juliol 2013 /informació freqüent · 2013-09-19 · vs93 infeccions de transmissió sexual vs92...
TRANSCRIPT

97juliol2013
MALALTIES RARES, INFORMACIÓ FREQÜENT ■ ANOMALÍAS CONGÉNITAS, CONOCER PARA AVANZAR ■ ELPAPEL DE LOS MÉDICOS DE FAMILIA EN EL ABORDAJE DE LOS PACIENTES CON ENFERMEDADES RARAS. LASCEBRAS NO SON TAN RARAS ■ ATENCIÓN ESPECIALIZADA DE LAS ENFERMEDADES DE BAJA PREVALENCIA.EXPERIENCIA DE LA UNIDAD DE ENFERMEDADES DE BAJA PREVALENCIA DEL HOSPITAL GENERALUNIVERSITARIO DE ALICANTE ■ MEDICAMENTOS HUÉRFANOS ■ EL DIAGNÓSTICO GENÉTICO DE LASENFERMEDADES RARAS ■ NO ES RARO TENER UNA ENFERMEDAD POCO FRECUENTE
Malalties rares,informació freqüent/
++o

vS 93Infeccions�de
transmissió�sexual
vS 92Desigualtats�ensalut�i�gènere
vS 91Dades�de�la�nutrició�a�laComunitat�Valenciana
vS 88Resistències�aantimicrobians
vS 90Salut�bucodentalen�edat�infantil
vS 96Educació�per�a
la�salut 2.0
vS 94Salut
laboral
vS 95Trastorns
musculoesquelètics
EDITADirecció General de Salut Pública.Conselleria de Sanitat. Generalitat
COORDINADORSValentín Esteban Buedo i
Joan Quiles i Izquierdo
EDITOR�TÈCNICJavier Parra Gasent
ASSESSORAMENT�LINGÜÍSTICServei de Gestió Administrativa
Secretaria General Administrativa. SubsecretariaConselleria de Sanitat
COORDINADOR�CIENTÍFIC�DEL�MONOGRÀFICÓscar Zurriaga Llorens
COL·LABORADORS�DEL�MONOGRÀFIC�Almudena Amaya Rubio, Clara Cavero
Carbonell, María Dolores Edo Solsona, MiguelGarcía-Ribes, Silvia Gimeno Martos, Rosario
Guaita Calatrava, Zaira Martín-Villalba,Carmen Martos Jiménez, José M. Millán
Salvador, Lucía Páramo Rodríguez, ManuelPosada de la Paz, José Luis Poveda Andrés,
Rosario Sánchez Martínez, José Vicente SorlíGuerola i Óscar Zurriaga Llorens.
IMPRESSIÓ�I�FOTOCOMPOSICIÓGrafo Impresores SL
DISTRIBUCIÓMeydis
DIPÒSIT�LEGALV-1063-1988
ISSN�1888-6833
Inclosa�en�el�directori�depublicacions�seriades�LATINDEX
VIURE EN SALUT s’envia gratuïtament a tots els col·legis,ajuntaments i centres sanitaris de la ComunitatValenciana. També es tramet a les associacions, entitatso persones que ens ho sol·liciten.
Agrairíem que se'ns comunicara qualsevol anomaliaobservada en la recepció, per tal de corregir-la, i tambéels canvis de domicili.
La Conselleria de Sanitat, entitat editora de VIURE EN
SALUT, no s'identifica necessàriament amb les opinionsexpressades pels redactors i col·laboradors de la publicació.
VIURE EN SALUT autoritza la reproducció dels seus tex-tos, sempre que se'n cite la procedència. Alhora, dema-nem que ens feu arribar dos exemplars de la publicacióon s'incloguen els seus continguts.
Us convidem a fer una revista cada vegada més vostra. Nodubteu a donar-nos la vostra opinió sobre la revista,expressar el vostre acord o desacord amb els articles d'opi-nió que hi apareixen, aportar suggeriments, plantejar dub-tes o peticions, etcètera. Esperem les vostres aportacions.
Adreceu-vos a:
Direcció�General�de�Salut�PúblicaServici�de�Plans,�Programes�i�Estratègies�de�Salut
Avinguda�de�Catalunya,�2146020�València961�92�57�[email protected]
SUBSCRIPCIÓ�GRATUÏTA
1 nom i cognoms
2 Domicili
3 localitat
4 Província
5 Codi postal
6 telèfon
7 Correu electrònic
8 Data de naixement
9 Professió
Direcció General de Salut PúblicaServici de Plans, Programes iestratègies de Salutavinguda de Catalunya, 2146020 València
961 92 57 92
Adreça�postal
Correu�electrònic
Telèfon
Per a subscriure's a Viure en Salut cal comunicar les dades demanades, per a la qual cosa es pot utilitzar
qualsevol d’estos mitjans: correu postal, correu electrònic o telèfon. també es pot fer una subscripció en línia
en la web de la Direcció General de Salut Pública: www.sp.san.gva.es
vS 89Seguretatalimentària
2 vS 97
últimes pàginesMalalties rares, informació freqüent

Sumari vS 97
últimes�pàgines2 .......................Últims�números�publicats
editorial3 ..........................................................
informe4........Malalties�rares,�informació�freqüent
ÓSCAR�ZURRIAGA�LLORENS�I�MANUEL�POSADA�DE�LA�PAZ
6 .......................Anomalías�congénitas,conocer�para�avanzar
CARMEN�MARTOS�JIMÉNEZ,�CLARA�CAVERO�CARBONELL,
SILVIA�GIMENO�MARTOS,�ROSARIO�GUAITA�CALATRAVA�Y
LUCÍA�PÁRAMO�RODRÍGUEZ
8......El�papel�de�los�médicos�de�familiaen�el�abordaje�de�los�pacientes�con
enfermedades�raras.Las�cebras�no�son�tan�raras
JOS�VICENTE�SORL�GUEROLA�Y
MIGUEL�GARCÍA-RIBES
10...........Atención�especializada�de�lasenfermedades�de�baja�prevalencia.
Experiencia�de�la�Unidad�deEnfermedades�de�Baja�Prevalencia�del
Hospital�General�Universitario�deAlicante
ROSARIO�SÁNCHEZ�MARTÍNEZ
12 ...................Medicamentos�huérfanosMARÍA�DOLORES�EDO�SOLSONA�Y
JOSÉ�LUIS�POVEDA�ANDRÉS
15..........El�diagnóstico�genético�de�lasenfermedades�raras
JOSÉ�M.�MILLÁN�SALVADOR
17.........................No�es�raro�tener�unaenfermedad�poco�frecuente
ALMUDENA�AMAYA�RUBIO�Y
ZAIRA�MARTÍN-VILLALBA
recursos19 ............................Materials�temàtics
Que una cosa siga rara no vol dir que siga estranya: simplement que és poc fre-qüent. Això és el que passa amb les malalties rares. Estan definides per laescassa freqüència que tenen i això el que significa principalment és que
requerixen una atenció especial que fins fa poc no es prestava.
Des del punt de vista de la salut pública són diversos els aspectes que poden abordar-se per a millorar la situació dels afectats per les malalties rares, i millorar la informa-ció sobre estes és un aspecte crucial i fonamental. Una major i més exacta informacióen permet superar la invisibilitat i millorar la prevenció, l’atenció sanitària i la investi-gació. Per això, s’ha volgut utilitzar el títol d’este número per a ressaltar que encaraque les malalties siguen rares la informació ha de ser freqüent.
I en eixe sentit, s’aborden aspectes diferents que permeten incrementar la informació.Començant per quins són els problemes i quina és la situació dels registres de malal-ties rares i de la investigació hui en dia en diferents àmbits, Europa, Espanya i laComunitat Valenciana, i tenint clar des del primer moment que quan la freqüència ésbaixa és necessària la unió, en tots els camps, des de la investigació a l’associacionis-me.
Es dedica també tot un capítol a les anomalies congènites que, encara que no totessón malalties rares, sí que ocupen un espai dins d’estes malalties i on tan necessarissón els esforços per a conéixer-les millor per a poder actuar en la seua prevenció.
Atés que la majoria de les malalties rares són d’origen genètic, s’ha considerat impor-tant incloure un article sobre el diagnòstic genètic, en el qual s’expliquen els tipus dediagnòstic que hi ha, les seues dificultats, el marc legal i les recomanacions.
Des del punt de vista assistencial s’inclouen dos visions que no són antagòniques sinócomplementàries i necessàries ambdós: la de l’atenció primària i la de l’assistènciaespecialitzada. Es mostren els esforços que la Societat Espanyola de MedicinaFamiliar i Comunitària, i també la Valenciana, fan en este camp. I també els que desd’un hospital, com el General d’Alacant, s’han fet en l’atenció especialitzada. I es res-salta que, en ambdós casos, els aspectes més importants són l’actitud i la coordinaciód’esforços.
En l’àmbit terapèutic s’ha considerat igualment necessari considerar el paper delsmedicaments òrfens, en els quals es basen moltes esperances i també moltes frustra-cions. Per això és important tindre informació sobre ells: conéixer què són, quins sóni quan s’utilitzen.
Per a guanyar en dimensió, quan la freqüència és escassa, és la unió la que fa la forçai, per això, la tasca de les associacions de pacients és imprescindible: permeten alspacients, familiars i propparents superar la sensació de soledat, contribuïxen al suporti ajuda mútua, amplifiquen la seua veu, canalitzen les seues reivindicacions i els fanmés visibles. Les associacions, i la seua federació, contribuïxen també a la millora dela informació.
L’objectiu d’este número de VIURE EN SALUT és aportar un altre granet d’arena a eixaatenció especial que es reclama com a necessària i que s'estructura des de l’EstratègiaNacional de Malalties Rares i les successives actuacions que es realitzen a les comuni-tats autònomes, màximament enguany 2013 que s’ha declarat com a l'Any Espanyolde les Malalties Rares. Que esta declaració servisca per a donar impuls a totes lesactivitats que poden millorar la situació dels afectats per estes malalties. Malaltiesrares. Informació freqüent.
Malalties rares, informació freqüenteditorial
3vS 97

4 vS97
La�unió�fa�la�força. Habitualment esmesura la importància d’un problemade salut pel nombre de persones a lesquals afecta, de manera que si n’hi hapocs casos no es considera un proble-
ma prioritari. Això és el que succeïa amb malal-ties que afecten un nombre xicotet de persones,malalties amb noms difícils de recordar, sovintamb noms propis, però no amb noms comuns.Malalties que es tornen invisibles perquè els queles patixen moltes vegades ni tan sols ho saben,perquè els que poden atendre-les i tractar-lestampoc les coneixen en profunditat moltesvegades, perquè els seus familiars no saben ni aqui acudir, perquè els diagnòstics no arriben iperquè els tractaments no existixen o no sónrendibles.
Davant d’esta situació, la necessitat de l’agrupa-ció emergix com un aspecte fonamental per atots: per als que les patixen, per als que les podendiagnosticar, per als que les tracten, per als queinvestiguen, per als que han de desenrotllar elstractaments i per als que conviuen amb elsmalalts.
Per això, la definició de “grup de malalties” encompte de “malaltia” ha contribuït perquè enlloc de parlar de malalties concretes ara parlemdel grup de les malalties rares i que, a Europa, lesconsiderem com les malalties que tenen una pre-
valença inferior al 5 per 10.000 habitants. Açòha permés superar, en part, el problema delsnombres xicotets. Amb esta consideració, elsafectats ja no són tan invisibles.
I si comencem pel concepte de grup, en lloc deper l’individual, el normal és que l’agrupaciócontinue sent fonamental. I, així, els afectats(malalts, familiars i propparents) troben quel’associacionisme els ajuda i afavorix, i les asso-ciacions d’afectats s’agrupen i es fonen en federa-cions d’associacions. I els professionals i investi-gadors també necessiten agrupar-se per aaconseguir ser més efectius en el seu treball.
D’esta manera van nàixer i créixer les grans xar-xes d’associacions que hui en dia estan presents aEuropa, com ara EURORDIS, o també a Espanya,com ara FEDER (Federació d’Associacions deMalalties Rares). I les xarxes d’investigació, comva ser en el seu moment REpIER (XarxaEpidemiològica d’Investigació en Malalties Rares),o el CIBERER (Consorci d’Investigació Biomèdicaen Xarxa sobre Malalties Rares). I els grans con-sorcis d’investigació supranacionals, com és hui endia IRDiRC (International Rare Diseases ResearchConsortium).
De manera que cada vegada es concep menysl’esforç aïllat. I és que en este àmbit, com enaltres, es demostra que la unió fa la força.
La�informació�com�a�aspecte�essencialSi la definició segons la qual una malaltia raraestà basada en la prevalença, és absolutamentfonamental saber quantes persones patixen unamalaltia per a poder caracteritzar-la com a tal.
Per exemple, a la Comunitat Valenciana(5.129.266 d’habitants en 2012), si apliquem ladefinició de la Unió Europea, qualsevol malaltiaque, en un moment determinat, patisquen 2.565persones seria considerada una malaltia rara. Iquantes malalties complixen esta condició?
Açò, que podria paréixer una qüestió fàcil de res-pondre, no ho és tant. Encara que s’ha avançatmolt en els últims anys, probablement no es dis-posa encara de la suficient informació sobre ladistribució de les malalties rares a Espanya, itampoc a escala europea, i per això és necessariampliar els esforços per a disposar de ferramen-tes millors que permeten ampliar el coneixement.
En este sentit, els registres de malalties rares i depacients constituïxen un instrument clau per aldesenrotllament i la millora de l’atenció clínica,la investigació i les actuacions sanitàries.
Disposar d’informació sobre aspectes bàsicsd’una malaltia rara és una ferramenta fonamen-tal per a millorar l’abordatge d’esta i permet ofe-
rir una oportunitat als que la patixen, des delspunts de vista preventiu, diagnòstic, terapèutic isociosanitari.
En epidemiologia es definix vigilància, d’unamanera abreviada, com “disposar d’informacióper a possibilitar l’acció sanitària”: informacióper a l’acció. Eixe és l’objectiu.
Els�problemes�de�la�informació�en�lesmalalties�raresEn l’àmbit de les malalties rares són diverses, ivariades, les dificultats que és necessari superarperquè existisca eixa informació que pot ser tanútil. Entre altres tenim:a) El concepte de malaltia rara (la definició): allò
que és un avantatge per a superar el problemadels nombres xicotets es pot tornar un incon-venient. La definició de malaltia rara, basadaexclusivament en el criteri de la mesura epide-miològica de la prevalença, comporta inclouredins del mateix grup malalties amb causesmolt diverses (encara que les genètiques en sónuna part important), amb possibilitats de trac-tament molt diferents, amb expectatives decuració variades i amb possibilitats d’actuacióque, de vegades, poc tenen a veure entre si.Això dificulta a vegades el treball, tant a esca-la assistencial clínica com des del punt de vistade salut pública, perquè de vegades són consi-derades com si foren una única entitat malal-ties que no s’assemblen gens entre si, que notenen un mateix curs clínic, amb teràpies moltdiverses, i que des del punt de vista de la infor-mació compartixen poques característiques encomú.
b) La diversitat de les fonts de dades: la varietatde malalties fa que en l’abordatge d’estes inter-vinguen des del nivell més especialitzat possi-ble fins al més generalista, incloent-hi a més elsociosanitari. Per això, la informació s’had’obtindre de múltiples fonts que han de con-fluir per a poder tindre una visió completa.Esta multiplicitat de fonts complica el treball adesenrotllar perquè cada una d’estes pot tindreuna dependència i una estructura diferent, i ésnecessari poder fer-les convergir. I, ací, el con-cepte d’interoperabilitat (la capacitat de dos omés sistemes o components d’intercanviarinformació i d’utilitzar la informació que s’haintercanviat) emergix com a absolutamentnecessari.
c) La importància de la codificació: hi ha dife-rents formes d’entendre què és una malaltia, ies poden considerar com a tal una entitat clí-nica, o un fenotip evolutiu, o el mateix feno-tip però amb distinta etiologia, o una àmpliavarietat més de criteris. Per tant, és necessariprimer tindre clar que una entitat nosològica(entitat clinicosemiològica, independent iidentificable segons criteris idonis) és diferentd’altres i es caracteritza d’una determinada
Malalties�rares,informació�freqüentÓSCAR�ZURRIAGA�I�MANUEL�POSADA
Un�millor�coneixement�deles�malalties�rares,�de�laseua�història�natural,�del
nombre�de�malaltsexistents�i�de�quines�sónles�seues�característiques
clíniques�i�socials,�ésnecessari�per�a�avançar�enla�investigació�d’estes.�Al
mateix�temps,�la�informacióen�este�camp�es�convertixen�un�factor�que�redundaen�benefici�dels�malalts�i
dels�seus�familars.
informeMalalties rares, informació freqüent

5
Malalties rares, informació freqüentinforme
vS97
manera. Després, des del punt de vista de lainformació, és necessari poder assignar-li uncodi específic, que és el que possibilitarà quesiga reconeguda en els sistemes d’informaciócom una entitat diferent d’altres i el que, endefinitiva, la farà visible. Doncs bé, en elcamp de les malalties rares açò és una com-plicació important. Hi ha diferents sistemesde classificació i codificació i la majoriad’estos tenen molt poca especificitat pel quefa a les malalties rares, la qual cosa vol dirque no hi ha codis específics per a la majoriad’estes (en la Classificació Internacional deMalalties, CIE, en la desena revisió, només300 malalties rares poden ser identificadesamb un codi únic). Sense codi específic enuna classificació, una entitat nosològica no téreconeixement “oficial” i, per tant, és mésdifícil conéixer la prevalença, realitzar l’estu-di epidemiològic i la vigilància. I tot aixòcontribuïx perquè eixa entitat o malaltia siga“invisible”.
d) L’efecte dels nombres xicotets: per definició,com ja hem comentat, són pocs els casos quees produïxen de cada una de les malalties con-siderades rares. L’aplicació de qualsevol mesu-ra epidemiològica es veu afectada per esta cir-cumstància, ja que disminuïx la precisió d’estaen incrementar-se la seua variabilitat. Aixòcomporta que siga més difícil distingir varia-cions aleatòries de canvis verdaders, per laqual cosa les comparacions en el temps i enl’espai poden portar a conclusions errònies ofalses, sobre diferències en el risc.
Què�s’està�fent
1. A�EuropaL’informe d’Orphanet1 publicat al gener de2013 i titulat “Disease Registries in Europe”identifica 588 registres relacionats amb lesmalalties rares a Europa. Un nombre que pareixmolt elevat, màximament quan es referix que423 d’estos són de nivell nacional. Tal vegada lapregunta que cal fer-se és com és possible queamb un nombre de registres tan alt de nivellnacional no existisca prou informació sobre unampli grup de malalties?
El Consorci Internacional d’Investigació deMalalties Rares2 (IRDiRC), en el qual participaEspanya i col·laboren agències de finançamentd’uns quants països i també la Unió Europea através de la Direcció General d’Investigació, trac-ta de donar precisament resposta a esta preguntai a iniciativa seua s’estan finançant diversesactuacions que permetran disposar de més infor-mació i millor sobre malalties rares a escalanacional i internacional. El consorci IRDiRC técom a objectius aconseguir que l’any 2020 exis-tisquen 200 noves teràpies en el mercat per a lesmalalties rares i que totes estes (més de 6.000)disposen d’un biomarcador diagnòstic. En estecontext, IRDiRC està organitzat per comitéscientífics i penjant d’un d’estos, el ComitéInterdisciplinari, es desenrotllen grups de treballrelacionats amb registres i biobancs.
2. A�EspanyaLa Xarxa Espanyola de Registres de MalaltiesRares per a Investigació3 (SpainRDR) és un pro-jecte finançat per l’Institut de Salut Carles III(ISCIII) en l’àmbit del consorci IRDiRC subscritper este institut. El projecte llançat per l’ISCIII idenominat SpainRDR és un dels projectes
immersos en este gran consorci i té com a objec-tiu construir el Registre Nacional de MalaltiesRares a Espanya a partir de la unificació de dosestratègies metodològiques diferents: registres depacients, enfocats a la investigació de resultats, iregistres de base poblacional dirigits a la investi-gació epidemiològica, sociosanitària i a la plani-ficació en salut. El projecte té una duració inicialprevista fins a desembre de 2014 i té el finança-ment per a poder desenrotllar este treball deforma adequada, almenys en els primers passos.
SpainRDR s’ha organitzat com altres projecteseuropeus utilitzant àrees de treball que comprenendes de la metodologia (estandardització de dades,fonts d’informació, llistes de malalties, normati-ves, aspectes ètics, etc.) fins a la posada en marxade mecanismes d’intercanvi de fitxers de dades,criteris d’accessibilitat, qualitat i anàlisi i presenta-ció de resultats. La docència també està present através de mitjans telemàtics i la seua relació i coo-peració internacional amb altres projectesIRDiRC, com ara RD-CONNECT4, EPIRARE5 oel desenrotllat als Estats Units, GRDR6, estàgarantida.
En l’actualitat, SpainRDR està tancant l’etapad’harmonització de dades i procediments i inter-canviant els primers fitxers de dades, així comestablint el marc normatiu actual dels registres demalalties rares a Espanya.
3. A�la�Comunitat�ValencianaLes actuacions en l’àmbit de la informació sobremalalties rares a la Comunitat Valenciana tenenuna fita important amb la constitució en 2003 deRepiER, en la qual el node de la ComunitatValenciana va participar amb interés i esforç, i fruitd’això va ser la publicació de l’informe de salutnúm. 91 titulat “Aproximació a les malalties raresa la Comunitat Valenciana. Període 1999-2003”(disponible en la Biblioteca Virtual de la DireccióGeneral de Salut Pública). També va ser destacat elpaper del node valencià de REpIER en l’elaboracióde l’Atles Nacional Provincial de Malalties Rares1999-20037, publicació que per primera vegadaaportava xifres de malalties rares en l’àmbit de totel país amb desagregació provincial.
La creació i posada en marxa en 2009 de l’Àread’Investigació sobre Malalties Rares en el CentreSuperior d’Investigació en Salut Pública (CSISP-FISABIO) també va significar un importantavanç, perquè el treball conjunt amb la DireccióGeneral de Salut Pública va possibilitar el desen-rotllament del Registre d’Anomalies Congènitesde la Comunitat Valenciana, que va seguir desdel seu començament les directrius establides perEUROCAT8 (la Xarxa Europea de Vigilànciad’Anomalies Congènites), s’ha establit com amembre de ple dret en esta xarxa i és un delsregistres europeus amb més cobertura i un delspocs que operen a Espanya.
L’última fita, fins a l’actualitat, és la publicació en2012 en el Diari Oficial de la ComunitatValenciana de l’orde de la Conselleria de Sanitat perla qual es crea el Sistema d’Informació sobreMalalties Rares de la Comunitat Valenciana (SIER-CV). Esta normativa dóna carta de naturalesa atotes les activitats que, en matèria d’informaciósobre este grup de malalties, es realitzaven i possibi-lita a més incrementar el ventall d’activitats a realit-zar i, a més, permet donar cobertura normativa atotes estes. El SIER-CV integra en les seues activi-
tats tots els centres sanitaris ubicats a la ComunitatValenciana i permet una integració, amb totes lesgaranties legals, en l’àmbit nacional i internacionalde la informació sobre malalties rares.
Per això, la Comunitat Valenciana, des de laDirecció General de Salut Pública i de l’Àrea deMalalties Rares del CSISP-FISABIO, està integra-da en les actuacions del projecte SpainRDR, lide-ra, a més, un dels paquets de treball d’este pro-jecte i contribuïx, per tant, a la constitució delregistre nacional espanyol.
Informació�freqüentEncara que, com ja s’ha mencionat, hi ha proble-mes importants i encara que en l’actualitat quedemolt de tram del camí per recórrer, s’estan assen-tant les bases perquè, tant a la ComunitatValenciana com a Espanya, existisca informaciósobre malalties rares suficient i de qualitat quepermeta que en un futur, ja no tan llunyà,s’incrementen les oportunitats d’atenció,diagnòstiques, terapèutiques i preventives per alsafectats i els seus acostats.
Un millor coneixement de les malalties rares, dela seua història natural, de les seues característi-ques, possibilitarà un millor diagnòstic i més pre-coç. I també permetrà valorar, d’una millormanera, l’eficàcia de les terapèutiques (fàrmacs iprocediments).
Saber quants malalts hi ha, quants en cadamalaltia, quines són les seues característiques, clí-niques i socials, també permetrà millorar l’aten-ció sociosanitària, i adaptar-la al que és necessarii facilitar-ne la provisió.
I la investigació sobre malalties rares també potaprofitar-se d’una millora en la informació, i incre-mentar les seues capacitats i àmbits d’actuació.
La informació actual (i futura) ha de redundar enbenefici dels malalts i les seues famílies. Açò nopot oblidar-se. I és una tasca a la qual estan cri-dats a exercir la seua responsabilitat les adminis-tracions sanitàries, les associacions de malalts, elsclínics especialitzats, els professionals d’atencióprimària, els epidemiòlegs, els salubristes i elsinvestigadors interessats en les malalties rares.
Cal compensar la raresa. Encara que les malaltiessiguen rares, la informació no hauria de ser-ho ihauria d’estar disponible i actualitzada com a únicavia de promoció de la investigació i de la millora dela planificació sociosanitària per a estes malalties.
Óscar�Zurriaga�LlorensDirecció General de Salut Pública.
Conselleria de Sanitat. Comunitat Valenciana.
Manuel�Posada�de�la�Pazinstitut d’investigació de Malalties rares (iier).
institut de Salut Carles iii. Madrid.
1http://www.orpha.net/orphacom/cahiers/docs/GB/registries.pdf
2http://www.irdirc.org
3https://spainrdr.isciii.es
4http://www-rd-connect.eu
5http://www.epirare.eu
6http://www.grdr.info
7http://ec.europa.eu/health/ph_threats/non_com/atlas_nacional_Provincial_er_lD2_prot.pdf
8http://www.eurocat.org

6 vS97
Las anomalías congénitas (AC) represen-tan un importante problema de saludpública ya que tienen un efecto negati-vo en la calidad de vida de los pacien-tes y sus familias. Pueden dar lugar a
abortos espontáneos y son una de las principalescausas de mortalidad infantil. Además, con fre-cuencia producen graves discapacidades físicasy/o mentales, que en la mayoría de los casos afec-tan al paciente de por vida con importantesimplicaciones personales, familiares, sociales,sanitarias, educacionales y económicas.
La Organización Mundial de la Salud (OMS)estima que las AC son responsables cada año de272.940 muertes durante los primeros 28 días devida en todo el mundo y de 275 en España. En laComunitat Valenciana se registraron en 2011 untotal de 65 fallecimientos debidos a AC en meno-res de 28 días de vida, lo que representó el 27%del total de defunciones en estos niños/as.
Las AC forman un grupo heterogéneo de patolo-gías de origen prenatal, siendo la mayoría deellas de baja frecuencia, y por tanto consideradascomo enfermedades raras. La OMS define las ACcomo toda anomalía del desarrollo morfológico,estructural, funcional o molecular presente alnacimiento (aunque pueden manifestarse mástarde), externa o interna, familiar o esporádica,hereditaria o no, única o múltiple. La etiologíade la mayoría de las AC es desconocida, sospe-chándose una interacción de múltiples factoresambientales y genéticos.
Cuando a una familia se le informa de que suhijo/a tiene una AC, a veces asociadas a otras ACo síndromes poco frecuentes, se va a producir unchoque emocional en los padres. El nacimientopasa de ser un motivo de satisfacción a ser unmomento doloroso, dando lugar a una situaciónde aflicción o duelo, que puede conllevar a una
desorganización emocional en la pareja al enfren-tarse a una condición letal o incapacitante queles roba las expectativas que habían depositadoen ese hijo/a.
Aunque las AC estructurales externas suelendiagnosticarse en el momento del nacimiento,otras AC pueden diagnosticarse en edades másavanzadas con el consiguiente desasosiego de lospadres que no tienen respuesta al problema quepadece su hijo/a y con la incertidumbre de si elproblema se va a repetir en otros descendientes.
Conscientes de la necesidad de disponer de un sis-tema de información sobre AC en la ComunitatValenciana para la vigilancia de estas patologías yla base para la investigación clínica y epidemioló-gica y así avanzar hacia la prevención y controlde estas enfermedades, la Dirección General deSalud Pública junto con CSISP-FISABIO estántrabajando en el desarrollo del RegistroPoblacional de las Anomalías Congénitas de laComunitat Valenciana (RPAC-CV). Este registroforma parte de la Red Europea de RegistrosPoblacionales para la Vigilancia de las AnomalíasCongénitas, siguiendo la metodología consensua-da con el resto de registros europeos, lo que favo-rece la comparación de los datos.
El RPAC-CV recoge desde el 2007 todos loscasos de anomalías congénitas mayores (ACM)diagnosticadas en menores de un año residentesen la Comunitat Valenciana. Las ACM son aque-llas que ponen en riesgo grave la salud o la cali-dad de vida del recién nacido, generalmente tie-nen graves consecuencias estéticas o funcionalesy requiere atención médica especializada, amenudo quirúrgica.
Durante el periodo 2007-2010 se han registradoun total de 5.048 ACM lo que representa unaprevalencia de 232 menores de un año con almenos una ACM por 10.000 nacimientos. Lasanomalías congénitas cardiacas son el grupo másfrecuente, representando el 34% del total. Lesigue en frecuencia las AC cromosómicas, queconstituyen el 13% del total, siendo el síndromede Down el más frecuente en este grupo. En lafigura 1 se presenta la distribución porcentual delas AC en la Comunitat Valenciana en el periodo2007-2010.
De las 5.048 AC registradas, el 80,6% (4.068casos) correspondieron a nacidos vivos, el18,5% (936 casos) a interrupciones voluntariasdel embarazo y el 0,9% (44 casos) fallecieronantes del nacimiento.
La prevalencia obtenida en la ComunitatValenciana es superior a la media estimada en elconjunto de los registros europeos (EURO-
Anomalías�congénitas,conocer�para�avanzarCARMEN�MARTOS,�CLARA�CAVERO,�SILVIA�GIMENO,ROSANA�GUAITA�Y�LUCÍA�PÁRAMO
Las�anomalías�congénitas�forman�un�grupo�heterogéneo�depatologías�de�origen�prenatal,�siendo�la�mayoría�de�ellas�de
baja�frecuencia,�y,�por�tanto,�consideradas�comoenfermedades�raras.�A�pesar�de�que�en�la�mayoría�de�lasveces�no�se�conocen�las�causas�de�estas�anomalías,�hay
unas�medidas�de�prevención�a�diferentes�niveles�quepueden�contribuir�a�evitar�la�enfermedad,�a�controlarla�y�a
mejorar�la�calidad�de�vida�de�los�pacientes�y�de�sus�familias.
informeMalalties rares, informació freqüent

CAT), 232 en Comunitat Valenciana y 209 enEUROCAT, principalmente debido al grupo delas anomalías congénitas cardiacas. En la figura2 se presenta la prevalencia (número de casospor 10.000 nacimientos) de los diferentes gru-pos de ACM en la Comunitat Valenciana y enEUROCAT.
Aunque la prevalencia de las anomalías congéni-tas cardiacas son superiores en la ComunitatValenciana, el grupo más grave de estas patologí-as es ligeramente inferior en la ComunitatValenciana, con una prevalencia de 16,6 por10.000 nacimientos y 17,4 en EUROCAT.
¿Se�pueden�prevenir�las�AC?A pesar de que no se conocen la mayoría de lasveces las causas de las AC, hay unas medidasde prevención a diferentes niveles —como paraotras enfermedades— que pueden contribuir aevitar la enfermedad, a controlarla y a mejorarla calidad de vida de los pacientes y de susfamilias.
En el primer nivel está la prevención primaria, yse trataría de impedir que la AC se produzca.
Existen medidas generales que contribuyen aldesarrollo adecuado del feto, como hábitos devida saludables de la mujer antes y durante elembrazo, con dieta equilibrada y realizaciónmoderada de ejercicio físico, no consumo dealcohol y otras drogas, no fumar y evitar laexposición al humo del tabaco.
El uso racional del medicamento, tanto en muje-res en edad fértil como durante el embarazo,contribuye a la prevención de las AC, debiendoser prescrita la medicación necesaria por el médi-co y no automedicarse.
La suplementación de ácido fólico, bajo la super-visión del personal sanitario, durante el periodopreconcepcional y durante los primeros meses delembarazo se ha asociado a una disminución nosólo de anomalías congénitas del tubo neural,sino también a otras anomalías congénitas comolas cardiacas.
Una deficiencia de yodo durante el embarazo yprimera infancia puede producir una hipotiroxi-nemia que afecte negativamente al cerebro endesarrollo. Se sugiere la recomendación de suple-mentos en aquellas mujeres embarazadas, que no
alcanzan las cantida-des recomendadas deyodo con su dieta.
La exposición a radia-ciones ionizantesdurante el embarazo puede suponer un riesgopara el feto, este riesgo depende de la dosis y delmomento de la gestación en que se produzca laexposición. Por tanto, deben evitarse, al igualque la exposición a sustancias químicas tóxicas,extremando el cumplimiento de las normas deprevención laboral.
Las mujeres en edad avanzada (35-40 años) tie-nen mayor riesgo de anomalías cromosómicas,por tanto una planificación del embarazo en eda-des más tempranas, contribuye a la prevenciónde estas anomalías.
En mujeres con alguna enfermedad crónica y condeseo de gestación, también se debe planificar elembarazo en fase de compensación de la enfer-medad.
La visita preconcepcional en el centro de atenciónprimaria permite, además de valorar el estado desalud de la mujer, comprobar su estado inmunitariopara las enfermedades de alto riesgo de causar AC,como la toxoplasmosis, rubeola, citomegalovirus.Además, es necesario también comprobar el estadode la mujer y su pareja respecto a enfermedadesinfecciosas con riesgo de transmisión, tanto sexualcomo vertical materno-infantil (hepatitis C, sida...).
Por último, es deseable el asesoramiento genéticoa parejas con antecedentes de anomalías congéni-tas, consanguinidad...
El segundo nivel de prevención (prevención secun-daria) se aplica cuando la AC ya se ha producido. Sebasa en el diagnóstico precoz correcto de la enfer-medad y la aplicación inmediata del tratamientoadecuado y de medidas correctoras o paliativas.
El seguimiento adecuado del embarazo permite eldiagnóstico precoz de algunas de estas patologíasy, por tanto, también el tratamiento precoz.
Para aquellas AC que no es posible el diagnósticoprenatal, el seguimiento adecuado del reciénnacido contribuye al diagnóstico precoz de estaspatologías y la instauración del tratamiento inte-gral, tanto del paciente como de la familia.
Cuando han fracasado los dos niveles de preven-ción anteriormente citados, es necesario instaurarel tercer nivel de prevención (prevención tercia-ria). Esta prevención se centra en medidas socio-sanitarias que mejoren la autonomía y la calidadde vida del paciente y permitan la integraciónsocial y laboral de los pacientes.
Existe un cuarto nivel de prevención, especial-mente importante en el ámbito de las AC menosfrecuentes y asociadas a síndromes raros, queconsiste en evitar toda sobreactuación médica, enla que se someta al paciente a pruebas y trata-mientos innecesarios que no solo no le ayudansino que, además, pueden producirle efectosnegativos, tanto físicos como psicológicos.
En resumen, las AC son un problema de saludpública con repercusión sanitaria, social y econó-mica, que con la aplicación de medidas de pre-vención primaria, secundaria, terciaria y de cuar-to nivel pueden mejorar la calidad de vida de lospacientes y sus familias. Esto es un trabajo con-junto individual y del sistema sociosanitario, quecomienza con la planificación del embarazo res-ponsable y un seguimiento maternoinfantil ade-cuado, proporcionando a los pacientes los recur-sos sanitarios y sociales necesarios para prevenirlas AC y mejorar la calidad de vida de estospacientes y sus familias.
Carmen�Martos�JiménezClara�Cavero�CarbonellSilvia�Gimeno�Martos
Rosana�Guaita�CalatravaLucía�Páramo�Rodríguez
unidad de enfermedades raras. Centro Superior de
investigación en Salud Pública (CSiSP-FiSaBiO).
B i b l i o g r a f í a
Martínez-Frías Ml, rodríguez-Pinilla e, Bermejo e, Martínez-Fernández Ml. Pautas de prevención de defectos congénitos conespecial referencia a los niveles primario y secundario. Guías deactuación preventiva desde la atención primaria. Semergen.2011; 37 (8): 412-41.
7vS97
Distribución porcentual de las anomalías congénitas mayoresen menores de 1 año, Comunitat Valenciana 2007-2010
1 Prevalencia de las anomalías congénitas mayores en menoresde 1 año, en la Comunitat Valenciana y EUROCAT
2
Malalties rares, informació freqüentinforme
Grupos de anomalías congénitas
90
80
70
60
50
40
30
20
10
0
Fisura palatal y labio leporino
Sistema nervioso Ojo
Oído, cara y cuello
Digestivas
Pared abdominal
Urinarias
Genital
Miembros
Cromosómicas
Otras
Cardíacas
Respiratorio
Eurocat Comunitat Valenciana
Prevalencia x 10.000 nacimientos

¿Conocen� los�médicos�de� familiaque�son�las�enfermedades�raras?Si bien la relación entre los médicos defamilia (MF) y las enfermedades raras(ER) ha sido hasta la fecha muy pun-
tual, gran parte del problema reside en que lamayor parte de ellos desconocen realmente quése entiende cuando hablamos de una ER. Engeneral, los MF las consideran enfermedadesexcepcionalmente infrecuentes, de las que nuncatendrían que abordar en la consulta diaria, ymucho menos diagnosticar, ya que “para esoestán los especialistas”, con alguna característicaextravagante, peculiar, pintoresca: “el hombre degoma, el ojo seco, los huesos de cristal…”, y queen los últimos meses, aparecen de forma másinsistente en los medios de comunicación. Sisupieran que en nuestro país afectan a tres millo-nes de individuos y que patologías como la enfer-medad de Crohn, la esclerosis múltiple o la acon-droplasia se consideran ER, probablemente sedarían cuenta de que no son tan raras comoparecen y están representadas en la consulta casidiariamente.
¿Por�qué�parece�tan�difícil�abordar�eldiagnóstico�de�las�ER�desde�la�consultadel�MF?Como es bien conocido, la Atención Primaria (AP)es, en nuestro país, la puerta de entrada al SistemaNacional de Salud. En las consultas de los centrosde salud, los MF ven una media de 40-50 pacientesdiarios repartidos en consultas de 5-10 minutos,con un alto contenido burocrático. Del total depacientes que acuden por un problema de tipo clíni-co, más de la mitad presentan signos y síntomas maldefinidos, lo que hace que el diagnóstico a menudosea complejo. Normalmente, el MF dispone de pro-tocolos diagnósticos y guías de actuación para lasenfermedades más prevalentes, pero en el caso delas ER esto no es así. En ocasiones, por la compleji-dad de la sintomatología, otras veces por su infre-cuencia y desconocimiento, junto a la falta de tiem-po, suelen inducir a la derivación de estos pacientesde especialista en especialista durante meses o añoshasta que una idea feliz o una afortunada coinci-dencia propician el diagnóstico de la enfermedad.
¿Significa�esto�que�el�médico�decabecera�quede�relegado�a�segundoplano�en�la�atención�a�estos�pacientes?De ninguna manera. Antes del diagnóstico de la
ER, tanto los MF como los pediatras en APdeberían ser capaces de detectar signos precocesde alarma en los pacientes que sugieran estetipo de patología, así como de intentar acelerarlas interconsultas con los especialistas pertinen-tes para llegar lo antes posible al diagnóstico. Aeste respecto, no hay que olvidar que, aunquemás del 80% son de origen genético, las dosterceras partes de las ER se diagnostican por laclínica.
Tras el diagnóstico, tendrá que desarrollar unalabor de acompañamiento del paciente y su fami-lia, atento a sus necesidades y problemas asocia-dos, lo que incluye el asesoramiento genético pre-concepcional adecuado, y en conjunto unamejora de la calidad asistencial.
¿No�son�demasiadas�ER�para�un�MF?Evidentemente, es imposible que el MF conozcatodas las ER, el último listado de laOrganización Mundial de la Salud (OMS) abarcamás de 9.000 enfermedades distintas, de las cua-les unas 1.000 tienen casos diagnosticados ennuestro país. No obstante, una vez se ha diagnos-ticado la enfermedad en un paciente del cupo deun MF, es obligación del profesional conocerla afondo, desde su transmisión hereditaria a suscomplicaciones, necesidades o tratamientos, silos tuviese.
¿Esto�no�sobrecargará�demasiado�laconsulta�diaria?En un cupo estándar de un MF (1.500-2.000individuos) tendríamos unos 15-20 casos de ER.Cuando comparamos este número con el resto depacientes crónicos (una tercera parte del cupo),las diferencias saltan a la vista: ¿cuántas vecestomamos al año la tensión arterial a los hiperten-sos? ¿cuántas veces pesamos a los obesos? ¿cuán-tos diabéticos tienen los dedos amoratados detanto comprobar su glucemia capilar? Por lotanto, el tiempo necesario para dedicar a lospacientes con ER apenas supone un esfuerzo encomparación con el dado al resto de enfermeda-des crónicas.
¿Qué�aporta�la�AP�en�la�atención�de�lasER?�¿Acaso�los�pacientes�con�ER�noson�enfermos�crónicos�también?Y aún diríamos más, son “pacientes crónicoscon necesidades especiales”. Además de una
Antes�del�diagnóstico�deuna�enfermedad�rara,�tanto
los�médicos�de�familiacomo�los�pediatras�en
Atención�Primaria�deberíanser�capaces�de�detectar
signos�precoces�de�alarmaen�los�pacientes�quesugieran�este�tipo�depatología,�así�como�deintentar�acelerar�las
interconsultas�con�losespecialistas�pertinentes
para�llegar�lo�antes�posibleal�diagnóstico.�Tras�el
diagnóstico,�tendrán�quedesarrollar�una�labor�deacompañamiento�delpaciente�y�su�familia,
atentos�a�sus�necesidadesy�problemas�asociados.
informeMalalties rares, informació freqüent
8
El�papel�de�los�médicos�defamilia�en�el�abordaje�de�lospacientes�con�enfermedadesrarasLas�cebras�no�son�tan�raras
JOSÉ�VICENTE�SORLÍ�Y�MIGUEL�GARCÍA-RIBES
vS97

mala calidad de vida, los pacientes que laspadecen presentan numerosas necesidades dedistinta índole (sanitarias, sociales, psicológi-cas, económicas, educativas…), muchas de lascuales no están cubiertas por nuestro SistemaNacional de Salud. No obstante, la mayoría delos estudios españoles e internacionales sobrelas necesidades que los pacientes con ERdemandan a los profesionales sanitarios coinci-den en la solicitud de dos cosas muy sencillas:la coordinación entre los niveles asistenciales yel “acompañamiento” o “escucha activa”. Seda la circunstancia de que el MF es el profesio-nal más capacitado para realizar ambas. En elprimer caso por su puesto privilegiado comocoordinador o “director de orquesta” de lasalud del paciente y, en el segundo caso, por-que somos especialistas en “comunicación”aplicando el enfoque “biopsicosocial” a pacien-tes y familiares.
Entonces,�¿es�el�paciente�con�ER�elparadigma�del�paciente�crónico?Lo es, y no solo por las características específicasde su enfermedad, que los hacen mucho másconscientes de su condición de enfermos. Enestos tiempos en los que el paciente crónico estáen boca de todos, ya que suponen el 80% de lasconsultas en AP, damos una gran importancia almodelo de atención a enfermos crónicos, basadoen el apoyo del profesional, en la detección tem-prana de la cronicidad, en la prevención y en lacorrecta gestión de la enfermedad.
Los pacientes con ER y sus familiares seenfrentan a unos problemas de salud con unascaracterísticas propias y a la vez poco frecuen-tes de la consulta del MF, llevándoles a consul-tar fuentes de información de diferente proce-dencia que pueden sorprender al MF con datosacerca de nuevas pruebas genéticas de cribado,estudios de investigación sobre su enfermedad,nuevos tratamientos curativos o paliativos, etc.En la actualidad, el asociacionismo y las nue-vas tecnologías suponen una herramienta degran potencia que hace que asociaciones depacientes y especialistas de todo el mundoestablezcan redes de conocimiento que escapana la actividad diaria del médico de cabecera.Ante estas demandas, que en ocasiones puedencrear situaciones de conflicto, la obligación delMF siempre es investigarlas aprovechandoestos recursos, intentar ser lo más objetivoposible, exponiendo la realidad con todas lasalternativas disponibles, poniéndose en contac-to con las asociaciones de pacientes o profesio-nales sanitarios de referencia y animando alpaciente a que siempre que encuentre informa-ciones de estas características acuda a la con-sulta para ponerlas en común.
Todo�esto�está�muy�bien,�pero�¿acasono�hablamos�de�equipos�de�Primaria?¿Cuál�debería�ser�su�implicación?Obviamente, el MF o el pediatra no es el únicoimplicado en la atención de los pacientes con ERen los centros de salud. Los equipos de AP tienenque estar especialmente coordinados para tratara estos pacientes y sus familiares, siendo deseablela realización de reuniones periódicas en las quese realice el seguimiento de estos enfermos a nivelmultidisciplinar.
Enfermería de AP es el enlace idóneo para abor-dar la esfera biopsicosocial de estos pacientes y
detectar si aparecen signos de alarma a todos losniveles. Su accesibilidad para el paciente y elhabitual conocimiento de su entorno hacen de laenfermería la avanzadilla perfecta para detectarproblemas ocultos. Además, en pacientes delarga y mala evolución será la encargada de orga-nizar un programa de atención domiciliaria,reforzando, aún más si cabe, esta función de cen-tinela.
Otros de los profesionales vinculados a muchosde estos pacientes una vez diagnosticados son elfisioterapeuta, con quien conviene que tenganuna relación lo más precoz posible, y el asisten-te social, al que habrá que derivar para prevenirposibles problemas de la esfera social delpaciente.
¿Hay�alguna�forma�de�ayudar�a�los�MFen�el�abordaje�de�estos�pacientes?Por numerosas razones ya expuestas, el abordajede los pacientes con ER resulta complejo, ya desdesu diagnóstico, a menudo lento y complicado,hasta su seguimiento y control, que ha de ser unatarea compartida por el equipo de AtenciónPrimaria y la Atención Especializada. Su granheterogeneidad y desconocimiento hace muy difí-cil diseñar herramientas que permitan de formaestandarizada ayudar a los profesionales a abor-dar las necesidades de estos pacientes. El protoco-lo DICE-APER, desarrollado por el grupo de tra-bajo de la Sociedad Española de MedicinaFamiliar y Comunitaria (SEMFyC) sobre genéticaclínica y ER en colaboración con el Instituto deInvestigación de ER (IIER) del Instituto de SaludCarlos III (ISCIII) es, hasta la fecha, el único pro-tocolo conocido al respecto. Se trata de una plata-forma online de acceso libre y gratuito en la direc-ción http://dice-aper.semfyc.es/web/index.php,cuyos objetivos corresponden con el acrónimo delnombre del protocolo (DICE), junto a la suma delas iniciales de Atención Primaria y enfermedadesraras (APER):1. Diagnóstico: Identificar a las personas que tie-
nen un diagnóstico correspondiente a algunade las ER descritas, o bien están en estudiobajo sospecha de poder tenerla. Esta identifi-cación conlleva de forma inmediata la salva-guarda de esa información en el propio siste-ma de la consulta del médico (papel oaplicación informática de AP).
2. Información: Proporcionar una informaciónbásica y de soporte al paciente y al profesio-nal, partiendo de los recursos existentes enorganizaciones de pacientes y de laAdministración.
3. Coordinación: Contribuir a la coordinaciónasistencial que cada paciente necesite, estable-ciendo los lazos oportunos con el serviciomédico especialista de esa enfermedad.
4. Epidemiología: Proporcionar información alsistema sanitario sobre las dimensiones delproblema, facilitando que el paciente puedainscribirse en el registro de personas con enfer-medades raras del ISCIII, y en el futuro, en elcorrespondiente registro autonómico depacientes con enfermedades raras, y pudiendocontribuir a la investigación mediante la dona-ción voluntaria de una muestra de sangre parael biobanco del ISCIII.
Estos objetivos se deben desarrollar medianteunas actividades sencillas que lleven el mínimotiempo al médico, pero que a su vez le permitanser flexible, de manera que el propio clínico
asuma la gestión de las visitas y de los tiempos ydecida qué hacer en cada momento.Obviamente, el primer punto se considera clavey, por lo tanto, debe ser el primero y el eje detodo el resto de las actuaciones. Para alcanzarestos objetivos, la plataforma contiene lassiguientes herramientas:1. Un completo buscador de ER, con acceso a
resúmenes actualizados de cada una de laspatologías, la mayoría en castellano.
2. Una herramienta de diagnóstico diferencialintroduciendo síntomas y signos del paciente.
3. Un sistema de consulta online con los profe-sionales del IIER para orientar el diagnósticosobre un posible caso de ER.
4. Un listado de recursos de utilidad para profe-sionales y pacientes relacionados con las ER,incluyendo un listado actualizado de los cen-tros de referencia nacionales a donde derivar-los en caso de sospecha diagnóstica.
5 Un acceso directo al Registro Nacional dePacientes con Enfermedades Raras, a travésdel cuál se puede registrar a los pacientes.
6. Un enlace al Biobanco Nacional de Muestrasde Pacientes con Enfermedades Raras, desde elque se informa del procedimiento a seguir encaso de que el paciente estuviera interesado encolaborar donando una muestra de sangre.
7. Una herramienta para coordinar la atención alos pacientes con el resto de médicos especia-listas que se encargan de su caso.
¿Y�después?�¿Qué�iniciativas�existenen�la�actualidad�para�mejorar�laatención�de�pacientes�con�ER�en�elentorno�de�la�AP?La labor de las asociaciones de pacientes hacomenzado a dar frutos, entre los más destaca-dos está la visualización de las principales nece-sidades de este colectivo, ha puesto a los MF enprimera línea en lo que a la atención a estecolectivo se refiere. Por otra parte, la RedEspañola de Registros de ER (SPAIN-RDR), unproyecto europeo liderado a nivel nacional porel IIER y en el que participan todas las comuni-dades autónomas junto a numerosas asociacio-nes de pacientes y sociedades científicas, tieneentre sus objetivos la creación de un registro depacientes en cada comunidad autónoma, deforma que desde Atención Primaria, los MFregistren los casos de ER, como si de enfermeda-des de declaración obligatoria se tratasen, apro-vechando los programas de gestión de la historiainformatizada que manejan en sus consultas. Elpotencial de los MF es importante, y prueba deello es que, desde la creación de la especialidad,han estado a la cabeza de las principales refor-mas para mejorar la atención de los pacientes. Elcaso de las ER no será una excepción.
José�Vicente�Sorlí�GuerolaGrupo de trabajo SeMFYC “Genética Clínica y
enfermedades raras”.
Departamento de Medicina Preventiva y Salud Pública.
universitat de València.
Centro de Salud integrado de Xirivella (Valencia).
Miguel�García-RibesGrupo de trabajo SeMFYC “Genética Clínica y
enfermedades raras”.
Centro de Salud Cotolino ii, Castro urdiales (Cantabria).
9
Malalties rares, informació freqüentinforme
vS97

10 vS97
Se define enfermedad rara (baja preva-lencia, poco frecuente o huérfana)como aquella cuya prevalencia en lapoblación general es inferior a5/10.000 personas. Existen más de
6.000 enfermedades raras catalogadas, sujetas avariabilidad geográfica, social y temporal. Son,en su mayoría, graves, crónicas y/o progresivas, y
solo unas 200 tienen tratamiento. No existenregistros lo suficientemente amplios y serios querecopilen todas y cada una de ellas a nivel nacio-nal o internacional, aunque si existen registrospara alguna de ellas.
Con respecto a la prevalencia de estas enferme-dades, una cincuentena afectan a algunos milla-res de personas en España, unas 500 no afectan amás de unos centenares y miles solo afectan aunas decenas de personas, de ahí la dificultad dela atención general a estas enfermedades que ade-más son muy diferentes entre sí.
Las enfermedades raras partiendo desde unaexistencia silenciosa y casi invisible, pasaron aser una prioridad del Programa de Salud Pública2003-2008 de la Unión Europea, desde la consi-deración de un enfoque conjunto, dado que noera posible establecer una política de salud públi-ca específica para cada una de ellas.
El informe de la ponencia de estudio encargada deanalizar la especial situación de los pacientes conestas enfermedades, en el Senado, en febrero de2007, estipuló la puesta en marcha de un plan deacción para ellas, en el que interactuaban losMinisterios de Sanidad y Consumo, de Trabajo yAsuntos Sociales y de Educación y Ciencia, en coor-dinación con las comunidades autónomas y lassociedades científicas, contando con la participaciónde la Federación Española de Enfermedades Raras(FEDER); y todo ello, con la intención de abordar laproblemática que plantean dichas patologías.
Bajo este concepto, el 23 de enero de 2008, elMinisterio de Sanidad anunció la puesta en mar-cha de la Estrategia Nacional de EnfermedadesRaras, con la que se pretendía establecer unaserie de acciones específicas en el tratamiento deestas patologías.
Estas son las premisas de las que partíamos en el2008, antes de la creación de la Unidad deEnfermedades de Baja Prevalencia del HospitalGeneral Universitario de Alicante (HGUA), siendoen ese momento una de las pocas que existían enEspaña. Al igual que en las políticas sanitarias quese desarrollaron para el abordaje de las enfermeda-des minoritarias, tampoco existía un modelo claroestablecido para la asistencia de estas enfermedades.
Antecedentes�previos�a�la�creación�de�laUnidadNo es posible desarrollar una política sanitaria
específica para cada enfermedad, es una de lasconclusiones a la que se llegó antes de la puestaen marcha de este plan estratégico nacional, delmismo modo en ese mes de noviembre de 2008,nosotros nos preguntamos, ¿cómo podríamosplantearnos la asistencia de estas enfermedadesúnicas y diferentes entre sí?, ¿a qué enfermos yde que manera podríamos, sin aumentar sucarga emocional, ayudarles desde el punto devista sanitario y asistencial?, ¿cómo podíamosaportar algo al conjunto, partiendo de la basedel desconocimiento del que se parte de algunasde estas patologías? En cualquier caso, y a pesarde que teníamos de forma inicial más preguntasque evidencias, al no existir referencia algunaque consultar, continuamos con el desarrollo delproyecto.
Lo siguiente que tuvimos en cuenta fue preguntara pacientes que las padecían, qué era para ellosvivir con una enfermedad rara, ya que al serpoco frecuentes existe en general un mayor gradode incertidumbre y/o desinformación tanto parael paciente como para los profesionales sanita-rios. Todo ello produce dificultades operativaspara elaborar un diagnóstico certero, así comopara ofrecer un tratamiento adecuado, prevenireficazmente la aparición de complicaciones onuevos casos y complicaciones para desarrollarproyectos de investigación. En general, existe unafalta de experiencia en el manejo por parte de losprofesionales de la salud. Los pacientes se encon-traban perdidos en la organización asistencial,pero unidos por las mismas preocupaciones, ymuchos de ellos colaborando con su asistencia deforma activa, buscando profesionales sanitariosque tuvieran experiencia en su enfermedad, queles atendieran si sus médicos habituales o el siste-ma sanitario al que pertenecían desconocían esedato y, muchos familiares, formando parte deasociaciones de enfermos con esas patologíaspara luchar de forma conjunta y en la mismadirección.
Apertura�de�la�UnidadCon todos estos antecedentes, el 7 de noviembrede 2008, en el seno del IV Encuentro deEnfermedades Raras de la ComunitatValenciana, contando como asistentes con elsecretario autonómico de la Agencia Valencianade Salud, representantes de FEDER, pacientes,familiares, profesionales y medios de comunica-ción, se inauguró, dependiente del Servicio deMedicina Interna, la Unidad Multidisciplinariade Enfermedades de Baja Prevalencia del
Atención�especializada�de�lasenfermedades�de�baja�prevalenciaExperiencia�de�la�Unidad�de�Enfermedades�deBaja�Prevalencia�del�Hospital�GeneralUniversitario�de�Alicante
ROSARIO�SÁNCHEZ
La�Unidad�de�Enfermedadesde�Baja�Prevalencia�del
Hospital�General�Universitariode�Alicante�nace�con�la�misiónde�facilitar�la�accesibilidad�y
gestionar�la�asistenciasanitaria�integral�requerida�porlas�personas�afectadas�por
enfermedades�de�bajaprevalencia.�En�sus�cuatro
años�de�vida�se�hadesarrollado,
fundamentalmente,�la�red�decontactos�con�otros�hospitalescon�unidades�de�referencia�dedeterminadas�patologías,�asícomo�la�colaboración�con
proyectos�multicéntricos�deinvestigación�en
enfermedades�raras�y�eldesarrollo�del�consejo
genético.
informeMalalties rares, informació freqüent

Hospital General Universitario de Alicante, sien-do hasta el momento la única en la ComunitatValenciana y de las pocas que existen en Españacon estas características de asistencia general.
Hasta entonces existía un pool de enfermos conenfermedades raras, fundamentalmente con afecta-ción de un órgano, con un seguimiento adecuadopor cada una de las especialidades médicas de la quedependían de forma fundamental, pero existía otropool de enfermos con diagnóstico ya establecido ycon enfermedades con afectación multiorgánica sinpredominancia sintomática, de algún sistema concre-to. Este grupo de pacientes fue de forma inicial elque empezamos a seguir en la Unidad para ofrecerlesseguimiento clínico, información médica pertinente,programación de exploraciones complementarias,implicación multidisciplinar según lo requiriera lapatología y derivación a centro de referencia o con-sulta de especialista adecuado si no se encontraba ennuestra área, ya que la Unidad tenía característicasde atención especializada general.
Uno de los trabajos iniciales era el de contactarcon unidades específicas en España que atendie-ran algunas de estas patologías, por ejemplo laUnidad de Porfirias del Hospital Doce de Octubreen Madrid o la Unidad de Rendu-Osler deSierrallana/Torrelavega (Cantabria) que es unidadde referencia de Rendu-Osler a nivel nacional.
Desarrollo�del�proyectoLa Unidad, dependiente del Servicio de MedicinaInterna del Hospital General Universitario deAlicante, consta de una consulta y de un técnicopara gestión de los aspectos no asistenciales y decitación, además de la colaboración del jefe delServicio y del resto del Servicio de MedicinaInterna del Hospital General de Alicante, así comode los otros servicios intrahospitalarios, en espe-cial del Servicio de Laboratorio para la gestión yanálisis de los estudios genéticos, sin los cuales eltrabajo en la Unidad sería más difícil. Y, finalmen-te, cuenta con la colaboración de otros serviciosde la Comunitat Valenciana, como la Unidad deGenética del Hospital La FE y de la Unidad deMalformaciones Congénitas del Hospital La FE.Las funciones aistenciales y no asistenciales en laUnidad llevadas a cabo a través de la consulta deenfermedades raras se detallan en la tabla 1.
La Unidad nace con la misión de facilitar la accesi-bilidad y gestionar la asistencia sanitaria integralrequerida por las personas afectadas por enferme-dades de baja prevalencia en colaboración conFEDER-Comunitat Valenciana y con la visión dellegar a ser una unidad eficiente y resolutiva capazde prestar servicios sanitarios de calidad asisten-cial y evolucionar hacia un modelo de organiza-ción y atención integral que responda rápidamentea necesidades concretas, basada en valores de coo-peración, coordinación, adaptabilidad, eficiencia ytrabajo en equipo entre FEDER-CV y el HGUA.
El objetivo principal con el que nació era la aten-ción sanitaria integral ambulatoria a pacientesdel Departamento 19 con diagnóstico ya estable-cido y sin seguimiento adecuado por especialistadeterminado.
Durante estos cuatro años, fundamentalmente,hemos desarrollado la red de contactos con otroshospitales con unidades de referencia de determi-nadas patologías, así como la colaboración conproyectos de investigación en enfermedades raras
multicéntricos y el desarrollo del consejo genéti-co en colaboración con la Unidad de Genética deHospital La Fe y la Unidad de ReproducciónAsistida del Hospital La Fe, en los pacientes quelo han precisado. De la misma manera, se haminimizado la burocracia entre servicios y se haintentado tener en cada uno de los serviciosimplicados un profesional de referencia.
Experiencia�desde�la�apertura�de�laUnidadDesde noviembre de 2008, se han realizado 929visitas a consultas externas, con 185 primerasvisitas y 731 consecutivas en la consulta de enfer-medades minoritarias. Hasta mayo de 2012, hansido evaluados 165 nuevos pacientes, 69 eranvarones (41,8%) y 96 mujeres (58,2%) con unamedia de edad de 45,3 años.
Los pacientes fueron referidos en un 25% desde elServicio de Medicina Interna, en un 9,7% desdeFEDER, 6,1% desde Atención Primaria y el restodesde otras especialidades médicas o través delServicio de Atención e Información al Pacientecuando los pacientes eran enviados desde otra áreade salud. Aunque inicialmente se atendían solopacientes de nuestra área de salud, en la actualidadse atienden pacientes de toda la ComunitatValenciana y de otras comunidades autónomas.
Se han atendido 81 patologías diferentes, entreellas la más frecuente es el despistaje de enferme-dades del colágeno hereditarias, sobre todo porhábito marfanoide, seguido de síndrome deMarfan (11,5 %), síndrome de Ehler-Danlos(5,5%), neurofibromatosis tipo I (4,2%), RenduOsler y porfiria aguda intermitente (3,6%).
El 48,5% de las patologías atendidas son heredi-tarias. Se han realizado estudios genéticos a 91pacientes sobre todo del gen FBN1, TGFBR I y IIpara estudio de enfermedad de Marfan, deRendu Osler, de porfiria aguda intermitente, sín-drome de Noonan, fibrosis quística, neurofibro-matosis tipo 1 y COL3A (pacientes con sospechade Ehler Danlos tipo IV, siendo seguidos en laUnidad tres pacientes con esta patología). Lasconsultas que con más frecuencia se han realiza-do de estos pacientes, dado el perfil del pacienteatendido, fue a oftalmología (16,7%), cardiolo-gía (16,25%), digestivo 10,83%, neurología8,8% y reumatología.
Desde el inicio, muchas son las herramientas quenos han facilitado el trabajo asistencial enpacientes con enfermedades minoritarias. Poruna parte, la aparición de nuevas unidades tantomonográficas de determinadas patologías mino-ritarias, como genéricas, para aumentar el gradode conocimiento sobre estas patologías, así comolos especialistas interesados en su atención y conexperiencia en el manejo de algunas de ellas.Una herramienta fundamental que ha agilizado eltrabajo en la asistencia a estas enfermedades,incluye recursos informáticos de documentación,como puede ser la pagina de Orphanet(www.orpha.net ) con varias sedes internacionesy con sede española en Valencia, que ofreceinformación general sobre enfermedades minori-tarias, en forma de resumen, últimas revisiones yprotocolos de actuación, así como un listado delos centros y profesionales encargados tanto en laasistencia como en la investigación de cada unade ellas.
Para facilitar el intercambio de información entrelos profesionales que atendemos estas patologías,así como para la realización de registros y estu-dios de investigación coordinados se han forma-do varios grupos de trabajo, tanto en el seno dela Sociedad Española de Medicina Familiar yComunitaria como en la Sociedad Española deMedicina Interna, en nuestro caso.
También se han desarrollado nuevos fármacoshuérfanos. Los gobiernos y organizaciones depacientes de enfermedades raras como EUROR-DIS abogan por incentivos económicos para ani-mar a las compañías farmacéuticas a desarrollary comercializar medicamentos para el tratamien-to de enfermedades raras. Esto ha incrementadoel numero de medicamentos huérfanos aceptadospara el tratamiento de enfermedades raras, entorno a 400 en 2008 y más de 800 designacioneshuérfanas en el 2010.
Todas estas iniciativas han hecho que la asis-tencia de las enfermedades minoritarias sedesarrolle de forma exponencial y aumenten lasexpectativas de su evolución en el futuro, tantoen la asistencia como en el desarrollo de pro-yectos de investigación y desarrollo de fárma-cos huérfanos y, lo que es más importante, enlas vías de información tanto para los profesio-nales que las atienden como para los pacientesque las padecen.
Rosario�Sánchez�MartínezCoordinadora médica de la unidad de enfermedades de
Baja Prevalencia. Servicio de Medicina interna.
Hospital General universitario de alicante.
11vS97
® Recopilación de casos urgentes con diagnóstico yaestablecido, que precisen evaluación multidisciplinaria
® Creación de base de datos con historia clínica completay recopilación de historia antigua de cada paciente
® Protocolo de seguimiento individualizado
® Consejo de tratamiento y evaluación por especialistaspertinentes de forma coordinada
Funciones de la Unidad1
Asistenciales
® Medición y evaluación de resultados: identificación deindicadores de actividad y de resultado; elaboración ymantenimiento de documentación
® Gestión de calidad: mejora continua de los procesos;identificación de oportunidades de mejora
Funciones complementarias
® Doctora Rosario Sánchez, coordinadora médica de laUnidad y responsable Consulta ER. Servicio deMedicina Interna
® Cecilia Soriano, coordinadora de aspectos noasistenciales
Profesionales implicadas
® Gestión de citas: recogida sistemática de informa-ción, cumplimentación de documentación clínica,petición de citas…
® Coordinación de la visita en el hospital: recogida enel hall principal y acompañamiento a la consulta
® Comunicación con los servicios sanitarios: coordinarprofesionales, petición de necesidades, obtenciónde resultados, coordinación y gestión de recursos…
® Satisfacción del paciente: reuniones periódicas,atención telefónica, análisis de satisfacción…
No asistenciales (gestiones administrativas y organizativas)
Malalties rares, informació freqüentinforme

12 vS97
Los medicamentos huérfanos son aque-llos destinados a la prevención, diag-nóstico o tratamiento de enfermedadesraras o al tratamiento de una enferme-dad grave o incapacitante, cuando sea
improbable que sin incentivos, la comercializa-ción de dicho medicamento genere suficientesbeneficios para justificar la inversión necesaria.En el ámbito de la Unión Europea (UE) se defi-nen las enfermedades raras como aquellas pato-logías que representan un riesgo para la vida oconllevan una incapacidad grave y crónica sinque afecten a más de 5 personas cada 10.000habitantes. Estas patologías poco frecuentesafectan a un gran número de personas, ya quesegún la Organización Mundial de la Salud,existen cerca de 7.000 enfermedades raras queafectan al 7 % de la población mundial. Entotal, se estima que en España existen más detres millones de personas con enfermedadespoco frecuentes1.
Pese a que en su conjunto afectan a un grannúmero de personas, esta baja prevalencia des-crita ha sido durante años la principal barreraen el desarrollo de tratamientos farmacológicospara los pacientes afectados. El número limita-do de pacientes a tratar ha hecho que la indus-tria farmacéutica no considerara viables los pla-nes de desarrollo e investigación de fármacos enlas condiciones habituales por la falta de rendi-miento económico en relación con la inversiónrealizada. Se estima que el coste de desarrollode un medicamento se sitúa entre 250 y 500millones de dólares, y que el período medio dedesarrollo de un medicamento es de entre 10 y14 años. Obviamente, recuperar esta inversióninicial en el caso de medicamentos huérfanos escomplicado. Por este motivo, las diferentesagencias reguladoras del medicamento decidie-ron estimular el desarrollo de estas nuevasmoléculas por medio de la creación de legisla-ción específica e incentivación económica paralos medicamentos huérfanos. En este sentido,con la aprobación de la Orphan Drug Act, en1983, los Estados Unidos fueron los pioneros dela investigación en medicamentos huérfanos conla creación de un marco legislativo e impositivofavorable impulsando en gran medida el desa-rrollo de la legislación en el resto del mundo.
Política�sanitaria�europea�demedicamentos�huérfanos�La legislación que regula los medicamentos huér-fanos en la UE se encuentra recogida en elReglamento (CE) nº 141/2000 del ParlamentoEuropeo y del Consejo, de 16 de Diciembre de1999, sobre medicamentos huérfanos, y en elReglamento (CE) nº 847/2000 de la Comisión,de 27 de abril de 2000, por el que se establecenlas disposiciones de aplicación de los criterios dedeclaración de los medicamentos huérfanos y la
definición de los conceptos de medicamento simi-lar y superioridad clínica.
Los pasos que siguen estos fármacos hasta conse-guir la comercialización se enumeran a continua-ción.
Designación�de�medicamento�como�huérfanoEn el año 2000, a raíz del Reglamento (CE)nº 141/2000, se creó el Comité deMedicamentos y Productos Huérfanos (COMP)en el seno de la Agencia Europea delMedicamento (EMA). Este organismo es el res-ponsable de revisar las solicitudes para la desig-nación de medicamentos huérfanos.Normalmente es el laboratorio farmacéutico elque solicita la designación huérfana al COMP ysi se le concede disfrutará de una serie de incen-tivos, entre los que destacan la exclusividadcomercial durante 10 años, la asistencia en laelaboración de protocolos de acceso al procedi-miento centralizado de la EMA para solicitar lacomercialización, la exención de tasas y lainvestigación subvencionada por la UE. Ladesignación huérfana es posible en cualquierfase del desarrollo de un medicamento, siempreque se demuestre la justificación científica de laverosimilitud del producto en la indicación soli-citada.
En la evaluación que realizó la EMA diez añosdespués de poner en marcha el COMP(03/05/2010), de las 1.113 solicitudes de designa-ción de medicamentos huérfanos, el 68% reci-bieron una opinión favorable llegando a autori-zarse oficialmente sólo 62 medicamentoshuérfanos.
Y es que la designación de un medicamentocomo huérfano es sólo un paso preliminar y nogarantiza su uso en la condición designada niimplica necesariamente que el producto satisfagalos criterios de eficacia, y seguridad. Para la con-cesión de la autorización de comercializacióndeben realizarse los ensayos clínicos pertinentes,como en cualquier otro medicamento.
Realización�de�ensayos�clínicosLa puesta en marcha de un ensayo clínico parala valoración de un fármaco huérfano presentauna complejidad añadida con respecto al restode medicamentos. En primer lugar, el promotordebe hacer frente a los elevados costes de lainvestigación, puesta en marcha y obtención deautorizaciones para el desarrollo del estudio. Porotro lado, los estudios de enfermedades raras sediferencian sustancialmente del resto por el limi-tado número de pacientes al que van dirigidos;ello conlleva varias dificultades, como el recluta-miento de un número de individuos suficientepara realizar ensayos o la complicación paraasegurar la protección de datos cuando se trata
MedicamentoshuérfanosMARÍA�DOLORES�EDO�Y�JOSÉ�LUIS�POVEDA
El�número�limitado�depacientes�a�tratar�ha�hecho
que�la�industriafarmacéutica�no
considerara�viables�losplanes�de�desarrollo�e
investigación�de�fármacosen�las�condiciones
habituales,�aludiendo�lafalta�de�rendimiento
económico�en�relación�conla�inversión�realizada.
Obviamente,�recuperar�estainversión�inicial�en�el�caso
de�los�medicamentoshuérfanos�es�complicado.
Por�este�motivo,�lasdiferentes�agencias
reguladoras�delmedicamento�decidieronestimular�el�desarrollo�de
nuevas�moléculas�pormedio�de�la�creación�de
una�legislación�específica�yla�incentivación�económica
para�los�medicamentoshuérfanos.
informeMalalties rares, informació freqüent

13vS97
de estudios con escasos pacientes, necesaria paratodo estudio de investigación tal como se recogeen la ley. Este hecho diferenciador se ha recogi-do por la normativa de la UE y la española,aceptando la autorización de fármacos huérfa-nos con reservas, aún con ensayos en un númeroreducido de pacientes, y pudiéndose adoptarmedidas especiales en relación con su fabrica-ción, régimen económico, fiscal, de distribucióny dispensación.
Actualmente, de acuerdo con el Registro deEnsayos Clínicos de la Unión Europea(EudraPharm Portal), de los 14.369 ensayos enmarcha registrados en Marzo de 2013, el 5% secorresponde a ensayos en enfermedades raraspara fármacos con designación huérfana en laindicación. Restringiendo la búsqueda a aquellosensayos activos en los que participa España deltotal de 4.325, el 7% se corresponde a fármacoscon designación huérfana.
Autorización�y�comercializaciónUna vez completados los ensayos clínicos corres-pondientes, los medicamentos huérfanos tienenque someterse obligatoriamente al procedimientocentralizado de autorización de comercializaciónestablecido en el Reglamento (CE) nº 726/2004por el que se establecen procedimientos comuni-tarios para la autorización y el control de medi-camentos de uso humano y veterinario.
No obstante, que un medicamento huérfanotenga la autorización comunitaria de comercia-lización no significa que esté ya a disposiciónde los pacientes. Después de este primer paso,el medicamento debe pasar por los organismosadministrativos de cada Estado para gestionarsu puesta en el mercado (condiciones de finan-ciación y precio). Cada Estado negocia el pre-cio por separado con las compañías farmacéu-ticas. Hay una tendencia por parte de lascompañías a empezar a negociar con Estadosmiembros que les conceden el precio más alto,que después se utiliza como precio de referen-cia en las negociaciones con el resto de países.Esto supone una gran variabilidad intracomu-nitaria, ya que las condiciones de comercializa-ción de un mismo medicamento pueden sermuy diferentes en función del país en que nosencontremos.
Acceso�temprano�a�los�medicamentoshuérfanosEn algunos casos, el acceso temprano a un medi-camento es posible antes de que se conceda suautorización de comercialización siempre que elfármaco esté ya en la tercera fase del ensayo clí-nico y el medicamento haya demostrado seguri-dad y eficacia. Dos procedimientos lo hacenposible1:1. Que exista una solicitud de aprobación para la
comercialización que haya sido enviada por lacompañía farmacéutica encargada de desarro-llar el medicamento en el país pertinente. Eneste caso o bien la compañía pediría una apro-bación temporal de la autoridad administrativapara un grupo de pacientes concretos (TUA)como ocurre en Francia o Italia, o bien se tra-mitaría el uso compasivo de medicamentos enel resto de países europeos. Esta autorizaciónsólo tiene validez durante un periodo concreto.
2. Que el médico pida a las autoridades adminis-trativas una aprobación reguladora temporalnominativa para pacientes concretos identifi-
cados con su nombre y para un periodo deter-minado.
Disponibilidad�de�los�medicamentoshuérfanos�en�EspañaEn el estudio ENSERio realizado por laFederación Española de Enfermedades Raras en2009 se concluye que solo el 6% de los pacientescon este tipo de enfermedades accede a medica-mentos huérfanos, y de éstos, el 51 % tienemuchas dificultades para acceder a su tratamien-to2. Como dicen las cifras, el acceso a estos medi-camentos es realmente complicado. En primerlugar, porque solo un reducido grupo de labora-torios enfocan su investigación y actividad hacialas enfermedades raras por su baja rentabilidad,pese a todos los incentivos de los que disfrutanestos fármacos. Además, en el caso de medica-mentos que finalmente son comercializados, sudisponibilidad y financiación estará sujeta a lalegislación sanitaria de cada país. Y en España ala de cada comunidad autónoma.
A fecha de Abril de 2013, en España, de los 76medicamentos huérfanos designados y aproba-
dos en Europa3, 21 todavía no están comerciali-zados en España (27,6%). De éstos, 14 tienenla autorización de comercialización pero noestán todavía en el mercado y de los otros 7 noconsta autorizada su comercialización en laAEMPS4.
De los 55 medicamentos huérfanos comerciali-zados en España, hay que destacar los antineo-plásicos e inmunomoduladores (43,6%) y losdestinados a patologías de metabolismo(23,6%). En este último apartado, destacan lostratamientos de las metabolopatías congénitas,que eran enfermedades raras sin tratamientohasta la comercialización de este tipo de medi-camentos, lo que suposo una innovación tera-péutica de primer orden. Otras categorías en lasque están autorizados estos medicamentos son:sistema nervioso (7,3%), sistema cardiovascu-lar (7,3 %), sangre y órganos formadores(5,5%), antiinfecciosos (3,6%), sistema hormo-nal (3,6%), sistema genitourinario (1,8%) yotros (3,6%).
Impacto�presupuestario�de�losmedicamentos�huérfanosCuando se aprobó la legislación europea en elaño 2000 el impacto presupuestario de los medi-camentos huérfanos era muy pequeño. En laactualidad esto ha cambiado sustancialmente. Elinforme del Centre Fédéral d’Expertise des Soinsde Santeé de Bélgica estima que los fármacosconsumían el 5% de todo el presupuesto hospi-talario en 2008 y que esta cifra alcanzará el 10%antes del 20205.
En el Hospital Universitari i Politècnic La Fe deValencia el consumo de medicamentos huérfanosen 2012 supuso 18.500.000 euros lo que secorresponde con el 17,8% del gasto total demedicamentos, cifra que ha ido ascendiendo enlos últimos años. Además, los 15 medicamentoshuérfanos que presentan mayor coste oscilanentre los 3.500.000 y 300.000 euros anualespudiendo llegar a superar los 300.000euros/paciente/año si nos fijamos en las terapiasde sustitución enzimática empleadas en el trata-miento de las lisosomopatías.
El elevado coste tanto por año de vida gana-do, como por años de vida ajustados por cali-dad que presentan muchos de estos medica-mentos, junto con la alta incertidumbre sobresu eficacia derivada del reducido número depacientes en los ensayos clínicos, ha desatadocontroversia sobre la disponibilidad a pagarpor estos medicamentos. Lo que adquiere másrelevancia si cabe en el escenario económicoen el que nos encontramos actualmente dondela financiación selectiva se abre paso en elámbito español desde la aprobación del RealDecreto Ley 16/2012. En este sentido elMinisterio de Sanidad rechazó financiar el30% de los medicamentos que solicitaron serfinanciados con fondos públicos durante2012, haciendo hincapié en que sólo se finan-ciará la innovación que tenga resultados adi-cionales probados.
Dadas las dimensiones de las cifras con las quenos encontramos y las características intrínse-cas de este tipo de medicamentos (tecnologíasde alto coste y pruebas científicas escasas de sueficacia, seguridad, efectividad o coste-efectivi-dad) se han planteado diferentes estrategias
® Afinitor
® Aldurazyme
® Atriance
® Busilvex
® Carbaglu
® Cayston
® Cystadane
® Elaprase
® Evoltra
® Exjade
® Fabrazyme
® Firazyr
® Gliolan
® Glivec
® Ilaris
® Increlex
® Inovelon
® Kuvan
® Litak
® Lysodren
® Mepact
® Mozobil
® Myozyme
® Naglazyme
® Nexavar
® Nplate
® Orfadin
® Pedea
® Peyona
® Prialt
® Replagal
® Revatio
® Revlimid
® Revolade
® Savene
® Siklos
® Soliris
® Somavert
® Sprycel
® Sutent
® Tasigna
® Tepadina
® Tepadina
® Tobi podhaler
® Torisel
® Tracleer
® Trisenox
® Ventavis
® Vidaza
® Volibris
® Vprip
® Wilzin
® Xagrid
® Xyrem
® Yondelis
® Zavesca
Listado de 55 medicamentos huérfanoscomercializados en España (abril 2013)
1
Malalties rares, informació freqüentinforme

14 vS97
para la gestión de estos medicamentos. En estesentido, en los últimos años han irrumpido confuerza los contratos de riesgo compartido entreel financiador y la industria, estableciendo unsistema de pago en función de los resultadosclínicos obtenidos en los pacientes. Esta estra-tegia contribuye a fijar precios razonables,ajustados a la efectividad clínica de estos medi-camentos y a reducir la incertidumbre con elfin de mantener el acceso a la innovación y lasostenibilidad.
María�Dolores�Edo�Solsona.Farmacéutica especialista.
Hospital universitari i Politècnic la Fe.
José�Luis�Poveda�Andrés.Jefe del Servicio de Farmacia Hospitalaria.
Hospital universitari i Politècnic la Fe.
Presidente de la Sociedad española de
Farmacia Hospitalaria.
B i b l i o g r a f í a1. Medicamentos huérfanos. www.orpha.net
2. estudio sobre situación de necesidades Sociosanitarias de laspersonas con enfermedades raras en españa. estudio enSerio.FeDer. edición Octubre 2009. Depósito legal: aV-126 2009.https://www.obrasocialcajamadrid.es/Ficheros/CMa/ficheros/OSSolidaridad_Puenserio.PDF
3. list of medical products for rare diseases in europe. april 2013.http://www.orpha.net/orphacom/cahiers/docs/GB/list_of_orphan_drugs europe.pdf
4. Centro de información on line de Medicamentos (CiMa). www.agemed.es
5. Cost-effectiveness assessment of orphan drugs: a scientificand political conundrum. appl econ Health Policy. 2013 Feb11(1):1-3.
Especialidad Principio activo Indicación
Afinitor Everolimus Linfoma de Hodgkin CD30+, linfoma anaplásico de células grandesAldurazyme Laronidasa Mucopolisacaridosis Tipo IAtriance Nelarabina Leucemia linfoblástica aguda de células T, linfoma linfoblástico de células T Busilvex Busulfán Acondicionamiento previo a trasplante de progenitores hematopoyéticosCarbaglu Ácido carglúmico HipermamonemiaCayston Aztreonam Infecciones pulmonares por pseudomonas aeruginosa en pacientes con FQCystadane Betaina HomocisteinuriaElaprase Idursulfasa Síndrome de HunterEvoltra Clofarabina Leucemia linfoblástica agudaExjade Deferasirox Beta talasemia mayor, Sobrecarga férrica crónica Fabrazyme Agalsidasa beta Enfermedad de FabryFirazyr Icatibant Angioedema hereditarioGliolan Ácido 5-aminolevulínico Glioma maligno
Glivec Imatinib Leucemia mieloide crónica, leucemia linfoblástica aguda, enfermedades mielodisplásicas, tumores del estroma gastrointestinal, dermatofibrosarcoma protuberans
Ilaris Canakimumab Síndromes periódicos asociados a criopirinaIncrelex Mecasermina Déficit primario de IGF graveInovelon Rufinamida Síndrome de Lennox-GastautKuvan Sapropterin Fenilcetonuria, déficit de tetrahidrobiopterina (BH4)Litak Cladribina Leucemia de células pilosasLysodren Mitotane Carcinoma adrenocortical avanzadoMepact Mifamurtida OsteosarcomaMozobil Plerixafor Mobilización de células hematopoyéticas previo a trasplante autólogoMyozyme Alglucosidasa alfa Enfermedad de PompeNaglazyme Galsulfasa Mucopolisacaridosis VINexavar Sorafenib Carcinoma hepatocelular, carcinoma de células renalesNplate Romiplostim Purpura trombocitopénica inmune (idiopática)Orfadin Nitisinona Tirosinemia hereditaria tipo IPedea Ibuprofeno Ductus arteriosusPeyona Cafeina citrato Apnea primariaPrialt Ziconotida Dolor crónico en pacientes con analgesia intratecalReplagal Agalsidasa alfa Enfermedad de FabryRevatio Sildenafilo Hipertensión pulmonarRevlimid Lenalidomida Mieloma múltipleRevolade Eltrombopag Púrpura trombocitopénica idiopáticaSavene Dexrazoxano Extravasación por antraciclinasSiklos Hidroxicarbamida Anemia drepanocítica sintomáticaSoliris Eculizumab Hemglobinuria paroxística nocturna, síndrome urémico malignoSomavert Pegvisomant AcromegaliaSprycel Dasatinib Leucemia mieloide crónica, leucemia linfoblástica aguda Ph+Sutent Sunitinib Tumores del estroma gastrointestinal, cáncer renal, tumores neuroendocrinos pancreáticosTasigna Nilotinib Leucemia mieloide crónica Ph+Tepadina Tiotepa Acondicionamiento previo a trasplante de progenitores hematopoyéticosTobi podhaler Tobramicina Infecciones pulmonares por pseudomonas aeruginosa en pacientes con FQTorisel Temsirolmus Carcinoma de células renales, linfoma de células del mantoTracleer Bosentán Hipertensión pulmonar,esclerosis sistémicaTrisenox Trióxido arsénico Leucemia promielocítica agudaVentavis Iloprost Hipertensión pulmonar primariaVidaza Azacitidina Síndrome mielodisplásico, leucemia mielocítica crónica,leucemia mieloide agudaVolibris Ambrisentán Hipertensión pulmonar arterialVprip Velaglucerasa alfa Enfermedad de GaucherWilzin Zinc acetato Enfermedad de WilsonXagrid Anagrelida Trombocitopenia esencialXyrem Oxibato sódico Narcolepsia con cataplexiaYondelis Trabectedina Sarcoma de tejidos blandos,cáncer de ovarioZavesca Miglustat Enfermedad de Gaucher, enfermedad de Niemann-Pick
Medicamentos huérfanos comercializados en España (abril 2013)2
Medicamentos con designación huérfana europea y comercializados en España a fecha de abril de 2013.Se incluyen los medicamentos que obtuvieron la designación huérfana y que en la actualidad no la conservan por petición del titular o por haber transcurrido los 10 años desde la designación. Se han eliminado todos los medicamentos retirados de la comercialización por alertas de seguridad.
informeMalalties rares, informació freqüent

Se estima que alrededor de un 80% delas enfermedades raras (ER) son deorigen genético. La mayoría de ellassiguen un patrón de herencia mende-liano, es decir, se ajustan a las leyes
de herencia formuladas por Gregor Mendel,padre de la genética, en 1865. Estas enfermeda-des se conocen como monogénicas y en ellas lapresencia de una mutación, o dos mutaciones enlas formas autosómicas recesivas, confieren unriesgo muy elevado de padecer la enfermedaddeterminada (mutaciones de alta penetrancia).
Hay otras que presentan una forma de herenciamultifactorial, debida a la presencia de mutacio-nes en varios genes cada una de ellas con un ries-go relativamente bajo de causar la enfermedadpor sí sola (mutaciones de baja penetrancia),pero que en conjunto contribuyen a producir eltrastorno. Ejemplos de ER con este tipo deherencia son la esclerodermia, el lupus eritemato-
so sistémico o la espondilitis anquilosante. Enestos casos, el diagnóstico genético es complica-do, poco predictivo, por lo que no suele incluirseen la cartera de servicios del sistema nacional desalud.
Respecto a las enfermedades raras mendelianas,se pueden distinguir dos tipos: las enfermedadesmonogénicas sencillas o simples y las enfermeda-des monogénicas genéticamente heterogéneas.
Las enfermedades monogénicas simples son aque-llas que están causadas por mutaciones en unúnico gen. Ejemplos de éstas son la fibrosis quísti-ca, causada por mutaciones en el gen CFTR, ladistrofia muscular de Duchenne, debida a muta-ciones en el gen DMD, o la enfermedad deHuntington, cuyo gen resposable es IT-15.
Las enfermedades monogénicas heterogéneasestán causadas por mutaciones en un solo gen,pero a diferencia de las anteriores hay un númerovariable de genes que pueden ser responsables dela misma. Ejemplos paradigmáticos de este tiposon la retinosis pigmentaria, en la que mutacio-nes en cualquiera de los más de 50 genes descri-tos puede causar la enfermedad, o la neuropatíade Charcot-Marie-Tooth, con cerca de 40 genesidentificados.
El diagnóstico genético de una ER determinadadependerá de la naturaleza de la ER, el tipo dedefecto genético subyacente y el conocimientobiomédico disponible sobre la misma.
Diagnóstico�genéticoEl diagnóstico genético debe realizarse en uncontexto clínico y debe orientar siempre un diag-nóstico clínico, bien sea porque el paciente pre-senta síntomas de una ER, bien porque tieneantecedentes familiares de la misma. Las pruebasgenéticas constituyen la herramienta técnica delaboratorio que permite realizar este diagnósticoy son un elemento esencial para el mismo y parael asesoramiento genético.
La UNESCO define prueba genética como “unprocedimiento para detectar la presencia oausencia, o cambio en un gen particular o cro-mosoma, incluyendo una prueba indirecta paraun producto génico u otros metabolitos específi-cos que sean primariamente indicativos de uncambio genético específico”.
Ambos procesos, diagnóstico y asesoramientogenético deben ir siempre de la mano.
El asesoramiento genético, mal llamado consejogenético, pues no es tarea del profesional que lorealiza aconsejar a los pacientes lo que debenhacer, sino ofrecerles toda la información dis-ponible sobre su enfermedad, es el proceso por
el cual los enfermos o sus familiares con riesgode padecer una enfermedad hereditaria soninformados de la probabilidad de su aparición,la probabilidad de transmitirla a su descenden-cia, de las consecuencias de la misma y de losmedios que permiten prevenir o tratar dichaenfermedad.
Se recomienda realizar un asesoramiento pretest,es decir, anterior a la realización de la pruebagenética donde se explicarán los beneficios ylimitaciones de la prueba, posibles riesgos y con-secuencias previsibles de su realización, inicián-dose el proceso de consentimiento informado.Una vez realizada la prueba, se realizará un ase-soramiento postest en el que se explicarán losresultados y las posibilidades de manejo de laenfermedad derivadas de dichos resultados.
Existen varios tipos de diagnóstico genético basa-dos en sus objetivos:•Diagnóstico de sospecha. Tiene como propósitoconfirmar, matizar o descartar un diagnósticoclínico. Este es el diagnóstico más ampliamentesolicitado.
•Diagnóstico predictivo. Consiste en estimar elriesgo de desarrollar un trastorno genético en elfuturo para una persona asintomática. Hay dostipos de diagnósticos predictivos:•Diagnóstico presintomático. Pretende buscarel defecto genético que causa la enfermedaden un individuo sano, de modo que cuandoestá presente se sabe con certeza que el indi-viduo ahora sano padecerá el trastorno en elfuturo. Es habitual la aplicación de este tipode diagnósticos en enfermedades hereditariascuyos síntomas aparecen a una edad adulta yexisten antecedentes familiares de la enfer-medad.
•Diagnóstico de predisposición. Su fin es anali-zar si variaciones concretas en la secuencia deDNA pueden aumentar la susceptibilidad apadecer un trastorno. Es propio de enfermeda-des complejas y no se suele aplicar al diagnós-tico genético de ER por el bajo valor predicti-vo de las mutaciones.
•Diagnóstico de portadores. En las enfermedadescon una herencia autosómica recesiva o ligadasal cromosoma X el diagnóstico de portadorescobra especial relevancia, ya que estos sonsanos. Este tipo de diagnóstico resulta sobretodo útil para la toma de decisiones sobreaspectos reproductivos.
•Diagnóstico prenatal. El objetivo es averiguar siel feto es portador de una mutación y, portanto, susceptible de desarrollar la enfermedaden cuestión tras el nacimiento. Habitualmente,este tipo de diagnóstico suele realizarse cuandoexisten antecedentes en la familia de algunaenfermedad mendeliana.
•Diagnóstico genético preimplantatorio (DGP).Es un proceso diagnóstico que se practica en
15vS97
El�diagnóstico�genético�delas�enfermedades�rarasJOSÉ�M.�MILLÁN
Aunque�los�avances�en�eldescubrimiento�de�losgenes�responsables�deenfermedades�raras�han
sido�extraordinarios�en�lasúltimas�dos�décadas,�elconocimiento�biomédicoactual�todavía�no�puede
explicar�la�causa�genéticasubyacente�a�la�mayoría�de
estas�enfermedades.�Eladvenimiento�de�las�nuevastécnicas�genómicas�y�su
introducción�en�eldiagnóstico�asistencialpermitirá�el�abordaje�de
todas�estas�enfermedades,cuyo�manejo�es�actualmente
muy�complicado.
Malalties rares, informació freqüentinforme

embriones obtenidos mediante técnicas dereproducción asistida en parejas con anteceden-tes de enfermedad monogénica en ellos mismoso en la familia. Se realiza sobre una o dos célu-las del embrión en fase de blastocisto y se selec-cionan para su implantación aquéllos que noson portadores de la mutación en cuestión.
Dificultades�del�diagnóstico�genéticoSe estima que existen 7.000-8.000 ER cataloga-das como distintas entidades clínicas. Este con-junto de patologías es muy heterogéneo, tantopor el órgano u órganos que afectan como por suetiología, su expresión clínica, su gravedad o laedad de aparición de los síntomas. Todo estohace que en muchas ocasiones no se pueda llegara un diagnóstico clínico claro, ya que hay patolo-gías con síntomas solapantes o, en el momentodel diagnóstico de sospecha, el paciente todavíano ha desarrollado todos los síntomas de laenfermedad.
Entre las principales dificultades a las que seenfrentan los pacientes afectados de una ER es lafalta de acceso al diagnóstico correcto. En un estu-dio realizado por Eurordis (European Organisationfor Rare Diseases; www.eurordis.org) en 2004,centrado en ocho ER se revelaron problemasimportantes, como el retraso en el diagnóstico, losdiagnósticos incorrectos, la ausencia de asesora-miento o la no comunicación del carácter heredita-rio de la enfermedad. Otras características propiasde las ER, como la dispersión geográfica y la bajaprevalencia, magnifican los problemas de acceso aldiagnóstico genético.
Las consecuencias del retraso diagnóstico sontrágicas: otros niños nacidos con la misma enfer-medad en el seno de la familia y el empeoramien-to clínico de la salud de los pacientes en términosde condición intelectual, psicológica y física.
Aunque los avances en el descubrimiento de losgenes responsables de ER han sido extraordina-rios en las últimas dos décadas, el conocimientobiomédico actual todavía no puede explicar lacausa genética subyacente a la mayoría de lasER. Según el IRDiRC (International RareDiseases Research Consortium) en la actualidadse estima que hay pruebas genéticas disponiblespara cerca de 1.900 ER.
Aún más, para muchas ER para las que se cono-ce la causa genética y las pruebas son técnica-mente factibles, el alto número y el tamaño delos genes responsables hace que en algunos casosel estudio sea inabordable por un laboratorioasistencial. El advenimiento de las nuevas técni-cas genómicas y su introducción en el diagnósti-co asistencial permitirá el abordaje de todas estasenfermedades cuyo manejo es actualmente muycomplicado.
La vía hacia el acceso a diagnósticos más preco-ces pasa por la potenciación de una serie deactuaciones que van desde el fomento de la inves-tigación aplicada al diagnóstico hasta garantizarla accesibilidad a las pruebas. El objetivo es tantoel de prevenir la transmisión de enfermedadesgenéticas hereditarias como el de acelerar losdiagnósticos de forma que el abordaje terapéuti-co sea más temprano.
En este sentido, la meta del consorcio IRDiRCpara el año 2020 es el desarrollo de pruebas
diagnósticas para todas las ER y el tratamientopara al menos 200 de ellas.
Marco�legal�y�recomendaciones�decalidadEn España no existe un sistema específico para laacreditación de centros o laboratorios que reali-cen diagnóstico genético. Algunos de ellos for-man parte del directorio de EDDNAL (EuropeanDirectory of DNA Diagnostic Laboratories), quelleva a cabo actividades que tienen como fingarantizar la calidad de las pruebas realizadas.
Muchos laboratorios pasan controles de calidadpara determinadas técnicas o diagnósticos deenfermedades concretas a través de EuroGentest,un proyecto financiado por el 7º ProgramaMarco de la Comisión Europea, con el fin dearmonizar las pruebas genéticas en Europa,desde la toma de la muestra hasta el asesora-miento genético. Su objetivo es garantizar la cali-dad y las buenas prácticas en los laboratoriosdedicados al diagnóstico genético.
La incorporación de los laboratorios a estasredes europeas de calidad es voluntaria. No ocu-rre lo mismo en los Estados Unidos donde loslaboratorios que realizan pruebas genéticas nece-sitan obtener un certificado que asegure la cali-dad y las buenas prácticas (Clinical LaboratoryImprovement Amendments; CLIA)
Todas estas cuestiones evidencian la necesidad deadoptar una estrategia reguladora común de laactividad de los centros de diagnóstico genéticoen Europa.
Información�sobre�diagnóstico�genéticode�las�ER�Ningún Estado miembro de la UE cuenta con uncentro nacional de información sobre ER, si bienhay portales web mantenidos por agencias guber-namentales, por asociaciones de pacientes o pororganizaciones profesionales en distintos paísesde la UE.
Orphanet (www.orpha.net) es un portal de inter-net gratuito de referencia para la informaciónsobre ER. Su infraestructura y coordinaciónestán financiadas por el INSERM, la DirecciónGeneral para la Salud de Francia y la ComisiónEuropea. Su objetivo es mejorar el diagnóstico,cuidado y tratamiento de los pacientes con ER yestá formado por más de 40 países asociadosentre los que se encuentra España.
El socio español de Orphanet es el Centro deInvestigación Biomédica en Red de EnfermedadesRaras (CIBERER; www.ciberer.es), una iniciati-va del Instituto de Salud Carlos III para fomentarla investigación en ER.
Entre los servicios que ofrece Orphanet están uninventario de ER basado en clasificaciones reali-zadas por expertos, un inventario de medicamen-tos huérfanos, una enciclopedia de ER, un direc-torio de centros y laboratorios de diagnóstico,proyectos científicos en marcha, ensayos clínicos,registros, asociaciones de pacientes, plataformastecnológicas y redes de investigación dedicadas alas ER en cada uno de los países que formanparte del consorcio, una herramienta de ayuda aldiagnóstico que permite una búsqueda basada ensignos y síntomas de las ER, guías para emergen-cias médicas y una revista electrónica con noti-
cias sobre los acontecimientos, médicos, de inves-tigación y asuntos políticos de interés en ER.
Otro punto de información sobre el diagnósticode ER se puede encontrar en el portal de laAsociación Española de Genética Humana(AEGH; www.aegh.org). En su página web sepuede consultar un listado de centros de diagnós-tico de cada una de las comunidades autónomasy un listado de patologías y centros donde se rea-liza el diagnóstico de cada una.
La�genómica�en�el�diagnóstico�genéticoLas técnicas utilizadas en las pruebas genéticashan ido cambiando conforme se ha ido desarro-llando la tecnología. En el campo de la citogené-tica, el cariotipo convencional se apoya en otrastécnicas genómicas como los arrays de hibrida-ción genómica comparada (aCGH) que permi-ten detectar variaciones en el número de copiasen todo el genoma. El Colegio Americano deGenética Médica ya recomienda el aCGH comoprueba diagnóstica de primera opción antecasos de retraso mental con malformacionescongénitas.
El desarrollo de la secuenciación masiva enparalelo o secuenciación de nueva generación(NGS) ha revolucionado la investigación genéti-ca y desde 2009 se emplea en el diagnósticogenético como alternativa a la amplificación deregiones genómicas por PCR y secuenciaciónpor el método de Sanger. Esta tecnología permi-te obtener una visión panorámica del genoma deun paciente o parte de éste en función de laestrategia que se utilice: secuenciación del geno-ma completo, secuenciación del exoma (regióncodificante del genoma que supone un 1,5% delmismo, pero acumula el 85% de las mutacionespatogénicas) o secuenciación dirigida a panelesde genes conocidos por su relación con la sospe-cha clínica.
La creciente reducción de los costes de esta tec-nología y su potencia y rapidez la sitúan como laherramienta de análisis genético del futuro cerca-no (quizá ya del presente). La NGS es muy útilpara las enfermedades mendelianas, especialmen-te las ER genéticamente heterogéneas ya que per-mite el análisis de un alto número de genes paravarios pacientes a la vez a un coste y en un tiem-po inimaginable pocos años atrás.
Sin embargo, todavía hay que resolver una seriede problemas para la incorporación definitiva dela NGS a la rutina asistencial.
Los complejos análisis bioinformáticos, la valida-ción analítica de la NGS, la correcta interpreta-ción de los resultados y los riesgos derivados dela ingente información obtenida (hallazgos aje-nos al objetivo primario de la prueba, posiblepérdida de privacidad, sobrecarga del sistemasanitario, etc) son aspectos que requieren la aten-ción de los profesionales y su resolución seráesencial para la incorporación de esta tecnologíaa la práctica clínica. Una vez superados estosproblemas, la NGS supondrá un beneficio extra-ordinario para el diagnóstico genético en generaly de las ER en particular.
José�M.�Millán�Salvadorunidad de Genética. Hospital universitari i Politècnic la Fe.
CiBer de enfermedades raras (CiBerer).
16
informeMalalties rares, informació freqüent
vS97

En el año 2013, Año Nacional de lasEnfermedades Raras, decretado asípor el Gobierno español, desde laFederación Española de EnfermedadesRaras (FEDER) exigimos de este que
exprese su compromiso, solidaridad y responsa-bilidad a través de la publicación del plan de tra-bajo basado en las siguientes 13 propuestas: 1. Que las enfermedades raras (ER), siendo enfer-
medades crónicas y de interés prioritario ensalud pública, tengan un marco jurídico y admi-nistrativo que garantice la protección de losderechos (sociales y sanitarios) de las personas.
2. Que se exima del copago a las familiascon ER.
3. Que se impulsen medidas concretas para ase-gurar el acceso en equidad a medicamentos deuso vital para las familias con ER (medica-mentos huérfanos, medicamentos coadyuvan-tes, productos sanitarios y cosméticos).
4. Que se publique el Mapa de unidades clínicasde experiencia en enfermedades raras que exis-ten en España.
5. Que se acrediten al menos diez centros, servi-cios y unidades de referencia (CSUR) paraER en el Sistema Nacional de Salud en 2013,dotando a estas unidades de financiaciónsuficiente para garantizar su calidad y soste-nibilidad.
6. Que se impulse la investigación en ER a tra-vés de los centros, servicios y unidades dereferencia.
7. Que se establezca y publique la ruta de deriva-ción para garantizar la efectiva atención de lasfamilias con ER, en cualquier punto de la geo-grafía española.
8. Que se establezcan en todas las comunidadesautónomas unidades multidisciplinares deinformación, seguimiento, control y atencióngeneral a las personas con ER, a fin de garan-tizar la atención real y efectiva de todos losafectados por estas enfermedades, eliminandolas situaciones de desigualdad existentes hoyen día.
9. Que se incluyan en la Estrategia Nacional deEnfermedades Raras las propuestas e indica-dores del Informe EUROPLAN, a fin de mejo-rar su incidencia real en la atención de las per-sonas con ER.
10.Que se garantice la atención a las personascon ER que requieran su traslado a otroEstado miembro, cuando sea preciso, a travésde la Directiva de movilidad sanitaria trans-fronteriza
11.Que se utilice por parte del Gobierno espa-ñol la Clasif icación Internacional delFuncionamiento de la Discapacidad y de laSalud, liderando el IMSERSO la homogenei-zación de los criterios de valoración a la dis-capacidad en las ER en las comunidadesautónomas. Por otro lado, es urgente que setomen medidas que agilicen los trámites enmateria de dependencia para las personasafectadas por ER.
17
No�es�raro�tener�unaenfermendad�pocofrecuenteALMUDENA�AMAYA�Y�ZAIRA�MARTÍN-VILLALBA
Ante�la�falta�de�investigación�y�políticas�unificadas,�lasasociaciones�de�pacientes�de�enfermedades�pocofrecuentes�se�unen�con�el�único�objetivo�de�que�las
personas�que�sufren�este�tipo�de�patologías�tengan�unamejor�calidad�de�vida,�no�solo�en�el�plano�sanitario�sinotambién�en�el�aspecto�social.�Nacen�con�la�vocación�dedefender�a�afectados�y�familiares�de�todas�las�injusticias
que�provoca�el�sistema.�En�el�caso�concreto�de�laFederación�Española�de�Enfermedades�Raras�se�aboga
por�la�creación�de�nuevas�redes�que�favorezcan�elintercambio�de�información,�y�así�romper
con�el�aislamiento�social.
Malalties rares, informació freqüentinforme
vS97

12.Que se impulse la escolarización del alumna-do con necesidades educativas especiales enlos centros ordinarios, incorporando recursoseducativos y de asistencia sanitaria y sefomente la creación de protocolos de actua-ción conjunta entre centros educativos y hos-pitales.
13.Que se asegure la inclusión laboral mediantela flexibilización de los horarios, la adapta-ción de las condiciones laborales y de la ubi-cación del puesto de trabajo.
Estos 13 compromisos exigidos son el fruto dela incansable labor de las asociaciones de pacien-tes con enfermedades poco frecuentes (en adelan-te EPF).
Sentimientos encontrados, emociones, todo untorrente de incertidumbres ante un diagnóstico,que llega tarde... o no llega.
Cuando una familia —no olvidemos que enfermatodo el entorno familiar del afectado— se enfren-ta a una EPF, debemos añadir otros hechos rea-les, como el aislamiento, el dolor físico (en lamayoría crónico), discapacidad física, orgánica,sensorial, intelectual que genera la patología. Esahí, donde se hace imprescindible la concurrenciaactiva de las asociaciones de pacientes.
Este tipo de asociaciones de pacientes, se unenante la falta de investigación y políticas unifica-das, con el único objetivo de que las personasque sufren este tipo de patologías tengan unamejor calidad de vida, no solo en el plano sanita-rio sino también en el terreno social.
Nacen con la vocación de defender a afectados yfamiliares de todas las injusticias que provoca elsistema. Ya que hemos podido comprobar cómoen tiempos de bonanza los pacientes de EPFhemos estado al final de la lista de derechos y entiempos de crisis nos hallamos en un doloroso einjusto primer lugar a la hora de copagos, reduc-ción de prestaciones, enlentecimiento de la aten-ción, y así mediante la unión, creamos un bloquecada día más sólido para romper todas aquellasbarreras a las que tenemos que hacer frente deforma continua.
Nuestra entidad aboga por la creación de nuevasredes que favorezcan el intercambio de informa-ción, y así romper con el aislamiento social, sien-do facilitadores de contacto entre asociaciones dela misma patología.
Nacimiento�de�FEDERFEDER surge, ante la falta de investigación ypolíticas unificadas, de la unión de seis asociacio-nes con el principal objetivo de que las personasque sufren este tipo de patología tengan unamejor calidad de vida y para romper todas aque-llas barreras a las que tienen que hacer frente ensu día a día.
Actualmente, FEDER está constituida por 241entidades que trabajan en beneficio de este colec-tivo.
No solo representamos a asociaciones de patolo-gías concretas o entidades, sino que tambiéndamos cabida a personas individuales que no tie-nen asociación de referencia en España y queposiblemente sean un caso único en nuestro país.Este hecho genera la creación de asociaciones
que aglutinan a afectados de EPF, las cuales tie-nen diferentes características entre sí, pero que,quizá, éste sea el único camino que encuentranpara que se oiga su voz.
No olvidemos que las asociaciones de pacientesactúan tanto de altavoz como de portavoz de losque se hallan silentes.
Nuestra entidad lucha para que se reconozcanlos derechos, tanto sociales, sanitarios, educati-vos y laborales, de estas personas y aboga por lacreación de nuevas redes que favorezcan el inter-cambio recíproco de información, romper con elaislamiento social y aportar un importante apoyoemocional. Estas redes se forman a través de lacreación de asociaciones específicas de enferme-dades de baja prevalencia.
Desde los inicios, el papel de las asociacionessiempre ha sido muy importante, proporcionan-do orientación, apoyos, mediación, además defacilitar atención a todos los niveles y de ser unposibilitador de una mayor inclusión social.
El movimiento asociativo de las EPF en Españatiene signos evidentes de expansión y consolida-ción, ya que es el mejor camino que encuentran,tanto afectados como familiares, para apoyarse eir mejorando. Es la forma de adquirir estrategiasde afrontamiento tanto en sentido individualcomo colectivo.
Esta imparable expansión, tiene que servir paraque las administraciones públicas nos considerenno solo interlocutores válidos ante cualquierorganismo, sino un movimiento de gran poten-cial social con una fuerza capaz de cambiar polí-ticas sociales.
Poco a poco vamos avanzando para ocupar nosolo el lugar que nos corresponde, sino el que senos tiene que otorgar desde los organismospúblicos
En la Comunitat Valenciana ya contamos con 28socios y asociaciones integradas en nuestra dele-gación, repartidos de la siguiente manera:•Castellón, 1•Valencia, 22•Alicante, 5
Panorama�actual�de�estas�patologíasFruto de la designación de este año como AñoNacional de las Enfermedades Raras, podemosresaltar una vía de trabajo muy importante quenos marca el camino para conseguir algunas delas prioridades reflejadas en nuestras propuestasanteriormente mencionadas. Recientemente, elconsejo asesor de Sanidad ha creado un grupo detrabajo dedicado a las ER, con el objetivo pri-mordial de ampliar conocimientos y analizar lasituación actual de estas patologías en continuocambio dadas las incesantes novedades que veni-mos observando en el sistema sanitario, como hapodido ser el copago.
La situación que están viviendo muchas familiases preocupante. El ambiente de una familia conalgún miembro afectado por ER queda quebran-tado cuando sufre la pérdida de su trabajo acausa de la enfermedad, de forma que los ingre-sos descienden de manera vertiginosa frente a losgastos que le supondrá hacer frente a la enferme-dad. Según el estudio ENSERIO I, realizado por
FEDER en 2009 donde se estudió la situación denecesidades sociosanitarias de las personas conER en España, el 20% de la economía familiarse destina a la enfermedad; gastos que se van enmedicamentos que no cubre el Sistema Nacionalde Salud, desplazamientos a otras comunidadesautónomas para tener una segunda opinión orecibir el tratamiento adecuado, médicos priva-dos, etc. Este porcentaje actualmente se ha vistoincrementado, llegando a gastarse de media unafamilia 720 euros por cada miembro afectadopor una ER, viéndose en familias cuyos miem-bros afectados son 2 o 3.
Es de vital importancia que en este año las ERsean declaradas como patologías crónicas, puestoel 85% de los casos son enfermedades discapaci-tantes e invalidantes, como hacemos referenciaen nuestra primera propuesta. Esto daría la posi-bilidad de poder optar a una serie de beneficios,sobre todo sanitarios, que hasta ahora no tienen,como sería poder dispensar medicamentos de lafarmacia hospitalaria o acceso a financiar medi-camentos.
Estas patologías no tienen cura en el tiempo, porlo que un buen diagnósstico es la base para queposteriormente tengan acceso a un tratamientofarmacológico correcto que sea paliativo y quepermita al paciente tener una vida calidad devida.
Otra prioridad planteada en nuestras propuestases la investigación. Muchas de las líneas de inves-tigación actualmente abiertas han empezado poriniciativa de las propias asociaciones de pacientesque trabajan y buscan fondos privados para quese investigue su enfermedad. Si se trabaja paraconocer más de la enfermedad se puede trabajarpara que en el futuro estas enfermedades se pue-dan evitar desde el nacimiento o incluso antes.Tenemos que tener muy claro que investigar en elpresente es prevenir en el futuro.
Recientemente, FEDER ha constituido un comitéasesor, órgano cuya finalidad es el asesoramientoa la Federación en tres áreas fundamentales:1. Apoyo en la resolución de consultas de tipo
clínico recibidas por el Servicio deInformación y Orientación sobre ER.
2. Contribuir con artículos e informes, valora-ción de documentos, posicionamientos con elObservatorio de FEDER.
3. Sugerir tendencias u orientaciones en materiacientífica y profesional que ayuden a FEDER aestablecer cada año las líneas estratégicas desu plan de acción.
Sabemos que la sociedad valenciana está muy sen-sibilizada con este tipo de patologías, pero aúnnos queda hacer escuchar nuestra voz en muchosotros ámbitos, especialmente en el terreno escolary laboral. Siempre debemos aprovechar cualquierforo para dar a conocer que existen asociacionesde pacientes con patologías poco frecuentesdonde podrán acudir y ser escuchados.
Almudena�Amaya�Rubio.Delegada de la Federación española de enfermedades
raras CV (FeDer CV).
Zaira�Martín-Villalba.trabajadora social de la Federación española de
enfermedades raras CV (FeDer CV).
18
informeMalalties rares, informació freqüent
vS97
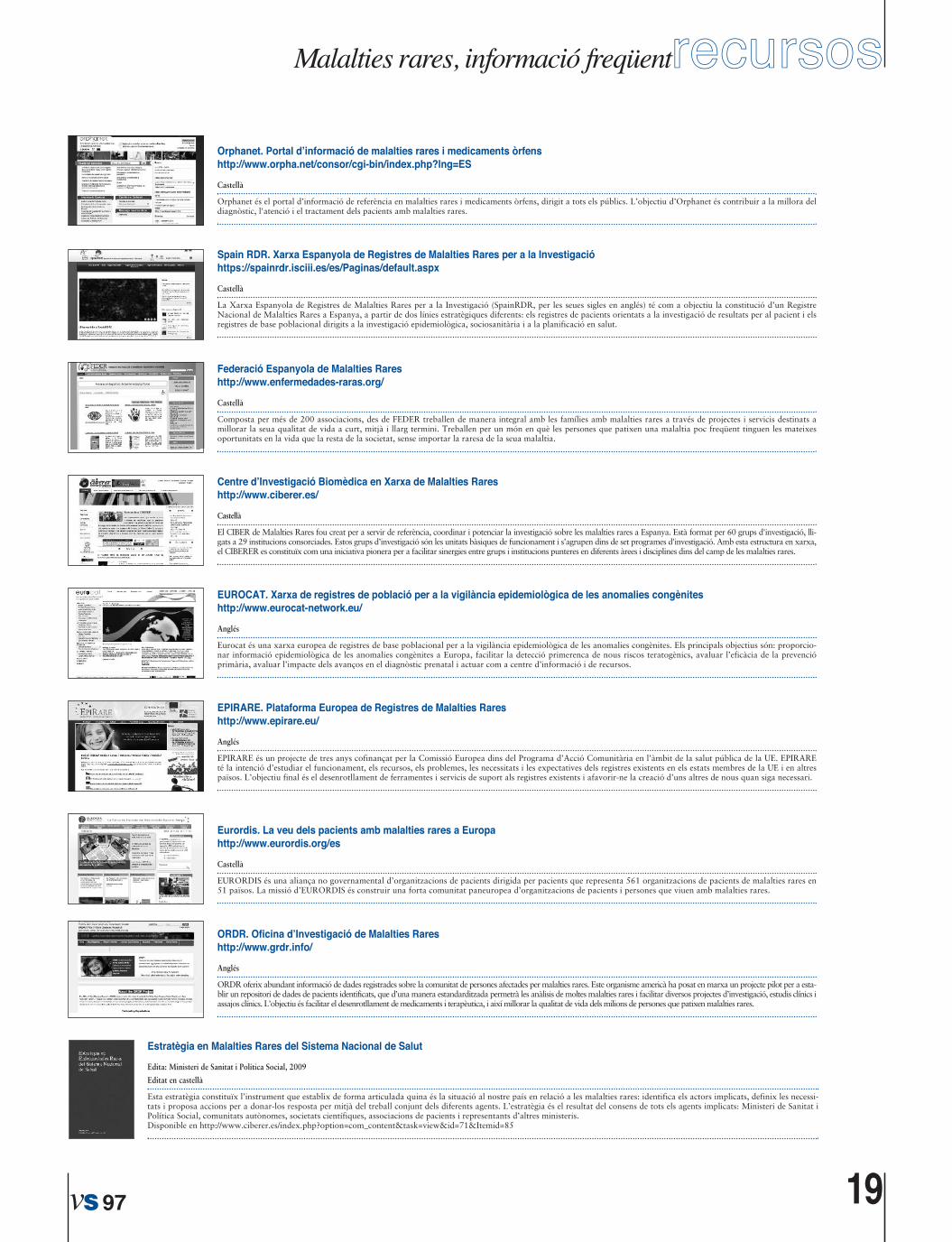
19
EUROCAT.�Xarxa�de�registres�de�població�per�a�la�vigilància�epidemiològica�de�les�anomalies�congèniteshttp://www.eurocat-network.eu/
Anglés
Eurocat és una xarxa europea de registres de base poblacional per a la vigilància epidemiològica de les anomalies congènites. Els principals objectius són: proporcio-nar informació epidemiològica de les anomalies congènites a Europa, facilitar la detecció primerenca de nous riscos teratogènics, avaluar l’eficàcia de la prevencióprimària, avaluar l’impacte dels avanços en el diagnòstic prenatal i actuar com a centre d’informació i de recursos.
ORDR.�Oficina�d’Investigació�de�Malalties�Rareshttp://www.grdr.info/
Anglés
ORDR oferix abundant informació de dades registrades sobre la comunitat de persones afectades per malalties rares. Este organisme americà ha posat en marxa un projecte pilot per a esta-blir un repositori de dades de pacients identificats, que d’una manera estandarditzada permetrà les anàlisis de moltes malalties rares i facilitar diversos projectes d’investigació, estudis clínics iassajos clínics. L’objectiu és facilitar el desenrotllament de medicaments i terapèutica, i així millorar la qualitat de vida dels milions de persones que patixen malalties rares.
Orphanet.�Portal�d’informació�de�malalties�rares�i�medicaments�òrfenshttp://www.orpha.net/consor/cgi-bin/index.php?lng=ES
Castellà
Orphanet és el portal d’informació de referència en malalties rares i medicaments òrfens, dirigit a tots els públics. L’objectiu d’Orphanet és contribuir a la millora deldiagnòstic, l'atenció i el tractament dels pacients amb malalties rares.
Centre�d’Investigació�Biomèdica�en�Xarxa�de�Malalties�Rareshttp://www.ciberer.es/
Castellà
El CIBER de Malalties Rares fou creat per a servir de referència, coordinar i potenciar la investigació sobre les malalties rares a Espanya. Està format per 60 grups d'investigació, lli-gats a 29 institucions consorciades. Estos grups d’investigació són les unitats bàsiques de funcionament i s’agrupen dins de set programes d'investigació. Amb esta estructura en xarxa,el CIBERER es constituïx com una iniciativa pionera per a facilitar sinergies entre grups i institucions punteres en diferents àrees i disciplines dins del camp de les malalties rares.
Spain�RDR.�Xarxa�Espanyola�de�Registres�de�Malalties�Rares�per�a�la�Investigacióhttps://spainrdr.isciii.es/es/Paginas/default.aspx
Castellà
La Xarxa Espanyola de Registres de Malalties Rares per a la Investigació (SpainRDR, per les seues sigles en anglés) té com a objectiu la constitució d’un RegistreNacional de Malalties Rares a Espanya, a partir de dos línies estratègiques diferents: els registres de pacients orientats a la investigació de resultats per al pacient i elsregistres de base poblacional dirigits a la investigació epidemiològica, sociosanitària i a la planificació en salut.
Federació�Espanyola�de�Malalties�Rareshttp://www.enfermedades-raras.org/
Castellà
Composta per més de 200 associacions, des de FEDER treballen de manera integral amb les famílies amb malalties rares a través de projectes i servicis destinats amillorar la seua qualitat de vida a curt, mitjà i llarg termini. Treballen per un món en què les persones que patixen una malaltia poc freqüent tinguen les mateixesoportunitats en la vida que la resta de la societat, sense importar la raresa de la seua malaltia.
EPIRARE.�Plataforma�Europea�de�Registres�de�Malalties�Rareshttp://www.epirare.eu/
Anglés
EPIRARE és un projecte de tres anys cofinançat per la Comissió Europea dins del Programa d’Acció Comunitària en l’àmbit de la salut pública de la UE. EPIRAREté la intenció d’estudiar el funcionament, els recursos, els problemes, les necessitats i les expectatives dels registres existents en els estats membres de la UE i en altrespaïsos. L’objectiu final és el desenrotllament de ferramentes i servicis de suport als registres existents i afavorir-ne la creació d’uns altres de nous quan siga necessari.
Eurordis.�La�veu�dels�pacients�amb�malalties�rares�a�Europahttp://www.eurordis.org/es
Castellà
EURORDIS és una aliança no governamental d’organitzacions de pacients dirigida per pacients que representa 561 organitzacions de pacients de malalties rares en51 països. La missió d’EURORDIS és construir una forta comunitat paneuropea d’organitzacions de pacients i persones que viuen amb malalties rares.
Estratègia�en�Malalties�Rares�del�Sistema�Nacional�de�Salut
Edita: Ministeri de Sanitat i Política Social, 2009
Editat en castellà
Esta estratègia constituïx l’instrument que establix de forma articulada quina és la situació al nostre país en relació a les malalties rares: identifica els actors implicats, definix les necessi-tats i proposa accions per a donar-los resposta per mitjà del treball conjunt dels diferents agents. L’estratègia és el resultat del consens de tots els agents implicats: Ministeri de Sanitat iPolítica Social, comunitats autònomes, societats científiques, associacions de pacients i representants d’altres ministeris.Disponible en http://www.ciberer.es/index.php?option=com_content&task=view&id=71&Itemid=85
Malalties rares, informació freqüentrecursos
vS97

/Jornades/
Viure en
Salut