universidad de guayaquil facultad de...
TRANSCRIPT

UNIVERSIDAD DE GUAYAQUIL
FACULTAD DE CIENCIAS QUÍMICAS
MODALIDAD: INVESTIGACIÓN
TEMA:
ESTUDIO DE LOS PERFILES DE DISOLUCIÓN DE LAS TABLETAS DE
DILTIAZEM 60 mg COMERCIALIZADOS EN ECUADOR
TRABAJO DE TITULACIÓN PRESENTADO COMO REQUISITO PREVIO PARA
OPTAR AL GRADO DE QUÍMICO FARMACÉUTICO.
AUTORES:
CALDERÓN RENGEL KATHERINE YULIANA
GONZÁLEZ CARRILLO KASTUSCA CECILIA
TUTORA:
Q.F. PILAR ASUNCIÓN SOLEDISPA CAÑARTE, MsC.
GUAYAQUIL - ECUADOR.
2017- 2018



II

III

IV

V

VI

VII

VIII

IX
AGRADECIMIENTO
A Jehová Dios por ser la guía en nuestro camino. A nuestros padres por ser
nuestro apoyo incondicional con sus consejos. A nuestros esposos quienes nos
brindaron su amor, paciencia y apoyo constante en el transcurso de este
proyecto de vida. A nuestros hijos quienes son la felicidad en nuestras vidas y a
quienes dedicamos este proyecto de graduación.
A la Universidad de Guayaquil por brindarnos este espacio de formación, la
Facultad de Ciencias Químicas donde se cultivan las enseñanzas que todo
Químico Farmacéutico necesita para ejercer en el campo profesional; a nuestros
queridos maestros que aportaron con sus conocimientos a esta formación.

X

XI

XII
ÍNDICE APROBACIÓN DEL TUTOR ................................. ¡Error! Marcador no definido.
CERTIFICADO DEL TRIBUNAL ........................... ¡Error! Marcador no definido.
CARTA DE AUTORÍA DE TITULACIÓN ............................................................. VI
AGRADECIMIENTO ........................................................................................... IX
ÍNDICE .............................................................................................................. XII
ÍNDICE DE TABLAS ........................................................................................ XIV
ÍNDICE DE FIGURAS ....................................................................................... XV
ABREVIATURAS ............................................................................................. XVI
RESUMEN ...................................................................................................... XVII
ABSTRACT ................................................................................................... XVIII
INTRODUCCIÓN ................................................................................................. 1
PROBLEMA ......................................................................................................... 3
HIPÓTESIS ......................................................................................................... 3
OBJETIVOS ........................................................................................................ 3
CAPÍTULO I: REVISIÓN BIBLIOGRÁFICA .......................................................... 4
I.1. Antecedentes ............................................................................................. 4
I.2 Base Teórica ......................................................................................... 7
I.2.1.1 Diagnóstico ..................................................................................... 8
I.2.1.2 Mediciones de la presión arterial .................................................... 8
I.2.1.3 Síntomas ........................................................................................ 9
I.2.2.1 Efecto farmacológico y Mecanismo de Acción ................................ 9
I.2.2.2 Reacciones Adversas ................................................................... 10
I.2.2.3 Contraindicaciones ....................................................................... 10
I.2.3 Medicamento original, innovador o de patente. ................................... 10
I.2.3.1 Medicamento genérico ..................................................................... 10
I.2.4 Biodisponibilidad .............................................................................. 11
I.2.4.1 Bioexención .................................................................................. 11
I.2.4.2 Pruebas in Vitro ............................................................................ 11
I.2.5 Equivalencia farmacéutica ............................................................... 11
I.2.6 Farmacodinamia .............................................................................. 12
I.2.7 Farmacocinética: .............................................................................. 12
I.2.8 Sistema de Clasificación Biofarmacéutica ........................................... 12

XIII
I.2.8.1 Perfiles de Disolución ................................................................... 12
I.2.8.2 Comparación y evaluación de perfiles de disolución ........................ 13
I.2.8.3 Categorías de especificaciones de pruebas y perfiles de disolución 14
I.2.9 Enfoque independiente de modelo utilizando un factor de Diferencia y
Similitud f1 y f2 ........................................................................................... 15
I.2.9.1 Aparatos .......................................................................................... 15
I.2.9.2 Aparato 1 ......................................................................................... 16
I.2.9.3 Aparato 2 ......................................................................................... 16
I.2.10 Reglamento a la ley de medicamentos genéricos de uso humano .... 16
I.2.10.1 De la adquisición de medicamentos genéricos .............................. 16
I.2.10.2 Del registro sanitario y homologación ............................................ 17
CAPÍTULO II. MATERIALES Y METODOS ....................................................... 18
II.1 Materiales ........................................................................................ 18
II.2 Métodos ........................................................................................... 19
II.2.3 Métodos de análisis e interpretación de los resultados..................... 20
II.3 Variables .......................................................................................... 20
II.4 Muestreo .......................................................................................... 21
II.4.1 Tipo de Muestreo ............................................................................. 21
II.5 Población y muestra ......................................................................... 21
II.6 Criterios de inclusión y exclusión ..................................................... 21
CAPITULO III. RESULTADOS Y DISCUSIÓN ................................................... 23
III.1 Comprobación del modelo independiente a través del factor de similitud f2
y diferencia f1 para cada lote de los genéricos en estudio, de los diferentes
medios de disolución. ..................................................................................... 23
CAPITULO IV. CONCLUSIONES Y RECOMENDACIONES ............................. 37
IV.1 CONCLUSIONES ................................................................................... 37
IV.2 RECOMENDACIONES .............................................................................. 39
Bibliografía......................................................................................................... 40

XIV
ÍNDICE DE TABLAS
Tabla I Valores de Presión Arterial ............................................................................... 8
Tabla II Materiales usados para la investigación ...................................................... 18
Tabla III Operacionalización ......................................................................................... 20
Tabla IV Concentración y porcentaje disuelto del medicamento innovador y
medicamento genérico a pH 1.2 .................................................................................. 24
Tabla V Comprobación del Factor de similitud y diferencia para cada lote del
medicamento genérico a pH 1.2 .................................................................................. 27
Tabla VI Concentración y porcentaje disuelto del medicamento innovador y
medicamento genérico a pH 4.5 .................................................................................. 29
Tabla VII Comprobación del Factor de similitud y diferencia para cada lote del
medicamento genérico a pH4.5 ................................................................................... 31
Tabla VIII Concentración y porcentaje disuelto del medicamento innovador y
medicamento genérico a pH 6.8 .................................................................................. 33
Tabla IX Comprobación del Factor de similitud y diferencia para cada lote del
medicamento genérico a pH 6.8 .................................................................................. 35

XV
ÍNDICE DE FIGURAS
Figura 1 Estructura Química de Diltiazem ................................................................... 9
Figura 2 Sistema de Clasificación Biofarmacéutica (Guzmán, 2013). .................. 12
Figura 3 Categorías de especificaciones y perfiles de disolución (FDA, 2015). . 14
Figura 4 Muestra la curva de concentración Vs tiempo de genérico frente al de
referencia ......................................................................................................................... 28
Figura 5 Muestra la curva de concentración vs Tiempo de genérico frente al de
referencia ......................................................................................................................... 32
Figura 6 Muestra la curva de concentración vs Tiempo de genérico frente al de
referencia ......................................................................................................................... 36

XVI
ABREVIATURAS
Agencia Europea de Medicamentos (EMA)
Consejo Nacional de Salud (CONASA)
Eficiencia de Disolución (EF)
Encuesta nacional de salud (ENSANUT)
Factor de Diferencia (F1)
Factor de riesgo cardiovascular (FRCV)
Farmacopea de Estados Unidos (USP)
Factor de Similitud (F2)
Food and Drug Administration (FDA)
Hipertensión Arterial (HTA)
Instituto Nacional de Estadística y Censos (INEC)
Ministerio de salud pública (MSP)
Manufacturing Equipment Addendum – FDA (SUPAC)
New Drug Application (NDA)
Organización Mundial de la Salud (OMS)
Sistema de Clasificación Biofarmacéutica (SCB)
Tiempo Medio de Disolución (MDT)

XVII
RESUMEN
Los perfiles de disolución permiten demostrar, si un principio activo tiene el
mismo desempeño farmacocinético en un medicamento genérico, con respeto al
medicamento de referencia. Diltiazem es una benzotiazepina considerada
antagonista de los canales del calcio, es usado para el tratamiento en pacientes
que presentan angina de pecho e hipertensión. Para la elaboración de este
trabajo se emplearon tres lotes de diltiazem tabletas de 60 mg y un lote del
medicamento de referencia; para la comparación de los perfiles de disolución se
utilizó un disolutor (aparato 2) y como medios de disolución buffer de Ácido
Clorhídrico 0.1 N pH 1.2, o simulación de jugo gástrico, Acetato de Potasio a pH
4.5 solución amortiguadora y Monofosfato Básico de Potasio a pH 6.8 o jugo
intestinal simulado fueron cuantificados a través de técnica UV VIS. Los cálculos
se realizaron a través de un modelo Independiente, determinando el factor de
similitud f2 y el factor de diferencia f1. El análisis comparativo de los lotes,
permitió evidenciar marcadas diferencias en cuanto a la liberación in vitro del
principio activo, a los 180 minutos que es el tiempo máximo de la toma de
muestra el lote A1, a pH 1.2 libera 60.57 mg de principio activo, a pH 4.5 libera
61.33 mg de principio activo, y a pH 6.8 libera 61.07 mg de principio activo. Se
concluye que, de los tres lotes del medicamento genérico, solo el lote A1 cumple
con el valor de f1 y f2 y tiene un mejor comportamiento con la velocidad de
disolución y porcentaje de eficiencia.
Palabras Claves: Diltiazem, Equivalentes, Intercambiables, Perfiles de
Disolución

XVIII
ABSTRACT
The dissolution profiles allow to demonstrate, if an active ingredient has the
same pharmacokinetic performance in a generic medicine, with respect to the
reference medicine. Diltiazem is a benzothiazepine considered to be an
antagonist of calcium channels, it is used for the treatment of patients with
angina pectoris and hypertension. For the elaboration of this work, three
batches of diltiazem tablets of 60 mg and a batch of the reference medicine
were used; For the comparison of the dissolution profiles, a dissolutor
(apparatus 2) was used as dissolution media. Buffer of Hydrochloric Acid 0.1 N
pH 1.2, or simulation of gastric juice, Potassium Acetate at pH 4.5 buffer
solution and Basic Potassium Monophosphate a pH 6.8 or simulated intestinal
juice were quantified through the UV VIS technique. The calculations were
made through an independent model statistical method, determining the
Similarity factor F2 and the Difference factor F1. The comparative analysis of
the lots allowed to show marked differences regarding the in vitro release of
the active principle, the batch, at 180 minutes which is the maximum time of
the A1 sample, at pH 1.2 it releases 62.36 mg of principle active,at pH 4.5
releases 61.33 mg of active principle, to pH 6.8 liberi 61.07 mg of active
principle. It is concluded that, of the three batches of the generic drug, only lot
A1 has a better behavior with dissolution speed and percentage of efficiency.
Keywords: Diltiazem, Equivalent, Interchangeable, Profiles of Dissolution

1
INTRODUCCIÓN
La hipertensión es un factor de riesgo cardiovascular muy prevalente en
el mundo, y especialmente abrumador en los países de bajos y de medianos
ingresos. Informes recientes de la Organización Mundial de la Salud (OMS) y del
banco mundial destacan la importancia de las enfermedades crónicas tales como
la hipertensión, como obstáculo al logro de un buen estado de salud (Salud,
2015).
La OMS afirma que, de cada 100.000 ecuatorianos, 1.373 sufren de esta
afección. Esta cifra coincide con la Encuesta Nacional de salud (ENSANUT); una
investigación realizada por el Ministerio de Salud Pública (MSP) y el Instituto
Nacional de Estadística y Censos (INEC), la cual indica que más de un tercio de
los habitantes mayores de 10 años (3’187.665) es pre hipertenso y 717.529
ciudadanos de 10 a 59 años sufre de hipertensión arterial (ENSANUT, 2012).
Para el tratamiento de esta patología, uno de los medicamentos utilizado
es diltiazem, que es un derivado de la benzotiazepina, bloqueante de los canales
de calcio, utilizado en la farmacoterapia de la hipertensión y consta en el Cuadro
Nacional de Medicamentos Básicos y Registro Terapéutico del Ecuador (CNMB),
(Consejo Nacional de Salud, 2013).
Debido a que el incremento de los costos de los medicamentos
innovadores se ha convertido en un aspecto trascendente en la política sanitaria,
comprometiendo los presupuestos de los sistemas de salud pública y privada; en
el Ecuador se estimula la producción de medicamentos genéricos con la finalidad
de limitar el gasto farmacéutico ya que es deber del estado garantizar la salud de
los ecuatorianos. Dentro de la oferta del mercado farmacéutico, encontramos
medicamentos innovadores como genéricos y estos últimos cada vez continúan
teniendo mayor participación en el mercado mundial por su bajo costo e
intercambiabilidad (Bejarano, 2014).

2
Uno de los principales objetivos de las ciencias farmacéuticas es
asegurar la calidad de los productos farmacéuticos durante todo su ciclo de vida,
calidad que se evalúa mediante estudios de laboratorio como identificación,
potencia, pureza, disolución, estabilidad de la forma farmacéutica (Bejarano,
2014).
No obstante, la evaluación de calidad fisicoquímica comparativa entre dos
productos farmacéuticos de administración oral sólo permite demostrar la
equivalencia farmacéutica entre los medicamentos, pero no es suficiente para
asegurar la intercambiabilidad durante la práctica clínica (Bejarano, 2014).
Actualmente se acepta la intercambiabilidad entre medicamentos,
asegurando que los mismos sean bioequivalentes con el medicamento que ha
demostrado ser eficaz y seguro en los estudios clínicos. Dentro de los estudios
aceptados por las Agencias Regulatorias, para demostrar bioequivalencia están
los estudios in vitro, sin embargo, la introducción de medicamentos genéricos
inicialmente ha estado acompañada de problemas de calidad, medicamentos de
baja concentraciones o pobres disoluciones que podrían afectar la efectividad
del tratamiento, dentro del marco legal se respalda que se cumplan con análisis
de calidad post- registro. (Bejarano, 2014)
De acuerdo a la Guía de la Food and Drug Administration (FDA), los
ensayos de disolución son necesarios y en consecuencia requeridos porque son
utilizados como una herramienta para identificar los factores que influyen en la
biodisponibilidad, en el control de calidad y para garantizar que los perfiles de
disolución siguen siendo similares a los obtenidos con los lotes del ensayo
clínico; además puede ser utilizado para demostrar similitud entre diferentes
formulaciones de una sustancia activa y el medicamento de referencia (Jordi,
2011). Por consecuencia los estudios comparativos de Disolución in vitro son
útiles cuando la disolución como tal es un paso limitante de la absorción (Jung et
al, 2012).
Debido a la naturaleza de los factores mencionados con anterioridad es
imprescindible realizar pruebas in vitro como es la Disolución y los Perfiles de

3
Disolución con el afán de interpolar o proyectar el comportamiento del fármaco
en el organismo vivo, asegurando su calidad (Jung et al, 2012).
PROBLEMA
¿Las tabletas de Diltiazem 60 mg comercializado en Ecuador serán
equivalente terapéutico con el medicamento innovador Incoril®?
HIPÓTESIS
Las tabletas de 60 mg genérico comercializado en Ecuador, analizados
mediante el empleo de los perfiles de disolución son equivalentes con el
innovador Incoril®.
OBJETIVOS
Objetivo general
Estudiar los perfiles de disolución de las tabletas de diltiazem 60mg
comercializadas en Ecuador.
Objetivos específicos
•Obtener el perfil de disolución de la tableta de Incoril® como referencia
para el diltIasem genérico.
•Demostrar la equivalencia in vitro entre el fármaco de referencia y el
genérico, mediante los perfiles de disolución y la determinación de los factores
de diferencia f1 y de similitud f2

4
CAPÍTULO I: REVISIÓN BIBLIOGRÁFICA
I.1. Antecedentes
La calidad de los medicamentos genéricos debe estar garantizada por
estudios de bioequivalencia, en conjunto con las directrices de la OMS. Con el fin
de mejorar su eficiencia, se fortaleció las políticas de medicamentos genéricos.
En 1981, la Organización Mundial de la Salud estableció el Programa de Acción
para Medicamentos esenciales, para apoyar a los países en el desarrollo de su
política nacional de medicamentos y para trabajar hacia un uso racional de
medicamentos (Pilon, 2016).
A partir del estudio comparativo en países latinoamericanos, mediante la
metodología de documentos oficiales normativos y reglamentarios, en América
latina, México y Brasil aplican estudios de bioequivalencia como requisito de
calidad e intercambiabilidad de genérico, Argentina, Colombia, Chile lo aplican
en fármacos de riesgo sanitario elevado, en Perú actualmente ha sido
incorporado para el registro de medicamentos genéricos. Como resultado se
discute sobre los requisitos legales y el impacto sobre la bioequivalencia sobre
las prestaciones farmacéuticas, favoreciendo la incorporación de medicamentos
genéricos, seguros, eficaces y de bajo costo (Placencia, 2010).
En Ecuador actualmente el organismo regulador que es la Agencia
Nacional de Control y Vigilancia Sanitaria (ARCSA) en el año 2017 hizo la
reforma al reglamento de registro sanitario para medicamentos en general
realizo la inclusión de estudios de equivalencia in vitro y ensayos de disolución
(ARCSA, 2017).
El impacto sanitario de los estudios de Bioequivalencia (BE) en países
desarrollados es la garantía de calidad de los medicamentos genéricos; en
países tales como Francia y España. En Estados Unidos (EE.UU.) en 1970, en
Canadá en 1992, y Australia en 1993 se incorporó formalmente la exigencia de

5
estas evaluaciones, además de las de seguridad y efectividad para incluir un
medicamento en el sistema de financiamiento público (Placencia, 2010).
En el 2008 Mendoza y Rodriguez evaluaron la capacidad del método de
disolución farmacopéico para discernir entre lotes de diferentes fabricantes de
albendazol. Los estudios fueron realizados seleccionando 6 medicamentos
genéricos conteniendo albendazol y el Innovador (Zentel®). Se empleó el
aparato 2 a 50 rpm y 900 ml de HCL 0.1N como medio de disolución. Se
determinó el porcentaje liberado a los 10, 20, 30, 45, 60 y 90 minutos. Los
resultados mostraron diferencias en la velocidad de disolución, así como en la
cantidad disuelta entre los productos estudiados. Los perfiles de disolución
fueron comparados usando la prueba de f2, se demostraron que solo dos de los
medicamentos fueron similares al innovador (Mendoza et al, 2008).
La OMS ha establecido pautas en relación de evaluación de
medicamentos genéricos en cuanto a la calidad, materia prima, etiquetado, datos
toxicológicos y farmacológicos e intercambiabilidad, siendo requisito el estudio
de bioequivalencia.
Según Ramírez (2014), los estudios in vitro son métodos que han sido
empleados para la comparación de perfiles de disolución de formas
farmacéuticas sólidas de liberación inmediata, así, sin embargo, todos los
medicamentos, sean estos genéricos o no, deben someterse a sistemas de
garantía de calidad, cuando se han realizado estudios de bioequivalencia, y más
aún, estudios de biodisponibilidad sobre algún medicamento multiorigen
esperando que no haya diferencias en su eficacia y seguridad.
En el estudio de ketoprofeno, realizado por Lobos (2013) con el fin de
demostrar la equivalencia terapéutica, realizó pruebas in vitro, entre dos
formulaciones farmacéuticas que contienen este principio activo y a
medicamentos de la Clase II (según Clasificación Biofarmacéutica) el objetivo
general del estudio se asocia con la posibilidad de simplificar el procedimiento de
determinación de estudios de bioequivalencia entre formulaciones farmacéuticas
que cumplan una serie de características establecidas por las agencias
regulatorias a nivel mundial (Lobos, 2013).

6
De igual manera Lobos (2013) evaluó la hipótesis de que el ketoprofeno
en cápsulas de 50 mg (Doloketason®) cumple con los requisitos propuestos por
la OMS para ser eximido de la realización de estudios in vivo para demostrar la
equivalencia terapéutica con el innovador. Para este estudio, se realizaron
pruebas de solubilidad mediante el método de agitación en matraz considerando
una serie de pH elegidos conforme a las características fisicoquímicas del
medicamento, además de la evaluación del perfil de disolución en base al factor
de similitud f2 y la revisión bibliográfica acerca de su permeabilidad (Lobos,
2013).
Un estudio realizado por Perez (2013) además de los perfiles de
disolución, se validó y demostró el cumplimiento de los criterios de linealidad,
precisión, exactitud y especificidad de la metodología analítica en tres medios
de disolución de pH de 1.2, 4.5 y 6.8 para la cuantificación del principio activo
metformina en pruebas de disolución in vitro, los resultados mostraron que solo
uno de los cuatro medicamentos liberaron más del 80% del fármaco en 30
minutos, concluyendo que no todos los medicamentos de estudio son
equivalentes terapéuticos (Pérez, 2013).
Lobos (2013) evaluó las características físicas, químicas y
biofarmacéuticas como variación de peso, dureza, desintegración, test de
disolución, perfil de disolución, eficiencia de la disolución y valoración de
principio activo, esta última, a partir de metodologías optimizadas y validadas.
Los medicamentos en estudios fueron de 19 marcas comerciales de captopril y
losartan, expendidas en droguerías y farmacias de cuatro principales ciudades
del país de Colombia: Bogotá, Cartagena, Cali y Barranquilla (Lobos, 2013).
Según Terán (2017), el proceso de evaluación de la calidad de los
medicamentos se da mediante el proceso estándar en la realización de las
pruebas de bioequivalencia; en la actualidad en el Ecuador las mencionadas
pruebas requieren de laboratorios especiales, de los cuales no se dispone en el
país. Entonces, si es que no se pueden hacer los análisis respectivos, como se
pueden validar efectivamente los medicamentos o ver si caen en la categoría de
medicamento genérico. Por esta razón, desafortunadamente en el país, existe un

7
vasto número de medicamentos que se denominan copia, que es el que se
fabrica sin haber demostrado que son bioequivalentes (Terán, 2017).
En la facultad de Ciencias Químicas de la Universidad de Guayaquil de
acuerdo con el contexto mencionado y como aporte al país se han realizado
análisis de Bioexención a través de estudios de fármacos tales como losartán,
ciprofloxacino, entre otros.
I.2 Base Teórica
I.2.1 Hipertensión Arterial
La Encuesta Nacional de Salud de Ecuador (ENSANUT-ECU) permite
contar con información del comportamiento de la presión arterial sistólica y
diastólica, a partir de las cuales se define la hipertensión arterial, como uno de
los grandes retos en salud. La presión arterial es una medición que refleja el
funcionamiento del sistema cardiovascular y es un indicador que se constituye
en uno de los primeros pasos para generar grupos de riesgo en el ámbito de las
enfermedades cardiovasculares (ENSANUT, 2012).
Según OMS, las enfermedades cardiovasculares son las principales
causas de morbilidad y mortalidad en los adultos de los países industrializados y
países en vías de desarrollo. La Hipertensión Arterial (HTA) representa por sí
misma una enfermedad y también un importante factor de riesgo cardiovascular
y su prevalencia aumenta con la edad, puede provocar cardiopatías, accidentes
cerebrovasculares, insuficiencia renal, mortalidad y discapacidad prematura
(Rivero, 2017).
La hipertensión constituye una de las principales causas de muerte en el
mundo, ya que junto a otros factores de riesgo son precedentes de un mal estilo
de vida en donde influye una mala alimentación, falta de actividad física,
consumo de alcohol, tabaco, entre otros malos hábitos (OMS, 2015).
La hipertensión, también conocida como presión arterial alta o elevada,
es un trastorno en el que los vasos sanguíneos tienen una presión
persistentemente alta, lo que puede dañarlos. Cada vez que el corazón late,

8
bombea sangre a los vasos, la cual es llevada a todas las partes del cuerpo, por
medio de la presión arterial que ejerce la sangre contra las paredes de los vasos
(arterias) al ser bombeada por el corazón. Cuanto más alta es la presión, más
esfuerzo tiene que realizar el corazón para bombear (OMS, 2015).
I.2.1.1 Diagnóstico
Esta enfermedad vascular generalizada se detecta de manera indirecta a
través de las mediciones con el esfigmomanómetro, que no siempre identifica a
los sujetos con enfermedad, he aquí la importancia de determinar la prevalencia
de los factores y riesgo que llevan de padecer de hipertensión y su relación con
los malos hábitos (Rivero, 2017).
I.2.1.2 Mediciones de la presión arterial
Estudios recientes en América Latina corroboraron la prevalencia de la
hipertensión arterial, el colesterol elevado en plasma, los trastornos metabólicos
y otros factores de riesgo cardiovascular (FRCV), todo esto sumado a la falta de
control de la enfermedad, la escasa adhesión al tratamiento y el uso de pocas
medidas preventivas, lo que derivó en la necesidad de educar tanto al médico
como al paciente (Pulido et al, 2016).
La presión arterial normal en adultos es de 120 mm Hg cuando el corazón
late (presión sistólica) y de 80 mm Hg cuando el corazón se relaja (presión
diastólica). Cuando la presión sistólica es igual o superior a 140 mm Hg y/o la
prensión diastólica es igual o superior a 90 mm Hg, la presión arterial se
considera alta o elevada (OMS, 2015).
Tabla I Valores de Presión Arterial
Baja Normal Alta
< 120 – 80
mmHg
120– 80
mmHg
140 – 90
mmHg
9
I.2.1.3 Síntomas
La hipertensión arterial es una enfermedad asintomática, por esta razón
es conocida como el "asesino silencioso". En ocasiones, la hipertensión causa
síntomas como dolor de cabeza, dificultad respiratoria, vértigos, dolor torácico,
palpitaciones del corazón y hemorragias nasales (OMS, 2015).
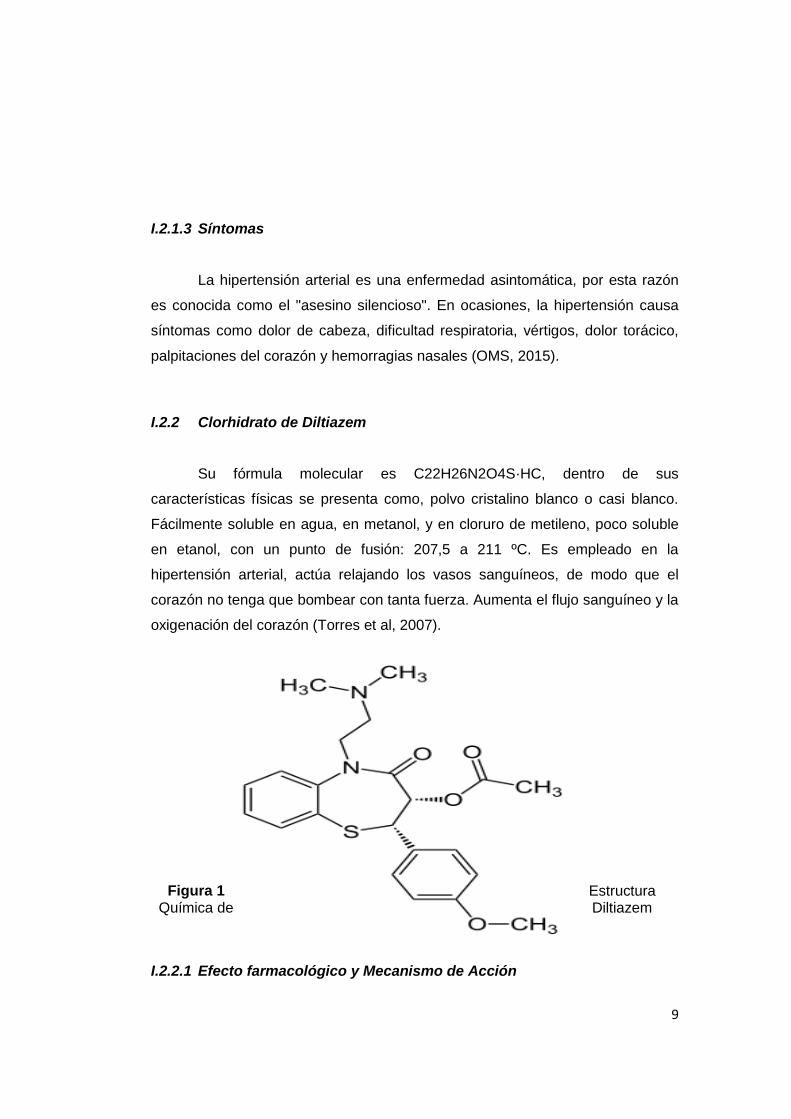
I.2.2 Clorhidrato de Diltiazem
Su fórmula molecular es C22H26N2O4S·HC, dentro de sus
características físicas se presenta como, polvo cristalino blanco o casi blanco.
Fácilmente soluble en agua, en metanol, y en cloruro de metileno, poco soluble
en etanol, con un punto de fusión: 207,5 a 211 ºC. Es empleado en la
hipertensión arterial, actúa relajando los vasos sanguíneos, de modo que el
corazón no tenga que bombear con tanta fuerza. Aumenta el flujo sanguíneo y la
oxigenación del corazón (Torres et al, 2007).
Figura 1 Estructura Química de Diltiazem
I.2.2.1 Efecto farmacológico y Mecanismo de Acción

10
El Diltiazem se usa para tratar la presión arterial alta y control de angina
(dolor en el pecho). Pertenece a una clase de medicamentos llamados
bloqueadores de los canales de calcio. Tiene acción anti arrítmica de tipo IV,
antihipertensiva, anti anginosa, y vasodilatadora periférica (Torres et al, 2007).
I.2.2.2 Reacciones Adversas
Las reacciones adversas más comunes son: inflamación de la cara, ojos,
labios, lengua, manos, brazos, pies, tobillos y pantorrillas. Dificultad al respirar o
tragar, desvanecimiento, sarpullido, coloración amarillenta en la piel y ojos
(Torres et al, 2007).
I.2.2.3 Contraindicaciones
Está contraindicado en caso de shock cardiogénico, hipotensión, bloqueo
cardiaco de grado II o III, en embarazo, lactancia y menores de 18 años (Torres
et al, 2007).
I.2.3 Medicamento original, innovador o de patente.
Se define como medicamento innovador, original o de patente aquel que
contiene un principio activo nuevo con el cual se ha realizado una investigación
única y un desarrollo completo desde su síntesis química hasta su utilización
clínica. Se lo denomina también medicamento de referencia y es el que se
emplea como patrón en estudios de bioequivalencia dado que aporta datos de
seguridad y eficacia (Miranda, 2015).
I.2.3.1 Medicamento genérico
Según la Organización Mundial de la Salud, un medicamento genérico es
un medicamento con las mismas características que otro medicamento cuya
patente ha caducado y que se denomina “medicamento de referencia”. Se deben
realizar una serie de estudios tales como: pre clínicos y clínicos para demostrar
que el medicamento original y el medicamento genérico son equivalentes e
intercambiables (Imira, 2013).

11
I.2.4 Biodisponibilidad
Desde el punto de vista farmacéutico se denomina a la biodisponibilidad
como la fracción y la velocidad a la cual la dosis administrada de un fármaco
alcanza su diana terapéutica tales como los canales, transportadores,
receptores, siendo estas moléculas proteicas, lo que implica llegar hasta el tejido
donde actúa. Esta cuantificación es posible determinando el porcentaje del
fármaco en el plasma sanguíneo (Luján, 2013).
I.2.4.1 Bioexención
La bioexención es el término al cual hace referencia a la exención de los
estudios de biodisponibilidad in vivo para demostrar la bioequivalencia de las
formulaciones orales de liberación inmediata de un mismo principio activo
mediante un estudio comparativo de los perfiles de disolución in vitro (Delgado et
al, 2016).
I.2.4.2 Pruebas in Vitro
Las pruebas in vitro están constituidas por estudios comparativos de
perfiles de disolución, en donde se determina la cantidad o porcentaje del
principio activo disuelta en función del tiempo bajo condiciones controladas y
validadas (Saavedra 2013).
I.2.5 Equivalencia farmacéutica
Propiedad que señala que dos medicamentos, siendo equivalentes
farmacéuticos, presentan perfiles de concentración plasmática versus tiempo
estadísticamente iguales (Delgado et al, 2016).

12
I.2.6 Farmacodinamia
Ciencia que estudia el efecto que produce un fármaco en un organismo
vivo (Delgado et al, 2016).
I.2.7 Farmacocinética:
Ciencia que estudia la evolución del fármaco en el organismo (absorción,
distribución y eliminación) (Delgado et al, 2016).
I.2.8 Sistema de Clasificación Biofarmacéutica
Este sistema se basa principalmente en la solución acuosa y
permeabilidad intestinal, para la clasificación del principio activo de un
medicamento, como muestra en la figura 2. Se puede utilizar esta clasificación
como base, para establecer las especificaciones de disolución in vitro y también
puede proveer una base para predecir la probabilidad de lograr una correlación
in vivo-in vitro exitosa.
Figura 2 Sistema de Clasificación Biofarmacéutica (Guzmán, 2013).
I.2.8.1 Perfiles de Disolución
Un perfil de disolución es un estudio que considera diversos tiempos de
muestreo, lo que permite establecer la velocidad de disolución, que se
CLASE 1
Fármacos de Alta
Solubilidad y Alta
Permiabilidad
CLASE 2
Fármacos de Baja
Solubilidad y Alta
Permiabilidad
CLASE 3
Fármacos de Alta
Solubilidad y Baja
Permiabilidad
CLASE 4
Fármacos de Baja
Solubilidad y Baja
Permiabilidad

13
representa mediante una curva del porcentaje disuelto en función del tiempo,
comparando dos productos, uno de referencia y otro de prueba, bajo un
procedimiento establecido. Como se muestra en la figura Figura 3
I.2.8.2 Comparación y evaluación de perfiles de disolución
La comparación de perfiles de disolución se puede llevar a cabo
empleando métodos de modelo dependiente o modelo independiente.
Actualmente se han utilizado especificaciones y pruebas de disolución, para
evaluar los aumentos en escala y cambios en la aprobación:
Aumento en escala
Cambios en el sitio de fabricación
Cambios en componentes y composición
Cambios en equipos y procesos (FDA, 2015)
La comparación de los perfiles de disolución in vitro de cápsulas, tabletas
y polvos le proporciona al tecnólogo la información necesaria para:
Discernir entre las formulaciones durante el desarrollo del producto
Evaluar su estabilidad
Evaluar el efecto producido en la
disolución por los cambios en las variables
del proceso
Mide la uniformidad de lote a lote y asegura la calidad (FDA, 2015).
14
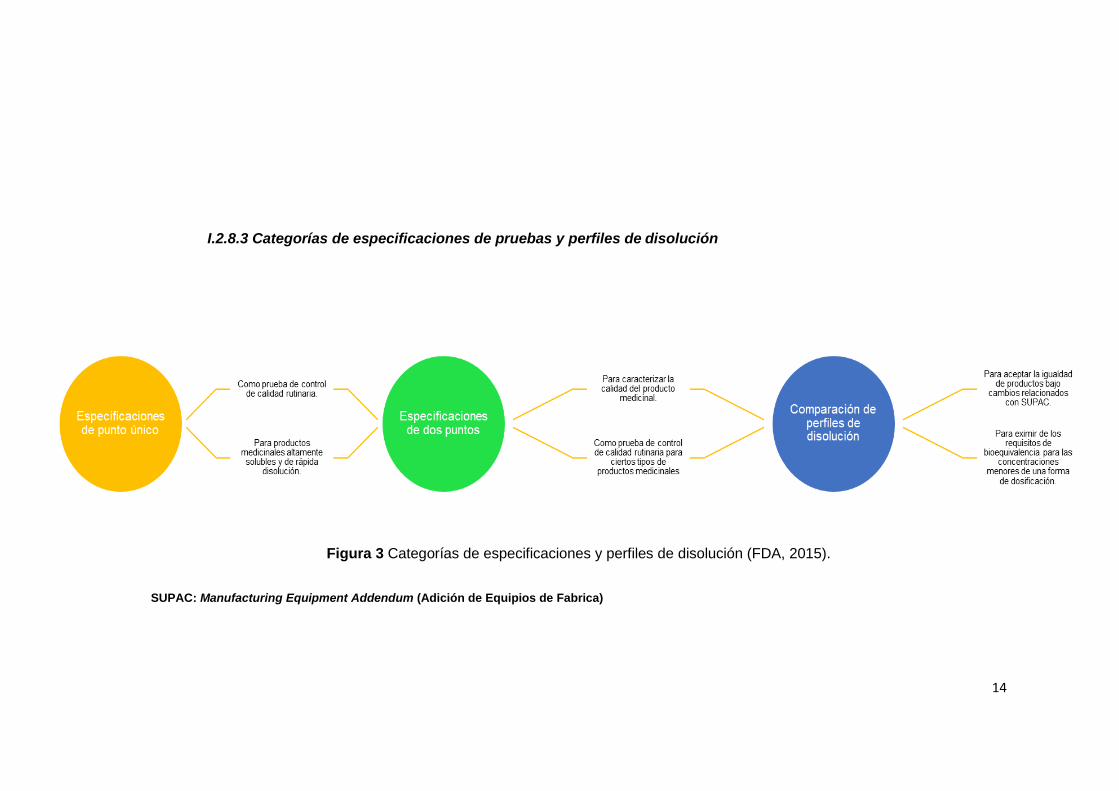
I.2.8.3 Categorías de especificaciones de pruebas y perfiles de disolución
Figura 3 Categorías de especificaciones y perfiles de disolución (FDA, 2015).
SUPAC: Manufacturing Equipment Addendum (Adición de Equipios de Fabrica)

15
I.2.9 Enfoque independiente de modelo utilizando un
factor de Diferencia y Similitud f1 y f2
Un enfoque independiente de modelo sencillo utiliza un factor de diferencia
(f1) y un factor de similitud (f2) para comparar los perfiles de disolución (Moore
1996). El factor de diferencia (f1) calcula la diferencia porcentual (%) entre las dos
curvas en cada punto temporal y es una medida del error relativo entre las dos
curvas (Guzmán, 2013).
f1 = {[_t=1n | Rt - Tt |]/ [_t=1n Rt]} _ 100
Rt: es el valor de disolución del lote de
referencia
t: tiempo
Tt: es el valor de disolución de la tanda de
prueba en el tiempo t
El factor de similitud (f2) es una transformación de raíz cuadrada recíproca
logarítmica de la suma del error cuadrado y es una medición de la similitud en la
disolución porcentual (%) entre las dos curvas (Guzmán, 2013).
f2 = 50 _ log {[1+(1/n) _t=1n (Rt - Tt )2]-0.5_ 100}
I.2.9.1 Aparatos
Los métodos de prueba de disolución utilizados más comúnmente son el
método de cesta (Aparato 1) y el método de paleta (Aparato 2) (Shah 1989). Los
métodos de cesta y paleta son sencillos, robustos, están bien normalizados y se
utilizan en todo el mundo. Estos métodos son lo suficientemente flexibles como
para permitir la realización de pruebas de disolución para una variedad de
medicamentos, por este motivo la Farmacopea Estadounidense (USP) sugiere
los estudios de disolución in vitro usando Aparato 1 y Aparato 2 salvo que se
pruebe que no son satisfactorios (USP NF, 2016).

16
I.2.9.2 Aparato 1
Llamado también método del canastillo consiste esencialmente en un vaso
de vidrio u otro material inerte y trasparente, con o sin tapa, un eje propulsor
metálico y una canastilla cilíndrica. El vaso está parcialmente sumergido en un
baño de agua adecuado de cualquier dimensión conveniente o recibe calor de un
dispositivo adecuado, como por ejemplo una camisa de calentamiento (USP NF,
2016).
I.2.9.3 Aparato 2
Llamado también método de la paleta emplea el mismo equipo, el cual está
compuesto por un aspa y un eje como elemento de agitación, que está a una
distancia máxima de 2 mm con respecto a cualquier punto del eje vertical del vaso
y rota suavemente sin fluctuaciones significativas que pudieran afectar los
resultados (USP NF, 2016).
I.2.10 Reglamento a la ley de medicamentos genéricos de uso humano
A partir de este reglamento, se da a conocer la necesidad e importancia
que tiene la legislación de medicamentos genéricos con respecto al proceso de
fabricación, almacenamiento, distribución y dispensación de medicamentos
(Bejarano, 2014).
I.2.10.1 De la adquisición de medicamentos genéricos
Dentro del Art. 15 menciona que, la adquisición de medicamentos
genéricos estará sujeta a lo que dispone la Ley de Producción, Importación,
Comercialización y Expendio de Medicamentos Genéricos de Uso Humano, con
el objetivo de llevar un control sanitario de los mismos (Bejarano, 2014).
El Consejo Nacional de Salud, (CONASA), es quien convoca a concurso
para seleccionar a los proveedores que suministrarán medicamentos genéricos a
las instituciones del sector público. Para tal efecto, el CONASA cuenta con el
apoyo de su Comisión Nacional de Farmacología (Bejarano, 2014).

17
I.2.10.2 Del registro sanitario y homologación
Para la obtención del registro sanitario de medicamentos de uso humano
e insumos, el Ministro de Salud Pública, mediante instructivo, establece los
requisitos y el procedimiento que se debe cumplir para que las universidades,
escuelas politécnicas y laboratorios, tanto públicos como privados, sean
acreditados y autorizados para la realización de los exámenes de calidad
(Bejarano,2014).

18
CAPÍTULO II. MATERIALES Y METODOS
II.1 Materiales
Para la elaboración de este trabajo se tomó como muestras tabletas de
diltiazem de 60 mg del laboratorio que comercializa en el Ecuador; se realizó la
adquisición de tres lotes diferentes del medicamento genérico y un lote del
medicamento de referencia para el desarrollo de los perfiles de disolución.
Tabla II Materiales usados para la investigación
Equipos Reactivos Material de vidrio Otros
Espectrofotómetro marca Shimadzu modelo UV 1700
Solución estándar de Diltiazem
Beackers
Marca Glasso
Jeringas de 20 mL
Balanza analítica Solución HCl 0.1 N pH 1.2
Matraz aforado
Marca Glasso
macrogotero
Disolutor marca Sotax tipo AT 7smart
Solución Monofosfato Básico de Potasio pH 6.8
Cubeta para espectrofotómetro
Papel Manteca
Solución Amortiguadora de Acetato pH 4.5
Pipeta volumétrica
Marca Glasso
Papel Filtro
Fiola
Marca Glasso
Toalla de Cocina
Embudo
Marca Glasso
Cuchareta del Estándar
Vidrio de Reloj
Marca Glasso
Probetas

19
II.2 Métodos
II.2.1 Tipo de Estudio
Esta investigación es de tipo experimental, de laboratorio y comparativo
II.2.2 Metodología
El perfil de disolución se realizó de acuerdo con el ensayo 711 de la USP
38, utilizando un equipo Disolutor, este equipo consta de 7 vasos y programado
con los parámetros establecidos, para el desarrollo de este estudio se utilizó 900
ml de los diferentes buffers las cuales corresponden a una solución de Ácido
Clorhídrico 0.1N a pH 1.2,o jugo gástrico simulado, solución amortiguadora de
Acetato pH 4.5, y solución de Mono fosfato Básico de Sodio pH 6.8, o jugo
intestinal simulado, con aparato 2 o de paletas (USP NF, 2016).
Una vez que el equipo está programado a 75 rpm y temperatura de 37ºC,
se introducen las 6 tabletas de diltiazem, quedando un vaso como blanco, se
procede a tomar muestras de 20 mL de cada vaso sin reposición de volumen, a
los tiempos de 5, 10, 15, 20, 30 minutos y una toma final a los 180 minutos (3
horas), es importante recalcar, que el volumen que no se repone, en cada toma
de muestra en los diferentes tiempos, es considerado al momento de realizar los
diferentes cálculos de perfiles de disolución (USP NF, 2016).
Este volumen extraído es filtrado, y con una pipeta volumétrica se toman
una alícuota de 10 mL, el cual es llevado a un matraz de 50 mL y es enrasado a
volumen con el medio de disolución. Una vez culminado este procedimiento se
realiza las lecturas de cada una de las seis muestras de sus respectivos tiempos
con un espectrofotómetro a una longitud de onda de 273 nanómetros, y este es
comparada con una solución estándar preparada a una concentración conocida
de diltiazem. Es importante recalcar que este método analítico UV-VIS fue
validado por las compañeras Jenifer Campusano y Carla Gutiérrez quienes
también realizan su trabajo de titulación (USP NF, 2016).

20
II.2.3 Métodos de análisis e interpretación de los resultados
Utilizando el modelo independiente de este estudio, y las
recomendaciones de la FDA, OMS, se compara los perfiles de disolución por
medio del factor f1 y f2; donde f1 está entre 0 y 15 y los valores de f2 están entre
50 y 100 (FDA, 2016).
II.3 Variables
II.3.1 Dependiente
Medicamento bioequivalente. II.3.2 Independiente Factor f1 (diferencia). Factor f2 (similitud).
II.3.2 Interviniente Excipientes utilizados. Laboratorio productor.
Tabla III Operacionalización
VARIABLES CONCEPTUALIZACIÓN INDICADOR
MEDICAMENTO BIOEQUIVALENTE (BIOEXENCIÓN)
Permite la demostración de la equivalencia terapéutica mediante estudios de disolución in vitro que compara la magnitud y velocidad de las formulaciones analizadas.
Disolución del principio activo en %
FACTOR DE SIMILITUD F2
Es una medida de la similitud en el % de disolución entre ambas curvas. Es una función reciproca de la transformación de la raíz cuadrada media de la suma de las distancias cuadradas en todos los puntos.
F2 (50 a 1000)
FACTOR DE DIFERENCIA F1
Refleja la diferencia acumulativa entre ambas curvas donde todos los puntos de muestreo es una medida del error relativo entre las dos curvas
F1 (0 a 15)
EXCIPIENTES
Pueden influir en la solubilidad y permeabilidad del tracto digestivo, si la formulación contiene diferentes

21
EMPLEADOS excipientes o cantidades las autoridades competentes pueden optar por no permitir la Bioexención.
Hidroxipropilmetilcelulosa Estereato de magnesio
II.4 Muestreo
Para la conformación y determinación del tamaño de la muestra se
diseñará un estudio estadístico no poblacional, es decir de aprobación y rechazo
de los productos nacionales, se tomarán muestras de las tabletas de diltiazem de
60 mg innovador y de los medicamentos genéricos, evaluando un lote para el
medicamento innovador y tres lotes para el medicamento genérico (Posada,
2009).
II.4.1 Tipo de Muestreo
El muestreo consistirá en la selección de las unidades de estudio según el
criterio establecido por la FDA y la OMS, siendo las tabletas las unidades
representativas para el análisis (FDA, 2015).
II.5 Población y muestra
Constituido por las tabletas de diltiazem 60 mg, de un laboratorio
extranjero que comercializa el producto farmacéutico en el Ecuador. Los
estudios se realizaron sobre tres lotes de cada producto genérico y un lote para
el innovador, se marcó con las letras A mayúscula para el producto genérico con
sus respectivas codificaciones numéricas para diferenciar cada lote y se marcará
con la letra R el producto innovador. Por motivos de protección los nombres de
los laboratorios no serán mencionados.
II.6 Criterios de inclusión y exclusión
El Diseño Experimental se desarrolló en el Laboratorio de Medicamentos
de la Facultad de Ciencias Químicas de la Universidad de Guayaquil, las muestras
fueron adquiridas en las diferentes Farmacias Mayoristas de la Ciudad de
Guayaquil pata obtener diferentes lotes del medicamento de diltiazem.

22

23
CAPITULO III. RESULTADOS Y DISCUSIÓN
III.1 Comprobación del modelo independiente a través del factor de
similitud f2 y diferencia f1 para cada lote de los genéricos en estudio, de los
diferentes medios de disolución.
Los estudios de perfiles de disolución en la actualidad son muy
importantes para realizar la comparación de la equivalencia terapéutica de un
medicamento de marca frente a un genérico (OMS, 2015).
Para comparar los estudios de los perfiles de disolución es necesario
hacer uso del modelo independiente, que son factor de similitud y factor de
diferencia. A la medida de similitud se la conoce como f2 oscila en el intervalo de
50 a 100, valor que representa la similitud en la disolución porcentual entre las 2
curvas. La medida de la diferencia conocida como f1 representa el error relativo
entre las dos curvas y su rango va entre 0 y 15 (Guzmán, 2013).
En este estudio experimental, para el medicamento diltiazem 60 mg se
realizaron las corridas en los diferentes medios de disolución (buffers), como lo
indica la guía de la FDA; en la Tabla IV, se puede observar la concentración en
miligramos y el porcentaje de disolución, del principio activo del medicamento
innovador y de los tres lotes del medicamento genérico en los diferentes tiempos
a pH 1.2

24
Tabla IV Concentración y porcentaje disuelto del medicamento innovador y medicamento genérico a pH 1.2
MEDICAMENTO IMNOVADOR pH 1,2
MEDICAMENTO 5 MINUTOS 10 MINUTOS 15 MINUTOS 20 MINUTOS 30 MINUTOS 180 MINUTOS
R Conc (%) 40 44,07 51,01 51,51 52,28 96,84 Conc mg 24 26,82 30,61 30,91 31,37 58,1
MEDICAMENTO GENERICO pH 1,2
MEDICAMENTO 5 MINUTOS 10 MINUTOS 15 MINUTOS 20 MINUTOS 30 MINUTOS 180 MINUTOS
A1 Conc (%) 24,19 31,81 40,86 55,02 60,57 115,59 Conc mg 14,51 19,09 24,52 33,01 36,34 62,36
A2 Conc (%) 23,57 30,03 39,87 40,19 51,72 84,87 Conc mg 14,14 12,02 23,92 24,11 31,03 50,92
A3 Conc (%) 18,55 27,59 36,39 43,49 49,12 82,92 Conc mg 11,13 16,55 21,83 26,09 29,47 49,75

25
Se observa dentro de este estudio experimental, que, en el buffer
de simulación de jugo gástrico, a pH 1.2, tanto la concentración en mg de
principio activo y el porcentaje de disolución del medicamento innovador,
van aumentando progresivamente versus el tiempo. Como resultado de
esto se obtuvo que a los 5 minutos se libera 24 mg de principio activo y
alcanza el 40% de disolución, dentro de los 15 a los 30 minutos hay una
disolución lenta del principio activo, encontrándose un promedio general
de 30.96mg y 51.6% de disolución dentro de este intervalo de tiempo,
mientras que a los 180 minutos la concentración es de 58.10 mg de
principio activo y 96.84% de disolución.
Al comparar el porcentaje disuelto, tanto del innovador versus los 3
lotes del medicamento genérico, se pudo demostrar, que el medicamento
de referencia tiene un mayor porcentaje de disolución a los 5 minutos,
mientras que el medicamento genérico, con valor promediado de 22.05%
de disolución; esto quiere decir que el medicamento de referencia es dos
veces más soluble que los tres lotes promediados del medicamento
genérico a los 5 minutos. Sin embargo, el promedio general entre los tres
lotes de genéricos alcanza mayor solubilidad a partir de los 30 y 180
minutos.

26

27
Tabla V Comprobación del Factor de similitud y diferencia para cada lote del medicamento genérico a pH 1.2
MEDICAMENTOS
TIEMPO
PORCENTAJE DISUELTO DE PRINCIPIO ACTIVO EN EL TIEMPO (%)
Factor de diferencia F1
Factor de similitud F2
Observación
5
10
15
20
30
180
Innovador R
R
40
44.07
51.01
51.51
52.28
96.84
Genérico A
A1
24.19
31.81
40.86
52.02
60.57
115.59
5.93
58.01
Si Cumple
A2
23.57
30.03
39.87
40.19
51.72
84.87
19.65
55.96
Cumple F2
A3
18.55
27.59
36.39
43.49
49.12
82.92
28.85
39.98
No Cumple
28
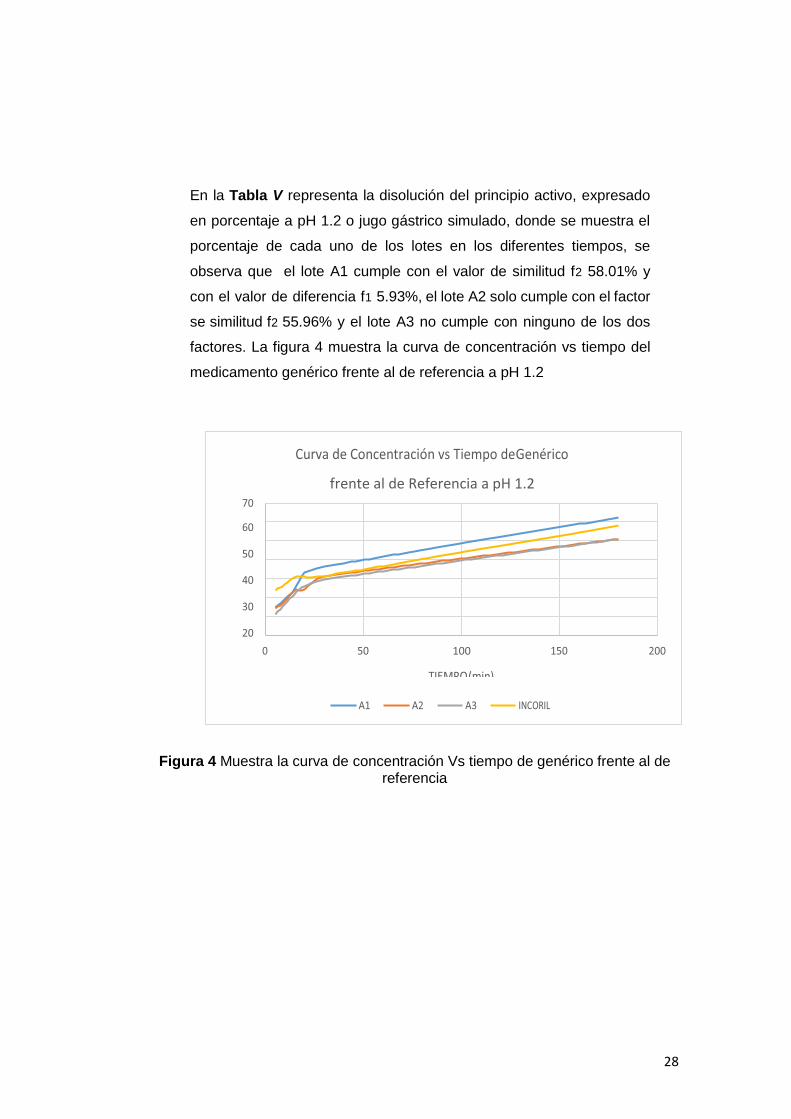
En la Tabla V representa la disolución del principio activo, expresado
en porcentaje a pH 1.2 o jugo gástrico simulado, donde se muestra el
porcentaje de cada uno de los lotes en los diferentes tiempos, se
observa que el lote A1 cumple con el valor de similitud f2 58.01% y
con el valor de diferencia f1 5.93%, el lote A2 solo cumple con el factor
se similitud f2 55.96% y el lote A3 no cumple con ninguno de los dos
factores. La figura 4 muestra la curva de concentración vs tiempo del
medicamento genérico frente al de referencia a pH 1.2
Figura 4 Muestra la curva de concentración Vs tiempo de genérico frente al de referencia
100
frente al de Referencia a pH 1.2
29
Tabla VI Concentración y porcentaje disuelto del medicamento innovador y medicamento genérico a pH 4.5
MEDICAMENTO IMNOVADOR pH 4,5
MEDICAMENTO 5 MINUTOS 10 MINUTOS 15 MINUTOS 20 MINUTOS 30 MINUTOS 180 MINUTOS
R Conc (%) 10,56 14,18 21,85 28,86 38,98 73,03
Conc mg 6,34 8,51 13,11 17,32 23,39 46,22
MEDICAMENTO GENERICO pH 4,5
MEDICAMENTO 5 MINUTOS 10 MINUTOS 15 MINUTOS 20 MINUTOS 30 MINUTOS 180 MINUTOS
A1 Conc (%) 17,6 21,4 25,86 30,96 43,49 102,21
Conc mg 10,3 12,84 15,51 18,58 26,09 61,33
A2 Conc (%) 22,74 29,52 35,08 39,26 53,01 98,4
Conc mg 13,64 17,71 21,01 23,56 31,81 59,04
A3 Conc (%) 19,18 34,11 36,1 53,56 53,56 124,39
Conc mg 11.51 26.46 21.66 32.13 32.13 74.63

30
En la Tabla VI se observa que el medio de disolución (buffer) a pH
4.5, es menos favorable para la disolución del principio activo, del
medicamento innovador, en comparación con el medio de disolución
anterior a pH 1.2; se observa que a los cinco minutos solo libera 6.34 mg de
principio activo y 10.56% de disolución, habiendo un valor creciente a partir
de los 10, 15, 20, se observa que a los 30 minutos, libera 23.39 mg de
principio activo y 38.98 % de disolución y a los 180 minutos alcanza a
liberar 46.22 mg de principio activo y 77.03% de disolución.
A diferencia del medicamento genérico, los tres lotes en estudio
tienen un mayor porcentaje de disolución a pH 4.5 en relación con el
medicamento innovador. Los lotes A1, A2 y A3 le corresponde el siguiente
porcentaje de disolución a los 5 minutos, 17.6%, 22,74%, 19.18%
respectivamente, en comparación al medicamento de referencia que libera
10.56% de principio activo a este mismo tiempo; se observa que ocurre lo
mismo en los tiempos restantes, donde el porcentaje liberado de principio
activo del medicamento genérico sigue siendo mayor al porcentaje de
disolución del medicamento de referencia.

31
Tabla VII Comprobación del Factor de similitud y diferencia para cada lote del medicamento genérico a pH4.5
MEDICAMENTOS
TIEMPO
PORCENTAJE DISUELTO DEL PRINCIPIO ACTIVO EN EL TIEMPO (%)
Factor de diferencia F1
Factor de similitud F2
Observación
5
10
15
20
30
180
Innovador R
R
10.56
14.18
21.85
28.86
38.98
73.03
Genérico A
A1
17.6
21.4
25.86
30.96
43.49
102.21
45.17
50.45
F2 cumple
A2
22.74
29.52
35.08
39.26
53.01
98.4
67.58
46.22
No Cumple
A3
19.18
34.11
38.1
53.56
53.56
124.39
25.91
56.20
Cumple F2

32
La Tabla VII representa la disolución del principio activo, expresado
en porcentaje a pH 4.5 o tampón amortiguador, donde se muestra el
porcentaje de cada uno de los lotes en los diferentes tiempos, observando
que el lote A1 cumple con el valor de similitud f2 50.45%, el lote A2 no
cumple con ninguno de los dos factores y el lote A3 solo cumple con el
factor se similitud f2 con un valor de 56.20. La Figura 5 muestra la curva de
concentración vs tiempo del medicamento genérico frente al de referencia
a pH 4.5.
Figura 5 Muestra la curva de concentración vs Tiempo de genérico frente al de referencia
frente al de Referencia a pH 4.5
100
33
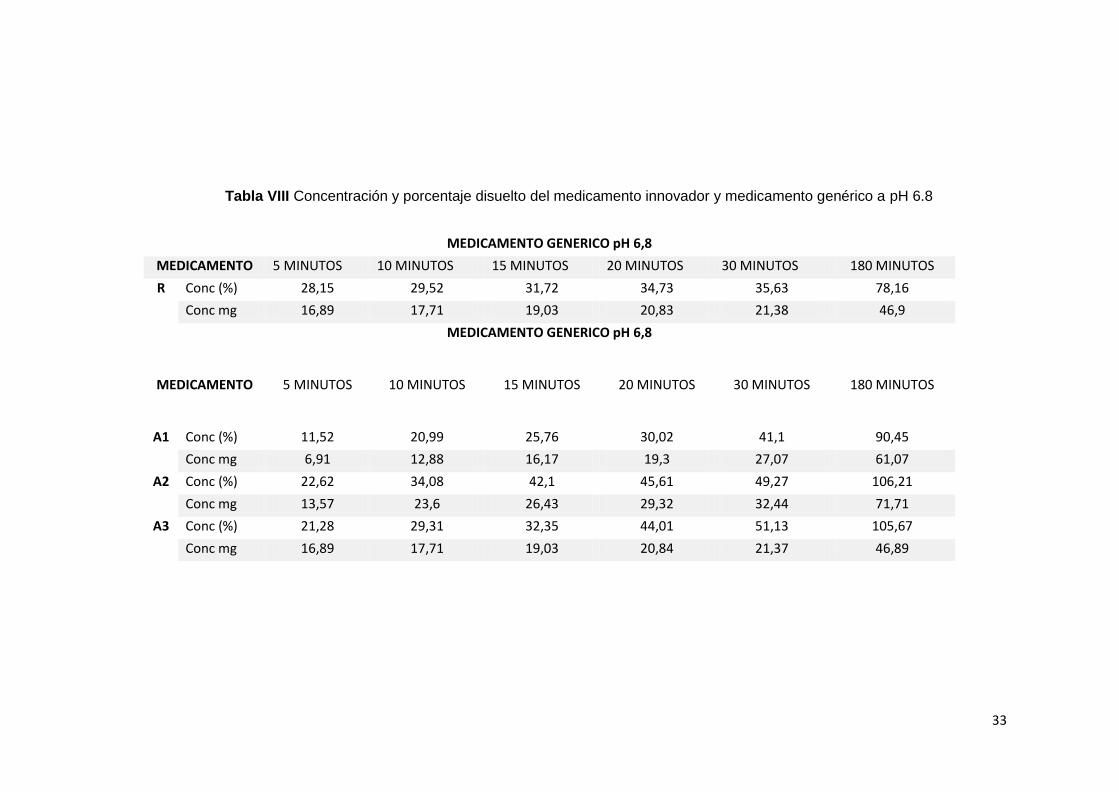
Tabla VIII Concentración y porcentaje disuelto del medicamento innovador y medicamento genérico a pH 6.8
MEDICAMENTO GENERICO pH 6,8
MEDICAMENTO 5 MINUTOS 10 MINUTOS 15 MINUTOS 20 MINUTOS 30 MINUTOS 180 MINUTOS
R Conc (%) 28,15 29,52 31,72 34,73 35,63 78,16
Conc mg 16,89 17,71 19,03 20,83 21,38 46,9
MEDICAMENTO GENERICO pH 6,8
MEDICAMENTO 5 MINUTOS 10 MINUTOS 15 MINUTOS 20 MINUTOS 30 MINUTOS 180 MINUTOS
A1 Conc (%) 11,52 20,99 25,76 30,02 41,1 90,45
Conc mg 6,91 12,88 16,17 19,3 27,07 61,07
A2 Conc (%) 22,62 34,08 42,1 45,61 49,27 106,21
Conc mg 13,57 23,6 26,43 29,32 32,44 71,71
A3 Conc (%) 21,28 29,31 32,35 44,01 51,13 105,67
Conc mg 16,89 17,71 19,03 20,84 21,37 46,89

34
En este medio de disolución a pH 6.8, se observa que la
disolución del principio activo del medicamento innovador es mejor
que en la anterior disolución a pH 4.5; a los 5 minutos se libera 16.89
mg de principio activo y 28.15 % de disolución, existe un valor creciente
a partir de los 10, 15, 20 se observa que a los 30 minutos, se libera
21.38 mg de principio activo y 35.63% de disolución
respectivamente y a los 180 minutos alcanza 46.90 mg de principio
activo y 78.16% de disolución.
Al comparar el porcentaje disuelto, tanto del innovador versus
los 3 lotes del medicamento genérico, se pudo demostrar, que el
medicamento de referencia tiene un mayor porcentaje de disolución
a los 5 minutos, el cual fes de 28.15% vs el genérico, con valores de
11.52% para A1, 22.62% para A2 y 21.28% de disolución para A3;
esto quiere decir que el medicamento de referencia es más soluble
que los tres lotes del medicamento genérico a los 5 minutos. Sin
embargo, el promedio general entre los tres lotes de genéricos
alcanza mayor solubilidad a partir de los 30 y 180 minutos.

35
Tabla IX Comprobación del Factor de similitud y diferencia para cada lote del medicamento genérico a pH 6.8
MEDICAMENTOS
TIEMPO
PORCENTAJE DISUELTO DEL PRINCIPIO ACTIVO EN EL TIEMPO (%) Factor de diferencia F1
Factor de similitud F2
Observación
5
10
15
20
30
180
Innovador R
R
28.15
29.52
31.72
34.73
35.63
78.16
Genérico A
A1
11.52
20.99
25.76
30.02
41.1
90.45
0.47
53.21
Si cumple
A2
22.62
34.08
42.1
45.61
49.27
106.21
38.07
49.98
No Cumple
A3
21.28
29.31
32.35
44.01
51.13
105.67
29.43
44.28
No Cumple

36
La Tabla IX representa la disolución del principio activo,
expresado en porcentaje a pH 6.8 o jugo intestinal simulado, donde
se muestra el porcentaje de cada uno de los lotes en los diferentes
tiempos, observándose que en el lote A1 cumple tanto el valor de
similitud F2 53.21 % como para el valor de diferencia F1 0.47%,
para el lote A2 y A3 no cumple con ninguno de los dos factores. En
la Figura 6 se muestra la curva de concentración vs tiempo del
medicamento genérico frente al medicamento de referencia.
Figura 6 Muestra la curva de concentración vs Tiempo de genérico frente al de
referencia
Genérico pH 6.8
100

37
CAPITULO IV. CONCLUSIONES Y RECOMENDACIONES
IV.1 CONCLUSIONES
Se obtuvo el perfil de disolución del medicamento innovador, y a través
de él, se demuestra que el porcentaje de disolución, a pH 1.2 a los 180 minutos
alcanza el 96.84% considerándose óptimo, ya que, en su mayoría, el principio
activo es liberado en el medio de simulación gástrica, llegando a ser disuelto a
los 180 minutos 58.10 mg de los 60 mg que corresponden.
En este estudio experimental se demuestra que en el medio de disolución
a pH 1.2 el lote A1 del medicamento genérico cumple con el valor de f1 y f2
Según los resultados obtenidos a pH 4.5 concluimos lo siguiente:
La liberación del principio activo, de los tres lotes del genérico es absoluta
a este pH, no siendo así para el innovador, que apenas se disuelve un 73.03 % a
los 180 minutos
Según los resultados obtenidos, en el buffer de simulación entérica a pH
6.8 concluimos qué, el innovador, apenas se libera en un 78.16%, sin embargo,
los tres lotes del genérico se desintegran bordeando el 100%. Además, se repite
que el lote A1, cumple con factor f1 y factor de Similitud f2. Probando que este
lote es equivalente terapéutico con el medicamento innovador en los medios de
disolución a pH 1.2 y 6.8

38
Se demuestra en el estudio experimental, que no todos los lotes del
medicamento genérico cumplen con el factor de diferencia y similitud, esto
puede ser por diferentes factores como proceso de formulación, excipientes,
entre otros.
De acuerdo, a lo señalado en la OMS, al cumplir el valor de similitud f2 en la parte
experimental de nuestro estudio, demostramos que hay una correlación in vitro in
vivo, por cuanto el medicamento de diltiazem 60 mg, comercializado en Ecuador
es equivalente, cumpliendo la misma función terapéutica que el innovador, sin
embargo, se concluye que, por los diversos procesos, de manufactura efectuados
entre lote y lote no todos pueden llegar a cumplirlo.

39
IV.2 RECOMENDACIONES
Realizar más estudios de Perfiles de disolución a los medicamentos
Genéricos que se encuentran en el Cuadro Nacional de Medicamentos Básicos,
para asegurar según el cumplimiento de nuestras leyes que éstos sean seguros y
eficaces.
Informar al laboratorio farmacéutico implicado de los resultados de este
estudio para que puedan mejorar sus procesos y controles de calidad, mejor
selección de materias primas y excipientes, que ayuden a la solubilidad del
fármaco en el organismo.
Tanto las políticas nacionales como los entes reguladores, empresas
privadas y Universidades deberían trabajar en conjunto con recursos tangibles
económicos y científicos para llevar a cabo estudios de esta índole por y para la
salud de los ecuatorianos.
Con el fin de contribuir a las siguientes generaciones de estudiantes de
Química y Farmacia, además de la sociedad ecuatoriana se recomienda promover
la apertura del primer Centro de Bioequivalencia en la Facultad de Ciencias
Químicas, ya que en el país no se cuenta con laboratorios de ésta índole; es
deseable dicha apertura para ampliar y profundizar más investigaciones, con más
estudios sobre los medicamentos genéricos, su efecto terapéutico, equivalencia
farmacéutica o estudios de Bioexcención.

40
Bibliografía
Bejarano, G. N. (2014). Reglamento a la ley de medicamentos genéricos de uso
humano. (págs. 1-5). Lexis Finder.
Bioquímicos, C. d. (22 de Junio de 2013). Bioequivalencia: Calidad, Seguridad y
Eficacia de los medicamentos., (págs. 1-10). Obtenido de
Bioequivalencia: Calidad, Seguridad y Eficacia de los medicamentos:
http://www.colegiofarmaceutico.cl/index.php/noticias-regionales/40-
regional-santiago/237-bioequivalencia-calidad-seguridad-y-eficacia-de-
los-medicamentos-en-chile
Consejo Nacional de Salud. (2013). Cuadro Nacional de Medicamentos Basicos
(Vol. 9). Quito.
Delgado, A. B. (2016). Medicamentos genéricos, genéricos intercambiables,
innovadores y el efecto terapéutico según la OMS y la legislación
peruana. Perú.
ENSANUT. (2012). Encuesta de salud y nutrición., (págs. 1-20).
Guzmán, M. R. (2013). Estudio de bioequivalencia in vitro de dos formas
farmacéuticas perorales multifuente de liberación inmediata con
metformina como principio activo., (págs. 10-100).
HURLÉ, A. D.-G. (2011). Avances en tecnología farmacéutica. Salamanca.
Imira. (2013 de Mayo de 2013). Notas de Prensa y Artículos . Obtenido de Notas
de Prensa y Artículos : http://www.notaes.net/medicamentos-genericos-y-
medicamentos-de-marca.html
Jordi, G. O. (19 de junio de 2011). Técnicas de remuestreo en la comparación de
curvas de disolución de fármacos. 1-19. Obtenido de
http://www.analesranf.com/index.php/aranf/article/view/1147/1202
Lecaros, M. F. (2013). Evaluación de la solubilidad y disolución. (págs. 2-5).
Chile: Repositorio Académico de la Universidad de Chile. Obtenido de
http://repositorio.uchile.cl/handle/2250/114222
Luján, M. (8 de mayo de 2013). Farmacología Clínica Aplicada. (págs. 1-50).
Open CourseWare.
Miranda, M. d. (2015). Fármacos y Medicamentos. (págs. 1-808). Editorial
UNED.
OMS, O. M. (septiembre de 2015). Hipertensiòn., (pág. 1). Obtenido de
http://www.who.int/es/
Pharmacists, A. S.-S. (15 de Abril de 2015). Medline Plus. Obtenido de Medline
Plus: https://medlineplus.gov/spanish/druginfo/meds/a684027-es.html

41
Pilon, S. (2016). Guía práctica de utilización destinado a médicos, farmacéuticos,
enfermeros y auxiliares sanitarios. (págs. 4-10). Médecins Sans
Frontiéres.
Pubchem. (19 de noviembre de 2017). National Center for Biotechnology
Information. Obtenido de
https://pubchem.ncbi.nlm.nih.gov/compound/39186
Pulido, P., Silva, H., Marques, J., & Pereira, G. (agosto de 2016). Manejo de
factores de riesgo cardiovascular. (págs. 2-5). Online ISSN.
Rivero, M. d. (marzo de 2017). Salud del barrio. (págs. 20-25). Biblioteca Mèdica
Nacional.
Salud, O. M. (Septiembre de 2015). Organizacion Mundia de la Salud., (págs. 5-
10). Obtenido de Organizacion Mundia de la Salud:
http://www.who.int/features/qa/82/es/