urgencias metabÓlicas en el recien nacido · con sospecha de enfermedad metabólica ... (alcalosis...
TRANSCRIPT

URGENCIAS METABÓLICAS
EN EL RECIEN NACIDO
Dra. María D. MarchioneMetabolismo InfantilHospital Italiano, Bs. [email protected]

Sexo: masculino, RNTPAEG Primer hijo de pareja no consanguínea, sin antecedentes familiares. Comienzo: 48hs de vida con mala actitud alimentaria, ictericia, aletargado. Cuadro compatible sepsis.Se ingresa a UCIN. Cultivos, ATB, flujo de glucosa a 4 mg/kg/min. con electrolitos a basales. Primer lab: EAB:7.56/27.8/+4.6/25, Glucemia 82 mg/dl. Urea: 10 mg% Cr:0.8mg%
Caso clínico 1

BB en grave estado general, se deriva al HILaboratorio de ingreso: Amonio: 5830ucg/dl Láctico: 5.6 mmol/lCon sospecha de enfermedad metabólica con hiperamonemia se toman muestras de suero, GSPF Y orina de 12hs.Se aumenta flujo a 12 mg/k/min. Se inicia benzoato de sodio a 500mg/k/día y luego se asocia fenilbutirato de sodio a 250mg/k/día c/u.

Mientras, se realiza primeraexanguinotrasfusión (en total 3) con leve descenso del amonio.
BB en grave estado general, en ARM y con requerimiento de inotrópicos.
A los 7 días de vida se realiza hemodiafiltración con descenso del amonio hasta llegar a 200 mcg/dl

Se reciben resultados
AA plasmáticos:glutamina 1220 (<709)alanina 863 (<710)citrulina 0 (10-45)
Ácido orótico urinario:1886 mg/gr.creat
Edad diagnóstico: 9 días Déficit de Ornitin trascarbamilasa (OTC)

Paciente continúa grave, con fallo multiorgánico fallece a los 12 días de vida. Se guarda tejido hepático (-70°) y muestra para aislamiento de ADN.Se obtiene la mutación Xp 21.1
Forma de presentación aguda y grave.Difícil manejo, generalmente fallecen.
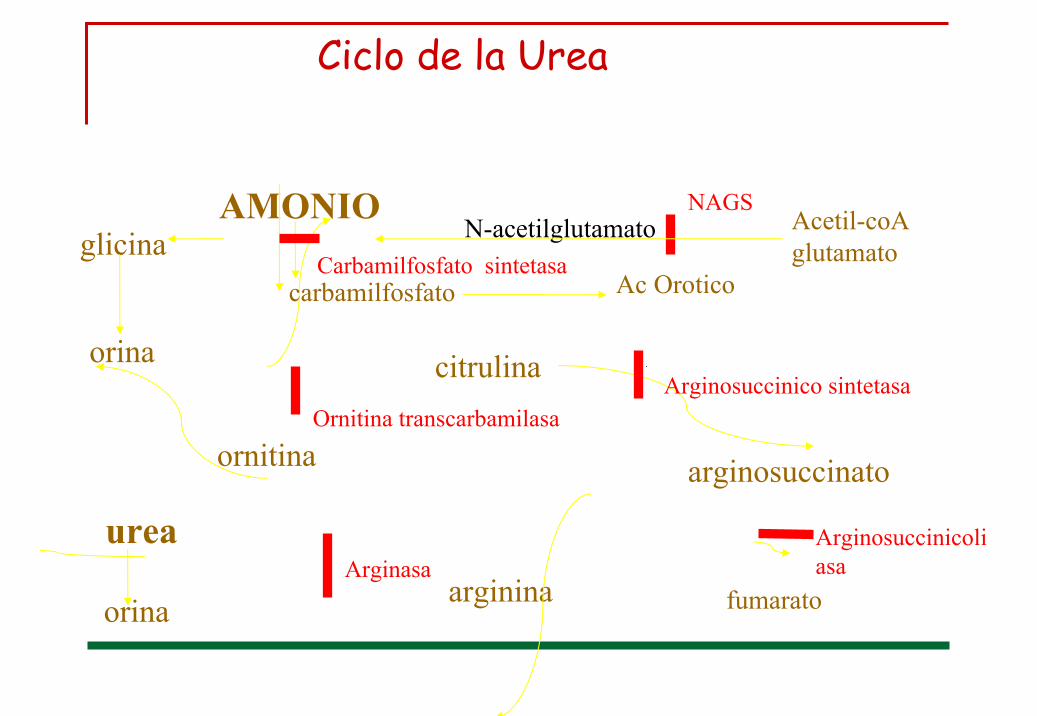
Ciclo de la Urea
citrulina
arginina
arginosuccinatoornitina
fumarato
urea
AMONIOglicina
orina
orina
carbamilfosfato
N-acetilglutamato Acetil-coA glutamato
Ac OroticoCarbamilfosfato sintetasa
Ornitina transcarbamilasaArginosuccinico sintetasa
ArginosuccinicoliasaArginasa
NAGS

Alteración ciclo de la urea
Prevalencia 1:30.000. Herencia autosómica recesiva, salvo OTC herencia ligada al cromosoma X.
Cuanto más proximal el defecto enzimático más severo el cuadro clínico y más difícil su manejo (OTC-CPS).
Gran heterogeneidad en su forma de presentación, dado por la ubicación del bloqueo enzimático y el grado de deficiencia enzimática.

Clínica
Presentación: casi a cualquier edad, más frecuencia en período neonatal, infancia tardía y pubertad.
Síntomas iniciales no son específicos (por pérdida del apetito, vómitos, letargo, irritabilidad, alteración de la conducta). Dato diagnóstico: hiperamonemia.
Forma neonatal: RNT PAEG, que comienza entre el 1-5 día de vida con somnolencia, hipotonía, vómitos, rechazo del alimento, apnea, convulsiones y coma.
Forma tardía: enfermedad neurológica crónica o encefalopatía aguda.

Laboratorio
Amonio:Brazo en posición relajada, no usar manguitoTubo heparinizado y frioProcesado en forma inmediata.Estable por 1hr a 4 °Tiempo de ayuno: 3-4 hs.
EAB (alcalosis respiratoria)Na-K-ClGlucosaUreaCetonas negativasFunción hepática

Laboratorios específicos
Aminoácidos plasmáticos cuantitativosÁcidos orgánicos urinariosÁcido orótico urinario
Dosaje enzimático en tejido hepático
Estudio de ADN
Identificación de portadores: prueba de allopurinol, prueba de sobrecarga protéica.

Diagnóstico de certeza
Dosaje enzimático por biopsia hepática.
Estudio genético ADN:-CPS: C2p-OTC: CXp21-ASS: C9q
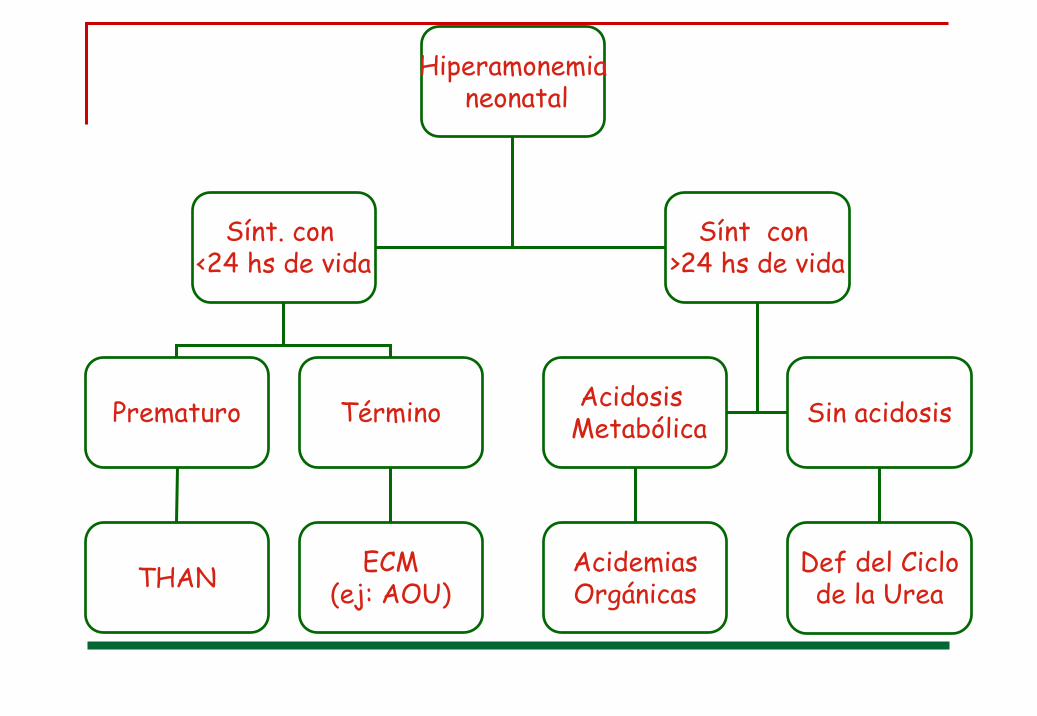
Hiperamonemianeonatal
Sínt. con <24 hs de vida
Sínt con >24 hs de vida
Acidosis MetabólicaPrematuro
THAN
Término
ECM(ej: AOU)
AcidemiasOrgánicas
Sin acidosis
Def del Ciclode la Urea
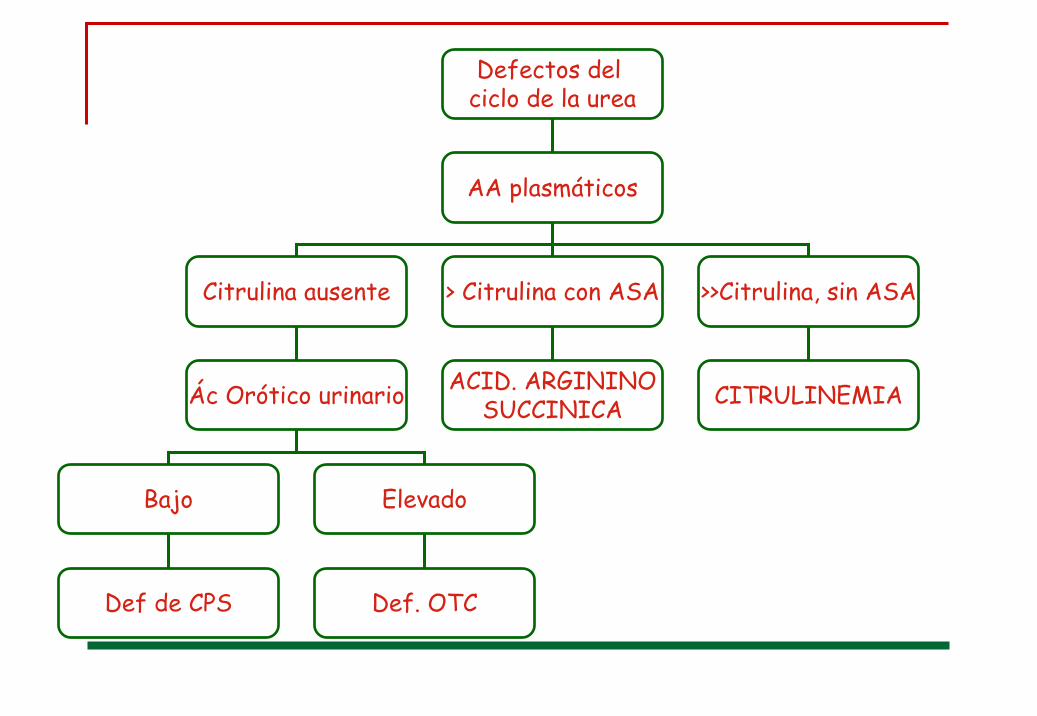
Defectos del ciclo de la urea
AA plasmáticos
Citrulina ausente > Citrulina con ASA >>Citrulina, sin ASA
Ác Orótico urinario
Bajo Elevado
Def de CPS Def. OTC
ACID. ARGININOSUCCINICA CITRULINEMIA

Tratamiento agudoTerapia de sostén:- acceso venoso seguro o via central multilumen- suplementación calórica (flujo de glucosa > 10) - suspender aporte protéico (24 - 48hs).- no sobrehidratar, recordar que las drogas para
scavenging tienen alto contenido de Na y CL.
Remosión de amonio:Diálisis: ECMO+HD. ECMO+HF.
Drogas: Benzoato: 500 mg/k/día. Fenilbutirato de sodio:250mg/k/díaArginina: 300 mg/k/día. L-Carnitina: 200 MG/K/día.

Seguimiento
Adecuado desarrollo pondoestaturalPrevención déficit hierro y zincAporte adecuado vitaminas (complejo B)Control seriado de:- Amonio- AA plasmáticos (glutamina estrecha relación
amonio plasmático)- Albúmina y pre-albúmina

Caso clínico 2
RNT PAEG, sexo femenino. Primera hija.Comienza con sintomatología a las 48hs vida. Rechazo del alimento, sensorio alternante, distress respiratorio, apneas, opistótono con movimientos mioclónicos. Encefalitis.Se realiza protocolo de sepsis con policultivos y atbterapia.Olor dulzón referido por enfermería al abrir incubadora. Se sospecha metabolopatía.

Laboratorio:EAB: 7,43/-5,8/17,1Na: 134 K: 5 Cl:102 AR: 15Amonio: 90 Ac. Láctico: 0,9Creat: 0,7 Urea: 2Got: 31 GPT: 13Con cetonuria en varias determinaciones rápidas
Sin alimentación.
Se ic a metabolismo infantil y se envían muestras para aa, acylcarnitinas, dosaje de carnitina t y l y AOU, junto con medidas iniciales de tratamiento.

Dosaje Carnitina Total: 32,6 umol/LLibre: 27 umol/L (83%)
AA plasmáticos (en mmol/L)Leucina: 2252 (48-160)Isoleucina: 331 (26-91)Valina: 980 (86-190)
Dinitrofenilhidrazina (DNPH): +Ac orgánicos urinarios: > Keto acidos
Deficiencia Keto-acid dehidrogenasa (BCKD

Se realiza diagnóstico de Leucinosis.Con mejoría clínica de la niña, se inicia tolerancia con fórmula libre de leucina, isoleucina y valina con dosajes seriados de aminoácidos. Se regula el aporte.
Se realiza botón de gastrostomía pre alta hospitalaria, por alteración de la succión y necesidad de SNG permanente.

Enfermedad Jarabe deArce
MSUD o Leucinosis

Leucinosis
Alteración en el catabolismo de los aa ramificados
En gral el debut neonatal no tiene grandes alteraciones de laboratorio. Sí cetonuria presente, debido a presencia de 2-oxoácidos.
Clínica: neonatal severa con distress metabólico; o agudo, intermitente, de inicio tardío o crónica progresiva como hipotonía, fallo de crecimiento y/o retraso madurativo

Tratamiento agudo
Reducir metabolitos tóxicos: suspender aporte de proteína natural.Evitar catabolismo: Disminuir horas de ayuno, colocar en alimentación enteral continua. Aumentar el aporte de HC. Colocar alto flujo de glucosa > 10mg/kg/minTratamiento de todo foco infecciosoLaboratorio metabólico de control en crisis para regular el aporte AA esenciales.

MSUD: sostén
DIETA: Restricción protéica con suplemento de AA libres de Leu/Isoleu/Val y con requerimientos de los ramificados según tolerancia.
Líquidos (ojo con hiperosmolaridad).
EVITAR CATABOLISMO: Hipercalórica con distribución porcentual normal.
EVALUACION: AA plasmáticos cuantitativos, dosaje carnitina, amonio y laboratorio de evaluación nutricional

Tratamiento
Evitar ayunos (alimentación nocturna contínua)
Ingesta de maicena cruda como hidrato de carbono de absorción lenta cada 3-4 hs.
Evitar ingesta de lactosa, galactosa, fructosa y sucrosa

Caso clínico 3
Paciente nacida de embarazo y parto sin particularidades. Peso Nac: 3.050 kg
Se realiza FEI: Phe: 24mg. Se repite para su confirmación.
Cromatografia de AA: fenilalanina 997umol/lTirosina 54 umol/l
Tratamiento: análogoXP + pechoSeguimiento : dosaje cuantitativo fenilalanina

Seguimiento
Clínico:1er mes: 1vez por sem 2do-3er mes: cada 15 dias. 1 Año de vida: mensual 3 años de vida: cada 3 meses. Continúa con seguimiento semestral.
Nivel de fenilalanina:Guthrie hasta los 18 meses semanal Método cuantitativo: 2 veces por semana hasta estabilizar Hasta 2 meses: semanal Hasta 1 año: quincenal Continúa con controles mensuales
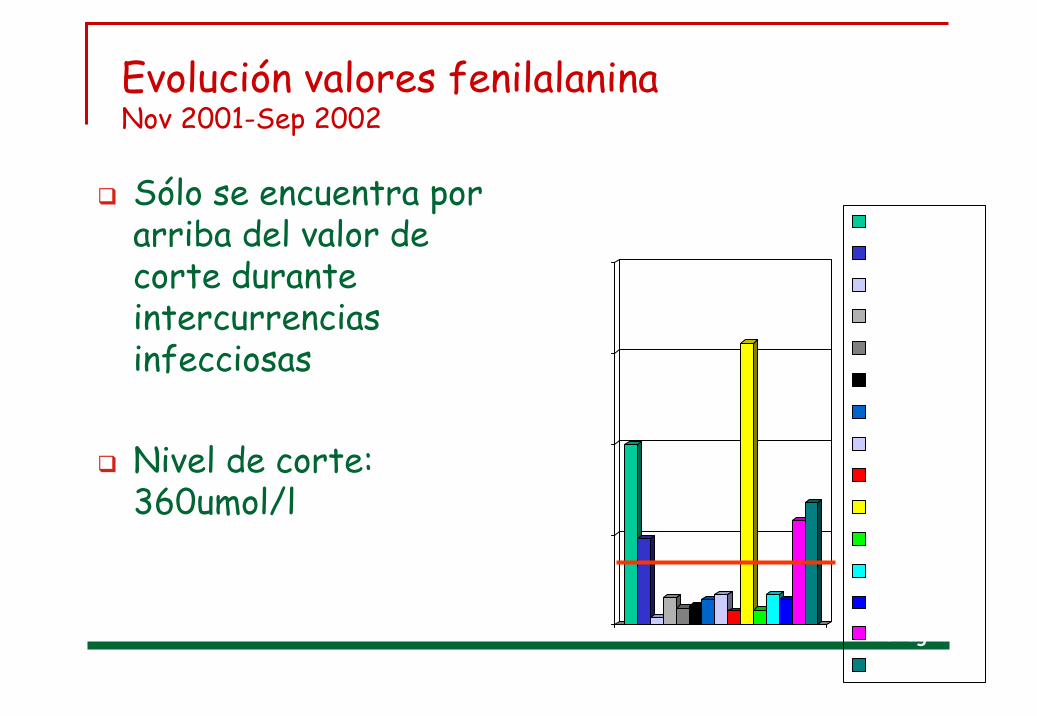
Evolución valores fenilalaninaNov 2001-Sep 2002
Sólo se encuentra por arriba del valor de corte durante intercurrencias infecciosas
Nivel de corte: 360umol/l
0500
1000
1500
2000
21-nov27-nov-0104-dic-0120-dic-0101-ene-0204-feb-0212-mar-0208-abr-0218-may-0225-may-0230-may-0204-jun-0205-jul-0220-ag-0209-sep-02
Fenil
Umol/L
meses

Fenilcetonuria
Diagnóstico por screeningDiagnóstico tardío
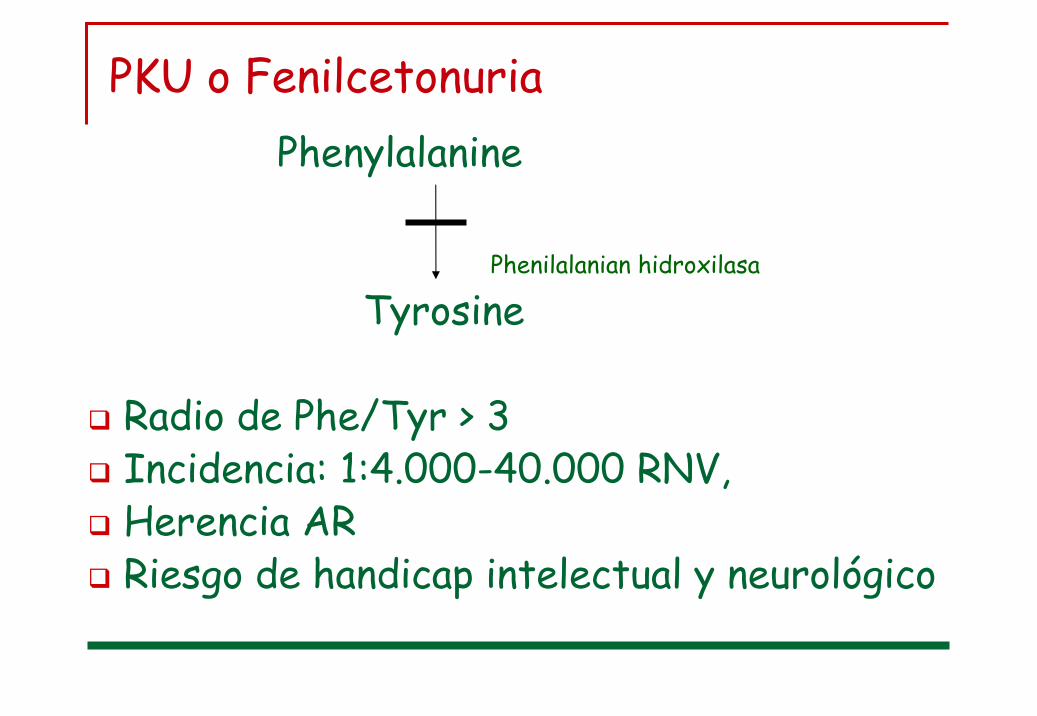
PKU o FenilcetonuriaPhenylalanine
Tyrosine
Radio de Phe/Tyr > 3Incidencia: 1:4.000-40.000 RNV, Herencia ARRiesgo de handicap intelectual y neurológico
phe
Phenilalanian hidroxilasa

PKU. Clínica
Espasmos infantiles con hipsarritmias en EEG, microcefalia, dentro de los primeros meses de vida en el 30% de los pacientes.Poca pigmentación (ojos,piel y cabellos claros), eczemas. Pacientes más grandes retraso mental, trastornos de la conducta, hiperactividad, autoinjuria, características autistas con episodios de excitación psicomotriz. Signos piramidales, temblor, parkinsonismo, anormalidades en la marcha, postura, tics.

PKU. Tratamiento crónico
Corregir el disbalance primario: restricción dietaria de la Phe en cantidad suficiente para mantener valores normalesSuplir los productos bloqueados: Tyr necesaria para mantener nivel normalAsegurar crecimiento adecuado

Recomendaciones para el tratamiento de PKU
Lactantes y Niños:Fenilalaninemia: 1.5 a 6 mg/dl
60 a 360 micromoles/dl1mg x 60.94 = micromoles
Adolescentes:Fenilalaninemia: hasta 10 mg/dl

Causas de hiperfenilalaninemias neonatales
Primarias: déficit de fenilalanina hidroxilasa (PKU
clásica o no clásica)
déficit de biopterinas

Otras causas
Secundarias, esporádicas:
con hipertirosinemia: transitoria neonatal (gral en prematuros)alta ingesta protéicaenfermedad hepática (incluso galactosemia o tirosinemia)
sin hipertirosinemia: transitoria neonatal, (gral prematuros)relac. a drogas (metotrexate, trimetropima)Rsta inflamatoria severaEnfermedad renal


Los errores congénitos del metabolismo son individualmente raros pero en su conjunto numerosos (10% de cuadros agudos en RN).
El neonato responde a las enfermedades con un repertorio limitado de síntomas, siendo éstos inespecíficos.

Los ECM se transmiten en forma recesiva pero, en la mayor parte de los casos aparecen como esporádicos dada la escasa población diagnosticada.
“Hereditario” no quiere decir “congénito”, por lo tanto debemos recordar las formas de presentación tardía.

Grupo 1: alteración en la síntesis o catabolismo de moléculas complejas.
Los síntomas son permanentes, progresivos e independientes de eventos intercurrentes o de alimentos ingeridos.Ej: Enfermedades lisosomales
Enfermedades peroxisomalesCGD (defectos en la glicosilación)

Grupo 2: errores del metabolismo intermedio que llevan a una intoxicación aguda o progresiva por acumulación de metabolitos tóxicos proximales al bloqueo metabólico.
Caracterizados por presentar un periodo libre de síntomas. Curso agudo o crónico y de presentación tardía e intermitenteej: Aminoacidopatias: PKU, MSUD
Acidemias orgánicas: MMA, PA, IVAErrores del ciclo de la ureaIntolerancia a los azúcares: galactosemia, fructosemia

Grupo 3: errores que comprometen el metabolismo energético.
Los síntomas se deben a un déficit energético ya sea por una falla en la producción o en su utilización.Síntomas: hipoglucemia, hiperlactacidemia, hipotonía, falla de crecimiento, síndrome de muerte súbita.Ej.: Glucogenosis tipo I
Acidemias lácticas congénitas: PC, PDHAlteración en la oxidación de los ácidosgrasos (FAO)Defectos en la cadena respiratoriamitocondrial.

Para lograr el diagnóstico debemos:
1) Pensar
2) Sospechar
3) Utilizar en forma adecuada las herramientas de diagnóstico
clínico-bioquímico.

¿Por qué?
Disponibilidad para realizar diagnóstico certero e instaurar un tratamiento eficaz.El diagnóstico y tratamiento a tiempo evitael daño neurológico y la muerte precoz.Posibilidad de realizar consejo genético.Aparición de nuevas opciones terapéuticas que cambian el pronóstico y evolución natural de la enfermedad.

Sospecha diagnóstica
Paciente que ingresa con mayor compromiso que el esperado por el relato.
Pensar en estas enfermedades al mismo tiempo que en otros diagnósticos más frecuentes (sepsis)
Interrogatorio: evolución del embarazo, muerte súbita o de causa poco clara en hermanos, hydrops fetalis, desencacenantes de los síntomas, etc.

Presentación neonatal
Repertorio clínico limitado con síntomas y signos no específicos: succión pobre, rechazo del alimento, distress respiratorio, apneas, hipotonía y/o mioclonías, vómitos, deshidratación, letargo, coma, convulsiones.Distress metabólico tipo intoxicación.Déficit de energía con compromiso multisistémico.

De inicio tardío
El 50% de las ECM son de inicio tardío.
Período libre de síntomas largo, durante los cuales el paciente tiene apariencia normal.
Precipitado por infecciones virales banales, fiebre, ayuno prolongado, ejercicio intenso u otra condición que provoque catabolismo.
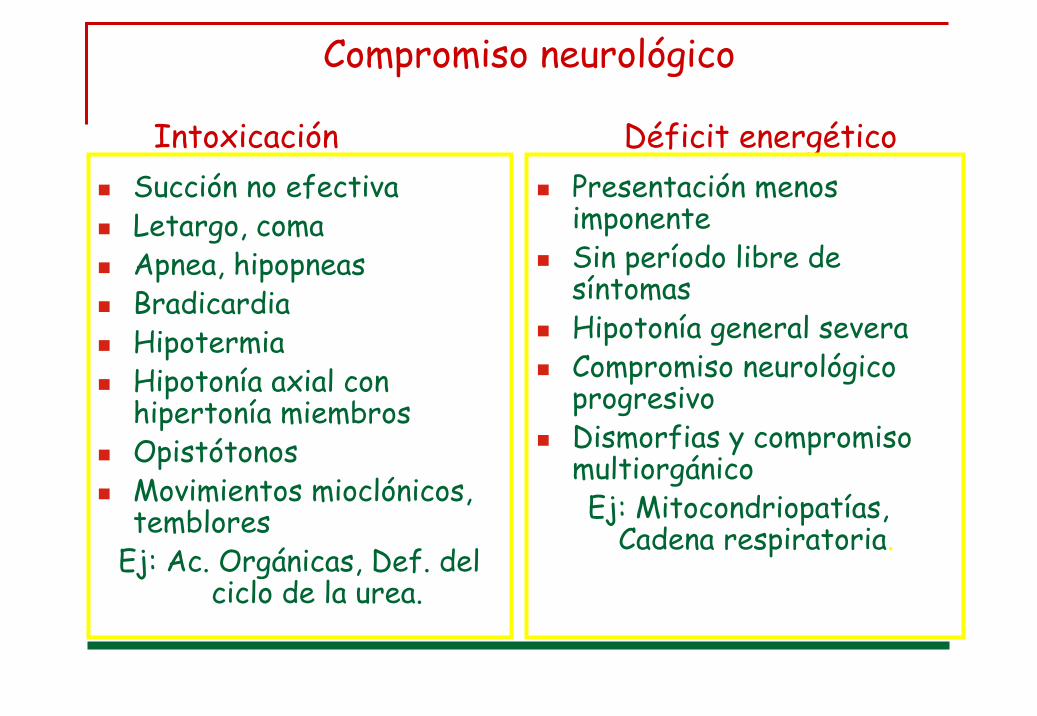
Compromiso neurológico
Intoxicación Déficit energéticoSucción no efectivaLetargo, comaApnea, hipopneasBradicardiaHipotermiaHipotonía axial con hipertonía miembrosOpistótonosMovimientos mioclónicos, temblores
Ej: Ac. Orgánicas, Def. del ciclo de la urea.
Presentación menos imponenteSin período libre de síntomas Hipotonía general severaCompromiso neurológico progresivo Dismorfias y compromiso multiorgánicoEj: Mitocondriopatías,
Cadena respiratoria.

Compromiso hepático
Hepatomegalia con hipoglucemiaEj: Glucogenosis
tipo1-3
Compromiso hepático:-ictericia-necrosis hepatocelularEj.: Fructosemia,
Galactosemia, Tirosinemia tipo1HemocromatosisCadena respiratoriaAlfa 1 antitripsina
Síndrome reye:esteatosis hepática>Tiempo protrombina> GOT GPTEj.: Defectos de la b
oxidaciónCiclo de la urea

Compromiso Cardíaco
Miocardiopatía: asociado a hipotonía Ej.: Enf Pompe
Defectos de la B oxidaciónDerrame pericárdicoEj.: CGD
Defectos conducciónEj.: Defectos de la b oxidación (long chain)


Screening neonatal
Pesquisa de enfermedades en la etapa neonatal, previo a la aparición de síntomas.
Ley nacional: 23413
Fenilcetonuria Hipotiroidismo congénitoEnfermedad fibroquísticaGalactosemiaBiotinidasa
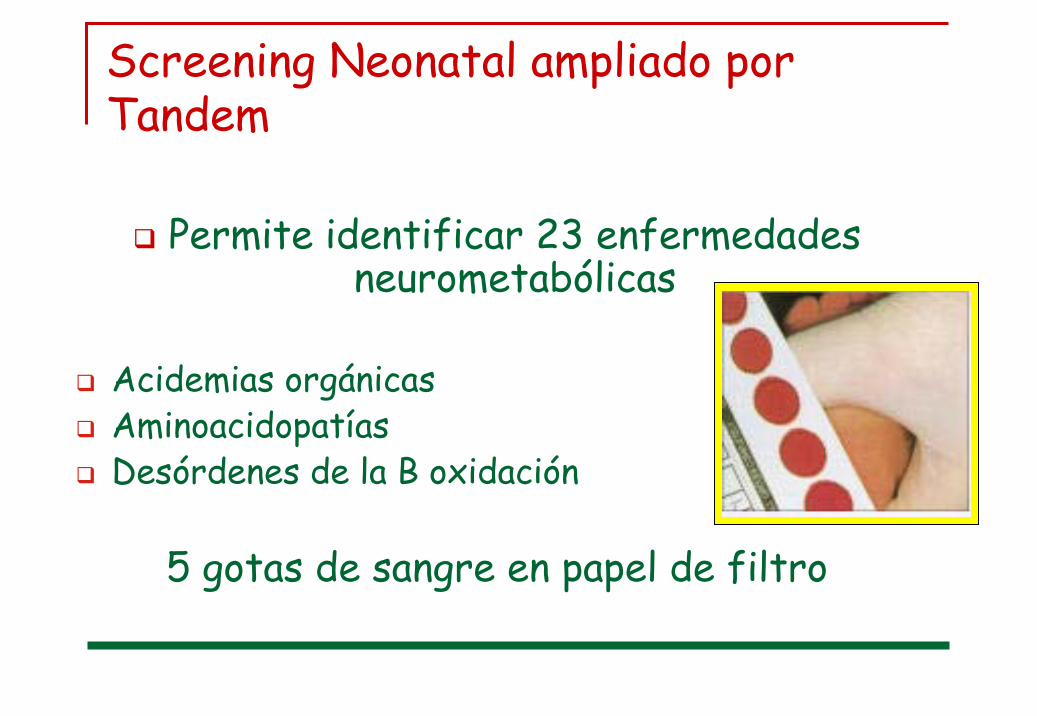
Screening Neonatal ampliado por Tandem
Permite identificar 23 enfermedades neurometabólicas
Acidemias orgánicasAminoacidopatíasDesórdenes de la B oxidación
5 gotas de sangre en papel de filtro

FenilcetonuriaGalactosemiaDef de BiotinidasaLeucinosisHomocistinuriaArgininemiaCitrulinemiaHiperglicinemiaHiperornitinemiaTirosinemiaAcidemia MetilmalónicaAciduria PropiónicaAciduria IsovaléricaAc Glutárica tipo I
Def. de HMG CoA liasaDef de Metil Crotonil CoA carboxilasaDef de carnitin palmitoil transferasa IIDef. de múltiples acyl CoA deshidrogenasaDef. de acyl CoA deshidrogenasa de cadena corta, media y largaDef de 3 OH acyl CoA DH de cadena larga
Cuáles son????

1. Intoxicación
2. Trastornos de la energía
3. Moléculas complejas
AminoacidopatiasAcidemias orgánicasDef. del ciclo de la ureaIntolerancia a azucares Metales- porfiriasNeurotransmisores
Acidemias lácticas primariasTrastornos de la cadena respiratoriaTrast. de beta oxidaciónGlucogenosis- def.de gluconeogenesisTrast. del metabolismo de creatina cerebralTrast. del metabolismo de las pentosas
LisosomalesPeroxisomalesCGD ( RE y Golgi)Colesterol

Siempre ante la sospecha de ECM...
...Extraer Muestras de Sangre y Orina ANTES de comenzar a tratar al paciente!!!

IMPORTANTE
Previo a la colocación de la VCL, debemos obtener 3-4 ml de suero congelado, gotas de sangre en papel de filtro a temperatura ambiente y 2-5 ml de orina congelada (luego de verificar la presencia de cetonas)

¿Cómo estudiarlas?Estudios de laboratorio:técnica y condiciones de la toma de
muestra: manguito, conservación, traslado, horas de ayuno, presencia de medicación, tipo de alimentación.
Los estudios específicos tienen un costo elevado. Sacados bajo condiciones no precisas pierden su valor. Siempre es conveniente asesorarse con el especialista antes de realizar el pedido.

Laboratorio Básico
HemogramaGlucemiaEstado Ac/base (Na K Cl)Ácido LácticoAmonio
Cetonuria y cuerpos reductores en orina Enzimas hepáticasEnzimas musculares Ac úricoPerfil lipídico

Laboratorios específicos
Ácido Láctico y Pirúvico (L/P <25) AmonioNefa y BOH butirato (Nefa/BOHb <1)Acilcarnitinas (gotas en papel secante)Carnitina total y libreAA plasmáticos y urinariosÁcidos orgánicos en orinaPuede ser necesario realizar estudios en LCR.
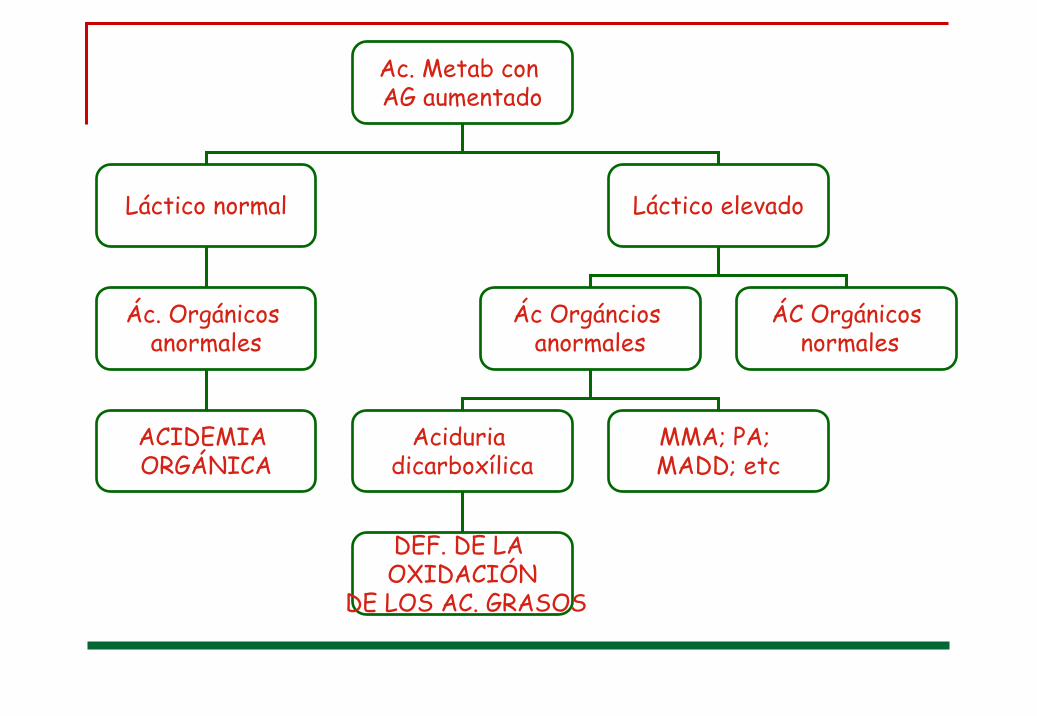
Ac. Metab con AG aumentado
Láctico normal Láctico elevado
Ác. Orgánicos anormales
ACIDEMIA ORGÁNICA
Ác Orgánciosanormales
Aciduriadicarboxílica
MMA; PA; MADD; etc
DEF. DE LA OXIDACIÓN
DE LOS AC. GRASOS
ÁC Orgánicosnormales
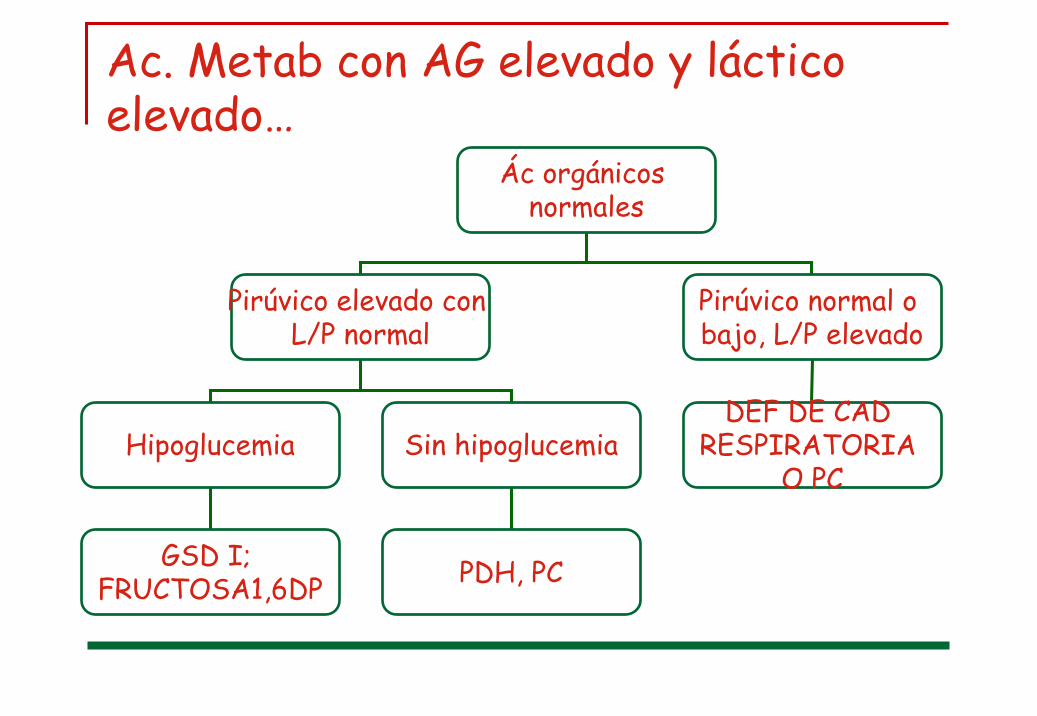
Ac. Metab con AG elevado y láctico elevado…
Ác orgánicos normales
Pirúvico elevado con L/P normal
Pirúvico normal o bajo, L/P elevado
DEF DE CAD RESPIRATORIA
O PCHipoglucemia Sin hipoglucemia
PDH, PCGSD I; FRUCTOSA1,6DP


Terapia de soporte
Evitar catabolismo: disminuir la temperatura, evitar ayunos, etc.Soporte ventilatorio o circulatorio.Rehidratación y corrección de electrolitos: sodio, potasio, calcio, fósforo, etc.Tratamiento de sepsis concomitante (ya que induce catabolismo persistente)

Tratamiento inicial de la emergencia
Intoxicación aguda por acumulación de metabolitos tóxicos
Inhibición del Catabolismo:Flujo de glucosa entre 6-8 mg/kg/min En las acidemias orgánicas pueden ser necesarios aportes aun más importantes. De ser necesario incorporar insulina.
Suspender la alimentación (máximo 48hs) Suspensión de todo aporte de azúcares que no sea
glucosa.Suspender aprote protéico

Anabolismo. NutriciónReinicio de la alimentación No superar las 48hs de ayuno
Cualquiera sea el diagnóstico hay que inhibir la producción de metabolitos tóxicos debido al catabolismo protéico endógeno
Inducción del anabolismo con dieta hipercalórica e hipoprotéica
Los micronutrientes, osmolaridad y la carga de solutos renales deben ser controlados para aportar RDA y prevenir diarrea y deshidratación

Anabolismo. Nutrición
Reinicio de la alimentación: no superar las 48hs de ayuno
Vía oral con ayuno controlado o enteral contínuo
Composición:-PROTEINAS: se comenzará con un aporte inicial 0,2 gr/kg/día, progresando lentamente con controles clínico y seriado de Eab y amonio (recordar hs de ayuno al tomar la muestra)Si hay sospecha diagnóstica: eliminar aminoácido ofensor (flas especiales)

TRATAMIENTO DIETARIO: A largo plazo
PROTEINAS (FEN, TIR, LEUC)CALORIASSUSTITUTO PROTEICO (AAEsenciales, fórmulas sin AAs Limitados)SUPLEMENTOS VIT/MINLIQUIDOS

Nutrición. Principales composiciones
Bajo contenido protéico (defectos del ciclo de la urea, acidurias orgánicas con mezcla de aa apropiada)Restringida en hidratos de carbono (glucogenosis, galactosemia, intolerancia hereditaria a la fructosa)Hiperhidrocarbonada con restricción de grasa (defectos de la ß oxidación)Normal o bajo contenido de azúcares e hipergrasa (PDH y enfermedades de cadena respiratoria)

Vías de alimentación
Oral: primera elección si el estado clínico lo permite.Enteral contínuaParenteral: altos requerimientos enegéticos o de glucosa. Inicial: glucosa 15-20% y lípidos 2-3 g/k/d, aa para lograr requerimientos mínimos y con controles bioquímicos.

Anabolismo. Nutrición
HIDRATOS DE CARBONO: no se utilizarálactosa, sacarosa o fructosa. Incorporar polisacáridos y/o almidones crudos según tolerancia.
LÍPIDOS: como fuente energética. Si no hay sospecha de alteración de la cetogénesis y/o cetolisis. En dicho caso en gral se aporta hasta un 20-25% del VCT
Glucogenosis tipo 1

Glucogenosis
Defectos enzimáticos en la degradación del glucógeno, incidencia 1:20.000
Síntomas y signos grales: fallo de crecimiento, hipoglucemia, hiperlactacidemia, hepatomegalia hipotonía generalizada, miopatía, cardiomiopatía, etc.

Glucogenosis tipo I
Clínica: Hipoglucemia frecuentes con ayunos cortos, en general
acompañadas de hiperventilación por la acidosis láctica.
Desarrollo psicomotor depende de la gravedad y frecuencia de las hipoglucemias en el primer año de vida
Abdomen prominente por la hepatomegalia, obesidad truncal, cara de muñeca, músculos hipotróficos.
Nefromegalia, no esplenomegaliaTalla baja Sangrados fáciles, epistaxis, etc. Diarreas frecuentes.
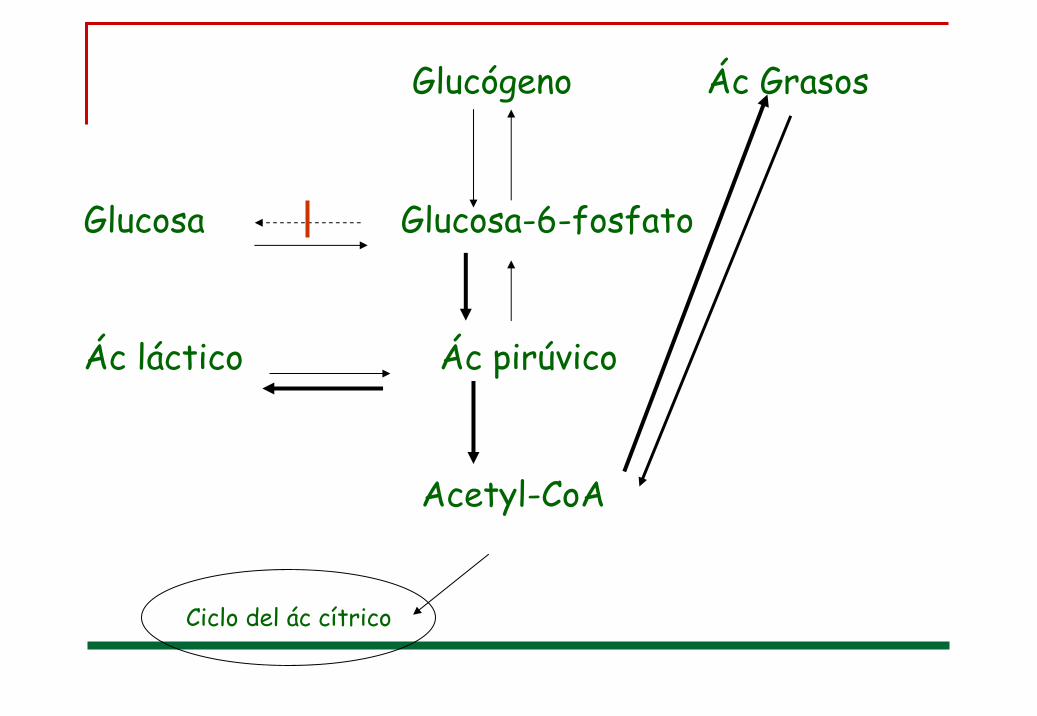
Glucógeno Ác Grasos
Glucosa Glucosa-6-fosfato
Ác láctico Ác pirúvico
Acetyl-CoA
Ciclo del ác cítrico

Diagnóstico
Datos clínicos y de laboratorios:
Hipoglucemia + hiperlactacidemia con hepatomegalia.
Además hiperlipemia, hiperuricemia, aumento de enzimas hepáticas.

Pruebas funcionales
Curva de tolerancia oral a la glucosa: del láctico normal alto inicial, cae a normal con el aumento de la glucosa (paradojal).
Test de glucagon: marcado descenso de la glucemia con aumento de la lactacidemia
Glu Lactb 42 3,1
30' 74 2,560' 89 2,1120' 92 1,8

Prueba de glucagonT Gluc. Ac.Láct.
mg% mg%
B: 38 61,210’: 25 20’: 1630’: 17 65,745’: 60’:



Defectos de la β oxidación
Se han descripto alrededor de 12 defectos en la vía de la β oxidación.La mayoría se presenta en la infancia temprana como un coma hipoglucémico hipocetósico de alta mortalidad inducido por el ayuno.En algunos casos puede haber historia de
debilidad muscular o rabdomiolisis inducida por ejercicio o cardiomiopatía crónica

MCADMás común, incidencia estimada 1:10.000Presentación hepática: 3-24 meses de edad, niños aparentemente sanos con un episodio de enfermedad que provoque catabolismo y un excesivo ayuno. Severa hipoglucemia con letargo, nauseas, vómitos y un rápido progreso al coma en 1-2 horas de evolución con severo edema cerebral y daño neurológico permanente o muerte.

MCAD: Intercurrencias
Glucosa: Aporte endovenoso con flujo alto (10 mg/k/min.)
Esto estimula la secreción de insulina a niveles que inhiben la oxidación de ácidos grasos hepática y muscular y bloquea la lipólisis en tejido graso.
Carnitina: 100 mg/k/día.

MCAD: sostén
Prevenir crisis ajustando la dieta para minimizar el stress del ayuno
Ajustar ingestas regladas de H de C.(fundamentalmente al acostarse y no ayunos mayores a 6-8 hs).Si apetito disminuido: agregar ingestas de hidratos de carbono.Restricción de grasas a 15-20% del total del valor calórico diario.