tema 2 estunon/qfiii/nuevo_tema_2.pdfqf iii – tema 2 4 r mecanismos de reacción más complejos,...
TRANSCRIPT

QFIII–TEMA2 1
QUÍMICA FÍSICA III
Tema 2
CINÈTICA MOLECULAR
Revisión: 12/01/2020

QFIII–TEMA2 2
Tema2.CinéticaMolecular
2.1.Introducción
2.2.TeoríadeColisiones
2.2.1.VelocidadesMoleculares
2.2.1.1.Funcionesdedistribucióndelavelocidad
2.2.1.2.Obtencióndelasfuncionesdedistribucióndelavelocidad
2.2.2.VelocidadesCaracterísticas
2.2.3.DistribucióndeEnergías
2.2.4.ColisionesconlasParedes.Efusión
2.2.5.ColisionesIntermolecularesyrecorridolibremedio
2.2.6.ColisionesyReactividadQuímica
2.3.SuperficiesdeEnergíaPotencial
2.4.TeoríadelEstadodeTransición
2.4.1.Hipótesisbásicasydesarrollo
2.4.2.FormulacióntermodinámicadelaTET
2.4.3.LimitacionesdelaTET
2.5.Bibliografía
Apéndices
Ejerciciosadicionales

QFIII–TEMA2 3
2.1.-IntroducciónComoseestudióenlalecciónanterior,elobjetivodelaTermodinámicaEstadísticaerael
dehacerdepuenteentreelmundomicroscópicoyelmacroscópico,permitiendoexplicarypredecir las propiedades macroscópicas de la materia a partir de las propiedades de susconstituyentes (átomos y moléculas), concretamente de predecir las propiedades deequilibrio(termodinámicas)desistemastantoidealescomonoideales.Enestalecciónsevanaabordarsituacionesdenoequilibrio,específicamentelamásimportanteenelcampodelaQuímica, lavelocidaddelastransformacionesoreaccionesquímicas,conunobjetivoqueesexplicar y predecir la velocidad de reacción mediante la Termodinámica Estadística y laTeoría Cinética deGases. Para abordar este objetivo, supóngase, en principio, una reacciónelemental(enunsolopaso)bimolecular,directayenfasegas:
B+C®ProductosExperimentalmente se encuentra que la velocidad de reacción (definida como la
variación con el tiempo de la concentración de uno de los componentes de la ecuaciónquímica)esdirectamenteproporcionalalproductodelasconcentracionesdelosreactivos:
(2.1)
La constante de proporcionalidad se denomina constante de velocidad (expresada
habitualmente en unidades M-1s-1, para una reacción de segundo orden, que no son delSistema Internacional) y se encuentra también experimentalmente que depende de latemperaturaatravésdelaecuacióndeArrhenius:
(2.2)
dondeA(factorpreexponencial)yEa (energíadeactivación)sondosconstantespropiasdecada reacción. De acuerdo con esta ecuación, la representación de ln kr frente a 1/T debeproporcionar una línea recta, como lamostrada en la Figura 2.1, de cuya pendiente puededeterminarselaEa:
(2.3)
Nuestro objetivo es pues dar una justificación teórica, desde un punto de vista
microscópico, a la dependencia de la ley de velocidad con las concentraciones y latemperatura,asícomoobtenerunaexpresiónquepermitacalcularconstantesdevelocidad,sin necesidad de determinarlas experimentalmente. Una vez comprendido a nivelmicroscópicoesteprocesobimolecularsencillo,sepodráabordar,asimismo,elestudiode
[ ] [ ][ ]BCkdtCdv r=-=
RTE
r
a
Aek-
=
dTklndRT
)T1(dklndRE r2r
a =-=

QFIII–TEMA2 4
mecanismos de reacción más complejos, pues éstos se pueden descomponer en etapaselementalescomolaqueesnuestroprimerobjetodeestudio.Esmás,desdelasexpresionesalas que lleguemos debería ser posible incorporar los efectos del medio: disolvente, fuerzaiónica,etc.
El estudio de las reacciones químicas se va a abordar desde tres aproximacionesdiferentes,perocomplementarias,concretamentedesdelaópticade:
a)LaTeoríadeColisionesquesebasaenlaTeoríaCinéticadeGases.b) Las Superficies de Energía Potencial y Dinámicas de Reacción: Constituyen una
aproximaciónalestudiodelasreaccionesapartirdelasherramientasdelaQuímicaCuánticaylaresolucióndelasecuacionesdemovimiento.
c) La Teoría del Estado de Transición o del Complejo Activado que utiliza laTermodinámicaEstadísticaaplicadasobrelaSuperficiedeEnergíaPotencial.
2.2.-TeoríadeColisiones
LaTeoríadeColisiones(TC)intentaexplicarlareactividadquímicadesdeelmovimientode lasmoléculasqueformanlamezclareactivay lascolisionesqueseproducenentreellas.Comoseverá,laenergíapuestaenjuegoenlacolisiónresultaráfundamentalparadeterminarsi una colisión puede ser efectiva o no. Para desarrollar esta TC primero necesitamos sercapacesdedescribirelmovimientodetraslaciónquetienenlasmoléculasenunafasefluida(gasolíquido)yapartirahíestudiarlascolisionesylascondicionesparaquedichacolisiónproduzcareacción.
LaTeoríaCinéticadeGases(TCG)explicaelmovimientodetraslacióndelasmoléculasdeunafasefluida.Estateoría,aunquehistóricamenteanterioralaTermodinámicaEstadística(TE),esdehechounapartemásdeésta.LaTCGcomparteobjetivosyprocedimientosconlaTE:explicarlaspropiedadesmacroscópicasapartirdelasmicroscópicashaciendousodelaestadística.LoqueparticularizaalaTCGeselhechodecentrarseenexplicarlaspropiedadesde los gases (en particular los gases ideales) a partir de las propiedades cinéticas de susconstituyentes (átomos omoléculas). La TCG tiene sus raíces en los trabajos de Bernouillellevadosacaboalrededorde1738yfuedesarrolladaporMaxwell,ClausiusyBoltzmannentre
1/T
ln kr
Figura2.1.-RepresentacióndeArrhenius

QFIII–TEMA2 5
los años1858y1868.La formulaciónde laTEquehemosestudiado surgeaprincipiosdelsigloXX con los trabajosdeGibbsy espor tantoposterior. Sinembargo, los conocimientosadquiridosdeTEnosvanapermitirhacerunapresentacióndelaTCGmássencillaydirectaquelaqueseciñealdesarrollohistórico.
PuestoquelaTCGsurgeparaexplicarlaspropiedadesdelosgases,dondelasenergíasde interacción intermolecularessuelenserpequeñas, secentraen laspropiedadescinéticasde las moléculas o átomos que los forman, es decir, en sus velocidades y en cómo éstasdeterminan laspropiedadesdelgas.A continuación, sevaaescribir la funcióndeparticióndesde un punto de vista clásico para poder, desde ella, deducir cuáles son las velocidadesmoleculares que presenta una muestra de gas en unas determinadas condiciones. Esadistribucióndevelocidadessepuedecaracterizarusandodiferentesfuncionesdedistribuciónreferidasalascomponentesdelavelocidad,elvectorvelocidadoelmódulodelavelocidad.Unavezsehayanobtenidolasfuncionesdedistribuciónsepodránestudiar,haciendousodeellas,lascolisionesintermolecularesyconlasparedesdeunamuestradegas.
2.2.1.-VelocidadesMoleculares
Para conocer las propiedades de una muestra macroscópica de gas a partir de lasvelocidadesdesusconstituyentesnoesnecesarioconocercuáleslavelocidaddecadaunadelasmoléculasquelacomponen,sinocómosedistribuyedichapropiedad(velocidad)entreloselementos que forman la población a estudiar (moléculas). Es decir, el problema decaracterizarlavelocidaddelasmoléculasdeungasse ‘reduce’aconocerlaprobabilidaddeque unamolécula tenga una determinada velocidad. En el Apéndice 2.1 de esta lección sepresentaunaintroducciónalconceptodefuncióndedistribución,quees la formaadecuadademanejar laprobabilidadcuandose tratademagnitudescontinuas.Sedeberecordarque,desdeunpuntodevistaclásico, lavelocidaddeunamoléculaesunamagnitudcontinua,yaquepuedeadquirircualquiervalor.
2.2.1.1.-FuncionesdedistribucióndelavelocidadSe pueden definir hasta tras funciones de distribución diferentes para conocer la
distribución de velocidades de unamuestra de moléculas, según se esté interesado en lascomponentesdelavelocidad(vx,vy,vz),elvectorvelocidad( )oelmódulodelavelocidad(v).
Para conocer la fracción de moléculas cuya componente x de la velocidad estécomprendida entre vx y vx+dvx o, lo que es equivalente, la probabilidad de encontrar unamoléculacuyacomponentexdelavelocidadestécomprendidaentreesosvalores,sedefinelacorrespondientefuncióndedistribucióng(vx)como:
v!
xxxv dv)v(g)v(dpN
dN x ==

QFIII–TEMA2 6
pudiendodefinirse,igualmente,lasfuncionesdedistribuciónparalascomponentesvyyvzdelavelocidad:
Sitodaslasdireccionesdelespaciosonequivalentes,loquesepodráasumirsielcentro
demasas de la nuestra de gas no se desplaza y si se puede despreciar la contribución delcampogravitatoriouotroscamposexternos,entonces las tres funcionesg(vx),g(vy)yg(vz)seránfuncionesidénticas,difiriendosóloenlavariabledelaquedependen.
La probabilidad de encontrar moléculas con una componente de la velocidad (porejemplo, la vy) comprendida entre vy y vy+dvy se puede representar gráficamente en unespacio de velocidades. En este espacio, la velocidad de cada molécula viene dada por unvector.Silapuntadelvectorestácomprendidaentreunplanoparaleloalvzvxsituadoenvyyotrosituadoenvy+dvyentoncesestamoléculatienelavelocidaddeseada.EstasituaciónvienerepresentadaenlaFigura2.2.
Figura2.2.-Localizacióndeunvectorvelocidadcuyacomponenteyestécomprendidaentrevyyvy+dvy
Porotraparte, sepuedeestar interesadoen ladistribucióndel vectorvelocidad.Paraobtenerlafraccióndemoléculasconvectorvelocidadcomprendidoentre y sedefinelafuncióndedistribucióndelvectorvelocidad :
Laprobabilidaddeencontrarunamoléculaconvectorvelocidadentre y esigual
a la probabilidad de encontrar una molécula cuyos componentes de la velocidad esténcomprendidosentrevxyvx+dvx,vyyvy+dvyyvzyvz+dvz.Silosmovimientosalolargodelos
yyyv
dv)v(g)v(dpN
dN y ==
zzzv dv)v(g)v(dpN
dN z ==
v! vdv !!+
)v( !f
zyxv dvdvdv)v(vd)v()v(dp
NdN !!!!!
ff ===
v! vdv !!+
vx
vy
vz dvy

QFIII–TEMA2 7
tres ejes son independientes, esta probabilidad podrá obtenerse como el producto de laprobabilidaddetressucesosindependientes:
Porlotanto,teniendoencuentalasdefinicionesdelasfuncionesdedistribuciónintroducidashastaahora:
Gráficamente,sobreelespaciodevelocidades loqueestamosobteniendoes laprobabilidadde encontrar un vector velocidad cuyo extremo (punta) esté situado en el interior de unelementodevolumendvxdvydvz,comoseilustraenlaFigura2.3.
Figura2.3.-Localizacióndelvectorsituadoenelelementodevolumendvxdvydvz
Si, como se hizo anteriormente, se supone que todas las direcciones del espacio son
equivalentes, entonces la probabilidad de encontrar un determinado vector velocidad nodependerádelaorientacióndelvector,sólodesumódulo,talycomoseveráposteriormente.
Cuando se está interesado en conocer la fracción de moléculas con un determinadomódulodevelocidad,o loquees lomismo, laprobabilidaddeencontrarunamoléculacuyomódulo de velocidad esté comprendido entre v y v+dv, se define una nueva función dedistribucióndelosmódulosdelavelocidadG(v):
Gráficamente, esta probabilidad puede representarse en el espacio de velocidades por laprobabilidad de encontrar vectores velocidad cuyo extremo esté comprendido entre unaesferaderadiovyotraderadiov+dv,comoseilustraenlaFigura2.4.
NdN
N
dN
NdN
NdN zyx vvvv =!
)v(g)v(g)v(g)v( zyx=!
f
dv)v(G)v(dpNdNv ==
vx
vy
vz
dvx
dvy
dvz

QFIII–TEMA2 8
Figura2.4.-Localizacióndeunvectorvelocidadconmódulocomprendidoentrevyv+dv
2.2.1.2.-Obtencióndelasfuncionesdedistribucióndelavelocidad
Talycomosehavistoenlalecciónanterior,laprobabilidaddeencontrarunamolécula
enundeterminadoestadoenergéticojvienedadapor:
En concreto, la probabilidad de que una molécula se encuentra en el estado traslacionalcaracterizadopor el número cuánticonx o fraccióndemoléculas que se encuentran en eseestado,vendrádadapor:
Desde un punto de vista clásico, el movimiento traslacional a lo largo del eje x vienecaracterizadonoporunnúmerocuántico,sinoporundeterminadovalordelavelocidadvx,la
cualpuedevariarcontinuamente.Laenergíanoestácuantizadayvale .Así,
haciendo la correspondencia entre la función de partición cuántica y clásica, la fracción demoléculasconcomponentexdelavelocidadcomprendidaentrevxyvx+dvxvendrádadapor:
å-
-
=
i
kT
kTj
i
j
e
epe
e
å¥
=
-
-
==
1n
kT
kTnn
x
xn
xn
xx
e
eNN
pe
e
m2pmv
21 2
x2xx ==e
ò¥+
¥-
--
--
=
xkT2mv
xkT2mv
x
dve
dve)v(dp 2x
2x
vy
vz
vx

QFIII–TEMA2 9
donde lasuma(integral)sobre todos losposiblesvaloresdevxseextiendedesde hasta
.Estaintegralesdeltipo ysusoluciónes .Asípues,sepodráescribir:
yteniendoencuentaladefinicióndelafuncióndedistribucióng(vx)=dp(vx)/dvx:
(2.4)
Análogamente, teniendo en cuenta la equivalencia entre las direcciones espaciales, para lasdemáscomponentesdelavelocidadsepuedeescribir:
(2.5)
(2.6)
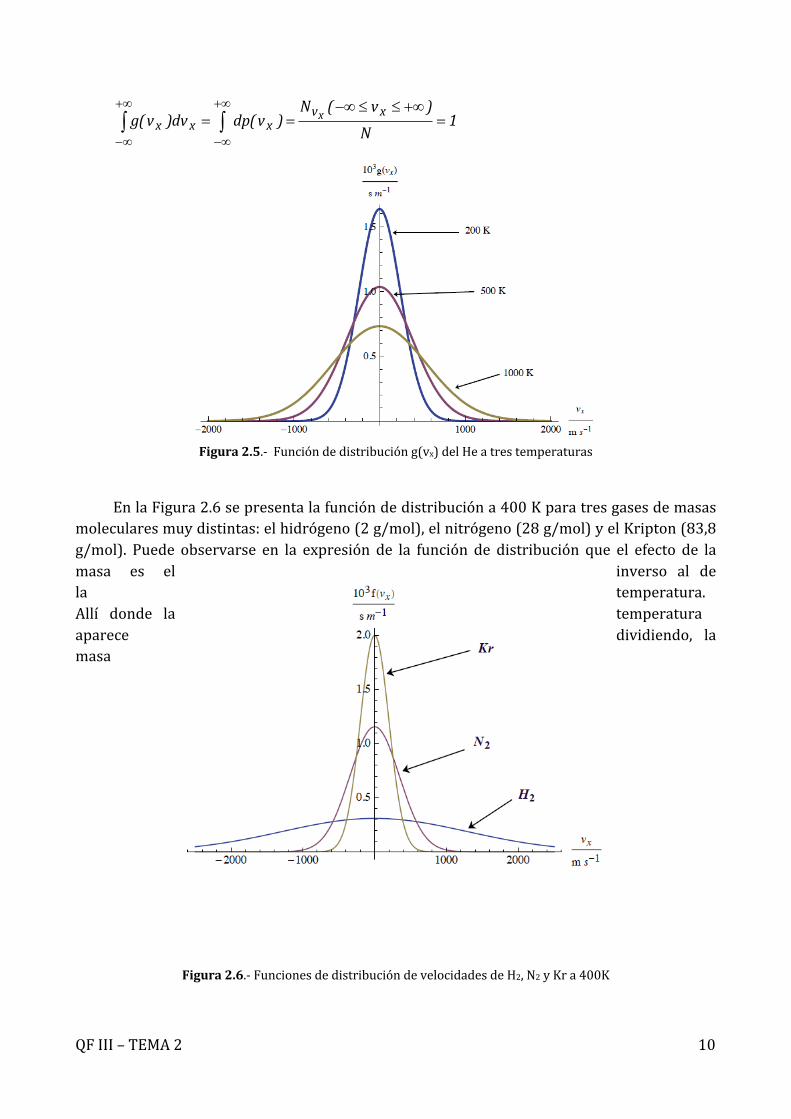
En la Figura 2.5 aparece representada la función g(vx) para el Helio a tres temperaturasdistintas. Siendo esta función una densidad de probabilidad, las unidades de la función dedistribuciónsonlasdevelocidadalamenosuno(s/menelsistemainternacional).Lafuncióndedistribuciónde lascomponentesde lavelocidadesuna funcióngaussianacentradaenelvalor vi=0. Se tratapor tantodeuna función simétrica respecto al origeny ladensidaddeprobabilidad de encontrarmoléculas con velocidad vi=c será lamisma que la de encontrarmoléculasconvelocidadvi=-c.Por tantoelvalormediode las componentesde lavelocidadserá<vi>=0(i=x,y,z).Puedeobservarsequeelvalormásprobabledelascomponentesdelavelocidad (el valor donde la función alcanza un máximo) es también el vi=0. Además lafunción se anula sólo para velocidades igual a ± . Es interesante destacar el efecto de latemperatura. Para unmismo gas, el aumentode la temperatura produce un aumento en ladensidaddeprobabilidaddeencontrarmoléculasconvaloresaltos(positivosonegativos)delas componentes de la velocidad, mientras que disminuye la probabilidad de encontrarmoléculas con valores pequeños. Obviamente, el área encerrada bajo la curva semantieneconstanteyaquelafuncióndedistribucióndebeestarnormalizada:
¥-
¥+ ò+¥
¥-
- dxe2ax
2/1
a÷øö
çèæp
2/1xkT2
mv
x
mkT2
dve)v(dp
2x
÷øö
çèæ
=
--
p
kT2mv2/1
x
2x
ekT2m)v(g
-÷øö
çèæ=p
kT2mv2/1
y
2y
ekT2m)v(g
-÷øö
çèæ=p
kT2mv2/1
z
2z
ekT2m)v(g
-÷øö
çèæ=p
¥
QFIII–TEMA2 10
Figura2.5.-Funcióndedistribucióng(vx)delHeatrestemperaturas
EnlaFigura2.6sepresentalafuncióndedistribucióna400Kparatresgasesdemasasmolecularesmuydistintas:elhidrógeno(2g/mol),elnitrógeno(28g/mol)yelKripton(83,8g/mol). Puede observarse en la expresión de la función de distribución que el efecto de lamasa es el inverso al dela temperatura.Allí donde la temperaturaaparece dividiendo, lamasa
Figura2.6.-FuncionesdedistribucióndevelocidadesdeH2,N2yKra400K
1N
)v(N)v(dpdv)v(g xv
xxxx =
+¥££-¥== òò
+¥
¥-
+¥
¥-

QFIII–TEMA2 11
molecular aparece multiplicando. Esto implica que el efecto es justo el inverso. Unadisminucióndelamasamolecular(comoocurrealpasardelRadónalHelio)tieneelmismoefectoqueunaumentodelatemperatura:lafuncióndedistribuciónseensancha,aumentandoparavaloresaltosdelavelocidadydisminuyendoparavalorespequeños(alrededordevi=0).
Ejercicio 2.1.- Calcular la densidad de probabilidad para la componente x de la velocidad de una muestra de moléculas de O2 a 300 K en el intervalo 0< |vx| <1000 ms-1. Representar la función de distribución resultante.
Solución.- La densidad de probabilidad o probabilidad por unidad de intervalo viene
expresada por y la fracción de moléculas con componente x de la velocidad entre vx y vx+dvx por:
con .
En nuestro caso:
con lo que
Teniendo en cuenta la relación encontrada anteriormente entre la función dedistribucióndelvectorvelocidadydelascomponentesdelavelocidad:
kT2mv2/1
x
2x
ekT2m)v(g
-÷øö
çèæ=p
yK300T,Kg10x3137,5gKg10
mol10x022,61molg0,32m 263
1231 === --
--
123 JK10x38066,1k --= ( ) ( ) 2x
226 vsm10x4145,613x esm10x4289,1)v(g
-----=
)v(g x
xdx
dx)x(f)x(dp =
xxxv dv)v(g)v(dpN
dN x ==

QFIII–TEMA2 12
(2.7)
enqueseobserva,talycómosehabíaargumentado,quelafuncióndedistribucióndelvectorvelocidadnodependedelaorientacióndeestevector,sinoexclusivamentedesumódulo.
Esta función de distribución no puede representarse de forma convencional ya quenecesitarían cuatro dimensiones (tres para los valores de vx, vy, vz y una para la función).Obviamente,larepresentaciónenfuncióndelmóduloresultaríaenunasemi-gaussianayaqueelmódulosólosedefineparavalorespositivos.Unaformacomúnderepresentarestafunciónesmedianteunanubedepuntossobreelespaciodevelocidades.Losmayoresvaloresde lafunción se corresponden con aquellas zonas conmayor densidad de puntos, tal y como seilustraenlaFigura2.7.Obviamente,yaquelasfuncionesg(vi)presentanelmáximoparavi=0,lafuncióndedistribucióndelvectorvelocidadtendrásuvalormáximoenvx=vy=vz=0.
Figura2.7.-Representacióndelafuncióndedensidaddeprobabilidaddelmódulodelavelocidadenelespaciodevelocidades
Paraencontrar la fraccióndemoléculasconundeterminadomódulode lavelocidady
portantosufuncióndedistribuciónG(v)sepuedepartirdelresultadoacabadodealcanzar.Lafuncióndedistribución nosproporciona la fraccióndemoléculascon undeterminadovectorvelocidad. Si loquesequierees la fracciónconundeterminadomódulo,se tendránque sumar todas aquellas moléculas cuyo vector velocidad tenga ese módulo, aunquepresentendistintasorientaciones:
kT2mv2/3)vvv(
kT2m2/3
kT2mv2/1
kT2mv2/1
kT2mv2/1
zyx
22z
2y
2x
2z
2y2
x
ekT2me
kT2m
ekT2me
kT2me
kT2m)v(g)v(g)v(g)v(
-++-
---
÷øö
çèæ=÷
øö
çèæ=×
=÷øö
çèæ
÷øö
çèæ
÷øö
çèæ==
pp
pppf!
)v( !f
òò ==norientació
zyxnorientació
vv dvdvdv)v(NdN
NdN !!
f
vx
vy
vz

QFIII–TEMA2 13
EstasumavienegráficamenterepresentadaenlaFigura2.8,quemuestraquelaprobabilidaddeencontrarmoléculasconundeterminadomóduloseobtienesumandoprobabilidadesdeencontrar moléculas con vectores velocidad que presenten ese módulo y distintasorientaciones.
Figura2.8.-Laprobabilidaddeencontrarunvectorvelocidadconundeterminadomódulosólodependedelvalordeésteynodelaorientacióndelvector
Para realizar la integral es conveniente expresar el elemento de volumen utilizando
coordenadasesféricas,yaque la integralquevamosa realizarconsisteexactamenteenunasumadeelementosdiferencialessobrelasuperficiedeunaesfera:
Así, la integral sobre toda orientación posible del vectorvelocidad seconvierteenunaintegralsobretodovalorposibledelosángulos y :
conloquelafuncióndedistribuciónbuscadaes:
dvddsenvdvdvdv 2zyx jqq=
j q
dvekT2mv4ddsendvve
kT2m
dvddsenvekT2mddsenv)v(dv)v(G
NdN
kT2mv2/3
22
0 0
2kT2mv2/3
2
0 0
2kT2mv2/32
0 0
2v
22
2
--
-
÷øö
çèæ=÷
øö
çèæ=
=÷øö
çèæ===
ò ò
ò òò ò
ppjqq
p
jqqp
jqqf
p p
p pp p!
vx
vy
vz
v
j
q
vy
vz
vx

QFIII–TEMA2 14
(2.8)
Estafuncióncontieneunadobledependenciaconelmódulovdelavelocidad.Porunaparte,
presentaeltérminoparabólicov2yporotraeltérminoexponencial .Elprimeroesuntérmino creciente convmientrasqueel segundoesdecreciente.El resultadoapareceen laFigura 2.9 donde se ha representado la función G(v) para el Helio a tres temperaturas. LafunciónG(v)notieneelmáximoenv=0,apesardequesehavistoqueelvectormásprobableesaquelenquevx=vy=vz=0.Elmotivoeselsiguiente.Elvectormásprobableesefectivamenteel (0,0,0) pero sólo hay uno de estos vectores. Sin embargo, si nos preguntamos por laprobabilidaddequeunamoléculatengaunmódulodevelocidaddigamosde100m/s,existenmuchosvectoresquecumplenestacondición: (100,0,0); (0,100,0); (70,40,59,2); (57,7 ,57,7,57,7);….Cadaunodeestosvectorestieneunaprobabilidadmenorqueel(0,0,0)peroal habermuchos conmódulo 100, resulta que estemódulo esmuchomás probable que elmódulo0.Así,engeneral,lafuncióndedistribucióndemódulosdevelocidadeselresultadode la combinacióndedos factores: el númerode vectores quepuedendarundeterminadomódulomultiplicadoporlaprobabilidaddecadaunodeesosvectores(quesedeberecordarquedependesólodelmódulo).Eltérmino (queeslasuperficiedelaesferaderadiov)nosdaelnúmerodevectoresquepodemoscombinarparaobteneresemódulo,mientrasque
eltérmino nosdalaprobabilidaddecadaunodeesosvectores.
Figura2.9.-FunciónG(v)delHeatrestemperaturas
Como se observa en la Figura, a valores pequeños del módulo v domina el términoparabólico(v2)yG(v)crececuadráticamente.Sinembargo,avaloresgrandesdevdominael
término exponencial y G(v) decae rápidamente hasta anularse en el infinito. Alaumentar la temperatura se observa un ensanchamiento de la función de distribución,
kT2mv2/3
22
ekT2mv4)v(G
-÷øö
çèæ=p
p
2ve-
2v4p
2ve-
2ve-

QFIII–TEMA2 15
aumentando la probabilidad de encontrar moléculas con valores altos del módulo de lavelocidadyelmáximo(módulomásprobable)sedesplazaavaloresmayores.Elefectodelamasa sobre la función G(v) es el inverso al de la temperatura, igual que ocurría para lascomponentesdelavelocidad.Unaumentodemasa(alpasardeHelioaradón,ódeHidrógenoaKriptóncomoelejemplográficode laFigura2.10)se traduceenunestrechamientode lafunciónG(v)yaundesplazamientodelmáximoavaloresmáspequeños.Esdecir,aumentalaprobabilidad de encontrarmoléculas con un valor pequeño delmódulo de la velocidad. Esimportanterecordarqueeláreadelasuperficiebajolafunciónessiemprelamisma,yaquelaprobabilidaddeencontrarmoléculasconcualquiervalordelmódulo(entre0e )tienequesersiemprelaunidad:
Figura2.10.-FuncionesG(v)deH2,N2yKra400K
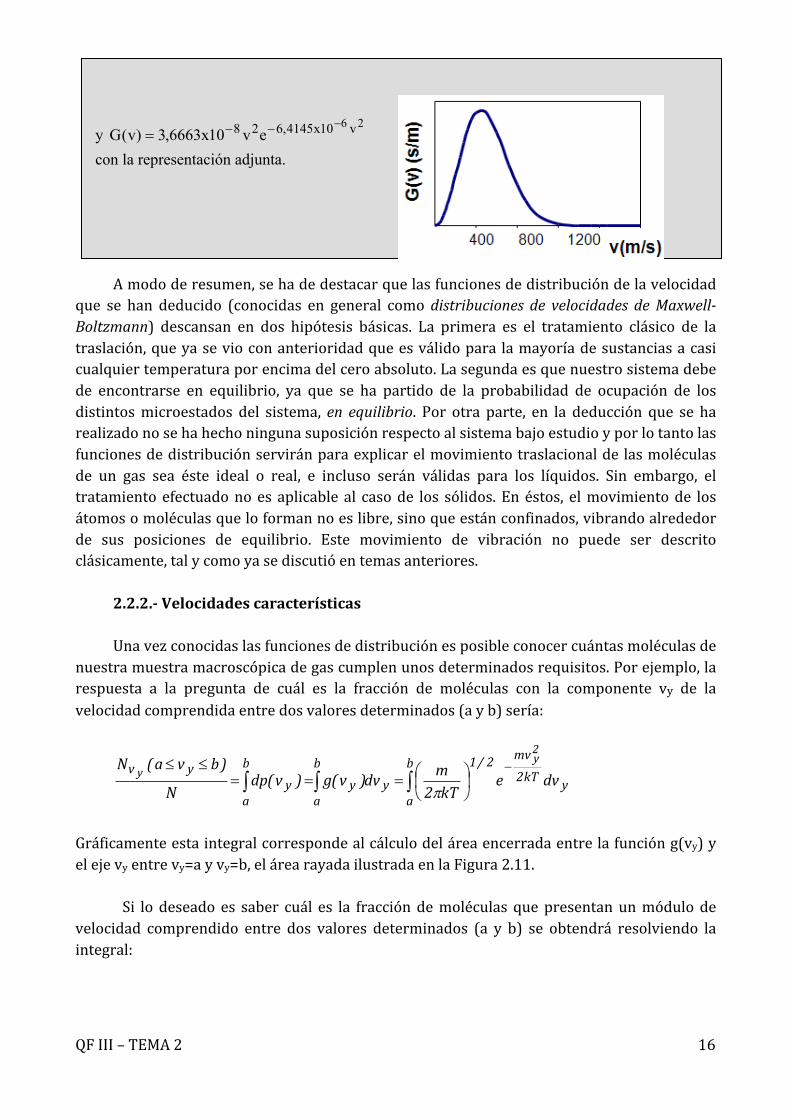
Ejercicio 2.2.- Calcular la densidad de probabilidad para la velocidad de una muestra de moléculas de O2 a 300 K en el intervalo 0 < v < 1000 m s-1. Representar la función de distribución resultante
Solución.- La fracción de moléculas con módulo de velocidad entre v y v + dv :
con .
Con los datos: se tiene:
;
¥
òò¥¥
===¥££
00
v 1dv)v(G)v(dpN
)v0(N
( )dvvGNdNv = kT2
mv2/32
2
ekT2mv4)v(G
-÷øö
çèæp
p=
3392/32/3
sm10x9175,2RT2M
kT2m --=÷
øö
çèæp
=÷øö
çèæp
226 sm10x4145,6RT2M
kT2m --=÷
øö
çèæ=÷
øö
çèæ
1113 molJK3145,8R;K300T;molKg10x32M ---- ===
QFIII–TEMA2 16
y con la representación adjunta.
Amododeresumen,sehadedestacarquelasfuncionesdedistribucióndelavelocidad
que se han deducido (conocidas en general comodistribuciones de velocidades deMaxwell-Boltzmann) descansan en dos hipótesis básicas. La primera es el tratamiento clásico de latraslación,queyasevioconanterioridadqueesválidopara lamayoríadesustanciasacasicualquiertemperaturaporencimadelceroabsoluto.Lasegundaesquenuestrosistemadebede encontrarse en equilibrio, ya que se ha partido de la probabilidad de ocupación de losdistintosmicroestados del sistema, en equilibrio. Por otra parte, en la deducción que se harealizadonosehahechoningunasuposiciónrespectoalsistemabajoestudioyporlotantolasfuncionesdedistribuciónserviránparaexplicarelmovimientotraslacionaldelasmoléculasde un gas sea éste ideal o real, e incluso serán válidas para los líquidos. Sin embargo, eltratamientoefectuadonoes aplicable al casode los sólidos.Enéstos, elmovimientode losátomosomoléculasqueloformannoeslibre,sinoqueestánconfinados,vibrandoalrededorde sus posiciones de equilibrio. Este movimiento de vibración no puede ser descritoclásicamente,talycomoyasediscutióentemasanteriores.
2.2.2.-Velocidadescaracterísticas
Unavezconocidaslasfuncionesdedistribuciónesposibleconocercuántasmoléculasdenuestramuestramacroscópicadegascumplenunosdeterminadosrequisitos.Porejemplo,larespuesta a la pregunta de cuál es la fracción de moléculas con la componente vy de lavelocidadcomprendidaentredosvaloresdeterminados(ayb)sería:
Gráficamenteestaintegralcorrespondealcálculodeláreaencerradaentrelafuncióng(vy)yelejevyentrevy=ayvy=b,elárearayadailustradaenlaFigura2.11.
Si lo deseado es saber cuál es la fraccióndemoléculasquepresentanunmódulodevelocidad comprendido entre dos valores determinados (a y b) se obtendrá resolviendo laintegral:
26 v10x4145,628 ev10x6663,3)v(G---=
yb
a
kT2mv2/1
yb
ay
b
ay
yvdve
kT2mdv)v(g)v(dp
N
)bva(N2y
yòòò
-÷øö
çèæ===
££
p

QFIII–TEMA2 17
Figura2.11.-Fraccióndemoléculasconvycomprendidaentreayb
Tambiénesposiblecaracterizarlamuestradegasporalgunosvalorescaracterísticosde
lavelocidad.Así,yasabemosqueelvalormediodelascomponentesx,y,zdelavelocidadescero(yaqueexistelamismaprobabilidaddequelasmoléculasrecorranelejeenunsentidoque en otro), sin embargo, la velocidad media (entendida como módulo) no es cero. Estavelocidad media, como cualquier valor medio, se puede obtener a partir de la función dedistribución:
Esta integral puede resolverse con ayuda de tablas de integrales. En general, cuando lavariableaparececonexponenteimparantesdelaexponencial:
yutilizandoestarelación:
dvekT2mv4dv)v(G)v(dp
N)bva(N b
a
kT2
mv2/3
2b
a
b
a
v
2
òòò-
÷øö
çèæ===
££p
p
dvevkT2m4
dvekT2mv4dv)v(vG)v(vdpv
kT2
mv
0
32/3
0 0
kT2
mv2/3
3
02
2
-¥
¥ ¥ -¥
ò
ò òò
÷øö
çèæ=
=÷øö
çèæ===
pp
pp
1n0
ax1n2
a2!ndxex
2
+
¥-+ =ò

QFIII–TEMA2 18
(2.9)
Por lo tanto, la velocidadmediadeunamuestrade gas es tantomayor cuantomayor es latemperatura y cuanto menor sea la masa de las moléculas. Para hacernos una idea, lavelocidadmediaa298Kdelhelioesde1256m/s,ladelnitrógeno475m/syladeldióxidodecarbono379m/s.
También es posible caracterizar la velocidad de la muestra a través de la velocidadcuadrática media <v2>, la cual se obtiene siguiendo la misma técnica que para obtenercualquierpropiedadpromediodependientedelmódulo:
Para resolver esta integral, en la que la variable aparece conun exponente par antes de laexponencial,sedebeemplearlarelación:
conloquelavelocidadcuadráticamediaresultavaler:
ylaraízdelavelocidadcuadráticamedia(ovelocidad‘root-meansquare’,vrms)es:
(2.10)
Ademásdelasvelocidadesmediassepuedeusarlavelocidad(módulo)másprobable
paracaracterizar lamuestrademoléculasbajoestudio.Lavelocidadmásprobableesaquelvalordelmóduloparaelquelaprobabilidadsehacemáximao,loqueesequivalente,sehacemáximalafuncióndedistribuciónG(v).MatemáticamenteestosignificaencontrarelvalordevquehacenulalaprimeraderivadadeG(v)respectodevynegativalasegundaderivada.Sisetomalaprimeraderivada:
2/1
2
2/3
0
kT2
mv
32/3
mkT8
kT2m2
1kT2m4dvev
kT2m4v
2
÷øö
çèæ=
÷øö
çèæ
÷øö
çèæ=÷
øö
çèæ= ò
¥ -
ppp
pp
dvevkT2m4dve
kT2mv4dv)v(Gv)v(dpvv kT2
mv
0
42/3
0 0
kT2
mv2/3
42
0
22
22
-¥¥ ¥ -¥
òò òò ÷øö
çèæ=÷
øö
çèæ===
pp
pp
2/1n1n2
2/1
0
axn2
a!n2!n2dxex
2
++
¥- =ò
p
÷øö
çèæ=
÷øö
çèæ
÷øö
çèæ=÷
øö
çèæ=
-¥
ò mkT3
kT2m!22
!4kT2m4dvev
kT2m4v
2/55
2/12/3kT2
mv
0
42/3
2
pp
pp
p
2/12/12rms m
kT3vv ÷øö
çèæ==

QFIII–TEMA2 19
hay tres valores distintos de v tales que . Estos valores son v=0; v= y
. El único que es un verdadero máximo es el tercero, lo que se puede
comprobargráficamente(sobrelarepresentacióndeG(v)frenteav)ocalculandoladerivadasegunda.Asípues,elvalormásprobabledelavelocidad(vp)resultaser:
(2.11)
Lastresvelocidades:velocidadmedia,laraízdelavelocidadcuadráticamediaylavelocidadmásprobablemantienenunarelaciónconstanteparacualquiertipodegasytemperatura:
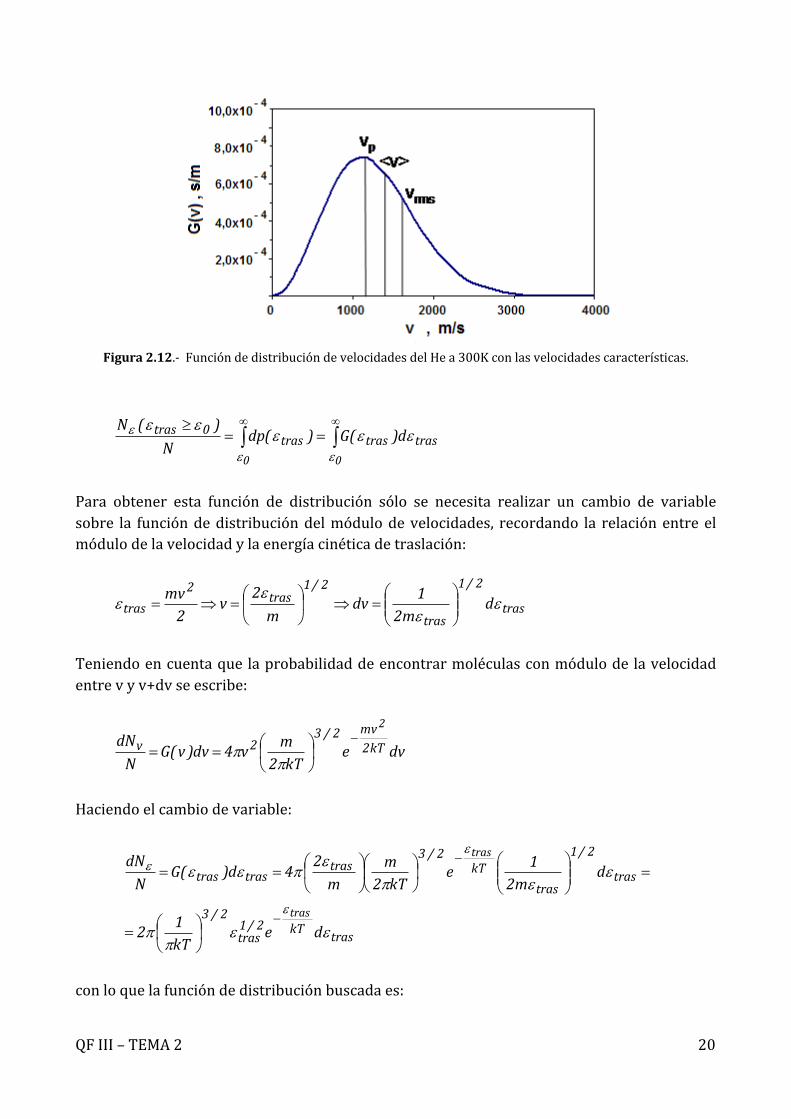
Porejemplo,paraeloxígenomoleculara298Klavelocidadmásprobableesde394m/s,lavelocidadmediaesde444m/sylaraízdelacuadráticamedia482m/s.Siempreaparecenenesteorden,talycomoseindicaesquemáticamenteenlaFigura2.12.
2.2.3.-DistribucióndeEnergíasA veces puede ser más interesante caracterizar la muestra de gas en lugar de por ladistribucióndevelocidadesporladistribucióndeenergíacinéticasdetraslación( ).Estadistribución, que puede ser obtenida de forma inmediata a partir de las que conocemos,resultaserútilcuandoloqueinteresasabereslaenergíacinéticadetraslaciónquetienenlasmoléculasdelamuestradegasyquepuedenponerenjuegoenelcasodeunacolisión.Comoseverá,unrequisitotípicoparaconsiderarqueunacolisiónentremoléculaspuededarlugarareacciónesquetengalugarconunaenergíamínimaumbral( ).Siseconocelafuncióndedistribucióndeenergíacinéticasde traslación, , sepodrácalcularrápidamentecuáleslafraccióndemoléculasquecumpleesacondición:
úúû
ù
êêë
é-÷
øö
çèæ=
=
úúúú
û
ù
êêêê
ë
é
-÷øö
çèæ=
úúúú
û
ù
êêêê
ë
é
÷øö
çèæ
¶¶
=¶
¶
-
---
kTmv2ve
kT2m4
ekTmvve2
kT2m4e
kT2mv4
vv)v(G
2kT2
mv2/3
kT2
mv3
kT2
mv2/3
kT2
mv2/3
2
2
222
pp
pp
pp
0v)v(G=
¶¶
¥
( ) 2/1m
kT2v =
2/1
p mkT2v ÷
øö
çèæ=
225,1:128,1:13:8:2v:v:v rmsp ºº p
trase
0e)(G trase
QFIII–TEMA2 20
Figura2.12.-FuncióndedistribucióndevelocidadesdelHea300Kconlasvelocidadescaracterísticas.
Para obtener esta función de distribución sólo se necesita realizar un cambio de variablesobre la funcióndedistribucióndelmódulodevelocidades, recordando la relaciónentreelmódulodelavelocidadylaenergíacinéticadetraslación:
Teniendoencuentaquelaprobabilidaddeencontrarmoléculasconmódulodelavelocidadentrevyv+dvseescribe:
Haciendoelcambiodevariable:
conloquelafuncióndedistribuciónbuscadaes:
òò¥¥
==³
00trastrastras
0tras d)(G)(dpN
)(N
ee
e eeeee
tras
2/1
tras
2/1tras
2tras d
m21dv
m2
v2
mv ee
ee ÷÷
ø
öççè
æ=Þ÷
ø
öçè
æ=Þ=
dvekT2mv4dv)v(G
NdN kT2
mv2/32v
2-
÷øö
çèæ==p
p
traskT2/1tras
2/3
tras
2/1
traskT
2/3tras
trastras
dekT12
dm21e
kT2m
m2
4d)(GNdN
tras
tras
eep
p
eep
epee
e
ee
-
-
÷øö
çèæ=
=÷÷ø
öççè
æ÷øö
çèæ÷ø
öçè
æ==

QFIII–TEMA2 21
(2.12)
Unapropiedadinteresantedeestafunciónesquenodependedelamasadelasmoléculasqueformanelgasyporlotantolamismafunciónvaleparacualquiergas.Lafraccióndemoléculasconenergíascinéticascomprendidasenundeterminadointervaloeslamismaparadosgasesque se encuentren a lamisma temperatura. La Figura 2.13muestra una representación deesta función a dos temperaturas distintas. La función recuerda a la G(v) excepto en quepresentaunaasíntotaverticalenelorigenenlugardehorizontalcomoocurríaparaG(v).Porlodemás la funciónpresentaunmáximoydecaehastaanularse sóloparavalores infinitos.Por supuesto la función se ensancha y su máximo se desplaza a valores mayores cuandoaumenta la temperatura, con lo que la fraccióndemoléculas que alcanzanundeterminadoumbralenergéticoaumentaconlatemperatura(estoexplica,comoseverámásadelante,quelavelocidaddeunareacciónelementalaumenteconlatemperatura).Figura2.13.-FuncióndistribucióndelaenergíacinéticadetraslaciónparaátomosdeHeadostemperaturas
Ejercicio 2.3.- Obtener la energía de traslación molecular media a partir de la distribución de Maxwell expresada como distribución de energías.
Solución.-
; ;
(a)
Llamando , se tiene y que sustituido en (a)
kT2/1tras
2/3
tras
tras
ekT12)(G
e
ep
pe-
÷øö
çèæ=
( ) eee dGNdN
= ( ) traskT21
tras23
dekT12G
tras
eep
pee
-÷øö
çèæ=
( ) == ò¥
tras0
trastrastras dG eeee( )
tras0
kTtras
23 de
kT
2 tras23
eep
pe
ò¥ -
( ) x21
tras =e 2tras x=e xdx2d tras =e
función G(e )
0,0E+00
2,0E+19
4,0E+19
6,0E+19
8,0E+19
1,0E+20
1,2E+20
1,4E+20
0 2E-20 4E-20 6E-20 8E-20 1E-19
e (J)
G( e
) (1/
J)
300 K1000 K

QFIII–TEMA2 22
pues en la integral n=2 y a=(1/kT) Ejercicio 2.4.- Determinar la proporción entre (a) las velocidades medias y (b) las energías
cinéticas traslacionales medias de las moléculas de H2 y los átomos de Hg a 20 °C Solución.- a) ; Independiente de T b) con lo que , también independiente de T
2.2.4.-Colisionesconlapared
Paracalcularelnúmerodecolisionesqueseproducenentrelasmoléculasdeungasylas paredes del recipiente que lo contiene, considérese un recipiente cúbico como el de laFigura2.14,devolumenVycuyasparedestienenáreaA.EsterecipientecontieneNmoléculasdeunmismotipoaunatemperaturaT.Paraelcálculosevaaconsiderarunadelascaras,laqueaparecerayadaenlafigura,aunque,obviamente,todassonequivalentes.
Considérenselascolisionesquepuedenproducirseenunciertotiempodt.Comolacara
considerada esperpendicular al eje y, el hechodequeunamolécula golpeeono sobre esacaradependerádelacomponentevydelavelocidaddedichamolécula.Loprimeroquedebehacerse es fijarse en aquellas moléculas que presentan un determinado valor de lacomponente,entrevyyvy+dvy.¿Cuántasmoléculassetieneconestavelocidad?:
trase( )
== ò¥ -
xdx2exkT
2
0
kTx
23
2
3
p
p
( )=
p
pò¥ -
dxexkT
4
0
kTx
4
23
2
( )kT23
kT1!22
!4
kT
4
25
5
21
23 =
÷øö
çèæ
p
p
p
yyv
dv)v(gN
dN y =
21
MRT8v ÷
øö
çèæp
= ( )( ) 975,9
016,26,200
HMHgM
v
v 21
21
2Hg
H2 =÷ø
öçè
æ=÷÷
ø
öççè
æ=
2kT3
mkT3m
21vm
21 2
tras ===e 1Hgtras
Htras2 =
e
e

QFIII–TEMA2 23
Figura2.14.-Representacióndelmodeloderecipienteescogidoparaelcálculodecolisionesconlasparedes.
¿Cuántaspuedencolisionarconlaparedconsideradaenunintervalodt?Puesaquellasque estén viajando hacia la pared (es decir, ) y se encuentren a una distancia losuficientementepequeña( ).Sehandeconsiderarpuessóloaquellasmoléculasqueseencuentrandentrodelvolumen comprendidoentrelaparedconsideradayel
plano punteado de la figura anterior. De todas las posibles moléculas, la fracción que seencuentra en ese volumen es justamente V’/V. Así pues, en un intervalo de tiempo dt lasmoléculas con componente vy comprendida entre vy y vy+dvy que colisionan con la paredconsideradaserán:
(si )
Paraobtenerelnúmero totaldemoléculasquegolpean laparedenun intervalodtse
han de sumar las contribuciones de todos los posibles valores de la componente vy de lavelocidad, loqueseconsigueintegrandoparatodoslosvaloresdeestacomponente,enestecasoentre0y (yaquelasmoléculasconcomponentenegativanogolpeanlaparedeneldtconsiderado):
Elnúmerodemoléculasquecolisionanconunapareddependedeltamañodelapared
(A) y del tiempo considerado (dt). Unamagnitudmás útil es la frecuencia de colisión porunidaddeárea(ZP)quesecalculacomo:
(2.13)
0v y ³
dtvd y£
dtvA'V y×=
yyy
vv,P dv)v(NgVdtAv
dNV'VdN yy == 0v y ³
¥+
2/12/1
0y
2y
y
2/1
0y
2y
2/1
y0
yyy
0yv,PP
m2kT
VNAdt
mkT
kT2m
VNAdtdv
kT2mv
expvkT2m
VNAdt
dvkT2
mvexp
kT2mv
VNAdtdv)v(Ng
VdtAv
)v(dNdN y
÷øö
çèæ=÷
øö
çèæ=÷
÷
ø
ö
çç
è
æ-÷
øö
çèæ=
=÷÷
ø
ö
çç
è
æ-÷
øö
çèæ===
ò
òòò
¥
¥¥¥
ppp
p
VNv
41
VN
mkT8
41
dtdN
A1Z
2/1P
P =÷øö
çèæ==p
x
y
z vydt

QFIII–TEMA2 24
Lafrecuenciadecolisióndependepuesdeladensidaddelgas(N/V)ydesuvelocidadmedia(<v>). Cuantomayores sean estasmagnitudes,mayor será la frecuencia de colisión con lasparedes.Teniendoencuentaqueparaungasideal(N/V)=(p/kT),larelaciónanteriorsepuedeescribirtambiéncomo:
(2.14)
Unaideadelvalordeestamagnitudlapuededarelcálculodelnúmerodecolisionespor
unidadde tiempo y unidadde área para un gas como el oxígenomolecular en condicionesestándar (298 K y 105 Pa), con un resultado de 2,7x1023 cm-2s-1, es decir, ¡se producenalrededordeunmoldecolisionesporsegundosobrecadacentímetrocuadrado!
Ejercicio 2.5.- La presión de vapor de la plata a 2000 °C es de 170 torr. Calcular los
gramos de plata que colisionan por unidad de área (cm2) y de tiempo (s) en las paredes de un recipiente que contiene plata en equilibrio con su vapor a 2000 °C.
Solución. - Número de partículas que colisionan por m2 y por s: , donde
con lo que .
Ya que , , ,
resulta para , que
teniendo en cuenta que da
2.2.5.-ColisionesIntermolecularesyRecorridoLibreMedio
Para considerar las colisiones intermoleculares se van a tratar las moléculas del gascomoesferasrígidascaracterizadasporunciertodiámetro(d).Elcasodeestudioeseldeunamezcladedosgases,ocupandounvolumenValatemperaturaT,formadaporN1moléculasdetipo1,conmasam1ydiámetrod1yN2moléculasdetipo2,conmasam2ydiámetrod2.Además,sevaasimplificareltratamientosuponiendoquetodaslasmoléculassemuevenconmódulode lavelocidad iguala lavelocidadmedia, lasde tipo1<v1>y lasde tipo2<v2>. Setratadeunmodelosimplificadoparalamezcladedosreactivosgaseosos.
Calculemosprimerolascolisionesdeunamoléculadetipo1conlasdetipo2.Paraello,enlugardetratarelmovimientosimultáneodelosdostiposdepartículas,consideremosaladetipo2enreposo,mientrasqueladetipo1semoveráconunavelocidadigualalavelocidad
( ) 2/1PmkT2
pZp
=
=÷øö
çèæp
= 21
P MRT8
kTp
41Z
( )21
A
MRT2
pN
p11molJK31451,8R --= K15,2273T = 123
A mol10x02214,6N -=
Pa8,22664atm1Pa101325
Torr760atm1Torr170p == 1226
p sm10x2060,1Z --=
MN1Z)Ag(mAv
P= 12scmg16,2 --=13Ag molKg10x868,107M --=
kTp
RTnN
VN A ==
vVN
41ZP =

QFIII–TEMA2 25
relativamedia<v12>.EstaestrategiapermitedescribirnuestroproblematalycomoaparecerepresentadoenlaFigura2.15.Enuntiempodtlapartícula1recorreráunadistancia<v12>dt
Figura 2.15.- Volumen barrido por unamolécula de tipo 1 en su desplazamiento libre en un recipiente quecontieneunamezcla
ychocarácontodasaquellasmoléculasdetipo2queseencuentrenaunadistanciamenordela sumade los radiosen cualquiermomentode la trayectoriadescritapor la1.Esdecir, lamoléculatipo1puedecolisionarconcualquiermoléculadetipo2cuyocentroseencuentre
enelinteriordeuncilindrodealturaiguala<v12>dtybaseconáreaiguala .El
númerodemoléculasdetipo2quecumplenestacondiciónes:
(2.15)
dondesehaintroducidoelconceptodediámetrodecolisión(d12)comosumadelosradiosdelasmoléculasimplicadas,consideradascomoesferasrígidas.Enalgunoscontextosaláreadela basedel cilindro se le denomina secciónde colisión y se representapor el símbolo
.Conesteresultadosepuedeyacalcularelnúmerodecolisionesquesufreunapartículadetipo1conlasdetipo2porunidaddetiempo,ofrecuenciadecolisiónz12como:
(2.16)
Quedaporencontrarelvalorde lavelocidadrelativamedia.Estavelocidaddependerá
dedosfactores,elmódulodelasvelocidadesconquecolisionanlasmoléculasdetipo1y2ysuorientación.Respectoalmódulo, seha supuestoque lasmoléculas semueven todas convelocidadesigualalavelocidadmedia.Respectoalaorientación,lacolisiónpuedeproducirseconcualquieránguloentrelosvectoresvelocidadentre0ºy180º.Sinuestrasmoléculassonesféricas, no hay en principio ninguna orientación privilegiada y todas las situaciones son
221
2dd
÷ø
öçè
æ +p
VNdtvd
VNdtv
2dd
VNV 2
12212
212
2212
cil pp =÷ø
öçè
æ +=
212dp
12s
VN
vVN
vdz 21212
212
21212 sp ==
1•
2•2•
2•2•
2•2•
d1+d2
d2
d1<v12>
<v12>dt

QFIII–TEMA2 26
igualmenteprobables.Enesascircunstanciaselángulopromedioentrelosvectoresvelocidadenlacolisiónseráde90º.O,dichodeotraforma,lacolisiónpromedio1-2tienelugarentre
Figura2.16.-Cálculodelavelocidadrelativamediaentredosmoléculas.Deizquierdaaderecha:definicióndelvectorvelocidadrelativa,posibleorientaciónentre losvectoresvelocidadycálculodelmóduloen lasituaciónpromedio
moléculasconvelocidadesconmódulos<v1>y<v2>yformandounángulorecto.Enesecaso,lavelocidadrelativamediasepuedeobtenermuyfácilmente:
Esdecir,quelavelocidadrelativamediaeslavelocidadmediademoléculasconmasaigualalamasareducida ( ). Sustituyendoestaexpresiónde lavelocidaden lade la frecuenciadecolisión,queda:
(2.17)
Si se quiere conocer la frecuencia total de colisiones de tipo 1-2, sólo se tendrá que
multiplicarlafrecuenciadecolisionesdeunamoléculadetipo1porelnúmerodemoléculasquesetiene,esdecir, .Estamagnitudsueletomarvaloresmuygrandes,porloqueesmáshabitualexpresarlafrecuenciatotaldecolisiónporunidaddevolumen(Z12):
(2.18)
Nóteseque,engeneral,paraunamezclacualquiera pero .
22
21
212 vvv +=
pµpppkT8
m1
m1kT8
mkT8
mkT8v
2121
212 =÷÷
ø
öççè
æ+=+=
µ
VNkT8dz 2
2/121212 ÷÷
ø
öççè
æ=
pµp
121zN
VN
VN
M1
M1RT8
VN
VNkT8d
VzN
Z 122/1
2112
122/1
212
12112 ú
û
ùêë
é÷÷ø
öççè
æ+=÷÷
ø
öççè
æ==
ps
pµp
2112 zz ¹ 2112 ZZ =
vx
vy
vz
v2
v12
v1
0º 180º 90º
v2
v1 v12

QFIII–TEMA2 27
Si se quieren calcular las colisionesde tipo1-1, es decir, entremoléculasdelmismotipo,lasexpresionescorrespondientessepuedendeducirapartirdelasanteriores.Así,paralafrecuenciadecolisióndeunamoléculasepodríaescribir:
donde,deacuerdoconlasdefiniciones y lavelocidadrelativamediasería:
quesustituidaenlaexpresiónanteriordafinalmente:
(2.19)
Elnúmerototaldecolisionesdetipo1-1porunidaddetiempoydevolumensepuede
obtenermultiplicandolafrecuenciaz11porelnúmerodemoléculasdetipo1ydividiendoporelvolumen.Sinembargo,deestaformasecontaríandosvecescadacolisiónentremoléculasdelmismotipo(laaconlabylabconlaa)porloqueesnecesariodividirentredos:
(2.20)
Como puede observarse en esta fórmula y en la de Z12, la frecuencia total de colisión porunidaddevolumendependedelproductodelasconcentracionesdelasmoléculasimplicadasen la colisión.Esteeselorigenmicroscópicodequeen las leyesdevelocidaddeunaetapaelementalaparezcaelproductodelasconcentracionesdelasespeciesreactivas.
Sepuedeadquirirunaideadelamagnituddeestasvariablesrealizandoloscálculosdefrecuenciadecolisióndeunamoléculayfrecuenciatotalporunidaddevolumenparaelcasodeloxígenomolecular(d=2,4Å)a298Ky105Padepresión,obteniéndosez11=2.8x109s-1yZ11=3,4x1034s-1m-3.Esdecir, cadamoléculaencondicionesestándar, sufredelordendemilmillonesdecolisionesporsegundo,mientrasqueencadacentímetrocúbicodegassepodríancontardelordendediezmilmolesdecolisionesporsegundo.
Hay que tener en cuenta, a la hora de emplear estas expresiones, algunas de laslimitacionesmásnotables.Yasehamencionadoquenuestrasmoléculassehanconsideradocomo esferas rígidas, las cuales no presentan interacciones específicas entre ellas. Lapresencia de interacciones podría favorecer las colisiones en determinadas orientaciones.
VNvdz 1
1121111 p=
11111 d2/)dd(d =+=
1
21
21
211 m
kT82vvvp
=+=
VN
mkT8d2z 1
2/1
1
2111 ÷÷
ø
öççè
æ=
pp
21
2/1
1
2111
111 V
NmkT8d
21z
VN
21Z ÷
ø
öçè
æ÷÷ø
öççè
æ==
pp

QFIII–TEMA2 28
Además, sehan tratado todas lasmoléculascomosi tuvieran lamismavelocidad(iguala lavelocidadmedia).Porúltimo, sólosehanconsideradocolisionesbinarias, locualpuedeserunabuenaaproximaciónengasesdiluidos,perohayquerecordarquetantoenlíquidoscomoengasesconcentradoslascolisionesternariaspuedencobrarimportancia.
Ejercicio 2.6.- La temperatura media de la superficie de Marte es 220 K y la presión 4,7 torr. La atmósfera marciana está compuesta principalmente por CO2 y N2, con pequeñas cantidades de Ar, O2, CO, H2O y Ne. Considerando sólo los dos componentes principales, se puede aproximar la composición de la atmósfera marciana como x(CO2) ≈ 0,97 y x(N2) ≈ 0,03. Los diámetros de colisión son 4,6 Å para el CO2 y 3,7 Å para el N2. Para la atmósfera de la superficie marciana, calcular:
a) La frecuencia de colisión de una determinada molécula de CO2 con otras moléculas de CO2
b) La frecuencia de colisión de una determinada molécula de N2 con moléculas de CO2 (c) el número de colisiones por segundo de una determinada molécula de N2 (d) el número de colisiones CO2-N2 por segundo en 1,0 cm3 (e) el número total de colisiones por segundo en 1 cm3. Solución. -
a) donde : ,
, , , , con lo
que
b) , habida cuenta que:
de donde , que
, , ,
y
c)
=11z VN
MRT8d2 12
1
121 ÷÷
ø
öççè
æp
p 13CO molKg10x00,44M 2
--=
11molJK31451,8R --= =VN1 =
RTNp A1
RTpNx A1 Pa6,626p = 97,0x 2CO = K220T =
( ) 17A21
1
21011 s10x12,6
RTNxPa6,626x97,0
MRT8m10x6,42z -- =÷÷
ø
öççè
æp
p=
=÷÷ø
öççè
æ÷÷ø
öççè
æ+=
RTNp
MMRTdz A2
21
21
21221
118p
p 17s10x65,5 -
oN
oCO A7,3d,A6,4d 22 == ( )
o12 A15,47,36,4
21d =+=
13CO molKg10x00,44M 2
--= 13N molKg10x00,28M 2
--= 97,0x2CO = 11molJK31451,8R --=
=+ 1211 zz =÷÷ø
öççè
æp
pRTNp
MRT8d2 A22
1
222
1776 s10x80,510x65,510x54,1 -=+
RTpNx
RTNp
VN A2A22 ==

QFIII–TEMA2 29
d)
e)
Unconceptorelacionadoconlafrecuenciadecolisiónyqueresultamuyútilalahorade
entenderelcomportamientode losgaseseselderecorrido libremedio: ladistanciaqueentérmino medio recorre una molécula entre dos colisiones sucesivas. Esta magnitud,normalmente designada mediante la letra l, se puede calcular como el cociente entre elespaciorecorridoenunintervalodetiempoyelnúmerodecolisionesquehaefectuadoo,loqueeslomismo,elcocienteentrelavelocidad(espacioporunidaddetiempo)ylafrecuenciadecolisión(colisionesporunidaddetiempo).Así,paraungaspuro:
(2.21)
expresiónenlaquesehatenidoencuentaqueparaungasideal(N/V)=(p/kT).
Paranuestroejemplo,eloxígenomoleculara298Ky105Pa,elrecorridolibremedioresulta ser Å, un valormuchomayor que el tamañomolecular, peromucho más pequeño que el tamaño del recipiente que puede encerrar a una muestramacroscópica del gas. Este valor indica que antes de poder recorrer la distancia entre dosparedesdelrecipiente(delordendevarioscentímetros)nuestramoléculaefectuaráungrannúmerodecolisionesconotrasmoléculas,Esdecir,lascolisionesintermoleculares(enestascondiciones)sonmuchomásfrecuentesquelascolisionesconlasparedesdelrecipiente.Porotra parte, si tenemos el gas en condiciones de alto vacío ( ) el recorrido libremedio resulta serdelordendel=160m.Esdecir, encondicionesdealtovacío lamolécularecorredelordende160metrosentreunacolisiónconotramoléculaylasiguiente.Dichodeotramanera,colisionaráconmásfrecuenciaconlasparedesqueconelrestodemoléculas.
Sinuestrogasresultaserunamezcladedostiposdemoléculassetendráunrecorrido
libremediodistintoparacadaunadeellas:
=12Z === 21A2
212
121 z
RTNPz
VNz
VN 13297
23sm10x51,310x65,5
220x31451,810·022,6x6,626x03,0 --==
132411
A111 scm10x12,6z
RTNp
21Z -==
=++ 221211 ZZZ 132312
A212 scm10x51,3z
RTNpZ -== 1324 scm10x48,6 --=
132122
A222 scm10x78,4z
RTNp
21Z -==
pkT
d21
NV
d21
VN
mkT8d2
mkT8
zv
2112
112/1
21
2/1
11
11
ppp
p
pl ==
÷øö
çèæ
÷øö
çèæ
==
1600m10x6,1 7 == -l
atm10p 9-»

QFIII–TEMA2 30
Ejercicio 2.7.- Para el N2(g) con un diámetro de colisión de 3,7 Å, calcular el recorrido
libre medio a 300 K y: a) 1,00 bar; b) 1,00 torr; c) 1,0 ·10-6 torr (presión típica de “vacío”).
Solución.-
a) , , con y b) ,
c) , , más choques con el recipiente que entre las moléculas.
2.2.6.-ColisionesyReactividadQuímica
LaTeoríadeColisiones,desarrolladahacia1920,partedelossiguientessupuestospara
encontrarunaexpresiónparalaconstantedevelocidad:i)lasmoléculassetratancomoesferasrígidas,caracterizadasporundiámetrod.ii)paraqueseproduzcareacciónentreByCdebeproducirseunacolisiónentreambas
moléculas.iii) durante la reacción semantiene en todomomento la distribución de velocidades
moleculardeMaxwell-Boltzmann(quecorrespondeaunasituacióndeequilibrio).
ApartirdelaTeoríaCinéticadeGases,ecuación(2.18),elnúmerodecolisionesB-Cporunidaddetiempoyvolumenes:
dondes es la sección de colisión , , vBC la velocidad relativaentrelamoléculadetipoByladetipoCyNielnúmerodemoléculasdetipoi.
Si cada colisión produjera una transformación de B y C en productos (si todas lascolisiones fuesen eficaces) entonces el número de moléculas de C que desaparecerían porunidaddetiempoyvolumenseríaigualaZBC
1211
11 zz
v+
=l
2221
22 zz
v+
=l
÷÷ø
öççè
æ
p=l
A21pNRT
d21
Pa10x00,1bar00,1 5= m10x81,6 8-=l recipientelongitud<<l
Pa32,133torr00,1 = m10x11,5 5-=l
Pa10x32,133torr10x00,1 66 -- = m1,51=l
VN
VN
M1
M1RT8
VN
VNvZ CB
2/1
CB
CBBCBC
úúû
ù
êêë
é÷÷ø
öççè
æ+=><=
pss
2BCdps = 2/)dd(d CBBC +=
moléculaladetamaño>>l

QFIII–TEMA2 31
queexpresadoenmoles(ni)oenconcentracionesmolaresqueda:
La última expresión explica la dependencia de la ley de velocidades con el producto de lasconcentraciones de B y C. Comparando con la ley experimental (ecuación (2.1)) se tendrá,además,unaexpresiónparalaconstantedevelocidad:
(2.22)
Esta ecuación implica, erróneamente, una dependencia de la constante de velocidad con latemperatura como T1/2 y conduce a sobreestimaciones muy severas de las velocidades dereacción.Porejemplo,paralareacciónentreH2yI2paradarHI,ladensidaddefrecuenciadecolisionesencondicionesestándaresdeaproximadamente1035m-3s-1,loqueconduceaunavelocidaddelordende108molL-1s-1,esdecirquereaccionarían100millonesdemolesporlitroysegundo!!!
Parecepuesevidentequesólounapequeñafraccióndecolisionesdalugarareacción.PorellosehadeañadirunsupuestoadicionalanuestraTeoríadeColisiones:
iv)Sóloaquellascolisionesconenergíacinéticasuficientedanlugarareacción.Esterequerimientoenergéticopuedeincorporarsedeunamanerasencillaatravésdel
concepto de sección eficaz. La sección de colisión, constante para dosmoléculas dadas, dacuenta de todas las colisiones que pueden producirse entre ellas. La idea es sustituir estaseccióndecolisiónsporunaseccióneficazs(e)quedécuentasólodelascolisioneseficaces,demaneraquesilacolisiónseproduceconunaenergíaemenorquelamínimanecesaria(e0oenergíaumbral)entoncesaefectosdereacciónlasmoléculasnoseveny laseccióneficazseríanula.Deestamanerasecontaríansólolascolisionescapacesdedarlugarareacción.Sepuedeasícalcularunaconstantedevelocidadparacadavalordelaenergíadecolisión:
(Paraunadeterminadaenergíadecolisión<vBC> indicaelpromediode lavelocidadrelativasobre todas las posibles orientaciones durante la colisión). La constante de velocidad
VN
VNvZ
dtdN
V1 CB
BCBCC ><==- s
Vn
VnNv
dtdn
VN CB2
ABCCA ><=- s
[ ] [ ][ ]CBNvdtCd
ABC ><=- s
A
2/1
CBABCr N
M1
M1RT8Nvk
úúû
ù
êêë
é÷÷ø
öççè
æ+=><=
pss
><= BCAr vN)()(k ese

QFIII–TEMA2 32
experimental, como toda propiedadmacroscópica, corresponderá al promedio de todos losvaloresposiblesdelaenergíadecolisióne:
(2.23)
dondeG(e)eslafuncióndedistribucióndelaenergíacinéticatraslacionalproporcionadaporlaTeoríaCinéticadeGases(ecuación(2.12)):
Además,pararesolverlaintegralqueapareceenlaecuación(2.23)sehadetenerencuentaque la energía de la colisión y la velocidad relativa están relacionadas mediante la masareducidaµ:
Sustituyendoenlaecuación(2.23):
(2.24)
Pararesolverlaintegralsenecesitaunaexpresiónparalaseccióneficazs(e).LafuncionalidadmássencillaseríalarepresentadaenlaFigura2.17,conlosvaloresindicadosenlamisma:
Figura2.17.-Modelosencillodeseccióneficaz.Todacolisiónquesuperaunaciertaenergíaumbraldalugarareacción
Es decir, toda colisión con energía superior a un cierto umbral dará lugar a reacción ycualquiercolisiónconenergíamenorseránoreactiva.Enestecasolaintegraldelaecuación(2.24)queda:
eees d)(Gv)(Nk BC0
Ar ><= ò¥
kT2/12/3
ekT12)(G
e
ep
pe-
÷øö
çèæ=
2/1
BC2
BC2vv
21
÷÷ø
öççè
æ>=Þ<><=
µeµe
eesepµ
eeese
de)(kT
NkT8d)(Gv)(Nk
0
kTA
2/1
BC0
Ar òò¥ -¥
÷÷ø
öççè
æ=><=

QFIII–TEMA2 33
Estaexpresióntiene laventajade incorporarya ladependenciaexponencialde lavelocidadcon 1/T. Ahora bien, suponer que toda colisión con la energía suficiente produce reacciónconduce también a una sobreestimación importante de la constante de velocidad. A esterespecto,supónganselasdoscolisionesqueaparecenenlaFigura2.18,ambasconunamismavelocidadrelativatalquelaenergíapuestaenjuegoseasuperioralumbralmínimonecesario.Laprimeradeestascolisiones(izqda)seproduciríade‘refilón’,mientras
Figura2.18.-Diferentestiposdecolisiónenfuncióndelparámetrodeimpacto
que la segunda (dcha) es una colisión ‘de lleno’. Ambas colisiones se diferencian en elparámetrodeimpactob,técnicamenteladistanciaentrelaslíneasquepasanporloscentrosdelasmoléculascondirecciónigualaladelavelocidadrelativa.Evidentemente,paraqueseproduzcacolisiónb≤dBC.Peroademáscuantomenorseabmayorprobabilidadhabrádequela colisión con e≥e0 produzca reacción. La diferencia entre ambas colisiones es que cuantomenorseaelparámetrodeimpacto,mayoreslacomponentedelavelocidadalolargodelalíneaqueuneloscentrosdelasmoléculas;esdecirmayoreslaenergíacinéticaacumuladaenesalíneayportantomayorserálaprobabilidaddeproducirreacción,yaquedeestaformalaenergíacinéticasecomunicamejoralosenlacesquedebenromperse.
La componente de la velocidad relativa a lo largo de la línea que une los centros es<vBC>cosqyporlotantolaenergíacinéticaacumuladaenesadirecciónserá
Se puede dar un paso más en la Teoría de Colisionesintroduciendounsupuestoadicional:
( ) kT02BCA
2/1
kTA
2/1
0
kTA
2/1
r
0
0
ekTdNkT8
dekT
NkT8de)(
kTN
kT8k
e
e
ee
eppµ
eespµ
eesepµ
-
¥ -¥ -
+÷÷ø
öççè
æ=
=÷÷ø
öççè
æ=÷÷
ø
öççè
æ= òò
( ) ( )
÷÷
ø
ö
çç
è
æ-=
÷÷
ø
ö
çç
è
æ-><=
=-><=><=
2BC
2
2BC
22
BC
22BC
2BCc
db1
db1v
21
sen1v21cosv
21
eµ
qµqµe
b<vBC>
<vBC>b
dBCb
q<vBC>

QFIII–TEMA2 34
iv.b)Unacolisiónseráreactivasiseproduceconenergíacinéticasuficienteacumulada
enladireccióndelalíneaqueuneloscentrosdelasmoléculasquecolisionan.Esdecir,paraque se produzca reacción ec≥e0. Se ha de encontrar pues la sección eficazs(e) que recojaadecuadamente este requisito para poder realizar la integral que aparece en la ecuación(2.24).Laenergíacinéticaacumuladaenlalíneadeloscentroseces,evidentemente,menoroigualalaenergíatotaldelacolisióneporloquesiestaúltimaesmenorquelaenergíaumbrale0laseccióneficazdebesernula:
•Si ,porloque
¿Peroquéocurresi ?Enesecasohabráreacciónsi ,esdecir,
habrá reacción si la colisión se produce con un parámetro de impacto .
Podemosdecirpor lo tanto, queparauna cierta energíade colisióne, la seccióneficazquepresentanlasmoléculasyquedacuentadelnúmerodecolisionesreactivasserá:
•Si ,
LafunciónresultanteeslamostradaenlaFigura2.19conlosvaloresenellaincluidos
Figura 2.19.- Modelo de sección eficaz que considera la energía cinética acumulada en la línea que une loscentrosFijémonosenloquerepresentaestafunción.Evidentementesilaenergíapuestaenjuegoenla colisiónno alcanza el valorumbral ninguna colisión será reactiva. Si, por el contrario, laenergía es infinita, toda colisión será reactiva. ¿Qué pasa entre ambos límites? Pues que lacolisiónesefectivasólosiseacumulasuficienteenergíaenlalíneaqueuneloscentrosdelasmoléculas. Lógicamente, a medida que aumenta la energía total de la colisión aumenta laprobabilidaddetenerenergíasuficienteacumuladaenesadirecciónyaumentalaproporcióndecolisionesreactivas.Evidentementelafraccióndecolisionesefectivasesmenorqueconelmodeloanterior(línearojadelaFigura2.19).
Sustituyendolaexpresiónencontradaenlaecuación(2.24):
0c0 eeee <Þ< 00)( eees <"=
0ee ³ 02BC
2c
db1 eee ³÷
÷
ø
ö
çç
è
æ-=
÷ø
öçè
æ -£ee02
BC2 1db
0ee ³ 002
BC2max 1db)( ee
ee
ppes ³"÷ø
öçè
æ -==

QFIII–TEMA2 35
Unanálisisdeesteresultadomuestraquelaconstantedevelocidadpuedeversecomodebidaadostérminos:
(2.25)
Elprimerodeellos, ,esdebidoalnúmerototaldecolisionesdetipoB-C,
mientras que el segundo, , da cuenta de la fracción de colisiones que cumplen con elrequisitoenergético.
Apartirdeestaexpresiónparalaconstantedevelocidadproporcionadaporlateoríadecolisionessepuedenencontrarlasexpresionescorrespondientesalaenergíadeactivaciónyalfactorpreexponencial.
DeacuerdoconlaecuacióndeArrhenius(ecuaciones(2.2)y(2.3)):
,
Sustituyendolaecuación(2.25)enestasexpresiones:
(2.26)
dondeE0representalaenergíaumbralnecesariaparaqueunmoldecolisionesseaefectiva.Normalmente (½)RTespequeñocomparadocon laenergíadeactivación (alrededorde0,5
( ) kT2BCA
2/1kT02
BCA
2/1
kT02BCA
2/10
kTA
2/1
r
0
0
0
edNkT8dekT
dNkT8
de1kT
dNkT8
de)(kT
NkT8k
e
e
e
e
e
e
ppµ
eee
ppµ
eeeep
pµ
eesepµ
-¥ -
¥ -
¥ -
÷÷ø
öççè
æ=
-÷÷ø
öççè
æ=
=÷ø
öçè
æ -÷÷ø
öççè
æ=
=÷÷ø
öççè
æ=
ò
ò
ò
kT2BCA
2/1
r
0
edNkT8ke
ppµ
-÷÷ø
öççè
æ=
2BCA
2/1dNkT8 p
pµ ÷÷ø
öççè
æ
kT0
ee
-
dTklndRTE r2
a = RTE
eke/kA arRT
E
r
a
==-
00A
20202
BCA
2/12
a
ERT21NRT
21
kTT21RT
kTTln
21dNk8ln
dTdRTE
+=+=
=úû
ùêë
é+=
úú
û
ù
êê
ë
é-+÷÷
ø
öççè
æ=
e
eep
pµ

QFIII–TEMA2 36
kcal/mola300K,mientrasquelasenergíasdeactivacióntípicamenteimplicanvariasdecenasdekcal/mol)porloqueprácticamenteEa»E0.ElproblemaesquelaTeoríadeColisionesnoproporciona ningún método para calcular E0 por lo que debe tomarse directamente de laenergíadeactivaciónexperimental.Encambio, laTeoríadeColisionessípermitecalcularelfactorpreexponencial:
(2.27)
Para la mayoría de reacciones los valores calculados de A utilizando la Teoría de
Colisiones son mucho mayores que los determinados experimentalmente, como se puedeobservar en algunos ejemplos mostrados en la Tabla 2.1, lo que implica la necesidad deintroduciralgúnrequisitoadicionalenlaTeoríadeColisiones:
Tabla2.1ComparaciónentrevaloresdeAcalculadosyexperimentales
Reacción Ea(kJmol-1) Aexp(M-1s-1) Acalc(M-1s-1)
2NOCl®2NO+2Cl 102,0 9,4x109 5,9x10102ClO®Cl2+O2 0,0 6,3x107 2,5x1010C2H4+H2®C2H6 180,0 1,24x106 7,3x1011
v) Para que una colisión sea eficaz se ha de producir además con una determinadaorientaciónentrelosreactivos.
Esterequisitoenelfondosuponeadmitirquenuestrasmoléculasnosonesféricas,sinoquetienenunadeterminadaestructura,loquelashacesusceptiblesdereaccionarsolamentesilosreactivosseencuentranenlaorientaciónadecuada.Porejemplo,lareacciónO2+CO®O+CO2 sólo puede tener lugar si la molécula de oxígeno colisiona con la demonóxido decarbonoporelladodelátomodecarbono,nosilohaceporelladodelátomodeoxígeno.Sepuedeintroduciresterequisitoatravésdel llamadofactorestéricop,quees la fraccióndecolisiones que se producen con la orientación precisa. De esta manera la constante develocidadseríaelproductodetresfactores:unoquetieneencuentaelnúmerodecolisionesB-C, otro que introduce el requisito energético y finalmente otro para el requisito deorientación:
(2.28)
2/12BCA
2/1
)RTE
21(
kT2BCA
2/1RTE
r
edNkT8
eedNkT8ekA00a
ppµ
ppµ
e
÷÷ø
öççè
æ=
=÷÷ø
öççè
æ==
+-
kT2BCA
2/1
r
0
edNkT8pke
ppµ
-÷÷ø
öççè
æ=

QFIII–TEMA2 37
Enlaprácticapsedeterminacomoelcocienteentreelfactorpreexponencialexperimentalyelcalculadocon(2.27)(Aexp/Acalc)conloquetieneescasovalorpredictivo.
Nosencontramospuesconunateoríaquepermiteinterpretarladependenciadelaleydevelocidadconelproductodelasconcentraciones,asícomolavariacióndelasconstantesdevelocidadconlatemperatura(leydeArrhenius).Básicamentelareacciónsedaapartirdeunacolisiónquehadecumplirunaseriederequisitos.Sinembargo,estateoría,enlaformaexpuesta hasta aquí, tiene escaso valor predictivo pues no permite calcular el valor de laenergía de activación y conduce, habitualmente, a sobreestimaciones notables del factorpreexponencial.EstosdefectosdelaTeoríadeColisionessondebidosfundamentalmentealaconsideración de las moléculas como esferas rígidas. De hecho, la introducción del factorestérico es un intento de considerar en el modelo la estructura molecular. La teoría decolisiones podrá puesmejorarse considerando en elmodelo la estructura de lasmoléculas(enlaces, vibraciones, …) y las interacciones intermoleculares. A este respecto, un ejemploclaroparailustrarlaimportanciadelasinteraccionesintermoleculareseslareacciónK+Br2®KBr+Br.EnestecasolaTeoríadeColisionesprediceunfactorpreexponencialmenorqueelexperimental,loquellevaaunfactorestéricomayorquelaunidad(p=4,8).Larazóndeestecomportamientoesquealgunastrayectoriasqueenprincipioseríannoreactivas(confactorde impacto b>dBC) pueden dar lugar finalmente a colisión por la aparición de fuertesinteraccionesatractivas.Efectivamenteunelectróndelpotasiopuedesaltarhastalamoléculadebromo,quedandoambasespeciescargadas(K+yBr-).Estodalugaralaaparicióndeunafuerza electrostática que puede conducir a la colisión entre las dos moléculas, como seesquematizaenlaFigura2.20.
Figura2.20.-Lasinteraccionesintermolecularesinfluyensobrelastrayectoriasmoleculares.
Ejercicio 2.8.- Calcular, haciendo uso de la teoría de colisiones, la constante de velocidad para la reacción H2+I2 ®2HI a 700 K; utilizando un diámetro de colisión de 1 Å y una energía de activación de 40 kcal mol-1. Comparar el resultado con el valor experimental (6,42x 10-2 l mol-1s-1). Solución.- En su versión más sencilla la teoría de colisiones proporciona la siguiente
expresión para la constante de velocidad donde: kT2BCA
2/1r
0
edNkT8ke
-p÷÷
ø
öççè
æpµ
=
b
K
Br2
b
K
Br2-
+
b
K
Br2-
+e-

QFIII–TEMA2 38
, , ,
, resultando:
Si se calcula el factor estérico comparando el resultado obtenido frente al valor experimental se
comprueba que se obtiene un resultado extraño, . El factor estérico representa,
en principio, la fracción de colisiones que tiene lugar con la orientación requerida. De acuerdo con esta definición, el factor estérico debería ser menor que la unidad. Sin embargo, al calcularse como cociente entre el valor experimental y el calculado recoge cualquier limitación en la teoría y podría darse el caso de ser mayor a la unidad cuando las interacciones intermoleculares hacen, por ejemplo, que las moléculas se atraigan y tengan lugar más colisiones que en ausencia de dichas interacciones (véase el ‘efecto arpón’). Este no parece ser, sin embargo, este caso de la reacción entre hidrógeno y iodo donde las interacciones no deben ser muy fuertes. La explicación del resultado obtenido para el factor estérico es mucho más sencilla. El diámetro de colisión propuesto es excesivamente pequeño, por lo que se están calculando muchas menos colisiones que las que tienen lugar realmente, motivo por el que se ha subestimado la constante de velocidad. Si se emplea un diámetro de colisión más lógico, derivado de los diámetros de las moléculas de hidrógeno y iodo: y
, resulta un , obteniéndose con este valor
y un factor estérico , menor que la unidad, como cabría
esperar.
2.3.-SuperficiesdeEnergíaPotencial
Lamejorformadeincorporarlasinteraccionesintermoleculares,asícomolaestructurainterna de las moléculas reaccionantes, es mediante el planteamiento y solución de lacorrespondiente ecuaciónde Schrödingerdel sistema reaccionante (si se quiere estudiar lareaccióndehidrogenacióndeleteno,elsistemacuánticobajoestudioseráelformadoporlasumadelasdosmoléculas:H2+C2H4).Elhamiltonianoquedescribeunsistemadeestetipoestá compuesto por términos que se refieren a los núcleos (n), a los electrones (e) y a lasinteraccionesentreambos(en):
dondeTeeslaenergíacinéticadeloselectrones,Tn ladelosnúcleos,Venlaenergíapotencialde interacción electrostática entre los electrones y los núcleos,Vee la energía de repulsiónelectrostática entre electrones y Vnn la energía de repulsión electrostática entre núcleos.Normalmente,pararesolverelhamiltonianodeunamolécula(oenestecasodeunconjuntode moléculas) se utiliza la aproximación de Born-Oppenheimer que permite separar elmovimientodeloselectronesdeldelosnúcleossobrelabasedesusmuydiferentesmasas.De
m102dd
d 10CBBC
-=+
= µBC =mB ·mC
mB +mC
= 3,295x10−27Kg K700T =
1a0 molKcal3,39
2RTEE -=-= 1121135
r sM10x77,2smolm10x77,2k ------ ==
32,2kk
pcalc,r
exp,r==
m10x9,2d 10H2
-=
m10x2,6d 10I2
-= m10x5,42dd
d 10CBBC
-=+
=
111r sM10x61,5k ---= 11,0
kk
pcalc,r
exp,r==
nneeenne VVVTTH ++++=

QFIII–TEMA2 39
unamanerasimplificadasepuededecirqueyaque lamasade losnúcleosesmuchomayorque la de los electrones (para el átomo de hidrógeno mn~1000 me), se puede resolver lafunción de onda electrónica suponiendo fijos los núcleos. Esta aproximación lleva pues atomarcomoconstantelaenergíaderepulsiónnúcleo-núcleo(Vnn=cte)ycomocerosuenergíacinética(Tn=0)alahoraderesolverlaestructuraelectrónica.Así,quedapuesporresolverelhamiltonianoelectrónico:
La ecuación de Schrödinger correspondiente proporciona el valor de la energía electrónica(energía cinética de los electrones + energía de repulsión interelectrónica + energíainteracción electrón-núcleo) y la función de onda que describe el comportamiento de loselectronesenelcampocreadoporlosnúcleos:
Siaestaenergíaelectrónicaselesumalaenergíaderepulsióninternuclear(Vnn)setendrálaenergía total que corresponde a una configuración fija de los núcleos, es decir, la energíapotencialdenuestramoléculaoconjuntodemoléculas:
V(R)=Eele+Vnn (2.29)
Paratenerlaenergíatotaldelamoléculasetendráqueañadiralaenergíapotenciallaenergíacinéticadelosnúcleos:
ET=V(R)+Tn
EnprincipioestaenergíapotencialdependedetodaslascoordenadasnuclearesR,esdecirde3Nvariables,siNeselnúmerodeátomos.Sinembargo,ni latraslación,ni larotaciónde lamolécula o conjunto de moléculas afectan a su energía potencial (ya que en ese caso lasdistancias entre los núcleos no cambian), por lo que ésta dependerá de 3N-6 coordenadas(3N-5 si nuestro sistema es lineal). Esta energía potencial es de suma importancia puespermite conocer cuáles son las fuerzasque actúan sobre los átomosdenuestrasmoléculascuandoéstascomienzanareaccionar,esdecir,cuandoseproducencambiosenladistribuciónde los electrones rompiéndose y formándose enlaces. Así, la fuerza que actuaría sobre unátomoicualquierasería:
V(R) (2.30)
eneeeele VVTH ++=
eleeleeleele EH YY =
iiF -Ñ=!

QFIII–TEMA2 40
ConsidéresecomounejemplolareacciónH2®2H.Nuestrosistemaaestudiarsonlosdos átomos de hidrógeno. La energía potencial en este caso depende de 3x2 – 5 =1coordenada,quees ladistancia internuclearR.Paraobtener la funciónenergíapotencial serealizael cálculode la funcióndeondaelectrónicaadistintosvaloresdeRy se leañade larepulsión internuclear ( ). El resultado, si los cálculos son correctosdebeserunafuncióncomolarepresentadaenlaFigura2.21.LacurvanaranjarepresentalavariacióndelaenergíapotencialdelamoléculadeH2conladistanciainternuclear.Tienela
Figura2.21.-Variacióndelaenergíapotencialdelamoléculadehidrógenoconladistanciainteratómicaforma característica de un proceso disociativo. En general este tipo de representaciones seconocencomoSuperficiesdeEnergíaPotencial(SEP),siendoenestecasounidimensional.Loprimeroquehayquedestacaresquetodaenergíapotencialtieneceroarbitrario,queenestecaso se ha escogido en el mínimo de la curva (R0). El valor de la energía de disociación,energía necesaria para llegar desde este mínimo hasta la disociación completa (átomos adistanciainfinita),esde4,74eVparalamoléculadehidrógeno.Imaginemosqueenunciertoinstante se tiene congelada nuestra molécula de hidrógeno (sin energía cinética en losnúcleos)aunadistanciaRa.¿Quéocurriríacuandosedescongeleelmovimientonuclear?Deacuerdoconlaecuación(2.30),vaaaparecerunafuerzapositivasobrelosnúcleos(yaqueladerivadadeVconladistanciainternuclearesnegativaenestepunto)quetenderáporlotantoaaumentarladistanciainternuclear.AlcabodeunciertotiempolosnúcleosseencontraránaunaciertadistanciaRb>Ra.Eneseinstantelaenergíapotencial(representadacomounalíneavertical azul) ha disminuido ya que la repulsión internuclear esmenor. Por supuesto, si lamoléculanocolisionaconotraoconlasparedes,éstadebedeconservarlaenergía.¿Quéhapasadoconlaenergíapotencial?Sehatransformadoenenergíacinética(representadacomouna líneavertical roja),de formaque laenergía totalTnn+Vpermanezcaconstantee iguala
R)4/(qqV 0HHnn pe=
R
E
De=4.74 eV
RoRa
E1
Rb RcRdRe
E2
R¥

QFIII–TEMA2 41
E1). La fuerza sigue empujando a nuestra molécula a aumentar la distancia internuclear.CuandosellegaaR0todalaenergíapotencialsehaconvertidoencinética.Enestaposiciónlafuerzaesnula( ),perolainerciaadquiridasiguealejandolosátomos.Pocoapocolaenergía cinética va convirtiéndose ahora en energía potencial, hasta llegar a Rd donde laenergíacinéticaesnula.Enestaposición,lasfuerzasactúanensentidocontrario(lafuerzaesnegativa pues la derivada de V con R ahora es positiva) y el sistema comenzará ahora elprocesocontrario,disminuyendosudistanciainternuclear.Elmovimientoquehemosdescritonoesmásqueunavibraciónmolecular.Pero, ¿quépasa si anuestramolécula se ledaunaenergíainicialigualaE2?Laenergíapuedellegarledeunacolisión,porejemplo.SupongamosquetenemoslamoléculaconunadistanciaigualaReyenergíacinéticanula.Lafuerzahaceque la molécula comience a separarse, transformando energía potencial en cinética, peroahora,alllegaraR¥nohayningunafuerzarecuperadora(lafunciónpotencialeshorizontal,conloqueladerivadaconResnula).Por lotanto,elsistemacontinuaríamoviéndosehaciadistanciasmayoresproduciéndoseladisociacióncompletayporlotantotenemosahoraunareacciónquímicadescritasobrelaSEP.
El conocimiento pues de la SEP resulta fundamental para abordar el estudio de lasreaccionesquímicas:
- NosinformadecuáleslaestructuraestableparanuestramoléculadeH2.Enestecaso
la estructura estable aparece cuando R=R0, que para lamolécula de hidrógeno valealrededorde0,7Å.
- Nos informa del cuál es la energía umbral o mínima necesaria para producir lareacción(enestecaso4,74eVparadisociarlamoléculadeH2).
Elproblemasurgealpasarasistemasconunmayornúmerodeátomos.Porejemplo,si
se tiene unamolécula de agua, con 3 átomos, la superficie de energía potencial dependeráahora de 3x3-6=3 variables o grados de libertad. En este caso se podrían elegir las dosdistanciasOHyel ánguloHOH,obien lasdosdistanciasOHy ladistanciaHH.Este tipodesuperficiesdeenergíapotencialnopuedenrepresentarsepuesnecesitaríamos4dimensiones(lastresvariablesgeométricasmás laenergía).Sehablaenestecasodehipersuperficiesdeenergía potencial. Aunque no existe ninguna restricción en cuanto al cálculo, el máximonúmerodevariablesquesepuederepresentaresdos.Afortunadamente,lagranmayoríadeprocesosquímicospuedenseguirsebienutilizandounaodosvariables,esdecir trabajandoconrepresentacionesbiotridimensionales.Veamosalgunosejemplos.
•Estudiodelavelocidadalaquesedaelequilibrioentrelasformastransygauchedel1,2-dicloroetano:
0R/V =¶¶

QFIII–TEMA2 42
En este caso se tiene un sistema con un total de 8 átomos y por lo tanto con 3x8-6=18variables que afectan a la energía potencial del sistema. Sin embargo, sólo una de estasvariables cambia significativamente durante la transformación trans⇌ gauche: el ángulodiedroquedalarotaciónalrededordelenlaceC-C.Efectivamente,elángulodiedroClCCClvale180gradosenlaformatransyaproximadamente60enlaformagauche.Elrestodevariables(distanciasdeenlaceCH,CC,CCl,ángulosinternocomoelClCC…)apenassufrenvariacionesdurante el cambio conformacional del dicloroetano, por lo quepara esteproceso, sepuedereducirelestudiodelasuperficiedeenergíapotencialasudependenciaconelángulodiedroClCCCl. Se tiene, por tanto, un problema representable en dos dimensiones (la energía y eldiedro).RealizandoloscálculosdelaenergíapotencialseobtieneunarepresentacióncomoladelaFigura2.22.Enestafigurahayvariosaspectosimportantesadestacar:
-Existenvariasestructurasparalasquelasfuerzasseanulan,esdecir,enlasquesehaceceroladerivadadelaenergíapotencial.Estasestructurassonlasqueaparecenavaloresdeldiedro de 180,±60, 0 y±120 grados. Estas estructuras se denominanpuntoso estructurasestacionarias, pues en ellas se anulan las fuerzas. Sin embargo, no todas las estructuras
0
2
4
6
8
10
-180 -120 -60 0 60 120 180
ClCCCl (grados)
DEconfDE‡
Cl
Cl
ClCl
Figura2.22.-Energíapotencialdel1,2-dicloroetanoenfuncióndelángulodetorsiónClCCCl

QFIII–TEMA2 43
estacionarias son estructuras estables. Las estructuras que aparecen a 180 grados(confórmerotrans)y±60(confórmerosgauche)sonestables.Siserealizacualquierpequeñodesplazamientoaderechaoaizquierdalaenergíapotencialaumenta,o, loqueeslomismo,apareceunafuerzaque intentarestituirelpuntodepartida(las flechasazulesde la figura).Estas estructuras estables son mínimos sobre la SEP. Sin embargo, las estructuras queaparecen a 0 y ±120 grados no son estables. Cualquier desplazamiento conduce a unadisminución de la energía potencial o, lo que es lo mismo, ante cualquier desplazamientoaparecenfuerzasquealejanlaestructuradelpuntodepartida.EstasestructurassonmáximossobrelaSEP.
- Las energías que aparecen en la figura también son relevantespara el equilibriodelprocesoqueseestáestudiando.Ladiferenciadeenergíaentreelconfórmerogaucheyeltrans(DEconf) determina el equilibrio entre dichas formas, la poblaciónde cadaunode ellos queexistiráenunamuestradedicloroetano(yasabemosqueparatenerlaconstantedeequilibrioademás de las diferencias de energía nos hace falta conocer las funciones de particiónmoleculardelasformasgaucheytrans).
-Parapasardelaformatransalagauchesedebesobrepasarunabarreraenergéticaen
cuya cima aparece uno de los máximos de la SEP. La diferencia entre la energía de estemáximo (con un diedro de ±120 grados) y la forma trans (DE‡) da el mínimo de energíanecesarioparapasardesde la formatransa la formagauche,esdecir,determinael tránsitoentrelasdosformasestablesdeldicloroetano.Esporello,queestospuntosestacionariosquesonmáximos en la dirección que conecta entre dosmínimos se denominan estructuras detransición.Estadiferenciadeenergíaentrelaestructuradetransiciónyelreactivo(energíadetransición)determinarálavelocidaddelprocesoquímico.
• El ejemplo de reacción más sencillo con rotura y/o formación de enlaces es la deintercambio de hidrógeno Ha + HbHc® HaHb + Hc. En principio la SEP para este procesodependeríade3x3–6=3variablesgeométricasquepodríanserladistanciaentrelosátomosayb(Rab),ladistanciaentrelosátomosbyc(Rbc)yelánguloentrelosátomosabc(qabc).Sinembargo, la aproximación del átomo Ha para dar lugar a la reacción suele ocurrirpreferentementealolargodelalíneaqueuneloscentros(esdecir,conqabc=1800),yaqueesteángulodeaproximacióneseldemenorenergíaalpermitirestablecerunnuevoenlaceHa-Hbmásfácilmente.Asípues,sueleestudiarselaSEPconestevalorfijo,pasandoentoncesadepender laenergíaúnicamentededosvariables(RabyRbc).Se tieneasíunproblemade3dimensiones(lasdosdistanciasylaenergía).LaSEPseobtendríacalculandolaenergíaparacada par de valores de las distancias. Punto a punto obtendríamos una representación 3-DcomolaqueapareceenlaFigura2.23.Estetipoderepresentaciones3-Dpuedenserdifícilesdever conclaridad,por loque frecuentemente seprefiereuna representaciónen formadecurvasdenivel.Aligualquelosmapastopográficosmuestrancurvasqueindicanlaalturadelterreno para cada punto (latitud y longitud), en una SEP las curvas indican el valor de laenergíaparacadapunto(coordenadasRabyRbc).ParaestareacciónelcorrespondientemapadecurvasdenivelseríaelrepresentadoenlaFigura2.24.

QFIII–TEMA2 44
Figura2.23.-EnergíapotencialdelsistemacolinealH2+Henfuncióndedosdistanciasentreátomosdehidrógeno
Estado InicialHa + HbHc
Estado FinalHaHb + Hc
Energía Potencial
Atomos disociadosHa + Hb + Hc
Estructura Transición(RAB=RBC)
‡
Camino de Reacción
Figura2.24.-RepresentacióndecurvasdenivelisoenergéticascorrespondientealaFigura2.23

QFIII–TEMA2 45
SobreestaSEP,dondelaslíneasisoenergéticasaparecencada10Kcalmol-1,sepuedenidentificardiferentesestructurasparalasqueseanulanlasfuerzas,esdecirlasderivadasdelaenergíapotencialrespectoalascoordenadaselegidas( ).Estasestructurassonlasqueaparecenseñaladascomo‘x’,‘z’,‘s’,‘y’enlafiguradecurvasdenivel.Sin embargo, la naturaleza de estas cuatro estructuras estacionarias (estructuras congradienteofuerzanulo)esmuydiferente:
- Las estructuras ‘x’ y ‘z’ son mínimos sobre la superficie de energía potencial. Encualquierdirecciónquenosmovamosapartirdeestospuntoslaenergíapotencialaumenta(matemáticamente diríamos que las derivadas segundas son positivas). En un mapatopográfico correspondería al fondodeun valle. Losmínimos sobre la SEP corresponden aestructurasestables,yaqueantecualquierdeformaciónde lageometríaapareceuna fuerzarecuperadora.Ennuestrocaso lospuntos ‘x’ y ‘z’ correspondenal complejo formadopor lamoléculadehidrógenoconelátomodehidrógenoenproductosyreactivos:
‘x’: Ha--------HbHc Rab»2,7ÅyRbc»0,74Å‘z’: HaHb--------Hc Rab»0,74ÅyRbc»2,7Å
En este caso particular el valle esmuy poco profundo en una de las direcciones ya que elcomplejo formado por la molécula y el átomo de hidrógeno presenta una energía deinteracciónmuypequeña.
-MáximosyPuntosAsintóticos-Enlafigurapuedeobservarseque,siaumentamoslasdosdistanciasRabyRbcsimultáneamentelaenergíadelsistemacrececontinuamentehastallegar,asintóticamente,atenerlostresátomosdehidrógenoporseparado.Estaeslaestructuraalaquedenominamos‘y’:
‘y’: Ha--------Hb--------Hc Rab=Rbc®¥Este tipodeestructuras,nosonrelevantesparaelestudiode la reacciónquímicaen laqueestamos interesados, ya que la reacción puede transcurrir sin que el sistema visite estasregiones tan energéticas. Es decir, es posible formar una nuevamolécula de hidrógeno sinnecesidad de pasar por el estado correspondiente a tener los átomos completamenteseparados.
-Laestructura‘s’presentaunacaracterísticamuysingular.Esunmáximoúnicamenteenunadirección,mientrasqueesunmínimoenelrestodedirecciones(ennuestrocaso,sólohaydos coordenadas, por lo tanto, es máximo en una y mínimo en la otra). En un mapatopográfico correspondería a un collado entre montañas. Este tipo de estructuras sedenominanpuntosdesilla,yaquesurepresentaciónrecuerdaaunasillademontar.
Paraelsistemabajoestudioelpuntodesillaaparececonlasiguientegeometría:
0R/V;0R/V bcab =¶¶=¶¶

QFIII–TEMA2 46
s:Ha---Hb---Hc Rab=Rbc»0,93ÅEsta estructura es fundamental en el estudio de la reactividad química, pues da lamínimaenergía necesaria para transformar nuestros reactivos en productos. Para pasar desde ‘x’hasta ‘z’ se ha de alcanzar almenos la energía dada por la estructura s. Cualquier caminotrazadode‘x’a‘z’requierecomomínimoquelosreactivosalcancenunaenergíaigualalade‘s’.Estaesportantolaestructuradetransiciónde lareacción.Engeneral,unaestructuradetransiciónhadeverificaralmenoslassiguientescondiciones:
- Debedetratarsedeunpuntoestacionario.- Debedesermáximosóloenunadirecciónymínimoenlasdemás.- Laestructurade transicióndebe serelpuntomásaltodeuna línea trazada sobre la
SEPqueconecteentresíreactivosyproductos.
Figura2.25.-Representacióndelospuntossingularesqueaparecenenunasuperficiebidimensional:mínimos,
máximosypuntosdesillaEn principio, durante una reacción química los reactivos pueden transformarse en
productossiguiendocualquierrutasobrelaSEP.Sinembargo,detodaslasrutasposibles,lamás favorableesaquellaquepresente lamenorenergíapotencialposible.Estecamino,quedebe pasar por la estructura de transición, se conoce como camino de reacción o tambiéncoordenadadereacciónyaparecedibujadoconlíneasdiscontinuassobrelarepresentacióndelascurvasisoenergéticasdelaFigura2.24yquepasaporlasestructurasx-t-s-u-z.Deacuerdoconestecaminolareacciónpuededescribirsedelasiguientemanera:inicialmenteelátomode hidrógeno se va acercando a la molécula, permaneciendo la distancia de enlaceprácticamente constante (transformación de ‘x’ a ‘t’). A continuación, la distancia Rab siguedisminuyendo, pero ahora la distancia Rbc aumenta simultáneamente (de ‘t’ a ‘s’). En laestructura‘s’lasdosdistanciassoniguales.Acontinuación(de‘s’a‘u’)seacabadeformarelenlaceab y de romperse elbc, demanera que, en ‘u’, ya se ha formado la nuevamolécula.Finalmente (de ‘u’ a ‘z’) el átomo c se aleja quedando ya estabilizada la molécula ab. LarepresentacióndelcambiodeenergíaalolargodeestecaminoapareceenlaFigura2.25yes

QFIII–TEMA2 47
eltipoderepresentacionesquesueledarseparaesquematizarloscambiosenergéticosenunareacción química. Ahora se puede ver que este camino corresponde a uno de los posiblescaminossobrelaSEPqueconducendereactivosaproductos.Peronounocualquiera,sinoalde menor energía. Este perfil energético recoge la esencia de la reacción química quequeremosestudiar:enélaparecenlosreactivosyproductos(comomínimosdelacurva)ylaestructura de transición (como el punto de máxima energía), así como las diferencias deenergíaentreellos,loquedapieaconocerelequilibrioquímicoylaconstantedevelocidad.
Figura2.26.-VariacióndelaenergíapotencialalolargodelcaminodereaccióndemínimaenergíaparaelsistemacolinealH2+H
Figura 2.27.- Movimiento con punto de silla de mínima energía. Representación de los movimientosdetensiónenlaestructuradetransicióndelareacciónH2+H
Volviendodenuevoalaestructuradetransición,enconcretoyparalareacciónquese
sigueestudiandocorrespondeaunaestructuradondelasdistanciasdelenlacequeserompeyque se forma son iguales. Esto no es una característica general de las estructuras de
Rbc(Å)
Rab(Å)
80
60
40
20
0
‡
x
z1.0
2.0
1.0 2.0
y
su
t
v
w
reactivos
t u
productos
E(kcal/mol)
20
40‡
coord. reacción (Å)
®¬------ HHH
¬®¬------ HHH

QFIII–TEMA2 48
transición, sino que se da en este caso por la simetría entre reactivos y productos. Laestructuradetransiciónespuesunaestructuradetresátomoslinealysimétrica.Enprincipioestaestructuradeberíatener4vibracionestensiónasimétrica,tensiónsimétricayflexión,queestádoblementedegenerado).Sinembargoalobligaraqueelánguloseasiempreqabc=180ºlaflexióndesapareceyquedandosposiblesvibraciones.EstavibracióncorresponderíaenlaSEPaunmovimientoalolargodelarectadependienteunidadquepasaporelpuntodesilla(líneaw-s-venlaSEP,talycomosedestacaenlaFigura2.27).Alolargodeestemovimientoelpuntodesillaeseldemínimaenergíaporloquelaestructuradetransiciónesunmínimoenestadirección.
Esta vibración correspondería en la SEP a un movimiento a lo largo de la recta de
pendientemenosunoquepasaporelpuntodesilla.Alolargodeestemovimientoelpuntodesillaesunmáximodeenergía.Fijémonosqueestavibraciónenlaqueunenlacesealargayelotroseacortaeselmovimientoquepermitepasardereactivosaproductos,esdecirdeunmínimo a otro mínimo. El punto de silla es justamente el máximo en la coordenada quetransformaelreactivoenproducto.Estavibraciónespecialde laestructuradetransiciónsedenominavectordetransiciónyesespecialporqueenellanuestraestructuranounmínimo,no hay fuerza recuperadora. Cualquier desplazamiento a lo largo de esta coordenada haríacaer nuestra estructura de transición hacia el valle de los reactivos o hacia el valle de losproductos.
LaSEPesporlotantolapiedraangularquepermiteaccederalestudiodelasreaccionesquímicas.Paraquelosreactivospuedantransformarseenproductosdeberánteneralmenosunaenergíacinéticaigualaladiferenciadeenergíapotencialentrelaestructuradetransicióny los reactivos. En ese caso, los reactivos podrían llegar hasta el punto de silla a base deinvertir toda su energía cinética. Una vez allí podrían caer hacia el valle de productostransformandoahoralaenergíapotencialencinética.Paraconocerlavelocidaddelareacciónquímica no es por tanto suficiente con conocer la SEP, se han de resolver además lasecuacionesdelmovimientoparaanalizarconquéfrecuenciapuedenconvertirselosreactivosen productos. Para ello se escoge un estado inicial de los reactivos (geometría, energíacinética traslacional, rotacional, estado vibracional). Las fuerzas se obtienen de la SEP y apartirdeellasseresuelvenlasecuacionesdeNewtondelmovimiento.
ElcaminoquevatrazandoelsistemasobrelaSEPseconocecomotrayectoria.Deestamanera, siguiendo la evolución del sistema (su trayectoria) se podría determinar si lascondiciones iniciales conducen o no a la transformación de los reactivos en productos.Calculando las trayectorias para un conjunto representativo de condiciones iniciales ypromediando para todas ellas se puede obtener la constante de velocidad. La Figura 2.28ilustradosposiblestrayectoriasadicionales(a)y(b)delafigura)sobrelaSEP.Laprimera,

QFIII–TEMA2 49
Figura2.28.-Ejemplosdetrayectorias:reactiva(a),noreactiva(b)
la(a),esunatrayectoriareactiva.ComienzaenreactivosHa+HbHcyterminaenproductos.Sepuede observar que la trayectoria no sigue exactamente el camino de reacción, ni pasaexactamenteporelpuntodesilla,esoocurriríasilaenergíacinéticafueranulaalllegaraesepunto.AlprincipiodelatrayectorialalíneamuestraunadisminucióndeladistanciaRab(losreactivosseaproximan)mientrasladistanciaRbcoscila(loqueescaracterísticodeunenlace).Unavezsepasaporelpuntodesillayseentraenelvalledeproductos,ahoraseobservaquela distancia Rbc crece, los productos se alejan, mientras aparecen las oscilaciones en ladistanciaRab(sehaformadoelenlace).
Lasegundadeellas,la(b)delaFigura2.28,esunejemplodetrayectorianoreactiva.Ladistancia Rab primero disminuye (el átomo Ha se acerca a la molécula HbHc), colisionan yluegovuelveaalejarseHadelamoléculaHbHc.Silaenergíacinética(traslacional+rotacional+vibracional)noessuficienteparasuperarlabarreradeenergíapotenciallatrayectorianopuedeserreactiva(almenosenmecánicaclásica).
Otra forma de dibujar las trayectorias consiste en representar las distancias frente altiempo,talycomosemuestraenlaFigura2.29.Enestecaso,elejemplodeladerechailustrauna trayectoria colineal no reactiva. La distancia Rbc está oscilando (vibración del enlace)mientrasquedisminuyenlasdistanciaRabyRbc(lasegundaessiempremayorquelaprimeraloqueindicaqueHaseacercaalamoléculaporelladodeHb).Unavezproducidalacolisión,el enlaceHbHc sigue formado (distancia corta vibrando)mientras que el átomoHa se alejamanteniendolacolinealidad(laslíneasRabyRacsemantienenconunaseparaciónconstante).

QFIII–TEMA2 50
Figura2.29.-Representacióndelavariacióndelasdistanciasentreátomosdehidrógenosfrentealtiempoparaunatrayectoriareactiva(a)yotranoreactiva(b)
El ejemplo de la izquierda ilustra una trayectoria reactiva pero donde el ángulo qabcpuede cambiar. Al principio se tiene al átomoHa acercándose a lamoléculaHbHc que estávibrando.Cuandocolisionan seproduce reacción (ladistanciaRab sehacemás cortaque laRbc).ElátomoHc sealejade lamoléculaHaHbsiendoprimeromás larga ladistanciaconHaqueconHb.Sinembargo,ahorapartedelaenergíacinética liberadahasidotransferidaa larotación,lamoléculaHaHbgira,haciéndosemáspequeñaladistanciaRacquelaRbc.Sepodríailustraresquemáticamenteelprocesocomo: giro
Ha----HbHc®Ha--Hb—Hc®HaHb----Hc®HbHa----HcLadeterminacióndelaconstantedevelocidadmedianteelcálculodetrayectoriasdereaccióneslaboriosaycomplicada.PrimeroexigeconocerlaSEPydespuésresolverlasecuacionesdelmovimiento para un gran número de casos. Además, se ha de tener en cuenta que losmovimientosatómicosobedecenrealmente lasreglasde lamecánicacuánticayno lasde lamecánica clásica. Cuanto más ligeros sean los átomos (como es el caso del hidrógeno)mayores serán lasdesviacionesque cabeesperar entre el tratamiento cuánticoy el clásico.Por supuesto el tratamiento cuántico es mucho más complejo, ya que se debe resolver laecuacióndeSchrödingerdependientedeltiempo.Unadelasconsecuenciasmásimportantesesquedesapareceelconceptodetrayectoria,quehadesustituirseporeldeprobabilidaddeencontraralsistemaenundeterminadoestado.Anivelprácticoeltratamientocuánticollevaaqueesposibleencontrar trayectoriasde reactivosque sindisponerde laenergía cinéticanecesaria son capaces de atravesar la barrera de energía potencial y transformarse enproductos(efectotúnel).
Esportodoello,porloqueseríadeseabledisponerdeunateoríaque,incorporandolarigurosidadenladescripcióndelasestructuraseinteracciones(lasmoléculasnosonesferas
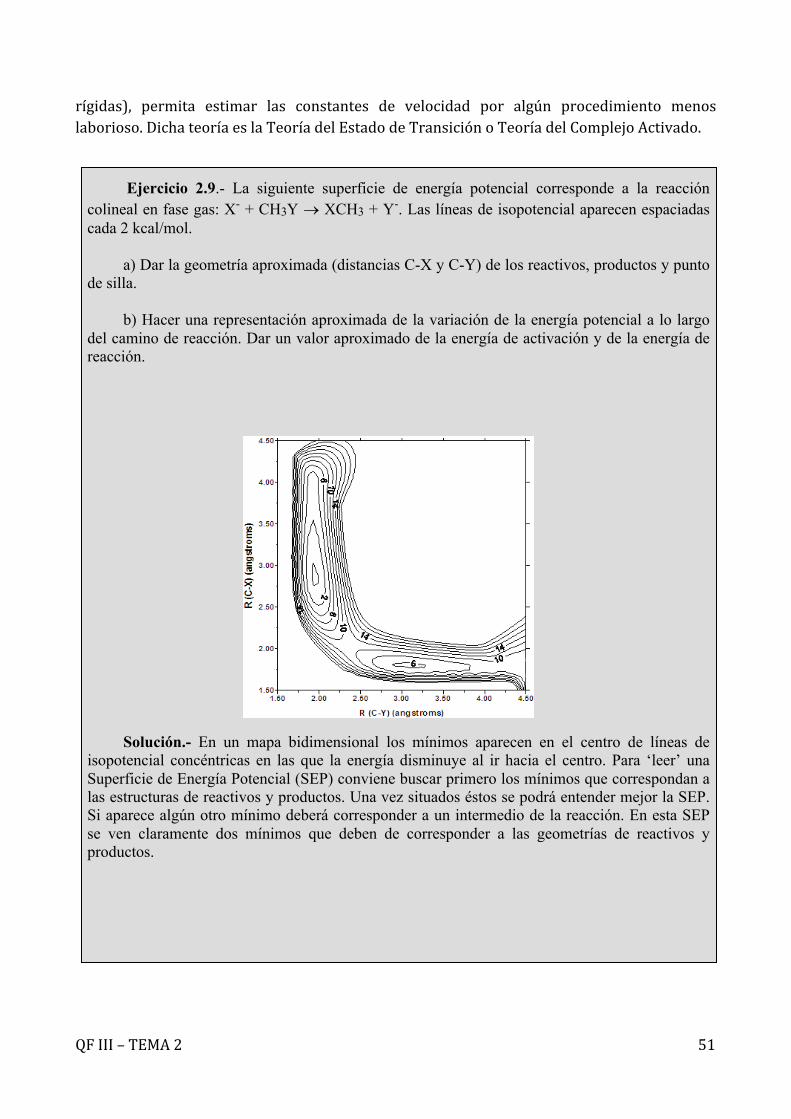
QFIII–TEMA2 51
rígidas), permita estimar las constantes de velocidad por algún procedimiento menoslaborioso.DichateoríaeslaTeoríadelEstadodeTransiciónoTeoríadelComplejoActivado.
Ejercicio 2.9.- La siguiente superficie de energía potencial corresponde a la reacción colineal en fase gas: X- + CH3Y ® XCH3 + Y-. Las líneas de isopotencial aparecen espaciadas cada 2 kcal/mol.
a) Dar la geometría aproximada (distancias C-X y C-Y) de los reactivos, productos y punto
de silla. b) Hacer una representación aproximada de la variación de la energía potencial a lo largo
del camino de reacción. Dar un valor aproximado de la energía de activación y de la energía de reacción.
Solución.- En un mapa bidimensional los mínimos aparecen en el centro de líneas de
isopotencial concéntricas en las que la energía disminuye al ir hacia el centro. Para ‘leer’ una Superficie de Energía Potencial (SEP) conviene buscar primero los mínimos que correspondan a las estructuras de reactivos y productos. Una vez situados éstos se podrá entender mejor la SEP. Si aparece algún otro mínimo deberá corresponder a un intermedio de la reacción. En esta SEP se ven claramente dos mínimos que deben de corresponder a las geometrías de reactivos y productos.

QFIII–TEMA2 52
A continuación, se busca el ET que una los dos valles de reactivos y productos. El ET debe
ser máximo en una dirección (la que va de un valle al otro) y mínimo en la otra. Una vez localizado el ET se puede trazar el camino de mínima energía (en azul) que va desde el ET a los valles de reactivos y productos.
A medida que se va avanzando por el camino de mínima energía se pueden ir leyendo los
valores de la energía (las líneas de isopotencial que corta) y trazando la siguiente representación unidimensional.

QFIII–TEMA2 53
La diferencia de energía potencial entre ET y reactivos está relacionada con la energía de
activación. Realmente sería necesario incluir energía de punto cero y las contribuciones térmicas, además del término n·R·T que aparece al relacionar la entalpía de activación con la energía de activación.
2.4.-TeoríadelEstadodeTransiciónodelComplejoActivado
LaenormedificultadqueentrañaelgenerarlaSEPyunnúmerosuficientementegrande
detrayectorias,paradespuésrealizarunpromedioestadístico,hacedeseabledisponerdeunateoríamássimpleparaobtenerlasconstantesdevelocidad,peroqueincorporealgunasdelasventajas obtenidas al estudiar la SEP. Esta es la Teoría del Estado de Transición (TET)desarrollada por Eyring y colaboradores a partir de 1930. La TET elimina la necesidad derealizartrayectoriasysólorequieredelconocimientodelaSEPenlaregióndelosreactivosydelestadodetransición.
EnlaTETsedefineunasuperficiequecontieneelpuntodesillayquedividelaSEPendos zonas, la correspondiente a reactivos y la de productos. Esta superficie se denominasuperficiedivisoriaocríticayparalareacciónH+H2aparecerepresentadaenlaFigura2.30.La superficie divisoria en este ejemplo recoge el conjunto de especies Ha—Hb—Hc condistancias Rab=Rbc. De todo el conjunto de estructuras de la superficie crítica la demenorenergíaesladelpuntodesilla.Cualquierestructurasituadaaunadistanciaarbitrariamentepequeña(d)delasuperficiedivisoriasedenominacomplejoactivado.Elconjuntodetodosloscomplejosactivadossedenominaestadodetransición.
2.4.1.-Hipótesisbásicasydesarrollo
ParaestudiarlareaccióndirectaB+C®Productossepartedelossiguientessupuestos:
i) Se suponeque todos los reactivosque cruzan la superficie críticahacia losproductosnopuedenvolveratrás.Estasuposiciónparecerazonablepues,apartirdelasuperficiecrítica,lapendienteescontinuamentedescendentehastaelvalledelosproductos,porloqueeslógico

QFIII–TEMA2 54
admitirquelamayoríadereactivoscontinuarán,unavezcruzadalasuperficiedivisoria,hastallegaralazonadeproductos.Sinembargo,convieneseñalarquepuedenllegaraobservarsealgunas trayectorias en que una vez superada la superficie divisoria, vuelven hacia atrás(recruzanlasuperficie),hechoquesecomentaráposteriormente.
Figura2.30.-Representaciónde la superficiedivisoriaentre reactivosyproductospara laSEPde la reacciónH2+H
ii)SesuponequelosreactivosmantienenunadistribucióndeBoltzmann(deequilibrio),correspondiente a la temperatura del sistema, durante toda la reacción. Obviamente lasmoléculasdereactivosquesuperanunadeterminadaenergíasonlasquepuedenconvertirseenproductos y por tanto los niveles energéticos correspondientes se despoblarían. Se va asuponer,sinembargo,quelafrecuenciadecolisiónconlasparedesyconotrasmoléculaseslosuficientementealtacomopararecuperarrápidamenteladistribucióndeequilibrioentodoslosgradosdelibertadmoleculares(traslación,rotación,vibraciónyelectrónicos).
iii)SesuponequelasmoléculasquealcanzanlasuperficiecríticatambiénmantienenladistribucióndeBoltzmanncorrespondientea lamisma temperaturaque los reactivos.Aquíconvienetenerpresentequecualquierestructuradelasuperficiecrítica,cualquiercomplejoactivado,tieneuntiempodevidamediamuycorto(delordendelosfemtosegundos).Porello,no parece lógico llegar a considerar que experimenta un número suficiente de colisiones,oscilaciones o rotaciones como para redistribuir su energía y alcanzar el equilibrio. Sinembargo, se puede admitir que el conjunto de todos los complejos activados que aparecendurantelareacciónsíquerespondenaunadistribucióndeequilibrio.Esdecir,consideradossimultáneamente, loscomplejosactivadosquese formandurante la reaccióndarían lugaraunadistribucióndeequilibrioparaelestadodetransición.Estoquieredecirque,puestoqueelpuntodesillaeseldemenorenergíade todos loscomplejosactivados,habráunamayor

QFIII–TEMA2 55
probabilidad de que el complejo activado presente una geometría cercana a él que a la decualquierotropunto.
iv)Sesuponequeelmovimientodelosreactivosalolargodelacoordenadadereaccióncuandoatraviesanlasuperficiedivisoriapuedeserseparadodelosdemásgradosdelibertady tratado clásicamente. Como ya se ha discutido, el tratamiento clásico de unmovimientopuedesermásomenosválidodependiendodelascaracterísticasdelmovimiento(traslación,rotación o vibración), de la masa de la molécula y de las condiciones de temperatura yvolumen. La principal limitación de esta hipótesis se encontrará en la descripción dereacciones que impliquen transferencias de átomos ligeros (como el hidrógeno) donde losefectoscuánticospuedensernotables.
Deacuerdoconestosupuestos,nuestrareacción,enlaTETpuederepresentarsecomo
B+C⇆X‡®Productos dondeX‡representaalcomplejoactivadoyladobleflechaindicanounequilibrioensentidoquímico(elcomplejoactivadonoesunaespecieaislableymensurable,exceptoenelrégimende los femtosegundos), sino que de acuerdo con los supuestos ii) y iii) estas especies semantendránconladistribuciónquecorrespondaalequilibriotérmicoentreellas.
Elnúmerodemoléculasdeproducto(NP)queseformaránporunidaddetiempopuedeexpresarse como el número de moléculas de complejo activado (N‡) que atraviesan la
superficie divisoria por unidad de tiempo: , donde t es el tiempo que el
complejoactivadotardaenatravesarlasuperficiedivisoria.Cuando nuestra molécula atraviesa la superficie divisoria podemos decir que se ha
transformadodereactivosaproductos.Sielespesordeestasuperficieesd,estetiempopuedecalcularsesabiendolavelocidadconlaquesemueveelcomplejoalolargodelacoordenadadereacción(c.r.): .Asíelnúmerodemoléculasdeproductoqueseformanes:
queexpresadoenmolesydividiendoporelvolumenV:
(2.31)
Asípues,paracalcularlavelocidaddereacciónsenecesitaconocer,enprincipio,<vcr>,[X‡]yd.Detodasformas,yasepuedeanticiparque,puestoqueelespesordelasuperficiecríticaesarbitrario,elresultadonodeberádependerdeestamagnitud.
t
‡PC N
dtdN
dtdN
==-
><= crv/dt
d><
=-= cr‡
CP vNdtdN
dtdN
[ ] [ ] [ ]dd
><=
><=-= cr‡cr
‡
A
vX
vNVN1
dtCd
dtPd

QFIII–TEMA2 56
•Paraobtenerlaconcentracióndecomplejoactivadosepuedehacerusodelashipótesis
de equilibrio térmico entre reactivos y complejo activado. Así, la relación entre lasconcentracionesdeambasespeciesvendrádadapor:
(2.32)
dondeqrepresentalasfuncionesdeparticióny esladiferencia:
ladiferenciadeenergíaentrelosestadosfundamentalesdereactivosyestadodetransición,ypor tantonoesdirectamente ladiferenciadeenergíaobtenidade laSEPsinoque sehadetenerencuentalaenergíadelosnivelesvibracionalesfundamentales(energíadepuntocero,EPC),talycomosedestacaenlaFigura2.31.
Elcálculodelasfuncionesdeparticióndereactivosyproductos(ByC)serealizacomodecostumbre,teniendoencuentalasdistintascontribucionesalaenergía(normalmentelostérminosnuclearessedescartanpuespermanecenconstantesdurantelareacciónquímica):
Para el estado de transición se tendrán las mismas contribuciones de los diferentes
gradosde libertad, perohayque recordarqueunode esos gradosde libertad correspondeprecisamente a la coordenada de reacción. Efectivamente, una de las vibraciones de laestructuradetransición,elvectordetransición,correspondealacoordenadaquetransforma
reactivosenproductos: ,paralareacciónH+H2.Comolosgradosdelibertadseconsideranindependientes,paracalcularlafuncióndeparticióndelestadodetransiciónsevaasepararestacontribución(qcr):
[ ][ ][ ]
kT
AC
AB
A
‡‡
kTCB
‡
CB
‡‡0
‡0
e
VNq
VNq
VNq
CBXe
qqq
NNN
eDeD--
==
‡0eD
( ) ( ) ( ) EPC)CB(EPC)CB()X(EPC)X()CB()X( ‡‡‡0
‡0
‡0 DeDeeeeeD +=+++-+=+-=
elevibrottrasB qqqqq =
¬®¬------ HHH

QFIII–TEMA2 57
Figura2.31.-RepresentacióndelasmagnitudesenergéticasrelevantesparalaTeoríadelEstadodeTransición.EPCeslaenergíadepuntocero.
donde indica la función de partición para todos los grados de libertad excepto para el
movimiento (de vibración) a lo largo de la coordenada de reacción. El cálculo de noimplicaningúnproblemaadicionalalasfuncionesdeparticióndelosreactivos.Senecesitaránconocerlamasa,elmomentodeinercia(geometría),lasfrecuenciasdevibraciónylosniveleselectrónicosdelestadodetransiciónparapoderrealizarelcálculo.Sinembargo,elcálculode
supone una novedad respecto a lo realizado hasta ahora. Efectivamente este grado delibertadcorrespondeaunavibración,peroconelpozoinvertido.Laestructuradetransiciónesunmáximoenestacoordenaday,formalmente,estecasocorresponderíaaunmovimientovibracionalconlaconstantedefuerza(kF)negativa,yaquelaenergíadisminuyecuandonosdesplazamos del punto de silla. La frecuencia que podríamos calcular a partir de estaconstantedefuerzaseríaportantoimaginaria:
donde es la masa reducida asociada al movimiento a lo largo de la coordenada dereacción. Así pues, no se pueden emplear las fórmulas habituales basadas en el osciladorarmónicoparaelcálculodelafuncióndeparticiónvibracional.Paratratarestemovimiento,puestoqueenelpuntodesilla lasderivadasde laenergíarespectoacualquiercoordenadason cero, se puede suponer que en el entorno inmediato apenas cambia la energía. Elmovimiento del complejo activado a lo largo de la coordenada de reacción se tratará puescomoelmovimientodetraslacióndeunapartículaenunacajaunidimensionalcuyotamañovienedadoporelespesordelasuperficiedivisoriad,como
‡‡cr
‡ele
‡vib
‡rot
‡tras
‡cr
‡ele
‡vib
‡rot
‡tras
‡ qqqqqqqqqqqq =úûù
êëé==
‡q‡q
‡crq
‡cr
F
m
k21p
n =
‡crm
‡
Reactivos
Productos
EPC
EPC
Deo‡
De‡

QFIII–TEMA2 58
seilustraenlaFigura2.32.Portanto,asumiendoellímiteclásico((separaciónentreniveles<<kT),laexpresiónparalafuncióndeparticióntraslacionalasociadaalmovimientoalolargodelacoordenadadereacciónpuedeescribirsecomo:
Así,laconcentracióndecomplejoactivado[X‡],deacuerdoconlaecuación(2.32),quedará:
(2.33)
•Paraelcálculodelavelocidadmediaalolargodelacoordenadadereacciónsepuede
emplear la distribución de Maxwell-Boltzmann para una componente de la velocidad(supuestosiii)yiv)delaTET):
Puesto que se está calculando el proceso directo de conversión de reactivos en productos,paraevaluarelvalormediodeestacomponentedelavelocidadsólosetomaránlosvalorespositivos (o negativos). Los valores negativos (o positivos) corresponderían al procesoinverso:
(2.34)
dp
2/1
2
‡cr‡
crh
kTm2q
÷÷
ø
ö
çç
è
æ=
[ ] [ ][ ]CBe
VNq
VNq
VNq
h
kTm2X kT
AC
AB
A
‡2/1
2
‡cr‡
‡0eD
dp -
÷÷
ø
ö
çç
è
æ=
kT2vm2/1‡
crcr
2cr
‡cr
ekT2
m)v(g
-
÷÷
ø
ö
çç
è
æ=
p
2/1
‡cr0
crkT2vm2/1‡
crcr
0crcrcrcr
m2kTdve
kT2m
vdv)v(gvv
2cr
‡cr
÷÷
ø
ö
çç
è
æ=
÷÷
ø
ö
çç
è
æ=>=< òò¥ -¥
pp
d d
=Punto de Silla
Figura 2.32.- En la TET el movimiento a lo largo de la coordenada de reacción en el ET se asemeja a unatraslaciónclásica

QFIII–TEMA2 59
Sustituyendo las ecuaciones (2.33) y (2.34) en la (2.31), se obtendrá la expresión queproporcionalaTETparalaleydevelocidad:
(2.35)
Expresión que refleja correctamente la dependencia de la velocidad con el producto de lasconcentraciones. Comparando con la ley de velocidad experimental (ecuación (2.1)), laconstantedevelocidadpredichaporlaTETvale:
(2.36)
Parael cálculopuesde la constantedevelocidadsenecesitan las funcionesdeparticióndereactivos y del estado de transición, así como la diferencia de energía entre los estadosfundamentales. La información de los reactivos puede obtenerse experimentalmente. Demomento, para el estado de transición la información necesaria para obtener su energía yfuncióndepartición(masa,geometría,frecuencias,estadoselectrónicos…)debedeobtenersemediante cálculos demecánica cuántica. El conocimiento de la SEP, almenos alrededor dereactivos y estructura de transición, proporciona los datos necesarios. Sin embargo, elreciente desarrollo de la espectroscopia de femtosegundos está empezando a cambiar estehecho,alpermitirlaobservacióndelestadodetransición.
Ejercicio 2.10.-Calcular, mediante el uso de la TET, la constante de velocidad para la reacción D + H2 ® DH + H a T= 450 K y compararlo con el valor experimental kr= 9x109 cm3 mol-1 s-1. Datos:
a) Reactivos • H-H dHH = 0,741 Å n = 4400 cm-1 Mr = 2,0156 g mol-1; I = 4,5945x10-41 g cm2 • Atomo de Deuterio M r = 2,0014 g mol-1 b) Estructura de Transición • D----H----H dDH = dHH = 0,930 Å
[ ] [ ] [ ][ ]
[ ][ ]CBe
VNq
VNq
VNq
hkT
CBe
VNq
VNq
VNq
hkTm2
m2
kT1Xv1dtCd
kT
AC
AB
A
‡
kT
AC
AB
A
‡2/1
2
‡cr
2/1
‡‡cr
‡cr
‡0
‡0
eD
eD
dp
pdd
-
-
=
=÷÷ø
öççè
æ
÷÷÷
ø
ö
ççç
è
æ=><=-
kT
AC
AB
A
‡
r
‡0
e
VNq
VNq
VNq
hkTk
eD-
=

QFIII–TEMA2 60
Vibraciones: 1) D¬®H---H® Tensión Asim. 2) ¬D---H---H® Tensión Sim. n = 1764 cm-1 3 y 4) D---H---H Flexión n = 870 cm-1 (degenerada) ¯ ¯ M r = 4,0297 g mol-1; I = 3,9788x10-40 g cm2
c) La Diferencia de energía entre estados fundamentales de reactivos y Estado de
Transición es 8,33 Kcal mol-1 = 5,79x10-20 J Solución.- Según la Teoría del Estado de Transición, la constante de velocidad de una
reacción del tipo B+C® Productos puede expresarse como
Comenzando con el cálculo de la función de partición de los reactivos. Para el átomo de deuterio se tiene qD=qD,trasqD,ele , ya que se trata de un átomo y donde:
y . El átomo de D tiene un electrón
desapareado, el valor de S=1/2 y L=0. Existen dos estados electrónicos degenerados, que corresponden a ms=1/2 y ms=-1/2.
Para la molécula de hidrógeno qHH = qHH,tras qHH,rot qHH,vib qHH,ele con:
, (s=2) ¡OJO! El valor
obtenido indica que la aproximación de alta temperatura no será válida. Realmente habría que usar el sumatorio sobre los niveles rotacionales para evaluar correctamente la función de partición, aunque aquí daremos por buena esta aproximación. Continuando se tiene que
y finalmente , pues es una molécula con todos los electrones
apareados y S=0. Para el estado de transición hay que calcular la función de partición dejando fuera el vector
de transición, es decir la vibración que permite la transformación hacia el valle de reactivo y productos, que en este caso es la tensión asimétrica (esta vibración en el ET tiene una frecuencia imaginaria). Se tiene pues con:
, (s=1 , molécula lineal) ,
(descontando el vector de transición sólo quedan 3
vibraciones en el ET) y (molécula con un electrón desapareado). Sustituyendo todos los valores en la expresión que proporciona la TET:
kT
AHH
AD
Ar
0
e
VNq
VNq
VNq
hkTk
¹eD-
¹
=
V10x0797,5VhkTm2q 30
2/3
2D
tras,D =÷÷ø
öççè
æ p= 2q ele,D @
V10x1338,5q 30tras,HH = 5668,2
hIkT8
hBkTq 2
2rot,HH =
s
p=
s=
1
e1
1q
kTvhcvib,HH @
-
=-
1q ele,HH @
ele,DHHvib,DHHrot,DHHtras,DHHDHH qqqqq =
V104513.1q 31tras,DHH ×=
140,1e1
1q kT/vhc
3
1ivib,DHH
i=
-=
-=P
2q ele,DHH »
456,44q rot,DHH =

QFIII–TEMA2 61
queda:
El resultado obtenido es prácticamente la mitad del experimental. Sin embargo, se ha de
recordar que se está haciendo un tratamiento clásico de la dinámica de los núcleos. En este caso, al tratarse de hidrógeno y deuterio (átomos ligeros) los efectos cuánticos pueden ser importantes. En concreto, el efecto túnel permite que situaciones sin la suficiente energía como para alcanzar el complejo activado den lugar a reacción química. Por lo tanto, debe ser un factor que aumente la velocidad de la reacción. Un cálculo aproximado de este factor (ver Bibliografía) da un valor de 2,0 para esta reacción a esta temperatura. Así el valor final sería bastante similar al valor experimental.
Bibliografía:
*Sobre el problema: I. N. Levine, Fisicoquímica, 4ªed., Mc. Graw Hill, Vol.2, páginas 916-917, 1996.
*Sobre estudios teóricos de la reacción: D. G. Truhlar, R. E: Wyatt, Ann. Rev. Phys. Chem. Vol. 27, p 1, 1976.
*Sobre el efecto túnel: B. C. Garret, D. G. Truhlar, J. Chem. Phys. Vol. 72, p 3460, 1980.2.4.2.-FormulaciónTermodinámicadelaTET
Paraanalizar(2.36)desdeunpuntodevistainterpretativosepuedeutilizarlallamada
Formulación Termodinámica de la TET. Efectivamente, fijándose en la ecuación se puedeobservarqueapartedel factorkT/h,elrestoseparecemuchoaunaconstantedeequilibrioentreel complejoactivadoy los reactivos.Taly comosevioen (2.32), la relaciónentre lasconcentracionesdeestadodetransiciónyreactivosvienedadapor:
kT
AC
AB
Ar
0
e
VNq
VNq
VNq
hkTk
¹eD-
¹
=
1139113352936
kT10x79,5
3030
31
kT
ele,HHvib,HHrot,HHtras,HH
ele,Dtras,D
ele,DHHvib,DHHrot,DHHtras,DHH
Ar
smolcm10x5,5smolm10x5,510968,810x098,110x646,5
e115668,210x1338,5210x0797,5
2140,1456,4410x4513,1hRT
eqqq
Vq
qV
q
qqqV
q
NhkTk
20
0
------
-
eD-
===×´´=
=´´´´´
´´´=
==
-
¹
[ ][ ][ ]
kT
AC
AB
A
‡‡
‡0
e
VNq
VNq
VNq
CBX
eD
=

QFIII–TEMA2 62
Dividiendo todas las concentraciones por c0 (concentración estándar, 1M) se tendrá laconstantedelequilibrioenescaladeconcentraciones:
Laexpresióndelaconstantedevelocidaddadapor(2.36)contieneuntérminoprácticamenteidéntico excepto por el hecho de que la función de partición del estado de transición nocontienelacontribucióndelacoordenadadereacción.Alaconstantedeequilibriocalculadasinestetérminoladenominaremos conloquelaexpresióndelaconstantedevelocidadquedaríacomo:
(2.37)
Laconstantedevelocidadpuedeversepuescomountérminodeequilibrioentrelosreactivosyelestadodetransición(equilibrioenelquesedescuentalacontribucióndelacoordenadade reacción en el estado de transición) y otro término (kT/h), con unidades de s-1, queprovienede lacoordenadadereacciónyda la frecuenciacon laqueelestadode transiciónatraviesalasuperficiedivisoria.Laconstantedevelocidaddependepuesdelaconcentraciónque se logre de complejos activados y de la frecuencia con que éstos se transformen enproductos.
Continuandocon la formulación termodinámica, sepuedenobteneralgunas relacionesútiles para analizar los datos de constantes de velocidad. Efectivamente si es unaconstante de equilibrio a la que se ha descontado un grado de libertad en el estado detransición,sepodrárelacionarconunavariacióndeenergíalibreestándarenlaquesehayadescontadoesamismacontribución:
(2.38)
donde eslaenergíalibredeactivaciónestándarenescalademolaridades.EstaenergíalibresecalculatalycomoseestudióeneltemadeTermodinámicaEstadística,exceptoporelhechodequeenelestadodetransiciónnosehadecontarelgradodelibertadasociadoalacoordenadadereacción.Esdecir,paraelcálculodelaenergíalibredelestadodetransiciónsecontarían 3 traslaciones, 3 rotaciones (2 si fuera lineal) y 3N-7 vibraciones (3N-6 si fueralineal).Lavibraciónqueseomitiríasería lacorrespondientealvectordetransición,aquellaque presenta una constante de fuerza negativa.Utilizando las ecuaciones (2.37) y (2.38) laconstantedevelocidadquedacomo:
[ ][ ] [ ]
[ ][ ][ ]
kT
AC
AB
A
‡
0‡
0
00
0‡
c
‡0
e
VNq
VNq
VNq
cCB
Xc
cC
cB
cX
KeD
-===
‡cK
0
‡c
rc
KhkTk =
‡cK
( )‡c‡0c KlnRTG -=D
‡0cGD

QFIII–TEMA2 63
(2.39)
Si se descompone la energía libre en sus contribuciones entrópica y entálpica
sepuedeescribir:
(2.40)
dondeaparecenlaentalpíaylaentropíadeactivaciónestándar(1M),queseobtienendejandoapartelacontribucióndelgradodelibertadvibracionalasociadoalacoordenadadereacciónenelcomplejoactivado.(Nota:sienlugardedosreactivoshubiesen,entonceselfactor1/c0apareceríaelevadoan-1).
Las ecuaciones (2.37)-(2.40) están basadas en la selección del estado estándar 1M,aunquepara gasesmuchas veces se prefiere el estado estándarp0=1bar. La conversión essencilla teniendo en cuenta que, para nuestro equilibrio entre dos reactivos para dar unestadodetransición,larelaciónentrelasconstantesdeequilibrioenescalasdeconcentraciónydepresiónes:
(2.41)
Empleandop0comoestadoestándarlasexpresionesanterioresquedaríanpues:
(2.42)
(2.43)
(2.44)
Nota:sienlugardedosreactivoshubiesen,entonceselfactorRT/p0apareceríaelevadoan-1.
A partir de estas últimas expresiones se pueden encontrar relaciones útiles para elanálisisyprediccióndelaenergíadeactivaciónyelfactorpreexponencial.Así,porejemplo,paralaenergíadeactivación,deacuerdoconlaecuación(2.42):
RTG
0r
‡0c
ec1
hkTk
D-
=
( )‡0c‡0c
‡0c STHG DDD -=
RTH
RS
0r
‡0c
‡0c
eec1
hkTk
DD-
=
p00c K
pRT
cK
=
‡p0r K
pRT
hkTk =
RTG
0r
‡0p
epRT
hkTk
D-
=
RTH
RS
0r
‡0p
‡0p
eepRT
hkTk
DD-
=

QFIII–TEMA2 64
deacuerdoconlaecuacióndevan’tHoff,dequeladerivadaconrespectoalatemperaturadeuna constante de reacción está relacionada con la entalpía de reacción. Puesto que se estáderivandolaconstantedelequilibriodeactivación,suderivadaserálaentalpíadeactivaciónestándar, que se calcula dejando fuera la contribución de la coordenada de reacción en elestadodetransición:
(2.45)
Utilizando esta relación se puede también obtener una expresión para el factorpreexponencial:
(2.46)
Paranespeciesreactivaslasecuaciones(2.43),(2.45)y(2.46)quedarían:
YdeformasimilarparalasecuacionesexpresadasutilizandoC0comoestadodereferencia.
Se tienen, por tanto, unas expresiones que relacionan la energía de activación y elfactor preexponencial con las entalpías y entropías de activación. Respecto a estasexpresionessehandereseñartresanotacionesimportantes:
- Laenergíadeactivaciónyelfactorpreexponencialsonmagnitudesexperimentales,la
entalpíaylaentropíadeactivaciónnosonderivablesúnicamentedelaexperiencia.Paradarvaloresdelaentalpíayentropíadeactivaciónhayqueusarlasrelaciones(2.45)y(2.46),quesebasanenlaTETyporlotantoenlavalidezdesussuposiciones.Lasentalpíasyentropías
dTKlnd
RTRT2dTKlnd
T2RTK
pRT
hkTln
dTdRT
dTklndRTE
‡p2
‡p2‡
p02r2
a +=÷÷
ø
ö
çç
è
æ+=÷
÷ø
öççè
æ==
0‡p
‡p2
a HRT2dTKlnd
RTRT2E D+=+=
RS
20
RTH
2RTH
RS
0RTHRT2
RTH
RS
0RTE
r
‡0p
‡0p
‡0p
‡0p
‡0p
‡0p
‡0pa
eepRT
hkT
eeeepRT
hkTeee
pRT
hkTekA
D
DDDDDD
=
====-
+-
RTG1n
0r
‡0P
epRT
hkTk
D-
-
÷÷ø
öççè
æ=
0‡pa HRT·nE D+=
RS
n1n
0
‡0p
eepRT
hkTA
D-
÷÷ø
öççè
æ=

QFIII–TEMA2 65
de activación que se dan muchas veces como ‘experimentales’ contienen por tanto untratamientodelosdatosbasadoenlateoría.
- Sepuededarlavueltaalargumentoanterior.Sedisponeahoradeunasrelacionesque
permiten calcular la energía de activación y el factor preexponencial, pero para ello senecesitan la entalpía y entropía de activación que se sabe calcular de acuerdo con lasexpresionesvistasenTermodinámicaEstadística.Evidentementeparacalcularestosvaloressonnecesariaslasfuncionesdeparticióndereactivosydecomplejoactivado.Lainformaciónde los reactivos (masa,momentode inercia, frecuenciasde vibración, estados electrónicos)puedeobtenerseexperimentalmente.LainformacióndelcomplejoactivadodebeextraersedecálculosdelaSEP,esdecir,utilizandolamecánicacuántica.
- Estasrelacionespermiteninterpretarelsentidodelaenergíadeactivaciónyelfactor
preexponencial. Laenergíadeactivaciónvienedeterminadaporel cambiodeentalpíaparapasar de los reactivos al complejo activado. Es decir, depende, fundamentalmente, de laenergía que se necesita para reorganizar los enlaces y llegar al punto de silla de la SEP. Elfactorpreexponencialestá ligadoencambioa laentropíadeactivación,esdecir, aldistintogradodedesordenquesevaa teneralpasarde reactivosal estadode transición.Comosesabe,estodependefundamentalmentedelnúmerodeestadosaccesiblesquesetenganenunasituaciónoenotra.Normalmente,parareaccionesdondedosreactivosseunenparadaruncomplejo activado, la entropía de activación es negativa, ya que el proceso implica unaordenacióndelsistema.
Ejercicio 2.11.- A partir de los valores de la constante de velocidad para la descomposición térmica unimolecular en fase gas del exodiciclopentadieno, calcular los parámetros de Arrhenius así como la entalpía y entropía de activación a 543,6 K.
T (K) 473,.7 494,.8 516,2 536,5 600,3 628,1 kr 104 (s-1) 0,947 4,50 27,2 116,0 5640 17430 Solución.-
De acuerdo con la ecuación de Arrhenius: se tiene y
realizando la oportuna representación
RTE
r
a
Aek-
=RTEAlnkln a
r -=
+

QFIII–TEMA2 66
Del ajuste por mínimos cuadrados, comparando con la ec. de Arrhenius se obtiene:
y (NOTA: Las unidades de A son las mismas que las de la constante de velocidad, tal y
como se observa en la ecuación de Arrhenius) De acuerdo con la TET y teniendo en cuenta que en esta reacción n=1:
;
Sustituyendo los valores proporcionados por el ajuste se encuentra:
;
El valor positivo de la entropía de activación está de acuerdo con el hecho de que se trata de una reacción unimolecular en la que en el ET los enlaces empiezan a romperse. Es decir, el ET está más ‘desordenado’ que los reactivos.
2.4.3.-LimitacionesdelaTET
Las pruebas más sólidas sobre la bondad de la TET provienen de experimentos deefectos cinéticos isotópicos, es decir de estudios del cambio de las constantes de velocidadcomo consecuencia de la sustitución de un isótopo por otro. Este efecto se explica en elApéndice 2.2. Por supuesto, la TET presenta también algunas limitaciones. Las limitacionesmásimportantesdelaTETson:
1a molKJ2,160R19272E -== 113 s10·278,4A387,31Aln -=Þ=
÷÷
ø
ö
çç
è
æ D
=RS ‡0p
eehkTA RTHE ‡0
pa +D=
1a
‡0p molKJ7,155RTEH -=-=D 11‡0
p molJK73,2eTkhAlnRS --==D

QFIII–TEMA2 67
i) Elmovimiento a lo largo de la coordenada de reacción se ha tratado clásicamente,
tantoalusarlafuncióndedistribucióndevelocidadesdeMaxwell-Boltzmannparaelcálculode lavelocidadmedia, comoal tomarel límitedealta temperaturaal calcular la funcióndepartición de la coordenada de reacción. Para reacciones que impliquen el movimiento deátomos muy ligeros (por ejemplo, hidrógeno) es necesario describir este movimientocuánticamente, lo que lleva a considerar la posibilidad de efecto túnel. Ignorar este efectoconduce a subestimar la constante de velocidad cuando se utiliza la TET, ya que lassituaciones que dan lugar a reacción sin tener energía suficiente para pasar la barreraenergéticanosontenidasencuentaenlaTET,situacionescontempladasesquemáticamenteenlaFigura2.33a).
Experimentalmente,elefectotúnelseponeenevidenciaalobservarseefectoscinéticos
isotópicosmayoresquelosobtenidosmediantelaTETyaquealsereldeuteriomáspesadoque el hidrógeno sufremenos efecto túnel, por lo que la constantede velocidaddisminuyetambiénporesteefectoynosóloporelaumentoenladiferenciadeenergíasfundamentales.
ii) Otra limitación importante es el hecho de que se ha supuesto que todas las
trayectorias que alcanzan la superficie divisoria son reactivas. Sin embargo, en estudios dedinámicasdereacciónseobservanavecesrecruzamientos,esdecir,trayectoriasqueunavezatraviesan la superficie divisoria vuelven de nuevo hacia los reactivos. Habitualmente estetipo de trayectorias pueden suponer alrededor de un 10% del total, pero pueden llegar asignificarhastaun50%omás,sobretodoparareaccionesendisolución.Imagíneseelcaso
Figura2.33.-a)Esquemadeefectotúnel;b)Trayectoriadeidayretroceso
deunareaccióndetipoSN2( )dondeenundeterminadocomplejoactivadoel grupo saliente (Y-)no se encuentra correctamente solvatado,mientrasque si loestáelnucleófilo.Enesecaso,latrayectoriapodríavolverdenuevoalreactivoparaformardenuevo el anión X-. Ignorar los recruzamientos conduce a sobreestimar la constante de
-- +®+ YXCHYCHX 33

QFIII–TEMA2 68
velocidadalutilizarlaTET.Puedecorregirseesteefectomultiplicandolaexpresiónobtenidapor un factor que da cuenta de la proporción entre trayectorias que llevan de reactivos aproductos y el número total de trayectorias que llegan al estado de transición. Este factor(menor o igual a la unidad) se denomina coeficiente de transmisión (k), con lo que laexpresiónparalaconstantedevelocidadquedaría:
iii)Porúltimo,en laTETsehasupuestounadistribucióndeequilibrioen losdistintos
grados de libertad de los reactivos y del estado de transición. Lógicamente, reaccionaránaquellas moléculas que alcancen una determinada energía mínima (si se ignora el efectotúnel)porloquelareaccióniríadespoblandolosnivelesenergéticosquesupereneseciertonivel umbral en los reactivos. Para que se pueda mantener la hipótesis del equilibrio esnecesarioqueestosniveles se repueblen rápidamente, loque se consiguepormediode lascolisiones con las paredes o entre las moléculas de reactivos. Así pues, la TET dejará decumplirse en aquellas condiciones en que la frecuencia de colisión disminuyadramáticamente, comoenelcasodereaccionesapresionesmuybajas.Enesascondicionesexperimentaleslahipótesisdelequilibrionosecumpliría.
Ejercicio 2.12. Mediante métodos cuánticos se ha estudiado la reacción de Diels-Alder
entre el eteno y el cis-butadieno: A partir de la localización de las estructuras correspondientes a los reactivos y estructura
de transición se han determinado las propiedades que aparecen en la siguiente tabla:
Cis-butadieno Eteno E.T. Mr (g/mol) 54 28 82 qrot(350) 4,65x104 3,34x103 1,71x106 qvib(350) 10,54 1,11 13,92*
Energía Potencial (kcal mol-1) 30,62 16,44 70,05
Energía Punto Cero (kcal mol-1) 53,87 32,00 88,86*
*Excluida la coordenada de reacción
Considerando que la mezcla reactiva puede tratarse como un gas ideal y que no existen estados electrónicos de baja energía, calcular:
kT
AC
AB
A
‡
TETrr
‡0
e
VNq
VNq
VNq
hkTkk
eD
kk-
==

QFIII–TEMA2 69
a) La constante de velocidad de la reacción (en M-1 s-1) a 350 K utilizando la teoría del estado de transición (TET).
b) La energía libre de activación estándar a dicha temperatura. Solución.- Para calcular la constante de velocidad se utiliza la expresión de la TET:
La diferencia de energía entre los estados fundamentales no es directamente la diferencia de energía potencial, hay que añadir las contribuciones de la energía de punto cero
Con este valor ya se puede calcular el término exponencial de la expresión de la TET,
, y las funciones de partición: - con ; ; ; - con
; ; ;
- con
; ; ; Sustituyendo en la expresión de la TET y cambiando las unidades del resultado:
kT
AA
A
‡
r
‡0
eVN
)Butc(qVN
q(Et)VN
q
hkTk
eD-
-=
17kT 10x013,6e
‡0
-eD
-=
)Et(q)Et(q)Et(q)Et(qq(Et) elevibrottras=
3rot 10x34,3)Et(q = 11,1)Et(qvib =
1)Et(qele =
)Butc(q)Butc(q)Butc(q)Butc(qBut)-q(c elevibrottras ----=
V10x883,4)Butc(q 32tras =- 4
rot 10x65,4)Butc(q =- 54,10)Butc(qvib =-1)Butc(qele =-
‡ele
‡vib
‡rot
‡tras
‡ qqqqq =
V10x137,9q 32‡tras =
6‡rot 10x71,1q = 92,13q‡vib = 1q‡ele =
V10x823,1Vh
kTm2)Et(q 322/3
2Et
tras =÷÷ø
öççè
æ p=

QFIII–TEMA2 70
=
Para calcular la energía libre de activación se puede emplear la relación
. Para n=2,
2.5.-ApéndicesApéndice2.1.-FuncióndedistribuciónCuandosedescribelavelocidaddesdeunpuntodevistaclásicoseestápensandoenuna
variablequecambiadeformacontinua,nodiscreta,yportanto,estrictamente,laprobabilidaddeencontrarundeterminadovalorexactodelavelocidadescero.Cuandosetienenvariablescontinuas, su distribución entre la población no se caracteriza por un conjunto discreto devaloresdelaprobabilidad,sinoporunafunciónqueproporcionaladensidaddeprobabilidado función de distribución. Piénsese en el caso de un examen de 10 preguntas donde losalumnospuedenobtener,paracadaunadeellas,lacalificaciónde0ó1.Lanotadelexamenpuedeserportantocualquiervalordelconjuntodiscreto[0,1,2,…,9,10].Ladistribucióndelapropiedadentrelapoblacióndeestudiantes(digamosN=50)vienedadaporelnúmerodeestudiantesquehaobtenidocadaunodelosposiblesvalores[N0,N1,…N10].Evidentemente,estadistribuciónpuededarsetambiénenformadeprobabilidad,sinmásquedividircadaunodelosNiporelnúmerototalN.
Nota(xi) Ni pi0 0 01 1 0,022 1 0,023 2 0,044 3 0,065 7 0,146 8 0,167 10 0,208 8 0,169 6 0,1210 4 0,08
kT
AA
A
‡
r
‡0
eVN
)Butc(qVN
q(Et)VN
q
hkTk
eD-
-= 111111314 sM10x540,3smolm10x540,3 ------ =
RTG1n
0r
‡0p
epRT
hkTk
D-
-
÷÷ø
öççè
æ= 1
2r
0‡0p molKJ0,166
TRkkphlnRTG -=-=D
50Ni
i =å 1pi
i =å

QFIII–TEMA2 71
Larepresentacióndeladistribucióndela
propiedad es en este caso el conjunto devalores de la probabilidad de obtener cadaunadelasposiblesnotasenelexamen.
En cambio, si se trata de una variablecontinua no es posible hablar de laprobabilidad de encontrar en nuestrapoblación un determinado valor. ¿Cuál es laprobabilidaddequeunamoléculatengacomo
módulo de la velocidad 100 m/s?. ¿Qué quiere decir esta pregunta? ¿Exactamente100,000…00m/s?Entonceslarespuestaseríacero.Cuandosetieneunavariablediscretaesnecesariohablardelaprobabilidaddequelavariablextomeundeterminadovalorentrexyx+∆x.Evidentemente,elresultadodependerádelaamplituddelintervaloconsiderado(∆x).
Sinuestramuestraesmuygrandesepuedeobviaresteproblemaconsiderandointervalosdetamañoinfinitesimalypreguntándonosentoncescuáleslaprobabilidaddequelavariablextomeundeterminadovalorentrexyx+dx.Evidentementeenesecasolaprobabilidadseráundiferencial,yaqueelnúmerodecasospresentandovaloreseneseintervaloseríainfinitesimaltambién(dN):
Paraevitareltrabajarcondiferencialesypodertenerunafunciónfinitasedefinelafuncióndedistribución de la variable x (f(x)) como la densidad de probabilidad o probabilidad porunidaddeintervalo,deformaque:
Teniendo en cuenta que la probabilidad es una magnitud adimensional, la función dedistribucióntendráunidadesdex-1.Estafunciónesrepresentableysepuedetrabajarconelladeformaanálogaacomosehaceconlaprobabilidad.
NN)x(p xxx D+-=
NdN
)x(dp dxxx +-=
dx)x(f)x(dp =
0,00
0,05
0,10
0,15
0,20
0,25
0 1 2 3 4 5 6 7 8 9 10
Nota
Probabilidad
f(x)
x

QFIII–TEMA2 72
Así, si estamos interesados en obtener la nota media de nuestro caso anterior se puedesimplementemultiplicarcadaunodelosposiblesresultados(xi)porsuprobabilidadysumar.Estoesequivalenteasumartodaslasnotasydividirporelnúmerototaldecasos(N):
Pues bien, en el caso de una variable continua, el valor medio también se obtienemultiplicandolavariableporlaprobabilidad(dp(x))ysumando(integrando)paratodoslosposiblesvalores:
De igual manera se calcular el valor promedio de cualquier propiedad que dependa de x(h(x)):
Ejercicio 2.13.- La distribución gaussiana se utiliza muy frecuentemente en distintos
ámbitos para caracterizar funciones de distribución. Su forma genérica es:
. Representar esta función tomando:
a) s=0,6 y d=0, 1 y 2 b) d=0 y s=0,6 , 1,0 y 1,5
Solución.- a)
b)
åå ==i
iii
ii xpxNN1x
òò""==
xxdx)x(xf)x(xdpx
òò""==
xxdx)x(f)x(h)x(dp)x(h)x(h
( )2
2
2)x(
2/12e
2
1)x(f s
d--
ps=
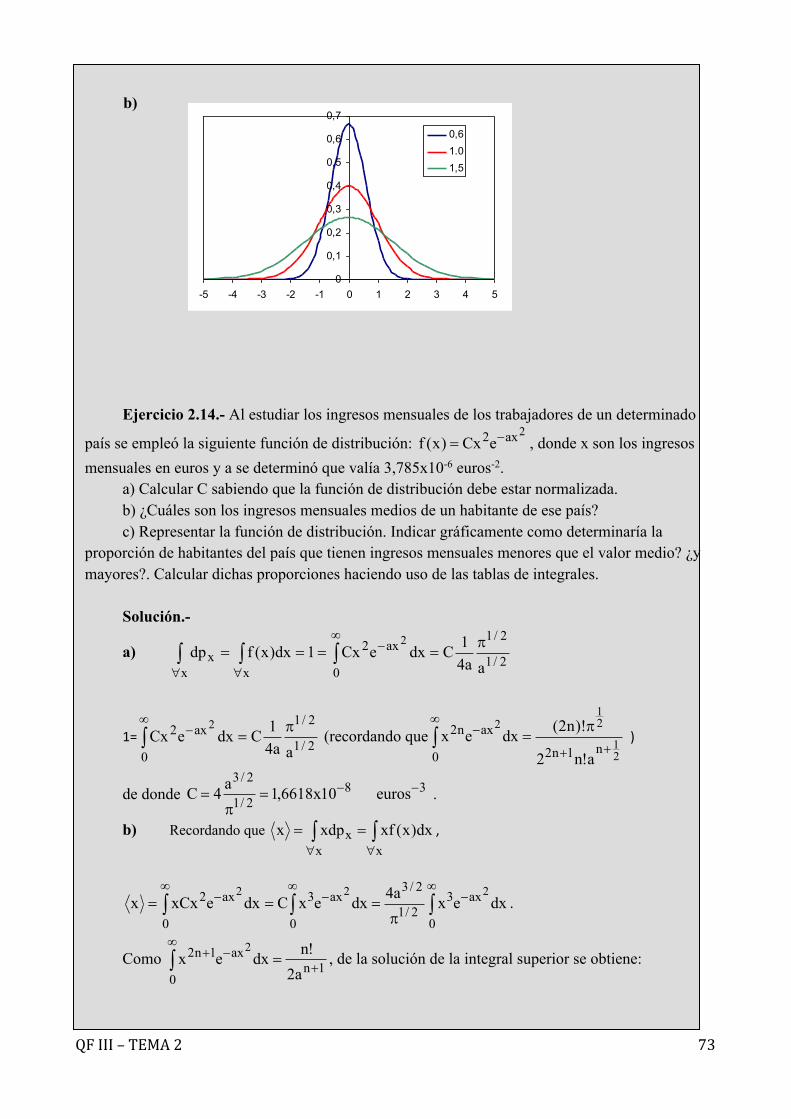
QFIII–TEMA2 73
b)
Ejercicio 2.14.- Al estudiar los ingresos mensuales de los trabajadores de un determinado
país se empleó la siguiente función de distribución: , donde x son los ingresos mensuales en euros y a se determinó que valía 3,785x10-6 euros-2.
a) Calcular C sabiendo que la función de distribución debe estar normalizada. b) ¿Cuáles son los ingresos mensuales medios de un habitante de ese país? c) Representar la función de distribución. Indicar gráficamente como determinaría la
proporción de habitantes del país que tienen ingresos mensuales menores que el valor medio? ¿y mayores?. Calcular dichas proporciones haciendo uso de las tablas de integrales.
Solución.-
a)
1= (recordando que )
de donde .
b) Recordando que ,
.
Como , de la solución de la integral superior se obtiene:
2ax2eCx)x(f -=
===ò ò" "x x
x 1dx)x(fdp 2/1
2/1
0
ax2
aa41CdxeCx
2 p=ò
¥-
2/1
2/1
0
ax2
aa41CdxeCx
2 p=ò
¥-
a!n2
)!n2(dxex21
21
2
n1n20
axn2++
¥- p
=ò
382/1
2/3euros10x6618,1a4C --=
p=
òò""==xx
x dx)x(xfxdpx
ò¥
-=0
ax2 dxexCxx2
òò¥
-¥
-
p==
0
ax32/1
2/3
0
ax3 dxexa4dxexC22
1n0
ax1n2
a2!ndxex
2
+
¥-+ =ò
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
-5 -4 -3 -2 -1 0 1 2 3 4 5
0,61.01,5
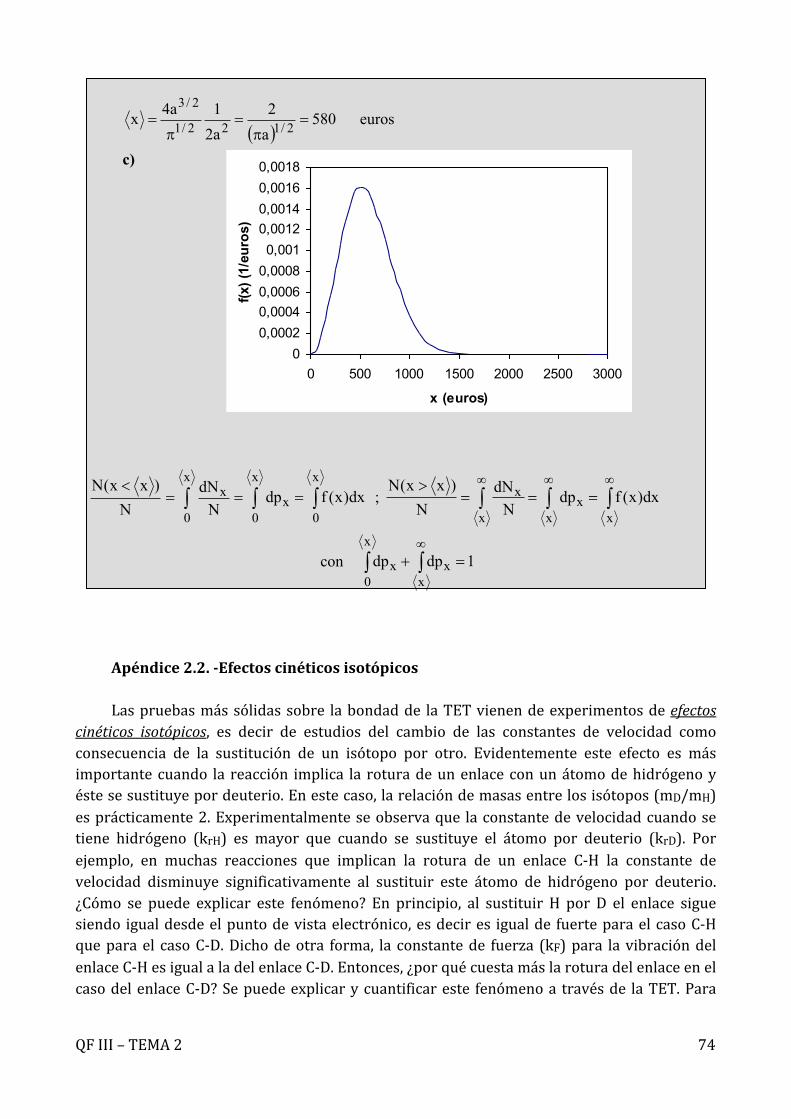
QFIII–TEMA2 74
c)
;
con
Apéndice2.2.-Efectoscinéticosisotópicos
Laspruebasmássólidassobre labondadde laTETvienendeexperimentosdeefectos
cinéticos isotópicos, es decir de estudios del cambio de las constantes de velocidad comoconsecuencia de la sustitución de un isótopo por otro. Evidentemente este efecto es másimportantecuando la reacción implica la roturadeunenlaceconunátomodehidrógenoyéstesesustituyepordeuterio.Enestecaso,larelacióndemasasentrelosisótopos(mD/mH)esprácticamente2.Experimentalmenteseobservaque laconstantedevelocidadcuandosetiene hidrógeno (krH) es mayor que cuando se sustituye el átomo por deuterio (krD). Porejemplo, en muchas reacciones que implican la rotura de un enlace C-H la constante develocidad disminuye significativamente al sustituir este átomo de hidrógeno por deuterio.¿Cómo se puede explicar este fenómeno? En principio, al sustituir H por D el enlace siguesiendo igualdesdeelpuntodevistaelectrónico,esdecires igualde fuerteparaelcasoC-Hqueparael casoC-D.Dichodeotra forma, la constantede fuerza (kF)para lavibracióndelenlaceC-HesigualaladelenlaceC-D.Entonces,¿porquécuestamáslaroturadelenlaceenelcasodelenlaceC-D?Sepuedeexplicarycuantificareste fenómenoa travésde laTET.Para
( )euros580
a2
a21a4x 2/122/1
2/3=
p=
p=
N)xx(N <
ò=x
0
xNdN
ò ò==x
0
x
0x dx)x(fdp ò òò
¥ ¥¥===
>
x xx
x
x dx)x(fdpNdN
N)xx(N
òò¥
=+x
x
x
0x 1dpdp
00,00020,00040,00060,00080,0010,00120,00140,00160,0018
0 500 1000 1500 2000 2500 3000
x (euros)
f(x) (
1/eu
ros)

QFIII–TEMA2 75
ello se ha de tener en cuenta que, aunque los enlaces son químicamente idénticos, lasfrecuenciasdevibraciónnoseránigualesyaquelasmasascambian:
;
dondelasmasasreducidas(µ)puedencalcularseasimilandolavibracióndelenlaceC-H(oC-D)aunproblemadedoscuerpos,unoconmasaigualaladelhidrógeno(odeuterio)yotroconmasaigualaladelrestodelamolécula(mR):
(yaquemR>>mH)
Asípues,dividiendolasecuacionesdelasfrecuenciasysustituyendolasmasasreducidassepuedevercuántocambialafrecuenciadevibraciónalcambiarelátomodeHporD:
Esdecir,lafrecuenciadevibracióndelenlaceC-Hes~1,4vecesmayorqueladelenlaceC-D.¿Cómo influye este hecho sobre la constante de velocidad? Fundamentalmente a través delcambioenlasenergíasdepuntocerodelosreactivos.Lasenergíasdepuntocerodebidasalavibración del enlace C-H o C-D pueden calcularse teniendo en cuenta que la del nivelvibracionalfundamental(v=0)es(½)hn:
Comoseobserva,laenergíadepuntoceroenelcasoC-HesmayorqueladelC-Den0,146hveceslafrecuenciadevibracióndelenlaceC-H.¿Quépasaenelestadodetransición?ParaunprocesoderoturadelenlaceC-H(oC-D)estavibracióndefine justamente lacoordenadadereacción,porloquenocontribuyealaenergíadepuntocero(recuérdesequeenelestadodetransiciónestavibraciónnoesunavibraciónnormalpueselpuntodesillaesunmáximoparaestemovimiento,nounmínimo).Asípues,setendríaunesquemaenergéticocomoelquesemuestraenlafigurasiguiente:
CH
FCH
k21
µpn =
CD
FCD
k21
µpn =
HR
HR
HR
HRCH m
mmm
mmmm
=»+
=µ
DDR
DRCD m
mmmm
»+
=µ
2mm 2/1
H
D2/1
CH
CD
CD
CH »÷÷ø
öççè
æ»÷÷
ø
öççè
æ=
µµ
nn
CD0
CH0
h21)DC(
h21)HC(
ne
ne
=-
=-
( ) CHCHCH
CHCDCH00 h146,0211h
21
2h
21h
21)DC()HC( nn
nnnnee =÷÷
ø
öççè
æ-=÷÷
ø
öççè
æ-=-=---

QFIII–TEMA2 76
Resumiendo,ladiferenciaentrelasenergíasdelosestadosfundamentalesdereactivosyestado de transición aumenta cuando se cambia H por D. Evidentemente, al aumentar laenergíanecesariaparallegaralestadodetransición,disminuirálaconstantedevelocidad.Deacuerdoconlaexpresión(2.35)eignorandolosefectosdelasustituciónsobrelasfuncionesdepartición(elcambiodemasaafectaa latraslación, larotaciónytambiénpuedeafectaraotrasvibracionesmoleculares),sepuedecalcularelefectodelasustituciónisotópicasobrelaconstantedevelocidad:
TeniendoencuentaquelosenlacesC-Hpresentanunnúmerodeondasnormalmentecercanoa 2800 cm-1 esta expresión predice que la constante de velocidad para el hidrógeno atemperaturaambienteesaproximadamente8vecesmayorqueparaeldeuterio.Estarelaciónestáenmuybuenacuerdoconlosdatosexperimentalesparamuchosprocesosdeestetipo.
Eltratamientoanteriordelefectocinéticoisotópicoesmuysimplificado.Hayquetenerencuentaque,engeneral,laenergíadepuntocerodelestadodetransiciónsíquepuedeversealterada por la sustitución isotópica, ya que la masa del átomo sustituido afecta a otrasvibraciones.Si,porejemplo,seestudiaunprocesodetransferencia,comolareacciónH---H---Hyavistaenunapartadoanterior,lasustitucióndeunHporDcambialafrecuencianosólode la tensión asimétrica (que es la coordenada de reacción) sino también de la tensiónsimétrica y de las flexiones. El efecto de la sustitución isotópica sobre las funciones departiciónesfundamentalparaexplicarlosllamadosefectoscinéticosisotópicossecundarios.Enestecasoseproduceuncambioenlaconstantedevelocidadalsustituirunátomoquenoparticipadirectamenteenlareacción(nointervienenienloroturaniformacióndeenlaces)porotroisótopo.Sucálculorequiereevaluarlaconstantedevelocidadutilizandolaexpresióncompleta(ecuación(2.36)).
kTh146,0
kT)HC()DC(
kT)DC(
kT)HC(
CD,r
CH,rCH
‡0
‡0
‡0
‡0
ee
e
ekk
neDeD
eD
eD
==»
---
--
--
C-DC-H
De0‡(C-H)De0‡(C-D)
‡
0.146hnCH

QFIII–TEMA2 77
2.6.-Ejerciciosadicionales
Ejercicio2.15.-Calcular el valor de la energía cinética de traslaciónmásprobable enfuncióndelatemperaturaparaungasdemasamolecularMr.
Solución.-
Ejercicio 2.16.-La velocidad de la reacciónH2 + I2→ 2HI depende de las colisiones
entre las distintas especies en la mezcla de reacción. Calcular las frecuencias totales decolisiónporunidaddevolumenparalosencuentros:(a)H2+H2;(b)I2+I2;(c)H2+I2,paraungasa400Ky1atmconcantidadesequimolecularesdeamboscomponentes.Lasseccioneseficacesdecolisiónsons(H2)≈0,27nm2ys(I2)≈1,2nm2.
Solución.- a) ; b) ;
c)
Ejercicio 2.17.- Se ha estudiado la siguiente reacción trazando para ello la superficie de
energía potencial en función de las dos distancias que parecen punteadas en el dibujo. La superficie de energía potencial se representa mediante curvas isopotenciales trazadas cada 1 kcal/mol y las distancias están en angstroms.
2/kTp =e
1334H,H sm10x29,3Z 22
--= 1334I,I sm10x30,1Z 22
--=
1335I,H sm10x13,1Z 22
--=
1.4 1.6 1.8 2 2.2 2.4 2.6 2.8
d(C-N)
1
1.2
1.4
1.6
1.8
2
2.2
d (O
-H)
34

QFIII–TEMA2 78
a). Describe todos los puntos estacionarios relevantes desde el punto de vista de reactividad
que aparecen sobre la SEP indicando su naturaleza (reactivos, productos, estructuras de transición, intermedios), el valor de las distancias seleccionadas, así como su energía.
b). Representa la variación de la energía potencial con la coordenada de reacción para los
posibles mecanismos de reacción, indicando los valores aproximados de la energía de activación y de reacción. Describe los mecanismos indicando cuál se dará preferentemente.
Solución.- a) dos posibles mecanismos, uno concertado y otro por etapas b) concertado 35,5
kcal mol-1; por etapas 38,5 kcal·mol-1 . Energía reacción 13,5 kcal mol-1
Ejercicio 2.18.- La reducción enzimática de piruvato a lactato tiene lugar por la transferencia de dos átomos de hidrógeno (un protón y un hidruro) tal y como aparece representado en el siguiente esquema:
La transferencia puede tener lugar por medio de dos mecanismos distintos que aparecen reflejados en la superficie de energía potencial obtenida en función de las distancias C-H y O-H- En la figura las líneas de isopotencial se representan espaciadas cada 5 kcal/mol y las distancias en angstroms.
a) Dibujar los perfiles de energía potencial a lo largo del camino de reacción indicando los valores aproximados de las energías de activación y de reacción para cada mecanismo. ¿Qué mecanismo estará favorecido cinéticamente y cuál termodinámicamente?
b) La determinación experimental del efecto cinético isotópico, como cociente entre la constante de velocidad original y la obtenida cuando los hidrógenos transferidos se substituyen por deuterio, condujo a un valor mucho mayor del predicho mediante la Teoría del Estado de Transición. Explicar el posible origen de esta discrepancia.

QFIII–TEMA2 79
c) Representar claramente sobre la superficie los caminos de reacción correspondientes a los dos posibles mecanismos. Identifica los diferentes puntos estacionarios indicando si son reactivos, productos, intermedios o estructuras de transición y dando el valor de las distancias C-H y O-H correspondiente a cada una de ellas. Explica brevemente en qué se diferencian ambos mecanismos.
Solución.- a) Dos mecanismos, uno concertado y otro por etapas; b) siendo favorecido
cinéticamente el concertado (energía de activación entre 20-25 kcal·mol-1) frente al por etapas (energía de activación entre 30-35 kcal·mol-1); c) efecto túnel.
Ejercicio 2.19.- Para la reacción entre el isocianato de m-tolueno y el alcohol n-butílico se
han obtenido las siguientes constantes de velocidad de segundo orden: T(°C) 0,0 7,0 15,0 25,0 105 kr (l mol-1s-1) 4,04 7,72 12,9 25,0 Calcular la energía de activación y el factor de frecuencia. Usando la teoría del estado de transición calcular la entropía de activación a 298 K.
Solución.-
Ejercicio 2.20.- Calcular la energía libre, la entalpía y la entropía de activación para la reacción de hidrogenación del eteno a 628 K sabiendo que A=1,24x106 M-1s-1 y Ea=180 kJ mol-1.
Solución.-
Ejercicio 2.21.- La molécula de hidrógeno (H2) reacciona con un átomo de cloro (Cl) para dar ClH + H a T=500 K pasando a través de un estado de transición lineal. Para este sistema:
a) Calcular la función de partición molecular electrónica de reactivos y estado de transición a
500 K. Téngase en cuenta que el átomo de cloro neutro posee un estado electrónico fundamental 2P3/2 y un estado excitado de baja energía 2P1/2 a 881 cm-1 y que el estado de transición posee un electrón desapareado.
b) Calcular la constante de velocidad para dicha reacción sabiendo que la diferencia de
energía entre los niveles fundamentales del estado de transición y los reactivos es de 4,9 kcal/mol
Datos:
Cl H2 E.T. (Cl—H—H) M (g/mol) 35,5 2,0 37,5 B (s-1) --- 1,823x1012 1,888x1011 n(cm-1) --- 4400 1360; 540; 540*
*excluida la coordenada de reacción
Solución.- b)
Ejercicio 2.22.- La reacción de substitución nucleofílica en fase gas entre el fluoruro de metilo y el anión cloruro transcurre desde un complejo ion-dipolo (un sólo reactivo) hasta productos a través de un estado de transición, tal y como viene representado en la siguiente figura:
1‡0p
11‡0p
1‡0p molkJ1,285G;molKJ0,184S;molkJ6,169H ---- =D-=D=D
1135r smolm10x262,1k --=
11‡0p
1311a molKJ9,193S;smmol71,84A;molkJ64,48E ----- -=D==

QFIII–TEMA2 80
Mediante cálculos teóricos se han determinado las siguientes propiedades de reactivos y estado de transición a 298 K:
Complejo de reactivos Estado de transición Mr (uma) 69,5 69,5 qrot 1,393x104 1,082x104 qvib 23,51 3,703* Energía potencial, kcal/mol 0 27,01 Energía punto cero, kcal/mol 24,93 23,66* Entalpía, kcal/mol 0 25,32
* Excluida la contribución de la coordenada de reacción a) Calcular, mediante la teoría del estado de transición, la constante de velocidad a 298 K,
expresándola en unidades del sistema internacional. Suponer que no existen estados electrónicos de baja energía ni en reactivos, ni en el estado de transición
b) Calcular la entalpía, entropía y energía libre de activación, así como el factor
preexponencial y la energía de activación a 298 K.
Solución.- a) b) ; ;
; ;
Ejercicio 2.23.- Considere la aplicación de la teoría del estado de transición a dos moléculas
sin estructura interna, caracterizadas por tener masas m1 y m2 y diámetros d1 y d2.
a) Calcule la función de partición de los reactivos (considerando únicamente la contribución traslacional)
b) Calcule la función de partición considerando que la estructura de transición presenta
contribuciones traslacionales, rotacionales y un sólo modo vibracional que es justamente el asociado a la coordenada de reacción (stretching del átomo formado entre los átomos 1 y 2). Para el cálculo del momento de inercia considere que en la estructura de transición las moléculas de reactivos están en contacto.
c) Compare la expresión obtenida para la constante de velocidad utilizando la TET con la
correspondiente a la Teoría de Colisiones. ¿Qué conclusiones puede extraer? Solución.- a) b)
17r s10x017,1k --= mol/kcal98,26G‡ =D mol/kcal32,25H‡ =D
)Kmol/(cal57,5S‡ -=D mol/kcal91,25Ea =112s10x023,1A -=
‡q
VhkTm2q
2/3
21
1 ÷øö
çèæ p
=
2
212
21
212
rot h
dmmmmkT8
qúû
ùêë
é+
p=

QFIII–TEMA2 81
c) Para ‘moléculas’ sin estructura la teoría de colisiones y del estado de transición conducen a la misma expresión.
2.7.-Bibliografía
-Levine,I.N.Fisicoquímica;5aed.,McGraw-Hill,2004.-Bertrán,J.;Núñez,J.QuímicaFísica;Ariel,2002.-Moore,J.W.;Pearson,R.G.KineticsandMechanism;3aed.,WileyIntescience,1981.-Tuñón,I.;Silla,E.QuímicaMolecularEstadística;Síntesis,2008.-Engel,T.;Reid,P.QuímicaFísica;Pearson,2006.