materiales unidad 1.iv - frlp.utn.edu.arfrlp.utn.edu.ar/materias/protecmat/solventes.pdf · debe...
TRANSCRIPT

PROTECCIÓN DE MATERIALES
UNIDAD 1.IV SOLVENTES Y DILUYENTES
1. Introducción Se define una pintura, desde el punto de vista fisicoquímico, como una dispersión de un sólido finamente dividido (pigmento) en un medio fluido (ligante), el que a su vez estará compuesto por un material formador de película (resina) y una mezcla solvente, objeto del presente trabajo. Los solventes se incorporan a fin de facilitar la elaboración, almacenaje y aplicación de la pintura. Debe mencionarse que no todas las pinturas poseen solvente, tal es el caso de las pinturas en polvo. La mezcla solvente, en general, no está constituida por una única sustancia, sino que por el contrario suele contener varias. Es oportuno diferenciar, dentro de esta mezcla, al solvente verdadero (que es el que posee la capacidad de disolver a la resina), de los diluyentes (que si bien no son capaces de disolver por sí mismos a la resina, permiten disminuir la cantidad del disolvente real). La utilización de diluyentes permite la disminución de costos (ya que suelen ser más baratos que los disolventes), y el ajuste de otras propiedades. El presente trabajo expone las propiedades básicas de las mezclas solventes, y su influencia sobre la pintura terminada.
2. Poder solvente
2.1 Índices de solubilidad Es indispensable que la mezcla solvente tenga la capacidad de disolver a la resina, este es su fin principal y es por esta razón que es la primera y más importante propiedad que se estudiará. Existen diversos parámetros para evaluar el poder solvente de una determinada sustancia. Por ejemplo el número Kauri-butanol, para hidrocarburos, que se define como la cantidad máxima de solvente que puede agregarse a una solución de resina Kauri (una resina que se extrae de una especie de coníferas: Agathis australis) en butanol sin producir turbidez. Dado que la resina es insoluble en hidrocarburos, mientras mayor cantidad de solvente se pueda agregar, mayor será el poder solvente del mismo. Así los solventes “fuertes” como el xileno tienen un alto número Kauri-butanol. En la tabla 1 se ejemplifican algunos valores. Este método esta estandarizado por ASTM. Otro índice similar es el número de cera, que indica la cantidad máxima de solvente que puede agregarse a una solución de cera en benceno. En punto de anilina, en cambio, es la mínima temperatura a la que volúmenes iguales de solvente a ensayar y anilina son completamente miscibles.

Por otro lado, para estimar la solubilidad de un polímero, se utiliza el grado de solubilidad, como el mínimo porcentaje de tolueno que debe agregarse a una mezcla del polímero y n-dodecano (en proporción 10-90) para obtener una solución límpida. Cuanto más soluble sea el polímero, menor la cantidad de tolueno que deberá agregarse. En la tabla 2 se muestran algunos ejemplos.
�������� ��� �����
Ciclohexano 54.3
Diclorometano 136
n-Heptano 25.4
n-Hexano 26.5
D-Limoneno 68
n-Octano 24.5
n-Pentano 33.8
Isopropanol >100
1,1,2,2-Tetracloroetileno 90
Tolueno 105
1,1,1-Tricloroetano 124
Tricloroetileno 129
1,1,2-Triclorotrifluoroetano 32
Xileno (mexcla de isómeros) 95
Tabla 1- Índices Kauri-butanol (2)
Polímero Grado de solubilidad Poli acetato de vinilo 89 Poli metil metacrilato 87 Poli n-butil metacrilato 25 Poli i-butil metacrilato 23
Tabla 2 – Grado de solubilidad
Para mezclas solvente polímero también existe una estimación de la solubilidad mutua. El número de heptano representa la cantidad máxima de n-heptano que puede agregarse a una solución polímero/solvente sin presentar turbidez. Estos índices tienen la ventaja de aportar información concisa acerca de los disolventes, pero carecen de una base teórica e ignoran el hecho que cada solvente o tipo de solvente es más o menos afín a determinada resina o tipo de resina.(un solvente que puede disolver fácilmente una resina, quizá no sea tan eficiente con otra, y un segundo solvente que sea incapaz de solubilizar a la primer resina, sí pueda hacerlo con la segunda). Será entonces necesario estudiar la termodinámica de la solvatación, que en este trabajo se centrará en los análisis de Hildebrand, Hansen y Teas.

2.2 El parámetro de Hildebrand
2.2.1 Densidad de energía de Cohesión Para que se produzca un proceso espontáneo, como el de disolución, la variación de energía libre debe ser negativa. Dicho de otro modo el proceso debe producir una disminución de energía libre del sistema.
∆G = ∆H - T∆S (I)
Los polímeros en general se encuentran en estado amorfo (no cristalino) y su disolución puede considerarse como una simple mezcla. Por otro lado, los solventes líquidos presentan un gran desorden (elevada entropía) y una gran libertad que les permite, incluso, escapar de la fase líquida hacia la fase vapor. Los polímeros en cambio, a causa de su gran tamaño y forma tienen muy poca libertad. Al mezclarse estos últimos con las pequeñas moléculas de solvente, adquieren una mayor libertad y la posibilidad de reordenarse, lo que significa un aumento de entropía. Esto hace que el segundo término de la ecuación (I) tienda a hacer negativa a la varación de energía libre. Por otro lado para separar las moléculas del solvente debe emplearse una determinada cantidad de energía, la que tenderá a aumentar (y a hacer positiva) la ecuación (I). De este modo la disolución será posible si y sólo si la energía necesaria para separar las moléculas del solvente es pequeña a comparación de la variación entrópica. La densidad de energía de cohesión (CED) es una medida de la energía necesaria para separar las moléculas. Dado que durante la evaporación las moléculas de un solvente también deben separarse (como en la solvatación), estas dos propiedades deberán estar relacionadas. La relación propuesta es la siguiente:
m
vap
V
TRHCED
.−∆= (II)
Donde: ∆Hvap = Calor de vaporización R = Constante de los gases ideales T = Temperatura Vm =Volumen molar Hildebrand propone un parámetro de solubilidad δ, igual a la raíz cuadrada de la energía necesaria para separar las moléculas de determinado tipo. Es decir:
m
vap
V
TRHCED
.−∆==δ (III)
Según esta teoría, mientras más similares sean los parámetros de solubilidad de dos sustancias, más posibilidad hay de que sean miscibles entre si. De este modo, la variación de entalpía involucrada en el proceso de solvatación será:
( )22121 δδ −ΦΦ=∆H (IV)
Donde Φ es el volumen parcial de cada componente y los subíndices 1 y 2 se refieren al solvente y al soluto. Esto implica que cuanto menor sea la diferencia de los parámetros de solubilidad del

soluto y el solvente, más posibilidades hay de que estos resulten miscibles (dependiendo de la variación entrópica, que ya se había establecido positiva).
2.2.2 Valores del parámetro de Hildebrand El parámetro de Hildebrand (δ) será de enorme utilidad para la estimación de solubilidades mutuas, y está presente en numerosa bibliografía. En la tabla 3 se listan algunos valores. Las unidades características de este serán:
[ ] [ ][ ] ( ) 5,03. −== cmcalVolumenEnergíaδ (V)
[ ] [ ][ ]
[ ][ ][ ]
[ ][ ] [ ] 5,0
23Pr
.MPaesión
Longitud
Fuerza
Longitud
LongitudFuerzaVolumenEnergía =====δ (VI)
Las unidades establecidas por la ecuación (V) toman el nombre de Hildebrans, en honor a quién desarrolló esta teoría. Las unidades del SI, están determinadas por la ecuación (VI): la unidad de presión del SI es el Pascal, y el parámetro se expresará en MegaPascales (106 Pa). La relación entre ambos esta dada por:
� [(cal.cm-3)0,5] = 0,48888 x � [MPa0,5] (VII)
� [MPa0,5] = 2,0455 x � [(cal.cm-3)0,5] (VIII)
Solvente � �(SI) n-Pentano (7.0) 14.4 n-Hexano 7.24 14.9 Freon® TF 7.25 - n-Heptano (7.4) 15.3 Dietil eter 7.62 15.4
1,1,1 Tricloroetano 8.57 15.8 n-Dodecano - 16.0 Coclohexano 8.18 16.8
Tetracloruro de Carbono 8.65 18.0 Xileno 8.85 18.2
Acetato de Etilo 9.10 18.2 Tolueno 8.91 18.3
Tetrahidrofurano 9.52 18.5 Benceno 9.15 18.7
Cloroformo 9.21 18.7 Tricloroetileno 9.28 18.7
Metil etil cetona (MEK) 9.27 19.3 Acetona 9.77 19.7
Alcohol n-propílico 11.97 24.9 Alcohol Etílico 12.92 26.2
Alcohol n-butílico 11.30 28.7 Alchol metílico 14.28 29.7 Propilen glicol 14.80 30.7

Etilen glicol 16.30 34.9 Agua 23.5 48.0
Tabla 3 – Parámetros de Hildebrand de algunos solventes. (3) En el caso de los solventes que no estén listados en la bibliografía, el parámetro de Hildebrand podría estimarse con la ecuación (III), pero para los polímeros resulta imposible calcular el calor de vaporización, y es por este motivo que Small propone una estimación a través de las constantes de atracción molar (G):
M
GD�=.
δ (IX)
Donde: D = Densidad del polímero M = Peso Fórmula del polímero G= Constante de atracción molar (ver tabla 4)

Grupo G (cal.cm3)0,5.mol-1
214
133
28
-93
190
111 19
285 Fenilo 735
Fenileno 658 80-100
410 250-270
340
150
225 Tabla 4 – Constantes de atracción molar de Small a 25ºC (4)
2.2.3 Relación del parámetro de Hildebrand con otros índices de solubilidad El parámetro de Hildebrand se puede relacionar con los índices de solubilidad que se trataron al principio del trabajo. En la figura 1 se puede observar la relación con el número de Kauri-butanol, la recta punteada corresponde a la ecuación:
δ(MPa0,5) = 0,04 KB + 14,2 (X)
Esta relación se cumple aproximadamente para solventes con números de Kauri-butanol mayores a 35. Para aquellos con valores menores se deberían realizar correcciones por el tamaño molecular.
Figura 1 – Relación Kauri-Butano/ parámetro de Hildebrand (3)

Por otro lado, la tabla 5 muestra una comparación entre el grado de solubilidad de distintos polímeros y el parámetro de Hildebrand correspondiente.
Polímero Grado de solubilidad
δ (MPa0,5)
Poli acetato de vinilo 89 18.05 Poli metil metacrilato 87 18.00
Poli n-butil metacrilato 25 16.58 Poli isobutil matacrilato 23 16.53
Tabla 5 – Comparación de grado de solubilidad/ Parámetro de Hildebrand
2.2.4 Aplicaciones y limitaciones del parámetro de Hildebrand Este parámetro, al igual que los índices enumerados al principio del presente trabajo, tiene la gran ventaja de ser un valor único que brinda información concisa acerca de un solvente. Pero cuenta con la ventaja adicional de brindar también información acerca de los solutos, en un mismo formato, lo que permite estimar rápidamente la solubilidad mutua: si los respectivos δ no son muy distintos, probablemente el solvente y el soluto formarán una solución homogénea. También tiene un gran interés desde el punto de vista conceptual, ya que tiene una base teórica termodinámica, y no sólo sirve como parámetro en si mismo, sino también para el desarrollo de otras teorías. Como se verá a continuación es la base de otras teorías más complejas y exactas. Sin embargo tiene las mismas limitaciones que los otros índices, en cuanto a que no predice la selectividad de los solventes. La figura 2 presenta un gráfico que relaciona el grado de hinchamiento de una película de aceite de lino con el parámetro de Hildebrand: en el mismo se puede observar que el cloroformo es seis veces mejor solvente del aceite de lino que el dicloruro de etileno y más de diez veces mejor que el tolueno, a pesar que tienen un δ muy similar. Es por este motivo que deben buscarse mejores modelos para predecir la solubilidad de los polímeros en diversos solventes.
Figura 2 – Grado de hinchamiento de una película de aceite
de lino respecto del parámetro de Hildebrand.

2.3 Fuerzas de atracción intermoleculares y su relación con la solubilidad.
El hecho de que existan discrepancias entre la teoría de Hildebrand y los datos experimentales llevó a pensar a varios autores que en realidad las fuerzas de atracción intermoleculares no correspondían a un parámetro único, sino que por el contrario tienen naturaleza diferente y por lo tanto no podían agruparse dentro de un único parámetro unidimensional. Como se sabe, las fuerzas de atracción intermoleculares, o fuerzas de van der Walls, corresponden a fenómenos diferentes y se las puede clasificar en tres grupos:
• Fuerzas de London, o fuerzas de dispersión, originadas por la interacción de dipolos transitorios.
• Fuerzas polares, ya sea por interacción dipolo-dipolo (fuerzas de Kenson) o dipolo-dipolo inducido (fuerzas de Debye).
• Puentes de Hidrógeno, producidas en las moléculas que posean un átomo de hidrógeno unido a un elemento muy electronegativo como ser O, N o F.
La inclusión de esta idea trae a colación el axioma que ya promulgaban los alquimistas “lo similar disuelve lo similar”. Los solventes en que las fuerzas de atracción sean de un determinado tipo, disolverán a aquellos polímeros que posean el mismo tipo de interacción intermolecular. Un primer intento para corregir las inconsistencias del modelo de Hildebrand fue propuesto por Harry Burrel, que consistia en dividir los solventes en tres grupos de acuerdo a la posibilidad del mismo de formar puentes de Hidrógeno. Crowley,Teague y Lowe crearon posteriormente el primer modelo tridimensional, que utilizaba el parámetro de Hildebrand como un eje cartesiano y las fuerzas polares y de puente de hidrógeno en los otros dos. En el espacio tridimensional así obtenido ubicaba los solventes y los polímeros, y estimaba la solubilidad de acuerdo a la distancia entre ambos.
2.4 El modelo de Hansen Fue Hansen quien propuso que en realidad el parámetro de Hildebrand se debía a la suma de los tres tipos de interacciones. De este modo:
δ2 = δp2 + δd
2 + δh2 (XI)
Donde: d = Parámetro total de Hildebrand. dp = Componente polar. dd = Componente de dispersión. dh = Componente de puentes de Hidrógeno. Esto permite ubicar a los solventes y los polímeros en un espacio tridimensional, de coordenadas (δp, δd, δh), como se observa en la figura 3.

Figura 3 – Coordenadas de Hansen
La posible solubilidad ente un polímero y un solvente esta dada por la distancia entre los puntos correspondiente a uno y al otro en este espacio tridimensional: si esta distancia D es menor que un radio R, característico del polímero, este par podrá ser miscible, si es igual o similar será parcialmente miscible y si es mayor serán insolubles. La ecuación propuesta por Hansen es la (XII). Nótese el 4 que afecta el término de fuerzas de dispersión, fue propuesto para aproximar los volúmenes de solubilidad a esferas.
( ) ( ) ( )22,1,
22,1,
22,1,4 hhppddD δδδδδδ −+−+−= (XII)
Donde los subíndices 1 y 2 representan al polímero y al solvente respectivamente. Cabe destacar el método por el cuál se determinaron los componentes de este sistema. En primer lugar se define el término homomorfo, que es una sustancia que tiene igual estructura molecular que las que se desea estudiar. Al homomorfo no polar se le asigna el total de las fuerzas de dispersión. Por ejemplo para estudiar el n-octanol se elige como homomorfo no polar al n-octano, y el valor del parámetro de Hildebrand para el hidrocarburo se lo considera como el valor de la componente de dispersión de Hansen. El resto de las componentes se obtiene posteriormente por métodos de ensayo y error sobre numerosos polímeros y solventes, y de acuerdo a la evidencia experimental. Las tablas 6 y 7 listan los parámetros para distintos solventes y polímeros
�/MPa1/2 Polímero (nombre comercial, proveedor)
�d �p �h R Acetato de Celulosa (Celliclore® A, Bayer) 18.6 12.7 11.0 7.6 Propileno Clorado (Parlon® P- 10, Hercules) 20.3 6.3 5.4 10.6 Epoxy (Epikote® 1001, Shell) 20.4 12.0 11.5 12.7 Elastómero de Isopreno (Ceriflex® IR305, Shell) 16.6 1.4 -0.8 9.6 Nitrato de Celulosa (1 /2 sec, H-23, H forn) 15.4 14.7 8.8 11.5 Poliamida, termoplastico (Versamid® 930, General Mills) 17.4 -1.9 14.9 9.6 Poli isobutileno (Lutonal® IC-123, BASF) 14.5 2.5 4.7 12.7 Poli Etil Metacrilato (Lucite® 2042, DuPont) 17.6 9.7 4.0 10.6 Poli Metil Matacrilato (Rohm and Haas) 18.6 10.5 7.5 8.6 Poliestireno (Polystyrene LO, BASF) 21.3 5.8 4.3 12.7 Poli Acetato de Vinilo (Mowilith® 50, Hoechst) 20.9 11.3 9.6 13.7 Poli Cloruro de Vinilo (Vilpa® KR, k=50, Montecatini) 18.2 7.5 8.3 3.5 Poliéster saturado (Desmophen® 850, Bayer) 21.5 14.9 12.3 16.8
Tabla 6 – Parámetros de Hansen y radio de interacción de polímeros .
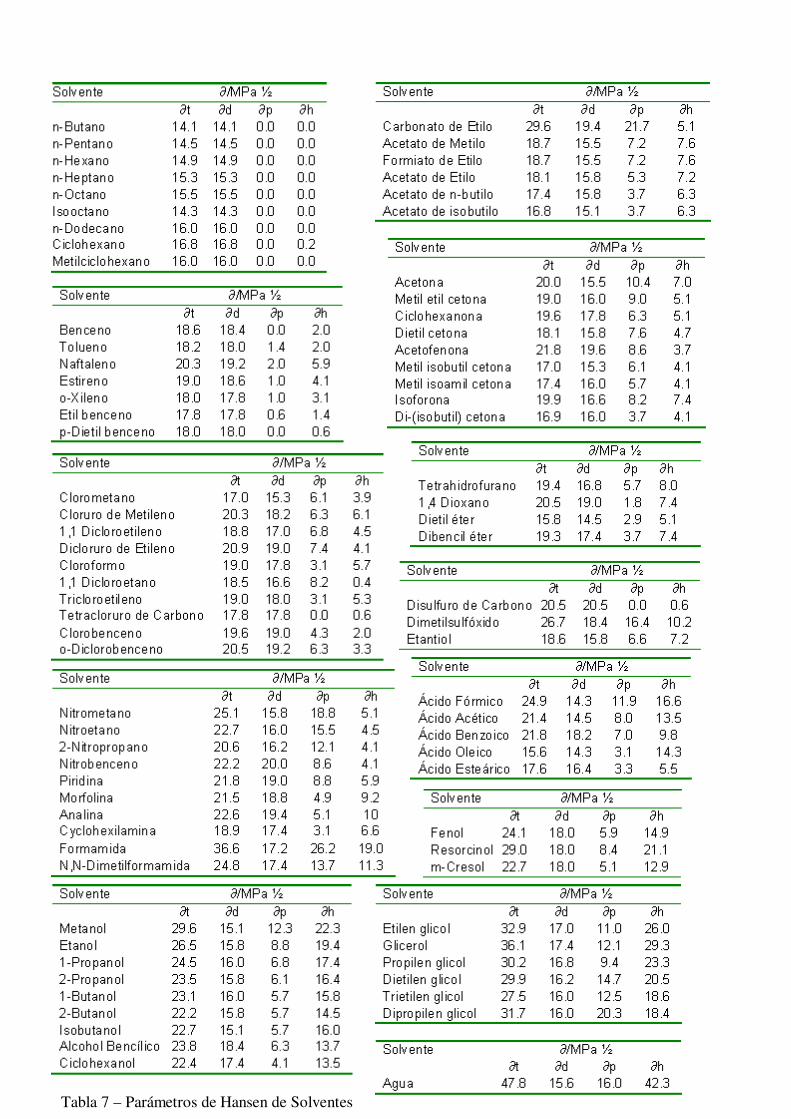
Tabla 7 – Parámetros de Hansen de Solventes

La estimación de solubilidad usando los parámetros de Hansen es, según la bibliografía, una de las más exactas y posee una base teórica interesante, lo cual lo constituye en un excelente método. Sin embargo resulta poco práctico en cuanto a la dificultad de realizar y más aún de visualizar los gráficos en 3 dimensiones.
2.5 Parámetros fraccionales. El triángulo de solubilidad de Teas. Teas intentó solucionar la deficiencia del método de Hansen mediante el uso de parámetros fraccionales, definidos según la ecuación (XIII):
phd
pp
phd
hh
phd
dd fff
δδδδ
δδδδ
δδδδ
++=
++=
++=
.100.100.100 (XIII)
Aprovechando las propiedades de los triángulos equiláteros, estos parámetros (cuya suma es siempre 100) se pueden ubicar en un sistema de coordenadas triangulares (figura 4), donde también se grafican las ventanas de solubilidad de los polímeros.
Figura 4 - Triángulo de solubilidad.
Para las mezclas de solventes se pueden estimar estos parámetros como la media aritmética de los solventes puros, considerando las proporciones en que están presentes. Por ejemplo para una mezcla de metanol y 1,2-Dicloroetano:
Fd Fp Fh Metanol 30 22 48 1,2-Dicloroetano 67 19 14 Mezcla 40:60 0,4 x 30 + 0,6 x 67
= 52,2 0,4 x 22 + 0,6 x 19
= 20,2 0,4 x 48 + 0,6 x 14
= 27,6

Otra posibilidad para calcular los parámetros de mezclas es mediante un método gráfico consistente en unir los puntos correspondientes a los solventes puros, y dividir dicho segmento en dos segmentos menores cuya longitud sea proporcional a la cantidad de cada solvente. El punto que divide dichos segmentos es el correspondiente a la mezcla.
Tabla 8- Parámetros de Teas de algunos solventes.

3 Viscosidad. La viscosidad es una propiedad de fundamental importancia en la pintura. Una viscosidad excesiva puede traer aparejado (según el método de aplicación) problemas relacionados con un alto espesor de capa (curado lento o deficiente) y con la aplicación en si misma (atomización deficiente). Por otro lado una viscosidad baja podria relacionarse con bajo espesor de pintura por capa (bajo poder cubritivo, alta permeabilidad) y problemas de chorreado. La viscosidad de las pinturas es finalmente ajustada con aditivos reológicos, por lo que la influencia del solvente es relativa. En realidad la viscosidad final de una pintura resulta de la influencia de varios factores: disolventes, resina (tipo, concentración, peso molecular), carga de pigmentos, incorporación de aditivos. En general la viscosidad de una pintura aumenta con el peso molecular del solvente de una serie homóloga, a la vez que disminuye el poder solvente. Las comparaciones entre solventes de diferentes series, por otro lado, no parecen correlacionar el poder solvente con la viscosidad. Se observa sin embargo que cuando un solvente (o una mezcla) tiene los parámetros de solubilidad centrados en el área de solubilidad de una resina, esta mezcla tiene una alta viscosidad; para lograr una baja viscosidad se debería trabajar en el borde del área de solubilidad. Es por esta razón que el agregado de solventes no polares y con baja afinidad a la resina disminuye la viscosidad de la pintura, aún con el riesgo de producir turbiedad. En cuanto a la relación entre la viscosidad y la concentración de resina, existen estudios realizados para soluciones de polímeros. La relación de las viscosidades de una disolución de un polímero y la del disolvente se denomina viscosidad relativa (ηr). Este valor menos 1 se llama viscosidad específica (ηsp), y la viscosidad reducida (ηred), o índice de viscosidad, se obtiene dividiendo ηsp por la concentración de la disolución (c). La viscosidad intrínseca, o índice de viscosidad límite, se obtiene extrapolando ηred a una concentración cero. Staudinger demostró que la viscosidad intrínseca de una disolución ([η]), como la viscosidad del producto fundido (η), estaba relacionada con el peso molecular medio del polímero (M). La forma actual de esta relación viene dada por la ecuación de Mark-Houwink, en la que la constante de proporcionalidad K es característica del polímero y del disolvente y el exponente a es una función de la forma de la hélice del polímero en la disolución. En un disolvente θ1, el valor de a para la hélice estadística ideal es 0.5. Este valor que es en realidad una medida de la interacción entre disolvente y polímero, aumenta a medida que la hélice se expande en disolventes más adecuados, tomando a un valor entre 1.8 y 2.0 para una cadena de polímero rígida estirada en toda su longitud y 0 para las formas esféricas. El valor de a se encuentra normalmente entre 0.5 y 0.8 en disoluciones de los polímeros, K tiene valores que generalmente van de 10-2 a 10-4 ml/g (Tabla 9).
[η] = KMa (XIV)
La viscosidad intrínseca de una disolución, como la viscosidad de un producto fundido, depende de la temperatura y disminuye a medida que aumenta la temperatura como se observa en la siguiente ecuación de Arrhenius (ecuación XV). Sin embargo, si la temperatura original se halla por debajo de la temperatura θ, la viscosidad se incrementará cuando la mezcla del polímero y del disolvente se calienta a una temperatura ligeramente superior a la temperatura θ. : [η] = Ae E/(RT) (XV)
1 Disolvente theta: disolvente en el que el polímero existe como hélice estadística siendo la segunda constante
virial nula a temperatura θ , es decir la temperatura a la que un polímero de peso molecular infinito empieza a precipitar en una disolución.

Polímero Disolvente Temp. (K) K x 10 5 dl g -1
LDPE (polietileno de baja densidad)
Decalina 343 39
HDPE (polietileno de alta densidad)
Decalina 408 68
Polipropileno (isotáctico) Decalina 408 11 Poliestireno Decalina 373 16
Policloruro de vinilo Clorobenceno 303 71 Poliacetato de vinilo Acetona 298 11 Poliacrilato de metilo Acetona 298 6
Poliacrilonitrilo Dimetilformamida 298 17 Polimetacrilato de metilo Acetona 298 10 Politereftalato de etileno m-Cresol 298 1
Nilón-66 90 % ácido fórmico acuoso
298 110
Tabla 9- Valores típicos de K en la ecuación de Mark-Houwink La viscosidad relativa está relacionada con [η] mediante una ecuación de virial como sigue (ecuación XVI):
ηsp / c = [η] + k1[η] 2 c + k´ [η] 3 c 2 + …. (XVI)
Para la mayoría de los sistemas, la ecuación XVI se reduce a la ecuación de viscosidades de Huggins (ecuación XVII):
ηsp / c = [η] + k1[η] 2 c (XVII)
Que permite determinar [η] a partir del corte de la curva ηsp / c en función de c, siendo la base de la curva que se da en la parte superior de la figura 5.
Figura 5 – Viscosidades en función de la concentración.
Otra relación usada a menudo para determinar [η] es la ecuación que se llama de la viscosidad inherente y es como sigue (ecuación XVIII):
(log ηr ) / c = [η] - k2[η] 2 c (XVIII)

4 Evaporación La velocidad de evaporación de un solvente es una de las dudas más comunes y más difíciles de resolver por los químicos. Esta velocidad de evaporación depende de un gran número de factores a veces contradictorios entre sí.
4.1 Presión o tensión de vapor de saturación Un líquido ubicado en un recipiente abierto o extendido en una superficie, se evapora progresivamente hasta que todas las moléculas en estado líquido hallan pasado al estado vapor. Si se cierra herméticamente el recipiente, se establece un equilibrio entre el líquido y su vapor. Después de cierto tiempo, el vapor alcanza una presión característica para cada líquido a una temperatura determinada: Es la presión de vapor de saturación, que corresponde al número de moléculas que pasa del estado vapor al estado líquido e inversamente se equilibran para mantener esta presión constante a temperatura determinada Mientras más elevada es la presión de vapor de saturación, más moléculas en estado de vapor existirán en un recipiente cerrado. En un recipiente abierto, el líquido se evaporará por lo tanto mucho más rápido.
Figura 6 - Tensión de vapor
2.6 Ebullición La presión (o tensión) de vapor aumenta con la temperatura. Así la temperatura de ebullición de un líquido corresponde al valor de la tensión de vapor, que es igual a la presión exterior sobre este líquido, cual sea el valor de esta presión. La ebullición es en efecto, una forma particular de evaporación que se produce en el interior de un líquido.
Figura 7 – Ebullición.

Si se tiene una burbuja de aire a profundidad h dentro de un líquido. Esta burbuja existe porque la presión existente al interior compensa la presión exterior, cuya parte más importante es la presión atmosférica H. Cuando se calienta, la presión al interior de la burbuja aumenta. En el momento donde alcanza el valor de la presión atmosférica, sube a la superficie donde revienta suficiente aire para que así se formen otras burbujas: Es la ebullición. Para un cuerpo puro, la temperatura de ebullición se mantiene constante durante toda la ebullición, mientras por supuesto la presión exterior no varíe. En una mezcla de solventes, por el contrario, la temperatura a la cual la presión de vapor alcance el valor de la presión exterior, dependerá de la composición de la mezcla. Normalmente, es el compuesto más volátil el que se evapora más rápido y el líquido se enriquece con el compuesto menos volátil. La t° de ebullición entonces aumenta progresivamente. En una mezcla, puede ocurrir que debido a fuertes interacciones intermoleculares, se forme lo que se denomina azeótropos. Es una mezcla que hierve a temperatura constante y que por lo tanto es imposible de separar por destilación. Corresponde a proporciones muy determinadas de cada solvente. Algunas de estas mezclas pueden ser utilizadas en conservación; sobre todo cuando hay necesidad de un solvente que contiene agua pero del cual necesitamos sea más volátil que esta.
Constituyentes Tº de ebullición Tº ebullición azeótropos Composición % Ácido acético 118.1 76.6 3 Agua 100.0 97 Acetona 56.5 56.08 88.5 Agua 100.0 11.5 Etanol 78.5 76.7 68.0 Tolueno 110.6 32.0 Metiletilcetona 79.6 73.4 88.0 Agua 100.0 12.0 Isopropanol 82.3 76.3 38.2 Tolueno 110.6 48.7 Agua 100.0 13.1
Tabla 10- Azeótropos La temperatura de ebullición es una de las primeras características para clasificar los solventes. Se distinguen entre los de bajo punto de ebullición (< a 100°C), los de mediano punto de ebullición (entre 100°C y 150°C) y los solventes de alto punto de ebullición (más de 150°C). Al igual que con las presiones de vapor, no se puede relacionar directamente la temperatura de ebullición a la velocidad de evaporación. Un ejemplo característico es el del propanol que hierve a 97°C. Se evapora sin embargo, dos veces menos rápido que el acetato propileno que hierve a 101°C. Otros factores intervienen y alteran esta relación directa.
2.7 Calor latente de evaporación Si se piensa en un espacio mantenido a una T° constante. Si se pone ahí un recipiente que contiene un líquido sellado por un pistón (bien hermético). Si se levanta lentamente el pistón, el líquido pasará progresivamente al estado de vapor y en un momento determinado no quedará ninguna gota de líquido. A todo lo largo de esta evaporación, el termostato ha tenido que aportar el calor necesario para mantener la T° constate, ya que la evaporación conlleva un enfriamiento. La cantidad de calor aportado para que un gramo de líquido sea convertido totalmente al estado de vapor constante es llamado calor latente de evaporación de ese líquido. Varía según la presión atmosférica.

Figura 8 – Calor Latente
Se ha constatado que los valores más altos de T° de vaporización son los del agua y alcoholes. Esto se explica por las muy fuertes interacciones intermoleculares que existen en estado líquido para estos compuestos, y particularmente a los enlaces de hidrógeno. Los alcoholes forman agregados de deferentes tipos.

En la fase vapor solo hay monómeros a menos que sea en condiciones de presión muy elevadas. Por lo tanto hay que aplicar al líquido suficiente calor y así romper las cadenas de polímeros para transformarlos en monómeros susceptibles de pasar al estado vapor. El resultado es un valor elevado (alto) del calor latente de vaporización. El consumo de calor en la evaporación de un líquido provoca que en algunos casos se alcance la T° de condensación del agua haciendo que la humedad contenida en el ambiente, en las inmediaciones de la superficie de evaporación, se condense. Este fenómeno puede traer inconvenientes si la superficie contiene substancias sensibles al agua, como las resinas terpénicas naturales. Se observan entonces superficies mates, blanqueamientos y otros defectos ópticos. Es casi siempre el caso cuando se utilizan solventes que se evaporan muy rápido como el isooctano o éter. Para poder ser volátil, un solvente debe presentar a la vez: a.- Una presión de vapor de saturación elevada. b.- Un bajo punto de ebullición. c.- Un bajo Calor latente de vaporización. Estas condiciones se reúnen en solventes como el éter, el diclorometano, el sulfuro de carbono, etc....
Solvetes Presión de vapor en Mg. a 20 ºC
Temperatura de ebullición a 760 mmHg
Calor latente de vaporización en cal
Agua 17.5 100 540 Éter 440 34.6 86 Diclorometano 440 41 78.7 Sulfuro de carbono 360 46 82 Formiato de etilo 200 54 97 Acetona 178 56.2 125 Cloroformo 160 61.3 59 Metanol 100 65 262.8 Benceno 74 80 94 Acetato de etilo 73 77 87 Etanol 40 78 204.3 Heptano 40 98.2 76 Isooctano 40 a 38 99.3 Tolueno 37 110.6 83 Isopropanol 33 82.4 159 p-xileno 10 138 81 Diacetonalcohol 1 168 111 Etilenglicol 0.06 197 191 Glicerol 0.0025 210
Tabla 11-Parámetros que influencian la “volatilidad” de un solvente

4.4 La conductividad térmica El calor se transmite de un cuerpo caliente a un cuerpo frío o de una zona más caliente a una zona más fría de un mismo cuerpo. Este proceso ocurre más o menos rápido según la capacidad de ese o esos cuerpos de ser más o menos conductores del calor. Ciertas substancias como los metales son my conductores, otros como el vidrio y la madera lo son menos y pueden servir de aislante térmico.
Aire 0.0000014 Cera 0.00009 Lino 0.0002 Madera 0.0003 Papel 0.0003 Cal 0.002 Yeso 0.003 Acetona 0.0004 Etanol 0.0004 Tolueno 0.00038 Isopropanol 0.000336 Éter 0.000328 Diclorometano 0.000291 Cloroformo 0.000246
Tabla 12-Coeficientes de conductibilidad térmica a 20°C
Un solvente que para evaporarse necesita un aporte calor, lo tomara mucho más fácilmente del medio ambiente si es conductor del calor. Es necesario tomar en cuenta que si la evaporación ocurre muy bruscamente, esto provocará un fuerte y brusco enfriamiento que puede frenar la continuación del proceso.
4.5 Descripción de la evaporación El secado de las capas de pintura por pérdida del solvente ha sido investigado por C.M. Hansen y sobre lo cuál ha sido recientemente publicado un artículo por Newman y Nunn. Hansen ha establecido que el secado de una película de pintura puede dividirse en dos fases: La primera fase (1) está marcada por la resistencia de la superficie al paso del solvente hacia el aire: La superficie todavía está mojada por el solvente y se deben tomar en cuenta varios factores como la T° latente de evaporación del solvente, su tensión de vapor, el coeficiente de difusión del solvente en el aire, el movimiento del aire en la superficie, etc. La segunda fase (2) consiste esencialmente en la resistencia de las capas internas de la película para liberar el solvente. Esta etapa estaría controlada sobre todo por el coeficiente de difusión del solvente en el polímero. Es la etapa de retención del solvente y puede durar varios años. En ciertos casos, la primera fase presenta una importancia preponderante. Es lo que plantea Sletmoe para el secado de películas de resinas alquídicas y etil celulosa que contengan solventes que no formen enlaces de hidrógeno (hidrocarburos saturados, aromáticos y clorados). El paso a la superficie pareciera ser la etapa más lenta. Es la que impone una velocidad límite al conjunto. Es denominada determinante.

Una de las explicaciones a tomar en cuenta sería que el enfriamiento en la superficie como consecuencia del consumo de calor latente de vaporización reduciría a la vez la presión de vapor del solvente y el coeficiente de difusión. Se describe de la siguiente manera la evaporación de un solvente a partir de una solución. 1.- Difusión a través de la masa de líquido de A a B. 2.- Paso a la superficie líquida B. 3.- Difusión a través de la masa de aire laminar estático a la superficie del líquido de B a C. 4.- Evacuación en la región de aire turbulento D.
Figura 9 – El proceso de evaporación.
Dauchot y De Witte han estudiado el secado del barniz paraloid B72 que se utiliza para barnizar las pinturas en el IRPA. Soluciones en tolueno y xileno al 10% fueron respectivamente consideradas. Las muestras fueron depositadas sobre una copela metálica y sobre una muestra de pintura. Haciendo un seguimiento de la evaporación por registro radioactivo, se constató que el tolueno puro se evapora más rápido que el p-xileno puro y que el solvente se evapora menos rápido sobre el metal que sobre la muestra de pintura. Se explica este fenómeno haciendo notar que el barniz, siguiendo las irregularidades de la pintura, es delgado en ciertas partes, lo que favorecería la evaporación del solvente. Se podría también explicar con el hecho de que el enfriamiento local es en este caso sin duda más sensible en el metal que es conductor, que en la capa de pintura. Por otro lado, después de la formación de la película, el tolueno se evapora menos rápido que el P-xileno. Una explicación sería que hay sin duda más interacción de tipo polar entre el tolueno y paraloid ya que el p-xileno presenta momento dipolar nulo. Yaseen y Ashton muestran que durante la fase 2, de difusión del solvente en la capa sólida, la eliminación del solvente es directamente proporcional a la diferencia de tensión de vapor del solvente a través de la película e inversamente proporcional al espesor de esta. Como no hay prácticamente solvente en la atmósfera después de la fase 1, la difusión va a continuar hasta que no haya más solvente en la película. Sin embargo la salida del porcentaje restante es muy lenta, por un lado porque se trata de una película en formación, por lo que la resistencia a la difusión aumenta. Por otro lado (lo que es también así en un película seca), porqué la diferencia de presión de vapor a través de la película es muy pequeña.

No es raro encontrar solvente aún en películas pictóricas que han sido expuestas a la intemperie por más de dos años. La evaporación de solventes después de la aplicación de estos sobre un cuerpo poroso, puede dividirse en dos etapas: La primera, rápida, donde la evaporación del solvente que quedó en la superficie predomina, la segunda, más lenta, donde el solvente que penetró en el sustrato sale poco a poco. Antiguamente se determinaba la velocidad de evaporación depositando 0.5 ml de solvente en un papel filtro y midiendo el tiempo de evaporación en un lugar sin corrientes de aire. El éter dietílico era tomado como referencia, con un tiempo de evaporación determinado como igual a 1. En los Estados Unidos se utilizó durante largo tiempo un método que medía el tiempo de evaporación de 5 ml. de solvente contenidos en una superficie de porcelana de aproximadamente 4 centímetros de diámetro usando el acetato de butilo como estándar. Métodos como este y otros no arrojan resultados comparables, lo que los hace más aleatorios. En efecto, estas velocidades de evaporación corresponden bastante certeramente a la fase 1 pero no toman en cuenta la fase 2. No se pueden utilizar, ni en mezclas de solventes, ni para prever la evaporación post impregnación de un cuerpo poroso. M. Dauchot ha estudiado con ayuda de registro radioactivo la evaporación de solventes utilizados en una pintura. Durante los 3 o 4 primeros minutos, es la capa de la superficie la que se evapora, comportándose como solvente aislado. Más tarde, se observa una disminución de la evaporación que puede explicarse por la penetración en los poros. Según el tiempo que dura el fenómeno, se puede deducir en qué medida un solvente es penetrante. M. Dauchot clasifica por orden creciente de penetración, sucesivamente el isopropanol, el etanol, el metanol y la acetona. Cabe hacer notar que este orden corresponde a valores en aumento de la tensión superficial y a valores decrecientes de viscosidad. Estos dos parámetros podrían entonces contribuir a prever el poder de penetración. Los datos obtenidos demuestran también que la cantidad de solvente retenido es muy diferente si se aplica con gota o hisopo, en el caso del etanol e isopropanol, pero no en el caso del metanol y de la acetona. El autor interpreta este fenómeno de la siguiente forma: Para solventes de poca difusión, hay una gran diferencia ya que el hisopo hace penetrar una cantidad de solvente que con una gota no penetra, lo que no ocurre en el caso de los solventes muy penetrantes que tienen rápida difusión en el sustrato. Schoeller estudia fenómenos muy similares que acompañan la evaporación del agua en los suelos. Él considera sucesivamente la evaporación a partir de una napa de agua, la evaporación por un suelo saturado de agua y luego la evaporación por un suelo no saturado de agua. Estas 3 etapas pueden ser comparadas a lo que ocurre en el curso de una limpieza de un cuerpo poroso con ayuda de solventes. Para la evaporación de agua del suelo, hay que tomar en cuenta el déficit de saturación del aire en agua, la presión atmosférica, la agitación del aire, y las dimensiones (extensión y profundidad) de la capa líquida. Un suelo completamente impregnado de agua se evapora tanto como una superficie de agua y a veces más. El agua que se evapora así puede ser renovada por subida capilar, sobre todo cuando los capilares afloran en gran cantidad, pero este reaprovisionamiento es muy lento, y aún más lento si el suelo está compuesto de elementos de mayor tamaño (más toscos). En un suelo no saturado de agua, el agua libre o el agua capilar ha descendido por debajo de la superficie del suelo. El movimiento del agua en forma de vapor ocurrirá si el aire no está saturado con vapor de agua, desde las regiones de fuerte tensión de vapor hacia regiones de menor tensión de vapor. Para el agua capilar, el paso de agua líquida a agua vapor depende del tamaño de los capilares. Mientras más estrechos son los capilares, más pequeño es el radio de curvatura y por lo tanto más débil es la presión de vapor de saturación, según la formula de Lord Kelvin:

Pr es la tensión de vapor de saturación bajo la superficie de radio r P� es la tensión de vapor bajo una superficie plana del radio � T es la tensión superficial H una constante que depende de la densidad del líquido La evaporación será más lenta si los capilares son más estrechos. El agua que se ha evaporado hasta la superficie del menisco debe difundirse hacia la superficie del suelo. Esta difusión resultará más fácil si los conductos son más cortos y más abiertos. Aunque así sea, esta difusión es extremadamente lenta: Una capa de 70 cm de arena, con granos de 2mm de diámetro evapora alrededor de 6 lts por m2 en un año, o sea un espesor de 6mm de agua.
3 Seguridad Un factor ineludible a la hora de la selección de un solvente o mezcla solvente es considerar su toxicidad y su inflamabilidad, dos factores que son en general comunes a los solventes utilizados en pinturas. La toxicidad de los solventes en general y los hidrocarburos aromáticos en particular es ampliamente reconocida en la bibliografía. La razón radica en la afinidad de estos con los tejidos grasos del organismo, en especial con el sistema nervioso central, aunque también afecta el sistema nervioso periférico. Los solventes orgánicos se absorben principalmente por el tracto respiratorio, aunque también pueden ingresar al organismo a través de la piel. Una vez que penetran en el organismo se distribuyen por los tejidos y órganos internos donde son sujetos a biotransformación, principalmente en el hígado con participación en las reacciones de oxidación y conjugación. Además se acumulan en tejidos con abundantes lípidos como el sistema nervioso. La baja tensión de vapor, y el efecto sinérgico causado por su aplicación por atomización favorecen la absorción por vía respiratoria si no se usa la protección adecuada. En muchos casos los operarios utilizan barbijos completamente inútiles para retener los vapores de solventes. Algunas de las afecciones relacionadas con solventes son alteraciones del sistema nervioso central, hígado, sistema pulmonar, sistema hematopoyético. Por ejemplo el benceno es un agente mutagénico reconocido, embriotóxico y carcinogénico. La tendencia mundial es hacia el uso de pinturas sin solvente, acuosas, o con solventes menos agresivos hacia el ser humano y el medio ambiente.

3.1 Toxicidad - Valores TLV Una buena guía para observar la toxicidad de los solventes son los valores TLV (Thresold limite value, valores límite umbral), publicados por la ACGIH (American Conference of Governmental Industrial Hygienists) y se refieren a las concentraciones de los compuestos peligrosos en el aire. Establecen una concentración límite por debajo de la cual, según se cree, prácticamente todos los trabajadores pueden sufrir una exposición repetida día tras día y, sin embargo, no causarles efectos adversos. Los TLV están sometidos a una revisión/corrección periódica, según van apareciendo nuevos datos. Se publican 3 categorias: TLV - TWA (Valor límite umbral - Media ponderada en el tiempo): es la concentración, como media ponderada temporal, durante una jornada laboral de ocho horas (40 horas a la semana) a la cual pueden estar expuestos de manera repetida los trabajadores sin sufrir efectos adversos. TLV - STEL (Valor límite umbral - Límite de exposición a corto plazo): es la concentración a la cual pueden estar expuestos durante un periodo breve (normalmente, 15 minutos) los trabajadores sin sufrir irritación, daños hísticos crónicos o irreversibles o un deterioro susceptible de aumentar daños por accidente, perjudicar la capacidad de autoprotección o reducir el rendimiento en el trabajo. TLV - C (Valor límite umbral - Límite superior) :es la concentración que jamás debe superarse durante la exposición laboral. Más allá de la toxicidad en sí misma de cada solvente, es importante estimar la concentración que es factible encontrar en el ambiente debida a la evaporación del mismo. Algunos autores proponen un índice de toxicidad resultante de la razón entre el valor TLV y la presión de vapor del disolvente a la temperatura ambiente. Hidrocarburos Compuestos Halogenados n-Hexano 20 Cloroformo 10 Ciclohexano 200 Tetracloruro de C 5 n-Heptano 500 Clorobenceno 5 Hidrocarburos aromáticos Éteres Benceno 10 Tetrahidrofurano 50 Tolueno 50 Dietil Eter 400 Xileno 50 Cetonas Estireno 20 Acetona 500 Alcoholes Metil Etil Cetona 200 Metanol 200 Ésteres Etanol 1000 Acetato de Metilo 200 i-Propanol 400 Acetato de Etilo 400 n-Propanol 200 Compuestos Nitrogenados Acetonitrilo 40 Anhilina 2
Tabla 13- Valores TLV
3.2 Inflamabilidad – Riesgo de Explosión Los solventes orgánicos suelen presentar riesgo de inflamabilidad e incluso de explosión. Estos riesgos están relacionados con la temperatura de ignición y con la tensión de vapor del disolvente. Más allá del peligro mismo causado por el fuego, algunos solventes pueden eliminar sustancias especialmente tóxicas, como NOx, SO y SO2, Dioxinas en el caso de los compuestos nitrogenas, azufrados y halogenados respectivamente. El medio de extinción es otro factor a tener en cuenta en caso de incendio. Es inútil el uso de agua cuando se trata de fuego sobre solventes. Los matafuegos a usarse deben indicar que son útiles para fuegos tipo B (por ejemplo matafuegos de dióxido de carbono, de polvo químico, de derivado de halon)

3.3 Medidas de Seguridad En cuanto a las medidas de seguridad personales se pueden citar:
1) Se debe conocer la sustancia con la que se trabaja. Una fuente importante de datos son las Hojas de Datos de Seguridad (MSDS), que las debe brindar el proveedor.
2) Uso de protección respiratoria adecuada. Cabe destacar que el uso de barbijos no es efectivo contra solventes y debe usarse máscaras diseñadas para tal fin.
3) Vestimenta, anteojos y guantes adecuados. Del mismo modo que en el ítem anterior, es práctica común el uso de guantes de latex, material soluble en muchos solventes, y a menudo muy permeable.
4) Ventilación adecuada, a fin de evitar riesgos de inhalación y de explosión. 5) Se debe alejar toda fuente de calor o de chispa de los lugares donde se almacenan o se
trabaja con disolventes. 6) Almacenamiento apropiado, con extracción de vapores y sistema colector de
derrames. Separación de las sustancias de modo que no queden juntas sustancias que pueden reaccionar entre si.
7) Cuando se pueda evitar el uso de solventes y de sustancias tóxicas, debe hacerse. El triángulo de solubilidad explicado en el presente trabajo es una herramienta muy útil a tal fin: se puede reemplazar un solvente tóxico por una mezcla de solventes no tóxicos, simplemente asegurando que los parámetros de solubilidad de la mezcla sea similar los del solvente (también debe hacerse notar que la taa de evaporación y la viscosidad variarán)

4 Bibliografía
1. Wikipedia. Articulo: Kauri-butanol
2. VWR. Tecnical tables.
3. John Burke. Solubility Parameters: Theory and Application.
4. Raimond B. Seymour y Charles E. Carraher Jr. Introducción a la química de los polímeros.
5. Max Doerner y Thomas Hoppe. Los materiales de pintura y su empleo.
6. INP sector activo. Exposición a los solvents.
7. Documento técnico PPG. Diluyentes.
8. IBM Research Almaden Research Center. Teas Solubility Parameter Map
9. Dieter Stoye, Werner Freitag. Paints, Coatings and Solvents.
10. Fichas de seguridad (www.fichasdeseguridad.com). Hojas de datos de seguridad.
11. Foro: Instituto de Estudios Superiores. Seguridad e higiene industrial.

5 Índice
1 INTRODUCCIÓN 1
2 PODER SOLVENTE 1
2.1 Índices de solubilidad 1
2.2 El parámetro de Hildebrand. 3 2.2.1 Densidad de energía de Cohesión 3 2.2.2 Valores del parámetro de Hildebrand 4 2.2.3 Relación del parámetro de Hildebrand con otros índices de solubilidad 6 2.2.4 Aplicaciones y limitaciones del parámetro de Hildebrand 7
2.3 Fuerzas de atracción intermoleculares y su relación con la solubilidad. 8
2.4 El modelo de Hansen 8
2.5 Parámetros fraccionales. El triángulo de solubilidad de Teas. 11
3 VISCOSIDAD. 13
4 EVAPORACIÓN 15
4.1 Presión o tensión de vapor de saturación 15
4.2 Ebullición 15
4.3 Calor latente de evaporación 16
4.4 La conductividad térmica 19
4.5 Descripción de la evaporación 19
5 SEGURIDAD 22
5.1 Toxicidad - Valores TLV 23
5.2 Inflamabilidad – Riesgo de Explosión 23
5.3 Medidas de Seguridad 24
6 BIBLIOGRAFÍA 25
7 ÍNDICE 26