organoclorados en suero: nuevas metodologÍas …
TRANSCRIPT

ORGANOCLORADOS EN SUERO: NUEVAS METODOLOGÍAS ANALÍTICAS E INVESTIGACIÓN DE LOS NIVELES EN LA
COHORTE EPIC-ESPAÑA
Memoria para optar al grado de Doctor en Ciencias Químicas presentada por: Fernando Goñi
Donostia-San Sebastián, 2011


AUTORIZACION DEL/LA DIRECTOR/A DE TESIS
PARA SU PRESENTACION
Dra. Esmeralda Millán Martín con N.I.F. 17.847.378 E
como Directora de la Tesis Doctoral: ORGANOCLORADOS EN SUERO: NUEVAS
METODOLOGÍAS ANALÍTICAS E INVESTIGACIÓN DE LOS NIVELES EN LA
COHORTE EPIC-ESPAÑA
realizada en el Departamento de Química Aplicada
por el Doctorando Don José Fernando Goñi Irigoyen,
autorizo la presentación de la citada Tesis Doctoral, dado que reúne las condiciones
necesarias para su defensa.
En San Sebastián a cuatro de marzo de 2011
LA DIRECTORA DE LA TESIS
Fdo.: Esmeralda Millán Martín


CONFORMIDAD DEL DEPARTAMENTO
El Consejo del Departamento de QUÍMICA APLICADA
en reunión celebrada el día de de 2011 ha acordado dar la conformidad a
la admisión a trámite de presentación de la Tesis Doctoral titulada:
Organoclorados en suero: nuevas metodologías analíticas e investigación de los
niveles en la cohorte EPIC-España
dirigida por la Dra. Esmeralda Millán Martín
y presentada por Don José Fernando Goñi Irigoyen
ante este Departamento.
En San Sebastián a de de 2011
Vº Bº DIRECTORA DEL DEPARTAMENTO SECRETARIA DEL DEPARTAMENTO
Fdo.: Mª Angeles Garralda Hualde Fdo.: Rosa Garcia Arrona


ACTA DE GRADO DE DOCTOR
ACTA DE DEFENSA DE TESIS DOCTORAL DOCTORANDO: Don José Fernando Goñi Irigoyen TITULO DE LA TESIS: Organoclorados en suero: Nuevas metodologías analíticas e investigación de los niveles en la cohorte EPIC-España designado por la Subcomisión de Doctorado de la UPV/EHU para calificar la Tesis Doctoral arriba indicada y reunido en el día de la fecha, una vez efectuada la defensa por el doctorando y contestadas las objeciones y/o sugerencias que se le han formulado, ha otorgado por___________________la calificación de: unanimidad ó mayoría
Idioma/s defensa: _______________________________________________________________
En a de de
EL/LA PRESIDENTE/A, EL/LA SECRETARIO/A,
Fdo.: Fdo.:
Dr/a: ____________________ Dr/a: ______________________
VOCAL 1º, VOCAL 2º, VOCAL 3º,
Fdo.: Fdo.: Fdo.:
Dr/a: Dr/a: Dr/a: EL DOCTORANDO, Fdo.: _____________________


A mi mujer e hijos, sujetos pacientes
de esta locura y a mis padres, que
aunque la vida no les dio la ocasión,
habrían estado orgullosos de serlo.


Este trabajo es fruto de la colaboración entre el Laboratorio de Salud
Pública (sedes de Gipuzkoa y Bizkaia) y el servicio de Epidemiología de la
Subdirección Territorial de Salud Pública de Gipuzkoa, ambos pertenecientes al
Departamento de Sanidad y Consumo del Gobierno Vasco, el Instituto Catalán
de Oncología, la cohorte EPIC-España, la Sección Bioquímica del Laboratorio
Unificado del Hospital Donostia y el Grupo de Química Analítica del
Departamento de Química Aplicada (Facultad de CC.QQ. de Donostia, UPV-
EHU). Mi más sincero agradecimiento a todos sus integrantes y en particular a
Raúl, Arsenio, Agustín, Javier, Asun, Pilar, Toni y Paula por su esfuerzo y
dedicación.
También quiero mostrar un agradecimiento especial a Miren Dorronsoro
por ser la promotora y principal impulsora de esta empresa y a la Dra.
Esmeralda Millán por su dirección, magisterio y colaboración.
Por último, deseo agradecer igualmente la financiación de este proyecto
al Departamento de Sanidad y Consumo del Gobierno Vasco (proyecto
200311014) y al Fondo de Investigación Sanitaria del Ministerio de Sanidad,
Política Social e Igualdad (proyecto PI 02/1598).


ÍNDICE GENERAL
INTRODUCCIÓN 3
Descripción de los compuestos investigados 3
Heptacloro 4
Hexaclorociclohexano (HCH) 5
Hexaclorobenceno (HCB) 7
Diclorodifeniltricloroetano (DDT) 9
Bifenilos Policlorados (PCBs) 11
Problemática medioambiental 15
Efectos sobre la salud 24
Introducción 24
Factores comunes en los compuestos investigados 32
Heptacloro epóxido 33
HCH 35
HCB 37
DDT 39
PCBs 42
Consideraciones analíticas 49
Referencias 52
OBJETIVOS 57
CAPÍTULO 1. Metodología analítica basada en placas de extracción
63
1.1 Introducción 65
1.2 Parte experimental 67
1.2.1 Reactivos y materiales 67
I

1.2.2 Muestras de suero 68
1.2.3 Equipamiento instrumental 68
1.2.4 Procedimiento analítico 69
1.2.5 Calibración y validación 70
1.3 Resultados y discusión 73
1.3.1 Optimización del método 73
1.3.2 Evaluación del método 74
1.3.3 Aplicación a muestras de suero 78
1.4 Conclusiones 80
1.5 Referencias 81
CAPÍTULO 2. Metodología analítica basada en microextracción en fase sólida
83
2.1 Introducción 85
2.2 Parte experimental 88
2.2.1 Reactivos y material 88
2.2.2 Muestras de suero 89
2.2.3 Equipamiento instrumental 89
2.2.4 Procedimiento analítico 90
2.3 Resultados y discusión 91
2.3.1 Diseño de selección 91
2.3.2 Diseño de optimización 94
2.3.3 Evaluación del método 98
2.3.4 Aplicación a muestras de suero 101
2.4 Conclusiones 105
2.5 Referencias 106
II

CAPÍTULO 3. Metodología analítica basada en limpieza de extractos por cromatografía de permeabilidad en gel
109
3.1 Introducción 111
3.2 Parte experimental 114
3.2.1 Reactivos y materiales 114
3.2.2 Muestras de suero 115
3.2.3 Equipamiento instrumental 115
3.2.4 Procedimiento analítico 117
3.2.5 Calibración y validación 118
3.3 Resultados y discusión 120
3.3.1 Optimización de la limpieza de extractos 120
3.3.2 Ventana de tiempos de recolección y
evaluación del sistema GPC
125
3.3.3 Evaluación del método 127
3.3.4 Aplicación a muestras de suero 130
3.4 Conclusiones 134
3.5 Referencias 135
CAPÍTULO 4. Niveles séricos de PCBs en la cohorte EPIC-España
137
4.1 Introducción 139
4.2 Metodología experimental 143
4.2.1 Población y diseño del estudio 143
4.2.2 Procedimientos analíticos 143
4.2.3 Dieta, estilo de vida y medidas
antropométricas
145
4.2.4 Análisis estadístico 145
III

4.3 Resultados y discusión 147
4.3.1 PCBs y principales variables demográficas 148
4.3.2 PCBs y otras variables no dietéticas 149
4.3.3 PCBs y dieta 151
4.3.4 Modelo final 153
4.3.5 Comparación con otros estudios 155
4.4 Conclusiones 158
4.5 Referencias 160
CAPÍTULO 5. Niveles séricos de plaguicidas organoclorados en la cohorte EPIC-España
163
5.1 Introducción 165
5.2 Metodología experimental 167
5.2.1 Población estudiada 167
5.2.2 Procedimientos analíticos 167
5.2.3 Cuestionarios de dieta, estilo de vida y
medidas antropométricas
168
5.2.4 Análisis estadístico 169
5.3 Resultados y discusión 171
5.3.1 Plaguicidas organoclorados y variables no
dietéticas
173
5.3.2 Plaguicidas organoclorados y dieta 177
5.3.3 Comparación con otros estudios 180
5.4 Conclusiones 183
5.5 Referencias 186
CONCLUSIONES 189
APÉNDICE. Comunicaciones y publicaciones 195
IV

INTRODUCCIÓN
Descripción de los compuestos investigados
Problemática medioambiental
Efectos sobre la salud
Consideraciones analíticas
Referencias

Introducción
2

Introducción
Descripción de los compuestos investigados
Todos ellos son compuestos orgánicos que poseen átomos de cloro en su
molécula. Desde el punto de vista de su aplicación podemos distinguir entre
plaguicidas y productos de uso industrial. Como veremos más adelante, tienen
en común su resistencia química y su liposolubilidad, lo que conlleva una alta
persistencia en el medio ambiente y su acumulación en los tejidos grasos de
los seres vivos. Esto, que supone un grave problema, unido a la evidencia de
que son lesivos para los organismos, ha sido la causa de que, desde hace
algunos años, prácticamente se haya renunciado a su aplicación. Aun así, en
general siguen siendo los compuestos organoclorados más habituales y
mayoritarios en las fracciones grasas de las muestras biológicas en España
[Olea y Fernández 2001, Porta et al. 2009]. Sus estructuras químicas se
muestran en la Figura 1.
Figura 1 Estructuras químicas de los compuestos estudiados. Se incluyen también el PCB 46 y el PCB 143, utilizados como patrones internos en la analítica.
A continuación se procede a una descripción más detallada de los
mismos.
3

Introducción
Heptacloro
El heptacloro [WHO 1988, Sullivan and Krieger 2001, WHO 2003, ATSDR
2007, Tomlin 2009, UNIDO 2010] o 1,4,5,6,7,8,8-heptacloro-3a,4,7,7a-
tetrahidro-4,7-metanoindeno es un plaguicida organoclorado que pertenece a la
familia de los diciclopentadienos. Fue descubierto en 1946 al ser aislado a
partir del producto técnico que se obtenía en la fabricación del clordano, otro
plaguicida de la misma familia, en el que estaba presente a una concentración
aproximada de un 10%. Sus propiedades insecticidas se describieron por
primera vez en 1951 por Rogoff y Metcalf, resultando ser, para muchas
aplicaciones, entre tres y cinco veces más potentes que las del clordano.
Registrado en 1952, la producción industrial y comercialización se inició en
1953 en EE.UU. de la mano de la compañía Velsicol bajo el nombre de Velsicol
104. Como otros plaguicidas de esta familia, industrialmente se obtiene a partir
del clordeno, que a su vez proviene de la condensación Diels-Alder del
hexaclorociclopentadieno con ciclopentadieno. El clordeno se disuelve en
benceno conteniendo entre un 0,5-5% de tierra Fuller (tierra de batán) y se
somete a cloración radicalaria. El proceso es lento y puede llevar hasta 8
horas. Se obtiene así un producto técnico que es un sólido marrón con aspecto
de cera, funde a 46-74 ºC y contiene alrededor de un 72% de heptacloro. El
resto son impurezas, entre las que destacan el clordano y el nonaclor. Una vez
purificado es un sólido blanco cristalino con olor alcanforado que funde a 95-96
ºC y tiene una presión de vapor de 53 mPa (25 ºC). Es prácticamente insoluble
en agua y soluble en la mayor parte de disolventes orgánicos. Aunque se le
considera estable a la luz, la humedad, el aire, el calor (aguanta temperaturas
de hasta 160 ºC) y los álcalis débiles, se metaboliza con relativa facilidad por
oxidación a heptacloro epóxido en el medio ambiente o en los organismos
vivos. El heptacloro epóxido es un polvo blanco químicamente más estable que
el heptacloro y es una buena traza de la exposición a este compuesto y
también al clordano, ya que uno de los principales metabolitos de éste último
es el heptacloro.
El heptacloro es un insecticida persistente y no sistémico que actúa por
ingestión o contacto. Durante los años 60 y 70 del pasado siglo se utilizó
masivamente en aplicaciones sobre suelos (de cultivo y urbanos), cultivos
4

Introducción
(principalmente maíz) y foliares, y en el tratamiento de semillas de cereales.
Los insectos diana eran hormigas, gusanos, gorgojos, etc. Otra de sus
principales aplicaciones fueron los tratamientos estructurales antitermitas.
También encontró uso como insecticida doméstico y como antiparasitario en
personas y animales, así como para el control de la malaria. Además de
Velsicol 104, se han utilizado otros nombres en su comercialización: Velsicol
Heptachlor, Heptagran, Basaklor, Drinox, Termide, Gold Crest H-60, Aahepta,
Agroceres, Clorahep, H-34 Heptamul, Heptox, Rhodiachlor, Eptacloro, E 3314,
GPKh, Heptacloran, Heptasol, Soleptax, Hepta, Heptagranox, etc. En 1970 el
reparto aproximado de su consumo anual a nivel global era: África 5%, Asia
15%, Norteamérica (incluyendo Canadá) 5%, Sudamérica 15% y Europa 60%.
Se estima que la producción de 1971, tan sólo en EE.UU., fue de 2700
toneladas [WHO 1988].
Debido a problemas medioambientales a partir de los años 70 del pasado
siglo surgieron disposiciones legales en numerosos países, que fueron
restringiendo paulatinamente su uso. Sin embargo, en los últimos años se ha
registrado un ligero repunte al emplearse como termicida en la construcción de
grandes infraestructuras viarias en África y en estructuras residenciales en Asia
y el Noreste de Australia; también en la protección de ciertos cultivos en
América del Sur. Incluso en EE.UU. su agencia de protección ambiental (EPA)
aprobó en septiembre de 2005 su uso en transformadores instalados bajo tierra
para luchar contra las hormigas de fuego, especie prácticamente desconocida
en España y catalogada como invasora que, proveniente de América del Sur,
se está convirtiendo en una auténtica plaga en el sureste de EE.UU. y el caribe.
Aunque estos datos están sujetos a constantes cambios, a título orientativo
cabe decir que a principios de 2009 era un producto prohibido o severamente
restringido en 38 países y otros 93 prohibían su importación [UNIDO 2010].
Hexaclorociclohexano (HCH)
El hexaclorocicloxano [Ortiz-Martínez 1991, Sullivan and Krieger 2001,
WHO 2003, ATSDR 2005, UNEP 2006, Tomlin 2009] fue sintetizado por
primera vez por Faraday, quien, tras descubrir el benceno, lo hizo reaccionar
con cloro a la luz del sol en 1825, obteniendo un polvo blanco con tonos
marrones de fórmula molecular C6H6Cl6, fórmula que dio lugar a que durante
5

Introducción
algún tiempo se le llamara también hexacloruro de benceno (BHC). A finales
del siglo XIX Menieur demostró que no se trataba de un compuesto sino de una
mezcla de distintos isómeros, logrando aislar dos de éstos. En 1912 el químico
holandés Van der Linden confirmó los hallazgos anteriores y aisló otros dos
isómeros más. Se trataba de estereoisómeros que se diferenciaban por la
posición relativa de cada uno de los cloros con respecto al plano del anillo de
ciclohexano, pudiendo situarse bien por encima o bien por debajo de éste.
Aunque el número potencial de estereoisómeros del HCH es de ocho, a los que
se denomina por medio de las letras griegas α, β, γ, δ, ε, ζ, η, θ (Menieur aisló
los dos primeros y Van der Linden los dos siguientes), en el HCH técnico sólo
están presentes de forma apreciable los cinco primeros. A principios de los
años cuarenta del pasado siglo los franceses Dupire y Racourt y el inglés Slade
describen sus propiedades insecticidas. En 1943 los laboratorios de la empresa
inglesa ICI (Imperial Chemical Industries) demostraron que era el isómero γ-
HCH el que aportaba el poder insecticida, isómero que recibe también el
nombre de Lindano en honor a Van der Linden, su descubridor y que, para la
mayoría de los insectos, es entre 100 y 1000 veces más tóxico que el resto.
Tras la segunda guerra mundial comienza la producción de HCH a escala
industrial mediante reacción fotoquímica entre benceno y cloro. El cloro se
disolvía en benceno y la mezcla se hacía pasar a través de tubos iluminados
con luz UV a 15-20 ºC. El HCH técnico así obtenido contenía: 55-70% α-HCH,
5-14% β-HCH, 10-18% γ-HCH, ≥5% de δ-HCH, ≥1% de ε-HCH, trazas del resto
de isómeros e impurezas como heptaclorociclohexano y octaclorociclohexano.
Aunque el HCH técnico se utilizó comercialmente, resultaba más interesante el
uso de formulaciones a base de Lindano. El Lindano se obtenía a partir del
HCH técnico aislándolo mediante cristalización selectiva. El Lindano es un
sólido cristalino, incoloro o con tonos blancos, que funde a 112-113 ºC y
presenta una presión de vapor de 5,6 mPa (20 ºC). Es prácticamente insoluble
en agua y soluble en la mayoría de disolventes orgánicos. Resiste al oxígeno
del aire, a la luz y al calor (<180 ºC), es estable en medio ácido pero se
descompone en medio alcalino por deshidrocloración para dar triclorobenceno.
No obstante, de todos los isómeros del HCH el más estable es el β-HCH.
6

Introducción
El Lindano es un insecticida no sistémico de amplio espectro, que actúa
por ingestión o contacto. Se comercializó bajo una gran cantidad de nombres,
entre ellos: Gammexane, Gamma-col, Hexagon, Lindacol, Gammasan, Litexa,
Lintox, Gammexine, Aalindan, Agrocide, Ameisentod, Gamacid, Gamalin,
Lendine, Lindafor, Lindatox, Hexicide, Hexachloran, etc. Ha sido uno de los
insecticidas más utilizados, tanto en preparados fitosanitarios, como en
preparados zoosanitarios contra moscas, piojos, pulgas, garrapatas y otros
ácaros del ganado, o en preparados para combatir los vectores de la malaria y
el mal de Chagas. Durante mucho tiempo se ha utilizado además en los
hogares como aerosol y ha sido el ingrediente principal en las lociones contra
los piojos; también ha encontrado uso en preparados contra otros parásitos
corporales de los seres humanos como la sarna y las ladillas. Cabe destacar
que este tipo de aplicaciones no solían tener restricciones en cuanto a la edad
o condición, por lo que no era extraño que se utilizaran en ancianos y niños, o
en mujeres embarazadas. El período de mayor utilización se sitúa entre los
años 60 y 70 del pasado siglo. Se estima que entre 1950 y el 2000 se habrían
usado unas 600.000 toneladas, la mitad de ellas en Europa [UNEP 2006]. Si
tenemos en cuenta que por cada tonelada de lindano se producían de 4,5 a 9
toneladas de otros isómeros de HCH sin valor comercial, esto ha supuesto la
generación adicional de 2,6 a 5,4 millones de toneladas de residuos de HCH.
Razones medioambientales y sanitarias llevaron a restringir su uso en
diversos países: Canadá en 1970, Japón en 1971, Suecia y Finlandia en 1974,
EE.UU. en 1977, Colombia 1978, etc. Ya en fechas más recientes, la Unión
Europea, en su Reglamento 850/2004 sobre contaminantes orgánicos
persistentes [EPCEU 2004], establecía la práctica prohibición de la fabricación,
comercialización y uso del HCH, incluido el Lindano, a partir de 2008 [UNIDO
2010]. Estas restricciones y prohibiciones han llevado a que actualmente la
utilización del Lindano se limite a aplicaciones muy específicas y en áreas muy
localizadas [UNEP 2006].
Hexaclorobenceno (HCB)
El hexaclorobenceno, también conocido como perclorobenceno dado que
todas las posiciones del anillo se encuentran sustituidas con cloro en lugar de
hidrógeno, inicialmente se produjo por cloración del benceno en fase vapor con
7

Introducción
aire y ácido clorhídrico como agente de cloración [PPU 1990]. Como este
proceso no resultaba suficientemente rentable, la industria optó posteriormente
por la cloración del benceno en fase líquida mediante un mecanismo de
sustitución electrofílica con cloro como agente de cloración y cloruro férrico
como catalizador.
El HCB [PPU 1990, Sullivan and Krieger 2001, ATSDR 2002b, WHO
2003, Tomlin 2009, UNIDO 2010] es un sólido cristalino con tonalidades que
van de incoloras a blanquecinas. Su punto de fusión es de 226-229 ºC y tiene
una presión de vapor de 1,45 mPa (20 ºC). Aunque se inflama a partir de 242
ºC, a temperatura ambiente resulta muy estable desde el punto de vista
químico y es prácticamente insoluble en agua y soluble en la mayoría de
disolventes orgánicos, con excepción de los muy polares. Sus propiedades
fungicidas se describieron por primera vez por el francés H. Yersin en 1945.
Durante varios años se utilizó como tal dada su efectividad contra diversas
“caries” del trigo de origen fúngico, como la Tilletia caries o la Tilletia
controversa. Más adelante su uso se extendió a la protección antifúngica de
semillas o granos preparados para siembra de cebolla, sorgo, trigo y otros. Su
empleo como plaguicida empezó a decaer tras una intoxicación masiva que
tuvo lugar en Turquía entre 1955 y 1959, al utilizarse granos de trigo tratados
con HCB y destinados a siembra, para la fabricación de pan. Éste hecho llevó a
su prohibición como plaguicida en Turquía y, en 1965, en EE.UU. Fuera del
entorno de los plaguicidas y, aunque existe un cierto oscurantismo al respecto,
hay indicios de aplicaciones del HCB en el ámbito industrial: en pirotecnia y
munición de artillería hasta los años 70 del pasado siglo, en la preservación de
la madera, como agente dispersante en la fabricación de neumáticos y goma
sintética, etc.
Sin embargo, a pesar de sus diversas aplicaciones, gran parte de la
producción de HCB se podría calificar de “no deseada”. En efecto, aparece o
aparecía como subproducto en multitud de procesos industriales relacionados
con la síntesis química. Uno de los más importantes, por la cantidad total de
HCB generada, es la fabricación de plaguicidas conteniendo átomos de cloro,
llegando a detectarse su presencia en la síntesis de más de 135 de ellos,
donde alcanzaba valores de hasta el 11%. Esto llevó a la promulgación de
8

Introducción
distintas disposiciones legales que obligaron a la industria a perfeccionar sus
procesos para minimizar la producción de HCB, ya que, en muchos casos, se
aplicaba como impureza junto con el principio activo al utilizarse como base de
la formulación el producto técnico. Otro proceso industrial no menos importante
es la fabricación de disolventes clorados, donde surge como subproducto en
las fases de cloración o de oxicloración y en ciertas operaciones de
fragmentación química (cracking). Los casos más conocidos son: la producción
de tetracloruro de carbono, tricloroeteno, tetracloroeteno, y, ya en menor
medida, otros disolventes como dicloroetenos, tricloroetanos y algunos
clorobencenos. También se le ha relacionado con las industrias siderúrgica y
electroquímica. Un ejemplo de este último caso era la fabricación de cloro a
partir de la electrolisis del cloruro sódico sobre electrodos de grafito, donde la
aparición del HCB como subproducto fue una de las causas que obligó, a
finales de los años 70 del pasado siglo, a sustituir dicho tipo de electrodos por
los metálicos. Por último, otro de los procesos con los que se le ha relacionado
es la incineración de residuos, bien sean sólidos urbanos o de cualquier otro
tipo.
Problemas medioambientales y, como ya se ha comentado, de salud
pública, hacen que su utilización esté muy restringida o prácticamente
prohibida en todo el mundo. No obstante, aunque en la mayoría de los países
industrializados las restricciones y prohibiciones llevan muchos años en vigor,
el HCB ha sido un producto difícil de controlar debido principalmente a la
fabricación “no deseada”.
Diclorodifeniltricloroetano (DDT)
El DDT [Calva y Torres 1998, Olea y Fernández 2001, Sullivan and
Krieger 2001, ATSDR 2002a, WHO 2003, Sanz-Gallardo 2006, Porta et al.
2009, Tomlin 2009, UNIDO 2010] es quizás el plaguicida organoclorado más
popular. Se obtiene por condensación del clorobenceno con
tricloroacetaldehido, reacción que da lugar a un producto técnico de aspecto
parecido a la vaselina y punto de fusión indefinido. El componente mayoritario
de esta mezcla es el 1,1,1-tricloro-2,2-bis(4-clorofenil) etano, más conocido
como p,p’-DDT o 4,4’-DDT. En menor proporción (< 30%), está el 1,1,1-tricloro-
2-(2-clorofenil)-2-(4-clorofenil) etano, también llamado o,p’-DDT o 2,4’-DDT y, a
9

Introducción
nivel de trazas, el 1,1,1-tricloro-2,2-bis(2-clorofenil) etano u o,o’-DDT o 2,2’-
DDT. Dado que todos tienen propiedades insecticidas en mayor o menor
medida, ha sido muy habitual el uso del producto técnico, sin purificar. El p,p’-
DDT puro es un sólido que forma cristales incoloros con un punto de fusión de
108,5 ºC y una presión de vapor de 0,025 mPa (20 ºC). Es insoluble en agua,
moderadamente soluble en disolventes orgánicos polares y soluble en el resto
de disolventes orgánicos. Es el más relevante en cuanto a propiedades
insecticidas y en general es bastante estable desde el punto de vista químico,
especialmente a la oxidación, aunque se deshidroclora a temperaturas
superiores a su punto de fusión para dar el 1,1-dicloro-2,2-bis(4-clorofenil)
etileno o p,p’-DDE. Esta reacción tiene lugar a temperaturas <100 ºC si es
catalizada por trazas de metales, luz UV, o medios alcalinos. El p,p’-DDE no
tiene propiedades insecticidas pero al ser todavía más estable que el p,p’-DDT
se convierte en la principal traza de su utilización. Aunque menos significativo,
otro producto de degradación es el 1,1-dicloro-2,2-bis(4-clorofenil) etano o p,p’-
DDD.
El p,p’-DDT es un potente insecticida no sistémico que actúa por ingestión
o contacto. Su estabilidad química y su liposolubilidad hacen que sea muy
persistente sobre superficies sólidas y que se acumule en las grasas del
insecto, lo que incrementa sobremanera su efectividad. Aunque Ziedler lo
sintetizó por primera vez en 1874, no fue hasta 1939 cuando el químico suizo
Müller descubrió sus propiedades insecticidas (descubrimiento por el que
obtuvo el premio Nobel en 1948) y se empezó a utilizar para proteger la lana
contra la polilla. Su producción a escala industrial comenzó en 1943 y fue
introducido en el mercado por la empresa Geigy (más tarde Ciba-Geigy) bajo
los nombres comerciales de Gesarol, Guesarol y Neocid. Durante la segunda
guerra mundial fue muy efectivo para combatir los vectores del tifus y la
malaria, lo que evitó la proliferación de epidemias. Posteriormente fue
empleado para combatir plagas de todo tipo de artrópodos e insectos y en 1946
comenzó su empleo profuso como fitosanitario con más nombres comerciales,
como Genitox, Anofex, Detoxen, Pentachlorin, Dicophane o Chlorophenotene.
Además de su efectividad, otro factor que contribuyó a su éxito fue su bajo
precio, llegándose a utilizar en los años 60 en todo el mundo cerca de 400.000
10

Introducción
toneladas por año. La Organización Mundial de la Salud (OMS) estima que
hasta 1971 más de 1000 millones de personas se habrían salvado de contraer
malaria gracias a él. Por ejemplo en la India, tras un uso masivo de DDT, se
pasó de 75 millones de casos en 1952, a 100.000 en 1964. En EE.UU. el pico
de utilización se alcanzó en 1962, con 80.000 toneladas de DDT, usadas
mayoritariamente en labores agrícolas. A nivel mundial, la máxima producción
se alcanzó en 1970.
Debido también a problemas medioambientales su declive comienza a
principios de la década de los 70 del pasado siglo. Ya en 1969 fue excluido de
la lista de sustancias activas a utilizar en preparados fitosanitarios. Suecia, en
1970, fue el primer país en prohibir su utilización. En EE.UU. la Agencia de
Protección Ambiental (EPA) prohibió su uso en 1972, excepto en casos de
emergencia para la salud pública, lo que llevó a que los principales productores
redujeran drásticamente su fabricación. Ese mismo año Alemania aprobaba
medidas similares. Esta postura fue adoptada también por la mayoría de países
del primer mundo a lo largo de esa década aunque, paradójicamente, en
muchos de ellos se dejó la puerta abierta a la fabricación y exportación,
siempre que se cumplieran determinados requisitos administrativos. En España
las restricciones comenzaron en 1971 y posteriormente se ampliaron entre
1975 y 1977, llegándose a la práctica prohibición de su uso y comercialización
en 1994. Actualmente su utilización a nivel global está muy restringida y
principalmente se circunscribe a la lucha contra la malaria en aquellos países
donde esta enfermedad es endémica. Para ello se emplea en impregnación de
mosquiteras y tratamientos de superficies dentro del hogar, donde, dada su
persistencia y la escasa superficie a tratar, las cantidades a emplear son
mucho menores que las que se utilizaban para uso fitosanitario en cultivos
extensivos. También se sigue usando como base de síntesis para otros
productos, como el herbicida dicofol.
Bifenilos Policlorados (PCBs)
Los Bifenilos Policlorados [Kimbrough and Jensen 1989, Erickson 1997,
ATSDR 2000, Sullivan and Krieger 2001, WHO 2003, Porta et al. 2009, UNIDO
2010], más conocidos por sus siglas en inglés como PCBs, son hidrocarburos
aromáticos con un grado de sustitución variable de átomos de cloro. La
11

Introducción
estructura básica son dos moléculas de benceno unidas por un carbono de
cada anillo, mediante un enlace sencillo (bifenilo), quedando cinco posiciones
en cada anillo con enlaces C-H susceptibles de cloración (ver Figura 1). Estas
posiciones se numeran, desde el carbono contiguo al que sirve de enlace, en
uno de los anillos en el sentido contrario a las agujas del reloj del 2 al 6 y en el
otro en el sentido de las agujas del reloj del 2’ al 6’. La fórmula general es pues
C12H10-xClx, pudiendo variar “x” entre 1 y 10. El número de potenciales isómeros
por cada grado de cloración es el siguiente:
• 1 Cl = 3 isómeros • 2 Cl = 12 “ • 3 Cl = 24 “ • 4 Cl = 42 “ • 5 Cl = 46 “ • 6 Cl = 42 “ • 7 Cl = 24 “ • 8 Cl = 12 “ • 9 Cl = 3 “ • 10 Cl = 1 “
Lo cual hace un total de 209 posibles compuestos distintos o
“congéneres”. Dada la dificultad de nombrar estas sustancias siguiendo las
reglas clásicas aplicadas por la IUPAC (p.e. 2,3,3’,4,5,5’,6,7’-octaclorobifenilo,
ó 2,3,4,4’,5,5’,6,7’-octaclorobifenilo y así hasta 209 compuestos distintos),
Ballschmiter y Zell, en un trabajo publicado en 1980 [Ballschmiter and Zell
1980], idearon una sistemática para nombrarlos mediante números del 1 al
209. Dicha nomenclatura sistemática, ligeramente corregida por Schulte y
Malisch en su trabajo publicado en 1983 [Schulte and Malisch 1983], ha sido
aceptada por la IUPAC y es la que actualmente se emplea.
Los PCBs como compuestos individuales son sólidos a 25 ºC y 1 atm. con
un punto de fusión que va de 54 ºC para el PCB 1 (2-clorobifenilo), hasta 310
ºC para el PCB 209 (2,2’,3,3’,4,4’,5,5’,6,6’-decaclorobifenilo). Generalmente el
punto de ebullición aumenta con el grado de cloración. Paradójicamente las
mezclas de un número apreciable de ellos dan lugar a líquidos incoloros o
ligeramente amarillentos a temperatura ambiente y presión atmosférica,
aunque eso sí, muy viscosos. Desde el punto de vista químico son muy
estables y resistentes a la degradación, con la cualidad adicional de ser, no
solo ininflamables, sino capaces de extinguir la llama al producir grandes
12

Introducción
cantidades de HCl gas en la pirólisis. Otra característica a resaltar es que, dada
su baja polaridad, son insolubles en agua e inmiscibles con ella, siendo
solubles en compuestos orgánicos en general y en particular en aceites y
grasas; además presentan un muy acusado comportamiento dieléctrico con
altos valores de las correspondientes constantes. También poseen una
capacidad calorífica bastante aceptable así como propiedades lubricantes.
Se descubrieron allá por el año 1881 y su producción y explotación
comercial empezó en EE.UU. en 1929 de la mano de la Swan Chemical
Company (que en 1935 pasaría a pertenecer a la Monsanto), bajo el nombre
comercial de Aroclor. Desde entonces se generalizó su producción y uso bajo
distintas denominaciones en diversos países, entre otras: la mencionada
Aroclor, Chloretol, DyKanol, Inerteen y Pyranol en EE.UU., Phenochlor y
Pyrelene en Francia, Chlophen en la antigua Alemania Federal, Kaneclor y
Sataterm en Japón, Fenclor en Italia y Soval en la extinta URSS.
La fabricación comercial de PCBs se llevaba a cabo mediante la cloración
del bifenilo con cloro anhidro en presencia de catalizadores, normalmente
virutas de hierro o cloruro férrico. Esta reacción discurría a temperaturas altas
(~140 ºC) y 1-2 atm., dando lugar a un producto en bruto que debía ser
refinado posteriormente mediante lavados alcalinos o destilación, siendo
inevitable la presencia de impurezas como dibenzofuranos policlorados
(PCDFs), naftalenos policlorados (PCNs) y tetrafenilos (o cuaterfenilos)
policlorados (PCQs). El producto final era un líquido aceitoso que consistía
básicamente en una mezcla de congéneres de PCBs cuyo grado de cloración o
porcentaje de cloro-sustitución se podía ajustar jugando con las condiciones de
reacción (principalmente el tiempo de contacto de los reactivos). Generalmente
este grado de cloración se indicaba, directa o indirectamente, mediante un
número que acompañaba a las denominaciones comerciales antes citadas. Así,
por ejemplo, para el Aroclor existían el 1232 (32-33% de Cl), el 1248 (48% de
Cl), el 1254 (52-54% de Cl), el 1260 (60% de Cl), etc. Para algunas
aplicaciones estos productos se presentaban disueltos en triclorobenceno o
similares. Se calcula que en total, desde 1929, se llegaron a producir en todo el
mundo alrededor de 1,5 millones de toneladas, aunque, por diversas razones,
es difícil estimar esta cifra con exactitud.
13

Introducción
Su rápido éxito comercial se debió a las características físico-químicas
antes mencionadas, que les llevaron a sustituir con enormes ventajas a otros
productos existentes, en aplicaciones donde se requerían ininflamabilidad, gran
resistencia al calor y a los ataques químicos, o alta capacidad dieléctrica y
resistividad. Ejemplos clásicos de su aplicación fueron: fluidos hidráulicos,
fluidos trasmisores de calor, lubricantes de alta presión, aditivos de pinturas y
tintas, plastificantes, recubrimientos para prendas antiinflamables, fijadores de
tintas en papeles de autocopia, aislamiento de material eléctrico, cables,
motores y generadores, y mayormente como fluidos dieléctricos refrigerantes
en transformadores y condensadores.
Debido a problemas medioambientales su declive comercial comienza en
1971, cuando los principales productores reducen su fabricación. Después
llegarían las regulaciones legales de 1976, 1979 y 1985 en EE.UU., que serían
imitadas por otros países y organismos internacionales, y que llevaron a la
práctica prohibición de la fabricación y uso comercial de estos productos en
nuestros días.
14

Introducción
Problemática medioambiental
No hay evidencias de que todos estos compuestos aparezcan en el
planeta tierra de forma natural, por lo que su presencia en la naturaleza hay
que achacarla a la actividad del ser humano [UNEP 2006, UNIDO 2010]. Nos
encontramos pues, como en muchas otras ocasiones, con unos productos
químicos que, bien por ser utilizados en medios abiertos, bien tras desecharlos
una vez utilizados o por ser subproductos sin valor comercial, o bien como
consecuencia de pérdidas en los sistemas cerrados donde estaban siendo
utilizados, acaban indefectiblemente por recalar en el medio natural. Una vez
ahí, una de las cualidades que originaron su auge comercial en los dos cuartos
centrales del siglo XX, es también la causa de que se hayan convertido en un
gran problema medioambiental. En efecto, su gran resistencia química hace
prácticamente inviable, a corto y medio plazo, su eliminación mediante los
clásicos mecanismos de foto-oxidación, hidrólisis ácido-base o
biometabolización (o una combinación de ellos). Como consecuencia, desde
los puntos donde hayan sido abandonados, puedan ser poco a poco
trasportados por el aire o el agua hasta llegar prácticamente a la totalidad del
planeta [ATSDR 2000, ATSDR 2002a, ATSDR 2002b, ATSDR 2005, ATSDR
2007].
El ciclo medioambiental empieza en el suelo, el agua o el aire donde
hayan podido ser vertidos. En estos medios permanecen largo tiempo al ser de
difícil degradación. Pueden circular fácilmente entre los tres medios, por
ejemplo evaporarse desde el suelo o el agua al aire. En el aire, pueden ser
trasportados largas distancias sobre las partículas en suspensión o en forma de
vapor, y volver a caer luego al suelo o al agua junto con el polvo, la nieve o la
lluvia. En general cuanto más volátil es el compuesto (en el caso de los PCBs a
menor grado de cloración mayor volatilidad) a mayor distancia de la fuente
contaminante podrá ser trasladado por el aire. En el agua pueden ser
trasportados por corrientes para seguir el ciclo continental o marino, pueden
adherirse a los sedimentos del fondo (preferentemente los menos volátiles) o
pueden evaporarse al aire (preferentemente los más volátiles). En el suelo, se
adhieren firmemente y pueden permanecer años, aunque los más livianos
tienen más probabilidad de volverse a evaporar. Desde el suelo, el aire o las
15

Introducción
aguas continentales pasan a los vegetales y a las cosechas. De las plantas
pasan a los animales y desde estos, al morir, a otros animales o de nuevo al
suelo, al agua o al aire. En el caso del medio marino pasan del agua al
plankton y las algas y de ahí, al resto de organismos superiores.
Tras incorporarse a los seres vivos, debido a la unión de su estabilidad
química con su carácter liposoluble, se acumulan en sus tejidos grasos al no
poder metabolizarse una vez ingeridos. Esta acumulación no es uniforme sino
que, cualquiera que sea el medio, se acrecienta a lo largo de la cadena
alimentaria (biomagnificación) [Calva y Torres 1998, Norstrom 2002, WHO
2003, Porta et al. 2009], de manera que los organismos que ocupan los lugares
más altos de la misma (carnívoros y en última instancia el ser humano),
presentan mayores concentraciones de estos compuestos en sus tejidos
grasos (ver Figura 2). La concentración de ppm en agua se refiere a mg L-1 y
en la biota a mg kg-1.
Figura 2 Ejemplo de biomagnificación del DDT a lo largo de la cadena alimentaria. Los valores deben tomarse a título ilustrativo ya que dependen del tiempo y del lugar. (Fuente: Universidad de Sevilla. Escuela Técnica Superior de Ingenieros. Diapositivas del Máster en Tecnología Química y Ambiental).
Dada su estabilidad química, sería de esperar que encontráramos estos
compuestos tal como fueron depositados en el medio ambiente. Sin embargo,
16

Introducción
algunos de ellos sufren trasformaciones físico-químicas o bioquímicas en los
distintos medios, cuyo efecto global es asimilable a un proceso de
metabolización ambiental que da lugar, con el tiempo, a compuestos diferentes
y más estables. Por ejemplo, las mezclas comerciales de PCBs que causaron
la contaminación original, tienen una composición (perfil) distinta a la que nos
encontramos en las muestras ambientales. La metabolización ambiental
trasforma los perfiles originales, cualesquiera que estos hayan sido, dando
lugar a perfiles ambientales mucho más uniformes a lo largo de toda la
biosfera. Los perfiles ambientales llegan a perder totalmente algunos de los
congéneres de las mezclas comerciales originales, enriqueciéndose en otros
de ellos. Algunas de las reglas que sigue esta transformación son [Zell and
Ballschmitter 1980, Faugère et Chaurial 1989, Erickson 1997, ATSDR 2000,
Sullivan and Krieger 2001]:
• Los congéneres más volátiles tienen mayor tendencia a pasar a la
fase vapor y por tanto al aire, mientras que los más pesados se
encontrarán en suelo y agua.
• Los compuestos con mayor grado de cloración son por lo general
más estables y presentan mayores tiempos de vida media.
• Los compuestos con 4-sustitución o con 2,5-sustitución en
cualquiera de los anillos (para-sustitución) son más resistentes a la
degradación.
• Cuanto mayor sea el grado de sustitución en la posición orto (2, 2’,
6, ó 6’), mayor resistencia a la degradación.
• Los compuestos sin ninguna sustitución en alguno de los anillos
son más fácilmente degradables.
• La metabolización en animales de sangre caliente es más intensa
que en los de sangre fría o en las plantas.
En el caso del HCH [Sullivan and Krieger 2001, ATSDR 2005, Porta et al.
2009], aunque el isómero mayoritario en el producto técnico es el α-HCH, por
sus características físico-químicas presenta mayor persistencia y capacidad de
17

Introducción
bioacumulación el β-HCH. Por ello, en la mayoría de muestras biológicas el
isómero predominante, si no el único detectable, es el β-HCH. Dado que
últimamente no se usaba el producto técnico sino el γ-HCH en las
formulaciones, en algunas muestras éste también aparece en cantidades
apreciables.
En cuanto al DDT [Sullivan and Krieger 2001, ATSDR 2002a, Porta et al.
2009], el isómero más abundante en las formulaciones es el p,p’-DDT, que se
metaboliza principalmente a p,p’-DDE, producto todavía más persistente y de
gran capacidad de bioacumulación que por ello es fácil de detectar en la
mayoría de las muestras biológicas. No obstante, todavía hoy en día el
compuesto original es apreciable en un número significativo de muestras.
Por lo que respecta al heptacloro [Sullivan and Krieger 2001, ATSDR
2007], se metaboliza con cierta facilidad a heptacloro epóxido, que es más
persistente y de mayor capacidad de acumulación. No es de extrañar, pues,
que sea este último el que aparezca mayoritariamente en las muestras
biológicas de los organismos expuestos a heptacloro. Aunque no todo el
heptacloro epóxido tiene su origen en los productos formulados con heptacloro.
Las formulaciones a base de clordano dan lugar a heptacloro como uno de los
metabolitos mayoritarios, con lo que indirectamente también son fuente de
heptacloro epóxido.
Por último, el HCB [ATSDR 2002b, Porta et al. 2009] no presenta
transformaciones demasiado acusadas y tiene gran persistencia y capacidad
de acumulación como tal, con lo que lo habitual es encontrarlo inalterado en las
muestras biológicas.
A pesar de que estos compuestos se han aplicado en áreas concretas del
planeta, los incontables estudios ambientales existentes sobre ellos
demuestran su presencia en la totalidad de la biosfera. Es posible encontrarlos
desde las nieves de la Antártida a los hielos del Ártico, pasando por todos los
océanos y mares. Están presentes tanto en las aguas de lagos y ríos, como en
todo tipo de suelos. En lo referente a la atmósfera, no hay ningún rincón de la
capa baja de la misma que se libre de ellos. Y como era de prever también los
seres vivos los hemos ido incorporando a nuestros organismos, sin que el lugar
18

Introducción
de residencia haya supuesto una barrera infranqueable, sino tan sólo una
diferenciación en las cantidades acumuladas.
No es fácil hacer un cálculo acertado de su distribución relativa en la
biosfera, debido sobre todo a la cantidad de datos que sería necesario manejar
y a que los niveles de concentración van variando con el tiempo. No obstante,
existe una estimación realizada en 1982 sobre el reparto de los residuos de
PCBs en los distintos compartimentos medioambientales a fecha de 1981
[Erickson 1997]. Los resultados vienen reflejados en la Tabla 1:
Tabla 1 Reparto de residuos de PCBs en compartimentos ambientales
UBICACIÓN % del TOTAL de PCBs
Atmósfera 0,003
Agua continental 0,004
Sedimentos y lodos de aguas continentales 0,702
Fauna y flora de aguas continentales 0,003
Suelos de cualquier tipo 0,175 Fangos de depuradoras y de vertidos en general 0,842
Vegetación y cultivos 0,175
Fauna terrestre salvaje 0,00005
Ganado y animales domésticos 0,0001
Seres humanos 0,0009
Agua, fauna y flora de océanos y mares 1,053
Sedimentos de océanos y mares 0,175
Depósitos controlados y vertederos 30,7
En uso en equipamientos eléctricos 13,0
En otros usos industriales o comerciales 13,2 Destruidos, degradados o en paradero desconocido 40,0
Aunque los valores cuantitativos hay que tomarlos con ciertas
precauciones por las razones antes aducidas, estos datos sí que reflejan
19

Introducción
claramente la ubicuidad de su presencia; ubicuidad que no sólo se da en los
PCBs, sino en la totalidad de estos compuestos.
Como consecuencia de su expansión a la totalidad del medio ambiente
planetario, tenemos una exposición globalizada de la fauna y la flora a los
mismos. Teniendo en cuenta que se trata de compuestos ajenos a la
bioquímica de su organismo y que algunos de ellos, concretamente los
plaguicidas, fueron diseñados como tóxicos para ciertas especies, no es
disparatado esperar que esta exposición tenga efectos negativos en la biota.
De hecho está ya aceptado por organismos internacionales como Naciones
Unidas [UNIDO 2010], que estas sustancias afectan al desarrollo y crecimiento
de plantas y animales, que pueden causar una reducción en el éxito
reproductor, defectos congénitos, cambios conductuales e incluso la muerte,
así como que son disruptores del sistema inmunológico y endocrino.
Ya a mediados del siglo XX se empezó a estudiar y conocer mejor su
capacidad de persistencia y bioacumulación, lo que llevó a algunos científicos a
percatarse del peligro latente que ello suponía, máxime cuando su utilización
iba en aumento. A principios de los años 60 se atribuyó a algunos de estos
plaguicidas organoclorados, principalmente al DDT [ATSDR 2002a], el
debilitamiento en la cáscara de los huevos de las aves, especialmente las
silvestres, que llevaba a una disminución de sus tasas de reproducción. La
preocupación por estos temas indujo a la bióloga Rachel L. Carson, técnica al
servicio de la Administración de Pesca y Vida Salvaje de EE.UU., a escribir el
libro Silent Spring (Primavera Silenciosa), cuyo título hacía referencia a un
futuro con una hipotética primavera sin pájaros [Carlson 2002]. El libro, con un
carácter divulgativo más que científico, fue publicado en 1962 y versaba sobre
la problemática medioambiental de los plaguicidas organoclorados y sus
efectos perjudiciales sobre la biota y el ser humano, a la vez que denunciaba
por ello a las grandes empresas químicas. Provocó una gran controversia con
encendidos debates entre partidarios y detractores, pero fue un gran éxito de
ventas y sirvió para concienciar del problema a la opinión pública. Tal fue la
repercusión, que la presión social obligó al gobierno de los EE.UU. a regular e
incluso prohibir algunos de estos productos. Estas acciones legales crearon un
precedente que a su vez condujo a acciones similares en distintos países.
20

Introducción
Pero se trataba de un problema global y con el tiempo se vio que las
acciones unilaterales, en forma de normativas nacionales muchas veces
contradictorias, incompletas o divergentes, no eran suficientes. Había que
buscar soluciones conjuntas a nivel internacional en base a tratados o
convenios de obligado cumplimiento, la mayoría de ellos auspiciados por
Naciones Unidas, en los que estuviera implicado el mayor número de países
posible [UNIDO 2010].
En 1989 surge el Convenio de Basilea dentro del Programa
Medioambiental de Naciones Unidas (UNEP), que se ratifica en 1992. El
objetivo de este convenio es el control del movimiento transfronterizo de los
productos químicos peligrosos y su gestión. El 14 de junio de 1992 se adopta
en Río de Janeiro la Agenda 21, cuyos capítulos 19 y 20 tratan sobre la gestión
de productos químicos tóxicos y desechos peligrosos, respectivamente. El
convenio de Rótterdam, establecido en 1998 y que entró en vigor en 2004,
tiene por objeto la vigilancia y control del comercio de ciertos productos
químicos peligrosos. Aunque todos ellos tratan sobre productos químicos
tóxicos o peligroso en general, dentro estos epígrafes están incluidos los
compuestos aquí estudiados.
Además de estos acuerdos, con un enfoque general en cuanto a
productos concernidos, existen otros cuya tema específico son estos
compuestos y otros similares agrupados todos ellos bajo el calificativo de
Contaminantes Orgánicos Persistentes o, según su acrónimo en inglés, POPs
(Persistent Organic Pollutants). Todos estos compuestos tienen en común que
son:
• Muy tóxicos para los seres humanos y la biota.
• Persistentes en el medio ambiente y resistentes a la
biodegradación.
• Ingeridos y bioacumulados en ecosistemas terrestres y acuáticos.
• Susceptibles de transporte atmosférico transfronterizo de largo
alcance y posterior deposición.
21

Introducción
El 24 de junio de 1998 la Comisión Económica de Naciones Unidas para
Europa (UNECE), introduce en Aarhus, Dinamarca, el Protocolo sobre POPs
como parte del Convenio sobre Contaminación Atmosférica Transfronteriza de
Largo Alcance (CLRTAP o Convenio de Ginebra). Este protocolo abarca una
lista de 16 POPs y su objetivo último es eliminar cualquier vertido, emisión o
pérdida de estos productos en el medio ambiente. Para ello prohíbe la
producción y uso de: Aldrín, Clordano, Clordecona, Dieldrín, Endrín,
Hexabromobifenilo, Mirex y Toxafeno; dispone para un siguiente estadio la
eliminación de: Heptacloro, HCB, DDT y PCBs, restringiendo severamente el
uso de estos dos últimos y del HCH (incluido el Lindano), y demanda a los
firmantes que reduzcan sus emisiones de Dibenzodioxinas Policloradas
(PCDDs o simplemente dioxinas), PCDFs, Hidrocarburos Aromáticos
Policíclicos (PAHs) y HCB. El protocolo entró en vigor el 23 de octubre de 2003
y hasta la fecha cuenta con 36 firmantes y 19 ratificaciones.
El Convenio de Estocolmo se firmó en la capital sueca el 23 de mayo de
2001 por 92 países y la Unión Europea, bajo la coordinación del UNEP. Entró
en vigor el 17 de mayo de 2004, 90 días después de que se alcanzaran las 50
primeras ratificaciones. Hoy en día pasan de 150 los países firmantes y las
ratificaciones (España lo ratificó el 28 de mayo de 2004). Es un tratado global
cuyo fin último es la protección de la salud humana y el medio ambiente frente
a los POPs y, en lo referente a ellos, es el vínculo legal más importante a nivel
internacional. Los países firmantes se comprometen a tomar las medidas
contempladas en él para eliminar o reducir la liberación de estos productos en
el medio ambiente. Inicialmente la lista se reducía a 12 compuestos (the dirty
dozen): Aldrín, Clordano, DDT, Dieldrín, Endrín, HCB, Heptaclor, PCDDs,
PCDFs, PCBs, Mirex y Toxafeno. El articulado del convenio abre la posibilidad
e indica el procedimiento a seguir para la introducción de nuevos compuestos.
En aplicación del mismo la lista ya se ha ampliado e incluye también, entre
otros, el α, β y γ-HCH desde mayo de 2009 [Stockholm Convention 2010].
La Unión Europea ha elaborado el Reglamento 850/2004 [EPCEU 2004],
en vigor desde el 20 de mayo de 2004, con objeto de establecer un marco
jurídico común para llevar a efecto las principales disposiciones del Convenio
de Estocolmo y del Protocolo sobre POPs del Convenio de Ginebra y asegurar
22

Introducción
la coordinación y la coherencia al aplicar, a nivel de la UE, las disposiciones de
los mismos y de los Convenios de Rótterdam y Basilea. En cumplimiento de
estos acuerdos y del Reglamento de la Unión Europea, España ha establecido
un Plan Nacional de Actuación, redactado por la Subdirección General de
Calidad del Aire y Prevención de Riesgos del Ministerio del Medio Ambiente
[MMA 2007], que fue aprobado el 2 de febrero de 2007 y remitido a los
organismos competentes el 20 de marzo de 2007.
Por último, para completar este capítulo medioambiental, decir que hay
que dejar la puerta abierta al optimismo, pues, fruto de la aplicación de todas
las medidas legales y acuerdos antes descritos, la mayor parte de estudios
ambientales realizados en los últimos años coinciden en señalar un marcado
descenso en los niveles de estos compuestos en matrices ambientales
[Sullivan and Krieger 2001]. Así por ejemplo, según recoge el informe
monográfico 10/2005 de la Agencia Europea del Medio Ambiente, los niveles
de PCBs en leche materna a esa fecha supondrían alrededor de un 30% de los
máximos alcanzados el pasado siglo [EEA 2005].
23

Introducción
Efectos sobre la salud
Introducción
El estudio de los efectos nocivos de los productos químicos sobre los
organismos vivos entra en el campo de la toxicología [Timbrell 2008, Galiano
2010]. Dentro de ella, a la capacidad de un producto químico para generar una
respuesta nociva sobre los organismos vivos se le conoce como toxicidad y al
efecto nocivo tras la exposición a un producto se le denomina también
respuesta tóxica del organismo. En la toxicología subyace latente un fin último
de preservar a los seres humanos de tales efectos nocivos.
Uno de los grandes problemas con que se tropieza al plantear el estudio
toxicológico de un determinado producto químico es que la respuesta tóxica
varía sobremanera dependiendo de varios factores [Timbrell 2008, CCOHS
2009, Galiano 2010]:
• Vía de exposición o penetración
• Dosis o nivel de la exposición
• Frecuencia y duración de la exposición
• Vehiculización de la exposición
• Especie expuesta e individuo expuesto (variación biológica)
Las principales vías de penetración en el organismo son:
− Por contacto, principalmente cutáneo u ocular
− Por inhalación, es decir, a través del sistema respiratorio
− Por ingestión, es decir, a través del aparato digestivo
− Parenteral, que a su vez tiene distintas variantes: intravenosa,
intramuscular, subcutánea, intraperitoneal, etc.
Los efectos causados difieren dado que las barreras a franquear para
introducirse en el organismo, así como los órganos a los que inicialmente se
accede, son distintos para cada tipo de vía de penetración. Una vez el producto
24

Introducción
dentro del organismo el efecto causado puede ser local, si se produce
directamente en los órganos donde recala, o sistémico, si se ven afectados
órganos más alejados.
En cuanto a la dosis, el principio general es que a mayor cantidad de
producto mayor es el efecto producido o la probabilidad de que se produzca. La
curva dosis-respuesta suele tener forma sigmoidal, con un tramo recto en la
zona central cuya pendiente es una medida de la toxicidad del producto.
Ante la presencia de sustancias extrañas los organismos desencadenan
mecanismos de defensa que buscan su neutralización o eliminación. Como
este proceso requiere un tiempo, un aumento en la frecuencia o en la duración
de la exposición supone nuevos aportes que mantienen la presencia del
producto dentro del organismo, posibilitando así la aparición de efectos
adversos de desarrollo más lento. Por ello se distingue entre exposición aguda,
como aquella que se produce en una sola vez o en un corto espacio de tiempo
(días), exposición sub-aguda (semanas), sub-crónica (meses) y crónica (años).
En cualquiera de los casos la respuesta tóxica puede ser: reversible (cesada la
exposición el efecto desaparece al cabo del tiempo) o crónica (cesada la
exposición el efecto se prolonga durante varios años o toda la vida).
A la hora de exponerse al organismo, el producto suele ir acompañado de
distintas sustancias que lo disuelven o vehiculizan. Pues bien, el pH, el carácter
lipídico o hídrico del vehículo, el que esté disuelto o emulsionado, etc., en
muchos casos influyen en la cantidad absorbida y la distribución del producto
dentro del organismo.
Es un hecho que en multitud de ocasiones un mismo producto químico
afecta de forma distinta a diversas especies. Baste recordar que un insecticida
tiene un efecto rápido y fatal para determinados insectos y sin embargo el
grado de afectación es mucho menor o incluso prácticamente nulo en plantas o
mamíferos. También que un herbicida elimina algunas especies vegetales a la
vez que permite el desarrollo de otras. Esta diversidad de respuestas tóxicas
tiene su base en la diferencia que existe en los órganos y en los complejos
sistemas metabólicos de cada una de las especies. Pero no sólo varía la
respuesta entre distintas especies, sino que dos individuos de una misma
especie pueden verse afectados de distinta manera ante idéntica exposición a
25

Introducción
un producto. Las razones de esto son principalmente, el peso, la edad, el sexo,
la etapa de desarrollo o el estado en que se encuentran, pero también la
susceptibilidad genética (pequeñas diferencias genéticas que generan mayor
vulnerabilidad) y la sensibilización (efecto memoria de rechazo creado tras
exposiciones pasadas).
Lo primero que se estudia al investigar la toxicidad de un compuesto es
qué le sucede dentro del organismo, o lo que es lo mismo, su disposición, que,
a su vez, consta de cuatro fases [Timbrell 2008, Galiano 2010]:
• Absorción. Es el proceso mediante el cual el producto penetra en el
interior del organismo. Implica atravesar la barrera de las
membranas (bicapas semipermeables fosfolípido/proteína) que
sirven de protección y aíslan al organismo del exterior.
• Distribución. El producto es transportado y repartido, por medio de
los sistemas sanguíneo o linfático, a tejidos y órganos. En el
transporte sanguíneo de los productos juegan un papel importante
las proteínas vehiculizadoras, a los que se ligan para poder
compatibilizarse mejor con el medio. La más común es la albúmina
pero, en el caso de compuestos liposolubles, también son
importantes las lipoproteínas. Para atravesar membranas existen
proteínas vehiculizadoras específicas, como la glicoproteína P.
• Metabolización o biotransformación. El producto sufre una serie de
transformaciones químicas que lo convierte en otra especie
química (generalmente de mayor polaridad) más fácil de eliminar.
En una primera fase las reacciones tienden a romper la molécula o
a introducir grupos funcionales más polares mediante oxidación (N-
oxidación, S-oxidación, desalquilación), reducción
(deshalogenación), hidrólisis (esteres, amidas) o hidratación
(epóxidos). Estas reacciones se catalizan mayormente mediante el
sistema enzimático citocromo P-450, localizado en el retículo
endoplasmático liso (fracción microsomal) de las células y es
especialmente abundante en las células hepáticas. En una
segunda fase los productos resultantes se unen a moléculas
26

Introducción
endógenas (conjugación) buscando la formación de compuestos
mas fáciles de excretar y menos tóxicos. Las reacciones de
conjugación más habituales son: glucuronación, sulfatación,
acilación (no da lugar a productos más polares), glicinación,
glutationación. Los enzimas implicados son transferasas
localizadas mayormente en las células hepáticas, intestinales y
renales. A veces se da una tercera fase en la que alguno de estos
conjugados vuelve a sufrir reacciones de las contempladas en la
primera fase. También se da el caso de productos que se conjugan
directamente, sin pasar por la primera fase. Aunque el organismo
busca justo lo contrario, puede ocurrir que alguno de los
metabolitos formados sea más tóxico que el producto original,
sobre todo en la primera fase.
• Eliminación o excreción. Es el proceso por el cual el producto o sus
metabolitos son expulsados del organismo. En general, la principal
vía de salida es la orina. Otras vías utilizadas son la bilis y, en
menor grado, la sudoración, el lagrimeo, el semen y la saliva. El
aire exhalado por los pulmones supone también una vía importante
para compuestos volátiles. En el caso de compuestos muy
liposolubles o de difícil absorción una ruta a tener en cuenta son las
heces. También para compuestos liposolubles la leche materna
supone una vía de eliminación significativa que se debe tener muy
presente, ya que a través de ella se pueden trasladar los efectos
nocivos a la siguiente generación, al igual que ocurre con la
transmisión a través de la placenta.
Conocida la disposición de un compuesto es más fácil entender o predecir
la respuesta tóxica, que se puede encuadrar en alguno de los siguientes tipos
[Timbrell 2008]:
− Efectos tóxicos directos, es decir, lesiones en los tejidos celulares de
los órganos directamente expuestos al producto químico.
− Efectos o alteraciones fisiológicas o bioquímicas.
27

Introducción
− Disrupción endocrina, provocada por interacciones con el sistema
hormonal.
− Anomalías en el desarrollo, entre las que destaca la teratogénesis o, lo
que es lo mismo, alteraciones en el desarrollo del embrión o el feto que
se manifiestan posteriormente en anormalidades, malformaciones o
muerte.
− Inmunotoxicidad o alteraciones del sistema inmunológico, como:
inmunodepresión, inmunoestimulación, o hipersensibilidad.
− Alteraciones genéticas o genotoxicidad, con tres principales variantes:
mutagénesis (pérdida, adición, o cambio de bases con la consiguiente
alteración de secuencias de ADN), clastogénesis (pérdida, adición o
reagrupamiento de partes de cromosomas) y aneugénesis (adquisición
o pérdida de cromosomas completos).
− Carcinogénesis.
El hecho de clasificarlos por separado no implica que se tengan que dar
de forma independiente ya que, más bien al contrario, es muy habitual que
unos actúen como desencadenante de otros.
Para conocer la respuesta tóxica concreta atribuible a un producto se
pueden utilizar varias opciones [Timbrell 2008]:
a) Estudios in vitro: se exponen al producto células, partes de células, u
organismos unicelulares y se observan los resultados.
b) Estudios in vivo: organismos superiores se exponen a dosis
conocidas del producto bajo condiciones controladas para investigar
su disposición y qué efectos tienen lugar, en qué condiciones y a qué
dosis.
c) Estudios epidemiológicos: se investiga un conjunto suficientemente
grande (población) de individuos de una misma especie y mediante
herramientas estadísticas se intenta establecer la relación entre el
grado de exposición a un producto y la frecuencia de aparición de
determinados efectos nocivos.
28

Introducción
Para el caso particular de los seres humanos es obvio que los estudios in
vivo hay que hacerlos de forma muy restrictiva. Cuando lo que se quiere probar
es la ausencia de toxicidad a dosis efectivas de uso, por ejemplo para los
medicamentos, aditivos y compuestos alimentarios, o productos de uso
cosmético, los ensayos in vivo se realizan antes con otras especies y se
extrapolan los resultados para establecer potenciales dosis sin efecto nocivo
aparente que fijen el margen de seguridad para realizar los ensayos en
humanos. Cuando lo que se pretende establecer son los efectos y las dosis
tóxicas es evidente que los estudios in vivo como norma general quedan
restringidos a especies distintas a la humana, lo que genera un déficit de
información. Este déficit se intenta subsanar con información procedente de
exposiciones accidentales (normalmente son exposiciones agudas o sub-
agudas a dosis elevadas) y de ámbitos laborales concretos con niveles de
exposición superiores a los de la población general (normalmente son
exposiciones sub-crónicas o crónicas a dosis medias).
Tanto en los estudios in vivo como en los in vitro, debido a la inmediatez
de la relación causa-efecto, es más fácil obtener resultados con experimentos a
corto plazo y dosis altas [Olea y Fernández 2001]. Esto hace que sea bastante
accesible la información toxicológica de la mayoría de los productos químicos
en exposición aguda a niveles altos. Conforme baja la dosis y aumenta el
tiempo de exposición, crece la dificultad, la complejidad y el coste del estudio
por lo que generalmente, a partir de un cierto punto ni se plantea. Lo que
implica que para muchas especies químicas la información toxicológica de
exposición crónica a largo plazo (varios años) y dosis bajas, procedente de
esta clase de estudios experimentales, sea muy escasa o inexistente. Para
paliar este problema se recurre a estudios epidemiológicos observacionales.
Una de las grandes ventajas de estos estudios epidemiológicos es que se
pueden llevar a cabo con personas, por lo que no es necesario extrapolar los
resultados de una especie a otra y las conclusiones obtenidas son
directamente aplicables a los seres humanos.
Algunos conceptos básicos en epidemiología son [Vaquero y Asensio
1992, Sullivan and Krieger 2001, Bhopal 2008, Piédrola 2008]:
29

Introducción
− Población. Conjunto de individuos sujetos a estudio. También se
emplea el término de muestra poblacional para definir el conjunto de
individuos con los que se realiza el estudio, como contrapunto al
concepto de población diana, que sería el conjunto de individuos a los
que se pretende extrapolar los resultados del estudio.
− Morbilidad. Aparición de una respuesta tóxica o enfermedad. Si el
resultado es la muerte hablaremos de Mortalidad.
− Prevalencia. cociente entre el número de individuos que presentan
morbilidad y el número total de individuos de la población, en un
momento dado. Se puede expresar en tanto por uno o en % y
representa la extensión de la morbilidad en la población en ese
momento.
− Incidencia (tasa). cociente entre el número de nuevos individuos que
presentan morbilidad y el número total de individuos de la población,
en un período de tiempo especificado. Se puede expresar en tanto por
uno o en %, por unidad de tiempo, y representa la velocidad de
incremento de la morbilidad en la población. En caso de muerte
hablamos de tasa de mortalidad. De alguna manera representa
también la probabilidad o el riesgo de que un individuo sano contraiga
la enfermedad en ese espacio de tiempo.
− Odds. En términos genéricos es el cociente entre la probabilidad de un
suceso y la de su contrario. En epidemiología representa el cociente
entre las probabilidades de estar o no expuesto, o bien de estar o no
enfermo.
Desde el punto de vista de la estrategia de diseño, en epidemiología se
trabaja con dos modelos de enfoque [Vaquero y Asensio 1992, Sullivan and
Krieger 2001, Bhopal 2008, Piédrola 2008]:
• Estudios de Casos y Controles. Se trata de coger dos muestras
poblacionales: un grupo de personas que presentan la enfermedad
(o el efecto) esperable y que serían los casos, y otro que no lo
30

Introducción
presenta y que serían los controles. Una vez obtenidas las
muestras poblacionales, se estudia la exposición a la posible causa
o factor de riesgo (sustancia química, hábito no saludable, etc.) en
ambos grupos, para ver si es significativamente mayor en los casos
que en los controles. De ser así concluiremos que existe una
relación causa/efecto entre el factor de riesgo y la enfermedad
estudiados.
• Estudios de Cohortes. Al igual que antes se cogen dos muestras
poblacionales, pero en este caso un grupo es de personas
expuestas al factor de riesgo y el otro de personas no expuestas.
Una vez obtenidas ambas muestras poblacionales se investiga si la
probabilidad de enfermedad es significativamente mayor en el
grupo de expuestos que en el de no expuestos. De ser así, en este
caso también podremos concluir que existe una relación
causa/efecto entre el factor de riesgo y la enfermedad estudiados.
Ambos modelos admiten diversas variantes como: estudio de casos y
controles apareados, estudio de cohorte única, estudio caso-cohorte, etc.
La intensidad de la relación causa/efecto se mide a través de diversos
parámetros. Los más utilizados son [Vaquero y Asensio 1992, Sullivan and
Krieger 2001, Bhopal 2008, Piédrola 2008]:
− Riesgo Relativo (RR). Es la razón entre la incidencia de la enfermedad
en personas expuestas y la incidencia de la enfermedad en personas
no expuestas. Representa las veces que se multiplica el riesgo debido
al factor de riesgo.
− Riesgo Atribuible (AR). Se puede cuantificar como cantidad absoluta:
diferencia de la incidencia de la enfermedad en personas expuestas y
personas no expuestas, en cuyo caso representa el exceso de riesgo
achacable al factor de riesgo; o como cantidad relativa o proporción
(por lo que se le suele denominar también proporción de riesgo
atribuible), normalmente referida a la población expuesta: diferencia de
la incidencia de la enfermedad en personas expuestas y personas no
31

Introducción
expuestas, dividida entre la incidencia de la enfermedad en personas
expuestas, en cuyo caso representa la fracción del riesgo ocasionada
por el factor de riesgo en las personas expuestas.
− Razón de Mortalidad Normalizada (SMR). Es la razón entre la tasa de
mortalidad en personas expuestas y la tasa de mortalidad esperable
en el grupo de población estudiado.
− Odds Ratio (OR). Es la razón entre los Odds de exposición en
enfermos y no enfermos, o bien la razón entre los Odds de
enfermedad en los expuestos y no expuestos.
− Coeficiente de Correlación (r). Se realiza una regresión (normalmente
lineal) entre la incidencia de la enfermedad (variable dependiente) y el
nivel de exposición (variable independiente). El grado de dependencia
de la primera con la segunda se mide a través de “r” que en valor
absoluto va de 0 (total independencia) a 1 (dependencia total). La
intensidad del efecto estaría representada por la pendiente.
Todos estos parámetros deben ir acompañados de su correspondiente
significación estadística o intervalo de confianza.
Factores comunes en los compuestos investigados
Hay una serie de características que comparten todos los compuestos
investigados [ATSDR 2000, ATSDR 2002a, ATSDR 2002b, ATSDR 2005,
UNEP 2006, ATSDR 2007, Porta et al. 2009, UNIDO 2010]:
− Su origen es antropogénico.
− Aunque fueron muy utilizados en el pasado, hoy en día prácticamente
están fuera de uso.
− Son muy persistentes.
− Son liposolubles y muy poco solubles en agua.
− Aunque su tensión de vapor permite que una pequeña fracción pueda
pasar a la atmósfera, no son compuestos volátiles.
− A lo largo del siglo XX se han extendido por todo el planeta.
32

Introducción
− Presentan biomagnificación al avanzar en la cadena trófica.
Como consecuencia de todas ellas, estos compuestos muestran factores
toxicológicos comunes respecto a la exposición de los seres humanos:
− Dadas su solubilidad y relativamente baja volatilidad, se dan
concentraciones mucho mayores en la grasa de los alimentos que en
el agua o en la atmósfera, por lo que su principal vía de exposición es
oral. Esta exposición, además, es de índole global. Las exposiciones
por inhalación o por contacto es más probable que alcancen valores
apreciables en situaciones puntuales relacionadas, en la mayoría de
los casos, con vertederos o depósitos residuales de estos compuestos,
o con procesos, tanto industriales en los que aparezcan como
subproductos, como de tratamiento y destrucción de residuos.
− Las concentraciones en la grasa de los alimentos son del orden de ng
g-1, por lo que la exposición es a dosis bajas.
− Consumimos alimentos varias veces al día y a lo largo de toda nuestra
vida, por lo que la exposición es continua y crónica.
− La exposición se vehiculiza a través de los alimentos, especialmente
con contenido notable de grasa y particularmente de origen animal, por
ocupar éstos un lugar más alto en la cadena trófica.
Heptacloro Epóxido
La información disponible sobre sus efectos tóxicos proviene
mayoritariamente de estudios in vivo con animales y estudios in vitro con
cultivos de procedencia animal o humana. A esta información se añaden, en
humanos, estudios de exposición laboral y de exposición accidental a altas
dosis por consumo de alimentos derivados de animales criados con piensos
fabricados con vegetales contaminados [WHO 2003, ATSDR 2007].
De ella se deduce que tras una exposición por vía oral el compuesto se
adsorbe en el tracto intestinal. De ahí se distribuye, a través de la sangre, a
hígado, riñones, músculos y otros órganos. Al cabo del tiempo las mayores
concentraciones se encuentran en el tejido graso. Diversos estudios de
monitorización en humanos muestran concentraciones apreciables en grasa,
33

Introducción
suero, hígado, pulmones, cerebro, riñones, bazo y corazón, sin que se
discierna si son resultado de exposición a heptacloro epóxido, heptacloro o
clordano. Aunque no hay estudios in vivo del metabolismo en humanos,
estudios in vivo en ratas e in vitro en microsomas hepáticos de humanos y
ratas, sugieren que el principal metabolito es un derivado deshidrogenado del
1-exo-hidroxi-2,3-exo epoxiclordeno. No obstante, el grueso del heptacloro
epóxido permanece inalterado y acumulado en el tejido graso, si bien, dentro
de la pequeña fracción que se excreta, la mayor parte lo hace sin metabolizar,
siendo la principal vía las heces y, en el caso de las mujeres, también la leche
materna.
Como efectos tóxicos en animales, además de la letalidad asociada a
determinadas dosis, se han descrito alteraciones en el hígado, en el sistema
nervioso, en el sistema reproductivo y en el organismo en desarrollo, así como
alteraciones menores en el tracto gastrointestinal [WHO, 2003, ATSDR 2007,
UNIDO 2010]. Por lo que se refiere al hígado las principales afecciones son:
alteración de la actividad enzimática, alargamiento y aumento de tamaño de los
hepatocitos, necrosis, hepatomegalia y hepatitis. Como alteraciones
neurológicas están: excitabilidad, espasmos, aumento de la tensión nerviosa y
dificultades para caminar y mantener la verticalidad. En cuanto al sistema
reproductivo se mencionan: disminución de la fertilidad, aumento de las
reabsorciones de las células reproductivas y disminución de la cantidad de
esperma. Por último, en lo tocante a los organismos en desarrollo tenemos:
disminución del peso y aumento de la mortalidad de las crías y alteraciones en
los sistemas neurológico e inmunológico, sin al parecer afectar a su sistema
reproductivo o que aumenten el número de anomalías y malformaciones. Así
mismo, en animales, se ha reportado formación de granulomas renales y
alteración del metabolismo de los hidratos de carbono. Por lo que se refiere al
cáncer, se ha detectado un incremento del carcinoma hepatocelular. En cuanto
a la actividad genotóxica, por lo menos en mamíferos, se puede concluir que es
escasa o cuestionable.
Por su parte, en humanos [WHO, 2003, ATSDR 2007, UNIDO 2010],
parece confirmarse el efecto sobre los organismos en desarrollo con: aumento
de riesgo de muerte del feto o del neonato, así como alteraciones
34

Introducción
neuroconductuales que algunos relacionan con la gran afinidad de este
compuesto por los receptores GABAA (ácido γ-amino butírico), afinidad que
también se observa en animales. En cuanto a la carcinogénesis, los datos son
confusos y difíciles de interpretar, existiendo estudios con resultados
contradictorios. No obstante, con los datos existentes, la Agencia Internacional
para la Investigación sobre el Cáncer (IARC en sus siglas en inglés) clasifica el
heptacloro dentro de su grupo 2B, es decir, como “posiblemente cancerígeno
para los humanos” [IARC 2009]; por su parte, la Agencia para la Protección
Medioambiental de los EE.UU. (USEPA ó EPA en sus siglas en inglés) lo
clasifica como “probablemente cancerígeno para los humanos”, aunque dentro
del subgrupo B2 [EPA 2010].
HCH
Además de estudios in vitro, la información toxicológica sobre el HCH
proviene de estudios experimentales con animales y de estudios con humanos
expuestos como consecuencia de su trabajo (fabricación de HCH o aplicación
de sus formulaciones), a los que se añaden estudios en humanos expuestos a
formulaciones de HCH de uso doméstico (insecticidas y lociones
antiparasitarias) o que las habían ingerido intencionadamente [Sullivan and
Krieger 2001, WHO 2003, ATSDR 2005].
De sus resultados se desprende que, tras ingestión, se absorbe
rápidamente en el tracto gastrointestinal, desde donde la mayor parte pasa a la
sangre y sólo una pequeña porción accede al sistema linfático. A través de la
sangre se distribuye a tejidos grasos, riñón, pulmones, hígado, músculos,
corazón, bazo y cerebro, siendo el isómero β-HCH el de mayor poder de
acumulación y el de metabolización más lenta. Los principales metabolitos son
clorofenoles y, en menor escala, clorobencenos, clorociclohexanos,
clorociclohexanoles y clorociclohexenos. Muchos de ellos se conjugan
posteriormente para dar glucurónidos, sulfatos, o ácidos mercaptúricos. Todos
estos metabolitos de eliminan mayormente por la orina. Sin embargo, para el β-
HCH, dada su resistencia a la metabolización, esta no supone su principal vía
de eliminación y la mayor fracción excretada lo es, en forma inalterada, a través
de las heces y la leche materna. No obstante, la eliminación es muy lenta y
35

Introducción
gran parte del β-HCH permanece como tal, en los tejidos grasos del organismo,
durante años.
Por lo que se refiere a efectos tóxicos en animales, las principales
afecciones se han detectado en el sistema nervioso, en el circulatorio, en el
inmunológico, en el reproductivo y en el hígado [WHO 2003, ATSDR 2005,
UNEP 2006]. El sistema nervioso central parece ser el órgano diana en
exposición aguda a grandes cantidades, provocando alteraciones
neuroquímicas del cerebro, así como alteraciones del comportamiento en
adultos y crías. En sangre se ha detectado disminución de glóbulos rojos,
glóbulos blancos y hemoglobina. En el sistema inmunológico el principal efecto
es inmunosupresión, que se detecta a exposición intermedia. En el sistema
reproductivo da lugar a embriotoxicidad, alteraciones testiculares, del esperma
y del ciclo ovulatorio. En cuanto al hígado, se produce un aumento del
citocromo P-450 y de la actividad de las enzimas a él asociadas, necrosis y
degeneración grasa. También tumores hepáticos con algunos isómeros (entre
ellos el β-HCH) a dosis crónicas. Tumores a los que pueden contribuir algunos
de los metabolitos como el 2,4,6-triclorofenol o un epóxido clorado estable
derivado del pentaclorociclohexeno.
En humanos [Sullivan and Krieger 2001, WHO 2003, ATSDR 2005, UNEP
2006], también es el sistema nervioso el órgano diana a dosis altas en
exposición aguda, apareciendo hiperexcitabilidad, espasmos, convulsiones e
incluso la muerte. A dosis más bajas, se han detectado vómitos y nauseas en
trabajadores expuestos durante tiempo prolongado. Además se le relaciona
con efectos hematológicos y aumento de enzimas hepáticos. En cuanto al
cáncer, aunque en algún estudio se le asocia con el linfoma no-Hodgkin, en
general los estudios de relación con el cáncer no son concluyentes. No
obstante, y dadas las evidencias en animales, la IARC lo clasifica dentro de su
grupo 2B, es decir, como “posiblemente cancerígeno para los humanos” [IARC
2009] y la EPA como “probablemente cancerígeno para los humanos”, aunque
dentro del subgrupo B2 [EPA 2010].
36

Introducción
HCB
Los datos toxicológicos sobre el HCB surgen de estudios experimentales
in vitro e in vivo, en este último caso con animales, así como de estudios en
humanos involuntariamente expuestos [ATSDR 2002b]. Entre estos últimos
destacan los estudios hechos en Flix, localidad tarraconense en la ribera del
Ebro, donde una fábrica de disolventes organoclorados produjo durante
décadas HCB como subproducto [Sala et al. 1999, ATSDR 2002b, Porta et al.
2009] y en Anatolia (Turquía), donde miles de personas sufrieron exposición
crónica (1955-1959) a elevadas dosis (0,05-0,2 g/día) de HCB por vía oral, al
ingerir pan fabricado con cereales contaminados por este compuesto [PPU
1990, Sullivan and Krieger 2001, ATSDR 2002b, WHO 2003, UNIDO 2010].
En exposición por vía oral se absorbe a través del tracto intestinal, con
fuerte influencia de la vehiculización: < 20% en solución acuosa y > 80% en
solución grasa. La capacidad de absorción decrece al aumentar los niveles en
sangre. Del tracto intestinal pasa al sistema linfático o al riego sanguíneo, por
medio de los cuales se distribuye a: tejido adiposo, nódulo linfático
mesentérico, glándulas endocrinas, hígado, médula ósea, fluido folicular,
riñones, cerebro, bazo, corazón, pulmones y músculos. Se metaboliza muy
lentamente, a través del citocromo hepático P-450, dando como productos
mayoritarios pentaclorobenceno (mediante deshidrocloración reductiva) que se
convierte parcialmente en tetraclorohidroquinona, y pentaclorofenol, que se
conjuga a pentaclorotiofenol. Otros metabolitos más minoritarios son
clorobencenos, clorofenoles y clorotiofenoles de menor grado de cloración.
Como vías principales de eliminación están las heces (excreción intestinal y
biliar) y la leche materna (convirtiéndose en fuente de exposición para el
lactante), donde la forma que predomina es la no metabolizada y en menor
medida la orina, donde predominan las formas metabolizadas. También se
elimina una parte, en forma inalterada, a través de la placenta y el cordón
umbilical, lo que supone una vía de exposición para el feto. No obstante, dado
su poder de acumulación, la mayor parte se almacena sin metabolizar en el
tejido adiposo y en órganos con alto contenido en grasa.
Los principales efectos tóxicos en animales [ATSDR 2002b, WHO 2003,
UNIDO 2010] tienen como órganos diana el hígado, el aparato reproductor
37

Introducción
(ovarios) y el sistema nervioso central. Destacan: la porfiria (causada por la
formación excesiva de porfirinas debido a una alteración enzimática en la
síntesis del grupo hemo), descenso de la fertilidad, aumento de mortinatos,
alteraciones y lesiones en los ovarios (cambios de peso, degeneración,
alteración en la génesis de estrógenos y progesterona) y alteraciones del
desarrollo de las facultades locomotrices en crías. También se asocia a cáncer
de hígado, conducto biliar, riñones, sistema linfático y tiroides.
En humanos [Sullivan and Krieger 2001, ATSDR 2002b, WHO 2003,
UNIDO 2010] se han detectado efectos sistémicos (en hígado, piel, huesos,
riñones y tiroides), efectos en el organismo en desarrollo, neurológicos,
endocrinos, inmunológicos e incluso la muerte. En hígado destaca la alteración
enzimática, cuya consecuencia más notable es el incremento de porfirinas
(porfiria), origen a su vez de siderosis, hiperplasia, necrosis y cirrosis. En piel
fototoxicidad, alteración de la pigmentación y ulceraciones, ocasionadas todas
ellas por el incremento de porfirinas en el hígado. En huesos artritis. En riñones
microproteinuria y fallos renales debidos a las porfirinas. En tiroides descenso
del nivel de tiroxina, hipotiroidismo y bocio. Los efectos neurológicos más
importantes se detectan en el organismo en desarrollo, con la alteración de
ciertas facultades locomotrices en niños. Por lo que respecta al sistema
inmunológico se han referido: descenso de la actividad neutrófila, incremento
de inmunoglobulinas y susceptibilidad de infección.
En el caso concreto del episodio de Anatolia [Sullivan and Krieger 2001,
ATSDR 2002b, WHO 2003, UNIDO 2010], los niños menores de 2 años
(expuestos a través de la leche materna) padecían lesiones en la piel en forma
de eritema anular de color rosa que se conocían como pembe yara, seguidas
de debilidad y convulsiones, cuadro clínico que derivó en la mortandad del 95%
de ellos. En niños entre 6 y 15 años (también en el resto de la población
aunque con una morbilidad mucho menos acusada) se dio un cuadro clínico
que denominaron kara yara, caracterizado por la aparición en la piel de
fotosensibilidad, fragilidad, hiperpigmentación e hirsutismo, que ocasionó la
muerte al 10% de los que la padecieron. Se trataba de porfiria cutánea tarda,
originada por el incremento de porfirinas en el hígado. También se refirieron:
pérdida de apetito, debilidad, artritis, hepatomegalia, alargamiento de tiroides y
38

Introducción
pérdida de habilidad motrices, síntomas que permanecieron en adultos
expuestos en la niñez.
Por lo que se refiere al cáncer en humanos, algunos estudios han
encontrado débiles asociaciones con cáncer de hígado, tiroides y cerebro, pero
no se consideran datos concluyentes. Por ello y por los resultados obtenidos en
animales, la IARC lo clasifica dentro de su grupo 2B, es decir, como
“posiblemente cancerígeno para los humanos” [IARC 2009 ] y la EPA como
“probablemente cancerígeno para los humanos”, aunque dentro del subgrupo
B2 [EPA 2010].
DDT
La información toxicológica proviene de estudios experimentales in vitro
(animales y humanos) e in vivo (animales), estudios epidemiológicos con
humanos expuestos en su ámbito laboral (operarios de fábricas y aplicadores
del producto), estudios epidemiológicos de humanos con exposición ambiental
por encima de lo normal (por proximidad a fábricas, vertederos, campos
tratados, etc.) y estudios con humanos que han ingerido, accidental o
intencionadamente, grandes cantidades en exposición aguda [Sullivan and
Krieger 2001, ATSDR 2002a, WHO 2003, UNIDO 2010]. En el caso del DDT
también se dispone de estudios experimentales con personas voluntarias
sometidas a exposición sub-crónica (6 meses) y crónica (18 meses) de dosis
de hasta 35 mg/día [ATSDR 2002a].
De los datos recogidos se desprende que se absorbe en el tracto
gastrointestinal para pasar al sistema linfático y, de forma menos acusada, al
sistema sanguíneo. La absorción se ve favorecida por el acompañamiento de
grasas alimenticias. A través de estos sistemas se distribuye a casi todos los
tejidos corporales, aunque en función de su contenido en grasa. En el hígado
se metaboliza primeramente a DDD y DDE. Estos dos compuestos siguen rutas
de metabolización independientes que llevan a un mismo compuesto, el ácido
2,2-bis(p-clorofenil) acético (DDA), que resulta ser a la postre el principal
metabolito del DDT. Esta metabolización tiene lugar en el hígado y en el riñón e
incluye diversas reacciones en la parte alifática de la molécula, como:
decloración, deshidrocloración, reducción, hidroxilación y oxidación. La ruta del
DDE es mucho más lenta que la del DDD, lo que explica el alto poder de
39

Introducción
acumulación del primero. Dos de los productos intermedios detectados, el
epóxido de 1-cloro-2,2-bis(p-clorofenil) eteno (DDMU epóxido) y el cloruro de
2,2-bis(p-clorofenil) acetilo (DDA-Cl), dan lugar a la formación de aductos de
DDA que podrían ser causa de carcinogénesis. Tanto el DDA como otros
metabolitos se pueden conjugar con glicina, ácidos biliares, serina, ácido
aspártico o ácido glucurónico. El DDT también da lugar a metil-sulfonil
derivados entre los que destacan los metil-sulfonil-DDE que tras activación
metabólica se convierten en potentes tóxicos, especialmente en la glándula
suprarrenal.
El DDT se excreta mayormente por vía urinaria y, en menor medida, por
las heces (vía excreción biliar). Lo hace metabolizado (principalmente como
DDA) y sólo en pequeñas cantidades como DDT. La proporción excretada en
heces aumenta a dosis altas de DDT. La principal ruta de eliminación de la
fracción metabolizada a DDE o del DDE ingerido son las heces y sólo una
porción menor se elimina por la orina. Una parte sustancial del DDE eliminado
en heces lo hace en forma inalterada mientras que todo el eliminado por la
orina está metabolizado (mayormente a DDA). Otras rutas de eliminación
significativas, tanto para el DDT como para el DDE, son a través de la placenta
y la leche materna, donde se trasmiten en gran medida sin metabolizar. Estas
rutas, a su vez, suponen vías de exposición muy importantes para el feto y el
lactante respectivamente. El tiempo de permanencia del DDT en el organismo
es notablemente inferior al del DDE, por lo que este último se convierte en la
principal traza de exposición tanto a DDT como a DDE, acumulándose una
buena parte, en forma inalterada, en los tejidos grasos.
En cuanto a efectos toxicológicos primeramente hay que decir que en
animales sometidos a dosis extremas en exposición aguda, la muerte se
produce por parada respiratoria y que en humanos se han registrado
exposiciones de hasta 285 mg/Kg de peso sin resultado de muerte [ATSDR
2002a]. Todos los casos de muerte registrados en humanos lo han sido como
consecuencia de exposición combinada con otros compuestos de similar índole
de aplicación. Por otra parte, los principales efectos son neurológicos,
reproductivos, sobre el organismo en desarrollo y hepáticos [Longnecker et al.
1997, Sullivan and Krieger 2001, ATSDR 2002a, WHO 2003, Sanz-Gallardo
40

Introducción
2006, UNIDO 2010]. Una buena parte de ellos se deben a su carácter de
disruptor endocrino que le lleva a interferir con el sistema hormonal del
organismo. Los isómeros del DDT y sus metabolitos poseen propiedades
estrogénicas que junto con las antiandrogénicas del p,p’-DDE llevan a un
incremento general de la actividad estrogénica. El estudio de las propiedades
antiandrogénicas del p,p’-DDE en animales sugiere varios mecanismos de
actuación: unión directa sobre el receptor andrógeno, incremento de los
sistemas enzimáticos ZYP del hígado que hidroxilan la testosterona facilitando
su eliminación y aumento de la actividad aromatasa del hígado en adultos
machos, que cataliza la conversión de los C19 andrógenos en estrógenos. El
resultado global es una feminización del individuo y notables efectos
reproductivos y en el desarrollo del organismo. Hay suficiente información en
animales de que esta disrupción endocrina puede ser especialmente notoria si
actúa durante el desarrollo, produciendo: reducción de la distancia anogenital,
alteración de la formación de pezones en crías, retraso de la pubertad, etc. En
humanos se ha achacado a ella la relación entre altas cargas de DDT/DDE con
alteraciones de procesos cuyo punto final está controlado por hormonas:
duración de la lactancia, fertilidad, duración del embarazo, aborto espontáneo,
etc.
Por lo que respecta a los efectos sobre el hígado, existe fuerte evidencia
en animales, aunque no concluyente en humanos, de que el producto da lugar
a: inducción tipo fenobarbital de enzimas microsomales (lo que altera el
metabolismo de sus sustratos entre los que se incluyen las hormonas
esteroideas), aumento de la actividad transaminasa, hipertrofia, hiperplasia,
necrosis y cáncer.
El principal y más conocido efecto neurológico del DDT es prolongar
drásticamente la corriente de sodio en los axones, lo que provoca un deterioro
de la conducción del impulso nervioso afectando a funciones básicas como la
respiración y la actividad cardiovascular [ATSDR 2002a]. Además, tanto en
humanos como en animales expuestos a altas dosis, se han relatado:
parestesia de la lengua, labios y cara, aprensión, hipersusceptibilidad a
estímulos externos, irritabilidad, pérdida de atención verbal y velocidad
visomotora, mareos, vértigo, temblores y convulsiones. También, en el caso de
41

Introducción
animales, la exposición del feto o del recién nacido provoca cambios
neuroquímicos y del comportamiento en la edad adulta, como: alteración de los
receptores de los neurotrasmisores del cerebro y alteración del comportamiento
motriz.
En lo referente al cáncer, existe una clara evidencia de asociación en
animales. Además del ya mencionado de hígado, está también el de próstata.
En humanos, aunque no existe evidencia concluyente, se apunta una posible
asociación entre niveles altos de DDT/DDE y la agresividad en el cáncer de
mama [Demers et al. 2000, Woolcott et al. 2001]. Por todo ello la IARC lo
clasifica dentro de su grupo 2B, es decir, como “posiblemente cancerígeno para
los humanos” [IARC 2009] y la EPA como “probablemente cancerígeno para
los humanos”, aunque dentro del subgrupo B2 [EPA 2010].
PCBs
La información disponible hoy en día deriva de estudios in vitro (animales
y humanos) y estudios in vivo (animales) realizados con mezclas comerciales
de PCBs, mezclas ex profeso y congéneres individuales concretos. A éstos se
unen estudios de exposición laboral, estudios de exposición ambiental y
estudios de exposición alimentaria a través de productos de origen animal
[ATSDR 2000, Sullivan and Krieger 2001, WHO 2003, UNIDO 2010], sobre
todo los procedentes de zonas contaminadas, como en los casos de consumo
de pescado procedente de los lagos Michigan y Ontario. Pero han sido en
concreto dos exposiciones accidentales a grandes dosis por vía oral, las que
han hecho que para estos compuestos predominen los datos toxicológicos en
humanos. Se trata de dos episodios de intoxicación masiva, uno en Fukuoka
(Japón) en 1968 y otro en Taiwan en 1979, conocidos por la traducción a los
respectivos idiomas de lo que podríamos llamar “mal o enfermedad del aceite”
(Yusho en japonés y Yucheng en chino), y en ellos unas 1600 y 2000 personas
respectivamente, resultaron intoxicadas al consumir aceite de arroz
contaminado con grandes cantidades de PCBs [Erickson 1997, Yoshimura
2003, Guo et al. 2004]. Existe mucha literatura científica al respecto, pero hay
que añadir, no obstante, que en estos aceites de arroz contaminados se
detectaron también cantidades notables de PCDFs y otros derivados
42

Introducción
(seguramente fruto del tratamiento térmico al cocinarlos) y que muchos autores
achacan a estos compuestos parte importante de los efectos producidos.
En exposición por vía oral los PCBs se absorben en el tracto
gastrointestinal por difusión pasiva, por lo que la absorción es función de la
diferencia entre los niveles intestinales y los niveles en suero o corporales. Esta
absorción es también congénere-dependiente. Del intestino pasa al sistema
sanguíneo o linfático, a través de los cuales se distribuye por un amplio abanico
de localizaciones del organismo, en función de su contenido en grasa: tejido
adiposo, hígado, riñón, músculo, glándula suprarrenal, pulmones, tiroides,
bazo, timo, médula ósea, testículos, cerebro, piel, etc. La distribución de los
distintos congéneres (perfil) a lo largo del organismo depende también de la
especie, el grado de cloración y la estereoquímica de la sustitución. Por
ejemplo, en el pulmón se encuentran las mayores concentraciones de los
congéneres de menor grado de cloración (más volátiles) y en el cerebro las
menores concentraciones relativas a grasa ya que ésta tiene mayor porcentaje
de fosfolípidos (lípidos más polares) [ATSDR 2000]. Debido a que tanto la
absorción, como la distribución, el metabolismo y la excreción son congénere-
dependientes, el perfil de PCBs se altera de la exposición a la acumulación.
Por eso, el perfil detectado en la mayoría de los casos es un perfil de estado
estacionario que difiere del perfil de las mezclas comerciales. La mayoría de los
seres vivos presenta un perfil similar en los 4-6 congéneres dominantes (entre
los que cabe destacar los PCBs 138, 153 y 180) y se observan diferencias en
el resto de los congéneres.
La metabolización se inicia en el sistema mono-oxigenasa microsomal del
hígado, a través del citocromo P-450 (enzymas CYP1A1, CYP1A2, CYP2B1,
CYP2B2 y otros de la familia 3A). Inicialmente se obtienen óxidos de areno que
a su vez se transforman en compuestos aromáticos mono y dihidroxilados (OH-
PCBs) o en metil-sulfonil derivados (MeSO2-PCBs). Las posiciones meta y para
no sustituidas son las más favorables a la oxidación. En los no-orto o mono-
orto sustituidos la hidroxilación se produce en la posición para del anillo menos
sustituido. En el resto en la posición meta. La velocidad de metabolización
decrece al aumentar el grado de sustitución y al disminuir el número de
posiciones meta-para adyacentes no sustituidas. Por lo tanto, PCBs con alto
43

Introducción
grado de cloración y pocas posiciones meta-para adyacentes no sustituidas,
son los más bioacumulables. La forma y velocidad de metabolización depende,
además, de la especie. Los óxidos de areno son muy reactivos y se les ha
relacionado con necrosis celular, mutagénesis, carcinogénesis y también
efectos genotóxicos vía formación de aductos de DNA. Los OH-PCBs se
pueden conjugar a glucurónidos o sulfatos. Algunos OH-PCBs son también
bioacumulables (en pulmones, hígado o riñón) bien por su liposolubilidad o por
ser capaces de ligarse a proteínas. En humanos el más abundante es el 4-OH-
2,2’,3,4’,5,5’,6-heptaclorobifenilo, que proviene de la metabolización de los
PCBs 183 y 187. Los principales MeSO2-PCBs en humanos derivan de los
PCBs 87, 101, 132 y 149. Dominan los 4-MeSO2 seguidos de los 3-MeSO2.
Algunos pueden ligarse a proteínas y bioacumularse. A los MeSO2-PCBs se les
achaca los efectos tóxicos sobre el aparato respiratorio y algunos son fuertes
inductores tipo fenobarbital de enzimas CYP2B del citocromo P-450 del hígado.
Los PCBs se eliminan por las heces (la mayor parte proviene de excreción
biliar), la orina y cantidades minoritarias por los pulmones. Por las heces se
excretan los PCBs no metabolizados y los metabolitos poco polares. Por la
orina los metabolitos polares, mayormente conjugados. Se elimina más
cantidad por heces que por orina y, en general, en lo excretado predominan las
formas metabolizadas. Otras formas de eliminación son la leche materna y a
través de la placenta, con fuerte presencia de PCBs inalterados y suponiendo
una fuente de exposición para el lactante y para el feto, respectivamente. Los
congéneres más resistentes a la metabolización tienden a bioacumularse en
tejidos y fluidos con alto contenido en grasa: tejido adiposo, hígado, piel, leche,
etc.
La información aportada por los distintos estudios toxicológicos revela
principalmente efectos en hígado, pulmones, ojos y piel, disrupción endocrina,
efectos inmunológicos y neurológicos, efectos en el desarrollo del organismo,
efectos en el sistema reproductivo y cáncer [George et al. 1988, Longnecker et
al. 1997, ATSDR 2000, Faroon et al. 2000, Faroon et al. 2001, Sullivan and
Krieger 2001, Kimbrough and Krouskas 2003, WHO 2003, Ulbrich and
Stahlmann 2004, UNIDO 2010]. Algunos estudios con animales concluyen que
en monos (especie más próxima a la humana) se inducen efectos a dosis más
44

Introducción
bajas que en otras especies. No se sabe a ciencia cierta qué congéneres son
los responsables de los episodios tóxicos estudiados en humanos ni la posible
contribución de otras sustancias (PCDFs). Desde el punto de vista de la
relación estructura-actividad, se distingue entre dos tipos de congéneres: los
no-orto o mono-orto sustituidos, en los que los dos anillos de benceno están en
el mismo plano (de ahí que se les denomine también congéneres coplanares) y
los poli-orto sustituidos, en los que por efecto estérico un anillo se encuentra
parcialmente girado con respecto al otro (por lo que se les denomina también
congéneres no-coplanares). Desde hace tiempo se conocía la facultad de
ciertos hidrocarburos aromáticos policíclicos para inducir la actividad de un
enzima que se denominó aril hidrocarburo hidroxilasa (AHH). Esta inducción se
realiza a través de un receptor que se conoció desde entonces como Receptor
Ah (AhR) [Okey 2007]. Más adelante se encontró que la 2,3,7,8-
tetraclorodibenzo-p-dioxina (TCDD), potente tóxico de estructura coplanar, es
todavía un inductor más potente de la AHH a través de dicho receptor,
achacándose a este hecho gran parte de su toxicidad. Pues bien, también se
ha demostrado que los PCBs no-orto y, en menor extensión, los mono-orto
sustituidos, es decir, los coplanares, manifiestan igualmente esta afinidad por el
AhR. Esto les confiere un carácter tóxico similar al de la TCDD, aunque menos
intenso. Por eso son conocidos como PCBs similares a la dioxina o Dioxin-Like
PCBs. La Organización Mundial de la Salud (WHO), en base a diversos
estudios, ha creado para los más activos una tabla con valores de toxicidad
relativos a la TCDD a la que se le asigna el valor 1. A estos valores se les
conoce como Factores de Equivalencia Tóxica (TEFs) y están reflejados en la
Tabla 2 [Van den Berg et al. 1998, Van der Berg et al. 2006]:
45

Introducción
Tabla 2 Factores de equivalencia tóxica de PCBs relativos a la TCDD
WHO TEFs CONGÉNERE MAMÍFEROS y
HUMANOS PECES AVES
PCB 77 0,0001 0,0001 0,05 PCB 81 0,0003 0,0005 0,1 PCB 105 0,00003 <0,000005 0,0001 PCB 114 0,00003 <0,000005 0,0001 PCB 118 0,00003 <0,000005 0,00001 PCB 123 0,00003 <0,000005 0,00001 PCB 126 0,1 0,005 0,1 PCB 156 0,00003 <0,000005 0,0001 PCB 157 0,00003 <0,000005 0,0001 PCB 167 0,00003 <0,000005 0,00001 PCB 169 0,03 0,00005 0,001 PCB 189 0,00003 <0,000005 0,00001
Sin embargo, los TEFs no son suficientes para predecir la toxicidad de los
perfiles ambientales de PCBs, ya que se ha demostrado que [ATSDR 2000]:
• Los efectos basados en el AhR no son aditivos.
• Los congéneres mayoritarios son no-coplanares y por lo tanto no
son agonistas del AhR (algunos incluso presentan cierto
comportamiento antagonista).
• Existen otros mecanismos de actuación tóxica independientes del
AhR.
• Son mezclas complejas de congéneres que interactúan entre ellos
dando respuestas AhR dependientes, AhR independientes o
ambas.
Por lo que a respuesta tóxica se refiere, está bien documentada en
animales la inducción de enzimas microsomales en el hígado, lo que provoca
aumento de enzimas hepáticos en suero, cambios bioquímicos en tejidos y
alteración de niveles de lípidos, colesterol, porfirinas y vitamina A como
consecuencia de la disfunción hepática, alargamiento del hígado, porfiria y
cambios histopatológicos como deposición grasa, fibrosis, necrosis o cáncer
[ATSDR 2000]. En humanos los efectos no son tan claros pero se ha
46

Introducción
encontrado cierta asociación con aumento de enzimas hepáticos en suero, así
como de lípidos y colesterol, aumento de porfirinas en orina y porfiria hepática
tipo B. La principal respuesta tóxica en el aparato respiratorio de los humanos
es la disminución de capacidad pulmonar. En cuanto a piel, se ha relatado
irritación dérmica, cloroacné y pigmentación anormal de piel y uñas con
deformación e incluso pérdida de estas últimas. En ojos, hipersecreción de la
glándula de Meibomio, pigmentación anormal de la conjuntiva e hinchazón de
párpados y cloroacné. En monos también se dan este tipo de efectos dérmicos
y oculares.
Entre los animales, los visones y los monos son las especies más
sensibles en cuanto a efectos tóxicos de los PCBs en el aparto reproductivo,
registrándose alteraciones de los ciclos menstruales o de celo, retraso de la
pubertad, descenso de la tasa de implantación, descenso de la fertilidad,
muerte fetal, descenso de la libido y alteraciones en el sistema de producción
de esperma. En humanos se asocian con alteraciones de la menstruación en
mujeres y de la morfología, producción y movilidad del esperma en hombres.
En lo tocante a efectos inmunológicos, se dan principalmente en exposición a
través del útero y de leche materna. Los más destacables, tanto en humanos
como en animales, son susceptibilidad a enfermedades infecciosas (sobre todo
en el tracto respiratorio y el oído medio) e inmunodepresión por descenso de
inmunoglobulinas, monolitos y linfocitos T. En animales también se observan
alteraciones de timo, bazo y nódulos linfáticos.
Como efectos más destacables de disrupción endocrina están el efecto
estrogénico de los congéneres no-coplanares y los OH-PCBs, el efecto
antiestrogénico de los congéneres coplanares y la alteración del sistema
hormonal de la glándula tiroides [ATSDR 2000, Alverez-Pedrerol 2009]. En
animales la exposición en el útero o durante la lactancia provoca estados de
hipotiroidismo con sus consiguientes desórdenes en el desarrollo neurológico
que ocasionan déficits motores, auditivos e intelectuales. En humanos la
evidencia es más limitada y se les ha asociado con casos de bocio. Por lo que
se refiere a efectos neurológicos [ATSDR 2000, Faroon et al. 2000], están muy
documentados en humanos y animales. En niños nacidos de madres expuestas
se ha encontrado asociación con diversas alteraciones neuronales como:
47

Introducción
reflejos anormales, déficits de memoria y de capacidad de aprendizaje, y bajos
coeficientes de inteligencia. También entre hipotonía en niños y niveles de
PCBs coplanares en leche materna. Aunque ambos tipos de PCBs parecen
inducir efectos neurológicos, de los estudios se desprende que son más activos
los no-coplanares. Además de los derivados de la disrupción endocrina y los
neurológicos ya descritos, otros efectos sobre el organismo en desarrollo
[ATSDR 2000, Ulbrich and Stahlmann 2004] serían descenso de peso al nacer
y ganancia de peso ralentizada, ambos constatados en los estudios con
animales pero con resultados contradictorios en humanos.
Los estudios de asociación con cáncer [ATSDR 2000, Faroon et al. 2001]
dan resultados concluyentes para el de hígado en animales. En humanos se
han estudiado múltiples localizaciones: mama, riñón y conductos urinarios,
páncreas con mutación K-Ras, linfoma no-Hodgkin y otros hemáticos,
endometrio, próstata, piel (melanoma), cerebro y sistema nervioso, hígado,
vesícula y conductos biliares, sistema digestivo e intestinal, etc. En unos casos
los resultados han sido contradictorios o negativos y en otros, sugestivos
aunque no concluyentes. Por todo ello la IARC los clasifica, de forma conjunta,
dentro de su grupo 2A, es decir, como “probablemente cancerígenos para los
humanos” [IARC 2009] y la EPA como “probablemente cancerígenos para los
humanos”, aunque dentro del subgrupo B2 [EPA 2010].
48

Introducción
Consideraciones analíticas
Para la mayoría de los seres humanos la principal vía de exposición a
estos compuestos es oral y vehiculizada a través de los alimentos,
especialmente con contenido notable de grasa y particularmente de origen
animal. Esta exposición tiene lugar de forma crónica y a dosis bajas. Pero,
además, actualmente afecta a toda la población y durante toda su vida. Estas
dos últimas características, unidas a su potencial toxicidad, los convierte en
objeto de especial interés desde el punto de vista de la Salud Pública
[Longnecker et al. 1997, Feeley and Brouwer 2000, Koppen et al. 2002,
Kimbrough and Krouskas 2003, WHO 2003, Bates et al. 2004, LaKind et al.
2004, Ross 2004, EEA 2005]. En este tipo de exposiciones los síntomas a corto
plazo suelen ser sub-clínicos y, por lo tanto, imperceptibles. Sin embargo, a
largo plazo se pueden manifestar en forma de diversas alteraciones o
enfermedades que en muchas ocasiones son difícilmente asociables, a primera
vista, al agente causante (relación causa/efecto difícil de establecer) [Olea y
Fernández 2001]. El estudio directo experimental de este tipo de toxicología es
muy complicado. En muchos casos estamos hablando de efectos que se
manifiestan a los 10, 20, 30 ó más años. Diseñar un estudio concluyente sobre
la relación/causa efecto en estas condiciones sería un auténtico ejercicio de
dedicación y paciencia. Empezando porque la vida media de la mayoría de los
animales que habría que emplear sería inferior a la duración del estudio,
siguiendo porque en plazos tan largos y como consecuencia de la posible
exposición a otras sustancias son esperables sinergias o efectos depresores
difíciles de controlar, y terminando por las consecuencias que tendría cualquier
replanteamiento del mismo (dosis, forma de administración, tipo de animal
utilizado, etc.) que obligara a cambiar las condiciones iniciales una vez
trascurridos varios años desde el comienzo. Por ello se recurre a estudios
epidemiológicos observacionales. Aún así se detecta un déficit de información
sobre los niveles que alcanza en la población general esta exposición crónica
por vía oral a bajas dosis y los efectos que causa, especialmente los
carcinogénicos [ATSDR 2000, ATSDR 2002a, ATSDR 2002b, ATSDR 2005,
Porta et al 2006, ATSDR 2007, Porta et al. 2009, UNIDO 2010].
49

Introducción
Es difícil establecer con exactitud la exposición a estos compuestos a
través de la ingesta, partiendo de los cuestionarios al uso que recogen los
hábitos alimentarios. Por eso la utilización de biomarcadores se convierte en
una alternativa razonable. De hecho la monitorización biológica de estos
compuestos o sus metabolitos en tejidos y fluidos biológicos se ha revelado
como una herramienta útil para valorar el riesgo de exposición en los estudios
epidemiológicos. Concretamente el suero es uno de los materiales más usados
por su facilidad de obtención y la constatación de la presencia de estos
compuestos en el mismo [Koppen et al. 2002, Cruz et al. 2003, Axmon et al.
2004, Bates et al. 2004]. Por otra parte, en este tipo de estudios
epidemiológicos existe una relación positiva entre la fiabilidad de las
conclusiones finales y el tamaño de la muestra poblacional elegida, de forma
que se busca trabajar con un número elevado de individuos, por lo que no es
extraño que se llegue a reclutar más de 1000 o incluso de 10000 voluntarios en
los más ambiciosos. Esto supone un esfuerzo considerable y un nutrido equipo
de trabajo para llevar a cabo las diferentes tareas: captación de voluntarios,
confección y cumplimentación individualizada del formulario de datos,
confección y alimentación de la base de datos, toma de muestras
individualizada, creación y mantenimiento del banco de muestras, seguimiento
de la evolución de los voluntarios durante el tiempo necesario (en muchos
casos a lo largo de toda su vida), etc. Por eso y con miras a un
aprovechamiento eficiente de recursos, se tiende a emplear todo ello en el
mayor número de estudios posible.
Este conjunto dibuja un panorama muy exigente para las metodologías
analíticas a emplear en este tipo de estudios, con requerimientos como [Reiner
et al. 2010]:
• Alta sensibilidad. La exposición es a dosis bajas y los analitos se
encuentran a una concentración del orden de ng mL-1 o por debajo.
Además, el tamaño de la alícuota de muestra disponible suele ser
pequeño (en muchos casos < 1 mL), ya que sólo se puede extraer
una cantidad limitada de sangre y hay que optimizar el uso de la
muestra para disponer del mayor número posible de alícuotas que
posibiliten otros estudios [Laden et al. 1997, Lindh et al. 2008].
50

Introducción
• Alta selectividad. Las matrices biológicas, entre ellas el suero, son
de por sí muy complejas, con una gran variedad de componentes
que se encuentran a concentraciones muy por encima de las de los
analitos: grasas, proteínas, hidratos de carbono, sales, etc.
• Economía de medios y alta productividad. Aunque muchos
individuos a estudiar implican muchas muestras a analizar, la etapa
analítica no debe ir más allá de un coste y un tiempo razonables.
51

Introducción
Referencias
♦ Alvarez-Pedrerol M, Guxens M, Ibarluzea J, Rebagliato M, Rodríguez A, Espada M, Goñi F, Basterrechea M, Sunyer J, 2009. Organochlorine compounds, iodine intake, and thyroid hormone levels during pregnancy. Environ. Sci. Technol. 43, 7909-7915.
♦ ATSDR of U.S. Department of Health and Human Services, 2000. Toxicological profile for polychlorinated biphenyls (PCBs). http://www.atsdr.cdc.gov/toxprofiles/tp17.pdf. Último acceso: 15-04-2010.
♦ ATSDR of U.S. Department of Health and Human Services, 2002a. Toxicological profile for DDT, DDE and DDD. http://www.atsdr.cdc.gov/toxprofiles/tp35.pdf. Último acceso: 15-04-2010.
♦ ATSDR of U.S. Department of Health and Human Services, 2002b. Toxicological profile for Hexachlorobenzene. http://www.atsdr.cdc.gov/toxprofiles/tp90.pdf. Último acceso: 15-04-2010.
♦ ATSDR of U.S. Department of Health and Human Services, 2005. Toxicological profile for alpha-, beta-, gamma-, and delta-hexachlorocyclohexane. http://www.atsdr.cdc.gov/toxprofiles/tp43.pdf. Último acceso: 15-04-2010.
♦ ATDSDR of U.S. Department of Health and Human Services, 2007. Toxicological profile for heptachlor and heptachlor epoxide. http://www.atsdr.cdc.gov/toxprofiles/tp12.pdf. Último acceso: 15-04-2010.
♦ Axmon A, Rylander L, Strömberg U, Jonson B, Nilsson-Ehle P, Hagmar L, 2004. Polychlorinated biphenyls in serum and time to pregnancy. Environ. Res. 96, 186-195.
♦ Ballschmiter K, Zell M, 1980. Analysis of polychlorinated biphenyls (PCB) by glass capillary gas chromatography: Composition of technical Aroclor and Clophen-PCB mixtures. Fresenius Z. Anal. Chem. 302, 20-31.
♦ Bates MN, Buckland SJ, Garrett N, Ellis H, Needham LL, Patterson Jr. DG, Turner WE, Russell DG, 2004. Persistent organochlorines in the serum of the non-occupationally exposed New Zealand population. Chemosphere 54, 1431-1443.
♦ Bhopal RS, 2008. Concepts of Epidemiology: Integrating the Ideas, Theories, Principles and Methods of Epidemiology. Oxford University Press, Oxford, UK.
♦ Calva LG, Torres MR, 1998. Plaguicidas organoclorados. Contactos 30, 35-46.
♦ Carlson RL, 2002 (1ª edición 1962). Silent Spring. Mariner Books (Houghton Mifflin), New York, USA.
♦ CCOHS (Canadian Centre for Occupational Health and Safety), 2009. What makes chemicals poisonous. http://www.ccohs.ca/oshanswers/chemicals/poisonou.html. Último acceso: 22-04-2010.
♦ Cruz S, Lino C, Silveira MI, 2003. Evaluation of organochlorine pesticide residues in human serum from an urban and two rural populations in Portugal. Sci. Total Environ. 317, 23-35.
♦ Demers A, Ayotte P, Brisson J, Dodin S, Robert J, Dewailly E, 2000. Risk and aggressiveness of breast cancer in relation to plasma organochlorine concentrations. Cancer Epidemiol. Biomarkers Prev. 9, 161-166.
♦ EEA (European Environment Agency), 2005. Report 10/2005: Environment and Health. http://www.eea.europa.eu/publications/eea_report_2005_10. Último acceso: 21-04-2010.
♦ EPA, 2010. USEPA Integrated Risk Information System (IRIS), A-Z list of substances. http://cfpub.epa.gov/ncea/iris/index.cfm?fuseaction=iris.showSubstanceList. Último acceso: 23-04-2010.
♦ EPCEU (European Parliament and Council of the European Union), 2004. Regulation (EC) No 850/2004 of the European Parliament and of the Council of 26 April 2004 on persistent organic pollutants and amending Directive 79/117/EEC.
52

Introducción
http://ec.europa.eu/environment/waste/studies/pdf/annex_1_pop_regul_850_2004.pdf. Último acceso: 27-04-2010.
♦ Erickson MD, 1997. Analytical Chemistry of PCBs, 2nd Ed. Lewis Publishers (CRC Press), Boca Ratón, FL, USA.
♦ Faugère JF, Chaurial B, 1989. Methode d’appreciation de la biodegradation des polychlorobiphenyles (PCB) dans les sols. Ann. Fals. Exp. Chim. 82, 19-27.
♦ Faroon O, Jones D, de Rosa C, 2000. Effects of polychlorinated biphenyls on the nervous system. Toxicol. Ind. Health 16, 305-333.
♦ Faroon OM, Keith S, Jones D, De Rosa C, 2001. Carcinogenic effects of polychlorinated biphenyls. Toxicol. Ind. Health 17, 41-62.
♦ Feeley M, Brouwer A, 2000. Health risks to infants from exposure to PCBs, PCDDs and PCDFs. Food Addit. Contam. 17, 325-333.
♦ Galiano A, 2010. IQB: farmacología, principios de toxicología. http://www.iqb.es/cbasicas/farma/farma05/toc05a.htm. Último acceso: 22-04-2010.
♦ George CJ, Bennett GF, Simoneaux D, George WJ, 1988. Polychlorinated biphenyls a toxicological review. J. Hazard. Mater. 18, 113-144
♦ Guo YL, Lambert GH, Hsu CC, Hsu MM, 2004. Yucheng: health effects of prenatal exposure to polychlorinated biphenyls and dibenzofurans. Int. Arch. Occup. Environ. Health 77, 153-158.
♦ IARC, 2009. Agents reviewed by the IARC monographs, volumes 1-100A (alphabetical order). http://monographs.iarc.fr/ENG/Classification/Listagentsalphorder.pdf. Último acceso: 23-04-2010.
♦ Kimbrough RD, Jensen AA, 1989. Halogenated biphenyls, terphenyls, naphthalenes, dibenzodioxins and related products, 2nd Ed. Elsevier, Amsterdam, the Netherlands.
♦ Kimbrough RD, Krouskas CA, 2003. Human exposure to polychlorinated biphenyls and health effects: a critical synopsis. Toxicol. Rev. 22, 217-233.
♦ Koppen G, Covaci A, Van Cleuvenbergen R, Schepens P, Winneke G, Nelen V, Van Larebeke N, Vlietinck R, Shoeters G, 2002. Persistent organochlorine pollutants in human serum of 50-65 years old women in the Flanders Environmental and Health Study (FLEHS). Part 1: concentrations and regional differences. Chemosphere 48, 811-825.
♦ Laden F, Wolff MS, Niguidula NJ, Spiegelman D, Hankinson SE, Hunter DJ, 1997. Reduced aliquot size for a plasma organochlorine assay for use in epidemiological studies. Cancer Epidem. Biomar. 6, 333-338.
♦ LaKind JS, Amina Wilkins A, Berlin CM Jr., 2004. Environmental chemicals in human milk: a review of levels, infant exposures and health, and guidance for future research. Toxicol. Appl. Pharmacol. 198, 184-208.
♦ Lindh CH, Littorin M, Johannesson G, Jönsson BAG, 2008. Analysis of ethylenethiourea as a biomarker in human urine using liquid chromatography/triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 22, 2573-2579.
♦ Longnecker MP, Rogan WJ, Lucier G, 1997. The Human health effects of DDT (dichlorodiphenyltrichloroethane) and PCB (polychlorinated biphenyls) and an overview of organochlorines in public health. Annu. Rev. Public. Health 18, 211-44.
♦ MMA (Ministerio de Medio Ambiente, Subdirección General de Calidad del Aire y Prevención de Riesgos), 2007. Plan Nacional de Aplicación del Convenio de Estocolmo y el Reglamento 850/2004, sobre Contaminantes Orgánicos Persistentes. http://www.mma.es/secciones/calidad_contaminacion/quimicos/pops/pdf/pn_estocolmo_nov06.pdf. Último acceso: 21-04-2010.
♦ Norstrom RJ, 2002. Understanding bioaccumulation of POPs in food webs. Environ. Sci. Pollut. Res 9, 300-303.
53

Introducción
♦ Okey AB, 2007. An aryl hydrocarbon receptor odyssey to the shores of toxicology: the Diechmann lecture, International Congress of Toxicology-XI. Toxicol. Sci. 98, 5-38.
♦ Olea M, Fernández MF, 2001. Plaguicidas persistentes. Congreso sobre la Implementación del Convenio de Contaminantes Orgánicos Persistentes. Madrid, 26-27 noviembre.
♦ Ortiz-Martínez A, 1991. Tesis doctoral: Incidencia del lindano en el organismo animal. Facultad de Ciencias Biológicas, Universidad Complutense, Madrid.
♦ Piédrola G, 2008. Medicina preventiva y salud pública 11ª Ed. Elsevier España S.L. (Masson S.A.), Barcelona.
♦ Porta M, Ballester F, Ribas-Fitó N, Piugdoménech E, Selva J, Llop S, 2006. Concentraciones de compuestos tóxicos persistentes en la población general española. Criterios para un diagnóstico de la situación actual. Gac. Sanit. 20, 233-238.
♦ Porta M, Puigdomènech E, Ballester F, 2009. Nuestra contaminación interna. Editorial Catarata, Madrid.
♦ PPU (Promociones y Publicaciones Universitarias S.A.), 1990. Libro de Actas de las 1as Jornadas Nacionales sobre Hexaclorobenceno, (Barcelona 23-24 mayo de 1988). http://www.recercat.net/bitstream/2072/5191/1/spm41.pdf. Último acceso: 19-04-2010.
♦ Reiner EJ, Boden AR, Chen T, MacPherson KA, Muscalu AM, 2010. Advances in the analysis of persistent halogenated organic compounds. LC GC Eur. 23, 60-70.
♦ Ross G, 2004. The public health implications of polychlorinated biphenyls (PCBs) in the environment. Ecotoxicol. Environ. Saf. 59, 275-291.
♦ Sala M, Sunyer J, Otero R, Santiago-Silva M, Camps C, Grimalt JO, 1999. Organochlorine compound concentration in the serum of inhabitants living near an electrochemical factory. Occup. Environ. Med. 56, 152-158.
♦ Sanz-Gallardo MI, 2006. Tesis doctoral: Determinantes de DDE en tejido adiposo y asociación con factores reproductivos en mujeres. Facultad de Medicina, Universidad Complutense, Madrid.
♦ Schulte E, Malisch R, 1983. Berechnung der Vahren PCB-Gehalte in Umweltproben I: Ermittlung der Zusammensetzung Zweier Technischer PCB-Gemische. Fresenius J. Anal. Chem. 314, 545-551.
♦ Stockholm Convention, 2010. Stockholm Convention on Persistent Organic Pollutants (POPs). http://chm.pops.int/. Último acceso: 20-04-2010.
♦ Sullivan Jr. JB, Krieger GR, 2001. Clinical environmental health and toxic exposures 2nd Ed. Lippincott Williams & Wilkins, Philadelphia, PA, USA.
♦ Timbrell JA, 2008. Principles of Biochemical Toxicology 4th Ed. Informa Healthcare USA Inc., New York, USA.
♦ Tomlin CDS, 2009. The Pesticide Manual: A World Compendium, 15th Ed. British Crop Protection Council, Surrey, UK.
♦ Ulbrich B, Stahlmann R, 2004. Developmental toxicity of polychlorinated biphenyls (PCBs): a systematic review of experimental data. Arch. Toxicol. 78, 252-268.
♦ UNEP (United Nations Environment Programme), 2006. Persistent Organic Pollutants (POPs) Review Committee, 2nd meeting; Draft risk profile: Lindane. http://www.pops.int/documents/meetings/poprc_2/meeting_docs/en/K0652380%20POPRC-2-10.pdf. Último acceso: 16-04-2010.
♦ UNIDO (United Nations Industrial Development Organization). UNIDO POPs Portal: Facts and Figures. http://www.unido.org/index.php?id=5763. Último acceso: 15-04-2010.
♦ Van der Berg M, Birnbaum L, Bosveld ATC y otros 21 autores, 1998. Toxic Equivalency Factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Persp. 106, 775-792.
54

Introducción
♦ Van der Berg M, Birnbaum LS, Denison M y otros 16 autores, 2006. The 2005 World Health Organization reevaluation of human and mammalian Toxic Equivalency Factors for dioxins and dioxin-like compounds. Toxicol. Sci. 93, 223-241.
♦ Vaquero JL, Asensio P, 1992. Manual de medicina preventiva y salud pública. Ediciones Pirámide, Madrid.
♦ WHO (World Health Organization), 1988. Heptachlor: Health and Safety Guide (nº 14). http://www.inchem.org/documents/hsg/hsg/hsg014.htm. Último acceso: 15-04-2010.
♦ WHO (World Health Organization), 2003. Health risks of persistent organic pollutants from long-range transboundary air pollution. http://www.euro.who.int/document/e78963.pdf. Último acceso: 15-04-2010.
♦ Woolcott CG, Aronson KJ, Hanna WM, SenGupta SK, McCready DR, Sterns EE, Miller AB, 2001. Organochlorines and breast cancer risk by receptor status, tumor size, and grade (Canada). Cancer Causes Control 12, 395-404.
♦ Yoshimura T, 2003. Yusho in Japan. Ind. Health 41, 139-148.
♦ Zell M, Ballschmiter K, 1980. Baseline studies of the global pollution III: trace analysis of polychlorinated biphenyls (PCB) by ECD glass capillary gas chromatography in environmental samples of different trophic levels. Fresenius Z. Anal. Chem. 304, 337-349.
55


OBJETIVOS

Objetivos
58

Objetivos
En este estudio se plantearon los siguientes objetivos generales:
• Desarrollar nuevas metodologías analíticas sencillas y de alta
productividad, capaces de determinar las concentraciones de
organoclorados mayoritarios en suero (PCBs 28, 52, 101, 118, 138, 153 y
180, HCB, β-HCH, heptacloro epóxido, p,p’-DDE y p,p’-DDT) partiendo de
volúmenes inferiores a 1 mL y el empleo de instrumental versátil,
asequible e implantado en los laboratorios, con miras a su utilización en
estudios epidemiológicos.
• Aplicar una de estas metodologías a la investigación de los niveles
séricos de dichos compuestos en una sub-cohorte de unos 1000
individuos, con una estructura de edad y sexo semejante a la población
española de 35-64 años, seleccionada al azar dentro de la cohorte EPIC-
España.
• Caracterizar la exposición de la población estudiada a partir de los niveles
séricos obtenidos e investigar su relación con diversas variables
sociodemográficas, antropométricas y dietéticas.
Estos objetivos generales se concretaron en los objetivos específicos de
cada una de las partes de este estudio que se contemplan en los capítulos 1, 2,
3, 4, y 5. Dichos objetivos específicos fueron los siguientes:
En el capítulo 1:
Desarrollar una metodología analítica basada en discos de extracción en
fase sólida C18, dispuestos en placas de 96 pocillos.
Optimizar varios aspectos de la etapa de extracción y limpieza de
extractos.
Validar dicho método y considerar su posible utilización en estudios
epidemiológicos.
59

Objetivos
En el capítulo 2:
Desarrollar una metodología analítica basada en microextracción en fase
sólida en espacio de cabeza (HS-SPME).
Fijar las condiciones de extracción, empleando el diseño experimental, en
dos fases: en la primera estableciendo las variables significativas y en la
segunda optimizando las mismas.
Validar dicho método y considerar su posible utilización en estudios
epidemiológicos.
En el capítulo 3:
Desarrollar una metodología analítica basada en una limpieza de
extractos por cromatografía de exclusión (GPC).
Optimizar y fijar las condiciones de limpieza con la ayuda del diseño
experimental.
Validar dicho método y considerar su posible utilización en estudios
epidemiológicos.
En el capítulo 4:
Emplear la metodología que se considere más adecuada para la
determinación de los niveles séricos de los PCBs 28, 52, 101, 118, 138,
153 y 180, en una sub-cohorte de unos 1000 individuos, con una
estructura de edad y sexo semejante a la población española de 35-64
años, seleccionada al azar dentro de la cohorte EPIC-España.
Estudiar su relación con diversos factores sociodemográficos,
antropométricos y dietéticos como: edad, sexo, localización geográfica,
ingesta de los distintos alimentos, índice de masa corporal, lactancia, etc.
Comparar estos niveles con los de otros estudios llevados a cabo, tanto
en España como en otros países.
60

Objetivos
En el capítulo 5:
Emplear la misma metodología para la determinación de los niveles
séricos de HCB, β-HCH, heptacloro epóxido, p,p’-DDE y p,p’-DDT en la
sub-cohorte anterior.
Estudiar su relación con diversos factores sociodemográficos,
antropométricos y dietéticos como: edad, sexo, localización geográfica,
ingesta de los distintos alimentos, índice de masa corporal, lactancia, etc.
Comparar estos resultados con los de otros estudios llevados a cabo,
tanto en España como en otros países.
61


CAPÍTULO 1
Metodología analítica basada en placas de extracción
1.1 Introducción
1.2 Parte experimental
1.3 Resultados y discusión
1.4 Conclusiones
1.5 Referencias

Método de placas de extracción
64

Capítulo 1
65
1.1 Introducción
Los métodos tradicionales para la determinación de plaguicidas
organoclorados y PCBs en suero incluyen extracciones líquido-líquido (LLE)
con disolventes orgánicos [Dale et al. 1970; Franken and B.Luyten 1976; Burse
et al. 1991; Najam et al. 1999; Rogers et al. 2004, Zwir-Ferenc and Biziuk 2004]
o extracciones en fase sólida (SPE) con columnas o cartuchos [Bucholski et al
1996; Lino et al. 1998; Pitarch et al 2003; Zwir-Ferenc and Biziuk 2004, Conka
et al. 2005] seguidas de una etapa de limpieza de extractos para eliminar las
interferencias potenciales que suponen los co-extraídos. Una vez limpio el
extracto se analiza por cromatografía de gases en columna capilar con detector
de captura de electrones (GC-ECD) [Najam et al. 1999; Covaci and Schepens
2001; Rogers et al. 2004; Zwir-Ferenc and Biziuk 2004, Conka et al. 2005,
Reiner et al. 2010] o detector de espectrometría de masas (GC-MSD) [Najam et
al. 1999; Covaci and Schepens 2001; Pitarch et al 2003; Barr et al. 2003;
Focant et al. 2004, Zwir-Ferenc and Biziuk 2004, Reiner et al. 2010].
Generalmente los métodos basados en LLE son complejos, largos, tediosos y
utilizan grandes volúmenes de disolventes, resultando inadecuados para el alto
número de muestras que hay que analizar en los estudios epidemiológicos
[Bucholski et al 1996; Brock et al. 1996]. Los métodos SPE con columnas o
cartuchos suponen una mejora por su simplicidad, ahorro de disolvente y
productividad [Bucholski et al 1996; Lino et al. 1998; Brock et al. 1996, Zwir-
Ferenc and Biziuk 2004]. Sin embargo, requieren no menos de 5-10 mL de
disolvente para completar la etapa de extracción, generalmente emplean más
de 2 mL de suero y la productividad raramente pasa de 15 muestras por
analista y día.
Para la determinación rutinaria de PCBs y plaguicidas organoclorados
mayoritarios en suero humano todos estos datos se pueden mejorar utilizando
discos de SPE dispuestos en placas de 96 pocillos. El procedimiento analítico
desarrollado en este capítulo combina las ventajas de los discos de SPE
(reducidos volúmenes de elución y buen rendimiento) [Zwir-Ferenc and Biziuk
2004] con la alta productividad derivada del uso de placas de 96 pocillos,
obteniéndose un método capaz de satisfacer los requisitos necesarios para la
monitorización biológica de la exposición a PCBs y plaguicidas organoclorados

Método de placas de extracción
66
mayoritarios en estudios epidemiológicos. Para la optimización del método se
han estudiado diferentes formas de desnaturalización de proteínas y
condiciones de elución y limpieza de extractos. El método se ha optimizado
para la determinación de PCBs 28, 52, 101, 118, 138, 153 y 180, HCB, β-HCH,
heptacloro epóxido, p,p’-DDE y p,p’-DDT, en suero humano.

Capítulo 1
67
1.2 Parte experimental
1.2.1 Reactivos y materiales
La disolución patrón conjunta de PCBs 28, 52, 101, 118, 138, 153 y 180,
cada uno a 10 μg mL-1 en isooctano, así como las disoluciones patrón de PCB
46 y PCB 143 a 100 μg mL-1 en hexano y el material de referencia certificado
NIST SRM 1589a (PCBs, pesticidas y dioxinas/furanos en suero humano),
fueron suministradas por LGC-Promochem (Middlesex, Reino Unido). Los
patrones de HCB, β-HCH, heptacloro epóxido, p,p’-DDE y p,p’-DDT, se
adquirieron a Dr. Ehrenstorfer (Augsburg, Alemania). El acetonitrilo y el
metanol, ambos calidad HPLC gradiente, fueron de Scharlab (Barcelona,
España). El n-hexano, diclorometano, tolueno y ciclohexano, calidad análisis de
trazas, junto con la gel de sílice 60 para cromatografía en columna y la lana de
vidrio, los suministró Merck (Darmstadt, Alemania). El H2SO4 concentrado
calidad para análisis y el Na2SO4 anhidro calidad para análisis de residuos,
fueron de Panreac (Barcelona, España). Las placas de 96 pocillos con discos
de SPE Empore (C18, 1mL) y su correspondiente dispositivo de extracción a
vacío fueron de 3M (St. Paul, MN, EE.UU.). Las gradillas de recolección con 96
tubos de vidrio las suministró Marsh Bio Products (Rochester, NY, EE.UU.). El
evaporador Turbo-Vap LV era de la casa Zymark Corp. (Hopkinton, MA,
EE.UU.).
Las disoluciones conjuntas intermedias de PCBs se prepararon en
ciclohexano y las de plaguicidas en tolueno, conservándose a -20 ºC. A partir
de éstas se prepararon las de calibración en ciclohexano y las de adición en
acetonitrilo, conservándose a 4 ºC. La mezcla de gel de sílice/ácido sulfúrico
para la limpieza de extractos, se preparó añadiendo, gota a gota y con
agitación manual, 5 mL de ácido sulfúrico a 10 g de gel de sílice. Una vez
añadido el ácido, la mezcla se mantuvo bajo agitación mecánica hasta
ausencia de grumos. La lana de vidrio, el Na2SO4 anhidro y la gel de sílice se
estufaron a 300 ºC toda una noche antes de su uso. El material de vidrio se
lavó con detergente, se aclaró abundantemente con agua Milli-Q, se estufó
toda una noche a 300 ºC y se enjuagó con diclorometano o hexano antes de su
uso.

Método de placas de extracción
68
1.2.2 Muestras de suero
El suero utilizado para el desarrollo y la validación del método se obtuvo
del banco de muestras de la cohorte EPIC-España y de voluntarios de los
Laboratorios de Salud Pública de Gipuzkoa y Bizkaia, utilizándose siempre bajo
total anonimato. La sangre se extrajo por punción venosa y se recogió en tubos
Vacutainer. El suero se separó por centrifugación. Se preparó una mezcla de
suero a partir de 35 muestras individuales a la que se denominó Suero de
Laboratorio (LS). Todas las muestras de suero se almacenaron a -20 ºC hasta
su análisis. Para no someterlo a excesivos ciclos de congelado y
descongelado, el LS se alicuotó en tubos de vidrio de 10 mL provistos de tapón
de rosca y junta de teflón.
1.2.3 Equipamiento instrumental
La cuantificación se realizó en un cromatógrafo de gases 5890 series II,
equipado con inyector con/sin división de flujo, detector de captura de
electrones (ECD) y muestreador automático 7673, todo ello de Agilent
Technologies (Wilmington, DW, EE.UU.). Se inyectó 1 μL de muestra en modo
sin división de flujo, abriéndose la válvula de división de flujo 1 min después de
la inyección. La temperatura del portal de inyección era de 250 ºC con Helio
como gas portador a 135 kPa. La temperatura del detector era de 300 ºC con
N2 como gas de apoyo. La separación cromatográfica se realizó con dos
columnas: una DB-XLB de 30m × 0,25 mm × 0,25 µm (J&W Scientific, Folsom,
CA, EE.UU.) para los PCBs y una DB-5MS de 50 m × 0,25 mm × 0,25 µm (J&W
Scientific) para los plaguicidas organoclorados. El horno estaba programado a
60 ºC (1 min), seguido de 20 ºC min-1 hasta 180 ºC, 1,5 ºC min-1 hasta 240 ºC y
30 ºC min-1 hasta 300 ºC (8 min).
La confirmación, cualitativa y cuantitativa, se llevó a cabo en un
cromatógrafo de gases 6890 equipado con inyector con/sin división de flujo con
control electrónico de presión, detector de espectrometría de masas (MSD)
5973 de cuadrupolo y muestreador automático 7683 (Agilent Technologies). Se
inyectaron 2 μL de muestra en modo sin división de flujo, abriéndose la válvula
de división de flujo 1,2 min después de la inyección. La temperatura del portal
de inyección era de 270 ºC con Helio como gas portador a 140 kPa. La

Capítulo 1
69
separación cromatográfica se realizó con una columna DB-5MS de 30 m × 0,25
mm × 0,25 µm (J&W Scientific). El horno estaba programado a 65 ºC (1,5 min),
seguido de 20 ºC min-1 hasta 180 ºC (9 min), 10 ºC min-1 hasta 230 ºC (7 min) y
3 ºC min-1 hasta 275 ºC (7 min). La temperatura de la interfaz era de 300 ºC, la
de la fuente de ionización (EI, 70 eV) de 230 ºC y la del cuadrupolo de 150ºC.
El MSD se utilizó en modo Monitorización Selectiva de Iones (SIM) con un
tiempo de permanencia de 40 ms por ión. Para la cuantificación e identificación
se usaron las masas mostradas en la Tabla 1.1. Como criterio de identificación
se consideró aceptable una desviación porcentual relativa del ión cualificador,
menor del ±20% con respecto a su abundancia relativa teórica en relación al
ión cuantificador.
Tabla 1.1. Masas (m/z) utilizadas para la confirmación por GC-MSD.
Analito Ión cuantificador (m/z) Ión cualificador(m/z)
Triclorobifenilo 258 260
Tetraclorobifenilo 290 294
Pentaclorobifenilo 326 324
Hexaclorobifenilo 360 290
Heptaclorobifenilo 394 396
HCB 284 282
β-HCH 183 217
Heptacloro epóxido 353 355
p,p’-DDE 246 318
p,p’-DDT 235 237
1.2.4 Procedimiento analítico
Se tomaron 500 μL de cada muestra de suero y se depositaron en sendos
viales de vidrio de 2 mL. A cada muestra se le añadieron 500 μL de una
disolución acuosa de Na2SO4 al 5% y 250 μL de la disolución de adición de

Método de placas de extracción
70
patrón interno que contenía 5 ng mL-1 de PCB 46 y 143 en acetonitrilo. Los
viales se sellaron con tapones de rosca y junta septum revestida de teflón y se
sometieron a baño de ultrasonidos durante 20 min. Mientras tanto se colocó
una placa de discos de SPE en el dispositivo de extracción y se acondicionó
cada disco C18 con, sucesivamente, 2×300 μL de diclorometano, 300 μL de
metanol y 2×300 μL de agua Milli-Q, con la ayuda del vacío generado por una
trompa de agua. Una vez acondicionados los discos y terminado el tratamiento
de ultrasonidos, el contenido de cada vial se depositó en un pocillo de la placa
y se eluyeron las muestras suavemente a vacío, desechándose el eluato. En
aquellos discos que presentaron indicios de colmatación, la elución se
completó con presión positiva de N2. Tras la elución, los pocillos se enjuagaron
con 2×500 μL de agua Milli-Q y se sometieron a secado, primero con N2 a 200
kPa durante 15 segundos y luego por centrifugación a 1500×g durante 15 min.
A continuación se colocó la gradilla de 96 tubos de vidrio de recolección en el
dispositivo de extracción y en la parte superior la placa de discos SPE. Cada
uno de los discos se eluyó con 2×300 μL de hexano y 300 μL de
diclorometano/hexano (1:1), recolectándose todo el eluato de cada disco en su
correspondiente tubo de vidrio.
Por cada muestra se preparó una columna de vidrio rellena, de abajo a
arriba, con: lana de vidrio, 300 mg de la mezcla de gel de sílice/ácido sulfúrico
(preparada en el día) y 100 mg de Na2SO4 anhidro. Cada columna se
acondicionó con 2 mL de diclorometano, 2 mL de diclorometano/hexano (1:1) y
2 mL de hexano. Se colocaron sendos tubos de evaporador TurboVap debajo
de las columnas y se añadieron a éstas, de forma secuencial, los
correspondientes 900 µL de extracto de cada muestra y 2×1 mL de
diclorometano. Tras añadir a cada tubo de evaporación 500 μL de ciclohexano,
los eluatos se concentraron en el TurboVap (25 kPa de N2, 30 ºC) hasta 50 µL,
que se transfirieron a viales con inserto de 200 μL para su posterior análisis por
cromatografía de gases.
1.2.5 Calibración y validación
La calibración GC-ECD se llevó a cabo con disoluciones patrón de los
analitos a 0,75, 2, 10 y 50 ng mL-1 en ciclohexano (1,5, 4, 20 y 100 ng mL-1

Capítulo 1
71
para el p,p’-DDE). Para la calibración del GC-MSD se usaron las disoluciones
de 2, 10 y 50 ng mL-1. La cuantificación se llevó a cabo utilizando el PCB 46
(para HCB, β-HCH, heptacloro epóxido, PCBs 28 y 52) y el PCB 143 (para p,p’-
DDE, p,p’-DDT, PCBs 101, 118, 138, 153 y 180) como patrones internos (IS), a
25 ng mL-1. Se emplearon regresiones del cociente entre las áreas de pico de
los analitos con respecto a las de sus respectivos ISs, frente al cociente de las
concentraciones, con una ponderación del inverso del cuadrado de las
concentraciones.
La repetibilidad y la recuperación se determinaron adicionando el LS a
dos niveles: 0,15 y 1,5 ng mL-1 para los PCBs y 0,3 y 3 ng mL-1 para los
plaguicidas organoclorados (n = 8 para cada nivel).
En la aplicación del método en estudios epidemiológicos, cada tanda
analítica incluía dos blancos, un duplicado del LS y un duplicado del material de
referencia certificado NIST SRM 1589a, como controles internos. Para el
blanco se utilizaron 500 μL de agua Milli-Q en lugar de suero. Se realizó una
hoja de cálculo y un gráfico de control para cada analito con los resultados del
LS (ver Figura 1.1). Tras un año de aplicación, a partir de los datos del LS se
calculó la reproducibilidad intralaboratorio y a partir del SRM 1589a, la
veracidad.
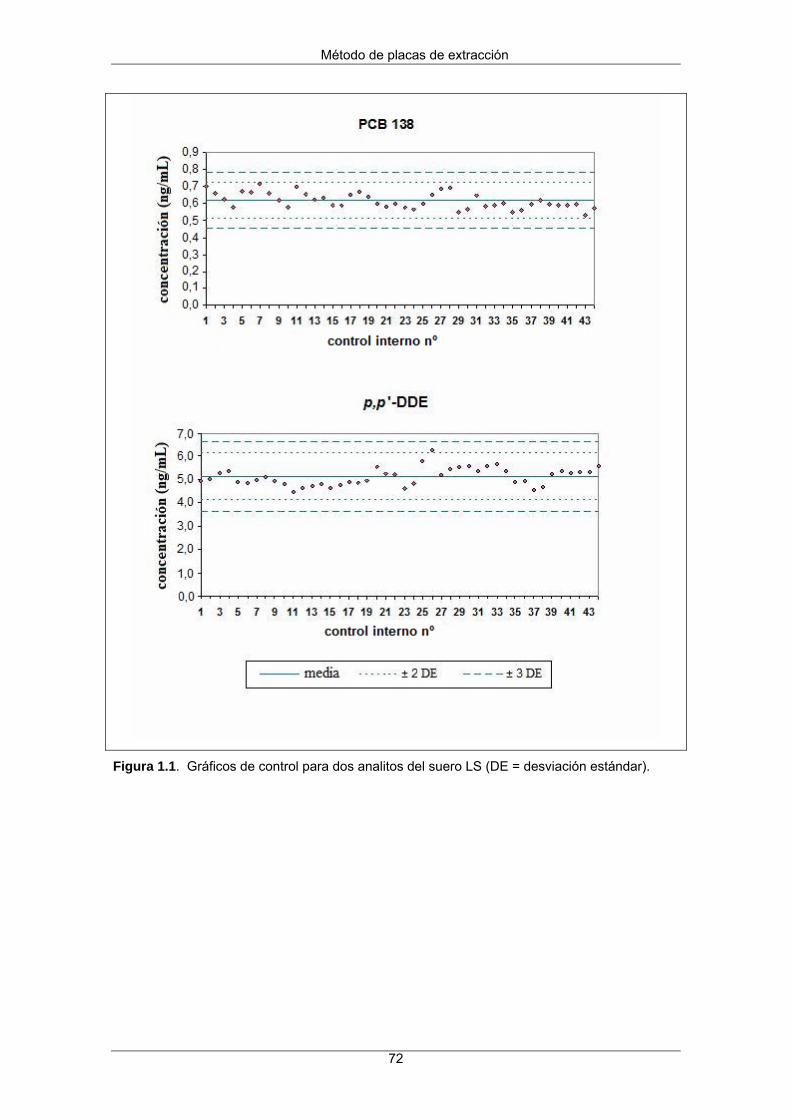
Método de placas de extracción
72
Figura 1.1. Gráficos de control para dos analitos del suero LS (DE = desviación estándar).

Capítulo 1
73
1.3 Resultados y discusión
1.3.1 Optimización del método
En el desarrollo de este método de determinación de PCBs y plaguicidas
organoclorados en suero, mediante placas de discos de extracción de 96
pocillos, fue necesario considerar y optimizar varios parámetros que afectaban
a los procesos de extracción y limpieza de extractos.
La elección del desnaturalizante fue crucial, ya que era el responsable de
destruir las interacciones proteína-analito y evitar la vehiculización micelar en
medio acuoso, circunstancias ambas que disminuyen la recuperación de los
analitos. Por otra parte, si la acción del desnaturalizante era demasiado
intensa, podría causar excesiva precipitación de proteínas y se colmataría el
disco de extracción, lo que supondría la pérdida de la muestra o, como mal
menor, pérdida de recuperación por co-precipitación. Por ello se probaron
diversos desnaturalizantes con vistas a obtener la mejor recuperación. El ácido
fórmico tradicionalmente viene siendo uno de los más usados [Brock et al.
1996; Pauwels et al. 1999; Sandau et al. 2003]. En nuestro caso daba
excelentes resultados para los PCBs y la mayoría de los plaguicidas
organoclorados, pero las recuperaciones finales del heptacloro epóxido y del β-
HCH eran sistemáticamente <50%. También se probaron el ácido
tricloroacético y algunos surfactantes como Tween 20, Tween 80 y Tritón X-
100. Todos ellos daban lugar a fuertes interferencias en la señal
cromatográfica. En las pruebas que se hicieron con disoluciones de Carrez, las
recuperaciones eran muy bajas. El metanol causaba excesiva precipitación de
proteínas y se colmataban los discos de extracción. Los mejores resultados se
alcanzaron con la acción combinada del acetonitrilo y una disolución de
Na2SO4 anhidro al 5% en agua, ya que proveían de la suficiente
desnaturalización sin causar excesiva precipitación de proteínas.
Las columnas de adsorción o el ácido sulfúrico han sido profusamente
utilizados para eliminar lípidos y otros co-extractivos en extractos no polares
[Amakura et al. 2002, Pan et al. 2004, Zwir-Ferenc and Biziuk 2004, Saito et al.
2005]. Algunos autores [Covaci and Schepens 2001; Sjödin et al. 2004] han
descrito el uso de columnas combinadas de gel de sílice/ácido sulfúrico,

Método de placas de extracción
74
eluídas con hexano, relatando resultados muy prometedores. Por anteriores
experiencias con ácido sulfúrico sabíamos que diversos plaguicidas
organoclorados: heptacloro epóxido, o,p’-DDT, α-endosulfán, aldrín, endrín,
dieldrín, y metoxiclor, eran sensibles a este ácido y se degradaban. Por ello,
para minimizar cualquier posible degradación, el tiempo de elución debía ser lo
más corto posible. Esto se podía conseguir empleando menos volumen de un
disolvente más polar (mayor poder eluotrópico) y columnas de limpieza de
extractos lo más pequeñas posible. Con ese fin se ensayó con diclorometano y
se encontró que 2 mL eran suficientes para completar la elución sin alterar el
rendimiento de la limpieza del extracto. En el mismo sentido se probó con
diferentes cantidades de relleno: 200, 300, 400 y 500 mg, para optimizar el
tamaño de las columnas y resultó que 300 mg era el tamaño que garantizaba
buenas recuperaciones sin comprometer el resultado de la limpieza.
1.3.2 Evaluación del método
Tras la relatada optimización de varias condiciones de extracción y
limpieza de extractos, se estudiaron el límite de cuantificación, la recuperación
y la repetibilidad del método.
El límite de cuantificación de cada compuesto (LQ) se calculó como la
concentración del analito que producía una señal 10 veces mayor que el ruido
de fondo de un blanco sometido al proceso analítico. Para los PCBs el LQ iba
de 0,08 a 0,13 ng mL-1 y para los plaguicidas organoclorados de 0,16 a 0,40 ng
mL-1 (ver Tabla 1.2).

Capítulo 1
75
Tabla 1.2. Límites de cuantificación (LQ).
Compuesto LQ (ng mL-1)
PCB 28 0,10
PCB 52 0,13
PCB 101 0,10
PCB 118 0,10
PCB 138 0,10
PCB 153 0,08
PCB 180 0,08
p,p’-DDT 0,40
p,p’-DDE 0,33
Heptacloro epóxido 0,32
HCB 0,16
β-HCH 0,38
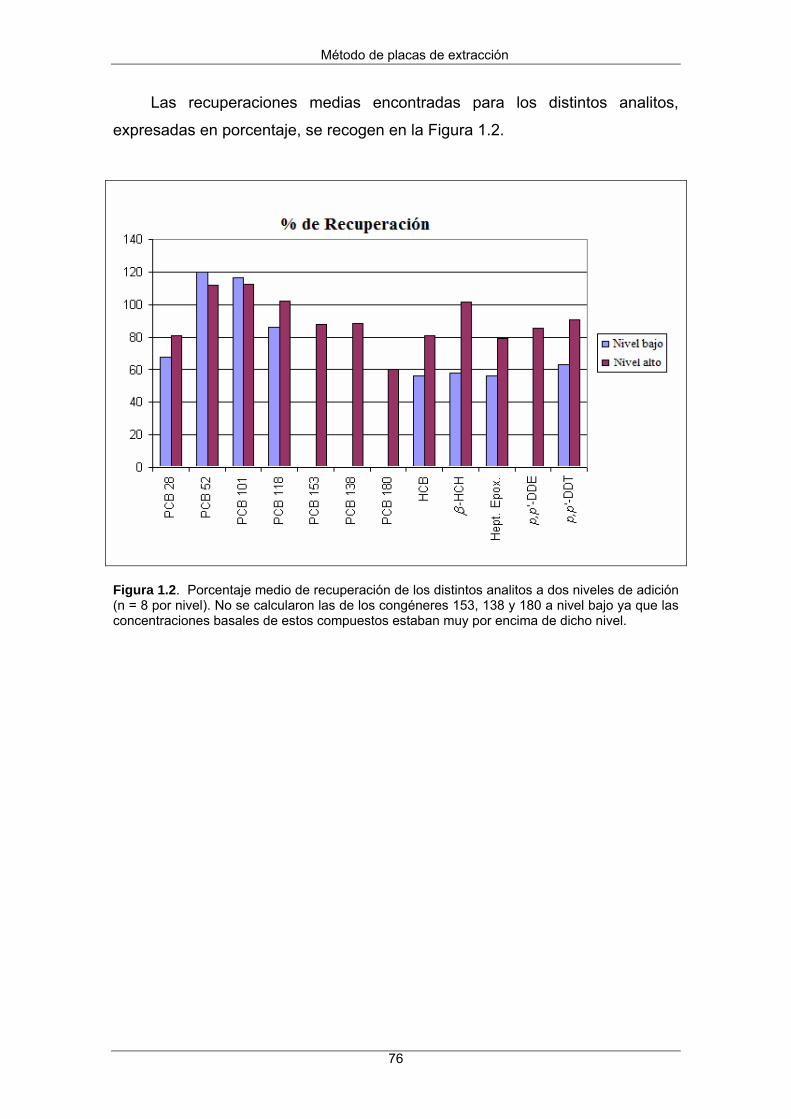
Método de placas de extracción
76
Las recuperaciones medias encontradas para los distintos analitos,
expresadas en porcentaje, se recogen en la Figura 1.2.
Figura 1.2. Porcentaje medio de recuperación de los distintos analitos a dos niveles de adición (n = 8 por nivel). No se calcularon las de los congéneres 153, 138 y 180 a nivel bajo ya que las concentraciones basales de estos compuestos estaban muy por encima de dicho nivel.

Capítulo 1
77
La repetibilidad de cada uno de ellos, medida como porcentaje de
desviación estándar relativa (DER), se recoge en la Figura 1.3.
Figura 1.3. Repetibilidad, en porcentaje de desviación estándar relativa (DER), para cada uno de los analitos a dos niveles de adición (n = 8 por nivel). No se calcularon las de los congéneres 153, 138 y 180 a nivel bajo ya que las concentraciones basales de estos compuestos estaban muy por encima de dicho nivel.
La reproducibilidad intralaboratorio, calculada también como % de DER a
partir de los controles internos del LS, cuyos resultados para algunos analitos
se muestran gráficamente en la Figura 1.1, fue < 15% para todos ellos (n= 42).
En cuanto a la veracidad, las concentraciones medias (n=42) obtenidas
con el NIST SRM 1589a, no diferían significativamente de los valores
certificados (rev. 4/05/2007) para el p,p’-DDE y los PCBs 118 y 180 y estaban
un 15,6% y un 13,4% por debajo, respectivamente, para los PCBs 138 y 153.
El resto de analitos estaba por debajo de nuestro LQ, lo que también coincidía
con los valores certificados.
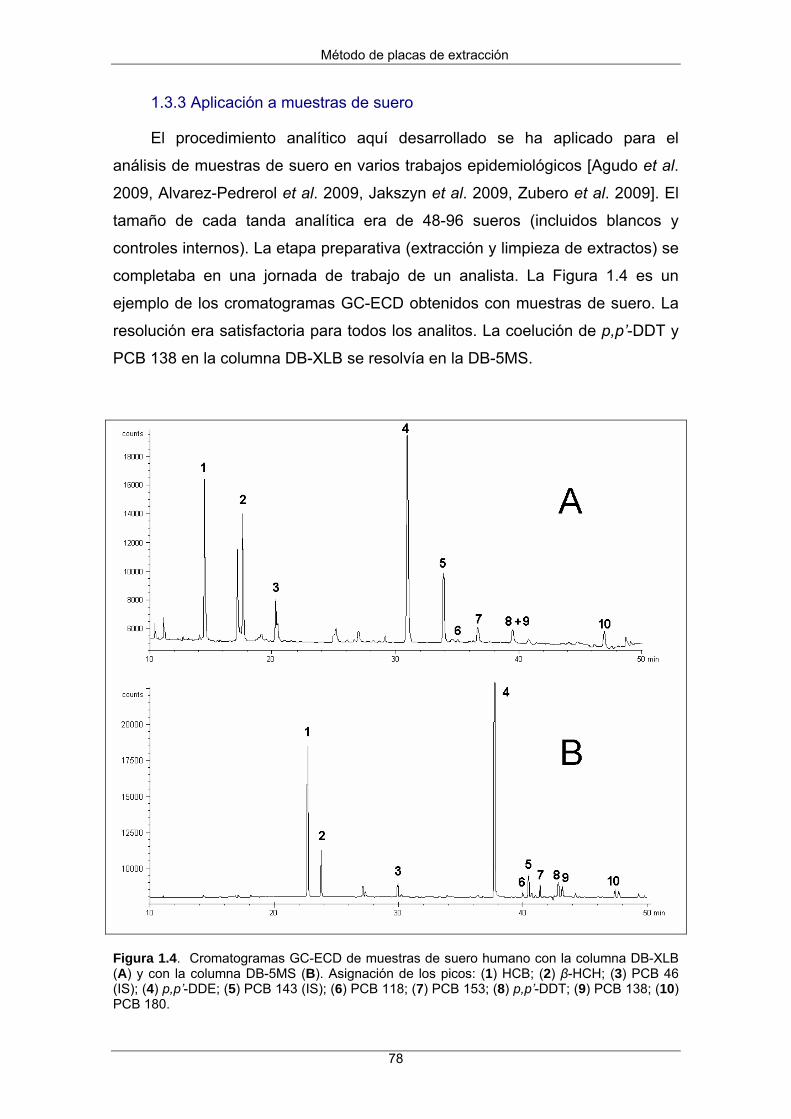
Método de placas de extracción
78
1.3.3 Aplicación a muestras de suero
El procedimiento analítico aquí desarrollado se ha aplicado para el
análisis de muestras de suero en varios trabajos epidemiológicos [Agudo et al.
2009, Alvarez-Pedrerol et al. 2009, Jakszyn et al. 2009, Zubero et al. 2009]. El
tamaño de cada tanda analítica era de 48-96 sueros (incluidos blancos y
controles internos). La etapa preparativa (extracción y limpieza de extractos) se
completaba en una jornada de trabajo de un analista. La Figura 1.4 es un
ejemplo de los cromatogramas GC-ECD obtenidos con muestras de suero. La
resolución era satisfactoria para todos los analitos. La coelución de p,p’-DDT y
PCB 138 en la columna DB-XLB se resolvía en la DB-5MS.
Figura 1.4. Cromatogramas GC-ECD de muestras de suero humano con la columna DB-XLB (A) y con la columna DB-5MS (B). Asignación de los picos: (1) HCB; (2) β-HCH; (3) PCB 46 (IS); (4) p,p’-DDE; (5) PCB 143 (IS); (6) PCB 118; (7) PCB 153; (8) p,p’-DDT; (9) PCB 138; (10) PCB 180.

Capítulo 1
79
En la Figura 1.5 se presentan los diferentes extractos iónicos de un
cromatograma GC-MSD, adquirido en modo SIM, de una muestra de suero.
Figura 1.5. Extractos iónicos del cromatograma GC-MSD SIM de una muestra de suero. Asignación de los picos: (1) HCB; (2) β-HCH; (3) PCB 46 (IS); (4) p,p’-DDE; (5) PCB 118; (6) PCB 143 (IS); (7) PCB 153; (8) p,p’-DDT; (9) PCB 138; (10) PCB 180.

Método de placas de extracción
80
1.4 Conclusiones
El método de discos de extracción SPE dispuestos en placas de 96
pocillos descrito en este capítulo, permite la determinación simultánea de los
plaguicidas organoclorados y PCBs más relevantes en suero humano.
Comparado con otras técnicas presenta varias ventajas que lo hacen adecuado
para su empleo en estudios de riesgo de exposición o epidemiológicos, donde
se requiere el análisis de un elevado número de muestras. Estas ventajas son:
aceptable LQ con baja cantidad de muestra (500 µL), recuperación, precisión y
veracidad satisfactorias y alta productividad (se pueden preparar 48-96
muestras por jornada de trabajo y analista sin requerir especiales dispositivos
de automatización). Como resultado de todo ello, se ha aplicado ya en varios
trabajos epidemiológicos. Además, este método podría ser ampliado a otros
compuestos contaminantes ya que se emplean adsorbentes y condiciones de
extracción no específicos.

Capítulo 1
81
1.5 Referencias
♦ Agudo A, Goñi F, Etxeandia A y otros 16 autores, 2009. Polychlorinated biphenyls in Spanish adults: determinants of serum concentrations. Environ. Res. 109, 620-628.
♦ Alvarez-Pedrerol M, Guxens M, Ibarluzea J, Rebagliato M, Rodríguez A, Espada M, Goñi F, Basterrechea M, Sunyer J, 2009. Organochlorine compounds, iodine intake, and thyroid hormone levels during pregnancy. Environ. Sci. Technol. 43, 7909-7915.
♦ Amakura Y, Tsutsumi T, Sasaki K, Toyoda M, Maitani T, 2002. Comparison of sulfuric acid treatment and multi-layer silica gel column chromatography in cleanup methods for determination of PCDDs, PCDFs and dioxin-like PCBs in foods. J. Food Hyg. Soc. Jpn. 43, 312-321.
♦ Barr JR, Maggio VL, Barr DB, Turner WE, Sjödin A, Sandau CD, Pirkle JL, Needham LL, Patterson DG Jr., 2003. New high-resolution mass spectrometric approach for the measurement of polychlorinated biphenyls and organochlorine pesticides in human serum. J. Chromatogr. B 794, 137-148.
♦ Brock JW, Burse VW, Ashley DL, Najam AR, Green VE, Korver MP, Powell MK, Hodge CC, Needham LL, 1996. An improved analysis for chlorinated pesticides and polychlorinated biphenyls (PCBs) in human and bovine sera using solid-phase extraction. J. Anal. Toxicol. 20, 528-536.
♦ Bucholski KA, Begerow J, Winneke G, Dunemann L, 1996. Determination of polychlorinated biphenyls and chlorinated pesticides in human body fluids and tissues. J. Chromatogr. A 754, 479-485.
♦ Burse VW, Korver MP, McClure PC, Holler JS, Fast DM, Head SL, Miller DT, 1991. Problems associated with interferences in the analysis of serum for polychlorinated biphenyls. J. Chromatogr. A 566, 117-125.
♦ Conka K, Drobná B, Kocan A, Petrík J, 2005. Simple solid-phase extraction method for determination of polychlorinated biphenyls and selected organochlorine pesticides in human serum. J. Chromatogr. A 1084, 33-38.
♦ Covaci A, Schepens P, 2001. Simplified method for determination of organochlorine pollutants in human serum by solid-phase disk extraction and gas chromatography. Chemosphere 43, 439-447.
♦ Dale WE, Miles JW, Gaines TB, 1970. Quantitative method for determination of DDT and DDT metabolites in blood serum. J. AOAC Int. 53, 1287-1292.
♦ Franken JJ, Luyten BJ, 1976. Comparison of dieldrin, lindane, and DDT extractions from serum, and gas-liquid chromatography using glass capillary columns. J. AOAC Int. 59, 1279-1285.
♦ Focant JF, Cochran JW, Dimandja JM, DePauw E, Sjödin A, Turner WE, Patterson DG Jr., 2004. High-throughput analysis of human serum for selected polychlorinated biphenyls (PCBs) by gas chromatography-isotope dilution time-of-flight mass spectrometry (GC-IDTOFMS). Analyst 129, 331-336.
♦ Jakszyn P, Goñi F, Etxeandia A y otros 16 autores, 2009. Serum levels of organochlorine pesticides in healthy adults from five regions of Spain. Chemosphere 76, 1518-1524.
♦ Lino CM, Ferreira CB, Valente DS, Rocha JM, Noronha MI, 1998. Methods for the determination of organochlorine pesticide residues in human serum. J. Chromatogr. B 716, 147-152.
♦ Najam AR, Korver MP, Williams CC, Burse VW, Needham LL, 1999. Analysis of a mixture of polychlorinated biphenyls and chlorinated pesticides in human serum by column fractionation and dual-column capillary gas chromatography with electron capture detection. J AOAC Int 82, 177-185.

Método de placas de extracción
82
♦ Pan B, Liu WX, Shi Z, Cao J, Shen WR, Qing BP, Sun R, Tao S, 2004. Sample purification for analysis of organochlorine pesticides in sediment and fish muscle. J. Environ. Sci. Heal. B 39, 353-365.
♦ Pauwels A, Wells DA, Covaci A, Schepens PJ, 1999. Improved sample preparation method for selected persistent organochlorine pollutants in human serum using solid-phase disk extraction with gas chromatographic analysis. J. Chromatogr. B 723, 117-125.
♦ Pitarch E, Serrano R, López FJ, Hernández F, 2003. Rapid multiresidue determination of organochlorine and organophosphorus compounds in human serum by solid-phase extraction and gas chromatography coupled to tandem mass spectrometry. Anal. Bioanal. Chem. 376, 189-197.
♦ Reiner EJ, Boden AR, Chen T, MacPherson KA, Muscalu AM, 2010. Advances in the analysis of persistent halogenated organic compounds. LC GC Eur. 23, 60-70.
♦ Rogers E, Petreas M, Park J, Zhao G, Charles M, 2004. Evaluation of four capillary columns for the analysis of organochlorine pesticides (OCPs), polychlorinated biphenyls (PCBs), and polybrominated diphenyl ethers (BDEs) in human serum for epidemiologic studies. J. Chromatogr. B 813, 269-285.
♦ Saito K, Ogawa M, Takekuma M, Ohmura A, Kawaguchi M, Ito R, Inoue K, Matsuki Y, Nakazawa H, 2005. Systematic analysis and overall toxicity evaluation of dioxins and hexachlorobenzene in human milk. Chemosphere 61, 1215-1220.
♦ Sandau CD, Sjödin A, Davis MD, Barr JR, Maggio VL, Waterman AL, Preston KE, Preau JL Jr, Barr DB, Needham LL, Patterson DG Jr., 2003. Comprehensive solid-phase extraction method for persistent organic pollutants. Validation and application to the analysis of persistent chlorinated pesticides. Anal. Chem. 75, 71-77.
♦ Sjödin A, Jones RS, Lapeza CR, Focant JF, McGahee EE 3rd, Patterson DG Jr., 2004. Semiautomated high-throughput extraction and cleanup method for the measurement of polybrominated diphenyl ethers, polybrominated biphenyls, and polychlorinated biphenyls in human serum. Anal. Chem. 76, 1921-1927.
♦ Zubero MB, Ibarluzea JM, Aurrekoetxea JJ, Rivera J, Parera J, Abad E, Goñi F, López R, Etxeandia A, Rodríguez C, Sáenz JR, 2009. Serum levels of polychlorinated dibenzodioxins and dibenzofurans and PCBs in the general population living near an urban waste treatment plant in Biscay, Basque Country. Chemosphere 76, 784-791.
♦ Zwir-Ferenc A, Biziuk M, 2004. An analysis of pesticides and polychlorinated biphenyls in biological samples and foods. Crit. Rev. Anal. Chem. 34, 95-103.

CAPíTULO 2
Metodología analítica basada en microextracción en fase sólida
2.1 Introducción
2.2 Parte experimental
2.3 Resultados y discusión
2.4 Conclusiones
2.5 Referencias

Capítulo 2
84

Método de microextracción en fase sólida
2.1 Introducción
Los métodos de determinación de PCBs y plaguicidas organoclorados en
suero basados en extracción líquido-líquido (LLE) o extracción en fase sólida
(SPE) incluyen pasos de acondicionamiento, lavado, elución, limpieza de
extractos, etc., en las etapas de preparación de la muestra. Todo ello supone
mano de obra, gasto de disolvente y empleo de equipos auxiliares. A esto se
añaden otros posibles inconvenientes como: formación de emulsiones,
saturación de fases de extracción, colmatación de filtros previos, inapropiada
desnaturalización de proteínas, creación de canales preferentes en la fase
extractante, pérdida de analitos por evaporación y demás. Y así, aunque se
consiguen extractos razonablemente limpios, el riesgo de error analítico se
incrementa como consecuencia del gran número de pasos en las etapas de
preparación de la muestra. Una vez limpios, los extractos se analizan por GC-
ECD [Najam et al. 1999, Covaci and Schepens 2001, Rogers et al. 2004,
Conka et al. 2005], GC-MSD [Covaci and Schepens 2001, Pitarch et al. 2003,
Rantakokko et al. 2009], cromatografía de gases y espectrometría de masas de
alta resolución con dilución isotópica (GC-IDHRMS) [Barr et al. 2003, Focant et
al. 2004] o cromatografía de gases y espectrometría de masas de tiempo de
vuelo con dilución isotópica [Focant et al. 2004].
Hoy en día la microextracción en fase sólida (SPME) está considerada
una útil alternativa a la LLE o la SPE. La SPME no requiere disolventes y se
puede llevar a cabo directamente sobre la muestra en estado líquido
(inmersión) o sobre el espacio de cabeza (HS) de la misma. Además, la SPME
requiere menos pasos y menos manipulación de la muestra. El muestreo en HS
está recomendado cuando la matriz pudiera afectar a la determinación de los
analitos. En adición, el muestreo en HS supone una reducción importante del
tiempo de extracción frente al muestreo por inmersión [Pawliszyn 1997].
Existen trabajos donde se contempla la aplicación de la SPME, con sus
ventajas y desventajas, a fluidos biológicos [Mills and Walker 2000, Ulrich
2000]. De ellos se concluye que con fluidos biológicos debería aplicarse
siempre que fuera posible el muestreo en HS ya que, entre otras ventajas, la
carga de proteínas sobre la fibra es considerablemente menor y se incrementa
el tiempo de vida de la misma [Ulrich 2000]. Respecto a los compuestos de
85

Capítulo 2
este estudio, se han reportado métodos SPME para la determinación de PCBs
y plaguicidas organoclorados en matrices ambientales, incluidos agua y tejidos
biológicos [Fidalgo-Used et al. 2003, Li et al. 2003, Shu et al. 2003, Dong et al.
2005], aunque sólo unos pocos de ellos se han centrado en la determinación
de plaguicidas organoclorados en suero humano [Beltrán et al. 2001, López et
al. 2001]. Sin embargo, en la literatura revisada no existen referencias sobre el
potencial de la técnica HS-SPME para la determinación simultánea de PCBs y
plaguicidas organoclorados en suero humano.
Hay algunas variables experimentales que afectan a la técnica HS-SPME,
tales como: el tipo de fibra, la velocidad de agitación, la temperatura, el tiempo
de extracción, el volumen de espacio de cabeza y el contenido en electrolitos
salinos. Su optimización global, trabajando con las variables una a una, se
muestra en varios estudios sobre el análisis de compuestos volátiles en fluidos
biológicos [Zuba et al. 2002, Poli et al. 2005], PCBs en agua [Shu et al. 2003] y
plaguicidas organoclorados en agua [Dong et al. 2005] y en suero humano
[Beltrán et al. 2001]. El inconveniente es que este proceso requiere un gran
número de ensayos y lleva mucho tiempo. Un diseño experimental estructurado
que tuviera en cuenta dichas variables de forma simultánea parece, a priori,
una forma más cómoda y más rápida de abordar dicha tarea [Brereton 2003,
Bezerra et al. 2008, Stalikas et al. 2009]. Por ejemplo, una metodología basada
en superficies de respuesta (Doehlert design) ya se ha utilizado para la
optimización de las condiciones de extracción SPME en la determinación de
plaguicidas organoclorados en medio acuoso [Aguilar et al. 1999]. Esta
metodología usa un modelo de espacio multifactor donde una serie de
experimentos se lleva a cabo de forma sistemática para estimar el punto óptimo
y los efectos de interacción. También un diseño con los pasos de cribado y
optimización se ha aplicado para encontrar las mejores condiciones
experimentales para la determinación de alquil-éteres, además de benceno,
tolueno, etilbenceno y xilenos (BTEX), en agua por HS-SPME [Arambarri et al.
2004]. Por lo tanto, el empleo de este tipo de metodologías estructuradas
podría ser una forma útil y corta para encontrar las mejores condiciones
experimentales trabajando en HS-SPME con suero humano.
86

Método de microextracción en fase sólida
Este capítulo se centra en el trabajo realizado para la obtención de un
método adecuado para la determinación simultánea de plaguicidas
organoclorados y PCBs en suero humano usando HS-SPME y GC-ECD. Dicho
método cuenta con las ventajas inherentes a la HS-SPME, como son:
eliminación de la etapa de limpieza de extractos y evitar el uso de disolventes
orgánicos, la mayoría de ellos nocivos o peligrosos. Para delimitar las variables
experimentales significativas de la HS-SPME se ha utilizado un diseño factorial
reducido Plackett-Burman y la optimización de las mismas se ha realizado
mediante un diseño central compuesto. Finalmente, el procedimiento se ha
aplicado al análisis de 33 muestras de suero humano.
87

Capítulo 2
2.2 Parte experimental
2.2.1 Reactivos y material
La disolución patrón conjunta de PCBs 28, 52, 101, 118, 138, 153 y 180,
cada uno a 10 μg mL-1 en isooctano, así como las disoluciones patrón de PCB
46 y PCB 143 a 100 μg mL-1 en hexano, fueron suministradas por LGC-
Promochem (Middlesex, Reino Unido). Los patrones de HCB, β-HCH,
heptacloro epóxido, p,p’-DDE y p,p’-DDT, se adquirieron a Dr. Ehrenstorfer
(Augsburg, Alemania). El ácido H3PO4 y el Triton X-100 fueron de Fluka
(Buchs, Suiza). El NaH2PO4 (99% de pureza) fue suministrado por Carlo Erba
(Milán, Italia). El acetonitrilo y el n-hexano calidad HPLC, el ácido HCl (37%), el
hidróxido sódico y el Na2SO4 (99% de pureza) fueron obtenidos de Panreac
(Barcelona, España).
Se prepararon dos disoluciones patrón intermedias en ciclohexano
conteniendo, respectivamente, 2,5 mg L-1 de los PCBs y 0,1 mg L-1 de los
plaguicidas organoclorados; ambas se almacenaron a -20 ºC. A partir de estas
disoluciones se preparó una disolución conjunta de adición a 20 μg L-1 en
acetonitrilo. Así mismo se preparó una disolución 0,1 M de H3PO4 y otra 0,074
M de NaH2PO4, que se almacenaron en refrigeración a 4 ºC. A partir de ellas se
preparó una disolución tampón a pH=3.
Los contenedores (holders) de SPME y las fibras (polidimetilsiloxano o
PDMS de 100 µm de espesor de fase y polidimetilsiloxano/divinilbenceno o
PDMS-DVB de 65 µm), los viales de 5 mL para muestras y los tapones (septa)
de silicona laminada de PTFE, se obtuvieron de Supelco (Bellefonte, PA,
EE.UU.). Antes de su uso, los viales se hornearon a 300 ºC durante 10 h.
Se utilizó un baño termostatizado Lauda RE 104 (Lauda Dr. R. Wobser
GMBH & Co. KG, Lauda-Königshofen, Alemania) para mantener la temperatura
y un agitador Heidolph MR 303 (Heidolph Elektro GMBH & Co. KG, Kelheim,
Alemania) a 900 rpm. para mantener la homogeneidad de las muestras. En los
viales de 5 mL se introdujeron imanes agitadores de 10 mm recubiertos de
PTFE antes de comenzar el proceso de extracción. Su limpieza incluía lavado
con agua destilada y enjuague con n-hexano calidad HPLC.
88

Método de microextracción en fase sólida
2.2.2 Muestras de suero
El suero utilizado para el desarrollo y la validación del método se obtuvo
de voluntarios vinculados al Laboratorio de Salud Pública de Gipuzkoa,
utilizándose siempre bajo total anonimato. La sangre se extrajo por punción
venosa y se recogió en tubos Vacutainer. El suero se separó por
centrifugación. Se preparó una mezcla de suero a partir de 35 muestras
individuales a la que se denominó Suero de Gipuzkoa (GS). También se
reservaron 33 alícuotas individualizadas, que posteriormente fueron analizadas
por el método. Todas las muestras de suero se almacenaron a -20 ºC hasta su
análisis. Para no someterlo a excesivos ciclos de congelado y descongelado, el
GS se alicuotó en tubos de vidrio de 10 mL provistos de tapón de rosca y junta
de teflón.
2.2.3 Equipamiento instrumental
La cuantificación se realizó en un cromatógrafo de gases 6890N,
equipado con inyector con/sin división de flujo y detector de captura de
electrones (ECD), todo ello de Agilent Technologies (Wilmington, DW, EE.UU.).
En el portal de inyección se instaló un alineador (liner) de 0,75 mm de d.i.
(Supelco). Se trabajó en modo sin división de flujo, abriéndose la válvula de
división 4 min después de la inyección. La temperatura del portal de inyección
era de 270 ºC con Helio como gas portador a una presión de 20 psi y un flujo
de 5 mL min-1. La temperatura del detector era de 300 ºC con N2 como gas de
apoyo a 20 mL min-1. La separación cromatográfica se realizó con una columna
DB-XLB de 30m × 0,32 mm × 0,50 µm (J&W Scientific, Folsom, CA, EE.UU.)
cuya fase estacionaria es equivalente en polaridad a una de fenil(12%)-metil
polisiloxano [Rogers et al. 2004]. El horno estaba programado a 60 ºC (1 min),
seguido de 20 ºC min-1 hasta 180 ºC, 1,5 ºC min-1 hasta 240 ºC y 30 ºC min-1
hasta 300 ºC (8 min). Para la adquisición y procesado de datos se utilizó un
ordenador conectado al cromatógrafo, operado con el programa Chemstation
de Agilent.
Los diseños experimentales de matriz central compuesta y Plackett-
Burman se desarrollaron y evaluaron con el programa informático STATISTICA
(StatSoft, Tulsa, OK, EE.UU.).
89

Capítulo 2
2.2.4 Procedimiento analítico
En un vial de 5 mL se puso 1 mL de suero diluído (0,5 mL de suero:0,5
mL de agua Milli-Q) y se añadió 1 mL de tampón pH=3 y el imán agitador.
Seguidamente se procedió a la extracción de los analitos exponiendo la fibra
PDMS al espacio de cabeza de la muestra sometida a una velocidad de
agitación de 900 rpm. La extracción se realizó a 85 ºC y se prolongó durante 50
min. Tras la extracción la fibra se retrajo dentro de la aguja del holder y se llevó
inmediatamente al inyector del cromatógrafo. La desorción se llevó a cabo a
270 ºC durante 10 min. Con este tiempo de desorción no se observó
contaminación cruzada entre muestras.
Los plaguicidas organoclorados y los PCBs se cuantificaron por el
procedimiento de adición estándar. Tras la determinación de cada muestra se
preparó una adición estándar lo más cercana posible a los valores de la
muestra, añadiendo al vial 4, 7, 10, 15 ó 20 µL de un patrón conjunto de PCBs
y plaguicidas organoclorados de una concentración de 20 μg L-1 (p,p’-DDE 40
μg L-1) en acetonitrilo. La adición estándar se analizó en las mismas
condiciones que la muestra.
90

Método de microextracción en fase sólida
2.3 Resultados y discusión
En el desarrollo de un método adecuado para la determinación de
plaguicidas organoclorados y PCBs en suero por HS-SPME es necesario
considerar y optimizar varios parámetros que afectan al proceso de extracción.
Para encontrar las mejores condiciones experimentales se utilizó un diseño con
dos etapas: selección (screening) y optimización. La evaluación de los
resultados se llevó a cabo con los PCBs 46 y 143, que se suelen utilizar como
patrones internos (ISTDs) [Covaci and Schepens 2001]. En todas las pruebas
se añadían 2,5 ng de cada ISTD por mL de suero GS. También se ponía en el
vial 1 mL de la solución ácida o básica. La solución anterior podía contener o
no aditivos (sal o surfactante), en función de que así lo requiriera el diseño de
selección.
2.3.1 Diseño de selección
La selección es el primer paso en una eficiente estimación de los factores
que afectan al sistema analítico a estudiar. Si hay un gran número de factores
involucrados, se suelen aplicar diseños factoriales reducidos, como, por
ejemplo, el Plackett-Burman. Este tipo de diseños es muy útil ya que, con
pocos experimentos, es posible detectar los factores o variables más
significativas. Por otro lado, su principal limitación es que los efectos principales
se confunden con los términos de interacción. En el diseño Plackett-Burman se
asume que las interacciones son despreciables frente a los efectos principales.
A pesar de ello se puede utilizar para detectar los efectos o factores más
determinantes, que luego se pueden estudiar más a fondo con otro tipo de
diseños.
En base a la literatura [Pawliszyn 1997, Aguilar et al. 1999, Mills and
Walker 2000, Ulrich 2000, Beltrán et al. 2001, López et al. 2001, Valor et al.
2001, Zuba et al. 2002, Fidalgo-Used et al. 2003, Li et al. 2003, Shu et al. 2003,
Arambarri et al. 2004, Dong et al. 2005, Poli et al. 2005] y a la experiencia del
laboratorio se seleccionaron siete variables para definir el campo experimental
(una cualitativa o categórica y seis cuantitativas o continuas): tipo de fibra,
temperatura de extracción, tiempo de extracción, volumen de espacio de
cabeza, pH, concentración de sulfato de sodio anhidro y concentración de
91
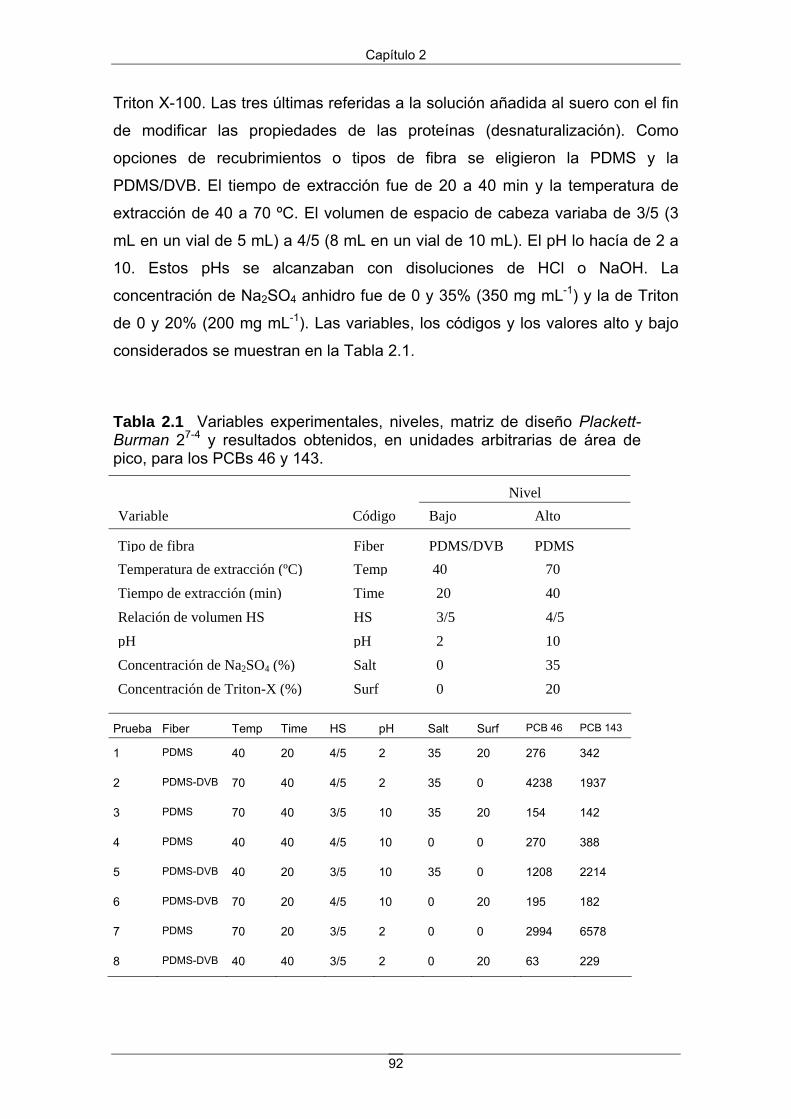
Capítulo 2
Triton X-100. Las tres últimas referidas a la solución añadida al suero con el fin
de modificar las propiedades de las proteínas (desnaturalización). Como
opciones de recubrimientos o tipos de fibra se eligieron la PDMS y la
PDMS/DVB. El tiempo de extracción fue de 20 a 40 min y la temperatura de
extracción de 40 a 70 ºC. El volumen de espacio de cabeza variaba de 3/5 (3
mL en un vial de 5 mL) a 4/5 (8 mL en un vial de 10 mL). El pH lo hacía de 2 a
10. Estos pHs se alcanzaban con disoluciones de HCl o NaOH. La
concentración de Na2SO4 anhidro fue de 0 y 35% (350 mg mL-1) y la de Triton
de 0 y 20% (200 mg mL-1). Las variables, los códigos y los valores alto y bajo
considerados se muestran en la Tabla 2.1.
Tabla 2.1 Variables experimentales, niveles, matriz de diseño Plackett-Burman 27-4 y resultados obtenidos, en unidades arbitrarias de área de pico, para los PCBs 46 y 143.
Nivel Variable Código Bajo Alto
Tipo de fibra Fiber PDMS/DVB PDMS Temperatura de extracción (ºC) Temp 40 70 Tiempo de extracción (min) Time 20 40 Relación de volumen HS HS 3/5 4/5 pH pH 2 10 Concentración de Na2SO4 (%) Salt 0 35 Concentración de Triton-X (%) Surf 0 20
Prueba Fiber Temp Time HS pH Salt Surf PCB 46 PCB 143
1 PDMS 40 20 4/5 2 35 20 276 342
2 PDMS-DVB 70 40 4/5 2 35 0 4238 1937
3 PDMS 70 40 3/5 10 35 20 154 142
4 PDMS 40 40 4/5 10 0 0 270 388
5 PDMS-DVB 40 20 3/5 10 35 0 1208 2214
6 PDMS-DVB 70 20 4/5 10 0 20 195 182
7 PDMS 70 20 3/5 2 0 0 2994 6578
8 PDMS-DVB 40 40 3/5 2 0 20 63 229
92
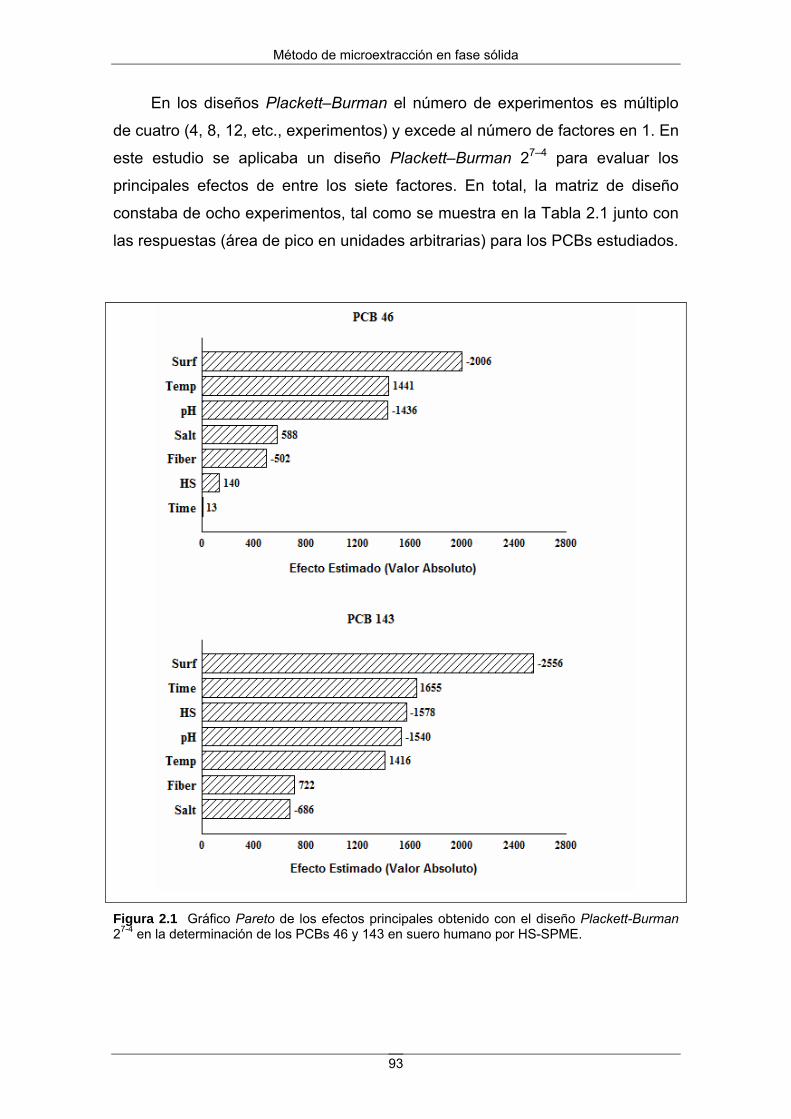
Método de microextracción en fase sólida
En los diseños Plackett–Burman el número de experimentos es múltiplo
de cuatro (4, 8, 12, etc., experimentos) y excede al número de factores en 1. En
este estudio se aplicaba un diseño Plackett–Burman 27–4 para evaluar los
principales efectos de entre los siete factores. En total, la matriz de diseño
constaba de ocho experimentos, tal como se muestra en la Tabla 2.1 junto con
las respuestas (área de pico en unidades arbitrarias) para los PCBs estudiados.
Figura 2.1 Gráfico Pareto de los efectos principales obtenido con el diseño Plackett-Burman 27-4 en la determinación de los PCBs 46 y 143 en suero humano por HS-SPME.
93

Capítulo 2
Los datos obtenidos se evaluaron mediante una prueba ANOVA y los
efectos principales se visualizaron en un gráfico Pareto (Figura 2.1). En el
gráfico, la longitud de las barras es proporcional a los efectos proporcionales
estimados. El signo de los efectos muestra si la respuesta aumentaba o no al
pasar del nivel bajo al nivel alto del efecto. La concentración del Triton fue la
variable más importante con un efecto negativo para la concentración más alta,
por lo que se eliminó para el siguiente paso de optimización. Como el pH
mostraba un efecto negativo a valores básicos, se eligió continuar trabajando a
un pH de 3. Ambos analitos mostraban un efecto positivo con el tiempo y la
temperatura, por lo que ambos factores se tuvieron en cuenta en el siguiente
paso de optimización. Se encontraron tres factores con resultados opuestos.
Para el PCB 46, la fibra PDMS/DVB, el aumento de la concentración de sal y el
incremento del volumen del espacio de cabeza eran condiciones favorables. En
cambio, para el PCB 143 la fibra PDMS, la ausencia de sal y volúmenes de
espacio de cabeza bajos eran condiciones experimentales más favorables.
Como existían antecedentes de uso de la fibra PDMS para trabajar con PCBs
en agua [Shu et al. 2003], plaguicidas organoclorados en pescado [Fidalgo-
Used et al. 2003] y plaguicidas organoclorados en tejidos biológicos [López et
al. 2001] se eligió ésta para el siguiente paso de optimización, al igual que las
otras condiciones favorables para el PCB 143 (menos volátil que el PCB 46), ya
que era de esperar que los PCBs más relevantes en muestras fueran el 138, el
153 y el 180, igual o menos volátiles que el 143. En resumen, para la siguiente
etapa se eliminaron la sal y el surfactante, se fijaron tres variables: fibra PDMS,
relación de volumen de espacio de cabeza 3/5 y pH=3 y se consideraron otra
dos para su optimización en la siguiente etapa: el tiempo y la temperatura de
extracción.
2.3.2 Diseño de optimización
Esta segunda etapa tenía por objeto la optimización de las variables
significativas elegidas mediante un diseño central compuesto (CCD). Las dos
variables y sus valores bajo, central y alto fueron los siguientes: temperatura de
extracción (temperatura, 35-60-85 ºC), tiempo de extracción (tiempo, 15-30-45
min). Estos valores están incluidos en la Tabla 2.2.
94

Método de microextracción en fase sólida
Tabla 2.2 Variables experimentales, niveles, matriz de diseño central compuesto (CCD) y resultados obtenidos, en unidades arbitrarias de área de pico, para los PCBs 46 y 143.
Nivel Variable Código Bajo Medio Alto Temperatura de extracción (ºC) Temp 35 60 85 Tiempo de extracción (min) Time 15 30 45
Prueba Bloque Temp Time PCB 46 PCB 143
1 1 35 15 889 453
2 1 35 45 2686 2459
3 1 85 15 5332 2725
4 1 85 45 5549 3743
5 (C)a 1 60 30 1464 1119
6 (C)a 1 60 30 1794 814
7 (C)a 1 60 30 1444 835
8 2 35 30 544 462
9 2 85 30 4618 3063
10 2 60 15 1767 826
11 2 60 45 3066 1014
12 (C)a 2 60 30 1793 848
13 (C)a 2 60 30 2475 1338
14 (C)a 2 60 30 1744 805 a (C), punto central
El CCD consta de los puntos de un diseño factorial (2N) más los puntos de
un diseño de estrella (2N+1), siendo N el número de variables. En nuestro caso
22 + (2×2 + 1), es decir 8 puntos, estando los de la estrella centrados en las
caras (α±1). Se crearon dos bloques ya que se necesitaban dos días para
llevar a cabo todas las pruebas. Además, se incorporaron dos repeticiones
adicionales de las pruebas en el punto central. Por lo tanto, la matriz de diseño
CCD constaba de 14 experimentos en total, a realizar en orden aleatorio. En la
95

Capítulo 2
Tabla 2.2 se pueden ver también los resultados de los experimentos para los
dos PCBs, expresados en unidades arbitrarias de área de pico.
Para el CCD se utilizó un modelo polinómico de segundo grado que
incluía los efectos principales de los dos factores, su interacción y sus
componentes cuadráticas. Con este modelo de segundo orden la prueba
ANOVA mostraba una falta de ajuste no significativa para ambos PCBs, así
como buenos valores de R2 (0,9732 para el PCB 46 y 0,9166 para el PCB 143)
y de R2 ajustado (0,9502 para el PCB 46 y 0,8452 para el PCB 143). R2 es la
proporción de la varianza justificada por el modelo en las medidas de la
variable dependiente. El R2 ajustado se obtiene al aplicar a R2 un coeficiente de
ajuste en función del número de términos del modelo. Ambos datos se
consideran una indicación de la adecuación del modelo.
Tras comprobar la adecuación del modelo, el siguiente paso fue utilizar
los coeficientes de regresión para encontrar las condiciones de los factores o
variables independientes (tiempo y temperatura de extracción) que
maximizaban la respuesta de la variable dependiente (áreas de pico de los
PCBs 46 y 143) ya que los coeficientes de regresión obtenidos con este
modelo polinómico de segundo grado permitían predecir los valores de la
variable dependiente en función de las combinaciones de valores de las
variables independientes. Lo más habitual es realizar una primera aproximación
visual por medio de un gráfico 3D denominado superficie de respuesta. En este
estudio se podía obtener la superficie de respuesta de cada uno de los dos
PCBs utilizados, pero en lugar de dos superficies independientes preferimos
trabajar con una global, mediante la fusión de las respuestas obtenidas para
ambos PCBs en una única variable dependiente. Para ello se utilizó una
aplicación del programa STATISTICA que permitía transformar la respuesta
cromatográfica en una nueva variable denominada deseabilidad. Esta nueva
variable se creó asignando para cada PCB el valor 0 a la respuesta menos
deseable (en nuestro caso la más baja) y el valor 1 a la más deseable (en
nuestro caso la más alta). A partir de aquí el programa utilizaba un algoritmo
lineal para asignar valores entre 0 y 1 al resto de las respuestas. Seguidamente
se fusionaban las deseabilidades individuales en una variable conjunta,
denominada deseabilidad global, mediante el cálculo de la media geométrica.
96

Método de microextracción en fase sólida
Tras efectuar el cambio de variable se obtuvo el gráfico 3D de la
superficie de respuesta para la deseabilidad global en función del tiempo y la
temperatura de extracción (Figura 2.2).
Figura 2.2 Superficie de respuesta para la deseabilidad global, obtenida a partir del CCD, en función del tiempo y la temperatura de extracción empleados en la determinación de los PCBs 46 y 143 en suero humano mediante HS-SPME.
Dicho gráfico es útil para interpretar visualmente el efecto de las variables
independientes sobre la respuesta conjunta de deseabilidad. Como se puede
ver en la Figura 2.2, en el rango de tiempos y temperaturas de extracción
investigados en este estudio las mejores respuestas se obtenían a valores altos
de ambas variables. Sin embargo, por razones prácticas, el techo de dichas
variables estaba limitado: la temperatura porque se trabajaba con baño de
97

Capítulo 2
agua y además se trataba de evitar que la muestra pudiera ebullir y alcanzar la
fibra, que estaba posicionada cerca de la superficie líquida, ocasionándole un
daño irreparable, y el tiempo de extracción porque prolongarlo hasta intentar
alcanzar el equilibrio podría llevar horas, con lo que disminuiría drásticamente
la productividad del método. Por todo ello se optó por trabajar a 85 ºC y 50 min
de exposición ya que estos valores eran suficientes para extraer cantidades
detectables de los analitos y además el tiempo era similar a la duración del
cromatograma, lo que permitía simultanear ambas etapas. En algún otro
trabajo se optaba por 90 ºC y 30 min para plaguicidas organoclorados en suero
por HS-SPME con fibra PDMS [Beltrán et al 2001]. Sin embargo, era de
esperar que compuestos de mayor peso molecular, como los PCBs de alto
grado de cloración, requirieran mayor tiempo de extracción debido a sus
menores coeficientes de difusión. Además, el aumento en el grado de cloración
da lugar a una disminución en el coeficiente de partición, tal como se
demuestra en un estudio sobre eficiencias de extracción SPME (en modo
inmersión) en la determinación de plaguicidas y PCBs en agua [Valor et al.
2001], donde se proponía un tiempo de extracción de 150 min, trabajando a
temperatura ambiente.
Finalmente, a tenor de los resultados y las consideraciones anteriores, se
decidió que las mejores condiciones de trabajo eran: no utilización de sal ni
surfactante, fibra PDMS, relación de volumen de espacio de cabeza 3/5 (2 mL
de líquido en un vial de 5 mL), temperatura de extracción 85 ºC, tiempo de
extracción 50 min y pH ácido (adición de 1 mL de solución tampón pH=3 a 1
mL de suero diluido).
2.3.3 Evaluación del método
Tras establecer las condiciones de extracción, se evaluó la linealidad,
límites de detección (LD) y precisión del método HS-SPME/GC-ECD. A tal
propósito se utilizó una disolución en acetonitrilo de los distintos analitos a
estudiar, que contenía HCB, β-HCH, heptacloro epóxido, p,p’-DDE, p,p’-DDT y
PCBs 28, 52, 101, 118, 138, 153 y 180 a una concentración de 13,2 ng g-1. Se
prepararon cinco niveles diferentes añadiendo 10, 16, 24, 32 y 40 µL de la
disolución anterior a un vial de 5 mL con un blanco consistente en 2 mL
98
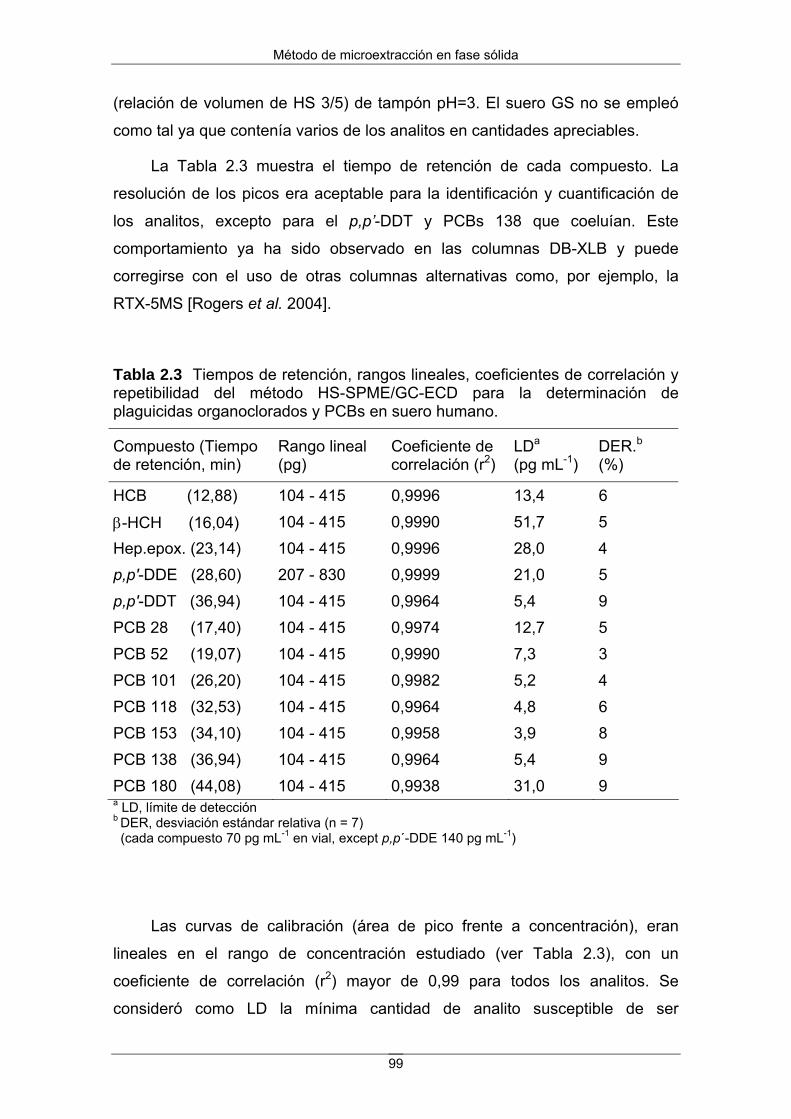
Método de microextracción en fase sólida
(relación de volumen de HS 3/5) de tampón pH=3. El suero GS no se empleó
como tal ya que contenía varios de los analitos en cantidades apreciables.
La Tabla 2.3 muestra el tiempo de retención de cada compuesto. La
resolución de los picos era aceptable para la identificación y cuantificación de
los analitos, excepto para el p,p’-DDT y PCBs 138 que coeluían. Este
comportamiento ya ha sido observado en las columnas DB-XLB y puede
corregirse con el uso de otras columnas alternativas como, por ejemplo, la
RTX-5MS [Rogers et al. 2004].
Tabla 2.3 Tiempos de retención, rangos lineales, coeficientes de correlación y repetibilidad del método HS-SPME/GC-ECD para la determinación de plaguicidas organoclorados y PCBs en suero humano.
Compuesto (Tiempo de retención, min)
Rango lineal (pg)
Coeficiente de correlación (r2)
LDa
(pg mL-1) DER.b(%)
HCB (12,88) 104 - 415 0,9996 13,4 6
β-HCH (16,04) 104 - 415 0,9990 51,7 5 Hep.epox. (23,14) 104 - 415 0,9996 28,0 4 p,p'-DDE (28,60) 207 - 830 0,9999 21,0 5 p,p'-DDT (36,94) 104 - 415 0,9964 5,4 9 PCB 28 (17,40) 104 - 415 0,9974 12,7 5 PCB 52 (19,07) 104 - 415 0,9990 7,3 3 PCB 101 (26,20) 104 - 415 0,9982 5,2 4 PCB 118 (32,53) 104 - 415 0,9964 4,8 6 PCB 153 (34,10) 104 - 415 0,9958 3,9 8 PCB 138 (36,94) 104 - 415 0,9964 5,4 9 PCB 180 (44,08) 104 - 415 0,9938 31,0 9 a LD, límite de detección b DER, desviación estándar relativa (n = 7) (cada compuesto 70 pg mL-1 en vial, except p,p´-DDE 140 pg mL-1)
Las curvas de calibración (área de pico frente a concentración), eran
lineales en el rango de concentración estudiado (ver Tabla 2.3), con un
coeficiente de correlación (r2) mayor de 0,99 para todos los analitos. Se
consideró como LD la mínima cantidad de analito susceptible de ser
99

Capítulo 2
diferenciada del ruido de fondo. En algunos trabajos se calcula aplicando un
factor de 3 a la relación señal/ruido [Najam et al. 1999, López et al. 2001,
Conka et al. 2005]. En otros, se obtiene a partir del gráfico de calibración
[Arambarri et al. 2004]. En este caso se utilizó esta última opción. El LD de
cada compuesto se calculó tomando la ordenada en el origen de las
respectivas curvas de calibración (yB) como señal atribuible al blanco y se
estimó su desviación estándar como Sy/x (en lugar de usar la desviación
estándar del blanco, SB) [Miller and Miller 2005]. Los LD, tal como se puede ver
en la Tabla 2.3, iban de 3,9 a 51,7 pg mL-1. Estos valores eran similares a los
encontrados en otros trabajos donde se determinaban estos compuestos en
agua por SPME/GC-ECD utilizando fibras PDMS comerciales [Shu et al. 2003]
y mayores que los de otros trabajos (donde eran del orden de magnitud de
unos pocos pg mL-1) en los que se trabajaba en la determinación de
plaguicidas organoclorados en las condiciones anteriores pero con fibras más
selectivas [Li et al. 2003, Dong et al. 2005].
La precisión, en condiciones de repetibilidad, se estimó analizando 7
veces 0,5 mL de muestra adicionada con 140 pg (70 pg mL-1 en el vial, p,p-
DDE 140 pg mL-1). La repetibilidad para cada compuesto, medida como
porcentaje de desviación estándar relativa (DER%), iba del 3 al 9%. Estos
valores eran similares a los encontrados en otros trabajos sobre
determinaciones de plaguicidas organoclorados en agua por HS-SPME [Li et al.
2003, Dong et al. 2005] o en suero por SPME en inmersión [López et al. 2001].
Finalmente este método HS-SPME se comparó con el de placas descrito
en el capítulo 1, que ya estaba validado, procesando el suero GS por ambas
técnicas. Los resultados mostraban una buena concordancia entre
procedimientos para HCB, p,p’-DDE, y PCBs 118, 138, 153 y 180. Los
porcentajes de concordancia con respecto al método de placas iban del 86 al
105%, con una excepción, el β-HCH, en que era menor del 40%. Por ello los
valores obtenidos para dicho compuesto por el método aquí descrito, deben
tomarse con precaución.
100

Método de microextracción en fase sólida
2.3.4 Aplicación a muestras de suero
Se analizaron 33 muestras de suero por este método HS-SPME/GC-ECD.
Para cuantificar se empleó la técnica de la adición estándar (según se ha
descrito en el apartado 2.2.4) con objeto de compensar, en la medida de lo
posible, la influencia de la matriz en el rendimiento de la extracción. Esta
técnica ya se ha usado en la cuantificación de compuestos volátiles en agua
[Arambarri et al. 2004] y de plaguicidas organoclorados en pescado [Fidalgo-
Used et al. 2003].
La Figura 2.3 muestra los cromatogramas obtenidos para una muestra de
suero humano antes y después de la adición del patrón conjunto conteniendo
los plaguicidas organoclorados y los PCBs. En todas las muestras analizadas
se detectaron: HCB, p,p’-DDE, y PCBs 118, 138, 153 y 180, siendo mayoritario
el p,p’-DDE y, entre los PCBs, el 153.
101

Capítulo 2
Figura 2.3 Cromatogramas obtenidos por HS-SPME/GC-ECD de: (a) muestra de suero humano y (b) muestra de suero humano adicionada con plaguicidas organoclorados y PCBs (0,3 ng mL-1; p,p’-DDE 0,6 ng mL-1). Asignación de picos: (1) HCB, (2) β-HCH, (3) PCB 28, (4) PCB 46 (ISTD), (5) PCB 52, (6) heptacloro epóxido, (7) p,p’-DDE, (8) PCB 143 (ISTD), (9) PCB 118, (10) PCB 153, (11) PCB 138, (12) PCB 180.
Las 33 muestras se analizaron también por el método de placas de discos
SPE descrito en el capítulo 1 (usado como referencia) y se compararon los
resultados mediante regresión lineal. El análisis de correlación de Pearson
mostraba que los dos conjuntos de datos estaban alta y significativamente
correlacionados con coeficientes (r) que iban desde 0,8875 (PCB 138) hasta
0,9953 (HCB). Las pendientes de la recta de regresión estaban entre 0,773
102
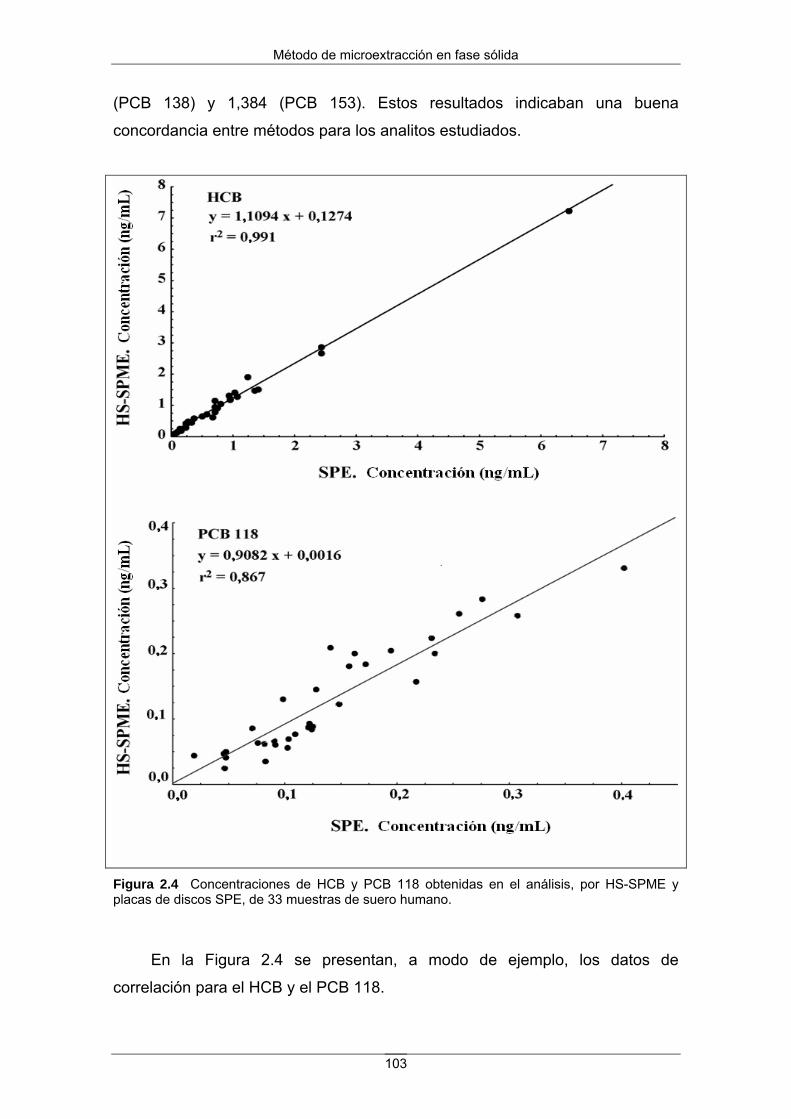
Método de microextracción en fase sólida
(PCB 138) y 1,384 (PCB 153). Estos resultados indicaban una buena
concordancia entre métodos para los analitos estudiados.
Figura 2.4 Concentraciones de HCB y PCB 118 obtenidas en el análisis, por HS-SPME y placas de discos SPE, de 33 muestras de suero humano.
En la Figura 2.4 se presentan, a modo de ejemplo, los datos de
correlación para el HCB y el PCB 118.
103

Capítulo 2
Desde el punto de vista de la productividad, el método, tal cual se llevó a
cabo (extracción y desorción manual, cuantificación por adición estándar), sólo
permitía analizar unas 5 muestras por día, con lo que no se podía calificar
como de alta productividad. Aun así, consideramos la técnica HS-SPME muy
prometedora e interesante de cara a su uso en estudios epidemiológicos por
varias razones:
• Desde la perspectiva instrumental es sencilla de realizar y no precisa
gran equipamiento, ni que este sea caro, sofisticado o de difícil manejo.
• Por la razón anterior y por no requerir prácticamente el empleo de
disolventes o fungibles desechables, es una técnica muy económica y
muy “limpia” desde el punto de vista medioambiental y de condiciones
laborales.
• En este momento existen dispositivos que permiten automatizar los
procesos de extracción y desorción para que puedan realizarse de forma
continua y desatendida [Zwir-Ferenc and Biziuk 2004, Vuckovic et al.
2010]. El empleo de estos dispositivos redundaría en un enorme
aumento de la productividad sin un incremento excesivo de los costes o
la dificultad de manejo.
• El uso de detectores de espectrometría de masas de alta sensibilidad
permitiría utilizar la dilución isotópica como técnica de cuantificación
[Sjödin et al. 2004], en lugar de la adición estándar. Aunque
incrementaría sustancialmente los costes (sobre todo de inversión y
mantenimiento del equipamiento) y la complejidad de la etapa
instrumental, supondría una notable mejora de la productividad ya que
se evitaría la multiplicidad de inyecciones sobre una misma muestra.
104

Método de microextracción en fase sólida
2.4 Conclusiones
En este capítulo se ha desarrollado un método para la determinación
simultánea de plaguicidas organoclorados y PCBs en suero humano, basado
en HS-SPME. También se ha visto la utilidad del diseño experimental en la
optimización de las condiciones de extracción del método. El único instrumental
requerido fue la fibra PDMS y su holder para la extracción y el equipo GC-ECD
para la medida final. El método presentaba notables ventajas con respecto a
los basados en otras técnicas, como: eliminación de la etapa de limpieza de
extractos y evitación del uso de disolventes. Un tiempo de extracción de 50 min
era suficiente para detectar y cuantificar de forma satisfactoria los plaguicidas
organoclorados y los PCBs más relevantes. Dada la complejidad del suero
como matriz, se vio conveniente el empleo de la adición estándar para la
cuantificación. Sin embargo, al realizarse esta operación de forma manual, el
proceso se dilataba en exceso en el tiempo con lo que la productividad no
parecía la adecuada para su empleo en estudios epidemiológicos aunque,
considerando los resultados, el método sí se demostró útil para el análisis de
un número limitado de muestras de suero. No obstante, la productividad se
podría mejorar de forma muy notable con el empleo de dispositivos que
permiten automatizar los procesos de extracción y desorción para que puedan
realizarse de forma continua y desatendida, y el uso de detectores de
espectrometría de masas que posibilitan la sustitución de la adición estándar
por la dilución isotópica, evitando así la multiplicidad de inyecciones sobre una
misma muestra. Por todo ello consideramos que tiene un gran potencial de
cara a su uso en estudios epidemiológicos. Además, al emplearse sorbentes y
condiciones de extracción no específicos, este método podría ser ampliado a
otros compuestos contaminantes.
105

Capítulo 2
2.5 Referencias
♦ Aguilar C, Peñalver A, Pocurull E, Ferré J, Borrull F, Marce RM, 1999. Optimization of solid-phase microextraction conditions using a response surface methodology to determine organochlorine pesticides in water by gas chromatography and electron-capture detection. J. Chromatogr. A 844, 425-432.
♦ Arambarri I, Lasa M, García R, Millán E, 2004. Determination of fuel dialkyl ethers and BTEX in water using headspace solid-phase microextraction and gas chromatography-flame ionization detection. J. Chromatogr. A 1033, 193-203.
♦ Barr JR, Maggio VL, Barr DB, Turner WE, Sjödin A, Sandau CD, Pirkle JL, Needham LL, Patterson DG Jr., 2003. New high-resolution mass spectrometric approach for the measurement of polychlorinated biphenyls and organochlorine pesticides in human serum. J. Chromatogr. B 794, 137-148.
♦ Beltrán J, Pitarch E, Egea S, López FJ, Hernández F, 2001. Gas chromatographic determination of selected pesticides in human serum by head-space solid-phase microextraction. Chromatographia 54, 757-763.
♦ Bezerra MA, Santelli RE, Oliveira EP, Villar LS, Escaleira LA, 2008. Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta 76, 965-977.
♦ Brereton RG, 2003. Chemometrics: Data Analysis for the Laboratory and Chemical Plant. John Wiley & Sons Ltd., Chichester, West Sussex, UK.
♦ Conka K, Drobná B, Kocan A, Petrík J, 2005. Simple solid-phase extraction method for determination of polychlorinated biphenyls and selected organochlorine pesticides in human serum. J. Chromatogr. A 1084, 33-38.
♦ Covaci A, Schepens P, 2001. Simplified method for determination of organochlorine pollutants in human serum by solid-phase disk extraction and gas chromatography. Chemosphere 43, 439-447.
♦ Dong CZ, Zeng ZR, Yang M, 2005. Determination of organochlorine pesticides and their derivations in water after HS-SPME using polymethylphenylvinylsiloxane-coated fiber by GG-ECD. Water Res. 39, 4204-4210.
♦ Fidalgo-Used N, Centineo G, Blanco-González E, Sanz-Medel A, 2003. Solid-phase microextraction as a clean-up and preconcentration procedure for organochlorine pesticides determination in fish tissue by gas chromatography with electron capture detection. J. Chromatogr. A 1017, 35-44.
♦ Focant JF, Sjödin A, Turner WE, Patterson DG Jr., 2004. Measurement of selected polybrominated diphenyl ethers, polybrominated and polychlorinated biphenyls, and organochlorine pesticides in human serum and milk using comprehensive two-dimensional gas chromatography isotope dilution time-of-flight mass spectrometry. Anal. Chem. 76, 6313-6320.
♦ Li HP, Li GC, Jen JF, 2003. Determination of organochlorine pesticides in water using microwave assisted headspace solid-phase microextraction and gas chromatography. J. Chromatogr. A 1012, 129-137.
♦ López FJ, Pitarch E, Egea S, Beltrán J, Hernández F, 2001. Gas chromatographic determination of organochlorine and organophosphorus pesticides in human fluids using solid phase microextraction. Anal. Chim. Acta 433, 217-226.
♦ Miller JN, Miller JC, 2005. Statistics and Chemometrics for Analytical Chemistry, 5th Ed. Pearson Education Ltd, Harlow, Essex, UK.
♦ Mills GA, Walker V, 2000. Headspace solid-phase microextraction procedures for gas chromatographic analysis of biological fluids and materials. J. Chromatogr. A 902, 267-287.
♦ Najam AR, Korver MP, Williams CC, Burse VW, Needham LL, 1999. Analysis of a mixture of polychlorinated biphenyls and chlorinated pesticides in human serum by column fractionation
106

Método de microextracción en fase sólida
and dual-column capillary gas chromatography with electron capture detection. J AOAC Int 82, 177-185.
♦ Pawliszyn J, 1997. Solid-Phase Microextraction. Theory and Practice. Wiley-VCH Inc., New York, USA.
♦ Pitarch E, Serrano R, López FJ, Hernández F, 2003. Rapid multiresidue determination of organochlorine and organophosphorus compounds in human serum by solid-phase extraction and gas chromatography coupled to tandem mass spectrometry. Anal. Bioanal. Chem. 376, 189-197.
♦ Poli D, Manini P, Andreoli R, Franchini I, Mutti A, 2005. Determination of dichloromethane, trichloroethylene and perchloroethylene in urine samples by headspace solid phase microextraction gas chromatography-mass spectrometry. J. Chromatogr. B 820, 95-102.
♦ Rantakokko P, Kiviranta H, Rylander L, Rignell-Hydbom A, Vartiainen T, 2009. A simple and fast liquid-liquid extraction method for the determination of 2,2´,4,4´,5,5´-hexachlorobiphenyl (CB-153) and 1,1-dichloro-2,2-bis(p-chlorophenyl)-ethylene (p,p´-DDE) from human serum for epidemiological studies on type 2 diabetes. J. Chromatogr. A 1216, 897-901.
♦ Rogers E, Petreas M, Park J, Zhao G, Charles M, 2004. Evaluation of four capillary columns for the analysis of organochlorine pesticides (OCPs), polychlorinated biphenyls (PCBs), and polybrominated diphenyl ethers (BDEs) in human serum for epidemiologic studies. J. Chromatogr. B 813, 269-285.
♦ Shu YY, Wang SS, Tardif M, Huang YP, 2003. Analysis of polychlorinated biphenyls in aqueous samples by microwave-assisted headspace solid-phase microextraction. J. Chromatogr. A 1008, 1-12.
♦ Sjödin A, Jones RS, Lapeza CR, Focant JF, McGahee EE 3rd, Patterson DG Jr., 2004. Semiautomated high-throughput extraction and cleanup method for the measurement of polybrominated diphenyl ethers, polybrominated biphenyls, and polychlorinated biphenyls in human serum. Anal. Chem. 76, 1921-1927.
♦ Stalikas C, Fiamegos Y, Sakkas V, Albanis T, 2009. Developments on chemometric approaches to optimize and evaluate microextraction. J. Chromatogr. A 1216, 175-189.
♦ Ulrich S, 2000. Solid-phase microextraction in biomedical analysis J. Chromatogr. A 902, 167-194.
♦ Valor I, Pérez M, Cortada C, Apraiz D, Moltó JC, Font G, 2001. SPME of 52 pesticides and polychlorinated biphenyls: Extraction efficiencies of the SPME coatings poly(dimethylsiloxane), polyacrylate, poly(dimethylsiloxane)-divinylbenzene, carboxen-poly(dimethylsiloxane), and carbowax-divinylbenzene. J. Sep. Sci. 24, 39-48.
♦ Vuckovic D, Cudjoe E, Musteata FM, Pawliszyn J, 2010. Automated solid-phase microextraction and thin-film microextraction for high-throughput analysis of biological fluids and ligand–receptor binding studies. Nat. Protoc. 5, 140-161.
♦ Zuba D, Parczewski A, Reichenbächer M, 2002. Optimization of solid-phase microextraction conditions for gas chromatographic determination of ethanol and other volatile compounds in blood. J. Chromatogr. B 773, 75-82.
♦ Zwir-Ferenc A, Biziuk M, 2004. An analysis of pesticides and polychlorinated biphenyls in biological samples and foods. Crit. Rev. Anal. Chem. 34, 95-103.
107


CAPÍTULO 3
Metodología analítica basada en limpieza de extractos por cromatografía de permeabilidad en gel
3.1 Introducción
3.2 Parte experimental
3.3 Resultados y discusión
3.4 Conclusiones
3.5 Referencias

Método basado en limpieza por GPC
110

Capítulo 3
3.1 Introducción
Es muy habitual que en estudios epidemiológicos la exposición a
plaguicidas organoclorados y PCBs se estime a partir de sus niveles en tejidos
o fluidos biológicos, siendo el suero uno de los materiales más empleados
[Koppen et al. 2002, Cruz et al. 2003, Axmon et al. 2004, Bates et al. 2004]. En
el Capítulo 1 se discutían las ventajas y los inconvenientes de diferentes
procedimientos para la medida sérica de plaguicidas organoclorados y PCBs
en este tipo de estudios, donde se suele disponer de poca cantidad de muestra
(≤ 1 mL) [Laden et al. 1997, Lindh et al. 2008] y se requiere el análisis de un
elevado número de ellas (alta productividad). La mayoría de los métodos
utilizados en este campo constan de una etapa de extracción, una etapa de
limpieza de extractos y una etapa final de cuantificación [Zwir-Ferenc and
Biziuk 2004]. Tal como se vio en el Capítulo 1, actualmente existen técnicas de
extracción en fase sólida que permiten procesar muestras de pequeño tamaño
(≤ 0,5 mL) con una aceptable capacidad de producción. Por otra parte, la
cromatografía de gases con detector de captura de electrones o de
espectrometría de masas es, por el momento, la opción indiscutible para la
etapa de cuantificación [Reiner et al. 2010], pudiéndose automatizar gracias a
la incorporación de los populares sistemas robotizados de inyección, también
conocidos como automuestreadores o inyectores automáticos. Sin embargo, la
mayoría de técnicas usadas para la limpieza de extractos, tales como la
partición líquido-líquido, el tratamiento con ácido sulfúrico concentrado, las
columnas de adsorción de sílice y/o alúmina, etc., requieren mucha mano de
obra y son difíciles de automatizar, convirtiendo a esta etapa en la más
laboriosa de todo el proceso.
El uso de la cromatografía de permeabilidad en gel (GPC o cromatografía
de exclusión molecular) para la limpieza de extractos, fue introducido por
primera vez por Stalling y sus colaboradores en 1972 [Stalling et al. 1972] y
actualmente existen métodos estandarizados desarrollados para este
propósito, siendo su principal campo de aplicación la determinación de residuos
de contaminantes orgánicos en muestras ambientales o de alimentos [EPA
1994, Schenzler and Their 2001, AOAC 2007]. La GPC es una técnica no
destructiva, que separa en base a la diferencia de tamaño molecular, cuya
111

Método basado en limpieza por GPC
principal ventaja es que permite que la grasa y otros compuestos de relativo
alto peso molecular (pigmentos vegetales, restos de proteínas, etc.) puedan
diferenciarse y aislarse de los analitos de interés [Patterson 1991, Van Rhijn
and Tuinstra 1991, EPA 1994, Pauwels and Schepens 1998, Woodman and
Stone 2003]. A lo que hay que añadir que estos coextractivos eliminados, lejos
de quedar retenidos permanentemente, son los primeros compuestos en ser
eluidos y, por consiguiente, la vida útil de la columna es relativamente larga
[Pauwels et al. 1999]. Además, dado que se trata de una técnica instrumental,
puede automatizarse simplemente incorporando un automuestreador y un
colector de fracciones [Patterson 1991, Pauwels et al. 1999, Woodman and
Stone 2003]. En general, todos estos métodos de limpieza basados en GPC
deben satisfacer unos requisitos de separación entre compuestos de una
mezcla sintética representativa de los principales componentes que se pueden
encontrar en extractos de muestras ambientales: grasa, plastificante, plaguicida
organoclorado, hidrocarburo aromático policíclico y sulfuro [EPA 1994].
Su mayor inconveniente es que casi todos ellos están diseñados para
trabajar con muestras de tamaño apreciable (≥ 10 g), alto consumo de
disolvente (flujos de 5 mL min-1) y tiempos largos (hasta 60 min por muestra),
ya que utilizan columnas de baja resolución, de más de 1 cm de diámetro. Sin
embargo, algunos estudios [Patterson 1991, Van Rhijn and Tuinstra 1991
Woodman and Stone 2003] han sugerido la posibilidad de miniaturizar el
sistema usando columnas de menor diámetro y mayor resolución. Esto permite
procesar muestras de menor tamaño con un ahorro sustancial de disolvente
(flujos de 0,5 a 1 mL min-1) y tiempo (< 30 min por muestra). Obviamente, la
cantidad de coextractivos, particularmente grasa, que se puede eliminar bajo
estas condiciones es menor que con los sistemas GPC tradicionales, pero con
estos últimos se llega a volúmenes finales de extracto relativamente grandes (≥
1 mL) de los cuales sólo se inyecta en el cromatógrafo una mínima parte (0,5-2
µL), desechándose el resto.
En relación a otras técnicas se encontraron pocos trabajos sobre la
aplicación de la GPC al análisis de compuestos organoclorados en matrices
humanas [Janák et al. 1994, Pauwels and Schepens 1998, Pauwels et al. 1999,
112

Capítulo 3
Hovander et al. 2000, Smeds and Saukko 2001, Furberg et al. 2002] y la
mayoría se centraban en tejido adiposo.
En este capítulo se presenta el trabajo realizado en el desarrollo de un
método basado en la limpieza de extractos por GPC compaginando las
ventajas de la miniaturización con la automatización de la etapa. El método
está pensado para la determinación de residuos de plaguicidas organoclorados
y PCBs en muestras de suero humano. Usando menos de 1 mL de suero la
cantidad esperada de grasa es menor de 40 mg y, por lo tanto, dentro de los
márgenes que un sistema de este tipo es capaz de eliminar [Woodman and
Stone 2003]. A pesar del reducido tamaño de la muestra, si el extracto se lleva
a un volumen final ≤100 µL el factor de concentración es suficientemente
elevado como para poderse determinar los plaguicidas organoclorados y PCBs
mayoritarios. Este procedimiento de limpieza de extractos se optimizó con
respecto a las variables de mayor interés: volumen de inyección, flujo de
elución y cantidad de grasa inyectada y proporciona sustanciales ahorros de
disolvente y tiempo de procesado en comparación con otras limpiezas por
GPC. Los compuestos estudiados fueron: PCBs 28, 52, 101, 118, 138, 153 y
180, HCB, β-HCH, γ-HCH, heptacloro epóxido, p,p’-DDE y p,p’-DDT.
113

Método basado en limpieza por GPC
3.2 Parte experimental
3.2.1 Reactivos y materiales
La disolución patrón conjunta de PCBs 28, 52, 101, 118, 138, 153 y 180,
cada uno a 10 μg mL-1 en isooctano, así como las disoluciones patrón de PCB
46 y PCB 143 a 100 μg mL-1 en hexano y el material de referencia certificado
NIST SRM 1589a (PCBs, pesticidas y dioxinas/furanos en suero humano),
fueron suministradas por LGC-Promochem (Middlesex, Reino Unido). Los
patrones de HCB, β-HCH, γ-HCH, heptacloro epóxido, p,p’-DDE y p,p’-DDT, se
adquirieron a Dr. Ehrenstorfer (Augsburg, Alemania). El acetonitrilo y el
metanol, ambos calidad HPLC gradiente, fueron de Scharlab (Barcelona,
España). El n-hexano, diclorometano, tolueno y ciclohexano, calidad análisis de
trazas, los suministró Merck (Darmstadt, Alemania). El Na2SO4 anhidro calidad
para análisis de residuos, era de Panreac (Barcelona, España). Las placas de
96 pocillos con discos de SPE Empore (C18, 1mL) y su correspondiente
dispositivo de extracción a vacío fueron de 3M (St. Paul, MN, EE.UU.). Las
gradillas de recolección con 96 tubos de vidrio las suministró Marsh Bio
Products (Rochester, NY, EE.UU.). El evaporador Turbo-Vap LV y sus tubos de
evaporación eran de la casa Zymark Corp. (Hopkinton, MA, EE.UU.). La mezcla
sintética en diclorometano para calibración del GPC (aceite de maíz 62,5 mg L-
1, bis 2-etilhexil ftalato 2,5 mg L-1, metoxicloro 0,5 mg L-1, perileno 0,05 mg L-1 y
sulfuro 0,2 mg L-1) y los viales formato recuperación total para automuestreador
HPLC 717 fueron suministrados por Waters Corporation (Milford, MA, EE.UU.).
Los viales de 2 mL para automuestreador GC, los insertos de 100 µL, los
tapones y las juntas revestidas de teflón eran de Agilent (Agilent Technologies,
Santa Clara, CA, EE.UU.).
Las disoluciones conjuntas intermedias de PCBs se prepararon en
ciclohexano y las de plaguicidas en tolueno, conservándose a -20 ºC. A partir
de éstas se prepararon las de calibración en ciclohexano y las de adición en
acetonitrilo, conservándose a 4 ºC. El Na2SO4 anhidro se estufó a 300 ºC toda
una noche antes de su uso. El material de vidrio se lavó con detergente, se
aclaró abundantemente con agua Milli-Q, se estufó toda una noche a 300 ºC y
se enjuagó con diclorometano o hexano antes de su uso.
114

Capítulo 3
3.2.2 Muestras de suero
El suero utilizado para el desarrollo y la validación del método se obtuvo
del banco de muestras de la cohorte EPIC-España y de voluntarios de los
Laboratorios de Salud Pública de Gipuzkoa y Bizkaia, utilizándose siempre bajo
total anonimato. La sangre se extrajo por punción venosa y se recogió en tubos
Vacutainer. El suero se separó por centrifugación. Se preparó una mezcla de
suero a partir de 38 muestras individuales a la que se denominó Suero de
Laboratorio (LS). Todas las muestras de suero se almacenaron a -20 ºC hasta
su análisis. Para no someterlo a excesivos ciclos de congelado y
descongelado, el LS se alicuotó en tubos de vidrio de 10 mL provistos de tapón
de rosca y junta de teflón.
3.2.3 Equipamiento instrumental
El sistema de limpieza de extractos por GPC consistía en un dispositivo
de bombeo para HPLC modelo 1515, un automuestreador modelo 717, un
detector UV/VIS para HPLC modelo 2487 operando a 254 nm, un colector de
fracciones modelo Collector III y una columna Ultrastyragel THF 100 Å de 7,8 ×
300 mm de gel de estireno-divinilbenceno reticulado con un tamaño de
partícula de 7 µm, todo ello de Waters (Waters Corporation, Milford, MA,
EE.UU.). El sistema trabajaba a temperatura ambiente con diclorometano como
fase móvil.
La cuantificación se realizó en un cromatógrafo de gases 5890 series II,
equipado con inyector con/sin división de flujo, detector de captura de
electrones (ECD) y muestreador automático 7673, todo ello de Agilent
Technologies. Se inyectó 1 μL de muestra en modo sin división de flujo,
abriéndose la válvula de división de flujo 1 min después de la inyección. La
temperatura del portal de inyección era de 250 ºC con Helio como gas portador
a 135 kPa. La temperatura del detector era de 300 ºC con N2 como gas de
apoyo. La separación cromatográfica se realizó en una columna DB-5MS de 50
m × 0,25 mm × 0,25 µm (J&W Scientific, Folsom, CA, EE.UU.). El horno estaba
programado a 60 ºC (1 min), seguido de 18 ºC min-1 hasta 180 ºC, 1,1 ºC min-1
hasta 220 ºC y 5 ºC min-1 hasta 300 ºC (13 min).
115

Método basado en limpieza por GPC
La confirmación, cualitativa y cuantitativa, se llevó a cabo en un
cromatógrafo de gases 6890 equipado con inyector con/sin división de flujo con
control electrónico de presión, detector de espectrometría de masas (MSD)
5973 de cuadrupolo y muestreador automático 7683 (Agilent Technologies). Se
inyectaron 2 μL de muestra en modo sin división de flujo, abriéndose la válvula
de división de flujo 1,2 min después de la inyección. La temperatura del portal
de inyección era de 250 ºC con Helio como gas portador a 140 kPa. La
separación cromatográfica se realizó con una columna DB-5MS de 30 m × 0,25
mm × 0,25 µm (J&W Scientific). El horno estaba programado a 60 ºC (1 min),
seguido de 20 ºC min-1 hasta 180 ºC (9 min), 10 ºC min-1 hasta 230 ºC (6 min) y
10 ºC min-1 hasta 280 ºC (4 min). La temperatura de la interfaz era de 280 ºC,
la de la fuente de ionización (EI, 70 eV) de 230 ºC y la del analizador
cuadrupolo de 150ºC. El MSD se utilizó en modo Monitorización Selectiva de
Iones (SIM) con un tiempo de permanencia de 40 ms por ión. Para
cuantificación e identificación se usaron las masas mostradas en la Tabla 3.1.
Tabla 3.1. Masas (m/z) utilizadas para la confirmación por GC-MSD.
Analito Ión cuantificador (m/z) Ión cualificador(m/z)
Triclorobifenilo 258 260
Tetraclorobifenilo 290 294
Pentaclorobifenilo 326 324
Hexaclorobifenilo 360 290
Heptaclorobifenilo 394 396
HCB 284 286
HCH 219 217
Heptacloro epóxido 353 355
p,p’-DDE 246 318
p,p’-DDT 235 237
116

Capítulo 3
Como criterio de identificación se consideró aceptable una desviación
porcentual relativa del ión cualificador, menor del ±20% con respecto a su
abundancia relativa teórica en relación al ión cuantificador.
El diseño experimentale de matriz central compuesta se desarrolló y
evaluó con el programa informático STATISTICA (StatSoft, Tulsa, OK, EE.UU.).
3.2.4 Procedimiento analítico
Se tomaron 500 μL de cada muestra de suero y se depositaron en sendos
viales de vidrio de 2 mL. A cada muestra se le añadieron 500 μL de una
disolución acuosa de Na2SO4 al 5% y 250 μL de la disolución de adición de
patrón interno que contenía 5 ng mL-1 de PCB 46 y 143 en acetonitrilo. Los
viales se sellaron con tapones de rosca y junta revestida de teflón y se
sometieron a baño de ultrasonidos durante 20 min. Mientras tanto se colocó
una placa de discos de SPE en el dispositivo de extracción y se acondicionó
cada disco C18 con, sucesivamente, 2×300 μL de diclorometano, 300 μL de
metanol y 2×300 μL de agua Milli-Q, con la ayuda del vacío generado por una
trompa de agua. Una vez acondicionados los discos y terminado el tratamiento
de ultrasonidos, el contenido de cada vial se depositó en un pocillo de la placa
y se eluyeron las muestras suavemente a vacío, desechándose el eluato. En
aquellos discos que presentaron indicios de colmatación, la elución se
completó con presión positiva de N2. Tras la elución, los pocillos se enjuagaron
con 2×500 μL de agua Milli-Q y se sometieron a secado, primero con N2 a 200
kPa durante 15 segundos y luego por centrifugación a 1500×g durante 15 min.
A continuación se colocó la gradilla de 96 tubos de vidrio de recolección en el
dispositivo de extracción y en la parte superior la placa de discos SPE. Cada
uno de los discos se eluyó con 2×300 μL de hexano y 300 μL de
diclorometano/hexano (1:1), recolectándose todo el eluato de cada disco en su
correspondiente tubo de vidrio.
La capa orgánica de cada tubo se transfirió a sendos tubos de
evaporación TurboVap, evitando añadir la más mínima traza de agua. Cada
tubo de recolección se enjuagó con 500 μL de diclorometano que se
transvasaron seguidamente a los tubos TurboVap que contenían el eluato. A
continuación se colocaron estos últimos tubos en el sistema de evaporación
117

Método basado en limpieza por GPC
TurboVap (25 kPa de N2, 30 ºC) para concentrar su contenido hasta 0,1 mL. Se
añadieron 500 μL de diclorometano al concentrado y se llevó la mezcla hasta
un volumen final de 275 µL. Este extracto se transfirió a un vial formato
recuperación total para automuestreador HPLC y se introdujeron 250 µL en el
sistema GPC, que operaba a un flujo de elución de 1 mL min-1. El tiempo total
de elución era de 15 min por muestra y la ventana de recolección aplicada a los
plaguicidas organoclorados y PCBs era de 3 min (entre 9 min 18 s y 12 min 18
s), lo que suponía un volumen de recolección de 3 mL que se recogían en un
tubo de evaporación TurboVap. Tras añadir 500 µL de ciclohexano la fracción
recolectada se concentraba en el sistema de evaporación TurboVap (25 kPa de
N2, 30 ºC) hasta 50 µL, que se tranferían a un vial de 2 mL para
automuestreador GC provisto de inserto de 100 µL para su posterior análisis.
3.2.5 Calibración y validación
La calibración GC-ECD se llevó a cabo con disoluciones patrón de los
analitos a 0,75, 2, 10 y 50 ng mL-1 en ciclohexano (1,5, 4, 20 y 100 ng mL-1
para el p,p’-DDE). Para la calibración del GC-MSD se usaron las disoluciones
de 2, 10 y 50 ng mL-1. La cuantificación se llevó a cabo utilizando el PCB 46
(para HCB, β-HCH, γ-HCH, heptacloro epóxido, PCBs 28 y 52) y el PCB 143
(para p,p’-DDE, p,p’-DDT, PCBs 101, 118, 138, 153 y 180), a una
concentración de 25 ng mL-1, como patrones internos (IS). Se emplearon
regresiones del cociente entre las áreas de pico de los analitos con respecto a
las de sus respectivos ISs, frente al cociente de las concentraciones, con una
ponderación del inverso del cuadrado de las concentraciones.
La ventana de tiempos de recolección para el sistema GPC se estableció
inyectando tres veces la disolución patrón de 50 ng mL-1 y recolectando
fracciones de 30 s entre los minutos 8 y 14. La repetibilidad y recuperación de
dicho sistema se evaluó procesando 5 veces la disolución patrón de 10 ng mL-1
y comparando las señales obtenidas en el GC con las del mismo patrón sin
pasar por el GPC.
La repetibilidad (calculada como desviación estándar relativa) y la
recuperación del método global se determinaron adicionando al LS dos niveles:
0,15 y 1,5 ng mL-1 para los PCBs y 0,3 y 3 ng mL-1 para los plaguicidas
118

Capítulo 3
organoclorados (n = 8 para cada nivel). La veracidad y comparabilidad se
evaluaron usando el NIST SRM 1589a y Material de Referencia procedente de
varias rondas del ejercicio interlaboratorios AMAP (Artic Monitoring and
Assessment Programme) Ring Test for Persistent Organic Pollutants in Human
Serum, organizado por el Centro de Toxicología del Instituto Nacional de Salud
Pública de Quebec (Canadá), en el que se participaba con regularidad [AMAP
2007].
Finalmente, una serie de muestras de suero individuales se analizaron por
este método y por el descrito en el capítulo 1, con la subsiguiente comparación
de resultados mediante regresión lineal.
119

Método basado en limpieza por GPC
3.3 Resultados y discusión
3.3.1 Optimización de la limpieza de extractos
El grado de separación entre los distintos componentes de la mezcla para
calibración del GPC, expresado como resolución, se usó como indicador de la
capacidad de limpieza del sistema. El primer parámetro a optimizar fue el
volumen de inyección dado que mayor volumen de inyección permitía procesar
mayor cantidad de muestra pero, al ser el GPC un sistema cromatográfico de
elución isocrática, también implicaba mayor ensanchamiento de picos con la
consiguiente pérdida de resolución. Con objeto de establecer el volumen de
inyección óptimo se prepararon varias diluciones de la mezcla para calibración
del GPC (aceite de maíz 62,5 mg L-1, bis 2-etilhexil ftalato 2,5 mg L-1,
metoxicloro 0,5 mg L-1, perileno 0,05 mg L-1 y sulfuro 0,2 mg L-1), en
diclorometano: 1/2, 1/4, 1/8, 1/12, 1/16 y 1/22, de las cuales se inyectaron
respectivamente en el GPC 60, 120, 240, 360, 480 y 660 μL, de forma que las
cantidades de los componentes de la mezcla eran las mismas
independientemente del volumen inyectado (aceite de maíz 1,875 mg, bis 2-
etilhexil ftalato 0,075 mg, metoxicloro 0,015 mg, perileno 0,0015 mg y sulfuro
0,006 mg). En la Figura 3.1 se observa el cromatograma de la inyección de 240
μL.
Figura 3.1 Cromatograma de la mezcla de calibración del GPC en una columna Ultrastyragel THF 100 Å de 7,8 × 300 mm. Volumen de inyección 240 µL. Compuestos : (1) aceite de maíz, (2) bis 2-etilhexil ftalato, (3) metoxicloro, (4) perileno, (5) sulfuro 0,006 mg.
120
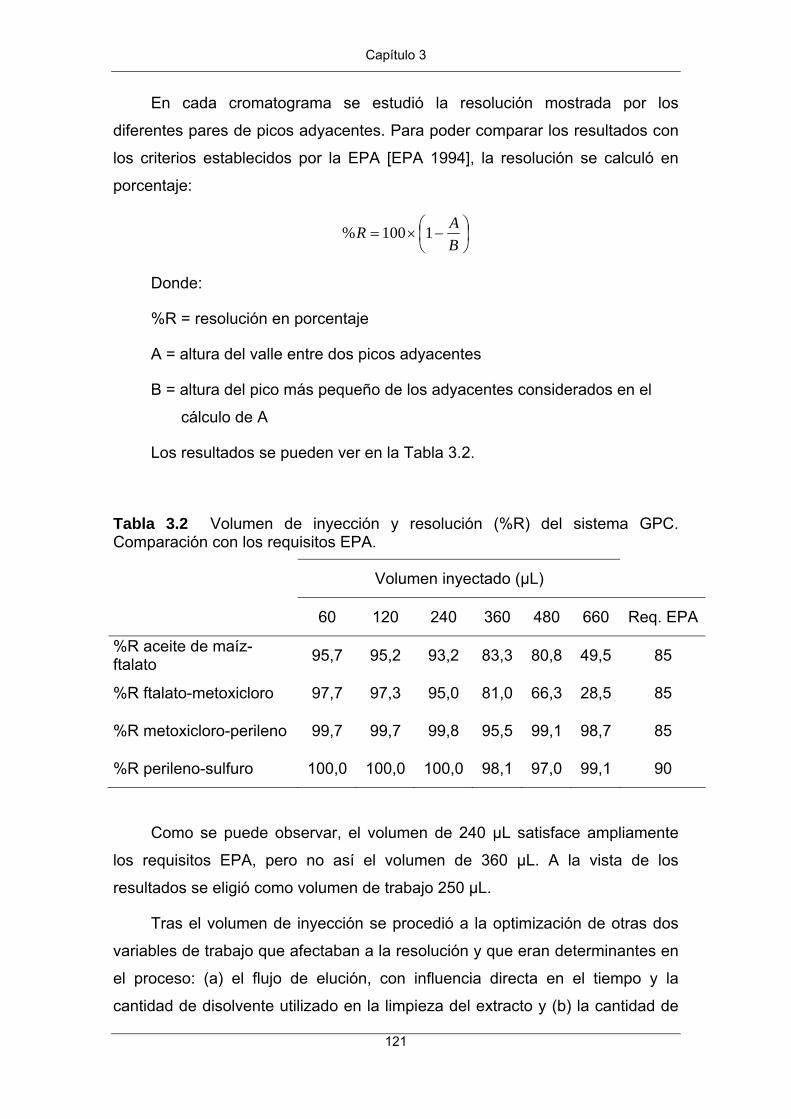
Capítulo 3
En cada cromatograma se estudió la resolución mostrada por los
diferentes pares de picos adyacentes. Para poder comparar los resultados con
los criterios establecidos por la EPA [EPA 1994], la resolución se calculó en
porcentaje:
⎟⎠⎞
⎜⎝⎛ −×=
BAR 1100%
Donde:
%R = resolución en porcentaje
A = altura del valle entre dos picos adyacentes
B = altura del pico más pequeño de los adyacentes considerados en el
cálculo de A
Los resultados se pueden ver en la Tabla 3.2.
Tabla 3.2 Volumen de inyección y resolución (%R) del sistema GPC. Comparación con los requisitos EPA.
Volumen inyectado (µL)
60 120 240 360 480 660 Req. EPA
%R aceite de maíz-ftalato 95,7 95,2 93,2 83,3 80,8 49,5 85
%R ftalato-metoxicloro 97,7 97,3 95,0 81,0 66,3 28,5 85
%R metoxicloro-perileno 99,7 99,7 99,8 95,5 99,1 98,7 85
%R perileno-sulfuro 100,0 100,0 100,0 98,1 97,0 99,1 90
Como se puede observar, el volumen de 240 μL satisface ampliamente
los requisitos EPA, pero no así el volumen de 360 µL. A la vista de los
resultados se eligió como volumen de trabajo 250 μL.
Tras el volumen de inyección se procedió a la optimización de otras dos
variables de trabajo que afectaban a la resolución y que eran determinantes en
el proceso: (a) el flujo de elución, con influencia directa en el tiempo y la
cantidad de disolvente utilizado en la limpieza del extracto y (b) la cantidad de
121

Método basado en limpieza por GPC
grasa inyectada que, al ser el componente mayoritario del extracto,
determinaba la cantidad de muestra que podía ser procesada. Para la
optimización de estas variables se utilizó el diseño experimental y, más
concretamente, un diseño central compuesto (CCD) tomando la cantidad de
grasa y el flujo de elución como variables independientes y la resolución entre
el pico del aceite de maíz (representativo del contenido en grasa) y el
metoxicloro como variable dependiente. Se eligieron los picos de estos dos
componentes porque la grasa era el componente mayoritario de los extractos
de suero y el metoxicloro un plaguicida organoclorado de alto peso molecular
que marcaba el límite a partir del cual era esperable la elución de nuestros
analitos, que eran de menor tamaño molecular. Como ambos picos no eran
adyacentes se calculó la resolución en la forma habitual de los manuales de
cromatografía (cociente entre la diferencia de tiempos de elución y la anchura
media de la base de los picos expresada en unidad de tiempo) [Hübschmann
2009].
El CCD consistía en los puntos de un diseño factorial (2N) añadidos a los
puntos de un diseño estrella (2N + 1) más R réplicas, siendo N el número de
variables independientes (en nuestro caso N = 2 y R = 3). Los puntos de la
estrella estaban centrados en las caras (α±1) y las réplicas se llevaron a cabo
en los puntos centrales del diseño. Por lo tanto, la matriz de diseño CCD
constaba de 12 experimentos en total, a realizar en orden aleatorio. Los
factores experimentales y sus niveles, la matriz de diseño y sus resultados se
pueden ver en la Tabla 3.3. Para el CCD se utilizó un modelo polinómico de
segundo grado que incluía los efectos principales de los dos factores, su
interacción y sus componentes cuadráticas. Con este modelo de segundo
grado la prueba ANOVA mostraba una falta de ajuste no significativa (p > 0,1),
así como un buen valor de R2 (0,9795). Estos datos se consideraron una
indicación de la adecuación del modelo. Los coeficientes de regresión
obtenidos se utilizaron para estimar los valores de la variable dependiente en
función de los valores de las variables independientes.
122

Capítulo 3
Tabla 3.3 Variables experimentales del sistema GPC, niveles, matriz de diseño central compuesto (CCD) y resultados de resolución obtenidos.
Variable Nivel
Bajo Medio Alto
Grasa (aceite de maíz, mg) 1,042 15,625 30,208 Flujo (mL min-1) 0,500 1,000 1,500 Prueba (orden de ejecución) Grasa (mg) Flujo (ml/min) Ra
grasa/ metoxicloro
1 30,208 0,500 2,30 2 1,042 1,000 2,20 3 (C)b 15,625 1,000 1,87 4 30,208 1,500 2,67 5 (C)b 15,625 1,000 1,52 6 (C)b 15,625 1,000 1,91 7 15,625 1,500 2,22 8 1,042 1,500 2,97 9 1,042 0,500 1,75 10 15,625 0,500 2,34 11 (C)b 15,625 1,000 2,67 12 30,208 1,000 2,20 a R, resolución entre el pico del aceite de maíz y el del metoxicloro b (C), punto central
La forma más común de resumir los resultados de un CCD es un gráfico
3D de superficie de respuesta. Para la variable dependiente aquí estudiada
(resolución) dicho gráfico se muestra en la Figura 3.2. Como se puede ver, el
modelo predecía un incremento de la resolución al disminuir la cantidad de
grasa y el flujo, sin ningún máximo relativo en el rango estudiado que permitiera
establecer una combinación óptima de valores para estas dos variables de
trabajo.
123

Método basado en limpieza por GPC
Figura 3.2 Superficie de respuesta, obtenida a partir del CCD, para la resolución en el sistema GPC entre el pico del aceite de maíz y el metoxicloro, en función del flujo y la cantidad de grasa inyectada.
Sin embargo, el modelo era útil para predecir la resolución una vez fijados
los valores de flujo y cantidad de grasa inyectada. Así, por ejemplo, si
partíamos de 1 mL de suero (aproximadamente 6 mg de grasa) y trabajábamos
a un flujo de 1 mL min-1 (que permitía realizar la limpieza del extracto en 15
min), el modelo predecía una resolución de 2,64. Con el mismo flujo y 500 μL
de suero, la resolución predicha llegaba a 2,76. Por otra parte, asumiendo que
los dos componentes de una mezcla separada cromatográficamente tienen el
mismo factor de respuesta y que sus picos son gaussianos, es posible estimar
teóricamente, por medio de cálculos estadísticos, la composición final en cada
uno de los picos en función de la composición inicial de la mezcla y de la
resolución [Laitinen and Harris 1975]. En el caso de un suero con una
concentración de analito de 0,1 ng mL-1 y 6 mg mL-1 de grasa, la concentración
de analito referida a grasa en el extracto inicial sería de aproximadamente
1,7×10-6 % (17 ng g-1). Con una resolución de 2,64, tras la limpieza del extracto
124

Capítulo 3
por separación cromatográfica en el GPC, el porcentaje final de analito en la
fracción recolectada, estimado teóricamente y referido a grasa, sería del 20 %.
Trabajando con una resolución de 2,76 llegaría al 50 %. En cualquiera de los
dos casos la grasa residual ya no sería una interferencia a tener en cuenta en
la posterior cuantificación por cromatografía de gases.
Tras examinar todos estos datos se decidió trabajar con 500 μL de suero,
ya que podía haber sueros con contenidos en grasa > 6 mg mL-1, y usar un
flujo de 1 mL min-1 en el GPC, ya que permitía combinar rapidez en la limpieza
del extracto (15 min por muestra) con una aceptable resolución y bajo consumo
de disolvente (15 mL por muestra).
3.3.2 Ventana de tiempos de recolección y evaluación del sistema GPC
Una vez conocidos el volumen de inyección y el flujo de elución del
sistema GPC se establecieron las ventanas de recolección tal como se
describe en el apartado 3.2.5. Los resultados se presentan en la Tabla 3.4.
Como se puede ver, entre los minutos 9,5 y 12 se recogía prácticamente el
100% de cada compuesto. La ventana de recolección se amplió a 3 min (de 9
min 18 s a 12 min 18 s) para compensar posibles fluctuaciones y se utilizó para
estudiar la precisión y la recuperación del sistema GPC siguiendo lo descrito en
el apartado 3.2.5. Los resultados están reflejados en la Tabla 3.5. La media del
porcentaje de recuperación de cada compuesto (n=5) iba de 89,5 a 111,7%,
con un porcentaje de desviación estándar relativa (DER%) entre el 0,8 y el
5,8%.
125
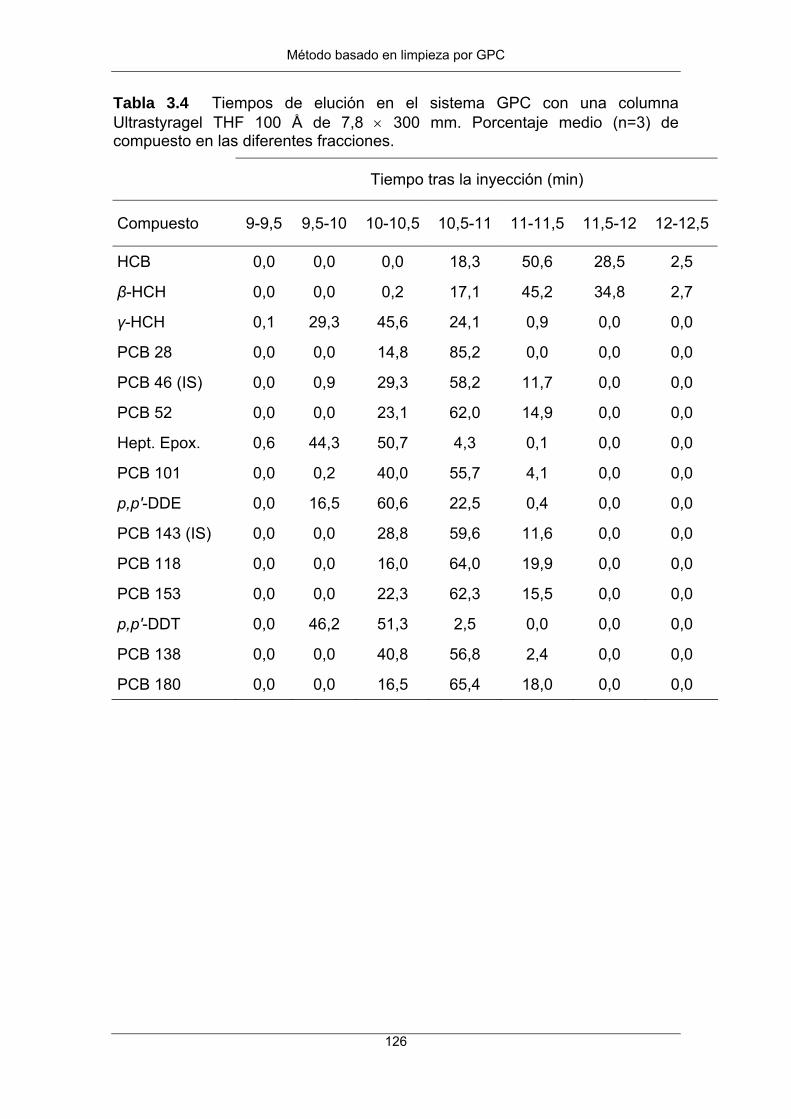
Método basado en limpieza por GPC
Tabla 3.4 Tiempos de elución en el sistema GPC con una columna Ultrastyragel THF 100 Å de 7,8 × 300 mm. Porcentaje medio (n=3) de compuesto en las diferentes fracciones.
Tiempo tras la inyección (min)
Compuesto 9-9,5 9,5-10 10-10,5 10,5-11 11-11,5 11,5-12 12-12,5
HCB 0,0 0,0 0,0 18,3 50,6 28,5 2,5
β-HCH 0,0 0,0 0,2 17,1 45,2 34,8 2,7
γ-HCH 0,1 29,3 45,6 24,1 0,9 0,0 0,0
PCB 28 0,0 0,0 14,8 85,2 0,0 0,0 0,0
PCB 46 (IS) 0,0 0,9 29,3 58,2 11,7 0,0 0,0
PCB 52 0,0 0,0 23,1 62,0 14,9 0,0 0,0
Hept. Epox. 0,6 44,3 50,7 4,3 0,1 0,0 0,0
PCB 101 0,0 0,2 40,0 55,7 4,1 0,0 0,0
p,p'-DDE 0,0 16,5 60,6 22,5 0,4 0,0 0,0
PCB 143 (IS) 0,0 0,0 28,8 59,6 11,6 0,0 0,0
PCB 118 0,0 0,0 16,0 64,0 19,9 0,0 0,0
PCB 153 0,0 0,0 22,3 62,3 15,5 0,0 0,0
p,p'-DDT 0,0 46,2 51,3 2,5 0,0 0,0 0,0
PCB 138 0,0 0,0 40,8 56,8 2,4 0,0 0,0
PCB 180 0,0 0,0 16,5 65,4 18,0 0,0 0,0
126

Capítulo 3
Tabla 3.5 Media del porcentaje de recuperación (n=5) y porcentaje de desviación estándar relativa (DER%) de los analitos en el sistema GPC con una columna Ultrastyragel THF 100 Å de 7,8 × 300 mm.
Compuesto Media DER%
PCB 28 100,7 1,2
PCB 52 105,9 1,2
PCB 101 89,5 2,8
PCB 118 97,0 1,9
PCB 138 105,4 1,3
PCB 153 96,8 1,4
PCB 180 107,5 0,8
p,p’-DDT 92,5 5,8
p,p’-DDE 95,4 2,5
Heptacloro epóxido 111,7 3,2
HCB 101,9 1,5
β-HCH 102,9 3,4
γ-HCH 108,1 3,1
3.3.3 Evaluación del método
Tras establecer las condiciones de limpieza de extractos se procedió a la
validación de la totalidad del método.
El límite de cuantificación de cada compuesto (LQ) se calculó como la
concentración del analito que producía una señal 10 veces mayor que el ruido
de fondo de un blanco sometido al proceso analítico. Para los PCBs el LQ iba
de 0,06 a 0,16 ng mL-1 y para los plaguicidas organoclorados de 0,12 a 0,36 ng
mL-1 (ver Tabla 3.6).
127

Método basado en limpieza por GPC
Tabla 3.6. Límites de cuantificación (LQ) de los distintos compuestos
Compuesto LQ (ng mL-1)
PCB 28 0,11
PCB 52 0,16
PCB 101 0,16
PCB 118 0,09
PCB 138 0,07
PCB 153 0,06
PCB 180 0,06
p,p’-DDT 0,25
p,p’-DDE 0,36
Heptacloro epóxido 0,36
HCB 0,12
β-HCH 0,21
γ-HCH 0,21
La recuperación y la precisión se evaluaron según se describe en el
apartado 3.2.5. Los valores obtenidos se muestran en la Figura 3.3.
128

Capítulo 3
Figura 3.3 Porcentaje medio de recuperación y repetibilidad, en porcentaje de desviación estándar relativa (DER), de los distintos analitos a dos niveles de adición (n = 8 por nivel). No se calcularon las del congénere 153 a nivel bajo ya que la concentración basal de este compuesto estaba muy por encima de dicho nivel.
Los resultados de aplicar el método al NIST SRM 1589a mostraron una
aceptable concordancia con los valores certificados (rev. 4/05/2007). La media
(n=3) de la concentración obtenida no difería significativamente del valor
certificado para el PCB 118 y el p,p’-DDE, era un 15% más alta para el PCB138
y un 19 y 16% más baja para el PCB 153 y el PCB 180 respectivamente. El
resto de compuestos estaba por debajo del LQ, lo que también coincidía con
los valores certificados.
129

Método basado en limpieza por GPC
Por lo que respecta a los Materiales de Referencia provenientes de las
rondas del AMAP Ring Test, todos los valores obtenidos para los analitos
detectados (HCB, β-HCH, p,p’-DDE, p,p’-DDT y PCBs 28, 118, 138, 153 y 180),
estaban dentro del rango satisfactorio (ver Tabla 3.7).
Tabla 3.7 Porcentaje de recuperación con respecto a los valores asignados para varios Materiales de Referencia (MR) provenientes del AMAP Ring Test for Persistent Organic Pollutants in Human Serum. Rango satisfactorio: 60-140% del valor asignado.
Ronda de origen del MR
Compuesto W-05-07
W-07-04
W-07-05
W-07-06
W-07-07
W-07-08
W-07-09 Media DER%
PCB 28 105 98 106 102 95 96 97 99 4.5 PCB 118 98 96 97 110 98 96 95 98 5.2 PCB 138 80 100 122 94 105 101 106 101 12.6 PCB 153 117 84 97 93 85 103 86 95 12.6 PCB 180 92 74 98 73 71 89 94 84 13.5
β-HCH 130 121 102 126 130 119 111 119 8.6 p,p’-DDE 97 98 94 100 102 92 94 96 3.7 p,p’-DDT 129 123 120 120 126 129 125 124 3.0 HCB 82 90 98 84 91 92 101 91 7.5
3.3.4 Aplicación a muestras de suero
El procedimiento analítico basado en la limpieza de extractos por GPC
desarrollado en este estudio se aplicó a 38 muestras de suero humano. Un
cromatograma representativo se muestra en la Figura 3.4. En todas las
muestras analizadas se detectaron: HCB y PCBs 138, 153 y 180, mientras que
HCB, p,p’-DDE y PCB 118 se detectaron en el 74, 97 y 82% de las muestras
respectivamente. En la casi totalidad de todas ellas el compuesto mayoritario
era el p,p’-DDE y, entre los PCBs, el 153.
130

Capítulo 3
Figura 3.4 Cromatograma GC-ECD de una muestra de suero humano sobre una columna DB-5 MS. Asignación de los picos: (1) HCB; (2) β-HCH; (3) PCB 46 (IS); (4) p,p’-DDE; (5) PCB 118; (6) PCB 143 (IS); (7) PCB 153; (8) p,p’-DDT; (9) PCB 138; (10) PCB 180.
Estas muestras se analizaron también por el método basado en la
limpieza de extractos por adsorción sobre columnas de sílice/ácido sulfúrico
descrito en el capítulo 1 (usado como referencia) y se compararon los
resultados mediante regresión lineal. El análisis de correlación de Pearson
mostraba que los dos conjuntos de datos estaban alta y significativamente
correlacionados con coeficientes (r) que iban desde 0,957 (PCB 180) hasta
0,996 (β-HCH). Las pendientes de la recta de regresión estaban entre 0,894
(PCB 118) y 1,193 (β-HCH). Estos resultados indicaban una buena
concordancia entre métodos para los analitos encontrados. En la Figura 3.5 se
presentan, a modo de ejemplo, los datos de correlación para el p,p’-DDE y el
PCB 153.
131

Método basado en limpieza por GPC
Figura 3.5 Concentraciones de p,p’-DDE y PCB 153 obtenidas en el análisis, por limpieza con GPC y limpieza por sílice ácida, de 38 muestras de suero humano.
Un dato a tener en cuenta al aplicar este método a series de muestras es
que el extracto a procesar por GPC está disuelto en diclorometano. Si los viales
132

Capítulo 3
no se tapan convenientemente, dada la volatilidad de este solvente es de
esperar que se evapore de forma notable durante la permanencia de los viales
a temperatura ambiente en el carrusel del automuestreador del GPC, pudiendo
llegarse hasta casi sequedad en intervalos relativamente cortos de tiempo (1 a
2 h). Pero la mayoría de los materiales plásticos utilizados para la fabricación
de los tapones son potenciales fuentes de contaminación del extracto al entrar
en contacto con el reflujo del diclorometano. Por lo tanto, si se quiere trabajar
con el equipo de forma automática y desatendida habría que recurrir a alguna
de estas dos opciones: utilizar tapones de teflón (que son caros y no siempre
están como opción de catálogo, en cuyo caso habría que solicitarlos por
encargo), ó dejar los viales sin tapar e incorporar al carrusel un sistema de
refrigeración que la mayoría de fabricantes ofrecen como opción. Aunque la
solución ideal sería la conjunción de ambos factores: tapones de teflón y
refrigeración del carrusel.
133

Método basado en limpieza por GPC
3.4 Conclusiones
El método basado en limpieza de extractos por GPC desarrollado en este
capítulo, permite la determinación simultánea de los plaguicidas
organoclorados y PCBs más relevantes en suero humano. Comparado con
otras técnicas presenta varias ventajas que lo hacen adecuado para su empleo
en estudios de riesgo de exposición o epidemiológicos. Estas ventajas
incluyen: aceptable LQ con baja cantidad de muestra (500 µL), recuperación,
precisión y veracidad satisfactorias, alta productividad y menos mano de obra
ya que la etapa de limpieza de extractos está automatizada, y menor costo
puesto que el uso de un sistema de GPC miniaturizado reduce el tiempo de
análisis por muestra y la cantidad de disolvente utilizado. La única
consideración a tener en cuenta es que su empleo de forma automática y
desatendida requiere el uso de tapones de teflón y/o un sistema de
refrigeración del carrusel del automuestreador del GPC. Además, este método
podría ser ampliado a otros compuestos contaminantes ya que se emplean
adsorbentes y condiciones de extracción no específicos junto con un
procedimiento de limpieza de extractos no destructivo, lo que lo hace muy
interesante para analitos sensibles al ácido sulfúrico.
134

Capítulo 3
3.5 Referencias
♦ AMAP, 2007. Artic Monitoring and Assessment Programme Ring Test for Persistent Organic Pollutants in Human Serum. http://www.inspq.qc.ca/ctq/paqe/amap/. Ultimo acceso: 12-07-2010.
♦ AOAC, 2007. Official Method 984.21. Official Methods of Analysis, 18th ed. Rev. 2, Secs. 984.21 A-F. AOAC, Arlington, VA, USA.
♦ Axmon A, Rylander L, Stromberg U, Jonsson B, Nilsson-Ehle P, Hagmar L, 2004. Polychlorinated biphenyls in serum and time to pregnancy. Environ. Res. 96, 186-195.
♦ Bates MN, Buckland SJ, Garrett N, Ellis H, Needham LL, Patterson DG, Turner WE, Russell DG, 2004. Persistent organochlorines in the serum of the non-occupationally exposed New Zealand population. Chemosphere 54, 1431-1443.
♦ Cruz S, Lino C, Silveira MI, 2003. Evaluation of organochlorine pesticide residues in human serum from an urban and two rural populations in Portugal. Sci. Total Environ. 317, 23-35.
♦ EPA (United States Environmental Protection Agency), 1994. Gel-permeation Cleanup. Method 3640A, Rev. 1. http://www.epa.gov/epawaste/hazard/testmethods/sw846/pdfs/3640a.pdf. Último acceso: 25-05-2010.
♦ Furberg AS, Sandanger T, Thune I, Burkow IC, Lund E, 2002. Fish consumption and plasma levels of organochlorines in a female population in Northern Norway. J. Environ. Monitor. 4, 175-181.
♦ Hovander L, Athanasiadou M, Asplund L, Jensen S, Wehler EK, 2000. Extraction and cleanup methods for analysis of phenolic and neutral organohalogens in plasma. J. Anal. Toxicol. 24, 696-703.
♦ Hübschmann HJ, 2009. Handbook of GC/MS. Fundamentals and applications.2nd. ed. Wiley-VCH, Weinheim, Germany.
♦ Janák K, Grimvall E, Östman C, Colmsjö A, Athanasiadou M, Beergman A, 1994. Gas chromatography-atomic emission detection (GC-AED) set-up for bio-monitoring of PCBs and methylsulfonyl-PCBs. J. Microcolumn. Sep. 6, 605-616.
♦ Koppen G, Covaci A, Van Cleuvenbergen R, Schepens P, Winneke G, Nelen V, Van Larebeke N, Vlietinck R, Shoeters G, 2002. Persistent organochlorine pollutants in human serum of 50-65 years old women in the Flanders Environmental and Health Study (FLEHS). Part 1: concentrations and regional differences. Chemosphere 48, 811-825.
♦ Laden F, Wolff MS, Niguidula NJ, Spiegelman D, Hankinson SE, Hunter DJ, 1997. Reduced aliquot size for a plasma organochlorine assay for use in epidemiological studies. Cancer Epidem. Biomar. 6, 333-338.
♦ Laitinen HA, Harris WE, 1975. Chemical Analysis. McGraw-Hill, New York, USA.
♦ Lindh CH, Littorin M, Johannesson G, Jönsson BAG, 2008. Analysis of ethylenethiourea as a biomarker in human urine using liquid chromatography/triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 22, 2573-2579.
♦ Patterson JR, 1991. Reducing solvent consumption in automated gel permeation chromatographic cleanup for pesticide residue analysis: a modified GPC AutoPrep 1002. J. AOAC 74, 1016-1018.
♦ Pauwels A, Schepens PJC, 1998. Simultaneous separation and determination of polychlorinated biphenyl congeners and other chlorinated hydrocarbon residues in human matrices using gel permeation or adsorption chromatographic clean-up and GC-MS quantification. Int. J. Environ. Anal. Chem. 71, 105-118.
♦ Pauwels A, David F, Schepens P, Sandra P, 1999. Automated gel permeation chromatographic clean-up of human adipose tissue for multiresidue analysis of organochlorine compounds. Int. J. Environ. Anal. Chem. 73, 171-178.
135

Método basado en limpieza por GPC
♦ Reiner EJ, Boden AR, Chen T, MacPherson KA, Muscalu AM, 2010. Advances in the analysis of persistent halogenated organic compounds. LC GC Eur. 23, 60-70.
♦ Schenzler C, Thier HP, 2001. European standardization of methods for pesticide residue analysis in foods—current status. Food Addit. Contam. 18, 875-879.
♦ Smeds A, Saukko P, 2001. Identification and quantification of polychlorinated biphenyls and some endocrine disrupting pesticides in human adipose tissue from Finland. Chemosphere 44, 1463-1471.
♦ Stalling DL, Tindle RC, Johnson JL, 1972. Cleanup of pesticide and polychlorinated biphenyl residues in fish extracts by gel permeation chromatography. J. AOAC 55, 32-38.
♦ Van Rhijn JA, Tuinstra LGMTh, 1991. Miniaturisation of size-exclusion chromatography as a powerful clean-up tool in residue analysis. J. Chromatogr. 552, 517-526.
♦ Woodman M, Stone P, 2003. Optimizing sample loading in automated size exclusion chromatography sample preparation for small molecule analysis from complex matrices. Agilent Application Note 5989-0181EN. http://www.chem.agilent.com/Library/applications/5989-0181EN.pdf. Último acceso: 25-05-2010.
♦ Zwir-Ferenc A, Biziuk M, 2004. An analysis of pesticides and polychlorinated biphenyls in biological samples and foods. Crit. Rev. Anal. Chem. 34, 95-103.
136

CAPÍTULO 4
Niveles séricos de PCBs en la cohorte EPIC-España
4.1 Introducción
4.2 Metodología experimental
4.3 Resultados y discusión
4.4 Conclusiones
4.5 Referencias

PCBs en la cohorte EPIC-España
138

Capítulo 4
139
4.1 Introducción
Recordando algunas consideraciones que se realizaban en el capítulo
introductorio:
− La principal exposición a PCBs tiene lugar por vía oral, de forma
crónica y a dosis bajas, afectando a toda la población y durante toda
su vida. Estas dos últimas características, unidas a su potencial
toxicidad, los convierte en objeto de especial interés desde el punto de
vista de la salud pública.
− En este tipo de exposiciones los síntomas a corto plazo suelen ser
sub-clínicos y, por lo tanto, imperceptibles. Sin embargo, a largo plazo
se pueden manifestar en forma de diversas alteraciones o
enfermedades que en muchas ocasiones son difícilmente asociables, a
primera vista, al agente causante (relación causa/efecto difícil de
establecer).
− El estudio directo experimental de este tipo de toxicología es muy
complicado. Por ello se recurre a estudios epidemiológicos
observacionales.
− Se detecta un déficit de información sobre los niveles que alcanza esta
exposición crónica en la población general y los efectos que causa.
− La monitorización biológica de estos compuestos o sus metabolitos en
tejidos y fluidos biológicos se ha revelado como una herramienta útil
para valorar el riesgo de exposición en los estudios epidemiológicos.
Concretamente el suero es uno de los materiales más usados.
Se comprende el interés que tiene determinar los niveles séricos de PCBs
en amplias bases de población para valorar su nivel de exposición e incluso,
posteriormente, realizar comparaciones con otras poblaciones, estudiar su
evolución con el tiempo, o establecer su posible asociación con algún efecto
tóxico.
El Estudio Prospectivo Europeo sobre Cáncer y Nutrición (EPIC)
[Bingham and Riboli 2004, González et al. 2004] es un macroestudio
epidemiológico observacional de carácter multicéntrico, cuyo reclutamiento se

PCBs en la cohorte EPIC-España
140
inició en 1993 con la recogida de datos y muestras en 23 centros de 10 países
europeos: Alemania, Dinamarca, España, Francia, Grecia, Holanda, Italia,
Noruega, Reino Unido y Suecia. Está coordinado por la Agencia Internacional
de Investigación del Cáncer (IARC) de la Organización Mundial de la Salud. Se
diseñó y desarrolló con el convencimiento de que, para profundizar en el
conocimiento científico sobre la relación entre la dieta y el cáncer, se requieren
amplios estudios epidemiológicos prospectivos sobre personas sanas,
combinados con importantes estudios de laboratorio, incluyendo marcadores
bioquímicos y de susceptibilidad genética. Sus objetivos finales eran, y siguen
siendo, mejorar el conocimiento científico sobre factores nutricionales
implicados en el cáncer [Bueno de Mesquita and González 1997] y,
consecuentemente, poder aportar las bases científicas para intervenciones de
salud pública dirigidas a promover una dieta y estilos de vida saludables [Riboli
1992]. Entre sus principales ventajas están el amplio rango de variabilidad de la
ingesta alimentaria contemplada, fruto de las grandes diferencias todavía
observadas entre la dieta mediterránea de Grecia, Italia y España y los
patrones alimenticios del norte de Europa, así como su gran potencia
estadística, siendo uno de los estudios epidemiológicos de cohorte
prospectivos más amplios que se esté realizando hasta la fecha. Tras una fase
piloto entre 1990 y 1993, el trabajo de campo para el reclutamiento y la
recogida de información y muestras se realizó entre 1993 y 1998, dependiendo
del país. En total la macrocohorte inicial incluía a 519.978 personas, reclutadas
de forma voluntaria entre población general, donantes de sangre, afiliados a
mutuas y empleados de ciertas empresas, de las que 366.521 eran mujeres y
el resto hombres. Sus edades iban de 21 a 83 años, siendo la franja principal la
comprendida entre 35 y 69 años. A cada individuo se le realizaron varias
medidas antropométricas como altura, peso y circunferencia de cintura y
cadera. A todos ellos se les realizó también un cuestionario recabando
información sobre su dieta y otros hábitos de vida: consumo de tabaco,
actividad física ocupacional, actividad deportiva y de ocio, consumo de
anticonceptivos y uso de terapia hormonal sustitutiva, historial reproductivo,
exposición a ocupaciones de riesgo y antecedentes médicos y quirúrgicos. A la
mayoría de ellos (385.719) se les extrajo muestras de sangre para posteriores
análisis de laboratorio. Las muestras se fraccionaron en 28 partes que se

Capítulo 4
141
introdujeron en pequeños recipientes de plástico termosellados de 0,5 mL
(pajuelas): 12 de plasma, 8 de suero, 4 de eritrocitos y 4 de leucocitos para la
obtención de ADN. Se hicieron dos partes iguales de 14 pajuelas, de forma que
una de ellas se quedaba en el propio centro de recogida y la otra se mandaba a
Lyon a la IARC. En ambos casos las muestras se almacenaron en
contenedores de nitrógeno líquido a -196 ºC. Además, se realizan seguimientos
periódicos de toda la macrocohorte al objeto de identificar los nuevos casos de
cáncer producidos, los fallecimientos y sus causas y conocer el estado vital del
resto así como posibles cambios en sus hábitos.
Dado que hay una gran diversidad de congéneres, es importante elegir
cuáles utilizar en los estudios de exposición a PCBs. Por supuesto, la decisión
a tomar sobre qué compuestos medir en un escenario específico dependerá del
propósito de la investigación, así como de las concentraciones y perfiles
esperables en la población estudiada. Sin embargo, algunos perfiles parecen
ser casi universales. Como ya se comentaba en el capítulo introductorio, la
mayoría de los seres vivos presenta un perfil similar en los 4-6 congéneres
dominantes (entre los que cabe destacar los PCBs 138, 153 y 180). Además,
en humanos, toda la evidencia hasta la fecha sugiere que puede asumirse que
el congénere 153 es el mayoritario, altamente correlacionado con los otros dos,
en todas las poblaciones. Por ello, ha sido elegido en muchas ocasiones como
marcador de la exposición a PCBs. Algunos estudios han enfocado el tema de
los perfiles de PCBs en muestras biológicas humanas. En un estudio sueco
[Glynn et al. 2000], donde se medían 10 congéneres en suero y en otro
canadiense [Gladen et al. 2003], donde se determinaban 38 congéneres en
leche, se encontró que los congéneres 118, 138, 153 y 180 aparecían en el
90% de las muestras y que sumaban alrededor del 80% de la concentración
total de PCBs analizados, mostrando todos ellos una fuerte correlación con
dicha concentración total. En otro estudio en Corea [Park et al. 2007] estos
cuatro PCBs suponían aproximadamente el 50% de la suma de las
concentraciones de los 78 congéneres medidos, siendo también muy altas
(>0.90) las correlaciones respectivas del congénere 153 con el total de PCBs y
del congénere 118 con el total de PCBs similares a la dioxina. A efectos
prácticos y para economizar medios, en Europa normálmente se monitorizan

PCBs en la cohorte EPIC-España
142
siete congéneres (28, 52, 101, 118, 138, 153 y 180), conocidos habitualmente
como congéneres indicadores o descriptores (key congeners), para establecer
la exposición global a PCBs por considerarse que son estables en el medio
ambiente y buenos marcadores de la exposición humana a través de la dieta
[Wingfors et al. 2006]. Otro dato a tener en cuenta al estudiar la exposición a
PCBs es que, dado su carácter lipofílico, su distribución entre los distintos
tejidos está sometida a un equilibrio dinámico que depende de las trasferencias
de grasa entre uno y otro, lo que implica que, a efectos normalizadores, es
conveniente referir las concentraciones al contenido total de grasa [Phillips et
al. 1989].
En España, hasta fechas recientes, sólo un estudio basado en una
muestra representativa de población había consignado concentraciones de
contaminantes orgánicos persistentes en sangre pero, aunque incluía
plaguicidas organoclorados, no contemplaba PCBs [Porta et al. 2008]. Es por
ello que los niveles de exposición de PCBs en España, así como su distribución
geográfica o social son, en gran medida, desconocidos. Por tanto, el propósito
de esta parte del estudio fue establecer dichos niveles en la población
española, a través de una muestra representativa de la cohorte EPIC-España
de unas 1000 personas adultas sanas residentes en cinco provincias
diferentes, cuya muestra de sangre se recolectó entre 1992 y 1996.

Capítulo 4
143
4.2 Metodología experimental
4.2.1 Población y diseño del estudio
La cohorte EPIC-España está formada por cinco centros de la
macrocohorte EPIC que se corresponden con cinco áreas geográficas
españolas, tres en el norte: Asturias, Guipúzcoa y Navarra, y dos en el sur:
Granada y Murcia. Cuenta con la participación de: la Consejería de Salud y
Servicios Sanitarios del Principado de Asturias, la Escuela Andaluza de Salud
Pública de Granada, la Subdirección Territorial de Salud Pública de Guipúzcoa
del Departamento de Sanidad del Gobierno Vasco, la Consejería de Sanidad
de la Región de Murcia y el Departamento de Salud del Gobierno de Navarra,
siendo coordinada desde el Servicio de Epidemiología y Registro del Cáncer
del Instituto Catalán de Oncología. En su inicio estaba constituida por 41.438
voluntarios sanos (15.632 hombres y 25.806 mujeres), con edades
comprendidas entre 29 y 69 años. Todos fueron reclutados entre 1992 y 1996 y
procedían de diferentes niveles sociales y educativos.
Para este estudio se seleccionaron al azar 1000 sujetos de la cohorte
EPIC-España (200 de cada centro) con edades comprendidas entre 35 y 64
años (media 49 años) en el momento del reclutamiento de la cohorte, por
medio de un muestreo estratificado de acuerdo a la estructura edad-sexo de la
población española. De todos ellos se disponía de información sobre su dieta
habitual, estilo de vida y hábitos, ocupación y medidas antropométricas y se
disponía de una muestra de suero de al menos dos pajuelas almacenadas en
tanques de nitrógeno líquido a -196 ºC.
Una vez realizada la selección e iniciado el estudio, no fue posible
recuperar el material biológico de 14 individuos y no se pudo realizar el análisis
de dicho material por motivos técnicos (principalmente poca cantidad de
muestra en las pajuelas o mala calidad de la muestra de suero), para otros 33
individuos. Por tanto, el tamaño final de la muestra de población estudiada fue
de 953 sujetos (479 hombres y 474 mujeres).
4.2.2 Procedimientos analíticos
El análisis de PCBs en suero se llevó acabo según el método de placas
de extracción descrito en el capítulo 1 de este trabajo. Se determinaron los

PCBs en la cohorte EPIC-España
144
niveles séricos de los congéneres 28, 52, 101, 118, 138, 153 y 180. Por
razones prácticas se tomó 0,1 μg L-1 como límite de cuantificación (LQ) único
para todos ellos. La concentración de los congéneres 28, 52 y 101 estaba por
debajo del LQ en todas las muestras, a diferencia de los congéneres 138, 153 y
180, que superaban dicho límite en todas ellas. El congénere 118 presentaba
concentraciones superiores al LQ en todas las muestras menos una, a la que
se le asignó un valor de 0,05 µg L-1 (la mitad del LQ) a efectos estadísticos.
De las dos formas que existen para determinar el contenido en grasa total
del suero: gravimétricamente o sumando el contenido individual de cada uno de
los componentes de la fracción grasa (triglicéridos, fosfolípidos y colesterol
total, compuesto, a su vez, de colesterol libre y esterificado) [Akins et al. 1989],
se optó por la segunda, debido a que la primera es cara y laboriosa. En
cambio, la segunda se lleva a cabo de forma rutinaria a través de la
determinación enzimática de varios componentes. Existen diversas fórmulas
que permiten calcular el contenido total de grasa a partir de componentes
individuales [Phillips et al. 1989]. Para este trabajo se eligió una de las
denominadas “fórmulas cortas”:
623,0TGTC27,2TL ++=
donde:
TL = lípidos totales
TC = Colesterol total
TG = triglicéridos
En la aplicación de esta fórmula todos los componentes estaban
expresados en g L-1. El colesterol total y los triglicéridos se determinaron
enzimáticamente, desarrollando compuestos coloreados que después se
midieron espectrofotométricamente. [Wahlefeld 1974, Wiebe and Bernert 1984].
Por simple cociente entre el nivel sérico referido a suero (ng mL-1) y el
contenido en lípidos totales del suero (g mL-1), se obtenía el nivel sérico
referido a grasa (ng g-1).
Aunque se han propuesto nuevas fórmulas alternativas para el cálculo del
contenido en grasa a partir de los componentes individuales [Covaci et al.

Capítulo 4
145
2006, Rylander et al. 2006], un trabajo reciente [Bernert et al. 2007] indicaba
que la “fórmula corta” utilizada en este estudio proporciona una buena
estimación del contenido total en lípidos del suero.
4.2.3 Dieta, estilo de vida y medidas antropométricas
La dieta habitual durante el año anterior al reclutamiento, incluyendo las
variaciones estacionales, se estimó mediante entrevista personal llevada a
cabo por un nutricionista cualificado al respecto. Para ello se usó una versión
informatizada de un cuestionario validado anteriormente [EPIC Group of Spain
1997].
Otro cuestionario, también administrado por un entrevistador, se utilizó
para recoger información sobre características socio-demográficas y varios
factores relacionados con la salud y el estilo de vida. El nivel educativo se basó
en el grado de estudios alcanzado (universitario, bachiller, técnico o
profesional, graduado escolar, ninguno o sin alcanzar el graduado escolar). El
historial sobre actividad física, bien laboral o bien de ocio, se combinó en una
variable de cuatro niveles: inactivo, moderadamente inactivo, moderadamente
activo y activo [Wareham et al. 2003]. Para las mujeres, el cuestionario también
incluía información sobre el historial reproductivo, donde se demandaba, entre
otros datos, el número y la fecha de los nacimientos y el tiempo de lactancia.
El índice de masa corporal (IMC), en kg m-2, se calculó a partir de la altura
y el peso medidos en el reclutamiento. Los sujetos con un IMC ≤ 25 se
consideraron como de “peso normal”, los de un IMC entre 25 y 30 como “con
sobrepeso” y los de un IMC ≥ 30 como “obesos”.
4.2.4 Análisis estadístico
Los niveles séricos de cada PCB (congéneres 118, 138, 153 y 180), así
como la suma de PCBs, se expresaron en ng g-1 referidos a grasa aunque, por
razones de comparabilidad, en ocasiones también se utilizaron los valores
originales referidos a volumen de suero en µg L-1. Las distribuciones de los
niveles séricos presentaban un perfil sesgado hacia la derecha por lo que para
normalizar la distribución se procedió a la transformación logarítmica de los
valores, de forma que los resultados se expresaron como medias geométricas
con los correspondientes intervalos de confianza (IC) al 95%. Todas las medias

PCBs en la cohorte EPIC-España
146
presentadas se considerarían estandarizadas respecto a la estructura edad-
sexo de la población española entre 35 y 64 años. Otros ajustes estimados
relevantes se especifican en las correspondientes tablas de datos.
La comparación de medias se realizó mediante análisis de varianza y
regresión lineal, tras comprobar la ausencia de interacciones binarias, usando
el nivel sérico de los PCBs (log-transformado) como variable dependiente
[Armitage et al. 2002]. La tendencia lineal para las variables ordenadas se
evaluó por el grado de significación del coeficiente calculado al incluir en el
modelo cada una de dichas variables usando el valor numérico asignado a
cada categoría. El modelo de asociación de niveles séricos de PCBs con la
dieta se construyó mediante regresión paso a paso (stepwise regression)
[Armitage et al. 2002]. Se tomó 0,05 como nivel de significación general para el
valor p (test bilateral), excepto para la regresión paso a paso en que fue 0,1.
Todos los análisis estadísticos se llevaron a cabo con la versión 10 del
paquete estadístico STATA (StataCorp LP, College Station, TX, EE.UU.).

Capítulo 4
147
4.3 Resultados y discusión
La Tabla 4.1 describe los niveles séricos de los cuatro PCBs medidos en
la población de 953 adultos sanos seleccionados a partir de la cohorte EPIC-
España. En conjunto, la media geométrica de la suma de concentraciones fue
de 459 ng g-1 (referido a grasa) ó 3,1 μg L-1 (referido a suero); la mediana fue
de 450 ng g-1 y los percentiles 25 y 75 fueron 346 y 597 ng g-1 respectivamente.
El congénere más abundante fue el PCB 153 (media geométrica 186 ng g-1),
representando casi el 40% de la suma total, mientras que la menor
concentración fue para el mono-orto-sustituído PCB 118. Las concentraciones
medias de colesterol y triglicéridos fueron de 215,7 y 132,1 mg dL-1
respectivamente. Los niveles séricos de los cuatro congéneres estaban
altamente correlacionados entre sí, al igual que con la suma total, por lo que se
usó esta última para los posteriores análisis de datos.
Tabla 4.1 Niveles séricos de PCBs (congéneres 118, 138, 153, 180 y suma total) en 953 adultos sanos procedentes de la cohorte EPIC-España.
Media Aritmética
(DE)a Mediana (p25 – p75)b Media Geométrica (IC
95%)
PCB 118 ng g-1 grasa 39,6 (21,5) 34,4 (24,9 - 49,2) 34,9 (33,8 - 36,0)
μg L-1 0,27 (0,18) 0,23 (0,16 - 0,34) 0,23 (0,22 - 0,24)
PCB 138 ng g-1 grasa 117,5 (56,4) 104,6 (80,0 - 141,1) 106,7 (103,8 - 109,7)
μg L-1 0,81 (0,47) 0,70 (0,51 - 0,99) 0,71 (0,69 - 0,74)
PCB 153 ng g-1 grasa 205,2 (101,4) 182,6 (137,9 - 248,4) 186,0 (181,0 - 191,2)
μg L-1 1,42 (0,83) 1,22 (0,88 - 1,71) 1,25 (1,21 - 1,29)
PCB 180 ng g-1 grasa 136,4 (68,2) 119,5 (92,8 - 164,2) 123,3 (120,0 - 126,8)
μg L-1 0,94 (0,55) 0,80 (0,59 - 1,12) 0,83 (0,80 - 0,85)
Suma de ng g-1 grasa 498,8 (223,9) 449,6 (346,1 - 596,7) 458,8 (447,3 - 470,6)
PCBs μg L-1 3,44 (1,88) 2,96 (2,27 - 4,14) 3,07 (2,99 - 3,16)
a DE, desviación estándar. b p25, p75, percentiles 25 y 75, respectivamente.

PCBs en la cohorte EPIC-España
148
4.3.1 PCBs y principales variables demográficas
Las concentraciones totales de PCBs (media geométrica, IC 95%) según
sexo, edad y área de procedencia, se muestran en la Tabla 4.2. En promedio,
los hombres tenían un nivel sérico de PCBs alrededor de un 30% mayor que
las mujeres, siendo esta diferencia estadísticamente significativa. En ambos
sexos, la concentración de PCBs aumentaba, también de forma significativa,
con la edad, lo que es consistente con el efecto de bioacumulación de estos
compuestos. El perfil geográfico tendía a seguir un gradiente norte-sur, aunque
no de una forma totalmente clara: las mayores concentraciones se observaban
en Guipúzcoa, seguidas de las de Navarra, ambas localizadas en el norte; sin
embargo, Murcia, en la costa mediterránea sudeste, tenía un nivel ligeramente
superior al de Asturias, situada en el norte. Las menores concentraciones en
ambos géneros se registraban en Granada.
Tabla 4.2 Nivel sérico de PCBs referido a grasa (suma de congéneres 118, 138, 153 y 180 en ng g-1) en adultos sanos de la cohorte EPIC-España, según sexo edad y centro.
Hombresa Mujeresa Totalb
N Media Geométrica (IC 95%) N Media Geométrica (IC 95%) N Media Geométrica (IC 95%)
Edad c 35-44 años 185 477,3 (452,9 - 503,1) 190 390,5 (373,0 - 408,8) 375 431,4 (416,6 - 446.7) 45-54 años 159 542,8 (512,9 - 574,5) 161 393,0 (373,9 - 413,1) 320 461,5 (444,4 - 479.3) 55-64 años 135 563,4 (529,8 - 599,1) 123 434,1 (410,0 - 459,7) 258 497,8 (477,3 - 519.3)
Centro c Asturias 98 480,2 (446,7 - 516,1) 92 379,6 (355,4 - 405,5) 190 428,7 (408,2 - 450.3) Guipúzcoa 94 635,8 (590,6 - 684,4) 97 524,7 (492,1 - 559,5) 191 577,2 (549,6 - 606.1) Navarra 99 577,9 (537,8 - 620,9) 95 429,7 (402,7 - 458,5) 194 499,9 (476,3 - 524.8) Granada 89 414,9 (284,6 - 447,5) 94 334,8 (313,6 - 357,3) 183 371,3 (353,2 - 390.3) Murcia 99 519,8 (483,8 - 558,5) 96 367,1 (344,1 - 391,7) 195 438,0 (417,3 - 459.8)
Total c 479 522.2 (506,3 - 538,6) 474 402,4 (390,1 - 415,1) 953 458,8 (447,3 - 470,6)
a Datos ajustadas por edad y centro. b Datos ajustadas por sexo, edad y centro. c Valores de p en la comparación de medias. Comparación en conjunto, bajo la hipótesis nula
de que todas son iguales: sexo (p<0,001), centro (p<0,001); comparación evaluando la tendencia: edad (p<0,001).

Capítulo 4
149
4.3.2 PCBs y otras variables no dietéticas
La relación del nivel sérico de PCBs con factores no alimenticios se
presenta en la Tabla 4.3.
Tabla 4.3 Variables no alimenticias y nivel sérico de PCBs referido a grasa (suma de congéneres 118, 138, 153 y 180 en ng g-1) en adultos sanos de la cohorte EPIC-España.
N (%) Media Geométricaa (IC 95%) valor p
Nivel educativo Sin graduado escolar 302 (31,8%) 454,4 (437,0 - 472,5) Graduado escolar 370 (39,0%) 462,5 (446,5 - 479,1) 0,42 Preuniversitario / F. profesional 159 (16,8%) 460,4 (436,3 - 485,8) (conjunto) Universitario 118 (12,4%) 458,2 (430,5 - 487,7)
Año de toma de la muestrab 1993 301 (31,6%) 497,8 (478,9 - 517,4) 1994 364 (38,2%) 443,5 (428,2 - 459,3) 0,03 1995 288 (30,2%) 439,7 (422,7 - 457,4) (tendencia)
Actividad física Inactivo 151 (15,9%) 472,6 (447,4 - 499,2) Moderadamente inactivo 222 (23,3%) 487,5 (465,9 - 510,0) 0,10 Moderadamente activo 273 (28,7%) 475,8 (456,8 - 495,6) (conjunto) Activo 306 (32,1%) 419,7 (403,8 - 436,2)
IMC (kg m-2) ≤ 25 (de peso normal) 208 (21,8%) 446,8 (426,4 - 468,1) 25-30 (con sobrepeso) 473 (49,7%) 483,1 (468,4 - 498,3) 0,005 ≥ 30 (obeso) 271 (28,5%) 427,9 (410,8 - 445,8) (tendencia)
Lactancia Nunca 106 (22,6%) 378,6 (356,2 - 402,4) 0,06 Alguna vez 362 (77,4%) 407,9 (394,6 - 421,6) (conjunto)
Duración acumulada de los distintos períodos de lactancia Menos de 6 meses 130 (36,0%) 427,8 (404,9 - 452,0) De 6 a 12 meses 97 (26,9%) 405,1 (380,1 - 431,8) 0,011 Más de 12 meses 134 (37,1%) 392,4 (371,6 - 414,4) (tendencia)
a Datos ajustados por edad, sexo y centro. b 13 sujetos cuyas muestras se recogieron el último trimestre de 1992 se han incluido en la categoría de 1993 y 36 sujetos con muestras recogidas en el primer trimestre de 1996 se han incluido en 1995.
Como se ve, no hay un perfil claro de asociación con el nivel educativo o
la actividad física. Sin embargo, aunque el período de recolección de las
muestras de sangre fue relativamente corto (poco más de 3 años), se observa
que el nivel de PCBs tiende a decrecer conforme más cercano es el año en que

PCBs en la cohorte EPIC-España
150
se tomó la muestra. Así, en las muestras de 1995 es un 12% menor que en las
de 1993 (un 15% para el congénere 153), siendo la tendencia estadísticamente
significativa. La mayoría de resultados provenientes de otros estudios están de
acuerdo con esta observación y llevan a la conclusión general de que los
niveles séricos de PCBs están decreciendo desde la pasada década de los 80.
Algunos han analizado la variación temporal del congénere 153 mediante
monitorizaciones sucesivas sobre los mismos sujetos. Por ejemplo, en Suecia,
se recogieron muestras de sangre de 39 varones en 1991 y en 2001 [Hagmar
et al. 2006], siendo las respectivas concentraciones medias 360 y 240 ng g-1, lo
que suponía un descenso del 33% en un período de 10 años. También en
EE.UU. se ha evaluado esta tendencia temporal analizando muestras conjuntas
de suero almacenado para su utilización como controles de calidad analítica
[Sjödin et al. 2004]. Los niveles encontrados para el congénere 153 en las 36
muestras conjuntas fueron (mediana en ng g-1): 90 en las del período 1985-
1989, 66 en las de 1990-1994, 52 en las de 1995-1999 y 35 en las de 2000-
2002. Con estos datos se estimó que la vida media del congénere 153 era de
11 años.
Otra observación del presente estudio es que hay una significativa
asociación negativa entre el nivel de PCBs y el IMC tomado como variable
continua. A pesar de ello, tal como se ve en la Tabla 4.3, si se toma esta
variable de forma categorizada, la dirección de la asociación varía entre las
diferentes categorías. Mientras aumenta al pasar de sujetos normales a sujetos
con sobrepeso, disminuye al pesar a sujetos obesos. A este respecto cabe
decir que, aunque las correlaciones de los compuestos organoclorados con el
IMC varían considerablemente entre los diferentes estudios existentes, la
imagen general sugiere que las correlaciones son preferentemente positivas
para los plaguicidas, tales como el p,p’-DDT o su metabolito p,p’-DDE, mientras
que para los PCBs la preferencia es la inversa [Wolff et al. 2005].
Por lo que respecta a la lactancia, aunque está considerada una vía
importante de eliminación de PCBs, no se encontraron diferencias significativas
en dicho sentido asociadas a la misma. No obstante, entre aquellas mujeres
que la habían practicado existía una tendencia significativa a disminuir el nivel
sérico conforme mayor era el tiempo de lactancia acumulado. Otro dato a

Capítulo 4
151
añadir es que el tiempo medio trascurrido desde la última vez que se practicó la
lactancia hasta que se recogió la muestra de sangre era de 17 años.
4.3.3 PCBs y dieta
Seguidamente se exploró la asociación entre factores relacionados con la
dieta y el nivel sérico de PCBs, por medio de una regresión paso a paso. Como
variables se utilizaron las ingestas (g día-1) de los distintos grupos de alimentos,
que se calcularon a partir de los cuestionarios de dieta habitual. Esto permitió
una selección preliminar de cinco grupos, todos ellos formados por alimentos
de origen animal: derivados lácteos (excluida la leche), carne (carne roja y
aves), pescado, mariscos y, por último, aceites y grasas. Sin embargo, tras
elaborar un nuevo modelo ajustado por el resto de variables asociadas al nivel
sérico de PCBs (sexo, edad, centro, año de toma de la muestra de sangre e
IMC), sólo uno resultaba significativo y, eso sí, con una fuerte asociación
(p<0,001): el pescado. No obstante, con el límite de significación utilizado en la
regresión paso a paso (p=0,1), otro grupo presentaría también asociación: los
derivados lácteos (excluida la leche), por lo que se incluyó en los posteriores
modelos. Dada la fuerte asociación con el pescado, se construyó un nuevo
modelo para explorar si esta asociación era debida a algún tipo de pescado en
concreto. Para ello se sustituyó este grupo por otros tres subgrupos: pescado
graso (contenido en grasa ≥ 4% en peso), pescado no graso (contenido en
grasa < 4% en peso) y resto de pescado no clasificado en los dos subgrupos
anteriores. Resultó que la asociación era significativa con cualquiera de los tres
subgrupos. Todos estos resultados quedan reflejados en la Tabla 4.4.
La relación encontrada con las variables dietéticas coincide en buena
parte con un estudio entre mujeres americanas que usaba un planteamiento
similar [Laden et al. 1999] aunque, en dicho estudio, además del pescado, los
huevos presentaban también una fuerte asociación y no así los derivados
lácteos. Varios estudios de dieta total han estimado la ingesta de PCBs a
través de la determinación de sus niveles en alimentos en: Holanda [Baars et
al. 2004], Suecia [Darnerud et al. 2006], Bélgica [Baeyens et al. 2007],
Eslovaquia [Salgovicová and Pavlovicová 2007] y España [Llobet et al. 2008].
En conjunto, carne, pescado, derivados lácteos y grasas contribuyen al 80-90%
de la ingesta total de PCBs. La mayor contribución es del pescado en Suecia y

PCBs en la cohorte EPIC-España
152
en España, de las grasas en Eslovaquia y de la carne en Bélgica. En Holanda,
alrededor de la mitad de la ingesta total viene, a partes iguales, de la carne y
del pescado. En lo referente a los congéneres, el 153 es el mayoritario, seguido
del 138, el 180 y el 118.
Tabla 4.4 Grupos de la dieta predictores del nivel sérico de PCBs referido a grasa (suma de congéneres 118, 138, 153 y 180 en ng g-1) en adultos sanos de la cohorte EPIC-España.
Grupos de alimentos Ingesta diaria (g) valor p
media (DE)
Dieta sin ajustara Lácteos (excepto leche) 71,75 (79,2) 0,051 Carne (carne roja y aves) 90,78 (47,8) <0,001 Pescado 57,10 (41,1) <0,001 Mariscos 7,43 (10,1) 0,092 Aceites y grasas 31,77 (15,3) <0,001
Dieta ajustadab Lácteos (excepto leche) 71,75 (79,2) 0,095 Carne (carne roja y aves) 90,78 (47,8) 0,29 Pescado 57,10 (41,1) <0,001 Mariscos 7,43 (10,1) 0,92 Aceites y grasas 31,77 (15,3) 0,37
Ajuste por tipos de pescadoc
Pescado no graso 29,20 (31,3) <0,001 Pescado graso 19,22 (23,5) <0,001 Pescado no clasificado 8,68 (19,9) 0,014
a Grupos seleccionados por medio de una regresión lineal paso a paso (p<0,1), en la que sólo se consideraban como variables independientes del modelo los distintos grupos de alimentos de la dieta. b Modelo en el que, además de los grupos de alimentos seleccionados en la regresión lineal paso a paso, se incluían el resto de variables asociadas al nivel sérico de PCBs: sexo, edad, centro, año de toma de la muestra de sangre e IMC. c Modelo análogo al anterior, en el que el grupo de pescado se divide en tres subgrupos.
En España, tres estudios recientes han aportado datos sobre las
concentraciones de PCBs en muestras de pescado recogidas en diferentes
períodos entre 2001 y 2005 [Gómara et al. 2005, Bordajandi et al. 2006, Bocio

Capítulo 4
153
et al. 2007]. En el perfil de PCBs predominaba el congénere 153, apareciendo
también a menudo el 138 y el 180. Las especies con mayor concentración de
PCBs eran: atún, sardina, salmón, salmonete y anchoa. En menor
concentración también aparecían en moluscos y crustáceos como: ostras,
mejillones, almejas y gambas. Sin embargo, en un estudio llevado a cabo en
Guipúzcoa en 1994 [Goñi et al. 1994] las concentraciones del congénere 153
eran todavía mayores, con valores que iban de 10 - 250 ng g-1 frente a los 4 -
34 ng g-1 encontrados por Gómara y colaboradores en muestras de pescado
recogidas en diferentes mercados españoles entre 2000 y 2003 [Gómara et al.
2005]. A su vez, este último estudio estimaba que el contenido de PCBs en
pescado se había visto reducido en España por un factor entre 2 y 5 durante el
período de 1995 a 2003. Hay que remarcar también que España registraba la
ingesta de pescado más alta entre los 10 países europeos incluidos en el
proyecto EPIC [Welch et al. 2002]. La ingesta media diaria (g día-1) iba de 55 a
72 entre las mujeres y de 65 a 120 entre los hombres, mientras que en el resto
de países los valores estaban entre 10 y 50, con un perfil similar en cuanto a
consumo de pescado blanco y azul. Para pescados muy grasos (> 14% en
grasa) los mayores consumos se registraban en zonas costeras del norte de
Europa (Noruega, Suecia, Dinamarca).
4.3.4 Modelo final
Por último se elaboró un modelo multivariable con los siete principales
determinantes del nivel sérico de PCBs encontrados. En este modelo final las
dos variables asociadas a la dieta (ingesta de lácteos y pescado) se
categorizaron en cuartiles. Los resultados de este modelo final se presentan en
la Tabla 4.5. En ella se observa que los perfiles para el sexo, la edad, el centro,
el año de toma de la muestra y el IMC, son muy similares a los encontrados en
los primeros modelos (Tablas 4.2 y 4.3). En el caso de las variables
relacionadas con la dieta, se ve claro en la Tabla 4.5 que, al igual que en los
modelos de la Tabla 4.4, la influencia de la ingesta de pescado es mayor que la
de derivados lácteos. Para el pescado, el nivel sérico de PCBs se incrementa
alrededor de un 26% al pasar del primer al último cuartil mientras que, en el
caso de la ingesta de derivados lácteos, este incremento es tan sólo de
aproximadamente un 5%.

PCBs en la cohorte EPIC-España
154
Tabla 4.5 Nivel sérico de PCBs referido a grasa (suma de congéneres 118, 138, 153 y 180 en ng g-1) en adultos sanos de la cohorte EPIC-España, obtenido con un modelo multivariable a partir de los principales determinantes encontrados.
Media geométricaa (IC 95%) valor pb
Sexo Hombre 522,5 (507,0 - 538,5) <0,001 Mujer 402,5 (390,4 - 414,9) (conjunto)
Edad 35-44 años 431,4 (416,9 - 446,4) 45-54 años 461,7 (445,0 - 479,1) <0,001 55-64 años 498,0 (477,9 - 518,9) (tendencia)
Centro Asturias 428,7 (408,7 - 449,8) Guipúzcoa 577,2 (550,2 - 605,4) Navarra 499,9 (476,8 - 524,2) <0,001 Granada 371,1 (353,4 - 389,7) (conjunto) Murcia 438,3 (418,0 - 459,5)
Año de toma de 1993 498,2 (479,5 - 517,6) la muestra 1994 443,5 (428,4 - 459,1) 0,080
1995 439,7 (422,9 - 457,1) (tendencia)
IMC (kg m-2) ≤ 25 (de peso normal) 446,8 (426,8 - 467,7) 25-30 (con sobrepeso) 483,2 (468,7 - 498,1) 0,005 ≥ 30 (obeso) 427,9 (411,1 - 445,4) (tendencia)
Derivados lácteos 1er cuartil (≤ 16,5) 444,6 (426,0 - 464,0) (ingesta en g día-1, 2º cuartil (16,6-48,9) 446,5 (427,8 - 466,1) 0,046 categorizada en 3er cuartil (49,0-104,3) 476,5 (456,5 - 497,4) (tendencia) cuartiles) 4º cuartil (≥ 104,4) 468,5 (448,9 - 489,0)
Pescado 1er cuartil (≤ 28,9) 418,6 (401,0 - 436,9) (ingesta en g día-1, 2º cuartil (30,0-48,8) 429,1 (411,1 - 447,8) <0,001 categorizada en 3er cuartil (48,9-75,1) 469,4 (449,7 - 489,9) (tendencia) cuartiles) 4º cuartil (≥ 75,2) 526,3 (504,2 - 549,4)
a Datos mutuamente ajustados por todas las variables del modelo b Valores de p en la comparación de medias. Comparación en conjunto bajo la hipótesis nula de que todas son iguales (conjunto) para variables no numéricas, o comparación evaluando la tendencia (tendencia) para variables numéricas categorizadas.
Hay que recalcar que se alcanzaba mejor ajuste si las variables dietéticas
se empleaban como continuas en lugar de categorizadas, en cuyo caso el
modelo llegaba a explicar casi un tercio de la varianza del nivel sérico de PCBs
(R2=0,323). De hecho, un modelo similar se aplicó también para el nivel sérico
de cada congénere individual. El comportamiento de los distintos congéneres
frente a las variables predictoras era idéntico al de la suma de PCBs, aunque

Capítulo 4
155
se observaban algunas excepciones, casi todas relacionadas con el PCB 118:
el nivel sérico de PCB 118 no variaba con el sexo, presentaba asociación
negativa con la ingesta de derivados lácteos y se asociaba positivamente al
IMC, comportamiento, éste último, que compartía con el congénere 138.
4.3.5 Comparación con otros estudios
Realizar una comparativa de concentraciones de PCBs entre regiones o
países no es una tarea sencilla ya que diversos factores pueden ser
determinantes en la evaluación de los niveles séricos de PCBs y hay que
tenerlos en cuenta. Entre ellos se pueden citar: la edad de los individuos
sujetos a estudio, el año de toma de las muestras, los congéneres concretos
analizados y otras consideraciones técnicas derivadas de la metodología
analítica. Dado que los PCBs se bioacumulan, su concentración tiende a
incrementarse con la edad. Por otra parte, como se prohibió su producción y se
ha restringido su uso hasta casi la prohibición, se asume que la exposición
ambiental lleva ya tiempo disminuyendo. Como consecuencia, tanto la edad del
individuo como el punto temporal de la recogida de la muestra son cruciales. A
la hora de determinar el contenido de PCBs, no en todos los estudios se
utilizan los mismos perfiles de congéneres ni se expresan los resultados de la
misma manera, lo que dificulta las comparativas. Y en lo que respecta a la
metodología analítica, con el paso del tiempo y la incorporación de nuevas
técnicas y equipamientos, han ido mejorando mucho los resultados analíticos,
sobre todo en sensibilidad y especificidad.
Para realizar la comparativa de los niveles séricos de PCBs aquí
presentados se utilizó como unidad el ng g-1 y se seleccionaron estudios que
reportaban la media geométrica o la mediana de la concentración del
congénere 153, referida al contenido en grasa. La comparación se restringió a
estudios llevados a cabo bien sobre muestras representativas reales o bien
sobre grupos de población general no sujetos a fuentes de exposición
específicos, que aportaran datos sobre el año de toma de las muestras, la edad
y el sexo de los participantes. En este estudio, la media geométrica del nivel
sérico del congénere 153, referida a grasa, era de 186 ng g-1. Este valor es
más alto que los encontrados en estudios realizados en otros países europeos
como: 41 en Reino Unido [Thomas et al. 2006], 99 en Bélgica [Voorspoels et al.

PCBs en la cohorte EPIC-España
156
2002], 102 en Rumanía [Dirtu et al. 2006], 141 en Eslovaquia [Petrik et al.
2006] y 168 en Noruega [Sandanger et al. 2006]. También son marcadamente
mayores que los 33 ng g-1 observados en EE.UU. [CDC 2005], los 25 de Nueva
Zelanda [Bates et al. 2004] o los 97 de Japón [Masuda et al. 2005]. Aunque, a
excepción de las de Nueva Zelanda (1996-1997) y las de Japón (1999), el resto
de las muestras de sangre de estos estudios estaban recogidas entre 2001 y
2005, es decir, varios años después que las nuestras.
Otros estudios presentaban resultados específicos de cada sexo. Con
respecto a hombres, en el presente estudio el nivel sérico medio del congénere
153 era de 216 ng g-1, por supuesto referido a grasa, que resultaba menor que
los obtenidos en otros dos estudios con hombres llevados a cabo en Suecia:
296 ng g-1 con muestras de sangre recogidas en 1995 [Glynn et al. 2000], o
340 ng g-1 con sangre recogida en 1991 [Hagmar et al. 2006]; en este último
caso el nivel decreció hasta 170 ng g-1 cuando las muestras se volvieron a
recoger en 2001. Por otra parte, para las mujeres de nuestro estudio el nivel
era de 160 ng g-1, que es un valor intermedio entre los 147 ng g-1 encontrados
en Bélgica con muestras de 1999 [Koppen et al. 2002] y los 210 ng g-1 de
Suecia con muestras de 1991 [Grimvall et al. 1997]. Nuestros niveles también
eran mayores que la mediana de 106 ng g-1 hallada en EE.UU. con muestras
de 1989-1990 [Laden et al. 2001] o los 42 ng g-1 determinados en Canadá con
muestras de 2003-2004 [Sandanger et al. 2007].
Por lo que respecta a España, una revisión reciente [Porta et al. 2008]
incluía 18 estudios con resultados de concentraciones de PCBs. Las
concentraciones medias del congénere 153, referidas a grasa, iban de 11,6 a
230 ng g-1 (0,05 a 1,18 µg L-1 referidas a volumen de suero), mientras que la
media geométrica de nuestro estudio era de 186 ng g-1 referida a grasa (1,25
µg L-1 referida a volumen de suero). Hay que decir que ninguno de los estudios
recogidos en la revisión se llevó a cabo con muestras representativas de
población general, sino con sujetos hospitalizados o grupos de gente
potencialmente expuestos por ocupación laboral o zona de residencia.
Además, algunos utilizaron muestras de leche materna o tejido adiposo en
lugar de suero, aunque se asume que existe una buena correlación entre
niveles en tejido adiposo y niveles séricos referidos a grasa. Dos estudios

Capítulo 4
157
adicionales, no incluidos en la mencionada revisión, midieron PCBs en tejido
adiposo de mujeres hospitalizadas. En el primero de ellos [Costabeber and
Emanuelli 2003] se encontró una media de 121 ng g-1 para el congénere 153 y
en el segundo [Fernández et al. 2008] una mediana de 168 ng g-1 que
concordaba bastante con nuestra media geométrica de 160 ng g-1 en mujeres.
Todas estas comparaciones están basadas en el nivel sérico del
congénere 153, tomado como un buen indicador de la exposición total a PCBs.
Sin embargo, puede darse el caso de que poblaciones con similares niveles de
exposición total tengan diferentes perfiles. Por ejemplo, un estudio realizado en
España y Suecia con un amplio abanico de congéneres, encontró unos niveles
totales (suma de 33 congéneres) y una concentración de congénere 153 muy
similares en poblaciones de ambos países, pero mayores valores de
congéneres con alto grado de cloración (PCBs 182, 187, 194 y 201) en las
muestras de España que en las de Suecia [Wingfors et al. 2000].

PCBs en la cohorte EPIC-España
158
4.4 Conclusiones
Se ha determinado la concentración de cuatro PCBs (congéneres 118,
138, 153 y 180) del grupo de los indicadores en 953 muestras de suero
recogidas durante los años 1992-1996 en cinco provincias españolas. Las
muestras provenían de adultos sanos con edades comprendidas entre los 35-
64 años en el momento del reclutamiento en la cohorte EPIC-España, cuya
selección no estaba relacionada con ninguna exposición específica. La
concentración total (media geométrica de la suma de los cuatro congéneres)
era de 459 ng g-1 referida a grasa y 3,1 µg L-1 referida a volumen de suero.
Para el congénere 153 estos valores eran de 186 ng g-1 y 1,25 µg L-1,
respectivamente. Estas concentraciones presentaban una asociación
significativa con el sexo, la edad, el centro de procedencia, el año de
recolección de la muestra, el IMC, la ingesta de pescado, la ingesta de
derivados lácteos y, en el caso de mujeres que habían practicado la lactancia,
con el tiempo acumulado de lactancia. Los hombres presentaban valores más
altos que las mujeres. El nivel de PCBs aumentaba con la edad y era mayor en
las provincias del norte, presentando los residentes en Guipúzcoa un valor
medio 50% más alto que el de los de Granada. Los niveles de PCBs
descendían conforme más reciente era la fecha de recolección de la muestra.
La asociación con el IMC era negativa. La ingesta de pescado era el factor
dietético que presentaba mayor asociación con el nivel sérico de PCBs.
Aunque, en general, las provincias con mayor consumo de pescado eran
también las que presentaban mayores valores de PCBs, el ajuste por la ingesta
de pescado no llegaba a justificar las diferencias regionales, por lo que éstas
permanecen inexplicadas. La asociación con el tiempo acumulado de lactancia
resulta negativa, lo que está en concordancia con la consideración general de
que la lactancia es una forma importante de eliminación de PCBs.
Aunque la cohorte EPIC-España no es una muestra representativa de la
población general española, hay que tener en cuenta que se seleccionó a partir
de un amplio estudio base, en diferentes áreas geográficas e incluía sujetos de
diferentes niveles educativos. En adición, para este estudio se eligieron al azar
1000 sujetos de la cohorte por medio de un muestreo estratificado de acuerdo
a la estructura edad-sexo de la población española. Por lo tanto, las medias

Capítulo 4
159
reportadas pueden considerarse una estimación de los niveles séricos de PCBs
en población española de 35-64 años en el período 1992-1996. Las
concentraciones encontradas eran similares a las consignadas en otros países
europeos con muestras recogidas en el mismo período, aunque relativamente
mayores que las medidas en América del Norte o en estudios europeos más
recientes.

PCBs en la cohorte EPIC-España
160
4.5 Referencias
♦ Akins JR, Waldrep K, Bernert Jr. JT, 1989. The estimation of total serum lipids by a completely enzymatic ‘summation’ method. Clin. Chim. Acta 184, 219-226.
♦ Armitage P, Berry G, Matthews JNS, 2002. Statistical Methods in Medical Research 4th Ed. Blackwell Science Ltd., Oxford, UK.
♦ Baars AJ, Bakker MI, Baumann RA, Boon PE, Freijer JI, Hoogenboom LA, Hoogerbrugge R, van Klaveren JD, Liem AK, Traag WA, de Vries J, 2004. Dioxins, dioxin-like PCBs and non-dioxin-like PCBs in foodstuffs: occurrence and dietary intake in the Netherlands. Toxicol. Lett. 151, 51–61.
♦ Baeyens W, Leermakers M, Elskens M, Van Larebeke N, De Bont R, Vanderperren H, Fontaine A, Degroodt JM, Goeyens L, Hanot V, Windal I, 2007. PCBs and PCDD/FS in fish and fish products and their impact on the human body burden in Belgium. Arch. Environ. Contam. Toxicol. 52, 563–571.
♦ Bates MN, Buckland SJ, Garrett N, Ellis H, Needham LL, Patterson Jr. DG, Turner WE, Russell DG, 2004. Persistent organochlorines in the serum of the non-occupationally exposed New Zealand population. Chemosphere 54, 1431–1443.
♦ Bernert JT, Turner WE, Patterson Jr. DG, Needham LL, 2007. Calculation of serum “total lipid” concentrations for the adjustment of persistent organohalogen toxicant measurements in human samples. Chemosphere 68, 824-831.
♦ Bingham S, Riboli E, 2004. Diet and Cancer – The European Prospective Investigation into Cancer and Nutrition. Nat. Rev. Cancer 4, 206-215.
♦ Bocio A, Domingo JL, Falcó G, Llobet JM, 2007. Concentrations of PCDD/PCDFs and PCBs in fish and seafood from the Catalan (Spain) market: estimated human intake. Environ. Int. 33, 170–175.
♦ Bordajandi LR, Martín I, Abad E, Rivera J, González MJ, 2006. Organochlorine compounds (PCBs, PCDDs and PCDFs) in seafish and seafood from the Spanish Atlantic Southwest Coast. Chemosphere 64, 1450–1457.
♦ Bueno de Mesquita BH, González CA, 1997. Main hypotheses on diet and cancer investigated in the EPIC study. Eur. J. Cancer Prev. 6, 107-117.
♦ CDC (Centers for Disease Control), 2005. Third National Report on Human Exposure to Environmental Chemicals. Centers for Disease Control, Atlanta, GA, USA.
♦ Costabeber I, Emanuelli T, 2003. Influence of alimentary habits, age and occupation on polychlorinated biphenyl levels in adipose tissue. Food Chem. Toxicol. 41, 73–80.
♦ Covaci A, Voorspoels S, Thomsen C, van Bavel B, Neels H, 2006. Evaluation of total lipids using enzymatic methods for the normalization of persistent organic pollutants levels in serum. Sci. Total Environ. 366, 361-366.
♦ Darnerud PO, Atuma S, Aune M, Bjerselius R, Glynn A, Grawé KP, Becker W, 2006. Dietary intake estimations of organohalogen contaminants (dioxins, PCB, PBDE and chlorinated pesticides, e.g. DDT) based on Swedish market basket data. Food Chem. Toxicol. 44, 1597–1606.
♦ Dirtu AC, Cernat R, Dragan D, Mocanu R, Van Grieken R, Neels H, Covaci A, 2006. Organohalogenated pollutants in human serum from Iassy, Romania and their relation with age and gender. Environ. Int. 32, 797–803.
♦ EPIC Group of Spain, 1997. Relative Validity and reproducibility of a diet history questionnaire in Spain. I. Foods. EPIC Group of Spain. European prospective investigation into cancer and nutrition. Int. J. Epidemiol. 26 (Suppl. 1), S91-S99.
♦ Fernández MF, Kiviranta H, Molina-Molina JM, Laine O, López-Espinosa MJ, Vartiainen T, Olea N, 2008. Polychlorinated biphenyls (PCBs) and hydroxy- PCBs in adipose tissue of women in Southeast Spain. Chemosphere 71, 1196–1205.

Capítulo 4
161
♦ Gladen BC, Doucet J, Hansen LG, 2003. Assessing human polychlorinated biphenyl contamination for epidemiologic studies: lessons from patterns of congener concentrations in Canadians in 1992. Environ. Health Perspect. 111, 437-443.
♦ Gómara B, Bordajandi LR, Fernández MA, Herrero L, Abad E, Abalos M, Rivera J, Gonzálea MJ, 2005. Levels and trends of polychlorinated dibenzo-p-dioxins/furans (PCDD/Fs) and dioxin-like polychlorinated biphenyls (PCBs) in Spanish commercial fish and shellfish products, 1995–2003. J. Agric. Food Chem. 53, 8406–8413.
♦ Goñi F, Serrano E, Ibarluzea JM, Doronsoro M, Galdeano LG, 1994. Polychlorinated biphenyl residues in various fatty foods consumed in Guipúzcoa, the Basque Country (Spain). Food Addit. Contam. 11, 387–395.
♦ Grimvall E, Rylander L, Nilsson-Ehle P, Nilsson U, Strömberg U, Hagmar L. Ostman C, 1997. Monitoring of polychlorinated biphenyls in human blood plasma: methodological developments and influence of age, lactation, and fish consumption. Arch. Environ. Contam. Toxicol. 32, 329–336.
♦ Glynn AW, Wolk A, Aune M, Atuma S, Zettermark S, Maehle-Schmid M, Darnerud PO, Becker W, Vessby B, Adami HO, 2000. Serum concentrations of organochlorines in men: a search for markers of exposure. Sci. Total Environ. 263, 197-208.
♦ González CA, Navarro C, Martínez C y otros 11 autores, 2004. El Estudio Prospectivo Europeo sobre Cáncer y Nutrición (EPIC). Rev. Esp. Salud Pública 78, 167-176.
♦ Hagmar L, Wallin E, Vessby B, Jönsson BA, Bergman A, Rylander L, 2006. Intra-individual variations and time trends 1991–2001 in human serum levels of PCB, DDE and hexachlorobenzene. Chemosphere 64, 1507–1513.
♦ Koppen G, Covaci A, Van Cleuvenbergen R, Schepens P, Winneke G, Nelen V, van Larebeke N, Vlietinck R, Schoeters G, 2002. Persistent organochlorine pollutants in human serum of 50–65 years old women in the Flanders Environmental and Health Study (FLEHS). Part 1: Concentrations and regional differences. Chemosphere 48, 811–825.
♦ Laden F, Hankinson SE, Wolff MS, Colditz GA, Willett WC, Speizer FE, Hunter DJ, 2001. Plasma organochlorine levels and the risk of breast cancer: an extended follow-up in the Nurses’ Health Study. Int. J. Cancer 91, 568–574.
♦ Laden F, Neas LM, Spiegelman D, Hankinson SE, Willett WC, Ireland K, Wolff MS, Hunter DJ, 1999. Predictors of plasma concentrations of DDE and PCBs in a group of US women. Environ. Health Perspect. 107, 75–81.
♦ Llobet JM, Martí-Cid R, Castell V, Domingo JL, 2008. Significant decreasing trend in human dietary exposure to PCDD/PCDFs and PCBs in Catalonia, Spain. Toxicol. Lett. 178, 1117–1126.
♦ Masuda Y, Haraguchi K, Kono S, Tsuji H, Päpke O, 2005. Concentrations of dioxins and related compounds in the blood of Fukuoka residents. Chemosphere 58, 329–344.
♦ Park H, Lee SJ, Kang JH, Chang YS, 2007. Congener–specific approach to human PCB concentrations by serum analysis. Chemosphere 68, 1699-1706.
♦ Petrik J, Drobna B, Pavuk M, Jursa S, Wimmerova S, Chovancova J, 2006. Serum PCBs and organochlorine pesticides in Slovakia: age, gender, and residence as determinants of organochlorine concentrations. Chemosphere 65, 410–418.
♦ Phillips DL, Pirkle JL, Burse VW, Bernet JT, Henderson LO, Needham LL, 1989. Chlorinated hydrocarbon levels in human serum: effects of fasting and feeding. Arch. Environ. Contam. Toxicol. 18, 495-500.
♦ Porta M, Puigdomènech E, Ballester F, Selva J, Ribas-Fitó N, Domínguez-Boada L, Martín-Olmedo P, Olea N, Llop S, Fernández M, 2008. Estudios realizados en España sobre concentraciones en humanos de compuestos tóxicos persistentes. Gac. Sanit. 22, 248-266.
♦ Riboli E, 1992. Nutrition and cancer: background and rationale of the European Prospective Investigation into Cancer and Nutrition (EPIC). Ann. Oncol. 3, 783-791.

PCBs en la cohorte EPIC-España
162
♦ Rylander L, Nilsson-Ehle P, Hagmar L, 2006. A simplified precise method for adjusting serum levels of persistent organohalogen pollutants to total serum lipids. Chemosphere 62, 333-336.
♦ Sandanger TM, Sinotte M, Dumas P, Marchand M, Sandau CD, Pereg D, Bérubé S, Brisson J, Ayotte P, 2007. Plasma concentrations of selected organobromine compounds and polychlorinated biphenyls in postmenopausal women of Que´ bec, Canada. Environ. Health Perspect. 115, 1429–1434.
♦ Sandanger TM, Brustad M, Sandau CD, Lund E, 2006. Levels of persistent organic pollutants (POPs) in a coastal northern Norwegian population with high fish-liver intake. J. Environ. Monit. 8, 552–557.
♦ Salgovicová D, Pavlovicová D, 2007. Exposure of the population of the Slovak Republic to dietary polychlorinated biphenyls. Food Chem. Toxicol. 45, 1641–1649.
♦ Sjödin A, Jones RS, Focant JF, y otros 8 autores, 2004. Retrospective time-trend study of polybrominated diphenyl ether and polybrominated and polychlorinated biphenyl levels in human serum from the United States. Environ. Health Perspect. 112, 654–658.
♦ Thomas GO, Wilkinson M, Hodson S, Jones KC, 2006. Organohalogen chemicals in human blood from the United Kingdom. Environ. Pollut. 141, 30–41.
♦ Voorspoels S, Covaci A, Maervoet J, Schepens P, 2002. Relationship between age and levels of organochlorine contaminants in human serum of a Belgian population. Bull. Environ. Contam. Toxicol. 69, 22–29.
♦ Wahlefeld AW, 1974. Triglycerides determination after enzymatic hydrolysis. In: Bergmeyer HU (ed.), Methods of Enzymatic Analysis 2nd Ed. Academic Press Inc., New York, USA, 1831-1835.
♦ Wareham NJ, Jakes RW, Rennie KL, Schuit J, Mitchell J, Hennings S, Day NE, 2003. Validity and repeatability of a simple index derived from the short physical activity questionnaire used in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Public Health Nutr. 6, 407-413.
♦ Welch AA, Lund E, Amiano P, y otros 22 autores, 2002. Variability of fish consumption within the 10 European countries participating in the European Investigation into Cancer and Nutrition (EPIC) study. Public Health Nutr. 5 (6B), 1273–1285.
♦ Wiebe DA, Bernert Jr. JT, 1984. Influence of incomplete cholesteryl ester hydrolysis on enzymic measurements of cholesterol. Clin. Chem. 30, 352-356.
♦ Wingfors H, Seldén AI, Nilsson C, Haglund P, 2006. Identification of markers for PCB exposure in plasma from Swedish construction workers removing old elastic sealants. Ann. Occup. Hyg. 50, 65-73.
♦ Wingfors H, Lindström G, van Bavel B, Schuhmacher M, Hardell L, 2000. Multivariate data evaluation of PCB and dioxin profiles in the general population in Sweden and Spain. Chemosphere 40, 1083–1088.
♦ Wolff MS, Britton JA, Teitelbaum SL, Eng S, Deych E, Ireland K, Liu Z, Neugut AI, Santella RM, Gammon MD, 2005. Improving organochlorine biomarker models for cancer research. Cancer Epidemiol. Biomarkers Prev. 14, 2224–2236.

CAPÍTULO 5
Niveles séricos de plaguicidas organoclorados en la cohorte EPIC-España
5.1 Introducción
5.2 Metodología experimental
5.3 Resultados y discusión
5.4 Conclusiones
5.5 Referencias

Plaguicidas organoclorados en la cohorte EPIC-España
164

Capítulo 5
165
5.1 Introducción
Como ya se recogía en el capítulo introductorio, los plaguicidas
organoclorados comparten con los PCBs unas características y problemática
similares:
− Exposición principal por vía oral, de gran interés en salud pública
debido a carácter global, duradero, crónico y a dosis bajas, con
potencial toxicidad.
− Exposición que presenta síntomas sub-clínicos y, por lo tanto,
imperceptibles a corto plazo y que a largo plazo se pueden manifestar
en formas difícilmente asociables al agente causante.
− Exposición con estudio toxicológico directo y experimental muy
complicado, por lo que se recurre a estudios epidemiológicos
observacionales.
− Exposición de la que se detecta un déficit de información sobre los
niveles que alcanza y los efectos que causa.
− Exposición en la que la monitorización biológica en suero constituye
una herramienta útil en estudios epidemiológicos para valorar su
riesgo.
Por tanto y, por idénticas razones y fines, también comparten ser objeto
del mismo interés por la determinación de sus niveles séricos en amplias bases
de población.
Entre los plaguicidas organoclorados, el p,p’-DDT y su principal metabolito
el p,p’-DDE, el HCB, el heptacloro epóxido y el isómero más persistente del
grupo HCH, el β-HCH, son de los que aparecen más frecuentemente cuando
se realiza este tipo de monitorización [WHO 2003]. Todos ellos se consideran
estables en el medio ambiente y buenos marcadores de la exposición humana
a través de la dieta. De todos ellos destacan por su persistencia el p,p’-DDE
(principal marcador del grupo DDT), el HCB (principal marcador de sí mismo), y
el β-HCH (principal marcador del grupo HCH). Algo menos persistentes
resultan el p,p’-DDT y el heptacloro epóxido (principal marcador del heptacloro
y marcador secundario del clordano).

Plaguicidas organoclorados en la cohorte EPIC-España
166
Otro dato a tener en cuenta, al igual que ocurría con los PCBs, es que,
dado el carácter lipofílico de estos compuestos, su distribución entre los
distintos tejidos está sometida a un equilibrio dinámico que depende de las
trasferencias de grasa entre uno y otro, lo que implica que, a efectos
normalizadores, es conveniente referir las concentraciones al contenido total de
grasa [Phillips et al. 1989].
En España, aunque la mayoría de los plaguicidas organoclorados se
prohibieron durante la década 1970-1980 y, como consecuencia, parece que
sus niveles están decreciendo, estos compuestos aparecen a menudo al
analizar los alimentos y concentraciones detectables de ellos se observan al
monitorizar la población, incluso a veces en proporciones verdaderamente altas
[Porta et al. 2008, Porta et al. 2009].
A pesar de ello, no se ha realizado una monitorización exhaustiva de
estos compuestos. Además, casi todos los estudios se han centrado en grupos
de población específicos con una exposición laboral o por razones de
residencia y sólo uno estaba basado en una muestra representativa de
población general [Porta et al. 2008, Porta et al. 2009]. Es por ello que los
niveles de exposición de estos plaguicidas organoclorados en España, así
como su distribución geográfica o social son, en gran medida, desconocidos.
Por tanto, el propósito de esta parte del estudio fue establecer dichos niveles
en la población española, a través de una muestra representativa de la cohorte
EPIC-España de unas 1000 personas adultas sanas residentes en cinco
provincias diferentes, cuya muestra de sangre se recolectó entre 1992 y 1996.

Capítulo 5
167
5.2 Metodología experimental
5.2.1 Población estudiada
Como en el caso de los PCBs, para este estudio se seleccionaron al azar
1000 sujetos de la cohorte EPIC-España (200 de cada centro: Asturias,
Granada, Guipúzcoa, Navarra y Murcia) con edades comprendidas entre 35 y
64 años en el momento del reclutamiento de la cohorte, por medio de un
muestreo estratificado de acuerdo a la estructura edad-sexo de la población
española. De todos ellos se disponía de información sobre su dieta habitual,
estilo de vida y hábitos, ocupación y medidas antropométricas y se disponía de
una muestra de suero de al menos dos pajuelas almacenadas en tanques de
nitrógeno líquido a -196 ºC.
Una vez realizada la selección e iniciado el estudio, no fue posible
recuperar el material biológico de 14 individuos y no se pudo realizar el análisis
de dicho material por motivos técnicos (principalmente poca cantidad de
muestra en las pajuelas o mala calidad de la muestra de suero), para otros 33
individuos. Por tanto, el tamaño final de la muestra de población estudiada fue
de 953 sujetos (479 hombres y 474 mujeres).
5.2.2 Procedimientos analíticos
El análisis de plaguicidas organoclorados en suero se llevó acabo según
el método de placas de extracción descrito en el capítulo 1 de este trabajo. Se
determinaron los niveles séricos de los compuestos HCB, β-HCH, heptacloro
epóxido, p,p’-DDE y p,p’-DDT. Por razones prácticas se tomó 0,4 µg L-1 como
límite de cuantificación (LQ) único para todos ellos. La concentración de
heptacloro epóxido estaba por debajo del LQ en todas las muestras. El p,p’-
DDT presentaba valores iguales o mayores que el LQ en aproximadamente
una cuarta parte de las muestras. Para el resto de los compuestos (HCB, β-
HCH y p,p’-DDE) se superaba dicho límite de forma mayoritaria (> 75% de las
muestras). Para estos últimos compuestos, en aquellas muestras con niveles
por debajo del LQ se les asignó un valor de 0,2 μg L-1 (la mitad del LQ) a
efectos estadísticos.

Plaguicidas organoclorados en la cohorte EPIC-España
168
El contenido en grasa total del suero se determinó a partir de
componentes individuales (colesterol total y triglicéridos) empleando la misma
fórmula que en el caso de los PCBs [Phillips et al. 1989].
623,0TGTC27,2TL ++=
donde:
TL = lípidos totales
TC = colesterol total
TG = triglicéridos
En la aplicación de esta fórmula todos los componentes estaban
expresados en g L-1. El colesterol total y los triglicéridos se determinaron
enzimáticamente, desarrollando compuestos coloreados que después se
midieron espectrofotométricamente. [Wahlefeld 1974, Wiebe and Bernert 1984].
El nivel sérico referido a grasa (ng g-1) se obtenía mediante el cociente entre el
nivel sérico referido a suero (ng mL-1) y el contenido en lípidos totales del suero
(g mL-1).
5.2.3 Cuestionarios de dieta, estilo de vida y medidas antropométricas
La ingesta habitual durante el último año, incluyendo las variaciones
estacionales, se estimó mediante entrevista personal realizada por un
nutricionista con ayuda de la versión informatizada de un cuestionario de dieta
previamente validado y desarrollado específicamente para la parte del proyecto
EPIC que se lleva a cabo en España [EPIC Group of Spain 1997]. El
cuestionario contenía una lista de más de 600 alimentos y bebidas.
Otro cuestionario, también administrado por un entrevistador, se utilizó
para recoger información sobre características socio-demográficas y varios
factores relacionados con la salud y el estilo de vida. El nivel educativo se basó
en el grado de estudios alcanzado (universitario, bachiller, técnico o
profesional, graduado escolar, ninguno o sin alcanzar el graduado escolar). El
historial sobre actividad física, bien laboral o bien de ocio, se combinó en una
variable de cuatro niveles: inactivo, moderadamente inactivo, moderadamente
activo y activo [Wareham et al. 2003]. Existía un apartado relacionado con la
exposición laboral pasada o actual, en el que, entre otras cosas, se preguntaba

Capítulo 5
169
al sujeto si había trabajado alguna vez en la agricultura y, en caso afirmativo, si
su trabajo implicaba también la manipulación de plaguicidas. Para las mujeres,
el cuestionario también contenía una sección sobre el historial reproductivo,
que recababa información sobre el número y la fecha de los nacimientos, si se
practicó la lactancia (sola o complementada de otro tipo de alimentación) y la
duración de la misma. A partir de estos datos se calculó la duración acumulada
de la lactancia, mediante la suma de las semanas o meses de los períodos de
lactancia en los distintos nacimientos, así como los años transcurridos desde
que cesó la última lactancia.
El índice de masa corporal (IMC), en kg m-2, se calculó a partir de la altura
y el peso medidos en el reclutamiento. Los sujetos con un IMC ≤ 25 se
consideraron como de “peso normal”, los de un IMC entre 25 y 30 como “con
sobrepeso” y los de un IMC ≥ 30 como “obesos”.
5.2.4 Análisis estadístico
Los niveles séricos de cada compuesto (HCB, β-HCH, p,p’-DDE y p,p’-
DDT), se expresaron en ng g-1 referidos a grasa aunque, por razones de
comparabilidad, en ocasiones también se utilizaron los valores originales
referidos a volumen de suero en µg L-1. Las distribuciones de estos niveles
séricos presentaban un perfil sesgado hacia la derecha por lo que las variables
se transformaron usando logaritmos naturales, de forma que los resultados se
expresaron como medias geométricas con los correspondientes intervalos de
confianza (IC) al 95%. Además de la media geométrica, a efectos descriptivos
se calcularon también la mediana y el percentil 75 con sus correspondientes IC
al 95%. Dada la forma de selección de la muestra, todas las medias
presentadas se considerarían estandarizadas respecto a la estructura edad-
sexo de la población española entre 35 y 64 años. Otros ajustes estimados
relevantes se especifican en las correspondientes tablas de datos.
La comparación de medias se llevó a cabo mediante análisis de varianza
y regresión lineal, tras comprobar la ausencia de interacciones binarias, usando
el nivel sérico de cada compuesto (log-transformado) como variable
dependiente [Armitage et al. 2002]. La tendencia lineal para las variables
ordenadas se evaluó por el grado de significación del coeficiente calculado al

Plaguicidas organoclorados en la cohorte EPIC-España
170
incluir en el modelo cada una de dichas variables usando el valor numérico
asignado a cada categoría. Como nivel de significación general para el valor p
(test bilateral) se tomó 0,05.
Todos los análisis estadísticos se llevaron a cabo con la versión 10 del
paquete estadístico STATA (StataCorp LP, College Station, TX, EE.UU.).

Capítulo 5
171
5.3 Resultados y discusión
Las principales características socio-demográficas y otros factores de la
población estudiada se muestran en la Tabla 5.1.
Tabla 5.1 Principales características de la población estudiada.
N (%)a N (%)a N (%)a
CentroAsturias 98 (20,5%) 92 (19,4%) 190 (19,9%)Guipúzcoa 94 (19,6%) 97 (20,5%) 191 (20,0%)Navarra 99 (20,7%) 95 (20,0%) 194 (20,4%)Granada 89 (18,6%) 94 (19,8%) 183 (19,2%)Murcia 99 (20,7%) 96 (20,3%) 195 (20,5%)
Edad (años)35-44 185 (38,6%) 190 (40,1%) 375 (39,3%)45-54 159 (33,2%) 161 (34,0%) 320 (33,6%)55-64 135 (28,2%) 123 (25,9%) 258 (27,1%)
IMC (kg m -2 )≤25 72 (15,1%) 136 (28,7%) 208 (21,8%)25-30 280 (58,6%) 193 (40,7%) 473 (49,7%)≥30 126 (26,4%) 145 (30,6%) 271 (28,5%)
Año de toma de la muestra b
1993 160 (33,4%) 141 (29,7%) 301 (31,6%)1994 178 (37,2%) 186 (39,2%) 364 (38,2%)1995 141 (29,4%) 147 (31,0%) 288 (30,2%)
Nivel educativoSin graduado escolar 125 (26,3%) 177 (37,4%) 302 (31,8%)Graduado escolar 164 (34,5%) 206 (43,6%) 370 (39,0%)Bachiller 67 (14,1%) 28 (5,9%) 95 (10,0%)F. profesional 39 (8,2%) 25 (5,3%) 64 (6,7%)Universitario 81 (17,0%) 37 (7,8%) 118 (12,4%)
Trabajó en agricultura 145 (30,3%) 76 (16,0%) 221 (23,2%)
Manipuló plaguicidas 68 (14,2%) 17 (3,6%) 85 (8,9%)
LactanciaNunca 106 (22,6%)Alguna vez 362 (77,4%)
Hombres (479) Mujeres (474) Total (953)
a Porcentajes calculados sobre el total de sujetos con información válida. b 13 sujetos cuyas muestras se recogieron el último trimestre de 1992 se han incluido en la categoría de 1993 y 36 sujetos con muestras recogidas en el primer trimestre de 1996 se han incluido en 1995.

Plaguicidas organoclorados en la cohorte EPIC-España
172
La edad media era de 49 años (48,7 las mujeres y 49,4 los hombres).
Menos de la cuarta parte de la población estaba dentro de lo considerado como
“peso normal”. Alrededor de una tercera parte carecía de educación formal (sin
graduado escolar). Casi una cuarta parte de los sujetos había trabajado alguna
vez en agricultura, aunque menos de un 10% había manipulado plaguicidas,
siendo sustancialmente más altas, ambas proporciones, para los hombres. Más
de tres cuartas partes de las mujeres habían practicado alguna vez la lactancia.
Aunque algunos sujetos se reclutaron durante el último trimestre de 1992 o el
primer trimestre de 1996, la mayoría de los sujetos se reclutó entre los años
1993-1995, con una distribución relativamente homogénea.
En cuanto a los plaguicidas organoclorados determinados en suero, el
compuesto más frecuente fue el p,p’-DDE, presente en el 98% de las muestras,
seguido del HCB y el β-HCH, hallados en el 89% y el 77% de las muestras,
respectivamente. Sin embargo, el p,p’-DDT sólo pudo ser cuantificado en el
26% de las muestras por lo que, a efectos descriptivos, las medianas y medias
geométricas, calculadas a partir de estos datos, no resultaban tan significativas
como en el caso de los otros tres compuestos. Las medias geométricas de los
niveles séricos hallados de p,p’-DDE, HCB y β-HCH fueron, respectivamente,
822, 379 y 167 ng g-1 (referidos a grasa). Excepto para el p,p’-DDE, las
concentraciones eran mayores en mujeres que en hombres. Todos estos
resultados están reflejados en la Tabla 5.2.
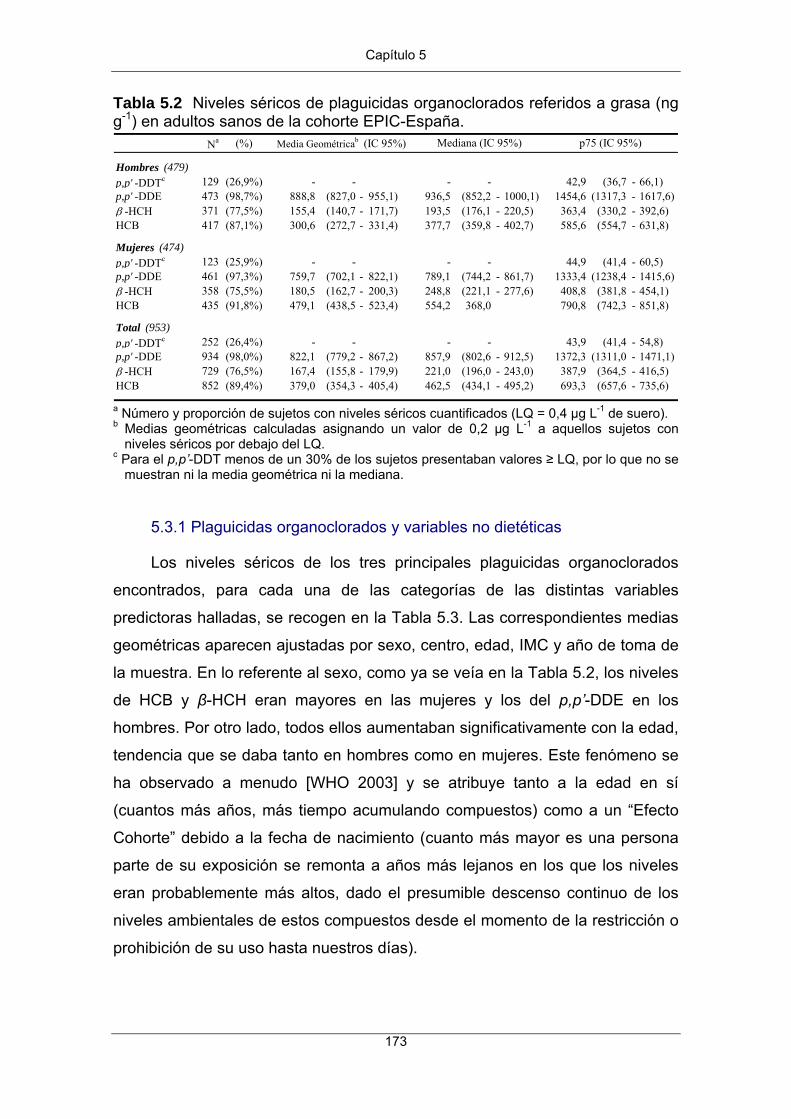
Capítulo 5
173
Tabla 5.2 Niveles séricos de plaguicidas organoclorados referidos a grasa (ng g-1) en adultos sanos de la cohorte EPIC-España.
Na (%)
Hombres (479)p,p' -DDTc 129 (26,9%) - - - - 42,9 (36,7 - 66,1)p,p' -DDE 473 (98,7%) 888,8 (827,0 - 955,1) 936,5 (852,2 - 1000,1) 1454,6 (1317,3 - 1617,6)β -HCH 371 (77,5%) 155,4 (140,7 - 171,7) 193,5 (176,1 - 220,5) 363,4 (330,2 - 392,6)HCB 417 (87,1%) 300,6 (272,7 - 331,4) 377,7 (359,8 - 402,7) 585,6 (554,7 - 631,8)
Mujeres (474)p,p' -DDTc 123 (25,9%) - - - - 44,9 (41,4 - 60,5)p,p' -DDE 461 (97,3%) 759,7 (702,1 - 822,1) 789,1 (744,2 - 861,7) 1333,4 (1238,4 - 1415,6)β -HCH 358 (75,5%) 180,5 (162,7 - 200,3) 248,8 (221,1 - 277,6) 408,8 (381,8 - 454,1)HCB 435 (91,8%) 479,1 (438,5 - 523,4) 554,2 368,0 790,8 (742,3 - 851,8)
Total (953)p,p' -DDTc 252 (26,4%) - - - - 43,9 (41,4 - 54,8)p,p' -DDE 934 (98,0%) 822,1 (779,2 - 867,2) 857,9 (802,6 - 912,5) 1372,3 (1311,0 - 1471,1)β -HCH 729 (76,5%) 167,4 (155,8 - 179,9) 221,0 (196,0 - 243,0) 387,9 (364,5 - 416,5)HCB 852 (89,4%) 379,0 (354,3 - 405,4) 462,5 (434,1 - 495,2) 693,3 (657,6 - 735,6)
Media Geométricab (IC 95%) Mediana (IC 95%) p75 (IC 95%)
a Número y proporción de sujetos con niveles séricos cuantificados (LQ = 0,4 µg L-1 de suero). b Medias geométricas calculadas asignando un valor de 0,2 µg L-1 a aquellos sujetos con
niveles séricos por debajo del LQ. c Para el p,p’-DDT menos de un 30% de los sujetos presentaban valores ≥ LQ, por lo que no se
muestran ni la media geométrica ni la mediana.
5.3.1 Plaguicidas organoclorados y variables no dietéticas
Los niveles séricos de los tres principales plaguicidas organoclorados
encontrados, para cada una de las categorías de las distintas variables
predictoras halladas, se recogen en la Tabla 5.3. Las correspondientes medias
geométricas aparecen ajustadas por sexo, centro, edad, IMC y año de toma de
la muestra. En lo referente al sexo, como ya se veía en la Tabla 5.2, los niveles
de HCB y β-HCH eran mayores en las mujeres y los del p,p’-DDE en los
hombres. Por otro lado, todos ellos aumentaban significativamente con la edad,
tendencia que se daba tanto en hombres como en mujeres. Este fenómeno se
ha observado a menudo [WHO 2003] y se atribuye tanto a la edad en sí
(cuantos más años, más tiempo acumulando compuestos) como a un “Efecto
Cohorte” debido a la fecha de nacimiento (cuanto más mayor es una persona
parte de su exposición se remonta a años más lejanos en los que los niveles
eran probablemente más altos, dado el presumible descenso continuo de los
niveles ambientales de estos compuestos desde el momento de la restricción o
prohibición de su uso hasta nuestros días).

Plaguicidas organoclorados en la cohorte EPIC-España
174
Tabla 5.3 Niveles séricos de plaguicidas organoclorados referidos a grasa (ng g-1) en adultos sanos de la cohorte EPIC-España y variables predictoras no dietéticas.
Sexo Hombres 888,0 (828,4 - 952,0) 156,0 (142,7 - 170,5) 300,6 (278,0 - 325,0)Mujeres 759,7 (708,5 - 814,7) 180,5 (165,1 - 197,4) 479,1 (442,9 - 518,2)
valor p (conjunto) b
Centro Asturias 608,1 (544,6 - 679,0) 107,3 (93,2 - 123,5) 259,2 (229,0 - 293,5)Guipúzcoa 664,5 (595,3 - 741,8) 135,5 (117,7 - 156,0) 485,1 (428,7 - 549,0)Navarra 750,2 (672,6 - 836,7) 162,1 (141,0 - 186,4) 722,2 (638,8 - 816,5)Granada 957,4 (855,3 - 1071,6) 170,8 (147,9 - 197,3) 310,3 (273,4 - 352,2)Murcia 1287,2 (1154,4 - 1435,3) 325,2 (282,9 - 373,7) 273,8 (242,3 - 309,5)
valor p (conjunto) b
Edad 35-44 716,6 (662,5 - 775,1) 119,1 (107,7 - 131,6) 282,4 (258,6 - 308,5)(años) 45-54 835,5 (767,3 - 909,7) 179,9 (161,3 - 200,6) 402,8 (366,0 - 443,2)
55-64 981,9 (893,3 - 1079,4) 253,4 (224,5 - 286,0) 539,7 (485,2 - 600,3)valor p (tendencia) b
IMC ≤25 632,4 (569,1 - 702,7) 120,2 (105,1 - 137,6) 265,9 (236,1 - 299,3)(kg m -2 ) 25-30 840,9 (784,1 - 901,8) 159,1 (145,5 - 174,0) 364,4 (336,8 - 394,1)
≥30 964,7 (879,6 - 1058,0) 237,6 (211,1 - 267,3) 533,5 (480,9 - 591,8)valor p (tendencia) b
Año de toma 1993 887,8 (813,2 - 969,3) 201,9 (180,4 - 225,8) 477,2 (432,3 - 526,7)de la muestra c 1994 812,4 (750,1 - 879,7) 170,0 (153,6 - 188,3) 427,8 (391,1 - 467,8)
1995 768,9 (703,0 - 841,0) 136,0 (121,3 - 152,5) 256,1 (231,6 - 283,2)valor p (tendencia) b
Lactancia d Nunca 715,2 (614,7 - 832,2) 165,4 (136,8 - 200,1) 413,6 (358,1 - 477,6)Alguna vez 772,5 (711,7 - 838,4) 189,0 (166,8 - 204,9) 497,1 (459,1 - 538,2)
valor p (conjunto) b
Años trascurridosdesde la < 10 683,2 (559,5 - 834,2) 116,2 (90,4 - 149,2) 260,1 (215,1 - 314,5)lactancia e ≥ 10 791,9 (723,8 - 866,4) 203,1 (181,4 - 227,4) 566,8 (520,4 - 617,4)
valor p (conjunto) b
0,82 0,52 0,06
0,40 0,08 <0,001
Media Geométricaa (IC 95%)
0,005
<0,001
<0,001
p,p' -DDE β -HCH HCB
0,014
<0,001
<0,001
<0,001
0,001
<0,001
<0,001 <0,001
<0,001
<0,001
<0,001
<0,001
a Datos ajustados por sexo, centro, edad, IMC y año de toma de la muestra. b Valores de p en la comparación de medias. Comparación en conjunto bajo la hipótesis nula
de que todas son iguales (conjunto) para variables no numéricas, o comparación evaluando la tendencia (tendencia) para variables numéricas categorizadas.
c 13 sujetos cuyas muestras se recogieron el último trimestre de 1992 se han incluido en la categoría de 1993 y 36 sujetos con muestras recogidas en el primer trimestre de 1996 se han incluido en 1995.
d Nunca: 106 mujeres. Alguna vez: 362 mujeres. e A partir de los datos de 362 mujeres que alguna vez habían practicado la lactancia.
Al igual que ocurría con los PCBs, el nivel sérico de estos compuestos
descendía también de forma significativa con el año de toma de la muestra. En
efecto, aunque el reclutamiento se llevó a cabo en un período de tiempo inferior
a cuatro años, se registraba una sustancial disminución en los niveles séricos
de estos compuestos entre los sujetos que se incorporaron en el año 1993
(último trimestre de 1992 incluido) y los que lo hicieron en 1995 (primer

Capítulo 5
175
trimestre de 1996 incluido). Este descenso iba desde el 13% para el p,p’-DDE
hasta el 50% para el HCB, ocupando un lugar intermedio el β-HCH con el 33%.
Esto refuerza lo indicado al explicar el anteriormente mencionado “Efecto
Cohorte” en base a una disminución, ya constatada en diversas publicaciones
[Sullivan and Krieger 2001, Hagmar et al. 2006], de los niveles ambientales de
estos compuestos desde el momento de la restricción o prohibición de su uso
hasta nuestros días y pone de relieve la eficacia de las medidas que se han
tomado al respecto.
Otro resultado a tener en cuenta de este estudio es que, en función de los
compuestos considerados, se distinguían claramente dos perfiles geográficos
de corte norte-sur. Por lo que respecta al p,p’-DDE y β-HCH, los niveles más
bajos se encontraron en dos provincias del norte (Asturias y Guipúzcoa), con
mayor peso de la actividad industrial que la agrícola en su economía, mientras
los niveles más altos se registraban en Murcia, seguida de Granada, ambas en
la costa mediterránea sur de España y de larga tradición agrícola, ocupando
Navarra, una provincia del norte tradicionalmente agrícola pero con una fuerte
trasformación industrial en las últimas tres décadas, una posición intermedia.
En cambio, para el HCB el perfil de alguna forma se invertía, hallándose los
niveles más altos en dos provincias del norte colindantes (Navarra y
Guipúzcoa) y niveles más bajos en las dos provincias del sur (Murcia y
Granada), marcando Asturias de nuevo el mínimo. En la provincia de Murcia,
que registraba los mayores niveles de p,p’-DDE y β-HCH, la agricultura ha sido
la principal fuente de recursos económicos durante décadas debido a razones
geográficas y climatológicas, con un uso intensivo de insecticidas como el DDT
en el pasado y lindano algo más recientemente. Según un estudio [Molina et al.
2005] los niveles de DDT, lindano (γ-HCH) y β-HCH en tejido graso de
pacientes residentes en Murcia eran notables, siendo significativamente más
altos en áreas de cultivo hortícola intensivo con sistema de riego tradicional,
cultivo extensivo de secano y cultivo en invernadero, todos ellos con una
necesidad moderada o alta de plaguicidas. Respecto al HCB, cuyos niveles
más altos se daban en Navarra, hay que decir que aunque en el pasado tuvo
muchos usos en la agricultura y la industria, prácticamente han cesado desde
1970. Sin embargo, todavía puede aparecer como subproducto en la

Plaguicidas organoclorados en la cohorte EPIC-España
176
fabricación de compuestos clorados (disolventes, compuestos aromáticos
halogenados, plaguicidas, etc.) y puede estar presente como contaminante en
muchos de ellos y sus derivados, así como en emisiones provenientes de
incineradoras de residuos [WHO 2003]. Aún teniendo en cuenta lo anterior, no
hemos podido identificar fuentes específicas en dicha región que pudieran
explicar la diferencia de niveles con las otras zonas.
En la Tabla 5.3 también se observa una clara y significativa asociación
positiva de los niveles séricos de estos plaguicidas con el IMC. Esto está en
consonancia con los resultados de la mayoría de estudios trasversales, aunque
el perfil puede cambiar dependiendo de factores temporales asociados a la
exposición [Wolff et al. 2005]. Inicialmente las correlaciones tienden a ser
negativas por simple dilución, ya que valores más altos de IMC implican mayor
cantidad de tejido adiposo. Posteriormente, debido a que la farmacocinética de
los compuestos organoclorados se ve alterada por el IMC, en el sentido de que
su vida media es mayor en personas obesas que en delgadas, la eliminación
se ralentiza en los primeros y tras dos o más períodos de vida media la carga
corporal se vuelve más alta en los obesos que en los delgados, invirtiéndose el
sentido de la correlación a positivo [Wolff et al. 2005, Wolff et al. 2007]. De aquí
se deduce que la acusada correlación positiva observada en el este estudio
sugiere una exposición de larga trayectoria temporal.
Por lo que respecta a la lactancia, considerada vía importante de
eliminación, no se encontró asociación significativa con los niveles séricos.
Tampoco el tiempo de lactancia acumulado que, en promedio, era de
aproximadamente un año (mediana 7,5 meses). Sin embargo, los niveles eran
menores en aquellas mujeres que habían practicado la lactancia en los últimos
10 años, en comparación con las que llevaban 10 años o más sin hacerlo,
aunque esta diferencia sólo era significativa para el HCB, quedando bastante
cerca del límite el β-HCH. Lo que induce a pensar que la ingesta que se ha ido
produciendo tras el cese de la lactancia ha ido reduciendo la influencia de ésta
sobre los niveles séricos y que, trascurridos uno o dos períodos de vida media,
el efecto eliminador de la lactancia ha sido neutralizado por el efecto
acumulativo de la edad. A su vez, este argumento, junto con el hecho de que el
tiempo medio trascurrido desde que cesó la misma hasta que se recogió la

Capítulo 5
177
muestra de sangre era de 17 años, explicaría la ausencia de asociación
negativa con la lactancia.
Tampoco se encontraron diferencias en los niveles séricos de estos
plaguicidas organoclorados en función de haber trabajado, o no, en la
agricultura. Ni siquiera en función de haber manipulado, o no, plaguicidas.
Al estudiar las interacciones binarias de estas variables, la mayoría
resultaron no significativas. Sólo se detectó interacción significativa entre las
variables edad y sexo para el p,p’-DDE y el HCB. En el caso del p,p’-DDE los
niveles eran mayores en hombres que en mujeres para los grupos de 35-44 y
45-54 años y menores para el grupo de 55-64 años. En cuanto al HCB, la
tendencia a aumentar con la edad que se observaba tanto en el total de sujetos
como en las mujeres, se atenuaba mucho para los hombres, llegando a ser no
significativa (p = 0,08). No obstante, estas interacciones eran de menor entidad
que el resto de términos, por lo que no se tuvieron en cuenta en el modelo de la
Tabla 5.3.
5.3.2 Plaguicidas organoclorados y dieta
A continuación se estudió la posible asociación entre los niveles séricos
de p,p’-DDE, β-HCH y HCB, ajustados por sexo, centro, edad, IMC y año de
toma de la muestra, con los factores dietéticos. El análisis se efectuó tratando
los factores dietéticos tanto como variables continuas, como categorizadas en
cuartiles. Como medida de los mismos se utilizó la ingesta en g día-1. Se
investigaron todo los grupos alimenticios sin obtener ninguna relación
significativa.
En la Tabla 5.4 se plasman los resultados obtenidos con determinados
grupos de alimentos de origen animal, considerados las principales fuentes de
exposición de plaguicidas organoclorados en la población general. Las medias
geométricas de los distintos cuartiles de ingesta de pescado, carne o derivados
lácteos no mostraban un perfil de tendencia o de asociación significativa.
Tampoco se encontraron diferencias en los niveles séricos cuando se tuvo en
cuenta el tipo de pescado ni cuando se agruparon todos los alimentos de
origen animal en una única variable. También se intentó encontrar esa posible
asociación ajustando el modelo por la ingesta de energía e incluso

Plaguicidas organoclorados en la cohorte EPIC-España
178
incorporando términos de interacción para la edad y el sexo, pero los
resultados fueron los mismos.
Pocos estudios han analizado la relación entre niveles séricos de
plaguicidas organoclorados y dieta en la población general. Uno de ellos,
llevado a cabo en una muestra poblacional de personas mayores en Alemania,
reportaba una correlación positiva de la ingesta de carne de vacuno y ovino con
los niveles séricos de DDT (total), β-HCH y HCB, mostrando también el β-HCH
asociación negativa con el consumo de fruta [DeVoto et al 1998]. Otros dos, en
EE.UU., se realizaron con mujeres incluidas como controles en sendos
estudios caso-control sobre cáncer de mama. En el primero de ellos [Laden et
al. 1999], al igual que en nuestro caso ningún factor dietético, incluidos ingesta
de carne, pescado, ave, lácteos, frutas y cereales, resultaba predictor de la
concentración de DDE en suero, aunque la ingesta de pescado se asociaba
positivamente a la concentración de PCBs. En el otro [Moysich et al. 2002], se
encontró que el nivel sérico de DDE presentaba asociación positiva con la
ingesta de carne roja mientras que la del HCB era negativa. Ambos tenían
también una débil asociación positiva con la ingesta de frutas. Los autores
reconocían que era difícil dar una explicación a estos resultados y que se
necesitaba un estudio más profundo al respecto. Finalmente, otro estudio con
voluntarios de un área rural de Japón [Hanaoka et al. 2002], halló asociaciones
positivas, aunque débiles, entre la ingesta de arroz y de leche y la
concentración de HCB en suero, mientras que no encontró asociación entre
factores dietéticos y los niveles séricos de la suma de DDE más DDT.

Capítulo 5
179
Tabla 5.4 Niveles séricos de plaguicidas organoclorados referidos a grasa (ng g-1) en adultos sanos de la cohorte EPIC-España y variables dietéticas de origen animal categorizadas en cuartiles.
Ingesta
(g día -1) p,p' -DDE β -HCH HCB
Pescado y ≤ 34,7 847,6 187,3 403,8derivados 34,8-56,0 787,9 172,2 370,5
56,1-84,6 806,1 148,5 347,6≥ 84,7 846,6 165,3 397,1
valor p (tendencia) 0,64 0,46 0,82
Pescado blanco b ≤ 6,1 1081,4 209,7 316,36,2-21,8 767,4 164,9 331,621,9-41,2 760,3 143,8 441,3≥ 42,4 716,1 158,0 448,4
valor p (tendencia) 0,10 0,70 0,57
Pescado Azul b ≤ 2,3 819,7 189,5 444,72,4-12,8 865,6 179,8 374,712,9-27,3 790,5 144,7 349,3≥ 27,4 812,8 160,6 354,6
valor p (tendencia) 0,54 0,54 0,31
Carne y produc- ≤ 86,9 831,7 186,8 375,7tos cárnicos 87,0-123,9 826,1 177,9 404,7
124,0-172,6 806,0 149,6 403,1≥ 172,7 822,9 159,2 337,1
valor p (tendencia) 0,71 0,97 0,20
Productos ≤ 178,4 876,9 163,2 380,0lácteos 178,5-268,9 829,4 173,2 370,8
270,0-387,9 852,9 168,9 352,2≥ 388,0 734,3 165,9 416,3
valor p (tendencia) 0,10 0,85 0,90
Alimentos de ≤ 392,9 901,7 175,8 377,1origen animal c 393,0-497,8 797,0 166,0 382,2
497,9-622,5 843,7 182,9 402,4≥ 622,6 751,2 148,3 356,1
valor p (tendencia) 0,23 0,88 0,36
Media Geométricaa
a Datos ajustados por sexo, centro, edad, IMC y año de toma de la muestra. b Clasificación basada en el contenido de grasa: pescado blanco < 4 %, pescado azul ≥ 4%. c Incluye pescado y derivados, carne y productos cárnicos, productos lácteos y huevos.

Plaguicidas organoclorados en la cohorte EPIC-España
180
5.3.3 Comparación con otros estudios
Los resultados se compararon con los procedentes, en la medida de lo
posible, de estudios que reportaban concentraciones de estos compuestos
ajustadas por lípidos, que se hubieran realizado con muestras recogidas
aproximadamente durante el mismo período que las nuestras y que aportaran
una clara referencia a la edad de los sujetos. Además, se procuró restringir la
comparación a resultados obtenidos con muestras de población general y no
con seleccionadas en base a una potencial exposición proveniente de fuentes
relacionadas con la ocupación laboral o el lugar de residencia.
Dos estudios en Suecia reportaban niveles medibles de p,p’-DDT en
suero en el 96% de los hombres y el 80% de las mujeres [Glynn et al. 2000,
Glynn et al. 2007]. La concentración máxima registrada fue de 124 ng g-1. Otro
estudio en Japón [Masuda et al. 2005] relataba un nivel máximo de 157 ng g-1.
En ambos casos estaba por debajo del percentil 95% de nuestro estudio (256
ng g-1). Por lo que respecta al p,p’-DDE, el nivel sérico de 822 ng g-1 de media
geométrica en nuestra población era comparable a los 919 ng g-1 encontrados
en un estudio realizado en Nueva Zelanda entre 1996-1997 [Bates et al. 2004],
pero más alto que el rango de 200-600 ng g-1 en que se movían los estudios de
Suecia antes mencionados [Glynn et al. 2000, Glynn et al. 2007], el de Japón
[Masuda et al. 2005] y otro más en EE.UU [CDC 2005]. En lo tocante al β-HCH,
nuestra media geométrica de 167 ng g-1 estaba muy por encima de los niveles
encontrados en Suecia [Glynn et al. 2000, Glynn et al. 2007], EE.UU [CDC
2005] o Nueva Zelanda [Bates et al. 2004], todos ellos por debajo de 40 ng g-1,
pero era inferior a los 600 ng g-1 de Japón [Masuda et al. 2005]. Por último,
nuestra media geométrica de 379 ng g-1 para el HCB era mucho más alta que
los niveles prácticamente indetectables de EE.UU. [CDC 2005] y Nueva
Zelanda [Bates et al. 2004] y las medias por debajo de 80 ng g-1 de Suecia
[Glynn et al. 2000, Glynn et al. 2007] y Japón [Masuda et al. 2005].
Un estudio en Alemania realizado a principios de la década de los 90 con
297 personas mayores (65-74 años) de ambos sexos residentes en tres
ciudades representativas de áreas urbanas de más de un millón de habitantes

Capítulo 5
181
[DeVoto et al. 1998], reportaba concentraciones de estos compuestos en
plasma. Las medianas encontradas para β-HCH (2,4 µg L-1), HCB (10,5 µg L-1)
y suma p,p’-DDT + p,p’-DDE (13 µg L-1) estaban por encima de las de nuestro
estudio: 1,5 µg L-1, 3,1 µg L-1 y 6,1 µg L-1, respectivamente. Otro estudio,
realizado a finales de la década de los 90 con más de 2000 sujetos
representativos de la población general alemana entre los 18-69 años [Becker
et al. 2002], reportaba niveles de estos compuestos en sangre total. El β-HCH
era prácticamente indetectable y las medias geométricas registradas para HCB
(0,44 µg L-1) y p,p’-DDE (1,3 µg L-1 para la zona oeste y 3,4 µg L-1 para la zona
este) eran sensiblemente inferiores a las de nuestro estudio: 2,6 y 5,6 μg L-1,
respectivamente, aunque los valores en plasma o suero son generalmente
mayores que los de sangre total [Becker et al. 2002]. En ambos estudios los
resultados no estaban referidos al contenido en grasa, por lo que la influencia
del ayuno u otros factores relativos al contenido de lípidos en sangre se
desconocían.
En lo referente a España, hasta fechas recientes el único estudio con
muestra representativa de población general estaba realizado en las Islas
Canarias [Zumbado et al. 2005]. En él se medían los principales compuestos
asociados al DDT en muestras de suero recogidas durante 1998, procedentes
de sujetos con edades entre 6-75 años. El 43% de la población tenía niveles
detectables de p,p’-DDT y el 88%, de p,p’-DDE. Los niveles de p,p’-DDT
parecían ser mayores que los de nuestra población, ya que el p75 era de 242
ng g-1 (referido a grasa) y el nuestro de 42,9 ng g-1. Por el contrario observaban
valores menores de p,p’-DDE, con una mediana de 118 ng g-1 frente a los 858
ng g-1 de nuestro estudio, ambas cantidades referidas a grasa. También
encontraron que los niveles de DDT eran mayores en las islas con amplias
áreas de cultivo intensivo en invernaderos, que en el resto de las islas, donde
la agricultura era mayormente de tipo tradicional.
Otro estudio más restringido se llevó a cabo en 1993-1994 con muestras
de suero de 100 sujetos adultos sanos con edades comprendidas entre 13-82
años, con domicilio en el área metropolitana de Barcelona y no
específicamente expuestos por razones laborales o de residencia [To-Figueras
et al. 1995]. Para el HCB y el β-HCH, las medias geométricas referidas a

Plaguicidas organoclorados en la cohorte EPIC-España
182
volumen de suero (3,0 µg L-1 y 1,7 µg L-1, respectivamente) eran similares a las
nuestras (2,6 µg L-1 y 1,1 µg L-1, respectivamente), mientras que la del p,p’-
DDE (3,3 µg L-1) era ligeramente inferior a la de nuestro trabajo (5,6 µg L-1). Por
último, otro estudio en la localidad catalana de Flix (Tarragona) medía niveles
séricos de varios plaguicidas organoclorados en muestras recogidas a
habitantes del mismo [Sala et al. 1999]. La población estaba situada en la
vecindad de un complejo electroquímico dedicado a la fabricación de cloro y
derivados y el muestreo se realizó en 1994. El HCB se detectó en el 100% de
los sujetos siendo mayor la concentración entre los trabajadores de la fábrica,
aunque iba disminuyendo tras la jubilación. La mediana reportada era 16,5 µg
L-1 (referida a volumen de suero), sensiblemente mayor que los 3,1 µg L-1
(mediana referida a volumen de suero) encontrados en nuestro estudio e
incluso que los 4,7 µg L-1 de mediana que hallamos en Navarra. Para el p,p’-
DDE registraba una mediana (5,2 µg L-1) similar a la nuestra (5,9 µg L-1). Tanto
en el trabajo de Barcelona como en el de Flix los resultados estaban referidos a
volumen de suero y no a contenido en grasa, por lo que la influencia del ayuno
u otros factores relativos al contenido de lípidos en sangre se desconocían.

Capítulo 5
183
5.4 Conclusiones
Se han investigado los niveles séricos de los principales plaguicidas
organoclorados y metabolitos en 953 muestras de suero recogidas durante
los años 1992-1996 en cinco provincias españolas (Asturias, Granada,
Guipúzcoa, Murcia y Navarra). Las muestras provenían de adultos sanos
con edades comprendidas entre los 35-64 años en el momento del
reclutamiento en la cohorte EPIC-España, cuya selección no estaba
relacionada con ninguna exposición específica. Se ha determinado que
existían niveles detectables de tres plaguicidas organoclorados o metabolitos
(p,p’-DDE, β-HCH y HCB) en el 80-100% de las muestras, mientras que otro
más (p,p’-DDT) se detectó en el 30% de los sujetos. En general, los niveles
eran relativamente altos. Las medias geométricas de p,p’-DDT, p,p’-DDE, β-
HCH y HCB eran, respectivamente: 44 ng g-1, 822 ng g-1, 167 ng g-1 y 379 ng
g-1 referidas a contenido en grasa y 0,3 µg L-1, 6,1 µg L-1, 1,5 µg L-1 y 3,1 µg
L-1 referidas a volumen de suero. Estas concentraciones presentaban una
asociación significativa con el sexo, la edad, el centro de procedencia, el año
de recolección de la muestra, el IMC y, en el caso de mujeres que habían
practicado la lactancia, con el tiempo transcurrido desde que cesó la misma.
Los niveles de HCB y β-HCH eran mayores en las mujeres y los del
p,p’-DDE en los hombres. Todos ellos aumentaban significativamente con la
edad, tendencia que se daba tanto en hombres como en mujeres. Este
fenómeno se atribuye tanto a la edad en sí, como a un “Efecto Cohorte”
debido a la fecha de nacimiento. Se detectaban claramente dos perfiles
geográficos. Para el p,p’-DDE y el β-HCH las concentraciones eran
superiores en las regiones del sur (Murcia, Granada), hecho muy
probablemente asociado al uso intensivo de plaguicidas en el pasado
relacionado con prácticas agrícolas. Sin embargo, para el HCB los niveles
séricos eran particularmente elevados en una de las regiones del norte
(Navarra), lo que estaría probablemente más relacionado con actividades
industriales que con aplicaciones agrícolas de este compuesto, aunque no
se ha podido identificar fuentes específicas en dicha región que pudieran
explicar la diferencia de niveles con las otras zonas. El segundo lugar lo
ocupaba también otra provincia del norte limítrofe con la anterior

Plaguicidas organoclorados en la cohorte EPIC-España
184
(Guipúzcoa). El nivel sérico de estos compuestos descendía también de
forma significativa con el año de toma de la muestra, registrándose una
disminución de hasta de un 50% para el HCB aunque el período de tiempo
era inferior a cuatro años. Esto constata la disminución de los niveles
ambientales de estos compuestos desde el momento de la restricción o
prohibición de su uso hasta nuestros días y pone de relieve la eficacia de las
medidas que se han tomado al respecto. También existía una clara y
significativa asociación positiva con el IMC, en consonancia con los
resultados de la mayoría de estudios trasversales.
Ni la lactancia, considerada vía importante de eliminación, ni el tiempo
acumulado de lactancia, presentaban asociación significativa con los niveles
séricos de estos compuestos. Sin embargo, los niveles eran menores en las
mujeres que habían practicado la lactancia en los últimos 10 años, aunque la
diferencia sólo era significativa para el HCB, quedando cerca el β-HCH. La
explicación más probable de todo ello es que el efecto eliminador de la
lactancia se vería neutralizado por el efecto acumulativo de la edad y que el
tiempo medio trascurrido desde que cesó la lactancia hasta que se recogió la
muestra de sangre era de 17 años. Tampoco se encontraron diferencias en
los niveles séricos de estos plaguicidas organoclorados en función de haber
trabajado, o no, en la agricultura. Ni siquiera en función de haber
manipulado, o no, plaguicidas. Lo mismo ocurría al considerar los factores
dietéticos.
A pesar de que la cohorte EPIC-España no es una muestra
representativa de la población general española, hay que tener en cuenta
que se seleccionó a partir de un amplio estudio base, en diferentes áreas
geográficas e incluía sujetos de diferentes niveles educativos. En adición,
para este estudio se eligieron al azar 1000 sujetos de la cohorte por medio
de un muestreo estratificado de acuerdo a la estructura edad-sexo de la
población española. Por lo tanto, las medias reportadas pueden
considerarse una estimación de los niveles séricos de los principales
plaguicidas organoclorados y metabolitos en población española de 35-64
años en el período 1992-1996. En general, las concentraciones encontradas
eran mayores que las medidas en otros países europeos, EE.UU., Japón y

Capítulo 5
185
Nueva Zelanda, con la excepción de algunos datos concretos en Alemania,
el p,p’-DDE en Nueva Zelanda y el β-HCH en Japón, y del mismo orden que
las de otros estudios realizados en España.

Plaguicidas organoclorados en la cohorte EPIC-España
186
5.5 Referencias
♦ Armitage P, Berry G, Matthews JNS, 2002. Statistical Methods in Medical Research 4th Ed. Blackwell Science Ltd., Oxford, UK.
♦ Bates MN, Buckland SJ, Garrett N, Ellis H, Needham LL, Patterson Jr. DG, Turner WE, Russell DG, 2004. Persistent organochlorines in the serum of the non-occupationally exposed New Zealand population. Chemosphere 54, 1431–1443.
♦ Becker K, Kaus S, Krause C, Lepom P, Schulz C, Seiwert M, Seifert B, 2002. German Environmental Survey 1998 (GerES III): environmental pollutants in blood of the German population. Int. J. Hyg. Environ. Health 205, 297-308.
♦ CDC (Centers for Disease Control), 2005. Third National Report on Human Exposure to Environmental Chemicals. Centers for Disease Control, Atlanta, GA, USA.
♦ DeVoto E, Kohlmeier L, Heeschen W, 1998. Some dietary predictors of plasma organochlorine concentrations in an elderly German population. Arch. Environ. Health 53, 147–155.
♦ EPIC Group of Spain, 1997. Relative Validity and reproducibility of a diet history questionnaire in Spain. I. Foods. EPIC Group of Spain. European prospective investigation into cancer and nutrition. Int. J. Epidemiol. 26 (Suppl. 1), S91-S99.
♦ Glynn AW, Wolk A, Aune M, Atuma S, Zettermark S, Maehle-Schmid M, Darnerud PO, Becker W, Vessby B, Adami HO, 2000. Serum concentrations of organochlorines in men: a search for markers of exposure. Sci. Total Environ. 263, 197-208.
♦ Glynn A, Aune M, Darnerud PO, Cnattingius S, Bjerselius R, Becker W, Lignell S, 2007. Determinants of serum concentrations of organochlorine compounds in Swedish pregnant women: a cross-sectional study. Environ. Health 6, 2.
♦ Hagmar L, Wallin E, Vessby B, Jönsson BA, Bergman A, Rylander L, 2006. Intra-individual variations and time trends 1991–2001 in human serum levels of PCB, DDE and hexachlorobenzene. Chemosphere 64, 1507–1513.
♦ Hanaoka T, Takahashi Y, Kobayashi M, Sasaki S, Usuda M, Okubo S, Hayashi M, Tsugane S, 2002. Residuals of beta-hexachlorocyclohexane, dichlorodiphenyltrichloroethane, and hexachlorobenzene in serum, and relations with consumption of dietary components in rural residents in Japan. Sci. Total Environ. 286, 119–127.
♦ Laden F, Neas LM, Spiegelman D, Hankinson SE, Willett WC, Ireland K, Wolff MS, Hunter DJ, 1999. Predictors of plasma concentrations of DDE and PCBs in a group of US women. Environ. Health Perspect. 107, 75–81.
♦ Masuda Y, Haraguchi K, Kono S, Tsuji H, Päpke O, 2005. Concentrations of dioxins and related compounds in the blood of Fukuoka residents. Chemosphere 58, 329–344.
♦ Molina C, Falcón M, Barba A, Cámara MA, Oliva J, Luna A, 2005. HCH and DDT residues in human fat in the population of Murcia (Spain). Ann. Agric. Environ. Med. 12, 133–136.
♦ Moysich KB, Ambrosone CB, Mendola P, Kostyniak PJ, Greizerstein HB, Vena JE, Menezes RJ, Swede H, Shields PG, Freudenheim JL, 2002. Exposures associated with serum organochlorine levels among postmenopausal women from western New York State. Am. J. Ind. Med. 41, 102–110.
♦ Phillips DL, Pirkle JL, Burse VW, Bernet JT, Henderson LO, Needham LL, 1989. Chlorinated hydrocarbon levels in human serum: effects of fasting and feeding. Arch. Environ. Contam. Toxicol. 18, 495-500.
♦ Porta M, Puigdomènech E, Ballester F, Selva J, Ribas-Fitó N, Domínguez-Boada L, Martín-Olmedo P, Olea N, Llop S, Fernández M, 2008. Estudios realizados en España sobre concentraciones en humanos de compuestos tóxicos persistentes. Gac. Sanit. 22, 248-266.
♦ Porta M, Puigdomènech E, Ballester F, 2009. Nuestra contaminación interna. Editorial Catarata, Madrid.

Capítulo 5
187
♦ Sala M, Sunyer J, Otero R, Santiago-Silva M, Camps C, Grimalt J, 1999. Organochlorine in the serum of inhabitants living near an electrochemical factory. Occup. Environ. Med. 5, 152–158.
♦ Sullivan Jr. JB, Krieger GR, 2001. Clinical environmental health and toxic exposures 2nd Ed. Lippincott Williams & Wilkins, Philadelphia, PA, USA.
♦ To-Figueras J, Barrot C, Rodamilans M, Gómez-Catalán J, Torra M, Brunet M, Sabater F, Corbella J, 1995. Accumulation of hexachlorobenzene in humans: a long standing risk. Hum. Exp. Toxicol. 14, 20-23.
♦ Wahlefeld AW, 1974. Triglycerides determination after enzymatic hydrolysis. In: Bergmeyer HU (ed.), Methods of Enzymatic Analysis 2nd Ed. Academic Press Inc., New York, USA, 1831-1835.
♦ Wareham NJ, Jakes RW, Rennie KL, Schuit J, Mitchell J, Hennings S, Day NE, 2003. Validity and repeatability of a simple index derived from the short physical activity questionnaire used in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Public Health Nutr. 6, 407-413.
♦ WHO (World Health Organization), 2003. Health risks of persistent organic pollutants from long-range transboundary air pollution. http://www.euro.who.int/document/e78963.pdf. Último acceso: 15-04-2010.
♦ Wiebe DA, Bernert Jr. JT, 1984. Influence of incomplete cholesteryl ester hydrolysis on enzymic measurements of cholesterol. Clin. Chem. 30, 352-356.
♦ Wolff MS, Britton JA, Teitelbaum SL, Eng S, Deych E, Ireland K, Liu Z, Neugut AI, Santella RM, Gammon MD, 2005. Improving organochlorine biomarker models for cancer research. Cancer Epidemiol. Biomarkers Prev. 14, 2224–2236.
♦ Wolff MS, Anderson HA, Britton JA, Rothman N, 2007. Pharmacokinetic variability and modern epidemiology: the example of dichlorodiphenyltrichloroethane, Body Mass Index, and Birth Cohort. Cancer Epidemiol. Biomarkers Prev. 16, 1925-1930.
♦ Zumbado M, Goethals M, Alvarez-León EE, Luzardo OP, Cabrera F, Serra-Majem L, Domínguez-Boada L, 2005. Inadvertent exposure to organochlorine pesticides DDT and derivatives in people from the Canary Islands (Spain). Sci. Total Environ. 339, 49–62.


CONCLUSIONES

Conclusiones
190

Conclusiones
En el presente trabajo se han desarrollado tres métodos que permiten la
determinación simultánea de los plaguicidas organoclorados y PCBs más
relevantes en suero humano:
1º) Método basado en extracción en fase sólida (SPE) sobre placas de 96
discos, seguida de limpieza con sílice sulfúrica.
2º) Método basado en microextracción en fase sólida en espacio de cabeza
(HS-SPME). Supone una mejora respecto a la preparativa tradicional por:
la simplicidad instrumental (sólo fibra PDMS y su holder), la eliminación de
la etapa de limpieza de extractos y la evitación del uso de disolventes.
3º) Método basado en limpieza de extractos por cromatografía de
permeabilidad en gel (GPC). La automatización y la miniaturización
reducen considerablemente los costes (disminución de la necesidad de
mano de obra, del tiempo de análisis por muestra y de la cantidad de
disolvente utilizado) respecto a otras preparativas habituales o similares.
En el desarrollo del segundo y el tercer método se ha visto la utilidad del
diseño experimental en la optimización de las condiciones de las etapas
preparativas.
Comparados con otros métodos presentan varias ventajas que los hacen
adecuados para su empleo en estudios de riesgo de exposición o
epidemiológicos, donde se requiere el análisis de una elevada cantidad de
muestras del menor tamaño posible: alta productividad, aceptable LQ con baja
cantidad de muestra (500 µL), recuperación, precisión y veracidad
satisfactorias. En cuanto a la productividad, el primero (extracción SPE y
limpieza con sílice sulfúrica) permite preparar 48-96 muestras por jornada de
trabajo y analista sin requerir especiales dispositivos de automatización por lo
que ha sido el método elegido para su posterior aplicación en este estudio. El
segundo, aunque es medianamente productivo ya que se realiza de forma
manual (50 min de preparativa por muestra, duplicidad de procesamiento por
adición a matriz), mejoraría sustancialmente su rendimiento con el empleo de
dispositivos que permiten automatizar los procesos de extracción y desorción
para que puedan realizarse de forma continua y desatendida. Además, el uso
de detectores de espectrometría de masas posibilitaría la sustitución de la
191

Conclusiones
adición estándar por la dilución isotópica, evitando así la multiplicidad de
inyecciones sobre una misma muestra. De ahí, que se considere tiene gran
potencial de cara a su uso en estudios epidemiológicos. En lo que respecta al
tercero, su empleo de forma automática y desatendida requiere el uso de
tapones de teflón y/o un sistema de refrigeración del carrusel del
automuestreador del GPC, para evitar la evaporación del diclorometano.
Todos ellos podrían ser ampliados a otros compuestos contaminantes ya
que se emplean sorbentes y condiciones de extracción no específicos.
Además, el segundo y el tercero se basan en preparativas no destructivas, lo
que los hace muy interesantes para analitos sensibles al ácido sulfúrico.
Se han investigado los niveles séricos de los PCBs indicadores y de los
principales plaguicidas organoclorados y metabolitos en 953 muestras de suero
recogidas durante los años 1992-1996 en cinco provincias españolas (Asturias,
Granada, Guipúzcoa, Murcia y Navarra). Las muestras provenían de adultos
sanos con edades comprendidas entre los 35-64 años en el momento del
reclutamiento en la cohorte EPIC-España, cuya selección no estaba
relacionada con ninguna exposición específica. Se ha determinado que existían
niveles detectables de cuatro PCBs (congéneres 118, 138, 153 y 180)
prácticamente en el 100% de las muestras y de tres plaguicidas
organoclorados o metabolitos (p,p’-DDE, β-HCH y HCB) en el 80-100% de los
sueros, mientras que otro más (p,p’-DDT) se detectó en el 30% de los sujetos.
La concentración total de PCBs (media geométrica de la suma de los
cuatro congéneres) era de 459 ng g-1 -1 referida a grasa y 3,1 µg L referida a
volumen de suero. Para el congénere 153 estos valores eran de 186 ng g-1 y
1,25 µg L-1, respectivamente. Estas concentraciones presentaban una
asociación significativa con el sexo, la edad, el centro de procedencia, el año
de recolección de la muestra, el IMC, la ingesta de pescado, la ingesta de
derivados lácteos y, en el caso de mujeres que habían practicado la lactancia,
con el tiempo acumulado de lactancia. Los hombres presentaban valores más
altos que las mujeres. El nivel de PCBs aumentaba con la edad y era mayor en
las provincias del norte, presentando los residentes en Guipúzcoa un valor
medio 50% más alto que los de Granada. Los niveles de PCBs descendían
conforme más reciente era la fecha de recolección de la muestra. La asociación
192

Conclusiones
con el IMC era negativa. La ingesta de pescado era el factor dietético que
presentaba mayor asociación con el nivel sérico de PCBs. Aunque, en general,
las provincias con mayor consumo de pescado eran también las que
presentaban mayores valores de PCBs, el ajuste por la ingesta de pescado no
llegaba a justificar las diferencias regionales, por lo que éstas permanecen
inexplicadas. La asociación con el tiempo acumulado de lactancia resulta
negativa, lo que está en concordancia con la consideración general de que la
lactancia es una forma importante de eliminación de PCBs.
Las medias geométricas de p,p’-DDT, p,p’-DDE, β-HCH y HCB eran,
respectivamente: 44 ng g-1, 822 ng g-1, 167 ng g-1 y 379 ng g-1 referidas a
contenido en grasa y 0,3 µg L-1 -1 -1 -1, 6,1 µg L , 1,5 µg L y 3,1 µg L referidas a
volumen de suero. Estas concentraciones presentaban una asociación
significativa con el sexo, la edad, el centro de procedencia, el año de
recolección de la muestra, el IMC y, en el caso de mujeres que habían
practicado la lactancia, con el tiempo transcurrido desde que cesó la misma.
Los niveles de HCB y β-HCH eran mayores en las mujeres y los del p,p’-DDE
en los hombres. Todos ellos aumentaban significativamente con la edad. Las
concentraciones de p,p’-DDE y el β-HCH eran superiores en las regiones del
sur (Murcia, Granada), hecho muy probablemente asociado al uso intensivo de
plaguicidas en el pasado relacionado con prácticas agrícolas. Sin embargo,
para el HCB los niveles séricos eran particularmente elevados en una de las
regiones del norte (Navarra), lo que estaría probablemente más relacionado
con actividades industriales que con aplicaciones agrícolas de este compuesto,
aunque no se ha podido identificar fuentes específicas en dicha región que
pudieran explicar la diferencia de niveles con las otras zonas. El segundo lugar
lo ocupaba también otra provincia del norte limítrofe con la anterior
(Guipúzcoa). El nivel sérico de estos compuestos descendía también de forma
significativa con el año de toma de la muestra. Existía una clara y significativa
asociación positiva con el IMC, en consonancia con los resultados de la
mayoría de estudios trasversales. Ni la lactancia, considerada vía importante
de eliminación, ni el tiempo acumulado de lactancia, presentaban asociación
significativa con los niveles séricos de estos compuestos. Sin embargo, eran
menores en las mujeres que habían practicado la lactancia en los últimos 10
años, aunque la diferencia sólo era significativa para el HCB, quedando cerca
193

Conclusiones
el β-HCH. Tampoco se encontraron diferencias en función de haber trabajado,
o no, en la agricultura. Ni siquiera en función de haber manipulado, o no,
plaguicidas. Lo mismo ocurría al considerar los factores dietéticos.
Aunque la cohorte EPIC-España no es una muestra representativa de la
población general española, hay que tener en cuenta que se seleccionó a partir
de un amplio estudio base, en diferentes áreas geográficas e incluía sujetos de
diferentes niveles educativos. En adición, para este estudio se eligieron al azar
1000 sujetos de la cohorte por medio de un muestreo estratificado de acuerdo
a la estructura edad-sexo de la población española. Por lo tanto, las medias
reportadas pueden considerarse una estimación de los niveles séricos de PCBs
y de los principales plaguicidas organoclorados y metabolitos en población
española de 35-64 años en el período 1992-1996. Para los PCBs estos niveles
eran similares a los consignados en otros países europeos con muestras
recogidas en el mismo período, aunque relativamente mayores que los
medidos en América del Norte o en estudios europeos más recientes. Para los
plaguicidas organoclorados estaban por encima de los hallados en otros países
europeos, EE.UU., Japón y Nueva Zelanda, con la excepción de algunos datos
concretos en Alemania, el p,p’-DDE en Nueva Zelanda y el β-HCH en Japón, y
del mismo orden que los de otros estudios realizados en España.
194

APÉNDICE


El trabajo que se recoge en esta memoria ha dado lugar a las siguientes
publicaciones y comunicaciones:
Publicaciones
“High troughput method for the determination of organochlorine pesticides and polychlorinated biphenyls in human serum”
Journal of Chromatography B, 2007, 852, 15-21.
“Determination of organochlorine pesticides and polychlorinated biphenyls in human serum using headspace solid-phase microextraction and gas chromatography-electron capture detection”
Journal of Chromatography B, 2007, 846, 298-305.
“Method for the determination of selected organochlorine pesticides and polychlorinated biphenyls in human serum based on a gel permeation chromatographic clean-up”
Chemosphere, 2009, 76, 1533-1539.
“Polychlorinated biphenyls in Spanish adults: Determinants of serum concentrations”
Environmental Research, 2009, 109, 620-628.
“Serum levels of organochlorine pesticides in healthy adults from five regions of Spain”
Chemosphere, 2009, 76, 1518-1524.
Comunicaciones Nacionales
“High troughput method for determination of organochlorine compounds in low volume serum samples”
as11 Jornadas de Análisis Instrumental (JAI), Barcelona, 15-17 noviembre, 2005.












