presentación de powerpoint - canifarma.org.mx · ris capitulo ix artÍculo 82. los equipos...
TRANSCRIPT

NOM-241-SSA1-2011
1
TALLER DE REVISION DE LA
Comisión de Operación Sanitaria Comisión de Autorización Sanitaria
8 de abril de 2013

AGENDA • Proceso de certificación de Buenas Prácticas
• Solicitud de Visita de Verificación
• Estructura general y aspectos relevantes de la Norma
• Receso
• Hallazgos recurrentes en visitas de verificación
• Contestación a oficios de seguimiento al acta

3
3
OBJETIVO
Conocer los lineamientos y tareas que permitirán la implementación de los requerimientos establecidos en la NOM-241.
¿A QUIENES
APLICA?
¿POR QUE ES
IMPORTANTE
ESTA NORMA?
¿QUE HAY
QUE HACER
PARA
CUMPLIR?
¿COMO
MANTENER EL
CUMPLIMIENTO
DE LA NORMA? ¿QUE
GANAMOS?

4
4
¿POR QUÉ CUMPLIR? La salud es un factor de suma importancia para el bienestar y desarrollo social de la comunidad. Se han establecido requisitos que se deben cumplir durante el proceso de fabricación de los dispositivos médicos que garanticen su calidad y funcionalidad
La Secretaria de Salud a través de la COFEPRIS ejerce el control Sanitario de los establecimientos empleando como marco de referencia la LGS, RIS, las Normas Oficiales Mexicanas, la FEUM y sus suplementos

5
5
NOM-241-SSA1-2011, Buenas prácticas de fabricación para establecimientos dedicados a la fabricación de dispositivos médicos
Proyecto Publicado en DOF el 15 de Noviembre de 2011
Respuesta Respuesta a comentarios en DOF el 20 de Septiembre de 2012
Publicación DOF 11 de Octubre de 2012
Vigencia 9 de Abril de 2013

6
6
APLICA A TODOS: Los establecimientos dedicados a la fabricación de dispositivos médicos. Los almacenes de acondicionamiento y almacenes de deposito y
distribución de dispositivos médicos. Todas aquellos establecimientos que efectúen algún proceso/etapa de producción; obtención, preparación, mezclado, ensamblado, manipulación, envasado, acondicionamiento, estabilidad, análisis, control, almacenamiento y distribución de los dispositivos médicos comercializados en el país.

7
7
RIS Capitulo IX
ARTÍCULO 82. Los equipos médicos, prótesis, órtesis, ayudas funcionales, agentes de diagnóstico, Insumos de uso odontológico, material quirúrgico, de curación, productos higiénicos y otros dispositivos de uso médico, requieren para su producción, venta y distribución de registro sanitario. (Acuerdo publicado en DOF 31 de Diciembre de 2011)
Los Establecimientos en los que se realice el proceso de los Insumos que se mencionan en el párrafo anterior deberán presentar aviso de funcionamiento, con excepción de los dedicados al proceso de fuentes de radiación de uso médico, que requieren de licencia expedida en forma coordinada con la Comisión Nacional de Seguridad Nuclear y Salvaguardias.

8
8
RIS ARTÍCULO 83. La Secretaría clasificará para efectos de registro a los
Insumos señalados en el artículo anterior, de acuerdo con el riesgo que implica su uso, de la manera siguiente:
CLASE I. Aquellos Insumos conocidos en la práctica médica y que su seguridad y eficacia están comprobadas y, generalmente, no se introducen al organismo;
CLASE II. Aquellos Insumos conocidos en la práctica médica y que pueden tener variaciones en el material con el que están elaborados o en su concentración y, generalmente, se introducen al organismo permaneciendo menos de treinta días, y
CLASE III. Aquellos Insumos nuevos o recientemente aceptados en la práctica médica, o bien que se introducen al organismo y permanecen en él, por más de treinta días.

Un requisito indispensable para registrar un dispositivo medico:
OFICIO DE CERTIFICACIÓN DE BUENAS PRACTICAS DE FABRICACION
8

Proceso de Certificación de Buenas
Practicas de Fabricación
9
SOLICITUD DE VISITA DE VERIFICACION (ASPECTOS ADMINISTRATIVOS)

11
11
Solicitud Visita de
verificación
LGS
RIS
NOM-241
Acta de visita de verificación
Dictamen del acta
COS COFEPRIS-01-029
COS - DESyVS
CBPF: Certificación de Buenas Prácticas de Fabricación
CAS

12
X

Estructura General y Aspectos
Relevantes de la Norma NORMA: NOM-241-SSA1-2012
PUBLICADA DOF: 11 /OCT/2012
12

Título
INDICE
5. Clasificación de los dispositivos médicos
6. Organización de un establecimiento
7. Personal
8. Documentación
9. Diseño y construcción de un establecimiento
dedicado a la producción, acondicionamiento,
almacenamiento y distribución de dispositivos médicos
10. Control de la fabricación
11. Equipo de fabricación
12. Manejo de producto fuera de especificaciones
(producto no conforme
13

13. Devoluciones y quejas
14. Retiro de producto del mercado
15. Validación
16. Estudios de estabilidad
17. Control de cambios
18. Desviaciones
19. Auditorías técnicas
20. Destrucción y destino final de residuos
contaminantes y/o peligrosos
13

ALCANCE :
Establece los requisitos que deben reunir los procesos, desde el diseño de la instalación, desarrollo, obtención, preparación, mezclado, producción, ensamblado, manipulación, envasado, acondicionamiento, estabilidad, análisis, control, almacenamiento y distribución de los dispositivos médicos comercializados en el país, por el tipo de insumo de que se trate;
14

y tiene por objeto asegurar que éstos cumplan consistentemente con los requerimientos de calidad y funcionalidad para ser utilizados por el consumidor final o paciente.
Esta norma es de observancia obligatoria en el territorio nacional, para todos los establecimientos dedicados al proceso de dispositivos médicos comercializados en el país.
15

5. CLASIFICACION DE LOS DISPOSITIVOS MEDICOS :
16

ACTA: CLASIFICACION DEL ESTABLECIMIENTO
AVISO DE FUNCIONAMIENTO
AVISO DE RESPONSABLE
ACTIVIDADES
17

Organización de un establecimiento:
ORGANIGRAMA actualizado, identifique que el responsable de fabricación y el del área de calidad no dependan el uno del otro.
18

RESPONSABLE SANITARIO
Ocupar el mayor nivel jerárquico del área de calidad y reportar directamente al puesto más alto del mismo.
Mínimo estudios de licenciatura en el área farmacéutica, química, biológica, medicina, biomédica, bioquímica o afín al proceso.
19

Personal
CAPACITACION: Personal calificado, incluyendo el temporal
DOCUMENTO: Que indique obligaciones, responsabilidades y nivel de autoridad
20

EXPEDIENTE MEDICO
Examen médico al ingreso y periódicos a todo el personal de las áreas de fabricación y calidad, incluyendo surtido de los insumos.
Reportar cualquier condición de enfermedad que pueda tener efectos adversos sobre los procesos de fabricación.
21

INDUMENTARIA Y EQUIPO DE SEGURIDAD DE ACUERDO AL AREA DE TRABAJO Y NIVEL DE RIESGO DEL D.M.
22

8. Documentación
MANUAL DE CALIDAD
Descripción del sistema de gestión de calidad y de los procesos que llevan a cabo en el establecimiento
23

LISTAS
( PNO´s, equipos de fabricación, equipos e instrumentos analíticos. Relación de dispositivos que comercializan )
PLANOS
Actualizados arquitectónicos, flujos y sistemas críticos
24

DOCUMENTOS MAESTROS
ESPECIFICACIONES
Actualizados, de acuerdo a Farmacopeas vigentes y correspondan a lo autorizado
PROCEDIMIENTOS NORMALIZADOS DE OPERACIÓN
25

EXPEDIENTE LEGAL DEL DISPOSITIVO MEDICO
(REGISTRO SANITARIO, ETIQUETA, INFORMACIÓN SOMETIDA PARA LA OBTENCIÓN DEL REGISTRO SANITARIO )
26

Diseño y construcción
REQUERIMIENTOS DE CONSTRUCCIÓN, AMBIENTALES, SEGURIDAD Y BUENAS PRÁCTICAS DE FABRICACIÓN.
Las dimensiones de las diferentes áreas deben estar en función de la capacidad de producción, al nivel de seguridad y al tipo de operaciones al que se destine cada una.
27

ANALISIS DE RIESGO QUE DEFINA LOS REQUERIMIENTOS PARA LA CLASIFICACION
(PROCESO /INSTALACION/ SISTEMAS CRITICOS)
Clasificación de las áreas de producción de acuerdo a la naturaleza del proceso y producto, el análisis de riesgo y el control de la contaminación
28

Control de la fabricación
(de acuerdo a PNO´s)
GENERALIDADES
Identificación de insumos
Limpieza y sanitación
Monitoreo ambiental
Acceso controlado de personal e insumos 29

ADQUISICION
Proveedores aprobados
Especificaciones vigentes
RECEPCION
Certificado de análisis
Envases que no interactúen
30

ALMACENAMIENTO
Áreas identificadas, separadas, por medios físicos y sistema de control
Primeras entradas/caducidades-primeras salidas
31

SURTIDO
Insumos y productos aprobados, cuantificados
y registrados
Medidas para prevenir mezclas o contaminación cruzada
Rastreabilidad
32

PRODUCCION
ORDEN MAESTRA
PROCEDIMIENTO DE PRODUCCION
ORDEN DE PRODUCCION/REGISTROS
CONTROLES DE PROCESO
RENDIMIENTOS
33

ACONDICIONAMIENTO
ORDEN MAESTRA
PROCEDIMIENTO DE ACONDICIONAMIENTO
ORDEN DE ACONDICIONAMIENTO/REGISTROS
CONCILIACION DE MATERIALES
RENDIMIENTOS 34

Área controlada para lotificación/codificación de materiales, registros y destrucción de remanentes
35

MAQUILAS
Las responsabilidades entre el maquilador y el titular del registro estar claramente establecidas en un documento que debe contener las etapas técnicas requeridas.
Transferencia de tecnología
Etapas a maquilar validadas
36

LABORATORIO ANALITICO
Métodos de análisis validado
Registros de limpieza, análisis, calibración, mantenimiento y operación de instrumentos y equipos.
Muestras de retención (área/conservación)
37

Manejo, identificación, preparación, valoración y revaloración (cuando aplique), almacenamiento y disposición final de sustancias o materiales de referencia, reactivos, soluciones, cepas y medios de cultivo empleados en el laboratorio.
38

LIBERACIÓN DE PRODUCTO TERMINADO
Expediente de fabricación de cada lote fabricado
Consideraciones y requisitos para la liberación de producto
Conservación de expedientes 39

DISTRIBUCION
Sistema y registros
Condiciones de almacenamiento en la cadena de distribución
40

EQUIPO DE FABRICACIÓN
(de acuerdo a PNO´s)
Operación, Limpieza
Mantenimiento
Calibración
Programas de los equipos computarizados validados
41

MANEJO DE PRODUCTO FUERA DE
ESPECIFICACIONES (PRODUCTO NO CONFORME)
Contar con PNO que describa las acciones a tomar para el tratamiento de producto no conforme y su dictamen. (Reacondicionado, retrabajado, reprocesado, rechazado, aprobado por concesión)
42

Los retrabajos o reprocesos no están permitidos en dispositivos médicos inyectables
Todos los productos rechazados deben ser identificados y segregados hasta su destrucción o su disposición final, debe llevarse a cabo de acuerdo a un PNO.
43

DEVOLUCIONES Y QUEJAS
Procedimientos
Registros
Acciones correctivas y preventivas para quejas
Retención temporal de devoluciones
44

RETIRO DE PRODUCTO DEL MERCADO
Contar con un sistema y procedimiento para retirar productos del mercado de manera oportuna y efectiva
Reporte final, conciliación, acciones y destino final de producto
45

Validación
46

POLITICA:
Requerimientos establecidos por una organización para realizar la calificación, validación y el mantenimiento de la validación de los equipos, áreas, procesos, limpieza, métodos analíticos y sistemas computacionales, que conllevan a demostrar que los procesos críticos de sus operaciones particulares se encuentran bajo control.
47

PLAN MAESTRO DE VALIDACION (PMV)
Documento en que esquematiza las actividades a
desarrollar para calificar los elementos del proceso y
posteriormente validar.
El PMV debe incluir todos los equipos, instalaciones,
sistemas críticos, procesos de fabricación y
acondicionamiento, limpieza, métodos analíticos,
sistemas computacionales y todas las operaciones que
impacten la calidad del producto.
48

CALIFICACION:
CD: Evidencia documentada que demuestra que el diseño propuesto de las
instalaciones, sistema y equipos es conveniente para el propósito proyectado
CI: Evidencia documentada de que las instalaciones propuesto de las
instalaciones, sistema y equipos se han instalado de acuerdo a las
especificaciones de diseño previamente establecidas.
CO: Evidencia documentada que demuestra que el equipo , instalaciones y
sistemas operan consistentemente de acuerdo a las especificaciones de
diseño establecidas
CE: Evidencia documentada de que las instalaciones, sistema y equipos se
desempeñan cumpliendo con los criterios de aceptación previamente
establecidos. 49

VALIDACIÓN DE PROCESOS:
Previo a la validación de los procesos se debe contar con la calificación de equipos, áreas, personal, calibración de instrumentos, protocolo de validación aprobado, etc.
La validación de los procesos se puede realizar de tres formas:
Retrospectiva
Concurrente
Recurrente
50

RETROSPECTIVA
Se puede realizar tomando en cuenta las siguientes consideraciones:
Número de lotes en base a análisis de riesgos.
El proceso no haya sufrido cambios.
Resultados satisfactorios de los lotes utilizados para la
validación.
Justificar y documentar el uso de validación retrospectiva.
NO APLICA A SISTEMAS Y PROCESOS CRITICOS
51

CONCURRENTE
Se puede realizar en casos excepcionales, tomando en
cuenta las siguientes consideraciones:
Quedar documentado y justificado.
Se requiere de al menos 3 corridas o lotes
consecutivos con resultados aprobatorios.
Es de mencionar que se pueden liberar los lotes
individualmente si se cumplen con las especificaciones.
52

RECURRENTE
Se requiere al menos 3 corridas o lotes consecutivos con
resultados aprobatorios, cuando aplique.
En el caso de productos nuevos y procesos críticos.
53

VALIDACIÓN DE LIMPIEZA
Se realiza para confirmar la eficiencia del proceso de
limpieza de áreas, equipos que se encuentran en
contacto directo con el producto
En caso de equipos multiproductos en los que el
procedimiento de limpieza es el mismo se puede
realizar la validación de limpieza con el peor de los
casos. 54

VALIDACIÓN DE LIMPIEZA Previo a realizar la validación de limpieza se debe considerar lo
siguiente:
El método analítico para determinar trazas debe estar
validado y demostrar la sensibilidad, especificidad y
precisión para la cuantificación del límite máximo
permisible de trazas.
El personal que realiza la validación del método, muestreo y cuantificación de trazas debe estar calificado.
Contar con el protocolo de validación aprobado.
55

VALIDACIÓN DE LIMPIEZA
Durante la validación de limpieza se debe tomar en
consideración lo siguiente:
Realizar mínimo tres corridas o lotes con
resultados aprobatorios.
Debe reflejar los patrones actuales de uso del
equipo.
Seguir el protocolo de validación para la ejecución
de la validación. 56

VALIDACIÓN DE MÉTODOS ANALÍTICOS
Se deben validar los siguientes métodos analíticos:
Evaluación de materia prima, producto a granel, en
proceso y terminado.
Métodos para el análisis de estabilidad, los cuales
deben ser indicativos de estabilidad.
57

Para los métodos analíticos farmacopeicos, normativos,
validados internamente en casa matriz o corporativo, se debe
demostrar la aplicabilidad del método antes de utilizar el
método de rutina.
Cualquier cambio en los métodos analíticos validados o
métodos farmacopeicos, se debe someter a control de
cambios y dependiendo del cambio puede aplicar una
revalidación o validación completa.
58

SISTEMAS COMPUTACIONALES/FIRMAS ELECTRÓNICAS
Deben validarse los sistemas computacionales que impacten la
calidad del producto:
Transferencia de materiales y producto
Disposición de materiales y producto
Control de procesos e instrumentos analíticos
Control de sistemas críticos
Sistema de documentación 59

En la validación de los sistemas computacionales y firmas
electrónicas se debe evaluar:
Diseño
Instalación
Operación
Ejecución
60

Es importante asegurar durante la validación de sistemas
computacionales y firmas electrónicas
Seguridad del sistema mediante la creación de usuarios y
clave de acceso.
Contar con perfil de usuarios acorde con la descripción de
actividades del usuario.
61

Sistema audit trail con el que se de rastreabilidad de los
movimientos realizados por cada usuario.
Comprobación de la adecuada secuencia de los proceso
realizados con el sistema.
Vinculación de firmas electrónicas con firmas autógrafas
en registros electrónicos.
Entre otros.
62

SISTEMAS Y PROCESOS CRITICOS
Se deben validar los siguientes sistemas críticos
Agua purificada y para fabricación de inyectables
Aire (comprimido y HVAC)
Vapor limpio
Esterilización ( por medios físicos y químicos)
Llenado simulado.
63

PROVEEDORES
Se considera calificado a un proveedor si se cumple con lo
siguiente:
Existe evidencia del desempeño adecuado del
proveedor en cuanto a la calidad de los insumos
suministrados.
Contar con auditoría en sitio.
Haya sido aprobado acorde con el numeral 10.2 de la
NOM 64

MANTENIMIENTO DEL ESTADO VALIDADO
Se verifica mediante el cumplimiento de los siguientes sistemas y programas:
Control de cambios
Calibración
Mantenimiento preventivo
Calificación de personal
Auditorías técnicas
65

MANTENIMIENTO DEL ESTADO VALIDADO Sistema de acciones preventivas y correctivas
Cuando haya cambio en los sistemas y programas se debe realizar
una revalidación o recalificación
Al menos cada cinco años se debe realizar una verificación de los
procesos, equipos, sistemas o instalaciones en las mismas
condiciones que fueron validados o calificados
para asegurar el estado de validación. 66

Estudios de estabilidad
Protocolos, informe y programa de estabilidades avalados por el responsable sanitario
67

Control de cambios
Contar con:
Sistema y PNO para control de cambios
Comité Técnico integrado
Aprobación por el área de Calidad
68

DESVIACIONES
Contar con:
Sistema y PNO para Desviaciones
Comité Técnico integrado
Evaluación de la efectividad de las acciones resultantes de una desviación.
69

AUDITORIAS TECNICAS
Procedimiento y programa
Evidencia documental
70

DESTRUCCIÓN Y DESTINO FINAL DE RESIDUOS CONTAMINANTES Y/O PELIGROSOS
Procedimiento y registros
71

72
HALLAZGOS RECURRENTES EN
VISITAS DE VERIFICACION

• El responsable sanitario, es el propietario
del establecimiento no afín al proceso
• Programas de capacitación incompletos o
no los cumplen, no hay evidencia
• No hay rótulos de identificación de la
empresa o con datos del responsable
sanitario 73

• No hay identificación ni segregación de áreas.
• No cuentan o no están actualizados los
planos con flujos de materiales, personal,
sistemas críticos
74

• Validación de procesos en base a
protocolos pocas empresas los tienen
• No cuentan con validación de sistemas
críticos, métodos analíticos.
75

• Análisis de materias primas y producto
terminado no se realizan completos de
acuerdo a FEUM, suplemento
actualizados o normas
• No cuentan con infraestructura para los análisis requeridos
• No analizan por cada lote de producción
76

No se cuenta con control de uso y vigencia
de estándares, cepas, medios de cultivo y
reactivos
No se cuenta con control y evaluación de
sanitizantes
77

• Identificación de materias primas y materiales de acondicionamiento, producto intermedio con datos incompletos, no indican condición (aprobado, cuarentena) de acuerdo a procedimiento
78
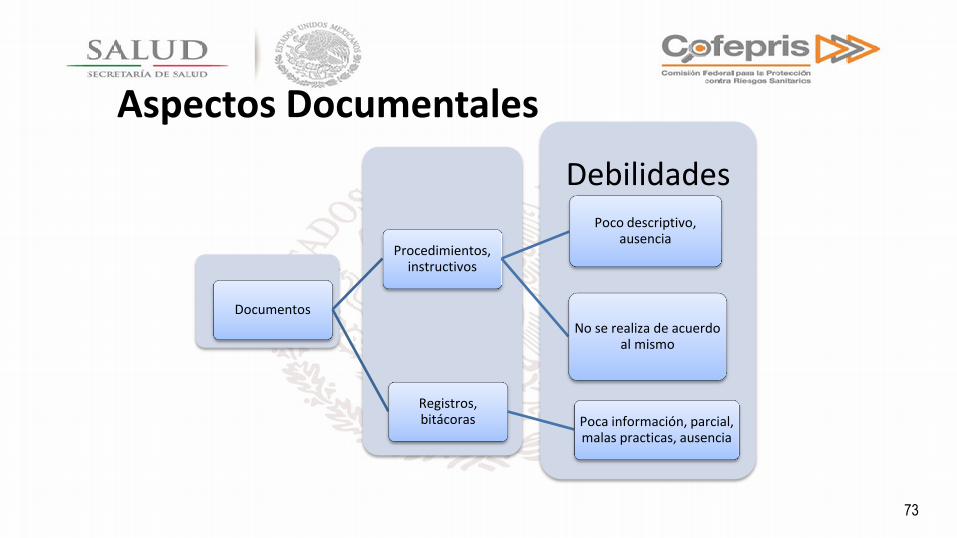
Aspectos Documentales
Debilidades
Documentos
Procedimientos, instructivos
Poco descriptivo, ausencia
No se realiza de acuerdo al mismo
Registros, bitácoras Poca información, parcial,
malas practicas, ausencia
73

Programas incompletos de:
• Mantenimiento
• Calibración
• Limpieza
• Capacitación
• Control de plagas
80

Ordenes de fabricación y acondicionamiento:
• No tienen
• Están incompletas
• No se realiza los registros al momento.
• No tienen el control de proceso
• Son diferentes al documento maestro
81

Retención de muestras:
• Sin identificación
• Insuficientes
• No esta documentada la cantidad requerida
• Envases inadecuados
• Control de condiciones del área no es
adecuado
82

ALMACENAMIENTO ADECUADO
83

INDUMENTARIA
84

DELIMITACION AREA
85

DELIMITACION AREA
LINEA ACONDICIONAMIENTO
86

AREA DE RECHAZOS
87

MUSEO DE MUESTRAS
88

LABORATORIO FISICOQUIMICOS
89

EQUIPO DE PRECISION
90

ALMACEN REACTIVOS ANALITICOS
91

CAMPANA DE FLUJO LAMINAR
92

Gracias
93

Contestación a Oficios de
95
Seguimiento al Acta de Verificación

97
97
APLICA A TODOS: Los establecimientos dedicados a la fabricación de dispositivos médicos. Los almacenes de acondicionamiento y almacenes de deposito y
distribución de dispositivos médicos. Todas aquellos establecimientos que efectúen algún proceso/etapa de producción; obtención, preparación, mezclado, ensamblado, manipulación, envasado, acondicionamiento, estabilidad, análisis, control, almacenamiento y distribución de los dispositivos médicos comercializados en el país.

98
98
Solicitud Visita de
verificación
LGS
RIS
NOM-241
Dictamen del acta de visita
Dictamen de evidencia CBPF
COS Respuesta a
Requerimiento
Desecho de
trámite
COFEPRIS-01-029
COS - DESyVS
Requerimiento
CBPF: Certificación de Buenas Prácticas de Fabricación
CAS

Acta de Verificación Sanitaria
A partir de la fecha de cierre del acta de verificación
• Revisar si se subsanaron las anomalías.
• CUMPLE, Emitir Oficio de Certificación.
• NO CUMPLE, Se emite Oficio de 1er Requerimiento.

Acta de Verificación Sanitaria
A partir de la fecha de cierre del acta, Con fundamento en el Art. 68 de la LEFEPA, el Interesado tiene derecho a manifestar lo que a su derecho le conviene en un plazo de 5 días hábiles

Evidencia documental en alcance al acta
Documentación que subsane lo indicado en el acta de verificación.
La remisión de programas o plan de acciones correctivas y preventivas, no proporcionan evidencia de Buenas practicas durante la fabricación

Dictamen de acta
• El dictamen del acta de verificación se efectuara en conjunto con la documentación enviada en alcance a esta, si hubiere
Resultando……

Si, con la documentación enviada se subsanan las desviaciones asentadas en acta, existe evidencia de cumplimiento a las buenas prácticas de fabricación.
Se emite OFICIO DE CERTIFICACION DE BUENAS PRACTICAS DE FABRICACION
por planta de fabricación

Oficio de 1er Requerimiento.
Si, con la documentación enviada NO se subsanan las desviaciones asentadas en acta, se emitirá oficio de Requerimiento con plazo 180 días naturales; con base al art. 223 del RIS
¿Responsabilidad?
Respuesta en tiempo y forma, No responde EN TIEMPO Y FORMA Se emite oficio de Desecho.

Una vez que presentan evidencia documental, se efectúa su revisión a fin de dictaminar:
Si, con la documentación enviada se subsanan las desviaciones asentadas en acta, existe evidencia de cumplimiento a las buenas prácticas de fabricación.
Se emite OFICIO DE CERTIFICACION

Si, no subsanan todas las desviaciones se emite un
Oficio de 2º Requerimiento. Con plazo 90 días naturales con base al art. 223 del RIS
¿Cumple? SI: Se emite Oficio de Certificación.
NO: Se emite Oficio de Desecho

Aspectos importantes
Visitas de verificación en condiciones dinámicas, es decir mientras están fabricando
• NO ES POSIBLE EMITIR OFICIO DE CERTIFICACION DE BUENAS PRACTICAS DE FABRICACIÓN, si no están FABRICANDO

Aspectos importantes • La implementación de las Buenas Prácticas de
Fabricación (BPF) es parte fundamental de un sistema de gestión de la calidad:
Auditorias
Sistema CAPAS
Manejo de desviaciones, quejas y devoluciones
Control de cambios
Evaluación de proveedores

Aspectos importantes
• Análisis a materias primas; independientemente de que cuenten con certificado de proveedor
Numerales:
8.5.2.1, 8.5.2.2

Aspectos importantes Deben generar:
• Orden de producción/acondicionamiento: que es la copia de la orden o fórmula maestra de producción/acondicionamiento a la cual se le asigna un número de lote, se utiliza como guía y registro de las operaciones efectuadas en la producción y/o acondicionamiento de un lote de dispositivo médico.

Aspectos importantes • Procedimiento de producción, al documento que
contiene las instrucciones detalladas para transformar las materias primas, materiales o componentes en dispositivos médicos a granel previo a su acondicionamiento en el empaque destinado para su comercialización.
• El procedimiento de acondicionamiento
Numerales: 8.3.1.2, 8.3.4.1

Aspectos importantes Es un requerimiento la emisión del Certificado analítico de producto terminado (numeral 8.5.1.2.).
Entiéndase por Certificado de producto, en algunos casos como certificado de conformidad (pruebas de funcionalidad)

Aspectos importantes “documento que avala que el producto ha sido probado antes de su liberación de la planta para garantizar su seguridad, eficacia, calidad y funcionalidad una vez que ha demostrado el cumplimiento con los parámetros de aceptación establecidos con base al tipo de producto y su nivel de riesgo. Debe incluir el número de lote o de serie, las especificaciones y resultado del producto terminado emitido por el fabricante, o copia de un certificado de análisis emitido por un laboratorio autorizado y firmado por el responsable de aseguramiento de la calidad o por el responsable sanitario”

Aspectos importantes • Calificación de equipos y sistemas críticos de
fabricación. Las diferentes etapas de calificación son secuenciales no es posible continuar a la siguiente si la etapa previa no se ha concluido de forma satisfactoria.
• Clasificación y Calificación de áreas

Aspectos importantes • Validación de proceso de esterilización; aun cuando
este proceso se envíe a un maquilador debe realizarse y documentarse su validación considerando los requerimientos mínimos según el método que se efectúe, por ejemplo:
Esterilización por Oxido de Etileno
ISO 11135-1:2007; ISO 11135-2:2008

• Clasificación y Calificación de áreas
GESTION DE RIESGO
Análisis de riesgo • ICH Q9; QUALITY RISK MANAGEMENT.
• ISO 14971:2009; Medical devices-Application of risk management to medical devices
Aspectos importantes

• 3.7 Análisis de riesgo, al método para evaluar con anticipación los factores que pueden afectar la funcionalidad de: sistemas, equipos, procesos o calidad de insumos y producto.
• 7.4.1 Los requerimientos de indumentaria para cada área de fabricación dependerán de la clasificación del área con base al nivel de riesgo del dispositivo médico y deben estar definidos por escrito, incluyendo la disposición de indumentaria desechable.
117
Enfoque con análisis de riesgo

• 8.5.7. Debe existir un PNO que permita llevar a cabo el análisis de riesgo del producto y la evidencia de su aplicación para su correcta administración, el cual debe contener como mínimo lo siguiente:
• 8.5.7.1 Metodología de Análisis de Riesgo empleado.
• 8.5.7.2 Determinación de puntos críticos de control.
• 8.5.7.3 Parámetros y límites críticos.
• 8.5.7.4 Monitoreo de los puntos críticos de control.
• 8.5.7.5 Acciones correctivas a realizar cuando indique que un punto crítico de control se encuentra fuera de control.
• 8.5.7.6 Plan de evaluación para garantizar que el análisis de riesgos y puntos críticos de control estén funcionando efectivamente.
Enfoque con análisis de riesgo

• 9.1.1 El establecimiento debe ser diseñado, construido y conservado de acuerdo con las operaciones que en él se efectúen tomando como base el nivel de riesgo del dispositivo médico.
• Su diseño y construcción debe permitir su limpieza, orden, mantenimiento y prevención de la contaminación, así como los flujos unidireccionales del personal y materiales.
119

• 9.1.2 Debe existir un plan para definir los requerimientos del dispositivo médico con base a su clasificación de riesgo, que incluya los procesos empleados, los sistemas críticos y el alcance de la instalación.
• 9.2.9 De acuerdo a la naturaleza del proceso y producto, el análisis de riesgo y el control de la contaminación; deben clasificarse las áreas de producción (véase Apéndice normativo A)
120
Enfoque con análisis de riesgo

15. Validación: los fabricantes de dispositivos médicos deben determinar qué actividades deben validar considerando que:
• Deben demostrar el control de los aspectos críticos de sus operaciones particulares.
• Los procesos de fabricación MANUALES no se validan.
• Debe utilizarse un enfoque de análisis de riesgos del dispositivo médico para evaluar el ámbito y grado de validación.
• Todas las instalaciones, equipos, sistemas críticos que impacten en la calidad del dispositivo médico, deben estar calificados y los procesos de producción, métodos de limpieza y analíticos deben validarse.
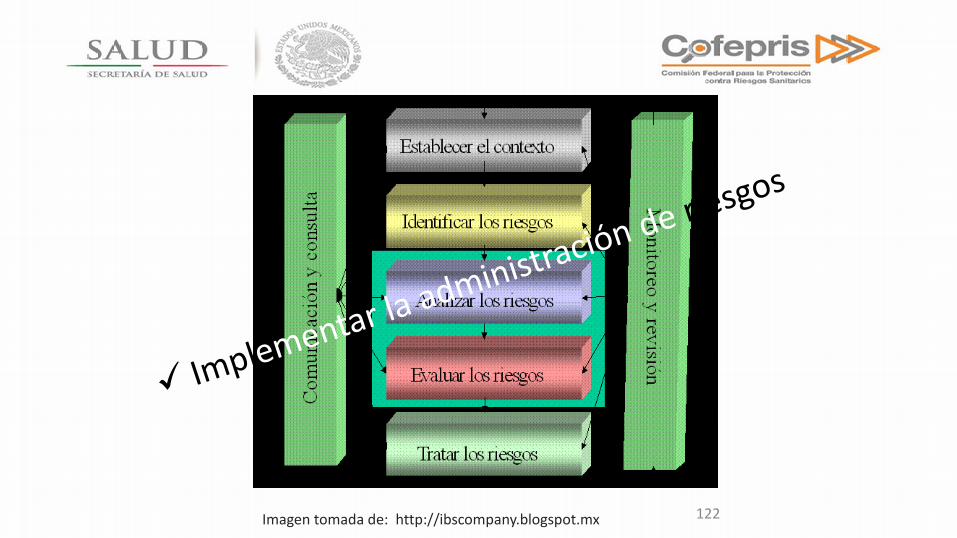
122 Imagen tomada de: http://ibscompany.blogspot.mx

Ejercicios
Trabajar en equipos de 10 personas.
Lea cuidadosamente las actividades que realiza el establecimiento, si existen dudas sobre el tipo de dispositivo medico asignado, no dude en preguntar.
Respete la opinión de los demás y trabajen en equipo.

Ejercicios
1. Plantee una metodología para la identificación y evaluación de los posibles riesgos.
2. Determine requerimientos de la NOM aplicables al tipo de establecimiento descrito.
3. Defina requisitos de instalación.
4. Determine cuales serán las actividades de calificación y validación que deberán realizar.

125
Iván V. Cruz Barrera :[email protected]
Norah M. Silva Morlaes :[email protected] Ma. del Rosario Delgadillo Martínez :[email protected]
Ma. De Lourdes López Torres : [email protected]
Arminda López Alquicira : [email protected] Llanet Bolaños Valerio :[email protected]
¡Muchas gracias!