mòdul 8.1. seqüenciació de proteïnes. mètodes de...
TRANSCRIPT

Módulos 8.3. (T) y 8.4. (P)Análisis de secuencias de proteínas.
Alineamiento de secuencias de proteínas, simples y múltiples: BLAST. Análisis de secuencies de proteínas: motivos y perfiles
(perfiles HMM): Prosite, Pfam, Prints, etc. Matrices de puntuación (PAM, ...). Familias proteicas. Dominios estructurales.
Curs d’Especialista Universitari en Bioinformàtica
Abril 2007

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 2
ContenidosAnálisis de secuencias de ácidos nucleicos
Traducción de secuenciasAnálisis de secuencias de proteína - fuentes primarias
Recuperación de secuencias completas de proteínaBúsqueda rápida de la identidad de una secuenciaBúsqueda de semejanzas de una secuencia
Alineamiento de secuencias: Global vs. LocalAlineamiento de secuencias de proteínas, simples i múltiples: BLAST
Matrices de puntuación (PAM, BLOSUM)Análisis de secuencias de proteínas - fuentes secundarias
Búsqueda de patrones, perfiles y Hidden Markov Models (HMM, Modelos de Markov ocultos)
Análisis de secuencias de proteínas: motivos y perfiles (perfilesHMM): Prosite, Pfam, Prints, etc.
Búsqueda de patronesBúsqueda de perfiles y HMMsBúsqueda de bloques.
Familias proteicasDominios estructurales

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 3
Estudio a partir de la secuencia de aminoácidos
Clasificación de los aminoácidos de acuerdo a sus propiedades físicas: aminoácidos apolares
Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 4
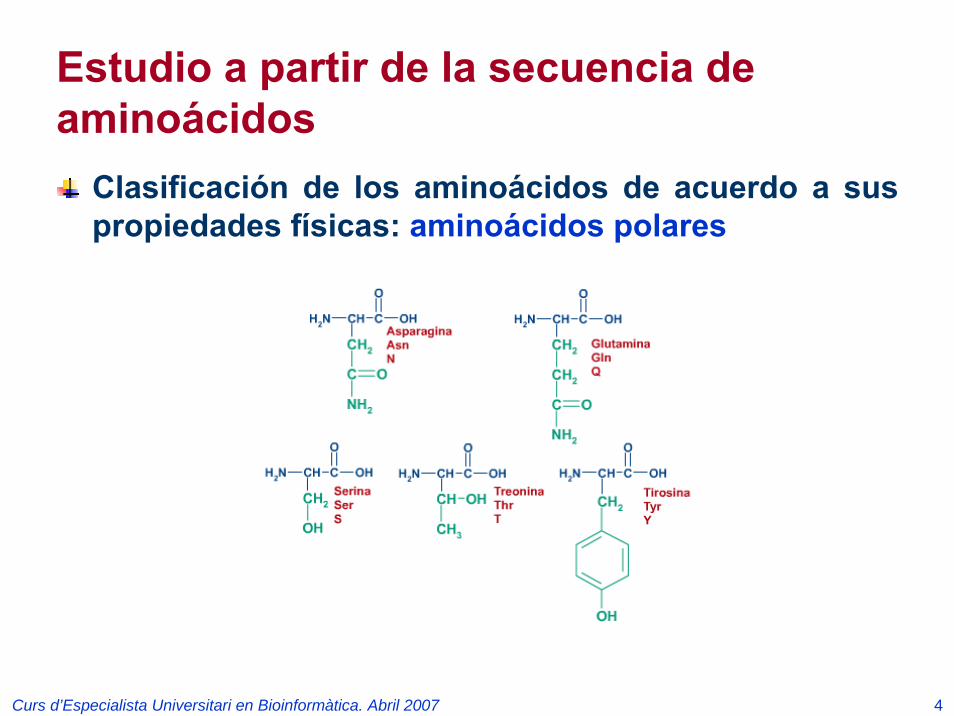
Estudio a partir de la secuencia de aminoácidos
Clasificación de los aminoácidos de acuerdo a sus propiedades físicas: aminoácidos polares

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 5
Estudio a partir de la secuencia de aminoácidos
Clasificación de los aminoácidos de acuerdo a sus propiedades físicas: aminoácidos básicos
aminoàcidos ácidos

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 6
Alineamiento de secuencias de proteínas
¿Por qué comparar secuencias?
Cambios en las secuencias :Reemplazo puntual
Sinónimos/No-SinónimosInserción y Delección (InDels)
Afecta pautas de lecturaTraslocaciónDuplicación
En todos los niveles y escalas de organización
ATG GAC CCA CGT TCG GAGCódigo estándar
AAA K ACA T AGA R ATA IAAC N ACC T AGC S ATC IAAG K ACG T AGG R ATG MAAT N ACT T AGT S ATT ICAA Q CCA P CGA R CTA LCAC H CCC P CGC R CTC LCAG Q CCG P CGG R CTG LCAT H CCT P CGT R CTT LGAA E GCA A GGA G GTA VGAC D GCC A GGC G GTG VGAG E GCG A GGG G GTG VGAT D GCT A GGT G GTT VTAA . TCA S TGA . TTA LTAC Y TCC S TGC C TTC FTAG . TCG S TGG W TTG LTAT Y TCT S TGT C TTT F
MDPRSE…
GAT
D
GCA
A
TAG
*

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 7
Alineamiento de secuencias de proteínasSignificado biológico:
Similitud de secuencia = relación evolutiva¿Porqué secuencias similares se supone que son homólogas?
Estructura
Función
SecuenciaLa secuencia de aminoácidos de una proteína
determina su estructura
La estructura de una proteína determina su función (exp. de desnaturalización, interacciones molec.,
modelos de sitios activos de enzimas)
La función es el objetivo de la selección natural (ni la secuencia ni la estructura). En ese caso, los cambios
en la secuencia y en la estructura son retringidos.

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 8
Alineamiento de secuencias de proteínas
La divergencia está restringidaDebe existir alguna presión para mantener ciertas funciones
Estructura
Función
SecuenciaSecuencias similares de aminoácidos deben plegarse
dando estructuras también similares
Y proteínas con similares estructuras deben realizar funciones también similares
Devos and Valencia (2000)

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 9
Alineamiento de secuencias de proteínas
Objetivos:Para determinar el grado de semejanza entre ellasPara deducir si tienen un antepasado comúnPara identificar las regiones conservadasque pueden ser de importancia estructural o funcionalPara hacer predicciones sobre la función y estructuraPara identificar
secuencias similares en bases de datos
Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 10
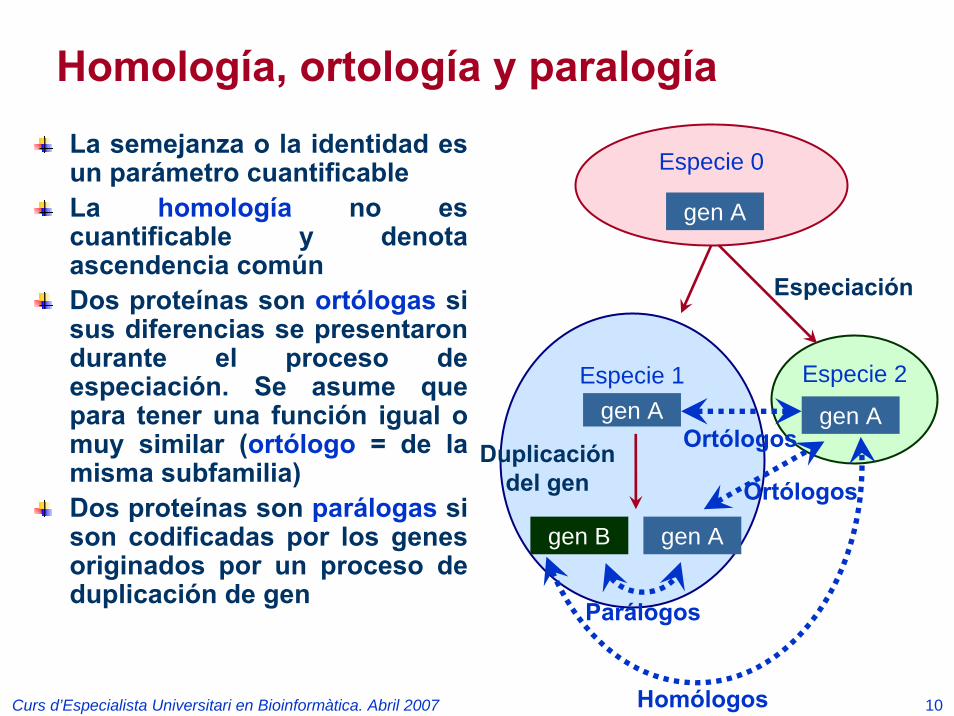
Homología, ortología y paralogíaLa semejanza o la identidad es un parámetro cuantificableLa homología no es cuantificable y denota ascendencia comúnDos proteínas son ortólogas si sus diferencias se presentaron durante el proceso de especiación. Se asume que para tener una función igual o muy similar (ortólogo = de la misma subfamilia)Dos proteínas son parálogas si son codificadas por los genes originados por un proceso de duplicación de gen
Especie 1gen A
Especie 2
gen A
Especie 0
gen A
Especiación
gen Agen B
Duplicacióndel gen
Ortólogos
Ortólogos
Parálogos
Homólogos

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 11
Generalidades
Secuencia: Cadena lineal finita y ordenada de símbolos pertenecientes a un alfabeto
Alfabeto: Conjunto de símbolos básicos de las secuenciasADN: A= {a,c,g,t|u}Proteínas= A={a,c,d,e,f,g,h,I,k,l,m,n,p,q,r,s,t,v,w,y}
ADN y Proteínas son cadenas co-linaelesExisten otros alfabetos diversos
Comparación: Encontrar la posición relativa entre dos secuencias que maximice su parecido

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 12
Comparación básica por identidades
Algoritmo: Desplazar una secuencia debajo de la otra anotando el número de coincidencias que ocurren, seleccionando como resultado la posición de mayor valor
Seq X = TCAGACGATTG (n=11)Seq Y = ATCGGAGCTG (m=10)

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 13
Comparación básica por identidades TCAGACGATTG ATCGGAGCTG (0)
TCAGACGATTG ATCGGAGCTG (1)
TCAGACGATTG ATCGGAGCTG (0)
TCAGACGATTG ATCGGAGCTG (2)
TCAGACGATTG ATCGGAGCTG (0)
TCAGACGATTG ATCGGAGCTG (0)
TCAGACGATTG ATCGGAGCTG (3)
TCAGACGATTG ATCGGAGCTG (1)
TCAGACGATTG ATCGGAGCTG (4)
TCAGACGATTG ATCGGAGCTG (3)
TCAGACGATTG ATCGGAGCTG (2)
TCAGACGATTG ATCGGAGCTG (3)
TCAGACGATTG ATCGGAGCTG (2)
TCAGACGATTG ATCGGAGCTG (2)
TCAGACGATTG ATCGGAGCTG (0)
TCAGACGATTG ATCGGAGCTG (1)
TCAGACGATTG ATCGGAGCTG (3)
TCAGACGATTG ATCGGAGCTG (1)
TCAGACGATTG ATCGGAGCTG (0)
TCAGACGATTG ATCGGAGCTG (0)

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 14
Comparación básica por identidadesTCAGACGATTG (r=4)ATCGGAGCTG
0 1 2 3 4 5 6 7 8 9 10 11┌───┬───┬───┬───┬───┬───┬───┬───┬───┬───┬───┬───┐
0 │ │ T │ C │ A │ G │ A │ C │ G │ A │ T │ T │ G │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-1 │ A │ │ │ 1 │ │ 1 │ │ │ 1 │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-2 │ T │ 1 │ │ │ │ │ │ │ │ 2 │ 1 │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-3 │ C │ │ 2 │ │ │ │ 1 │ │ │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-4 │ G │ │ │ │ 1 │ │ │ 2 │ │ │ │ 3 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-5 │ G │ │ │ │ 3 │ │ │ 1 │ │ │ │ 1 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-6 │ A │ │ │ 1 │ │ 4 │ │ │ 2 │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-7 │ G │ │ │ │ 2 │ │ │ 2 │ │ │ │ 2 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-8 │ C │ │ 1 │ │ │ │ 1 │ │ │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-9 │ T │ 1 │ │ │ │ │ │ │ │ 3 │ 1 │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-10│ G │ │ │ │ 2 │ │ │ 3 │ │ │ │ 2 │└───┴───┴───┴───┴───┴───┴───┴───┴───┴───┴───┴───┘
Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 15
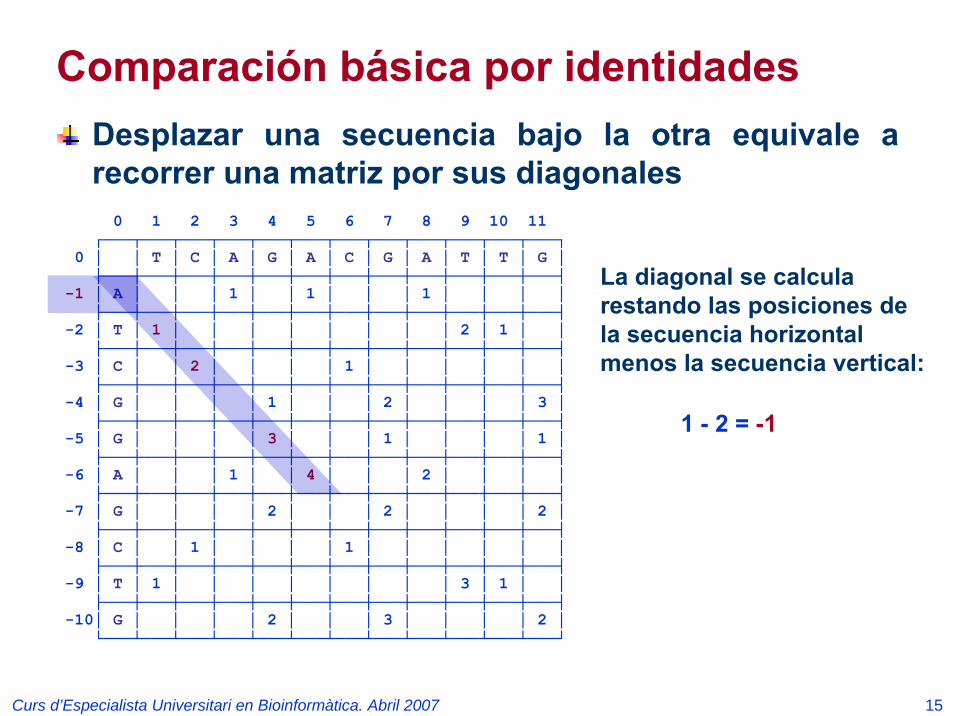
Comparación básica por identidadesDesplazar una secuencia bajo la otra equivale a recorrer una matriz por sus diagonales
0 1 2 3 4 5 6 7 8 9 10 11┌───┬───┬───┬───┬───┬───┬───┬───┬───┬───┬───┬───┐
0 │ │ T │ C │ A │ G │ A │ C │ G │ A │ T │ T │ G │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-1 │ A │ │ │ 1 │ │ 1 │ │ │ 1 │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-2 │ T │ 1 │ │ │ │ │ │ │ │ 2 │ 1 │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-3 │ C │ │ 2 │ │ │ │ 1 │ │ │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-4 │ G │ │ │ │ 1 │ │ │ 2 │ │ │ │ 3 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-5 │ G │ │ │ │ 3 │ │ │ 1 │ │ │ │ 1 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-6 │ A │ │ │ 1 │ │ 4 │ │ │ 2 │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-7 │ G │ │ │ │ 2 │ │ │ 2 │ │ │ │ 2 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-8 │ C │ │ 1 │ │ │ │ 1 │ │ │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-9 │ T │ 1 │ │ │ │ │ │ │ │ 3 │ 1 │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-10│ G │ │ │ │ 2 │ │ │ 3 │ │ │ │ 2 │└───┴───┴───┴───┴───┴───┴───┴───┴───┴───┴───┴───┘
La diagonal se calcula restando las posiciones de la secuencia horizontal menos la secuencia vertical:
1 - 2 = -1

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 16
Comparación básica por identidadesDesplazar una secuencia bajo la otra equivale a recorrer una matriz por sus diagonales
0 1 2 3 4 5 6 7 8 9 10 11┌───┬───┬───┬───┬───┬───┬───┬───┬───┬───┬───┬───┐
0 │ │ T │ C │ A │ G │ A │ C │ G │ A │ T │ T │ G │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-1 │ A │ │ │ 1 │ │ 1 │ │ │ 1 │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-2 │ T │ 1 │ │ │ │ │ │ │ │ 2 │ 1 │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-3 │ C │ │ 2 │ │ │ │ 1 │ │ │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-4 │ G │ │ │ │ 1 │ │ │ 2 │ │ │ │ 3 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-5 │ G │ │ │ │ 3 │ │ │ 1 │ │ │ │ 1 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-6 │ A │ │ │ 1 │ │ 4 │ │ │ 2 │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-7 │ G │ │ │ │ 2 │ │ │ 2 │ │ │ │ 2 │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-8 │ C │ │ 1 │ │ │ │ 1 │ │ │ │ │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-9 │ T │ 1 │ │ │ │ │ │ │ │ 3 │ 1 │ │├───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┼───┤
-10│ G │ │ │ │ 2 │ │ │ 3 │ │ │ │ 2 │└───┴───┴───┴───┴───┴───┴───┴───┴───┴───┴───┴───┘
Complejidad O (N x M)N y M son las longitudes de las secuencias a comparar
O = 11 x 10 = 110
Si consideramos N y M iguales (como cuando hay muchas comparaciones):
O = (N)2
Complejidad algorítmicaRelacionado con el número de operaciones que se requiere para resolver el problema

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 17
Comparación básica por identidades
Los fragmentos alineados aparecen como diagonales
en la matriz
Si se representan las identidades como puntos de una cuadrícula se obtiene una matriz de puntos o DotPlot.

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 18
Matriz de puntos o DotPlot

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 19
Dimensiones del problema
EMBL Nucleotide Sequence Database (versión 90, marzo 2007) (http://www.ebi.ac.uk/embl/index.html)
Nº entradas > 87 millones Nº bases > 159 GigaBytes90 archivos, 73 GB comprimido, 395 GB expandido
Recursos ComputacionalesCPU: 4,77 MHz (1983) Intel 8088
> 500 MHz (1999) Pent III, Celeron, Itanium,...> 1 GHz (2001) Pentium IV> 3 GHz (2007) Core 2 Duo

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 20
Métodos de reducción de la complejidad algorítmicaOptimización para reducir el tiempo de cálculo
Tabla de Dispersión (Hash)Lista con las posiciones de todos los elementos posibles de la secuencia
pos: 12345678901 Seq X: TCAGACGATTG n=11
Tabla (seqX)A 3, 5, 8C 2, 6G 4, 7, 11T 1, 9, 10
Secuencia a compararpos: 1234567890
Seq Y: ATCGGAGCTG m=10Se deben acumular las identidades en cada diagonal (d= h-v, si xh alinea con yv)
y1 (A) en d2 (3-1), d4 (5-1) y d7 (8-1)y2 (T) en d-1 (2-1), d7 (9-2) y d8 (10-2)…

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 21
Métodos de reducción de la complejidad algorítmica
Optimización para reducir el tiempo de cálculo
Complejidad: Será igual a cada elemento de la secuencia Y vs. el número de elementos de la tabla hash para ese símbolo, por tanto
O (N x media elementos celdas)media elementos celdas = M/longitud tabla
siendo (N, M = long de sec X y sec Y)
Proteínas: a más entradas (longitud de tabla) menos elementos por entrada dimensión de la tabla = Lk
Para proteínas: L alfabeto = 20 aa, k = 2Lk = 202 = 400 entradasProteínas: longitud media = 400 aaPor tanto equivale a 1 elemento por entradaComplejidad O (N + M)

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 22
Comparación por semejanzaLimitaciones (Doolittle, 1981):
Código genético degenerado o redundante (1 codón ≠ 1 aa, excepto Met, Trp)Se pueden dar substituciones sin influencia (aa con las propiedades parecidas)Hay que considerar las inserciones y deleccionesLos símbolos no son todos iguales: hay que considerar la cantidad de información que acarrea cada uno (= asociada a la frecuencia)Tener en cuenta el conocimiento biológico en el método de valoración
Proteínas: coincidencias de aa raros más importantes que de aanormales (Cys vs. Ala)DNA: purinas A-G y pirimidina C-T mejor que purina-pirimidina)
Distancia: El coste de transformar una secuencia en otra por medio de la aplicación de una serie de operaciones (sustitución, inserción, borrado).

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 23
Matrices de puntuaciónCon ellas se intenta valorar los cambios evolutivos para captar el significado biológico de las semejanzasSe deben considerar los reemplazos conservativos y las diferencias en las frecuencias observadas
Matrices PAM. Dayhoff (1972) Compara frecuencias en alineamientos relacionados con alineamientos no relacionados: proporcional a log(fAB/f'AB)
fAB frecuencia AB en alineamientos relacionadosf'AB frecuencia en los alineamientos no relacionados (f'AB=fAxfB)
fAB Depende de la semejanza entre las proteinaspara secuencias muy relacionadas = bajaa mayores distancias evolutivas = aumentapara grandes distancias = al azar
Se calcula a partir de alineamientos múltiples con no más de un 15% de diferencias por identidad con las tablas de las frecuencias de sustitución entre residuos y se normaliza (en 100 residuos 1 mutación) => PAM, 1% PercentAccepted Mutation, se extrapola a diferentes distancias (120, 250, 320, etc) PAMs.

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 24
Matrices de puntuaciónMatrices BLOSUM. Altschul (1991)
No se conoce a priori lo semejante que son las secuencias a buscarPAM extrapola las relaciones lejanas a partir de relaciones cercanas (esta hipótesis favorece las posiciones más mutables)
Se calcula a partir de alineamientos de bloques de secuencias. Bloque: Matriz cuyas filas representan segmentos de secuencias alineadas sin interrupciones
BLOSUMnn (BLOcks SUbstitution Matrices): (nn: umbral de identidad utilizado para la seleccionar los bloques)
Un BLOSUM bajo (umbral bajo de identidad) se corresponde con un número alto de PAM (distancia evolutiva grande)
EKPRKVMLMVRAGDVVDQFIEALLPHLEEGEKPRKIFLMVTAGKPVDSVIQSLKPLLEEGETPRKILLMVKAGTATDATIQSLLPHLEKDETPRKILLMVKAGTATDATIQSLLPHLEKDETPRRILLMVKAGAGTDAAIDSLKPYLDKGETPRRILLMVKAGSGTDSAIDSLKPYLDKG

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 25
Matrices de puntuaciónpam250 (inferior) y BLOSUM62 (superior)
A R N D C Q E G H I L K M F P S T W Y V B Z X *--+-------------------------------------------------------------------------+---| 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4 | A | 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4 | R
A | 2 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4 | NR | -2 6 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4 | DN | 0 0 2 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4 | CD | 0 -1 2 4 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4 | QC | -2 -4 -4 -5 12 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 | EQ | 0 1 1 2 -5 4 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4 | GE | 0 -1 1 3 -5 2 4 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4 | HG | 1 -3 0 1 -3 -1 0 5 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4 | IH | -1 2 2 1 -3 3 1 -2 6 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4 | LI | -1 -2 -2 -2 -2 -2 -2 -3 -2 5 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4 | KL | -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4 | MK | -1 3 1 0 -5 1 0 -2 0 -2 -3 5 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4 | FM | -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6 7 -1 -1 -4 -3 -2 -2 -1 -2 -4 | PF | -4 -4 -4 -6 -4 -5 -5 -5 -2 1 2 -5 0 9 4 1 -3 -2 -2 0 0 0 -4 | SP | 1 0 -1 -1 -3 0 -1 -1 0 -2 -3 -1 -2 -5 6 5 -2 -2 0 -1 -1 0 -4 | TS | 1 0 1 0 0 -1 0 1 -1 -1 -3 0 -2 -3 1 2 11 2 -3 -4 -3 -2 -4 | WT | 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 3 7 -1 -3 -2 -1 -4 | YW | -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -6 -2 -5 17 4 -3 -2 -1 -4 | VY | -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10 4 1 -1 -4 | BV | 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4 4 -1 -4 | ZB | 0 -1 2 3 -4 1 2 0 1 -2 -3 1 -2 -5 -1 0 0 -5 -3 -2 2 -1 -4 | XZ | 0 0 1 3 -5 3 3 -1 2 -2 -3 0 -2 -5 0 0 -1 -6 -4 -2 2 3 1 | *X | 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 |--+-------------------------------------------------------------------------+---
A R N D C Q E G H I L K M F P S T W Y V B Z X *
Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 26
Comparación Global vs. Local
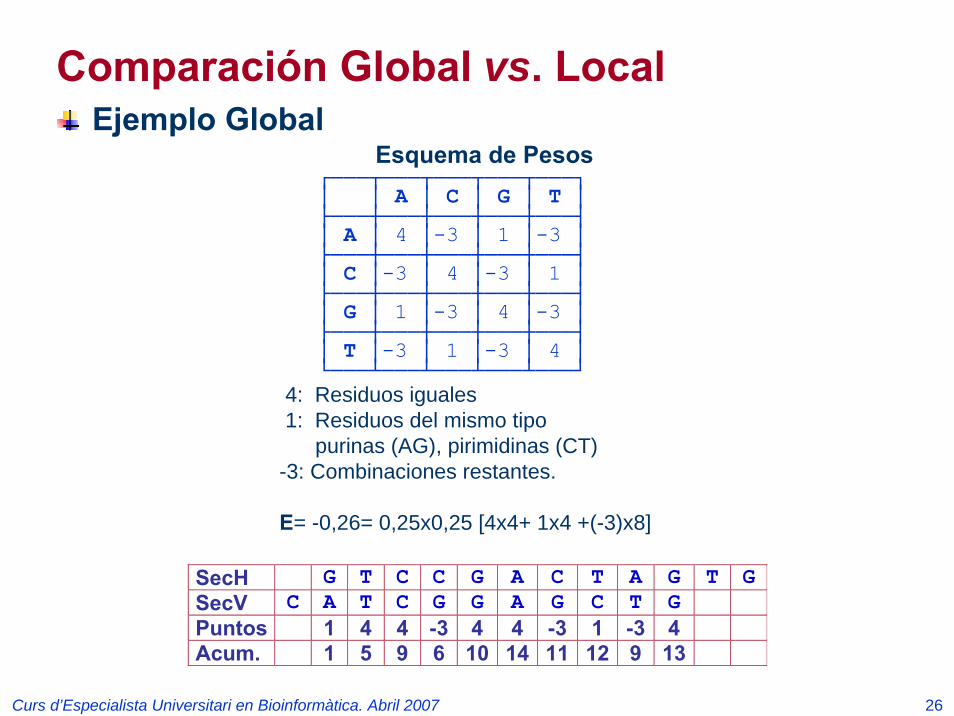
Esquema de Pesos┌───┬───┬───┬───┬───┐│ │ A │ C │ G │ T │├───┼───┼───┼───┼───┤│ A │ 4 │-3 │ 1 │-3 │├───┼───┼───┼───┼───┤│ C │-3 │ 4 │-3 │ 1 │├───┼───┼───┼───┼───┤│ G │ 1 │-3 │ 4 │-3 │├───┼───┼───┼───┼───┤│ T │-3 │ 1 │-3 │ 4 │└───┴───┴───┴───┴───┘
4: Residuos iguales1: Residuos del mismo tipo
purinas (AG), pirimidinas (CT)-3: Combinaciones restantes.
E= -0,26= 0,25x0,25 [4x4+ 1x4 +(-3)x8]
SecH G T C C G A C T A G T GSecV C A T C G G A G C T G Puntos 1 4 4 -3 4 4 -3 1 -3 4 Acum. 1 5 9 6 10 14 11 12 9 13
Ejemplo Global

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 27
Comparación Global vs. LocalEsquema de Pesos┌───┬───┬───┬───┬───┐│ │ A │ C │ G │ T │├───┼───┼───┼───┼───┤│ A │ 4 │-3 │ 1 │-3 │├───┼───┼───┼───┼───┤│ C │-3 │ 4 │-3 │ 1 │├───┼───┼───┼───┼───┤│ G │ 1 │-3 │ 4 │-3 │├───┼───┼───┼───┼───┤│ T │-3 │ 1 │-3 │ 4 │└───┴───┴───┴───┴───┘
Ejemplo LocalRecorrido exhaustivo de las diagonales
Fragmento con puntuación acumulada máximaNo se puede incrementar estirando ni recortando por los extremos
Inicia en Celda positiva Avanza acumulando valores (mientras acum.>0). Fin: Fin de diagonal: Acumulado <0 => regresar al máximoNo incorpora zonas no-conservadas
G T C C G A C T A G T GC (1) (4) (4 (4) (1 (1A (1 1 (4 4 (1 1)=2T 4 (1 (1) -3 1 (4 -3 4)=5C 1 4 4 4)=6 1 -3 1G (4) -3 4 (1) 1 4)=5 4)=7G (4) 4 1)=10 (1 4 (4)A (1) (1 4)=14 (4 1)=2 -3 (1)G (4 (4) 1 (1) 4 4-)=1
2C 1 (4 (4) 4 (1) 1)=9T (4) 1)=6 1 (1) 4 (4G (4) 4)=9 (1) 1)=11 (4) 4)=8

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 28
Comparación Global vs. LocalSecH G T C C G A C T A G T G SecV C A T C G G A G C T G Puntos 1 4 4 -3 4 4 -3 1 -3 4 Global 1 5 9 6 10 14 11 12 9 13 Local 1 5 9 6 10 14 1 4 Sem. G 3 <- -- -- -- -- -> <> <>
Las interrupciones o GapsPodemos aumentar el parecido deformando lo que comparamos¿Penalizarlos?
X: TCAG-ACG-ATTG TCAGACGATTG|| | | | | | || || | | |
Y: ATC-GGA-GC-T-G ATCGGA-GCT-G

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 29
Comparación Global vs. LocalCada continuidad de las coincidencias a lo largo de cualquier diagonal es un alineamiento LOCALLOCALPara un alineamiento GLOBAL se deben conectar los diversos alineamientos locales y definir una trayectoria que representa el alineamiento a lo largo de toda las secuencias. Esto implica definir explícitamente inserciones y delecciones.Pueden ser posible varias trayectorias, cada una de las cuales representa una solución distinta y posible del alineamiento

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 30
Comparación Global vs. LocalCada continuidad de las coincidencias a lo largo de cualquier diagonal es un alineamiento LOCALPara un alineamiento GLOBALGLOBAL se deben conectar los diversos alineamientos locales y definir una trayectoria que representa el alineamiento a lo largo de toda las secuencias. Esto implica definir explícitamente inserciones y delecciones.Pueden ser posible varias trayectorias, cada una de las cuales representa una solución distinta y posible del alineamiento

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 31
FASTA
Lipman & Pearson, 1985, Pearson & Lipman 1988
Reducir el espacio de búsqueda¿En qué diagonales es más probable que se encuentre el mejor alineamiento?Método:
Etapa I: Buscar regiones (diagonal) por identidad y sin GapsUso de k-tuplas para acelerar (palabras long. k iguales)Resultado: Las mejores diagonales (10)Complejidad: de O (N2) a O (N+M)
Etapa II: Re-evaluación de las regiones por semejanzaUnión de regiones con Gaps
Etapa III: Evaluación exhaustiva de las mejores secuencias

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 32
BLASTBasic Local Alignment Search Tool (BLAST): herramienta de alineamiento local básica (Altschul, S.F., Gish W., Miller W., MyersE.W., and Lipman D.J. J. Mol. Biol. (1990) 215:403-10)
Identificación de las mejores diagonales utilizando criterios de semejanza
Método:Uso de k-tuplas por semejanza (amplía el espacio de búsqueda de FASTA)Corte estadístico (por baja probabilidad de ocurrencia):
1. Identificación rápida de segmentos (MSP: maximal segment pair)Segmento: sub-secuencia continua de cualquier longitudPuntaje: Suma de la semejanza de cada par
2. Análisis detallado de los MSPs con mas probabilidad de formar parte del alineamiento final (umbral T para eliminar efecto debido al azar)
Tabla Hash (long=w) de Sq con Score > T (umbral de SS)La long de la tabla es función de w y T

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 33
Nuevas herramientas
Gapped BLAST y PSI-BLAST
A new Generation of Protein DB search Programs (Altschul, S.F., Madden T.L., Schaffer A.A., Zhanng J., Zhang Z., Miller W., and LipmanD.J. Nucleid Acids Research (1997) v.25, n.17 3389-3402)
Mejoras sobre la versión originalMétodo de dos coincidencias.
Exige la presencia de dos palabras (no solapen, misma diagonal, distancia A entre ellas) para recuperar sensitividad, disminuir T
Segmentos interrumpidos ¿Dos fragmentos forman el mismo alineamiento?Programación dinámica limitando el ancho de ventanaAlineamientos alternativos que no disminuyen la puntuación más de Xg
Búsqueda iterativa Construcción de una matriz de pesos específica por posición

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 34
Clasificación de las aplicaciones
n=1, m=1: comparación de parejas de secuencias (pairwisecomparison) para determinar la semejanza entre ellas. Exhaustiva. Programas: GAP, BESTFIT, BLAST2SEQ
n=1, m>>1: búsqueda en bases de datos para estudiar una secuencia problema obtenida en el laboratorio e inferir algunas propiedades. Rápida, consume tiempo de calculo. Programas: FASTA, BLAST
n>1, m>1: alineamiento múltiple, varias contra varias para tener el caso intermedio a los dos anteriores. Primer paso en los procesos de clasificación de secuencias en familias y grupos o para estudios de filogenia. Programas: PILEUP, CLUSTALW, T-COFFEE.

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 35
Alineamiento múltiple de secuencias
Para tres secuencias

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 36
Alineamiento múltiple de secuencias. ClustalW
Improving the sensitivity of progressive multiple sequence alignment through sequence weighting position specific gap penalties and
weight matrix choice. Thomson JD, Higgins DG, Gibson TJ (1994) Nucleid Acids Research 22 (22) 4673-4680
El algoritmo comienza realizando una comparación todos contra todos en parejasEl par más similar de secuencias se alinea y se considera desde entonces como un grupo o clusterSe trata ese cluster como si fuera una sola secuencia, y se repite el proceso hasta que se alinean todas las secuenciasDisponible como servidor o en modo local

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 37
A partir de este tipo de estudios
El uso de este tipo de comparaciones múltiples permite agrupar proteínas en base a su semejanza de secuencia formando cluster o familiasObservación: En proteínas de la misma familia podemos detectar pequeñas regiones conservadas llamadas motivos motivos ((motifsmotifs)), a menudo asociadas a su función. Ej. sitios de unión, centros activos de enzimas, etc.La semejanza de la secuencia puede ser perceptible solamente en bloques de alineamiento múltiple de secuencias en fragmentos que son importantes desde el punto de vista funcional o estructuralCaracterísticas: La conservación no es perfecta. No son detectables mediante técnicas de homología de secuencia completa (BLAST, FASTA, etc).

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 38
A partir de este tipo de estudios
La divergencia está restringidaDebe existir alguna presión para mantener ciertas funciones
Estructura
Función
SecuenciaSecuencias similares de aminoácidos deben plegarse
dando estructuras también similares
Y proteínas con similares estructuras deben realizar funciones también similares
Devos and Valencia (2000)

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 39
Motivos proteicos
Definición: pequeñas zonas conservadasLos motivos se conservan incluso a grandes distancias evolutivas debido a restricciones estructurales o funcionalesEstán relacionados con la función por tanto se conservanPermiten hacer predicciones al ser detectar homólogos remotosSe han desarrollado bases de datos de motivos y herramientas para búsqueda de motivos en secuencias o búsqueda de secuencias con un cierto motivo.

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 40
Motivos proteicos

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 41
Secuencias consenso y patrones
De la información obtenida en el alineamiento múltipleSecuencias consenso
AGTVATVSCAGTSATHACIGRCARGSCIGEMARLACIGDYARWSC.........IGTVARVSC <= Ejemplo de secuencia consenso
Patrones o expresiones regulares (para caracterizar motivos)ALRDFATHDDFSMTAEATHDSIECDQAATHEASA-T-H-[DE]

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 42
Sintaxis de expresiones regulares o patrones
Cualquier aminoácido: xAmbigüedades:
A o B o …: [AB…]Ni A, ni B, ni …: {AB…}
Repeticiones: A(2,4) el paréntesis indica el rango de repeticiones que pueden darse, en este caso, AA, AAA o AAAAExtremo N-terminal: <Extremo C-terminal: >
Ejemplo: [A,C]-x-V-x(4)-{E,D}Ala o Cys-cualquiera-Val-cualquiera-cualquiera-cualquiera-cualquiera-cualquiera menos Glu o Asp

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 43
Otro ejemploAGTVATVSCAGTSATHACIGRCARGSCIGEMARLACIGDYARWSC.........IGTVARVSC <= Ejemplo de secuencia consenso
[AI]-G-x-x-A-[RT]-x-[AS]-C <= patrón
Problemas y condicionantesSe construyen de manera más o menos subjetivamente, comprobación por ensayo-errorDeben tener una alta sensibilidad y especificidad
Equilibrio: suficientemente cortos para sensibilidadsuficientemente largos para especificidad
Hay algunos métodos automáticos (PRATT del EBI)Bases de datos: PrositeCualquier región que no se ajuste no se detectará

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 44
Perfiles o matrices de pesoSon tablas de pesos o puntuaciones de aa en posiciones específicas y del coste de deleccionesMás sensibles que las expresiones regularesSe da información de todos los aas posibles no sólo de los más probables
A -18 -10 -1 -8 8 -3 3 -10 -2 -8 C -22 -33 -18 -18 -22 -26 22 -24 -19 -7 D -35 0 -32 -33 -7 6 -17 -34 -31 0 E -27 15 -25 -26 -9 23 -9 -24 -23 -1 F 60 -30 12 14 -26 -29 -15 4 12 -29 G -30 -20 -28 -32 28 -14 -23 -33 -27 -5 H -13 -12 -25 -25 -16 14 -22 -22 -23 -10 I 3 -27 21 25 -29 -23 -8 33 19 -23 K -26 25 -25 -27 -6 4 -15 -27 -26 0 L 14 -28 19 27 -27 -20 -9 33 26 -21 M 3 -15 10 14 -17 -10 -9 25 12 -11 N -22 -6 -24 -27 1 8 -15 -24 -24 -4 P -30 24 -26 -28 -14 -10 -22 -24 -26 -18 Q -32 5 -25 -26 -9 24 -16 -17 -23 7 R -18 9 -22 -22 -10 0 -18 -23 -22 -4 S -22 -8 -16 -21 11 2 -1 -24 -19 -4 T -10 -10 -6 -7 -5 -8 2 -10 -7 -11 V 0 -25 22 25 -19 -26 6 19 16 -16 W 9 -25 -18 -19 -25 -27 -34 -20 -17 -28 Y 34 -18 -1 1 -23 -12 -19 0 0 -18
Usa pesos discriminatorios no solo para los aas que aparecen. Para los que no aparecen se basa en las frecuencias observadas en proteínas que poseen el dominio y en la probabilidad de que el aa sea sustituido
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E C I I Q F K L L G N V L V C
A tiene más baja probabilidad que M que, aunque no aparece, sabemos que M es fisicoquímicamente más similar a L, I, V y F que síaparecen
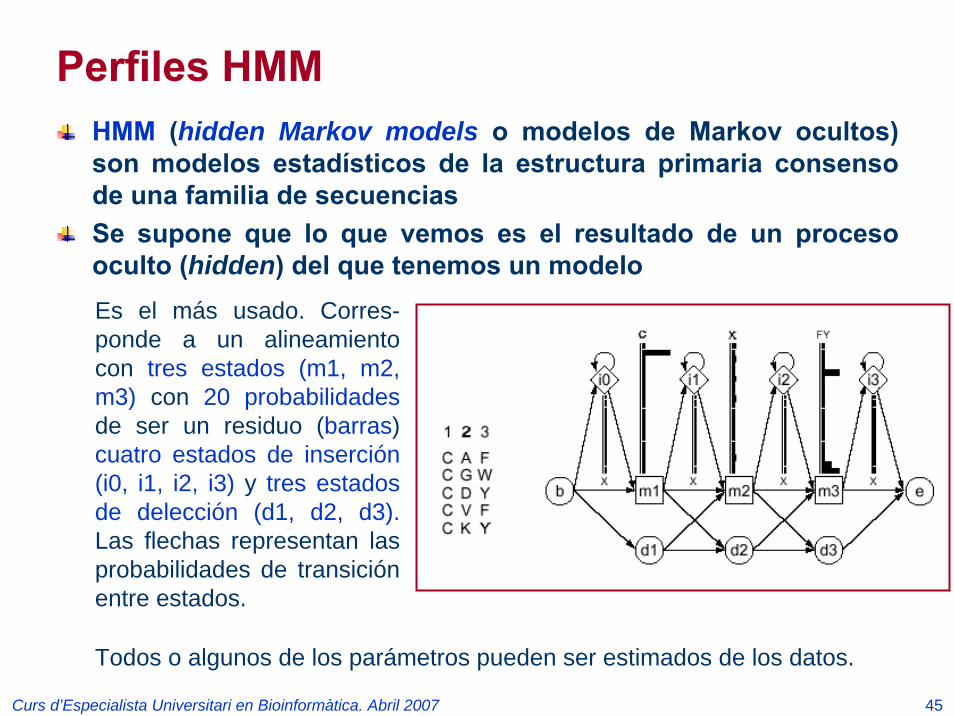
Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 45
Perfiles HMMHMM (hidden Markov models o modelos de Markov ocultos) son modelos estadísticos de la estructura primaria consenso de una familia de secuenciasSe supone que lo que vemos es el resultado de un proceso oculto (hidden) del que tenemos un modeloEs el más usado. Corres-ponde a un alineamiento con tres estados (m1, m2, m3) con 20 probabilidadesde ser un residuo (barras) cuatro estados de inserción (i0, i1, i2, i3) y tres estados de delección (d1, d2, d3).Las flechas representan las probabilidades de transición entre estados.
Todos o algunos de los parámetros pueden ser estimados de los datos.

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 46
Bases de datos de motivos
PROSITE: Expresiones regulares y perfiles. Basada en motivos conocidos (SwissProt) [PPSearch]BLOCKS: Perfiles. Basada en PROSITE.PRINTS: Perfiles. Basada en motivos conocidos.Pfam: Perfiles HMM. Generación automática de motivos. SwissProt + SP-TrEMBL. Obtenidos directamente de alineamientos. En algunos casos no se sabe su significado.
PROSITEBLOCKS PRINTS
PfamInformación
Swis
sPro
t SP-
TrEM
BL
Expresionesregulares
PrecisiónPerfiles
simples múltiples HMMs

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 47
ID Identification (Begins each entry; 1 per entry) AC Accession number (1 per entry)DT Date (1 per entry)DE Short description (1 per entry)PA Pattern (>=0 per entry)MA Matrix/profile (>=0 per entry)RU Rule (>=0 per entry)NR Numerical results (>=0 per entry)CC Comments (>=0 per entry)DR Cross-references to SWISS-PROT (>=0 per entry)3D Cross-references to PDB (>=0 per entry)DO Pointer to the documentation file (1 per entry)// Termination line (Ends each entry; 1 per entry)
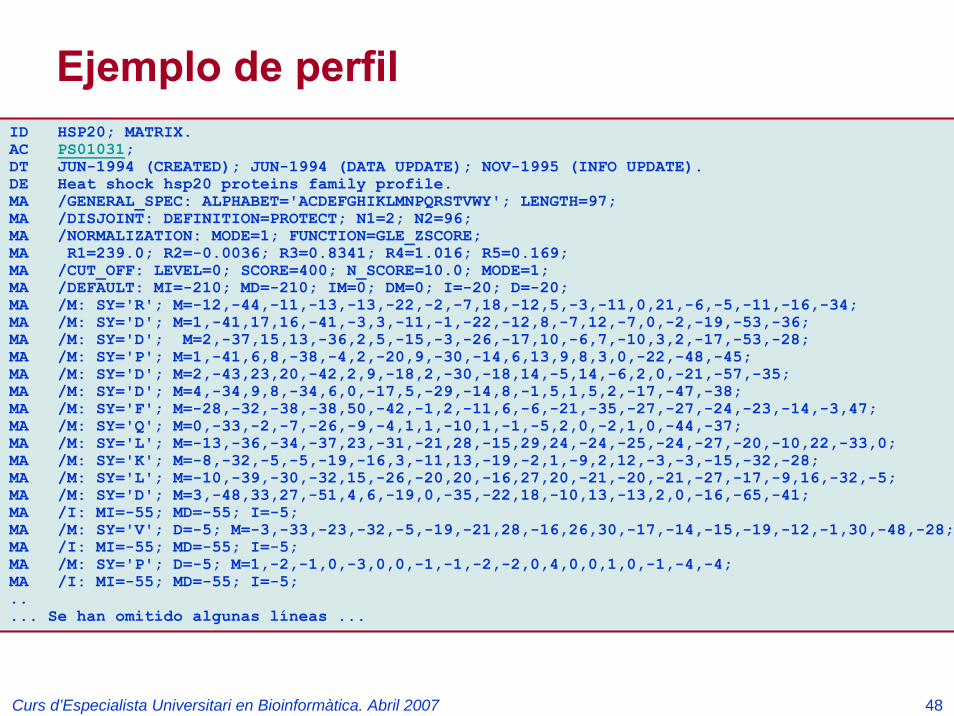
Ejemplo de patrón
ID T4_DEIODINASE; PATTERN.AC PS01205;DT NOV-1997 (CREATED); JUL-1999 (DATA UPDATE); JUL-1999 (INFO UPDATE).DE Iodothyronine deiodinases active site.PA R-P-L-[IV]-x-[NS]-F-G-S-[CA]-T-C-P-x-F.NR /RELEASE=40.7,103373;NR /TOTAL=16(16); /POSITIVE=16(16); /UNKNOWN=0(0); /FALSE_POS=0(0);NR /FALSE_NEG=0; /PARTIAL=0;CC /TAXO-RANGE=??E??; /MAX-REPEAT=1;CC /SITE=12,active_site;DR P49894, IOD1_CANFA, T; O42411, IOD1_CHICK, T; P49895, IOD1_HUMAN, T;DR Q61153, IOD1_MOUSE, T; O42449, IOD1_ORENI, T; P24389, IOD1_RAT , T;DR P79747, IOD2_FUNHE, T; Q92813, IOD2_HUMAN, T; Q9Z1Y9, IOD2_MOUSE, T;DR P49896, IOD2_RANCA, T; P70551, IOD2_RAT , T; O42412, IOD3_CHICK, T;DR P55073, IOD3_HUMAN, T; P49898, IOD3_RANCA, T; P49897, IOD3_RAT , T;DR P49899, IOD3_XENLA, T;DO PDOC00925;//
Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 48
Ejemplo de perfilID HSP20; MATRIX.AC PS01031;DT JUN-1994 (CREATED); JUN-1994 (DATA UPDATE); NOV-1995 (INFO UPDATE).DE Heat shock hsp20 proteins family profile.MA /GENERAL_SPEC: ALPHABET='ACDEFGHIKLMNPQRSTVWY'; LENGTH=97;MA /DISJOINT: DEFINITION=PROTECT; N1=2; N2=96;MA /NORMALIZATION: MODE=1; FUNCTION=GLE_ZSCORE;MA R1=239.0; R2=-0.0036; R3=0.8341; R4=1.016; R5=0.169;MA /CUT_OFF: LEVEL=0; SCORE=400; N_SCORE=10.0; MODE=1;MA /DEFAULT: MI=-210; MD=-210; IM=0; DM=0; I=-20; D=-20;MA /M: SY='R'; M=-12,-44,-11,-13,-13,-22,-2,-7,18,-12,5,-3,-11,0,21,-6,-5,-11,-16,-34;MA /M: SY='D'; M=1,-41,17,16,-41,-3,3,-11,-1,-22,-12,8,-7,12,-7,0,-2,-19,-53,-36;MA /M: SY='D'; M=2,-37,15,13,-36,2,5,-15,-3,-26,-17,10,-6,7,-10,3,2,-17,-53,-28;MA /M: SY='P'; M=1,-41,6,8,-38,-4,2,-20,9,-30,-14,6,13,9,8,3,0,-22,-48,-45;MA /M: SY='D'; M=2,-43,23,20,-42,2,9,-18,2,-30,-18,14,-5,14,-6,2,0,-21,-57,-35;MA /M: SY='D'; M=4,-34,9,8,-34,6,0,-17,5,-29,-14,8,-1,5,1,5,2,-17,-47,-38;MA /M: SY='F'; M=-28,-32,-38,-38,50,-42,-1,2,-11,6,-6,-21,-35,-27,-27,-24,-23,-14,-3,47;MA /M: SY='Q'; M=0,-33,-2,-7,-26,-9,-4,1,1,-10,1,-1,-5,2,0,-2,1,0,-44,-37;MA /M: SY='L'; M=-13,-36,-34,-37,23,-31,-21,28,-15,29,24,-24,-25,-24,-27,-20,-10,22,-33,0;MA /M: SY='K'; M=-8,-32,-5,-5,-19,-16,3,-11,13,-19,-2,1,-9,2,12,-3,-3,-15,-32,-28;MA /M: SY='L'; M=-10,-39,-30,-32,15,-26,-20,20,-16,27,20,-21,-20,-21,-27,-17,-9,16,-32,-5;MA /M: SY='D'; M=3,-48,33,27,-51,4,6,-19,0,-35,-22,18,-10,13,-13,2,0,-16,-65,-41;MA /I: MI=-55; MD=-55; I=-5;MA /M: SY='V'; D=-5; M=-3,-33,-23,-32,-5,-19,-21,28,-16,26,30,-17,-14,-15,-19,-12,-1,30,-48,-28;MA /I: MI=-55; MD=-55; I=-5;MA /M: SY='P'; D=-5; M=1,-2,-1,0,-3,0,0,-1,-1,-2,-2,0,4,0,0,1,0,-1,-4,-4;MA /I: MI=-55; MD=-55; I=-5;..... Se han omitido algunas líneas ...

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 49
Ejemplo de perfil (cont.)
..MA /M: SY='K'; M=-11,-52,1,-1,-1,-17,2,-18,43,-28,3,9,-10,8,33,-2,-1,-23,-33,-43;MA /I: MI=*; MD=*; I=0;NR /RELEASE=40.7,103373;NR /TOTAL=181(180); /POSITIVE=176(175); /UNKNOWN=5(5); /FALSE_POS=0(0);NR /FALSE_NEG=0; /PARTIAL=4;CC /MATRIX_TYPE=protein_domain;CC /SCALING_DB=reversed;CC /AUTHOR=P_Bucher;CC /TAXO-RANGE=A?EP?; /MAX-REPEAT=2;DR P30223, 14KD_MYCTU, T; P46729, 18K1_MYCAV, T; P46730, 18K1_MYCIT, T;DR P46731, 18K2_MYCAV, T; P46732, 18K2_MYCIT, T; P12809, 18KD_MYCLE, T;DR P80485, ASP1_STRTR, T; O30851, ASP2_STRTR, T; P02497, CRA2_MESAU, T;DR P24622, CRA2_MOUSE, T; P24623, CRA2_RAT , T; P15990, CRA2_SPAEH, T;..... Some lines omitted....DR P96193, IBPB_AZOVI, T; P29210, IBPB_ECOLI, T; P29778, OV21_ONCVO, T;DR P29779, OV22_ONCVO, T; Q06823, SP21_STIAU, T; P34328, YKZ1_CAEEL, T;DR P12812, P40_SCHMA , T;DR P81083, HS11_PINPS, P; P81161, HS2M_LYCES, P; P30220, HS3E_XENLA, P;DR Q9QUK5, HSB7_RAT , P;DR Q29438, ODFP_BOVIN, ?; Q14990, ODFP_HUMAN, ?; Q61999, ODFP_MOUSE, ?;DR Q29077, ODFP_PIG , ?; P21769, ODFP_RAT , ?;DO PDOC00791;
PS01031
Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 50
Homología, ortología y paralogíaLa semejanza o la identidad es un parámetro cuantificableLa homología no es cuantificable y denota ascendencia comúnDos proteínas son ortólogas si sus diferencias se presentaron durante el proceso de especiación. Se asume que para tener una función igual o muy similar (ortólogo = de la misma subfamilia)Dos proteínas son parálogas si son codificadas por los genes originados por un proceso de duplicación de gen
Especie 1gen A
Especie 2
gen A
Especie 0
gen A
Especiación
gen Agen B
Duplicacióndel gen
Ortólogos
Ortólogos
Parálogos
Ortólogos

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 51
In-paralogs y out-paralogs
Muchas veces las duplicaciones no dan lugar a nuevas proteínas con nuevas funciones, sino que los genes duplicados conservan su función y siguen perteneciendo al grupo original. in-parálogos: se usa para referirnos a las relaciones entre genes que proceden de duplicaciones recientes y conservan las características de la proteína de la que proceden. out-parálogos: en este caso ya no se conservan las características de la proteína de la que proceden.
Ejemplo: en el ser humano hay diversas copias del gen de ras, implicado en transducción de señales. Estas proteínas conservan más o menos la función original, aunque cada una se expresa en distintos tejidos, bajo distintas condiciones. Son in-parálogos. Por contra, las proteínas rab son parientes de las ras, y son out-parálogos.

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 52
Homólogos: superfamilias, familias y subfamilias.
Una superfamilia es un conjunto de proteínas con un origen evolutivo común, un conjunto de homólogos. Las superfamilias se pueden dividir, más o menos arbitrariamente, según lo grandes que sean, en familias y subfamilias. Son conceptos paralelos a los de ortólogos y parálogos: las proteínas de una misma subfamilia son ortólogas entre sí (también puede haber in-paralogs), mientras que son parálogas de las de otras subfamilia que pertenezca a la misma superfamilia.
Superfamilia: grupo de proteínas con un origen común
Familia/Subfamilia: grupo de proteínas con una función común (jerarquía subjetiva)

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 53
Ejemplo
F. Abascal. UCM-CNB

Curs d’Especialista Universitari en Bioinformàtica. Abril 2007 54
Superfamilias y familias
Suflé de dominios (domain shuffling)
Las proteínas homólogas pueden tener diferente organización de dominiosEl dominio y no el gen es la unidad evolutiva básicaLa función de una proteína es el resultado de las funciones de sus dominiosLas propiedades de una proteína pueden ser explicadas pero no deducidas a partir de sus dominios