28 octubre 2008 - transcripciones cuartos 2008 | just ... · web viewquímica medicinal i....
TRANSCRIPT

28 OCTUBRE 2008 Química medicinal I
SEMINARIO 1: RECEPTOR DE HISTAMINA H3Gabriela Leal, Juan José Mora
Objetivos
Señalar la ubicación de los RH3 Mencionar ejemplos de antagonistas y agonistasMostrar secuencia en 3D del receptorEvaluar la actividad biológica y posibles enfermedades en las que participan
Histamina actúa en 4 receptores distintos acoplados a proteínas G (GPCRs): H1, H2, H3, H4. El H3 fue descubierto en 1983 Arranz y un grupo de colaboradores.
Los dos primeros (H1 y H2) han sido comprobados como excelentes blancos para drogas, al contrario, el último (4H) se descubrió hace 7 años y actualmente es materia de estudios químicos y farmacológicos. Este receptor H3 va a estar principalmente acumulado en el SNC, donde permite la modulación de biosíntesis y liberación de histamina, además de la regulación y liberación de otros neurotransmisores (Ach, DA, SE, EP, GABA).
Receptor H3
Este receptor puede actuar como autoreceptor o heteroreceptor. En este caso lo vamos a ver principalmente como un autoreceptor.
El receptor H3 está ubicado mayoritariamente en las neuronas presináptica histaminérgicas, acá su acción es como autoreceptor donde regula la síntesis y liberación de Histamina por retroalimentación negativa. No permite que se forme la histamina a partir de la L-Histamina. Al no liberarse, no va a actuar en los receptores H1 y H2.
En esta imagen el receptor H3 actúa como un heteroreceptor, la histamina puede unirse a receptores en neuronas adyacentes y disminuir la liberación de otros tipos de neurotransmisores.

En el SNC va a estar ubicado principalmente en: el Putamen, Núcleo acumbens, Sustancia nigra, Córtex insular, Hipotálamo, Amígdala, Ganglios basales. En estos tres últimos va ser muy importante para la elaboración de técnicas terapéuticas.
RH3 también se encuentra en otros tejidos periféricos, incluyendo corazón, vasos, tejido linfoide (adenoides), aparato digestivo y vía aérea
Vía aérea: Va a estar ubicado en los nervios colinérgicos posganglionares → función es la de mediar la transmisión colinérgica en el bronquio humano, evitando un efecto de “exceso de broncoconstricción”.
RH3
En 1999 Lovenberg y colegas lograron la clonación ADNc (ADN cíclico) que codifica para el RH3 en humanos. Desde entonces se ha clonado el gen que codifica para el RH3 en varias especies, revelando considerables diferencias entre las especies (esta diferencia es muy notoria a nivel de antagonistas) lo que implica un descubrimiento de verdadera complejidad farmacológica.
Mecanismo celular
Ligandos específicos de RH3 (principalmente agonistas inversos) muestran gran potencial terapéutico en distintas funciones cerebrales, como procesos de aprendizaje y regulación de la ingesta de comida, formando parte de modelos de obesidad, epilepsia, narcolepsia, rinitis alérgica y enfermedades cognitivas o neurodegenerativas, tal como el síndrome de déficit atencional con hiperactividad, enfermedad de Alzheimer, enfermedad de Parkinson y esquizofrenia
Por medio de la clonación del gen del RH3 se ha permitido la realización de estudios detallados de las propiedades moleculares del receptor y se ha indicado que el RH3 puede activar diversos mecanismos de transducción de señales, incluyendo Gi/o- inhibición dependiente de la AC (adenilato ciclasa), la activación de PLA2 y MAP kinasa, así como la inhibición del intercambiador de Na+/H+ y la inhibición de K+-movilización inducida de Ca2+. Está involucrado en diversos sistemas y diversos mecanismos.
Agonistas

(R)--metilhistamina: Ocurre una metilación en el carbono , se reportó una alta potencia sin embargo surgieron muchos problemas debido a que por su polaridad y otras características se metabolizaba muy rápido.
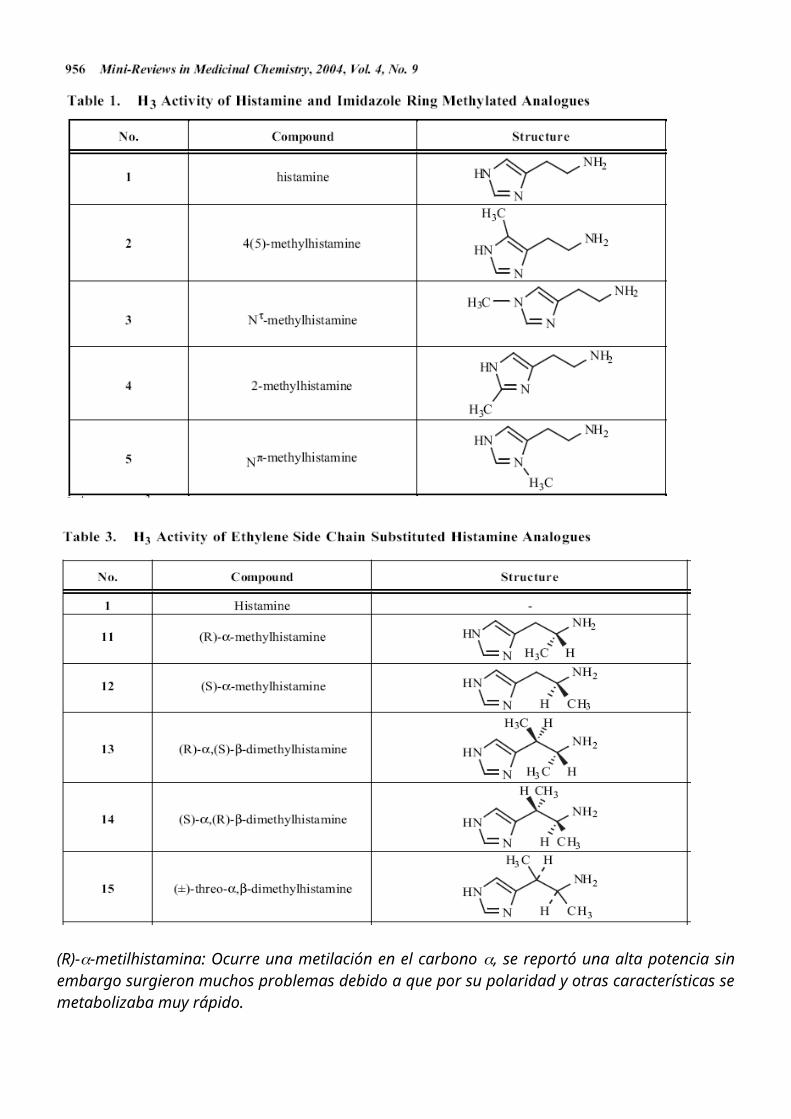
Posteriormente se buscaron formas de sintetizar análogos más lipofílicos, uno de estos productos obtenidos fue el BP2.94 que actualmente esta en estudios clínicos de fase 2 para el tratamiento del asma.
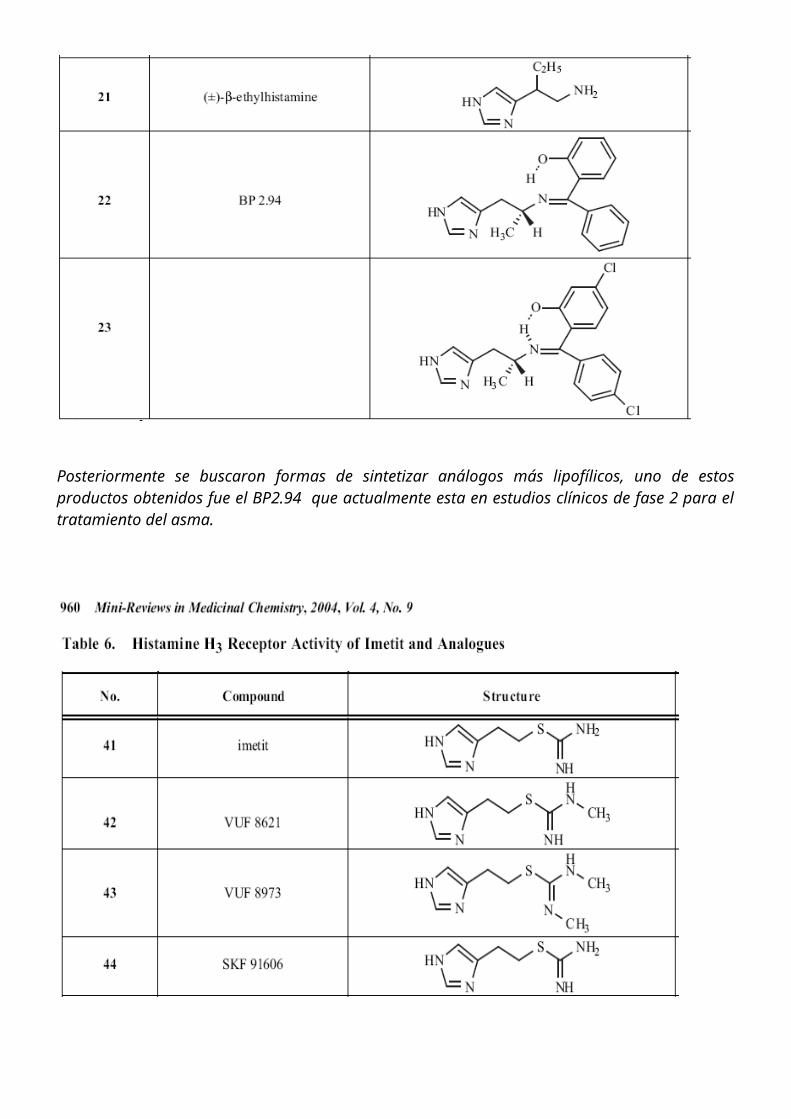
Otro compuesto obtenido fue el imetit donde lo que ocurre es una sustitución de la amina por una isotiourea, o también otra estrategia llevada a cabo como en el caso del immepip donde se hace la ciclación de la cadena lateral evitando su rápido metabolismo.
Antagonistas
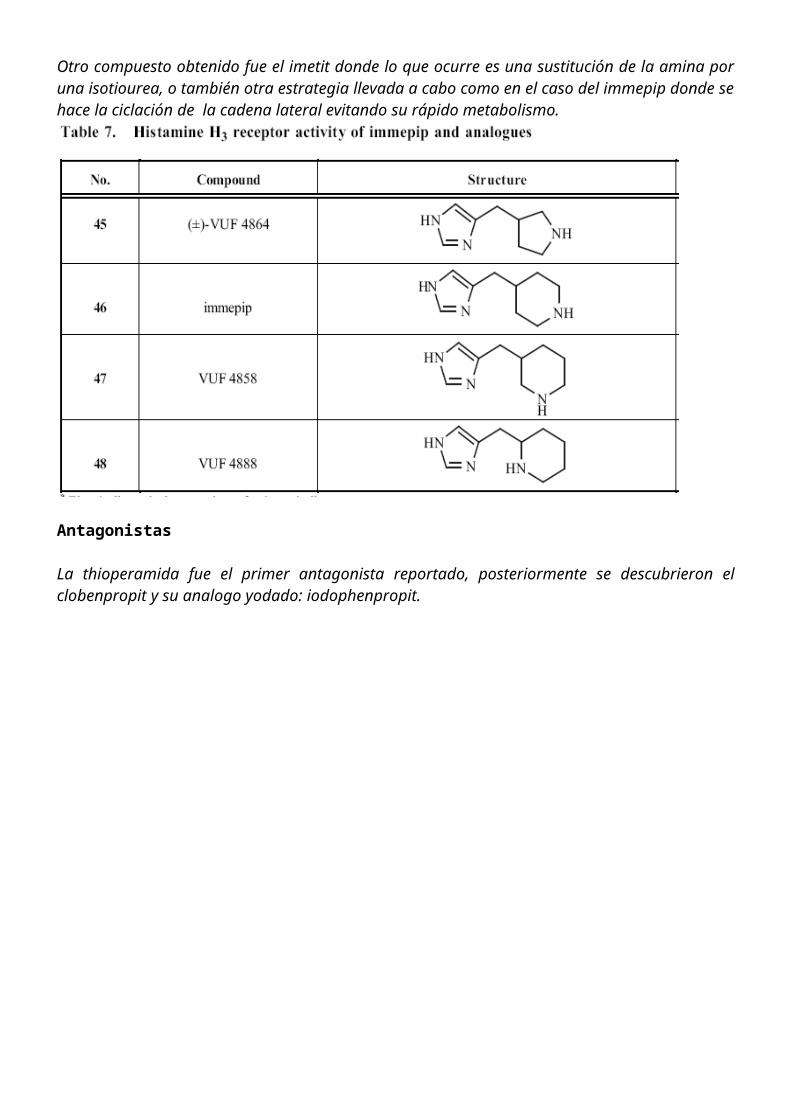
La thioperamida fue el primer antagonista reportado, posteriormente se descubrieron el clobenpropit y su analogo yodado: iodophenpropit.
Antagonista Neutral: No va a inhibir la actividad del receptor como tal, pero tampoco permite que otros agonistas y agonistas inversos puedan ejercer su acción.
THREE-DIMENSIONAL MODELS OF HISTAMINE H3 RECEPTOR ANTAGONIST COMPLEXES AND THEIR PHARMACOPHORE.
Frank U. Axe, Scout d. Bembenek, Sándor Szalma.
Es un estudio donde hay un farmacóforo y se determina la estructura donde se forma. Emplearon antagonistas para ayudarse en la determinación del farmacóforo.
Posteriormente (imagen siguiente) se muestra la secuencia donde sale no solo el receptor H3 sino también otros receptores. En rojo aparecen las secuencias muy conservadas y están en todos los receptores. Es un estudio de mutagénesis con base en los otros receptores, donde se determinaron los sitios de unión que van a estar principalmente en la hélice III y en la hélice V, en la imagen se muestran en color amarillo. Estas hélices son las que van a estar interactuando con los ligandos.

Arriba están el farmacóforo y su ligando. Abajo la imagen de su unión.
Recordemos que los receptores de H3 van a estar acoplados a proteínas G, van a tener la región transmembrana y aproximadamente tres dominios hélice. Ocurren interacciones con residuos específicos; se dice que principalmente va a ser con el Aspartato 114 en la hélice III y con la Glutamina 206 en la hélice V.
Los receptores H1 se dice que principalmente va a ser con un residuo de Tiramina.
VALIDATION OF HISTAMINE H3 RECEPTOR MODEL THROUG STRUCTURE-ACTIVITY FOR CLASSICAL H3 ANTAGONIST.
Simona Lorenzi et all.
Es otro estudio acerca de antagonistas, acá esta la piperamida y …(no se escucha), se muestran los “pockets” el 1 y el 2 donde ocurren las interacciones. Los puntitos rojos son los puentes de hidrógenos que se forman. Lo café es lo lipofílico y lo más azul es lo hidrofílico.
Posibles tratamientos farmacoterapéuticos.

Involucrando principalmente el receptor H3 tenemos: Alimentación, Ciclo sueño-vigilia, Epilepsia, Procesos cognitivos (Alzheimer), Trastornos neurodegenerativos (Parkinson, Huntington), Ansiedad y depresión, Esquizofrenia, Abuso de drogas.
Alimentación
Niveles de Histamina correlacionados inversamente con alimentación. Se cree que este efecto anoréxico puede deberse a la estimulación de los receptores H1 y H3 del hipotálamo. En la rata, el bloqueo de los RH3 ↓ consumo de alimentos inducido por NPY (neuropeptido Y) y potenció la anorexia inducida por el bloqueo del catabolismo de Histamina.
Agonistas: ↓ saciedad y ↑ consumo agua.
INFLUENCE OF SELECTIVE HISTAMINE H3 RECEPTOR ANTAGONIST ON HYPOTHALAMIC NEURAL ACTIVITY, FOOD INTAKE AND BODY WEIGHT.
K. Malmlöf et all.
Al utilizar un antagonista selectivo de RH3 se redujo la inhibición, aumentando la [ ] sináptica de Histamina junto con la transmisión de RH1
Se llegó a la conclusión de que el uso de antagonistas es una promisoria posibilidad para el desarrollo de anorexígenos. En contra parte el uso de agonistas puede permitir también el desarrollo de sustancias que permitan incrementar la ingesta de alimento.Enfermedades neurodegenerativas
Se ha observado cambios en la densidad de RH3. Ratas tratadas con la neurotoxina dopaminérgica 6-hidroxidopamina (6-OHDA, la cual tiene la capacidad de ocasionar parkinson en diferentes especies incluyendo el ser humano). Se observó la unión del receptor con regulación ascendente (aumento en la densidad de RH3) en la sustancia nigra y el estrato ventral. Mientras tanto hubo una regulación descendente (disminución de la densidad de RH3) en estos mismos receptores en casos con la enfermedad de Huntington, un desorden degenerativo que afecta el ganglio basal.
MODULATION OF HISTAMINE H3 RECEPTOR IN THE BRAIN OF 6-HYDROXYDOPAMINE-LESIONED RATS.
Oleg V. Anichtchik et all.
Su objetivo fue examinar cambios del sistema histaminérgico en ratas luego de la lesión con 6-OHDA, para posteriormente detectar cambios en las densidades de Receptor y su expresión por el ARNm (los receptores fueron expresados en mayor cantidad por parte del ARN mensajero en las ratas lesionadas).
DISTRIBUTION OF HISTAMINE H3-RECEPTOR BINDING IN THE NORMAL HUMAN BASAL GANGLIA: COMPARISON WITH HUNTINGTON´S AND PARKINSON´S DISEASE
CASESR.E Goodchild et all.

Buscó un mapeo detallado de RH3 en los ganglios basales y localización de los RH3 en los seres humanosLos RH3 en neuronas de proyección estratonigral confieren control histaminérgico sobre la actividad de los circuitos de ambas enfermedades
Ciclo de sueño-vigilia
Proyecciones histaminérgicas del núcleo tuberomamiliario del hipotálamo posterior importantes para el estado de vigilia y las lesiones en esta área promueven el sueño en ratas, gatos y monos. Los antagonistas de los receptores H3 podrían afectar el sueño y la vigilia, mediante el cambio del tono histaminérgico central
Los agonistas incrementan las ondas lentas del sueño y sorprendentemente, afectan el sueño REM (movimiento rápido de los ojos) en menor extensión con respecto a los bloqueadores de los receptores H1. Esto es muy importante porque lo que se busca con este tipo de fármacos es alterar lo menos posible la estructura del sueño.
Los antagonistas podrían utilizarse en el tratamiento de narcolepsia y los agonistas como hipnóticos.
HISTAMINERGIC CONTROL OF SLEEP-WAKE CYLCLES: RECENT THERAPEUTIC ADVANVE FOR SLEEP AND WAKE DISORDERS.
A.J. Barbier and M.J. Bradbury.
Actualización del desarrollo de ligandos H3 como medicación potencial para el sueño o la promoción del estado de vigilia.
Fue posible establecer:Agonistas de RH3: sedantes-hipnóticosAntagonistas de RH3: narcolepsia
PREGUNTAS/COMENTARIOS.
Esto fue una revisión bibliográfica.Recordar: antagonista pueden ser usados como anorexígenos y los agonistas pueden relacionarse con desordenes donde hay una ingesta de alimentos aumentada.
Profesor: Esto puede ser un poco complicado porque acuerdo con su estructura algunos pueden funcionar como agonistas o antagonistas, dependiendo de donde interactúe.
R/ Están evaluando la posibilidad de definirlos como agonistas, antagonistas y agonistas inversos. El agonista inversos es más complicado, ya que dependiendo del caso puede funcionar como agonista o antagonista. El receptor H3 es un poco nuevo, han salido muchos estudios para elucidar sus acciones.
El caso de la …(no se escucha) el cual en un inicio salió como un antagonista pero en ocasiones puede actuar como agonista. No se ha logrado investigar totalmente, y se han establecido posibles tratamientos y aplicaciones.
SEMINARIO 2: Jonathan Rojas, Tatiana Saénz.
4-BENZYL-1H-IMIDAZOLES WITH OXAZOLINE TERMINI AS HISTAMINE H3 RECEPTOR AGONISTS

Wijtmans, M.; et al. Journal of American Chemical Society 2008, 51, 2944–2953
Son conocidos 15 modelos para el enlace con los receptores H3, que involucran una interacción ácido/base entre un grupo básico protonado y un residuo de aspartato en los receptores acoplados a proteínas GPRCs para la neurotransmisión de aminas.
Algunos ejemplos de agonistas que actúan sobre receptores H3 y poseen este grupo básico son:
Interacción molecular
Los ligandos del receptor H3 se colocan entre la Región Transmembrana 2 (TM2), TM3, TM6 y TM7.
Todos los ligandos interaccionan con el residuo de Aspartato 114 en TM3, y los agonistas tambien con el Asp 80 en TM2.
El agonista de histamina RAMH ((R)-a-methylhistamine) forma enlaces en la parte amina protonada con el residuo Asp 114 (TM3), y la parte imidazol con el Asp 80 (TM2).
Hipótesis
Compuestos con una cadena sustituyente menos básica como la immethridine o sin el grupo básico como el caso de proxifan, o aún en casos en donde solo hay presencia de cadenas alifáticas como por ejemplo en los compuestos GT-2331 y VUF5523 pueden funcionar como agonistas H3.
Objetivos
Revelar según la investigación (química combinatoria y no combinatoria) hallazgos en el desarrollo de nuevos agonistas H3.

Analizar como afecta el grupo funcional en los receptores H3 y mediante esto proveer datos moleculares importantes de cómo agonistas carentes de un grupo básico en la cadena lateral, pueden activar el receptor H3.
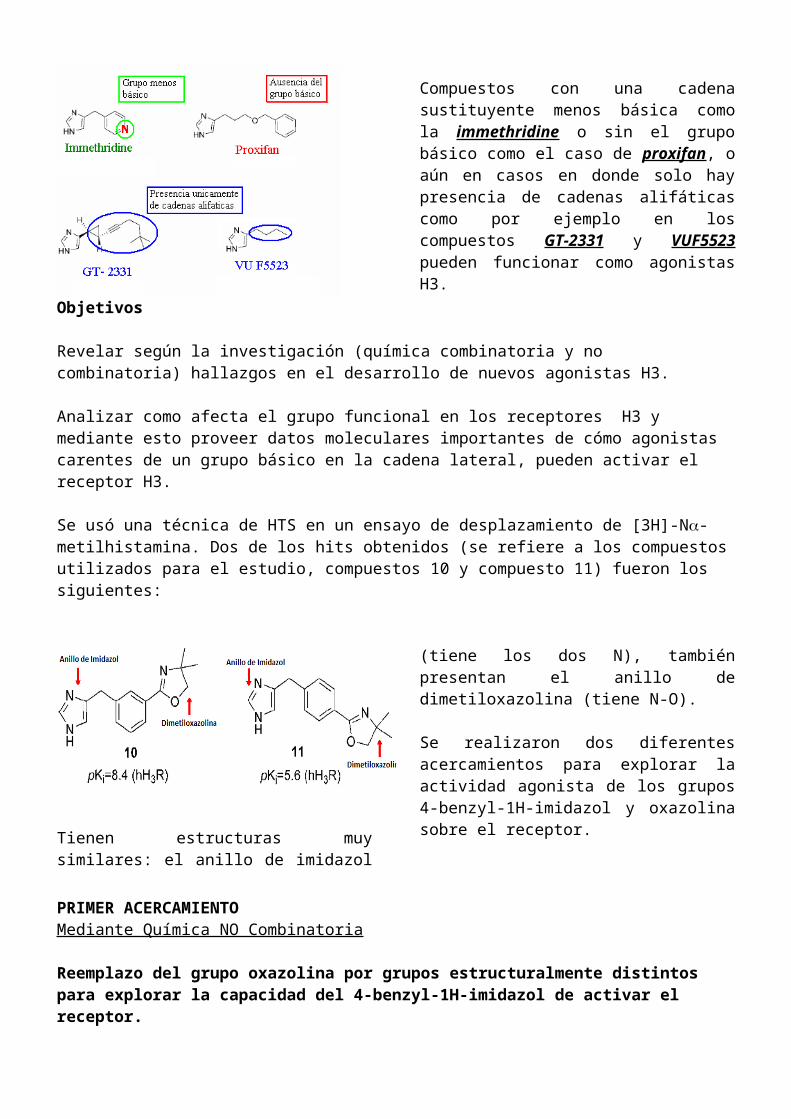
Se usó una técnica de HTS en un ensayo de desplazamiento de [3H]-N-metilhistamina. Dos de los hits obtenidos (se refiere a los compuestos utilizados para el estudio, compuestos 10 y compuesto 11) fueron los siguientes:
Tienen estructuras muy similares: el anillo de imidazol (tiene los dos N), también presentan el anillo de dimetiloxazolina (tiene N-O).
Se realizaron dos diferentes acercamientos para explorar la actividad agonista de los grupos 4-benzyl-1H-imidazol y oxazolina sobre el receptor.
PRIMER ACERCAMIENTO Mediante Química NO Combinatoria
Reemplazo del grupo oxazolina por grupos estructuralmente distintos para explorar la capacidad del 4-benzyl-1H-imidazol de activar el receptor.
Se sustituyó el anillo oxazolínico por otros grupos funcionales para entender cuáles variaciones son toleradas para ejercer la actividad agonista.
Sustituyendo este grupo oxazolínico se logro medir la actividad del grupo restante (4-benzyl-1H-imidazol) para activar el receptor.
La afinidad del receptor (pKi) se midió mediante el desplazamiento de 4-benzyl-1H-imidazol marcado radioactivamente en la membrana de células de ovario de hámster chino que expresan el receptor H3R.
Compuestos con pKi mayores de 6.5 fueron usados para evaluar su actividad funcional. Es decir, se tomaron en cuenta dos cosas la afinidad con el receptor y la del grupo funcional.
Los efectos de los sustituyentes meta fueron los siguientes:

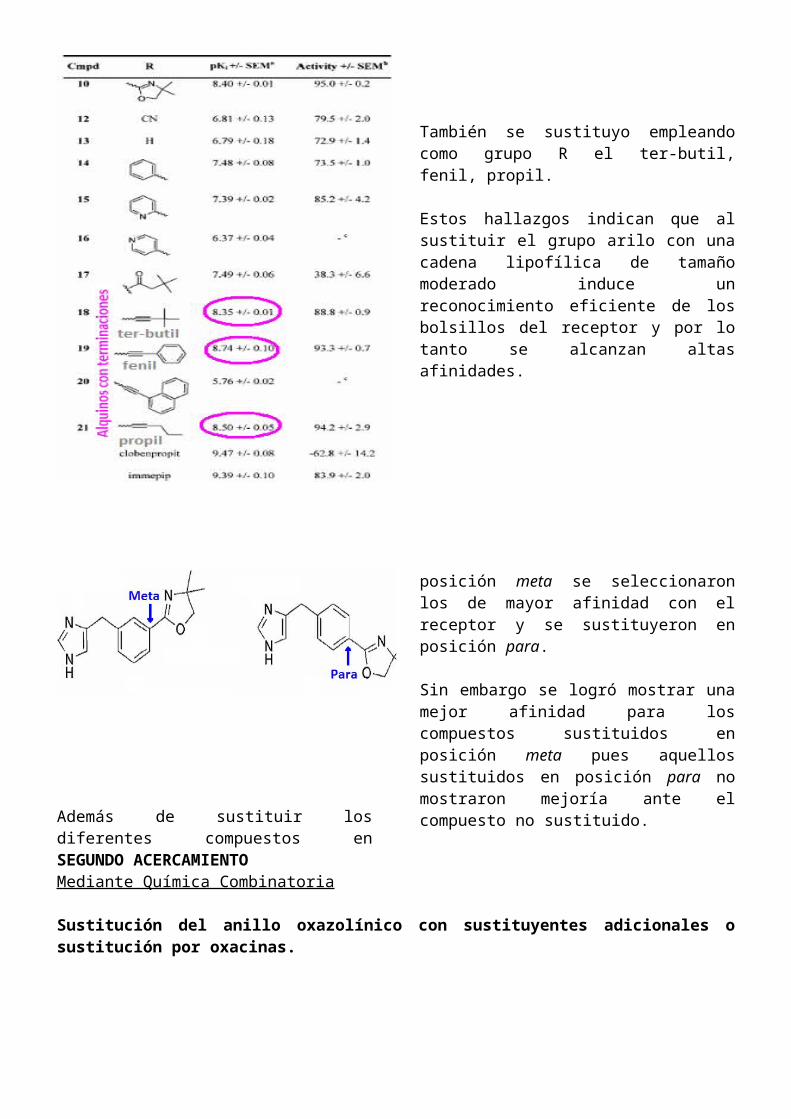
El grupo R se fue sustituyendo por cada uno de los grupos mostrados en la figura. Lo importante
acá es notar los valores de pKi.
La lipofilicidad que imparte la cadena arilo fue favorable a la hora de determinar afinidad con el receptor. Esto nos da una idea de que este grupo puede estar interaccionando con un “bolsillo lipofílico”.
Al sustituir con el 2-piridil y con el 4-piridil vemos pki diferentes, esto nos indica que el isosterismo en la posición 2 y 4 en el anillo entre C y N indica que la presencia de un N no juega un papel importante en la afinidad con el receptor sino que un grupo polar (Nitrogeno) más expuesto puede ser desfavorable.

También se sustituyo empleando como grupo R el ter-butil, fenil, propil.
Estos hallazgos indican que al sustituir el grupo arilo con una cadena lipofílica de tamaño moderado induce un reconocimiento eficiente de los bolsillos del receptor y por lo tanto se alcanzan altas afinidades.
Además de sustituir los diferentes compuestos en posición meta se seleccionaron los de mayor afinidad con el receptor y se sustituyeron en posición para.
Sin embargo se logró mostrar una mejor afinidad para los compuestos sustituidos en posición meta pues aquellos sustituidos en posición para no mostraron mejoría ante el compuesto no sustituido.
SEGUNDO ACERCAMIENTO Mediante Química Combinatoria
Sustitución del anillo oxazolínico con sustituyentes adicionales o sustitución por oxacinas.
No vamos a sustituir todo el grupo tiazina, sino simplemente en los carbonos 1 y 2. Esto en los anillos.
Se usó química combinatoria para:
Optimizar la subestructura oxazolínica.Explorar los efectos generales de sustituir los carbonos del anillo de oxazolina en su estructura de anillo de 5 carbonos y en la de 6 carbonos (oxazinas).
Se seleccionaron 42 amino alcoholes con ayuda de un modelo farmacóforo. Un total de 6 compuestos presentaron una alta afinidad por el receptor hH3R mientras que el resto presentaron valores bajos de pKi.
La sustitución dimetílica en el C1 es la que más beneficia la afinidad del compuesto.
Se demostró que:
De todo esto se puede apreciar que la presencia de un sustituyente oxazolina en posición meta a la vez sustituido en una posición cercana al nitrógeno es beneficioso para una alta afinidad.
Actividades funcionales en hH3R y gpH3R
Se monitoreó la actividad funcional de los compuestos asociados a proteínas Gi en el receptor H3R. Recordemos que para valores de pKi mayores a 6.5 se inhibe la actividad funcional. Este rastreo reveló que, sin alguna excepción, todos los compuestos fueron agonistas del receptor hH3R. La misma conclusión se derivó del acercamiento B (en el que se empleó química combinatoria) aunque se usó un tipo diferente de método de rastreo (ensayo GTPS).
pEC50= -log (concentración que alcanza el 50% de la respuesta máxima)
Diferentes compuestos derivados de los acercamientos 1 y 2 fueron seleccionados para investigación de sus propiedades funcionales sobre el receptor.
Todos los compuestos mostraron valores de pEC50 que correspondieron aceptablemente con sus valores de pKi.
La actividad funcional más alta fue la obtenida por los compuestos 10, 18 y 26.
Un alto valor de pki es posible relacionarlo con un alto valor de pEC50.

Mediante diseño de drogas asistido computacionalmente se explicó la fijación y las propiedades de los compuestos, los cuales son relativamente rígidos y poco básicos a excepción del grupo imidazol.
La observación clave es que el agregar una cadena lateral hidrofóbica es tolerada y en algunos casos aumenta la afinidad hacia el receptor hH3R y esto demuestra que el bolsillo en el cual interactúa el compuestos es lipofílico.
Algunos estudios de modelaje molecular revelan dos tipos distintos de bolsillos lipofílicos para fijación con antagonistas.
Estas son diferentes técnicas de análisis utilizadas:
Sybil16.8 = obtener la geometría de los ligandos H3R. CONFEX = determinar los espacios conformacionales. DISCOtec y FlexS = para encontrar características farmacóforas en común con los diferentes resultados.
Además se probaron los compuestos de la figura para asegurar máxima cobertura de las características farmacóforas.
Comparando las características de los diferentes compuestos se obtiene que uno de los bolsillos están disponible para ligar agonistas.
Este acople entre el bolsillo y el grupo lipofílico sitúa al anillo imidazol en superposición con la amina primaria de la histamina.
Los antagonistas y los agonistas comparten ciertas características como el sistema aromático de deslocalización de electrones , el grupo polar y el lipofílico encontrado en varios antagonistas.
CONCLUSIONES
Se exploró sistemáticamente el uso de un núcleo 4-benzyl-1H-imidazol y una cadena lateral de oxazolina buscando agonistas hH3R.
Se usó síntesis orgánica clásica y química combinatoria para obtener alrededor de 55 compuestos.
Se prefiere sustitución meta del grupo benzyl a la para.
Todos los compuestos presentaron actividad agonista.
Se usó la técnica de modelaje molecular por medio de la cual se conoció que el grupo lipofílico periférico de los agonistas ocupan el bolsillo hidrófobo del receptor para el cual era previamente conocida su actividad ante antagonistas.
La potencia clínica de los compuestos está comprometida por la afinidad hacia los receptores H3 y H4 (es la relación entre la pKi y la pEC50).

PREGUNTAS/COMENTARIOS.
Al inicio del artículo se menciona que es importante el H3 en tratamientos farmacológico (enfermedades mencionadas en el primer seminario de esta transcripción). El campo de los antagonistas tiene un área de aplicaciones más amplio que en el caso de los agonistas que es un área más puntual, por eso no se ha visto tanto auge de los agonistas.
La pki me indica la afinidad.
¿El bolsillo?
R/ Ok tenemos una secuencia de aminoácidos que interaccionan con un compuesto. Cuándo la proteína toma su configuración y es posible que interaccione con el fármaco, no se refiere específicamente a la interacción…es como una bolsita, es el bolsillo.
SEMINARIO 3: Pamela Meléndez, Melissa Salazar.
SYNTHESIS, STRUCTURE-ACTIVITY RELATIONSHIPS, AND BIOLOGICAL PROFILES OF A QUINAZOLINONE CLASS OF HISTAMINE H3
RECEPTOR INVERSE AGONISTSTsuyoshi Nagaseet all
Receptores H3
Están acoplados a Proteínas G, su efecto principalmente es a nivel de Gi donde inhiben la adenilato ciclasa y por lo tanto el AMPc intracelular. Se expresa predominantemente en SNC (acá es donde más nos va a interesar). Además puede actuar como autoreceptor permitiendo una regulación negativa de la liberación-síntesis de histamina y modula liberación de otros NT.
Agonista Inverso H3
Disminuye señalización constitutiva de H3
Bloqueo de inhibición de liberación de Histamina
Potenciación de efectos histaminérgicos
Objetivo de la Publicación
Obtener nuevos fármacos no-imidazólicos agonistas inversos del receptor H3, para ser utilizados posteriormente en el manejo de trastornos a nivel de SNC.
Los autores se refieren a la capacidad de los antagonistas para bloquear la retroalimentación negativa de histamina. Usan agonistas inversos H3 no imidazólicos. El problema con los agonistas imidazólicos es que afectan inhibitoriamente el CTP450 (mal aclaración hepático).
Hipótesis
Los compuestos no imidazólicos derivados de quinazolinonas presentan actividad agonista inverso/antagonista de los receptores histamínicos H3, por lo tanto son candidatos a desarrollo clínico para usarse en tratamiento de padecimientos del SNC.
Metodología
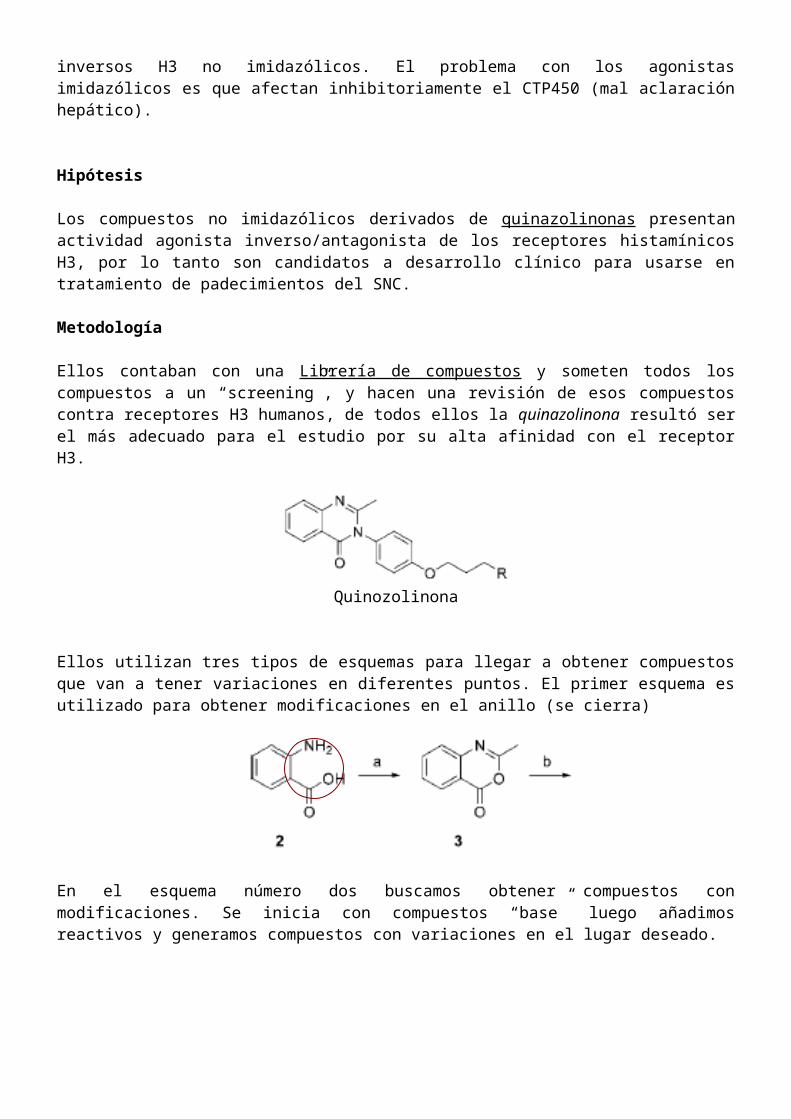
Ellos contaban con una Librería de compuestos y someten todos los compuestos a un “screening”, y hacen una revisión de esos compuestos contra receptores H3 humanos, de todos ellos la quinazolinona resultó ser el más adecuado para el estudio por su alta afinidad con el receptor H3.
Quinozolinona
Ellos utilizan tres tipos de esquemas para llegar a obtener compuestos que van a tener variaciones en diferentes puntos. El primer esquema es utilizado para obtener modificaciones en el anillo (se cierra)

En el esquema número dos buscamos obtener compuestos con modificaciones. Se inicia con compuestos “base” luego añadimos reactivos y generamos compuestos con variaciones en el lugar deseado.
Quinazolinona
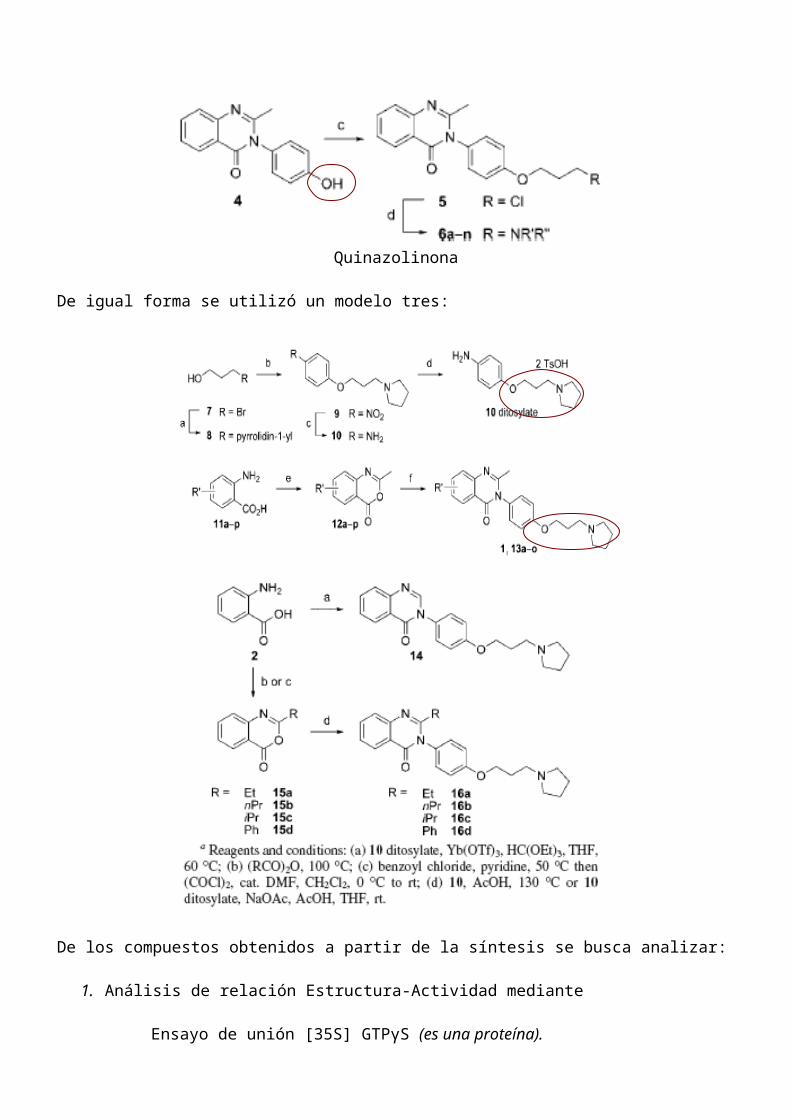
De igual forma se utilizó un modelo tres:

De los compuestos obtenidos a partir de la síntesis se busca analizar:
1. Análisis de relación Estructura-Actividad mediante
Ensayo de unión [35S] GTPγS (es una proteína).Evaluación de actividad inhibitoria del canal de potasio hERG
2. Perfiles in Vitro e in Vivo.
Ensayo de unión [35S] GTPγS
Lo que hacemos es una medición del nivel de activación de Proteína G, luego de la ocupación de un agonista en un receptor acoplado a Proteína G. Se determina la unión del [35S] GTPγS (análogo no hidrolizable) a las subunidades Gα..
El asunto esta así: llega el agonista y se une al receptor H3 (el cual es acoplado a proteínas G), ocurre la separación de subunidades. Si hay activación y separación, hay unión del compuesto a la subunidad Gα (una vez disociada), esto es lo que cuantificamos. Un valor bajo me indicaría poca unión de la proteína a la subunidad y por lo tanto menor fue la activación del receptor por parte de los compuestos estudiados, estaría actuando como un agonista inverso.
Este ensayo es utilizado para determinar parámetros farmacológicos como potencia, eficacia y afinidad antagonista.
Evaluación de actividad inhibitoria del canal de potasio hERG
El gen hERG codifica la subunidad α del canal de potasio. Este canal se encarga de coordina la repolarización eléctrica del potencial de acción en miocitos. Este canal de potasio va a estar susceptible a unión con otros fármacos (y lo bloquean) debido a que tiene un vestíbulo interno de gran tamaño, provee amplio espacio para que los fármacos se unan y bloqueen el canal prolongación del intervalo QT.
Este ensayo lo hacen para verificar que los fármacos que vayan a desarrollar no tengan este problema. Prolongaciones en el intervalo QT puede desencadenar arritmias y un paro cardíaco.

Resultados y Discusión
Ellos establecieron como compuesto más afín el primero de la imagen siguiente, el resto de estructuras son posibles sustituyentes a la cadena R.
Hay otros tipos de compuestos que provocan bloqueo del canal de potasio, como conclusión de las expositoras (considerando información del avendaño) probablemente la prolongación del intervalo QT este asociada la presencia de nitrógeno, el cual puede llegar a formar iones de amonio cuaternario que como ya sabemos puede bloquear el canal.
Ellos eligieron el compuesto de arriba como el más apto, seguidamente van a estudiar la relación de estructura-actividad de cada uno de los compuestos formados al sustituir la cadena R. Ellos hacen modificaciones del segmento terminal, donde está el R Estos cambios lo hacen más que todo formando ciclos con diferente número de carbono (el resto de estructuras en la imagen de arriba son los sustituyentes). Las estructuras formadas con los sustituyentes 6b (anillo de 5) y 6c (anillo de 6) demostraron tener la mayor relación estructura-afinidad con el receptor de H3.
Luego a estos dos compuestos le generan modificaciones en los sustituyentes. Sustituyen el hidrógeno por un grupo metilo en el caso de este anillo de 5. De manera similar para el anillo de 6. Resulta ser que luego de estos cambios el compuesto 6f y 6k tienen mejor relación estructura actividad.
Ellos evalúan potencia por medio de la unión a la proteína G y también evalúan la concentración inhibitoria 50 (IC50). La potencia la establecen con la concentración de la subunidad alfa unida a la proteína (columna a la izquierda de la tabla), la cual debería ser pequeña, ya que de esa manera a una menor concentración yo esperaría ver resultados (menor concentración mayor potencia).
La IC50 para el canal de potasio, deseo que sea mayor. Así, solo a concentraciones altas esperaría este resultado no deseado.

Seguidamente sustituyen en el punto R1 y R2 para evaluar también la relación estructura actividad. Ellos concluyen que en la posición R1 el mejor grupo que puede estar es un metilo y en la R2 es posible tener varios grupos. Presentan una potencia elevada y poca interferencia con el canal.
R1: MeR2: 5-CF3, 5-MeO, 6-MeO, 8-F
Otra prueba realizada fue con respecto al aclaramiento. Con los compuestos iniciales sustituyeron la cadena R1 por grupos metilo y la R2 con otros grupos (ver tabla de abajo). De igual forma se evaluó si son sustratos de la proteína P. Todos demuestran tener un buen aclaramiento en ratas. Con respecto a ser sustratos de proteína P, esto lo analizan mediante un estudio de transporte celular (una tasa de transporte celular). Al final los autores dicen que sí los compuestos tienen una tasa de transporte igual o menor a 3, NO son buenos sustratos de la proteína P. No queremos que sean sustratos de la protéina P porque los sacaría del SNC y no los dejaría realizar su acción.
El compuesto 1 y el 13f además de tener un buen aclaramiento, no son sustratos de la proteína G.

En otro estudio que ellas leyeron, a partir de este estudio y con los resultados acá mostraron los autores se dedicaron darle rigidez a la cadena. Para ello ciclaron el segmento y esa parte es la que podemos establecer como el farmacóforo. Perfiles in Vitro e in Vivo para el compuesto 1
Con estos estudios se determina que el compuesto 1 tiene:
Potente afinidad a receptores H3 en humanos, ratas y monos rhesus.
Unión basal reducida de GTPγS indican agonismo inverso.
Antagoniza potentemente R-methylhistamina (agonista selectivo del receptor H3) ydesplaza curva dosis-respuesta a la derecha.
Excelente potencia, selectividad y perfil farmacocinético (biodisponibilidad).
El compuesto 1 eleva significativamente los niveles de histamina en el cerebro de ratas.
Estudios de potencia indican un alto grado de ocupación en receptores H3 cerebrales y bajos niveles plasmáticos en humanos.
Se realiza un estudio de ocupación del receptor ex Vivo, observándose que más del 90% esta ubicado en cerebro de las ratas. El sustrato importante a nivel de la proteína P en roedores tiene una limitada eficacia, ya que está limitada al flujo de esa proteína. En humanos, esto no es tan bueno por lo tanto se espera que haya una ocupación mayor al 90% (mayor penetrabilidad y mejor ocupación).
Efectos Adversos.
Perros anestesiados: Administración IV de 3 mg/kg, no se observaron efectos cardiovasculares relacionados con el tratamiento.
Ratones: Administración oral de 100 mg/kg no registró cambios en actividad psicomotora, tono muscular, excitación del SNC, respuesta autónoma ni reflejos.
Conclusiones
El compuesto 1 tiene potente actividad agonista inverso del receptor H3 y excelente selectividad sobre otros subtipos de receptores de histamina.
El compuesto 1 mostró perfiles farmacocinéticos y penetrabilidad cerebral satisfactorios en estudios preclínicos.
El compuesto 1 fue seleccionado como candidato a desarrollo clínico para ser utilizado como potencial tratamiento de varios trastornos del SNC.
PREGUNTAS/COMENTARIOS.
Los antagonistas imidazólicos generan problemas por disminución del CYP450, el aclaramiento hepático se ve disminuido. Acá buscamos otras opciones.
La librería de compuestos les permitió notar la afinidad por el receptor H3. Es una librería de muchos compuestos (no solo de posibles agonistas de H3).
La potencia aunque no se menciono también se evalúa con la pKi.
La movilidad de la cadena se limitó ciclando la cadena, de esta manera se restringió la rotación de la cadena.
La quetiapina (fármaco imidazólico) altera el canal de potasio. Los que alteran esto son los imidazólicos y algunos antiarrítmicos. Además, algunos fármacos (otros no necesariamente imidazólicos) que por sus características a bajas dosis pueden alterar el canal.
En la presentación se menciona el nombre de un antiarritmico pero no logro escuchar bien.
SEMINARIO 4:Octavio Masís.
FRAGMENT BASED DESIGN OF NEW H4 RECEPTOR-LIGANDS WITH ANTI-INFLAMMATORY PROPERTIES IN VIVO
NOTA: De este seminario solo se vio la mitad, además existieron muchos problemas por la instalación del programa en la computadora. En fin, en la grabación solo se habla de figuras superponibles (utilizando el programa) pero no se comenta mucho diferente a la presentación.
Este artículo es sobre H4, el cual es muy nuevo y se conoce poco. El articulo abarca diseño, síntesis, y actividad.
La histamina es una amina endógena, su receptor está acoplado a proteína G.
Receptores de Histamina
H1R. Inflamación y Vasodilatación. Antagonista: tx de alergias (loratadina).
H2R. Regulación en la secreción de H+ gástrico. Antagonista: tx úlceras gástricas (cimetidina, famotidina)
H3R. Procesos cognitivos, alimentación, sueño y cansancio. Antagonista: ADHD, demencia y narcolepsia
H4R.
Difiere respecto a H1R y H2R
Considerable identidad con H3R (31% global y 54% transmembrana).
Procesos inmunológicos e inflamatorios
Expresa en células hematopoyéticas, Eosinófilos, Mastocitos y macrófagos.
También en células periféricas: timo, médula ósea y bazo
Ligandos de H4R
Los autores presentan dos moléculas como ligandos:
1. VUF6884Análogo tricíclico de la ClozapinaAlta afinidad por H4RpKi 7.6, agonista completo α=1 (alfa me indica la actividad intrínseca)Análisis de Saturación de enlaces
2. JNJ7777120Antagonista de H4RpKi 7.8, antagonista α=0Desplaza a VUF6884 de su sitio de enlace con H4R
VUF6884 VUF6884
¿Qué se busca?
Similitud entre 1 y 2 (estructura parecida); se propone crear un modelo para el futuro diseño de ligandos H4R. Descubrimiento de fármacos basado en “modelo de alineamiento flexible” de 1 y 2. Esto es lo importante del artículo, saber como hacer un fármaco con un “modelo de alineamiento flexible”.
Ellos traslapan las moléculas y se nota similitud. Obviamente, las moléculas no son flexibles pero se acomodan. Partiendo de ello sintetizan compuestos con esta estructura:
Síntesis

Noten la similitud de los compuestos.
Es diferente que tanto es afín (pKi), a que tanto se pega (Ligand Efficiency). La eficiencia del ligando se analiza con la fuerza del enlace, puede no ser muy afín pero si formar enlaces muy fuertes.
Luego a partir de todas las síntesis inician a formar muchos compuestos (ver todas las síntesis en la presentación)…se comentan todas las síntesis: zas! se pega y se forma…. zas! se pega y se forma…etc.
¿Qué hay hasta el momento?
El compuesto 3 (tabla arriba) resulto ser un buen ligando, este tiene un N en posición cuatro que funciona bien (en la posición 3 no funcionó). Sustituyen en el carbono 6 con un cloro, aumentando la actividad. La piperazina se mantiene. Optimizamos sustituyendo R3 por anillo aromático (18-19-20).
Al hacer un “screening” virtual notan que la molécula tiene un componente hidrofóbico, por lo tanto es necesario un bolsillo hidrofóbico.
TOC TOC…EL DOCTOR GRILLO INTERRUMPE Y SE DETIENE LA EXPOSICION….:)!