material complementario técnicasbiotecnologiaindustrial.fcen.uba.ar/wp-content/... ·...
TRANSCRIPT

- 1 -
Planta de Bioprocesos Industriales- INTI
2017
Material complementario
Técnicas
- 2 -
Cuantificación de proteínas totales por el método de BRADFORD
Objetivo y fundamento del método.
Se utiliza la técnica de Bradford con el objetivo de cuantificar las proteínas totales en
muestras como las de ruptura celular y las de fracciones cromatográficas, entre otras. Esta
técnica se ha convertido en el método preferido para la cuantificación de proteínas ya que
es más simple, rápido y más sensible que el método de Lowry.
El método de Bradford se basa en la unión del colorante Coomassie Blue G250 a las
proteínas. Donde la cantidad de proteína es proporcional a la coloración resultante.
Aunque el colorante libre puede existir en cuatro formas iónicas diferentes (cuyos
valores de pKa son 1,15, 1,82 y 12,4), las tres formas cargadas que predominan en la
solución acidificada del ensayo son las formas catiónicas roja y verde tienen una
absorbancia máxima a 470 nm y 650 nm respectivamente. En contraste, la forma más
aniónica del colorante, la que se una a las proteínas, tiene una absorbancia máxima a 590
nm. Así la cantidad de proteína puede ser estimada si se determina la cantidad de
colorante obtenido en su forma iónica azul. Esto se logra usualmente midiendo la
absorbancia de la solución a 595 nm.
Procedimiento.
Curva de calibración.
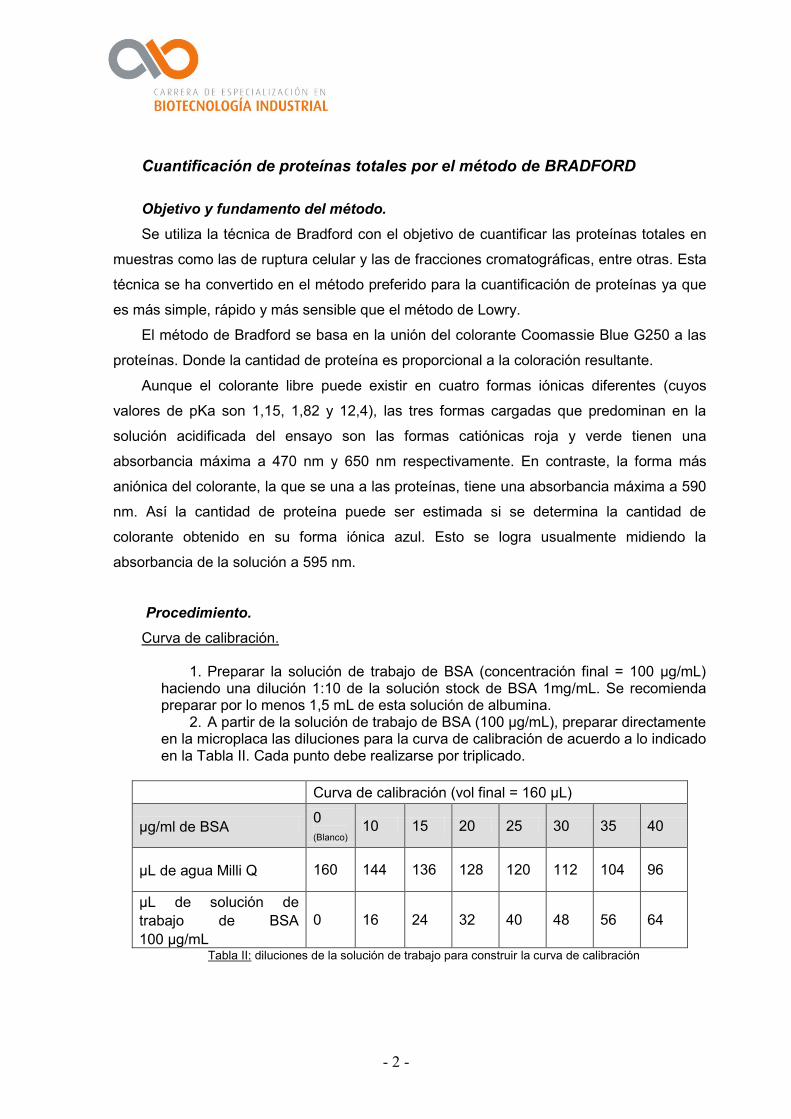
1. Preparar la solución de trabajo de BSA (concentración final = 100 µg/mL) haciendo una dilución 1:10 de la solución stock de BSA 1mg/mL. Se recomienda preparar por lo menos 1,5 mL de esta solución de albumina.
2. A partir de la solución de trabajo de BSA (100 µg/mL), preparar directamente en la microplaca las diluciones para la curva de calibración de acuerdo a lo indicado en la Tabla II. Cada punto debe realizarse por triplicado.
Curva de calibración (vol final = 160 µL)
µg/ml de BSA 0
(Blanco) 10 15 20 25 30 35 40
µL de agua Milli Q 160 144 136 128 120 112 104 96
µL de solución de
trabajo de BSA
100 µg/mL
0 16 24 32 40 48 56 64
Tabla II: diluciones de la solución de trabajo para construir la curva de calibración

- 3 -
Muestra.
Preparar varias diluciones de la muestra (con agua) de modo de obtener alguna cuya
concentración de proteínas totales quede incluida en el rango de la curva de calibración.
Realizar estas diluciones en un tubo eppendorf aparte y de cada una, alicuotar 160 µL
por pocillo; y a su vez, ensayar cada muestra por triplicado. Otra opción es realizar por
triplicado la dilución final directamente sobre la placa, considerando que el volumen final es
de 160 µL.
Reacción.
1. Agregar 30 µl del Reactivo de Bradford a cada well (a cada punto de la
curva de calibración, cada muestra y sus respectivas diluciones) utilizando la micropipeta multicanal.
2. Homogeneizar con la micropipeta multicanal evitando originar burbujas. 3. Incubar por 5 minutos a temperatura ambiente.
4. Leer en la lectora de microplacas a una longitud de onda de 595 nm. Cálculos.
Se realiza una curva de calibración con los datos obtenidos, graficando Abs 595 vs.
concentración de BSA. A partir de ésta, se obtiene la ecuación de la recta que mejor
ajusta, y luego se interpolan los valores de absorbancia obtenidos para las muestras. Si la
muestra fue diluida, será necesario multiplicar dicho resultado por el factor de dilución.
Criterios de aceptación.
- El ensayo se considera válido cuando el coeficiente de variación (C.V. %) de la
regresión de la curva es mayor al 98%.
- El C.V. % de cada punto de la curva patrón debe ser menor o igual a 15%
- Si alguna de estas condiciones no se cumpliera, se podrán eliminar hasta 2 puntos
correspondientes a triplicados independientes de la curva.
- El C.V. % de los triplicados de las muestras, debe ser menor o igual a 15%. Si esto
último no se cumpliera, se puede eliminar un punto de las tres lecturas independientes de
las muestras.

- 4 -
Electroforesis desnaturalizante en gel de Poliacrilamida (SDS-PAGE)
Objetivo y fundamento del método.
El SDS-PAGE es el método que se utiliza para analizar mezclas de proteínas
cuantitativamente y se basa en la separación de proteínas de acuerdo a su tamaño.
Existen varios métodos de electroforesis en geles de poliacrilamida (o PAGE del inglés
Polyacrylamide Gel Electrophoresis). Entre las diferentes variantes de ésta técnica, la
electroforesis en geles discontinuos de poliacrilamida con dodecil sulfato de sodio o SDS-
PAGE (del inglés de Sodium Dodecyl Sulphate-PolyAcrylamide Gel Electrophoresis)
descripta originalmente por Laemmli (1), es el sistema más comúnmente usado. Esta
técnica se utiliza para el análisis de proteínas mediante la separación según su relación
carga/masa dentro de una matriz de acrilamida.
Dado que las proteínas nativas tienen distintas formas, pesos moleculares y cargas,
se utiliza la desnaturalización mediante el tratamiento con agentes reductores (como el -
mercaptoetanol) y agentes caotrópicos (como el detergente dodecil sulfato de sodio ó
SDS) de modo que las proteínas conserven solamente su estructura primaria y queden con
carga negativa proporcional a su tamaño. Estas proteínas sometidas a un campo eléctrico
se desplazan a una velocidad proporcional a su relación entre carga y masa.
Cuando las proteínas se solubilizan en presencia del detergente aniónico SDS, éste se
une a las proteínas, rompiendo interacciones hidrofóbicas y desnaturalizándolas. Las
proteínas desnaturalizadas de la muestra adoptarán una estructura en forma de bastoncillo
con una serie de moléculas de SDS cargadas negativamente a lo largo de la cadena
polipeptídica.
Debido a que la cantidad de SDS que se une a las proteínas es prácticamente
proporcional a su tamaño, los complejos SDS-proteínas presentan un valor carga/masa
constante y por lo tanto se separan de acuerdo a su tamaño cuando migran desde el
cátodo al ánodo a una velocidad relacionada con su peso molecular.
El sistema aquí utilizado es discontinuo, donde la composición del gel concentrador y
el gel separador son heterogéneos. Es decir, se varían el tamaño de poro y el pH de modo
que se producir el apilamiento de la muestra, para lograr una mejor resolución de las
bandas.

- 5 -
Mediante esta técnica se puede determinar el peso molecular de las proteínas de una
muestra, comparando su movilidad relativa en el gel con la de un estándar (una mezcla de
proteínas de peso molecular conocido) o marcador de peso molecular.
En este Trabajo Práctico, se analizarán mediante esta técnica, muestras de cultivo
para verificar la inducción de la expresión de la proteína, muestras de la ruptura celular y
de la purificación mediante FPLC.
Procedimiento.
Armado del cassette de gel
1. Corroborar la limpieza de las placas de vidrio: la placa de vidrio corto (y fino) y la placa de vidrio espaciadora (mas gruesa y con los espaciadores). Repasar los vidrios con etanol 96% y papel.
2. Armar el molde del vidrio ubicando las placas de manera tal que la placa de vidrio espaciadora quede detrás de la placa corta, y en la placa espaciadora se pueda leer la palabra “UP” del lado superior.
3. Colocar el marco de molde hacia arriba con ambas aletas abiertas, es decir,
con ambas aletas en posición perpendicular con respecto al marco. Introducir ambas placas de vidrio.

- 6 -
4. Cerrar ambas aletas, llevándolas simultáneamente hacia afuera. Realizar
esta operación sobre una superficie plana, controlando que parte inferior de las placas de vidrio estén niveladas entre sí y con respecto a la base del marco de molde.
5. Colocar el marco de molde con los vidrios ensamblados en el soporte del
molde, asegurándose de que el espacio entre los vidrios quede sellado por la junta gris de la parte inferior del soporte.
6. Llenar los vidrios con agua y controlar de que no se produzca ninguna
pérdida por la junta. Si esto ocurriese, repetir el ensamblado del casette y volver a realizar esta prueba con agua.
7. Vaciar el molde de los vidrios sólo Si NO se observan pérdidas de agua, y secar el espacio entre ambas placas de vidrio con tiras de papel secante (procurar que no queden residuos del papel entre los vidrios).

- 7 -
Preparación de los geles
- Gel separador al 16%:
Las proteínas deben correr primero a través del gel concentrador y luego por el gel
separador, para ello, en primer lugar se prepara (y se coloca entre los vidrios del molde) el
gel separador; y por último el gel concentrador.
1. Preparar la mezcla del gel separador colocando los reactivos de a cuerdo a
los volúmenes y en el orden en que se presentan en la siguiente tabla.
Gel 16%
1 gel 2 geles 4 geles
1) Agua destilada 0,95 mL 1,9 mL 3,8 mL
2) Tris-HCl 1,5M pH 8,8 1,25 mL 2,5 mL 5 mL
3) SDS 10% 50 µL 100 µL 200 µL
4) Acrilamida 30% 2,7 mL 5,4 mL 10,8 mL
5) APS 10% 50 µL 100 µL 200 µl
6) TEMED 3,5 µL 7,0 µL 14,0 µL
Volumen final 5 mL 10 mL 20 mL
NOTA: Los geles se forman por la polimerización del monómero de acrilamida en presencia de pequeñas
cantidades de bis-acrilamida como cross-linker. Dicha polimerización es un ejemplo de catálisis por radicales libres. Es el TEMED el que cataliza la descomposición del ión persulfato (APS) para dar un radical libre. Por lo tanto, al agregar el TEMED comenzará la reacción de polimerización (reacción exotérmica). A partir de este momento se cuenta con tiempo limitado para el vaciado del gel entre los vidrios.
2. Colocar la mezcla del gel entre los vidrios utilizando una pipeta automática de 5 mL. Considerar que deben quedar 3 cm libres hasta el borde del vidrio corto (que es el espacio necesario para luego colocar el gel concentrador y el peine).
3. Agregar cuidadosamente aproximadamente 1 mL de alcohol isobutílico saturado en agua.
4. Dejar reposando hasta que concluya la reacción de polimerización (verificar la polimerización observando el resto de mezcla que quedó en el tubo).
5. Una vez que terminó la reacción, quitar la solución de alcohol de la parte superior del gel. Lavar con agua una o dos veces. Secar cuidadosamente la parte superior entre los vidrios con papel secante, sin dañar el borde superior del gel.
- Gel concentrador (o stacking):
6. Preparar los peines sobre la parte superior de los geles. 7. Preparar la mezcla del gel concentrador añadiendo los reactivos de a cuerdo
a los volúmenes y en el orden en que se presentan en la siguiente tabla.

- 8 -
Gel concentrador
1 gel 2 geles 4 geles
1) Agua destilada 0,95 mL 1,9 mL 3,8 mL
2) Tris-HCl 0,5M pH 6,8 375 µL 750 µL 1,5 mL
3) SDS 10% 15 µL 30 µL 60 µL
4) Acrilamida 30% 200 µL 400 µL 800 µL
5) APS 10% 17,5 µL 35 µL 70 µl
6) TEMED 3,5 µL 7,0 µL 14,0 µL
Volumen final aproximado 1,6 mL 3,1 mL 6,2 mL
NOTA: En este caso la reacción de polimerización es aún más rápida que la que sucede con el gel
separador dada la mayor concentración de catalizador, por lo cual se cuenta con menos tiempo para colocar el gel entre los vidrios.
8. Llenar el espacio entre los vidrios hasta el borde. Inmediatamente luego colocar los peines en la posición correcta. Cuidar que no queden burbujas atrapadas, ya que la base de las calles podrían no quedar rectas.
9. Dejar reposando hasta que concluya la reacción de polimerización (verificar la polimerización observando el resto de mezcla que quedó en el tubo).
Montaje de los geles en la cuba
1. Retirar los casettes del soporte del molde, moviendo simultáneamente las dos aletas hacia el centro.
2. Colocar ambos casettes en el montaje de los electrodos, esta vez con el
vidrio más corto hacia adentro. Registre la posición de cada gel dentro del montaje de electrodos.

- 9 -
3. Colocar los geles en el cuadro de abrazaderas. Cerrar las abrazaderas
presionando con los dedos índices de cada mano el montaje de los electrodos mientras que con los pulgares se cierran las aletas hacia el centro, tal como se observa en el lateral del cuadro de abrazaderas.
4. Colocar los geles y el cuadro de abrazaderas ensamblado en el tanque de
corrida.
5. Verter el buffer de corrida en el espacio que queda entre ambos casettes
hasta que desborde, permitiendo que el espacio entre el cuadro de abrazaderas y el tanque se llene hasta aproximadamente 3- 4 cm. desde del fondo del tanque.
6. Dejar reposar el buffer por unos minutos hasta que le nivel del buffer se estabilice entre los vidrios. Si el nivel del buffer no se estabiliza y no se mantiene

- 10 -
constante retirar el recuadro de abrazaderas sacar los geles y repetir todos los pasos desde el punto 2.
7. Retirar los peines desde parte superior deslizándolos hacia arriba cuidando de no deformar los pocillos de siembra (wells). Completar el nivel de buffer hasta el borde superior de los vidrios entre los geles.
Preparación y siembra de la muestra
1. Acondicionar cada muestra según la cuantificación de proteínas surgida de la cuantificación de Bradford.
Para una correcta visualización con tinción de Coomassie, debe sembrarse una masa de entre 4,5-9 µg de proteína; y el volumen de siembra final debería estar entre 15-20 µl, que es volumen que soporta el well.
2. Crackear las muestras, hirviendo la mezcla de proteínas con buffer muestra a 100° C durante 5 minutos. Pasar a hielo y centrifugar unos segundos de modo de bajar el agua condensada.
3. Colocar la guía para siembra y sembrar dentro de los pocillos. Registrar el orden de siembra para cada calle.
Preparación del equipo e inicio de la corrida
1. Coloque la cubierta del tanque, observando la correspondencia entre los colores de los bornes y los orificios de la cubierta.
2. Conectar los cables, considerando la misma correspondencia observada en el punto anterior.
3. Establecer el voltaje en 100 V e iniciar la corrida (botón “start”). 4. Dejar correr hasta que observar que el frente de corrida salga del gel.
Finalización de la corrida
1. Detener la corrida con el botón de “stop”. 2. Apagar y desconectar la fuente de poder. Retirar la cubierta. 3. Retirar el cuadro de abrazaderas del tanque. 4. Abrir el cuadro de abrazaderas y retirar el montaje de electrodos. 5. Retirar los geles del montaje de electrodos. Mantener identificado cada gel
durante todo el proceso. 6. Eliminar el gel concentrador con la espátula. Identificar el lado de cada gel
(muesca) 7. Lavar el gel con agua de la piseta. Despegarlo cuidadosamente del vidrio
con el chorro de agua y volcarlo en el recipiente para tinción. El gel debe quedar extendido y sin arrugas.

- 11 -
Tinción del gel con Coomassie Coloidal
Una vez finalizada la corrida electroforética se siguen los siguientes pasos:
1- Realizar dos lavados con agua de 3 minutos cada uno. 2- Incubar en un recipiente con solución de coomasie coloidal durante 18 horas a
temperatura ambiente en agitador orbital. 3- Realizar 3 lavados con agua de 5 minutos cada uno. 4- Fotografiar en equipo UVP con fondo de trasluz blanco Inmediatamente. 5- Secar.
Registro y análisis de los resultados
El registro del gel obtenido se realiza fotografiándolo o escaneándolo. Al tenerlo en
formato digital se puede realizar su análisis (por ejemplo: densitometría) por medio de
algún software adecuado.
Para analizar el peso molecular de las proteínas de la muestra, debe sembrarse en el
mismo gel una mezcla de proteínas de peso molecular conocido denominada calibrador o
“marcador de peso molecular”.
Para la mayoría de las proteínas un gráfico de log 10 de la masa molecular vs.
movilidad relativa arroja un grafico lineal, aunque se debe ser conciente para una
determinada concentración de gel esta relación es solamente lineal en un rango limitado a
masas moleculares1.
Para determinar el peso molecular de una proteína desconocida es necesario
establecer la relación entre la movilidad relativa (Rf) de las proteínas del estándar de
proteínas. Para ello, se grafica log del peso molecular (PM) vs. Rf donde:
Rf= distancia que recorrió la proteína
distancia recorrida por el frente
Una vez construido el gráfico y establecida la relación entre movilidad y masa,
conociendo el Rf de las proteínas desconocidas puede ser determinado el PM de la
proteína de interés.
1 Cabe mencionar que ésta relación se mantiene real para proteínas que unen SDS en una proporción constante. Esto es así para muchas proteínas pero algunas otras, por ejemplo aquellas altamente básicas, pueden correr diferente a lo esperado según lo que indica su peso molecular conocido. En el caso de las histonas, las cuales son altamente básicas, migran mas lentamente de lo esperado, presumiblemente por una reducción total de la carga negativa debido a la alta proporción de aminoácidos cargados positivamente. Las glicoproteínas también tienden a correr de forma anormal presumiblemente porque el SDS solo se une a la parte polipeptídica de la molécula.

- 12 -
Conservación
Dado que gel es muy frágil, y se hace difícil su manipulación, es muy conveniente
secar el gel obtenido. Para ello, luego de enjuagar el gel se lo monta sobre papel secante y
se cubre con papel de celofán. Se seca durante 1 hora a 80°C, en la secadora con vacío.
Método de elaboración de coomassie coloidal (g250)
Composición:
Sulfato de amonio 10%
Coomassie G-250 0,1%
Acido orto fosfórico 3,0%
Etanol absoluto 20,0%
Método de Elaboración:
Pesar en bandejas descartables separadas, limpias y secas la cantidad de materiales
según la fórmula del punto 1.1 para preparar el volumen de medio solicitado.
En un recipiente volumétrico de capacidad adecuada colocar la ¾ parte del volumen
de agua purificada necesaria según tabla para realizar la preparación.
Agregar las cantidades de Sulfato de amonio, acido orto fosfórico y etanol absoluto
medidas anteriormente.
Homogeneizar y luego adicionar la cantidad de coomassie G-250 indicada en la tabla
de volúmenes más usados.
Trasvasar la solución a una probeta graduada.
Completar a volumen con agua purificada PW según tabla de volúmenes más usados.
Volúmenes más usados:
Materiales Volumen (ml)
500 1000 2000
Sulfato de amonio (g) 50 100 200
Coomassie G-250(g) 0,5 1,0 2,0
Acido orto fosfórico (ml) 15 30 60
Etanol absoluto (ml) 100 200 400
Agua Purificada (PW) c.s.p 500 1000 2000