estudio atomÍstico de la propagaciÓn de grietas en
TRANSCRIPT

ESTUDIO ATOMÍSTICO DE LA PROPAGACIÓN DE GRIETAS EN MONOCRISTALES DE Fe–α AGRIETADOS
A. Irastorza, A. Luque, J. Aldazabal, J.M. Martínez–Esnaola, J. Gil Sevillano
CEIT y Tecnun (Universidad de Navarra)
Paseo de Manuel Lardizábal, 15. 20018 Donostia–San Sebastián, España E–mail: [email protected]
RESUMEN
Este trabajo recoge resultados preliminares de simulaciones de dinámica molecular (MD), realizadas para estudiar el efecto de la orientación de un monocristral pre–agrietado de hierro y la temperatura de simulación sobre el comportamiento a fractura. Se ha implementado el termostato de Nosé–Hoover para considerar el efecto de la temperatura. El método de MD empleado es el embedded–atom method (EAM). Tras un proceso de relajación, se ha procedido a la tracción de las nanoestructuras preagrietadas con control de desplazamiento. El fallo del material se produce por el crecimiento de la grieta mediante la emisión de dislocaciones. A las temperaturas más bajas, se observa un comportamiento de fractura frágil por la menor actividad de las dislocaciones. Además se ha medido la apertura de los labios de la grieta. Mediante las ecuaciones del continuo, se han estimado los valores del factor de intensidad de tensiones para la emisión de la primera dislocación.
ABSTRACT
This paper summarizes various molecular dynamics (MD) simulations, carried out to study the effect of the orientation of a cracked iron single crystal and the simulation temperature on the fracture behaviour. The Nosé–Hoover thermostat has been implemented to consider the effect of the temperature. The MD technique employed has been the embedded–atom method. After sample relaxation, traction of the cracked nanostructures with displacement control has been performed. The failure takes place with crack growth associated to dislocation emission. At lower temperatures, however, fracture behaviour is shown to be brittle, due to the reduced dislocation activity. Besides, the opening displacement of crack lips has been measured. By means of continuum equations, the values of stress intensity factor for the emission of the first dislocation have been calculated. PALABRAS CLAVE: Dinámica molecular, hierro, factor de intensidad de tensiones.
1. INTRODUCCIÓN
Desde que CEIT comenzó a trabajar en la modelización del comportamiento mecánico a través de simulaciones de tipo atomístico, el material que se ha empleado ha sido mayoritariamente el cobre (como se ha recogido en los Anales de Mecánica de la Fractura desde el 20º Encuentro del Grupo Español de Fractura). Con este material se ha ido refinando la técnica de simulación y se ha obtenido un conocimiento cuantitativo, con ciertas limitaciones y algunas consideraciones. Además, por analogía con el cobre, se ha logrado comprender de forma cualitativa otros materiales de tipo fcc (aluminio, níquel). El trabajo que aquí se presenta sigue esta línea de investigación, pero introduciendo un cambio de material. Este trabajo ha sido llevado a cabo considerando átomos de hierro dispuestos en una estructura cúbica centrada en el cuerpo (ferrita).
2. MÉTODO DE SIMULACIÓN El método empleado en estas simulaciones ha sido el embedded–atom method (EAM) [1, 2]. Esta técnica considera que cada átomo del sistema está embebido en la red formada por el resto de átomos del sistema. La energía asociada a un átomo, Ei, se debe a dos tipos de interacción interatómica:
( ) ( )iijij
i ρF+rV=E ∑≠2
1 (1)
La primera es de origen internuclear y está representada por el pair potential, V(rij), donde rij es la distancia entre dos átomos. La segunda es debida a la interacción entre el núcleo y los electrones, estando representada por el embedding potential, F(ρi), donde ρi es la densidad electrónica relativa total en la posición del átomo. Para el caso del Fe, estos potenciales han sido
Anales de Mecánica de la Fractura 26, Vol. 2 (2009)
388

obtenidos por Chamati et al. [3], para el estudio de la difusión de átomos de Fe [3] y vacantes [4] en Fe(001). Los resultados de este trabajo han de ser tomados con precaución en tanto que dichos potenciales, generados teniendo en cuenta las constantes elásticas del hierro, pueden no reproducir correctamente su comportamiento de fractura. Conociendo estos potenciales, es posible calcular las fuerzas que aparecen entre los átomos, y con esas fuerzas, se pueden calcular las trayectorias que seguirán los mismos. La integración numérica de las ecuaciones de movimiento requiere un incremento de tiempo, Δt, que se ha fijado en 2.5 fs para este trabajo. Para simular el efecto de la temperatura, se ha implementado el termostato de Nosé–Hoover [5, 6]. Como ya se ha mencionado en anteriores artículos [7], este método de control de la temperatura actúa sobre la velocidad de los átomos a través de la aceleración, para que la velocidad media del sistema corresponda a la velocidad media de un sistema a la temperatura objetivo, T0. En este trabajo, se han simulado las temperaturas de 4 K, 100 K y 300 K. 3. CARACTERÍSTICAS DE LOS ENSAYOS Se han generado probetas monocristalinas de hierro–α (ferrita, bcc) con dos orientaciones distintas. Si se hace referencia a la posición que ocupará la entalla, el primer tipo de probetas tiene una orientación tal que la entalla quedará incluida en el plano (001) del cristal (cuya normal coincide con el eje z del sistema) mientras que la punta de la entalla estará dispuesta a lo largo de la dirección [010] (que coincide con el eje y del sistema). El segundo tipo de probetas está orientado de forma que la entalla estará en el plano (011) (normal al eje z) y la punta de la entalla se encuentra a lo largo de la dirección [ 101 ] (coincidente con el eje y). Inicialmente, las dimensiones de estas probetas son 23.2 × 2.3 × 15.3 nm3. Sin embargo, se han sometido a un proceso de relajación de tensiones, durante el cual cambian ligeramente (menos del 0.5% en el rango de temperaturas simuladas [3]). La relajación de las probetas ha consistido en mantenerlas a 0 K durante 5 ps, aumentar progresivamente la temperatura del sistema hasta alcanzar la temperatura objetivo (4 K, 100 K ó 300 K) después de 20 ps, y mantener las probetas a esa temperatura durante 12.5 ps más. Durante este proceso, se han mantenido condiciones de contorno periódicas a lo largo de los ejes x e y del cristal, aunque permitiendo el cambio dimensional en estos ejes. Por otro lado, las superficies a lo largo del eje z quedan libres. Al final de este proceso, el sistema se encuentra libre de tensiones. Las tensiones han sido calculadas a través de la expresión del virial [8]:
∑ ∑ ⎟⎟⎠
⎞⎜⎜⎝
⎛⊗⊗−
≠
N
iij
ijijiii fr+vvm
Ω=σ 1
(2)
donde Ω es el volumen total del sistema, N es el número
de átomos del sistema, mi es la masa del átomo, vi es su velocidad, rij = rj – ri y fij es la fuerza que aparece entre dos átomos. El resultado del producto a ⊗ b es una matriz C, cuyos elementos se calculan como Cαβ = aα·bβ, con α, β = x, y, z. Antes de someter las probetas a un ensayo virtual de tracción, se han establecido sendas capas fijas de átomos de Fe, de 0.6 nm de espesor, en la parte superior e inferior de las muestras, a las que se les permite un movimiento de sólido rígido, pero impidiendo el movimiento relativo de los átomos de las mismas. Además, se ha procedido al pre–agrietamiento de las probetas. Se trata en todos los casos de una entalla periódica (de acuerdo con las condiciones de contorno impuestas), de 7.7 nm de longitud, pasante en el eje y y de 0.6 nm de espesor, como se puede ver en la figura 1. Los ensayos virtuales de tracción realizados han sido llevados a cabo en control de desplazamiento, con una velocidad de deformación, ε& , de 108 s–1. Esto equivale a desplazar las capas fijas mencionadas anteriormente 3.8 fm en cada etapa. Durante el proceso de tracción de las probetas se han registrado las tensiones aparecidas en el sistema en función del desplazamiento relativo de las tapas, con el que se ha realizado un análisis en términos de tensiones y deformaciones. También se han almacenado periódicamente las posiciones de los átomos del sistema [9], para realizar un estudio del proceso de fractura. Además, para algunas de las simulaciones realizadas, se ha podido estimar el factor
(a) Orientación (001)[010]
(b) Orientación (011)[ 101 ]
Figura 1. Configuración inicial de las probetas.
Anales de Mecánica de la Fractura 26, Vol. 2 (2009)
389

de intensidad de tensiones para la emisión de la primera dislocación, a partir de los campos de desplazamientos de los átomos de los labios de la entalla [10]. 4. RESULTADOS La figura 2 muestra las curvas de tensión–deformación, σ – ε, para las cinco probetas simuladas. 4.1. Régimen elástico A partir de ellas, se ha podido calcular el módulo elástico de la ferrita en las orientaciones estudiadas. Así, para el caso de las probetas con orientación (001)[010], se ha medido un módulo elástico en la dirección <001>, E001, igual a 181 GPa (245 GPa en una probeta sin entalla). Las probetas con orientación (011)[ 101 ] dan E011 = 231 GPa (310 GPa en el caso sin entalla). Como se puede ver, estas simulaciones sobreestiman los valores de módulo elástico del hierro–α, incluso descontando el efecto de la deformación plana. Los cálculos teóricos realizados considerando la anisotropía elástica de la ferrita [3, 11], resultan E001 = 144 GPa y E001 = 240 GPa en deformación plana (E001 = 132 GPa y E011 = 220 GPa en tensión plana). Para justificar esta desviación respecto de los valores teóricos, habría que señalar la restricción al movimiento que imponen las capas rígidas de las probetas. Por otro lado, las simulaciones han mostrado que este parámetro no es muy sensible a la temperatura (incremento de menos del 2% entre 4 K y 300 K), en línea con la variación dimensional de las probetas, observada durante el proceso de relajación de las mismas. 4.2. Régimen plástico Como se observa en la figura 2, la deformación y tensión, εmáx y σmáx, respectivamente, en que se abandona el comportamiento elástico, cambian en función de la orientación. Para el caso de las probetas orientadas según (011)[ 101 ], se han obtenido valores de σmáx entre 6.9 GPa y 6.3 GPa, a 100 K y 300 K, respectivamente. εmáx ≈ 3.1% a ambas temperaturas. Por su parte, los valores de σmáx medidos para las probetas
con orientación (001)[010] son mayores, variando entre 10.5 GPa y 9.1 GPa, entre 4 K y 300 K, respectivamente. También εmáx aumenta, siendo igual a 6.1% a 4 K y 100 K e igual a 5.5% a 300 K. Como se preveía [7], la energía cinética atómica debida a la temperatura adelanta el fin del comportamiento elástico de las probetas, por la aparición de mecanismos de deformación térmicamente activados, como la emisión de dislocaciones desde la punta de la entalla (ver figura 3). Esto se traduce en una reducción de los valores de σmáx y εmáx al aumentar la temperatura. El análisis de la dislocación emitida en la probeta mostrada en la figura 3 indica que el sistema de deslizamiento activado es el 1/2(211)[ 111 ] y que se trata de una dislocación total. Además de lo ya mencionado para el módulo elástico, la tensión tractiva resuelta en el sistema de deslizamiento, τSD, varía para una y otra orientación. El factor que relaciona la tensión aplicada, σ, y la tensión resuelta en el sistema de deslizamiento es el factor de Schmid, m: m = τSD / σ = cos (ϕ) cos (λ) (3) donde ϕ es el ángulo que forman la dirección de aplicación de la tensión y la normal al plano de deslizamiento, y λ es el ángulo que forma la dirección de aplicación de la tensión y la dirección de deslizamiento de la dislocación. Al calcular dicho factor para las dos orientaciones (suponiendo que el sistema de deslizamiento activado sea el (211)[ 111 ] en ambas; cabe señalar que este sistema de deslizamiento suele mostrar asimetría en el deslizamiento), se obtiene que m001 = 0.235 y m011 = 0.471. Si la tensión crítica, τc, del sistema de deslizamiento se alcanza con σ = σmáx, se obtendría, a 300 K, τc = 2.14 GPa para la orientación (001)[010] y τc = 2.97 GPa para la orientación (011)[ 101 ]. La diferencia entre estos valores puede indicar la activación de un mecanismo de fractura diferente en cada caso. Por esta razón, se analizará lo que ocurre en cada orientación por separado.
Figura 2. Curvas σ – ε de las probetas pre–agrietadas de ferrita simuladas a distintas temperaturas.
Figura 3. Momento de la emisión de la primera dislocación en la probeta de orientación (011)[ 101 ] simulada a 100 K (ε = 3.1%). En la imagen se muestran las superficies libres y la dislocación emitida.
Anales de Mecánica de la Fractura 26, Vol. 2 (2009)
390

4.3. Probetas orientadas según (011)[ 101 ] Haciendo un seguimiento del comportamiento mecánico de las probetas de orientación (011)[ 101 ] después de la emisión de la primera dislocación, se puede observar que la entalla se propaga mediante sucesivas emisiones de dislocaciones en el mismo sistema de deslizamiento (figura 4). Además, al interaccionar con las capas rígidas del sistema, las dislocaciones sufren cross–slip. Al aumentar la deformación de la probeta, tiene lugar la activación de un nuevo mecanismo de deformación: el maclaje. Varios autores han reportado este mecanismo en simulaciones estáticas con ferrita a baja temperatura [12] y hasta temperatura ambiente [13]. Este mecanismo es particularmente importante al deformar a bajas temperaturas (donde la actividad de dislocaciones es menor) y/o con elevadas velocidades de deformación (caso de las simulaciones atomísticas). La punta de la entalla es un lugar donde puede ocurrir la nucleación de
maclas pues allí se concentran las tensiones. La figura 5, que corresponde con una simulación a 300 K de una probeta (011)[ 101 ], muestra la formación de una macla a partir de la punta de la entalla. Esta macla se forma por el avance de dos dislocaciones parciales 1/6112<111> en planos adyacentes. La formación de esta macla y su interacción con las dislocaciones presentes y emitidas posteriormente, producen la transformación de fase de parte de la ferrita, dando lugar a la formación de un nuevo grano. Otros autores la han observado en las cercanías de la punta de la entalla, debido a las bajas temperaturas simuladas [12]. En este caso, las mayores temperaturas alcanzadas permiten su formación lejos de la entalla, aunque cerca de las capas rígidas (figura 6). El número de coordinación de los átomos de esa nueva fase es 12, a diferencia de los 8 primeros vecinos de la ferrita. Esto apunta a una fase compacta de átomos de Fe. Un análisis más detallado (figura 7) revela que se trata de una fase metaestable hexagonal compacta [12]. Podría tratarse de hierro–ε, estable a altas presiones, situación posible en estas simulaciones si se considera el apilamiento de dislocaciones emitidas desde la punta. Volviendo a la cuestión de la emisión de la primera dislocación (εmáx = 3.1%) en las probetas de orientación (011)[ 101 ], se ha medido el campo de desplazamientos de apertura de los átomos de las cercanías de la punta de la entalla, Δu2, en función de la distancia a la punta de la misma, r. Se trata de establecer una relación de Δu2 con r1/2, a través del factor de intensidad de tensiones, KI, como la propuesta para el continuo, y que se ha demostrado válida para sistemas atómicos [10]: Δu2 = u2(r, θ = ±π) – u2(0,0) = ±KI f r1/2 (3) donde θ es el ángulo formado por la coordenada radial, r, de un átomo y el plano de la grieta, y f es un
Figura 4. Probeta de orientación (011)[ 101 ] simulada a 100 K (ε = 6%). En la imagen se muestran las superficies libres y las dislocaciones emitidas.
Figura 5. Centro–simetría de la probeta de orientación (011)[ 101 ] simulada a 300 K (ε = 9.8%). En la imagen se muestran las superficies libres, las dislocaciones emitidas y la macla que se ha formado.
Figura 6. Probeta de orientación (011)[ 101 ] simulada a 100 K (ε = 12.5%). En la imagen se muestran las superficies libres, las dislocaciones emitidas y la región de fase compacta que se ha formado.
Anales de Mecánica de la Fractura 26, Vol. 2 (2009)
391
coeficiente dependiente de las propiedades elásticas del material. En deformación plana, f = 4(1–ν2)/(E011 2π ), donde ν es el módulo de Poisson del hierro. La figura 8 muestra que el ajuste propuesto es muy bueno para r1/2 < 1.2 nm1/2 (esto es, hasta 4 veces el parámetro de red). Considerando los valores de E011 y ν [15], se han calculado unos valores de KI para la emisión de la primera dislocación iguales a 0.596 MPa√m y 0.589 MPa√m, a 100 K y 300 K, respectivamente. Estos valores son inferiores al KIc de clivaje del hierro por el plano (211) (KIc = 0.904 MPa√m siendo la energía superficial γ211 = 1.66 J/m2 [15]) o el plano (001) (KIc = 0.667 MPa√m con γ001 = 1.54 J/m2 [15]). 4.4. Probetas orientadas según (001)[010] Cuando se observan las probetas orientadas según (001)[010], se puede ver (figura 9) que, mucho antes de llegar a σmáx, en el entorno de las puntas de la entalla, se ha activado un mecanismo de deformación distinto de la emisión de dislocaciones. Los átomos que se
encuentran delante de la entalla forman una nueva fase de tipo compacto. A diferencia de la observada anteriormente, se trata de austenita, al verificarse la relación de Kurdjumov–Sachs (111γ // 011α y <110>γ // <111>α). Algunos autores [16] han reportado una transformación reversible sin difusión de ferrita en austenita por efecto de la deformación plástica severa en materiales nano–estructurados. Esa región de fase distinta a la ferrita crece preferentemente en planos (011), sin aparente emisión de dislocaciones. Al llegar a σmáx (ver figura 10), se produce el fallo del material y comienza la fractura frágil de las probetas (la tensión de fractura σf = σmáx ≈ 0.08E001 [17]). La cuestión de la energía liberada en la fractura es importante en términos del termostato de Nosé–Hoover. Por esta razón, sólo se ha podido continuar la simulación hasta su finalización para la temperatura de 4 K. A esta temperatura, la velocidad de los átomos es muy baja y es posible observar el avance de la grieta mediante clivaje, preferentemente a lo largo de planos (001) y, en menor medida, (011), como se muestra en la
(a) (b) Figura 7. Detalle de la probeta de orientación (011)[ 101 ] simulada a 100 K (ε = 12.5%). (a) Vista de los planos (0001) de la fase hexagonal compacta, según el eje y (compárese con los planos (011) de la ferrita). (b) Vista de los planos (0001) de la fase hexagonal compacta, según el eje x.
Figura 8. Δu2 en función de la distancia radial, r, a la punta de la entalla, para las probetas de orientación (011)[ 101 ]. Se representa u2 para ambos labios de la grieta (negativos y positivos para θ = –π y θ = π, respectivamente), a 100 K y 300 K.
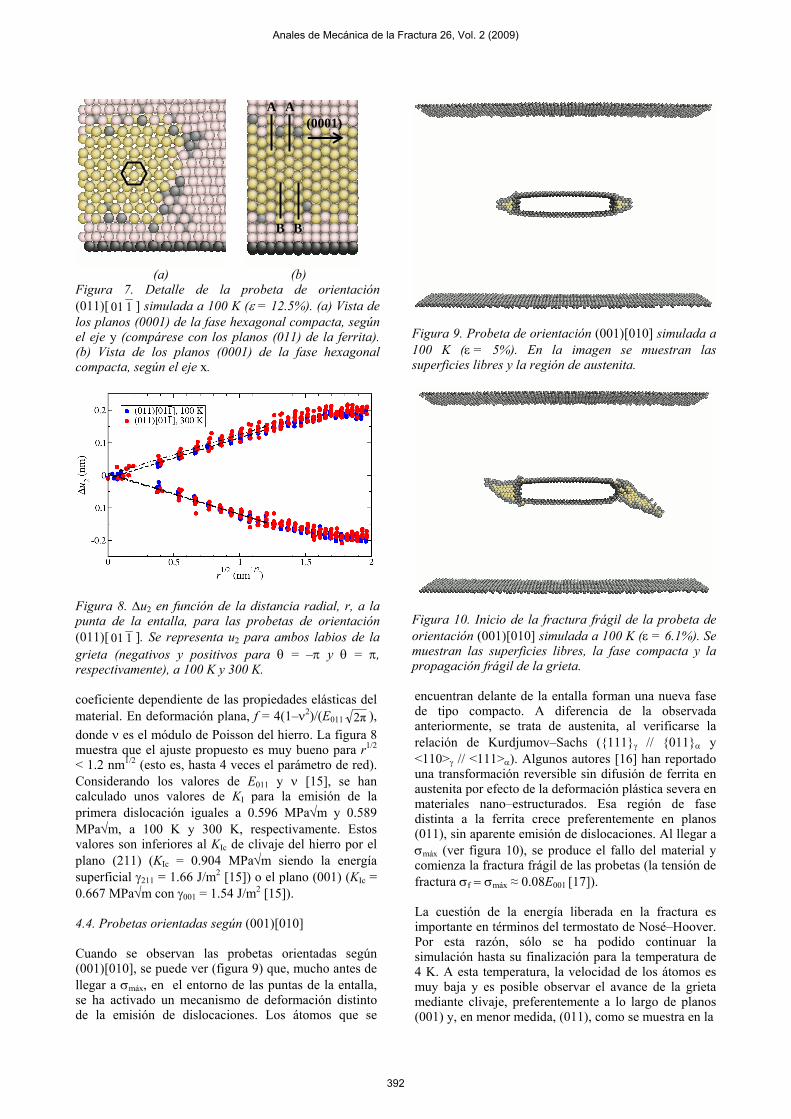
Figura 9. Probeta de orientación (001)[010] simulada a 100 K (ε = 5%). En la imagen se muestran las superficies libres y la región de austenita.
Figura 10. Inicio de la fractura frágil de la probeta de orientación (001)[010] simulada a 100 K (ε = 6.1%). Se muestran las superficies libres, la fase compacta y la propagación frágil de la grieta.
A A
B B
(0001)
Anales de Mecánica de la Fractura 26, Vol. 2 (2009)
392

figura 11. Se observa también cierta actividad de dislocaciones y el enromamiento de la punta de la entalla, aun a 4 K. La fractura frágil en la probeta simulada a 4 K continúa hasta que la entalla periódica se propaga horizontalmente e interseca consigo misma. 5. CONCLUSIONES Se ha simulado el comportamiento mecánico de probetas de hierro–α, de distinta orientación, (001)[010] y (011)[ 101 ], a diferentes temperaturas, 4 K, 100 K y 300 K. Se han observado comportamientos distintos para las dos orientaciones estudiadas, una vez que el régimen elástico se ha superado. Para las probetas de orientación (011)[ 101 ], se ha observado una mayor actividad de dislocaciones, de tipo total y de tipo parcial, que dan lugar al maclado de las probetas. Por interacción de las fronteras de macla con las dislocaciones presentes en el material, se forma una fase metaestable hexagonal de hierro, de la que ya han dado cuenta otros autores. Además, se ha calculado el factor de intensidad de tensiones para la emisión de la primera dislocación en esta orientación, que es inferior al valor de KIc de clivaje. Las probetas de orientación (001)[010], aunque también muestran nucleación y emisión de dislocaciones, se caracterizan por un comportamiento a fractura de tipo frágil, alcanzándose la tensión ideal del hierro antes de comenzar la propagación dúctil de la grieta.
AGRADECIMIENTOS Los autores desean agradecer al Departamento de Industria, Comercio y Turismo del Gobierno Vasco y a la Diputación Foral de Gipuzkoa la financiación parcial de este trabajo a través de los proyectos de investigación UET 1210/06 y ETORTEK inanoGUNE. A. Luque también quiere agradecer al Ministerio de Ciencia e Innovación y al Fondo Social Europeo por la financiación y la co–financiación, respectivamente, de su contrato a través del Programa Torres–Quevedo.
REFERENCIAS
[1] Daw, M.S. y Baskes, M.I., “Semiempirical, quantum
mechanical calculation of hydrogen embrittlement in metals”, Phys. Rev. Lett., 50, 1258–1288, 1983.
[2] Daw, M.S. y Baskes, M.I., “Embedded–atom method: derivation and application to impurities, surfaces and other defects in metals”, Phys. Rev. B, 29, 6443–6453, 1984.
[3] Chamati, H., Papanicolau, N.I., Mishin, Y. y Papaconstatopoulos, D.A., “Embedded–atom potential for Fe and its application to self–diffusion on Fe(001)”, Surf. Sci., 600, 1793–1803, 2006.
[4] Papanicolaou, N.I. y Chamati, H., “Diffusion of a vacancy on Fe(1 0 0): a molecular–dynamics study”, Comput. Mater. Sci., 44, 1366–1370, 2009.
[5] Nosé, S., “A unified formulation of the constant temperature molecular dynamics methods”, J. Chem. Phys., 81, 511–519, 1984.
[6] Hoover, W.G., “Canonical dynamics: equilibrium phase–space distributions”, Phys. Rev. A, 31, 1695–1697, 1985.
[7] Luque, A., Aldazabal, J., Martínez–Esnaola, J.M. y Gil Sevillano, J., “Propiedades termo–elásticas y de fractura de diversas estructuras de carbono simuladas mediantes dinámica molecular”, Anales de Mecánica de la Fractura, 25, 474–479, 2008.
[8] Sun, Z.H., Wang, X.X., Soh, A.K. y Wu, H.A., “On stress calculations in atomistic simulations”, Modelling Simul. Mater. Sci. Eng., 14, 423–431, 2006.
[9] Li J., “AtomEye: an efficient atomistic configuration viewer”, Modelling Simul. Mater. Sci. Eng., 11, 173–177, 2003.
[10] Luque, A., Aldazabal, J., Martínez–Esnaola, J.M. y Gil Sevillano, J., “Atomistic simulation of tensile strength of cracked Cu nanowires”, Fatigue Fract. Engng. Mater. Struct., 29, 615–622, 2006.
[11] Hirth J.P. y Lothe J., Theory of dislocations, John Wiley & Sons Ltd., Nueva York (EE.UU.), 1982.
[12] Farkas, D., “Twinning and recrystallisation as crack tip deformation mechanisms during fracture”, Philos. Mag., 85, 387–397, 2005.
[13] Guo, Y.–F., Wang, C.–Y. y Zhao, D.–L., “Atomistic simulation of crack cleavage and blunting in bcc–Fe”, Mater. Sci. Eng. A., 349, 29–35, 2003.
[14] Levy, M., Bass, H.E. y Stern, R.R., Handbook of elastic properties of solids, liquids and gases, vol. 2, Academic Press, San Diego (EE.UU.), 2001.
[15] Wen, Y.–N. y Zhang, J.–M., “Surface energy calculation of the bcc metals by using the MAEAM”, Comp. Mater. Sci., 42, 281–285, 2008.
[16] MacLaren, I., Ivanisenko, Y., Valiev, R.Z. y Fecht, H.J., “Reverse martensitic transformation of ferrite to austenite under severe plastic deformation”, J. Phys.: Conf. Ser., 26, 335–338, 2006.
[17] Clatterbuck, D.M., Chrzan, D.C. y Morris, J.W., “The inherent tensile strength of iron”, Philos. Mag. Letters, 82, 141–147, 2002.
Figura 11. Probeta de orientación (001)[010] simulada a 4 K (ε = 9.5%). En la imagen se muestran las superficies libres, la fase compacta, la extensión de la grieta (enromada) y la presencia de dislocaciones.
Anales de Mecánica de la Fractura 26, Vol. 2 (2009)
393