hemostasia
DESCRIPTION
Presentación hemostasia en powerpointTRANSCRIPT

Son los mecanismos por los cuales se detiene la hemorragia.
En fisiología, son los mecanismos que permiten a la sangre mantenerse como un tejido liquido en el interior de los vasos y formar un coagulo cuando ocurre una hemorragia
Es parte de la respuesta inflamatoriasistémica.

Tiene 3 fases:
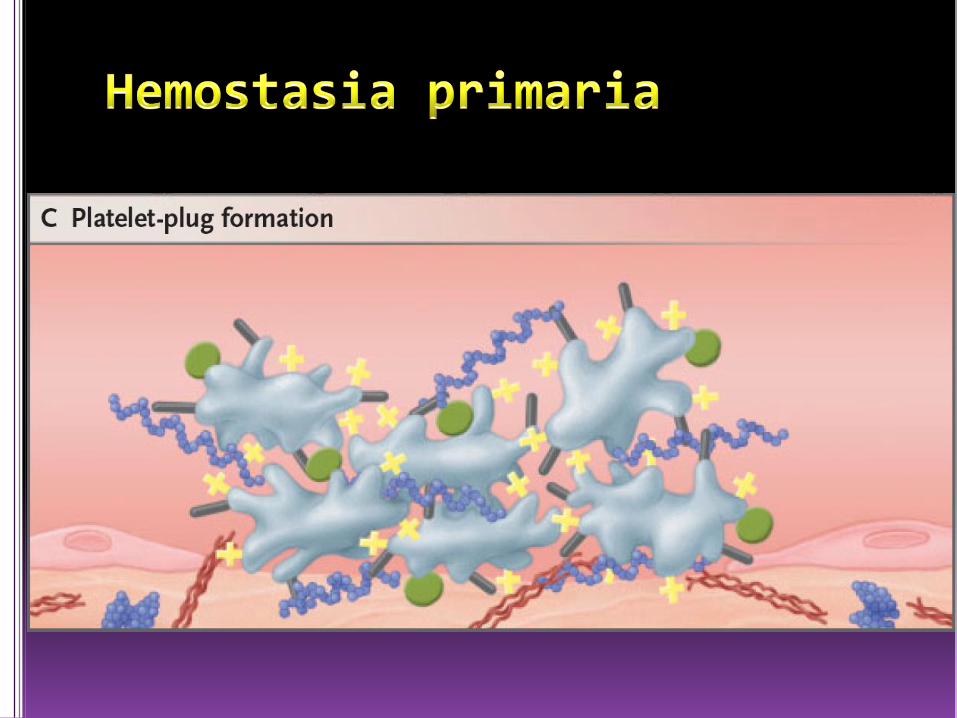
Primaria
• Su finalidad formar un tapón de plaquetas que se une alvaso dañado
Secundaria
• Su finalidad es formar fibrina en grandes cantidades paraestabilizar el tapón de plaquetas formando un coágulo
Fibrinólisis
•Limita la coagulación a un solo sitio y desintegra elcoágulo cuando ya no es útil



3 x 0.5 µm
6-10 fL
7 a 10 días de vida


Agonista Receptor Comentario
Adenosin difosfato (ADP)
P2Y1 y P2Y12 Liberado de los gránulos densos
Trombina PAR1 y PAR4 Formada por la activación de la cascada de la coagulación, el agonista mas potente in vivo
Colágeno GP Ia, GP IV y GP VI Presente en el subendotelio
Adrenalina (epinefrina) α2 Amina presente en el plasma
Calcio Incremento en el Ca++ intracelular
Acido araquidonico TPα y TPβ(receptores TxA2)
Liberado al activarse las plaquetas
Serotonina Liberado de los gránulos densos
Factor activador de plaquetas
Producido por otras células

Componente subendotelial Receptor plaquetario Otros nombres
Colagena GP Ia/IIaGP IVGP VI
α2β1, VLA-2GP IIIb, CD 36
Fibrinógeno ** GP IIb/IIIa αIIbβ3, CD41/CD61
Factor de Von Willebrand GP Ib/IX/V CD42b/CD42a
Fibronectina GP Ic/IIa α5β1, VLA-5
Trombospondina Receptor de la vitronectina αVβ3
Laminina GP Ic/IIa α6β1
** No es un componente subendotelial





Tipo de factor Nombre (Número) Producción Vida media
Proteína estructural
Fibrinógeno (I) Hígado /Megacariocito 70-120 h
Zimógenos GLA
No GLA
Protrombina (II)Factor VIIFactor IXFactor X
Factor XIFactor XIIFactor XIIIPrecalicreína
HígadoHígadoHígadoHígado
HígadoHígadoHígadoHígado
60-70 h6 h24 h40 h
52 h60 h240 h35 h
Cofactores Solubles
Celulares
Factor VFactor VIIICAPM
Factor Tisular (III)
Hígado/MegacariocitoEndotelioHígado
Endotelio/Monocitos
12 h8-12 h150 h
----
Iones Calcio (IV) ---- ----

Carboxilación de los factores K dependientes

X-asa intrínseco X-asa extrínseco II-asa
Factor K dependiente
IXa VIIa Xa
Cofactor VIIIa FT Va
Otros Fosfolípidos y Ca++ Fosfolípidos y Ca++ Fosfolípidos y Ca++


La coagulación se lleva a cabo sobre lasmembranas (fosfolipidos) de las células queexpresan factor tisular y de las plaquetas
La coagulación inicia por la exposición de FT(vía extrínseca)
La fase de contacto no inicia la coagulación,pero es la conexión con la fibrinólisis y lainflamación

Fases:
Iniciación
Formar pequeñas cantidades de trombina
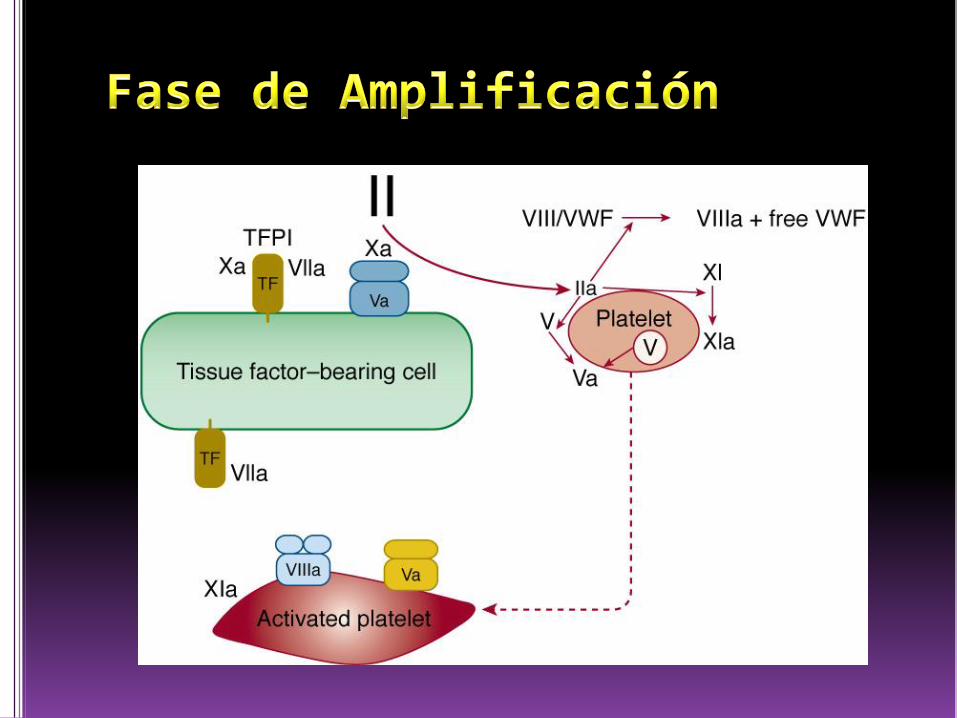
Amplificación
Activar plaquetas y factores V, VIII, XI y XIII
Propagación
Formar grandes cantidades de trombina y de fibrina

TFPI


Dominios D
La fibrina, inicialmente formada de uniones no
Covalentes sufre en presencia del FXIII la incorporación
De enlaces covalentes, que le dan una mayor resistencia

Inhibidor principal de latrombina, y de los FXa,FIXa, FXIa, XIIa y FVIIa
Su actividad aumenta1000 veces enpresencia de sulfato deheparan endotelial

La trombina se une a la TM endotelial y activa a la PC.
La PCA se une al REPC y actúa como anticoagulante degradando a los FV y FVIII, teniendo como cofactor a la PS


La plasmina es laenzima central de estesistema, circula comoproenzima(plasminogeno), latransformación selleva a cabo poractivadores (t-PA y u-PA)
La plasmina degradafibrina, fibrinógeno,FV, FVII, etc.La plasmina tiene como inhibidor a la α2
Antiplasmina y los activadores del plasminogeno
Tienen como reguladores a los inhibidores de
Los activadores del plasminogeno (PAI1, 2 y 3)


Tiempo de sangrado Técnica de Duke (lóbulo
de la oreja, en desuso)
Técnica de Ivy(antebrazo), normal 3 a 9minutos Evalúa pared vascular,
plaquetas y adhesión
Prolonga entrombocitopenia,trombocitopatias, defectosvasculares, enf. De VonWillebrand, etc

PFA-100: Mide el tiempo que tarda enocluirse un flujo de sangre en el que lasplaquetas son expuestas a agonistas
Agregometría: Registro grafico de larespuesta de agregación a distintos agonistas(epinefrina, ristocetina, ADP, etc)
Citometría de flujo: Determina lasglicoproteínas plaquetarias (CD 42,CD41/CD61)

Condiciones preanaliticas
Punción no traumática, para evitar la liberación detromboplastina
Tubo de tapón azul, de cristal siliconado o plásticocon citrato de sodio, relación 1:9 entreanticoagulante y sangre
En el laboratorio se centrifuga y separa el plasma,debe procesarse el mismo día o guardarse a -20grados

Tiempo de protrombina (TP)
Es el tiempo que tarda en formarse un coagulodespués de añadir al plasma citratado un reactivollamado tromboplastina cálcica (mezcla de factortisular, cefalina y calcio), evalúa la vía extrínseca ycomún (FVII, FX, FV, FII, fibrinógeno)
Normal: 10-15 seg

Causas de TP prolongado
Déficit de vitamina K
Hepatopatías
Uso de AO (Warfarina)
Hipofibrinogenemia

Tiempo de tromboplastina parcial activada(TTPa)
Es el tiempo que tarda en formarse un coagulodespués de añadir al plasma citratado el reactivode tromboplastina cálcica sin FT (solo cefalina ycalcio) y un activador de la fase de contacto (sílice,caolín o acido elagico) evalúa la vía intrínseca ycomún (todos los factores excepto VII y XIII)
Normal: 25-35 seg

Causas de un TTPa prolongado
Presencia de heparina
Anticoagulante lupico
Hemofilias
Deficiencia de factores de la fase de contacto
Uso de AO
Hepatopatía

Causas de TP y TTPa prolongado
Falla hepática
Deficiencia de vitamina K
Uso de AO
Deficiencia de los factores de la vía común (FX, FV, FII)
CID
Anticoagulante lupico

Causas de sangrado con tiempos normales
Déficit de FXIII
Déficit de alfa 2 antiplasmina
Déficit de PAI-1
Defectos vasculares
Enfermedad de Von Willebrand
Anticoagulantes como HBPM, fondaparinux, Rivaroxaban o Dabigatran

Fibrinógeno: 200 a 400 mg/dL
Tiempo de trombina (TT)
Es el tiempo que tarda en coagular un plasmacitratado al que se agrega trombina. Valora laconversión de fibrinógeno a fibrina. Normal 15 a20 seg
Se prolonga en disfibrinogenemias,hipofibrinogenemia, heparina, PDF altos, etc

Tiempo de reptilasa
Es un TT sin trombina, usando en su lugar venenode la serpiente Bothrops atrox. No se afecta porheparina.
Determinación de factores
Investigación de ACL
Prueba de Bethesda para búsqueda deinhibidores

PDF: Mide productos de la degradación delfibrinógeno y la fibrina.
DD: Detecta productos específicos de ladegradación de la fibrina.
Lisis de euglobulinas