hΨ=eΨ h t v - universitat de valència · - ángulo diedro formado por cuatro átomos molécula...
TRANSCRIPT

LQFII Mod. Mol. (01/03/05) pag. 1
Superficie de energía potencial • Energía de una molécula (E).
) H Ψ = EΨ
) H =
) T N +
) T e +
) V ee +
) V Ne +
) V NN
• Aproximación de Born-Oppenheimer.
) H ele (R)Ψele (R) = Eele (R)Ψele (R)
) H ele =
) T e +
) V ee +
) V Ne
• Energía potencial es la energía de una molécula para una posición relativa dada (fija) de los núcleos (R son las coordenadas de los átomos).
V (R) = Eele (R) + VNN (R)
No se puede resolver
La superficie de energía potencial es el conjunto de valores de la energía potencial para todas posiciones relativas de todos los átomos y corresponde a la función V(R).

LQFII Mod. Mol. (01/03/05) pag. 2
Coordenadas internas.
• Hacen uso de variables como:
Ejemplo: H2O
103.720.9421H3
0.9421H2
O1
ánguloRef.
distanciaRef.TipoN. Orden
HH
r31o
r21
Θ312
3 2
1
- distancia entre dos átomos.- ángulo formado por tres átomos- ángulo diedro formado por cuatro átomos
Molécula de H2O
HH
r1o
r2
Θ
La energía potencial dependerá de tres coordenadas

LQFII Mod. Mol. (01/03/05) pag. 3
El ángulo diedro formado por cuatro átomos i-j-k-n se define como el ángulo entre dos planos, uno conteniendo los átomos i,j,k y el otro los átomos j,k,n.
1
2 3
4 Φ = 60º Φ = 120º Φ = 180º Φ = 0º

LQFII Mod. Mol. (01/03/05) pag. 4
4
1
5
23
106.1010.9722H3
C1
-90.03110.4621.1131H5
90.003124.6821.2061O4
1.3361O2
diedroRefánguloRefdistanciaRefTipoOrden
Ejemplo: HCOOH
• ¿Cuántas coordenadas internas son necesarias para definir una molécula de N átomos?
moléculas no lineales 3N-6lineales 3N-5
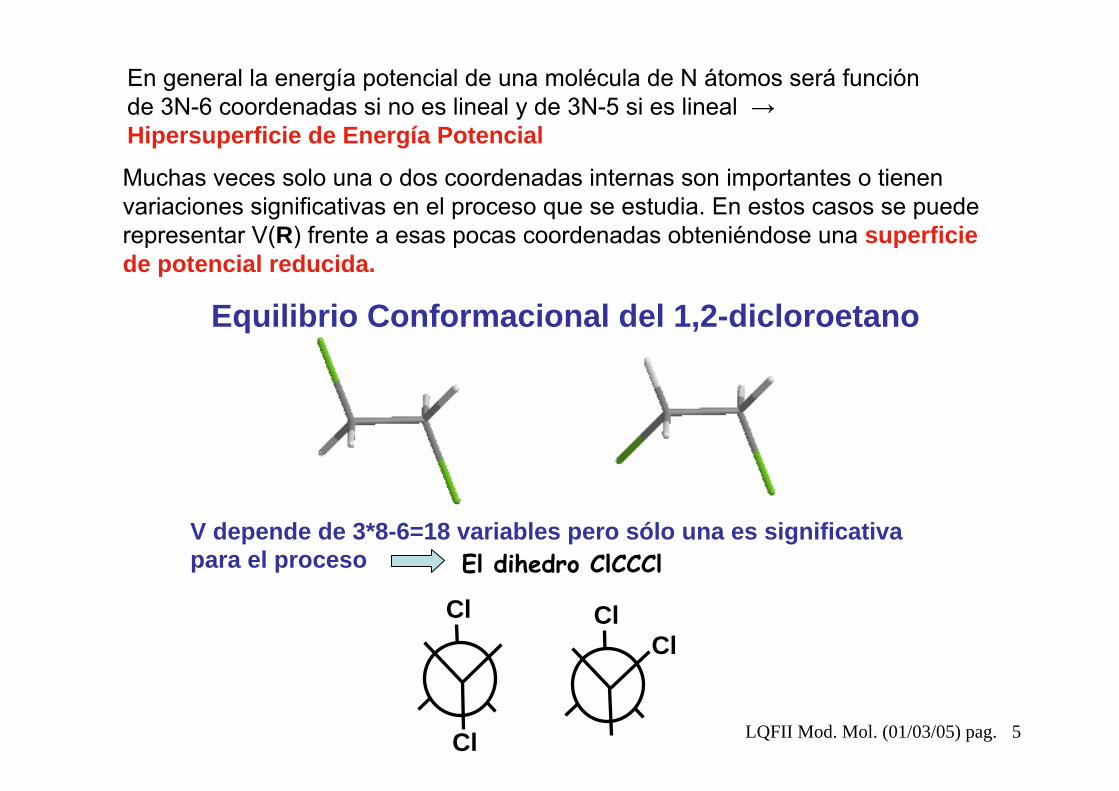
LQFII Mod. Mol. (01/03/05) pag. 5
Muchas veces solo una o dos coordenadas internas son importantes o tienen variaciones significativas en el proceso que se estudia. En estos casos se puede representar V(R) frente a esas pocas coordenadas obteniéndose una superficie de potencial reducida.
V depende de 3*8-6=18 variables pero sólo una es significativa para el proceso El dihedro ClCCCl
Cl
Cl
ClCl
Equilibrio Conformacional del 1,2-dicloroetano
En general la energía potencial de una molécula de N átomos será función de 3N-6 coordenadas si no es lineal y de 3N-5 si es lineal →Hipersuperficie de Energía Potencial

LQFII Mod. Mol. (01/03/05) pag. 6
0
2
4
6
8
10
-180 -120 -60 0 60 120 180
ClCCCl (grados)
Cl
Cl
ClCl
Cl
Cl
ClCl

LQFII Mod. Mol. (01/03/05) pag. 7
Puntos estacionarios de la superficie.
ii R
VF rr
∂∂
−=)(R
De la superficie de energía potencial se puede deducir las fuerzas que sienten los átomos.
• Hay puntos (un conjunto de coordenadas determinado) en la superficie para los que las fuerzas que sienten todos los átomos es cero
ola derivada de V con respecto a cada una de las coordenadas es cero .
A estos puntos se les denomina puntos estacionarios.
* Mínimos
* Máximos
* Puntos de Silla

LQFII Mod. Mol. (01/03/05) pag. 8
0
2
4
6
8
10
-180 -120 -60 0 60 120 180
ClCCCl (grados)
ΔEconfΔE‡
Cl
Cl
ClCl
Cl
Cl
ClCl

LQFII Mod. Mol. (01/03/05) pag. 9
HaHb + HcHa + HbHc
Rbc≈0.74 ÅRab≈2.2 ÅReactivos
Rab≈0.74 ÅRbc≈2.2 ÅProductos

LQFII Mod. Mol. (01/03/05) pag. 10
HaHb + HcHa + HbHc Rab →∞Rbc→∞
Rab ≈0.93 ÅRbc≈0.93 Å

LQFII Mod. Mol. (01/03/05) pag. 11
HaHb + HcHa + HbHc Camino de Reacción
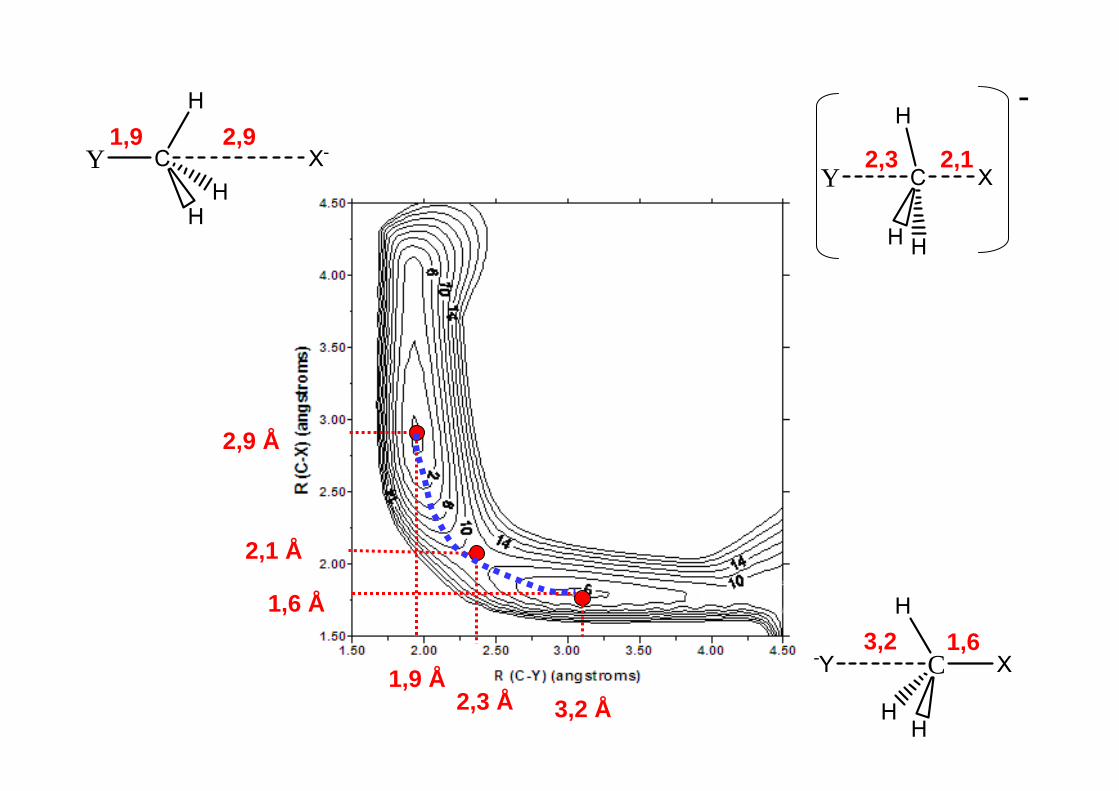
2,9 Å
1,9 Å
Y C
H
HH
X-2,9 1,9
2,1 Å
2,3 Å
-
2,3 2,1 Y C
H
HH
X
1,6 Å
3,2 Å
C
H
HH
X-Y1,6 3,2

LQFII Mod. Mol. (01/03/05) pag. 13
c.r.
E (Kcal/mol)
Er ∼ 6 kcal/mol
Ea ∼ 11 kcal/mol

LQFII Mod. Mol. (01/03/05) pag. 14
Los mínimos nos indican las coordenadas para las que tenemos estructuras estables.
Importancia de los mínimos
Importancia de los puntos silla
• Para pasar de reactivos a productos hay que pasar cerca del punto silla.
• La estructura del punto silla corresponde a la del estado de transición
• La velocidad de una reacción va a depender de la diferencia de energía entre el mínimo correspondiente a reactivos y el punto silla.
• La diferencia de energía entre el mínimo y el punto silla es la barrera de potencial.
Une los reactivos con los productos a través del estado de transición.
Camino de reacción:Camino a través de la superficie de potencial que une dos mínimos y pasa por un punto silla.

LQFII Mod. Mol. (01/03/05) pag. 15
V (R) = Eele (R) + VNN (R)
Cálculo de la energía potencial.
• Se puede obtener dentro de dos metodologías generales
- Mecánica Cuántica (QM)
- Mecánica Molecular (MM)
Mecánica Cuántica (QM)
- Se resuelve la ecuación de Schrödinger
- Hay muchos métodos y se diferencian en las aproximaciones utilizadas para resolver la ecuación de Schrödinger
- En general son más exactos que los métodos MM.
- En general son más costosos computacionalmente que los métodos MM
) H ele (R)Ψele (R) = Eele (R)Ψele (R)

LQFII Mod. Mol. (01/03/05) pag. 16
Mecánica Molecular (MM)
- Calculan cuanto cambia la energía de la molécula cuando modificamos las coordenadas de los átomos.
- Dependiendo del método los términos energéticos pueden ser diferentes, pero en general los más usados son:
V = Venlenlaces∑ + Vang
ángulos∑ + Vdie
diedros∑ + Vele
electrostaticas∑ + Vvdw
vdw∑

LQFII Mod. Mol. (01/03/05) pag. 17
Venl =12
kenl (b − benl )2
kenl y benl son parámetros que dependen de tipo de enlace.Por ejemplo todos los enlaces CC tendrán los mismos valores de estos parámetros
C Cb
El enlace es como un muelle con una constante de fuerza igual a kenly una distancia de equilibrio de benl
Vdie =12
kdie 1+ cos nφ − γ( )( )
Vang =12
kang (θ −θang )2
Vele =qiq j
4πε0rij
Vvdw = 4εij
σ ij
rij
⎛
⎝ ⎜ ⎜
⎞
⎠ ⎟ ⎟
12
−σ ij
rij
⎛
⎝ ⎜ ⎜
⎞
⎠ ⎟ ⎟
6⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
rij(distancia entre dos átomos i y j)
εij σij
rij (distancia entre dos átomos i y j)
qi qj
φ (ángulo diedro formado por 4 átomos)
kdie n γ
θ (ángulo formado por tres átomos)
kang θang
VariableParametros

LQFII Mod. Mol. (01/03/05) pag. 18
• Es un método muy aproximado
• No se puede utilizar para estudiar reacciones químicas.
• Es un método barato computacionalmente.
• Se utiliza mucho en el estudio de proteínas.

LQFII Mod. Mol. (01/03/05) pag. 19
Para ver esta película, debedisponer de QuickTime™ y deun descompresor TIFF (LZW).
5. Uso básico de Chem3D
Preparar programa:Ir al menu View => SettingsSeleccionar Building en el panel desplegable.No debe estar selccionado rectify
Cómo construir una molécula. Forma 1- Enlace simple, doble y triple de barra de herramientas.- Seleccionar átomo, átomos, enlaces.-Tools => Rectify (añade hidrogénos)-Tools => Show H’s and Lp’s (muestra hidrógenos y pares solitarios)-Tools => Clean Up Structure ( aplica valores estándar para distancias, ángulos..-Tools => Fit (ajusta molécula a la ventana)
Cómo construir una molécula. Forma 2-Seleccionar de la barra de herramientas=> Construcción por texto (A).-Picar en la ventana.-Escribir la fórmula de la molécula.

LQFII Mod. Mol. (01/03/05) pag. 20
Cómo modificar el tipo de átomo.-Seleccionar de la barra de herramientas => Construcción por texto (A).-Picar en un átomo.-Escribir el nombre del átomo.-Los diferentes tipos de átomos con sus nombres se pueden ver en:- View ==> Atom Types
Explicar como hacer.-Trasladar toda la molécula-Trasladar parte de la molécula.-Girar toda la molécula.-Girar toda la molécula a través de un enlace.-Girar parte de la molécula a través de un enlace.

LQFII Mod. Mol. (01/03/05) pag. 21
C. RESULTADOS Experimento 1Experimento 2Experimento 3Experimento 4

LQFII Mod. Mol. (01/03/05) pag. 22
Experimento 1
Localizar todos los mínimos de la superficie de potencial para las dos siguientes moléculas indicando cual es el mínimo global:
ICH2CH2ICiclohexano
Calculo de la energía potencial ==> mecánica molecular==> método MM2
Comando para localizar mínimos ==> menu “MM2” ==> Minimize Energy.
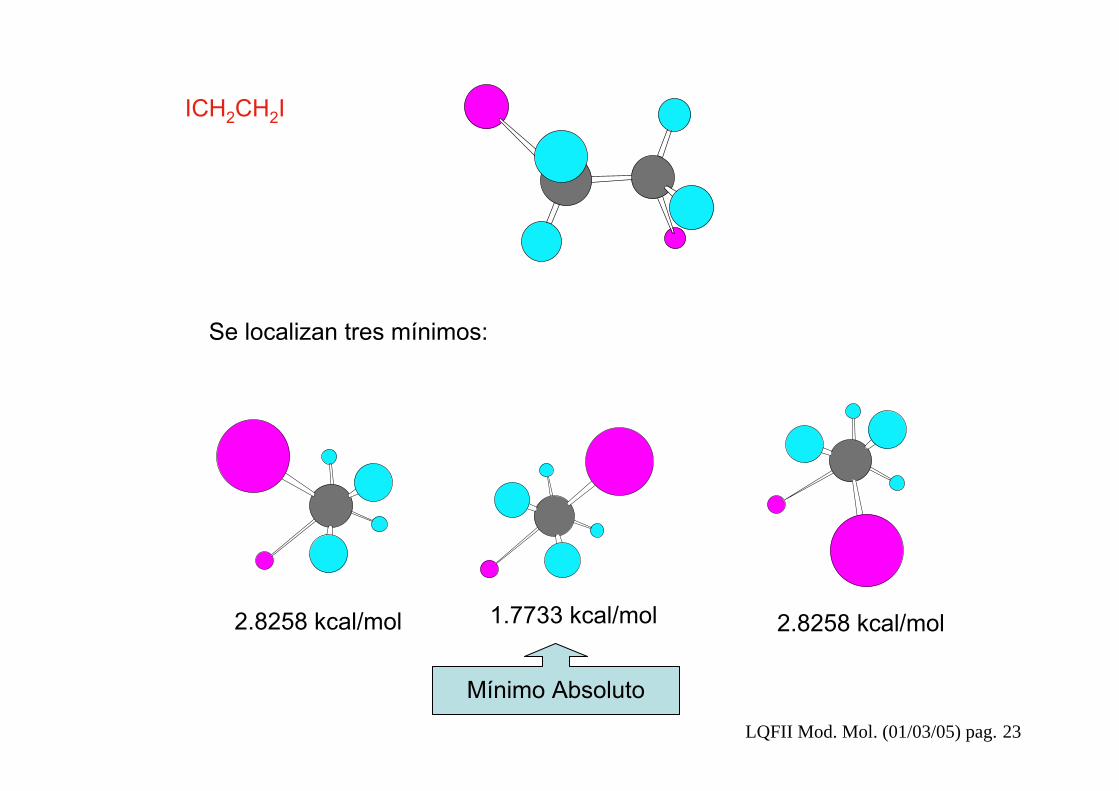
LQFII Mod. Mol. (01/03/05) pag. 23
ICH2CH2I
2.8258 kcal/mol 1.7733 kcal/mol 2.8258 kcal/mol
Se localizan tres mínimos:
Mínimo Absoluto

LQFII Mod. Mol. (01/03/05) pag. 24
Ciclohexano. • Hay dos formas de construirlo: por partes y haciendo uso de las subestructuras predefinidas. Escribir Cy en cuadro de texto.
6.5566 kcal/mol
silla
11.9123 kcal/mol
Bote torcido
• La conformación bote no es un mínimo
Mínimo global

LQFII Mod. Mol. (01/03/05) pag. 25
Experimento 2
Obtener la energía potencial en función del ángulo de rotación para:a)CH3-CH3b)CH3-CH2Ic)ICH3-CH3I
Calculo de la energía potencial ==> mecánica molecular==> método MM2
Comando para obtener la curva ==> menu “MM2” ==> Dihedral Driver

LQFII Mod. Mol. (01/03/05) pag. 26
0
0,5
1
1,5
2
2,5
3
3,5
4
-180 -120 -60 0 60 120 180
ener
gía
(kca
l/mol
)
ángulo diedro
Ángulo Energía-180.00 0.81800-170.00 0.97200-160.01 1.4060-150.01 2.0420-140.01 2.7440-130.01 3.3210-120.00 3.5520-110.00 3.3200-99.991 2.7390-89.990 2.0350-79.992 1.4010-69.997 0.97000-59.999 0.81800-50.004 0.97000-40.008 1.4000-30.007 2.0300-20.007 2.7320-10.007 3.31500.0000 3.55200.0000 3.552010.007 3.315020.007 2.732030.008 2.030040.008 1.400050.003 0.9700059.999 0.8180069.997 0.9700079.992 1.401089.992 2.035099.991 2.7390110.00 3.3200120.00 3.5520130.01 3.3210140.01 2.7440150.01 2.0420160.01 1.4060170.01 0.97200180.00 0.81800
CH3CH3 átomos que definen el ángulo diedro

LQFII Mod. Mol. (01/03/05) pag. 27
0
1
2
3
4
5
-180 -120 -60 0 60 120 180
Ene
rgía
(kca
l/mol
)
Ángulo diedro
átomos que definen el ángulo diedro
CH3CH2I Ángulo Energía-180.00 1.0070-170.00 1.1980-160.01 1.7340-150.01 2.5130-140.01 3.3600-130.01 4.0360-120.00 4.2840-109.99 3.9840-99.990 3.2810-89.990 2.4380-79.990 1.6820-69.995 1.1730-59.999 1.0080-50.006 1.2150-40.010 1.7540-30.010 2.5210-20.010 3.3490-10.007 4.01900.0000 4.28500.0000 4.285010.007 4.019020.010 3.349030.010 2.521040.010 1.754050.004 1.215060.001 1.008069.995 1.173079.992 1.682089.990 2.438099.990 3.2810109.99 3.9840120.00 4.2840130.01 4.0360140.01 3.3600150.01 2.5130160.01 1.7340170.00 1.1980180.00 1.0070

LQFII Mod. Mol. (01/03/05) pag. 28
0
2
4
6
8
10
-180 -120 -60 0 60 120 180
Ene
rgía
(kca
l/mol
)
Ángulo diedro
ICH2CH2I átomos que definen el ángulo diedro
Ángulo Energía-180.00 1.7730-170.00 1.9900-160.01 2.5960-150.01 3.4660-140.01 4.3960-130.01 5.1150-120.00 5.3590-109.99 5.0420-99.990 4.3410-89.990 3.5620-79.995 2.9900-70.001 2.8350-60.008 3.1910-50.011 4.0160-40.017 5.1730-30.015 6.4800-20.015 7.7290-10.007 8.67600.0000 9.04300.0000 9.043010.007 8.676020.015 7.729030.015 6.480040.015 5.173050.011 4.016060.008 3.191070.001 2.835079.995 2.990089.990 3.562099.990 4.3410109.99 5.0420120.00 5.3590130.01 5.1150140.01 4.3960150.01 3.4660160.01 2.5960170.00 1.9900180.00 1.7730

LQFII Mod. Mol. (01/03/05) pag. 29
Comparación de las tres curvas
0
2
4
6
8
10
-180 -120 -60 0 60 120 180
CH3CH3ICH2CH3ICH2CH2I
Ene
rgía
(kca
lmol
)
Ángulo diedro
Energías absolutas
0
1
2
3
4
5
6
7
8
-180 -120 -60 0 60 120 180
CH3CH3ICH2CH3ICH2CH2I
Ene
rgía
rela
tivas
al d
iedr
o de
180
º (kc
alm
ol)
Ángulo diedro
Energías relativas.

LQFII Mod. Mol. (01/03/05) pag. 30
Experimento 3Localizar los mínimos y puntos silla que aparecen en la rotación interna del bitiofeno correspondiente al enlace que une los anillos.
Calculo de la energía potencial ==> Mecánica Cuántica==> método AM1
Comandos para localizar puntos estacionarios.Mínimos ==> menú “MOPAC” ==> Minimize Energy…Silla ==> menú “MOPAC” ==> Optimize to Transition State…
S
S
Como ver el valor del ángulo diedro SCCS.1.-Seleccionar los átomos SCCS2.-Poner el puntero encima de uno de los enlaces.
omenú “Analyze” ==> Show Measurements ==> Show Dihedral Angles
Factores que afectana la energía de los conformeros.
Estabilización por resonancia.Factores estéricos.

LQFII Mod. Mol. (01/03/05) pag. 31
c) Estado de transición plano Anti
Diedro = 180.0Calor de formación = 56.51 kcal/mol

LQFII Mod. Mol. (01/03/05) pag. 32
b) Mínimo Anti-Gauche
Diedro = 153.3Calor de formación = 56.47 kcal/mol

LQFII Mod. Mol. (01/03/05) pag. 33
d) Estado de transición Gauche
Diedro = 83.4Calor de formación = 56.91 kcal/mol

LQFII Mod. Mol. (01/03/05) pag. 34
f) Mínimo Syn-Gauche
Diedro = 37.1Calor de formación = 56.67 kcal/mol

LQFII Mod. Mol. (01/03/05) pag. 35
e) Estado de transición plano Syn
Diedro = 0.0Calor de formación = 56.88 kcal/mol

LQFII Mod. Mol. (01/03/05) pag. 36
56,4
56,5
56,6
56,7
56,8
56,9
57,0
-180 -120 -60 0 60 120 180
Cal
or d
e fo
rmac
ión
(kca
l/mol
)
Ángulo diedro S-C-C-S
La resonancia estabiliza formas planas.Los factores estéricos estabilizan las formas giradas

LQFII Mod. Mol. (01/03/05) pag. 37
Experimento 4Localizar el mínimo correspondiente a reactivos y el punto silla correspondiente al estado de transición para dos reacciones de tipo SN2.
Calculo de la energía potencial ==> Mecánica Cuántica==> método AM1
Nu X
R'
R'''R'
C XR'
R'''R''Nu Nu C
R'
R''R'''
X
C ClH
HHCl Cl C
H
HH
ClTipo I
Tipo II C ClH
HHH3N NH3 C
H
HH
Cl
Reactivos: polaresE. transición: neutroProductos: polares
Reactivos: neutrosE. transición: polarProductos: polares
Propiedades a calcular para reactivos y estado de transición:-Calor de formación.-Momento dipolar-Cargas atómicas

LQFII Mod. Mol. (01/03/05) pag. 38
Comandos para localizar puntos estacionarios.Mínimos ==> menu “MOPAC” ==> Minimize Energy…Silla ==> menu “MOPAC” ==> Optimize to Transition State…
OBSERVACIONES:1) En la lengüeta “general” escribir en la ventana “Additional keywords” la
palabra “XYZ”. De esta forma se utilizan coordenadas cartesianas en la exploración de la superficie de potencial y no internas.
2) En la lengüeta “Properties” seleccionar:Heat of formationGradient NormDipoleCharges
Para seguir la geometría de la molécula se puede utilizar:menú “Tools” ==> “Show Model Table” ==> “Internal Coordinates”
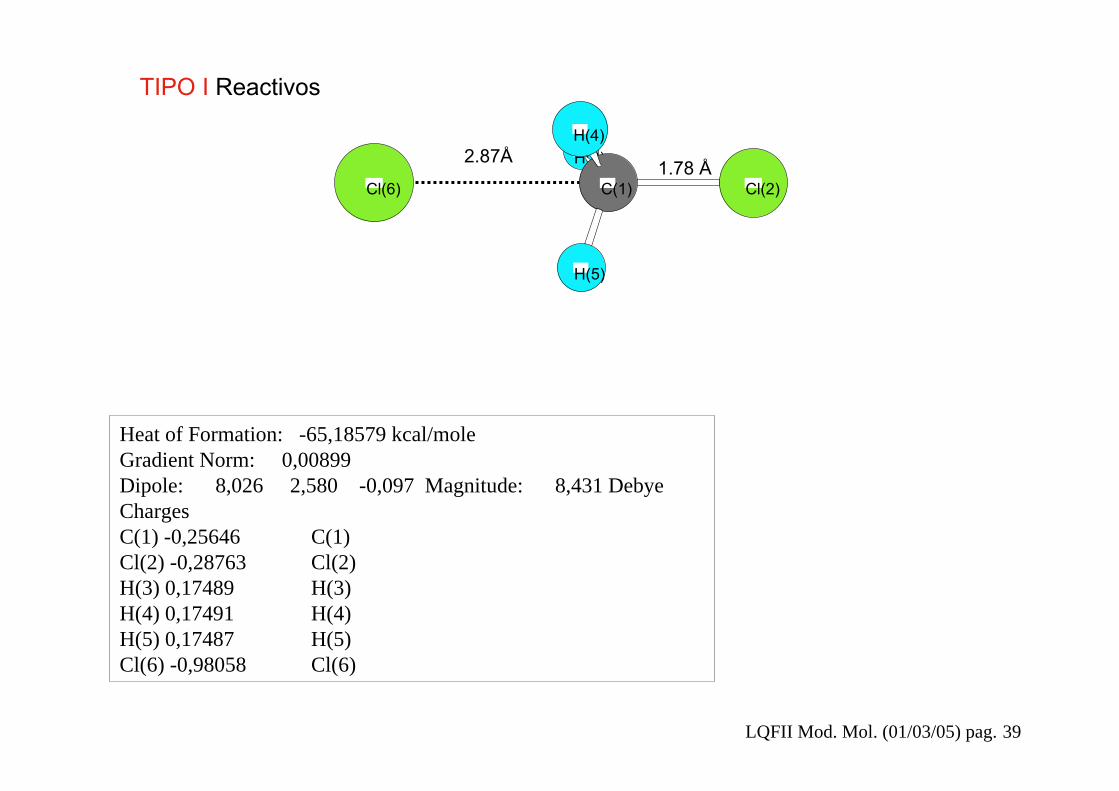
LQFII Mod. Mol. (01/03/05) pag. 39
Heat of Formation: -65,18579 kcal/moleGradient Norm: 0,00899Dipole: 8,026 2,580 -0,097 Magnitude: 8,431 DebyeChargesC(1) -0,25646 C(1)Cl(2) -0,28763 Cl(2)H(3) 0,17489 H(3)H(4) 0,17491 H(4)H(5) 0,17487 H(5)Cl(6) -0,98058 Cl(6)
H(3)
Cl(6) C(1) Cl(2)
H(5)
H(4)
1.78 Å2.87Å
TIPO I Reactivos

LQFII Mod. Mol. (01/03/05) pag. 40
Heat of Formation: -56,13535 kcal/moleGradient Norm: 0,00385Dipole: -0,000 -0,000 -0,000 Magnitude: 0,000 DebyeChargesC(1) -0,03268 C(1)Cl(2) -0,72068 Cl(2)H(3) 0,15802 H(3)H(4) 0,15803 H(4)H(5) 0,15797 H(5)Cl(6) -0,72066 Cl(6)
H(3)
C(1) Cl(2)Cl(6)
H(5)
H(4) 2.15 Å2.15 Å
TIPO I Estado de transición

LQFII Mod. Mol. (01/03/05) pag. 41
9.05 kcal/mol
μ = 0 D
μ= 8.4 D μ= 8.4 D
EN
ER
GÍA
COORDENADA DE REACCIÓN
GAS
FASEAQUOSA

LQFII Mod. Mol. (01/03/05) pag. 42
Heat of Formation: -27,10310 kcal/moleGradient Norm: 0,01876Dipole: -3,589 -0,101 0,080 Magnitude: 3,591 Debye
ChargesC(1) -0,30982Cl(2) -0,15217H(3) 0,15366H(4) 0,15411H(5) 0,15380N(6) -0,53530H(7) 0,17861H(8) 0,17842H(9) 0,17869
3,13 Å1.75 Å
H(5)
H(8)
H(7)
N(6) Cl(2)C(1)
H(4)
H(3)
H(9)
TIPO II Reactivos

LQFII Mod. Mol. (01/03/05) pag. 43
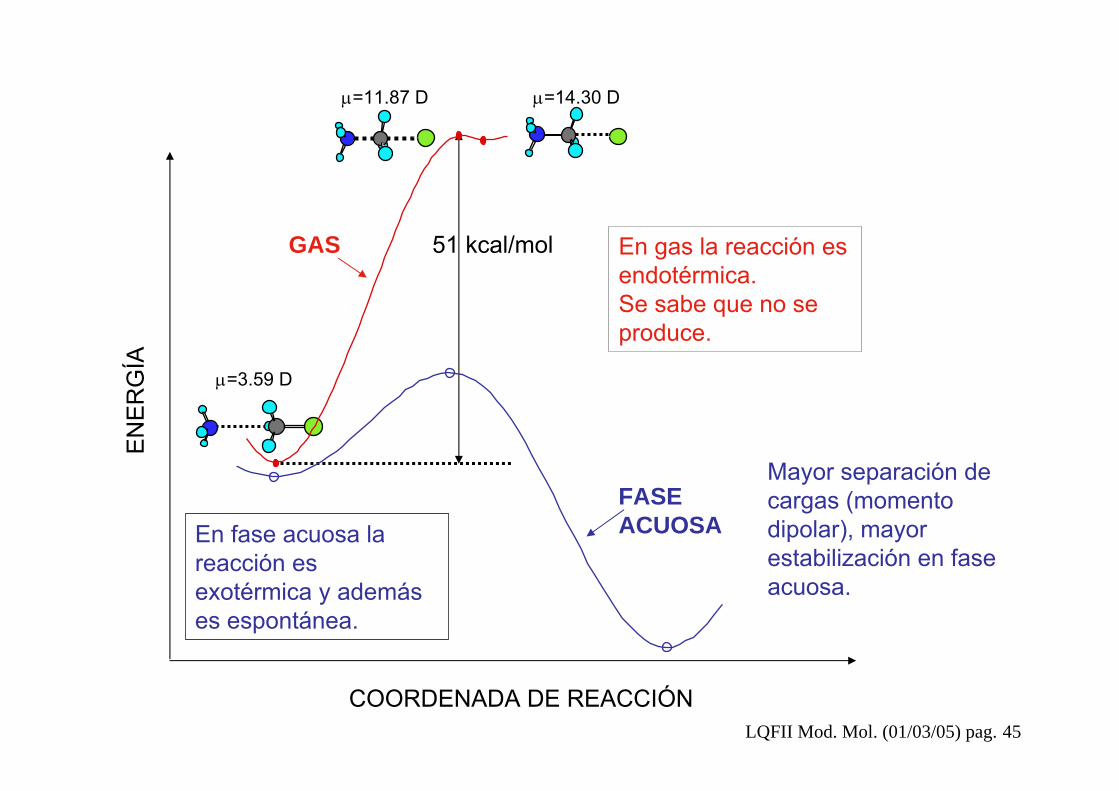
Heat of Formation: 23,78769 kcal/moleGradient Norm: 0,00351Dipole: -11,368 3,412 -0,018 Magnitude: 11,869 Debye
ChargesC(1) -0,15614Cl(2) -0,73453H(3) 0,18787H(4) 0,18788H(5) 0,18789N(6) -0,39422H(7) 0,24042H(8) 0,24041H(9) 0,24042
H(4)
H(9)
Cl(2)
H(8)
C(1)N(6)
H(5)
H(3)
H(7) 2.24 Å1.66 Å
TIPO II Estado de transición

LQFII Mod. Mol. (01/03/05) pag. 44
TIPO II Productos
Cl(2)
H(4)
H(9)
H(8)
C(1)N(6)
H(5)
H(3)
H(7) 1.54Å 2.43Å
Heat of Formation: 23,18114 kcal/moleGradient Norm: 0,01112Dipole: -14,288 0,424 0,509 Magnitude: 14,303 DebyeChargesC(1) -0,28335Cl(2) -0,84070H(3) 0,22114H(4) 0,22061H(5) 0,22181N(6) -0,32653H(7) 0,26244H(8) 0,26223H(9) 0,26235
LQFII Mod. Mol. (01/03/05) pag. 45
EN
ER
GÍA
COORDENADA DE REACCIÓN
μ=3.59 D
μ=11.87 D μ=14.30 D
51 kcal/molGAS
FASEACUOSA
En gas la reacción es endotérmica. Se sabe que no se produce.
En fase acuosa la reacción es exotérmica y además es espontánea.
Mayor separación de cargas (momento dipolar), mayor estabilización en fase acuosa.