tres
DESCRIPTION
FISIOLOGIA VEGETALTRANSCRIPT

TRESOsmosis
Figura 3-1 (a) Osmómetro analógico en un vaso de precipitados, (b) La célula como sistema osmótico.Es acto cotidiano abrir un grifo de agua o descargar un inodoro.
Estamos muy familiarizados con el movimiento del agua como
fenómeno de flujo masivo —nuestros sistemas de plomería se encargan
de éste—; pero en el mundo que nos rodea hay grandes cantidades de
agua que se mueven por difusión, casi siempre sin que podamos
advertirlo. En este capítulo y en los que le siguen veremos cómo es que
la difusión causa gradientes de presión que dan como resultado el flujo
masivo.
Se necesita de cierto esfuerzo mental para visualizar este
aspecto poco familiar del mundo real. Con los ojos de la mente (no hay
otra forma) debemos ver las moléculas de agua que saltan y vuelan
billones de veces cada segundo en el estado gaseoso, y que en el
estado líquido se retienen entre sí mediante puentes de hidrógeno —el
lado positivo de una se aferra al lado negativo de otra—, aun cuando
sus movimientos o efectos cinéticos hacen que algunas escapen. De
alguna manera debemos conceptualizar la entropía, las energías libres
y los potenciales químicos, y cómo estas propiedades pueden hacer
que las moléculas se difundan en un gradiente. Para ello, debe
entenderse que la presión aumenta las energías libres y los potenciales
químicos, mientras que las partículas de soluto y las superficies
mátricas las reducen.
Con estos modelos presentes se está en condiciones de
extrapolar tales conceptos a las células vegetales. Estamos listos ahora
para estudiar la osmosis y temas relacionados con ella.
3.1 Un Sistema OsmóticoUn dispositivo que cuantifica la osmosis se denomina osmómetro. Por
lo común es un aparato de laboratorio, pero una célula viva puede
considerarse un sistema osmótico (Fig. 3-1). En ambos casos, por lo
común se presentan dos situaciones: Primero, dos o más volúmenes de
solución o agua pura están aislados entre sí por una membrana que
restringe el movimiento de las partículas de soluto, más de lo que
restringe el de las partículas de solvente. Segundo, por lo común hay
manera de permitir que la presión se eleve en al menos uno de los
volúmenes. En un osmómetro, las presiones típicamente se elevan por

efecto hidrostático, elevando la solución en el tubo contra la gravedad.
Pero pueden usarse otros métodos, como un detector de volumen (por
ejemplo, un haz de luz y una fotocelda) capaz de incrementar la
presión en el sistema (con un émbolo
o un pistón, por ejemplo) tan pronto como el volumen del líquido
comienza a expandirse en respuesta al primer pequeño incremento en
la presión. En la célula vegetal, la rigidez de la pared celular es la causa
del incremento en la presión.Figura 3-2 Ilustración

esquemática de diversos factores que influyen en el potencial hídrico. Los rectángulos negros representan el potencial hídrico (eje de las ordenadas). Las flechas onduladas indican disminución en el potencial hídrico debida a solutos (el componente osmótico o de solutos del potencial hídrico). Las flechas rectas sugieren el efecto de la presión en el potencial hídrico. (Las flechas hacia arriba representan presión positiva, y hacia abajo, negativa.) Las líneas dentadas indican los efectos de las humedades relativas, por debajo del 100%, sobre el potencial hídrico de la atmósfera (lo cual se considera más adelante, en los Caps. 4 y 5). (a) Efectos básicos de solutos y presiones sobre el potencial hídrico, solos y en combinación, (b) Difusión del agua líquida en respuesta a gradientes de potencial hídrico, que muestra cómd cambia el potencial hídrico a medida que ocurre la difusión en un osmómetro. (c)Difusión del agua a favor de un gradiente del potencial hídrico más negativo, desde el suelo a la atmósfera, a través de un vegetal, y desde el agua de mar. Este último caso (nótese la tensión en la savia del xilema) se analiza en el Cap. 5 (véase la Fig. 5-17)tes del potencial hídrico son las fuerzas de solutos y mátricas, que
reducen dicho potencial, y la presión, que lo incrementa.
El potencial hídrico de un sistema expresa su capacidad de
realizar trabajo, comparada con la capacidad que hay en un sistema
con una cantidad comparable de agua pura a la misma temperatura y a
la presión atmosférica. El potencial osmótico de una solución es ne-
gativo porque el agua, como solvente en la solución, puede hacer
menos trabajo que el agua pura. A medida que aumenta la presión
sobre la solución, también aumenta la capacidad del solvente de
realizar trabajo (y por tanto el potencial hídrico de la solución).
4 Con unidades SI, la transformación es directa y simple:
Unidades de energía: J mol 1
Definición del joule en el SI: J = (newton)-(metro) = Nm

Por consiguiente: J mol 1 = N ni mol 1
Unidades para el volumen molar parcial de agua: m} mol 1
Unidades para el volumen específico de agua: m-' kg 1
Entonces:El trabajo lo efectúa el movimiento del agua pura hacia la
solución. En un osmómetro, una solución ideal 1.0 moIaP de azúcar
(glucosa, por ejemplo) a 28°C tiene un potencial osmótico de -2.5
kilojoules por kilogramo, o sea -45 J mol'1 (-10.75 cal mol'1), lo cual
significa que el trabajo máximo que puede realizarse mientras el agua
pura llega al equilibrio con la solución en el osmómetro es de 2.5 kj
kg'1, o 45 J mol ', de solución (véase la ecuación 3.2).
Para hacer la conversión usando el volumen molar parcial de agua (18 cm' mol 1 = J 8 x 10'’ m' mol ')Para el agua: 1 kg = 1,000 g: peso molecular = 18.015 g mol 1 Así: 1 kg = 55.5 mol
Es lógico utilizar unidades de energía cuando se estudia el
trabajo realizado por soluciones, pero tradicionalmente los especialistas
en fisiología vegetal expresan el concepto de potencial hídrico (que ha
recibido diversos nombres en los últimos cien años) en unidades de
presión. Resulta más sencillo medir la presión en la membrana de un
osmómetro de laboratorio (o calcularla, si se conoce la densidad de la
solución) que medir la cantidad de energía libre que se requiere para
elevar el agua en el tubo. Para una solución 1.0 molal, esta presión en
un osmómetro perfecto es igual a 2.5 megapascals (MPa; 2.5 MPa = 25
bars, 24.67 atmósferas, 18.75 metros de mercurio o 25-49 kg cnr2; el
pasca! se define como una fuerza de un newton por metro cuadrado).
En la célula, el trabajo se realiza por estiramiento de la pared celular.
Hay que recordar que el trabajo real lo efectúa el agua pura, que posee
el mayor potencial hídrico. Esto lo indica el hecho de que el potencial
hídrico de una solución tiene signo negativo.
Las unidades de presión y energía para el potencial hídrico son
1 más fría en una solución más fría.| . Como las ecuaciones de la termodinámica que se han presentado aquí sólo son válidas a temperatura constante, no es posible utilizarlas para calcular las fuerzas que pueden presentarse por termoósmosis, una situación \ en la cual el agua caliente se puede separar de la fría• mediante una membrana que permite que se acumule ; la presión en el agua fría, a medida que el agua caliente SC difunde en ella (véase la Fig. 2-4). Sin embargo, los : efectos de solutos, presión y temperatura sobre la pre-- Stónde vapor sugieren que las presiones altas podrían

fáciles de relacionar entre sí, como fue sugerido por Taylor y Slatyer en
un artículo presentado en 1962 (véase también Kramer, 1983; Slatyer,
1967).
Las unidades de presión se obtienen al dividir las unidades de energía
entre el volumen molar parcial (el volumen de 1 mol de H20, o sea
18,000 mm1 mol2), o el volumen específico del agua (103 mrn’ kg'1 = 1
cm? g'1).* Así, el potencial osmótico de una solución de glucosa 1.0
molal a 28°C (o el potencial hídrico de la misma solución, a la presión
atmosférica) puede expresarse en términos de energía como -2.5 kj
kg1, y en términos de presión como -2.5 MPa. Con unidades SI, los
kilojoules por kilogramo tienen el mismo valor numérico que los
megapascals.
En 1887, J. H. Vant’Hoff descubrió una relación empírica que
permite el cálculo aproximado de un potencial osmótico, a partir de la
concentración molal de una solución. Gráfico los potenciales osmóticos
obtenidos de lecturas directas de un osmómetro en función de la
concentración molal. El resultado fue la siguiente relación, cuya forma
es idéntica a la de la ley de los gases ideales:
1 Molalidad = moles de soluto por kilogramo de H¿0. Como se hi2zo notar en el Cap. 2, esto expresa las relaciones osmóticas de ma3el Cap. 2), y permaneceríamos incrédulos sólo si olvidáramos que los potenciales osmóticos que se calcularon se comparan con el agua pura a presión atmosférica ya la misma temperatura. El hecho de que la solu-ción más caliente tenga un potencial osmótico más negativo que la más fría, significa que el agua pura caliente, aúna atmósfera de presión y a la misma temperatura que la solución en el osmómetro, produciría una presión de equilibrio superior a la que podría causar el agua

progresiva un estrés hídrico, la presión de turgencia de las células foliares decrece hasta que alcanza un valor de cero, cuando el potencial hídrico foliar ¡guala al potencial osmótico. Argumenté que en este punto las hojas permanecerían marchitas de manera permanente y que, en consecuencia, era razonable esperar que cesara todo crecimiento. Sin embargo, aún con el cierre de estomas, podría esperarse que la transpiración continuada hiciese disminuir aún más el agua edàfica, hasta que la desecación de la planta en sí alcanzase niveles letales. De este argumento se seguía que el porcentaje de marchitez permanente no debería considerar se una constante del suelo, sino nada más una expresión del contenido de agua del suelo al que los potenciales hí- dricos de éste y del agua están balanceados, en un nivel igual al potencial osmótico de las células foliares, de manera que existe una presión de turgencia de cero.
De forma un tanto sorprendente estas consideraciones, publicadas en artículos tanto experimentales como de revisión, fueron aceptadas rápidamente por la comunidad científica, y desde entonces han sido confirmadas, en sus aspectos generales, por numerosos investigadores. Por su puesto que, mientras tanto, algunas de las afirmaciones más específicas han debido ser modificadas, pero en general ya estaban bien establecidas la naturaleza dinámica de la interacción suelo-planta-aire y el porcentaje de marchitez permanente.
Este punto de vista también pareció proporcionar una base mejor para la colocación entre especialistas en vegetales y en suelos, interesados en las interacciones planta- medio, y evidenció la necesidad de un término más integrativo para describir el estado del agua en plantas y suelos. Durante la década de 1950, para hablar de lo mismo ambosgrupos de científicos hacían uso de los términos "déficit en presión de difusión'', "estrés total de humedad en suelo" y otros relacionados. Este asunto finalmente maduró, de manera informal, en una comida en un restaurante de Madrid, durante una conferencia sobre relaciones hídricas en vegetales organizada por la UNESCO y precedida por, entre otros, Sterling Taylor, Wilford Gardner, Robert Hagan,Fred Milthorpe y yo. Propusimos el término "potencial hídrico'' (water potential) (ya sugerido años antes por edafólogos), basado en aspectos termodinámicos del potencial químico del agua, como un término único para especialistas en suelos y en plantas, divisible en sus potenciales componentes según fuera conveniente. En la reunión se nos pidió, a Sterling Taylor y a mí, redactar una carta a Nature y un artículo más definitivo sobre el tema, así que, a partir de este comienzo bastante informal y personal, se propuso la nueva terminología. Hasta donde yo sé, en la actualidad se usa de manera casi universal, aunque también ha sido mejorada mediante modificaciones y precisiones.
Comencé este breve ensayo haciendo referencia a la emoción del descubrimiento científico. Lo concluyo refiriéndome al espíritu de cooperación que ha existido en la parte de la comunidad científica a la que he estado asociado. Aparte de que siempre ha sido un reto el poder contribuir con un trabajo original, mi vida se ha visto enriquecida por las fraternales relaciones personales que se han desarrollado, tanto con mis colegas inmediatos como con los de campos relacionados, con quienes al principio uno entra en contacto por correspondencia, o en conferencias, y luego compartiendo espacios e instalaciones en un laboratorio común.acumularse del lado frío si se pudiera construir un ter- moosmómetro perfecto. En la Fig. 2-5 se muestran las magnitudes relativas de los efectos de solutos, presión y temperatura
sobre la presión de vapor y, por implicación, sobre otras propiedades de las soluciones que se relacionan con el potencial hídrico. El efecto de la temperatura es mucho mayor que
los efectos de los solutos o la presión.
Apliquemos la ecuación de van’t: Hoff (ecuación 3-2) a un ejemplo en el que la ionización causa que i tenga un valor diferente de 1.0. Considérese una solución 1.0 molal de
NaCl a 20°C. El cloruro de sodio se ioniza 100% en soluciones diluidas, pero el valor de i no es el que podría predecirse a partir de esto. Dos iones por cada fórmula (NaCl) podrían
sugerir que i = 2; cuanti- ficaciones reales revelan que i = 1.8:

en donde4>sl = potencial osmótico antes de la dilución
V, = volumen antes de la dilución
Vs2 = potencial osmótico después de la dilución
V2 = volumen después de la dilución
Por otra parte, la curva para el potencial de presión es más hipotética. Su forma depende del diámetro del tubo del osmómetro, o de las propiedades elásticas de la pared
celular; es pronunciada si el tubo es estrecho
o la pared es rígida, y más tendida s¡ el tubo es ancho
o la pared es menos rígida. En realidad, al principio las paredes celulares se estiran con facilidad; cuando la presión se incrementa, su resistencia aumenta algo, hasta que se
hace más bien constante (McClendon, 1982). Esto lo sugiere la curva para la presión en la Fig. 3-3. La curva para el potencial hídrico es la suma algebraica del potencial de presión y
el potencial osmótico (ecuación 3.1).
El diagrama de Hófler es un buen medio para visualizar los principios de la ecuación 3-1, junto con las complicaciones que trae la dilución. Describe lo que ocurriría si en
soluciones con diferentes potenciales osmóticos se colocaran células maduras, de forma que éstas no ganaran ni perdieran agua sino que la cantidad total de solutos en su interior
permaneciera constante. Algunos de estos aspectos se considerarán más adelante en este capítulo. Con el diagrama de Hófler también puede describirse lo que pasa en algunas
células vegetales en condiciones normales, al menos durante cortos intervalos de tiempo, aunque las células en crecimiento no actúan como sugiere el diagrama. Por un lado, el
potencial osmótico suele permanecer más bien constante en las células en crecimiento, a medida que absorben o producen solutos en su interior (o ambas cosas) (Cap. 16).
Además, cuando las células crecen, sus paredes se ablandan (Cap. 16 y 17), por lo que se estiran de manera irreversible (plástica), y con frecuencia la presión en ellas disminuye en
vez de aumentar (Rayle et al., 1982).
3.5 La membranaExiste una amplia variedad de membranas pero la osmosis se presenta cualquiera que sea el funcionamiento de la membrana, siempre que el movimiento de los solutos esté
restringido en comparación con el movimiento del agua (Fig. 5-4). La membrana puede consista* en una capa de alguna sustancia en la cual el solvente CS más soluble que el
soluto, de modo que permite el paso de más moléculas de solvente que de soluto. Una capa de aire entre dos soluciones acuosas constituye una barrera que restringe por completo
el movimiento de solutos no volátiles, mientras que permite el paso de vapor de agua. Un tercer modelo de membrana
Figura 3-4 Diagrama esquemático de cuatro posibles mecanismos membranales. Los puntos negros representan moléculas de agua, con diámetro aproximado de 0.3 nm; los círculos representan moléculas de sacarosa de alrededor de 1.0 nm de diámetro, y las membranas se trazan a la escala de casi todas las membranas celulares, con espesor de 7.5 nm. Nótese que la concentración del agua es casi la misma a ambos lados de las

membranas. Las moléculas de agua se desplaza con rapidez a través de las membranas en las células, quizá por mecanismos similares a los que se representan con los modelos a y c. Esto se analiza en el Cap. 7. El modelo d es un refinamiento del modelo c, como se estudia en el texto. El modelo b, para vapor de agua, es aplicable a plantas y suelos en diversas circunstancias, pero aún no se conoce una membrana que tenga poros llenos de gas.que podemos visualizar es un tamiz con orificios de tamaño tal que las moléculas de agua pueden pasar, pero no las partículas de soluto mayores. Veremos en el Cap. 7 que el agua
atraviesa con rapidez las membranas celulares —y por tanto es ligeramente soluble en ellas— y que las membranas celulares también actúan corno si tuviesen poros. Se ha
sugerido que, en suelos

secos, algunas veces el agua pasa en estado de vapor de las partículas del suelo a la raíz.
En 1960, Peter Ray llamó la atención de los especialistas en fisiología vegetal sobre un problema interesante. Cálculos del espesor de ciertas membranas, y de las velocidades
con que ocurre el movimiento osmótico del agua a través de ellas, mostraron que dicho movimiento no podía ocurrir sólo por difusión; las velocidades eran demasiado elevadas. Ray
sugirió que la zona de difusión puede ser muy delgada: una interfaz, digamos entre el agua que se encuentra en los poros de la ; membrana y la solución en el interior del sistema
os-
* ffiótlco. En esta interfaz el gradiente de potencial hí- drico podría ser muy pronunciado, lo que daría por f-tesultado una difusión rápida. Este movimiento rápido del agua a través
de la interfaz hacia la solución po- , dría crear tensión en el agua que queda en el poro, > Empujándola en un flujo masivo (Fig. 3-4d). Este cuar . lo mecanismo de membrana ilustra
una vez más la comicidad de la naturaleza. Debe hacerse notar que aún ion válidas las relaciones termodinámicas (sentido y , .equilibrio).
i El modelo vapor-membrana es un buen ejemplo de ¡ membrana semipermeable verdadera, pero todas las g membranas que existen en las plantas deben permitir el paso de
algunos solutos. Tales membranas son dife- «■dalmente permeables, más que semipermeables. Aun- ¡Sj que las membranas vivas son permeables tanto a ■¿frentes como a
solutos, lo son mucho más a los sol- B «Cates. La permeabilidad de las membranas a los solu- Bjbllptroduce una complicación en nuestro modelo de gj óimosis: determina la
velocidad a la cual un equilibrio, K elablecido por la concentración de soluto y la presión, Httffibia gradualmente a medida que cambian los poten- . - .¿les osmóticos a cada lado de
la membrana en res r 'puesta al paso de partículas de soluto.
3.6 Cuantificación de los Componentes i del Potencial HídricoPoco después de que Otto Renner formulara el concep- ; tOde potencial hídrico en 1915, se desarrollaron métodos para medir dicho potencial y sus componentes. Desde
entonces se han ideado métodos cada vez más novedosos, pero los métodos antiguos pueden ayudar nos a comprender las relaciones hídricas en las plantas. Los métodos recientes
son más útiles. Se resumirán ambos métodos, no tanto como un “recetario” de téc- ¿ nicas útiles, sino como ilustración y aplicación de los [ principios que se han venido
considerando.
R r ,Potencial hídrico
. Probablemente, la propiedad más significativa que pue- de medirse en el sistema suelo-planta-aire es el potencial hídrico. No es nada más el determinante tinal del movimiento
difusivo del agua, sino que con frecuencia es el determinante indirecto del movimiento masivo de ésta, que ocurre en respuesta a los gradientes de presión debidos a tal
movimiento difusivo, Además, en principio y en la práctica, el potencial hídrico es quizá el componente más simple que puede medirse en un sistema osmótico.
Debe recordarse que, en el equilibrio, AV = 0; esto es, V es igual en todas las partes del sistema. Así, una parte de la planta puede ser introducida en un sistema cerrado, y
después de que se ha alcanzado el equilibrio, es posible conocer o determinar V para cualquier otra parte del sistema, y por lo tanto para esa parte del vegetal. Hay varias
posibilidades de aplicación para este principio, de las cuales se muestran tres esquemas generales en la Fig. 3-5.
En el método de volumen dsular (Fig. 3-5b), se coloca una muestra del tejido en cuestión en cada una de una serie de soluciones de concentración variable pero conocida (por
lo común sacarosa, sorbitol, manitol o, mejor, polietilenglicol, PEG; véase la Fig. 3-6. El mejor soluto para tales mediciones es aquel que no pasa con facilidad a través de las
membranas ni daña el tejido. El objetivo es determinen■■ en cuál solución el volumen tisular no varía, lo que indica que no hay ganancia o
molalidadFigura 3-6 Potenciales osmóticos del PEG 4000 a diversas concentraciones. El PEG 6000 produce resultados casi idénticos. (Datos no publicados obtenidos por Cleon Ross con ayuda de un psicrómetro de termopar; los valores de H*s para soluciones de PEG no pueden medirse de manera correcta por depresión del punto de congelación.)
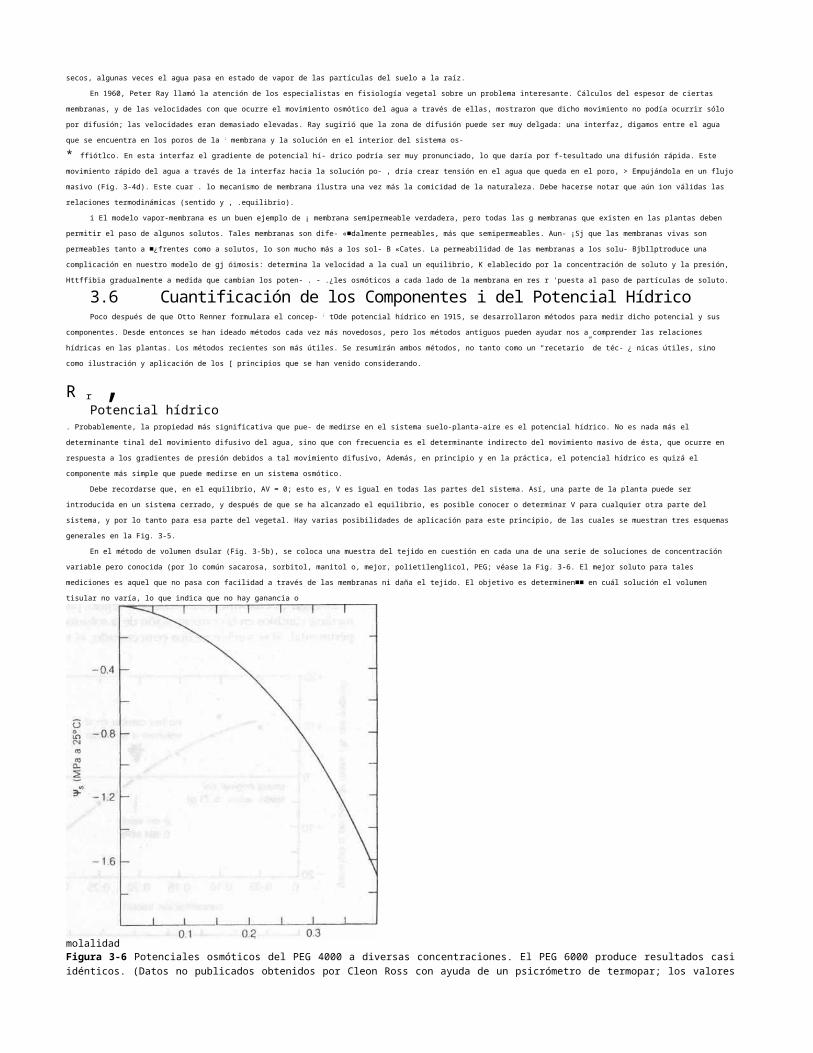
Figura 3-8 Relación entre la humedad en la atmósfera (expresada tanto en términos de densidad de vapor como de presión de vapor) y la temperatura de aire saturado y de aire con humedad relativa (HR) de 50 y 25%. Las líneas discontinuas son un ejemplo que muestra que aire con 100% de RH y a 10°C tiene la misma cantidad de humedad que aire a 21°C y 50% de RH, y que aire a 33°C y 25% de RH.
cada incremento de. 10°C. Si un volumen de aire que está saturado de vapor de agua (HR del 100%) se calienta 10°C más, puede contener alrededor del doble de agua que antes,
así que su humedad relativa es de un 50%. Esta relación se muestra en la Fig. 3-8.
Si se conocen la temperatura absoluta (Ten grados kel- vin), la densidad de saturación de vapor o la presión de agua pura (p°) a esta temperatura, la densidad o presión de vapor en
la cámara de prueba (p) y el volumen molar del agua (Vx en L mol'1), es posible calcular el potencial hídrico a partir de la fórmula siguiente, que se deduce de la ley de Raoult
(ecuación 2.13):
El cociente 100 plp° es la humedad relativa. Si se convierte a logaritmos comunes y se utilizan valores numéricos para R y Vit la ecuación 3 5 se simplifica como sigue:
Con esta ecuación se obtiene el potencial hídrico del aire, cuando se conocen su temperatura y humedad relativa.
En principio es sencillo cuantificar el potencial hídrico en muestras de tejido por este método, pero en la práctica se presentan ciertas dificultades. A éstas se les ha
encontrado solución sólo a partir de la década de 1950, y en la actualidad dicho método es uno de los más utilizados. Entre las dificultades, si se quiere tener la suficiente exactitud
en el método, la temperatura debe ser uniforme, cuando menos hasta un centésimo de grado Celsius. Esto se debe a que ligeras variaciones en la temperatura son causa de
grandes cambios en HR y potencial hídrico a una densidad de vapor constante.5
Otro problema se refiere a la medición de la humedad dentro de la cámara de experimentación. En 1951, en Inglaterra, D. C. Spanner desarrolló un método ingenioso para
solucionarlo, y desde entonces se le han hecho mejoras. Se instalan dos termopares en el interior de la cámara. Uno tiene masa relativamente grande, de modo que su temperatura
permanece cercana a la del aire presente en la cámara. El otro termopar es^ Él aire a un 100% de HR siempre tiene un potencial hídrico de cero. Si aire a 100% de HR y 20°C se calienta hasta 21 °C, su HR desciende a 94.02% y = -8.34 MPa, un valor más negativo que el observado en casi todas las células. De ahí que el intervalo de humedad relativa, en el cual las plantas “crecen bien" (a T constante) sea de 99 a 100%.

frecuencia esto conduce a un cambio en el potencial osmótico, por lo que los valores difieren de los que hay en una célula intacta, pero aun así el método es de uso frecuente.
El punto de congelación (y también la presión de vapor) depende de la fracción molar (ecuación 2.12), y por lo tanto del potencial osmótico. Las propiedades de las soluciones
que son funciones de la fracción molar se conocen como propiedades coligativas. Son punto de congelación, punto de ebullición, presión de vapor y potencial osmótico. El potencial
osmótico puede calcularse a partir de cualquiera de las otras propiedades. Cuando se mide el punto de congelación para calcular el potencial osmótico, se utiliza el método de punto
de congelación o crioscópico. Como se ha mencionado, el potencial osmótico de una solución molal no ionizada, a 0°C es idealmente -2.27 MPa. Su punto de congelación (Af) resulta
ser -1.86°C. El efecto del potencial osmótico sobre los puntos de congelación se mantiene aún para soluciones no ideales, como los líquidos vegetales, así que el potencial osmótico
(osmolalidad) de una solución desconocida diluida puede estimarse a partir de su punto de congelación mediante la re : lación:
La determinación del punto de congelación resulta ser relativamente sencilla: Existen en el comercio termómetros de mercurio con gran exactitud (hasta
0. 01 (>C) y termopares para este propósito, así como instrumentación completa para determinar con exactitud los puntos de congelación de las soluciones. Se obtiene una
curva de temperatura en función del tiempo (Fig. 3-10) para describir el enfriamiento extremo y el subsecuente calentamiento al llegar al punto de congelación, debido a la
liberación del calor de fusión. Los resultados se comparan con los del agua pura.
Obtener savia vegetal pura es más difícil. Es posible exprimir la planta con una prensa, congelar el tejido hasta romper las células y luego extraer el líquido, u homogenizar el
tejido en una mezcladora y filtrar la savia. Todos estos métodos, cuando se aplican al mismo tejido, por lo común clan valores distintos de V,, y la diferencia puede ser de hasta un
50%. Los valores que se obtienen con la mezcladora son casi siempre los más negativos (líquido más concentrado), y los menos negativos se obtienen cuando se extrae a mano la
savia, con ayuda de una manta de cielo (usada para envolver quesos). Un problema importante es que los diversos métodos implican distintos grados de mezcla de contenido
citoplásmico, agua de la pared celular (Markhart et al., 1980) y otras sustancias con el contenido vacuo- lar, que casi siempre es mucho mayor en volumen que el citoplasma. La
vacuola está en equilibrio osmótico con el citoplasma debido a que no hay una barrera rígida entre ellas, y además la gran vacuola proporciona estabilidad osmótica (capacidad de
amortiguamiento) a la célula, como se hizo notar en el Cap. 1. Podrían utilizarse vacuolas aisladas, pero no hay garantía de que el proceso de aislamiento no cambie el V s.
El problema que surge al prensar células vivas es que se obtiene agua casi pura por causa de la filtración osmótica. A pesar de sus limitaciones, el método crioscópico ha sido
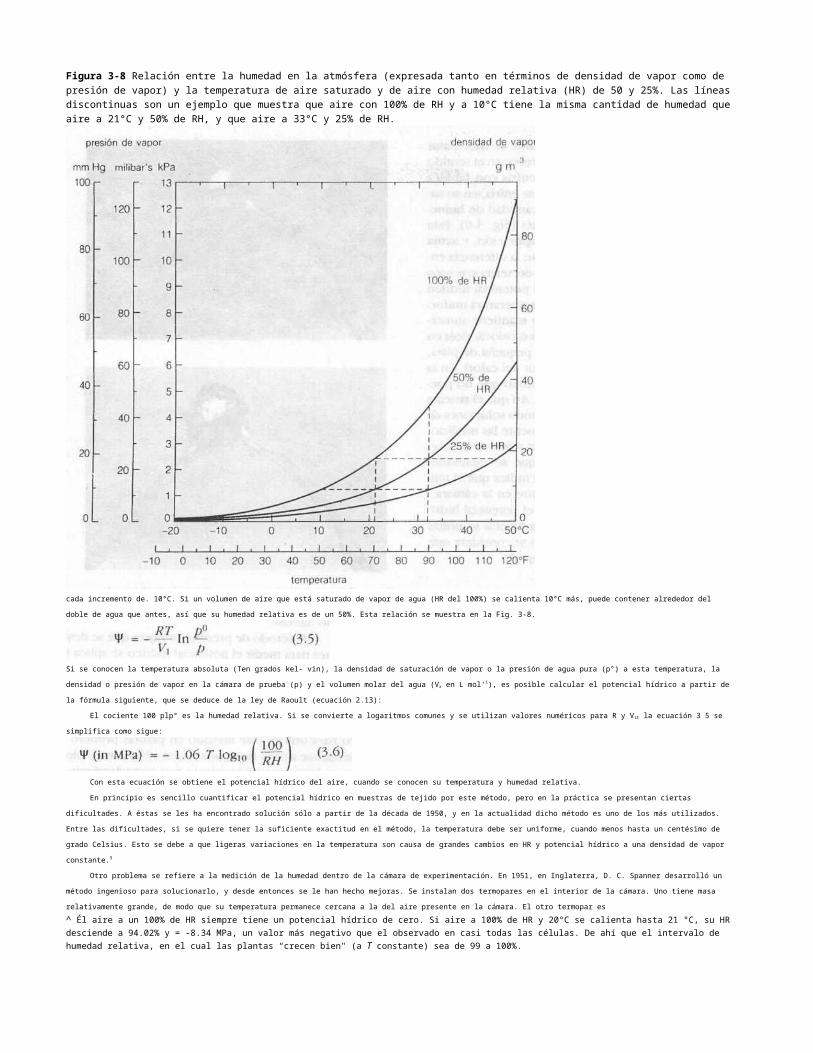
muy utilizado durante décadas.Figura 3-12 Esquema del empl&o de la bomba de presión como método para medir diversos parámetros de importancia para las relaciones hídricas de las plantas. Punto A: En este punto, el valor negativo de la presión equivale al volumen promedio del potencial osmótico del tejido hidratado, de unos -2.1 MPa. Punto B: El punto de pérdida de turgencia (comparable a una plasmólisis incipiente) señala un potencial osmótico aproximado de -3.1 MPa. Punto C: Volumen de agua libre en los tejidos hidratados, alrededor de 5.35 cm3. Punto D: Volumen total de agua tisular, unos 6.72 cm3. El agua confinada (incluyendo algo de agua apoplástica) es igual a D menos C, o sea 1.37 cm3 (véanse Tyree y Hammel, 1972, y Tyree et al.. 1973).
Si los líquidos de un tejido vegetal cualquiera están en equilibrio osmótico con alguna solución externa, circundante a presión atmosférica (AV = 0; P = 0) y no existe presión o
tensión alguna dentro del tejido, entonces el potencial osmótico de esa savia puede ser igual al potencial osmótico de la solución circundante. El problema con tal medición es
obtener una presión de cero en el interior del tejido, sin cambiar las demás propiedades osmóticas más de lo necesario (lo cual puede ser demasiado). Éste es el método de
medición del potencial osmótico por observación de plasmólisis incipiente. Se colocan muestras de tejido en una serie de soluciones con concentración escalonada y de potencial
osmótico conocido. (Pueden utilizarse soluciones de sacarosa o manitol, pero hay sustancias, como PEG, que tienen la ventaja de no penetrar el tejido y que éste no las altera
metabólicamente con facilidad.) Después de permitir un periodo para que se establezca el equilibrio (por lo común de 30 min a 1 h), se examina el teji-
etal., 1965). Para medir el potencial osmótico, se corta una hoja o rama de una planta y normalmente se hidrata (dependiendo de la especie), casi siempre CÓlocando la parte
con el corte en agua pura durante yarias horas o toda la noche, y después en una bolsa de plástico para asegurar un 100% de HR. Algunos es-
- tudios indican que el potencial osmótico por lo gene- ' ral cambia sólo un poco durante este tiempo (también dependiendo de la especie). La rama hidratada se colo- , caen la
bomba de presión (Fig. 3-12) con el extremo del corte afuera. Se aplica presión, y la savia empieza ■ entonces a exudar por dicho extremo. Esta savia es casi agua pura, ya que
exuda por osmosis inversa. (Las
I presiones que se presentan en la bomba se incrementan hasta valores positivos mayores en magnitud que los potenciales osmóticos negativos, por lo que el agua se difunde
hacia el exterior de las células.)
El recíproco de la presión de equilibrio (IIP) se gra-
* flca en función del volumen de savia exudada (V) para obtener una curva característica, como se muestra en1 fcl Fig. 3-12. Al principio, a medida que aumenta la pre- ¡ sjón, la gráfica es curva, pero después (en el punto B) | íehace lineal. En este punto de inflexión (B) las células , se
hallan en el punto de pérdida de turgencia (la presión : interna es igual a cero, y en esencia equivale a la plas- f JBÓlisis incipiente), y el valor negativo de la presión,
i. ; Cn la bomba, es igual al potencial osmótico en las célu las (véanse los artículos de Grant et al., 1981; Hellkvist et al., 1974; Millar, 1982; Pierce y Raschke, 1980; Tyree y
Dainty, 1973; Tyree et al., 1973; Tyree y Hammel, 1972; Tyree yjarvis, 1982; Waring y Cleary, 1967). Al extrapolar la línea hasta un volumen de cero, (punto A), se obtiene el
recíproco del potencial osmótico de las células del tejido original hidratado, y es posible entonces construir un diagrama de Hófler para calcular el volumen de agua en el tejido a esa
presión o cualquier otra. Extrapolar la línea hasta una presión infinita (punto C) da como resultado el volumen de líquido que estaba presente en las células al principio. El agua que
permanece en la muestra después de aplicar la máxima presión disponible puede cuantificarse si la muestra se pasa antes y después de secarla en un horno (punto D). Se supone
que el exceso respecto a la cantidad calculada para el punto C es el agua confinada, que se retiene con gran fuerza en las superficies hidrófilas, aunque también puede incluir
alguna cantidad de agua apoplástica.
Las mediciones de los potenciales osmóticos por estos métodos (en especial el uso de puntos de congelación) han dado resultados que varían entre valores aproximados de -
0.1 MPa en plantas acuáticas, hasta valores en extremo negativos de -20 MPa, o menores, en halofitas (plantas que crecen en suelos salinos). Tales valores pueden variar, pues
están sujetos a todos los pro-

Figura 3-13 Experimento de Green y Stanton. Después de equilibrarse en soluciones de azúcar con diversos potenciales osmóticos, las células de Nitella se punzan con tubos capilares (con un extremo cerrado), como se ilustra en la figura. En el experimento original, las unidades de medición fueron atmósferas, como en la ilustración (1 atm = 0.1013 MPa). (Datos de Green y Stanton, 1967).
■Étpresión en una solución no confinada, a presión at- E too? finca, es de cero. De ahí que la presión celular real nedida por el método de Creen y Stanton será alrede-
Pfoíde 1 atm (como 0.1 MPa) mayor que la presión de WKknencict convencional.■otencial MátricoBjtíuperficies hidrófitas (por ejemplo las de coloides »0 proteínas, almidón y arcilla; véase el ensayo) ad- Enicn agua, y la tenacidad con que lo hacen no sólo Hiende de la
naturaleza de la superficie, sino también ■i distancia entre la superficie y las moléculas de agua ^fesc adsorben: Las que están justo sobre la superfi- Htfsorbcntc serán retenidas
con gran fuerza; las que Bullan a cierta distancia de la superficie lo serán me- B. La adsorción de agua por superficies hidrófitas se Hoce como hidratación o imbibición.
H^potencial mátrico (t) es una medida de la tendencia Hl matriz celular a adsorber moléculas de agua adi- Katcsa la presión atmosférica. Esta tendencia es igual ■fcnacidad
promedio con que se adsorbe la capa me- Euertemente retenida (más distante) de moléculas Hjua. El potencial mátrico se expresa en las mismas Hkdcs de energía o presión que el
potencial hídri-
co, y puede contribuir al V. Una superficie coloidal o hidrófila seca, como papel filtro, madera, suelo, gelatina o el estípite de un alga parda, con frecuencia tiene potencial mátrico en
extremo negativo (tan bajo como -300 MPa), mientras que el mismo coloide, en un volumen grande de agua pura y a presión atmosférica, tiene potencial mátrico de cero (debido a
que está saturado y, por tanto, en equilibrio con el agua). En general, cuando cualquier coloide a la presión atmosférica se halla en equilibrio con sus alrededores, las moléculas de
agua menos fuertemente retenidas tienen la misma energía libre que las moléculas de agua en los alrededores, por lo que el potencial mátrico del coloide es igual al potencial
hídrico de los alrededores.
lín los trabajos modernos se menciona con frecuencia el radio de curvatura de las superficies de agua entre partículas coloidales (el menisco). El menisco es la base del
fenómeno de la capilaridad, que se considera en el Cap. 5 (véase la Fig. 5-3). Cuanto menor sea el radio en la curvatura del menisco, tanto más fuertemente son retenidas las
moléculas de agua por la superficie coloidal o alguna otra superficie hidrófila por hidratación, y más negativo es el potencial mátrico.
Será instructivo mencionar un método moderno de uso común para medir el potencial mátrico. Se coloca un coloide hidratado en una cámara de presión, con

Figura 3-14 Un aparato con placa (o membrana) de presión utilizado para medir potenciales métricos en suelos y otros materiales. Las muestras húmedas se colocan en los contenedores circulares sobre la placa de presión, una placa porosa que permite la difusión de agua. Después de que se coloca la tapa, asegurándola con tornillos de mariposa contra un anillo de caucho, se ejerce presión y el agua empieza a difundirse por la placa y hacia el pequeño tubo que hay dentro del vaso de precipitados (al centro de la fotografía). Cuando deja de salir agua por el tubo (con frecuencia después de 24 h), el potencial mátrico (un valor negativo) de la muestra es numéricamente igual a la presión (valor positivo) en el aparato, como se puede leer en las carátulas, en la parte inferior de la fotografía. Después puede aplicarse mayor presión para expeler más agua, lo que produce una curva de liberación de humedad como la que se muestra en la Fig. 3-15. (Placa de presión del laboratorio de Ray. W. Brown, de Forestry Sciences Laboratory, Logan, Utah; fotografía de Frank B. Salisbury.)
un filtro de membrana sostenido por un tamiz para resistir la alta presión (Fig. 3-14). Los poros de la membrana tienen de 2 a 5 nm de diámetro, por lo que son lo suficientemente
grandes para permitir el paso de solutos y agua, pero no del coloide. La tensión superficial impide el paso de aire a través-de la membrana húmeda. Supóngase (en el caso más
simple) que el coloide está humedecido sólo con agua pura (sin solutos). Entonces se introduce gas comprimido en la cámara de presión. Al aumentar la presión se incrementa hacia
cero el V del agua que se adsorbe en el coloide. Cuando el V de las moléculas que se hallan más lejos de la superficie del coloide alcanza el cero, dichas moléculas empiezan a
difundirse al exterior a través de la membrana. (También puede considerarse que el aire ejerce presión contra los meniscos y reduce los radios de cur vatura.) Posteriores
incrementos en la presión sobre e! coloide dan por resultado aumentos adicionales aunque menores en el desplazamiento del agua desde e¡ coloide a través de la membrana.
Cuando cesa todo movimiento de agua, la que permanece en el coloide a presión estará en equilibrio, a través de la membrana, con el agua pura a la presión atmosférica y a
la misma temperatura en el exterior de la membrana (MJ = 0; esto es, V de las moléculas de agua menos fuertemente adsorbidas en el coloide a presión = 0). Si M* = 0, y si los
potenciales mátrico y de presión son los únicos componentes que influyen en el potencial hídrico del lado del coloide de la membrana:
entonces el potencial mátrico negativo será igual al potencial de presión positivo que produce el aire comprimido (-t = P). Como se conoce la presión, también se conoce el potencial
mátrico negativo.
Usualmente una vez que se ha alcanzado el equilibrio a una presión dada el contenido de agua del coloide se determina pesándolo ante;» y después de secarlo en un horno.
Cuando el potencial mátrico del coloide se grafica como función de su contenido de agua, se Obtiene la curva de humedad liberada (Fig. 3-1“5). Tal curva es esencial para calcular el
flujo de agua en el suelo y ia capacidad de carga de agua.
Una forma de probar la suposición de que la presión final en el aparato con membrana pre.su rizada es una medida del potencial mátrico, consiste en medir el potencia)
hídrico del coloide a la presión atmosférica por el método de vapor, que se describió bajo el encabezado de “'Potencial Hídrico”. Las dos medidas coinci-Figura 3-16 (a) Doble capa de cargas eléctricas con distribución iónica alrededor de una partícula coloidal cargada. Las cargas cercanas a la superficie de la partícula representan cargas superficiales. Éstas son en su mayoría negativas, pero hay tres sitios en que se ven interrumpidas por cargas positivas. La distribución de la carga iónica es estadísticamente opuesta a la de la carga superficial, aunque se observan algunas pocas irregularidades, (b) La presión aumenta en el agua cuando se aproxima a una superficie hidratada con carga. Las líneas representan incrementos en isobars de presión, a intervalos de 30 MPa, a medida que el agua se acerca a una superficie. El incremento en la presión se debe a la atracción entre la superficie cargada y las moléculas de agua (por puentes de hidrógeno) y al incremento progresivo en la concentración del soluto, causado por la atracción mutua de solutos disueltos (iones) y la superficie cargada.

P = 0 (presión atmosférica!
los iones inorgánicos pequeños reducen la energía libre (y por lo tanto el potencial hídrico) de una cantidad de agua mucho mayor (de cientos a millones de veces) que la de una
sustancia hidróíila, como una proteína (ya que el efecto es proporcional a la fracción molar e independiente del peso molecular). Así, en el citosol la contribución del componente
mátrico al potencial hídrico final es probabablemente pequeña, y podría ser despreciable comparada con la de iones y moléculas como soluto. Aún así, el componente mátri- ; co es
muy significativo en la pared celular.
Los efectos mátricos son de especial importancia en las semillas secas y el suelo. Al principio, la captación
i de agua en las semillas depende sobre todo de fuerzas mátricas; las superficies de las proteínas y algunos po- : lisacáridos de la pared celular deben hidratarse antes deque
pueda comenzar la germinación. La madera se- J ca también es hidrófita, y por siglos se han hendido ro-
• cas con ayuda de una técnica que hace uso de esta propiedad: Se taladran agujeros a lo largo de una línea
* de fractura deseada en la roca, se introducen cuñas se-
cas de madera en los agujeros, y se mojan las cuñas, lo cual hace que se hidraten y expandan hasta que rompen la roca. Los hongos en crecimiento, y quizás las raíces y otros
sistemas, también desarrollan poderosas fuerzas de expansión que dependen de la hidración (y la osmosis).
Quizá algún día nuestro conocimiento avance hasta un grado tal que podamos calcular las contribuciones relativas de solutos (potencial osmótico), presión y adsorción de
agua en superficies hidrófilas (efectos mátricos) al estado hídrico real en los vegetales. Mientras tanto, hay que entender que los tres factores participan. y que no es válido (debido
a las complicaciones recién estudiadas) considerar al potencial mátrico como componente del potencial hídrico, en el mismo sentido en que se considera que el potencial osmótico y
la presión son componentes del potencial hídrico. De cualquier forma, cuando tales cálculos se aplican al suelo se obtienen resultados razonables, ya que las fuerzas mátricas
dominan el potencial hídrico de los suelos.nera un tanto más exacta que la molaridad = M = moles de solutopor litro de solución final. El símbolo para moled es m, pero como esta letra también es el símbolo de metro, aquí se escribirá siempre molal. Motar y mola! no son conceptos del SI, pero mol kg 1 (motil) es una combinación válida en uicho sistema. El litro no es una unidad SI, pero puede utilizarse con unidades del SI. El símbolo que se ha recomendado para litro es L(no 1 o t). Nótese que los símbolos literales para estas unidades; son de tipo redondo (vertical); y los símbolos para las cantidades se escriben con cursiva (inclinada) (por ejemplo, longitud = l, concentración = C).