459 * 474* 94.2 2,3-dihydroxypropyl octadecanoate 73/103/129/147/399/487 * 502 * 86.6 2,3-dihydroxypropyl eicosanoate 73/103/129/147/427/515 * 530 * 35.1 2,3-dihydroxypropyl docosanoate 73/103/129/147/455/543 * 558 * 43.0 2,3-dihydroxypropyl tetracosanoate 73/103/129/147/483/571 * 586 * 46.9 2,3-dihydroxypropyl hexacosanoate 73/103/129/147/511/599 * 614 * 56.2
179

5. Resultados y discusión
180
Diglycerides 47.6 dipalmitin, 1,2- (P2) 57/129/313/386/625 * 640 * 7.8 dipalmitin, 1,3- (P2) 57/129/314/371/385/625 * 640 * 12.1 palmitoylstearin (PS) 57/129/314/372/399/579 * 668 * 16.8 distearin, 1,2- and 1,3- (S2) 57/129/342/399/607 * 696 * 10.9 * as TMSi ether derivates; bold mass fragments indicate base peaks.
enriched in free fatty acids was eluted with diethyl ether-acetic acid (98:2). The identities and abundances of the main compounds identified are listed in Table 1. The most predominant lipid classes identified among the A. donax lipid extracts were series of n-fatty acids (41% of total lipids identified), sterols (19%), monoglycerides (13%), fatty alcohols (7%) and steryl glucosides (6%). Minor amounts of alkanes, aldehydes, tocopherols, steroid hydrocarbons, steroid and triterpenoid ketones and steryl/triterpenyl esters, were also present in these fibres. The structures of main and representative compounds are shown in Figure 3.
The series of free fatty acids were identified in A. donax fibres ranging from tetradecanoic (C14) to dotriacontanoic (C32) acids, with strong even-over-odd carbon atom predominance. Hexadecanoic acid (palmitic acid, I) was the most abundant fatty acid, however a bimodal distribution, with a second maximum for octacosanoic acid (C28) was observed. The unsaturated 9-octadecenoic (oleic acid, II) and 9,12-octadecadienoic (linoleic acid, III) acids were also present in important amounts. The series of n-alkanes was also identified in the A. donax fibre ranging from docosane (C22) to tritriacontane (C33), with a strong odd-over-even carbon atom number predominance, and nonacosane (IV) being the most predominant homolog. n-Fatty alcohols ranging from hexacosanol (C26) to dotriacontanol (C32) were present in the A. donax extracts with the presence of only the even carbon atom number homologues, triacontanol (V) being the most abundant. Significant amounts of a series of n-aldehydes ranging from hexacosanal (C26) to triacontanal (C30) were identified in the A. donax fibres with triacontanal (VI) predominating. Monoglycerides were also present in high amounts in A. donax fibres. The series of monoglycerides was identified in the range from C14 to C26, with maximum for monopalmitin, C16, (VII). Steroids and triterpenoids, including free sterols, steryl esters, steryl glucosides, steroid ketones and hydrocarbons are among the most predominant compounds in the lipophilic extract of A. donax fibre. Free sterols were the major compound class among steroids and triterpenoids, sitosterol (VIII) being the main sterol present. Other sterols, such as campesterol (IX), stigmasterol (X), stigmastanol (XI) and the oxidized 7-oxositosterol, were also present. Steryl esters were also present in A. donax extract, although in low amounts. The complete identification of the individual steryl esters by GC-MS was not possible since they only show fragments arising from the sterol moiety by electro-impact MS

5. Resultados y discusión
Figure 3. Structures of the main lipophilic compounds present in A. donax fibres. (I) palmitic acid, (II) oleic acid, (III) linoleic acid, (IV) nonacosane, (V) triacontanol, (VI) triacontanal, (VII) monopalmitin, (VIII) sitosterol, (IX) campesterol, (X) stigmasterol, (XI) stigmastanol, (XII) sitosteryl 3�-D-glucopyranoside, (XIII) �-amyrin, (XIV) �-amyrin, (XV) stigmasta-3,5-diene, (XVI) stigmasta-3,5,7-triene, (XVII) �-amyrenone, (XVIII) �-amyrenone, (XIX) cycloartenone, (XX) stigmasta-3,5-dien-7-one, (XXI) stigmast-4-en-3-one, (XXII) stigmasta-3,6-dione.
181

5. Resultados y discusión
182
and rarely give detectable molecular ions (Lusby et al. 1984, Evershed et al. 1989). By monitoring the ions corresponding to the different sterol moieties in the SPE fraction enriched in steryl esters, it was possible to identify series of sitosterol as well as �- and �-amyrin esters. Steryl glucosides, such as campesteryl, stigmasteryl and sitosteryl �-D-glucopyranosides (XII), were identified in significant amounts, the latter being the most predominant.
The identification of steryl glucosides was accomplished (after BSTFA derivatization of the lipid extract) by comparison with the mass spectra and relative retention times of authentic standards (Gutiérrez and del Río, 2001). Among triterpenols, �-amyrin (XIII) and �-amyrin (XIV) occurred in free and esterified form, with the latest being detected in low amounts. Finally, several steroid hydrocarbons, such as stigmasta-3,5-diene (XV) and stigmasta-3,5,7-triene (XVI) and triterpenoid and steroid ketones, such as �-amyrenone (XVII), �-amyrenone (XVIII), cycloartenone (XIX), stigmasta-3,5-dien-7-one (XX), stigmast-4-en-3-one (XXI) and stigmasta-3,6-dione (XXII), were also identified.
The different lipid classes present in A. donax fibres will have different behavior during pulping and bleaching and therefore the problematic of pitch will be different depending the type of pulping (i.e. mechanical, chemical) and bleaching (ECF, TCF) processes. The knowledge of the chemical composition of the lipophilic components of A. donax fibres shown here will assist to predict pitch problems during pulp and papermaking of this fibre and to establish appropriate methods for their control. Acknowledgements This study has been funded by the Spanish project AGL2005-01748. GM thanks the Spanish Ministry of Education and Science for a FPI fellowship. We thank M.J. Diaz (University of Huelva) for the Arundo donax fibres. References del Río, J.C., Gutiérrez A., Gonz.lez-Vila F.C., Martín F. and Romero J., 1998.
Characterization of organic deposits produced in kraft pulping of Eucalyptus globulus wood. J. Chromatogr. A. 823, 457-465.
del Río, J.C.; Romero, J.; Gutiérrez, A., 2000. Analysis of pitch deposits produced in Kraft pulp mills using a totally chlorine free bleaching sequence. J. Chromatogr. A 874, 235-245.
Driss, M., Rozmarin, G. and Chene, M., 1973. Some physicochemical properties of two xylans of reed (Phragmites communis and Arundo donax) in solution. Cell. Chem. Technol. 7, 703-713.

5. Resultados y discusión
Evershed, R.P., M.C. Prescott, N. Spooner and L.J. Goad. 1989. Negative ion ammonia chemical ionization and electron impact ionization mass spectrometric analysis of steryl fatty acyl esters. Steroids 53, 285–309.
Faix, O., Meier, D., Beinhoff, O., 1989. Analysis of lignocelluloses and lignins from Arundo donax and Miscanthus sinensis Anderss and hydroliquefaction of Miscanthus. Biomass 18, 109.
Freire, C.S.R., Silvestre, A.J.D. and Pascoal Neto, C., 2005. Lipophilic extractives in Eucalyptus globulus Kraft pulps. Behaviour during ECF bleaching. J. Wood Chem. Technol. 25, 67-80.
Gutiérrez, A., del Río, J.C., González-Vila F.J. and Martín F., 1998. Analysis of lipophilic extractives from wood and pitch deposits by solid-phase extraction and gas chromatography. J. Chromatogr. A. 823, 449-455.
Gutiérrez, A.; del Río, J.C., 2001. Gas chromatography/mass spectrometry demonstration of steryl glycosides in eucalypt wood, Kraft pulp and process liquids. Rapid Commun. Mass Spectrom., 15, 2515-2520.
Gutiérrez, A.; del Río, J. C., 2003. Lipids from flax fibers and their fate in alkaline pulping. J. Agric. Food Chem., 51, 4965-4971.
Gutiérrez, A., del Río, J.C. and Martínez, A.T., 2004. Chemical Analysis and Biological Removal of Wood Lipids forming Pitch deposits in paper pulp manufacturing. In: F.J.T. Spencer and A.L. Ragout de Spencer (Eds.), Protocols in Environmental Microbiology. In: Methods in Molecular Biology. Chapter 19, Humana Press, 2004, pp. 189-202.
Gutiérrez, A. and del Río, J.C., 2005. Chemical characterization of pitch deposits produced in the manufacturing of high-quality paper pulps from hemp fibers. Biores. Technol. 96, 1445-1450.
Joseleau, J.P. and Barnoud, F., 1974. Hemicelluloses of young internodes of Arundo donax. Phytochemistry 13, 1155-1158.
Joseleau, J.P. and Barnoud, F., 1975. Hemicelluloses of Arundo donax at different stages of maturity. Phytochemistry 14, 71-75.
Joseleau, J.P. and Barnoud, F., 1976. Cell wall carbohydrates and structural studies of xylan in relation to growth in the Arundo donax. Appl. Polym. Symp. 28, 983-992.
Joseleau, J.P., Miksche, G.E. and Yasuda, S., 1976. Structural variation of Arundo donax lignin in relation to growth. Holzforschung 31, 19-20.
183

5. Resultados y discusión
184
Lusby, W. R., M. J. Thompson and J. Kochansky. 1984. Analysis of sterol esters by capillary gas chromatography electron impact and chemical ionization-mass spectrometry. Lipids 19(11), 888–901.
McCubbin, N. and J. Folke. 1995. Significance of AOX vs. unchlorinated organics. Pulp Paper Can. 96, 43-48.
Moore, G., 1996. Nonwood Fibre Applications in Papermaking; Pira International, Leatherhead, Surrey, UK.
Pascoal Neto, C., Seca, A., Nunes, A.M., Coimbra, M.A., Domingues, F., Evtuguin, D., Silvestre, A.J.D, Cavaleiro, J.A.S., 1997. Variations in chemical composition and structure of macromolecular components in different morphological regions and maturity stages of Arundo donax. Ind. Crops Prod. 6, 51-58.
Rigol, A.; La Torre, A.; Lacorte, S.; Barceló, D., 2003. Bioluminiscence inhibition assays for toxicity screening of wood extractives and biocides in paper mill process waters. Environ. Toxicol. Chem., 23, 339-347.
Seca, A.M., Cavaleiro, J.A.S., Domingues, F.M.J., Silvestre, A.J.D, Evtuguin, D., Neto, C.P., 2000. Structural characterization of the lignin from the nodes and internodes of Arundo donax Reed. J.Agric.Food Chem. 48, 817-824.
Shatalov, A.A., Quilhó, T., Pereira, H., 2001. Arundo donax L. reed – new perspectives for pulping and bleaching. 2. Raw material characterization. TAPPI J. 84 (1), 1-12.
Shatalov, A.A., Pereira, H., 2002. Influence of stem morphology on pulp and paper properties of Arundo donax L. reed. Ind. Crops Prod. 15, 77-83.
Shatalov, A.A., Pereira, H., 2005. Kinetics of organosolv delignification of fibre crop Arundo donax L. Ind. Crops Prod. 21, 203-210.
Silvestre, A. J.D., C. L.C. Pereira, C. Pascoal Neto, A.C. Duarte, J. A. S. Cavaleiro and F. P. Furtado. 1999. Chemical composition of pitch deposits from an ECF Eucalyptus globulus bleached kraft pulp mill: Its relationship with wood extractives and additives in process streams. Appita J. 52(5), 375-382.
Tai, D., Cho, W. And Ji, W., 1987. Studies of Arundo donax lignins. Proceedings of the 4th ISWPC, Vol. II, April 1987, Paris, pp. 13-17.
van Dam, J. E. G.; van Vilsteren, G. E. T.; Zomers, F. H. A.; Shannon, W. B.; Hamilton, I. T., 1994. Industrial fibre crops - study on: increased application of domestically produced plant fibres in textiles, pulp and paper production

5. Resultados y discusión
and composite materials. Directorate-General XII, Science, Research and Development, European Commission.
Vecchiet, M., Jodice, R., Schenone, G., 1996. Agronomic research on giant reed (Arundo donax L.). Management system and cultivation of two different provenances. In: Chartier, Ph., Ferrero, G.L., Henius, U.M., Hultberg, S., Sachau, J., Wiinblab, M. (Eds.), Biomass for Energy and the Environment. In: Proceedings of the Ninth European Biomass Conference, Copenhagen, Pergamon, UK, pp. 644-648.
185

5. Resultados y discusión
Publicación VI: Marques G., Gutiérrez A. and del Río J.C. (2008) Chemical composition of lignin and lipids from tagasaste (Chamaecytisus proliferus spp. palmensis). Industrial Crops and Products, 28, 29-36.
186

5. Resultados y discusión
Chemical composition of lignin and lipids from tagasaste (Chamaecytisusproliferus spp. palmensis)
Gisela Marques, Ana Gutiérrez and José C. del Río
Instituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, P.O. Box 1052, 41080- Seville, Spain
Abstract The chemical characterization of trimming residues of tagasaste (Chamaecytisus proliferus spp. palmensis), a hardy leguminous shrub that has been recently explored for pulp and paper production, was performed with especial emphasis in the composition of lignin and lipophilic extractives. Tagasaste was characterized by a high content of holocellulose (81%) and �-cellulose (41%), while having a relatively low lignin content of 18.9%. The analysis of lignin was performed “in situ” by pyrolysis-gas chromatography/mass spectrometry (Py-GC/MS) and showed a composition with a guaiacyl:syringyl (G:S) molar proportion of 38:62 (S/G molar ratio of 1.6) and the absence of p-hydroxyphenyl (H) units. The high S/G ratio, together with its low lignin content makes tagasaste an adequate raw material for pulping. On the other hand, the relatively high acetone extractive content (1.4%) was mostly due to polar compounds and only 0.2% corresponded to lipophilic compounds. The lipophilic compounds, analyzed by GC and GC/MS, were mainly composed of fatty acids, including �-hydroxyfatty acids, and steroid compounds, such as free and conjugated (esters and glycosides) sterols. Keywords: Tagasaste; lipids; lignin; pyrolysis; paper pulp. 1. Introduction Tagasaste (Chamaecytisus proliferus spp. palmensis), also known as “tree lucerne”, is a hardy leguminous and fast-growing shrub of the Fabaceae (Genisteae) family. It is indigenous of the Canary Islands (Spain) but is now being cultivated in Australia, New Zealand and other countries (Francisco-Ortega et al., 1991). The shrub is being mainly exploited for high-protein fodder to maintain livestock (Borens and Poppi, 1990; Ventura et al., 2002) and also as N-fixing crops to improve soil fertility (Kindu et al., 2006). In order to encourage the formation of bushes with multiple stems the shrub must be grazed with regularity, which leads to a high accumulation of trimming residues. These residues are nowadays considered as agricultural waste since they cannot be converted to valuable products.
As attempts to reduce the adverse environmental impact and to use this renewable biomass, they have been recently explored as an alternative raw material for pulp production (Díaz et al. 2004; López et al., 2004; Jiménez et al.,
187

5. Resultados y discusión
188
2006; Jiménez et al., 2007; García et al., 2007). Tagasaste has been found to be an excellent raw material for paper pulp production, similar to eucalypt wood, with high hollocellulose and �-cellulose contents, and low lignin and extractives content, giving high yields (Díaz et al. 2004; López et al., 2004; Jiménez et al., 2006). However, despite all this previous work, a detailed chemical composition of tagasaste trimming residues has not been addressed so far, which is of high importance for optimizing the use of this raw material for paper pulp production.
The content and chemical structure of woody components, in particular the lignin content and its composition in terms of its p-hydroxyphenyl (H), guaiacyl (G) and syringyl (S) moieties are important parameters in pulp production in view of delignification rates, chemical consumption and pulp yields. The higher reactivity of the S lignin with respect to the G lignin in alkaline systems is known (Chang and Sarkanen 1973; Tsutsumi et al. 1995) and therefore, the lignin S/G ratio in hardwoods affects the pulping efficiency. It has already been shown for eucalypt woods that higher S/G ratios imply higher delignification rates, less alkali consumption and therefore higher pulp yield (González-Vila et al. 1999; del Río et al. 2005).
On the other hand, the composition of extractives, especially the lipophilic compounds, is also important for pulp and paper production. The different classes of lipids have different behavior during cooking and bleaching. The lipids can be classified into two principal groups, namely fatty acids and neutral components, the latter including waxes, long chain n-fatty alcohols, alkanes and steroids and triterpenoids. And the behavior of the fatty acids in an aqueous environment is quite different from that of the neutrals. In alkaline pulping, the acids dissociate and can dissolve in water to quite a high extent, forming fatty acid soaps. The neutrals, however, have a very low solubility in water and survive the cooking process and remain in the pulp being at the origin of the so-called pitch deposits, which are responsible of reduced product quality and higher operating costs due to production stops for cleaning the equipment (Hillis and Sumimoto 1989; Back and Allen 2000). The increasing trend in recirculating water in pulp mills to accomplish environmental demands is aggravating these problems.
Therefore, the main objective of this work is to perform a thorough chemical characterization of tagasaste trimming residues, with especial emphasis in the chemical composition of lignin and lipophilic extractives. In this work, the lignin composition of tagasaste was characterized “in situ” using analytical pyrolysis coupled to gas chromatography/mass spectrometry (Py-GC/MS), a powerful analytical tool for the rapid analysis of complex polymer mixtures including lignocellulosic materials (Ralph and Hatfield 1991; Faix et al. 1990; del Río et al. 2001; del Río et al. 2005) that can give information on the lignin composition in terms of the H, G and S moieties. On the other hand, the chemical characterization of the lipophilic extractives was performed by GC and GC/MS by using high-temperature short- and medium-length capillary columns,

5. Resultados y discusión
respectively. This method enables the elution and analysis of intact high molecular weight lipids (Gutiérrez et al. 1998). The knowledge of the chemical composition of the main components of tagasaste trimming residues will be useful for a better utilization of this agricultural waste. 2. Experimental 2.1. Samples Tagasaste trimming residues were supplied by University of Huelva, Spain. The dried samples were milled using a knife mill. For the isolation of lipids, the milled samples were extracted with acetone in a Soxhlet apparatus for 8 h. The acetone extracts were evaporated to dryness and resuspended in chloroform for chromatographic analysis of the lipophilic fraction. Two replicates were used for each sample, and all samples were subjected to GC and GC/MS analyses. For Klason lignin content estimation, the samples extracted with acetone were subsequently extracted with hot water (3 h at 100 ºC) to remove the water-soluble material. Holocellulose was isolated from the pre-extracted fibers by delignification for 4 hours using the acid chlorite method (Browning, 1967). The �-cellulose content was determined by removing the hemicelluloses from the holocellulose by alkali extraction (Browning, 1967). Klason lignin was estimated as the residue after sulfuric acid hydrolysis of the pre-extracted material according to Tappi rule T222 om-88 (Tappi, 1993). The acid-soluble lignin was determined, after filtering off the insoluble lignin, by spectrophotometric determination at 205 nm wavelength. Ash content was estimated as the residue after 6 h at 575 ºC. 2.2. Solid Phase Extraction (SPE) fractionation For a better characterization of the different homologous series, the lipid extracts were fractionated by a SPE procedure using aminopropyl-phase cartridges (500 mg) from Waters (Division of Millipore), as already described (Gutiérrez et al. 1998). Briefly, the dried chloroform extracts were taken up in a minimal volume (< 0.5 ml) of hexane:chloroform (4:1) and loaded into the cartridge column previously conditioned with hexane (4 ml). The cartridge was loaded and eluted by gravity. The column was first eluted with 8 ml of hexane and subsequently with 6 ml of hexane:chloroform (5:1), then with 10 ml of chloroform and finally with 10 ml of diethyl ether:acetic acid (98:2). Each isolated fraction was dried under nitrogen and analyzed by GC and GC/MS. 2.3. GC and GC/MS Analyses The GC analyses of the extracts were performed in an Agilent 6890N GC system using a short-fused silica capillary column (DB-5HT, 5 m × 0.25 mm I.D., 0.1 �m film thickness). The temperature program was started at 100 ºC
189

5. Resultados y discusión
190
with a 1-min hold and then raised to a final temperature of 350 ºC at 15 ºC/min, and held for 3 min. The injector and flame-ionization detector temperatures were set at 300 and 350 ºC, respectively. Helium was used as the carrier gas at a rate of 5 mL/min, and the injection was performed in splitless mode. Peaks were quantified by area, and a mixture of standards (octadecane, palmitic acid, sitosterol and cholesteryl oleate) was used to elaborate calibration curves.
The GC/MS analyses were performed with a Varian model Star 3400 GC equipped with an ion trap detector (Varian Saturn 2000) using a medium-length (12 m) capillary column of the same characteristics described above. The oven was heated from 120 (1 min) to 380 ºC at 10 ºC/min and held for 5 min. The transfer line was kept at 300 ºC. The injector was temperature programmed from 120 (0.1 min) to 380 ºC at a rate of 200 ºC/min and held until the end of the analysis. Helium was used as the carrier gas at a rate of 2 mL/min. Trimethylsilyldiazomethane methylation and BSTFA (bis(trimethylsilyl)-trifluoroacetamide) silylation, in the presence of pyridine, were used to produce the appropriate derivatives, when required. Compounds were identified by comparing their mass spectra with mass spectra in Wiley and NIST libraries, by mass fragmentography, and when possible, by comparison with authentic standards. 2.4. Py-GC/MS. The pyrolysis of tagasaste (1 mg) was performed in a micro-furnace pyrolyzer (model 2020, Frontier Laboratories Ltd) directly connected to a GC/MS system Agilent 6890 equipped with a fused silica capillary column HP 5MS (30 m × 0.25 mm × 0.25 �m I.D.). The detector consisted of an Agilent 5973 mass selective detector. The pyrolysis was performed at 500 °C. The final temperature was achieved at a rate of 20 °C/min. The GC/MS conditions were as follows: oven temperature was held at 50 °C for 1 min and then increased up to 100 °C at 30 °C/min, from 100 to 300 °C at 10 °C/min and isothermal at 300 °C for 10 min using a heating rate of 20 °C/min in the scan modus. The carrier gas used was helium with a controlled flow of 1 ml/min. The compounds were identified by comparing the mass spectra obtained with those of the Wiley and NIST computer libraries and that reported in the literature (Faix et al. 1990; Ralph and Hatfield 1991). Relative peak molar areas were calculated for carbohydrate and lignin pyrolysis products. The summed molar areas of the relevant peaks were normalized to 100%, and the data for two repetitive pyrolysis experiments were averaged. 3. Results and Discussion Tagasaste was characterized by a high content of holocellulose (81%) and �-cellulose (41%), and a lignin content of 18.9% (Klason lignin, 16.6%; acid-soluble lignin: 2.3%). Similar values have been reported by other authors (Díaz

5. Resultados y discusión
et al., 2004). This low lignin content is similar to that found in eucalypt wood (Rencoret et al. 2007), a widely raw material for pulp and papermaking, and together with the high holocellulose and �-cellulose contents makes tagasaste an interesting raw material for pulp and paper production, as already advanced by other authors (Díaz et al. 2004; López et al., 2004; Jiménez et al., 2006; Jiménez et al. 2007; García et al., 2007). On the other hand, the acetone extractives accounted for 1.4% but the chloroform-soluble lipids accounted for only 0.2%, which is a value lower than that found in most lignocellulosic materials (Back and Allen, 2000). However, while the content of the main organic fractions is an important parameter in wood processing for pulp and papermaking, the chemical composition of these organic fractions, in particular the lignin composition in terms of the relative proportions of the S- and G-units, and the lipid composition, in terms of the presence of saponifiable or unsaponifiable components, strongly influences the pulping and bleaching behavior of a raw material. 3.1. Lignin composition The lignin composition of tagasaste was characterized in situ by Py-GC/MS. The pyrogram of tagasate sample is shown in Figure 1 and the identities and relative molar abundances of the released compounds are listed in Table 1. Py-GC/MS of tagasaste wood trimming residues released compounds arising from carbohydrates (67%) and lignin-derived phenols (34%). The lignin-derived phenols arise from guaiacyl (G) and syringyl (S) lignin units whereas no traces of p-hydroxyphenyl (H) units were found in tagasaste. The main lignin-derived compounds identified were 4-vinylsyringol (38), syringol (24), 4-methylsyringol (29), 4-ethylsyringol (35) and 4-vinylguaiacol (23). Syringaldehyde (41), syringylacetone (47) and trans-sinapaldehyde (49) were also identified. Relative peak molar areas were calculated for carbohydrate, and lignin G- and S-type degradation products. The Py-GC/MS data showed a lignin composition in tagasaste with a predominance of the S units (S/G molar ratio of 1.6), typical of hardwoods. The predominance of S-lignin units in tagasaste makes this material advantageous for delignification due to the higher reactivity of the S-lignin in alkaline systems (Chang and Sarkanen, 1973; Tsutsumi et al. 1995; González-Vila et al., 1999) and its lower condensation degree. The G units have a free C-5 position available for carbon-carbon inter-unit bonds, which make them fairly resistant to lignin depolymerization in pulping, while the S lignin is relatively unbranched and has a lower condensation degree and therefore is easier to delignify. The high S/G ratio, together with its low lignin content, explains the excellent performances of tagasaste during alkaline pulping.
191

5. Resultados y discusión
192
Table 1.- Identification and relative molar abundance of the compounds released by Py-GC/MS of tagasaste. No. Compound Origin Mass fragments MW %1 acetic acid C 45/60 60 39.42 (3H) furan-3-one C 55/84 84 0.73 1,3-hydroxydihydro-2-(3H)-furanone C 58/102 102 3.14 (3H) furan-3-one C 55/84 84 1.05 2-furaldehyde C 67/95/96 96 3.86 2-(hydroxymethyl)-furan C 43/70/81/98 98 0.57 cyclopent-1-ene-3,4-dione C 54/68/96 96 0.68 (5H) furan-2-one C 55/84 84 2.99 2,3-dihydro-5-methylfuran-2-one C 55/69/98 98 3.1
10 5-methyl-2-furfuraldehyde C 53/109/110 110 0.411 4-hydroxy-5.6-dihydro-(2H)-pyran-2-one C 58/85/114 114 2.612 3-hydroxy-2-methyl-2-cyclopenten-1-one C 55/85/112 112 0.613 2-hydroxy-3-methyl-2-cyclopenten-1-one C 55/85/112 112 0.314 dihydroxypyran-1-one C 56/84/114 114 0.815 guaiacol LG 81/109/124 124 1.916 dimethyldihydropyranone C 55/70/83/111/126 126 0.317 4-methylguaiacol LG 95/123/138 138 1.218 catechol C 64/81/92/110 110 1.119 5-hydroxymethyl-2-tetrahydrofuraldehyde-3-one C 57/69/85/98/99 138 0.4
20 5-hydroxymethyl-2-furaldehyde C 69/97/109/126 126 0.321 methoxycatechol L 60/97/125/140 140 0.922 4-ethylguaiacol LG 122/137/152 152 0.423 4-vinylguaiacol LG 107/135/150 150 2.124 syringol LS 111/139/154 154 3.625 eugenol LG 131/149/164 164 0.426 4-propylguaiacol LG 122/136/166 166 0.127 vanillin LG 109/151/152 152 0.728 cis-isoeugenol LG 131/149/164 164 0.429 4-methylsyringol LS 125/153/168 168 1.830 trans-isoeugenol LG 131/149/164 164 1.231 homovanillin LG 122/137/166 166 0.232 4-propinylguaiacol LG 77/91/119/147/162 162 1.033 4-propinylguaiacol isomer LG 77/91/119/147/162 162 1.334 acetovanillone LG 123/151/166 166 0.235 4-ethylsyringol LS 167/182 182 1.536 guaiacylacetone LG 122/137/180 180 0.737 levoglucosane C 60/98 162 4.838 4-vinylsyringol LS 137/165/180 180 4.239 4-allylsyringol LS 167/179/194 194 0.740 cis-4propenylsyringol LS 167/179/194 194 0.541 syringaldehyde LS 167/181/182 182 1.2

5. Resultados y discusión
42 4-propynylsyringol LS 106/131/177/192 192 1.043 4-propynylsyringol isomer LS 106/131/177/192 192 0.844 trans-4-propenylsyringol LS 167/179/194 194 2.545 acetosyringone LS 153/181/196 196 0.646 trans-coniferaldehyde LG 107/135/147/178 178 0.847 syringylacetone LS 123/167/210 210 0.548 propiosyringone LS 151/181/210 210 0.349 trans-sinapaldehyde LS 137/165/180/208 208 1.3
%G 38 %S 62 S/G 1.6
C: cellulose; LG: lignin guaiacyl; LS: lignin syringyl 3.2. Lipid composition The lipid composition in tagasaste was analyzed by gas chromatography (GC) and gas chromatography/mass spectrometry (GC/MS), using short- and medium-length high-temperature capillary columns, respectively (Gutiérrez et al., 1998), which enable the elution and analysis of intact high molecular weight lipids such as waxes, sterol esters, and triglycerides. The GC/MS chromatogram of the lipids from tagasaste is shown in Figure 2. The identities and abundances
1
5
3
2
6
8
9
10
11
13
14
15
16
17
1821
22
23
24
25 2728
29
30
3233 3536
38
3940
4142
44
4546 47
48
49
2 4 6 8 10 12 14 16 18 20 22Retention time (min)
Figure 1. Py-GC/MS of tagasaste wood. The identities of the peaks are shown in Table 2.
193

5. Resultados y discusión
194
of the main lipid classes identified are summarized in Table 2. The structures of the main lipophilic compounds identified in the tagasaste extracts are shown in Figure 3. The main lipids identified in tagasaste were series of fatty acids, including �-hydroxy acids, and steroid compounds, including steroid hydrocarbons, steroid ketones, sterols, sterol esters and sterol glycosides. Other compounds, such as series of alkanes and monoglycerides were also found in minor amounts.
The series of free fatty acids (141.4 mg/kg) were present in the range from n-tetradecanoic (C14) to n-hexacosanoic (C26) acids, with a strong even-over-odd carbon atom predominance. Hexadecanoic (palmitic) acid (I, C16:0) and octadecanoic (stearic) acid (C18:0) were the most abundant fatty acids followed by n-tetracosanoic (C24) acid. Unsaturated fatty acids were also present, 9-octadecenoic (oleic) acid (II, C18:1) being especially abundant. Fatty acids also included a series of �-hydroxyfatty acids (51.8 mg/kg) that was present in the range from 2-hydroxyoctadecanoic acid (C18) to 2-hydroxyhexacosanoic acid (C26) with maximum at C24 (III), and the presence of exclusively the even carbon atom number homologs.
FA16
FA18:1
FA18
FA20 FA22 FA24
steroid hydrocarbons
steroid ketones
Retention time (min)5 10 15 20
1
2
34+5
67 8
sterol esters
Figure 2. GC/MS of the lipophilic extracts from tagasaste wood. Key labels are: Fn: fatty acids; 1: �-tocopherol; 2: campesterol; 3: stigmasterol; 4: sitosterol; 5: stigmastanol; 6: 7-oxocampesterol; 7: 7-oxostigmasterol; 8: 7-oxositosterol.

5. Resultados y discusión
Table 2.- Composition and abundance (mg/Kg) of lipophilic extractives in tagasaste trimming residues. compound mass fragments MW abundancen-alkanes 8.8n-eicosane 57/71/85/282 282 0.1n-heneicosane 57/71/85/296 296 0.2n-docosane 57/71/85/310 310 0.1n-tricosane 57/71/85/324 324 0.3n-tetracosane 57/71/85/338 338 0.2n-pentacosane 57/71/85/352 352 0.5n-hexacosane 57/71/85/366 366 0.3n-heptacosane 57/71/85/380 380 0.5n-octacosane 57/71/85/394 394 0.3n-nonacosane 57/71/85/408 408 1.2n-triacontane 57/71/85/422 422 0.2n-hentriacontane 57/71/85/436 436 4.8n-tritriacontane 57/71/85/464 464 0.1
fatty acids 141.4n-tetradecanoic acid 60/73/228 228 1.1n-pentadecanoic acid 60/73/242 242 1.09-hexadecenoic acid 55/69/236/254 254 2.2n-hexadecanoic acid 60/73/129/256 256 55.4n-heptadecanoic acid 60/73/129/270 270 2.99-octadecenoic acid 55/69/264 282 29.4n-octadecanoic acid 60/73/129/284 284 10.7n-nonadecanoic acid 60/73/129/298 298 1.4n-eicosanoic acid 60/73/129/312 312 7.4n-heneicosanoic acid 60/73/129/327 327 1.1n-docosanoic acid 60/73/129/340 340 5.4n-tricosanoic acid 60/73/129/354 354 6.0n-tetracosanoic acid 60/73/129/368 368 10.3n-pentacosanoic acid 60/73/129/382 382 2.6n-hexacosanoic acid 60/73/129/396 396 4.5
�-hydroxy fatty acids# 51.82-hydroxyoctadecanoic acid 73/327/371 386 0.62-hydroxyeicosanoic acid 73/355/399 414 0.92-hydroxydocosanoic acid 73/383/427 442 22.12-hydroxytetracosanoic acid 73/411/455 470 26.32-hydroxyhexacosanoic acid 73/439/483 498 1.9
steroid hydrocarbons 30.1ergostatriene 135/143/380 380 5.6ergostadiene 81/147/367/382 382 3.4stigmasta-3,5,22-triene 135/143/394 394 11.7
195

5. Resultados y discusión
196
stigmasta-3,5-diene 81/147/381/396 396 9.4
sterols 113.1campesterol 55/145/213/382/400 400 14.0stigmasterol 55/83/255/394/412 412 35.7sitosterol 145/213/396/414 414 30.7stigmastanol 215/416 416 2.97-oxocampesterol 382/397/414 414 10.17-oxostigmasterol 286/426 426 9.07-oxositosterol 396/413/428 428 10.7
�-tocopherol 165/205/430 430 2.1
steroid ketones 30.5stigmasta-7,22-dien-3-one 55/269/298/367/410 410 7.1stigmasta-3,5-dien-7-one 174/269/410 410 14.1stigmast-4-en-3-one 124/229/412 412 4.5stigmastadienone isomer 57/136/174/269/410 410 2.0stigmastane-3,6-dione 245/287/428 428 2.8
sterol glycosides* 13.2campesterol 3�-D-glucopyranoside 204/217/361/383 850 1.9stigmasterol 3�-D-glucopyranoside 204/217/361/413 880 4.3sitosterol 3�-D-glucopyranoside 204/217/361/397 864 7.0
sterol esters 28.3campesterol esters n.d 2.3stigmasterol esters n.d 9.2sitosterol esters n.d 16.8
monoglycerides* 8.11-monopalmitin 73/103/129/147/371/459 474 4.21-monostearin 73/103/129/147/399/487 502 3.9
* as TMS ethers; # as methyl esters and TMS ethers
Steroid compounds were the second most important lipid class found among
the tagasaste extractives and included steroid hydrocarbons, steroid ketones and sterols (in free and conjugated form). Sterols, in free and conjugated (esters and glycosides) form were the most important steroids identified, free sterols being present in important amounts (113.1 mg/kg). Stigmasterol (IV) was the most abundant among the free sterols with the presence of important amounts of campesterol (V) and sitosterol (VI) and minor amounts of stigmastanol (VII) as well as the presence of the oxidized 7-ketocampesterol, 7-ketostigmasterol and 7-ketostigmastanol. Lower amounts of sterols could also be found in ester form (28.3 mg/kg), with a predominance of sitosterol esters. Sterol glycosides, such as campesteryl, stigmasteryl and sitosteryl 3�-D-glucopyranosides (VIII) were

5. Resultados y discusión
Figure 3. Structures of the main lipophilic compounds present in tagasaste. I: hexadecanoic acid; II: 9-hexadecenoic acid; III: 2-hydroxyhexacosanoic acid; IV: stigmasterol; V: campesterol; VI: sitosterol; VII: stigmastanol; VIII: sitosteryl 3�-D-glucopyranoside; IX: stigmasta-3,5-dien-7-one; X: stigmasta-3,5,22-triene; XI: n-hentriacontane; XII: 1-monopalmitin.
also identified in lower amounts (13.2 mg/kg), the latter being the most predominant. The identification of steryl glycosides was accomplished, after BSTFA derivatization of the lipid extract, by comparison with the mass spectra and relative retention times of authentic standards (Gutiérez and del Río, 2001). It is important to point out the low content of free and conjugated (esters and glycosides) sterols, in comparison to that found in eucalypt wood (Rencoret et al., 2007), since these are the main compounds responsible for pitch deposition during kraft cooking of hardwoods, such as eucalypt wood (del Río et al. 1998, 2000; Silvestre et al. 1999; Gutiérrez and del Río 2001). On the other hand,
197

5. Resultados y discusión
198
steroid ketones were also found in important amounts (30.5 mg/kg), being mainly constituted by stigmasta-7,22-dien-3-one, stigmasta-3,5-dien-7-one (IX), stigmast-4-en-3-one and stigmasta-3,6-dione. Different steroid hydrocarbons (di- and triunsaturated) were also identified, although in low amounts (30.1 mg/kg), stigmasta-3,5,22-triene (X) being the most predominant.
Finally, a series of n-alkanes ranging from n-eicosane (C20) to n-tritriacontane (C33) with n-hentriacontane (X, C31) being the most prominent, was found in low amounts (8.8 mg/kg). And minor amounts of monoglycerides (8.1 mg/kg), such as 1-monopalmitin (XI) and 1-monostearin, were also present among tagasaste extractives. 4. Conclusions Tagasaste is characterized by a high content of holocellulose and �-cellulose and a relatively low lignin content. Moreover, the chemical composition of tagasaste lignin indicates a predominance of S-lignin units over the G-lignin ones (S/G molar ratio of 1.6) and the absence of H-lignin units. The total acetone extractives accounted for 1.4% but the content on lipophilic compounds is very low (only 0.2%). The main lipids identified in tagasaste were series of fatty acids, including �-hydroxyfatty acids, and steroid compounds, including steroid hydrocarbons, steroid ketones, sterols, sterol esters and sterol glycosides. Acknowledgements This study has been supported by the Spanish project AGL2005-01748. G. Marques acknowledges a FPI Fellowship from the Spanish Ministry of Education and Science. We thank Manuel J. Díaz (University of Huelva, Spain) for providing the tagasaste samples. References Back, E.L., Allen, L.H., 2000. Pitch Control, Wood Resin and Deresination.
Tappi Press, Atlanta.
Borens, F., Poppi, D.P., 1990. The nutritive and feeding value for ruminants of tagasaste (Chamaecytisus palmensis), a leguminous tree. Anim. Feed Sci. Technol. 28, 275–292.
Browning, B.L., 1967. Methods of Wood Chemistry, vol. II; Wiley-Interscience Publishers, New York.
Chang, H-M., Sarkanen, K.V., 1973. Species variation in lignin. Effect of species on the rate of kraft delignification. Tappi 56, 132-134.

5. Resultados y discusión
del Río, J.C., Gutiérrez, A., González-Vila, F.J., Martín, F., Romero, J., 1998. Characterization of organic deposits produced in the Kraft pulping of Eucalyptus globulus wood. J. Chromatogr. A 823, 457-465.
del Río, J.C., Romero, J., Gutiérrez, A., 2000. Analysis of pitch deposits produced in Kraft pulp mills using a totally chlorine free bleaching sequence. J. Chromatogr. A 874, 235-245.
del Río J.C., Gutiérrez A., Romero J., Martínez M.J. and Martínez A.T., 2001. Identification of residual lignin markers in eucalypt kraft pulp by Py-GC-MS. J. Anal. Appl. Pyrolysis 58/59, 425-439.
del Río, J.C., Gutiérrez, A., Hernando, M., Landín, P., Romero, J., Martínez, A.T., 2005. Determining the influence of eucalypt lignin composition in paper pulp yield using Py-GC/MS. J. Anal. Appl. Pyrolysis 74, 110-115.
Díaz, M.J., Alfaro, A., García, M.M., Eugenio, F.M.E., Ariza, J., López, F., 2004. Ethanol pulping from tagasaste (Chamaecytisus proliferus L.F. ssp. Palmensis). A new promising source for cellulose pulp. Ind. Eng. Chem. Res. 43, 1875-1881.
Faix, O., Meier, D., Fortmann, I., 1990. Thermal degradation products of wood. A collection of electron of electron-impact (EI) mass spectra of monomeric lignin derived products. Holz Roh- Werkst. 48, 351-354.
Francisco-Ortega, J., Jackson, M.T., Santos-Guerra, A., Fernández-Galván, M., 1991. Historical aspects of the origin and distribution of tagasaste (Chamaecytisus proliferus (L. fil.) Link ssp. palmensis (Christ) Kunkel), a fodder tree from the Canary Islands. J. Adelaide Botanical Garden 14, 67–76.
García, M.M., López, F., Alfaro, A., Ariza, J., Tapias, R., 2007. The use of tagasaste (Chamaecytisus proliferus) from different origins for biomass and paper production. Biores. Technol. (in press).
González-Vila, F.J., Almendros, G., del Río, J.C., Martín, F., Gutiérrez, A., Romero, J. 1999. Ease of delignification assessment of wood from different Eucalyptus species by pyrolysis (TMAH)-GC/MS and CP/MAS 13C NMR spectrometry. J. Anal. Appl. Pyrol. 49, 295-305.
Gutiérrez, A., del Río, J.C., González-Vila, F.J., Martín, F., 1998. Analysis of lipophilic extractives from Wood and pitch deposits by solid-phase extraction and gas chromatography. J. Chromatogr. A 823, 449-455.
199

5. Resultados y discusión
200
Gutiérrez, A., del Río, J.C., 2001. Gas chromatography/mass spectrometry demonstration of steryl glycosides in eucalypt wood, kraft pulp and process liquids. Rapid Commun. Mass Spectrom. 15, 2515-2520.
Hillis, W.E., Sumimoto, M., 1989. Effect of extractives on pulping. In: Rowe, J.W. (Ed.), Natural Products of Woody Plants II; Springer-Verlag, Berlin. pp. 880-920.
Jiménez, L., Pérez, A., Rodríguez, A., de La Torre, M.J., 2006. New raw materials and pulping processes for production of pulp and paper. Afinidad 63 (525), 362-369.
Jiménez, L., Pérez, A., de la Torre, M.J., Moral A., Serrano, L., 2007. Characterization of vine shoots, cotton stalks, Leucaena leucocephala and Chamaecytisus proliferus, and of their ethyleneglycol pulps. Biores. Technol. 98, 3487-3490.
Kindu, M., Glatzel, G., Tadesse, Y., Yosef, A., 2006. Tree species screened on nitosols of central Ethiopia: Biomass production, nutrient contents and effect on soil nitrogen. J. Trop. Forest Sci. 18, 173-180.
López, F., Alfaro, A., García, M.M., Diaz, M.J., Calero, A.M., Ariza, J., 2004. Pulp and paper from tagasaste (Chamaecytisus proliferus LF ssp palmensis). Chem. Eng. Res. & Des. 82, 1029-1036.
Ralph, J., Hatfield, R.D., 1991. Pyrolysis-GC/MS characterization of forage materials. J. Agric. Food Chem. 39, 1426-1437.
Rencoret, J., Gutiérrez, A., del Río, J.C., 2007. Lipid and lignin composition of woods from different eucalypt species. Holzforschung 61, 165-174.
Silvestre, A.J.D., Pereira, C.C.L., Pascoal Neto, C., Evtuguin, D.V., Duarte, A.C., Cavaleiro, J.A.S., Furtado, F.P., 1999. Chemical composition of pitch deposits from an ECF Eucalyptus globulus bleached kraft pulp mill: its relationship with wood extractives and additives in process streams. Appita J. 52, 375-382.
Technical Association of the Pulp and Paper Industry, 1993. Test methods, 1992-1993. TAPPI, Atlanta, GA..
Tsutsumi, Y., Kondo, R., Sakai, K., Imamura, H., 1995. The difference of reactivity between syringyl lignin and guaiacyl lignin in alkaline systems. Holzforschung 49, 423-428.

5. Resultados y discusión
Ventura, M.R., Castanon, J.I.R., Rey, L., Flores, M.P., 2002. Chemical composition and digestibility of tagasaste (Chamaecytisus proliferus) subspecies for goats. Small Ruminant Res. 46, 207-210.
201

5. Resultados y discusión
Publicación VII: Marques G., del Río J.C. and Gutiérrez A. (2010) Lipophilic extractives from several nonwoody lignocellulosic crops (flax, hemp, sisal, abaca) and their fate during alkaline pulping and TCF/ECF bleaching. Bioresource Technology, 101, 260-267.
202

5. Resultados y discusión
Lipophilic extractives from several nonwoody lignocellulosic crops (flax, hemp, sisal, abaca) and their fate during alkaline pulping and TCF/ECF
bleaching
Gisela Marques, José C. del Río and Ana Gutiérrez
Instituto de Recursos Naturales y Agrobiología, CSIC, PO Box 1052, E-41080, Seville, Spain Abstract The fate of lipophilic extractives from several nonwoody species (flax, hemp, sisal and abaca) used for the manufacturing of cellulose pulps, was studied during soda/anthraquinone (AQ) pulping and totally chorine free (TCF) and elemental chlorine free (ECF) bleaching. With this purpose, the lipophilic extracts from the raw materials and their unbleached and bleached industrial pulps, were analyzed by gas chromatography-mass spectrometry. Aldehydes, hydroxyfatty acids and esterified compounds such as ester waxes, sterol esters and alkylferulates strongly decreased after soda/AQ pulping while alkanes, alcohols, free sterols and sterol glycosides survived the cooking process. Among the lipophilic extractives that remained in the unbleached pulps, some amounts of free sterols were still present in the TCF pulps whereas they were practically absent in the ECF pulps. Sterol glycosides were also removed after both TCF and ECF bleaching. By contrast, saturated fatty acids, fatty alcohols and alkanes were still present in both bleached pulps. Keywords: pitch; lipophilic extractives; paper pulp; nonwoody fibers 1. Introduction Lipophilic extractives, i.e. the non-polar extractable fraction from wood and other lignocellulosic crops, include different classes of compounds, such as alkanes, fatty alcohols, fatty acids, free and conjugated sterols, terpenoids, triglycerides and waxes. These lipophilic compounds, even when present in low amounts in the raw material, may play an important role in industrial wood processing for bleached pulp and paper production since they are at the origin of the so-called pitch deposits along the pulp and paper manufacturing processes. Pitch deposition is a serious problem in the pulp and paper industry since it is responsible for reduced production levels, higher equipment maintenance costs, higher operating costs, and an increased incidence of defects in the finished products, which reduces quality and benefits (Back and Allen, 2000). The nature and severity of pitch deposition depend not only on the raw materials used (and hence on the nature of the lipophilic compounds), but also on the industrial processes of pulping and bleaching applied at the mill. In the manufacture of alkaline pulps, a large part of the lipids originally present in the
203

5. Resultados y discusión
204
raw material is removed during the cooking and bleaching stages. However, some chemical species survive these processes and are found as pulp extractives (Bergelin and Holmbom, 2003; Freire et al., 2005; 2006; Gutiérrez et al., 2001a), suspended in process waters (Gutiérrez et al., 2001b) or forming the so-called pitch deposits in circuits, equipments and final product (Bergelin et al., 2005; del Río et al., 1998; 2000; Gutiérrez and del Río, 2005; Silvestre et al., 1999). Growing pressure for closing water loops in pulp and paper mills leads to increasing build up of lipophilic compounds in the processes and therefore, to an increasing number of severe and costly pitch related problems. The different classes of lipids have different behavior during pulping and bleaching. Several studies have provided information on the behavior of wood lipids, such as fatty and resin acids, triglycerides and sterols and triterpenols during pulping and bleaching (Back and Allen, 2000; Bergelin and Holmbom, 2003; 2008; Freire et al., 2005; 2006; Gutiérrez et al., 2001a; Shin and Kim, 2006). However, the chemistry of lipids from nonwoody pulps in pulping and bleaching has not been examined much so far (Gutiérrez et al., 2004; Gutiérrez and del Río, 2003a; 2003b). Likewise, to the best of our knowledge, there is very little information available dealing with pitch problems on nonwoody pulps (Gutiérrez and del Río, 2005). In this context, the aim of this work was to identify the specific lipophilic constituents of different nonwoody fibers, which are used for the manufacturing of cellulose pulps for specialty papers, and to study their behavior during soda/anthraquinone (AQ) pulping and totally chorine free (TCF) and elemental chlorine free (ECF) bleaching. For this, a series of pulps (crude pulps taken after soda/AQ pulping and bleached pulps taken after TCF and ECF bleaching) from different nonwoody raw materials (flax, hemp, sisal, abaca) were selected for this study. The composition of the lipophilic compounds in the fibers and their respective pulps was analyzed by gas chromatography (GC) and gas chromatography-mass spectrometry (GC-MS) using short- and medium-length high temperature capillary columns, respectively, with thin films, which enables the elution and analysis of intact high molecular weight lipids such as waxes or sterol glycosides (Gutiérrez et al., 1998; Gutiérrez and del Río, 2001). The knowledge of the behavior of lipophilic extractives during pulping and bleaching is an important step towards understanding and predicting the pitch problems and designing effective solutions for its control. 2. Materials and methods 2.1. Samples Two bast fibers, flax (Linum usitatissimum) and hemp (Cannabis sativa), and two leaf fibers, sisal (Agave sisalana) and abaca (Musa textilis), were selected for this study. The raw materials and their respective crude (unbleached) pulps (pulps taken after soda/AQ pulping), as well as pulp samples after TCF and ECF

5. Resultados y discusión
bleaching, were supplied by CELESA pulp mill (Tortosa, Spain). General conditions of soda/AQ pulping included the use of sodium hydroxide and anthraquinone (up to 0.05%) as cooking chemicals, and 2-3 h of cooking time at a temperature of about 165ºC. These general conditions can be slightly modified depending on the raw material used. The TCF bleaching sequence used (QPoP) included a quelating stage (Q) and two hydrogen peroxide stages (P), the first one under pressurized oxygen (Po). The ECF bleaching sequence used (DPo) included a chlorine dioxide stage (D) followed by a hydrogen peroxide stage under pressurized oxygen (Po). 2.2. Lipid extraction Raw materials and pulps were air-dried until constant weight and the samples were Soxhlet-extracted with acetone for 8 h. All the extracts were evaporated to dryness and redissolved in chloroform for chromatographic analysis of the lipophilic fraction as described below. 2.3. GC and GC-MS analyses The GC analyses of the lipids from the raw materials and pulps were performed in an Agilent 6890N Network GC system using a short fused-silica DB-5HT capillary column (5 m x 0.25 mm internal diameter, 0.1 μm film thickness) from J&W Scientific, enabling simultaneous elution of the different lipid classes (Gutiérrez et al., 1998). The temperature program was started at 100°C with 1 min hold, and then raised to 350°C at 15°C/min, and held for 3 min. The injector and flame-ionization detector (FID) temperatures were set at 300°C and 350°C, respectively. Helium (5 ml/min) was used as carrier gas, and the injection was performed in splitless mode. The GC-MS analyses were performed with a Varian 3800 chromatograph coupled to an ion-trap detector (Varian 4000) using a medium-length (12 m) capillary column of the same characteristics described above for GC/FID. The oven was heated from 120°C (1 min) to 380°C at 10°C/min, and held for 5 min. The transfer line was kept at 300°C, the injector was programmed from 120°C (0.1 min) to 380°C at 200°C/min and held until the end of the analysis, and helium was used as carrier gas at a rate of 2 ml/min. The compounds were identified by mass fragmentography, and by comparing their mass spectra with those of the Wiley and NIST libraries and standards. Both underivatized and derivatized samples were analyzed by GC and GC-MS. Two derivatized samples, silylated samples and methylated and silylated samples, were analyzed. The silylation was peformed using bis(trimethylsilyl)trifluoroacetamide (BSTFA) in the presence of pyridine at 80ºC for 90 min (Gutiérrez and del Río, 2001). Trimethylsilyl-diazomethane methylation, in the presence of methanol (at room temperature for 10 min) followed by BSTFA silylation, in the presence of pyridine, were also performed.
205

5. Resultados y discusión
206
Fatty alcohols, fatty acids and sterol glycosides were determined as silylated derivatives in the derivatized samples. Hydroxyfatty acids were determined in the samples after methylation followed by silylation. The rest of compounds were determined in the underivatized samples. Peaks were quantified by area and a mixture of standards (octadecane, palmitic acid, sitosterol, cholesteryl oleate, and sitosteryl 3�-D-glucopyranoside) with a concentration range between 0.1 and 1mg/ml was used to elaborate calibration curves. The data from the two replicates were averaged. 3. Results and discussion The lipid content of the nonwoody fibers used in this study and their corresponding crude and TCF and ECF bleached pulps is shown in Table 1. Flax fibers have the highest content on lipophilic extractives followed by hemp, sisal and abaca fibers. Regardless the higher lipid content of flax fibers, the content of lipophilic extractives of its crude pulp was similar or even lower than in the other fibers. On the other hand, the lipid content of the pulps decreased further after TCF or ECF bleaching, ranging from 0.03 to 0.18%. The differences observed in the lipid content among the different crude and bleached pulps of the nonwoody fibers is related to the behavior of lipophilic extractives present in the raw materials in alkaline pulping and to their reactivity towards the bleaching agents used as described below. 3.1. Composition of lipophilic extractives in the nonwoody fibers selected for this study With the aim of studying the fate of the different lipophilic extractives during soda/AQ pulping, the lipid extracts from the different nonwoody fibers selected for this study were analyzed by GC and GC-MS and then compared with those in the crude pulps. The composition of the lipophilic extractives of the selected raw materials are listed in Table 2 and the chemical structures of the main compounds identified are represented in Fig. 1. Table 1. Lipophilic extractives content (%) in the selected nonwoody raw materials and pulps
Flax Hemp Sisal Abaca
Fiber 1.70 0.88 0.68 0.51
Crude pulp 0.21 0.26 0.30 0.20
TCF pulp 0.05 0.18 0.09 0.04
ECF pulp 0.13 0.18 0.14 0.03

5. Resultados y discusión
OH
O
OH
O
I II
III IV
HOV
HOHOHOV
O
O
VIO
O
O
O
VI
IXO
O
O
O
VIIO
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH
OHOH
H
O
H
O
H
O
HOVIII
HOVIII
Fig.1. Chemical structures of compounds representing the main classes of lipophilic extractives found in lignocellulosic fibers: I, pentacosane; II, octacosanol; III, octacosanal; IV, palmitic acid; V, sitosterol; VI, sitosteryl linoleate; VII sitosteryl 3�-D-glucopyranoside; VIII, �-amyrin; and IX, octacosyl hexadecanoate.
The main lipid classes present in the nonwoody fibers selected for this study were series of alkanes (I), fatty alcohols (II), aldehydes (III), fatty acids (IV), steroids, including free (V) and conjugated sterols (VI-VII), triterpenoids (VIII) and ester waxes (IX). The detailed composition of the lipophilic compounds present in these fibers have been previously addressed (del Río et al., 2004; del Río and Gutiérrez, 2006; Gutiérrez et al., 2006; 2008; Gutiérrez and del Río, 2003a; 2003b). In the case of flax bast fibers the predominant lipophilic compounds present were series of fatty acids and aldehydes, accounting for 34% and 23% of total extract, respectively, followed by ester waxes (18%), and fatty alcohols (13%). Fatty acids were also the predominant compounds (27% of total extracts) in hemp bast fibers, followed by alkanes (15%), free sterols (12%) and steroid hydrocarbons (12%). Among the selected leaf fibers, free sterols predominated in both sisal (20%) and abaca (45%), followed by alkanes (14%) and fatty acids (10%) in sisal, and by fatty acids (16%) in abaca fibers.
207

5. Resultados y discusión
208
These lipophilic compounds can be classified into two principal groups, namely organic acids and neutral components. Organic acids include fatty acids, and the neutrals include alkanes, aldehydes, fatty alcohols, and free sterols as well as esterified and conjugated compounds such as ester waxes, sterol esters, alkylferulates and sterol glycosides. Organic acids and neutral compounds, the latter including saponifiable and unsaponifiable compounds present different behavior during alkaline pulping, as shown below. 3.2. Fate of lipophilic extractives during soda/AQ pulping The fate of the different lipophilic extractives during soda/AQ pulping was studied by analyzing the respective crude pulps. This is exemplified in Figs. 2 and 3 for the hemp bast fibers and the sisal leaf fibers, respectively. The composition of main lipids present in the crude pulps from the studied fibers is listed in Table 2. It can be observed that the lipophilic compounds from the raw materials are modified to a different extent during alkaline (soda/AQ) pulping depending of their chemical nature. Fatty acids, which are among the main lipophilic extractives predominating in all these fibers, were also present in significant amounts in the crude pulps. The content of fatty acids decreased significantly after soda/AQ pulping of flax fibers whereas an increase in fatty acids content was produced after pulping of the other fibers, being especially evident in the case of sisal and abaca. In contrast, �-hydroxyfatty acids present in all the fibers and �-hydroxyfatty acids present in sisal and abaca were completely absent in crude pulps. On the other hand, esterified compounds such as ester waxes, sterols esters and ferulic acid esters, were completely hydrolyzed during soda/AQ pulping. This is especially evident in the case of flax fibers where ester waxes predominate. In contrast, other conjugated compounds, namely sterol glycosides, which are present in all nonwoody fibers studied here, resisted the alkaline cooking conditions and were present intact in the crude pulps, as already reported in the pulping of woody (Gutiérrez and del Río, 2001; Nilvebrant and Byström, 1995) and nonwoody plants (Gutiérrez and del Río, 2003a; 2003b; Gutiérrez et al., 2004). The importance of the presence of sterol glycosides after alkaline pulping is due to their high hydrophilic-lipophilic balance, high melting point and very low solubility in water, alkali and the usual organic solvents (Hillis and Sumimoto, 1989). Due to these properties, sterol glycosides constitute a part of protecting layers that prevent the cooking and bleaching chemicals to reach the resin and thereby keep them and other extractives in the pulp. Other neutral compounds, such as aldehydes, which were present in significant amounts in flax and hemp fibers, decreased largely during pulping and therefore were practically absent in the crude pulps. The content of steroid hydrocarbons and steroid ketones also decreased after soda/AQ cooking.

5. Resultados y discusión
5 10 15 20Retention time (minutes)
AQFa16 Fa18
25
5+6+7
43
Defoamer
111
Fa16 Fa18
AQ
1
7
Al30
11
TCF pulp
ECF pulp
Fa16
Fa18:1+
Fa18:2
Fa18
1
2
5+6+7
43
11
12 W42W44 W46
W48
Hemp bast fibers
Ak27+
Al24
AQFa18Fa16 1
5+6+7
43
11
Al30
Crude pulp
Ak29+
Al26
9+10
Fa18:1+
Fa18:2Ak27
+Al24
Ak29+
Al26
Ak31+
Al28
Ak27+
Al24
Ak29
+Al26
Ak31+
Al28
Al30
Ak27
+Al24
Al22
Ak29+
Al26
Ak31
+Al28
W50SE
8
Fig. 2. GC-MS chromatograms of underivatized lipophilic extractives from hemp fibers (raw material), and their crude, TCF and ECF pulps. Peak identification, 1: stigmasta-3,5,22-triene; 2: stigmasta-3,5-diene; 3: campesterol; 4: stigmasterol; 5: sitosterol; 6: stigmastanol; 7: �-amyrin; 8: stigmasta-3,5-dien-7-one; 9: �-amyrin acetate; 10: stigmast-4-en-3-one; 11: friedelan-3-one; 12: stigmastane-3,6-dione; AQ: anthraquinone; Fa(n): fatty acids; Ak(n): alkanes; Al(n): fatty alcohols; SE: sterol esters; W(n): ester waxes; n denotes the total carbon atom number.
209

5. Resultados y discusión
210
5+6+7
4
3
Al281
2
Ak27F24Ak25Ak23
Fa16
Ak21
8
Fe24Fe26 Fe28
5+6+7
8
AQ
Fa16
Al28
1
4
3Fa18
9
AQFa18
Fa16
5+6
914
AQ Fa18Fa16
5+6
91
4
3
Defoamer
TCF pulp
ECF pulp
Sisal leaf fibers
Crude pulp
Ak29+
Al26
Ak25+
Al22
Ak25
+Al22
Ak27+
Al24
Ak29
+Al26
Ak27
+Al24
Ak29+
Al26
Al28
Ak25+
Al22
Ak27+
Al24
Ak29
+Al26
Al28
5 10 15 20Retention time (minutes)
9
Al30
Al30
Al30
25
Fig. 3. GC-MS chromatograms of underivatized lipophilic extractives from sisal fibers (raw material), and their crude pulp, TCF pulp and ECF pulps. Peak identification, 1: stigmasta-3,5,22-triene; 2: stigmasta-3,5-diene; 3: campesterol; 4: stigmasterol; 5: sitosterol; 6: stigmastanol; 7: �-amyrin; 8: stigmasta-3,5-dien-7-one; 9: hecogenin; AQ: anthraquinone; Fa(n): fatty acids; Ak(n): alkanes; Al(n): fatty alcohols; Fe(n): n-alkylferulates; n denotes the total carbon atom number.

5. Resultados y discusión
Table 2. Composition of lipophilic extractives (mg/100 g) in nonwoody raw materials (M) and crude (C), TCF (T) and ECF (E) pulps
Flax Hemp Sisal Abaca Compound M C T E M C T E M C T E M C T E Fatty acids 552 96 29 39 78 60 12 80 9 50 10 42 9 57 19 28 n-hexadecanoic acid 121 27 4 18 34 15 4 29 4 24 8 27 4 20 9 10 9,12-octadecadienoic acid
1 tr - - 3 1 1 - - - - - 1 3 - -
9-octadecenoic acid 235 39 2 1 15 1 tr 1 - - - - 2 6 - - n-octadecanoic acid 52 22 8 12 6 10 3 20 <1 6 7 16 1 21 11 16 n-nonadecanoic acid tr 3 tr tr tr 5 1 tr - - - - tr - tr tr n-eicosanoic acid 20 2 4 4 4 6 <1 6 tr 1 <1 2 <1 2 tr 1 n-heneicosanoic acid 4 tr tr tr tr 1 1 1 - - - - tr - - tr n-docosanoic acid 36 2 4 2 5 7 1 8 tr tr 1 2 <1 tr tr tr n-tricosanoic acid 16 tr tr tr 2 3 1 4 1 tr <1 1 tr tr tr tr n-tetracosanoic acid 40 1 4 2 4 7 <1 6 1 1 1 3 1 3 <1 1 n-pentacosanoic acid 8 tr tr tr 2 1 <1 1 <1 2 tr - tr - tr tr n-hexacosanoic acid 19 - 3 tr 3 3 tr 1 1 10 1 2 tr 2 - 1 n-octacosanoic acid - - - - - - tr 3 2 17 tr tr - - - - �-Hydroxyfatty acids 11 - - - 9 tr - - 7 - - - 1 - - - 2-hydroxydocosanoic acid
3 - - - - - - - 1 - - - - - - -
2-hydroxytricosanoic acid
2 - - - - - - - 1 - - - - - - -
2-hydroxytetracosanoic acid
6 - - - 5 tr - - 4 - - - 1 - - -
2-hydroxypentacosanoic acid
tr - - - 4 - - - 1 - - - tr - - -
�-Hydroxyfatty acids - - - - - - - - 6 - - - 1 - - - 22-hydroxydocosanoic acid
- - - - - - - - 1 - - - 1 - - -
26-hydroxyhexacosanoic acid
- - - - - - - - 2 - - - tr - - -
28-hydroxyoctacosanoic acid
- - - - - - - - 3 - - - - - - -
Fatty alcohols 220 68 1 52 2 44 13 59 8 74 18 51 tr 6 1 <1 n-docosanol - - - - - 4 1 4 - 5 2 3 tr 2 <1 <1 n-tetracosanol 4 2 tr - <1 3 1 4 - 6 1 5 tr 1 <1 tr n-hexacosanol 49 15 <1 11 <1 8 2 12 2 20 5 14 tr 1 <1 tr n-octacosanol 116 38 1 29 1 21 6 28 5 35 9 25 tr 2 <1 tr n-triacontanol 51 13 <1 12 1 8 3 11 1 8 1 4 tr <1 - - Alkanes 27 22 1 3 43 44 14 23 15 18 4 22 - - - - n-heneicosane - - - - - - - - 2 - - - - - - - n-docosane - - - - - - - - 1 - - - - - - - n-tricosane - - - - - - - - 2 - <1 1 - - - - n-tetracosane - - - - - - - - 1 tr - tr - - - - n-pentacosane 2 2 <1 tr <1 1 - - 5 5 2 5 - - - - n-hexacosane - - - - - - - - 1 tr tr tr - - - - n-heptacosane 5 3 <1 1 3 5 1 2 3 3 1 4 - - - - n-nonacosane 20 17 <1 2 38 31 12 19 <1 10 1 12 - - - - n-hentriacontane - - - - 2 7 1 2 - tr tr tr - - - -
211

5. Resultados y discusión
212
Aldehydes 371 3 <1 8 25 <1 tr 6 1 tr tr 2 - - - - n-hexacosanal 58 tr tr 1 3 - - 1 tr tr tr - - - - - n-octacosanal 174 3 <1 5 9 <1 tr 3 1 tr 1 2 - - - - n-triacontanal 139 tr tr 2 13 tr - 2 tr tr 1 4 - - - - Steroid hydrocarbons 14 3 1 2 30 5 1 1 14 11 1 3 3 4 1 <1 stigmasta-3,5,22-triene 5 3 1 2 12 4 1 1 10 11 1 3 2 3 1 <1 stigmasta-3,5-diene 9 <1 tr tr 18 1 <1 tr 4 tr - - 1 1 <1 tr Steroid ketones 33 6 <1 tr 27 6 3 9 3 5 1 7 4 6 5 1 cycloartenone - - - - - - - - - - - - 2 1 <1 - �-amirenone - - - - tr 1 tr - - - - - - - - - stigmasta-3,5-dien-7-one 5 3 <1 tr 3 1 1 <1 2 3 1 5 1 3 3 1 stigmast-4-en-3-one 9 1 tr - 5 - - - 1 1 tr tr 1 1 1 <1 stigmastadienone isomer 3 1 - - 2 <1 <1 2 <1 tr <1 2 <1 1 1 tr friedelan-3-one - - - - 15 4 2 7 - - - - - - - - ergostane-3,6-dione 6 tr - - - - - - - - - - - - - - stigmastane-3,6-dione 10 1 - - 2 - - - <1 1 tr - - - - - Free sterols/triterpenols
92 33 3 - 36 78 15 6 20 107
14 14 25 20 5 <1
campesterol 27 8 1 - 3 9 2 - <1 9 tr - 2 3 1 tr stigmasterol 23 5 <1 - 3 9 2 - 3 15 2 2 2 3 1 <1 sitosterol 36 17 2 - 18 37 7 - 11 56 8 8 10 11 3 <1 stigmastanol 6 3 <1 - 2 6 1 - 1 6 1 1 tr 1 <1 <1 �-amyrin - - - - 9 15 3 5 1 6 tr tr - - - - �-amyrin - - - - 1 2 <1 1 tr 1 tr tr - - - - 7-oxositosterol - - - - - - - - 2 2 1 tr 11 2 <1 tr hecogenin - - - - - - - - 2 12 2 2 - - - - Sterol glycosides* 5 8 1 tr 13 16 <1 tr 2 25 2 1 2 8 <1 tr campesterol 3�-D-Glcp tr 2 <1 tr 2 3 tr - tr 6 <1 tr <1 3 <1 tr stigmasterol 3�-D- Glcp tr 1 tr tr 1 2 tr - <1 3 <1 tr tr 1 tr tr sitosterol 3�-D-Glcp 5 5 1 tr 10 11 <1 tr 2 16 2 1 2 4 <1 tr Sterol/triterpenol esters
6 - - - 7 - - - <1 - - - 1 - - -
�-amyrin acetate - - - - 6 - - - - - - - - - - - sitosterol esters 6 - - - 1 - - - <1 - - - 1 - - - Ester waxes* 284 - - - 17 - - - <1 - - - - - - - C40 8 - - - tr - - - - - - - - - - - C42 19 - - - 4 - - - tr - - - - - - - C44 68 - - - 5 - - - <1 - - - - - - - C46 130 - - - 4 - - - tr - - - - - - - C48 56 - - - 2 - - - <1 - - - - - - - C50 4 - - - 2 - - - <1 - - - - - - - Ferulates - - - - - - - - 5 - - - - - - - t-tetracosanil ferulate - - - - - - - - 1 - - - - - - - t-hexacosanil ferulate - - - - - - - - 2 - - - - - - - t-octacosanil ferulate - - - - - - - - 2 - - - - - - - * Abreviations: Glcp (glucopyranoside); C(n), n denotes the total carbon atom number; tr: traces

5. Resultados y discusión
Finally, alkanes, fatty alcohols and free sterols and triterpenols survived the alkaline conditions and therefore were the main lipophilic constituents in crude pulps. The behavior of the fatty acids in an aqueous environment is quite different from that of the neutrals. At alkaline pH, the acids dissociate and can dissolve in water to quite a high extent forming fatty acid soaps that can form micelles. Fatty acid soaps are effective solubilizing agents facilitating the removal from pulp of sparingly soluble neutral substances (Back, 2000). The ratio of saponifiables-to-unsaponifiables has been suggested to be a better index for predicting pitch problems than the total amount of lipids (Back and Allen, 2000). In fact, the higher abundances of unsaponifiable compounds with respect to the saponifiable ones is the main cause for pitch problems during pulping of some woods, such as aspen or eucalypt (Chen et al., 1995; del Río et al., 1998; 2000). This fact can explain why the lipid content of crude flax pulps is similar or even lower than the other pulps regardless flax fibers had the highest extractive content. As mentioned above, fatty acids are the predominat lipids in flax fibers and can help to solubilize other water-insoluble components such as fatty alcohols and sterols. In contrast, the lipid content of crude sisal pulp is higher than the other three pulps regardless the relatively low content of lipophilic extractives in the raw material. Sisal fibers have low fatty acid content whereas the content in neutrals such as alkanes, fatty alcohols and steroids is relatively high. Particularly, the high abundances of free sterols, which have a high propensity to form pitch deposits (del Río et al., 1998; 2000), in the crude pulps from hemp and sisal, would point to a high pitch deposition tendency of the lipophilics from these fibers. 3.3. Fate of lipophilic extractives during TCF and ECF bleaching The lipophilic extractives remaining in the crude pulp are carried over to the bleach plant, where they react with the bleaching agents used (Björklund-Jansson et al., 1995; Holmbom, 2000). Pulp bleaching technology radically changed in the 1990s when the previously used chlorine was replaced and several new bleaching chemicals were introduced (Sjöström, 1993) and ECF and TCF sequences were adopted. As a consequence, new pitch problems arose due to the different reactivity of pulp lipids with the new bleaching agents. With the aim of studying the behavior of the different classes of lipophilic extractives under different bleaching conditions, both TCF and ECF bleached pulps were analyzed and compared with their respective crude pulps.
In general terms, the qualitative composition of the lipophilic extractives from the TCF pulps was very similar to that of their respective crude pulps (Figs. 2 and 3) although the lipid content was significantly lower (Table 2). Fatty alcohols, alkanes, free sterols and triterpenols, and fatty acids were the main lipophilic compounds present in the TCF bleached pulps, although in lower amount than in their respective crude pulps. The low reactivity of most pulp
213

5. Resultados y discusión
214
lipids in TCF bleaching sequences (using hydrogen peroxide as a bleaching agent) has been reported (Freire et al., 2003; Gutiérrez et al., 2001a). The decrease in the content of these lipophilic compounds in the TCF pulps, regardless the low reactivity of hydrogen peroxide towards them, might be due to the extensive washing in alkaline conditions carried out in the industrial TCF bleaching sequences used. Moreover, it is interesting to note that sterol glycosides, which resisted soda/AQ pulping conditions, were removed to a high extent under the industrial TCF bleaching conditions used in this work. The high efficiency of hydrogen peroxide in degrading sterol glycosides was previously reported using model compounds (Nilvebrant and Byström, 1995). However, a lower extent in the removal of sterol glycosides were observed during TCF bleaching of eucalypt pulps (Gutiérrez et al., 2006). In addition to the lipophilic extractives arising from the raw materials, several non-resolved peaks corresponding to the defoamers used in the process could also be observed in TCF pulps (Figs. 2 and 3). The excessive use of defoamers can also produce problems of pitch deposition (Allen, 2000). The presence of defoamer in pitch deposits produced during manufacturing of paper pulp from hemp fibers has been reported (Gutiérrez and del Río, 2005).
On the other hand, the composition of the lipophilic extractives from ECF pulps was somewhat different compared to that from their respective crude pulps (Figs. 2 and 3). The main difference observed was the large removal of free sterols in all the ECF pulps, although some minor amounts of free sterols still remained in sisal ECF pulp. The complete degradation of unsaturated sterols, such as sitosterol, during ECF bleaching has been previously reported in pitch deposits, pulps and in reactions with pure compounds (Freire et al., 2003; Gutiérrez et al., 2001a; Björklund-Jansson et al., 1995). On the other hand, sterol glycosides were largely removed during the ECF bleaching and were practically absent in these pulps. A less reactivity of these compounds with chlorine dioxide compared to hydrogen peroxide has been reported (Nilvebrant and Byström, 1995). Therefore, the complete removal of sterol glycosides in the ECF pulps studied here may have been due to the use of hydrogen peroxide in the ECF sequence. The removal of lipophilic extractives in bleaching is mainly achieved by two mechanisms: by dissolving or dispersing the pulp lipids followed by removal with washing liquors, and by oxidation of lipids with bleaching chemicals resulting in more hydrophilic compounds, which then may be dissolved in bleaching liquors and subsequently removed in washing (Holmbom, 2000). The decrease in the content of the lipophilic compounds in the TCF pulps studied here might be due to the first mechanism as mentioned above, taking into account the low reactivity of these lipids towards hydrogen peroxide. In contrast, the removal of unsaturated compounds during ECF bleaching was due to the second mechanism (Holmbom, 2000). The lower reactivity of pulp lipids in TCF bleaching sequences using hydrogen peroxide as a bleaching agent

5. Resultados y discusión
compared to ECF bleaching using chlorine dioxide (Back and Allen, 2000; Gutiérrez et al., 2009) may cause pitch problems to be, in principle, more severe in the former bleaching sequences. This is specially evident in the case of unsaturated steroids and triterpenoids as well as unsaturated fatty acids, which are strongly modified by chlorine dioxide but remain practically unaltered by oxygen and hydrogen peroxide (Holmbom, 2000; Bergelin and Holmbom, 2003; Freire et al., 2005; 2006; Gutiérrez et al., 2001a). However, in the nonwoody pulps studied here, the major lipophilic compounds present are saturated fatty acids, fatty alcohols, alkanes, which do not show reactivity towards chlorine dioxide, and therefore there are not great differences in the composition of the lipophilic extractives between ECF and TCF bleached pulps, with the exception of abaca pulp which lacks fatty alcohols and alkanes and where unsaturated sterols are the predominant lipophilic compounds. Therefore, both ECF and TCF bleached pulps will undergo similar pitch problems. Fatty acids, fatty alcohols and alkanes have been reported to be the compounds responsible for pitch deposits formed during pulping of nonwoody plants (Gutiérrez and del Río, 2005). 4. Conclusions A thorough chemical characterization of the lipophilic extractives from different nonwoody fibers (flax, hemp, sisal and abaca) at different stages of pulp production (soda/AQ pulping and TCF/ECF bleaching) has been carried out. This study provides useful information into the extent of their removal along the cooking and bleaching processes. The soda/AQ pulping stage led to the removal of aldehydes, hydroxyfatty acids and the complete hydrolysis of esterified compounds such as ester waxes, sterol esters and alkyl ferulates. Among the bleaching processes, ECF bleaching showed high reactivity towards unsaturated sterols and both ECF and TCF bleaching were very effective in removing sterol glycosides from nonwoody pulps. In contrast, saturated fatty acids, fatty alcohols and alkanes, which are the main lipophilic compounds in most of the studied fibers, survived pulping and bleaching conditions and were the predominating compounds in both TCF and ECF bleached pulps. Acknowledgements This study has been supported by the Spanish projects BIO2007-28719-E and AGL2008-00709 and the EU project BIORENEW (NMP2-CT-2006-026456). We thank CELESA pulp mill (Tortosa, Spain) for providing the samples. G.M. thanks the Spanish MEC for a FPI fellowship.
215

5. Resultados y discusión
216
References Allen, L.H., 2000. Pitch control in pulp mills, in: Back, E. L., Allen, L. H.
(Eds.), Pitch control, wood resin and deresination. TAPPI Press, Atlanta, pp. 265-288.
Back, E.L., 2000. Deresination in pulping and washing, in: Back, E. L., Allen, L. H. (Eds.), Pitch Control, Wood Resin and Deresination. TAPPI Press, Atlanta, pp. 205-230.
Back, E.L., Allen, L.H., 2000. Pitch control, wood resin and deresination. TAPPI Press, Atlanta.
Bergelin, E., Holmbom, B., 2003. Deresination of birch kraft pulp in bleaching. J. Pulp Pap. Sci. 29, 29-34.
Bergelin, E., Holmbom, B., 2008. Reactions and distribution of birch extractives in kraft pulp oxygen delignification. J. Wood Chem. Technol. 28, 261-269.
Bergelin, E., Möller, R., Holmbom, B., 2005. Analysis of pitch and deposit samples in kraft pulp production. Pap. Puu-Pap. Tim. 87, 399-403.
Björklund-Jansson, M., Wormald, P., Dahlman, O., 1995. Reactions of wood extractives during ECF and TCF bleaching of kraft pulp. Pulp Pap. Can. 96, T134-T137.
Chen, T., Wang, Z., Zhou, Y., Breuil, C., Aschim, O.K., Yee, E., Nadeau, L., 1995. Using solid-phase extraction to assess why aspen causes more pitch problems than softwoods in kraft pulping. Tappi J. 78, 143-149.
del Río, J.C., Gutiérrez, A., 2006. Chemical composition of abaca (Musa textilis) leaf fibers used for manufacturing of high quality paper pulps. J. Agr. Food Chem. 54, 4600-4610.
del Río, J.C., Gutiérrez, A., González-Vila, F.J., Martín, F., Romero, J., 1998. Characterization of organic deposits produced in the kraft pulping of Eucalyptus globulus wood. J. Chromatogr. A 823, 457-465.
del Río, J.C., Rodríguez, I.M., Gutiérrez, A., 2004. Identification of intact long-chain p-hydroxycinnamate esters in leaf fibers of abaca (Musa textilis) using gas chromatography/mass spectrometry. Rapid Commun. Mass Sp. 18, 2691-2696.

5. Resultados y discusión
del Río, J.C., Romero, J., Gutiérrez, A., 2000. Analysis of pitch deposits produced in kraft pulp mills using a totally chlorine free bleaching sequence. J. Chromatogr. A 874, 235-245.
Freire, C.S.R., Silvestre, A.J.D., Neto, C.P., 2003. Oxidized derivatives of lipophilic extractives formed during hardwood kraft pulp bleaching. Holzforschung 57, 503-512.
Freire, C.S.R., Silvestre, A.J.D., Neto, C.P., 2005. Lipophilic extractives in Eucalyptus globulus kraft pulps. Behavior during ECF bleaching. J. Wood Chem. Technol. 25, 67-80.
Freire, C.S.R., Silvestre, A.J.D., Neto, C.P., Evtuguin, D.V., 2006. Effect of oxygen, ozone and hydrogen peroxide bleaching stages on the contents and composition of extractives of Eucalyptus globulus kraft pulps. Bioresource Technol. 97, 420-428.
Gutiérrez, A., del Río, J.C., 2001. Gas chromatography-mass spectrometry demonstration of steryl glycosides in eucalypt wood, kraft pulp and process liquids. Rapid Commun. Mass Sp. 15, 2515-2520.
Gutiérrez, A., del Río, J.C., 2003a. Lipids from flax fibers and their fate in alkaline pulping. J. Agr. Food Chem. 51, 4965-4971.
Gutiérrez, A., del Río, J.C., 2003b. Lipids from flax fibers and their fate in alkaline pulping (Vol 51, pg 4965, 2003). J. Agr. Food Chem. 51, 6911-6914.
Gutiérrez, A., del Río, J.C., 2005. Chemical characterization of pitch deposits produced in the manufacturing of high-quality paper pulps from hemp fibers. Bioresource Technol. 96, 1445-1450.
Gutiérrez, A., del Río, J.C., González-Vila, F.J., Martín, F., 1998. Analysis of lipophilic extractives from wood and pitch deposits by solid-phase extraction and gas chromatography. J. Chromatogr. A 823, 449-455.
Gutiérrez, A., del Río, J.C., Ibarra, D., Rencoret, J., Romero, J., Speranza, M., Camarero, S., Martínez, M.J., Martínez, A.T., 2006. Enzymatic removal of free and conjugated sterols forming pitch deposits in environmentally sound bleaching of eucalypt paper pulp. Environ. Sci. Technol. 40, 3416-3422.
Gutiérrez, A., del Río, J.C., Martínez, A.T., 2009. Microbial and enzymatic control of pitch in the pulp and paper industry. Appl. Microbiol. Biot. 82, 1005-1018.
217

5. Resultados y discusión
218
Gutiérrez, A., Rodríguez, I.M., del Río, J.C., 2004. Chemical characterization of lignin and lipid fractions in kenaf bast fibers used for manufacturing high-quality papers. J. Agr. Food Chem. 52, 4764-4773.
Gutiérrez, A., Rodríguez, I.M., del Río, J.C., 2006. Chemical characterization of lignin and lipid fractions in industrial hemp bast fibers used for manufacturing high-quality paper pulps. J. Agr. Food Chem. 54, 2138-2144.
Gutiérrez, A., Rodríguez, I.M., del Río, J.C., 2008. Chemical composition of lipophilic extractives from sisal (Agave sisalana) fibers. Ind. Crop. Prod. 28, 81-87.
Gutiérrez, A., Romero, J., del Río, J.C., 2001a. Lipophilic extractives from Eucalyptus globulus pulp during kraft cooking followed by TCF and ECF bleaching. Holzforschung 55, 260-264.
Gutiérrez, A., Romero, J., del Río, J.C., 2001b. Lipophilic extractives in process waters during manufacturing of totally chlorine free kraft pulp from eucalypt wood. Chemosphere 44, 1237-1242.
Hillis, W.E., Sumimoto, M., 1989. Effect of extractives on pulping, in: Rowe, J. W. (ed.), Natural products of woody plants. II. Springer-Verlag, Berlin, pp. 880-920.
Holmbom, B., 2000. Resin reactions and deresination in bleaching, in: Back, E. L., Allen, L. H. (Eds.), Pitch control, wood resin and deresination. Tappi Press, Atlanta, pp. 231-244.
Nilvebrant, N.-O., Byström, S., 1995. Demonstration of glucosidic linked sterols in birch. Proc. 8th Intern. Symp. Wood and Pulping Chem., Helsinki, 6-9 June 2, 135-140.
Shin, S.J., Kim, C.H., 2006. Residual extractives in unbleached aspen and pine kraft pulps and their fate on oxygen delignification. Nord. Pulp Pap. Res. J. 21, 260-263.
Silvestre, A.J.D., Pereira, C.C.L., Neto, C.P., Evtuguin, D.V., Duarte, A.C., Cavaleiro, J.A.S., Furtado, F.P., 1999. Chemical composition of pitch deposits from ECF Eucalyptus globulus bleached kraft pulp mill: Its relationship with wood extractives and additives in process streams. Appita J. 52, 375-382.

5. Resultados y discusión
Sjöström, E., 1993. Wood chemistry. Fundamentals and applications. Academic Press, San Diego.
219

5. Resultados y discusión
Publicación VIII: Marques G., Gutiérrez A., del Río J.C. and Evtuguin D.V. (2010) Acetylated heteroxylan from Agave sisalana and its behavior in alkaline pulping and TCF/ECF bleaching. Carbohydrate Polymers, 81, 517-523.
220

5. Resultados y discusión
Acetylated heteroxylan from Agave sisalana and its behavior in alkaline pulping and TCF/ECF bleaching
Gisela Marquesa, Ana Gutiérreza, José C. del Ríoa and Dmitry V. Evtuguinb
aInstituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, P.O. Box 1052, 41080-Seville, Spain
bCICECO and University of Aveiro, Department of Chemistry, 3810-193 Aveiro, Portugal
Abstract The heteroxylan from sisal (Agave sisalana), an O-acetyl-(4-O-methylglucurono)xylan with a molecular weight (Mw) of 18 kDa, was isolated by extraction of peracetic holocellulose with Me2SO and thoroughly characterized by wet chemistry, and NMR spectroscopy. The heteroxylan backbone is composed of (1�4)-linked �-D-xylopyranosyl units (Xylp) partially branched with terminal (1�2)-linked 4-O-methyl-�-D-glucuronosyl (MeGlcpA, 9%mol.) and a small proportion of �-D-glucuronosyl (GlcpA, <1%mol.) residues. Roughly 61%mol. of Xylp residues are acetylated (DS =0.70). During soda/AQ pulping of sisal fibers, MeGlcpA and GlcpA are mostly removed or converted to 4-deoxy-�-L-threo-hex-4-enopyranosyluronic acid (HexA), although about 15% of the initially present MeGlcpA was maintained intact upon cooking. The major part of acetyl groups (95%) was hydrolyzed during pulping. It was proposed that during bleaching, a low molecular weight xylan fraction associated to residual lignin was removed from pulp and small proportion of MeGlcpA was additionally converted to HexA. The profiles of uronosyl residues in xylans from TCF and ECF bleached sisal pulps were rather different. Keywords: Agave sisalana; soda pulping; bleaching; black liquor; xylan; structural analysis; NMR spectroscopy 1. Introduction Sisal fibers are hard fibrous material isolated from the leaves of the sisal plant (Agave sisalana), a monocotyledonous plant from the Agavaceae family (Li et al. 2000; Gutiérrez et al. 2008). Originally from Central America and Mexico, sisal plant is widely cultivated in South America (e.g. Brazil), Australia and Africa (e.g. Kenya) (Gutiérrez et al, 2008; Mwaikambo, 2006). The sisal fibers find out numerous applications in the manufacture of ropes for boats, goods for the agricultural industry and for the reinforcement of polymeric matrices (Li et al., 2000; Gutiérrez et al., 2008; Mwaikambo, 2006; Megiatto et al., 2007). Sisal cellulosic pulp possesses such characteristics as high tear resistance, alpha cellulose content, porosity, bulk, moister absorbency and high folding endurance, that offer unique opportunities for the papermaking (Gutiérrez et al.,
221

5. Resultados y discusión
222
2008; Mwaikambo, 2006; Megiatto et al., 2007; Hurter, 2001; Idárraga et al., 2001). Easily bleachable sisal chemical pulp is industrially produced by soda pulping in the presence of athroquinone (AQ) as catalyst.
The basic knowledge of the chemical composition of sisal fibers, as well as the behavior of its components during pulping and bleaching, is essential for the better understanding and improving the pulping and bleaching operations and for the assessment of pulp and paper properties. Previous papers have reported the composition of the lipophilic compounds (Gutiérrez et al., 2008) and the structure of the lignin (del Río et al., 2007, 2008) of sisal fibers, but only limited work has been performed on the structural characterization of the carbohydrate fraction of this fiber (Das Gupta & Mukherjee, 1967; Stewart et al., 1997; Megiatto et al., 2007). In the present work, we report the structural characterization of a heteroxylan in sisal fibers, as well as in their soda/AQ pulps (unbleached and TCF/ECF bleached). The study of hemicelluloses is of fundamental and practical interest, since their partial degradation and dissolution during pulping is responsible for significant consumption of pulping chemicals, the decrease of pulp yield and the papermaking properties of bleached pulps (Evtuguin et al., 2003; Pinto et al., 2005; Lisboa et al., 2004).
Hemicelluloses provide a supporting function to the cell wall being bounded to cellulose fibrils. Hemicelluloses are mainly branched polymers of low molecular weight (DP � 80-200) and are composed by diverse sugar residues (D-xylose, L-arabinose, D-glucose, D-galactose, D-mannose, D-glucuronic acid, 4-O-methyl-D-glucuronic acid, D-galacturonic acid, and to a lesser extent, L-rhamnose, L-fucose, and various O-methylated neutral sugars) (Shimizu, 1991; Sun et al., 2000; Ebringerová et al., 2005). In particular, glucuronoxylan (GX) is the major hemicellulose in such important industrial crops produced by agro-industry as kenaf, bamboo, flax, sisal and jute and is structurally similar to hardwood xylans (Rowell et al., 1998; Gorshkova et al., 1996; Neto et al., 1996; Vignon & Gey, 1997; Stewart et al., 1997). Among the above mentioned plants, GX of sisal is one of the less investigated. Previous structural/compositional studies of GX from sisal were carried out mainly with alkali-extracted GX (Stewart et al., 1997; Das Gupta & Mukherjee, 1967). Hence, some essential structural information, such as substitution patterns with acetyl groups, was not assessed. It was suggested, however, that sisal GX is an O-acetyl-4-O-methylglucuronoxylan with moderate molecular weight, around 15-20 kDa (Stewart et al., 1997; Das Gupta & Mukherjee, 1967). According to data from methylation analysis of sisal xylan, its backbone is constituted by �-(1-4)-linked D-xylopyranose residues, carrying a low degree of substitution (8-10 % mol.) with terminal 4-O-methyl-D-glucopyranosyluronic acid residue linked through the O-2 position (Das Gupta & Mukherjee, 1967).
In this report, the heteroxylan from sisal fibers was isolated by extraction of peracetic holocellulose with dimethyl sulfoxide and thoroughly characterized by wet chemistry and NMR spectroscopy. This isolation procedure allowed to

5. Resultados y discusión
obtain a xylan sample with intact O-acetyl moieties. Simultaneously, the fate of this heteroxylan structure during soda/anthraquinone pulping and TCF and ECF bleaching was also studied. 2. Experimental 2.1. Samples Sisal (Agave sisalana) leaf fibers from Africa (Kenya), their soda/AQ chemical pulps (unbleached and ECF/TCF bleached) and cooking black liquor were supplied by CELESA pulp mill (Tortosa, Spain). The TCF bleaching sequence E(O)P-EP included two hydrogen peroxide stages at 90ºC, the first under oxygen pressure (E(O)P stage) and the second without oxygen (EP stage). The ECF bleaching sequence D-EP included a chlorine dioxide (D) at 60ºC and a hydrogen peroxide (EP) stages at 90ºC.
The samples were air-dried, milled using a knife mill (Janke and Kunkel, Analysenmühle), and extracted with acetone in a Soxhlet apparatus for 8 h. For estimation of the Klason lignin content, the acetone extracted samples were subsequently extracted with hot water (3 h at 100 ºC). Klason lignin was estimated as the residue after 72% sulfuric acid hydrolysis of the pre-extracted material according to Tappi standard procedures (Tappi Test Methods, 1993). Ash content was estimated as the residue after 6 h calcination at 575 ºC. 2.2. Preparation of holocellulose The holocelluloses of sisal fibers and their unbleached pulp were obtained from extractives-free sawdust (5.0 g) by delignification with 10% peracetic acid at pH 3.5 for 20 min at 85 ºC. After delignification, the holocellulose was filtered off on a porous glass filter, washed with acetone and further with warm water and air-dried. The holocellulose yield was 69.7% and 94.7% for the sisal fibers and unbleached pulp, respectively.
2.3. Isolation of xylans from pulps The isolation of acidic heteroxylans was carried out by two consecutive extractions with Me2SO (1.5 g of holocelluloses with 50 mL of Me2SO in each assay) at 50 ºC for 24 h under stirring and further precipitation of the resulted extracts in an excess of 7:2:1 EtOH-MeOH-water acidified by HCOOH. The complete precipitation of the heteroxylan was accomplished in 3 days at 4 ºC. The heteroxylan was isolated by centrifugation, washed four times with anhydrous MeOH and quickly dried under vacuum at room temperature. For the ECF and TCF bleached pulps the xylans were extracted directly from the pulp without previous preparation of holocelluloses.
223

5. Resultados y discusión
224
2.4. Isolation of xylan from the black liquor
The xylan of the black liquor was isolated following a procedure previously described, with minor modifications (Engström et al, 1995). 200 ml of 1,4-dioxane were slowly added with agitation to 100 ml of diluted with distilled water 1/2 black liquor, followed by the addition of glacial acetic acid until pH 2-3. The solution and resulting precipitate was kept at 5 ºC during 2 days. The black liquor precipitated polysaccharides (BLPS) were separated by centrifugation and the solution decanted off. The precipitate was sequentially washed up with 150 ml of a 1,4-dioxane-water (2:1) solution, 150 ml of 1,4-dioxane, 150 ml of methanol and 150 ml of acetone and, finally, dried under vacuum with phosphorus pentoxide. 2.5. Carbohydrate analysis The heteroxylan was subjected to hydrolysis with 72% H2SO4 at 20 ºC for 3 h, followed by 2.5 h hydrolysis with diluted 1 M H2SO4 at 100 ºC (Saeman hydrolysis) and the released neutral monosaccharides were determined as alditol acetate derivatives by gas chromatography (Selvendran et al., 1979). The quantitative analysis was carried out on a Varian 3350 gas chromatograph equipped with a FID detector and with a DB-225 J&W column (30 m×0.25 mm i.d. ×0.15 �m film thickness). The temperature program was started at 220 ºC with a 5 min hold, and then raised to a final temperature of 230 ºC at 2 ºC/min, and held for 5 min. The injector and detector temperatures were set at 230 ºC. The quantification was made using calibration curves with standards.
2.6. Acid methanolysis for analysis of sugars and uronic acids About 4.5 mg of freeze-dried sisal fibers and their pulps (unbleached, ECF and TCF bleached pulps) were subjected to acid methanolysis by the addition of 2 mL, 2 M solution of HCl in anhydrous methanol at 100 ºC for 4 hours (Sundberg et al., 1996). After cooling to room temperature, about 80 �L of pyridine was added to neutralize the acidic solution. Additionally, 1 mL of internal standard solution containing 0.1 mg/mL of sorbitol was added. To avoid fibers silylation, 2 mL of the supernatant reaction solution was separated from fiber suspension and evaporated in a rotary evaporator with a water bath kept at 40-50 ºC. The samples were dissolved by addition of 70 �L pyridine. For silylation, 150 �L hexamethyldisiloxane and 80 �L trimethylchlorosilane were added and the samples were shaken well. After 12 hours at room temperature, the samples were ready for analysis. GC-MS analysis were performed using a Hewlet-Packard Gas Chromatograph 5890 equipped with a mass selective detector MSD series II, using helium as carrier gas (35 cm/s), equipped with a DB-1 J&W capillary column (30 m×0.32 mm i.d. 0.25 �m film thickness). The column temperature program was 100 – 4 ºC/min – 175 ºC followed by 175 – 12 ºC/min – 290 ºC. The detector (FID) temperature was 290 ºC. The different

5. Resultados y discusión
peaks were identified by comparing their mass spectra with mass spectra in Wiley and NIST libraries and that reported in the literature (Sundberg et al., 1996; Bertaud et al., 2002; Bleton et al., 1996).
2.7. Size-exclusion chromatography (SEC) The xylan samples were dissolved in a small amount of 10% LiCL solution in N,N-dimethylacetamide (DMAC) at 70-80 ºC and further diluted with DMAC to a xylan concentration of about 0.5% (5mg/mL). The SEC analysis has been carried out on two PLgel 10 μm MIXED B 300 × 7.5 mm columns protected by a PLgel 10 μm pre-column (Polymer Laboratories, UK) using a PL-GPC 110 system (Polymer Laboratories). The columns, injector system and the detector (RI) were maintained at 70 ºC during the analysis. The eluent (0.1 M LiCl solution in DMAC) was pumped at a flow rate of 0.9 mL/min. The analytical columns were calibrated with pollulan standards (Polymer Laboratories) in the range 0.8-100 kDa. The injected volume was 100 μL.
2.8. NMR spectroscopy One-dimensional 1H NMR spectra of the xylan samples were recorded in D2O (30 ºC) on a Bruker Avance 300 spectrometer operating at 300.13 MHz. Sodium 3-(trimethylsilyl)-propionate-d4 was used as internal standard ( 0.00). The relaxation delay was 16 s, r.f. 90º-pulse width of 10.2 μs and about 400 pulses were collected. All 2D NMR spectra were recorded on a Bruker Avance 300 spectrometer operating at 300.1 MHz for proton and at 75.2 MHz for carbon. 2D 1H–1H COSY spectroscopy was performed at 50 °C using a standard COSY sequence (90° pulse, relaxation delay 2 s). Two-dimensional 1H-1H TOCSY (Total Correlation Spectroscopy) spectra (mix= 0.050 s) were acquired at a spectral width of 2185 Hz in both dimensions at 60 ºC. The relaxation delay was 2.0 s. For each FID, 128 transients were acquired; the data size was 1024 in t1 × 512 in t2. The phase sensitive 1H-detected HSQC (Heteronuclear Single Quantum Coherence) spectrum was acquired at 50ºC over a F1 spectral weight of 12,000 Hz and a F2 width of 2000 Hz with a 2048 × 1024 matrix and 128 transients per increment. The delay between scans was 2 s and the delay for polarization transfer was optimized for 1JCH= 148 Hz.
2.9. Hexenuronic acid content The amount of hexenuronic acids (HexA) was determined by acidic hydrolysis in sodium formate buffer at pH 3.0 followed by UV detection of furan derivatives at 245 nm (Vuorinen et al. 1999).
225

5. Resultados y discusión
226
3. Results and discussion 3.1. Chemical composition of sisal fibers The chemical composition of sisal (Agave sisalana) fibers is presented in Table 1. The sugar analysis confirmed the data previously reported by Stewart et al. (1997) indicating that xylan is the principal hemicellulose of sisal fibers and the second most abundant polysaccharide after cellulose. At the same time, taking into account the small amount of lignin (around 6%) and extractives (slightly more than 3%) in sisal fibers, the relatively low yield of peracetic holocellulose (70%) indicates a significant content of easily removable hemicelluloses, other than xylan. These may be pectins and, in particular, glucans that are known to be present in the leaves of the genus Agave in noticeable amounts (Nobel, 2003). The misbalance in cellulose content and the amount of glucose detected upon sugars analysis (Table 1) allows suggesting that non-cellulosic glucans may contribute to at least 15% (w/w) of sisal fibers. This fact was further confirmed by analysis of the hemicelluloses dissolved in the black liquor from soda pulping of sisal fibers.
3.2. Isolation and structural characterization of xylan from sisal fibers The heteroxylan (yield of about 60% w/w) from sisal fibers was isolated from peracetic holocellulose by two consecutive extractions with Me2SO followed by precipitation of the extracted polyose in 7:2:1 ethanol/methanol/water. Such procedure guaranteed the isolation of intact and representative xylan sample, which can be structurally characterized including the quantification and distribution of O-acetyl moieties (Evtuguin et al. 2003).
Table 1. Chemical composition of sisal fibers, unbleached soda pulp and ECF and TCF bleached pulps (% w/w). Component Sisal fibers Unbleached pulp TCF pulp ECF pulp Ash 1.0 1.0 0.4 0.4 Extractives (acetone) 0.8 0.3 0.1 0.1 Extractives (water) 2.3 0.7 0.6 0.4 Klason lignin 5.9 0.7 - - Holocellulose 70.0 95.0 - - Cellulose* 54.5 - - -
Neutral sugars Rha 0.7 0.7 tr tr Ara 1.3 tr tr tr Xyl 20.0 19.0 19.4 20.6 Man 0.8 - - - Gal 1.0 tr tr tr Fuc <0.5 - - - Glc 75.7 80.4 80.6 79.4 * Kürschner-Hoffer method of determination; tr: traces;

5. Resultados y discusión
The composition of the isolated xylan was assessed by analyses of neutral sugars and easily hydrolyzed sugars after methanolysis (Tables 2 and 3). The high purity of the isolated xylan was confirmed using neutral monosaccharides analysis, that showed the predominance of xylose (Xyl) and only small amounts of glucose (Glc), galactose (Gal), arabinose (Ara) and rhamnose (Rha) (Table 2). The presence of Gal, Ara and Rham may indicate the eventual small contamination of xylan with pectin compounds. This was confirmed by methanolysis studies (Table 3, Figure 1), which revealed a much higher amount of galacturonic acid than could be expected if arisen only from the terminal structural fragment [�3)-�-L-Rhap-(1�2)-�-D-GalpA-(1�4)-D-Xylp] suggested to be present in xylans (Shimizu, 1991). The presence of D-glucopyranosyluronic acid (GlcpA), besides the expected 4-O-methyl-D-glucopyranosyluronic acid (MeGlcpA), may indicate that, at least part of glucuronosyl moieties attached to xylan backbone, is not methylated. The ratio between internal xylopyranosyl units (Xylp) in the backbone and terminal attached glucuronosyl residues (MeGlcpA and GlcpA) was estimated to be around 9:1. The molecular weight (Mw) of sisal xylan was about 18 kDa, as revealed by SEC analysis (Figure 2).
Table 2. Neutral monosaccharide composition (% w/w) of xylans isolated from sisal fibers, pulps and black liquor.
Rha Ara Xyl Man Fuc Gal Glc Sisal fibers 0.9 0.7 93.7 tr - 1.2 3.5 Unbleached pulp tr tr 99.5 - - tr 0.5 TCF bleached pulp tr tr 99.0 - - tr 1.0 ECF bleached pulp tr tr 98.7 - - tr 1.3 Black liquor precipitated polysaccharides (BLPS)
0.7
3.6
10.9
2.6
1.1
27.1
54.0
tr: traces According to previously reported methylation analysis of alkali extracted
sisal xylan, its backbone is constituted by �-(1�4)-linked D-xylopyranose residues branched at O-2 with terminal 4-O-methyl-D-glucopyranosyluronic acid residues (Das Gupta & Mukherjee, 1967). These structural features were confirmed by 1D and 2D NMR techniques. Single (COSY) and multiple (TOCSY) bonds 1H-1H correlation analyses and 1H-13C (HSQC) correlations (Figure 3) allowed assignment of proton and carbon signals in sisal heteroxylan, as summarized in Table 4. Chemical shifts of protons and carbons were practically the same as those reported for acetylated heteroxylans from other plant sources (Teleman et al. 2002; Evtuguin et al. 2003). The anomeric region in the TOCSY spectrum (Figure 4) revealed the characteristic proton correlations that are normally found in heteroxylans containing attached to backbone non-methylated GlcpA residues (Vignon & Gey, 1997; Gonçalves et al. 2008). Hence, NMR results corroborates the data obtained by methanolysis
227

5. Resultados y discusión
228
Table 3. Neutral monosaccharide and uronic acids composition (% w/w) determined by methanolysis of xylans isolated from sisal fibers and their pulps.
Rha Ara Xyl Man Gal Glc GalA GlcA MeGlcpA Sisal fibers 0.8 0.6 83.0 - 0.8 1.3 2.8 0.3 10.5 Unbleached pulp - - 98.3 - - 0.2 - - 1.5 TCF bleached pulp - - 98.0 - - 0.8 - - 1.2 ECF bleached pulp - - 97.5 - - 1.2 - - 1.3 (Table 3), evidencing that a small proportion of glucuronic residues in sisal heteroxylan are not methylated. Therefore, it can be suggested that the backbone of sisal heteroxylan is composed of partially acetylated (1�4)-linked �-D-Xylp units O-2 ramified with terminal (1�2)-linked MeGlcpA and GlcpA. �
8 10 12 14 16 18 20 22 24 26
Retention Time (min)
Xyl
Rha
Xyl
Ara A
ra
Gal
A
Glc
4-O-MeGLCA
Gal
AG
al Gal Gal
A
Glc
Glc
A
Gal
AG
al
IS Dimers
Ara
SISAL FIBERS
SISAL UNBLEACHED PULP
SISAL TCF PULP
SISAL ECF PULP
Xyl
Xyl
Figure 1. Gas chromatogram of methylated and silylated sugars obtained by acid methanolysis of xylans isolated from sisal fibers and their pulps. Xyl: xylose, Gal: galactose, Glc: Glucose, Rha: rhamnose, GlcA: glucuronic acid, 4-O-MeGlcA: 4-O-methylglucuronic acid, GalA: galacturonic acid.

5. Resultados y discusión
Table 4. Proton/carbon chemical shifts () of heteroxylan from sisal (30ºC, D2O). Structural unit Assignments
H1/C1 H2/C2 H3/C3 H4/C4 H5/C5 ax eq
Xyl (isol) 4.47/102.9 3.28/73.7 3.55/74.5 3.80/77.4 3.40/63.9 4.10/63.9Xyl-3Ac 4.57/102.4 3.49/71.9 4.98/76.3 3.93/76.5 3.47/63.8 n.d/63.8 Xyl-2Ac 4.68/101.2 4.68/74.6 3.80/72.5 3.87/77.2 3.45/63.8 n.d/63.8 Xyl-2,3Ac 4.80/100.5 4.82/74.6 5.16/74.0 4.06/76.6 3.54/63.9 n.d/63.9 Xyl-3Ac-2GlcA 4.73/102.1 3.70/75.8 5.06/75.8 3.98/77.2 3.50/n.d n.d/n.d MeGlcA 5.27/98.9 3.57/72.3 3.88/73.7 3.27/83.2 n.d/n.d _a GlcA 5.25/n.d 3.60/n.d 3.82/n.d 3.52/n.d n.d/n.d _a
n.dnon determined, anot relevant. The following designations were used: Xyl (isol.), non-acetylated Xylp in the backbone isolated from other acetylated Xylp units; Xyl-3Ac, 3-O-acetylated Xylp; Xyl-2Ac, 2-O-acetylated Xylp; Xyl-2,3Ac, 2,3-di-O-acetylated Xylp; Xyl-3Ac-2GlcA, MeGlcA 2-O-linked and 3-O-acetylated Xylp; MeGlcA, 2-O-linked MeGlcpA; GlcA, 2-O-linked GlcpA.
The acetylation patterns in the heteroxylan backbone were assessed by 1H NMR spectroscopy based on signal assignment employing 2D NMR techniques (Table 4). The total 1H NMR spectrum of sisal xylan and its expanded anomeric region with specified groups of protons in particular substructures, are presented in Figure 5. According to previously published methodology (Evtuguin et al. 2003), internal non-acetylated, 3-O- and 2-O-acetylated xylose residues, MeGlcpA residues were assessed based on their anomeric proton resonances, whereas the amounts of 2,3-di-O-acetylated and 3-O-acetylated/ MeGlcpA O-2 substituted internal xylose residues were estimated based on H-3 resonances in corresponding structures (Figure 5). This allowed the integration of protons from particular structural fragments and their quantification (Table 5). Around 61%mol. of the Xylp residues were acetylated; among them, 39 %mol. corresponded to 3-O-acetylated, 13% mol. corresponded to 2-O-acetylated and
�
11.2 12.3 13.5 14.6 15.7
(A)
240
480
720
960
1200
Retention time (min)
Res
pons
e
0
(B)
(C)
(D)14 kDa
12 kDa
10 kDa
18 kDa
Figure 2. The GPC elution curves of xylans isolated from sisal fibers and their pulps. (A) sisal fibers, (B) unbleached pulp, (C) TCF bleached pulp, (D) ECF bleached pulp.
229

5. Resultados y discusión
230
9 %mol. corresponded to 2,3-di-O-acetylated residues. Accordingly, sisal heteroxylan possessed a substitution degree with acetyl groups of 0.70. Worth notably, Xylp units in backbone of sisal heteroxylan are predominantly 3-O-acetylated. The proportion of 3-O-acetylated Xylp units in backbone was much higher (almost twice) in sisal xylan than in xylans from woody sources such as birch and beech (Teleman et al. 2002), eucalypt (Evtuguin et al. 2003), paulownia (Gonçalves et al. 2008) and aspen (Teleman et al. 2000). Almost all Xylp linked at O-2 with MeGlcpA (9 mol%) were 3-O-acetylated (Table 5). 3.3. Changes in xylan structure during alkaline pulping During the soda/AQ pulping, around 40% of the xylan was dissolved in the black liquor. This conclusion was made based on the xylose content in delignified unbleached sisal fibers (Table 1) and the pulp yields (ca. 60%). The chemical changes in the xylan during pulping were examined comparing the composition and structural features of the xylan from sisal fibers and their soda pulp, both isolated by Me2SO extraction of the corresponding peracetic holocelluloses (Tables 2 and 3, Figure 6). The molecular weight of the xylan remaining in the pulp decreased to 10 kDa, reflecting significant alkali-induced depolymerization (Figure 2). The xylan suffered also a significant deacetylation (about 95%) and the major part of the uronic moieties (at least 75%) were converted to 4-deoxy-�-L-threo-hex-4-enopyranosyluronic acid (hexenuronic acid or HexA), as revealed from 1H NMR
�
5.5 5.0 4.5 4.0 3.5
100
90
80
70
60
ppm
X1
X1Ac3
X1Ac2
X1Ac3MeGlcA
MeGlcA1
X3Ac3MeGlcAX3Ac3
X2Ac2
X5Ac3MeGlcA
X5
-OCH3 in MeGlcA
X5’
X2X3
X2Ac3MeGlcA2
X2Ac3MeGlcAMeGlcA4
MeGlcA3 X3Ac2
X4X4Ac2X4Ac3MeGlcA
Figure 3. HSQC spectrum (D2O, 50 ºC) of heteroxylan from sisal fibers.

5. Resultados y discusión
Table 5. Relative content in acetyl groups in structural units of sisal heteroxylan.
Structural fragment and short designation Relative abundance (per 100 Xylp units)
�4)-�-D-Xylp-(1� (Xyl) 39 �4)[3-O-Ac]-�-D-Xylp-(1� (Xyl-3Ac) 30 �4)[2-O-Ac]-�-D-Xylp-(1� (Xyl-2Ac) 13 �4)[3-O-Ac][2-O-Ac]-�-D-Xylp-(1� (Xyl-2,3Ac) 9 �4)[4-O-Me-�-D-GlcpA-(1�2)][3-O-Ac]-�-D-Xylp--(1� (Xyl-3Ac-2MeGlcA)
9
4-O-Me-�-D-GlcpA-(1� (MeGlcA) 9 spectra (Figure 6). The presence of HexA, formed under alkaline pulping conditions via �-elimination of methoxyl group, was confirmed applying the total correlation spectroscopy (TOCSY), according to previously published proton chemical shifts (Teleman et al. 1995). The HexA residues may be easily detected in the anomeric region of 1H NMR spectra, which showed the appearance of new signals at 5.36 and at 5.82 ppm that were assigned to H-1 and H-4 in corresponding structures (Figure 6). The HexA content in sisal soda pulp
�
5.4 5.3 5.2 5.1 5.0 4.9 4.8
5.5
5.0
4.5
4.0
3.5
I III
II
IV’ V IV
ppm
H1/H4H1/H2 H1/H4
H1/H2H1/H3
H1
H3/H2
H3/H5
H3/H4
H3/H1
H3H3 H3
H3/H1 H3/H1
H1/H3
H1
I- MeGlcpA-(1�
II- GlcpA-(1�
III- 4)[3-O-Ac]-�-D-Xylp-(1�
IV, IV’- 4)[3-O-Ac][2-O-Ac]-�-D-Xylp-(1�
V- 4)[4-O-Me-�-D-GlcpA-(1�2][3-O-Ac]-�-D-Xylp-(1�
5.5
Figure 4. Anomeric region of the TOCSY spectrum (D2O, 60 ºC) of heteroxylan from sisal fibers
231

5. Resultados y discusión
232
was 60.6 mmol/kg of pulp as determined after pulp treatment under acidic conditions followed by detection of furoic acid derivatives by UV-spectroscopy at 245 nm (Vuorinen et al. 1999).
The balance of uronic moieties in the initial xylan and in the xylan remaining in the pulp was estimated based on the ratio of anomeric protons in uronic groups at 5.27 ppm and in internal xylopyranose units at 4.47 ppm (pulp xylan) or at 4.40-4.65 ppm (fiber xylan). This analysis indicated a removal of about 30% of all uronic units (MeGlcpA and HexA) from xylan during pulping.
The polysaccharides dissolved in the black liquor (BLPS) during pulping were isolated according to a previously published procedure (Engström et al, 1995) and chemically characterized (Table 2). The aim of this study was to compare the structure of the xylan remaining in the pulp with that dissolved in the pulping liquor. Surprisingly, the heteroxylan was a minor polysaccharide dissolved in the liquor and its purification by fractional precipitation failed. At the same time, the analysis of neutral sugars of BLPS revealed glucans as the major precipitated polysaccharides (glucose represents around 54 % of BLPS weight) followed by galactans. The preliminary study on BLPS using multiple bonds 1H-1H correlation NMR spectroscopy (TOCSY) gave additional insights �
CH3-CO-
*
*H2O
5.5 5.0 4.5 4.0 3.5 3.0 2.5 ppm
5.4 5.3 5.2 5.1 5.0 4.9ppm
4.8 4.7 4.6 4.5 4.45.5
H1 MeGlcA
H3Xyl-2,3Ac
H3 Xyl-3Ac-2GlcA
H3 Xyl-3Ac
H1/H2 Xyl-2Ac
H1 Xyl-3Ac
H1 Xyl
Figure 5. 1H NMR spectrum (D2O, 30 ºC) of heteroxylans from sisal fibers (top image) and the expanded region of anomeric protons (bottom image). The designations for the structural fragments are the same as in Table 5. * solvent impurities.

5. Resultados y discusión
ppm5.7 5.5 5.3 5.1 4.95.9 4.7 4.5 4.3
SISAL ECF PULP
H1HexA
H1MeGlcA
H1 Xyl
H4 HexA
SISAL UNBLEACHED PULP
SISAL TCF PULP
Figure 6. 1H NMR spectra (D2O, 30 ºC ) of heteroxylans isolated from sisal unbleached pulp and TCF and ECF bleached pulps.
into the type of glucans dissolved during pulping from sisal fibers (Figure 7). These were suggested to be mixture of �-glucans, in particular, �-(1�3)-glucans with a low degree of ramification at C6 (callose type), by comparison of the proton-proton correlations with previously published data on proton resonances in �-(1�3)-glucan (Torosantucci et al. 2005). However, a more detailed study is required to elucidate the exact structure of the �-glucans in sisal fibers. 3.4. Changes in xylan structure during TCF and ECF bleaching The structural changes in the heteroxylan from sisal soda pulp during industrial TCF and ECF bleaching were also investigated. The chemical composition and structural features were assessed in xylan samples isolated directly from bleached pulps by Me2SO extraction. The TCF pulp was obtained by E(O)P-EP bleaching and the ECF pulp was obtained by D-EP sequence. TCF pulp was bleached essentially by hydrogen peroxide under alkaline conditions and included two hydrogen peroxide stages, the first with oxygen and the second without oxygen, at 90 ºC,, whereas ECF pulp bleaching included a treatment with chlorine dioxide (D) at 60ºC followed by hydrogen peroxide stage under alkaline conditions (EP) at 90 ºC.
233

5. Resultados y discusión
234
�
ppm (t2)
3.504.004.50
3.50
4.00
4.50
ppm (t1)
H1 H3 H2 H4 H6ax H5
OO
OH
HOH
OH
H
H
O
H
O
OH
HOH
OH
H
H
O
H
ppm (t2)
3.504.004.50
3.50
4.00
4.50
ppm (t1)
H1 H3 H2 H4 H6ax H5
OO
OH
HOH
OH
H
H
O
H
O
OH
HOH
OH
H
H
O
H
Figure 7. TOCSY spectrum (D2O, 60 ºC) of BLPS fraction isolated from black liquor. Top image represents the fragment of �-(1-3)-D-glucan backbone.
The chemical analysis of the sugars of TCF and ECF bleached pulps did not
show significant changes when compared to the unbleached pulp (Table 1). This indicates that no specific removal of xylan from pulp took place during bleaching. The chemical composition of the xylans isolated from TCF and ECF bleached pulps was also similar to the xylan from unbleached pulp, although a very small decrease in MeGlcpA content in pulp xylans after bleaching was detected (Tables 2 and 3). This fact may be explained by a partial degradation of MeGlcpA to HexA under alkaline conditions, which are inaccessible for the analysis by methanolysis. This explanation was further supported by 1H NMR analysis, that showed a relative increase of the HexA content and a decrease of MeGlcpA moieties in the xylan from TCF bleached pulp (Figure 6). In contrast to the xylan from TCF bleached pulp, the xylan from ECF bleached pulp did not contain HexA residues, which were degraded upon bleaching with chlorine dioxide (Figure 6). Taking into account that uronic moieties strongly affect the papermaking properties (Lindström, 1992) and brightness stability (Buchert at al. 1997) of cellulosic pulps this knowledge may be important to explain the different response of pulps bleached employing TCF and ECF sequences.

5. Resultados y discusión
The xylans from bleached pulps (either TCF or ECF) did not show any acetyl groups, as revealed by 1H NMR analysis. Hence, alkaline bleaching stages favored the removal of residual acetyl groups from xylan of unbleached pulp. Xylans from bleached pulps possessed slightly higher molecular weight (12 kDa in TCF pulp and 14 kDa in ECF pulp), when compared to this in unbleached pulp (Figure 2). This fact may be explained by a predominant removal of low molecular weight xylan fractions structurally associated to residual lignin during bleaching. 4. Conclusions The structure of the heteroxylan isolated from sisal fibers has been characterized and its behavior during soda/AQ pulping and TCF/ECF bleaching has been studied. The data indicates that the heteroxylan backbone is composed by (1�4)-linked �-D-xylopyranosyl units (Xylp) partially ramified with terminal (1�2)-linked 4-O-methyl-�-D-glucuronosyl (MeGlcpA, 9 %mol.) and a small proportion of �-D-glucuronosyl (GlcpA) residues. Around 61mol% of the Xylp residues are acetylated, the major proportion of acetyl groups being attached at the O-3 position of the Xylp residues (39 %mol.), followed by acetylation at the O-2 position (13 %mol.) and diacetylation at both O-2 and O-3 positions (9%mol.). The molecular weight (Mw) of the heteroxylan was of 18 kDa. Around 40% of xylan was removed during soda pulping. However, the major polysaccharides found in the black liquor were �-glucans rather than xylans. Sisal xylan suffered a significant depolymerisation (Mw decreased to almost half) and deacetylation (95%) during pulping. Terminal MeGlcpA residues were partially removed (to about 30%) or converted to HexA in a large extent. HexA revealed to be relatively stable during TCF bleaching with hydrogen peroxide and were predominant among uronic moieties of xylan. Since all HexA were degraded during ECF bleaching with chlorine dioxide, the final pulp contained a xylan with rather small amount of uronosyls (MeGlcpA residues). A small proportion of MeGlcpA residues (15% from initial amounts), remaining intact during soda pulping, were additionally converted to HexA residues during alkali bleaching stages. After bleaching, the residual acetyl groups were completely removed from the pulp xylan. It was suggested that a low molecular weight fraction of xylan, probably associated to residual lignin, was removed from upon bleaching. Acknowledgements This study has been supported by the Spanish Projects AGL2005-01748 and AGL2008-00709 and the EU BIORENEW project (NMP2-CT-2006-26456). We thank CELESA (Tortosa, Spain) for providing the samples. G.M. thanks the Spanish Ministry of Education for a FPI fellowship
235

5. Resultados y discusión
236
References Bertaud, F., Sundberg, A., & Holmbom, B. (2002). Evaluation of acid
methanolysis for analysis of wood hemicelluloses and pectins. Carbohydrate Polymers, 48, 319-324.
Bleton, J., Mejanelle, P., Sansoulet, J., Goursaud, S., & Tchapla, A. (1996). Characterization of neutral sugars and uronic acids after methanolysis and trimethylsilylation for recognition of plant gums. Journal of Chromatography A, 720, 27-49.
Buchert, J., Bergnor, E., Lindblad, G., Viikari, L., & Ek, M. (1997). Significance of xylan and glucomannan in the brightness reversion of kraft pulps. Tappi Journal, 80(6), 165-170.
Das Gupta P. C., & Mukherjee P. P. (1967) The hemicellulose of sisal fibre (Agave sisalana). Journal of the Chemical Society C: Organic, 1179-1182.
del Río, J. C., Marques, G., Rencoret, J., Martínez, A. T., & Gutiérrez, A. (2007). Occurrence of naturally acetylated lignin units. Journal of Agriculture and Food Chemistry, 55, 5461-5468.
del Río, J. C., Rencoret, J., Marques, G., Gutiérrez, A., Ibarra, D., Santos, J. I., Jiménez-Barbero, J., & Martínez, A. T., (2008) Highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. Journal of Agriculture and Food Chemistry, 56, 9525-9534.
Ebringerová, A., Hromádková, Z., & Heinze, T. (2005). Polysaccharides I. Advances in. Polymer Science, 186, 1-67.
Engström, N., Vikkula, A., Teleman A., & Vuorinen, T. (1995). Structure of hemicellulose in pine kraft cooking liquors. Proceedings of the eighth International Symposium on Wood and Pulping Chemistry, (pp. 195-200), 6-9 June, Helsinki, Finland.
Evtuguin, D. V., Tomás, J. L., Silva, A. M. S., & Pascoal Neto, C. (2003). Characterization of an acetylated heteroxylan from Eucalyptus globulus labill. Carbohydrate Research, 338, 597-604.
Gonçalves, V., Evtuguin D. V., & Domingues, M. R. (2008). Structural characterization of acetylated heteroxylan from the natural hybrid Paulownia elongate/Paulownia fortunei. Carbohydrate Research, 343, 256-266.
Gorshkova, T. A., Wyat, S. E., Salnikov, V. V., Gibeaut, D. M., Ibragimov, M. R., Lozovaya, V. V., & Carpita, N.C. (1996). Cell-wall polysaccharides of developing flax plants. Plant Physiology, 110, 721-729.

5. Resultados y discusión
Gutiérrez, A., Rodríguez, I. M., del Río, J. C. (2008). Chemical composition of lipophilic extractives from sisal (Agave sisalana) fibers. Industrial Crops & Products, 28, 81-87.
Hurter, R. W. (2001). Sisal fibre: market opportunities in the pulp & paper industry. FAO/CFC Technical Paper, 14, 61-74.
Idárraga, G., Ramos, J., Young, R. A., Denes, F., & Zuñiga, V. (2001). Biomechanical pulping of Agave sisalana. Holzforschung, 55, 42-46.
Li, Y., Mai, Y.-W., & Ye, L. (2000). Sisal fibre and its composites: a review of recent developments. Composites Science and Technology, 60, 2037-2055.
Lindström, T. (1992). Chemical factors affecting the behaviour of fibres during papermaking. Nordic Pulp and Paper Research Journal, 4(7), 181-192.
Lisboa, S. A., Evtuguin, D. V., & Pascoal Neto, C. (2004). Isolation and structural characterization of polysaccharides dissolved in Eucalyptus globulus kraft black liquors. Carbohydrate Polymers, 60, 77-85.
Megiatto, J. D., Houreau, Jr. W., Gardrat, C., Frollini, E., Castellan, A. (2007). Sisal fibers: surface chemical modification using reagent obtained from renewable source, characterization of hemicellulose and lignin as model study. Journal of Agriculture and Food Chemistry, 55, 8576-8584.
Mwaikambo, L. Y. (2006). Review of the history, properties and application of plant fibers. African Journal of Science and Technology, 7, 120-133.
Neto, P. C., Seca, A., Fradinho, D., Coimbra, M. A., Domingues, F., Evtuguin, D., Silvestre, A., & Cavaleiro, J. A. S. (1996). Chemical composition and structural features of the macromolecular components of Hibiscus cannabinus grown in Portugal. Industrial Crops and Products, 5, 189-196.
Nobel, P. S. (2003). Environmental Biology of Agaves and Cacti. Cambridge: Cambridge Press.
Pinto, P. C., Evtuguin, D. V., & Pascoal Neto, C. (2005). Structure of hardwood glucuronoxylans: modifications and impact on pulp retention during wood kraft pulping. Carbohydrate Polymers, 60, 489-497.
Rowell R. M., Han J. S., & Rowell, J. S. (2000). Characterization and factors affecting fiber properties. In: Natural Polymers and Agrofibers Composites (Frollini E, Leão AL, Mattoso LHC eds), IQSC/USP, UNESP and Embrapa Instrumentação Agropecuária. São Carlos - Brazil, pp. 115-134.
237

5. Resultados y discusión
238
Selvendran, R. R., March, J. F. & Ring, S. G., (1979). Determination of aldoses and uronic acids content of vegetable fiber. Analytical Biochemistry, 96, 282-292.
Shimizu, K. (1991). Chemistry of hemicelluloses. In: Hon, D. H. -S., Shiraishi, N. (Eds.), Wood and Cellulosic Chemistry. (pp. 3-57), New York: Marcel Dekker.
Stewart, D., Azzini, A. Hall, A. T., & Morrison, I. M. (1997). Sisal fibres and their constituent non-cellulosic polymers. Industrial Crops & Products, 6, 17-26.
Sun, R. C., Tomkinson, J., Ma, P. L., & Liang, S. F. (2000). Comparative study of hemicelluloses from rice straw by alkali and hydrogen peroxide treatments. Carbohydrate Polymers, 42, 111-122.
Sundberg, A., Sundberg, K., Lillandt, C., & Holmbom, B. (1996). Determination of hemicelluloses and pectins in wood and pulp fibers by acid methanolysis and gas chromatography. Nordic Pulp and Paper Research Journal, 4, 216-226.
Tappi Test Methods (1993), Atlanta: TAPPI Press.
Teleman, A., Hajunpää, V., Tenkanen, M., Buchert, J., Hausalo, T., Drakenberg, T., & Vuorinen, T. (1995). Characterization of 4-deoxy-�-L-threo-hex-4-enopyranosyluronic acid attached to xylan in pine kraft pulp and pulping liquor by 1H and 13C NMR spectroscopy. Carbohydrate Research, 272, 55-71.
Teleman, A., Lundqvist, J., Tjerneld, F., Stalbrand, H., & Dahlman, O. (2000). Characterization of acetylated 4-O-methylglucuronoxylan isolated from aspen employing 1H and 13C NMR spectroscopy. Carbohydrate Research 329, 807-815.
Teleman, A.; Tenkanen, M.; Jacobs, A.; & Dahlman, O. (2002). Characterization of O-acetyl-(4-O-methylglucurono)xylan isolated from birch and beech. Carbohydrate Research, 337, 373-377.
Torosantucci, A., Bromuro, C., Chiani, P., De Bernardis, F., Berti, F., Galli, C., Norelli, F., Bellucci, C., Polonelli, L., Costantino, P., Rappuoli, R., & Cassone, A. (2005) A novel glyco-conjugate vaccine against fungal pathogens. The Journal of Experimental Medicine, 202, 597- 606.
Vignon, M. R., & Gey, C. (1997). Isolation, 1H and 13C NMR studies of (4-O-methyl-D-glucurono)-D-xylans from luffa fruit fibers, jute bast fibers and mucilage of quince tree seeds. Carbohydrate Research, 307, 107-111.

5. Resultados y discusión
Vuorinen, T., Fagerström, P., Buchert, J., Tenkanen, M., & Teleman, A. (1999). Selective hydrolysis of hexenuronic acid groups and its application in ECF and TCF bleaching in kraft pulps. Journal of Pulp and Paper Science, 25(5), 155-162.
239

5. Resultados y discusión
Publicación IX: Marques G., Gamelas J. A., Ruiz-Dueñas F.J., del Río J.C., Evtuguin D.V., Martínez A.T. and Gutiérrez A. (2010) Delignification of eucalypt kraft pulp with manganese-substituted polyoxometalate assisted by fungal versatile peroxidase. Bioresource Technology, 101, 5935-5940.
240

5. Resultados y discusión
Delignification of eucalypt kraft pulp with manganese-substituted polyoxometalate assisted by fungal versatile peroxidase
Gisela Marques a, José A.F. Gamelas b,, Francisco J. Ruiz-Dueñas c, José C. del Rio a, Dmitry
V. Evtuguin b, Angel T. Martínez c, Ana Gutiérrez a
aInstituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, PO Box 1052, E-41080
Seville, Spain bUniversity of Aveiro, CICECO, 3810-193, Aveiro, Portugal
c Centro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid Abstract Oxidation of the manganese-substituted polyoxometalate [SiW11MnII(H2O)O39]6- (SiW11MnII) to [SiW11MnIII(H2O)O39]5-, one of the most selective polyoxometalates for the kraft pulp delignification, by versatile peroxidase (VP) was studied. First, SiW11MnII was demonstrated to be quickly oxidized by VP at room temperature in the presence of H2O2 (Km= 6.4±0.7 mM and kcat= 47±2 s-1). Second, the filtrate from eucalypt pulp delignification containing reduced polyoxometalate was treated with VP/H2O2, and 95-100% reoxidation was attained. In this way, it was possible to reuse the liquor from a first SiW11MnIII stage for further delignification, in a sequence constituted by two polyoxometalate stages, and a short intermediate step consisting of the addition of VP/H2O2 to the filtrate for SiW11MnII reoxidation. When the first ClO2 stage of a conventional bleaching sequence was substituted by the two-stage delignification with polyoxometalate (assisted by VP) a 50% saving in ClO2 was obtained for similar mechanical strength of the final pulp. Keywords: Polyoxometalate, versatile peroxidase, oxidative delignification, pulp bleaching, eucalypt kraft pulp 1. Introduction The residual lignin remaining after wood pulping is the target of the bleaching process to produce high quality pulp for the papermaking. This plant polymer, which is responsible for the undesirable dark color and photoyellowing of pulp, must be attacked with minimal polysaccharides damage to preserve the physical properties of the bleached pulp. In the middle of the 1990's, polyoxometalates (POM) were proposed as an environmentally-friendly alternative to the chlorine-based bleaching reagents, as well as to conventional alkaline oxygen pre-bleaching (Evtuguin and Neto, 1997; Weinstock et al., 1997). POM have been evaluated for the bleaching/delignification of pulps both as reagents under anaerobic conditions (in this case a second stage is required for POM reoxidation and reuse) or as catalysts under aerobic conditions (Gamelas et al., 2008; Gaspar et al., 2007;
241

5. Resultados y discusión
242
Weinstock et al., 1997). Applied as catalysts, POM oxidizes the residual lignin in pulp, and the reduced form of POM is reoxidized by molecular oxygen at the same stage. Therefore, it is possible to reuse the POM solutions in a closed loop. The thermodynamic conditions required for lignin oxidation and reoxidation of the POM are related to the corresponding redox potentials as follows: E (Lignin) < E (POM) < E (O2) = 1.22 – 0.059 pH. Several POM types, mostly with the Keggin-type structure (Fig. 1), have been considered for kraft pulp delignification, such as [SiW11VO40]5-, [SiW10V2O40]6-, “SiW10.1Mo1.0V0.9O40”, and [SiW11Mn(H2O)O39]5- (SiW11Mn) (Gamelas et al., 2005a; 2008; Gaspar et al., 2003; 2007; 2009; Weinstock et al., 1997; 2001). However, some of them possessing high M(n+1)/n redox potentials (E = +0.7–0.8 V), although lower than oxygen redox potential, are hardly reoxidized even at extreme conditions of oxygen pressure and temperature (Gamelas et al., 2005a; Gaspar et al., 2003; Weinstock et al., 1997), thus, limiting their reuse for pulp delignification. In particular, SiW11Mn has been found to be highly selective in pulp delignification (Gamelas et al., 2005a; Gaspar et al., 2003). The SiW11Mn/O2 catalytic system has been compared to the conventional alkaline oxygen process already used by the pulp industry. In addition to lignin removal, an important advantage of the SiW11Mn-based process, when applied to eucalypt kraft pulps, is the higher reduction of kappa number than in the alkaline oxygen process, due to the degradation of hexenuronic acids at the low pH used in these reactions (Gamelas et al., 2005a). However, SiW11MnII is very slowly reoxidized under these conditions, limiting its practical application. Fig. 1. Structural representation of the Mn-substituted polyoxometalate, �-[SiW11MnIII(H2O)O39]5-. The dark octahedron represents the MnIIIO5(H2O) group with Mn at the centre of the octahedron. Enzymatic catalysis is a promising approach to regenerate some of the POM species that are difficult to be reoxidized by O2 and other chemical oxidizers. In this context, fungal laccase (from Trametes versicolor) has been assayed for the reoxidation of [SiW11VIVO40]6- and [SiW11MnII(H2O)O39]6- (SiW11MnII)

5. Resultados y discusión
(Gamelas et al., 2005b; Tavares et al., 2004). Although laccase easily oxidized VIV to VV in the former POM, the corresponding oxidation of MnII to MnIII in the manganese-based POM was slow (less that 50% after 4 h at 45 ºC, and under 0.3 bar oxygen pressure) (Gamelas et al., 2005b). This urged the search for alternative faster methods of oxidation of MnII-substituted POM. In contrast to laccase, versatile peroxidase (VP) produced by fungi of the genera Pleurotus and Bjerkandera is a high redox-potential enzyme able to oxidize a variety of substrates, including free MnII, due to the presence of different catalytic sites in its molecular architecture (Ruiz-Dueñas et al., 2009). VP is activated by H2O2 in a two-electron reaction yielding highly reactive intermediate states. Activated VP can oxidize two substrate molecules in two successive one-electron reactions. It has been demonstrated that MnIII, resulting from MnII oxidation by VP or related manganese peroxidase (Ruiz-Dueñas et al., 2007), is stabilized in solution by the chelation of dicarboxylic acids of small size produced by ligninolytic fungi. The manganic cation can, then, act as an oxidizer of lignin contributing to wood delignification in nature (Wariishi et al., 1992). In the present work, reoxidation of the MnII-containing POM, SiW11MnII, by the VP/H2O2 system, was studied for the first time. Based on the easy oxidation of MnII (as a free ion or in POM complexes) by the enzyme a novel approach for the delignification catalysis was developed. Reduced POM in the liquor from a first eucalypt pulp delignification stage was reoxidized by VP, and the resultant liquor mixed with the partially delignified pulp for a further delignification stage in a simple POM-VP-POM trial. In addition, delignification of eucalypt pulp by POM in a VP-assisted process was tested as a pre-bleaching stage to substitute the first Cl2O stage in a conventional elemental chlorine free (ECF) bleaching sequence. 2. Materials and methods 2.1. Pulp samples and POM synthesis The delignification assays were carried out with Eucalyptus globulus unbleached kraft pulp supplied by ENCE pulp mill (Spain). The pulp had a kappa number of 13.7, and an intrinsic viscosity of 1180 cm3/g. For the kinetic studies of SiW11MnII oxidation by VP/H2O2, the potassium salt of the MnII-containing POM, K6[SiW11MnII(H2O)O39].10 H2O, was prepared (Tourné et al., 1970). For the delignification experiments, a solution containing 2.8 � 0.1 mmol/L of [SiW11MnIII(H2O)O39]5- (SiW11MnIII) was prepared as previously reported (Galli et al., 2007). 2.2. VP expression, in vitro activation and purification Recombinant VP was obtained from E. coli W3110 transformed with the pFLAG-VPL2 expression vector as previously described (Pérez-Boada et al.,
243

5. Resultados y discusión
244
2002). The enzyme was activated in vitro after solubilization of inclusion bodies in 8 M urea. The folding conditions included 0.15 M urea, 5 mM Ca2+, 20 μM hemin, a 4:1 oxidized-glutathione/reduced-glutathione ratio and 0.1 mg/mL protein at pH 9.5. The active enzyme was purified in a single chromatographic step (Resource-Q column, Pharmacia-Biotech) using a 0–0.3 M NaCl gradient (2 mL/min) in 10 mM sodium tartrate (pH 5.5) containing 1 mM CaCl2. The concentration of the enzyme was determined by spectrophotometry (�407 150 000 M-1 cm-1) (Ruiz-Dueñas et al., 1999).
2.3. SiW11MnII oxidation by VP Oxidation of the MnII-substituted POM was followed at 20 ºC in a quartz cuvette (1 cm optical path) under stirring: 3.0 mL of 0.1 M acetate solution (pH 4.5) containing 2.7 mM SiW11MnII (K6[SiW11MnII(H2O)O39].10H2O), 0.56-1.26 �M VP and 0.57-2.24 mM H2O2 were mixed inside the cell. The increase of absorbance at 490 nm was followed for 1 min, until a constant value was reached. The oxidation degree was estimated using the molar extinction coefficients of the oxidized and reduced POM (SiW11MnIII �490 325 M-1 cm-1; and SiW11MnII �490 22 M-1 cm-1) (Tourné et al., 1970). For the assays with the delignification liquor, the H2O2 amount varied between 0 and 2.06 mM, with the amount of enzyme kept at 1.20 �M. The total absorbance was corrected for the liquor contribution. Steady-state kinetic constants were calculated during VP oxidation of increasing SiW11MnII concentrations in 0.1 M sodium tartrate, pH 5, containing 0.1 mM H2O2. The enzymatic activity at 20 ºC was measured as the initial velocity, taking linear increments. Mean values and standard errors for the apparent affinity constant (Michaelis constant, Km) and maximal enzyme turnover (catalytic constant, kcat) were obtained by non-linear least-squares fitting of the experimental measurements to the Michaelis-Menten model. Fitting of these constants to the normalized equation v = (kcat/Km)[S]/(1+[S]/Km) yielded the efficiency value (kcat/Km) with its corresponding standard error. 2.4. Pulp delignification experiments Pulp delignification was carried out in a PARR reactor, model 4843 (0.25 L) equipped with an automatic temperature control system, pressure control and mechanical stirring (220 rpm). 7.5g of pulp (dry weight), 67 mL of 0.2 M sodium acetate (pH 4.5), 13 mL of 28 mM POM (SiW11MnIII) solution, and water to make a final volume of 132 mL were put inside the reactor. The final concentration of POM was 2.7 mM. At the end of the reactions, the reactor was quickly cooled with water and degasified. In the two-stage experiments, including intermediate POM reoxidation with VP (POM-VP-POMreox), the pulp from the first stage was filtered and pressed, the required amounts of enzyme and H2O2 were added to the delignification

5. Resultados y discusión
liquor, and the solution was stirred at 20-25 ºC for 10 min. The liquor containing the reoxidized POM (verified by visible absorption spectroscopy) was mixed again with the filtered pulp and a second delignification stage was applied under the same experimental conditions of the first stage. A two-stage experiment not including the reoxidation step of POM by VP/H2O2 was also performed by adding fresh POM (SiW11MnIII), acetate buffer and water to the washed pulp obtained after the first stage. After each delignification sequence the pulps were filtered and washed with water until neutral filtrate. Alkaline extraction of pulps was carried out at 70 ºC during 1 h and NaOH charge of 2% (on the dry pulp weight). 2.5. Modified ECF bleaching sequence Bleaching with Cl2O was performed on untreated kraft pulp and with pulp delignified with POM, at 10% pulp consistency, in plastic bags in a Grant model Y28 thermostatic bath. Two bleaching sequences, D-Ep-D-Ep-D and POM-VP-POMreoxE-D-Ep-D, were studied (D refers to Cl2O stage; Ep to peroxide-reinforced alkaline extraction, POM-VP-POMreox corresponds to VP-assisted two stage [2 h + 2 h] POM treatment; and E to alkaline extraction). The bleaching conditions in the D-Ep-D-Ep-D sequence were as follows: first D stage at 50 ºC for 1 h; second D stage at 70 ºC for 2 h; third D stage at 70 ºC for 2.5 h; first Ep stage at 70 ºC for 1 h, using 2.0% NaOH and 0.2% H2O2; second Ep stage at 70 ºC for 1 h, using 1.5% NaOH and 0.1% H2O2. The pulp delignified with POM (2 h)-VP-POMreox (2 h) and extracted with NaOH was subjected to D-Ep-D bleaching (POM-VP-POMreoxE-D-Ep-D sequence). The conditions of the last stages in this sequence were as follows: first D stage at 50 ºC for 1 h; second D stage at 70 ºC for 2.5 h; Ep stage at 70 ºC for 1 h, using 1.5% NaOH and 0.2% H2O2. The loads of ClO2 for each stage in both sequences are discussed in the text. 2.6. Pulp characterization The treated pulps were characterized using TAPPI T236 cm–99 standard for the kappa number (Tappi, 2006), and the SCAN-CM 15:88 standard for the intrinsic viscosity (Scandinavian Pulp Paper and Board Committee, 1994). Hexenuronic acid content was determined by acid hydrolysis in sodium formate (pH 3.0) followed by spectrophotometric (245 nm) quantitation of the furan derivatives formed (Vuorinen et al., 1999). The acid hydrolysis treatment of pulp was carried out in the same PARR reactor mentioned above (see 2.4). The strength properties, brightness, and opacity of the bleached pulps were determined according to ISO (International Organisation for Standardization Documentation and Information, 2003) and TAPPI (Tappi, 2006) standards.
245

5. Resultados y discusión
246
3. Results and discussion 3.1. Kinetics of SiW11MnII oxidation by VP The ability of VP, a peroxidase acting on free MnII and other substrates, to oxidize this metal ion in Mn-substituted POM is demonstrated here for the first time. The steady-state kinetic constants for SiW11MnII oxidation, obtained by non-linear fitting of initial velocities vs substrate concentration (Fig. 2), revealed high VP turnover on SiW11MnII, with a kcat of 47 ± 2 s-1, and a moderate affinity for this compound, with a Km of 6.4 ± 0.7 mM. This VP activity was lower than on free MnII, with a reported kcat value near 300 s-1, but the main difference between both substrates concerned Km that was around 0.19 mM for free MnII, revealing over 30-fold higher affinity of VP on the free metal ion (Ruiz-Dueñas et al., 2007). As a result, the global catalytic efficiency of VP oxidizing SiW11MnII (7.36 ± 0.6 mM-1 s-1) was around 200-fold lower than that for oxidation of free MnII (1600 ± 100 mM-1 s-1).
0
10
20
30
40
0 5 10 15 20 25 30[SiW11MnII] (mM)
kob
s (s-1
)
K m (6.4 ± 0.7) mMk cat (47.3 ± 2) s-1Efficiency (7.36 ± 0.6) mM-1 s-1
Fig. 2. Michaelis-Menten kinetics of SiW11MnII oxidation by VP. The Km, kcat and efficiency values (means and standard errors) are shown inside the plot.
The high affinity of VP for free MnII is due to the existence of a specific catalytic site in this enzyme constituted by three acidic residues forming a small channel on the internal heme propionate, where the free metal cation is oxidized (Ruiz-Dueñas et al., 2007). The lower efficiency observed for SiW11MnII oxidation by VP was in the order of those reported both for veratryl alcohol oxidation taking place at a tryptophan residue exposed to the solvent (Pérez-Boada et al., 2005), and for oxidation of phenols at the edge of the main heme access channel (Ruiz-Dueñas et al., 2008). This suggested that SiW11MnII could be oxidized in one of these two easily accessible catalytic sites, and not in the narrow channel described for free MnII that most probably provides a limited access to the bulky SiW11MnII. A detailed kinetic study of different VP variants mutated at the three catalytic sites mentioned above would be necessary to definitively identify the SiW11MnII oxidation site in VP.

5. Resultados y discusión
3.2. Optimization of SiW11MnII oxidation by VP (in the presence of H2O2) A set of assays was carried out aiming to optimize the oxidation of SiW11MnII to SiW11MnIII by VP (in the presence of H2O2), either by using an aqueous solution of SiW11MnII buffered at pH 4.5, and the liquor from previous eucalypt kraft pulp delignification with POM (Table 1). The assays were performed at 20 ºC, with 2.7 mM POM concentration, and varying the H2O2/POM and POM/VP molar ratios. During the oxidation of POM by VP (and H2O2) no indication about the formation of other species besides SiW11MnIII was obtained.
Table 1. Oxidation of SiW11MnII by VP/H2O2 in two distinct reaction media using different H2O2, POM and VP molar ratiosa H2O2/POM H2O2/VP POM/VP Oxidation (%) Time (min) A) Oxidation of SiW11MnII in aqueous solution buffered at pH 4.5 0.19 446 2382 40 2 0.40 883 2226 78 2 0.50 1117 2226 95 5 0.61 1351 2226 100 5 0.81 1793 2226 57 22 0.50 2495 5009 55 29 0.50b 2495 5009 94 10 B) Oxidation of SiW11MnII in the filtrate from one-stage delignification with POM 0 0 2227 18 1 0.21 472 2227 57 2 0.40 896 2227 93 6 0.50 1116 2230 97 2 0.59 1321 2227 74 27 0.79 1769 2227 44 18 a The assays were performed at 20 ºC, with 2.6-2.8 mM POM concentration b H2O2 was added in five portions each of them including 20% of the total volume required
In the assays performed with the SiW11MnII solution (Table 1A), the extent
of POM oxidation (for a fixed amount of enzyme) increased with the H2O2/POM ratio until a 0.5-0.6 molar ratio, and then decreased at higher ratios. Using this H2O2/POM ratio (0.5-0.6), 95-100% POM oxidation was obtained in less than 5 min, with a POM/VP ratio ~2200. These values were in agreement with the stoichiometry of the global enzymatic reaction, which predicts that 0.5 moles of H2O2 will be needed to oxidize 1 mole of SiW11MnII. For the H2O2/POM ratio of 0.8, only 57% oxidation of SiW11MnII was obtained, indicating enzyme inactivation by the excess of H2O2 (Valderrama et al., 2002). If the amount of enzyme was reduced to about 50%, keeping the H2O2/POM ratio of 0.5, the oxidation extent also decreased (to only 55%) due to the increased H2O2/VP ratio. However, when the later assay was carried out by adding the H2O2 in several steps, without exceeding a 500-fold molar excess of H2O2 in each addition, the extent of oxidation (94%) was similar to that attained using a higher amount of enzyme. These data confirmed VP inactivation by H2O2 (even
247

5. Resultados y discusión
248
in the presence of enough amount of SiW11MnII to consume all the H2O2) and showed that the enzyme dose can be reduced by stepwise addition of H2O2 (to prevent VP inactivation). No POM oxidation was observed in the absence of H2O2 or without enzyme. In the assays with the liquor from POM delignification of eucalypt kraft pulp (Table 1B), which were performed using a POM/VP ratio ~2200, and varying the H2O2/POM ratio between 0 and 1, the highest POM oxidation degrees (over 90%) were obtained at the H2O2/POM ratios of 0.4-0.5. In the absence of H2O2, some POM oxidation occurred suggesting that some substances present in the delignification liquor may act as the enzyme oxidizing agents. In fact, for all the assays performed with a H2O2/POM ratio up to 0.5, the oxidation of POM was higher when the reactions were conducted in the delignification liquor than those in the SiW11MnII aqueous solutions. Again, the use of H2O2/POM ratios � 0.6 did not improve POM reoxidation in the delignification liquor, and lower rates were obtained. It was concluded that a H2O2/POM ratio around 0.5 and a POM/VP ratio of 2000-3000 should be used to obtain near complete oxidation of the manganese-substituted POM. 3.3. Two-stage POM delignification of pulp assisted by VP
As a continuation of the above studies, which showed easy oxidation of SiW11MnII by VP/H2O2, a novel approach for the paper pulp delignification was developed. A first delignification stage using chemically-prepared SiW11MnIII and O2 was followed by pulp filtration, and a short intermediate step consisting of the addition of VP and H2O2 to the filtrate. Fig. 3 shows the visible absorption spectra of the filtrate before and after the enzymatic treatment (resulting in the complete reoxidation of the previously reduced POM). Then, the filtrate with reoxidized POM was mixed again with the pulp, and a second delignification stage (under the same conditions of the first one) was applied. The results were compared with those obtained when the second delignification stage was performed by adding chemically-prepared SiW11MnIII, as well as when only one-stage POM delignification was performed (Table 2).
After one-stage POM delignification at 110 ºC, decreases of kappa number (a rough measure of the lignin content in pulp) of 40% and 50%, with viscosity losses of only 3% and 6%, were obtained after 1-h and 2-h reaction, respectively (Table 2). Besides residual lignin, hexenuronic acids contribute significantly to the kappa number in E. globulus kraft pulps and to the consumption of bleaching reagents (Furtado et al., 2001). In fact, a significant removal of hexenuronic acids (up to 70% after 2 h) was detected after the POM treatment. It is noteworthy that the POM/O2 system was highly selective for delignification when compared with the oxygen-delignification control, which showed a viscosity loss of 28% (near 10-fold higher than that obtained with POM delignification) (Gamelas et al., 2005a).

5. Resultados y discusión
0
0,5
1
1,5
300 400 500 600 700 800
Wavelength (nm)
Abs
orba
nce
a
b
Fig. 3. UV-vis spectra of the filtrate after 2-h treatment of eucalypt kraft pulp with POM/O2 (a), and after short incubation of this delignification liquor with VP and H2O2 at 20-25ºC (b), revealing the typical SiW11MnII and SiW11MnIII spectra, respectively.
After two-stage (2-h each) POM delignification including the intermediate reoxidation step with VP and H2O2, kappa number was reduced of 62% and the viscosity had a drop of 11% (Table 2). Interestingly, this treatment also degraded almost 90% of the hexenuronic acids present in the pulp. The delignification degree corrected for the hexenuronic acids content was of 51%. Similar results in terms of pulp kappa number, viscosity and hexenuronic acid degradation were obtained in parallel assays in which freshly-prepared SiW11MnIII was added after the first POM stage, revealing that the presence of the enzyme did not exert a negative effect on the performance and selectivity of the SiW11MnIII/O2 system.
Table 2. Delignification of eucalypt kraft pulp with SiW11MnIII/O2 assisted by VP/H2O2 (effect of different treatments on pulp kappa number, intrinsic viscosity and hexenuronic acid content (HexA))a
Kappa number
Viscosity (cm3/g)
Kappa decrease (%)d
Viscosity loss (%)
HexA (mmol/kg)
Initial kraft pulp 13.6 1215 - - 61.2 O2 (without POM, 2 h) 7.3 875 46 (33) 28 15.7 POM (1 h) 8.2 1180 40 (33) 3 28.5 POM (2 h) 6.8 1140 50 (40) 6 18.4 POM (1 h)-VP-(1 h)b 6.5 1130 52 (42) 7 16.8 POM (2 h)-VP-(2 h)b 5.2 1080 62 (51) 11 9.3 POM (2 h)-POM (2 h)c 5.2 1085 62 (50) 11 8.2 a Pulp consistency of 5.3%; 2.7 mM POM; pH 4.5; pO2 of 0.5 MPa; 110 ºC; and 220 rpm b The pulp after the first stage was filtered, and the POM in the filtrate reoxidized by VP/H2O2. c The pulp after the first stage was washed, and fresh POM (SiW11MnIII) was added d Kappa number reduction corrected for HexA (kappacor= kappa - 0.073 [HexA]) in parentheses
249

5. Resultados y discusión
250
3.4. Modified ECF bleaching including a VP-assisted POM stage Pulp treatment with the above VP-assisted two-stage (2-h each) POM delignification followed by an alkaline extraction (POM-VP-POMreox-E) was investigated to substitute the first Cl2O stage in a conventional D-Ep-D-Ep-D ECF bleaching sequence for eucalypt kraft pulp. Results from the conventional D-Ep-D-Ep-D bleaching sequence (see Materials and methods) and the sequence including VP-assisted two-stage POM delignification, (POM-VP-POMreoxE-D-Ep-D), were compared in terms of Cl2O savings for the same final brightness (~89% ISO). Pulp bleached by the sequence including VP and POM showed a Cl2O consumption 50% lower than the conventional ECF sequence (Table 3). The Cl2O oxidation equivalents (OXE) per kappa number unit in the modified sequence were higher than in the conventional sequence. Moreover, the main strength properties of the unbeaten pulps after the two bleaching sequences were very similar (Table 4). The results obtained suggest that VP-assisted continuous reutilization of SiW11MnIII in a two-reactor system (Gamelas et al., 2007) may be implemented in future industrial ECF sequences, with no apparent deterioration of the pulp strength properties, while significantly reducing the Cl2O consumption, and consequently lowering the environmental impact of the bleaching process.
Table 3. Cl2O consumption and oxidation equivalents (OXE) for eucalypt pulp bleaching in a conventional ECF sequence and after substituting the first D-stage by VP-assisted two-stage POM delignification (89% ISO final brightness)
D-Ep-D-Ep-D POM-VP-POMreoxE-D-Ep-D
ClO2 consumption a 25 + 9 + 6 15 + 5 OXE b 90 134 a As active chlorine in each D stage (kg/ton) b As moles of active chlorine per ton of dry pulp and kappa unit
4. Conclusions In this work, we demonstrate that the reduced Mn-substituted POM, SiW11MnII, can be oxidized by VP (in the presence of H2O2) following Michaelis-Menten kinetics. This POM, whose oxidized form is highly selective for delignification, was fully oxidized by VP/H2O2 at 20-25 ºC in less than 10 min. In this way, a two-stage POM delignification process, including a fast intermediate step consisting of the addition of VP (and H2O2) to the delignification filtrate for POM reoxidation, was performed, resulting in 62% reduction of the pulp kappa number and a viscosity loss of only 10%. The substitution of the first Cl2O stage by a POM-VP-POMreox treatment in a conventional ECF bleaching sequence allowed 50% Cl2O saving without decreasing the pulp strength properties.

5. Resultados y discusión
Hence, the continuous reutilization of SiW11MnIII in a two-reactor system may be implemented in the future.
Table 4. Physical properties of unbeaten bleached pulps (89% ISO, and 65 g/m2) from a conventional ECF sequence and after substituting the first Cl2O stage by VP-assisted two-stage POM delignification D-Ep-D-Ep-D POM-VP-POMreox-E-D-Ep-D Beating degree (ºSR) 19 20 Bulk density (g/cm3) 0.56 0.57 Burst index (kPa.m2/g) 1.39 1.45 Tensile strength (N.m/g) 30.4 28.9 Tear index (mN.m2/g) 5.2 6.0 Elongation (%) 2.2 2.1 Stiffness (kN/m) 409 396 Opacity (%) 75.8 77.2 Internal bonds (Scott test, J/m2) 106 114 Air resistance (Gurley-100 mL, s) 0.8 0.8 Acknowledgements This study has been supported by the EU project BIORENEW (NMP2-CT-2006-026456) and the Spanish projects AGL2008-00709 and BIO2008-01533. We thank ENCE (Pontevedra, Spain) for pulp samples. G. M. and F. J. R.-D. thank the Spanish MICINN for a FPI fellowship and a Ramon y Cajal contract, respectively. References Evtuguin, D.V., Neto, C.P., 1997. New polyoxometalate promoted method of
oxygen delignification. Holzforschung 51, 338-342.
Furtado, F.P., Evtuguin, D.V., Gomes, T.M., 2001. Effect of the acid stage in ECF bleaching on Eucalyptus globulus kraft pulp bleachability and strength. Pulp Paper Can. 102, 89-92.
Galli, C., Gentili, P., Pontes, A.S.N., Gamelas, J.A.F., Evtuguin, D.V., 2007. Oxidation of phenols employing polyoxometalates as biomimetic models of the activity of phenoloxidase enzymes. New J. Chem. 31, 1461-1467.
Gamelas, J.A.F., Evtuguin, D.V., Gaspar, A.R., 2008. Transition metal complexes in the delignification catalysis. In: Varga, B., Kis, L. (Eds.), Transition metal chemistry: New research. Nova Science Publishers, Inc., New York, pp. 15-57.
251

5. Resultados y discusión
252
Gamelas, J.A.F., Gaspar, A.R., Evtuguin, D.V., Neto, C.P., 2005a. Transition metal substituted polyoxotungstates for the oxygen delignification of kraft pulp. Appl. Cat. A:General 295, 134-141.
Gamelas, J.A.F., Pontes, A.S.N., Evtuguin, D.V., Xavier, A.M.R.B., Esculcas, A.P., 2007. New polyoxometalate-laccase integrated system for kraft pulp delignification. Biochem. Eng. J. 33, 141-147.
Gamelas, J.A.F., Tavares, A.P.M., Evtuguin, D.V., Xavier, A.M.B., 2005b. Oxygen bleaching of kraft pulp with polyoxometalates and laccase applying a novel multi-stage process. J. Mol. Catal. B Enzym. 33, 57-64.
Gaspar, A., Evtuguin, D.V., Neto, C.P., 2003. Oxygen bleaching of kraft pulp catalysed by Mn(III)-substituted polyoxometalates. Appl. Cat. A:General 239, 157-168.
Gaspar, A.R., Gamelas, J.A.F., Evtuguin, D.V., Neto, C.P., 2007. Alternatives for lignocellulosic pulp delignification using polyoxometalates and oxygen: a review. Green Chem. 9, 717-730.
Gaspar, A.R., Gamelas, J.A.F., Evtuguin, D.V., Neto, C.P., 2009. Polyoxometalate-catalyzed oxygen delignification process: kinetic studies, delignification sequences and reuse of HPA-5-MnII aqueous solution. Chem. Eng. Commun. 196, 801-811.
International Organisation for Standardization Documentation and Information, 2003. ISO Standards Collection on CD-ROM. Paper, board and pulps. ISO, Geneva.
Pérez-Boada, M., Doyle, W.A., Ruiz-Dueñas, F.J., Martínez, M.J., Martínez, A.T., Smith, A.T., 2002. Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimisation of in vitro folding. Enzyme Microb. Technol. 30, 518-524.
Pérez-Boada, M., Ruiz-Dueñas, F.J., Pogni, R., Basosi, R., Choinowski, T., Martínez, M.J., Piontek, K., Martínez, A.T., 2005. Versatile peroxidase oxidation of high redox potential aromatic compounds: Site-directed mutagenesis, spectroscopic and crystallographic investigations of three long-range electron transfer pathways. J. Mol. Biol. 354, 385-402.
Ruiz-Dueñas, F.J., Martínez, M.J., Martínez, A.T., 1999. Molecular characterization of a novel peroxidase isolated from the ligninolytic fungus Pleurotus eryngii. Mol. Microbiol. 31, 223-236.
Ruiz-Dueñas, F.J., Morales, M., García, E., Miki, Y., Martínez, M.J., Martínez, A.T., 2009. Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J. Exp. Bot. 60, 441-452.

5. Resultados y discusión
Ruiz-Dueñas, F.J., Morales, M., Pérez-Boada, M., Choinowski, T., Martínez, M.J., Piontek, K., Martínez, A.T., 2007. Manganese oxidation site in Pleurotus eryngii versatile peroxidase: A site-directed mutagenesis, kinetic and crystallographic study. Biochemistry 46, 66-77.
Ruiz-Dueñas, F.J., Morales, M., Rencoret, J., Gutiérrez, A., del Río, J.C., Martínez, M.J., Martínez, A.T., 2008. Improved peroxidases. Patent (Spain) P200801292.
Scandinavian Pulp Paper and Board Committee, 1994. SCAN Test Methods. Sweden.
Tappi, 2006. 2006-2007 TAPPI Test Methods. TAPPI Press, Norcoss, GA 30092, USA.
Tavares, A.P.M., Gamelas, J.A.F., Gaspar, A.R., Evtuguin, D.V., Xavier, A.M.R.B., 2004. A novel approach for the oxidative catalysis employing polyoxometalate-laccase system: application to the oxygen bleaching of kraft pulp. Catal. Commun. 5, 485-489.
Tourné, C.M., Tourné, G.F., Malik, S.A., Weakley, T.J.R., 1970. Triheteropolyanions containing copper(II), manganese(II), or manganese(III). J. Inorg. Nucl. Chem. 32, 3875-3890.
Valderrama, B., Ayala, M., Vázquez-Duhalt, R., 2002. Suicide inactivation of peroxidases and the challenge of engineering more robust enzymes. Chem. Biol. 9, 555-565.
Vuorinen, T., Fagerstrom, P., Buchert, J., Tenkanen, M., Teleman, A., 1999. Selective hydrolysis of hexenuronic acid groups and its application in ECF and TCF bleaching of kraft pulps. J. Pulp Paper Sci. 25, 155-162.
Wariishi, H., Valli, K., Gold, M.H., 1992. Manganese(II) oxidation by manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. Kinetic mechanism and role of chelators. J. Biol. Chem. 267, 23688-23695.
Weinstock, I.A., Atalla, R.H., Reiner, R.S., Moen, M.A., Hammel, K.E., Houtman, C.J., Hill, C.L., Harrup, M.K., 1997. A new environmentally benign technology for transforming wood pulp into paper. Engineering polyoxometalates as catalysts for multiple processes. J. Mol. Catal. A Chem. 116, 59-84.
Weinstock, I.A., Barbuzzi, E.M.G., Wemple, M.W., Cowan, J.J., Reiner, R.S., Sonnen, D.M., Heintz, R.A., Bond, J.S., Hill, C.L., 2001. Equilibrating metal-oxide cluster ensembles for oxidation reactions using oxygen in water. Nature 414, 191-195.
253

5. Resultados y discusión
Publicación X:
Marques, G., Molina, S., Babot, E.D., Lund, H., del Río, J.C. y Gutiérrez, A. Exploring the potential of a fungal manganese-containing for pitch control and pulp delignification. Bioresource Technology (in preparation).
254

5. Resultados y discusión
Exploring the potential of a fungal manganese-containing for pitch control and pulp delignification
Gisela Marquesa, Setefilla Molinaa, Esteban D. Babota, Henrik Lundb, José C. del Ríoa, Ana
Gutiérreza
aInstituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, PO Box 1052, E-41080, Seville, Spain
bNovozymes A/S, Krogshoejvej 36, DK-2880 Bagsvaerd, Denmark
Abstract The potential of the lipoxygenase from Gaeumannomyces graminis to remove lipophilic extractives from eucalypt and flax pulps is explored. Pulp treatments with the lipoxygenase were performed in the presence and absence of linoleic acid, and with and without a subsequent hydrogen peroxide stage. The highest removal of lipophilic extractives from eucalypt pulp, including conjugated sterols (about 40% removal), and free sterols (up to 10% removal), was attained in the lipoxygenase treatment with linoleic acid followed by the peroxide stage. Different degradation patterns were observed among the lipophilic compounds in flax pulp with the lipoxygenase treatment, although a high removal (from 55% to 70%) of all extractives classes, including alkanes, fatty alcohols, and free and glycosylated sterols, was attained in most cases. Reactions of the lipoxygenase with model lipid mixtures were carried out to better understand the degradation patterns observed in pulps. Pulp delignification by the lipoxygenase treatments was also evaluated. Keywords: Lipoxygenase, Gäumannomyces graminis, Fungal enzymes, Paper pulps, Pitch deposits, Lignin removal. 1. Introduction The non-polar extractable fraction from wood and nonwood fibers, commonly referred to as lipophilic extractives, includes fatty and resin acids, fatty alcohols, alkanes, sterols, sterol esters and triglycerides. These lipophilic compounds are the precursors of the so-called pitch deposits within the pulp and paper manufacturing processes (Back and Allen 2000). Pitch deposition results in low quality pulp, and can cause the shutdown of pulp mill operations. Specific issues related to pulps with high extractives content include runnability problems, spots and holes in the paper, and sheet breaks. In addition to physicochemical methods, biological methods including both enzymes and microorganisms (Gutiérrez et al. 2001a; 2009), have been investigated to control pitch problems in the pulp and paper industry. Lipases, which hydrolyze triglycerides, are applied with success in softwood (mainly pine) mechanical pulping at mill scale (Fujita et al. 1992) and find widespread
255

5. Resultados y discusión
256
use nowadays. However, pitch problems in most of the chemical and mechanical processes using other raw materials have not yet been solved. Other compounds, such as free and esterified sterols, resin acids, fatty alcohols and alkanes, are responsible for pitch problems in these processes (del Río et al. 1999; 2000; Gutiérrez and del Río 2005b). In addition to lipases, the use of sterol esterases has also been suggested (Barfoed 2000; Calero-Rueda et al. 2004; Kontkanen et al. 2004) because sterol esters are often at the origin of pitch deposits owing to their high tackiness and resistance to kraft cooking. However, free sterols released by the action of these esterases are as problematic as sterol esters. On the other hand, the modification of some lipophilic extractives by the use of laccases has been reported (Buchert et al. 2002; Molina et al. 2008; Zhang et al. 2000; 2005). In contrast to lipases and sterol esterases, laccases are oxidative enzymes whose action is directed toward some aromatic compounds (such as phenols and anilines), although their reactivity with some unsaturated lipids was demonstrated. The interest on laccases as industrial biocatalysts has increased after discovering the effect of some synthetic compounds (Bourbonnais and Paice 1990; Call 1994) expanding the action of laccases to non-phenolic aromatics and, therefore, increasing their potential in the degradation of lignin and other aromatic compounds (Riva 2006; Rodríguez-Couto and Toca 2006; Widsten and Kandelbauer 2008). Moreover, the use of laccases in the presence of redox mediators has recently been described for the removal of the lipophilic extractives responsible for pitch deposition from wood and nonwood paper pulps (Gutiérrez et al. 2006a; 2006b; 2007; Molina et al. 2008). In addition to laccases, other oxidative enzymes, such as soybean lipoxygenases have been tested for the degradation of extractives in softwood thermo-mechanical pulp (Zhang et al. 2007). Earlier work had also suggested the use of lipoxygenases to reduce model wood “pitch” mixtures in pulp and paper (Borch et al. 2003). Lipoxygenases are non-heme iron-containing dioxygenases which catalyze the oxidation of polyunsaturated fatty acids to hydroperoxides. Despite extensive studies on the properties, genetic information and physiological functions of this group of enzymes, there is a lack of utilization of these natural abundant enzymes in industrial processing. The specific activity of lipoxygenases to degrade linoleic acid leads to a potential application in papermaking processes to degrade wood extractives that have adverse effects on pulp and paper properties. In the present work, we study the capability of the lipoxygenase from the fungus Gaeumannomyces graminis, to remove lipophilic extractives from hardwood (eucalypt) and nonwoody (flax) pulps. This enzyme is a manganese-containing lipoxygenase with several exceptional features different from other lipoxygenases (Hamberg et al. 1998; Su and Oliw 1998). It has a broad pH range (being active between pH 5 and 11), and good heat stability (being active at temperatures up to 60ºC) (Su and Oliw 1998) which confer great potential of use under mill process conditions. Since the oxidation of polyunsaturated fatty acids by lipoxygenase leads to the

5. Resultados y discusión
formation of reactive fatty acid hydroperoxides and lipid radicals (Prigge et al. 1997) that can co-oxidize lignin in addition to lipids (Kapich et al. 2010), delignification of this pulp mediated by lipoxygenase treatment in the presence of linoleic acid was also investigated. 2. Materials and methods 2.1. Pulp samples Unbleached kraft pulp from eucalypt (Eucalyptus globulus) wood, with 44% ISO brightness and 13.5 kappa number was obtained from ENCE (Pontevedra, Spain). Unbleached soda/anthraquinone (AQ) pulp from flax (Linum usitatissimum) with 34% ISO brightness and 11 kappa number was provided by CELESA (Tortosa, Spain). 2.2. Model lipids Linoleic acid, cholesteryl linoleate, nonacosane and octacosanol (from Sigma-Aldrich) and sitosterol (from Calbiochem) were used. 2.3. Lipoxygenase The lipoxygenase (GLOX) used was provided by Novozymes (Bagsvaerd, Denmark) and obtained from the fungus G. graminis. GLOX activity was 130000 units per mg, where one unit will cause an absorbance increase at 234 nm of 0.001 units per min, at pH 7.0 and 30ºC, when linoleic acid is used as substrate in a reaction volume of 1.0 ml (light path 1 cm). 2.4. Enzymatic treatments of paper pulps Pulp treatments with GLOX (10 mg/g eucalypt pulp and 20 mg/g flax pulp) were carried out using 5 g pulp (dry weight) at 1% consistency (w:w) in 100 mM monosodium phosphate (pH 7), 30ºC, with oxygen bubbling, in a thermostatic shaker at 170 rev/min for 4 h. Pulp treatments were performed in the presence and absence of linoleic acid (0.1 mg/g pulp). In a subsequent step, pulps at 5% consistency (w:w) were submitted to a bleaching stage using 3% (w:w) H2O2 and 1.5% (w:w) NaOH, both referred to pulp dry weight, at 90ºC for 2 h. Controls without GLOX were performed. Treated pulp samples were Soxhlet extracted with acetone (8 h) and the extracts were evaporated to dryness and redissolved in chloroform for gas chromatography/mass spectrometry (GC-MS) and GC analyses. When required, bis(trimethylsilyl)trifluoroacetamide (BSTFA, from Supelco) in the presence of pyridine was used to prepare trimethylsilyl derivatives.
257

5. Resultados y discusión
258
2.5. Enzymatic reactions with model lipids Enzymatic reactions with mixtures of model compounds listed in section 2.2 (1 mg) were performed using GLOX (0.1 mg/mg lipid), and Tween 20 as dispersant (1% v/v) in 100 mM monosodium phosphate (pH 7), 30ºC, with oxygen bubbling, in a thermostatic shaker at 100 rev/min for 2h. In a subsequent step, the model mixtures after the enzymatic reaction were submitted to a hydrogen peroxide stage, adding 50 μl H2O2 at 30% (w:w) and 37.5 μl 5 N NaOH to each reaction flask, at 90ºC and 100 rev/min for 2 h. Controls without GLOX were performed. After the enzymatic treatments, the lipid dispersions were immediately evaporated, and the reaction products recovered with chloroform: methanol (1:1), dried and redissolved in chloroform for GC and GC-MS analyses. When required, BSTFA in the presence of pyridine was used to prepare trimethylsilyl derivatives. 2.6. GC and GC-MS analyses of lipids The GC analyses of lipids from both the enzymatic treatments of pulps and enzymatic reactions with model compounds were performed in an Agilent 6890N Network GC system using a short fused-silica DB-5HT capillary column (5 m x 0.25 mm internal diameter, 0.1 �m film thickness) from J&W Scientific, enabling simultaneous elution of the different lipid classes (Gutiérrez et al. 1998). The temperature program was started at 100°C with 1 min hold, and then raised to 350°C at 15°C/min, and held for 3 min. The injector and flame-ionization detector (FID) temperatures were set at 300°C and 350°C, respectively. Helium (5 ml/min) was used as carrier gas, and the injection was performed in splitless mode. Peaks were quantified by area, and data from replicates were averaged. In all cases the standard deviations were below 7% of the mean values. The GC-MS analyses were performed with a Varian 3800 chromatograph coupled to an ion-trap detector (Varian 4000) using a medium-length (12 m) capillary column of the same characteristics described above for GC/FID. The oven was heated from 120°C (1 min) to 380°C at 10°C/min, and held for 5 min. In all GC/MS analyses, the transfer line was kept at 300°C, the injector was programmed from 120°C (0.1 min) to 380°C at 200°C/min and held until the end of the analysis, and helium was used as carrier gas at a rate of 2 ml/min. Compounds were identified by mass fragmentography, and by comparing their mass spectra with those of the Wiley and NIST libraries and standards. 2.7. Pulp evaluation Pulp brightness, kappa number and intrinsic viscosity were analyzed following ISO 3688:1999, ISO 302:1981 and ISO 5351/1:1981 standard methods,

5. Resultados y discusión
respectively (International Organisation for Standardization Documentation and Information (ISO) 2003). 3. Results and discussion 3.1. Composition of lipophilic extractives in eucalypt and flax pulps The lipophilic extractives from eucalypt and flax pulps were analyzed by GC and GC-MS (Fig. 1). The compounds from unbleached eucalypt kraft pulp (Fig.1a) include sterols (predominantly sitosterol) in free (peak 8) and conjugated form, both as glycosides (peak 10) and esters (peak 11), as well as fatty acids, mainly palmitic acid (peak 1). The detailed composition of lipophilic extractives from eucalypt pulp has been published elsewhere (Gutiérrez et al. 2001b; Gutiérrez and del Río 2001). Among these compounds, free and esterified sterols have been reported to be the main responsible for pitch deposition during the manufacturing of eucalypt pulp (del Río et al. 1998; 1999; 2000). On the other hand, fatty alcohols including hexacosanol (peak 6), octacosanol (peak 7), and triacontanol (peak 9), and free sterols with sitosterol predominating (peak 8) and fatty acids, predominantly palmitic acid (peak 1), linoleic acid (peak 2), oleic acid (peak 3) and stearic acid (peak 4) are the main lipophilic extractives identified in flax pulp (Fig.1b). Minor amounts of alkanes such as nonacosane (peak 5) and sterol glycosides (peak 10) were also present. The detailed characterization of lipophilic extractives from flax pulp has already been reported (Gutiérrez and del Río 2003a; 2003b; Marques et al. 2010). Fatty alcohols and alkanes have been shown as the main responsible for pitch problems during manufacturing of nonwoody soda pulps (Gutiérrez and del Río 2005a). From the analysis of pulp extractives it would not be expected to see a significant change in their total content after treatment with GLOX given that the amount of extractives with the 1-cis, 4-cis-pentadiene (i.e. linoleic acid) is quite scarce. Still it was chosen to pursue testing of GLOX on both pulps and model substrates since other studies (Gutiérrez et al. 1999) had shown the presence of linoleic acid esterified with sitosterol as the main sterol ester among eucalypt extractives and, at the same time, other studies had surprisingly shown an ability of lipoxygenase to significantly impact other components in unbleached eucalypt kraft pulp (Borch et al. 2003). 3.2. Removal of pulp lipophilic extractives by lipoxygenase treatment The eucalypt and flax pulps were treated with the lipoxygenase from G. graminis (GLOX) to evaluate the potential of this enzyme to remove lipophilic extractives from these pulps. Additional assays adding linoleic acid to the enzymatic reactions were also performed to study the mediating effect of peroxidation products on the lipid removal. After the enzymatic treatments, the pulps were subsequently submitted to a hydrogen peroxide bleaching stage,
259

5. Resultados y discusión
260
which is a common stage in the bleaching of these alkaline pulps. The composition of the lipophilic extractives in pulps after the treatment with GLOX was analyzed by GC and GC-MS and compared with that of control pulp treated under the same conditions but without enzyme addition.
(a)
2 4 6 8 10 12 14 16 18 20
100%
Rel
ativ
e res
pons
e
Retention time (min)
1110
8
1
(b)
2 4 6 8 10 12 14 16 18 20
100%
Rel
ativ
e res
pons
e
Retention time (min)
9
86
1
5
10
7
2+3
4
2+3
4
Figure 1. GC chromatograms of silylated lipid extract from eucalypt kraft (a), and flax soda (b) pulps. Peak identification: 1, palmitic acid; 2, oleic acid; 3, linoleic acid; 4, stearic acid; 5, nonacosane; 6, hexacosanol; 7, octacosanol; 8, sitosterol; 9, triacontanol; 10, sterol glycosides; and 11, sterol esters 3.2.1. Eucalypt pulp treatments Table 1 shows the percentage of degradation of the main eucalypt pulp extractives after the enzymatic treatment with GLOX. GLOX treatment produced a removal of 22% and 20% of the sterol esters and sterol glycosides, respectively, but the amount of free sterols remained unchanged. A similar lack of degradation of free sterols was also observed after TMP pulp treatment with soybean lipoxygenase (Zhang et al. 2007).

5. Resultados y discusión
Table 1. Removal (percentage of reduction) of the main lipophilic extractives from eucalypt pulp after treatment with lipoxygenase (GLOX) in the absence and presence of linoleic acid (LA), without and with a subsequent H2O2 stage (P). Without H2O2 With H2O2 GLOX GLOX/LA GLOX-P GLOX/LA-P Free sterols 0 0 0 7 Sterol glycosides 22 24 20 39 Sterol esters 20 29 27 40
Accordingly it appears that the effect of the lipoxygenase treatment can extend well beyond the primary substrates of the enzyme via the fatty acid hydroperoxides. When the GLOX treatments were performed in the presence of linoleic acid, a higher decrease of sterol esters and glycosides (up to 24% and 29%, respectively) was observed but the content of free sterols was not modified. On the other hand, in the GLOX treatments followed by a hydrogen peroxide stage, the addition of linoleic acid to the enzymatic reaction caused a higher removal of sterol esters (from 20% to 39%) and sterol glycosides (from 27% to 40%). A slight decrease of free sterols (from 0 to 7%) was also observed. To get further insight into these enzymatic reactions, GLOX reactions with model compounds (Fig. 2) representative for the main lipophilic extractives in eucalypt pulp, including free and esterified sterols (sitosterol and cholesteryl linoleate, respectively), and free fatty acids (linoleic acid) were carried out. The reactivity of the different lipids was studied by GC and GC-MS. The content of cholesteryl linoleate decreased about 20% while the content of free sitosterol remained unchanged by the GLOX treatment. In addition, linoleic acid was completely degraded, as expected. When the GLOX reactions were performed with a mixture of the three model compounds, reductions of 83% and 25% of cholesteryl linoleate and sitosterol, respectively, were observed in the reactions of lipoxygenase, suggesting that peroxidation reactions could mediate the co-oxidation of these model lipids. The co-oxidation of sitosterol observed in the reaction of GLOX with the model mixture was not observed in the eucalypt pulp treatment. One potential reason for the limited effect of the treatment on the free sterols may be the predominant localization of these compounds inside the pulp elements (Speranza et al. 2002). In the reaction of lipoxygenase with mixtures of the three model lipids, oxidative derivatives of cholesteryl linoleate and free sitosterol were observed, and these were more evident after a hydrogen peroxide stage (Fig. 3). The chemical structures of the oxidation products identified are shown in Fig. 4. The reaction products of cholesteryl linoleate were, as mentioned above, cholesta-3,5-dien-7-one (peak 2) and the cholesteryl ester core aldehyde (peak 6). Oxidized derivatives of free sitosterol, namely 7�-hydroxysitosterol (peak 4)
261

5. Resultados y discusión
262
(and traces of 7�-hydroxysitosterol) and 7-ketositosterol (peak 5) can also be observed in Fig. 3. These oxidized derivatives were also identified in the enzymatic reactions of these model lipids with laccase in the presence of 1-hydroxybenzotriazole (HBT) as redox mediator (Molina et al. 2008).
OH
O
OH
O
HOHO
O
O
O
O
OO
O
a b
c
d eOHOH
Figure 2. Chemical structures of the model compounds representative for main paper pulp lipophilic extractives used in the enzymatic reactions: (a) linoleic acid; (b) sitosterol; (c) cholesteryl linoleate; (d) nonacosane and (e) octacosanol.
3.2.2. Flax pulp treatments The percentage of removal of lipophilic extractives from flax pulp by the GLOX treatment is shown in Table 2. All lipophilic extractives from flax pulp decreased significantly in the several GLOX treatments. Surprisingly, the content of free sterols in flax pulp decreased up to 55% after the GLOX treatments, contrasting with the lack of removal in eucalypt pulp where no effect on free sterols was observed. A high removal of alkanes (up to 48%), fatty alcohols (up 60%) and sterol glycosides (up to 65%) was also observed. The sterols present in flax is likely distributed throughout the surface layers of the fibers alongside the other lipophilic components and may be partially co-solubilized with them, following the reaction with the lipoxygenase on the cis-cis-pentadiene structures present in flax pulp. The addition of linoleic acid to these enzymatic reactions caused different effects upon the different lipophilic compounds. A higher removal of alkanes and sterol glycosides was observed by the addition of linoleic acid whereas the contrary happened with fatty alcohols and free sterols. In the GLOX treatments of flax pulp followed by a hydrogen peroxide stage, the addition of linoleic caused an increase in the removal of all the lipophilic extractives.

5. Resultados y discusión
Retention time (minutes)
100%
Retention time (minutes)
Rel
ativ
e res
pons
e
6 104 8 12 14 16
45
67
100%
Rel
ativ
e res
pons
e
6 104 8 12 14
1 3
7
2
3
(a)
(b)
16
Figure 3. Behaviour of a mixture of different model compounds (linoleic acid, sitosterol, cholesteryl linoleate) representative for eucalypt pulp lipophilic extractives after enzymatic treatment with lipoxygenase. Shown are the GC chromatograms of silylated model compounds before treatment (a), and products after reaction with lipoxygenase (b). Peak identification: 1, linoleic acid; 2, cholesta-3,5-dien-7-one; 3, sitosterol; 4, 7�-hydroxysitosterol; 5, 7-ketositosterol; 6, cholesteryl 9-oxononanoate; and 7, cholesteryl linoleate.
Reactions of GLOX with mixtures of four model compounds (Fig. 2) representative for the main lipophilic extractives in flax pulp, including alkanes (nonacosane), fatty alcohols (octacosanol), free sterols (sitosterol), and free fatty acids (linoleic acid) were also carried out. The GC and GC-MS analyses showed reductions of 100, 51, 65 and 55% of linoleic acid, nonacosane, octacosanol and sitosterol, respectively (Fig. 5) after the GLOX treatment in agreement with the results observed in pulps, suggesting that peroxidation reactions could mediate the co-oxidation of these lipids. Similar findings were reported in the removal of alkanes and fatty alcohols from flax pulps with laccase in the presence of HBT as mediator (Molina et al. 2008). The fact that lipid radicals generated from peroxidation of unsaturated lipids such as linoleic acid participate in the
263

5. Resultados y discusión
264
oxidation of the less reactive lipophilic compounds can be used as a way to remove lipophilic extractives from pulps where linoleic acid is present using GLOX.
O
hOO
h
HO O
fHO O
f
O
OO
H
iO
OO
H O
OO
H
i
Sitosterol oxidation products:
Cholesteryl linoleate oxidation products:
HO OH
g
Figure 4. Chemical structures of the main oxidized derivatives identified after the lipoxygenase reactions with free sterols (sitosterol) and sterol esters (cholesteryl linoleate): (f) 7-ketositosterol; (g) 7�-hydroxysitosterol; (h) cholesta-3,5-dien-7-one and (i) cholesteryl 9-oxononanoate.
Table 2. Removal (percentage of reduction) of main lipophilic extractives in flax pulp after treatment with the G. graminis lipoxygenase (GLOX) in the absence and presence of linoleic acid (LA) without and with a subsequent H2O2 stage (P) Without H2O2 With H2O2 GLOX GLOX/LA GLOX-P GLOX/LA-P Alkane (C27) 26 39 21 46 Alkane (C29) 48 54 35 55 Fatty alcohol (C26) 48 44 45 50 Fatty alcohol (C28) 55 44 42 49 Fatty alcohol (C30) 61 43 52 52 Free sterols 55 30 16 34 Sterol glycosides 65 51 45 71

5. Resultados y discusión
3.4. Pulp properties after lipoxygenase treatment In addition to the enzymatic removal of lipophilic extractives from pulps, the effect of the lipoxygenase treatment on some selected properties of the pulps were assessed, including kappa number (a rough estimation of the lignin content in pulp), brightness and intrinsic viscosity (an estimation of cellulose integrity). Table 3 shows the results for eucalypt pulp. Pulps with lower kappa number (1.2 points decrease) and increased brightness (2.6 and 3.4 points increase, in the absence and presence of linoleic acid, respectively) were obtained after treatment with GLOX and subsequent peroxide stage. No improvement of pulp brightness (and only a decrease of 0.7-0.8 points in kappa number) was observed when the enzymatic treatment was not followed by a hydrogen peroxide stage, revealing the need of a peroxide stage after the lipoxygenase treatment to improve pulp properties. Pulp viscosity was maintained after the enzymatic treatment although decreased after the peroxide stage.
Table 3. Properties of eucalypt pulp treated with the G. graminis lipoxygenase (GLOX) in the absence and presence of linoleic acid (LA) before and after a H2O2 stage (P), and control without enzyme Control GLOX GLOX/LA initial P initial P initial P Kappa number 13.5 10.7 12.8 9.5 12.9 9.5 Brightness (% ISO) 44.0 55.9 43.8 58.5 43.9 59.3
Intrinsic viscosity (mL/g) 1140 925 1148 800 1143 767
In the case of flax pulp (Table 4), an increase of 0.8 and 1.2 points of ISO brightness was achieved after the enzymatic treatment followed by the hydrogen peroxide stage, in the absence and presence of linoleic acid, respectively (no increase of brightness was observed before the hydrogen peroxide stage). No significant decrease of kappa number (lower than 1 point) was observed after the enzymatic treatment of flax pulp (before the peroxide stage). After peroxide, the kappa number did not decrease and the viscosity increased.
Table 4. Properties of flax pulp treated with the G. graminis lipoxygenase (GLOX) in the absence and presence of linoleic acid (LA) before and after a H2O2 stage (P), and control without enzyme Control GLOX GLOX/LA initial P initial P initial P Kappa number 9.3 5.1 9.1 5.2 8.6 5.5 Brightness (% ISO) 35.1 61.2 35.1 62 35.3 62.4
Intrinsic viscosity (mL/g) 787 535 762 648 779 602
265

5. Resultados y discusión
266
The better delignification values observed in the GLOX treatment of eucalypt pulp compared with those of flax pulp treatment can be related with the different composition and structure of lignin in these two types of pulp. The lignin from eucalypt pulp is characterized by a high abundance of syringyl units (Ibarra et al. 2005; 2007) and, therefore, it is easier to delignify than lignin from flax pulp that is mainly constituted by guaiacyl units (Camarero et al. 2004).
100%
Rel
ativ
e res
pons
e
Retention time (minutes)104 6 8 12 14
100%
Rel
ativ
e res
pons
e
Retention time (minutes)104 6 8 12 14
(a)
(b)
41
2
3
42
3
Figure 5. Behaviour of a mixture of different model compounds (linoleic acid, nonacosane, octacosanol, sitosterol) representative for flax pulp lipophilic extractives after enzymatic treatment with lipoxygenase. Shown are the GC chromatograms of silylated model compounds before treatment (a), and after reaction with lipoxygenase (b). Peak identification: 1, linoleic acid: 2, nonacosane; 3, octacosanol; and 4, sitosterol. 4. Conclusions The potential of the lipoxygenase from G. graminis to remove lipophilic extractives from a hardwood (eucalypt) and a nonwood (flax) pulp has been studied. A removal up to 40% of esterified and glycosylated sterols was

5. Resultados y discusión
achieved by the GLOX treatment of eucalypt pulps, while only a decrease of 10% of free sterols was observed. Higher decreases (up to 70%) of lipophilic extractives from flax pulp were produced by the lipoxygenase treatment, including free sterols that decreased 55%. In addition, some pulp properties were determined on the enzymatically treated pulps observing a significant increase of brightness and decrease of kappa number for eucalypt pulp following subsequent peroxide treatment, while only limited brightness enhancement was observed for flax pulp. Given the significant alkaline activity of lipoxygenases it is suggested that these may have potential use in brown stock treatment as a means for reducing down-stream deposition problems as well as reducing the amount of chemicals in the subsequent bleaching stages. Further studies are needed to gain insight into the chemistry of the reactions of the lipoxygenase with different lipids, and in the potential applicability of this treatment in the pulp and paper industry. Acknowledgements This study has been supported by the Spanish projects BIO2007-28719-E and AGL2008-00709 and the EU project BIORENEW (NMP2-CT-2006-026456). Novozymes (Bagsvaerd, Denmark) is acknowledged for GLOX supply and ENCE (Pontevedra, Spain) and CELESA (Tortosa, Spain) for eucalypt and flax paper pulp samples, respectively. J. Romero (ENCE) and T. Vidal (UPC, Barcelona, Spain) are acknowledged for pulp properties determination. References Back EL, Allen LH (2000) Pitch control, wood resin and deresination. TAPPI
Press, Atlanta Barfoed M (2000) Methods of hydrolyzing cholesterol esters by using a
Pseudomonas fragi cholesterol esterase. Patent (International) WO9423052: Borch K, Franks N, Lund H, Xu H, Luo J (2003) Oxidizing enzymes in the
manufacturing of paper materials. Patent (USA)US 2003-0124710 A1 Bourbonnais R, Paice MG (1990) Oxidation of non-phenolic substrates. An
expanded role for laccase in lignin biodegradation. FEBS Lett 267:99-102 Buchert J, Mustranta A, Tamminen T, Spetz P, Holmbom B (2002)
Modification of spruce lignans with Trametes hirsuta laccase. Holzforschung 56:579-584
Calero-Rueda O, Gutiérrez A, del Río JC, Prieto A, Plou FJ, Ballesteros A, Martínez AT, Martínez MJ (2004) Hydrolysis of sterol esters by an esterase from Ophiostoma piceae: Application for pitch control in pulping of Eucalyptus globulus wood. Intern J Biotechnol 6:367-375
267

5. Resultados y discusión
268
Call H-P (1994) Verfahren zur Veränderung, Abbau oder Bleichen von Lignin, ligninhaltigen Materialien oder ähnlichen Stoffen. Patent (International) WO 94/29510:
Camarero S, García O, Vidal T, Colom J, del Río JC, Gutiérrez A, Gras JM, Monje R, Martínez MJ, Martínez AT (2004) Efficient bleaching of non-wood high-quality paper pulp using laccase-mediator system. Enzyme Microb Technol 35:113-120
del Río JC, Gutiérrez A, González-Vila FJ (1999) Analysis of impurities occurring in a totally-chlorine free-bleached Kraft pulp. J Chromatogr 830:227-232
del Río JC, Gutiérrez A, González-Vila FJ, Martín F, Romero J (1998) Characterization of organic deposits produced in the Kraft pulping of Eucalyptus globulus wood. J Chromatogr 823:457-465
del Río JC, Romero J, Gutiérrez A (2000) Analysis of pitch deposits produced in Kraft pulp mills using a totally chlorine free bleaching sequence. J Chromatogr A 874:235-245
Fujita Y, Awaji H, Taneda H, Matsukura M, Hata K, Shimoto H, Sharyo M, Sakaguchi H, Gibson K (1992) Recent advances in enzymic pitch control. Tappi J 75 (4):117-122
Gutiérrez A, del Río JC (2001) Gas chromatography-mass spectrometry demonstration of steryl glycosides in eucalypt wood, kraft pulp and process liquids. Rapid Commun Mass Spectrom 15:2515-2520
Gutiérrez A, del Río JC (2003a) Lipids from flax fibers and their fate in alkaline pulping. J Agric Food Chem 51:4965-4971
Gutiérrez A, del Río JC (2003b) Lipids from flax fibers and their fate in alkaline pulping (Vol 51, pg 4965, 2003). J Agric Food Chem 51:6911-6914
Gutiérrez A, del Río JC (2005a) Chemical characterization of pitch deposits produced in the manufacturing of high-quality paper pulps from hemp fibers. Bioresource Technol 96:1445-1450
Gutiérrez A, del Río JC (2005b) Pitch episodes produced during the manufacturing of high-quality paper pulps from hemp fibers. Bioresour Technol (submitted):
Gutiérrez A, del Río JC, González-Vila FJ, Martín F (1998) Analysis of lipophilic extractives from wood and pitch deposits by solid-phase extraction and gas chromatography. J Chromatogr 823:449-455
Gutiérrez A, del Río JC, González-Vila FJ, Martín F (1999) Chemical composition of lipophilic extractives from Eucalyptus globulus Labill. wood. Holzforschung 53:481-486

5. Resultados y discusión
Gutiérrez A, del Río JC, Ibarra D, Rencoret J, Romero J, Speranza M, Camarero S, Martínez MJ, Martínez AT (2006a) Enzymatic removal of free and conjugated sterols forming pitch deposits in environmentally sound bleaching of eucalypt paper pulp. Environ Sci Technol 40:3416-3422
Gutiérrez A, del Río JC, Martínez AT (2009) Microbial and enzymatic control of pitch in the pulp and paper industry. Appl Microbiol Biotechnol 82:1005-1018
Gutiérrez A, del Río JC, Martínez MJ, Martínez AT (2001a) The biotechnological control of pitch in paper pulp manufacturing. Trends Biotechnol 19:340-348
Gutiérrez A, del Río JC, Rencoret J, Ibarra D, Martínez AT (2006b) Main lipophilic extractives in different paper pulp types can be removed using the laccase-mediator system. Appl Microbiol Biotechnol 72:845-851
Gutiérrez A, Rencoret J, Ibarra D, Molina S, Camarero S, Romero J, del Río JC, Martínez AT (2007) Removal of lipophilic extractives from paper pulp by laccase and lignin-derived phenols as natural mediators. Environ Sci Technol 41:4124-4129
Gutiérrez A, Romero J, del Río JC (2001b) Lipophilic extractives from Eucalyptus globulus pulp during kraft cooking followed by TCF and ECF bleaching. Holzforschung 55:260-264
Hamberg M, Su C, Oliw E (1998) Manganese lipoxygenase - Discovery of a bis-allylic hydroperoxide as product and intermediate in a lipoxygenase reaction. J Biol Chem 273:13080-13088
Ibarra D, Chávez MI, Rencoret J, del Río JC, Gutiérrez A, Romero J, Camarero S, Martínez MJ, Jiménez-Barbero J, Martínez AT (2007) Lignin modification during Eucalyptus globulus kraft pulping followed by totally chlorine free bleaching: A two-dimensional nuclear magnetic resonance, Fourier transform infrared, and pyrolysis-gas chromatography/mass spectrometry study. J Agric Food Chem 55:3477-3499
Ibarra D, del Río JC, Gutiérrez A, Rodríguez IM, Romero J, Martínez MJ, Martínez AT (2005) Chemical characterization of residual lignins from eucalypt paper pulps. J Anal Appl Pyrolysis 74:116-122
International Organisation for Standardization Documentation and Information (ISO) (2003) ISO Standards Collection on CD-ROM. Paper, board and pulps, second edn. ISO, Geneva
Kapich AN, Korneichik TV, Hatakka A, Hammel KE (2010) Oxidizability of unsaturated fatty acids and of a non-phenolic lignin structure in the manganese peroxidase-dependent lipid peroxidation system. Enzyme Microb Technol 46:136-140
269

5. Resultados y discusión
270
Kontkanen H, Tenkanen M, Fagerstrom R, Reinikainen T (2004) Characterisation of steryl esterase activities in commercial lipase preparations. J Biotechnol 108:51-59
Marques G, del Rio JC, Gutiérrez A (2010) Lipophilic extractives from several nonwoody lignocellulosic crops (flax, hemp, sisal, abaca) and their fate during alkaline pulping and TCF/ECF bleaching. Bioresource Technol 101:260-267
Molina S, Rencoret J, del Río JC, Lomascolo A, Record E, Martínez AT, Gutiérrez A (2008) Oxidative degradation of model lipids representative for main paper pulp lipophilic extractives by the laccase-mediator system. Appl Microbiol Biotechnol 80:211-222
Prigge ST, Boyington JC, Faig M, Doctor KS, Gaffney BJ, Amzel LM (1997) Structure and mechanism of lipoxygenases. Biochemie 79:629-636
Riva S (2006) Laccases: blue enzymes for green chemistry. Trends Biotechnol 24:219-226
Rodríguez-Couto S, Toca JL (2006) Industrial and biotechnological applications of laccases: A review. Biotechnol Adv 24:500-513
Speranza M, Martínez MJ, Gutiérrez A, del Río JC, Martínez AT (2002) Wood and pulp localization of sterols involved in pitch deposition using filipin fluorescent staining. J Pulp Paper Sci 28:292-297
Su C, Oliw EH (1998) Manganese lipoxygenase - Purification and characterization. J Biol Chem 273:13072-13079
Widsten P, Kandelbauer A (2008) Laccase applications in the forest products industry: A review. Enzyme Microb Technol 42:293-307
Zhang X, Nguyen D, Paice MG, Tsang A, Renaud S (2007) Degradation of wood extractives in thermo-mechanical pulp by soybean lipoxygenase. Enzyme Microb Technol 40:866-873
Zhang X, Renaud S, Paice M (2005) The potential of laccase to remove extractives present in pulp and white water from TMP newsprint mills. J Pulp Paper Sci 31:175-180
Zhang X, Stebbing DW, Saddler JN, Beatson RP, Kruus K (2000) Enzyme treatments of the dissolved and colloidal substances present in mill white water and the effects on the resulting paper properties. J Wood Chem Technol 20:321-335

5. Resultados y discusión
271

6

6. Conclusiones
CONCLUSIONES En la presente Tesis se ha estudiado la composición química de los
principales constituyentes (lignina, lípidos y hemicelulosas) de diferentes cultivos lignocelulósicos utilizados como materia prima para la fabricación de pastas de celulosa de alta calidad, así como su comportamiento durante los procesos de pasteado y blanqueo. Además, se han estudiado dos procedimientos biotecnológicos que incluyen el uso de enzimas fúngicas para la eliminación de lignina y lípidos residuales en pastas. Las principales conclusiones obtenidas se citan a continuación:
1. En general, las materias primas estudiadas se caracterizan por presentar un alto contenido en polisacáridos y bajo en lípidos y lignina, lo que es, en principio, favorable para el proceso de producción de pasta de celulosa.
2. El contenido y composición de las diferentes clases de lípidos varía entre las distintas fibras estudiadas. Las fibras provenientes de tallos se caracterizan por un alto contenido en ácidos grasos y, en particular, las fibras de lino, kenaf y yute poseen también un alto contenido en ceras. Por otro lado, las fibras provenientes de hojas además de tener un alto contenido en ácidos grasos, poseen un alto contenido en esteroles (sisal y abacá) y ceras (curauá).
3. Las diferentes clases de lípidos muestran distinto comportamiento durante los procesos de cocción y blanqueo. Durante la cocción sosa-AQ, los compuestos esterificados se hidrolizan, los ácidos grasos se disuelven y los aldehídos se eliminan, mientras que los compuestos neutros (esteroles, alcanos, alcoholes, cetonas) sobreviven a la cocción. Los esteroles libres que sobreviven a la cocción alcalina se degradan en el blanqueo ECF mientras que permanecen prácticamente inalterados en el blanqueo TCF, pudiendo originar problemas de deposición de pitch en estos procesos. Los glicósidos de esteroles, por otra parte, se eliminan tanto en el blanqueo TCF como ECF. Los ácidos grasos saturados, alcoholes y alcanos sobreviven tanto a la cocción como al blanqueo ECF y TCF, y por tanto pueden originar también problemas de pitch.
4. La composición de las ligninas de las fibras liberianas de kenaf y yute, así como las de todas las fibras de hojas (sisal, abacá y curauá) son fundamentalmente de tipo S. Por el contrario, la lignina de las fibras de cáñamo, lino y caña común tienen un predominio de unidades de tipo G. A pesar de que las fibras de lino y cáñamo presentan un bajo contenido
273

6. Conclusiones
en lignina (< 5%), son más difíciles de deslignificar puesto la lignina de tipo G es más condensada.
5. Los principales enlaces entre las unidades de lignina en todas las fibras estudiadas son de tipo aril-éter �-O-4�. También están presentes unidades de tipo resinol �-��, fenilcumarano �-5�/�-O-4� y espirodienona �-1�/�-O-��. La mayor proporción de enlaces �-O-4� se encuentra en las ligninas del kenaf, sisal, abacá y curauá, las cuales al tener también mayor proporción de unidades S, son más fácilmente deslignificables.
6. Las ligninas de kenaf, sisal, abacá y curauá están extensamente aciladas en el carbono � de la cadena lateral (con grupos acetatos y/o p-cumaratos) y principalmente sobre unidades S. Se demostró que la acilación de la lignina tiene lugar a nivel de monómero y que el sinapil acetato (y otros monómeros acilados) se comportan como auténticos monómeros de la lignina. Además, estas ligninas están muy enriquecidas en unidades S y en enlaces �-O-4�, y con poca presencia de enlaces condensados, lo que las hace extremadamente lineales.
7. Las hemicelulosas de las fibras liberianas presentan una mayor variabilidad en cuanto a su composición en azúcares neutros que las fibras procedentes de hojas. Así, mientras que en las fibras de lino y cáñamo predominan la manosa y la galactosa, en las de kenaf y yute el monosacárido predominante es la xilosa. Por otro lado, en todas las fibras de hojas estudiadas (sisal, abacá y curauá) se observó un predominio de xilosa.
8. Las hemicelulosas de sisal se caracterizan por estar constituidas fundamentalmente por un glucuronoxilano acetilado, cuya cadena principal está formada por unidades de �-D-xilopiranosa parcialmente ramificada con residuos glucuronosilos (MeGlcpA y GlcpA). El 61% mol. de las unidades de xilopiranosa están acetiladas, principalmente en la posición O-3 y O-2 (39% mol. y 13% mol. respectivamente) y di-acetiladas (9% mol.) también en esas posiciones.
9. El heteroxilano de sisal sufre una despolimerización y una desacetilación significativas durante el proceso de pasteado, en el que los residuos MeGlcpA y GlcpA se eliminan parcialmente o se convierten en HexA y los grupos acetilo se hidrolizan mayoritariamente. Durante el proceso de blanqueo, una pequeña fracción de los xilanos asociados a la lignina residual se eliminan y los grupos acetilo residuales se eliminan completamente. El comportamiento de los residuos de HexA durante los procesos de blanqueo TCF y ECF son diferentes. Durante el blanqueo TCF, una pequeña proporción de los residuos MeGlcpA
274

6. Conclusiones
existentes en la pasta se convierten en HexA mientras que durante el blanqueo ECF todos los HexA son eliminados por la acción del dióxido de cloro.
10. El procedimiento biotecnológico basado en la utilización de un POM y la peroxidasa versátil (VP) del hongo Pleurotus eryngii muestra una gran eficacia para eliminar la lignina residual en pastas de celulosa. El POM en su forma oxidada es altamente selectivo para la deslignificación y se reoxida completamente por la VP en tiempos muy cortos. De esta manera, es posible la sustitución de etapas de blanqueo que usan dióxido de cloro por tratamientos POM-VP-POMreox pudiendo utilizarse sistemas de este tipo en procesos industriales.
11. El procedimiento biotecnológico basado en el uso de la lipoxigenasa del hongo Gaeumannomyces graminis muestra una gran eficacia para eliminar los lípidos residuales responsables de los problemas de pitch durante la producción de pastas de celulosa, especialmente en el caso de pastas de lino. También se observa una mejora de algunas propiedades de las pastas (especialmente en el caso de pastas de eucalipto) como es el aumento de la blancura y la disminución del número kappa.
En conclusión, el estudio de la composición química de los cultivos lignocelulósicos utilizados como materia prima para la fabricación de pastas de celulosa así como la evolución de sus principales componentes durante los procesos de cocción y blanqueo, contribuye a optimizar su aprovechamiento industrial mediante tecnologías menos contaminantes. Este conocimiento contribuirá a un aprovechamiento industrial más sostenible de estos materiales lignocelulósicos así como al desarrollo de nuevas especies de interés socioeconómico.
275

7

7. Anexos
ANEXOS
Anexo 1. Peso de pasta ideal (c/ � 7,5% humedad) para la determinación del índice kappa. Adaptado de un documento realizado por Armindo Gaspar de la Universidad de Aveiro.
IK peso ideal/g peso seco/g 70,0 0,190 0,176 65,0 0,204 0,189 60,0 0,221 0,205 55,0 0,242 0,224 50,0 0,266 0,246 45,0 0,295 0,273 40,0 0,332 0,308 35,0 0,380 0,351 30,0 0,443 0,410 25,0 0,531 0,492 20,0 0,664 0,615 19,0 0,699 0,647 18,0 0,738 0,683 17,0 0,781 0,724 16,0 0,830 0,769 15,0 0,886 0,820 14,0 0,949 0,879 13,0 1,022 0,946 12,0 1,107 1,025 11,0 1,208 1,118 10,0 1,328 1,230 9,0 1,476 1,367 8,0 1,661 1,538 7,0 1,898 1,757 6,0 2,214 2,050 5,0 2,657 2,460 4,0 3,321 3,705 3,0 4,428 4,100 2,0 6,642 6,150 1,0 13,284 12,300
277

7. Anexos
278
Anexo 2. Factores f de corrección del consumo de permanganato usado en la determinación del índice kappa ( Tappi 2006).
f+ 0 1 2 3 4 5 6 7 8 930 0,958 0,960 0,962 0,964 0,966 0,968 0,970 0,973 0,975 0,99740 0,979 0,981 0,983 0,985 0,987 0,989 0,991 0,994 0,996 0,99850 1,000 1,002 1,004 1,006 1,099 1,011 1,013 1,015 1,017 1,01960 1,002 1,024 1,026 1,028 1,030 1,033 1,035 1,037 1,039 1,04270 1,004

7. Anexos
Anexo 3. Peso de pasta ideal (c/ � 7,5% humedad) para la determinación de la viscosidad intrínseca. Adaptado de un documento realizado por Armindo Gaspar de la Universidad de Aveiro.
Volumen del frasco/ml Viscosidad 56 58 61
1400 0,129 0,134 0,141 1350 0,134 0,139 0,146 1300 0,139 0,144 0,152 1250 0,145 0,150 0,158 1200 0,151 0,156 0,165 1150 0,158 0,163 0,172 1100 0,165 0,171 0,179 1050 0,173 0,179 0,188 1000 0,181 0,188 0,197 950 0,191 0,198 0,208 900 0,201 0,209 0,219 850 0,213 0,221 0,232 800 0,227 0,235 0,247 750 0,242 0,250 0,263 700 0,259 0,268 0,282
279

7. Anexos
280
Anexo 4. Valores del producto [�]C para diferentes valores de �rel (Scandinavian Pulp Paper and Board Committee 1994).
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,08 0,091,0 0,000 0,010 0,020 0,030 0,040 0,049 0,059 0,069 0,078 0,0881,1 0,097 0,107 0,116 0,125 0,134 0,144 0,153 0,162 1,171 0,1801,2 0,189 0,198 0,207 0,216 0,224 0,233 0,242 0,250 0,259 0,2681,3 0,276 0,285 0,293 0,302 0,310 0,318 0,326 0,335 0,343 0,3511,4 0,359 0,367 0,375 0,383 0,391 0,399 0,407 0,415 0,423 0,4311,5 0,438 0,446 0,454 0,462 0,469 0,477 0,484 0,492 0,499 0,5071,6 0,514 0,522 0,529 0,537 0,544 0,551 0,558 0,566 0,573 0,5801,7 0,587 0,594 0,601 0,608 0,615 0,622 0,629 0,636 0,643 0,6501,8 0,657 0,664 0,671 0,678 0,684 0,691 0,698 0,705 0,711 0,7181,9 0,725 0,731 0,738 0,744 0,751 0,757 0,764 0,770 0,777 0,783
2,0 0,790 0,796 0,802 0,809 0,815 0,821 0,827 0,834 0,840 0,8462,1 0,852 0,858 0,865 0,871 0,877 0,883 0,889 0,895 0,901 0,9072,2 0,913 0,919 0,925 0,931 0,937 0,943 0,949 0,954 0,960 0,9662,3 0,972 0,978 0,983 0,989 0,995 1,001 1,006 1,012 1,018 1,0232,4 1,029 1,035 1,040 1,046 1,051 1,057 1,062 1,068 1,073 1,0792,5 1,084 1,090 1,095 1,101 1,106 1,111 1,117 1,122 1,127 1,1332,6 1,138 1,143 1,149 1,154 1,159 1,164 1,170 1,175 1,180 1,1852,7 1,190 1,196 1,201 1,206 1,211 1,216 1,221 1,226 1,231 1,2362,8 1,241 1,246 1,251 1,256 1,261 1,266 1,271 1,276 1,281 1,2862,9 1,291 1,296 1,301 1,306 1,310 1,316 1,320 1,325 1,330 1,335
3,0 1,339 1,344 1,349 1,354 1,358 1,363 1,368 1,373 1,377 1,3823,1 1,387 1,391 1,396 1,401 1,405 1,410 1,414 1,419 1,424 1,4283,2 1,433 1,437 1,442 1,446 1,451 1,455 1,460 1,464 1,469 1,4733,3 1,478 1,482 1,487 1,491 1,496 1,500 1,504 1,509 1,513 1,5173,4 1,522 1,526 1,531 1,535 1,539 1,544 1,548 1,552 1,556 1,5613,5 1,565 1,569 1,573 1,578 1,582 1,586 1,590 1,595 1,599 1,6033,6 1,607 1,611 1,615 1,620 1,624 1,628 1,632 1,636 1,640 1,6443,7 1,648 1,653 1,657 1,661 1,665 1,669 1,673 1,677 1,681 1,6853,8 1,689 1,693 1,697 1,701 1,705 1,709 1,713 1,717 1,721 1,7253,9 1,729 1,732 1,736 1,740 1,744 1,748 1,752 1,756 1,760 1,764
4,0 1,767 1,771 1,775 1,779 1,783 1,787 1,790 1,794 1,798 1,8024,1 1,806 1,809 1,813 1,817 1,821 1,824 1,828 1,832 1,836 1,8394,2 1,843 1,847 1,851 1,854 1,858 1,862 1,865 1,869 1,873 1,8764,3 1,880 1,884 1,887 1,891 1,894 1,898 1,902 1,905 1,909 1,9124,4 1,916 1,920 1,923 1,927 1,930 1,934 1,937 1,941 1,944 1,948

7. Anexos
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,08 0,09
4,5 1,952 1,955 1,959 1,962 1,966 1,969 1,973 1,976 1,979 1,9834,6 1,986 1,990 1,993 1,997 2,000 2,004 2,007 2,010 2,014 2,0174,7 2,021 2,024 2,028 2,031 2,034 2,038 2,041 2,044 2,048 2,0514,8 2,054 2,058 2,061 2,064 2,068 2,071 2,074 2,078 2,081 2,0844,9 2,088 2,091 2,094 2,098 2,101 2,104 2,107 2,111 2,114 2,117
5,0 2,120 2,124 2,127 2,130 2,133 2,137 2,140 2,143 2,146 2,1495,1 2,153 2,156 2,159 2,162 2,165 2,168 2,172 2,175 2,178 2,1815,2 2,184 2,187 2,191 2,194 2,197 2,200 2,203 2,206 2,209 2,2125,3 2,215 2,219 2,222 2,225 2,228 2,231 2,234 2,237 2,240 2,2435,4 2,246 2,249 2,252 2,255 2,258 2,261 2,264 2,267 2,270 2,2735,5 2,276 2,280 2,283 2,286 2,288 2,291 2,294 2,297 2,300 2,3035,6 2,306 2,309 2,312 2,315 2,318 2,321 2,324 2,327 2,330 2,3335,7 2,336 2,339 2,342 2,345 2,347 2,350 2,353 2,356 2,359 2,3625,8 2,365 2,368 2,371 2,374 2,376 2,379 2,382 2,385 2,388 2,3915,9 2,394 2,396 2,399 2,402 2,405 2,408 2,411 2,413 2,416 2,419
6,0 2,422 2,425 2,427 2,430 2,433 2,436 2,439 2,441 2,444 2,4476,1 2,450 2,452 2,455 2,458 2,461 2,463 2,466 2,469 2,472 2,4756,2 2,477 2,480 2,783 2,485 2,488 2,491 2,494 2,496 2,499 2,5026,3 2,504 2,507 2,510 2,512 2,515 2,518 2,521 2,523 2,526 2,5296,4 2,531 2,534 2,537 2,539 2,542 2,545 2,547 2,550 2,552 2,5556,5 2,558 2,560 2,563 2,566 2,568 2,571 2,573 2,576 2,579 2,5816,6 2,584 2,587 2,589 2,592 2,594 2,597 2,599 2,602 2,605 2,6076,7 2,610 2,612 2,615 2,617 2,620 2,623 2,625 2,628 2,630 2,6336,8 2,635 2,638 2,640 2,643 2,645 2,648 2,651 2,653 2,656 2,6596,9 2,661 2,663 2,666 2,668 2,671 2,673 2,676 2,678 2,681 2,683
7,0 2,686 2,688 2,690 2,693 2,695 2,698 2,700 2,703 2,705 2,7087,1 2,710 2,713 2,715 2,718 2,720 2,722 2,725 2,727 2,730 2,7327,2 2,735 2,737 2,739 2,742 2,744 2,747 2,749 2,752 2,754 2,7567,3 2,758 2,761 2,764 2,766 2,768 2,771 2,773 2,775 2,778 2,7807,4 2,783 2,785 2,787 2,790 2,792 2,794 2,797 2,799 2,801 2,8047,5 2,806 2,809 2,811 2,813 2,816 2,818 2,820 2,823 2,825 2,8277,6 2,829 2,832 2,834 2,836 2,839 2,841 2,843 2,846 2,848 2,8507,7 2,853 2,855 2,857 2,859 2,862 2,864 2,866 2,869 2,871 2,8737,8 2,875 2,878 2,880 2,882 2,885 2,887 2,889 2,891 2,894 2,8967,9 2,898 2,900 2,903 2,905 2,907 2,909 2,911 2,914 2,916 2,918
281

7. Anexos
282
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,08 0,09
8,0 2,920 2,923 2,295 2,927 2,929 2,932 2,934 2,936 2,938 2,9408,1 2,943 2,945 2,947 2,949 2,951 2,954 2,956 2,958 2,960 2,9628,2 2,964 2,967 2,969 2,971 2,973 2,975 2,978 2,980 2,982 2,9848,3 2,986 2,988 2,991 2,993 2,995 2,997 2,999 3,001 3,003 3,0068,4 3,008 3,010 3,012 3,014 3,016 3,018 3,020 3,023 3,025 3,0278,5 3,029 3,031 3,033 3,035 3,037 3,040 3,042 3,044 3,046 3,0488,6 3,050 3,052 3,054 3,056 3,058 3,061 3,063 3,065 3,067 3,0698,7 3,071 3,073 3,075 3,077 3,079 3,081 3,083 3,085 3,087 3,0908,8 3,092 3,094 3,096 3,098 3,100 3,102 3,104 3,106 3,108 3,1108,9 3,112 3,114 3,116 3,118 3,120 3,122 3,124 3,126 3,128 3,130
9,0 3,132 3,134 3,136 3,138 3,340 3,142 3,144 3,147 3,149 3,1519,1 3,153 3,155 3,157 3,159 3,161 3,163 3,165 3,166 3,168 3,1709,2 3,172 3,174 3,176 3,178 3,180 3,182 3,184 3,186 3,188 3,1909,3 3,192 3,194 3,196 3,198 3,200 3,202 3,204 3,206 3,208 3,2109,4 3,212 3,214 3,216 3,218 3,220 3,222 3,223 3,225 3,227 3,2299,5 3,231 3,233 3,265 3,237 3,239 3,241 3,242 3,245 3,247 3,2499,6 3,250 3,252 3,254 3,256 3,258 3,260 3,262 3,264 3,266 3,2689,7 3,270 3,271 3,273 3,175 3,277 3,273 3,281 3,283 3,285 3,8879,8 3,288 3,290 3,292 3,294 3,296 3,298 3,300 3,302 3,303 3,3059,9 3,307 3,309 3,311 3,313 3,315 3,316 3,318 3,320 3,322 3,324
10 3,326 3,344 3,363 3,381 3,399 3,416 3,434 4,452 3,469 3,48711 3,504 3,521 3,538 3,554 3,571 3,588 3,604 3,620 3,636 3,65312 3,669 3,684 3,700 3,716 3,731 3,747 3,762 3,777 3,792 3,80713 3,822 3,837 3,852 3,866 3,881 3,895 3,910 3,924 3,938 3,95214 3,966 3,980 3,994 4,008 4,021 4,035 4,048 4,062 4,075 4,08815 4,101 4,115 4,128 4,141 4,153 4,166 4,179 4,192 4,204 4,21716 4,229 4,242 4,254 4,266 4,279 4,291 4,303 4,315 4,327 4,33917 4,351 4,362 4,374 4,386 4,397 4,409 4,420 4,432 4,443 4,45518 4,466 4,477 4,488 4,499 4,510 4,521 4,532 4,543 4,554 4,56519 4,576 4,586 4,597 4,608 4,618 4,629 4,639 4,650 4,660 4,670