· title: estudios de estabilidad fisicoquímica. vicente merino created date: 6/5/2019 10:50:56...
TRANSCRIPT


Agenda
Concepto de Estabilidad
Aspectos regulatorios y metodológicos
(ICH, FDA, Gerpac,…)
Metodología de desarrollo de EE
Resultados destacables de los EE del GT de FT

Estabilidad: concepto
• Capacidad que tiene un medicamento para mantener en el tiempolas propiedades y características propias, dentro de márgenes decalidad establecidos.
• El período de tiempo debe de contemplar desde su fabricación, y abarcar el tiempo de almacenamiento y de uso (vida útil)
• Estamos hablando de conservar las propiedades físicas, químicas, microbiológicas y biofarmacéuticas que definen a los medicamentos.

Tipos de estabilidad (USP-40)
Tipo de Estabilidad Condiciones mantenidas durante toda la Vida Útil delProducto
Química Cada ingrediente activo conserva su integridad química y la potenciadeclarada en la etiqueta, dentro de los límites especificados
Física Se conservan las propiedades físicas originales, entre ellas, aspecto, palatabilidad, uniformidad, disolución y capacidad de suspensión
Microbiológica Se conserva la esterilidad o resistencia a la proliferación microbiana según los requisitos especificados. Los agentes antimicrobianos (si lo están) conservan la eficacia dentro de los límites especificados
Terapéutica No se altera el efecto terapéutico
Toxicológica No se produce ningún aumento significativo de la toxicidad
The United States pharmacopeia, 40nd rev., and The national formulary, 35th ed. Rockville (MD):United States Pharmacopeial Convention; 2017:1662-66.

Factores que afectan a la estabilidad
Hidrólisis
Epimerización
Descarboxilación
Deshidratación
Oxidación
Descomposición fotoquímica
Fuerza iónica
Efecto del pH
Compatibilidad Inter-iónica
Estabilidad en estado sólido
Temperatura
The United States pharmacopeia, 40nd rev., and The national formulary, 35th ed. Rockville (MD):United States Pharmacopeial Convention; 2017:1662-66.

¿Estudio de estabilidad?: por dónde empezamos…
Aspectos Regulatorios y Metodológicos

Fuentes bibliográficas para desarrollo de estudios estabilidad
• ICH: International Conference On Harmonisation of Technical Requeriments ForRegistration of Pharmaceuticals For Human Use.
• Farmacopeas: RFE, EP, USP, JP, BP,…
• Guías de sociedades científicas GERPAC
(Groupe d’Evaluation et de Recherche sur la Protection en Atmosphère Contrôlée).
• FDA: Stability testing of drugs substances and drugs products.
• OMS: Guideline for stability testing of pharmaceutical ingredients and finishedpharmaceutical products.
• Bibliografía: American Journal Health-System Pharmacy, IJPC, Pharmaceutical Development and Technology,...

Guías ICH
Reúne a las autoridades reguladoras y la industria farmacéutica para discutir los aspectos científicos y técnicos del registro de medicamentos.
Hacer recomendaciones para armonización en la interpretación y aplicación de directrices técnicas el registro de productos (EEUU, Europa y Japón)
Diálogo constructivo sobre cuestiones científicas entre las autoridades reguladoras y la industria farmacéutica sobre la armonización de los requisitos técnicos para los productos farmacéuticos.
Supervisar y actualizar los requisitos técnicos armonizados que conduzcan a una mayor aceptación mutua de los datos de investigación y desarrollo.
Evitar los requisitos divergentes futuros, a través de la armonización de determinados temas necesarios como resultado de los avances terapéuticos y el desarrollo de nuevas tecnologías para la producción de medicamentos.
Facilitar la adopción de enfoques técnicos y de investigación nuevos o mejorados que actualicen o sustituyan las prácticas actuales.
https://www.ich.org

Condiciones de almacenamiento
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2__Guideline.pdf
https://www.fda.gov/downloads/drugs/guidances/ucm073369.pdf
http://apps.who.int/medicinedocs/documents/s19133en/s19133en.pdf

ICH: TEST DE FOTOESTABILIDAD Q1B
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1B/Step4/Q1B_Guideline.pdf

Farmacopeas
Real Farmacopea Española (RFE)
Farmacopea Europea (EP)
United States Pharmacopeia (USP)
Japanese Pharmacopiea (JP)
British Pharmacopeia (BP)
International Pharmacopeia (IP)
ASP
ECTO
S M
ETOD
OLÓ
GIC
OS
TECN
OFA
RM
AC
ÉUTIC
OS

Ejemplo USP: piridoxina

Guía de la GERPAC
• Grupo Francés de gran experiencia
• Se basan en normativos ICH y EP.
• Conceptos claros y bien definidos.
• Disponible en Francés e Inglés.
• Buen punto de partida.

Etapas en el desarrollo de estudios de estabilidad
Identificación de la formulación a estudiar
Busqueda bibliográfica de estudios previos
Especificar que queremos determinar.
Buscar la metodología para el análisis (HPLC,
pH, osmolalidad,…)
Desarrollo metodológico del estudio (ICH,
USP,otras fuentes,…)
Validación de técnicas analíticas
Estudio de estabilidad propiamente dicho
Interpretación de resultados
Realización del informe final
Publicación del estudio

El paso siguiente…
BUSCAR ALIANZAS
UNIVERSIDADES
INSTITUTOS DE BIOMEDICINA
LABORATORIOS DE ANÁLISIS PRIVADOS
OTROS (instrumental propio,…)

Ayudas a GT de la SEFH
Estudio de estabilidad de soluciones orales pediátricas
2014-2015: Clonidina, Hidralazina, Fenobarbital
2015-2016: Isoniazida (hidrazina) y Etambutol (aminobutanol)
2016-2017: Riboflavina, Tiamina, Piridoxina y Ácido Fólico

SOLUCIÓN DE PIRIDOXINA HCl 25 MG/ML
Ejemplo común metodológico

Objetivo
Estudio de la estabilidad fisicoquímica de piridoxina HCl 25 mg/mL solución
En diferentes condiciones de conservación durante un período de tiempo de 90 días siguiendo las recomendaciones de las guías ICH.
Las condiciones a estudiar son tres:
• Refrigeración (5±3ºC)
• Temperatura ambiente (25±2ºC)
• 40 ºC (±2ºC)
Estudio microbiológico en la condición más favorable según USP 61 y 62
• Envases cerrados
• Envases abiertos
http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html

HPLC y validación del método
Método cromatográfico:•Columna C18 4.6x250 mm, 5 µm
•Fase móvil: 1% ácido acético glacial (exceso)
• 1.40 mg/ml de hexanosulfonato de sodio en 400 mL de agua Milli-Q, 1 mL ácido acético glacial y 600 mL de Metanol (ajustándose a pH=7.2 con KOH)
•Flujo 1 mL/min
•Inyección 1 µL
•Longitud de onda= 254 nm
•Temperatura columna 25ºC.
•Tiempo de análisis: 10 minutos/muestra.
Validación del método:
• Recta de calibrado: 0.1—0.35 mg/mL
• Exactitud: 0.1, 0.2 y 0.35 mg/mL
• Precisión intra e interdía: 0.1, 0.2 y 0.35 mg/mL
• Especificidad y selectividad:análisis espectral2D-3D
• Límite de cuantificación y de detección
The United States pharmacopeia, 40nd rev., and The national formulary, 35th ed. Rockville (MD):United States Pharmacopeial Convention; 2017:5925-27.

Fundamentos HPLC
HPLC: técnica de separación de moleculas orgánicas en disolución
Propiedades hidrofóbicas diferentes entre moléculas (HPLC-RP)
Permite identificación: cada molécula aparece a un TIEMPO DE RETENCIÓN CARACTERÍSTICO
Permite cuantificación: relación Señal-Concentración
La separación se lleva a cabo en COLUMNA porosa de Octadecilsilano (C18)
Versatilidad/Universalidad
(multitud de aplicaciones)

Elaboración Piridoxina HCl 25 mg/mL
Fórmula 1 (CON conservante)
• Piridoxina HCl.......................................2.5 g
• Agua cons. sin propilenglicol……..…….20 mL
• Jarabe simple csp..…….…………………..100 mL
Fórmula 2 (SIN conservante)
• Piridoxina HCl.....................................2.5 g
• Agua purificada……..…………………….….20 mL
• Jarabe simple csp..…….…………………..100 mL
*Agua conservante sin propilenglicol:o Nipagin base…………………………………0.08 go Nipasol base…………………………………0.02 go Agua purifcada csp……………………….100 mL
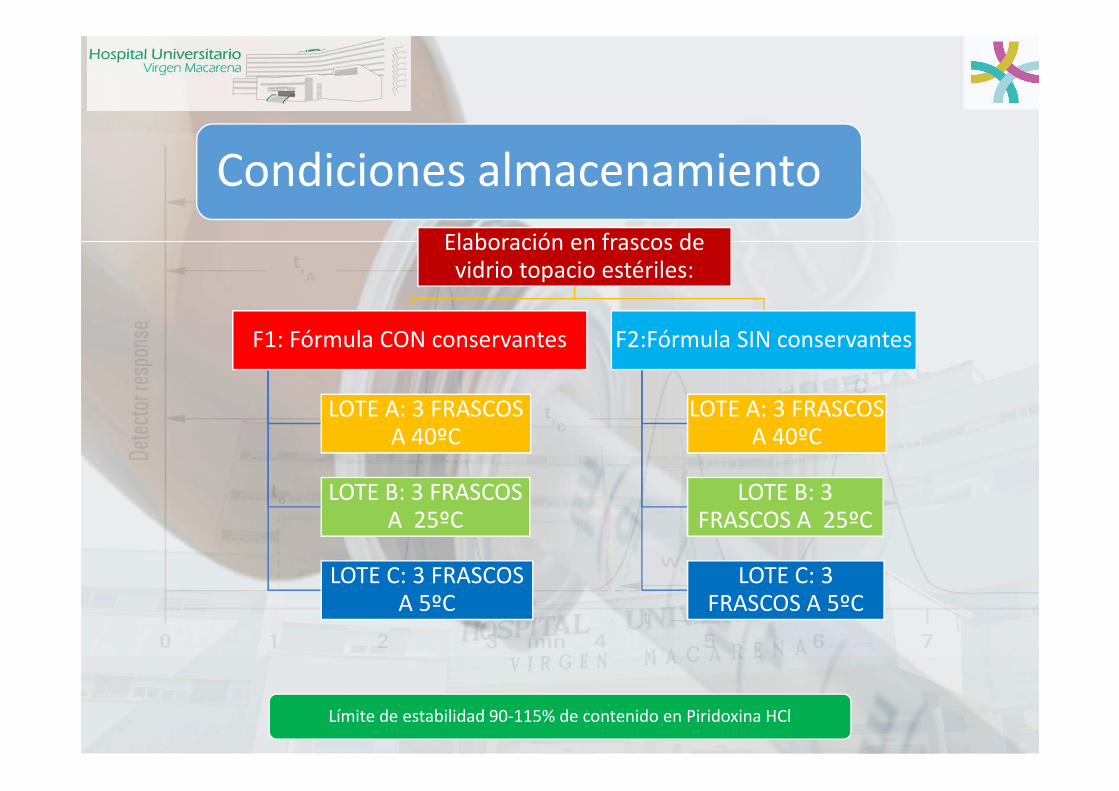
Condiciones almacenamiento
Elaboración en frascos de vidrio topacio estériles:
F1: Fórmula CON conservantes
LOTE A: 3 FRASCOS A 40ºC
LOTE B: 3 FRASCOS A 25ºC
LOTE C: 3 FRASCOS A 5ºC
F2:Fórmula SIN conservantes
LOTE A: 3 FRASCOS A 40ºC
LOTE B: 3 FRASCOS A 25ºC
LOTE C: 3 FRASCOS A 5ºC
Límite de estabilidad 90-115% de contenido en Piridoxina HCl

Metodología: control pH y visual
Control del pH en los días análisis:
• pHmetro CRISON GLP21+
• 3 puntos de calibración (pH 4.01, 7.00 y 9.21)
Control visual:
• Sobre fondo claro
• Sobre fondo oscuro
Control Osmolalidad
• Osmómetro OsmoSTATION® OM-6050 ARKRAY
• Análisis día 0 y 90.

Metodología: estudio microbiológico
Prueba de promoción del crecimiento y aptitud del método de recuento
•Se comprobó la capacidad de crecimiento de cepas patrón en cada formulación del estudio
•S. aureus ATCC 6538, P. aeruginosa ATCC 9027, B. subtilis ATCC 6633, C. albicans ATCC 10231, A. brasiliensis ATCC 16404, E. coli ATCC 8739, S. enterica ATCC 14028 y C. sporogenes ATCC 11437.
Prueba de recuento microbiano en placa
•Recuento total de microorganismos aerobios (RTMA) y el recuento total combinado de hongos filamentosos y levaduras (RTCHL) que podían desarrollarse en condiciones aerobias,
•Límite: RTMA de 102 y RTCHL 101 y debe haber ausencia de E. coli.
Prueba de microorganismos específicos
•Demostrar la ausencia o presencia limitada de microrganismos específicos:
Detección de bacterias gram negativas tolerantes a bilis
E. coli, Salmonella, P. aeruginosa, S.aureus, Clostridios, C. albicans.
Si hubiera crecimiento identificación final por MALDI-TOF
<USP-61> The United States pharmacopeia, 40nd rev., and The national formulary, 35th ed. Rockville (MD) United States Pharmacopeial Convention; 2017:57-61.
<USP-62>The United States pharmacopeia, 40nd rev., and The national formulary, 35th ed. Rockville (MD):United States Pharmacopeial Convention; 2017:61-7.

Metodología: Estabilidad microbiológica
Ensayo sobre envases cerrados:Seaplicaron los métodos antes
mencionados sobre las formulaciones (con conservante y sin conservante) en
la condición más estable desde el punto de vista FQ durante los días 0, 7,
14, 28, 42, 60 y 90.
Ensayo sobre envases abiertos:Paraemular unas condiciones de uso real y
evaluar la posible contaminación microbiana, los envases se abrieron y cerraron 1 vez al día durante los 42
días que duró este ensayo. El muestreo se realizó a los 0, 7, 14 y 28 y
42 días.

RESULTADOS DESTACABLES

Estudio de clonidina (interacción conservante)
Dihidropiridonas
Dihidropiridonas

Influencia del pH de los vehículos
Componentes/pH media±DE
A. Jarabe simple (64
partes sacarosa/36
partes de agua)
B. Jarabe simple con
conservantes
parahidroxibenzoato de
metilo y propilo base
(0.06% p/v-0.04%)
C. Jarabe simple con
conservantes
parahidroxibenzoato de
metilo sódico y propilo
sódico (0.06% p/v-0.04%
p/v)
D. Solución de sorbitol al
70% con sorbato potásico
0.15 % p/v
AGUA PURIFICADA PhEur
+ AZUCAR DOMÉSTICO6.89±0.050 6.32±0.017 8.79±0.005
6.44±0.005
(EN AGUA PURIFICADA PhEur)AGUA PURIFICADA PhEur
+ SACAROSA PhEur6.60±0.005 5.69±0.015 8.71±0.010
AGUA ULTRAPUFICADA
MILLI-Q + SACAROSA
PhEur
6.87±0.080 6.41±0.010 8.75±0.010 6.45±0.005
(EN AGUA ULTRAPURIFICADA
MILLI-Q)AGUA ULTRAPUFICADA
MILLI-Q + AZUCAR
DOMÉSTICO6.49±0.051 6.06±0.026 8.90±0.050
pH AGUA
ULTRAPURIFICADA
MILLI-Q6.58±0.010
pH AGUA PURIFICADA
PhEur 5.56±0.010
SOLUCIÓN DE MIDAZOLAM 1 mg/mL
A B C
Merino-Bohórquez et al. Comunicación Oral. 60 Congreso SEFH, Valencia 2015.

Fórmula 1 (CON conservante)
• Fenobarbital sódico...................1090 mg
• Glicerina...........................................10 g
• Sacarina sódica...........................100 mg
• Sorbitol 70%..................................30 mL
• Metilparaben sódico……...........….80 mg
• Propilparaben sódico………………...20 mg
• Agua purifcada csp……………….....100 mL
Fórmula 2 (SIN conservante)
•Fenobarbital sódico...................1090 mg
•Glicerina...........................................10 g
•Sacarina sódica...........................100 mg
•Sorbitol 70%..................................30 mL
•Agua purifcada csp……….……….....100 mL
Ejemplo pH: Fenobarbital
Merino-Bohórquez et al. Comunicación Póster Papel. 61 Congreso SEFH, Madrid 2017,
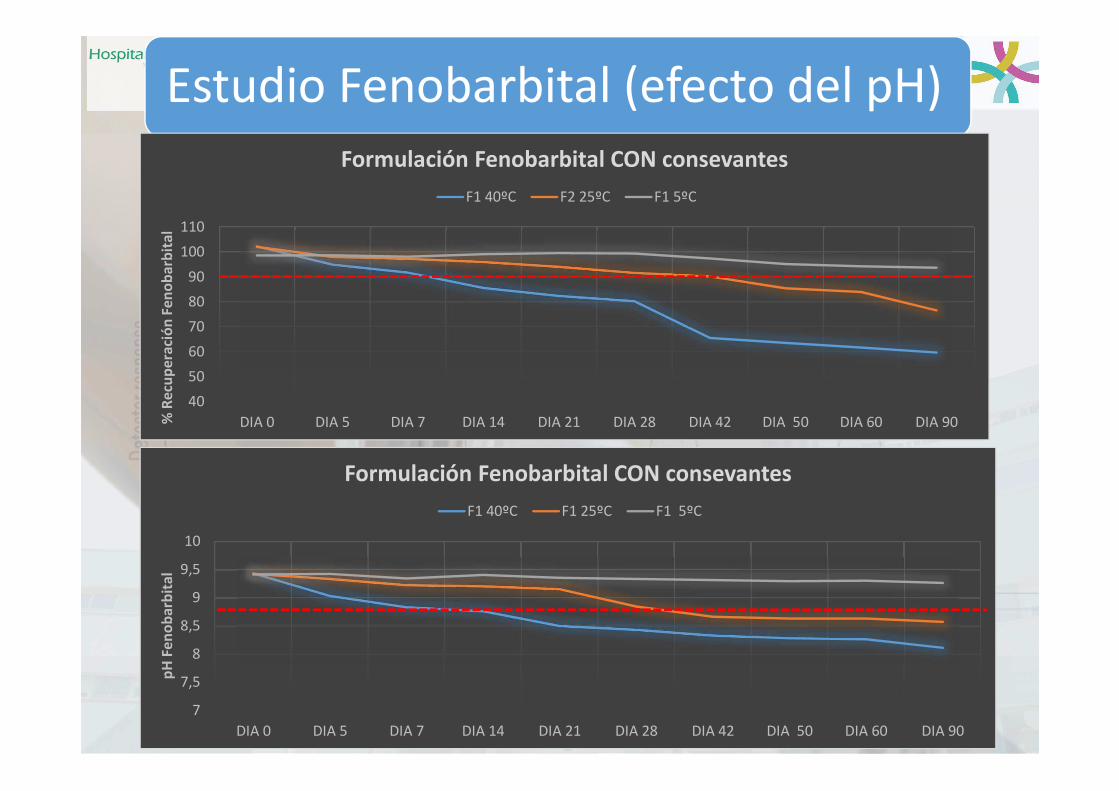
Estudio Fenobarbital (efecto del pH)
40
50
60
70
80
90
100
110
DIA 0 DIA 5 DIA 7 DIA 14 DIA 21 DIA 28 DIA 42 DIA 50 DIA 60 DIA 90% R
ecu
per
ació
n F
eno
bar
bit
alFormulación Fenobarbital CON consevantes
F1 40ºC F2 25ºC F1 5ºC
7
7,5
8
8,5
9
9,5
10
DIA 0 DIA 5 DIA 7 DIA 14 DIA 21 DIA 28 DIA 42 DIA 50 DIA 60 DIA 90
pH
Fen
ob
arb
ital
Formulación Fenobarbital CON consevantes
F1 40ºC F1 25ºC F1 5ºC

min2 4 6 8 10 12 14
mAU
0
20
40
60
80
100
120
140
3.186
13.04
5
min2 4 6 8 10 12 14
mAU
0
20
40
60
80
100
120
3.192
13.08
3
Día 0: Formulaciones CON y SIN conservante
min2 4 6 8 10 12 14
mAU
0
20
40
60
80
100
120
140
3.202
13.078
min2 4 6 8 10 12 14
mAU
0
20
40
60
80
100
120
3.195
13.121
min2 4 6 8 10 12 14
mAU
0
50
100
150
200
250
3.194
13.122
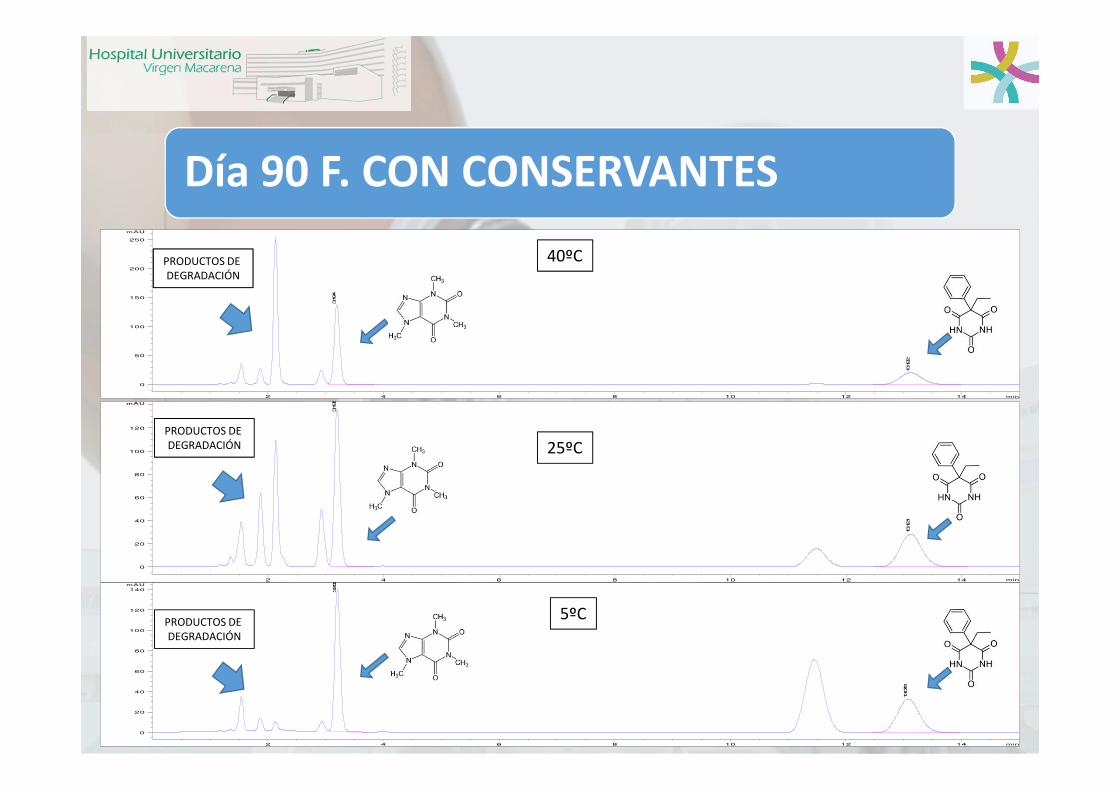
Día 90 F. CON CONSERVANTES
PRODUCTOS DE DEGRADACIÓN
40ºC
25ºC
5ºC
PRODUCTOS DE DEGRADACIÓN
PRODUCTOS DE DEGRADACIÓN

Día 90: F. SIN CONSERVANTES
min2 4 6 8 10 12 14
mAU
0
20
40
60
80
100
120
3.196
13.11
1
min2 4 6 8 10 12 14
mAU
0
20
40
60
80
100
120
3.195
13.102
min2 4 6 8 10 12 14
mAU
0
20
40
60
80
100
120
3.190
13.093
40ºC
25ºC
5ºC

Importancia de las impurezas: ISONIAZIDA e HIDRAZINA
ETAMBUTOL y AMINOBUTANOL
FDA ICH

Metodología: elaboración ISONIAZIDA
Fórmula 1 (CON conservante)
• Isoniazida.........................................5 g
• Agua conservante*……….............50 mL
• Sorbitol 70% csp.........................100 mL
Fórmula 2 (SIN conservante)
• Isoniazida.........................................5 g
• Agua purifcada……………….............50 mL
• Sorbitol 70% csp.........................100 mL
*Agua conservante sin propilenglicol:o Nipagin base…………………………………0.08 go Nipasol base…………………………………0.02 go Agua purifcada csp……………………….100 mL

Resultados: contenido en ISONIAZIDA (I)
50
60
70
80
90
100
110
DÍA 0 DIA 6 DÍA 10 DÍA 14 DÍA 21 DÍA 28 DÍA 50 DÍA 70 DÍA 90
% R
ECU
PER
AC
IÓN
CONTENIDO ISONIAZIDA FORMULACIÓN CON CONSERVANTES
F1 40ºC F1 25ºC F1 5ºC

Resultados: contenido en ISONIAZIDA(II)
50
60
70
80
90
100
110
DÍA 0 DIA 6 DÍA 10 DÍA 14 DÍA 21 DÍA 28 DÍA 50 DÍA 70 DÍA 90
% R
ECU
PER
AC
IÓN
CONTENIDO ISONIAZIDA FORMULACIÓN SIN CONSERVANTES
F2 40ºC F2 25ºC F2 5ºC

Hidrazina
Utilizado en síntesis de productos
farmacéuticos.
Producto de degradación Mutagénico, Genotóxico y
Carcinogénico.
Produce aductos de
ADN y alteraciones
en las cromátidas.
Potencial carcinogénico en roedores:
hígado y pulmones.
Otros fármacos que se degradan a
hidrazina: hidralazina, cisplatino,
oxaliplatino,…
M7(R1) Addendum to ICH M7: Assessment and Control of DNA Reactive(Mutagenic) Impurities in Pharmaceuticals to Limit Potential CarcinogenicRisk, International Consortium of
Harmonisation (ICH), (June 2015).

nm250 300 350 400 450 500 550
mAU
0
100
200
300
400
500
Metodología: HPLC y validación
HPLC: cuantificación de HIDRAZINA en los días de estudio:
• Método cromatográfico:
• Columna C18 4.6x250 mm, 5 µm
• Fase móvil: 50% ACN:50% AGUA.
• Tª columna: 30ºC
• Vol inyección=50 mcl
• Flujo=1,5 mL/min
• λ=310 nm (derivatización precolumna con benzaldehido)
• Tiempo de análisis: 8 minutos/muestra.
• Validación del método:
• Recta de calibrado: 50—300 ng/mL
• Exactitud:50, 200 y 300 ng/mL
• Precisión intra e interdía: 50, 200 y 300 ng/mL
• Límite de cuantificación y de detección
The United States pharmacopeia, 40nd rev., and The national formulary, 35th ed. Rockville (MD):United States Pharmacopeial Convention; 2017:4693-94.

Resultados: contenido en HIDRAZINA
0
5
10
15
20
25
30
DÍA 0 DIA 6 DÍA 10 DÍA 14 DÍA 21 DÍA 28 DÍA 50 DÍA 70 DIA 90
CO
NC
ENTR
AC
IÓN
(Μ
CG
/ML)
FORMACIÓN DE HIDRAZINAEN LAS DOS FORMULACIONES
F1 40ºC F1 25ºC F1 5ºC F2 40ºC F2 25ºC F2 5ºC

Día 0: Formulaciones CON y SIN conservante
min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000
min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000
HIDRAZINA

min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000
6.5
10
min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000
6.5
11
min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000
6.4
94
Día 90 F. CON CONSERVANTES HIDRAZINA
40ºC
25ºC
5ºC

min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000
6.5
22
min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000
6.5
05
min1 2 3 4 5 6 7
mAU
0
500
1000
1500
2000
2500
3000 6.5
08
Día 90 F. SIN CONSERVANTES HIDRAZINA
40ºC
25ºC
5ºC
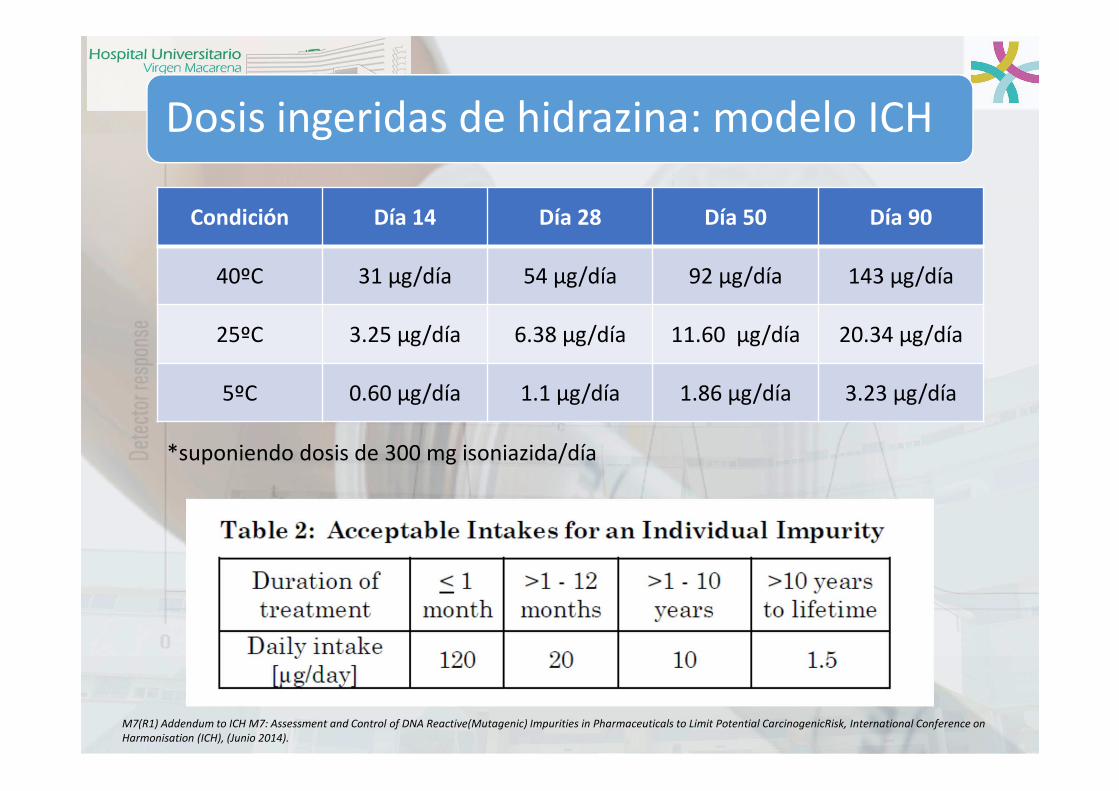
Dosis ingeridas de hidrazina: modelo ICH
Condición Día 14 Día 28 Día 50 Día 90
40ºC 31 µg/día 54 µg/día 92 µg/día 143 µg/día
25ºC 3.25 µg/día 6.38 µg/día 11.60 µg/día 20.34 µg/día
5ºC 0.60 µg/día 1.1 µg/día 1.86 µg/día 3.23 µg/día
*suponiendo dosis de 300 mg isoniazida/día
M7(R1) Addendum to ICH M7: Assessment and Control of DNA Reactive(Mutagenic) Impurities in Pharmaceuticals to Limit Potential CarcinogenicRisk, International Conference on
Harmonisation (ICH), (Junio 2014).

Dosis ingeridas de hidrazina: modelo FDA
Peso/Dosis Dosis Aceptable Ingerida
5 kg (50 mg INH) 3,87 µg/día
10 kg (100 mg INH) 7,74 µg/día
15 kg (150 mg INH) 11,61 µg/día
20 kg (200 mg INH) 15,48 µg/día
0
2
4
6
8
10
12
14
16
18
DIA 14 DÍA 28 DÍA 50 DÍA 90
LÍMITES DE HIDRAZINA ACEPTABLE POR PESO
5 kg
10 kg
15 kg
20 kg
25 ºC
5 ºC
U.S. Department of Health and Human Services, Food and DrugAdministration, Center for Drug Evaluation and Research (CDER), Center forBiologics Evaluation and Research (CBER), M7 Assessment and Control of DNAReactive (Mutagenic) Impurities in Pharmaceuticals to Limit PotentialCarcinogenic Risk, International Consortium of Harmonisation (ICH), (May2018)

Fórmula 1 (CON conservante)
• Etambutol HCl.........................................5 g
• Agua conservante sin propilenglicol...30 mL
• Jarabe simple csp..............................100 mL
Fórmula 2 (SIN conservante)
• Etambutol HCl....................................5 g
• Agua purificada….…………...............30 mL
• Jarabe simple csp.........................100 mL
Agua conservante sin propilenglicol:o Nipagin…………………………………………0.08 go Nipasol…………………………………………0.02 go Agua purificada csp……………………….100 mL
ETAMBUTOL y AMINOBUTANOL

AMINOBUTANOL
Utilizado en la síntesis de etambutol
Producto de degradación de etambutol
En grandes cantidades nocivo por ingestión, corrosivo y puede provocar quemaduras
Potencial mutagénico
Estructura del 2-amino-1-butanol
R. Hendrickson, et al. (eds.); Remington: The Science and Practice of Pharmacy 21th ed. Lippincott Williams and Wilkins, Baltimore, Maryalnd, p.1663 (2005)
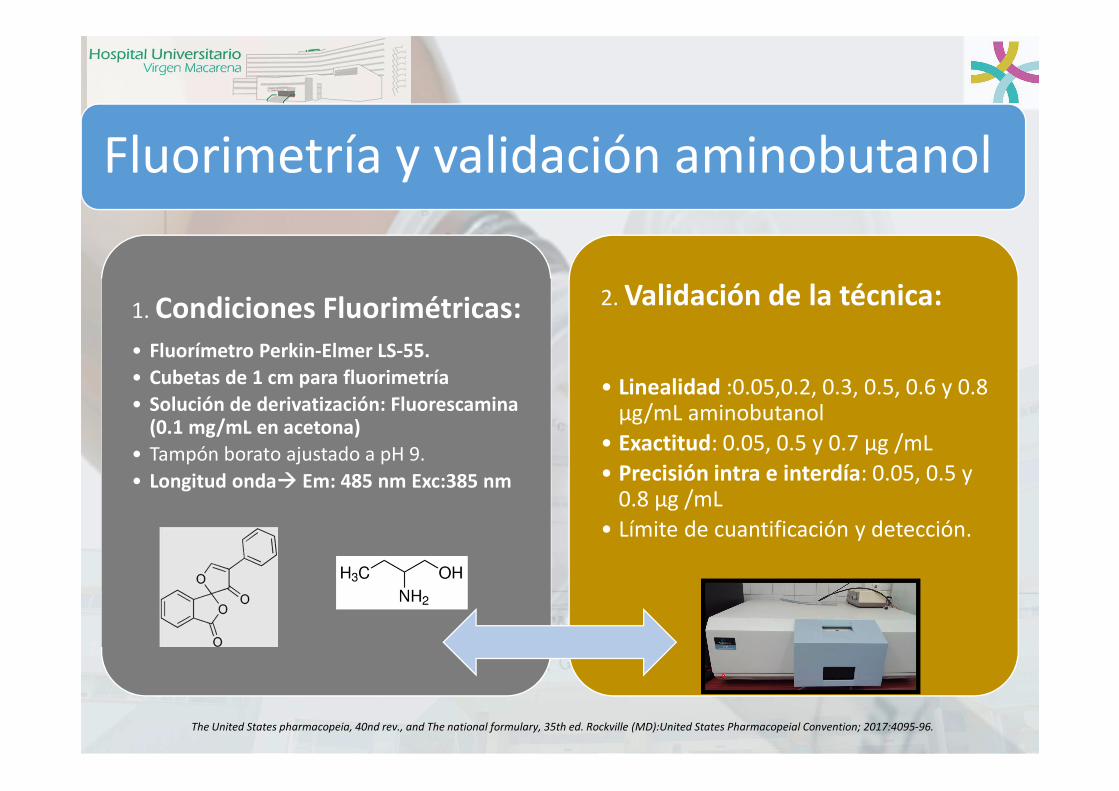
1. Condiciones Fluorimétricas:• Fluorímetro Perkin-Elmer LS-55.
• Cubetas de 1 cm para fluorimetría
• Solución de derivatización: Fluorescamina(0.1 mg/mL en acetona)
• Tampón borato ajustado a pH 9.
• Longitud onda Em: 485 nm Exc:385 nm
2. Validación de la técnica:
• Linealidad :0.05,0.2, 0.3, 0.5, 0.6 y 0.8 µg/mL aminobutanol
• Exactitud: 0.05, 0.5 y 0.7 µg /mL
• Precisión intra e interdía: 0.05, 0.5 y 0.8 µg /mL
• Límite de cuantificación y detección.
Fluorimetría y validación aminobutanol
The United States pharmacopeia, 40nd rev., and The national formulary, 35th ed. Rockville (MD):United States Pharmacopeial Convention; 2017:4095-96.
U.S. Department of Health and Human Services, Food and DrugAdministration, Center for Drug Evaluation and Research (CDER), Center forBiologics Evaluation and Research (CBER), M7 Assessment and Control of DNAReactive (Mutagenic) Impurities in Pharmaceuticals to Limit PotentialCarcinogenic Risk, International Consortium of Harmonisation (ICH), (Junio 2014)
0
0,2
0,4
0,6
0,8
1
1,2
D I A 0 D I A 5 D I A 7 D I A 1 4 D I A 2 1 D I A 2 8 D I A 5 0 D I A 7 0 D I A 9 0
CO
NC
ENTR
AC
IÓN
(M
CG
/ML)
FORMACIÓN DE AMINOBUTANOL EN LAS DOS FORMULACIONES
F1 40ºC F1 25ºC F1 5ºC F2 40ºC F2 25ºC F2 5ºC
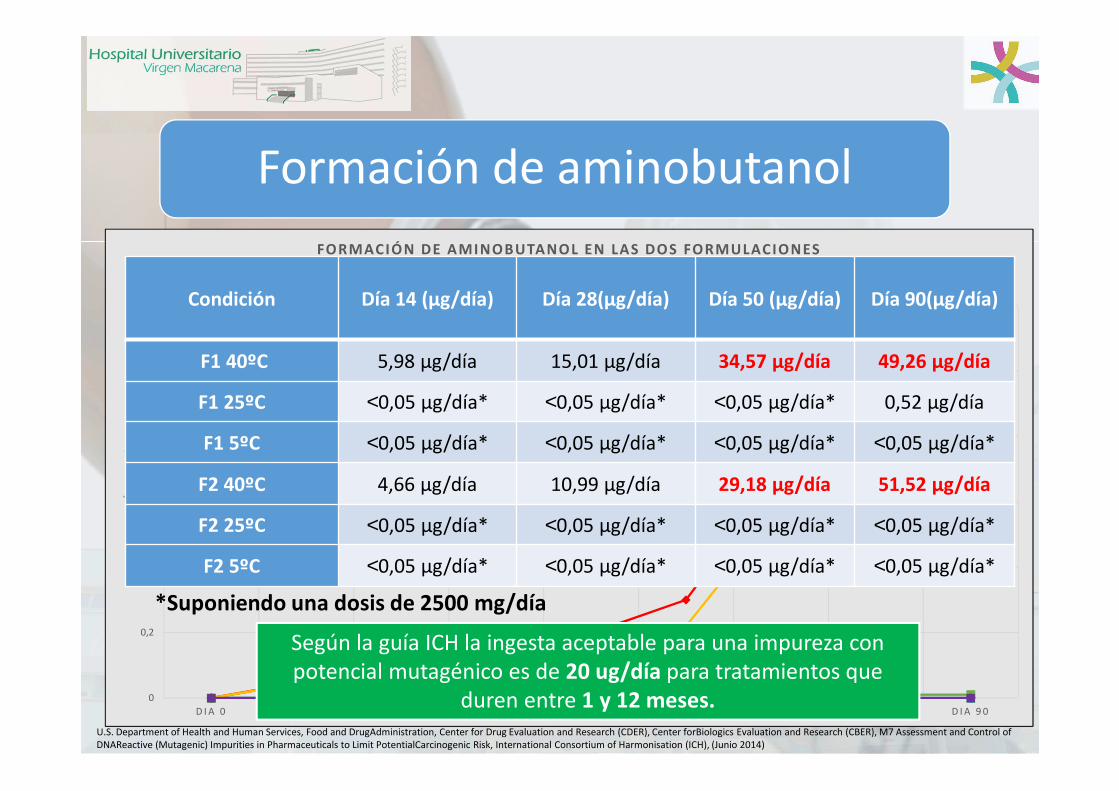
Formación de aminobutanol
Según la guía ICH la ingesta aceptable para una impureza con potencial mutagénico es de 20 ug/día para tratamientos que
duren entre 1 y 12 meses.
Condición Día 14 (µg/día) Día 28(µg/día) Día 50 (µg/día) Día 90(µg/día)
F1 40ºC 5,98 µg/día 15,01 µg/día 34,57 µg/día 49,26 µg/día
F1 25ºC <0,05 µg/día* <0,05 µg/día* <0,05 µg/día* 0,52 µg/día
F1 5ºC <0,05 µg/día* <0,05 µg/día* <0,05 µg/día* <0,05 µg/día*
F2 40ºC 4,66 µg/día 10,99 µg/día 29,18 µg/día 51,52 µg/día
F2 25ºC <0,05 µg/día* <0,05 µg/día* <0,05 µg/día* <0,05 µg/día*
F2 5ºC <0,05 µg/día* <0,05 µg/día* <0,05 µg/día* <0,05 µg/día*
*Suponiendo una dosis de 2500 mg/día
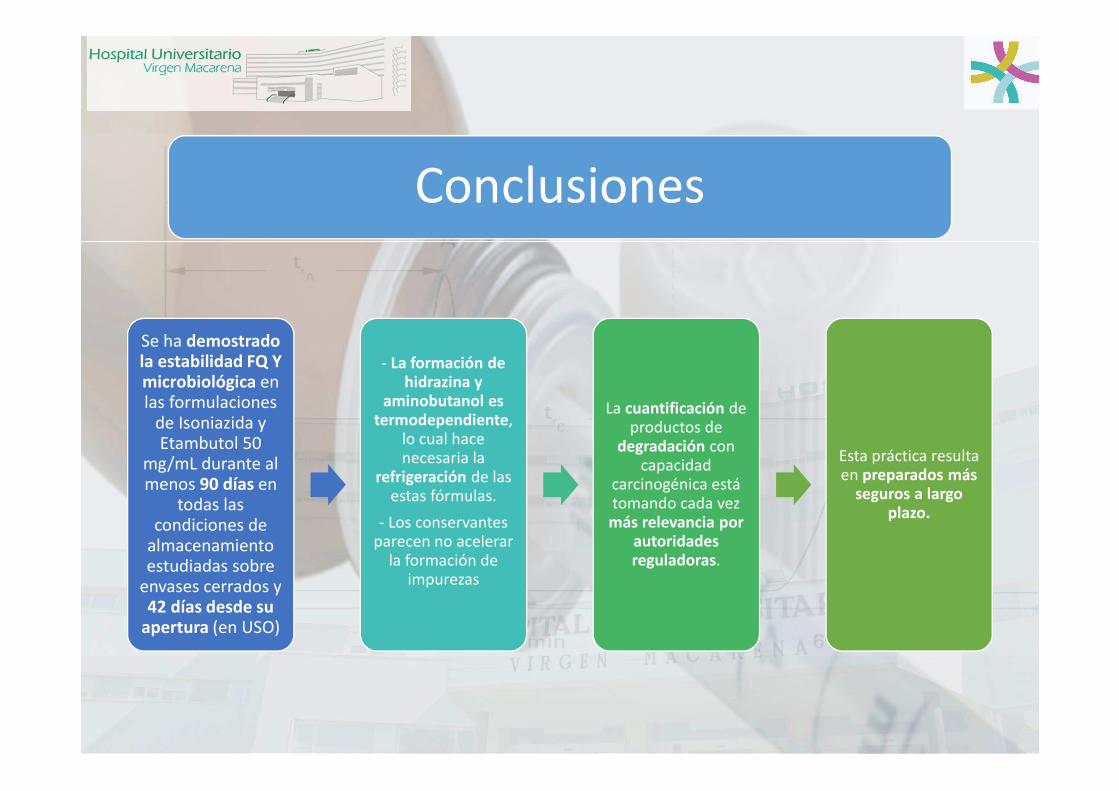
Conclusiones
Se ha demostrado la estabilidad FQ Y microbiológica en las formulaciones
de Isoniazida y Etambutol 50
mg/mL durante al menos 90 días en
todas las condiciones de
almacenamiento estudiadas sobre
envases cerrados y 42 días desde su
apertura (en USO)
- La formación de hidrazina y
aminobutanol es termodependiente,
lo cual hace necesaria la
refrigeración de las estas fórmulas.
- Los conservantes parecen no acelerar
la formación de impurezas
La cuantificación de productos de
degradación con capacidad
carcinogénica está tomando cada vez más relevancia por
autoridades reguladoras.
Esta práctica resulta en preparados más
seguros a largo plazo.

Oxidación
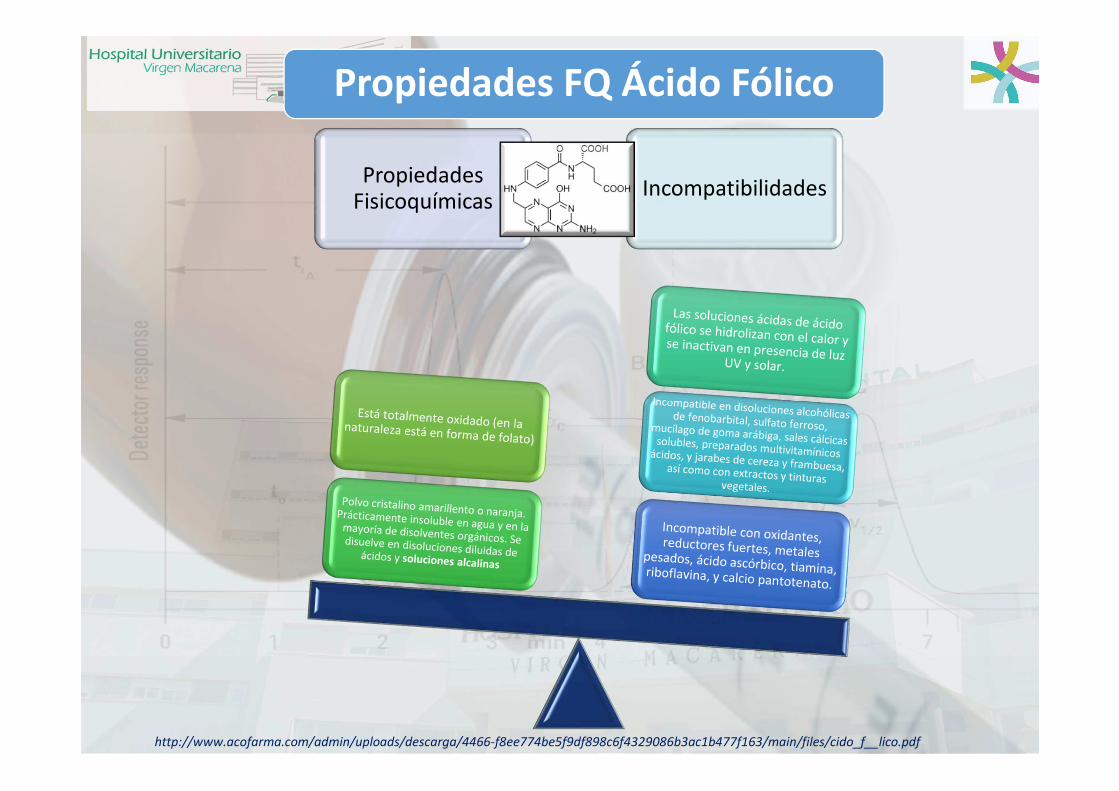
Propiedades FQ Ácido Fólico
Propiedades Fisicoquímicas
Incompatibilidades
http://www.acofarma.com/admin/uploads/descarga/4466-f8ee774be5f9df898c6f4329086b3ac1b477f163/main/files/cido_f__lico.pdf

Metodología: elaboración Ácido Fólico 1 mg/mL
Fórmula 1 (CON conservante)
• Ácido Fólico......................................100 mg
• Sorbitol 70%................……..................20 mL
• Agua cons. sin propilenglicol*csp….100 mL• Ajustar a pH=8.0-8.5 con NaOH 1N
Fórmula 2 (SIN conservante)
• Ácido Fólico......................................100 mg
• Sorbitol 70%................……..................20 mL
• Agua purificada csp……………………….100 mL
• Ajustar a pH=8.0-8.5 con NaOH 1N*Agua conservante sin propilenglicol:o Nipagin base…………………………………0.08 go Nipasol base…………………………………0.02 go Agua purifcada csp……………………….100 mL

Resultados: contenido en Ác. Fólico (I)
60
65
70
75
80
85
90
95
100
105
110
DÍA 0 DIA 5 DÍA 7 DÍA 1 4 DÍA 2 1 DÍA 2 8 DÍA 5 0 DÍA 7 0 DÍA 9 0
% R
ECU
PER
AC
IÓN
% RECUPERACIÓN DE ÁCIDO FOLICO FORMULACIÓN CON CONSERVANTES
F1A 40ºC F1B 25ºC F1C 5ºC
7
7,5
8
8,5
9
9,5
10
10,5
DÍA 0 DIA 5 DÍA 7 DÍA 14 DÍA 21 DÍA 28 DÍA 50 DÍA 70 DÍA 90
Evolución pH formulaciones CON conservantes
F1 40ºC F1 25ºC F1 5ºC

Resultados: contenido en Ác. Fólico (II)
60
70
80
90
100
110
DÍA 0 DIA 5 DÍA 7 DÍA 14 DÍA 21 DÍA 28 DÍA 50 DÍA 70 DÍA 90
% R
ECU
PER
AC
IÓN
% RECUPERACIÓN DE ÁCIDO FOLICO FORMULACIÓNSIN CONSERVANTES
F1A 40ºC F1B 25ºC F1C 5ºC
7
7,5
8
8,5
9
9,5
10
10,5
DÍA 0 DIA 5 DÍA 7 DÍA 14 DÍA 21 DÍA 28 DÍA 50 DÍA 70 DÍA 90
Evolución pH formulaciones SIN conservantes
F2 40ºC F2 25ºC F2 5ºC

Día 0: Cromatogramas Ácido Fólico CON y SIN conservantes
min1 2 3 4 5 6 7 8 9
mAU
0
10
20
30
40
50
8.174
min2 4 6 8
mAU
0
10
20
30
40
50
8.077
F2: Formulación SIN conservantes
F1: Formulación CON conservantes

min1 2 3 4 5 6 7 8 9
mAU
0
10
20
30
40
50
8.151
min1 2 3 4 5 6 7 8 9
mAU
0
10
20
30
40
50
8.111
min1 2 3 4 5 6 7 8 9
mAU
0
10
20
30
40
50
60
70
80
8.017
Día 90 F. CON CONSERVANTES Ácido Fólico
40ºC
25ºC
5ºC
Productos de degradación
Productos de degradación

min2 4 6 8
mAU
0
10
20
30
40
50
7.992
min2 4 6 8
mAU
0
10
20
30
40
50
7.974
min2 4 6 8
mAU
0
10
20
30
40
50
7.947
Día 90 F. SIN CONSERVANTES Ácido Fólico
40ºC
25ºC
5ºC
Productos de degradación

Resultados degradación acelerada
Las condiciones que más afectan:
Condición % Recuperación Impacto
Oxidantes (H202 15% v/v) 0% ++++
Ácidas (HCl 1N) 27.18% ++
Básicas (NaOH 1N) 72.96% +
Calor (90ºC) 100.8% -min1 2 3 4 5 6 7 8 9
mAU
0
50
100
150
200
250
2.6
95 2
.787
3.0
45
3.3
56
3.6
56
3.8
26
4.0
12
4.7
84
5.9
59 6.9
95
H2O2 15% v/v
min1 2 3 4 5 6 7 8 9
mAU
0
5
10
15
20
25
30
35
2.4
75
2.6
65 2.8
04
3.0
47
3.2
39
8.6
77
HCL 1N
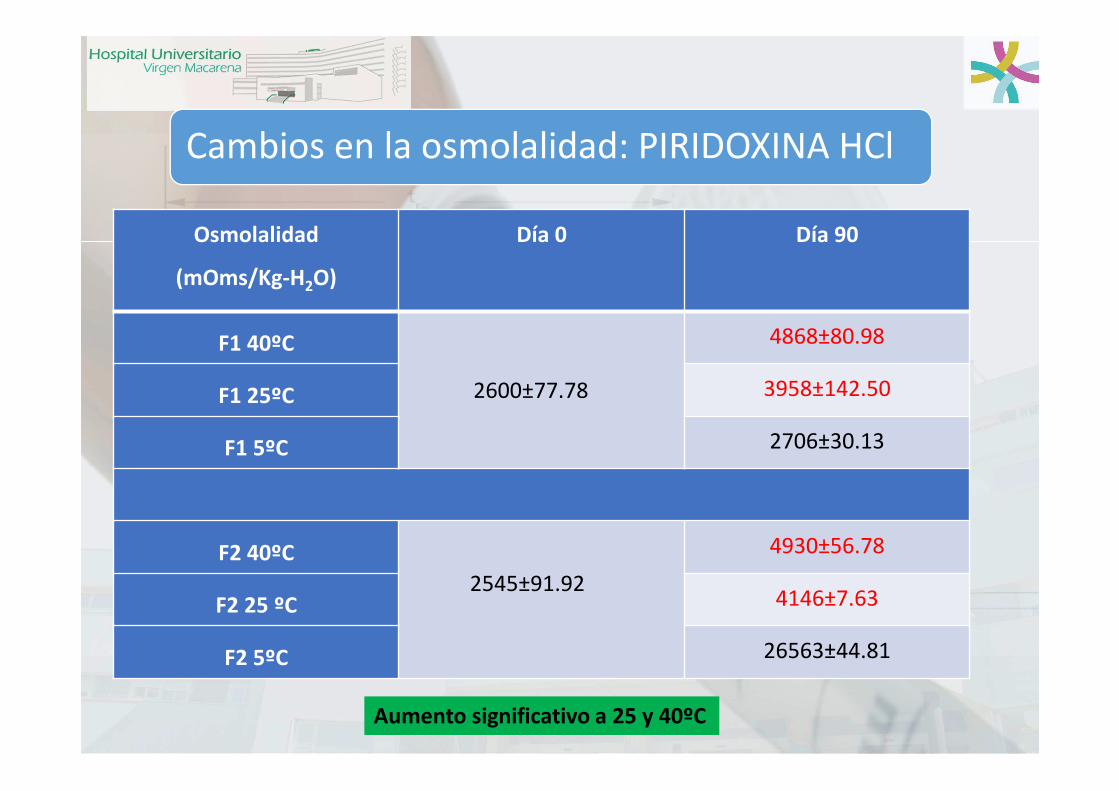
Cambios en la osmolalidad: PIRIDOXINA HCl
Osmolalidad
(mOms/Kg-H2O)
Día 0 Día 90
F1 40ºC
2600±77.78
4868±80.98
F1 25ºC 3958±142.50
F1 5ºC 2706±30.13
F2 40ºC
2545±91.92
4930±56.78
F2 25 ºC 4146±7.63
F2 5ºC 26563±44.81
Aumento significativo a 25 y 40ºC

Inestabilidades Microbiológicas
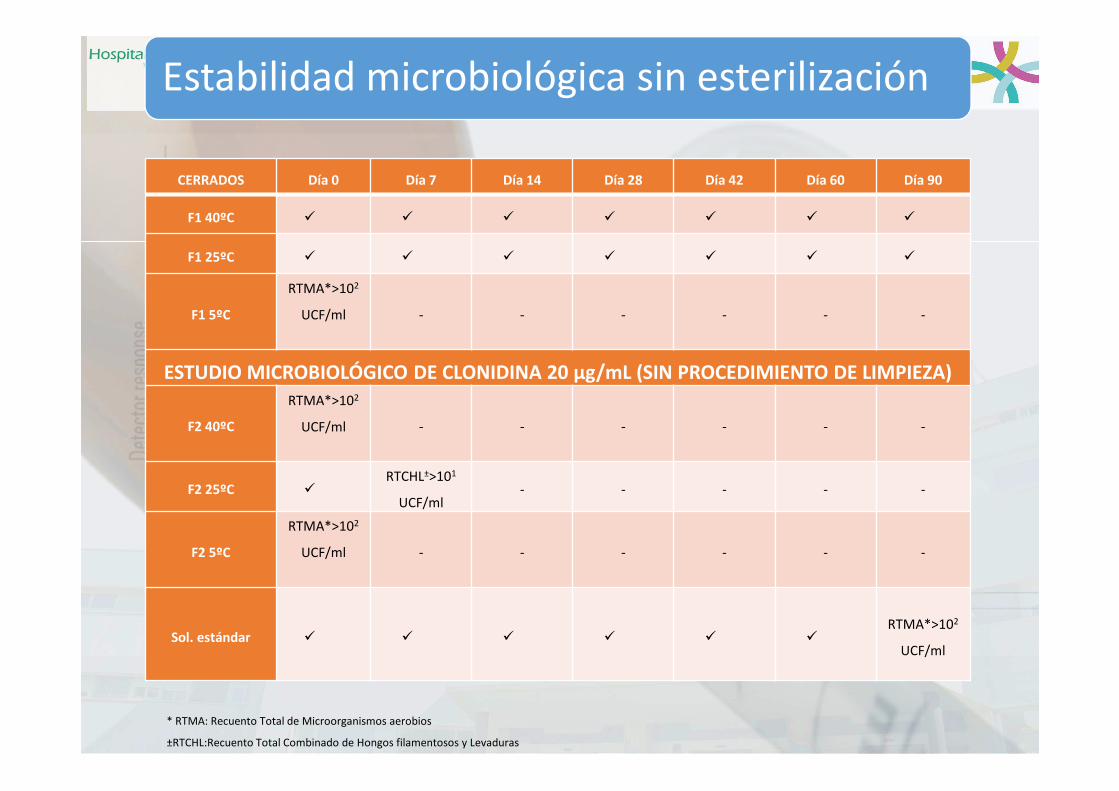
Estabilidad microbiológica sin esterilización
CERRADOS Día 0 Día 7 Día 14 Día 28 Día 42 Día 60 Día 90
F1 40ºC
F1 25ºC
F1 5ºC
RTMA*>102
UCF/ml - - - - - -
ESTUDIO MICROBIOLÓGICO DE CLONIDINA 20 µg/mL (SIN PROCEDIMIENTO DE LIMPIEZA)
F2 40ºC
RTMA*>102
UCF/ml - - - - - -
F2 25ºC RTCHL±>101
UCF/ml- - - - -
F2 5ºC
RTMA*>102
UCF/ml - - - - - -
Sol. estándar RTMA*>102
UCF/ml
* RTMA: Recuento Total de Microorganismos aerobios
±RTCHL:Recuento Total Combinado de Hongos filamentosos y Levaduras

Estabilidad microbiológica tras esterilización
CERRADOS Día 0 Día 7 Día 14 Día 28 Día 42 Día 60 Día 90
F1 40ºC
F1 25ºC
F1 5ºC
ESTUDIO MICROBIOLÓGICO DE CLONIDINA 20 µg/mL (ENVASES ESTERILIZADOS)
F2 40ºC
F2 25ºC
F2 5ºC
Sol. estándar RTMA*>102
UCF/ml
* RTMA: Recuento Total de Microorganismos Aerobios

Susceptibilidad a la contaminación microbiológica: Riboflavina
Fórmula 1 (CON conservante)
• Riboflavina Sodio Fosfato.................3,375 g
• Agua conservante sin propilenglicol...50 mL
• Jarabe simple csp..............................100 mL
Fórmula 2 (SIN conservante)
• Riboflavina Sodio Fosfato.................3,375 g
• Agua purificada………………………………..50 mL
• Jarabe simple csp..............................100 mL

Control Visual
FÓRMULA DIA 0 DIA 5 DIA 7 DIA 14 DIA 21 DIA 28 DIA 50 DIA 70 DIA 90
F1 40ºC √ √ √ √ √ √ √ √ √
F1 25ºC √ √ √ √ √ √ M M M
F1 5ºC √ √ √ √ √ √ √ √ √
F2 40ºC √ √ √ √ √ √ √ √ √
F2 25ºC √ √ √ M M M M M M
F2 5º C √ √ √ √ √ √ √ √ √
M: Se observan partículas en suspensión que impresionan de hongos mucilaginosos
NORMAL PARDEAMIENTO

CONTROL MICROBIOLÓGICO RIBOFLAVINA
CONDICIÓN/ENVASE DÍA 0 DÍA 7 DÍA 14 DÍA 28 DÍA 42 DÍA 60 DÍA 90
F1-F2 5ºC CERRADOS
F1-F2 5ºC CERRADOS
F1-F2 5ºC CERRADOS
F1-F2 25ºC CERRADOS
F1-F2 25ºC CERRADOS
Recuento Total Combinado de Hongos filamentosos patógenos y Levaduras (RTCHL)>101 UFC/mL

Resumen estabilidades estudiosFÓRMULAS PERÍODO DE VALIDEZ
(CON CONSERVANTES)PERÍODO DE VALIDEZ(SIN CONSERVANTES)
FACTOR LIMITANTE ESTABILIDAD
Clonidina 20 µg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertosInteracción con
conservante
Hidralazina 10 mg/mL solución90 días a 5ºC y a 25ºC
42 días abiertos90 días a 5ºC y a 25ºC
42 días abiertospH
Fenobarbital 10 mg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertospH
Isoniazida 50 mg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertos
Generación termodependiente de
Hidrazina
Etambutol 50 mg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertos
Generación termodependiente de
aminobutanol
Riboflavina 25 mg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertosContaminación
microbiológica a 25ºC
Ácido Fólico 1 mg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertosOxidación, pH y
temperatura
Tiamina 100 mg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertosAumento significativo de
osmolalidad
Piridoxina 25 mg/mL solución90 días a 5ºC
42 días abiertos90 días a 5ºC
42 días abiertosAumento significativo de
osmolalidad

TFG y TFM

PREMIOS Y NOTICIAS
