polipos y ca de colon
DESCRIPTION
REFERENCIA patologia de robbinsTRANSCRIPT

Universidad del MagdalenaPrograma De Medicina
Catedra Patología2014-2

TABLA DE CONTENIDO
POLIPOS..........................................................................................................................2
PÓLIPOS INFLAMATORIOS...............................................................................................2
PÓLIPOS HAMARTOMATOSOS..........................................................................................3
PÓLIPOS HIPERPLÁSICOS.................................................................................................7
ADENOMAS O POLIPOS NEOPLASICOS............................................................................7
SINDROMES FAMILIARES.................................................................................................7
ADENOCARCINOMA.........................................................................................................9
TUMORES DEL APÉNDICE..............................................................................................14
TUMORES DEL CANAL ANAL..........................................................................................15
BIBLIOGRAFÍA................................................................................................................15
1

POLIPOS
Los pólipos son más frecuentes en el colon, pero pueden presentarse en el esófago, estómago o intestino delgado. La mayoría de los pólipos, si no todos, comienzan como pequeñas elevaciones de la mucosa. En ese caso se denominan sésiles, un término que se ha tomado prestado de los botánicos, que lo usan para describir flores y hojas que crecen directamente del tronco, sin un tallo. A medida que los pólipos sésiles aumentan de tamaño, se combinan varios procesos, incluida la proliferación de células adyacentes a la masa y los efectos de tracción en la protrusión luminal, hasta crear un tallo. Los pólipos con tallos se denominan pedunculados. En general, los pólipos intestinales se pueden clasificar como de origen neoplásico o no neoplásico. El tipo más frecuente de pólipos neoplásicos es el adenoma, que puede progresar a cáncer. Los pólipos no neoplásicos se pueden clasificar como inflamatorio, hamartomatoso o hiperplásico.
PÓLIPOS INFLAMATORIOS
El pólipo que se forma dentro del síndrome de la úlcera rectal solitaria es un ejemplo de una lesión puramente infl amatoria. Los pacientes acuden con la tríada clínica de hemorragia rectal, emisión de moco y lesión infl amatoria de la pared rectal anterior. La causa subyacente es el deterioro de la relajación del esfínter anorrectal que crea un ángulo agudo en el cuerpo rectal anterior y provoca una abrasión recurrente y ulceración de la mucosa rectal suprayacente. Un pólipo inflamatorio se puede formar, en último término, como consecuencia de los ciclos crónicos de lesión y cicatrización. El atrapamiento de este pólipo en la masa fecal provoca el prolapso de la mucosa. Por tanto, las características histológicas propias son las típicas de un
2
Fig 1. Patología estructural y funcional de robbins Síndrome de úlcera rectal solitaria. A. Las glándulas dilatadas, el epitelio proliferativo, las erosiones superfi ciales y el infi ltrado infl amatorioson típicos de un pólipo infl amatorio. No obstante, la hiperplasia del músculo liso dentro de la lámina propia indica que también se ha producido el prolapso de la mucosa. B. Hiperplasia epitelial. C. Proliferación capilar similar al tejido de granulación dentro de la lámina propia

pólipo inflamatorio con prolapso superpuesto de la mucosa eincluyen la hiperplasia fi bromuscular de la lámina propia, infi ltrados infl amatorios mixtos, erosión e hiperplasia epitelial (fig 1).
PÓLIPOS HAMARTOMATOSOS
Los pólipos hamartomatosos aparecen esporádicamente y en el contexto de varios síndromes genéticamente determinados o adquiridos. No hay que olvidar que los hamartomas son proliferaciones tumorales compuestas por tejidos maduros que se presentan normalmente en el lugar en el que se desarrollan. Aunque los síndromes de poliposis hamartomatosa sean raros, es importante reconocerlos por las manifestaciones intestinales y extraintestinales asociadas y la posibilidad de que haya otros miembros de la familia afectados.
Pólipos juveniles
Los pólipos juveniles son malformaciones focales del epitelio de la mucosa y la lámina propia. Pueden ser esporádicos o sindrómicos, pero la morfología de las dos formas puede ser indistinguible. La inmensa mayoría de los pólipos juveniles se presenta en niños menores de 5 años de edad. Cuando aparecen en los adultos, los pólipos que tienen una morfología idéntica se denominan erróneamente pólipos infl amatorios. La mayoría de los pólipos juveniles se localiza en el recto y la mayoría se presenta con hemorragia rectal. En algunos casos se produce el prolapso y el pólipo hace protrusión a través del esfínter anal. Los pólipos juveniles esporádicos son normalmente lesiones solitarias y se pueden denominar pólipos de retención. Por el contrario, las personas que tienen un síndrome autosómico dominante de poliposis juvenil tienen entre 3 y más de 100 pólipos hamartomatosos y pueden requerir la colectomía para limitar la hemorragia crónica, y a veces intensa, asociada a la ulceración del pólipo. Una minoría de pacientes también tiene pólipos en el estómago y en el intestino delgado. Las malformaciones arteriovenosas pulmonares son manifestaciones extraintestinales del síndrome.
Morfología. La mayoría de los pólipos juveniles tienen menos de 3 cm de diámetro. Normalmente son lesiones pediculadas con una superfi cie lisa y color rojizo, con espacios quísticos característicos evidentes al corte. En el estudio con el microscopio se comprueba que esos quistes son glándulas dilatadas llenas con mucina y restos inflamatorios ( 2 ). El resto del pólipo consiste en la lámina propia expandida por un infi ltrado infl amatorio mixto. La muscular de la mucosa puede ser normal o adelgazada.
3

Aunque la morfogenia de los pólipos juveniles no se conoce con detalle, algunos autores han propuesto que la hiperplasia de la mucosa es el episodio desencadenante. Esta hipótesis es compatible con el descubrimiento de que las mutaciones de las vías que regulan el crecimiento celular provocan poliposis juvenil autosómica dominante. La mutación más frecuente identifi cada es la del gen SMAD4, que codifi ca un producto citoplasmático intermedio de la vía de señalización TGF- _ . BMPR1A, una cinasa que es miembro de la superfamilia TGF, puede estar mutada en otros casos (v. tabla 17-9 ). No obstante, esas mutaciones explican menos de la mitad de los pacientes, lo que indica que puede haber cambios en otros genes que también pueden causar la poliposis juvenil. La displasia aparece en una proporción de pólipos juveniles y el síndrome de poliposis juvenil se asocia a un aumento de riesgo de adenocarcinoma de colon.
Síndrome de Peutz-Jeghers
Se trata de un síndrome autosómico dominante raro que se presenta con una mediana de edad de 11 años con múltiples pólipos hamartomatosos digestivos e hiperpigmentación mucocutánea. Esta última adopta la forma de máculas azul oscuro o marrón alrededor de la boca, ojos, orificios nasales, mucosa bucal, superficies palmares, genitales y región perianal. Esas lesiones son similares a las pecas, pero se distinguen por su presencia en la mucosa bucal. Los pólipos de Peutz-Jeghers pueden dar lugar a una intususcepción, que en ocasiones resulta mortal. Más importante es que el síndrome de Peutz-Jeghers se asocia a un aumento de riesgo de varios procesos malignos, como los cánceres de colon, páncreas, mama, pulmón, ovarios, útero y testículos, así como otras neoplasias inusuales, como los tumores de los cordones sexuales.
Patogenia . Las mutaciones heterocigóticas de las células germinales con pérdida de función del gen LKB1/STK11 están presentes en la mitad de los sujetos con síndrome familiar de Peutz-Jeghers y también en un subgrupo de pacientes con síndrome de Peutz-Jeghers esporádico. La LKB1/STK11 es una cinasa que regula la polarización, crecimiento y metabolismo celular. La función de la segunda copia «normal» de LKB1/STK11 se pierde con frecuencia por la mutación somática de los
4
Fig 2. Patología estructural y funcional de robbins Poliposis juvenil. A. Pólipo juvenil. Obsérvese la erosiónde la superfi cie y las criptas con dilatación quística. B. El moco espesado,los neutrófi los y los restos infl amatorios se pueden acumular dentro de las

cánceres que aparecen en el síndrome de Peutz-Jeghers, lo que es compatible con la idea de que el LKB1/STK11 es un gen supresor tumoral y explica el alto riesgo de neoplasia en los pacientes afectados. Los adenocarcinomas digestivos surgen con independencia de los pólipos hamartomatosos, lo que indica que los hamartomas no son lesiones precursoras preneoplásicas.
Morfología. Los pólipos del síndrome de Peutz-Jeghers son los más frecuentes en el intestino delgado, si bien pueden aparecer en el estómago y en el colon, y, con mucha menor frecuencia, en la vejiga y en los pulmones. Macroscópicamente, los pólipos son grandes y pediculados, con un perfi llobulado. El estudio histológico demuestra una red arborizante característica de tejido conjuntivo, músculo liso, lámina propia y glándulas recubiertas por un epitelio intestinal deaspecto normal ( fig 3 ). La arborización y la presencia de músculo liso entremezclado con la lámina son útiles para distinguir los pólipos del síndrome de Peutz-Jeghers de los pólipos juveniles.
Características clínicas. Como la morfología de los pólipos de Peutz-Jeghers se superpone con la de los pólipos hamartomatosos esporádicos, la presencia de múltiples pólipos en el intestino delgado, hiperpigmentación mucocutánea y antecedentes familiares es clave para el diagnóstico. La detección de mutaciones LKB1/STK11 es útil para el diagnóstico en pacientes con pólipos sin hiperpigmentación mucocutánea asociada. No obstante, la ausencia de mutaciones LKB1/STK11 no excluye el diagnóstico, ya que puede haber mutaciones en otros genes, desconocidas en este momento, que también pueden causar este síndrome. Debido al aumento de riesgo de cáncer, se recomienda el seguimiento sistemático del tubo digestivo, pelvis y gónadas.
Síndrome de Cowden y síndrome de Bannayan-Ruvalcaba-Riley
El síndrome de Cowden y el síndrome de Bannayan-Ruvalcaba-Riley son síndromes
autosómicos dominantes de pólipos hamartomatosos asociados a mutaciones con pérdida de función del gen PTEN, un gen que codifi ca la fosfatasa lipídica que inhibe la señalización a través de la vía PI3K/AKT. El gen PTEN, un gen supresor tumoral de sobra conocido, también está
5
Fig 3. Patología estructural y funcional de robbins Pólipo de Peutz-Jeghers. A. Superfi cie del pólipo (parte superior) que recubre un estroma compuesto de haces de músculo liso que atraviesa la lámina propia. B. La arquitectura glandular compleja y la presencia de músculo liso son características que distinguen los pólipos de Peutz-Jeghers de los pólipos juveniles. Compárese con la fi gura 17-42.

mutado en un pequeño número de pacientes que acuden con poliposis juvenil. Hay muchos síndromes asociados a mutaciones PTEN que se pueden asociar bajo el encabezamiento «síndrome de hamartomas PTEN». No se conoce la base de las diferentes presentaciones de esos síndromes, aunque se sospecha una interacción de las mutaciones PTEN con pérdida de función con otros genes modifi cadores desconocidos.
El síndrome de Cowden se caracteriza por macrocefalia, pólipos hamartomatosos intestinales y tumores cutáneos benignos, típicamente triquilemomas, pápulas papilomatosas y queratosis acara.
También aparecen otras lesiones que derivan de las tres capas embriológicas, como son los lipomas subcutáneos, los leiomiomas y los hemangiomas. Aunque los sujetos con síndrome de Cowden no tienen un mayor riesgo de procesos malignos digestivos, están predispuestos a presentar carcinoma de mama, carcinoma folicular de tiroides y carcinoma endometrial.
El síndrome de Bannayan-Ruvalcaba-Riley se distingue del síndrome de Cowden por su cuadro clínico; por ejemplo, la defi ciencia mental y el retraso del desarrollo sólo aparecen en el síndrome de Bannayan-Ruvalcaba-Riley, que también parece asociarse a una incidencia de neoplasia menor que el síndrome de Cowden. Las características compartidas por esos dos síndromes son los pólipos hamartomatosos digestivos, lipomas, macrocefalia, hemangiomas y, en los varones, máculas pigmentadasen el glande del pene.
Síndrome de Cronkhite-Canada
El síndrome de Cronkhite-Canada es muy diferente a los demás síndromes con pólipos hamartomatosos porque no es hereditario, y con mayor frecuencia se desarrolla en sujetos mayores de 50 años de edad. Los síntomas clínicos son inespecíficos e incluyen diarrea, pérdida de peso, dolor abdominal y debilidad. La característica más identificativa es la presencia de pólipos hamartomatosos del estómago, intestino delgado y colon-recto que histológicamente son indistinguibles de los pólipos juveniles. No obstante, la mucosa no polipoide interpuesta también muestra dilataciones quísticas de las criptas y edema e infl amación de la lámina propia. Las anomalías asociadas consisten enatrofia y fisuras ungueales, pérdida del pelo y zonas con hiper- e hipopigmentación. La causa del síndrome de Cronkhite-Canada es desconocida y no contamos con tratamientos específi cos. El tratamiento nutricional de soporte, que alivia la caquexia y la anemia, puede inducir la remisión en ocasiones. No obstante, son mortales hasta el 50% de los casos.
6

PÓLIPOS HIPERPLÁSICOS
Los pólipos hiperplásicos del colon son proliferaciones epiteliales frecuentes, que se identifican principalmente en pacientes en la sexta o séptima década de la vida.Son los más frecuentes en el colon y el recto. Cuando son únicos, no tienen potencial maligno. Sin embargo, una lesión conocida como adenoma sésil serrado, que tiene algunas similitudes con los pólipos. Son los más frecuentes en el colon y el recto. Cuando son únicos, no tienen potencial maligno. Sin embargo, una lesión conocida como adenoma sésil serrado, que tiene algunas similitudes con los pólipos.
Morfología.Los pólipos hiperplásicos se localizan principalmente en el colon izquierdo y miden típicamente 5 mm de diámetro. Son protrusiones nodulares lisas de la mucosa, que suelen encontrase en las crestas de los pliegues mucosos. Pueden aparecer aislados, pero con mayor frecuencia son múltiples, sobre todo en el recto y en la sigma. Histológicamente los pólipos hiperplásicos están constituidos por células caliciformes y absortibas maduras. El retraso en el desprendimiento de estas células explica su apilamiento, que genera una estructura serrada en
la superficie, que representa la característica típica de estas lesiones.
ADENOMAS O POLIPOS NEOPLASICOS
Los adenomas son pólipos neoplásicos que van desde tumores pequeños, a menudo pediculados, hasta lesiones grandes habitualmente sésiles. Como la incidencia de adenomas en el intestino delgado es muy baja, este apartado se centra en los adenomas del colon. La prevalencia de éstos es del 20 al 30% antes de los 40 años, aumentando al 40-50% después de los 60 años de edad. Los hombres y las mujeres se afectan por igual. Hay una predisposición familiar bien definida ante adenomas nomas esporádicas, siendo responsable de un riesgo aproximadamente cuatro veces mayor de adenomas en los familiares de primer grado, y también es cuatro veces mayor el riesgo de carcinoma colonrectal en cualquier persona con adenomas.
Todas las lesiones adenomatosas surgen como resultado de proliferación y displasia epitelial, que puede variar desde leve hasta intensa, como presentar una transformación hacia un carcinoma. Además, existe una
7

fuerte evidencia de que la mayoría de los adenocarcinomas colonrectales invasivos esporádicos surgen a partir de lesiones adenomatosas preexistentes.
Los pólipos adenomatosos se dividen en cuatro subtipos,
Según la arquitectura epitelial:
• Adenomas tubulares: glándulas principalmente tubulares, repitiendo la topología de la mucosa.
• Adenomas vellosos: proyecciones vellosas.
• Adenomas tubulovellosos: una mezcla de los mencionados anteriormente.
• Adenomas serrados sésiles: epitelio serrado que tapiza las criptas.
Los adenomas tubulares son, con mucho, los más habituales; del 5 al 10% de los adenomas son tubulovellosos, y sólo el 1% es velloso. La mayoría de los adenomas tubulares son pequeños y pediculados; los adenomas vellosos tienden a ser grandes y sésiles. Por el contrario, la mayoría de los pólipos pediculados son tubulares, y los grandes pólipos sésiles habitualmente muestran características vellosas. El riesgo de malignidad en los pólipos adenomatosos se correlaciona con tres características interdependientes (localización del pólipo, arquitectura histológica e intensidad de la displasia epitelial) como sigue:
• El cáncer es raro en los adenomas tubulares de menos de 1 cm de diámetro.
• La probabilidad de cáncer es alta (aproximándose al 40%) en los adenomas vellosos sésiles de más de 4 cm de diámetro.
• La displasia intensa, cuando está presente, se encuentra a menudo en zonas vellosas.
• Entre estas variables, el diámetro máximo es el principal determinante del riesgo de albergar un carcinoma en un pólipo adenomatoso; la arquitectura no proporciona información independiente sustancial.
Morfología
Los adenomas tubulares pueden hallarse en cualquier sitio del colon, pero aproximadamente la mitad se encuentran en el recto-sigma,
8

aumentando la proporción con la edad. Aproximadamente en la mitad de los casos aparecen aisladamente, pero en el resto se distribuyen dos o más lesiones al azar. Los adenomas más pequeños son sésiles; las lesiones de 0,3 cm de tamaño pueden identificarse con endoscopia. Entre los adenomas tubulares más grandes, hasta de 2,5 cm de diámetro, la mayoría tiene tallos delgados, de 1 a 2 cm de largo, y «cabezas» en forma de frambuesa. Histológicamente, el tallo está cubierto por mucosa colónica normal, pero la cabeza está compuesta de epitelio neoplásico, formando glándulas ramificadas revestidas por células grandes, hipercromáticas, algo desordenadas, que pueden o no mostrar secreción de mucina. En algunos casos, existen pequeños focos de arquitectura vellosa. En la lesión claramente benigna, las glándulas ramificadas están bien separadas por la lámina propia, y el nivel de displasia o atipia citológica es leve. Sin embargo, pueden encontrarse todos los grados de displasia, hasta el cáncer confinado a la mucosa (carcinoma intramucoso) o el carcinoma invasor que se extiende a la submucosa del tallo. Un hallazgo frecuente en cualquier adenoma es la erosión superficial del epitelio como resultado de traumatismo mecánico. Los adenomas vellosos son los pólipos epiteliales más grandes y preocupantes. Tienden a aparecer en ancianos, más habitualmente en el recto y recto-sigma, pero pueden localizarse en otros sitios. Generalmente, son sésiles, hasta de 10 cm de diámetro, y son masas aterciopeladas o en forma de coliflor que se proyectan de 1 a 3 cm por encima de la mucosa normal circundante. La histología es la de extensiones viliformes frondosas de la mucosa cubierta por epitelio columnar displásico, a veces muy desordenado, otras veces apilado.
También Pueden encontrarse todos los grados de displasia, y se encuentra carcinoma invasivo hasta en el 40% de estas lesiones, correlacionándose a menudo su frecuencia con el tamaño de los pólipos. Los adenomas tubulovellosos están compuestos por una mezcla amplia de zonas tubulares y vellosas. Están en una situación intermedia entre las lesiones tubulares y vellosas en lo que se refiere a la frecuencia de tener un pedículo o ser sésiles, en su tamaño, el grado de displasia y el riesgo de albergar carcinoma intramural o invasivo.
Características clínicas.
Los adenomas más pequeños habitualmente son asintomáticos hasta el momento en que las hemorragias ocultas dan lugar a anemia clínicamente significativa. Los adenomas vellosos son sintomáticos
9

mucho más frecuentemente por su hemorragia rectal manifiesta u oculta.
Los adenomas vellosos más distales pueden secretar cantidades suficientes de material mucoide rico en proteínas y potasio como para producir hipoproteinemia o hipopotasemia. Cuando se descubren, todos los adenomas, independientemente de su localización en el tracto alimentario, deben considerar potencialmente malignos; así, en términos prácticos, es obligatoria la extirpación inmediata y adecuada.
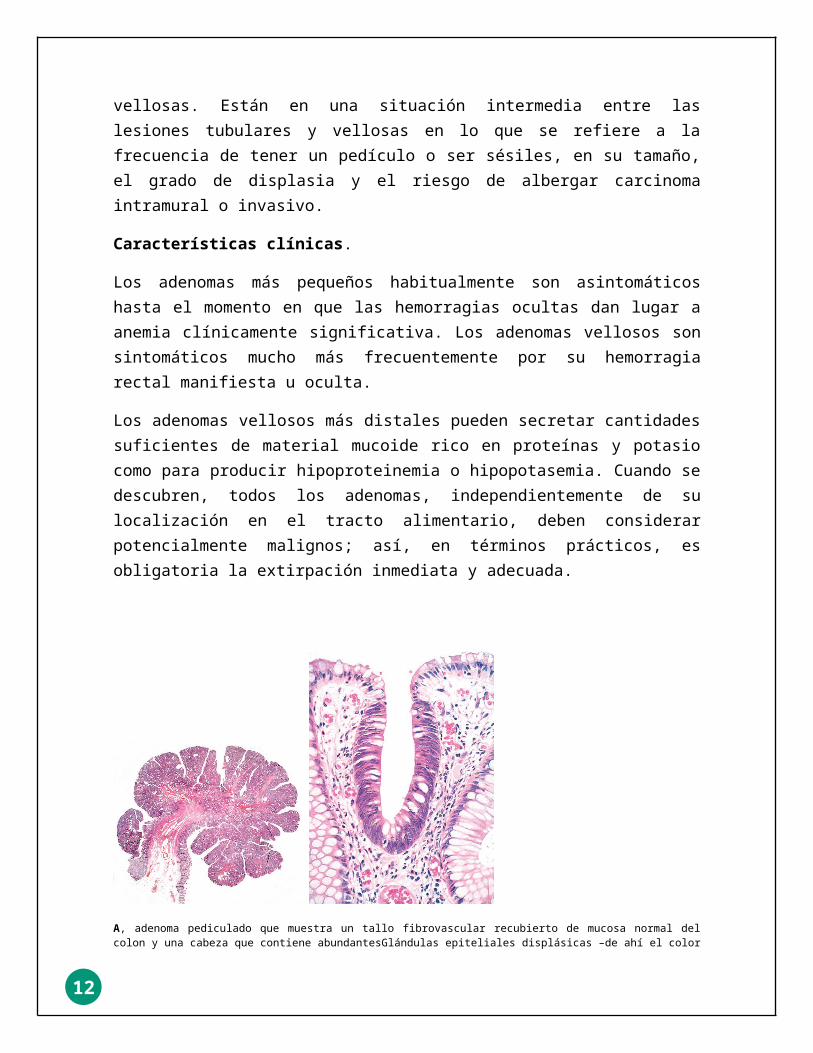
A, adenoma pediculado que muestra un tallo fibrovascular recubierto de mucosa normal del colon y una cabeza que contiene abundantesGlándulas epiteliales displásicas –de ahí el color azul–. B, un pequeño foco de epitelio adenomatoso en una mucosa colónica por lo demás Normal (secretora de mucina, clara) muestra cómo el epitelio columnar displásico (muy teñido) puede poblar una cripta del colon (arquitectura «Tubular»).
Adenoma sésil con arquitectura vellosa. Cada fronda está tapizada Por epitelio displásico. B, porción de una fronda vellosa con Epitelio columnar displásico a la izquierda y epitelio columnar normal Del colon a la derecha.
10

SINDROMES FAMILIARES
Existen varios síndromes asociados a pólipos de colon y a un aumento de la frecuencia de cáncer de colon, de los cuales se han establecido las bases genéticas, lo que implica que se han ampliado los conocimientos actuales acerca del cáncer de colon esporádico. Estos síndromes comprenden: Pólipos adenomatosos familiares (FAP):
Se trata de un trastorno autosómico dominante en el que se da la aparición de numerosos adenomas colorrectales en los adolescentes, debido a mutaciones del gen de la poliposis adenomatosa de colon (Adenomatous polyposis coli; CPA).El diagnostico de FAP clásica, se determina mediante la presencia de al menos 100 polipos , incluso pueden encontrarse algunos miles. A excepción de este importante dato cuantitativo, estas lesiones no se diferencian morfológicamente de los adenomas esporádicos.
El 100% de pacientes no tratados, desarrollan un adenocarcinoma colorrectal, con frecuencia antes de los 30 años; en consecuencia, la colectomía profiláctica representa el tratamiento convencional de los portadores de mutaciones del APC. Sin embargo los pacientes siguen teniendo riesgo de sufrir manifestaciones extraintestinales, incluidas neoplasias en otras localizaciones.
Las mutaciones específicas de APC, se asocian también al desarrollo de otras manifestaciones de la FAP y explican algunas variantes:
Síndrome de Turcot:
Es el síndrome menos frecuente, se caracteriza por Adenomas intestinales, tumores del SNC, las 2/3 parte de los pacientes presentan mutaciones del gen APC y desarrollan meduloblastomas; el tercio restante, sufre mutaciones en uno de los distintos genes implicados en la reparación de ADN y desarrolla glioblastomas Síndrome de Gardner:
Los pacientes con este síndrome, pueden sufrir pólipos intestinales, osteomas mandibulares, craneales, y de los huesos largos, quistes
11
epidermoides, tumores desmoides y tiroideos, y alteraciones dentales como la falta de erupción dentaria y dientes supernumerarios.
Los pacientes que padecen FAP sin pérdida de APC, tienen mutaciones en el gen reparador de la escisión de bases MUTYH.
PAPEL DE LOS GENES EN EL DESARROLLO DE TUMORES
El CA colorrectal hereditario no polipósico (CCRHNP), también llamado síndrome de Lynch se caracteriza por la presencia de acumulación familiar de canceres de distintos orígenes: CA colon y recto, endometrio, estómago, ovario, uréteres, cerebro, intestino delgado, vía hepatobiliar y CA piel.
CA de colon en pacientes CCRHNP suele aparecer a edades más tempranas que los canceres de colon esporádicos, y suelen afectar al colon derecho.
El CCRHNP se debe a mutaciones hereditarias en la línea germinal de los genes que codifican las proteínas responsables de la detección, rotura y reparación de errores que se producen durante la replicación del ADN.
Se han reconocido al menos cinco genes de reparación de errores de este tipo, aunque la mayor parte de los casos CCRHNP se asocian a : MSH2 y MLH1. Los pacientes CCRHNP heredan un gen reparador mutado y un alelo normal. Cuando se pierde la segunda copia por mutación o silenciamiento epigénico, los defectos en la reparación de errores permiten que se acumulen mutaciones a una velocidad hasta 1000 veces superior a lo normal, sobre todo en las regiones que contienen secuencias de ADN cortas repetidas, que se denominan ADN microsatélite.
El genoma humano, consta de aproximadamente 50.000-100.000 microsatélites que tienden a expandirse durante la replicación del ADN, y representan el lugar de mutación más frecuente en los CCRHNP.
ADENOCARCINOMA
El adenocarcinoma de colon es el proceso maligno más frecuente del tubo digestivo y es una causa mayor de morbilidad y mortalidad en todo el mundo. Por el contrario, el intestino delgado, que ocupa el 75% de
12

toda la longitud del tubo digestivo, es un lugar infrecuente de tumores benignos y malignos. Entre los tumores malignos del intestino delgado, los adenocarcinomas y los tumores carcinoides tienen una incidencia aproximadamente igual, seguidos por los linfomas y sarcomas. En consecuencia, nuestro comentario se centra en los adenocarcinomas colorrectales.
Epidemiología. Cada año se producen en EE. UU. Más de 130.000 casos nuevos y 55.000 muertes por un adenocarcinoma colorrectal, lo que representa casi el 15% de todas las muertes relacionadas con el cáncer, sólo después del cáncer de pulmón. La incidencia del cáncer colorrectal alcanza su máximo a los 60 o 70 años de edad, y menos del 20% de los casos se produce antes de los 50 años. Los varones se afectan con una frecuencia ligeramente mayor que las mujeres. El carcinoma colorrectal es más prevalente en EE. UU., Canadá, Australia, Nueva Zelanda, Dinamarca, Suecia y otros países desarrollados. La incidencia de este cáncer es hasta 30 veces menor en India, Sudamérica y África. En Japón, donde la incidencia era muy baja, las tasas han aumentado ahora hasta niveles intermedios (similares a los observados en el Reino Unido), presumiblemente como consecuencia de los cambios en su estilo de vida y su dieta.
Los factores alimentarios más estrechamente relacionados con el aumento de incidencia del cáncer colorrectal son una baja ingesta de fibras vegetales no absorbible y una ingesta rica en hidratos de carbono refinados y grasas. Aunque esas asociaciones son claras, la relación causal entre dieta y riesgo sigue siendo desconocida. Se ha propuesto que el menor contenido de fibra en la dieta disminuye el volumen de las heces y altera la composición de la microflora intestinal. Este cambio puede aumentar la síntesis de subproductos oxidativos del metabolismo bacteriano, potencialmente tóxicos, que se retendrían en contacto con la mucosa del colon durante períodos de tiempo más prolongados como consecuencia del menor volumen de las heces. Las deficiencias de vitaminas A, C y E, que actúan como eliminadores de radicales libres, complican el daño causado por los oxidantes. La ingestión de grandes cantidades de grasa mejora la síntesis hepática de colesterol y ácidos biliares, que pueden convertirse en productos carcinógenos por acción de las bacterias intestinales. Además de la modificación dietética, la prevención con farmacoterapia se ha convertido en un área de gran interés. En varios estudios epidemiológicos se ha propuesto que el ácido acetilsalicílico u otros AINE tienen efectos protectores, lo que concuerda
13

con estudios en los que se demuestra que algunos AINE causan la regresión de los pólipos en pacientes con PAF en los que el recto se conservó después de la colectomía. Se sospecha que este efecto está mediado por la inhibición de la enzima ciclooxigenasa 2 (COX-2), que se expresa con niveles altos en el 90% de los carcinomas colorrectales y en el 40-90% de los adenomas. La COX-2 es necesaria para la producción de prostaglandina E2, que favorece la proliferación epitelial, en particular después de una lesión. También es interesante que la expresión de la COX-2 esté regulada por el gen TLR4, que reconoce los lipopolisacáridos y también se sobreexpresa en adenomas y carcinomas.
Patogenia. En estudios de carcinogénesis colorrectal se han encontrado datos fundamentales sobre los mecanismos generales de la evolución del cáncer. A continuación se revisarán los conceptos relativos específi camente con la carcinogénesis colorrectal. La combinación de episodios moleculares que conducen al adenocarcinoma de colon es heterogénea e incluye anomalías genéticas y epigenéticas. Se han descrito al menos dos vías genéticas diferenciadas.
De forma simple, son la vía APC/ b-catenina, que se asocia a WNT y la secuencia clásica adenoma-carcinoma, y la vía de inestabilidad de los microsatélites, que se asocia a defectos de la reparación de los errores de apareamiento del ADN. Ambas vías implican la acumulación paulatina de varias mutaciones, pero los genes implicados y los mecanismos por los cuales se acumulan las mutaciones son diferentes. Los episodios epigenéticos, el más frecuente de los cuales es el silenciamiento génico inducido por metilación, inducen la progresión siguiendo ambas vías.
• La secuencia clásica adenoma-carcinoma, que es responsable de hasta el 80% de los tumores de colon esporádicos, se refi ere a la mutación APC al inicio del proceso neoplásico. Ambas copias del gen APC deben estar funcionalmente inactivadas, ya sea por una mutación o por episodios epigenéticos, para que se desarrollen los adenomas. El gen APC es un regulador negativo clave de la b -catenina, un componente de la vía de señalización WNT. La proteína APC se une normalmente y favorece la degradación de la _ -catenina. Con la pérdida de la función APC, la _ -catenina se acumula y se transloca en el núcleo, donde activa la transcripción de genes como los que codifican las proteínas MYC y ciclina D1, que favorecen la proliferación. Después, se produce una serie de mutaciones añadidas como las mutaciones que activan el KRAS, que también favorece el crecimiento y previene la
14

apoptosis. La conclusión de que la mutación del KRAS es un suceso tardío se apoya en la observación de que las mutaciones están presentes en menos del 10% de los adenomas menores de 1 cm de diámetro, pero se encuentran en el 50% de los adenomas mayores de 1 cm de diámetro y en el 50% de los adenocarcinomas invasivos. La progresión neoplásica también se asocia a mutaciones de otros genes supresores tumorales como los que codifi can las proteínas SMAD2 y SMAD4, que son efectoras de la señalización del TGF- _ . La señalización TGF- _ normalmente inhibe el ciclo celular, la pérdida de esos genes permite el crecimiento celular sin limitaciones. El gen supresor tumoral p53 está mutado en el 70-80% de los cánceres de colon, pero su afectación es infrecuente en los adenomas, lo que indica que las mutaciones p53 también aparecen en etapas posteriores de la progresión tumoral. La «pérdida de función» del gen p53 y de otros genes supresores tumorales se puede deber a deleciones cromosómicas, lo que señala a su vez que la inestabilidad cromosómica es una de las características de la vía APC/ _ -catenina. Otra posibilidad es que los genes supresores tumorales pueden estar silenciados por la metilación de la zona rica en CpG o del islote CpG, una región 5’ de algunos genes que con frecuencia incluyen el gen promotor y el locus de inicio de la transcripción. La expresión de la telomerasa también aumenta a medida que las lesiones son más avanzadas.
• En pacientes con deficiencias en la reparación de los errores de apareamiento del ADN (por la pérdida de genes de reparación de los errores de apareamiento, como ya hemos comentado) las mutaciones se acumulan en repeticiones de microsatélites, una situación denominada inestabilidad de microsatélites. Aunque esas mutaciones son silentes porque los microsatélites se encuentran normalmente en regiones no codifi cadoras, algunas secuencias de microsatélites se localizan en la región codifi cadora o promotora de genes implicados en la regulación del crecimiento celular, como los que codifi can el receptor tipo II del TGF- _ y la proteína proapoptósica BAX. Como el TGF- _ inhibe la proliferación de células epiteliales en el colon, los receptores tipo II del TGF- _ mutados contribuyen al crecimiento celular no controlado, mientras que la pérdida del gen BAX mejora la supervivencia de los clones genéticamente anormales. Las mutaciones del oncogén BRAF y el silenciamiento de grupos distintos de genes debido a la hipermetilación del islote CpG también son frecuentes en los cánceres que se desarrollan a través de los defectos de reparación de los errores de
15

apareamiento del ADN. Por el contrario, los genes KRAS y p53 no están normalmente mutados. Por tanto, la combinación de la inestabilidad de microsatélites, mutaciones BRAF y metilación de las dianas específicas, como MLH1, es la fi rma de esta vía de carcinogenia. También existe un tercer grupo de cánceres de colon con aumento de la metilación los islotes de CpG en ausencia de la inestabilidad de microsatélites. Muchos de esos tumores albergan las mutaciones KRAS pero las mutaciones p53 y BRAF son infrecuentes. Por el contrario, las mutaciones p53 son frecuentes en los cánceres de colon que no presentan el fenotipo metilador de los islotes CpG. Aunque la morfología no puede predecir con fi abilidad los episodios moleculares subyacentes que conducen a la carcinogenia, se han encontrado algunas correlaciones con la defi ciencia de la reparación de errores y la inestabilidad de los microsatélites. Esas alteraciones moleculares son frecuentes en los adenomas sésiles serrados. Además, los carcinomas invasivos con inestabilidad de los microsatélites sufren una diferenciación mucinosa prominente con infiltrados linfocíticos peritumorales. Esos tumores, así como los que tienen el fenotipo metilador de los islotes de CpG, se localizan con frecuencia en el colon derecho. Los tumores con inestabilidad de los microsatélites se pueden reconocer por la ausencia de tinción inmunohistoquímica de las proteínas de reparación de los errores o por los análisis de genética molecular de las secuencias de microsatélites. Es importante identifi car los casos con CCRHNP por las implicaciones que tiene la entidad en el consejo genético, el elevado riesgo de un segundo proceso maligno en el colon y otros órganos y, en algunos casos, por las diferencias en el pronóstico y en el tratamiento.
Morfología. En conjunto, los adenocarcinomas siguen una distribución aproximadamente igual en todo el colon. Los tumores del colon proximal crecen como masas exofíticas polipoides que se extienden por una pared del ciego y colon ascendente, unas zonas de gran calibre. Esos tumores raramente causan obstrucción. Por el contrario, los carcinomas en el colon distal tienden a ser lesiones anulares que producen constricciones «en servilletero» y estenosis luminal, a veces hasta el punto en que se produce la obstrucción. Ambas formas crecen en la pared intestinal con el tiempo y pueden ser palpables como masas firmes. Las características microscópicas generales de los adenocarcinomas del colon derecho e izquierdo son similares. La mayoría de los tumores están formados por células cilíndricas altas que se parecen al epitelio displásico encontrado en los adenomas. El
16

componente invasivo de esos tumores provoca una respuesta desmoplásica importante en el estroma que es responsable de su consistencia firme característica. Algunos tumores mal diferenciados forman pocas glándulas, otros producen abundante mucina que se acumula dentro de la pared intestinal y se asocian a un mal pronóstico. Los tumores también pueden estar formados por células en anillo de sello que son similares a las del cáncer gástrico. Otros muestran características de diferenciación neuroendocrina.
Características clínicas. La disponibilidad de la detección selectiva endoscópica combinada con el reconocimiento de que la mayoría de los carcinomas surge sobre adenomas representa una oportunidad única para la prevención del cáncer. Por desgracia, los cánceres colorrectales evolucionan insidiosamente y, por tanto, no se detectan durante períodos prolongados. Los cánceres de ciego y del colon derecho se estudian por la aparición de cansancio y debilidad por anemia ferropénica. En consecuencia, se aplica la máxima clínica de que la causa subyacente de una anemia ferropénica en un varón de edad avanzada o una mujer posmenopáusica es un cáncer digestivo hasta que se demuestre lo contrario. Los adenocarcinomas colorrectales izquierdos producen hemorragia oculta, cambios en los hábitos intestinales o dolores cólicos o molestias en el cuadrante inferior izquierdo. Aunque las histologías poco diferenciadas y mucinosas se asocian a un mal pronóstico, los dos factores pronósticos más importantes son la profundidad de la invasión y la presencia o ausencia de metástasis en los ganglios linfáticos. La invasión de la muscular propia confiere una reducción signifi cativa de la supervivencia que disminuye aún más por la presencia de metástasis en ganglios linfáticos. Esos factores fueron reconocidos por primera vez por Dukes y Kirklin y forman el núcleo de la clasificación TNM (tumor-ganglios-metástasis) ( tabla 17-11 ) y del sistema de estadificación ( tabla 17-12 ) del American Joint Committee on Cancer. Con independencia del estadio, hay que recordar que algunos pacientes con un pequeño número de metástasis evolucionan favorablemente durante años después de la resección de los nódulos tumorales a distancia. Una vez más, se resalta la heterogeneidad clínica y molecular de los carcinomas colorrectales. Las metástasis afectan a los ganglios linfáticos regionales, pulmó) y hueso, pero como consecuencia del drenaje portal del colon, el hígado es la localización más frecuente de las lesiones metastásicas El recto no
17

drena utilizando la circulación portal y los carcinomas de la región anal que metastatiza lo hacen a menudo evitando el hígado.
TUMORES DEL APÉNDICE
Los carcinoides, son los tumores más frecuentes del apéndice. Las únicas otras lesiones que merece
la pena mencionar son el mucocele del apéndice y las neoplasias de la mucosa.
Mucocele: Dilatación de la luz del apéndice por secreción mucinosa debido a una obstrucción no neoplásica de la luz y se asocia habitualmente con un fecalito en la luz que permite la acumulación
lenta de secreciones mucinosas estériles
Las neoplasias mucinosas van desde el cistadenoma mucinoso benigno, histológicamente idéntico a los tumores análogos en el ovario; hasta el cistoadenocarcinoma mucinoso que invaden la pared,
18

permitiendo que las células tumorales se implanten en la cavidad peritoneal, que puede llenarse de mucina dando lugar a una forma de cáncer intraperitoneal denominado seudomixoma peritoneal.
TUMORES DEL CANAL ANAL
El canal anal se divide en tercios. La zona superior está recubierta de un epitelio rectal cilíndrico, el tercio medio está formado por epitelio transicional y el tercio inferior está recubierto por epitelio escamoso estratifi cado. Los carcinomas del canal anal tienen patrones de diferenciación típicos glandulares o escamosos,que representan el epitelio normal de los tercios superior e inferior, respectivamente. Otro patrón de diferenciación más, denominado basaloide, está presente en los tumores poblados por células inmaduras derivadas de la capa basal del epitelio transicional. Cuando todo el tumor muestra un patrón basaloide, aún se aplica el término arcaico carcinoma cloacógeno. La diferenciación basaloide se puede mezclar con la diferenciación escamosa o mucinosa. Todas ellas se consideran variantes del carcinoma del canal anal. El carcinoma epidermoide puro del canal anal se asocia con frecuencia a infección por el VPH, que también causa lesiones precursoras como el condiloma acuminado.
BIBLIOGRAFÍA
Kumar. V, Abbas A., Fausto N., Richard N., Patología Humana Robbins Ed. 8a
Kumar. V, Abbas A., Fausto N., Richard N., Patología structural y functional de
Robbins y cotran Ed. 8a
19