universidad de guayaquil facultad de …repositorio.ug.edu.ec/bitstream/redug/17941/1/bcieq-t-0158...
TRANSCRIPT

UNIVERSIDAD DE GUAYAQUIL
FACULTAD DE CIENCIAS QUÍMICAS
MODALIDAD: INVESTIGACIÓN
TEMA:
ESTUDIO DE LA BIOEQUIVALENCIA IN VITRO DE LAS TABLETAS DE
CIPROFLOXACINO DE 500 mg, ELABORADAS POR DOS LABORATORIOS
FARMACÉUTICOS DEL ECUADOR, MEDIANTE EL EMPLEO DE LOS PERFILES
DE DISOLUCIÓN
TRABAJO DE TITULACIÓN PRESENTADO COMO REQUISITO PREVIO PARA
OPTAR AL GRADO DE QUÍMICO FARMACÉUTICO.
AUTORES:
CALLE BAYAS JUDIE NATHALY
COPETE VALENCIA CARLOS ENRIQUE
TUTORA:
Dra. PILAR ASUNCIÓN SOLEDISPA CAÑARTE, MsC.
CO-TUTORA:
Lic. MIGDALIA MIRANDA MARTÍNEZ, PhD.
GUAYAQUIL - ECUADOR.
2016

i
APROBACIÓN DEL TUTOR
En calidad de tutora del Trabajo de Titulación, Certifico: Que he asesorado, guiado y
revisado el trabajo de titulación en la modalidad de investigación cuyo título es
Estudio de la bioequivalencia in vitro de las tabletas de ciprofloxacino de 500
mg elaboradas por dos laboratorios farmacéuticos del ecuador, mediante el
empleo de los perfiles de disolución, presentado por Calle Bayas Judie Nathaly,
con cédula de ciudadanía N° 092810607-9, y Copete Valencia Carlos Enrique, con
cédula de ciudadanía N° 0803150432 con previo a la obtención del título de Química
y Farmacéutica, y Químico Farmacéutico.
Este trabajo ha sido aprobado en su totalidad y se adjunta el informe de Antiplagio del
programa URKUND. Lo Certifico. -
Guayaquil, 20 de diciembre del 2016
Dra. Pilar Soledispa, MSc. Lic. Migdalia Miranda, PhD FIRMA TUTOR DE TESIS FIRMA CO-TUTOR DE TESIS

ii
INFORME DE ANTI PLAGIO DEL SISTEMA URKUND
El plagio encontrado en el proyecto de titulación cuyo tema es Estudio de la
bioequivalencia in vitro de las tabletas de ciprofloxacino de 500 mg, elaboradas por
dos laboratorios farmacéuticos del ecuador, mediante el empleo de los perfiles de
disolución, fue del 5%, según lo certifica el sistema URKUND.
Dra. Pilar Soledispa, MSc. Lic. Migdalia Miranda, PhD FIRMA TUTOR DE TESIS FIRMA CO-TUTOR DE TESIS

iii
CERTIFICADO DEL TRIBUNAL
El Tribunal de Sustentación del Trabajo de Titulación la Srta. Calle Bayas Judie
Nathaly el Sr. Copete Valencia Carlos Enrique, después de ser examinados en su
presentación, memoria científica y defensa oral, da por aprobado el Trabajo de
Titulación.
Q.F. María Fernanda Vélez López
PRESIDENTE - MIEMBRO DEL TRIBUNAL
Dra. Alexandra López Barrera MSc Dra. María Elena Jiménez Heinert MSc
DOCENTE–MIEMBRO DEL TRIBUNAL DOCENTE–MIEMBRO DEL TRIBUNAL
Ing. Nancy Vivar Cáceres
SECRETARIA ENCARGADA

iv
CARTA DE AUTORÍA DE TITULACIÓN
Guayaquil, 20 de diciembre del 2016
Nosotros, CALLE BAYAS JUDIE NATHALY y COPETE VALENCIA CARLOS
ENRIQUE, autores de este trabajo declaramos ante las autoridades de la Facultad de
Ciencias Químicas de la Universidad de Guayaquil, que la responsabilidad del
contenido de este TRABAJO DE TITULACIÓN, nos corresponde a nosotros
exclusivamente; y el patrimonio intelectual de la misma a la Facultad de Ciencias
Químicas de la Universidad de Guayaquil.
Declaramos también es de nuestra autoría, que todo el material escrito, salvo el que
está debidamente referenciado en el texto. Además, ratifico que este trabajo no ha
sido parcial ni totalmente presentado para la obtención de un título, ni en una
Universidad Nacional, ni una Extranjera.
Calle Bayas Judie Nathaly Copete Valencia Carlos Enrique
C.I. 092810607-9 C.I. 080315043-2

v
AGRADECIMIENTO
Se le agradece a nuestros padres, hermanos y familiares quienes nos han brindado
su apoyo, a nuestra tutora: Q.F. Pilar Asunción Soledispa, MSc y Co- tutora: Lic.
Migdalia Miranda Martínez, PhD. quienes han sido nuestras guías para la realización
de este Proyecto, a Liliana Suarez y Abel Macías quienes han sido de apoyo, a
nuestros amigos de la Universidad por su amistad incondicional.

vi
ÍNDICE GENERAL
Página
INTRODUCCIÓN 1
Problema 4
Hipótesis 4
Objetivos 4
Objetivos Generales 4
Objetivos Específicos 4
CAPÍTULO I REVISIÓN BIBLIOGRÁFICA 5
I.1 Antecedentes 5
I.2 Base Teórica 8
I.2.1 Formas Medicamentosas 9
I.2.1.1 Presentación universal de las Formas Medicamentosas Sólidas 9
I.2.1.2 Clasificación 9
I.2.2 Ciprofloxacino 10
I.2.2.1 Mecanismo de Acción 10
I.2.2.2 Reacciones Desfavorables 11
I.2.3 Bioequivalencia 11
I.2.3.1 A qué se debe la elaboración de los ensayos de Bioequivalencia 11
I.2.3.2 En que radica la Bioexención 12
I.2.3.2.1 Cuándo debemos optar por realizar una Bioexención 12
I.2.4 Equivalentes Farmacéuticos 12
I.2.5 Equivalentes Terapéuticos 12
I.2.6 Innovador o de Referencia 13
I.2.7 Genérico o Copia 13
I.2.8 Metodologías para indicar la Bioequivalencia 13
I.2.8.1 Metodologías in vivo 14
I.2.8.2 Metodologías in vitro 14

Vii
Página
I.2.9 Sistema de Clasificación Biofarmacéutico 15
I.2.10 Perfiles de Disolución 15
I.2.10.1 Modelos de los Perfiles de Disolución 16
I.2.10.1.1 Modelo Independiente 16
I.2.10.1.2 Modelo Dependiente 18
I.2.11 Evaluación de Perfiles de Disolución 19
I.2.12 Instrumentos 19
CAPITULO II MATERIALES Y MÉTODOS 22
II.2.1 Materiales 22
II.2.2 Métodos 22
II.2.2.1 Tipo de Estudio 22
II.2.2 Metodología 22
II.2.2.3 Método de Análisis e Interpretación de los Resultados 23
II.2.3 Variables 23
II.2.3.1 Independiente 23
II.2.3.2 Dependiente 24
II.2.3.3 Interviniente 24
II.2.4 Muestreo 25
II.2.4.1 Tipo de Muestreo 25
II.2.5 Población y Muestra 25
II.2.6 Criterios de Inclusión y Exclusión 25
CAPITULO III RESULTADOS Y DISCUSIÓN 26
III. 1 Comportamiento de cada lote del medicamento de estudio frente a
la Disolución 26
III.2 Comprobación del modelo independiente a través del factor de
similitud para cada lote de los genéricos en estudio 30
III. 3 Comparación de los valores de Disolución promedio de cada uno de
los medicamentos analizados 32

Viii
Página
III.4 Demostración de la Bioequivalencia de los porcentajes de Disolución
promedios de los Genéricos A y B 33
IV CONCLUSIONES Y RECOMENDACIONES 35
IV.1 Conclusiones 35
IV.2 Recomendaciones 36
REFERENCIAS BIBLIOGRÁFICAS 37
ANEXOS 44
Anexo # 1 Materiales 44
Anexo # 2 Cálculos del factor de diferencia F1 y similitud F2 para cada
lote de los medicamentos genéricos de estudio 45
Anexo # 3 Cálculos del factor de diferencia F1 y similitud F2 para cada
medicamento genérico de estudio 47
Anexo # 4 Eficiencia de la disolución de los medicamentos a prueba 48
Anexo # 5 Tiempo medio de disolución MDT 49
Anexo # 6 Certificado del análisis del estándar 50

ix
ÍNDICE DE GRÁFICOS
Página
Gráfico I. Estructura del ciprofloxacino 10
Gráfico II. Fórmula del factor de diferencia 17
Gráfico III. Fórmula del factor de similitud 17
Gráfico IV. Elemento de agitación de la canastilla 20
Gráfico V. Esquema de las paletas 21
Gráfico VI. Comparación de los perfiles de disolución de cada lote de los
medicamentos A y B, versus el innovador R 29
Gráfico VII. Comparación de los perfiles de disolución de los promedios
de los medicamentos A y B, versus el innovador R 33

x
ÍNDICE DE TABLAS
Página
Tabla I. Operacionalización 24
Tabla II. Concentración y porcentaje disuelto del medicamento innovador
R 26
Tabla III. Porcentaje disuelto de cada uno de los lotes del medicamento
genérico A 27
Tabla IV. Porcentaje disuelto de cada uno de los lotes del medicamento
genérico B 28
Tabla V. Comprobación del factor de similitud y diferencia para cada lote
de los genéricos A y B 31
Tabla VI. Porcentaje promedio de cada medicamento de estudio 32
Tabla VII. Comprobación del factor de similitud y diferencia: Los
promedios de los medicamentos genéricos A y B vs innovador 34

xi
ABREVIATURAS
A: Medicamento Genérico 1
B: Medicamento Genérico 2
BE: Bioequivalencia
ClH: Ácido Clorhídrico
CV: Coeficiente de Variación
DCI: Denominación Común Internacional
ED: Eficiencia de Disolución
EFG: Especialidades Farmacéuticas Genéricas
EF %: Eficiencia de la Disolución
EMA: European Agency for the Evaluation of Medicinal Products
F1: Factor de Diferencia
F2: Factor de Similitud
FDA: Food and Drug Administration
ICH: International Conference on Harmonisation
Kd: Constante de Disolución
mg: Miligramo
MDT: Tiempo Medio de Disolución
MSD: Distancia Estadística Multivariada
nm: Nanómetros
N: Normalidad
OMS: Organización Mundial de la salud
R: Medicamento Innovador
Rt: Promedio del Porcentaje Disuelto del Medicamento Innovador
Scb: Sistema de clasificación Biofarmacéutico
Tt: Promedio de Porcentaje Disuelto del Medicamento de Estudio
USP: The United States Pharmacopeia

xii
RESUMEN
Introducción: La investigación y aplicación de los ensayos de Bioequivalencia ha
permitido indicar la posibilidad de que un principio activo cumple con la misma
actividad farmacológica en un medicamento genérico respecto al innovador.
Ciprofloxacino es un antibiótico de amplio espectro aprobado para el tratamiento de
infecciones bacterianas.
Materiales y Métodos: Se emplearon tabletas de ciprofloxacino de 500 mg de dos
laboratorios farmacéuticos y el medicamento innovador; un equipo disolutor (aparato
2) y como medio de disolución HCl 0.01N. Los cálculos se realizaron en base al
modelo independiente determinando el factor de similitud y diferencia.
Resultados: En el genérico A el valor de F1 fue 9,49 y F2 49,81 y en el caso del
Genérico B el valor de F1 fue de 7,06 y el de F2 48,76 incumpliendo con el valor de
F2 lo que demuestra que el producto no cumplió con el factor de similitud, ya que el
rango de aceptación para F1: 0 – 15; y F2: 50 -100.
Discusión: Los perfiles de disolución determinados para los diferentes lotes de los
medicamentos genéricos estudiados, presentaron diferencias en el tiempo de 5
minutos, existiendo diferencias entre lotes para ambos laboratorios y con respecto al
medicamento innovador.
Conclusión: Se determinó que los dos medicamentos genéricos no son
intercambiables en relación con el de referencia, esto puede ser debido a variaciones
con los excipientes o durante el proceso manufactura que pudieron afectar en la
disolución.
Palabras Claves: Ciprofloxacino, Bioequivalencia, Perfiles de disolución

xiii
ABSTRACT
Introduction: The research and application of the Bioequivalence tests has allowed
to indicate the possibility that an active principle fulfills the same pharmacological
activity in a generic drug with respect to the innovator. Ciprofloxacin is a broad
spectrum antibiotic approved for the treatment of bacterial infections.
Materials and Methods: Ciprofloxacin tablets 500 mg two pharmaceutical
laboratories and innovative drug were used; one disolutor equipment (apparatus 2)
and as 0.01N HCl dissolution medium. The calculations were made based on the
independent model determining the factor of similarity and difference.
Results: A generic value F1 and F2 were 9.49 and 49.81 in the case of Generic B
value was 7.06 F1 and F2 48.76 breach with the value F2 which It shows that the
product did not meet the similarity factor, since the acceptance range for F1: 0-15; and
F2: 50 -100.
Discussion: The dissolution profiles determined for different batches of generic drugs
studied, showed differences in the time of 5 minutes, with differences between batches
for both laboratories and regarding innovative drug.
Conclusion: It was determined that the two are not interchangeable generic regarding
the reference, this may be due to variations with excipients or during manufacturing
process that could affect the dissolution.
Keywords: Ciprofloxacin, bioequivalence, dissolution profiles

1
INTRODUCCIÓN
Los medicamentos en general cumplen una función muy importante, en la salud y
bienestar de las personas así también en las finanzas de los países, motivo por el
cual se hallan accesibles en la actualidad una gran diversidad de medicamentos
destinados a tratar las diversas dolencias de los pacientes. A pesar de esto el
incumplimiento de ciertas leyes y reglamentos esenciales para su comercialización,
permiso y vigilancia, perjudica el cumplimiento impuesto por los objetivos propuestos
en las mismas. (Moraga, 2008).
Motivo por el cual, en Ecuador al igual que en los demás países en desarrollo,
están preocupados por la gran demanda que representa los servicios de salud y han
buscado alternativas para tratar de disminuir los costos. Una de las principales
alternativas ha sido la producción de genéricos para su comercialización en los
sectores de salud tanto públicos como privados, por lo cual éstos deben cumplir con
los requisitos necesarios para garantizar su calidad, seguridad y eficacia (Posada y
Santos, 2009).
El desarrollo de nuevas formulaciones en los medicamentos, requiere la
elaboración de estudios tanto in vitro como in vivo que pongan de manifiesto que éstos
son capaces de aportar la misma cantidad de principio activo que el producto
innovador o el producto de referencia (Jung et al, 2012).
La disolución de un fármaco es prerrequisito para la mayoría de los fármacos
administrados por vía oral. La liberación in vitro de un fármaco a partir de la forma
farmacéutica que lo contiene depende de las características fisicoquímicas del
fármaco, de los excipientes empleados y de la tecnología utilizada para su fabricación
(Jung et al, 2012).

2
La necesidad de evidenciar la intercambiabilidad de medicamentos genéricos con
respecto a los innovadores (originales) se dio en el mundo avanzado por la década
de 1970. La aparición de un nuevo medicamento oral, que posee el mismo principio
activo, dosis y forma farmacéutica que un medicamento de referencia, se denomina
equivalente farmacéutico (Estévez et al, 2012).
La investigación y aplicación de los ensayos de Bioequivalencia ha permitido
indicar la posibilidad de que un principio activo cumple con la misma actividad
farmacológica en un medicamento genérico respecto al innovador y poder definirlos
como medicamentos intercambiables y respaldar la eficacia terapéutica del
medicamento analizado. (Moraga, 2008).
También se puede mostrar la equivalencia terapéutica de los medicamentos que
se van a analizar, por medio de la bioexención la cual consiste básicamente en que
los estudios de biodisponibilidad in vivo (animales, personas etc.), se los puede
indicar por medio de ensayos comparativos de los perfiles de disolución in vitro a las
diversas formulaciones orales de liberación inmediata, este tipo de ensayo y sus
bases teóricas están localizadas en el Sistema de Clasificación Biofarmaceutico (
SCB) (Moraga, 2008).
El ciprofloxacino pertenece al grupo de las quinolonas sintéticas de segunda
generación y es un agente bactericida de amplio espectro, frecuentemente prescrito
a nivel hospitalario y ambulatorio para el manejo de infecciones microbiológicas de
leves a persistentes. Químicamente es el ácido 1-ciclopropil-6-fluoro-1,4-dihidro-4-
oxo-7-(1-piperazinil)-3-quinolincarboxílico (Franco-Ospina et al, 2012).
El Sistema de Clasificación Biofarmacéutico es un marco científico para clasificar
sustancias farmacéuticas, basado en su solubilidad acuosa y permeabilidad intestinal,
permitiendo distinguir los fármacos según cuatro categorías: Clase I, Clase II, Clase
III, el medicamento de estudio pertenece a la Clase III por poseer una alta solubilidad
y baja permeabilidad (Van, 2012).

3
Este detalle es un factor importante para comprometer su calidad, por lo tanto, es
conveniente demostrar la bioequivalencia terapéutica entre el producto de referencia
y el genérico, mediante los perfiles de disolución (Martínez et al, 2010).

4
PROBLEMA
¿Las tabletas de ciprofloxacino de 500 mg elaboradas por dos laboratorios
farmacéuticos del Ecuador serán bioequivalentes con el innovador?
HIPÓTESIS
Las tabletas de ciprofloxacino de 500 mg genéricas elaborados por dos
laboratorios farmacéuticos del Ecuador, analizadas mediante el empleo de los perfiles
de disolución son bioequivalentes con el ciprofloxacino innovador.
OBJETIVOS
Objetivo general
Comprobar la bioequivalencia in vitro de las tabletas de ciprofloxacino de 500 mg
elaboradas por dos laboratorios farmacéuticos del Ecuador, mediante el empleo de
los perfiles de disolución.
Objetivos específicos
Obtener el perfil de disolución de la tableta de ciprofloxacino innovador como
referencia para el ciprofloxacino genérico.
Demostrar la bioequivalencia terapéutica entre el producto de referencia y los
genéricos, mediante los perfiles de disolución en base al factor de diferencia F1 y al
factor de similitud F2.

5
CAPÍTULO I. REVISIÓN BIBLIOGRÁFICA
I.1. ANTECEDENTES
Unión Europea
Los experimentos e investigaciones llevadas a cabo en el laboratorio Stada,
(2013), fueron ensayos de Bioequivalencia realizados en medicamentos genéricos
Montelukast 10 mg (comprimidos), frente al innovador Singulair 10 mg (comprimidos),
dando a conocer los resultados de los valores numéricos, variables etc., de la
actividad farmacológica, manifestando que los mismos están dentro del rango de
aceptación establecido, llegando a la conclusión que existe Bioequivalencia entre
genérico e innovador.
Los trabajos llevados a cabo por el laboratorio Stada, (2015), han sido presentados
últimamente en base a el medicamento Pregabalina, utilizado para aliviar el dolor en
el cuerpo producido por nervios dañados, finalmente se expuso, que existe
Bioequivalencia del medicamento genérico con relación al Lyrica (innovador),
mediante ensayos clínicos, dando como resultado que estos representan seguridad
y eficacia, observándose una mejoría en la sintomatología de la afección tras su
tratamiento en la primera semana.
Latino América
Fuentes, (2011), plasmó una bioexención del clorhidrato de metformina en base a
tres medicamentos genéricos producidos en Guatemala y el innovador; del resultado
de los tres medicamentos genéricos, fabricados por las 3 industrias farmacéuticas
guatemaltecas dos fueron consideradas equivalentes terapéuticos con respecto al
medicamento de referencia, así también los medicamentos genéricos A y B
cumplieron con F1 y F2 ya que mostraron valores que están dentro del rango de
aceptación.

6
Llerena, (2011), efectuó una equiparación de los perfiles de disolución de las
tabletas de ciprofibrato 100 mg coadyuvante para reducir la grasa en la sangre, utilizo
medicamentos genéricos de 3 laboratorios farmacéuticos con respecto al innovador,
y comprobó que el perfil de disolución del medicamento genérico B es similar al
medicamento de referencia, mientras que los genéricos A y C no cumplen con el F1
Y F2.
Medina et al, (2012), expusieron la equivalencia farmacéutica de las tabletas de
montelukast, utilizadas para el tratamiento de varias afecciones en el tracto
respiratorio el análisis se llevó acabo por medio de un ensayo comparativo in vivo, los
resultados fueron cumplidos a cabalidad siendo las tabletas seguras y eficaces.
Díaz, (2013), enfoco su tesis al ensayo y perfil de disolución de las tabletas de
loperamida de 2 mg coadyuvantes para el control de la diarrea, para la investigación
utilizo marcas comerciales producidas por 3 laboratorios farmacéuticos nacionales
distintos de la ciudad de Guatemala manifestando que las marcas A y C son
intercambiables, en cambio la marca B no cumplía con los rangos establecidos
obteniendo como resultado F1 (24) y F2 (31), por lo cual no eran aceptables.
Daza, (2013), concibió una investigación exhaustiva sobre la biodisponibilidad y
bioequivalencia in vitro en cápsulas de amoxicilina perteneciente a una clase de
antibióticos para contrarrestar las infecciones causadas por baterías, comercializadas
en Bolivia; en donde los resultados arribaron a que los medicamentos A, B, C y D, se
pueden considerar bioequivalentes e intercambiables, dicha confirmación, se obtuvo
a lo que se realizo los perfiles de disolución y los oportunos cálculos de F1 y F2.
Alvarado et al (2014), demostraron la equivalencia farmacéutica del medicamento
digoxina (medicamento multifuente) de 0,25 mg agente anti arrítmico, cuantificado en
cada lote estuvo dentro del rango de 90 – 110%; así como una estabilidad en el tiempo
de vida útil.

7
Ochaeta, (2014), formalizó su tesis sobre la intercambiabilidad terapéutica del
clorhidrato de propanolol genérico utilizado como betabloqueante el cual es
coadyuvante en las afecciones del corazón, elaborado por 2 laboratorios nacionales,
mediante el análisis de los perfiles de disolución, dando como resultado que los
medicamentos evaluados arrojaron valores de F1 Y F2 similares al medicamento
innovador, por lo que se confirmó su intercambiabilidad terapéutica.
Serón (2014), desarrolló un ensayo de bioexención en comprimidos de
atorvastatina, la cual es coadyuvante para la prevención de enfermedades
cardiovasculares y disminución de los niveles de colesterol, dándose a conocer que
las variedades de dosis de atorvastatina se liberan y diluyen en forma parecida entre
lotes.
León et al (2015), realizaron un ensayo de la actividad farmacológica de las
tabletas de acetaminofén 500 mg, cuya función es servir de analgésico y antipirético
para este análisis se emplearon medicamentos localizados en el mercado
colombiano, el análisis del medicamento, nos permitió tratar claras discrepancias con
respecto a la liberación in vitro del principio activo, dándonos como resultado final que
todos cumplen con este importante parámetro que es la Bioequivalencia.
Muñoz (2015), ejecutó una investigación sobre la bioexención de fluoxetina 20mg
comprimidos, medicamento utilizado básicamente para el tratamiento de la depresión
y el trastorno compulsivo, tras sus analices se entregaron como resultado que el
medicamento genérico es intercambiable con el innovador y por tanto posee
equivalencia terapéutica.
La Organización gubernamental de las Políticas Farmacéuticas (2015), realizó un
ensayo de Bioequivalencia a los medicamentos del cuadro básico evidenciando que
un total del 67% de los medicamentos investigados son Bioequivalentes con respecto
al innovador.
Saavedra et al, (2011), confeccionaron ensayos de bioexención en los que se llevó
a cabo los respectivos análisis para demostrar la Bioequivalencia de los

8
medicamentos genéricos con respecto al innovador. Además, se dio a conocer la
metodología usada en la bioexención a través de los resultados expuestos por los
perfiles de disolución, indicando los documentos necesarios para la aprobación de un
centro de Bioequivalencia a futuro.
En Ecuador
Robalino, (2013), desenvolvió su tema de tesis para titulación enfocándose en dos
temas principales como son los perfiles de disolución y la influencia de factores
ambientales en el almacenamiento de comprimidos (meloxicam) distribuidos en
Macas y Quito, comprobando que casi todos los medicamentos genéricos de estudio
eran bioequivalentes in vitro y por lo tanto intercambiables con el medicamento de
marca, excepto el genérico X.
Racines, (2013), estudió la posibilidad de utilizar el almidón pregelatinizado como
desintegrante para dar estabilidad a los comprimidos de acetaminofén; así como a
través de los perfiles de disolución manifestar que es intercambiable con comprimidos
fabricados con almidón de maíz.
Ramírez, (2014), introdujo un ensayo de Bioequivalencia con la Carbamazepina
cumpliendo con la función de anticonvulsivante, esto se dio entre medicamentos
comerciales y genéricos, dando como resultado que los genéricos producidos en
Ecuador en base a Carbamazepina son intercambiables ya que no se encuentra una
marcada diferencia entre los datos obtenidos entre el genérico Vs innovador.
I.2 BASE TEÓRICA
La aparición de los medicamentos genéricos en Ecuador ha creado dudas, es
decir, si en realidad son intercambiables con el innovador debido a que se ha dado
en unos casos efectos diferentes al que debería ser, motivo por el cual, los
laboratorios farmacéuticos nacionales y extranjeros se han interesado en garantizar
la calidad, seguridad y eficacia de los medicamentos. (Guerrero, 2013).

9
Para incrementar la confianza en las personas, se ha visto en la necesidad de
efectuar análisis y ensayos de Bioequivalencia en los medicamentos genéricos y así
incrementar la fabricación a nivel nacional. (Guerrero, 2013).
El enfoque a la Bioequivalencia es de suma importancia para lograr implementar
medicamentos que demuestren seguridad y eficacia por ende muchas organizaciones
internacionales como: la Administración de Drogas y Alimentos, la Farmacopea de los
Estados Unidos, Conferencia Internacional de Armonización, la Organización Mundial
de Salud y otros. Han indicado que los países deben implementar gradualmente
ensayos de Bioequivalencia para avalar la intercambiabilidad entre los medicamentos
genéricos y comerciales, pero sobre todo en países que se encuentran en vía de
desarrollo como el Ecuador. (Guerrero, 2013).
I.2.1 Formas Medicamentosas
Sustancias Medicamentosas compuestas de uno o más principios activos,
disponibles en diferentes presentaciones tanto físicas como químicas cuya finalidad
es ser administradas, conservadas y dosificadas al organismo. (Bernad, 2011).
I.2.1.1 Presentación universal de las formas medicamentosas sólidas
Las formas medicamentos pertenecientes al estado sólido al presentar ausencia
de agua posee una excelente estabilidad química por ende el tiempo de caducidad
es más extenso, así en la actividad farmacológica intervienen los procesos de
disolución, desintegración y propagación dependiendo si es un comprimido, tableta,
capsula etc. (Martínez, 2015).
I.2.1.2 Clasificación
Existe una variedad de presentaciones de esta forma medicamentosa entre los
cuales encontramos a los comprimidos no recubiertos obtenidos por el
amontonamiento de partículas del medicamento y fijado por medio de compresión,

10
comprimidos recubiertos obtenidos al realizarle múltiples compresiones, con
recubrimiento de azúcar: grageas, con recubrimiento o cubierta pelicular utilizada
para mejorar, proteger y modificar la función del medicamento, comprimidos
especiales como los bucales, sublinguales, vaginales, efervescentes. (Posada y
Santos, 2009).
I.2.2 Ciprofloxacino
El ciprofloxacino es un anti infeccioso bactericida de amplio espectro agente de la
familia de las fluoroquinolonas, está disponible en más de 100 países, en los que es
aprobado para el tratamiento de 14 tipos de infecciones, infecciones del tracto
urinario, especialmente como cistitis aguda y prostatitis bacteriana crónica, y las
infecciones respiratorias inferiores (Olivera et al ,2011).
Gráfico I. Estructura del ciprofloxacino (Martínez, 2014).
I.2.2.1 Mecanismo de acción
Este medicamento actúa inhibiendo la enzima ADN-girasa, al hacerlo bloquea la
reacción de superenrollamiento fijada en la ATP y catalizada por la girasa, esto ocurre
a lo que el fármaco ingresa a las bacterias Gram negativas y positivas por medio de
sus porinas sin dañar su pared celular siendo básicamente un inhibidor enzimático.
(Martínez, 2010).
El ciprofloxacino al ser administrados en dosis ascendentes a la indicada además
de inhibir a la ADN-girasa también inhabilita a la topoisomerasa bacteriana IV,
enzimas esenciales para la duplicación, reproducción y restitución del ADN.
(Laboratorios Stein, 2012).

11
Las bacterias Gram positivas y negativas han desarrollado un dispositivo de
fortaleza, avanzado hacia el ciprofloxacino básicamente efectúa una o varias
mutaciones en la enzima ADN-girasa, cuando reconoce la presencia del mismo u
otros principios activos que desempañaran la misma finalidad. (Martínez, 2010).
I.2.2.2 Reacciones desfavorables
En la actualidad se han presentado reacciones desfavorables consecutivas entre
las cuales incluyen: náuseas, vómitos, diarrea, dolor abdominal, erupción cutánea,
dolor de cabeza, y la inquietud, raras reacciones alérgicas como urticaria y anafilaxia
se han descrito y entre los efectos adversos graves incluyen psicosis inducida por
fármacos, inmunogénica reacción de hipersensibilidad, neuropatía periférica,
aumento de la presión intracraneal, convulsiones, tendinitis, rotura traumática o no
traumática del tendón (Olivera et al, 2011).
I.2.3 Bioequivalencia
Es un ensayo realizado in vitro, cuya finalidad es instituir si un medicamento
genérico posee seguridad y eficacia, tomando como referencia al medicamento
comercial. (Instituto Nacional de Vigilancia de Medicamentos y Alimentos, 2016).
I.2.3.1 A qué se debe la elaboración de los ensayos de Bioequivalencia
Esto comienza desde tiempos atrás, debido a la inseguridad que demostraban las
personas al consumir medicamentos genéricos ya que estos en unas ocasiones no
causaban el mismo efecto que el innovador, siendo este ensayo básico en la
actualidad para evaluar dos fármacos (genérico Vs Innovador) y demostrar así su
intercambiabilidad. (Ministerio de Salud y Protección Social, 2015).

12
I.2.3.2 En que radica la Bioexención
Método opcional a la elaboración de ensayos in vivo, logrando comprobarse por
medio de la Bioequivalencia, esto puede ser comprobado a través de perfiles de
disolución y corrobóralo por medio del factor de diferencia y similitud al analizar las
curvas expuestas (Ministerio de Salud y Protección Social, 2015).
I.2.3.2.1 Cuándo debemos optar por realizar una bioexención
Se opta por realizar la Bioexención a un fármaco cuando: Pertenece su principio
activo a los grupos I o III del sistema de Clasificación Biofarmaceutico, los mismos no
se encuentra enlistado al angosto margen terapéutico y finalmente que no posea
excipientes que alteren el proceso de absorción del medicamento. (Ministerio de
Salud y Protección Social, 2015).
I.2.4 Equivalentes Farmacéuticos
Son sustancias medicamentosas que están compuestas por los mismos principios
activos en igual forma farmacéutica, vías de administración, aunque no es obligatorio
que contengan los mismos excipientes ni que cumplan con las mismas o semejantes
detalles de calidad. (Gaete, 2014).
I.2.5 Equivalentes Terapéuticos
Un medicamento genérico es equivalente terapéuticamente con otro que posea el
mismo principio activo, acción terapéutica, seguridad y eficacia que el otro
medicamento (innovador), los cuales ya han sido demostrados a través de análisis
establecidos. (Agón, 2013).
Los medicamentos que cumplen con los siguientes puntos establecidos por la FDA
se consideran terapéuticamente equivalentes: seguros y eficaces, poseedores de
igual principio activo, forma medicamentosa y ruta de administración, cumplan con las

13
reglas técnicas establecidas por la farmacopea en fuerza, eficacia, decencia e
identidad, manifiesten Bioequivalencia, etiquetado adecuado y sean fabricados
cumpliendo con las políticas de las BPF vigentes. (Guerrero, 2013).
Un medicamento es terapéuticamente equivalente con el innovador cuando se ha
demostrado la intercambiabilidad existente con el genérico o copia. (Guerrero, 2013).
I.2.6 Innovador o de Referencia
Se le considera innovador a aquel que se le realizó estudios por primera vez y se
descubrió por ende la molécula otorgándole la patente al descubridor, se emplea
como patrón en ensayos de Bioequivalencia, dado que aporta datos propios de
seguridad y eficacia del principio activo (Leiva, 2011).
I.2.7 Genérico o Copia
Se le conoce como genérico a un medicamento “químicamente igual” al original
cuyo nombre es la molécula activa (principio activo), la definición de este nombre fue
admitido y divulgado por la Organización Mundial de Salud. (Leiva, 2011).
I.2.8 Metodologías para indicar la Bioequivalencia
Este tipo de estudio es básico para evitar la necesidad de realizar ensayos in vivo,
son la ruta por la cual los medicamentos genéricos, resultan aceptados para
comercializarse prolongando la seguridad y eficacia de los mismos; estos análisis son
básicos también a lo que se originan cambios significativos en su formulación o
elaboración. (Guerrero, 2013).
Los ensayos de Bioequivalencia involucra la semejanza que debe haber entre el
tiempo y velocidad de disolución y absorción del medicamento de referencia y
genérico, hasta que esto no se compruebe los fármacos no podrán ser considerados

14
intercambiables entre sí, aunque manifiesten ser equivalentes farmacéuticos.
(Guerrero, 2013).
La Bioequivalencia del medicamento genérico Vs innovador podrá ser demostrada
mediante varios métodos, entre los cuales señalamos a: Los métodos in vivo e in vitro.
(Guerrero, 2013).
I.2.8.1 Metodologías in vivo
Se toma como muestra a una cantidad establecida de voluntarios sanos a los que
se les administra el medicamento por la vía establecida, después se le realiza
extracciones de fluidos orgánico como: sangre, orina a distintos tiempos, durante el
ensayo se controlaran los signos vitales de los pacientes y se analizaran los posibles
efectos indeseados, a esto se lo conoce como Fase Clínica. (Guerrero, 2013).
Se utilizan métodos analíticos para conocer las distintas concentraciones del
principio activo presentes en los fluidos orgánicos, a este procedimiento se lo llama
Fase Analítica. (Guerrero, 2013).
Por ultimo utilizando los modelos estadísticos se confrontan los resultados
cinéticos de biodisponibilidad conseguidos del innovador y del genérico para
establecer si existe similitud y verificar su Bioequivalencia, conociéndolo a este
procedimiento como Fase Estadística. (Guerrero, 2013).
I.2.8.2 Metodologías in vitro
Los ensayos realizados in vivo pueden ser reemplazados por ensayos in vitro,
utilizados para indicar Bioequivalencia entre un medicamento genérico con respecto
a un innovador, mediante el empleo de los perfiles de disolución, en base al factor de
diferencia y similitud; pero solo cuando el fármaco pertenezca al grupo I o III del
sistema de clasificación Biofarmaceutico (SCB) siendo por ende una forma
medicamentosa de liberación inmediata. (Guerrero, 2013).

15
I.2.9 Sistema de Clasificación Biofarmacéutico
Básicamente es un cuadro irrefutable que nos permite ubicar los medicamentos en
cuatro grandes grupos, atendiendo a las características de solubilidad acuosa y
permeabilidad intestinal este sistema fue propuesto por Gordon Amidon (Iriarte,
2015).
Entre estos tenemos: Caso 1: Solubilidad alta – Permeabilidad alta, Caso 2: Baja
solubilidad - alta permeabilidad, Caso 3: Solubilidad alta - Baja permeabilidad
(Drogas), Caso 4: Baja solubilidad - baja permeabilidad (Drogas). (Food and Drug
Administration, 1997).
Estos casos son útiles para establecer que medicamento corresponde al grupo I o
III en la disolución in vitro, así como ayudar a predecir el posible éxito o fracaso en la
correlación in vivo – in vitro. (Food and Drug Administration, 1997).
Solubilidad: Se considera soluble a una sustancia medicamentosa cuando esta
presenta una concentración más soluble en 250 mL o menos soluble en un medio
acuoso de rango: 1,0 a 7,5 (Pérez, 2013).
Permeabilidad: Se considera permeable a un medicamento que permite que un
fluido atraviese su estructura compacta, permitiendo que pasa una cantidad
apreciable de fluido en él, así mismo se le considera altamente permeable cuando
este es absorbido en los humanos en una cantidad de 90% en la dosis administrada.
(Pérez, 2013).
I.2.10 Perfiles de disolución
Los resultados se expresan mediante curvas, las cuales nos indican el % disuelto
del medicamento en relación a los tiempos establecidos; relacionando 2

16
medicamentos uno el innovador y el otro el genérico esto se puede describir por medio
de los modelos independientes y dependientes. (Guerrero, 2013).
I.2.10.1 Modelos de los perfiles de disolución
Se puede realizar la comparación de perfiles de disolución utilizando:
I.2.10.1.1 Modelo independiente
Parámetros del modelo independiente
Los parámetros puntuales empíricos son magnitudes que se calculan o se deducen
de los datos experimentales como:
El tiempo que tarda en disolverse un determinado porcentaje de la dosis (t10% t 50
%, t80 %). (Montejo, 2015).
La Eficiencia de la Disolución (EF%): Se deduce partiendo de las curvas del
medicamento disuelto necesariamente debe disolverse como mínimo el 90% de la
cantidad. (Montejo, 2015).
El tiempo medio de disolución (MDT): Se obtiene al calcular las cantidades diluidas
del medicamento representadas por medio de una curva en función del tiempo.
(Montejo, 2015).
Modelo de acercamiento independiente a través del factor de similitud
Los perfiles de disolución los vamos a comparar en base al factor de diferencia y
similitud. Siendo F1 el responsable del cálculo diferencial porcentual y de las
cantidades del error relativo, entre cada punto temporal de las 2 curvas. (Food and
Drug Administration, 1997).

17
𝐹1 =∑ (𝑅𝑡 − 𝑇𝑡)𝑛
𝑡=1
∑ (𝑅𝑡)𝑛𝑡=1
𝑥100
Gráfico II. Fórmula del factor de diferencia
Siendo Rt la cantidad de disolución del turno del innovador en el tiempo t y Tt
vendría hacer la cantidad de disolución del turno del genérico en el tiempo t y n la
cantidad de sitios temporales. (Food and Drug Administration, 1997).
En cambio, F2 se obtiene de la metamorfosis de la raíz cuadrada alterna
logarítmica al sumar el error cuadrado; siendo una medida en la disolución porcentual,
entre ambas curvas. (Food and Drug Administration, 1997).
𝐹2 = 50𝑋𝑙𝑂𝐺{[1 + (1
𝑛) ∑(𝑅𝑡 − 𝑇𝑡)
𝑛
𝑡=1
2
]
−0.5
𝑋100}
Gráfico III. Fórmula del factor de similitud
Modelo independiente de la zona de confianza multivariada
Cuando el turno es más del 15% de CV, es preferible efectuar un medio
independiente de guía multivariado para la interpretación de los perfiles de disolución.
(Food and Drug Administration, 1997).
Para su desarrollo es necesario llevar a cabo lo siguiente: a) Establecer los
términos de F2 en técnicas del trayecto estadístico multivariado (MSD) tomando como
punto de partida las diferencias en disolución entre los turnos de referencia
(aceptadas por el estándar), b) Efectuar los cálculos establecidos para la MSD entre
las disoluciones genéricas y referencias medias, c) Obtener el cálculo del intervalo
de certidumbre del 90 % de la auténtica MSD entre los turnos de prueba y referencia,
d) Confrontar los términos superiores del intervalo de certidumbre con el límite de
similitud. (Food and Drug Administration, 1997).

18
Si el límite superior del intervalo de certeza es = o – al límite de similitud del turno
del genérico este vendría hacer similar al turno de referencia. (Food and Drug
Administration, 1997).
I.2.10.1.2 Modelo dependiente
Parámetros del modelo dependiente
Con base fisicoquímica: Cinética de orden cero, Cinética de primer orden, Cinética
de raíz cúbica, Cinética de raíz cuadrada.
Sin base fisicoquímica: Función de Weibull (Montejo, 2015).
Para obtener un valor acertado dentro de los perfiles de disolución, se han aplicado
diversos modelos matemáticos de los cuales se indica que debe seguir las siguientes
operaciones para el desarrollo de los mismos: a) Básicamente antes de escoger un
método se aconseja que este no posea más de 3 parámetros, por ende, elegir el más
apropiado para los perfiles de disolución tomando en cuenta los patrones que serán
expuestos al cambio y aprobados; b) Una vez obtenido los datos emparejar con el
modelo más indicado, utilizando los datos obtenidos por el perfil de cada unidad; c)
Una vez dada a conocer las medidas del modelo emparejado con las unidades del
experimento con respecto a los patrones aceptadas, se fija una región de similitud la
cual estará basada en estas medidas; d) En base a los turnos de prueba y referencia,
calcular la MSD tomando como consideración los parámetros del modelo. Las
medidas del modelo; e) Obtener la zona de certidumbre del 90% en base a la
auténtica disconformidad de los 2 turnos; f) Para establecer que el turno de prueba
posee un perfil de disolución similar al turno de referencia, es necesario confrontar los
límites de la zona de certidumbre Vs la zona de similitud. (Food and Drug
Administration, 1997).

19
I.2.11 Evaluación de los perfiles de disolución
Si al termino del análisis el CV obtenido en base al porcentaje disuelto ( calculado
con respecto a la dosis del medicamento), es ≤ al 20% en el primer tiempo de
muestreo y ≤ al 10% para los tiempos restantes se confrontan los perfiles de
disolución usando el factor de similitud, se deberá reportar el % disuelto a cada tiempo
de muestreo con el debido módulo de dosis; así también los % disueltos promedio,
CV y cantidades máximo y mínimo, finalmente graficar los % promedios disueltos de
cada módulo de dosis, Vs el tiempo. (Posada y Santos, 2009).
Ensayos de disolución: Son ensayos in vitro cuya función es valorar las
peculiaridades de la liberación del principio activo de un medicamento expuesto a un
medio de disolución adecuado con las debidas circunstancias experimentales
escrupulosamente establecidas (Vinueza, 2013).
I.2.12 Instrumentos
Para la realización de los ensayos de disolución los instrumentos más
comúnmente empleados son: La canastilla y la paleta se utilizan en todo el mundo
teniendo como características principales ser sencillos y robustos. Colaboran con la
elaboración de ensayos de disolución para una diversidad de medicamentos, por esta
razón es necesario que al realizar ensayos in vitro descritos en la Farmacopea
Estadounidense, el instrumento 1 y 2 estén establecidos, caso contrario que no este,
se indicarían que no son satisfactorios. (Food and Drug Administration, 1997).
Técnica del Instrumento 1: También conocida como técnica de la canastilla, está
compuesta básicamente por un vaso de plástico u otro material transparente sin tapa,
con su eje motriz y una canastilla cilíndrica. Esto se arma en un dispositivo establecido
en donde el vaso está parcialmente sumergido en un baño de agua cuya dimensión
es la indicada para que encaje y reciba el calor fijado. (Farmacopea de los Estados
Unidos de América, 2014).

20
Gráfico IV. Elemento de agitación de la canastilla (Farmacopea de los Estados
Unidos de América, 2014).
Técnica del instrumento 2: También conocida como técnica de la paleta, utiliza
un dispositivo establecido, el cual está compuesto por un aspa y un eje como
elemento de agitación, que está a una distancia máxima de 2 miligramos (mm) el cual
gira dócilmente sin provocar incertidumbres significativas que alcanzaran a perjudicar
los resultados, así el trayecto entre el fondo interno del vaso y el borde inferior del
aspa se mantiene a 25 ± 2 miligramos (mm) durante la prueba, las aspas son de un
material metálico idóneo que constituyen una unidad con el eje, en unas
circunstancias se usan instrumentos separables de 2 partes que deben estar
firmemente ajustadas durante la prueba, básicamente este tipo de instrumento es
utilizado en tabletas y comprimidos. (Farmacopea de los Estados Unidos de América,
2014).

21
Gráfico V. Esquema de las paletas (Farmacopea de los Estados Unidos de América,
2014).

22
CAPÍTULO II. MATERIALES Y MÉTODOS
“El factor de diferencia y similitud es un paso para la comparación de los perfiles
de disolución, pero del que pocas tabletas lograran aprobar”
II.1 MATERIALES
Para este estudio se empleó un lote de tabletas de ciprofloxacino innovador la cual
se marcó con la letra R y tres lotes de ciprofloxacino genérico de dos laboratorios
farmacéuticos diferentes que se rotularon con las letras A y B. Utilizando los
materiales descritos en el Anexo 1.
II.2 MÉTODOS
II.2.1 Tipo de estudio
Este estudio fue de carácter experimental y comparativo.
Experimental porque se realizaron pruebas de laboratorio para determinar la
bioequivalencia de los dos genéricos nacionales con respecto al innovador.
Comparativo porque se estableció la similitud y diferencia entre los productos
genéricos y el innovador (Posada y Santos, 2009).
II.2.2 Metodología
El ensayo número 711 de la Farmacopea de los Estados Unidos número 37 y el
Formulario Nacional número 32 indican como se efectuaron los ensayos de los
perfiles de disolución, utilizando un Disolutor marca Sotax tipo AT 7smart, el cual
posee control de temperatura y consta de 7 vasos, se utilizó el Equipo 2 de la USP
37, conocida como paletas y 900 mL de HCl 0,01 N como medio de disolución para

23
cada vaso a una velocidad de 50 rpm por 30 minutos (Farmacopea de los Estados
Unidos de América, 2014).
Se llenó con el volumen establecido del medio de disolución cada uno de los vasos
y se colocaron dentro del equipo, se encendió el equipo y cuando el medio alcanzó la
temperatura de 37º C, se añadieron las muestras en cada uno de los vasos del equipo
y se reguló a 50 rpm por 30 minutos. En este periodo de tiempo se tomaron 10 mL,
de cada vaso a los tiempos de: 5, 10, 15, 20 y 30 minutos reemplazando las
cantidades extraídas con la misma cantidad del medio de disolución. Las muestras
extraídas se filtraron y de este filtrado se tomó una alícuota de 1 mL con la pipeta
volumétrica que se colocó en una fiola de 100 mL se enrasó con HCl 0.01 N y por
ultimo mediante el espectrofotómetro marca Shimadzu modelo UV 1700, se procedió
a leer cada una de las diluciones de las muestra de estudio que correspondían a los
tiempos de 5, 10, 15, 20 y 30 minutos a una longitud de onda de 276 nm, los cuales
compararon con la solución estándar la cual se preparó a una concentración conocida
de ciprofloxacino (Leiva, 2011).
Finalmente, por medio del modelo independiente empleando el F2 se examinaron
las curvas de disolución, con las cuales se logra obtener una semejanza fiable de los
medicamentos. (Leiva, 2011).
II.2.2.3 Métodos de análisis e interpretación de los resultados
Los perfiles de disolución de los medicamentos se establecieron al utilizar el
modelo independiente según las indicaciones de la FDA, si los resultados expuestos
por los perfiles de disolución se encuentran dentro de los rangos establecidos como
F1: 0 - 15, F2: 50 – 100 se los consideran aceptables. (Pérez, 2013).
II.2.3 VARIABLES
II.2.3.1 Independiente
Factor F1 (diferencia)
Factor F2 (similitud)

24
II.2.3.2 Dependiente
Medicamento bioequivalente
II.2.3.3 Interviniente
Excipientes empleados
Laboratorio productor
Tabla I. Operacionalización
Variables Conceptualización Indicador Unidad
Medicamento
bioequivalente
(bioexención)
Permite la demostración de
equivalencia terapéutica mediante
estudios de disolución in vitro que
compara la biodisponibilidad en
magnitud y velocidad de las
formulaciones analizadas. El
ensayo realiza un estudio
comparativo de los perfiles de
disolución in vitro de las mismas.
Concentración
del principio
activo
mg
Disolución del
principio activo %
Factor de
diferencia (F1)
Es una dimensión del error relativo
con respecto a las 2 curvas e indica
la desemejanza presente en
ambas curvas.
F1: 0 - 15
%
Factor de
similitud (F2)
Se obtiene de la metamorfosis de
la raíz cuadrada mediante la suma
de las distancias cuadradas en
todos los puntos.
F2: 50 - 100 %
Excipientes
empleados
Pueden influir en la motilidad y/o la
permeabilidad en el tracto
gastrointestinal. Si el producto
contiene excipientes que han sido
utilizados antes en cantidades
similares en otras formulaciones del
mismo principio activo, se puede
concluir que los excipientes no
tendrán una influencia inesperada
sobre la biodisponibilidad del
producto. Sin embargo, si la
formulación contiene diferentes
excipientes o muy diferentes
Concentración y
tipo
g, Kg

25
II.2.4 MUESTREO
Se utilizó como base un análisis estadístico no poblacional para establecer el
tamaño del espécimen, enfocándose en la aprobación y rechazo de los productos
Nacionales, se tomaron muestras de las tabletas de ciprofloxacino de 500 mg
innovador y de los productos genéricos, valorando el medicamento innovador (en
base a un lote) y el medicamento genérico (en base a tres lotes) de dos laboratorios.
(Posada y Santos, 2009).
II.2.4.1 TIPO DE MUESTREO
El muestreo consistió en la selección de las unidades de estudio según el criterio
establecido, siendo las tabletas las unidades representativas para el análisis.
II.2.5 POBLACIÓN Y MUESTRA
Estuvo constituidas por las tabletas de ciprofloxacino de 500 mg, de los dos
laboratorios farmacéuticos del Ecuador. Los estudios se realizaron sobre tres lotes de
cada producto genérico y un lote para el innovador, se marcó con las letras A y B
mayúscula cada producto genérico con sus respectivas codificaciones numéricas
para diferenciar cada lote y se marcó con la letra R el producto innovador.
II.2.6 CRITERIOS DE INCLUSIÓN Y EXCLUSIÓN
Solo se utilizaron para este análisis las tabletas de ciprofloxacino de 500 mg,
innovador y de los dos laboratorios farmacéuticos del Ecuador, que estén dentro del
tiempo de vida útil y del mismo lote correspondiendo a su laboratorio farmacéutico.
cantidades de los mismos, las
autoridades competentes pueden
optar por no permitir la bioexención.
Laboratorio
productor Calidad del producto
Cumplimiento de
las BPM -
26
CAPÍTULO III. RESULTADOS Y DISCUSIÓN
III. 1 COMPORTAMIENTO DE CADA LOTE DEL MEDICAMENTO DE ESTUDIO
FRENTE A LA DISOLUCIÓN
El estudio comenzó con la determinación de los porcentajes de disolución del
medicamento innovador y de los tres lotes de los medicamentos genéricos.
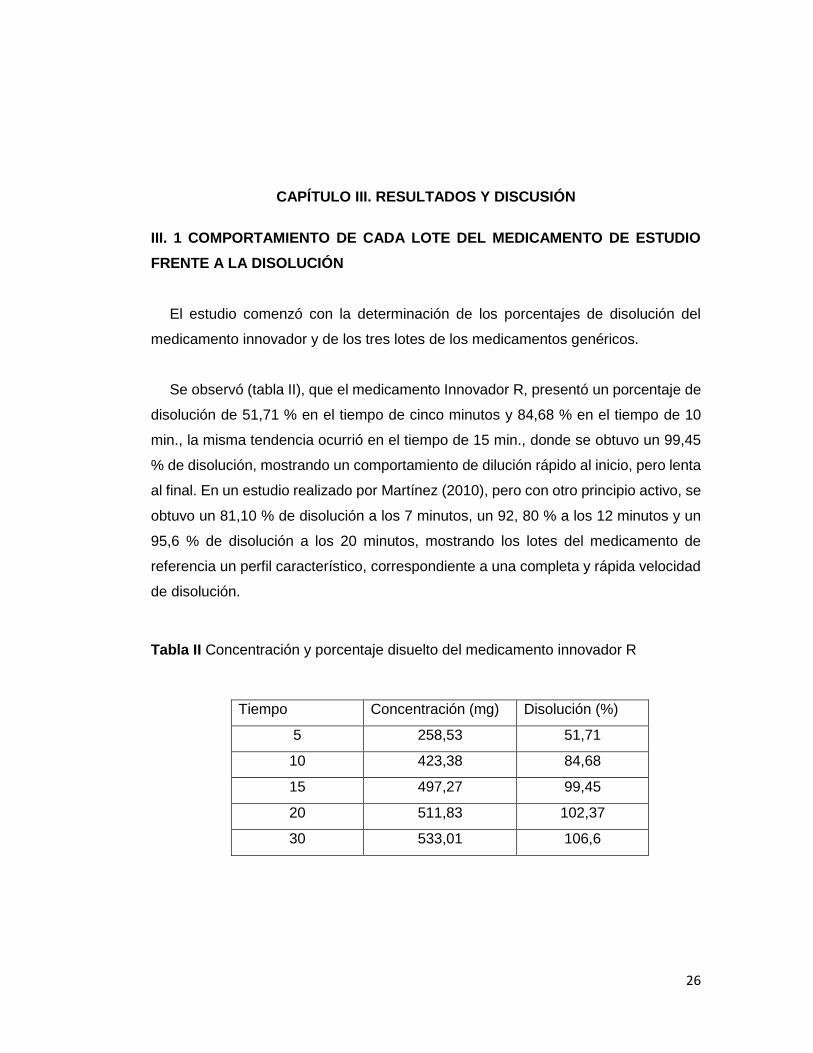
Se observó (tabla II), que el medicamento Innovador R, presentó un porcentaje de
disolución de 51,71 % en el tiempo de cinco minutos y 84,68 % en el tiempo de 10
min., la misma tendencia ocurrió en el tiempo de 15 min., donde se obtuvo un 99,45
% de disolución, mostrando un comportamiento de dilución rápido al inicio, pero lenta
al final. En un estudio realizado por Martínez (2010), pero con otro principio activo, se
obtuvo un 81,10 % de disolución a los 7 minutos, un 92, 80 % a los 12 minutos y un
95,6 % de disolución a los 20 minutos, mostrando los lotes del medicamento de
referencia un perfil característico, correspondiente a una completa y rápida velocidad
de disolución.
Tabla II Concentración y porcentaje disuelto del medicamento innovador R
Tiempo Concentración (mg) Disolución (%)
5 258,53 51,71
10 423,38 84,68
15 497,27 99,45
20 511,83 102,37
30 533,01 106,6

27
El análisis del comportamiento en la disolución del medicamento genérico A se
presenta en la tabla IIII.
Tabla III Porcentaje disuelto de cada uno de los lotes del medicamento genérico A
Tiempo
(min.)
Porcentaje disuelto (%)
A1 A2 A3
5 33,48 37,61 24,31
10 65,86 71,72 58,80
15 90,23 88,49 88,72
20 97,79 100,43 98,82
30 100,91 106,22 102,18
En ella se aprecia comportamiento de disolución, similares entre los diferentes
lotes de este genérico a los 5 minutos, donde se disuelve aproximadamente más del
30 % en A1 y A2, y menos del 30 % en A3. En un estudio realizado por Franco-Ospina
et al, (2012), al ciprofloxacino tabletas que se comercializa en Colombia como
genéricos, se observaron comportamientos de disolución similares para la mayoría
de los productos, liberando aproximadamente el 50 % del principio activo a los 10
minutos y más del 75 % a los 20 minutos, señalando, además, falta de homogeneidad
en cuanto a la liberación in vitro del fármaco entre las diferentes muestras evaluadas.
28
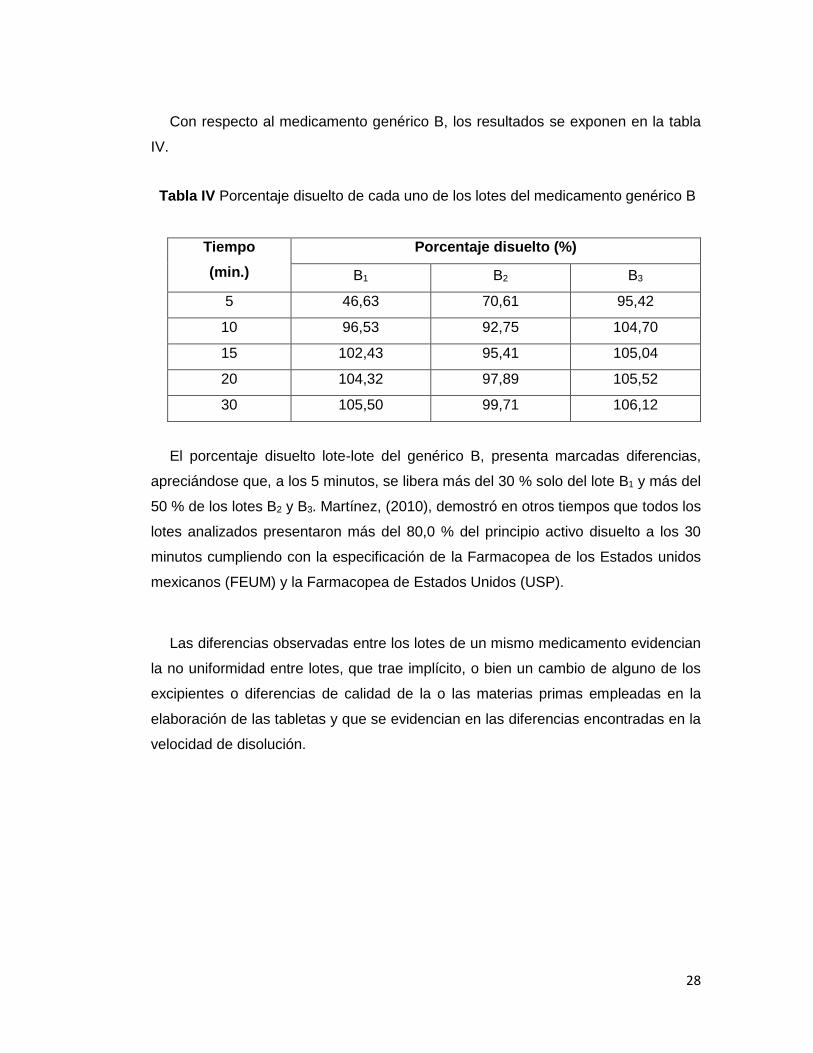
Con respecto al medicamento genérico B, los resultados se exponen en la tabla
IV.
Tabla IV Porcentaje disuelto de cada uno de los lotes del medicamento genérico B
Tiempo
(min.)
Porcentaje disuelto (%)
B1 B2 B3
5 46,63 70,61 95,42
10 96,53 92,75 104,70
15 102,43 95,41 105,04
20 104,32 97,89 105,52
30 105,50 99,71 106,12
El porcentaje disuelto lote-lote del genérico B, presenta marcadas diferencias,
apreciándose que, a los 5 minutos, se libera más del 30 % solo del lote B1 y más del
50 % de los lotes B2 y B3. Martínez, (2010), demostró en otros tiempos que todos los
lotes analizados presentaron más del 80,0 % del principio activo disuelto a los 30
minutos cumpliendo con la especificación de la Farmacopea de los Estados unidos
mexicanos (FEUM) y la Farmacopea de Estados Unidos (USP).
Las diferencias observadas entre los lotes de un mismo medicamento evidencian
la no uniformidad entre lotes, que trae implícito, o bien un cambio de alguno de los
excipientes o diferencias de calidad de la o las materias primas empleadas en la
elaboración de las tabletas y que se evidencian en las diferencias encontradas en la
velocidad de disolución.
29
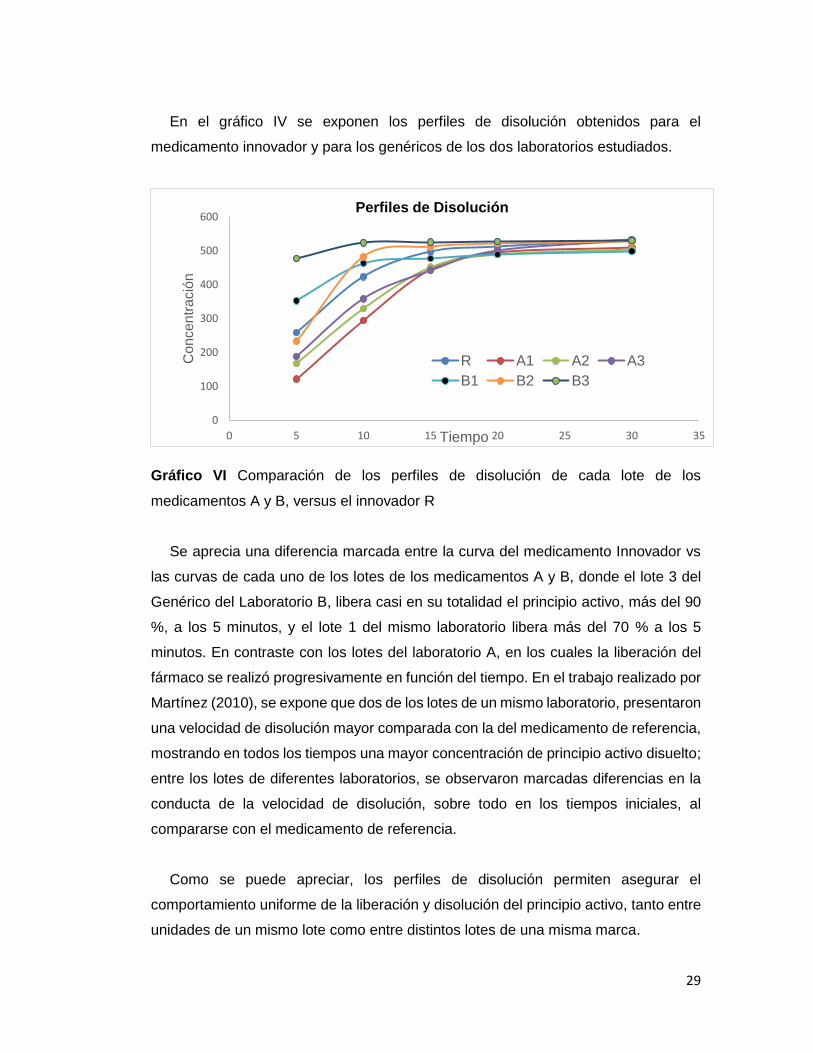
En el gráfico IV se exponen los perfiles de disolución obtenidos para el
medicamento innovador y para los genéricos de los dos laboratorios estudiados.
Gráfico VI Comparación de los perfiles de disolución de cada lote de los
medicamentos A y B, versus el innovador R
Se aprecia una diferencia marcada entre la curva del medicamento Innovador vs
las curvas de cada uno de los lotes de los medicamentos A y B, donde el lote 3 del
Genérico del Laboratorio B, libera casi en su totalidad el principio activo, más del 90
%, a los 5 minutos, y el lote 1 del mismo laboratorio libera más del 70 % a los 5
minutos. En contraste con los lotes del laboratorio A, en los cuales la liberación del
fármaco se realizó progresivamente en función del tiempo. En el trabajo realizado por
Martínez (2010), se expone que dos de los lotes de un mismo laboratorio, presentaron
una velocidad de disolución mayor comparada con la del medicamento de referencia,
mostrando en todos los tiempos una mayor concentración de principio activo disuelto;
entre los lotes de diferentes laboratorios, se observaron marcadas diferencias en la
conducta de la velocidad de disolución, sobre todo en los tiempos iniciales, al
compararse con el medicamento de referencia.
Como se puede apreciar, los perfiles de disolución permiten asegurar el
comportamiento uniforme de la liberación y disolución del principio activo, tanto entre
unidades de un mismo lote como entre distintos lotes de una misma marca.
0
100
200
300
400
500
600
0 5 10 15 20 25 30 35
Co
nce
ntr
ació
n
Tiempo
Perfiles de Disolución
R A1 A2 A3
B1 B2 B3

30
III.2 COMPROBACIÓN DEL MODELO INDEPENDIENTE A TRAVÉS DEL FACTOR
DE SIMILITUD PARA CADA LOTE DE LOS GENÉRICOS EN ESTUDIO.
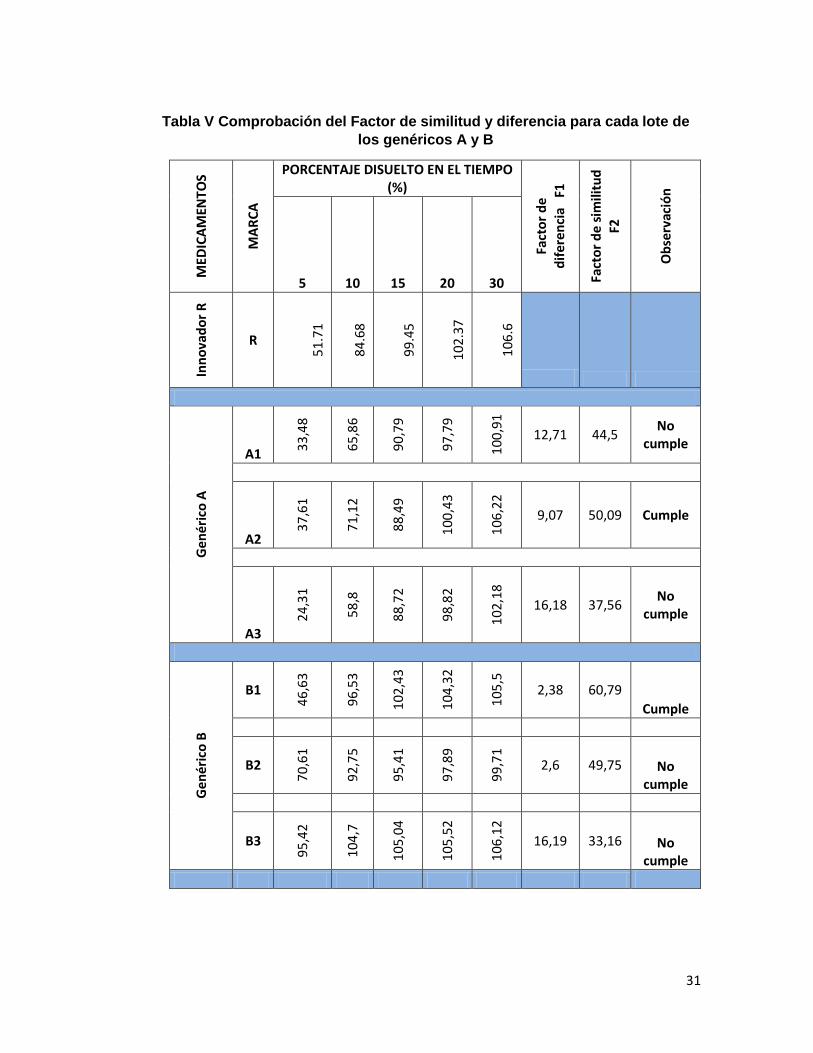
En la tabla V, se reflejan los valores obtenidos para la comprobación del modelo
independiente a través del factor de similitud de cada uno de los lotes de los
medicamentos genéricos en estudio.
Se pudo observar que sólo en los casos del genérico A2 y el genérico B1, se
cumplían con las especificaciones de aceptación de F1 (0 – 15) y F2 (50 – 100), pero
los demás lotes de los dos laboratorios farmacéuticos no cumplían con esta
disposición. Martínez (2010), demostró que al aplicar la prueba F2 los perfiles de
disolución de uno de los lotes, mostraba un valor menor a 50 con respecto al
medicamento de referencia; este comportamiento indicó, que pudo deberse a
diferencias en su formulación o a los procesos de manufactura.
Como se observa en este estudio, las diferencias entre lotes manifestadas en el
estudio de disolución pueden ser debidas a cambios de materias primas y procesos
de producción, incumpliéndose así con las buenas prácticas de manufactura de la
empresa.
31
Tabla V Comprobación del Factor de similitud y diferencia para cada lote de
los genéricos A y B
MED
ICA
MEN
TOS
MA
RC
A
PORCENTAJE DISUELTO EN EL TIEMPO (%)
Fact
or
de
dif
eren
cia
F1
Fact
or
de
sim
ilitu
d
F2
Ob
serv
ació
n
5 10 15 20 30
Inn
ova
do
r R
R
51
.71
84
.68
99
.45
10
2.3
7
10
6.6
Ge
né
rico
A
A1 33
,48
65
,86
90
,79
97
,79
10
0,9
1
12,71 44,5 No
cumple
A2
37
,61
71
,12
88
,49
10
0,4
3
10
6,2
2
9,07 50,09 Cumple
A3
24
,31
58
,8
88
,72
98
,82
10
2,1
8
16,18 37,56 No
cumple
Gen
éric
o B
B1
46
,63
96
,53
102
,43
104
,32
105
,5
2,38 60,79
Cumple
B2
70
,61
92
,75
95
,41
97
,89
99
,71
2,6 49,75 No cumple
B3
95
,42
104
,7
105
,04
105
,52
106
,12
16,19 33,16 No cumple
32
III. 3 COMPARACIÓN DE LOS VALORES DE DISOLUCIÓN PROMEDIO DE CADA
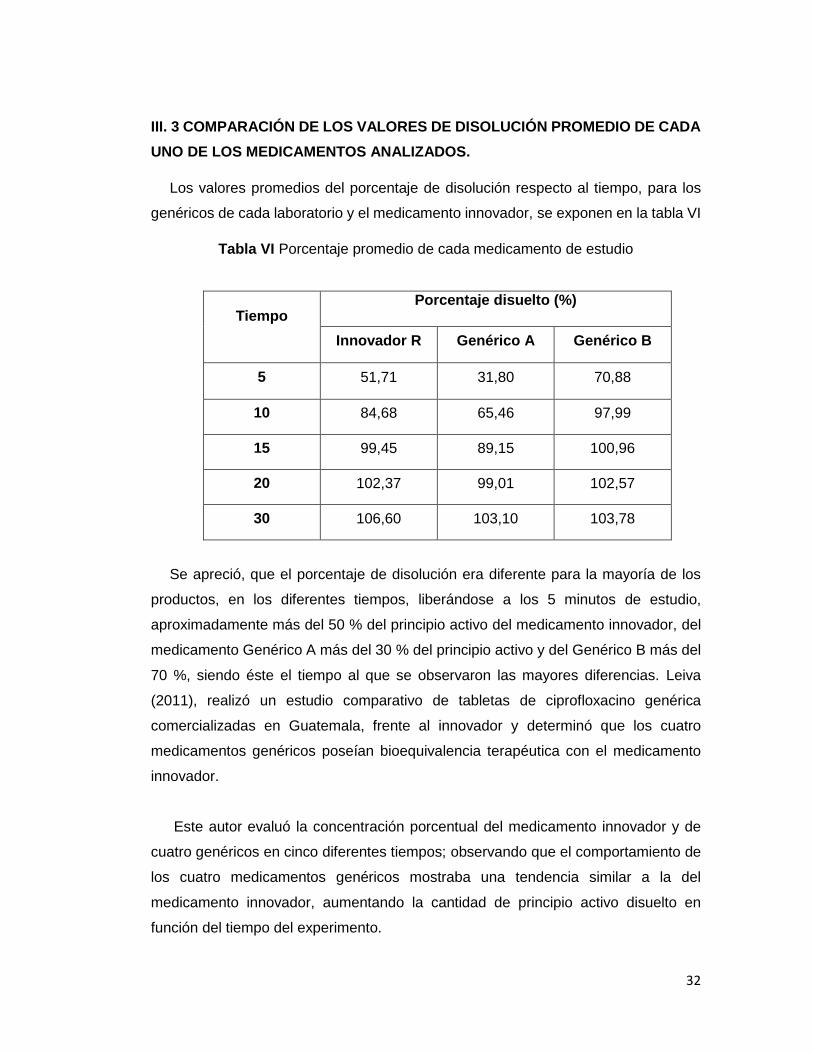
UNO DE LOS MEDICAMENTOS ANALIZADOS.
Los valores promedios del porcentaje de disolución respecto al tiempo, para los
genéricos de cada laboratorio y el medicamento innovador, se exponen en la tabla VI
Tabla VI Porcentaje promedio de cada medicamento de estudio
Tiempo Porcentaje disuelto (%)
Innovador R Genérico A Genérico B
5 51,71 31,80 70,88
10 84,68 65,46 97,99
15 99,45 89,15 100,96
20 102,37 99,01 102,57
30 106,60 103,10 103,78
Se apreció, que el porcentaje de disolución era diferente para la mayoría de los
productos, en los diferentes tiempos, liberándose a los 5 minutos de estudio,
aproximadamente más del 50 % del principio activo del medicamento innovador, del
medicamento Genérico A más del 30 % del principio activo y del Genérico B más del
70 %, siendo éste el tiempo al que se observaron las mayores diferencias. Leiva
(2011), realizó un estudio comparativo de tabletas de ciprofloxacino genérica
comercializadas en Guatemala, frente al innovador y determinó que los cuatro
medicamentos genéricos poseían bioequivalencia terapéutica con el medicamento
innovador.
Este autor evaluó la concentración porcentual del medicamento innovador y de
cuatro genéricos en cinco diferentes tiempos; observando que el comportamiento de
los cuatro medicamentos genéricos mostraba una tendencia similar a la del
medicamento innovador, aumentando la cantidad de principio activo disuelto en
función del tiempo del experimento.

33
Los perfiles de disolución de los valores de disolución promedio para cada tiempo,
se exponen en el gráfico VII. En él se observa que el promedio de disolución de los
medicamentos genéricos y del innovador, presentaron diferencias significativas en los
primeros tiempos de análisis, pero a medida que transcurría el tiempo, se observaba
semejanza entre los productos de estudio.
En su trabajo Franco-Ospina et al, (2012), probó que de los doce productos
evaluados once, incluyendo la referencia, cumplían con todas las especificaciones
establecidas en la USP- 33/NF28, siendo equivalentes farmacéuticos; sin embargo,
un análisis detallado del comportamiento cinético in vitro de la disolución, mostró
diferencias entre las formulaciones, lo que sugiere que su comportamiento in vivo
podría también ser diferente.
Gráfico VII Comparación de los perfiles de disolución de los promedios de los
medicamentos A y B, versus el innovador R.
III.4 DEMOSTRACIÓN DE LA BIOEQUIVALENCIA DE LOS PORCENTAJES DE
DISOLUCIÓN PROMEDIOS DE LOS GENÉRICOS A Y B
Al igual que para el análisis de los lotes independientes, se realizó el análisis de
los factores de similitud y diferencia para los valores promedios de disolución de los
lotes de los medicamentos genéricos y el medicamento innovador y los resultados se
exponen en la tabla VII.
0
20
40
60
80
100
120
5 10 15 20 30
po
rcen
taje
Tiempo
Perfiles de disolución
Innovador R Genérico A Genérico B

34
Se apreció que en el caso del genérico A, el valor de F1 fue 9,49 y el de F2 49,81
y para el Genérico B, el valor de F1 fue de 7,06 y el de F2 de 48,76.
Tanto el genérico A como el B, cumplieron con el factor de diferencia F1, pero
ambos incumplieron con el factor de similitud F2, ya que los valores obtenidos no
alcanzaron el rango de 50-100.
Jung Cook et al (2012), realizaron un estudio sobre los perfiles de disolución y el
impacto de los criterios de las diferentes agencias regulatorias en el cálculo de F2;
ellos demostraron que el valor de F2 fue mayor a 50 al utilizar el instrumento 1 (criterio
FDA), mientras que al usar el instrumento 2, el valor de F2 fue menor a 50.
Esto demuestra que el producto podría o no cumplir con el factor de similitud, en
dependencia del criterio utilizado.
Tabla VII Comprobación del factor de similitud y diferencia: Los promedios de
los medicamentos genéricos A y B vs Innovador
Medicamentos
PORCENTAJE DISUELTO EN EL TIEMPO
Fact
or
de
dif
ere
nci
a F
1
Fact
or
de
sim
ilitu
d F
2
Observación
5 10 15 20 30
Innovador R 51,71 84,68 99,45 102,37 106,6
Genérico A 36,78 70,26 91, 72 99,85 103, 98
9,4
9
49
,81
No cumple
Genérico B 70,88 97,99 100,96 102,57 103,78
7,0
6
48
,76
No cumple

35
IV. CONCLUSIONES Y RECOMENDACIONES
IV.1 CONCLUSIONES
Los resultados obtenidos permiten arribar a las siguientes conclusiones:
Los perfiles de disolución de los diferentes lotes de medicamentos genéricos
de los laboratorios estudiados presentaron diferencias entre lotes y en relación
al medicamento innovador, en los primeros tiempos analizados.
Los perfiles de disolución de valores promedios de disolución para los
medicamentos genéricos y el innovador, presentaron diferencias marcadas en
los tiempos iniciales del estudio.
Se comprobó que los medicamentos genéricos analizados, no presentaron
bioequivalencia terapéutica con el innovador en base al factor de diferencia F1
y al factor de similitud F2.

36
IV.2 RECOMENDACIONES
Realizar este estudio, con el producto elaborado por otros laboratorios
farmacéuticos.
Informar a los laboratorios farmacéuticos implicados, los resultados de este
estudio para que tomen las medidas pertinentes en el cumplimiento de las
BPM.
Publicar los resultados obtenidos.

37
REFERENCIAS BIBLIOGRAFÍCAS
Agón Banzo, P. J. (2013). Estudio comparativo entre estatinas genéricas y no
genéricas, en relación a su uso terapéutico, efectos farmacológicos y
reacciones adversas (Tesis para optar al Grado de Doctor). Universidad de
Zaragoza. Recuperado el 22 de octubre del 2016, de
https://zaguan.unizar.es/record/10342/files/TESIS-2013-031.pdf
Alvarado Yarasca, A., Lozada Colque, G. L., Llerena Benites, R. C., Sadia
Ximena, M. F., Marcos Eguizabal, C.E., Pisconte Campos, N.C., Poma
Ghiggo, J. A.,y YunisIsmodes Noor, H. S. (2014). . Determinación del margen
terapéutico y estudio de la equivalencia biofarmacéutica de las tabletas
multifuentes de digoxina de 0,25 mg. Horiz. Med, 14 (4): 48-52.
Recuperado el 22 de octubre del 2016, de
http://www.scielo.org.pe/pdf/hm/v14n4/a09v14n4.pdf
Bernad, M. (2011). Introducción a la tecnología Farmacéutica. Recuperado el
28 de junio del 2016, de http://depa.fquim.unam.mx/amyd/archivero/Tema1-
Parte2-Generalidades_14722.pdf
Daza Calderón, M. L. (2013). Biodisponibilidad y bioequivalencia in vitro en
cápsulas de amoxicilina de 500 mg comercializados en Bolivia. Revista
con-ciencia, 1(1), 103. Recuperado el 28 de junio del 2016, de
http://www.revistasbolivianas.org.bo/pdf/rcfb/v1n1/v1n1_a11.pdf
Díaz Marcos, A. J. (2013). Ensayo y perfil de disolución de tabletas de
Loperamida de 2 mg de marcas comerciales fabricadas por laboratorios
nacionales, que se expenden en farmacias comerciales de la ciudad capital
(Tesis para optar por el Título Profesional de Químico Farmacéutico).
Universidad de San Carlos de Guatemala. Recuperado el 28 de junio del
2016, de http://biblioteca.usac.edu.gt/tesis/06/06_3508.pdf

38
Estévez, F., Parillo. S., y Cedrés M. (2012). Estudios de bioequivalencia in
vivo para demostrar la intercambiabilidad de medicamentos.
Revista Médica de Uruguay, 28 (3), 165-167. Recuperado el 28 de junio del
2016, de http://www.rmu.org.uy/revista/2012v3/art2.pdf
Farmacopea de los Estados Unidos de América. (2014). Formulario Nacional.
Estados Unidos: Segunda Edición.
Franco-Ospina, L. A., Matiz-Melo, G. E. y Pájaro-Bolívar, I.B. (2012). Estudio
Biofarmacéutico comparativo de marcas comerciales de tabletas de
ciprofloxacina disponibles en el mercado colombiano. Revista de salud
pública, 14 (4), 697. Recuperado el 15 de marzo del 2016, de
http://www.scielosp.org/pdf/rsap/v14n4/v14n4a13.pdf
Food and Drug Administration. (1997). Centro de Evaluación e Investigación
de Fármacos CDER. Guía para la industria: Pruebas de disolución de formas
de dosificación oral sólidas de liberación inmediata.
Recuperado el 8 de marzo del 2016, de
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guida
nces/ucm200707.htm
Fuentes Ureta, M. F. (2011). Comparación de los perfiles de disolución de
clorhidrato de metformina tabletas de 850mg entre genéricos de producción
guatemalteca distribuidos por farmacias comerciales y el producto innovador.
(Tesis para optar por el Título Profesional de Químico Farmacéutico).
Universidad de San Carlos de Guatemala. Recuperado el 28 de junio del
2016, de http://biblioteca.usac.edu.gt/tesis/06/06_3208.pdf
Gaete Castro, F. J. (2014). Validación de la metodología analítica de
paracetamol y aplicación bioexención. (Tesis para optar al título de Químico
Farmacéutico). Universidad de Chile. Recuperado el 21 de octubre del 2016,
de http://repositorio.uchile.cl/bitstream/handle/2250/133822/Validacion-de-la-
metodologia-analitica-de-paracetamol-y-aplicacion-a-un-estudio-de-
bioexencion.pdf?sequence=3&isAllowed=y
Guerrero Segura, F. F. (2013). Determinación de la Intercambiabilidad
Terapéutica de cápsulas de Fluconazol comercializadas en el país en relación
a su innovador Diflucan. (Tesis para optar por el Título Profesional de Químico

39
- Farmacéutico). Universidad Central del Ecuador. Recuperado el 6 de marzo
del 2016, de http://www.dspace.uce.edu.ec/bitstream/25000/1881/1/T-UCE-
0008-26.pdf
Instituto Nacional de Vigilancia de Medicamentos y Alimentos. (2016).
invima.gov.co. Recuperado el 21 de octubre de 2016, de
https://www.invima.gov.co/bioequivalencia
Iriarte Ballesta, R. (2015). Desarrollo de medios de disolución biorrelevantes
para fármacos con problemas de bioequivalencia. (Trabajo de grado en
farmacia), Universidad Miguel Hernández.
Recuperado el 21 de octubre del 2016, de
http://dspace.umh.es/bitstream/11000/2115/1/TFG%20Iriarte%20Ballesta,%
20Ram%C3%B3n.pdf
Jung Cook, H., De Anda Jáuregui, G., Rubio Carrasco, K., Mayet Cruz, L.
(2012). Trabajo científico. Comparación de perfiles de disolución. Impacto de
los criterios de diferentes agencias regulatorias en el cálculo de F2, Revista
mexicana de ciencias farmacéuticas, 43 (3), 68 de. Recuperado el 23 de
marzo del 2016, de http://www.scielo.org.mx/pdf/rmcf/v43n3/v43n3a7.pdf
Laboratorio STADA. (2013). Montelukast stada 10 mg comprimidos efg,
resumen del estudio de Bioequivalencia. Recuperado el 21 de octubre del
2016, de http://www.stada.es/sites/default/files/productos/estudios/Estudio-
Bioequivalencia-Montelukast-10mg-STADA-EFG.pdf
Laboratorio Stada. (2015). STADA presenta Pregabalina STADA Genéricos
EFG bioequivalente a Lyrica de Pfize
Recuperado el 26 de octubre del 2016, de http://www.stada.es/prensa/stada-
presenta-pregabalina-stada-gen%C3%A9ricos-efg-bioequivalente-
lyrica%C2%AE-de-pfizer
Laboratorios Stein. (2012). labstein. Recuperado el 21 de octubre de 2016, de
http://www.labstein.com/wp-
content/uploads/2013/09/CIPROFLOXACINO.pdf
Leiva Anderson, L. R. (2011). Determinación de la intercambiabilidad
terapéutica de ciprofloxacina genérica de 500 mg en tableta recubierta
elaborada en Guatemala a través de perfiles de disolución. (Tesis para optar

40
por el Título de Químico Farmacéutico). Universidad de San Carlos de
Guatemala Recuperado el 10 de abril del 2016, de
http://biblos.usac.edu.gt/library/index.php?title=566268&query=@title=Speci
al:GSMSearchPage@process=@titulo=@autor=@subheadings=@keywords
=ciprofloxacina@material=@sortby=sorttitle@mode=&recnum=1
León Méndez, G., Osorio Fortich, M. R., y Matiz Melo, G. E. (2015)., Estudio
biofarmacéutico comparativo de tabletas de acetaminofén 500 mg disponibles
en el mercado colombiano. Revista Cubana de Farmacia, 49(4) 638-639.
Recuperado en 28 de junio de 2016, de
http://scielo.sld.cu/pdf/far/v49n4/far04415.pdf
Llerena Chang, A. L. (2011). Comparación de los perfiles de disolución de
tabletas de ciprofibrato 100mg genéricas de producción nacional con el
medicamento innovador. (Tesis para optar por el Título Profesional de
Químico Farmacéutico). Universidad de San Carlos de Guatemala.
Recuperado el 28 de junio del 2016, de
http://biblioteca.usac.edu.gt/tesis/06/06_3108.pdf
Martínez Terán, M. E. (2010). Estudio comparativo de la calidad de tabletas
de Ciprofloxacina que se comercializan en México. (Tesis para optar el Grado
de Maestría en Ciencias). Universidad Autónoma de Nuevo León.
Recuperado el 10 de marzo del 2016, de
http://eprints.uanl.mx/2189/1/1080190953.pdf
Martínez Carranza, B.P. (2015). Influencia del recubrimiento acuoso en el
método de lacado y en los perfiles de disolución de la Claritromicina. (Tesis
para optar por el Título Profesional de Química Farmacéutica), Universidad
Central del Ecuador. Recuperado el 21 de octubre del 2016, de
http://www.dspace.uce.edu.ec/bitstream/25000/6322/1/T-UCE-0008-063.pdf
Medina, A. P., Olaya, F. J., Navas, M. P., Tilano, A. M., & Muñoz, E. (2012).
Estudio de bioequivalencia de montelukast en tabletas masticables de 5
mg. Biomédica, 32(3), 399-407. Recuperado el 28 de octubre del 2016,
de http://www.scielo.org.co/pdf/bio/v32n3/v32n3a10.pdf
Ministerio de Salud y Protección Social. (2015). minsalud.gov.co.
Recuperado el 21 de octubre de 2016, de

41
https://www.minsalud.gov.co/sites/rid/Lists/BibliotecaDigital/RIDE/VS/MET/a
bece-biodisponiblidad-bioequivalencia.pdf
Montejo, M. (2015). Biofarmacia y Farmacocinética. España: 2º Edición
Moraga Frías, M. F. (2008). Estudio preliminar de bioequivalencia “in vitro” de
comprimidos genéricos de clorfenamina maleato comercializados en Chile.
(Tesis para optar al Título de Químico Farmacéutico), Universidad de Chile.
Recuperado el 15 de marzo del 2016, de
http://repositorio.uchile.cl/tesis/uchile/2008/qf-moraga_mf/pdfAmont/qf-
moraga_mf.pdf
Muñoz Ibarra, C. A. (2015). Estudio de bioexención de fluoxetina 20mg
comprimidos. (Practica prolongada para optar al Título de Químico
Farmacéutico). Universidad de Chile. Recuperado el 21 de octubre del 2016,
de http://repositorio.uchile.cl/handle/2250/133075
Ochaeta Palma, E. C. (2014). Determinación de la intercambiabilidad
terapéutica de Clorhidrato de Propranolol genérico 40 mg tabletas, producido
por laboratorios nacionales, por medio de la comparación de perfiles de
disolución. (Tesis para optar por el Título Profesional de Químico
Farmacéutico). Universidad de San Carlos de Guatemala. Recuperado el 28
de junio del 2016, de http://www.repositorio.usac.edu.gt/1699/1/06_3630.pdf
Olivera, M.E., Manzo, R.H., Junginger, H.E., Midha, K.K., Shah, V.P.,
Stavchansky, S., Dressman, J.B. & Barends, D.M. (2011). JOURNAL OF
PHARMACEUTICAL SCIENCES. Biowaiver Monographs for Immediate
Release Solid Oral Dosage Forms: Ciprofloxacin Hydrochloride, volumen 100
(1), pp. 23. Recuperado en 28 de junio de 2016, de
http://jpharmsci.org/article/S0022-3549(15)32338-8/pdf
ONG Políticas Farmacéuticas (2015). Bioequivalencia en Chile: Análisis y
recomendaciones. Recuperado el 22 de octubre del 2016, de
http://ciperchile.cl/wp-content/uploads/ONG-POL%C3%8DTICAS-
FARMAC%C3%89UTICAS-ESTUDIO-BIOEQ-ENERO-2014-vfinal-se.pdf
Pérez Guzmán, M. R. (2013). Estudio de Bioequivalencia in vitro de dos
formas farmacéuticas perorales multifuente de liberación inmediata con
Metformina como principio activo. (Tesis para optar al grado de Magister en

42
Ciencias Farmacéuticas). Universidad Nacional de Colombia.
Recuperado el 10 de marzo del 2016, de
http://www.bdigital.unal.edu.co/11201/1/tesismaestriamilenaperez.pdf
Posada Ventura, W. A. y Santos Cruz, P. A. (2009). Elaboración del perfil de
disolución de tabletas que contienen Tamoxifeno 20 mg (como citrato), de un
producto genérico Nacional con relación al producto innovador. (Tesis para
optar al grado de Licenciatura en Química y Farmacia). Universidad de el
Salvador. Recuperado el 10 de marzo del 2016, de
http://ri.ues.edu.sv/2829/1/TESIS%20ELABORACI%C3%93N%20%20DEL
%20PERFIL%20DE%20DISOLUCI%C3%93N%20DE%20TABLETAS%20Q
UE%20CONTIENEN%20%20TAMOXIFENO%2020%20mg%20%28COMO
%20CIT.pdf
Racines Bermeo, L. R. (2013). Influencia del almidón pregelatinizado como
desintegrante en el perfil de disolución y estabilidad de comprimidos de
acetaminofén. (Tesis para optar por el Título Profesional de Químico
Farmacéutico). Universidad Central del Ecuador. Recuperado el 28 de junio
del 2016, de http://www.dspace.uce.edu.ec/bitstream/25000/1058/1/T-UCE-
0008-08.pdf
Ramírez Gavidia, J.L. (2014). Estudio de bioequivalencia in vitro de
anticonvulsivantes en fármacos comerciales y genéricos con carbamazepina.
(Tesis para optar por el Título Profesional de bioquímico Farmacéutico).
Universidad superior politécnica de Chimborazo.
Recuperado el 28 de junio del 2016, de
http://dspace.espoch.edu.ec/bitstream/123456789/3795/1/56T00487%20UD
CTFC.pdf
Robalino Vizuete, P.J. (2013). Influencia de factores ambientales en el
almacenamiento y perfiles de disolución de comprimidos de meloxicam
comercializados en Quito y Macas. (Tesis para optar por el Título Profesional
de Químico Farmacéutico). Universidad Central del Ecuador. Recuperado el
28 de junio del 2016, de
http://www.dspace.uce.edu.ec/bitstream/25000/1699/1/T-UCE-0008-16.pdf

43
Saavedra S, I., Iturriaga V, V., Ávila M, L., y Quiñones S. L. (2011). Estudios
de bioexención (in vitro) para establecer equivalencia de medicamentos.
Revista Cuad Med Soc (Chile), volumen 51(2): 66-79.
Recuperado el 21 de octubre del 2016, de
http://repositorio.uchile.cl/bitstream/handle/2250/129058/Saavedra_et_al_20
11-articulo.pdf?sequence=1&isAllowed=y
Serón González, L. A. (2014). Desarrollo de un estudio de bioexención en
comprimidos de atorvastatina. (Unidad de práctica para optar al título de
Químico Farmacéutico). Universidad de Chile. Recuperado el 21 de octubre
del 2016, de http://repositorio.uchile.cl/handle/2250/133855
Van Arnum, P. (2012). Pharmaceutical Technology. Afrontando los retos de
la solubilidad. Primera plana: estrategias de solubilidad.
Revista farmacéutica-pharmaceutical technology en español, 10 (2), pp. 1.
Recuperado el 21 de octubre del 2016, de
http://www.pharmatechespanol.com.mx/articulo/596.afrontando_los_retos_d
e_la_solubilidad
Vinueza Oñate, K. R., (2013). Influencia del polimorfismo de la lactosa en los
perfiles de disolución en comprimidos de acetaminofén. (Tesis para obtener
el título profesional de Químico Farmacéutico). Universidad Central del
Ecuador. Recuperado el 21 de octubre del 2016, de
http://www.dspace.uce.edu.ec/bitstream/25000/4382/1/T-UCE-0008-49.pdf

44
ANEXOS
Anexo # 1 Materiales
Equipo Reactivos Cristalería Otros
Espectrofotómetro
marca Shimadzu
modelo UV 1700
solución estándar
de ciprofloxacino
Beackers Jeringas de
10 Ml
Balanza analítica HCl 0.01 N Matraz aforado Sondas
Disolutor marca Sotax
tipo AT 7smart
Agua destilada Cubeta para
espectrofotómetro
Papel
Manteca
Pipeta volumétrica Papel Filtro
Fiola Toalla de
Cocina
Embudo Cuchareta
del
Estándar
Vidrio de Reloj
Probeta
Pipeta Serológica

45
Anexo # 2 Cálculos del factor de Diferencia F1 y Similitud F2 para cada lote de los
medicamentos genéricos de estudio
Medicamento genérico A
Tiempo Innovador R Genérico A
A1 A2 A3
5 51,71 33,48 37,61 24,31
10 84,68 65,86 71,72 58,80
15 99,45 90,23 88,49 88,72
20 102,37 97,79 100,43 98,82
30 106,60 100,91 106,22 102,18
∑r 444.8
r-t A1 r-t A2 r-t A3
18,23 14,10 27,39
18,82 12,96 25,87
9,22 10,97 10,73
4,58 1,94 3,55
5,69 0,38 4,42
∑ 56.54 ∑40.35 ∑71.96
(r-t)2 A1 (r-t)2 A2 (r-t)2 A3
332,17 198,82 750,29
354,05 167,99 669,43
85,09 120,33 115,14
20,99 3,77 12,57
32,39 0,15 19,55
∑(r-t)2 824,68 ∑(r-t)2 491,05 ∑(r-t)2 1566,99
Lote F1 F2
A1 12,71 44.5
A2 9.07 50,09
A3 16.18 37.56
F1 A2= (40.35
444.8𝑥100) = 9.07
F2 A2= 50log (100
√(1+(1
5)𝑋491.04)
) = 50.09

46
Medicamento genérico B
Tiempo Innovador R Genérico B
B1 B2 B3
5 51,71 46,63 70,61 95,42
10 84,68 96,53 92,75 104,70
15 99,45 102,43 95,41 105,04
20 102,37 104,32 97,89 105,52
30 106,60 105,50 99,71 106,12
∑r 444.8
Lote F1 F2
B1 2.38 60.79
B2 2.6 49.75
B3 16.19 33.16
F1 B1= (10.59
444.8𝑥100) = 2.38
F2 B1= 50log (100
√(1+(1
5)X180.16)
) = 60.79
r-t B1 r-t B2 r-t B3
5,08 -18,90 -43,71
-11,85 -8,07 -20,03
-2,97 4,04 -5,59
-1,95 4,48 -3,15
1,10 6,90 0,48
∑= -10,59 ∑= -11,56 ∑= -72,00
(r-t)2 B1 (r-t)2 B2 (r-t)2 B3
25,81 357,38 1910,82
140,51 65,19 401,04
8,83 16,36 31,21
3,80 20,04 9,92
1,21 47,56 0,23
∑=180.16 ∑=506.52 ∑=2353.21

47
Anexo # 3 Cálculos del factor de Diferencia F1 y Similitud F2 para cada medicamento
genérico de estudio
Tiempo Innovador R Genérico A Genérico B
5 51,71 31,80 % 70,88 %
10 84,68 65,46 % 97,99 %
15 99,45 89,15 % 100,96 %
20 102,37 99,01 % 102,57 %
30 106,60 103,10 % 103,78 %
∑r 444.8
Tiempo (R-T) genérico A (R-T) genérico B
5 -19,91 19,18
10 -19,22 13,32
15 -10,31 1,50
20 -3,36 0,21
30 -3,50 -2,82
∑= -56.28 ∑= 31.38
Tiempo (R-T)2 genérico A (R-T) 2 genérico B
5 396,24 367,83
10 369,29 177,37
15 106,25 2,26
20 11,26 0,04
30 12,24 7,98
∑=895.28 ∑=555.49
Genérico F1 F2
A 12,65 43,61
B 7,06 48,76
F1 Genérico A= (56.28
444.8𝑥100) = 12.65
F2 Genérico A= 50log (100
√(1+(1
5)𝑋 895.28)
) = 43.61
F1 Genérico B= (31.38
444.8𝑥100) = 7.06
F2 Genérico B= 50log (100
√(1+(1
5)𝑋 555.49)
) = 48.76

48
Anexo # 4 Eficiencia de la disolución de los medicamentos a prueba
Eficiencia de la disolución medicamento innovador R
Tiempo Q (mg) Área (AUC T-O) Q100*T EF %
5 258,53 646,325
12399,675
15990,3
77,54
10 423,38 1704,775
15 497,27 2301,625
20 511,83 2522,75
30 533,01 5224,2
Eficiencia de la disolución medicamento genérico A
Tiempo disolución Área (AUC T-O) Q100*T EF %
5 159,00 397,5
10950,625
15465,6
70,81
10 327,30 1215,75
15 445,73 1932,575
20 495,05 2351,95
30 515,52 5052,85
Eficiencia de la disolución medicamento genérico B
Tiempo disolución área (AUC T-O) Q100*T EF %
5 354,42 886,05
13157,475
15390,3
85,49
10 489,97 2110,975
15 504,79 2486,9
20 512,87 2544,15
30 513,01 5129,4

49
Anexo # 5 Tiempo medio de disolución MDT
Medicamento Innovador R
Tiempo Q (mg) t1 ∆Qi t1∆Qi ∑(t1∆Qi) MDT (min)
5 258,53 5 258,53 1292,65
2770,95
5,54
10 423,38 5 164,85 824,25
15 497,27 5 73,89 369,45
20 511,83 5 14,56 72,8
30 533,01 10 21,18 211,8
Medicamento Genérico A
Tiempo Q (mg) t1 ∆Qi t1∆Qi ∑(t1∆Qi) MDT (min)
5 159,00 5 159,00 795
2679,95
5,36
10 327,30 5 168,30 841,5
15 445,73 5 118,43 592,15
20 495,05 5 49,32 246,6
30 515,52 10 20,47 204,7
Medicamento Genérico B
Tiempo Q (mg) t1 ∆Qi t1∆Qi ∑(t1∆Qi) MDT (min)
5 354,42 5 354,42 1772,1
2565,75
5,13
10 489,97 5 135,55 677,75
15 504,79 5 14,82 74,1
20 512,87 5 8,08 40,4
30 513,01 10 0,14 1,4

50
ANEXO # 6 Certificado de análisis del estándar