enfermedad de addison síndrome pluriglandular autoinmune hiperplasia adrenal congénita
DESCRIPTION
Enfermedad de Addison Síndrome pluriglandular autoinmune Hiperplasia adrenal congénita. ETIOLOGIA DE LA INSUFICIENCIA SUPRARRENAL. PRIMARIA: ENFERMEDAD DE ADDISON a) .- Lesión de la glándula por: - Infección: TBC, Meningococo (W -F), Hongos - Autoinmune o idiopática - PowerPoint PPT PresentationTRANSCRIPT

Enfermedad de AddisonSíndrome pluriglandular autoinmune
Hiperplasia adrenal congénita

PRIMARIA: ENFERMEDAD DE ADDISON
a) .- Lesión de la glándula por: - Infección: TBC, Meningococo (W -F), Hongos - Autoinmune o idiopática - Cirugía. - Metástasis (Ca broncógeno). - Hemorragia (tratamiento con heparina). - Metabólicas (hemocromatosis, amiloidosis)
b).- Alteración en la síntesis de cortisol: - Hiperplasia adrenal congénita.
- Tratamiento con inhibidores (Metopirona) c).- Otros alteraciones congénitas:
- Aplasia o hipoplasia adrenal. - Adrenoleucodistrofia. - Falta de respuesta adrenal a ACTH
ETIOLOGIA DE LA INSUFICIENCIA SUPRARRENAL

SECUNDARIA (falta ACTH) a).- Alteración hipotálamo hipofisaria:
- Adenoma hipofisario no productor (compresión). - Hipofisectomía. - Déficit aislado de ACTH b) Retirada brusca del tratamiento prolongado con corticoide
Insuficiencia adrenal relacionada con enfermedad aguda principalmente shock septico o distres respiratorio. Causa poco clara. Es reversible. Estos pacientes se benefician del tratamiento con corticoides iv /200-300 mg/día iv)
ETIOLOGIA DE LA INSUFICIENCIA SUPRARRENAL

ENFERMEDAD DE ADDISON
- La etiología autoinmune es la causa mas frecuente de insuficiencia adrenal en occidente (80%) (globalmente en el mundo es más frecuente por TBC) - La favorece el haplotipo DRB1, DQA, DQB1- Hay Acs contra la enzima 21 hidroxilasa en el 60-76% de los pacientes. (¿es el Ag responsable ?)- Destrucción de la glándula por linfocitos T citotoxicos- Es frecuente la presencia de otros Acs (tiroideos, contra la célula β pancreática, contra el factor intrínseco, ovarios)- Puede presentarse aislado o como parte de la enfermedad pluriglandular autoinmune - Se asocia con frecuencia (Acs contra melanocitos)

Se presenta de manera insidiosa y según la intensidad del déficit
a) Presentación aguda (crisis addisoniana) Por enfermedad intercurrente : Hipotensión, incluso shock hiponatremia, vómitos, dolor abdominal, diarrea (b) Larga evolución- Hipotensión , avidez por la sal- Pigmentación de las mucosas (boca y encías) y de la piel (zonas de roce y expuestas, pliegues, heridas). No en la 2ª- Astenia que aumenta con el ejercicio. - Anorexia con pérdida de peso- Impotencia, amenorrea. - Caida del vello axilar y púbico- Puede haber neuropatía: Calambres, paresia, paraplejia o cuadriplejia fláccida. - Hipoglucemias de ayuno
CLINICA DEL ADDISON

PIGMENTACIÓN
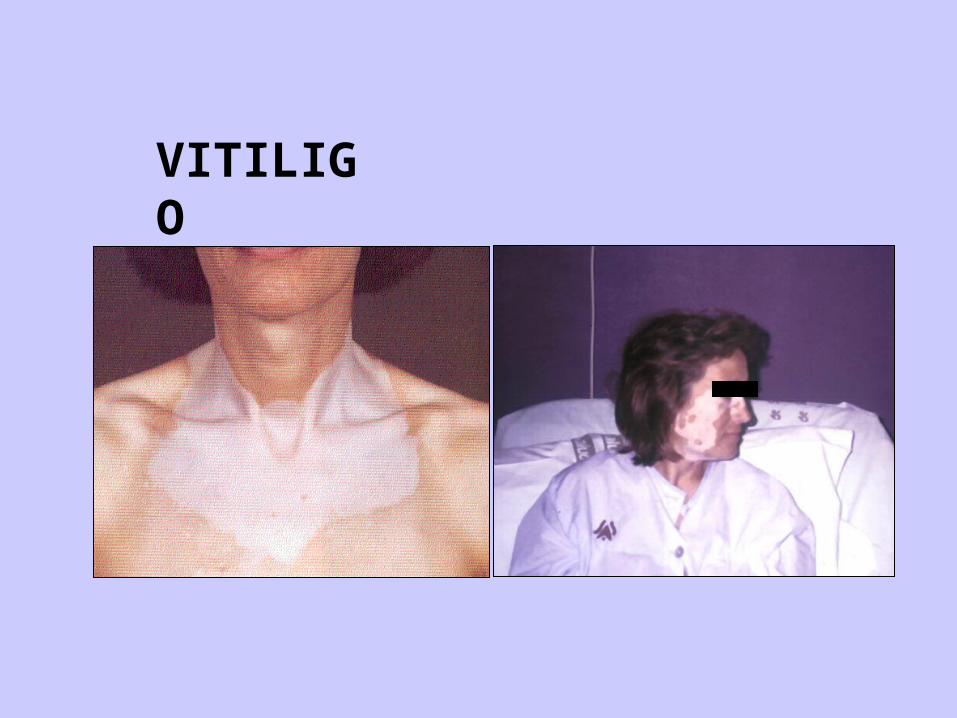
VITILIGO

Enfermedad de Addison Pigmentación
antes y después del tratamiento

- Laboratorio: Hiponatremia con hiperpotasemia. Con frecuencia hipoglucemia o hipocalcemia (10%)- Determinaciones hormonales basales: - Cortisol plasmático inferior a 10 µg/dl si el déficit es completo, entre 10 y 20 si es parcial. - Cortisoluria inferior a 60 mg/24 hs. - ACTH alto en el primario y bajo en el secundario
DIAGNOSTICO

Estimulo corto con ACTH (synacthen 250 μg im o iv). Determinar cortisol a los 0 y 30 minutos) Addison primario - Parcial: Cortisol sube 15 a 20 µg/dl (normal más de 20) - Completo: respuesta inferior a 10 µg/dl Estimulo prolongado con Sinacthen (depot o infusión 24-48 hs) - Primario: Cortisol sube > 36 μg/dl a las 4 hs y se mantiene - Secundario: Cortisol sube mas tarde Test de Insulina exógena (0.1 u/kg de insulina determinando cortisol a los 0,30,60,90 y 120 minutos: - Primario: Sube ACTH pero cortisol no - Secundario: No sube ni ACTH ni cortisol
Si el tratamiento ha de ser urgente, puede darse 10 mgrs de dexa o 20 mgrs de prednisona antes de hacer el test de ACTH
DIAGNOSTICO
Pruebas funcionales

A) Crisis Addisoniana: - Reposición hidroelectrolítica: Suero salino (con glucosa) 1 litro en la primera hora y completar hasta 6 litros en 24 hs. - Corticoides: Bolus iv de 100 mgrs de hidrocortisona (actocortina) y luego 10 mgrs/hora o 300 mgrs/día en perfusión. Profilaxis en cirugía: 300 mgrs en el 1 día y luego reducir la dosis 50% diariamente hasta llegar a la dosis de mantenimiento. Si la T/A no sube puede necesitar trasfusión o simpaticomiméticos- Tratar el factor desencadenante (antibióticos etc). B) Mantenimiento: - Hidrocortisona 20-30 mgrs/día ( 2/3 en desayuno y 1/3 en merienda). Hoy se considera preferible una dosis menor (15 – 25 mg/día) repartida en tres tomas (difícil de conseguir por la posología) En niños: 20 mg/m2 / día. En caso de enfermedad concomitante duplicar la dosis- Fludrocortisona 0.05 a 0.1 mgr /día si persiste la hiperpotasemia o la hipotensión a pesar de la hidrocortisona.- Tuberculostáticos si hay sospecha de TBC - Otras medidas: Dieta rica en sal
Tratamiento

SINDROME PLURIGLANDULAR AUTOINMUNE
Producida por anticuerpos múltiplesAsociación familiar. Más frecuente en mujeresTipo 1: Enfermedad de Addison Hipoparatiroidismo Moniliasis mucocutánea. Otras lesiones: Anemia perniciosa Hepatitis crónica activa Tipo 2: Enfermedad de Addison Tiroiditis de Hashimoto. Insuficiencia gonadal, Diabetes mellitus tipo 1 Enfermedad de Graves

Concepto: Defecto enzimático (autosómico recesivo) en la síntesis decortisol, con elevación de ACTH, aumento de la glándula, y elevación de los metabolitos previos al defecto enzimático.FisiopatologíaSegún el déficit puede haber:a) Exceso de andrógenos: - Produce en el hombre macrogenitosomía o pubertad precoz. - En la mujer puede producir: - Pseudohermafroditismo femenino (puede ir desde una clitoromegalia a seno urogenital o hipospadias). - Virilismo: Con o sin clitoromegalia - Hirsutismo (en heterozigotos o formas no clásicas)b) Defecto de andrógenos: Pseudohermafroditismo masculino (por ejemplo en el déficit de 17 hidroxilasa)c) Defecto de mineralocorticoides: Crisis adrenal a las dos semanas con shock y muerte (defecto completo de 21 hidroxilasa)d) Exceso de mineralocorticoides: Hipertensión
Hiperplasia adrenal congénita

Colesterol (1)
(3) Pregnenolona 17 OH Pregnenolona
(4) (2) DOCA Progesterona Dehidroepiandrosterona (5) (3)
Corticosterona 17 OH P Androstenediona Etiocolanolona (6) (4)
Aldosterona 11 deosicortisol Testosterona (5)
Cortisol Estrona Estradiol Androsterona
Estriol 1).- 20/22 desmolasa 2).- 3 hidroxiesteroide deshidrogenasa 3).- 17 hidroxilasa 4).- 21 hidroxilasa 5).- 11 hidroxilasa 6).- 18 hidroxilasa
Síntesis de esteroides adrenales

Alteración del gen P 450 c21 Clínica: - Forma clásica: Crisis adrenal (pierde sal), shock. Pseudohermafroditismo femenino y adrenarquia precoz en varones. - Forma virilizante en la mujer. Pubertad precoz con azoospermia en el hombre. - Forma no clásica: Adrenarquia precoz, alteración menstrual, infertilidad, hirsutismo, acné. - Puede presentarse sólo como hirsutismo tras la pubertad en la mujer Diagnóstico (extracción con suero) - Elevación de 17OHP basal con hiperrespuesta tras ACTH - Elevación de ACTH - Determinar la mutación y hacer estudio familiar. Consejo genéticoTratamiento: - Hidrocortisona (16 mg/m2/24hs) o prednisona 2.5 y 5 mg mañana y noche o dexametasona (0.25 a 0.5 mg nocturnos). - Fludrocortisona (0.1 mg/día) si persiste elevación de potasio o hipotensión
Déficit de 21 hidroxilasa

Hay elevación de DOCA y andrógenos
Clínica: Forma clásica: Pseudohermafroditismo femenino, virilización postnatal, hipertensión. Forma no clásica: adrenarquia precoz, hirsutismo, infertilidad, alteraciones menstruales, acné
Diagnóstico: Elevación de 11 deoxicortisol y DOCA basal o tras ACTH Elevación basal de ACTH. Hipopotasemia.
Tratamiento: Igual que el déficit de 21 hidroxilasa
Déficit de 11 hidroxilasa