anteproyecto de decision armonizacion de legislaciones en materia de...
TRANSCRIPT


PROPUESTA DE LA SECRETARÍA GENERAL SOBRE
ARMONIZACION DE LEGISLACIONES EN MATERIA DE REGISTROS SANITARIOS
DE PRODUCTOS FARMACEUTICOS
PRESENTACION
Desde el año 1997, la Secretaría General ha venido impulsando la negociación de un Proyecto de Decisión para reducir las posibles restricciones técnicas sanitarias que se puedan presentar en la comercialización de productos farmacéuticos en la Comunidad Andina, y que a la vez se garantice la seguridad, eficacia y calidad de los mismos.
Durante la VIII Reunión de Expertos llevada a cabo los días 13 y 14 de mayo de 2002, se llegó a un acuerdo total sobre el Proyecto de Decisión. No obstante, la delegación de Venezuela no participó, razón por la cual la Secretaría General adelantó consultas con dicho País, producto de las cuales se elaboró la presente Propuesta.
La Propuesta armoniza los requisitos técnicos, plazos y procedimientos necesarios para que las Autoridades Nacionales Competentes otorguen un Registro Sanitario, instrumento que permite a un producto farmacéutico comercializarse en la Subregión. Para efectos del registro, se clasifican los medicamentos en conocidos (literal “a” del artículo 6 de la Propuesta) y nuevos (literal “b” del artículo 6 de la Propuesta). Los primeros son los registrados en tres o más Países Miembros o cuyos principios activos constan en la farmacopea internacional y los nuevos son medicamentos registrados en uno o dos Países Miembros. Se establecen mayores exigencias y controles para los medicamentos nuevos.
La Propuesta de Decisión establece un mecanismo para el reconocimiento mutuo de los registros y certificaciones de Buenas Prácticas de Manufactura -BPM- otorgados a un medicamento producido y comercializado por cualquier País Miembro de la Comunidad Andina. El mecanismo se denomina Notificación Sanitaria. En esta Decisión se aplica el silencio administrativo positivo.
Asimismo, se incluyen elementos de orientación al consumidor tales como el etiquetado y prospecto. Se especifica las condiciones para la comercialización de los medicamentos que no necesitan prescripción médica.
La armonización de procedimientos y requisitos para obtención del Registro Sanitario, los compromisos en materia de Buenas Prácticas de Manufactura -BPM- y la Notificación Sanitaria, constituyen compromisos esenciales para el perfeccionamiento de la zona de libre comercio de la Comunidad Andina, al tiempo que contribuyen a mejorar la calidad de los medicamentos producidos y comercializados en los Países Miembros.

- 2 -
PROYECTO DE DECISION
Armonización de Legislaciones en materia de Registros Sanitarios de productos farmacéuticos
LA COMISION DE LA COMUNIDAD ANDINA,
VISTOS:
Los Artículos 51, 55 y 72 del Acuerdo de Cartagena, las Decisiones 418, 419 y 437 de la Comisión y la Propuesta Nº68 de la Secretaría General; y,
CONSIDERANDO:
Que dados los avances del proceso de integración subregional andino, y con la finalidad de suprimir las disparidades que puedan afectar el establecimiento del mercado común andino, es necesario iniciar los procesos de armonización de las normativas en materia de productos farmacéuticos, tomando en cuenta los nuevos desarrollos en el tratamiento de los temas que atañen al campo de los productos con riesgo sanitario;
Que toda regulación en materia de producción y distribución de productos farmacéuticos debe inspirarse, entre otros criterios, en la salvaguardia de la salud pública;
Que, no obstante, los medios que se utilicen para la consecución de este objetivo deben aplicarse de manera tal que no se constituyan en una forma de discriminación o restricción injustificada al comercio;
Que es conveniente, con miras a lograr una libre circulación de los productos farmacéuticos en los Países Miembros de la Comunidad Andina, armonizar los aspectos relativos al Registro Sanitario, principal requisito para el acceso de los medicamentos al mercado;
DECIDE:
CAPÍTULO I
DEFINICIONES Y ÁMBITO DE APLICACIÓN
Artículo 1.- Para los efectos de la presente Decisión se consideran como válidas las definiciones del Glosario de términos que se incluyen en el Anexo 1. Los términos no contemplados en el mencionado Anexo y que sean pertinentes en la Decisión serán los del glosario de términos de la Organización Panamericana de la Salud.
Artículo 2.- La presente Decisión establece un régimen común para el otorgamiento del Registro Sanitario de aquellos medicamentos fabricados en la Subregión Andina, así como las exigencias de calidad sanitaria, eficacia y seguridad que deberán cumplir estos medicamentos para circular libremente en el territorio de la Comunidad Andina.

Artículo 3.- Quedan excluidos del ámbito de esta Decisión los productos homeopáticos y naturales, los cuales serán regulados por decisiones específicas.
CAPÍTULO II
DISPOSICIONES GENERALES
Artículo 4.- Los medicamentos fabricados en la Comunidad Andina deberán garantizar un nivel óptimo de calidad sanitaria, eficacia y seguridad.
Artículo 5.- La Autoridad Nacional Competente en el sector de la salud, en cada País Miembro, es responsable de la vigilancia y el control sanitario de los medicamentos, así como de cumplir y velar por la aplicación de lo dispuesto en la presente Decisión.
CAPÍTULO III
DE LOS REQUISITOS PARA EL REGISTRO
Artículo 6.- Para los efectos de lo dispuesto en la presente Decisión y en desarrollo del Artículo 2, queda establecido que los Requisitos para el Registro Sanitario distinguen dos clases de medicamentos:
a) Aquellos registrados en tres o más Países Miembros o los Medicamentos cuyos principios activos consten en cualquiera de las siguientes farmacopeas:
- USP (Estados Unidos de América)- Farmacopea Británica- Farmacopea Internacional de la Organización Mundial de la Salud- Farmacopea Europea (Unión Europea)
b) Los medicamentos registrados en uno o dos Países Miembros y aquellos cuyos principios activos no consten en las farmacopeas aprobadas en el presente artículo.
Artículo 7.- Para la obtención del Registro Sanitario el interesado deberá presentar a la Autoridad Sanitaria Nacional Competente una solicitud acompañada de la siguiente información:
a) La formula cuali-cuantitativa del producto, identificando con el nombre genérico todas las substancias que formen parte del mismo, conforme a la nomenclatura internacional “International Union of Pure and Applied Chemistry” (IUPAC), con el nombre de marca del producto si lo tuviere y con la DENOMINACIÓN COMÚN INTERNACIONAL (DCI) para los principios activos, establecida por la Organización Mundial de la Salud. Dicha fórmula debe ser expresada en forma porcentual y por unidad posológica, esta última en el Sistema Métrico Decimal o en Unidades Internacionales cuando corresponda. Cuando se trate de aerosoles, además de las sustancias activas, se debe declarar los gases propulsores expresados en porcentaje de peso o volumen.
b) La fórmula cuali-cuantitativa deberá presentarse en dos partes que comprenda los principios activos y los excipientes.
- 3 -

c) Una muestra del envase o de la presentación, cuando se trate de innovaciones en el envase.
d) Método de análisis y especificaciones del producto terminado. Cuando corresponda a una de las farmacopeas aceptadas en la Comunidad Andina, el fabricante indicará el nombre de la farmacopea, su edición y la página correspondiente de la misma. Si el método no corresponde a ninguna de las farmacopeas reconocidas por la presente Decisión, que son las aceptadas en la Comunidad Andina, el fabricante o el interesado deberá presentar los documentos que acrediten los Análisis de Validación completos. Para las formas farmacéuticas sólidas, cuyo principio activo sea oficial en alguna Farmacopea, en la cual no se precisen las pruebas de disolución respectivas, se exigirán las correspondientes pruebas de Farmacopea de la USP.
e) Proyecto de rotulado del envase primario y del secundario cuando así proceda en idioma español, de acuerdo a lo dispuesto en el Capítulo correspondiente de la presente Decisión.
f) Forma farmacéutica y propiedades organolépticas del producto. Especificaciones y materiales del sistema envase-cierre y del material empaque secundario, cuando este exista.
g) Los resultados de las Pruebas Aceleradas de Estabilidad para la obtención del registro, sin perjuicio de la obligatoriedad de realizar las Pruebas de Envejecimiento Natural. Estas últimas, se regirán por la norma OMS/OPS para la zona climática IV.
h) Certificado de Buenas Prácticas de Manufactura pleno o condicionado
Artículo 8.- Para la obtención del registro sanitario de los productos incluidos en el literal a) del artículo 6, las Autoridades Nacionales Competentes deberán exigir lo contemplado en el artículo 7, tomando en cuenta las normas farmacológicas armonizadas por la Comunidad Andina, las cuales definen condiciones de uso (indicación, posología), restricciones de uso (reacciones adversas, precauciones, contraindicaciones, interacciones) y condiciones de comercialización (con prescripción o sin prescripción facultativa).
Artículo 9.- Para la obtención del registro sanitario de los productos incluidos en el literal b) del artículo 6, las Autoridades Nacionales Competentes deberán exigir lo contemplado en el artículo 7. Previo a ello la Autoridad Nacional Competente realizará una evaluación farmacotoxicológica, que garantice una relación beneficio riesgo positiva.
CAPÍTULO IV
DEL OTORGAMIENTO DEL REGISTRO SANITARIO
Artículo 10.- Los medicamentos fabricados en la Subregión deberán obtener el Registro Sanitario en el País donde son fabricados.
- 4 -

Artículo 11.- Las Autoridades Sanitarias Nacionales Competentes otorgarán el registro sanitario en los siguientes casos:
En el caso de los productos de que trata el literal a) del Artículo 6, la Autoridad Sanitaria Nacional Competente admitirá el expediente y emitirá el dictamen aprobando o negando el registro sanitario, dentro de los 30 días hábiles siguientes a la presentación de la solicitud acompañada de la documentación dispuesta en la presente Decisión. Si transcurrido dicho plazo, la Autoridad Nacional Competente no hubiese emitido su pronunciamiento, la solicitud se considerará aprobada.
En el caso de los productos de que trata el literal b) del artículo 6, la Autoridad Sanitaria Nacional Competente admitirá el expediente y emitirá el dictamen aprobando o negando el registro o inscripción sanitaria, dentro de los 90 días calendario siguientes a la presentación de la documentación a la que se refiere los artículos 7 y 9. Si transcurrido dicho plazo, la Autoridad Nacional Competente no hubiese emitido su pronunciamiento, la solicitud se considerará aprobada.
Artículo 12.- Para que un medicamento fabricado en un País Miembro que cuente con el respectivo Registro Sanitario pueda acceder al mercado subregional, se deberá cumplir con lo dispuesto en la presente Decisión y certificar en el País Miembro de fabricación el cumplimiento con la norma de Buenas Prácticas de Manufactura del Informe 32 de la Organización Mundial de la Salud.
Mediante la presente Decisión, los Países Miembros adoptan la Guía de Aplicación de la Norma de Buenas Prácticas de Manufactura, que figura como Anexo 2 de la presente Decisión. Dicha Guía de Aplicación será actualizada mediante acuerdos entre los Países Miembros los que serán incorporados al Ordenamiento Jurídico Andino mediante Resolución de la Secretaría General.
Las Autoridades Sanitarias Nacionales Competentes otorgarán la certificación de las Buenas Prácticas de Manufactura por área de fabricación plena o condicionada.
Artículo 13.- En aquellos casos en los que las empresas fabricantes no cumplan con las BPM plenamente, la Autoridad Nacional Competente del País Miembro de fabricación autorizará a dichas empresas el cumplimiento gradual de las Buenas Prácticas de Manufactura de acuerdo a un Programa previamente concertado con las mismas el cual no incluya puntos críticos indicados en la Guía de Aplicación señalada en el artículo anterior y que no podrá extenderse más allá del 31 de diciembre de 2005 Las Autoridades Nacionales Competentes de los Países Miembros reconocerán la certificación de BPM otorgada por el país miembro de fabricación, de conformidad con el artículo 12.
Artículo 14.- Los medicamentos fabricados y registrados en un País Miembro que cumplan con lo dispuesto en el artículo 12, serán registrados en los demás Países Miembros a través de una Notificación Sanitaria acompañada de la información prevista en la presente Decisión.
La Notificación Sanitaria consiste en la presentación por parte del interesado de la información a la Autoridad Nacional Competente del segundo País Miembro de comercialización. La Autoridad Nacional Competente tendrá como máximo hasta 20 días calendario para otorgar el código del Registro Sanitario.
- 5 -

Si transcurrido dicho plazo la Autoridad Nacional Competente no se hubiera pronunciado, el solicitante podrá comercializar el producto con el código de Registro Sanitario del primer País Miembro de comercialización.
Artículo 15.- El registro tendrá vigencia de 7 años y podrá ser renovado por períodos iguales. La renovación se entenderá efectuada automáticamente con la presentación de los siguientes documentos:
- Copia del acto administrativo mediante el cual fue concebido el registro sanitario,- Declaración juramentada del director técnico responsable de la elaboración del
producto, en la que se manifieste que el mismo no ha sido modificado en su composición, ni forma farmacéutica, si fuera el caso.
- Recibo de pago de los derechos correspondientes.- Información técnica contenida en el Artículo 7 de la presente Decisión.- Certificado de Buenas Prácticas de Manufactura actualizado.- Documentación relativa a la vigencia terapéutica del producto.
La renovación deberá ser solicitada dentro de los 30 días calendario previos a la expiración del plazo del Registro señalado en el primer párrafo.
Artículo 16.- Los fabricantes de productos farmacéuticos instalados en la Subregión Andina y que tengan el certificado de conformidad con la Norma de Buenas Practicas de Manufactura que se aprueba en la presente Decisión, están autorizados para fabricar cualquier producto farmacéutico que tenga el Registro Sanitario aprobado en el País que ha solicitado su elaboración. Las Autoridades Sanitarias Competentes, solicitarán al titular del registro, el contrato de fabricación entre las partes, para permitir el beneficio del derecho dispuesto en el artículo.
Artículo 17.- Cuando el titular de un Registro Sanitario solicite un nuevo registro por cambio de fabricante de los productos a que se refiere el artículo 16, y el mismo no haya sufrido cambios en su formulación y en el sistema envase-cierre, la Autoridad Nacional Competente deberá otorgar el Registro Sanitario en forma sumaria en términos no mayor de 10 días, previo a la presentación del certificado de BPM del nuevo fabricante.
CAPÍTULO V
DE LOS MEDICAMENTOS SIN PRESCRIPCIÓN FACULTATIVA
Artículo 18.- Al autorizar el Registro Sanitario de un medicamento, las Autoridades Sanitarias Nacionales Competentes especificarán la clasificación de los mismos:
- Medicamento con prescripción facultativa- Medicamentos sin prescripción facultativa
Artículo 19.- Se entiende por medicamento sin prescripción facultativa, aquellos productos farmacéuticos que puedan ser adquiridos sin mediación del prescriptor. Podrán ser considerados medicamentos sin prescripción facultativa, los que cumplan con las siguientes características:
- 6 -

19.1 Estar destinados al tratamiento de síntomas o enfermedades leves o comunes, de corta duración, ampliamente conocidas y que resulten fácilmente identificables
19.2 El medicamento debe ser de eficacia y seguridad reconocida; trayectoria terapéutica no menor a cinco años; sus efectos adversos deben ser de baja incidencia, escasa intensidad y que, en ningún caso, representen riesgos graves a la salud del consumidor. Se entenderá eficacia y seguridad reconocida que cumpla con las especificaciones técnicas para el cual fue diseñado y que sea manufacturado bajo condiciones exigidas para tal fin.
19.3 Tener un margen posológico amplio susceptible de ser adaptado a la edad y peso del paciente.
19.4 Tener un rango limitado de indicaciones. Se entenderá rango limitado de indicaciones aquel para el cual está principalmente diseñado o desarrollado técnicamente.
19.5 No deben ser susceptibles de desarrollar taquifilaxis o dependencias.
19.6 No deben enmascarar enfermedades serias.
19.7 Tener un amplio rango de seguridad, de tal modo que la administración voluntaria de dosis elevadas no represente un peligro grave a la salud del paciente.
19.8 No deben acumularse en el organismo.
19.9 Deben ser estables en condiciones extremas de almacenamiento.
19.10 Deben tener formulaciones diferentes para uso pediátrico.
19.11 No deben ser productos de administración parenteral.
19.12 Deben estar constituidos preferiblemente por un solo principio activo o por asociaciones a dosis fijas de reconocida trayectoria terapéutica.
Artículo 20.- Cuando un producto de venta con Prescripción Facultativa cumpla lo establecido en el Artículo 19 podrá solicitar a la Autoridad Nacional Competente del país donde obtuvo su registro sanitario la modificación de la clasificación de venta a venta sin Prescripción Facultativa.
Artículo 21.- Se adoptará el listado de Productos de Venta sin Prescripción Facultativa el cual será publicado mediante Resolución de la Secretaría.
La Autoridad Nacional Competente del país donde se expida un nuevo registro sanitario de venta sin Prescripción Facultativa ó se cambie la condición de venta a venta sin Prescripción Facultativa, lo comunicará a las Autoridades Nacionales Competentes de los otros Países Miembros y a la Secretaría General para la actualización del listado.
Artículo 22.- Las indicaciones terapéuticas de los medicamentos de venta sin Prescripción Facultativa serán las determinadas en el correspondiente registro sanitario
Artículo 23.- Las etiquetas, empaque e insertos de los medicamentos sin prescripción facultativa a que se refiere la presente Decisión, deberán cumplir con lo estipulado en los capítulos VI y VII, adicionando los siguientes requisitos:
a) Indicación (es) terapéutica(s)b) La frase venta sin prescripción facultativa
- 7 -

c) La frase “cumpla estrictamente las instrucciones anteriores y si los síntomas o carencias se intensifican o persisten, suspenda su uso y consulte a su médico”
Artículo 24.- El lenguaje utilizado en los empaques de los productos a que se refiere la presente Decisión, debe ser fácil de entender para cualquier persona y debe ser preciso, de suerte que no se preste para interpretaciones erradas y debe aparecer en idioma español.
Artículo 25.- En las etiquetas y empaque de medicamentos sin prescripción médica, se admite el uso de dibujos y gráficas que sean evocativas respecto del uso del producto.
Artículo 26.- La comercialización de Medicamentos de Venta sin Prescripción Facultativa que cuenten con el correspondiente Registro Sanitario, se realizará en establecimientos farmacéuticos, farmacias, droguerías y en cualquier establecimiento comercial, de acuerdo con las respectivas legislaciones nacionales, siempre y cuando se tengan presentes las condiciones de almacenamiento.
Las condiciones de almacenamiento deberán cumplir con criterios básicos como son la identificación clara del área para Venta de Medicamentos sin Fórmula Médica, donde se mantendrán separados de otros productos y el manejo de estos de acuerdo a las condiciones de temperatura y humedad propias de cada producto.
Artículo 27.- La Publicidad y promoción de los medicamentos sin prescripción facultativa se regirá por la legislación vigente en cada País Miembro.
CAPÍTULO VI
ETIQUETADO DE LOS MEDICAMENTOS
Artículo 28.- El empaque secundario o, a falta de este, el acondicionamiento primario de todo medicamento deberá llevar lo siguiente:
a) La marca del medicamento seguida de la Denominación Común Internacional con caracteres visibles, cuando contenga hasta dos principios activos. Si tuviera más de dos, los mismos se declararán solamente en la fórmula cuali-cuantitativa. Cuando se trate de medicamentos cuyo nombre es su Denominación Común Internacional, se debe agregar el nombre del laboratorio titular del Registro en el País.
b) Composición cualitativa y cuantitativa en principios activos por unidad posológica o según la norma de administración para un volumen o peso determinado, utilizando la Denominación Común Internacional;
c) La forma farmacéutica y contenido en peso, volumen o unidad posológica;
d) La lista de los excipientes que se incorporan en anexo a esta Decisión, Adicionalmente, deberá indicarse todos los excipientes cuando se trate de un producto inyectable, de una preparación tópica o de un colirio;
e) La forma de administración y la vía de administración;
- 8 -

f) Una advertencia especial que indique que el producto debe mantenerse fuera del alcance de los niños;
g) Una advertencia especial cuando el medicamento lo requiera;
h) La fecha de caducidad expresada claramente (mes/año);
i) Las precauciones particulares de la conservación;
j) Las precauciones especiales de eliminación de los productos no utilizados o de los residuos derivados de estos productos, si fuera el caso;
k) El nombre y dirección (ciudad y país) del titular de la autorización de comercialización;
l) El número de Registro Sanitario;
m) El número de lote de fabricación;
n) Indicación de que el medicamento que se expende lleva prescripción facultativa o es de venta sin prescripción.
El empaque secundario puede llevar signos o dibujos que permitan explicar determinadas informaciones contempladas en el presente artículo y otras informaciones relacionadas con las características del producto, que puedan resultar útil a la educación sanitaria, con exclusión de cualquier elemento que pueda tener carácter publicitario.
Artículo 29.- Los pequeños acondicionamientos primarios cuyas dimensiones hagan imposible incluir toda la información referida en el artículo anterior, deberán contener como mínimo lo establecido en los literales a), c), e), h), k) y m) del artículo 28, es decir: Nombre del producto, forma farmacéutica y concentración, vía de administración, fecha de expiración, laboratorio y lote. En todo caso, el resto de los datos deberá estar disponible en el empaque secundario o, en su defecto, en un prospecto.
Artículo 30.- Todas las informaciones que figuren en el empaque secundario y en los acondicionamientos primarios deberán ser fácilmente legibles, claramente comprensibles e indelebles.
Artículo 31.- Las informaciones que acompañan al medicamento podrán redactarse en varios idiomas, siempre y cuando uno de estos sea el español.
Artículo 32.- Los Países Miembros no podrán impedir la puesta en el mercado de los medicamentos provenientes de otro País Miembro, por motivos relacionados con el etiquetado, cuando éste se ajuste a las disposiciones contenidas en el presente capítulo.
CAPÍTULO VII
DEL PROSPECTO DE INFORMACIÓN
Artículo 33.- Cuando la información dispuesta en el Capítulo VI no figure directamente en el empaque secundario o en el acondicionamiento primario del medicamento, éste deberá incluir un prospecto de información.
- 9 -

Artículo 34.- El prospecto de información deberá elaborarse de acuerdo a los siguientes datos:
a) Para identificación del medicamento:
1. La marca del medicamentos seguida de la Denominación Común Internacional con caracteres visibles, cuando contenga hasta dos principios activos . Si tuviera más de dos, los mismos se declaran solamente en la fórmula cuali-cuantitativa. Cuando se trate de medicamentos con un solo principio activo cuya denominación sea con un nombre arbitrario, incluirá junto a este, el nombre del laboratorio titular del registro en el país. Lo anterior incluye la mención del usuario a que se dirige; lactantes, niños o adultos, en caso de ser necesario;
2. La composición cualitativa completa, en principios activos y los excipientes que se contemplan en anexo a esta Decisión, así como la composición cuantitativa en principios activos utilizando la Denominación Común Internacional para cada presentación del medicamento;
3. La forma farmacéutica y el contenido en peso, en volumen o en unidad posológica para cada presentación del medicamento.
4. Nombre y dirección (ciudad y país) del titular del Registro Sanitario y del fabricante (Importador o comercializador).
b) Enumeración de las informaciones necesarias previas a la toma del medicamento:
1. Contraindicaciones;
2. Precauciones de empleo;
3. Interacciones con otros medicamentos y otras interacciones que puedan afectar la acción del medicamento, principalmente con alcohol, alimentos, tabaco, entre otros;
4. Advertencias especiales en las cuales se deberá tener en cuenta la situación particular de ciertas categorías de usuarios, como niños, mujeres embarazadas o durante la lactancia, ancianos, personas con ciertas patologías específicas, entre otros. Mencionar, si es el caso, los posibles efectos del tratamiento sobre la capacidad para conducir un vehículo o manipular determinadas máquinas. Dentro de las advertencias deberá incluirse una lista de excipientes cuyo conocimiento sea importante para una utilización eficaz y sin riesgos del medicamento, este aspecto podrá regularse dentro del marco jurídico del Acuerdo de Cartagena.
c) Las instrucciones necesarias y habituales para una buena utilización, en particular:
1. Posología;
2. Indicaciones;
3. Forma de uso y vía de administración;
4. Frecuencia de administración, precisando, si fuera necesario, el momento en que deba o pueda administrarse el medicamento;
Según la naturaleza del producto:
- 10 -

1. Medidas que deben tomarse en caso de sobredosis, como síntomas o tratamiento de urgencia;
2. Acción que deba tomarse en caso de que se haya omitido la administración de una o varias dosis;
3. Indicación, si se aplica, del riesgo de síndrome de abstinencia.
d) Descripción de los efectos adversos que puedan observarse durante el uso normal del medicamento, y en cada caso, medidas que deban adoptarse; el usuario o su representante comunicará a su médico, a su farmacéutico o a la autoridad sanitaria respectiva cualquier efecto no deseado que no estuviese descrito en el prospecto.
e) Referencia a la fecha de caducidad que figura en el envase, con:
1. Una advertencia para no sobrepasar esta fecha;
2. Las precauciones especiales de conservación;
3. Cuando corresponda, una advertencia con respecto a ciertos signos visibles de deterioro.
f) Indicación de que el medicamento que se expende lleva prescripción facultativa o es de venta sin prescripción.
Artículo 35.- El prospecto podrá incluir signos o figuras destinadas a hacer explícito alguno de los datos contemplados en él, así como otras informaciones que sean compatibles con el resumen de las características del producto, con fines de educación sanitaria, excepto lo que pueda presentar carácter publicitario.
Artículo 36.- El prospecto deberá estar redactado en términos claros y comprensibles para el usuario en los idiomas que decida el fabricante, siempre y cuando uno de ellos sea el español. No obstante en todas las lenguas utilizadas deberá figurar la misma información.
Artículo 37.- Los Países Miembros no podrán impedir la puesta en el mercado de medicamentos en su territorio por motivos relacionados con el prospecto, si éste se ajusta a lo dispuesto en el presente capítulo.
CAPÍTULO VIII
DISPOSICIONES COMUNES
Artículo 38.- Cualquier proyecto de modificación de un elemento relativo al etiquetado o al prospecto de información regulado por la presente Decisión, que no esté relacionado con el resumen de las características del producto será presentado a las Autoridades Nacionales Competentes respectivas. Si las Autoridades Competentes no se pronuncian contra el proyecto de modificación en un plazo de treinta (30) días calendario, a partir de la fecha de aceptación de la petición, el solicitante podrá proceder a la realización de las modificaciones.
- 11 -

Artículo 39.- En caso de incumplimiento de lo dispuesto en los capítulos VI y VII, las Autoridades Nacionales Competentes de los Países Miembros, podrán, si sus requerimientos al responsable o interesado no dan respuesta, proceder a la suspensión de la comercialización del producto, hasta que el etiquetado y el prospecto del medicamento de que se trate se ajuste a lo dispuestos en los mencionados capítulos.
CAPITULO IX
DISPOSICION FINAL
UNICA.- La presente Decisión entrará en vigencia el primero de enero del 2003, excepto para Ecuador para quien entrará en vigencia el 01 de julio de 2003. Durante dicho lapso Ecuador podrá seguir confiriendo los registros sanitarios conforme lo estipulado en su legislación nacional a fin de permitir la comercialización dentro de su territorio.
Sin perjuicio de lo señalado en el párrafo precedente la Guía de Aplicación de BPM a que se refiere el artículo 12 podrá ser exigida a partir de la fecha de publicación de esta Decisión en la Gaceta Oficial del Acuerdo de Cartagena.
CAPITULO X
DISPOSICIONES TRANSITORIAS
PRIMERA.- Bolivia podrá mantener, dentro de los requisitos de registro a que se refiere el artículo 7, la solicitud de una muestra del medicamento que se desea registrar. Dicho requisito deberá eliminarse antes del 31 de diciembre del año 2005.
SEGUNDA.- Para los productos que cuentan con Registro Sanitario y que se comercialicen en la subregión al momento de la expedición de la presente Decisión, podrán continuar comercializándose dentro de la subregión cumpliendo con la legislación que en cada país hubiere estado vigente al momento de haberse otorgado dicho Registro. No obstante, la renovación del Registro se sujetará a lo dispuesto en esta Decisión.
TERCERA.- A más tardar el 31 de diciembre de 2001, los Países Miembros adelantarán las reuniones necesarias de especialistas que permitan contar con la norma farmacológica comunitaria a que se refiere el artículo 8, y las definiciones de criterios o especificaciones técnicas que orientarán la evaluación farmacotoxicológica a que se refiere el artículo 9, con el objeto de garantizar que la misma conduzca a resultados similares en todos los países.
- 12 -

A N E X O 1
GLOSARIO DE TÉRMINOS
1. Acción del Medicamento (Drug action)En un sentido estricto, dícese del estímulo que provoca la administración del medicamento o el mecanismo de acción bioquímico o farmacológico que media el efecto o la respuesta. No debe usarse como sinónimo de estos dos últimos términos.
2. Advertencia (Warning)Llamado de atención, generalmente incluido en el envase secundario y/o prospecto, sobre algún riesgo particular asociado al consumo del medicamento.
3. Buenas Prácticas de Manufactura (Good manufacturing practices)Conjunto de procedimientos y normas destinadas a garantizar, en todo momento, la producción uniforme de lotes de medicamentos que satisfagan las normas de identidad, actividad, pureza, etc. Otras definiciones hacen aún más explícito que las buenas prácticas de manufactura incluyen, además, los programas de control y garantía de calidad.
4. Calidad Medicamento (Drug Quality)1) Está determinada por su identidad, pureza, contenido o potencial cualesquiera
otras propiedades químicas, físicas, biológicas o del proceso de fabricación que influyen en su aptitud para producir el efecto para el cual se destina.
2) Aptitud del medicamento para el uso al cual se destina, la cual es determinada por: a) su eficacia ponderada en relación a su seguridad de acuerdo a la declaración rotulada o aquella promovida por el fabricante; y b) su conformidad a las especificaciones de identidad, concentración, pureza y otras características. Se entiende que estos dos grupos de factores son interdependientes, puesto que las especificaciones son establecidas para garantizar la eficacia y la seguridad.
5. Certificado de BPM Es la certificación que garantiza la calidad de todas las etapas del proceso productivo de un medicamento. Podrá certificarse en forma plena o condicionada, por planta o por áreas de fabricación.
6. Certificación plena de BPM Es la otorgada a las empresas que cumplen con las BPM en todas sus áreas de fabricación
7. Certificación condicionada de BPM Es la otorgada a las empresas que hayan concertado con la Autoridad Nacional Competente del País Miembro de fabricación el cumplimiento gradual de las BPM en aquellas áreas de fabricación que no las cumplen. La certificación condicionada de las BPM supone, en todo caso, el cumpliento de los puntos críticos.
8. Concentración (Concentration)1) La concentración de la sustancia medicamentosa o principio activo se expresa
generalmente de la siguientes formas: peso/peso, peso/volumen, dosis/unitaria (“unit dose”)/volumen.
- 13 -

9. Contenido (Contents)Característica del producto medicamentoso que, según su forma farmacéutica, describe la cantidad del principio activo en términos de su masa o peso, cuando es sólida, y su concentración, cuando es líquida. Cuando el contenido del principio activo no puede determinarse por un método físico o químico se expresa en términos de unidades definidas arbitrariamente por comparación con un patrón biológico de referencia. Es habitual en tales casos referirse al contenido como potencia.
10.Contraindicaciones (Contra-indication)Situación clínica o régimen terapéutico en el cual la administración de un medicamento determinado debe ser evitada.
11.Denominación Común Internacional DCI (International nonproprietary)Nombre común para los medicamentos recomendados por la Organización Mundial de la Salud, a objeto de lograra su identificación internacional. Las denominaciones empleadas se presentan en latín, español, francés, inglés y ruso.
12.Disolución, pruebas de (Dissolution test)Determinación de carácter farmacopeico de la velocidad de disolución de un medicamento empleando determinados aparatos (de cesta o canasta, de paleta, etc.) y en determinadas condiciones de temperatura, velocidad de agitación, naturaleza del disolvente, etc. La prueba requiere, generalmente, de una sola medición y sus resultados se expresan en términos del tiempo requerido para que una fracción específica del medicamento presente se disuelva.
13.Dosificación/posología (Dosage)Describe la dosis de un medicamento, los intervalos entre las administraciones y la duración del tratamiento. No debe confundirse con el término dosis.
14.Dosis (Dose)Cantidad total de medicamento que se administra de una sola vez o total de las cantidades fraccionarias administradas durante un período determinado.
15.Eficacia (Efficacy)Aptitud de un medicamento para producir los efectos propuestos, determinada por métodos científicos. La eficacia del medicamento generalmente se determina a partir de la fase II de los estudios clínicos, mediante la comparación de los tratamientos que emplean el medicamento problemas versus un grupo control (grupo que no recibe tratamiento o recibe un placebo).
16.Envase/empaque (Packing)Cualquier recipiente en el que un medicamento, cosmético o dispositivo terapéutico esté contenido total o parcialmente, o en el que haya sido colocado o empaquetado.
17.Envejecimiento (Aging)Cambios que ocurren en las características físicas, químicas y biológicas de la materia prima y el producto terminado, con el transcurso del tiempo.
- 14 -

18.Envejecimiento natural (Natural aging)Degradación química o física de un medicamento que ocurre en las condiciones normales o definidas de almacenamiento.
19.Estabilidad (Stability)Aptitud de un principio activo o de un producto medicamentoso de mantener sus propiedades originales dentro de las especificaciones establecidas, en relación a su identidad, concentración o potencia, calidad, pureza y apariencia física. Esta última característica es especialmente útil cuando es una manifestación de cambios en las primeras.
20.Estudios Acelerados de Estabilidad (Accelerated/stress stability studies)Estudios diseñados con el fin de aumentar la tasa de la degradación química o física de un medicamento, empleando condiciones extremas de almacenamiento. Estos estudios tienen como objeto determinar los parámetros cinéticos de los procesos de degradación y/o predecir la vida útil del medicamento, en condiciones normales de almacenamiento. El diseño de estos estudios puede incluir temperaturas elevadas, altas humedades y exposición a la luz intensa. Los resultados de estudios acelerados de estabilidad deben ser complementados por los estudios de estabilidad efectuados en las condiciones de almacenamiento normales o definidas.
21.Estabilidad, pruebas de (stability tests)Pruebas que se efectúan para obtener información sobre las condiciones en las que se deben procesar y almacenar las materias primas, los productos semielaborados y terminados, según sea el caso. Sirven, además, para determinar la vida útil del medicamento.
22.Estudios Clínicos (Clinical studies)En general, cualquier estudio que se efectúa en humanos.
23.Estudios Pre-clínicos (Preclinical studies)Todos aquellos estudios en el desarrollo de un medicamento que se efectúan in vitro o en animales de experimentación, diseñados con la finalidad de obtener la información necesaria para decidir si se justifican estudios más amplios en seres humanos, sin exponerlos a riesgos injustificados. Si bien muchos de los estudios preclínicos deben anteceder a los estudios clínicos, algunos de los primeros, especialmente aquellos que requieren de períodos prolongados para su ejecución o son estudios especiales en animales, se continúan durante las primeras fases de los estudios clínicos. Por ejemplo, estudios preclínicos de toxicidad crónica y estudios de teratogenicidad.
24.Evaluación Farmacotoxicológica Comprende el procedimiento mediante el cual la Autoridades Sanitaria se forma un juicio sobre la utilidad, conveniencia y seguridad de un medicamento. La evaluación se adelantará teniendo en cuenta las siguientes características del producto: Eficacia Seguridad Dosificación Indicaciones, contraindicaciones, interacciones y advertencias –Relación
beneficio-riesgo Toxicidad
- 15 -

Farmacocinética Condiciones de comercialización, y Restricciones especiales
25.Excipiente ( Excipient)1) Sustancia que, en las concentraciones presentes en una forma farmacéutica,
tiene actividad farmacológica nula. Se emplea a fin de dotar a dicha forma farmacéutica de aquellas características que aseguren la estabilidad, biodisponibilidad, aceptabilidad y facilidad de administración de uno o más principios activos. En la medida en la que los excipientes afectan la liberación del principio activo, ellos pueden afectar la actividad farmacológica del producto medicamentoso a través de cambios en su biodisponibilidad.
2) Producto farmacéutico auxiliar empleado para dar una forma o consistencia adecuada a una preparación. Ejemplos de tipo de excipientes: desintegrantes, emulsionantes, colorantes y saborizantes, aglutinantes, conservadores, espesantes, etc.
26.Fecha de Fabricación ( Manufacturing date)Fecha con la cual se distinguen los lotes individuales y que indica la fecha en la cual se terminó la fabricación, usualmente expresada por el mes y el año. Es práctica usual en la fabricación de lotes grandes, manufacturados a través de varios meses, emplear la fecha del análisis de control que autoriza la liberación del producto.
27.Fabricación/ manufactura (Manufacturing)1) Todas las operaciones que intervienen en la producción de un medicamento:
elaboración, mezcla, formulación, envase, empacamiento y etiquetado.2) Todas las operaciones involucradas en la producción de un producto
farmacéutico, desde la recepción de los materiales, a través del procesamiento y empacamiento, hasta su liberación como un producto terminado.
28.Farmacopea ( Pharmacopeia)Conjunto o colección de normas sobre principios activos, productos farmacéuticos auxiliares, productos medicamentosos o terminados y métodos recomendados a objeto de constatar si estos las cumplen y que ha sido publicado o reconocido por la autoridad sanitaria competente. Existen Farmacopeas nacionales, plurinacionales (Farmacopea Europea, Compendium medicamentorum) y la Farmacopea Internacional.
29.Farmacoterapéutico/farmacoterapia (Pharmacotherapeutics)La aplicación de los medicamentos a la prevención y tratamiento de la enfermedad.
30.Forma farmacéutica/forma de dosificación (Dosage form)Forma en la cual se expende el producto farmacéutico terminado, a saber, comprimidos, cápsulas, jarabes, supositorios, etc. En los últimos 20-30 años, con el desarrollo de la biofarmacia y específicamente con el reconocimiento de la importancia de la biodisponibidad, se ha enfatizado en papel que juegan las formas de dosificación como sistemas de liberación o de entrega de medicamentos o principios activos. Dicha concepción se traduce en la aceptación de la necesidad de evaluar su aptitud para liberar el principio activo, que es su principal característica.
- 16 -

31. Indicaciones (Indications)1) Se refiere a los estados patológicos o padecimientos a los cuales se aplica un
medicamento.
32. Interacción medicamentosa (Drug interaction)Influencia que tiene un medicamento, alimento u otra sustancia sobre el comportamiento o la eficacia de otro medicamento, cuando ambos son administrados simultáneamente.
33.Literatura interior, Prospecto (Package insert/enclosed leaflet)Información que se presenta en hoja aparte sobre las propiedades indicaciones, precauciones y usos de un determinado medicamento. Como tal, se debe considerar parte integral de la rotulación.
34.Lote (Lot)Cantidad de un medicamento que se produce en un ciclo de fabricación. La característica esencial del lote de fabricación es su homogeneidad. La OMS y la legislación de muchos países considera al lote como sinónimo de “batch” o partida o serie, sin embargo, en otros, como en EUA y Chile, se establecen ciertas diferencias entre ambos términos. En general, se considera a un lote como una fracción específica e identificada de un “batch” que muestra características de uniformidad y calidad dentro de límites especificados. Otra acepción aún más específica de lote es aquella aplicada a medicamentos producidos por un proceso continuo. En este caso, lote se define como una porción especificada e identificada de un “batch” producida durante una unidad de tiempo, en una forma que asegura sus características de uniformidad y calidad dentro de límites especificados. Dicho simplemente, el término lote no es equivalente a “batch” cuando éste se emplea para referirse a una porción de este último.
35.Lote, número de ( Lot number)Designación (en número y/o en letras) del medicamento, que permite identificar al lote al que éste pertenece y que, en caso de necesidad, permite localizar y revisar todas las operaciones de fabricación e inspección practicadas durante su producción.
36.Lote de Producción (Production lot)Lote destinado para los fines usuales de venta o distribución.
37.Marca de medicamento (drug name)Nombre que, en contraposición del nombre genérico o común, distingue un determinado medicamento, de propiedad o uso exclusivo de un laboratorio de producción y protegido por la ley por un período determinado de tiempo.
38.Medicamento (Drug)En el uso legal y técnico, éste término recibe dos acepciones1) En la primera, el término medicamento se emplea para describir al principio
activo o fármaco, que para su administración debe formularse.2) En la segunda de ellas, se entiende por medicamento todo producto
farmacéutico empleado para la prevención, diagnóstico y tratamiento de una enfermedad o estado patológico o para modificar sistemas fisiológicos en beneficio de la persona a quien le fue administrado. En esta acepción el término medicamento se refiere al producto farmacéutico o producto medicamentoso.
- 17 -

En la literatura técnica y legal anglosajona, se hace una distinción muy clara entre estas dos acepciones, al distinguirse entre medicamento como principio activo (drug, drug substance) y medicamento en un producto farmacéutico (drug product, medicine).
En este glosario se recomienda usar medicamento como un término genérico que abarque ambas acepciones y que, cuando sea necesario hacer una distinción entre ellas, se utilicen los términos principales activo y producto farmacéutico o sus sinónimos.
39.Medicamentos FabricadosBienes de la industria farmacéutica sometidos a un proceso productivo en el territorio de cualquiera de los Países Miembros de la Comunidad Andina. Para efectos de esta definición, no se consideran procesos productivos, las operaciones o procesos aplicables a la industria farmacéutica indicados en el artículo 11 de la Decisión 416.
40.Método Analítico (Analytical method)Descripción de una o más técnicas analíticas, en la cual se identifican los recursos materiales, la secuencia de actividades y los procedimientos normalizados de operación.
41.Muestra (Sample)1) Un conjunto o grupo de objetos o cosas seleccionadas de un grupo más
numeroso el cual, según el caso, se denomina “lote” o “población”. La característica esencial de una muestra es su aptitud de ser representativa del lote o población de la cual se obtuvo.
2) Parte o porción finita representativa de un lote de producción o de una cantidad de medicamento almacenada, transportada o en uso que se somete a análisis a efectos de verificar las características de calidad o su adecuación para el uso.
42.Nombre Genérico (Generic name)Nombre empleado para distinguir un principio activo que no está amparado por una marca de fábrica. Es usado comúnmente por diversos fabricantes y reconocido por la autoridad competente para denominar productos farmacéuticos que contienen el mismo principio activo. El nombre genérico se corresponde generalmente con la Denominación Común Internacional recomendada por la OMS.
43.Precaución (precaution)Información incluida en el rótulo del medicamento, dirigida al personal sanitario y al paciente sobre los cuidados que se deben tomar para evitar consecuencias indeseables que podrían resultar de su uso.
44.Prescripción (Prescription)El acto de expresar el o los medicamentos que debe recibir el paciente, la dosificación correcta y duración del tratamiento. En el caso de pacientes ambulatorios, el acto de prescripción se traduce en la elaboración de una receta médica; mientras que en los pacientes hospitalizados, la prescripción es consignada en el registro hospitalario.
- 18 -

45.Presentación (Package size)Se refiere a la naturaleza de los diferentes envases según sus volúmenes o número de unidades del producto farmacéutico que contienen. Por ejemplo: envases de 20 ó 40 comprimidos o envases de diversos volúmenes para productos farmacéuticos líquidos.
46.Principio Activo (Active principle)Dícese de una sustancia o mezcla de sustancias afines dotadas de un efecto farmacológico específico o que, sin poseer actividad, al ser administrados al organismo la adquieren, como es el caso de los profármacos. El término fármaco se utiliza como sinónimo. Sinónimo: ingrediente activo.
47.Producto Terminado (Finished product)Producto farmacéutico que ha pasado por todas las fases de producción y acondicionamiento (llenado, empacamiento y rotulación). Después de ser liberado, el producto terminado constituye el medicamento listo para la venta.
48.Prospecto (Leaflet)Información impresa que se adjunta al medicamento en forma separada y que, generalmente, trata sobre las propiedades, indicaciones y del uso del mismo. Se debe considerar como parte de la rotulación.
49.Reacción adversa (Adverse reaction)1) Reacción nociva o no intencionada y que ocurre en dosis usuales empleadas en
el hombre para la profilaxis, diagnóstico, o tratamiento de enfermedades o para modificar las funciones fisiológicas.
2) Evento clínico adverso atribuido al uso de un medicamento.
50.Registro (Drug lisensing/drug registration)Procedimiento de aprobación por la autoridad sanitaria competente de un país para la comercialización de un medicamento, una vez que el mismo ha pasado el proceso de evaluación. El registro debe establecer el uso específico del medicamento, las indicaciones y contraindicaciones de su empleo, de suerte tal que un cambio en ellas requiera de un nuevo registro. Generalmente, dicha autorización comprende, además, la información que sobre el medicamento se ofrece al cuerpo médico y al público.
51.Rótulo/etiqueta (Labeling/lable)Cualquier leyenda, esceito, marca o prospecto que se adjunta, se incluye dentro, se acompaña o pertenece a cualquier medicamento, cosmético o dispositivo terapéutico. Nótese que esta definición incluye, no solamente el material escrito sobe el envase primario o sobre el paquete que lo contiene, sino, además, material inserto. En otras palabras, no sólo el rótulo (“label”), sino lo que en inglés se describe colectivamente como “labeling”.
52.Seguridad/inocuidad (Safety)Dícese de la característica de un medicamento de poder usarse sin mayores posibilidades de causar efectos tóxicos injustificables. La seguridad de un medicamento, es por lo tanto, una característica relativa y en farmacología clínica su medición es problemática debido a la falta de definiciones operativas o por razones éticas y legales. Sin embargo, mediciones tales como el rango de concentraciones terapéuticas permiten, en ciertos casos, la comparación de la seguridad relacionada
- 19 -

con el uso de determinados medicamentos. Existen otros índices de seguridad que se establecen en animales de laboratorio y que ofrecen cierta utilidad, por ejemplo, dosis letal mediana; índice terapéutico; margen estándar de seguridad, factor de seguridad determinado. Muy acertadamente, se ha propuesto una distinción entre seguridad y toxicidad del medicamento, al indicarse que la toxicidad o inocuidad es una característica intrínseca del medicamento, en tanto que la seguridad es función tanto del medicamento como de las condiciones de su uso.
53.Sobredosis (Overdosage)Dícese de la administración de dosis mayores que lo usual o de dosis usuales administradas a intervalos de dosificación menores que los corrientes, lo cual resulta en la producción de efectos tóxicos del medicamento. La sobredosificación de un medicamento produce los mismos efectos que aquellos observados con dosificaciones normales en pacientes que muestran problemas en la eliminación del medicamento, como ejemplo, disfunción renal.
54.Uso medicamento (Drug use)1) Proceso total de transferencia de medicamentos desde el almacenamiento de
ellos hasta su empleo por el paciente. Incluye las actividades de información y promoción de medicamentos destinadas al paciente.
2) Empleo del medicamento por el paciente.En la bibliografía publicada en los EUA, a diferencia de la publicada por la OMS, se emplean los términos “drug use”, “drug usage” y “drug utilization” indistintamente.
55.Utilización del medicamento (Drug utilization)Comercialización, distribución, prescripción y uso de medicamentos en una sociedad, con acento especial sobre las consecuencias médicas, sociales y económicas resultantes. Recientemente se ha sugerido incluir dentro del concepto de utilización, los procesos de desarrollo de medicamentos. Conviene emplear el término consumo farmacéutico, cuando se desee hacer referencia específica a la demanda de medicamentos.
56.Validación (Validation)1) Acción de probar que cualquier material, proceso, procedimiento, actividad,
equipo o mecanismo empleado en la fabricación o control puede lograr y logrará los resultados para los cuales se destina.
2) La obtención y documentación de evidencia para demostrar la confiabilidad de un método de producir el resultado esperado, dentro de límites definidos.
57.Vencimiento, fecha de/fecha de caducidad (Expiry date/Expiration date)1) La fecha colocada en el empaque inmediato de un producto medicamentoso, el
cual designa la fecha hasta la cual se espera que el producto satisfaga las especificaciones. Esta fecha se establece, para cada lote, mediante la adición del período de vida útil a la fecha de fabricación.
2) Fecha proporcionada por el fabricante de una manera no codificada, que se basa en la estabilidad del producto farmacéutico y después de la cual el mismo no debe usarse.
- 20 -

58.Venta libre (Over the Counter)Un medicamento de venta libre no debe confundirse con aquel que es objeto de una certificación de libre venta dentro del Sistema de Certificación de la calidad de los Productos Farmacéuticos Objeto de Comercio Internacional.
59.Vigilancia (Surveillance)En salud pública, la recolección e interpretación de datos obtenidos a través de programas de monitorización y de cualquier otra fuente, los cuales sirven para detectar y evaluar cambios en la situación sanitaria de una o más poblaciones. La vigilancia requiere del análisis profesional y cuidadoso de los datos y debe resultar en recomendaciones sobre acciones de control.
- 21 -

- 22 -
ANEXO II
GUIA DE APLICACIÓN DE BUENAS PRACTICAS DE MANUFACTURA

- 23 -
INDICE
CAPITULO 1 DATOS INFORMATIVOS NACIONALES
CAPITULO 2 PERSONAL RELACIONADO A LOS PROCESOS PRODUCTIVOS 2.1 Generalidades 2.2 Capacitación 2.3 Higiene de Personal2.4 Dotación de Ropa de Trabajo
CAPITULO 3 INSTALACIONES Y SU MANTENIMIENTO 3.1 Generalidades3.2 Parte Exterior de la Planta3.3 Parte Interior de la Planta3.4 Areas Accesorias (de servicio al personal)
3.4.1 Servicios sanitarios3.4.2 Vestuarios
3.5 Areas de Mantenimiento 3.6 Areas Sociales (Cafetería, lugares de descanso y refrigerio) 3.7 Otras áreas
CAPITULO 4 SISTEMAS DE APOYO CRITICO4.1 Sistemas de Ventilación
Generalidades 4.2 Suministro y manejo de aire en las áreas de producción
4.2.1 A.- Producción Estéril4.2.2 B.- Producción no EstérilEnvasadoEmpaqueAlmacenesLaboratorio de Control Corredores de Circulación adyacentes a las áreas de producción
4.3 Sistema de Agua4.4 Sistema de Vapor industrial

- 24 -
4.5 Sistema de Vapor Limpio4.6 Aire Comprimido – Gases
CAPITULO 5 ALMACENAMIENTO5.1 Materias Primas 5.2 Dispensación o Pesada5.3 Almacenamiento de Materiales de Acondicionamiento y Envase5.4 Almacenamiento de Productos
5.4.1 A.- Intermedios o en Proceso 5.4.2 B.- A Granel
5.5 Productos Terminados
CAPITULO 6 PRODUCCION6.1 Producción no Estéril
6.1.1 Productos Sólidos 6.1.1.1 Areas6.1.1.2 Elaboración de Lotes6.1.1.3 Equipos
6.1.2 Líquidos y semi-sólidos6.1.2.1 Areas6.1.2.2 Equipos6.1.2.3 Elaboración de Lotes
6.2 Producción Estéril 6.2.1 Areas 6.2.2 Mantenimiento y Limpieza 6.2.3 Personal 6.2.4 Equipos y Filtros 6.2.5 Elaboración de Lotes6.2.6 Controles en Proceso
6.3 Productos Especiales6.3.1 Areas 6.3.2 Mantenimiento y Limpieza 6.3.3 Personal 6.3.4 Equipos y Filtros

- 25 -
6.3.5 Elaboración de Lotes6.3.6 Controles en Proceso
6.4 Productos Efervecentes
CAPITULO 7 QUEJAS, RETIROS, DEVOLUCIONES7.1 Quejas y Reclamos7.2 Retiro de Productos del Mercado7.3 Devoluciones
CAPITULO 8 AREA DE EMPAQUE
CAPITULO 9 CONTROL Y USO DE MATERIALES DE ETIQUETADO Y EMPAQUE
CAPITULO 10 DOCUMENTACION 10.1 Documentación 10.2 Documentos Exigidos
10.2.1 Etiquetas y Empaques .10.2.2 Materias Primas y Materiales de Envase Primario
CAPITULO 11 CONTROL DE CALIDAD
CAPITULO 12 GARANTIA DE CALIDAD
CAPITULO 13 SEGURIDAD INDUSTRIAL
CAPITULO 14 ESTABILIDAD
CAPITULO 15 VALIDACIONES15.1 Validación Area Estéril
CAPITULO 16 PROCESAMIENTO ELECTRONICO DE DATOS

- 26 -
GUÍA DE APLICACIÓN DE BUENAS PRÁCTICAS DE MANUFACTURA
CAPITULO 2 - PERSONAL RELACIONADO A LOS PROCESOS PRODUCTIVOS VALOR SI NO NP OBSERVACIONES
2.1 GENERALIDADES
1. Conoce el personal subalterno el organigrama de su departamento con las líneas de autoridad claramente definidas?
Mayor
2. Existen procedimientos que describan las funciones y responsabilidades de todo el personal?
Mayor
3. Todo el personal directamente relacionado con la producción tiene los niveles de educación u de capacitación requeridos por la empresa y adaptados a la ley del país?
Mayor
4. Si existe personal temporal bajo contrato esta disponible la información siguiente:
Listado del personal Tiempo de contratación Tipo de actividades a desempeñar
Menor
2.2 CAPACITACION
1. Existen programas anuales de cursos de capacitación continua del personal a todos los niveles de departamentos y secciones?
Mayor
2. El programa de entrenamiento está aprobado por personal calificado? Mayor
3. Existe un programa de capacitación para empleados nuevos y personal temporal? Mayor
4. Se capacita al personal sobre:
Normas BPF Comportamiento dentro de las áreas de producción Manejo de equipos e instrumentos de trabajo Uso correcto de ropas y accesorios de trabajo Aspectos básicos de microbiología Higiene personal
Mayor

- 27 -
CAPITULO 2 - PERSONAL RELACIONADO A LOS PROCESOS PRODUCTIVOS VALOR SI NO NP OBSERVACIONES
5. La presencia del personal en los cursos de entrenamiento está avalada con sus firmas?
Mayor
6. Se documentan estas actividades? Mayor
7. El personal nuevo es adecuadamente supervisado hasta comprobar que realiza el trabajo correctamente?
Mayor
8. Se dejan registros de su evaluación? Mayor
9. Existen procedimientos escritos para el ingreso de los visitantes a la planta? Mayor
2.3 HIGIENE DEL PERSONAL
1. Se realiza un examen médico y de laboratorio para el ingreso del personal a la empresa?
Crítico
2. Cuáles exámenes se realizan Informativo
3. Se documentan? Mayor
4. Se realiza un examen médico y de laboratorio al personal de planta de acuerdo a un procedimiento establecido, por lo menos una vez al año?
Mayor
5. Cuáles exámenes son realizados? Informativo
6. Con qué frecuencia? Informativo
7. Se documentan? Mayor
8. Existen normas escritas de higiene que incluyan la prohibición de comer, beber, y fumar dentro de las áreas de producción
Crítico
9. Se evita el contacto directo de las manos del operario con materias primas materiales de envase primario y productos intermedios y a granel?
Crítico
10. Existen carteles alusivos a las normas de higiene a observar especialmente referidas al lavado de las manos?
Menor
11. Las personas que muestran signos de enfermedades infecto - contagiosas o sufren lesiones abiertas son inhabilitadas para trabajar en las diferentes áreas de producción incluyendo el envasado en el acondicionamiento primario, hasta tanto se hayan restablecido?
Crítico

- 28 -
CAPITULO 2 - PERSONAL RELACIONADO A LOS PROCESOS PRODUCTIVOS VALOR SI NO NP OBSERVACIONES
12. Las normas de higiene establecidas son cumplidas por todas las personas que ingresan a las áreas de producción, se trate de empleados temporales o permanentes, o no empleados (contratistas, visitantes administradores , inspectores, etc.)?
Crítico
13. El personal de mantenimiento cumple las normas de higiene? Mayor
14. El personal de mantenimiento cumple las normas de comportamiento dentro de la áreas de producción?
Crítico
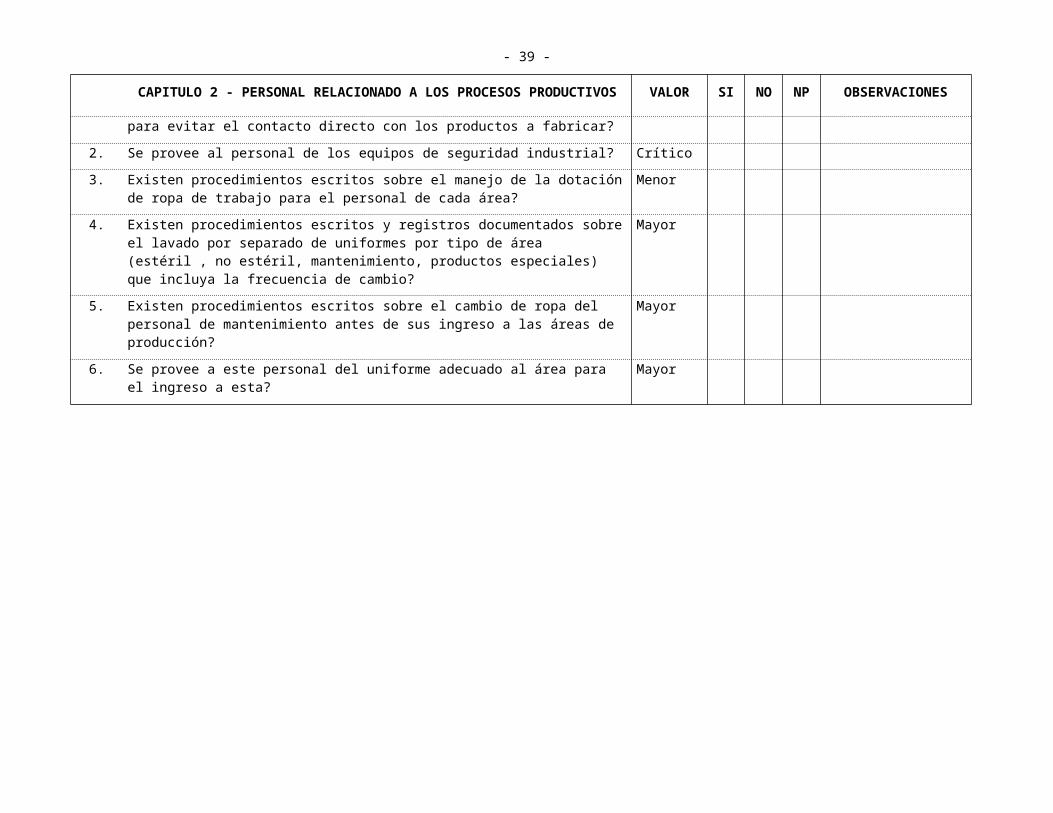
2.4 DOTACIÓN DE ROPA DE TRABAJO
1. Se provee al personal (temporal y fijo) de la vestimenta de trabajo adecuada para cada área, incluyendo los accesorios para evitar el contacto directo con los productos a fabricar?
Crítico
2. Se provee al personal de los equipos de seguridad industrial? Crítico
3. Existen procedimientos escritos sobre el manejo de la dotación de ropa de trabajo para el personal de cada área?
Menor
4. Existen procedimientos escritos y registros documentados sobre el lavado por separado de uniformes por tipo de área (estéril , no estéril, mantenimiento, productos especiales) que incluya la frecuencia de cambio?
Mayor
5. Existen procedimientos escritos sobre el cambio de ropa del personal de mantenimiento antes de sus ingreso a las áreas de producción?
Mayor
6. Se provee a este personal del uniforme adecuado al área para el ingreso a esta? Mayor

- 29 -
CAPITULO 3 – INSTALACIONES Y SU MANTENIMIENTO VALOR SI NO NP OBSERVACIONES
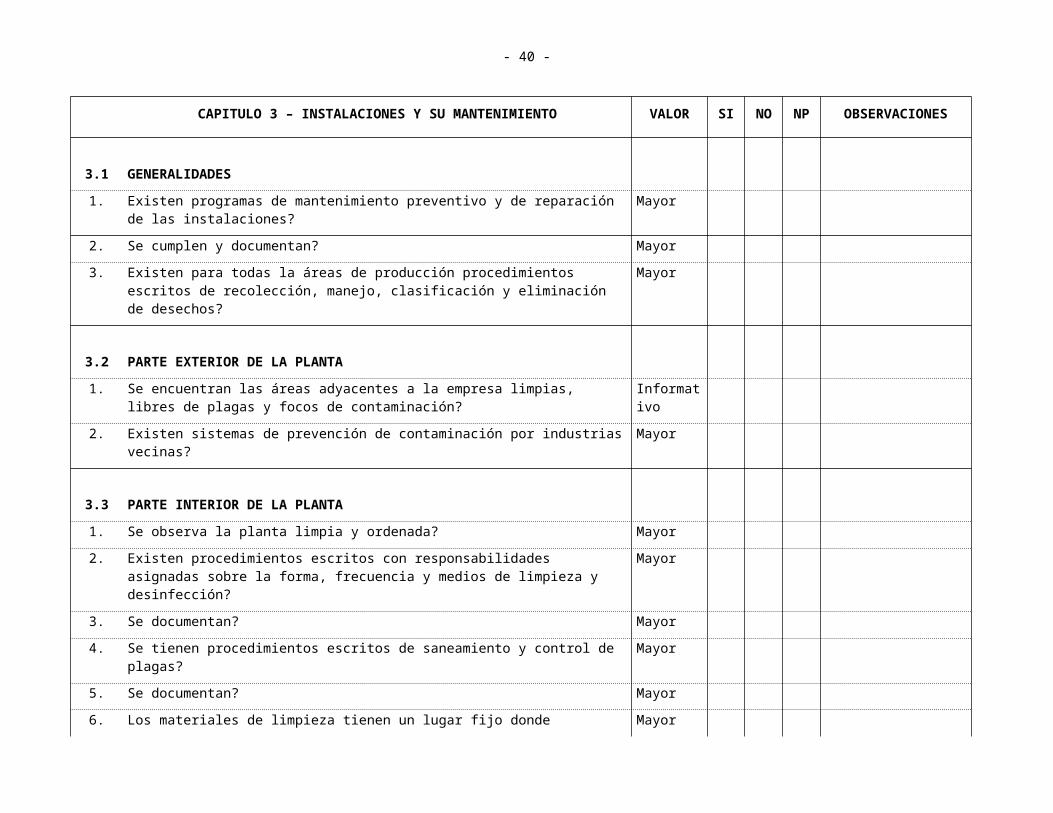
3.1 GENERALIDADES
1. Existen programas de mantenimiento preventivo y de reparación de las instalaciones?
Mayor
2. Se cumplen y documentan? Mayor
3. Existen para todas la áreas de producción procedimientos escritos de recolección, manejo, clasificación y eliminación de desechos?
Mayor
3.2 PARTE EXTERIOR DE LA PLANTA
1. Se encuentran las áreas adyacentes a la empresa limpias, libres de plagas y focos de contaminación?
Informativo
2. Existen sistemas de prevención de contaminación por industrias vecinas? Mayor
3.3 PARTE INTERIOR DE LA PLANTA
1. Se observa la planta limpia y ordenada? Mayor
2. Existen procedimientos escritos con responsabilidades asignadas sobre la forma, frecuencia y medios de limpieza y desinfección?
Mayor
3. Se documentan? Mayor
4. Se tienen procedimientos escritos de saneamiento y control de plagas? Mayor
5. Se documentan? Mayor
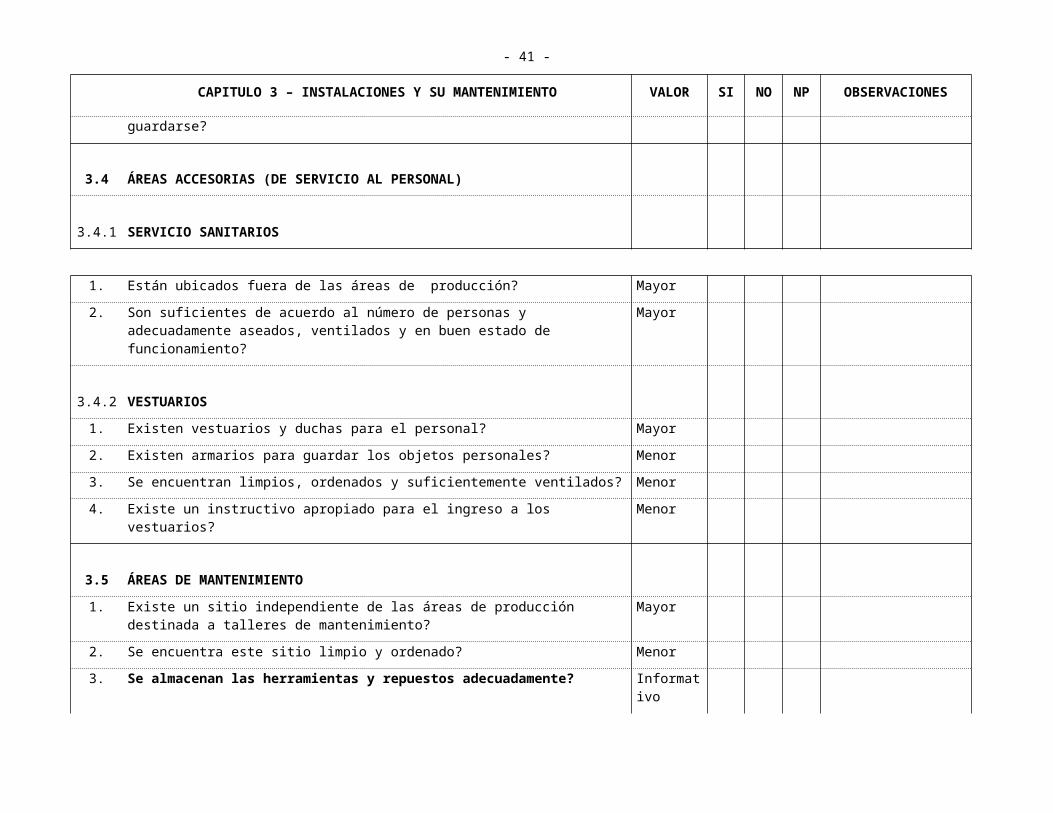
6. Los materiales de limpieza tienen un lugar fijo donde guardarse? Mayor
3.4 ÁREAS ACCESORIAS (DE SERVICIO AL PERSONAL)
3.4.1 SERVICIO SANITARIOS

- 30 -
CAPITULO 3 – INSTALACIONES Y SU MANTENIMIENTO VALOR SI NO NP OBSERVACIONES
1. Están ubicados fuera de las áreas de producción? Mayor
2. Son suficientes de acuerdo al número de personas y adecuadamente aseados, ventilados y en buen estado de funcionamiento?
Mayor
3.4.2 VESTUARIOS
1. Existen vestuarios y duchas para el personal? Mayor
2. Existen armarios para guardar los objetos personales? Menor
3. Se encuentran limpios, ordenados y suficientemente ventilados? Menor
4. Existe un instructivo apropiado para el ingreso a los vestuarios? Menor
3.5 ÁREAS DE MANTENIMIENTO
1. Existe un sitio independiente de las áreas de producción destinada a talleres de mantenimiento?
Mayor
2. Se encuentra este sitio limpio y ordenado? Menor
3. Se almacenan las herramientas y repuestos adecuadamente? Informativo
3.6 ÁREAS SOCIALES (CAFETERÍA, LUGARES DE DESCANSO Y REFRIGERIO)
1. Están separadas de las áreas de producción? Mayor
2. Se ingresa a esta áreas sin el uniforme de trabajo? Mayor
3.7 OTRAS ÁREAS
1. Los lugares destinados a los animales están aislados de las áreas de producción con entrada separada (acceso de animales exclusivamente) y cuentan con equipo aparte de suministro de aire?
Crítico

- 31 -
CAPITULO 4 – SISTEMAS DE APOYO CRITICO VALOR SI NO NP OBSERVACIONES
4.1 SISTEMAS DE VENTILACIÓNGENERALIDADES
1. Las áreas están adecuadamente ventiladas y si es necesario climatizadas? Mayor
2. Las áreas están clasificadas de acuerdo a la clase de limpieza del aire suministrado?
Mayor
3. Existen registros del mantenimiento de los equipos de climatización? Mayor
4. Existen procedimientos escritos para el manejo de posibles condiciones de riesgo en los sistemas de aire?
Mayor
5. Existen presiones diferenciales entre áreas adjuntas positiva / negativa? Mayor
6. Son monitoreados estos diferenciales de presión? (Lectura, registros) Mayor
7. El diseño de los filtros finales cumple los requerimientos de eficiencia establecidos para cada área de manufactura?
Mayor
8. La eficiencia de los filtros es la adecuada para cada tipo de área? Crítico
9. La ubicación de las entradas y salidas de aire permiten un flujo eficiente de aire en las áreas controladas?
Mayor
10. Se mide la velocidad del flujo en las áreas controladas? Mayor
11. Existen prefiltros instalados para proteger a los filtros finales? Informativo
12. Existe un sistema de advertencia que indique las faltas del sistema ( ejm., caída de presión)?
Mayor
13. En el caso de sustancias altamente activas (tóxicas) y beta – lactámicos, existe un adecuado sistema de retención de polvo (filtros, hepa, ciclones?
Crítico
4.24.2.1
SUMINISTRO Y MANEJO DE AIRE EN LA AREAS DE PRODUCCIÓNA. PRODUCCIÓN ESTÉRIL

- 32 -
CAPITULO 4 – SISTEMAS DE APOYO CRITICO VALOR SI NO NP OBSERVACIONES
1. Existe una unidad de manejo de aire (uma) exclusiva para esta área? Crítico
2. El aire es recirculado (tiene retorno)? Informativo
3. Cuál es el porcentaje de retorno? Informativo
4. Se han establecido los diferenciales de presión entre las áreas adjuntas? Informativo
4.2.2 B. PRODUCCIÓN NO ESTÉRIL
1. Existe una unidad de manejo de aire (UMA) individual o central para las áreas de producción no especial donde no se generen polvos?
Informativo
2. Indique cuáles áreas tienen una UMA común? Informativo
3. Se han establecido los diferenciales de presión entre las áreas adjuntas? Informativo
4. Si la UMA es central, se asegura la no contaminación cruzada? Crítico
ENVASADO
1. Tiene sistema central o individual? Informativo
2. Qué tipo de diferencial tiene respecto a las áreas adjuntas positivo / negativo? Informativo
3. El sistema empleado y los diferenciales de presión establecidos garantizan la no contaminación cruzada?
Crítico
EMPAQUE
1. Posee sistema central o individual? Informativo
2. Qué tipo de diferencial tiene respecto a las áreas adjuntas? Informativo
3. Se toman las medidas para evitar la contaminación cruzada hacia las áreas de producción?
Mayor
ALMACENES
1. Tiene ventilación con extracción? Informativo

- 33 -
CAPITULO 4 – SISTEMAS DE APOYO CRITICO VALOR SI NO NP OBSERVACIONES
2. Tiene aire acondicionado central o individual? Informativo
3. El sistema escogido permite alcanzar las condiciones adecuadas de almacenamiento?
Mayor
LABORATORIO DE CONTROL
1. Tiene sistemas de aire acondicionado central o individual en las áreas donde se requiera?
Mayor
2. El sistema escogido garantiza la no contaminación del personal ni de los productos? Mayor
CORREDORES DE CIRCULACIÓN ADYACENTES A LAS AREAS DE PRODUCCION.
1. Tienen sistema central o individual? Informativo
2. El sistema establecido garantiza los diferenciales de presión adecuados Mayor
4.3 SISTEMA DE AGUA
1. tipos de agua suministrada a la planta? De acueducto? De pozo? Red municipal? Otros?
Informativo
2. La calidad química y microbiológica del agua que entra es la apropiada? Informativo
3. Para qué utiliza cada tipo de agua? Informativo
4. Poseen tanques o cisternas de almacenamiento de agua? Informativo
5. Existen procedimientos documentados de limpieza y desinfección de los tanques y cisternas de agua?
Crítico
6. Qué clase de agua utilizan en producción? Potable? Purificada? Para inyectables?
Informativo

- 34 -
CAPITULO 4 – SISTEMAS DE APOYO CRITICO VALOR SI NO NP OBSERVACIONES
7. Qué procesos de purificación de agua utilizan? Desionización? Destilación? Bidestilación? Osmosis Inversa? Otros?
Informativo
8. El equipo es idóneo para la obtención del agua purificada? Crítico
9. El agua en los puntos curvos (loops) está en permanente recirculación, calentada o enfriada?
Informativo
10. Es adecuado el material de las tuberías de conducción del agua para los diferentes tipos de productos:
Productos estériles (acero inoxidable)? Preparaciones líquidas (inoxidable u otro material idóneo)? Preparaciones semisólidas (inoxidables u otro material idóneo)? Preparaciones sólidas (inoxidable u otro material idóneo)?
Crítico
11. Son sanitizados los puntos de salida de agua que se utiliza para la fabricación? Crítico
12. Si se sanitizan con luz UV se mide la actividad en ésta? Crítico
13. Con qué frecuencia? Informativo
14. Están debidamente identificados los puntos de muestreo? Mayor
15. Existe un plan de muestreo escrito que especifique cuándo y cómo tomar las muestras e incluya el diseño del equipo con los puntos de muestreo?
Mayor
16. Existe un programa documentado para el control del sistema de agua indicando los controles físico – químicos y microbiológicos, los cambios de filtro, las regeneraciones, desinfecciones y la esterilización?
Mayor
17. Se realizan los siguientes controles: Físico químicos? Microbiológicos?
Crítico
18. Se realizan controles Fisicoquímico, Microbiológicos y de Endotoxinas o piógenos para el agua PPI?
Crítico
19. Se registran? Crítico
20. Existe un registro de las acciones correctivas si se exceden los limites de alerta y de acción durante las actividades de monitoreo?
Crítico

- 35 -
CAPITULO 4 – SISTEMAS DE APOYO CRITICO VALOR SI NO NP OBSERVACIONES
4.4 SISTEMA DE VAPOR INDUSTRIAL
1. tienen suministro de vapor? Informativo
2. Que tipo de caldera usa? Informativo
3. Se efectúan controles al agua que alimenta la caldera? Informativo
4. En qué se utiliza el vapor? Informativo
5. Se efectúan controles de calidad al vapor generado que entra en contacto con el producto?
Crítico
6. Cuáles controles son realizados? Informativo
7. Qué tipo de combustible utiliza? Informativo
8. Qué manejo tienen los residuos de la caldera? Informativo
4.5 SISTEMA DE VAPOR LIMPIO
1. El equipo de producción de vapor limpio es adecuado? Crítico
2. El vapor limpio es obtenido a partir del sistema de agua PPI? Crítico
3. Existe un programa escrito de control para el agua de alimentación y para el vapor limpio?
Mayor
4. El programa incluye ensayos microbiológicos (endotoxinas) y de partículas? Crítico
5. El vapor limpio se utiliza para la esterilización de materiales y partes de máquinas usadas para la manufactura estéril y aséptica?
Informativo
6. Los puntos de agua (loops) desmineralizada y destilada son esterilizados por medio del vapor limpio?
Informativo
4.6 AIRE COMPRIMIDO – GASES
1. Existe un equipo adecuado para la obtención de aire comprimido? Informativo
2. En el aire comprimido y los gases se controlan periódicamente la ausencia de aceite, humedad, herrumbre?
Mayor

- 36 -
CAPITULO 5 – ALMACENAMIENTO VALOR SI NO NP OBSERVACIONES
5.1 MATERIAS PRIMAS1. Se aplica el sistema tradicional o el informático? Informativo
2. En caso de poner en práctica el sistema tradicional se cuenta con espacios suficientes debidamente separado e identificados para:
Recepción? Area de cuarentena? Almacenamiento de materias primas aprobadas Almacenamiento de insumos a conservar a bajas temperaturas (cámaras
frías)? Almacenamiento de sustancias estupefacientes y psicotrópicos (bajo llave)? Almacenamiento de sustancias inflamables y explosivas? Estos últimos están convenientemente alejados de los demás y construidos
idóneamente? Se dispone de un sistema de protección adecuado para manejar material
inflamables?
Mayor
3. En caso de usar sistema informático esta convenientemente validado para ofrecer condiciones de seguridad equivalente al sistema tradicional?
Mayor
4. Las materias primas sensibles a la humedad y temperatura se almacenan en condiciones controladas?
Crítico
5. Se registran los controles? Crítico
6. El área de recepción está equipada para permitir la limpieza de los recipientes antes de su almacenamiento?
Mayor
7. El acceso al almacén esta restringido a personal no autorizado? Mayor
8. Todas las áreas se encuentran limpias, ordenadas y sin deterioro:
Estanterías Paredes Pisos Techos Ventanas Puertas
Mayor

- 37 -
CAPITULO 5 – ALMACENAMIENTO VALOR SI NO NP OBSERVACIONES
Parihuelas, paletas, estibas?9. Existe ventilación e iluminación adecuadas? Mayor
10. Las materias primas se encuentran almacenadas en estanterías de tal manera que permitan la revisión y limpieza?
Mayor
11. Se usan parihuelas, paletas, estibas? Informativo
12. De qué material? Informativo
13. Las materias primas están identificadas en forma tradicional o mediante cualquier otro sistema que permita obtener la información siguiente:
Nombre? Código? Número de lote o análisis? Fecha de recepción? Fecha de análisis? Concentración porcentual (si procede)? Fecha de Vencimiento? Fecha de reanálisis? Número de recipientes?
Crítico
14. Las materias primas se utilizan de acuerdo al sistema?
FIFO/FEFO
Mayor
15. Están las materias primas y materiales de envase y empaque localizados e identificados de acuerdo al estado en que se encuentren (aprobado. Cuarentena, rechazado)?
Crítico
16. Existe un sistema que registre y controle la recepción y el manejo de las materias primas
Mayor
17. Existen procedimientos escritos para la recepción y el rotulado? Mayor
18. Se aplican las medidas que garanticen la inutilización de los rótulos de los envases de materias primas utilizadas, antes de reutilizarlos o desecharlos?
Menor
19. Si se reutilizan los envases se toman las medidas que aseguren su absoluta limpieza?
Crítico
20. Existen instructivo de almacenamiento y manipulación de cada una de las materias primas Estas fichas son conocidas y manejadas por el personal de almacenamiento y pesada?
Mayor

- 38 -
CAPITULO 5 – ALMACENAMIENTO VALOR SI NO NP OBSERVACIONES
21. Existen recipientes adecuados para la recolección de residuos? Menor
5.2 DISPENSACIÓN O PESADA
1. Existe un área de pesada independiente, limpia y ordenada? Crítico
2. Tiene ángulos sanitarios? Menor
3. Está iluminada y con sistema de extracción de aire debidamente ubicado? Mayor
4. Se dispensa contra orden de producción? Crítico
5. Existen procedimientos escritos para?
Manejo de insumos, utensilios empleados?Limpieza después de cada operación, se documenta?
Mayor
6. Se dispone de balanzas de rango y sensibilidad adecuados para las cantidades a pesar en ellas?
Crítico
7. Se cuenta con un programa de calibración de balanzas y se llevan los registros con calibraciones al menos una vez al año?
Crítico
8. Se verifica el estado de las balanzas diariamente antes de iniciar las labores de pesada y se registra?
Mayor
9. Se registran individualmente las materias primas dispensadas para cada orden de producción?
Crítico
10. Se pesan y disponen separadamente cada una de las órdenes de producción? Crítico
11. Se lleva un registro diario de las órdenes de producción pesadas y su secuencia? Menor
12. Utiliza el personal ropa y elementos de protección adecuados para el pesado de materias primas?
Mayor
13. Cuenta con un sitio especial para almacenar materias primas pesadas? Menor
14. Se transportan y entregan adecuadamente las materias primas dispensadas al área de producción en dispositivos fáciles de lavar y que no desprendan partículas?
Mayor
5.3 ALMACENAMIENTO DE MATERIALES DE ACONDICIONAMIENTO Y ENVASE
- 39 -
CAPITULO 5 – ALMACENAMIENTO VALOR SI NO NP OBSERVACIONES
1. Se encuentran las áreas debidamente identificadas? Menor
2. Son adecuadas las condiciones de limpieza, orden y mantenimiento de: Paredes? Pisos? Techos? Estanterías? Parihuelas, paletas, estibas? Máquinas?
Menor
3. Están iluminadas, ventiladas y con controles de humedad y temperatura si se requiere?
Menor
4. Se registran los controles? Menor
5. Existen procedimientos escritos para la recepción, manejo? Mayor
6. Los materiales de acondicionamiento y envase están identificados en forma tradicional o mediante cualquier otro sistema que permita obtener la información siguiente:
Código? Numero de análisis y / o lote? Fecha de recepción? Fecha de análisis? Número de unidades recibidas?
Mayor
7. Están los materiales identificados con etiquetas y localizados de acuerdo con el estado en que se encuentran:
Cuarentena? Aprobado? Rechazado?
Crítico
8. Están las etiquetas almacenadas en condiciones seguras, evitando el acceso a personas no autorizadas?
Crítico
5.4 ALMACENAMIENTO DE PRODUCTOS:
5.4.1 A - INTERMEDIOS O EN PROCESO.
1. Existen áreas separadas, por forma farmacéutica para este tipo de productos? Mayor
- 40 -
CAPITULO 5 – ALMACENAMIENTO VALOR SI NO NP OBSERVACIONES
2. Las áreas están correctamente identificadas? Menor
3. Están limpias y ordenadas? Mayor
4. Están iluminadas y ventiladas adecuadamente? Menor
5. Están construidas de materiales fáciles de limpiar? Mayor
6. Los productos se identifican correctamente colocando una etiqueta en el cuerpo del envase externo y otra en el envase que lo contiene?
Crítico
7. Existe una etiqueta que identifique el estado del producto: Muestreado? Aprobado? Bloqueado? En investigación? Otro
Crítico
5.4.2 B – A GRANEL
8. Los productos se identifican correctamente colocando una etiqueta en el cuerpo del envase externo y otra en el envase que lo contiene?
Crítico
9. Existe una etiqueta que identifique el estado del producto: Muestreado? Aprobado? Bloqueado? En investigación? Otro?
Crítico
10. Puede verificarse que los productos se almacenan de acuerdo a sus especificaciones?
Mayor
5.5 PRODUCTOS TERMINADOS
1. Los almacenes están debidamente identificados? Menor
2. Se encuentran limpios y ordenados? Menor
3. Las condiciones de almacenamiento son adecuadas a las especificaciones del producto impresas en el empaque?
Mayor
- 41 -
CAPITULO 5 – ALMACENAMIENTO VALOR SI NO NP OBSERVACIONES
4. Se realizan controles de temperatura y humedad cuando corresponda? Crítico
5. Se registran? Crítico
6. Son adecuadas las condiciones de limpieza, orden y mantenimiento de:
Paredes? Pisos? Techos? Estanterías? Parihuelas, paletas, estibas?
Menor
7. Los productos terminados se encuentran debidamente ordenados? Informativo
8. Se despachan los productos terminados de acuerdo a sus fecha de vencimiento (primero vence, primero sale??
Mayor
9. Existe un área especial para el almacenamiento de producto terminados de control especial? (psicotrópicos, estupefacientes, etc.)
Menor
10. Existen áreas separadas para el almacenamiento de:
Productos devueltos o retirados del mercado? Productos rechazados?
Mayor
11. Existe un área de despacho adecuadamente equipada? Menor
12. Existen instrucciones de almacenamiento, transporte y manipulación de cada uno de los productos terminados que requieran tratamiento especial?
Mayor
13. Estas instrucciones son conocidas y utilizadas por el personal de almacenamiento, despacho y transporte?
Mayor
14. Existen recipientes adecuados (resistentes, con tapa e identificados) para la recolección de residuos?
Informativo
15. Se cumplen los procedimientos escritos para el retiro y destrucción de productos vencidos?
Mayor
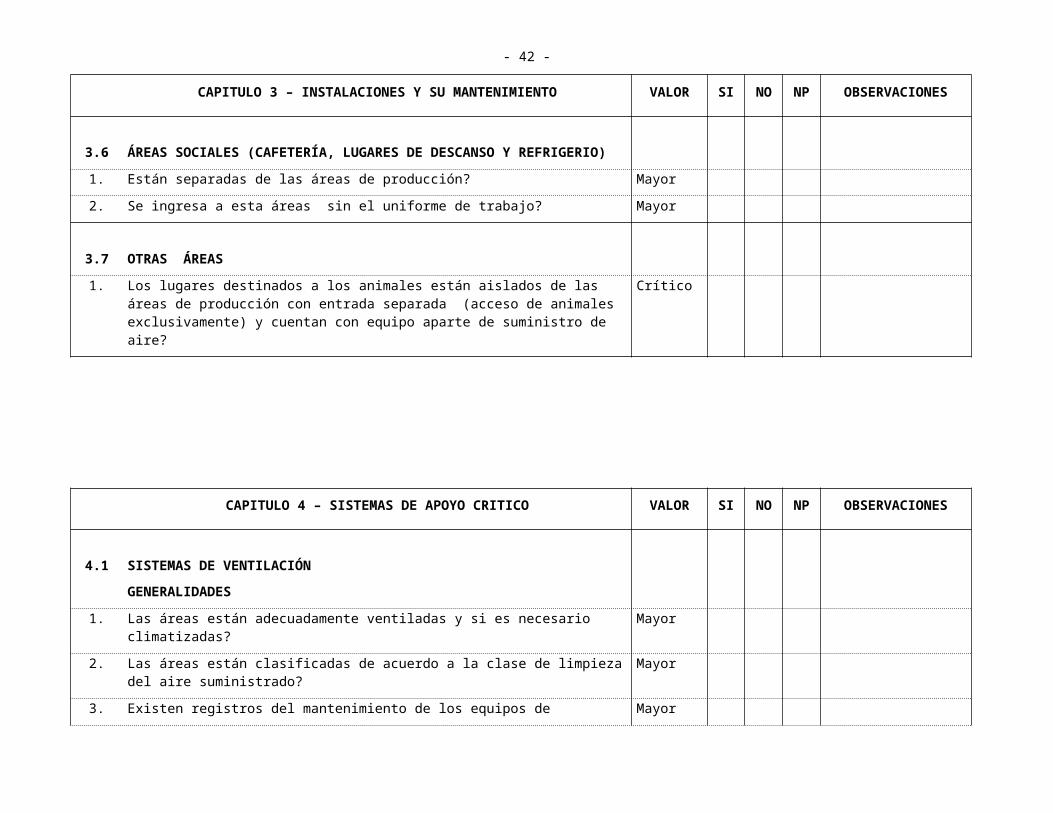
- 42 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
6.1 PRODUCCIÓN NO ESTERIL6.1.1
6.1.1.1PRODUCTOS SÓLIDOSÁREAS
1. Están las áreas definidas e identificadas para cada tipo de operación de manufactura:
Mezclado? Granulado? Secado? Tableteado? Grageado? Otros?
Mayor
2. Se procesa cada producto por vez y por área y se aplican mecanismos para evitar la contaminación cruzada?
Crítico
3. Las áreas antes del inicio de cada proceso, están limpias sanitizadas y ordenadas? Crítico
4. Existen procedimientos escritos de limpieza y sanitización? Mayor
5. Existen registros? Mayor
6. Dispone el área de sólidos de sitios destinados para:
Materias primas dispensadas? Materiales para uso de fabricación? Productos en proceso? Lavado de útiles y equipos de producción? Lavado y almacenamiento de material e implementos de aseo?
Mayor
7. Las áreas están construidas de materiales fáciles de limpiar:
Paredes? Puertas? Pisos? Techos?
Mayor
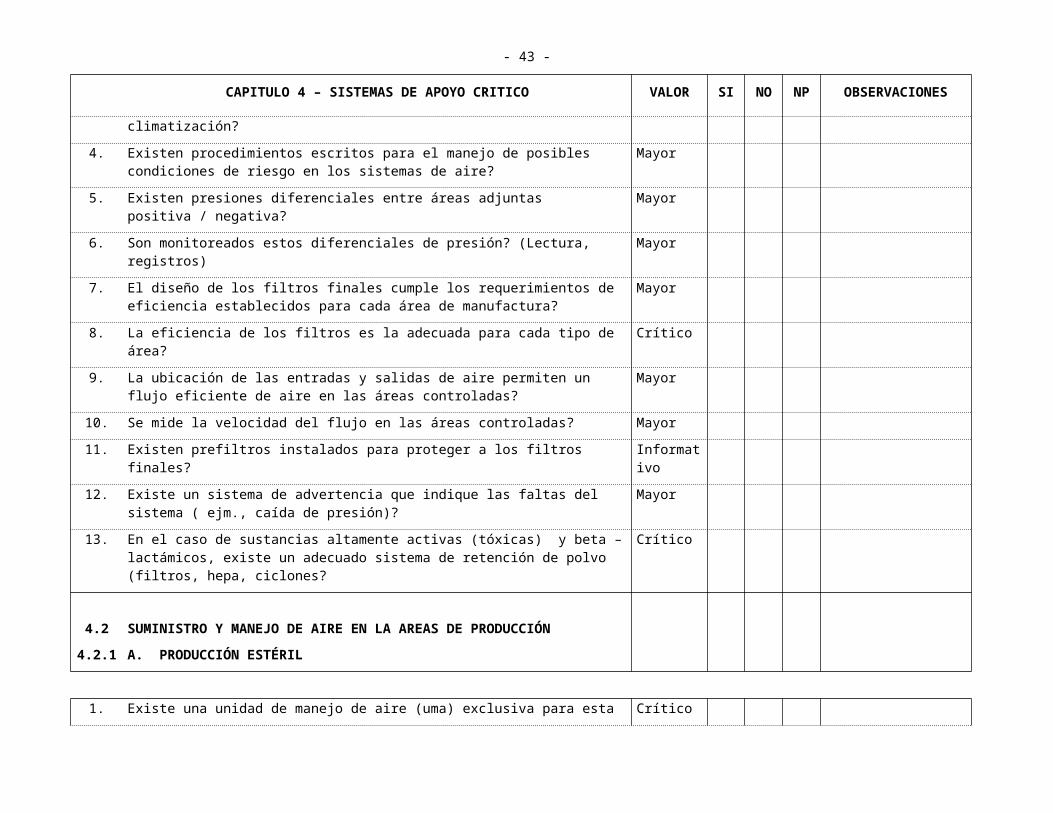
- 43 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
Ventanas? Lámparas? Rejillas de aire? Ductos y tuberías a la vista?
8. Disponen de drenajes y sifones protegidos? Mayor
9. Tienen ángulos sanitarios? Mayor
10. Están las áreas suficientemente iluminadas? Mayor
11. Si deben ser controladas (temperatura y humedad relativa), se tienen los registros? Mayor
12. Existe un sistema de medición de los diferenciales de presión? Mayor
13. Los diferenciales de presión son los adecuados? Mayor
6.1.1.2 ELABORACIÓN DE LOTES1. Se tienen órdenes de producción e instrucciones de manufactura para cada lote? Crítico
2. Se siguen las instrucciones de manufactura? Crítico
3. Se realizan los controles correspondientes, durante los procesos? Crítico
4. Se dispone de procedimientos escritos de estos controles? Crítico
5. Se deja registro escrito en las instrucciones de manufactura? Crítico
6. La producción tiene fases de campaña? Anexe listado de productos con fechas de fabricación.
informativo
6.1.1.3 EQUIPOS1. Son construidos de un material que no reacciona con el producto? Crítico
2. Tienen los dispositivos de seguridad en el caso que lo requieran? Mayor
3. Las partes en contacto con el producto pueden limpiarse fácilmente? Crítico

- 44 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
4. Existen los registros de limpieza? Mayor
5. Los equipos están debidamente identificados según su estado de limpieza (equipo limpio / equipo sucio)?
Mayor
6. La etiqueta de identificación menciona el producto anteriormente elaborado en él y su número de lote?
Mayor
7. Los operarios disponen de las instrucciones en español para el manejo y control de los equipos?
Mayor
8. Están las áreas de los equipos en uso identificados con:
Nombre del producto en elaboración? Número del lote? Fecha?
Mayor
9. Si se elaboran productos con sustancias inflamables o explosivas, se cuenta con equipos apropiados para prevenir accidentes?
Crítico
10. Las áreas para estas preparaciones tienen sistemas eléctricos y de extracción apropiados?
Mayor
11. Los sistemas de extracción son eficientes? Mayor
12. Se observa exceso de polvo en el ambiente? Informativo
13. El aire extraído es filtrado antes de salir al ambiente? Mayor
14. Se dispone de un programa escrito de calibración de equipos que lo requieran? Mayor
15. Existen procedimientos escritos? Mayor
16. Se registran los controles? Mayor
6.1.26.1.2.1
LÍQUIDOS Y SEMI – SÓLIDOSAREAS
1. Existen áreas definidas e identificadas para cada tipo de operación de manufactura: Líquidos / jarabes y suspenciones Cremas y ungüentos? Ovulos y supositorios?
Mayor

- 45 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
Otros?
2. Se procesa un producto por vez y se aplican mecanismos para evitar la contaminación cruzada?
Crítico
3. La producción tiene fases de campaña? Anexe listado de productos con la época del año que serán fabricados?
Informativo
4. Las áreas antes del inicio de cada proceso están limpias sanitizadas y ordenadas? Mayor
5. Existen procedimientos escritos de limpieza y sanitizacion? Mayor
6. Existe rotación de desinfectantes? Mayor
7. Se realizan controles microbiológicos del ambiente durante los procesos de fabricación según cronograma?
Mayor
8. Se registran? Mayor
9. Dispone el área de sitios destinados para? Materias primas dispensadas Materiales para uso de fabricación Productos en proceso? Lavado y almacenamiento de materiales e implementos de limpieza y
desinfección?
Mayor
10. Las áreas están construidas de material de fácil limpieza en Puertas? Pisos? Techos? Ventanas? Lámparas? Rejillas de aire? Ductos y tuberías?
Mayor
11. Disponen de drenaje y sifones protegidos? Mayor
12. Tienen ángulos sanitarios? Menor
13. Están las áreas suficientemente iluminados? Menor
6.1.2.2 EQUIPOS
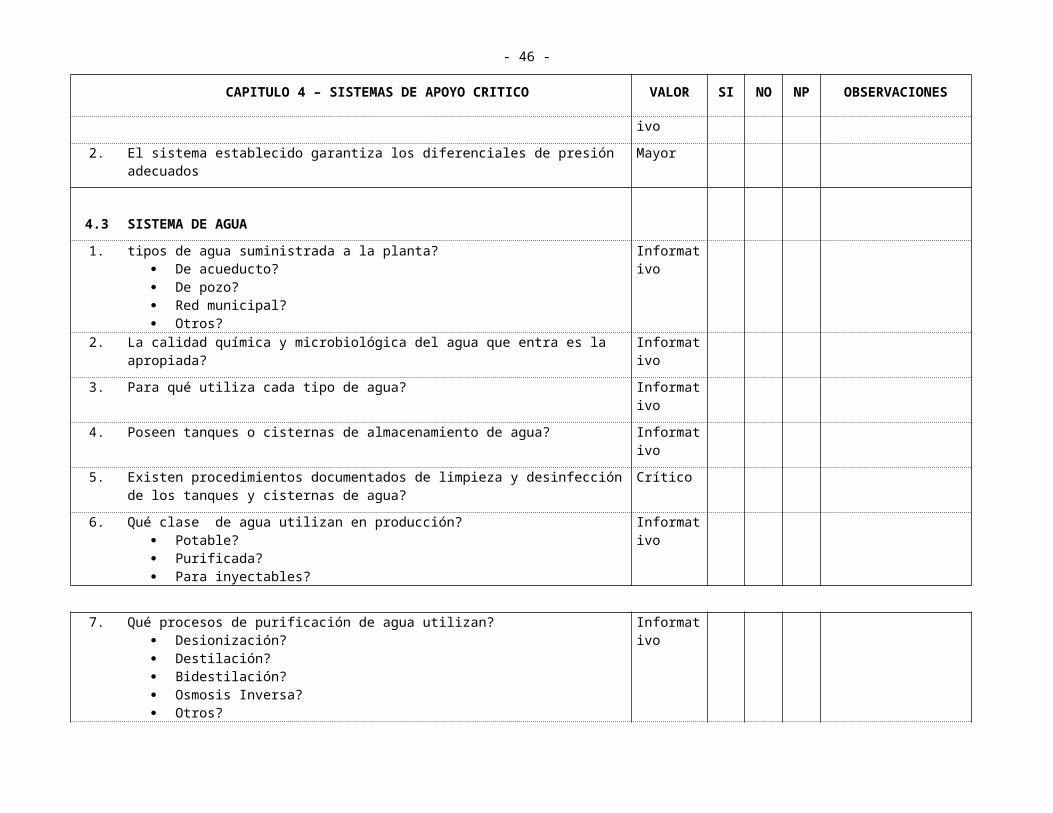
- 46 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
1. Están constituidos de un material que no reacciona con el producto? Crítico
2. Tienen los dispositivos de seguridad en caso que lo requieran? Mayor
3. Las partes en contacto con el producto pueden limpiarse completamente? Crítico
4. Existen los registros de limpieza? Mayor
5. Los equipos están identificados según su estado de limpieza (equipo limpio/equipo sucio)?
Mayor
6. La etiqueta de identificación menciona el producto anteriormente elaborado y su número de lote?
Mayor
7. Las áreas de los equipos donde se elabora el producto están identificados con: Nombre del producto? Número de lote? Fecha?
Mayor
8. Los tanques de preparación son cerrados con tapas adecuadas de acuerdo con el producto?
Mayor
9. Si se elaboran productos que requieran equipos a prueba de explosión, se cuenta con los equipos adecuados?
Crítico
10. Estos equipos están ubicados en áreas con sistemas eléctricos y de extracción apropiados?
Mayor
11. Se dispone de un programa escrito de calibración para equipos que lo requieran? Mayor
12. Existen los registros de calibraciones? Mayor
6.1.2.3 ELABORACION DE LOTES
1. Se tienen órdenes de producción e instrucciones de manufactura para cada lote? Crítico
2. Se siguen las instrucciones de manufactura? Crítico
3. Si la producción tiene fases de campaña, anexe listado de productos con las fechas de fabricación?
Informativo
4. Se realizan los controles correspondientes durante el proceso? Crítico
5. Se dispone de procedimientos escritos para estos controles? Crítico

- 47 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
6. Los resultados se dejan registrados en las instrucciones de manufactura? Crítico
7. Los ductos y las tuberías existentes en área, están identificadas de acuerdo a las normas de cada país?
Mayor
6.2
6.2.1
PRODUCCIÓN ESTÉRILÁREAS
1. El área dispone de sitios especialmente destinados para: Materias primas dispensadas? Materiales para uso de fabricación? Lavado de utensilios y equipos de producción? Lavado de envases primarios? Esterilización de envases primarios? Almacenamiento de envases primarios? Almacenamiento de envases limpios y estériles?
Crítico
Fabricación de productos’ Llenado de productos? Productos en proceso? Revisión y control óptico? desinfección de las áreas? Cuarentena de productos? Lavado y almacenamiento de material e implementos de limpieza
2. Están identificadas las áreas para cada operación de manufactura? Crítico
3. El área aséptica está clasificada en grados A, B, C y D de acuerdo a las características del aire?
Crítico
4. Son las instalaciones de diseño y materiales adecuados fáciles de limpiar:
Grado A – Crítico Grado B – Crítico Grado C – Mayor Grado D – Mayor Incluye ductos y tuberías
Crítico
5. Las áreas limpias carecen de drenajes? Crítico
6. Si existen drenajes en las áreas controladas, éstos están convenientemente Crítico

- 48 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
sellados?
7. Si no están sellados, están provistos de interruptor de aire adecuado para prevenir la contaminación microbiológica?
Crítico
8. Están las áreas suficientemente iluminadas, con presión y humedad relativa adecuadas, dependiendo del producto?
Crítico
9. Se toman las medidas necesarias para minimizar los riesgos de contaminación cruzada?
Crítico
10. Se elaboran productos a base de microorganismos vivos en el área limpia? Informativo
11. El área aséptica está dotada de esclusas para la entrada de personal y esclusas adicionales para la entrada de los materiales?
Crítico
12. El acceso al área limpia está restringido a no personal autorizado? Crítico
13. Existen instrucciones escritas para el ingreso y salida del área limpia respecto a: Personal? Materiales? Equipos?
Crítico
6.2.2 MANTENIMIENTO Y LIMPIEZA
1. Existen procedimientos escritos para la limpieza y desinfección de las áreas e instalaciones?
Crítico
2. Se han validado estos procedimientos? Crítico
3. Se realizan y registran controles microbiológicos del ambiente? Crítico
4. Existen procedimientos escritos que establezcan los limites de aceptación y la frecuencia de estas pruebas?
Crítico
5. Existen programas documentados de desinfección de las áreas? Crítico
6. Existe rotación de desinfectantes y se documenta? Crítico
7. Se efectúan mediciones y registros de partículas no viables del aire en las diferentes zonas del área?
Crítico
8. Los resultados están dentro de los límites establecidos? Crítico

- 49 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
9. Existen instrucciones escritas para el lavado y esterilización de materiales que ingresan al área limpia:
Uniforme? Materiales de envase primario? Filtros? Herramientas? Otros?
Crítico
6.2.3 PERSONAL
1. El personal usa ropa exclusiva que no desprenda partícula y adecuada para las áreas de preparación y llenado?
Crítico
2. Se realiza rotación periódica de los operarios? Mayor
3. Se realizan controles de higiene personal y están establecidos los límites de aceptación? Se documentan los controles de manos y de uniformes?
Crítico
6.2.4 EQUIPOS Y FILTROS1. Existe un sistema independiente de las demás áreas para el suministro y la
extracción de aire limpio a las áreas asépticas?Crítico
2. Existen procedimientos escritos sobre el cambio, limpieza y mantenimiento de:
Filtros de aire? Filtros usados en la producción Equipos?
Crítico
3. Se documentan los cambios y el mantenimiento de:
Filtros de aire? Filtros usados en la producción Equipos?
Crítico
4. Los equipos y partes que están en contacto con el producto pueden limpiarse completamente y esterilizarse?
Crítico
5. Se identifican las areas de los equipos de acuerdo a su estado de limpieza (equipo limpio/ equipo sucio)?
Crítico

- 50 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
6. La etiqueta de identificación menciona el último producto fabricado y su número de lote?
Crítico
7. Existen equipos de medición de diferenciales de presión y se registran las mediciones?
Crítico
8. Se dispone de un programa escrito de calibración de equipos que lo requieran y se documenta?
Crítico
6.2.5 ELABORACION DE LOTES
1. El agua utilizada para la fabricación de inyectables es calidad PPI? Crítico
2. Existe un equipo adecuado para la producción de agua destilada? Crítico
3. Se utiliza agua destilada o desionazada para el lavado de envases primarios y equipos de producción?
Crítico
4. El último enjuague se realiza con agua PPI? Crítico
5. Se registran en las instrucciones de manufactura? Crítico
6. Se tienen órdenes de producción e instrucciones de manufactura? Crítico
7. Se cumplen correctamente las instrucciones de manufactura? Crítico
8. Se fabrica un producto por vez dentro de cada área? Crítico
9. Si la producción se hace por campana, anexe listado de productos con las fechas probables de su fabricación?
Informativo
10. Clasifique la producción de acuerdo al siguiente cuadro: Informativo
11. Qué tipo de esterilización utiliza para los envases primarios? Informativo
Tipo producto Esteriliz. Terminal Filtración Esteriliz. Llenado aséptico
ENVASE CALOR SECO
CALOR HUMEDO
OXIDO DE ETILENO
RADIACION IONIZANTE
OTROS
AmpolletasFrasco vidrioFrasco PlásticoTubo Colapsib. Aluminio *PlásticoJeringasCárpulasOtros
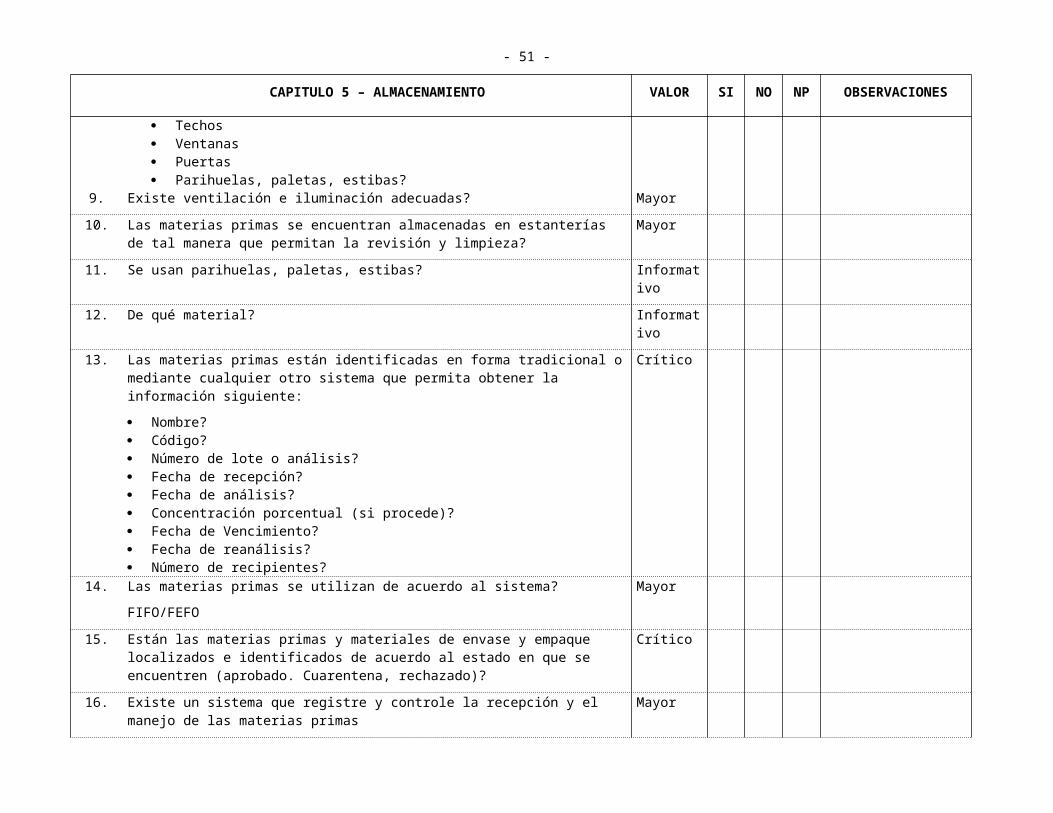
- 51 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
6.2.6 CONTROLES DE PROCESO
1. Se dispone de procedimientos escritos de los controles en proceso y sus especificaciones? ¿ Se documentan?
Crítico
2. Existen procedimientos escritos para controlar y validar los procesos de esterilización?
Crítico
3. Se controla y registra cada uno de los procesos de esterilización? Crítico
4. Se controla por métodos validados la ausencia de trazas de óxido de etileno en insumos, materiales, equipos y áreas donde se utiliza este agente esterilizante?
Crítico
5. Se realizan los controles fisicoquímico, microbiológicos y de endotoxinas al agua obtenida por:
Destilación? Filtración?
Crítico
6. Los resultados del control fisicoquímico y microbiológico de las aguas utilizadas están dentro de los límites establecidos?
Crítico
7. Con qué frecuencia se realizan? Informativo
8. Se han establecido los limites de aceptación? Informativo
9. Se realiza un control microbiológico y de partículas al vapor que se utiliza y los resultados están dentro de los limites de aceptación?
Crítico
10. Se manejan gases dentro del proceso de manufactura? Informativo
11. El gas se pasa a través de filtro esterilizantes de ponerse en contacto con el producto?
Crítico

- 52 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
12. Se realiza un control de partículas al gas que se utiliza directamente sobre el producto, si procede?
Crítico
13. En caso de utilizarse, los resultados están dentro de los limites establecidos? Crítico
14 Se controlan y documentan los resultados de la integridad de los filtros antes y después de la filtración del producto?
Crítico
15. Existen procedimientos escritos en caso de presentarse algún problema durante el proceso de filtración esterilizante?
Crítico
16. Se identifican y se separan convenientemente los materiales estériles y los no estériles?
Crítico
17. Se controla la carga microbiana de los productos antes de la filtración estéril (Bioburden)?
Crítico
18. Están fijados los límites de aceptación para cada producto? Crítico
19. Se controla y valida el llenado aséptico mediante el llenado de medios o caldos de cultivo estériles simulando las operaciones asépticas?
Crítico
20. Existen procedimientos escritos y se tiene fijados:
Número de unidades de ensayo? Limite de aceptación? Frecuencia?
Crítico
21. Se realiza controles de temperatura , humedad relativa dentro del área limpia y de iluminación, si el producto lo requiere, se registran?
Crítico
22. Existen los procedimientos escritos sobre estos controles? Crítico
6.3 PRODUCTOS ESPECIALES
6.3.1 ÁREAS
1. Además de los requerimientos generales correspondientes a cada área de producción, las áreas especiales cumplen con requisitos de acuerdo al tipo de productos elaborar?
Informativo
2. Indique cuáles productos elabora:
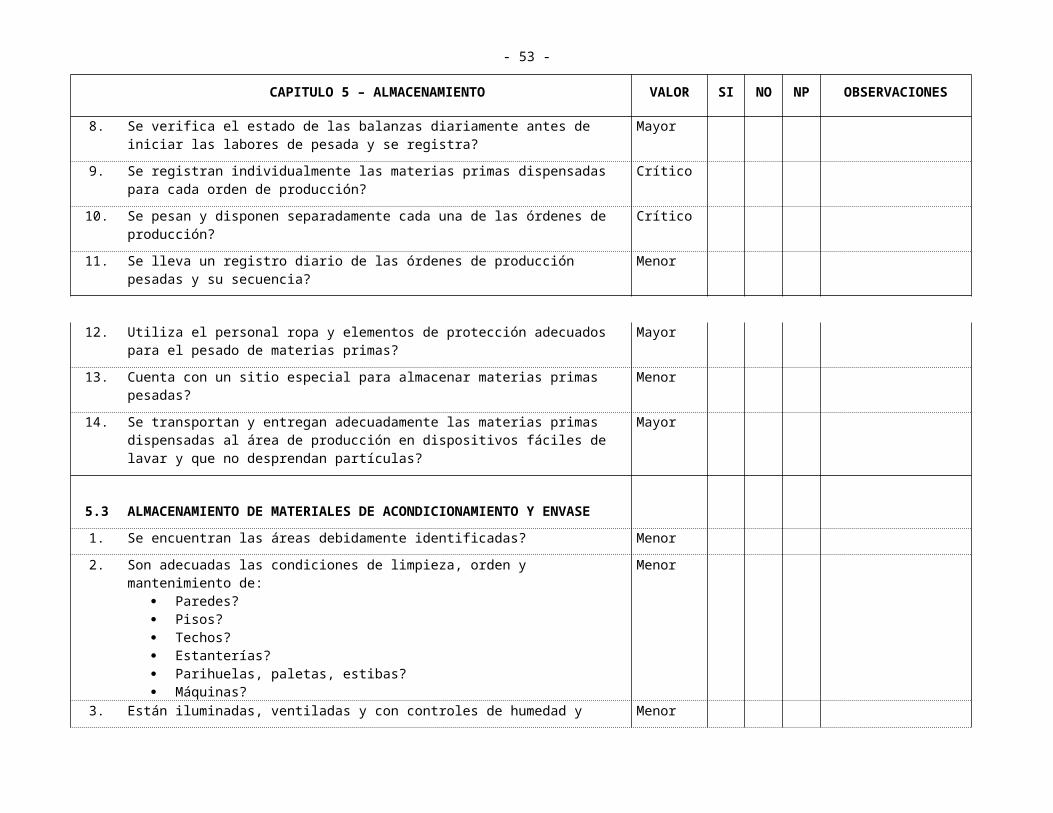
- 53 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
1. Antibióticos penicilinicos2. Antibióticos cefalosporínicos?3. Endocrinos?4. Biológicos?5. Citostáticos?6. Inmunosupresores?7. Otros?
3. Si fabrica esteroides, en áreas no especificas, lo hace por campañas? Crítico
4. Existen áreas separadas para la elaboración de los productos especiales? Crítico
5. Se toman las medidas necesarias para evitar la contaminación cruzada? Crítico
6. El área está dotada de esclusas y vestuarios independientes para el personal? Crítico
7. El área está dotada de esclusas para el ingreso de los materiales? Crítico
8. La entrada a las áreas está restringida al personal no autorizado? Crítico
9. Existen instrucciones escritas para el ingreso y salida del personal y de los materiales de las áreas:
1. Uniformes?2. Materiales de envase primario?3. Filtros?
Crítico
10. Se comprueba la inactivación y se dejan registros? Crítico
6.3.2 MANTENIMIENTO Y LIMPIEZA
1. Se evalúan las trazas del producto fabricado después de la limpieza de las áreas, instalaciones y equipos cuando se requiera:
1. En el área de manufactura?
2. En las áreas aledañas?
Mayor
2. Se efectúan las validaciones que demuestren que no existen riesgos de contaminación de los productos ni del personal, existen los registros?
Crítico
6.3.3 PERSONAL
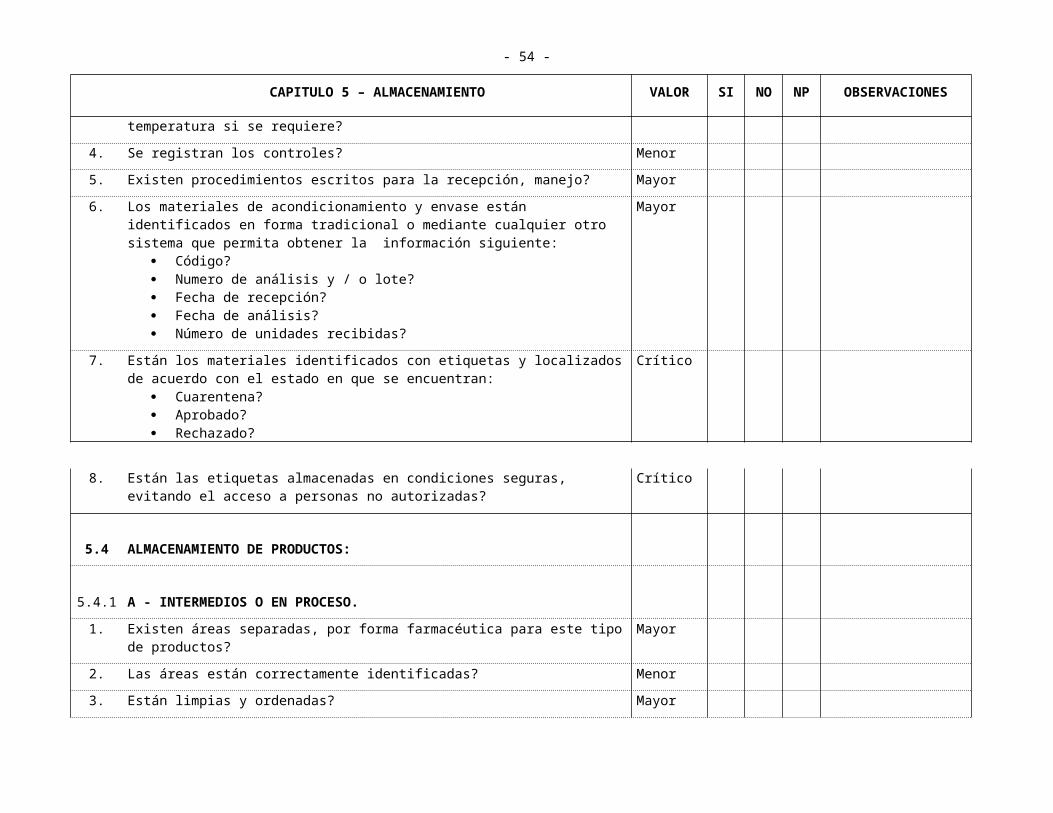
- 54 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
1. En caso de fabricar antibióticos penicilinicos y cefalosporinicos, se hace previamente una prueba de sensibilidad al personal que va a trabajar en estas áreas?
Crítico
2. Se toman las medidas necesarias para garantizar la no contaminación del personal:
1. Uso de máscaras con sistemas de aireación provisto de filtros eficientes?2. Uso de guantes y otros accesorios convenientes?
Crítico
3. Usa el personal ropa exclusiva y especial para estas áreas? Crítico
4. Especifique tipo de ropa y accesorios que usa: Informativo
6.3.4 EQUIPOS Y FILTROS
1. Los equipos que lo requieran tienen dispositivos de seguridad? Mayor
2. Los equipos y utensilios de limpieza son de uso exclusivo para cada área? Mayor
3. Las áreas de los equipos se identifican correctamente:
1. Nombre del producto?2. Número de lote?3. Fecha?
Mayor
4. Se identifican los equipos según su estado de limpieza? Crítico
5. Se menciona en la etiqueta el nombre y número de lote del producto anteriormente fabricado?
Crítico
6. Existe un sistema de suministro de aire filtrado independiente del usado para las demás áreas de producción?
Crítico
7. Existe un sistema de extracción de aire independiente con colector de particulas y filtros de eficiencia validada colocado justo a la salida al exterior?
Crítico
8. Existe un sistema de medición de diferenciales de presión, se cumplen los limites establecidos?
Crítico
9. Se efectúan las mediciones y registros de los cambios de aire en las áreas que lo requieran?. Se documentan?
Mayor
10. Existen procedimientos escritos sobre cambios y limpieza de: Crítico

- 55 -
CAPITULO 6 – PRODUCCION VALOR SI NO NP OBSERVACIONES
1. Filtros?2. Equipos?3. Areas?
6.3.5 ELABORACIÓN DE LOTES
1. Se tienen órdenes de producción e instrucciones de manufactura para cada lote de producto? Se siguen estrictamente las instrucciones de manufactura?
Crítico
2. Si la producción se hace por campaña, anexe listado de productos y fechas de su elaboración
Informativo
6.3.6 CONTROLES EN PROCESO
1. Se dispone de procedimientos escritos para los controles en proceso que incluya las especificaciones?
Crítico
2. Se registran los controles ambientales del área? Crítico
3. Existen procedimientos escritos para el manejo de desechos que salen de estas áreas?
Crítico
6.4 PRODUCTOS EFERVESCENTES
1. El área está dotada de temperatura y humedad relativa controlada? Crítico
2. Se comprueban las condiciones antes de comenzar el proceso de producción? Existen los registros?
Crítico

- 56 -
CAPITULO 7 – QUEJAS, RETIROS Y DEVOLUCIONES VALOR SI NO NP OBSERVACIONES
7.1 QUEJAS Y RECLAMOS
1. Existe un procedimiento escrito sobre quejas y reclamos que incluya al personal responsable del manejo de las mismas?
Mayor
2. Se registran detallada y totalmente las decisiones y las medidas tomadas como resultados de las quejas y reclamos?
Mayor
3. Estos registros se revisan periódicamente a fin de determinar si se trata de problemas especifico o recurrentes que pudieran ocasionar nuevas quejas y reclamos?
Menor
7.2 RETIRO DE PRODUCTOS DEL MERCADO
1. Existe un procedimiento escrito de operación que defina claramente el proceso de retiro y las responsabilidades de las personas involucradas y se redacta un informe?
Crítico
2. Se evalúa periódicamente la eficiencia del sistema de retiro de productos del mercado?
Mayor
3. Este procedimiento esta disponible en cualquier momento? Mayor
4. El procedimiento incluye la investigación de las causas? Mayor
5. Se registra el desarrollo del proceso de retiro de los productos y se redacta un informe con la conciliación entre las cantidades producidas, distribuidas y retiradas del mercado?
Mayor
6. Son informadas las autoridades competentes del retiro de los productos del mercado, por tener un defecto real o sospechado?
Mayor
7. Se lleva un registro de las destrucciones? Mayor

- 57 -
CAPITULO 7 – QUEJAS, RETIROS Y DEVOLUCIONES VALOR SI NO NP OBSERVACIONES
7.3 DEVOLUCIONES
1. Existe un procedimiento escrito que describa como manejar las devoluciones e incluya al personal responsable de la actividad?
Mayor
2. Los medicamentos devueltos están debidamente identificados y separados de los demás productos existentes?
Mayor
3. Existen y están disponibles procedimientos escritos de operación para los ensayos, reprocesos y destrucción de los productos devueltos?
Mayor
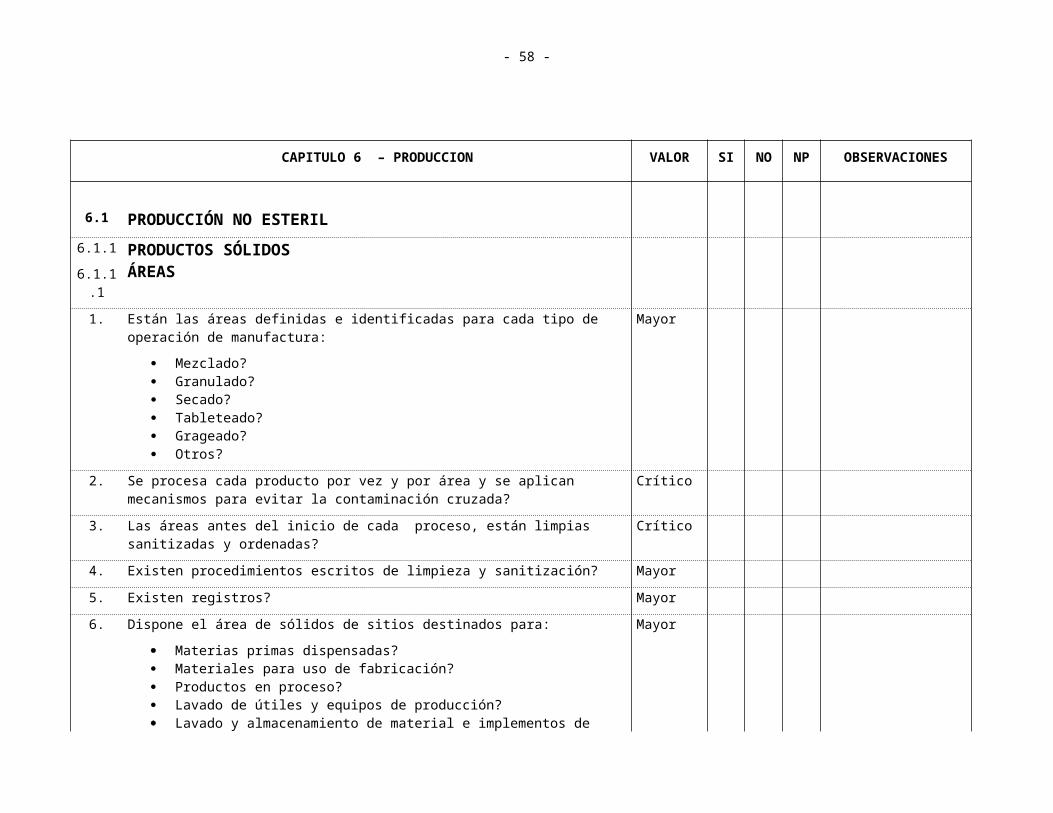
- 58 -
CAPITULO 8 – AREA DE EMPAQUE VALOR SI NO NP OBSERVACIONES
1. Dispone esta sección de sitios destinados para:
Etiquetas? Almacenamiento provisional o cuarentena ? Lavado de utensilios y equipos de envase y empaque? Lavado y almacenamiento de implementos de aseo ?
Mayor
2. Están los sitios anteriores limpios y ordenados previo al inicio del proceso? Mayor
3. Son fáciles de limpiar y de material adecuado:
Paredes? Pisos ? Techos ? Ventanas ? Lámparas ? Rejillas ? Drenajes y sifones ? Ductos y tuberías?
Menor
4. Están suficientemente iluminados y ventilados? Menor
5. Están protegidos del ambiente externo para que no ingresen las partículas a la zona?
Mayor
Cuando el producto está expuesto? Crítico
6. Existe un procedimiento escrito para ejecutar el despeje de línea, la identificación de productos en proceso y graneles, el manejo de materiales, para evitar confusiones?
Crítico

- 59 -
CAPITULO 9 – CONTROL Y USO DE MATERIALES DE ETIQUETADO Y EMPAQUE VALOR SI NO NP OBSERVACIONES
1. Existe un procedimiento escrito para la codificación de la fecha de expiración y el número de lote en las etiquetas y empaques y su almacenamiento posterior. Se documenta?
Crítico
2. Existe un área separada y con llave para el codificado de los materiales?
Está debidamente identificada esta área? Está limpia y ordenada? Está iluminada y ventilada adecuadamente? Existe una persona responsable de la misma?
Mayor
3. Son adecuados y de fácil limpieza los:
Pisos Paredes Techos Ventanas Puertas Lámparas Drenajes y Sifones Ductos y tuberias
Menor
4. Se mantiene un control cuidadoso de los inventarios de los materiales codificados con fecha de expiración y el número de lote, y no codificados?
Mayor
5. Se hace una conciliación entre el número de etiquetas y empaques marcados y los recibidos del almacén. Se documenta?
Crítico
6. Existen procedimientos escritos para la destrucción de los materiales sobrantes y deteriorados (etiquetas y empaques codificados), después de terminado el lote correspondiente. Se documenta?
Crítico
7. Se registra la aprobación del codificado de la fecha de expiración y número de lote. Se documenta?
Crítico
8. Existen procedimientos escritos para la devolución de las etiquetas y empaques sobrantes que no fueron codificados con la fecha de expiración y el número de lote. Se documenta?
Mayor

- 60 -
CAPITULO 10 –DOCUMENTACION VALOR SI NO NP OBSERVACIONES
10.1 DOCUMENTACION: (NO SE ACEPTAN TACHONES)
1. Existen procedimientos escritos para:
Manejo de etiquetas y empaques ? Manejo de materias primas y materiales de envase primario Manejo de Productos intermedios y a granel? Manejo de producto terminado? Manejo de devoluciones, quejas y reclamos? Pesar, manufacturar, envasar, empacar, controlar la calidad y liberar los
productos que fabrica la empresa?
Crítico
2. Existen procedimiento escritos para el muestreo y control de calidad de :
Materias Primas ? Materiales de envase y empaque ? Productos en proceso ? Productos intermedios ? Productos a granel ? Productos terminados? Agua, aire y otros?
Que contemple adicionalmente la manera de identificar, cerrar y sellar los componentes que han sido muestreados.
Crítico
3. Existen procedimientos Estándar de Operación para:
Limpieza y desinfección de áreas de trabajo ? Limpieza y desinfección de equipos ? Mantenimiento de Equipos? Mantenimiento de Sistemas de Apoyo Crítico ? Operación de equipos ?
Mayor
4. Existe un procedimiento para la preparación, la revisión y actualización periódica de los procedimientos escritos ?
Mayor
5. Retiran los procedimientos escritos no vigentes? Mayor
6. Cada procedimiento escrito se encuentra debidamente codificado y lleva la fecha de su emisión y vigencia, y el nombre, firma y cargo de la persona(s) responsable(s) y
Mayor

- 61 -
CAPITULO 10 –DOCUMENTACION VALOR SI NO NP OBSERVACIONES
autorizada?
7. Están los procedimientos escritos debidamente ubicados a disposición del respectivo operario, son conocidos por éste y escritos en lenguaje claro y concreto para su fácil comprensión?
Crítico
8. La empresa verifica que el personal operario conoce y entiende los procedimientos escritos. Se dejan registros?
Mayor
9. Son claros y legibles los originales y las copias del documento original ? Crítico
10. Se tiene un listado de Procedimientos Estándar de Operación con la indicación de a quienes se repartieron?
Anexar listado de los Procedimientos Estándar de Operación
Mayor
10.2 DOCUMENTOS EXIGIDOS
10.2.1 ETIQUETAS Y EMPAQUES
1. Existen criterios definidos escritos para el muestreo y la clasificación de defectos en las etiquetas y empaques?
Mayor
2. Existen las especificaciones escritas y sus tolerancias para la aceptación o rechazo de las etiquetas y empaques?
Crítico
3. Se documentan los resultados obtenidos en los controles realizados a las etiquetas y empaques?
Mayor
4. Se cierran, sellan e identifican debidamente los componentes que han sido muestreados?
Mayor
10.2.2 MATERIAS PRIMAS Y MATERIAL DE ENVASE PRIMARIO
1. Existen criterios definidos escritos para la clasificación de los defectos de material de envase y empaque primario?
Mayor
2. Existe un registro de proveedores de materias primas debidamente clasificados y calificados?
Mayor

- 62 -
CAPITULO 10 –DOCUMENTACION VALOR SI NO NP OBSERVACIONES
3. Tienen escritas las especificaciones y procedimientos oficiales o propios para la adquisición y análisis de materias primas?
Crítico
4. Se documentan y archivan adecuadamente los resultados de los ensayos? Mayor
5. Existe un inventario actualizado y completo de todas las materias primas y materiales?
Mayor

- 63 -
CAPITULO 11 –CONTROL DE CALIDAD VALOR SI NO NP OBSERVACIONES
1. El laboratorio de control de calidad es independiente en sus instalaciones físicas, de las demás áreas de la planta?
Mayor
2. Existen dentro de esta área los equipos e implementos de seguridad necesarios?
Extintores Duchas de seguridad Campanas de extracción Duchas de ojos Gafas de seguridad Máscaras de seguridad Otros :
Mayor
3. El personal de Control de Calidad utiliza estos implementos de seguridad, cuando lo requiere?
Informativo
4. Las condiciones de seguridad en el trabajo son aceptables? Informativo
5. Control de Calidad es independiente de Producción ? Crítico
6. El Laboratorio de Control de Calidad está bajo la responsabilidad de un profesional farmacéutico?
Crítico
7. El Responsable del Control de Calidad tiene autonomía en sus decisiones? Crítico
8. El personal de control de calidad recibe capacitación periódica. Se documenta? Mayor
9 Se tienen instalaciones y equipos adecuados propios o contratados, para la realización de los análisis requeridos de acuerdo a los productos que se elaboran?
Análisis Físicos? Análisis Fisicoquímicos? Análisis instrumental? Análisis microbiológico? Análisis biológico?
Crítico
10. Existen áreas para el almacenamiento de muestras de retención de:
Materias primas? Producto terminado?
Mayor
11. Existe un lugar con las condiciones adecuadas para almacenar los patrones de Referencia?
Crítico
12. Existe un sitio adecuado para guardar los documentos maestros, métodos analíticos Mayor

- 64 -
CAPITULO 11 –CONTROL DE CALIDAD VALOR SI NO NP OBSERVACIONES
procedimientos estándar de Operación y Registros de Lotes?
13. Existe bibliografía especializada para consulta?
Anexar listado completo de Equipos de análisis Anexar listado de libros de consulta disponibles
Informativo
14. Existen en forma escrita y clara especificaciones para Producto en Proceso (cuando se requiera), Producto a Granel y Producto terminado. Se tienen en cuenta estas especificaciones?
Crítico
15. Utilizan patrones primarios o secundarios certificados? Crítico
16. Existen en forma escrita y clara instructivos para el Manejo de equipos de análisis se cumplen estos instructivos?
Crítico
17. Existen programas de calibración y mantenimiento de los equipos de análisis, se registran?
Crítico
18. Existen en forma escrita y clara instructivos para el Manejo de reactivos y Patrones de Referencia?
Crítico
19. Existen en forma escrita y clara procedimientos para el Manejo de Muestras de retención. Se registran estos procedimientos?
Crítico
20. Control de calidad es el responsable de la evaluación de los productos devueltos. Se registra?
Mayor
21. Existen en forma escrita y clara procedimientos para la Validación de métodos analíticos. Se aplican y registran estos procedimientos?
Crítico
22. Existen en forma escrita y clara procedimientos para efectuar la Autoinspección. Se registran estos procedimientos?
Mayor
23. Se registran todos los datos y resultados de los ensayos analíticos? Crítico
24. Firman todos los resultados las personas responsables (Quien los efectuó y quien los reviso)?
Mayor
25. Presentan los recipientes de las soluciones reactivos y de los patrones de referencia una rotulación precisa?
Crítico
26. Se recopilan y revisan los documentos de la historia del lote por parte de la persona autorizada?
Crítico
27. Cada lote solamente es liberado por la persona autorizada? Crítico

- 65 -
CAPITULO 11 –CONTROL DE CALIDAD VALOR SI NO NP OBSERVACIONES
28. Se cuenta con procedimientos escritos que indiquen las medidas que deben ser aplicadas cuando hay desviaciones de los procedimientos estandarizados?
Crítico
29. Existen procedimientos escritos que establezcan los controles a seguir para que no se continúe un proceso crítico, sin haber sido autorizado por Control de Calidad?
Crítico
30. Existe un procedimiento escrito que establezca cómo, cuándo y quién puede autorizar la modificación de un procedimiento establecido?
Crítico
31. Dispone la empresa de Bioterio? Informativo
32. Es adecuada su ubicación dentro de la empresa? Crítico
33. Existe un procedimiento escrito para establecer el origen y proveedor de los animales de ensayo. Esta documentado?
Mayor
34. Se cuenta con un programa de limpieza y de desinfección apropiado del bioterio? Mayor
35. Existen procedimientos escritos para la limpieza, desinfección y mantenimiento en general del Bioterio?
Mayor
36. Se tienen sistemas de ventilación y temperatura adecuados? Mayor
37. Se documenta apropiadamente la humedad relativa, la temperatura y el nivel de ruido del Bioterio?
Mayor
38. Existen procedimientos escritos para la inspección de los animales en cuanto a salud, peso, alimentación y periodo de descanso. Se documenta esta actividad?
Mayor
39. Se realiza el ensayo de respuesta positiva a la endotoxina a los animales nuevos? Crítico

- 66 -
CAPITULO 12– GARANTIA DE CALIDAD VALOR SI NO NP OBSERVACIONES
1. Existe en la empresa un sistema de Garantía de Calidad. Existe una persona responsable del mismo?
Crítico
2. Están claramente definidas y escritas las funciones y responsabilidades del personal responsable de Garantía de Calidad?
Crítico
3. Reporta directamente a la Dirección de la Empresa? Crítico
4. Se divulga a todos los niveles de la empresa la política de garantía de calidad? Crítico
5. Se evalúa su comprensión por parte de todos los involucrados? Mayor
6. Se implementa la actualización o modificación de los procesos de fabricación y de los procedimientos operativos después de una completa evaluación y aprobación?
Crítico
7. Este departamento realiza al menos una vez al ano autoinspecciones? Mayor
8. Se efectúa un seguimiento de los resultados de las autoinspecciones y se realizan las acciones correctivas pertinentes?
Crítico

- 67 -
CAPITULO 13 – SEGURIDAD IINDUSTRIAL VALOR SI NO NP OBSERVACIONES
1. Existe un programa de Salud Ocupacional?
Existen programas permanentes de Seguridad Industrial?
Informativo
2. Existen programas de capacitación y equipos para control y prevención de incendios?
Mayor
3. Están vigentes los extinguidores, los accesos a estos se encuentran libres y delimitados?
Mayor
4. Existen planos y señales de evacuación? Mayor
5. Se realizan simulacros de evacuación?
Con qué frecuencia?
Informativo
6. Se verifica con frecuencia el correcto funcionamiento de:
Extinguidores? Hidrantes? Mangueras? Otros equipos para control de incendios?
Informativo
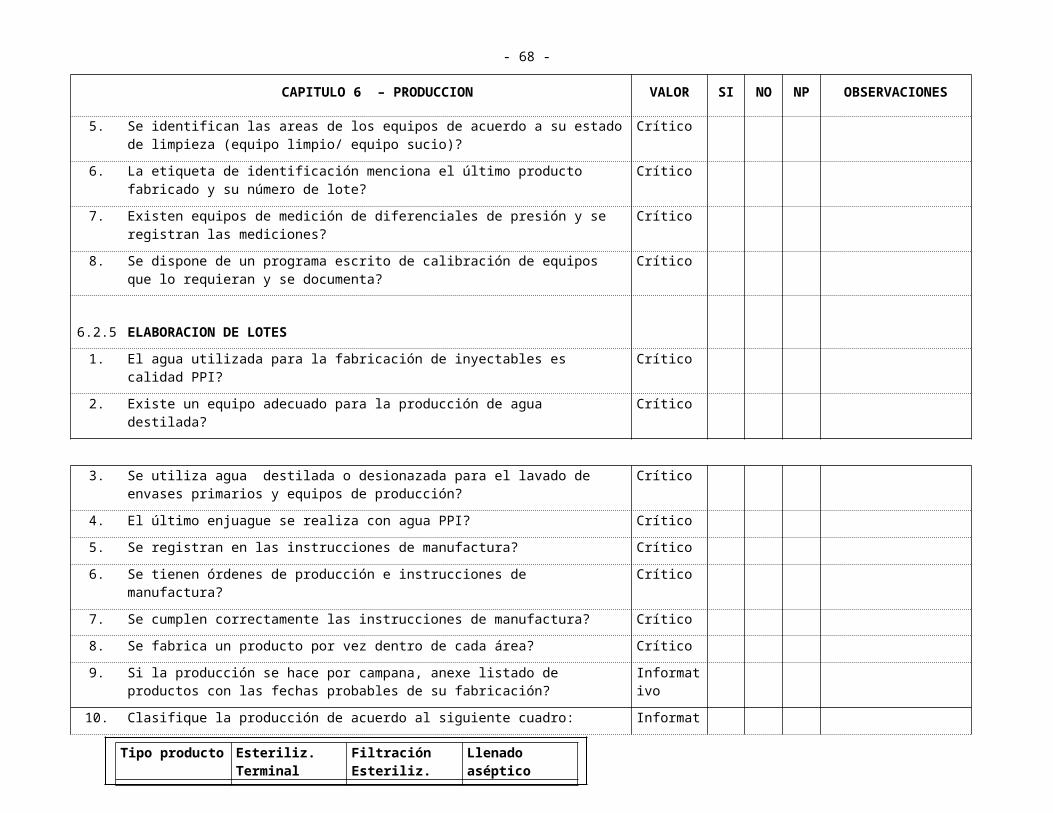
- 68 -
CAPITULO 14– ESTABILIDAD VALOR SI NO NP OBSERVACIONES
1. Existen procedimientos escritos para definir las características de estabilidad de los productos?
Crítico
2. Existen condiciones especiales de almacenamiento para las muestras de retención para estabilidad acelerada?
Cuáles? Son adecuadas estas condiciones? Se lleva un registro apropiado de las condiciones de humedad relativa y
temperatura de esta área de almacenamiento?
Crítico
3. Existe un protocolo escrito para el desarrollo de las pruebas de Estabilidad de cada producto?
Crítico
4. Existe un procedimiento escrito para establecer la vida útil de los productos? Crítico
5. Existe un procedimiento escrito para asignar la fecha de expiración de los productos?
Crítico
6. Cuenta con el área, los equipos, procedimientos y reactivos apropiados para el desarrollo de los estudios de estabilidad?
Crítico
Existe un contrato con un tercero para el desarrollo de estos estudios, en caso de no realizarlo en su propio laboratorio?
Crítico
7. En caso de presentarse una modificación significativa del proceso de manufactura, de los equipos y de las condiciones del área de fabricación, un reprocesamiento, un cambio de proveedor de materias primas y material de envase etc. se realizan nuevos estudios de estabilidad?
Crítico
8. Se documentan apropiadamente estos nuevos estudios ? Crítico
9. Se realizan evaluaciones y conclusiones de cada uno de los estudios de estabilidad?
Crítico

- 69 -
CAPITULO 15 –VALIDACIONES VALOR SI NO NP OBSERVACIONES
1. Existen programas y personal responsable de Validación de equipos, procesos productivos, procedimientos operativos y métodos analíticos?
Crítico
2. Se tienen procedimientos escritos establecidos sobre validación? Crítico
3. Los procesos de importancia crítica se validan:
Retrospectivamente? Prospectivamente?
Informativo
4. Se registra y se valida toda modificación importante del proceso de fabricación, incluyendo equipos o materiales que puedan influir en la calidad del producto y/o reproducibilidad del proceso?
Crítico
5. Se tienen registrados, evaluados y archivados los resultados de las validaciones realizadas?
Crítico
15.1 VALIDACIÓN ÁREA ESTÉRIL
1. Existen procedimientos escritos y registros de validación para los siguientes procesos:
Crítico
2. Esterilización por calor seco? Crítico
3. Esterilización por Vapor? Crítico
4. Esterilización por óxido de etileno? Crítico
5. Esterilización por radiaciones? Crítico
6. Existe procedimiento escrito y registros de validación de los procesos de llenado aséptico?
Crítico
7. Existen procedimientos escritos y registros de validación de:
El sistema de filtración del aire estéril?
Se validan las normas establecidas para el cambio de filtros?
____________________________________________________________________________________________________________________________________
Crítico
8. Existen programas de validación y registros para el recuento de: Crítico

- 70 -
CAPITULO 15 –VALIDACIONES VALOR SI NO NP OBSERVACIONES
Partículas No Viables
Partículas Viables
MEDIDA TRANSITORIA
Todas las validaciones del área estéril, tendrán un plazo mayor para su cumplimiento a excepción del proceso de validación del llenado aséptico, el cual debe cumplirse desde la vigencia de esta guía y garantizar un NAE(Nivel de Aseguramiento de Esterilidad), de 10 –3.
------------------------------------------------------------------------------------
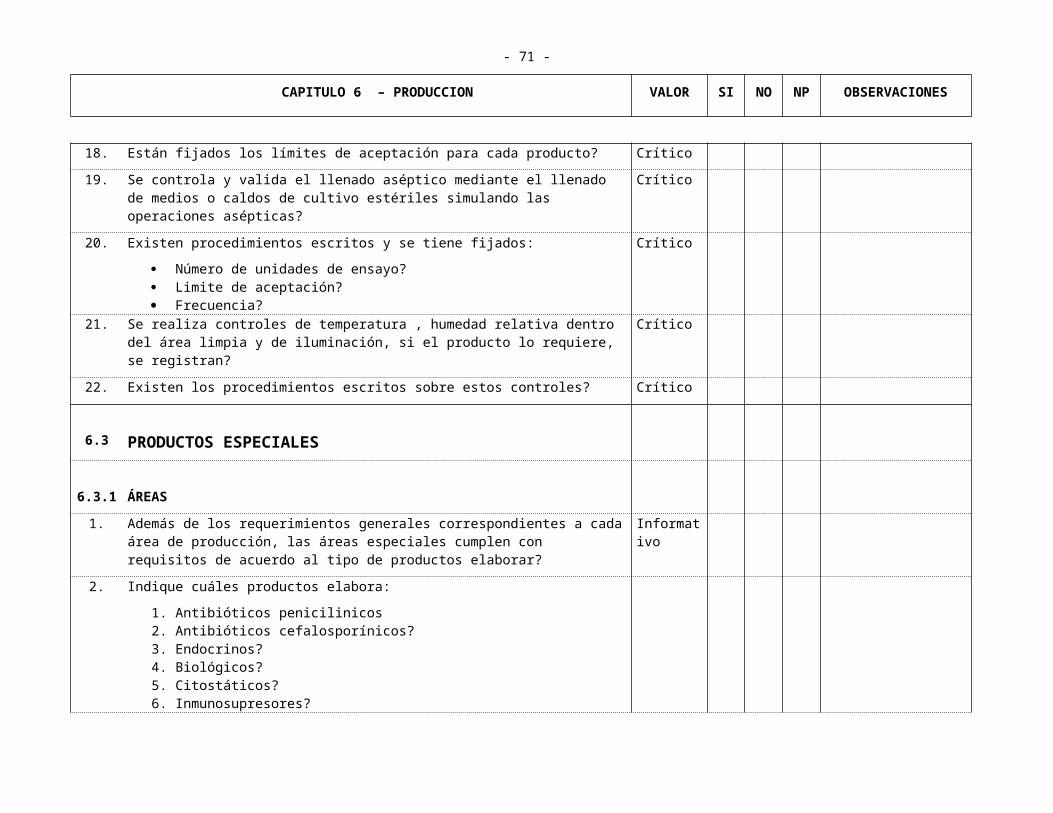
- 71 -
CAPITULO 16 –PROCESAMIENTO DE DATOS VALOR SI NO NP OBSERVACIONES
16. PROCESAMIENTO ELECTRÓNICO DE DATOS (INFORMÁTICA)
1. Existe un sistema de procesamiento y registro electrónico de datos? Informativo
2. Existen personas capacitadas responsables y autorizadas para el acceso al sistema ( entrada de datos, consultas y modificaciones)?
Crítico
3. Hay un procedimiento validado que indique las medidas a tomar en caso de una falla del sistema?
Crítico
4. Existe un procedimiento de seguridad que únicamente permita el acceso de personal autorizado al sistema?
Crítico
5. Se editan los datos y archivan en el historial del lote? Informativo
6. Son de fácil acceso y disponibilidad por parte del personal autorizado los datos correspondientes al registro de producción de cada lote?
Mayor
7. Se tiene grabación de reserva de los registros de la información archivados electrónicamente?
Crítico
8. Está convenientemente almacenada esta información? Mayor
* * * * *