tubulopatÍas perdedoras de sal€¢ alcalosis metabólica. • hipocaliemia y hipocloremia. • ↑...
TRANSCRIPT

TUBULOPATÍAS PERDEDORAS DE SAL
Maria Goretti Moreira Guimarães Penido Professora Associada do Departamento de Pediatria
Faculdade de Medicina da Universidade Federal de Minas Gerais Unidade de Nefrologia Pediátrica do Hospital das Clínicas da Faculdade de
Medicina da UFMG
2014

INTRODUCCIÓN
Tubulopatias perdedoras de sal:
• Primarias • Síndrome de Bartter
• Tipo I
• Tipo II
• Tipo III
• Tipo IV
• Tipo IVb
• Tipo V
• Síndrome de Gitelman
• Pseudohipoaldosteronismo Tipo I • Primarias: AR, AD
• Secundarias: uropatias e ITU
• Secundarias • Síndrome de Sjogren, Doença de Dent,
• Síndrome de Kearns-Sayre, EAST syndrome,
• Sarcoidose, Cistinose.
• Drogas: aminoglicosideos, diuréticos, agentes citotóxicos

INTRODUCCIÓN
Tubulopatias perdedoras de sal:
Causadas por mutaciones en las proteinas transportadoras de iones del epitelio tubular:
• Rama gruesa ascendente del asa de Henle
• Porción mas alta del túbulo contorneado distal (TCD1)
Causadas por reabsorción del sodio comprometida al largo
del nefrón distal aldosterona sensible:
• Porción mas baja del túbulo contorneado distal (TCD2)
• Túbulo conector
• Porción cortical del ducto coletor
• Porción medular del ducto coletor
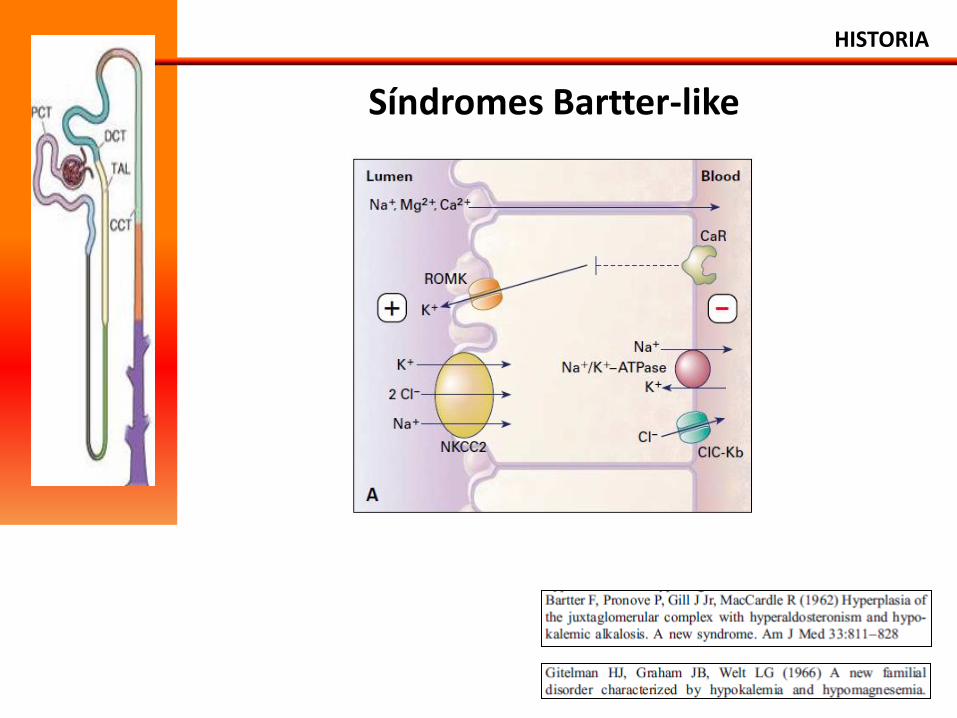
Síndromes Bartter-like
HISTORIA

En los años 80:
• Formas graves de la SB en recén-nacidos
• Papel del prostaglandina E Síndrome de la Hiperprostaglandina E
HISTORIA

HISTORIA

SB tipo I e II
SB clássica (tipo III) e SG
SB antenatal com surdez
neurosensorial (tipo IV e IVb)
HISTORIA

8
Síndrome de Bartter Mutaciones de NKCC2, ROMK , BSND (bartina), CLCNKB e CLCNKA, SaSR
• Alcalosis metabólica.
• Hipocaliemia y hipocloremia.
• ↑ de renina (hiperplasia del aparato yuxtaglomerular) y aldosterona.
• ↑ de excreción urinaria de prostraglandinas, potasio.
• ↑ excreción urinaria de cálcio: Hipercalciuria.
• Normo o hipotensión.
• Poliuria, polidipsia, episodios de desidratación.
• Retardo pondero-estatural.
• Diagnóstico neonatal o no lactente.
Apresentación:
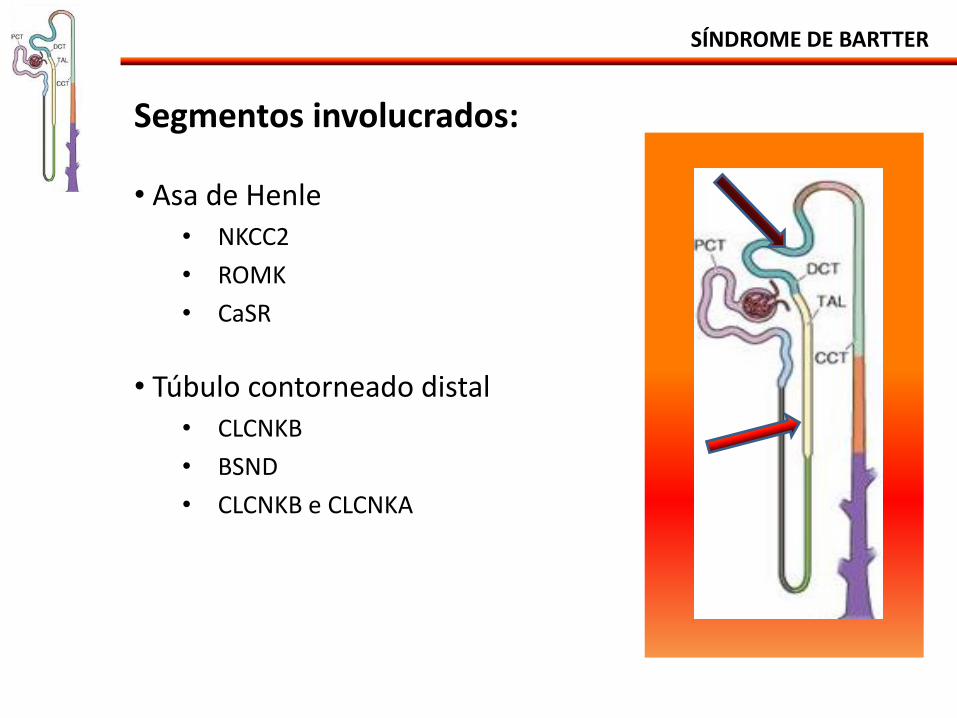
SÍNDROME DE BARTTER
Segmentos involucrados: • Asa de Henle
• NKCC2
• ROMK
• CaSR
• Túbulo contorneado distal
• CLCNKB
• BSND
• CLCNKB e CLCNKA
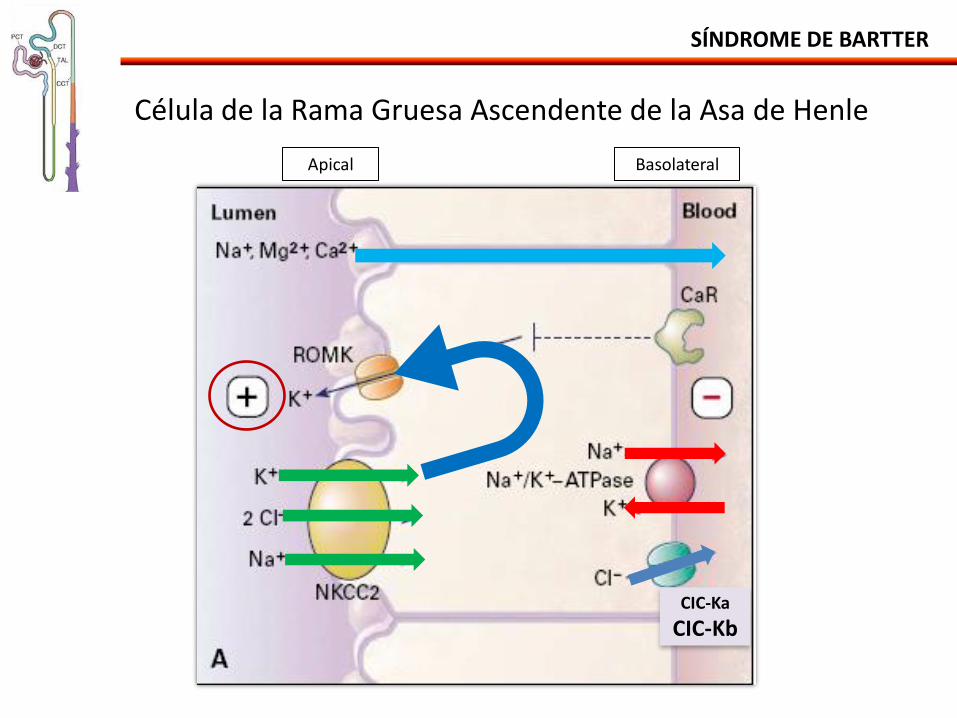
SÍNDROME DE BARTTER
Célula de la Rama Gruesa Ascendente de la Asa de Henle
CIC-Ka
CIC-Kb
Apical Basolateral

SÍNDROME DE BARTTER
Fisiopatología:
Anormalidades electrolíticas similares a las causadas por la utilización crónica de diuréticos de asa.
•Reducción de la reabsorción de NaCl hipovolemia SRAA.
•Reducción del Cl intracelular (Ap. Yustaglomerular) SRAA.
•Aumento de la produción de PGs.
- Antagonismo de la ADH poliuria.
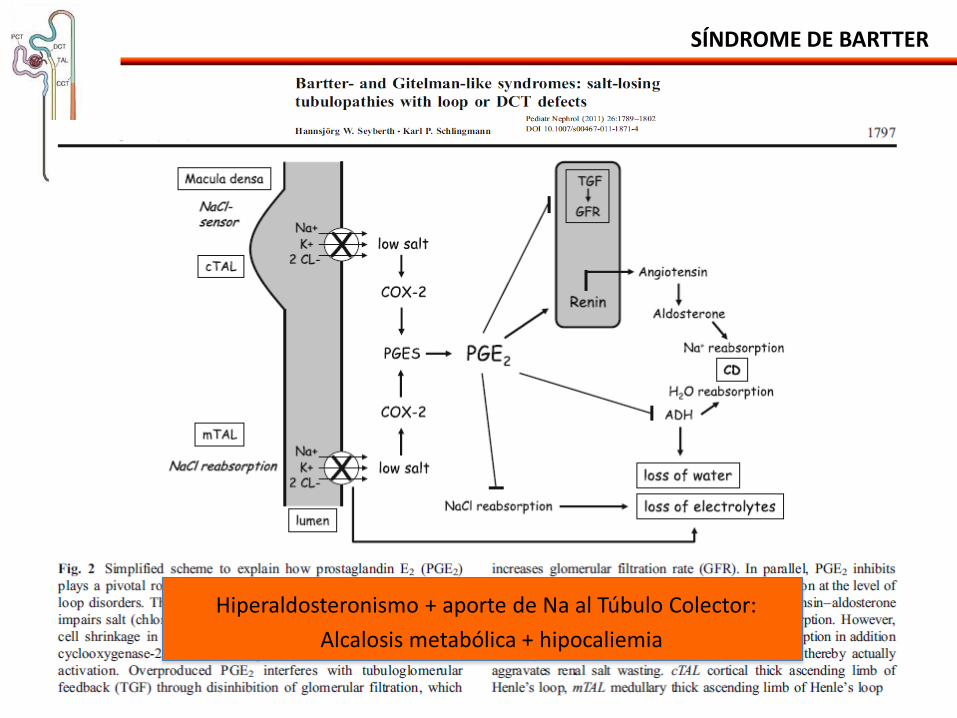
SÍNDROME DE BARTTER
Hiperaldosteronismo + aporte de Na al Túbulo Colector:
Alcalosis metabólica + hipocaliemia

SÍNDROME DE BARTTER
• Reducción de la absorción paracelular de Ca y Mg:
- Hipercalciuria nefrocalcinosis.
- Magnesiuria menos afectada: no ha hipomagnesemia.
• Hipovolemia ↑angiotensina ↑ reabsorción de Mg en el túbulo contorneado proximal.
• Hiperaldosteronismo estimula la reabsorción de Mg en el túbulo contorneado distal.
• Incapacidad de concentración urinaria:
- Fracaso en el mecanismo de contracorriente: poliuria y polidipsia.
Fisiopatología:

SÍNDROME DE BARTTER
Tipos:
Adaptado de Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol 2008; 4:560-7.
SÍNDROME DE BARTTER
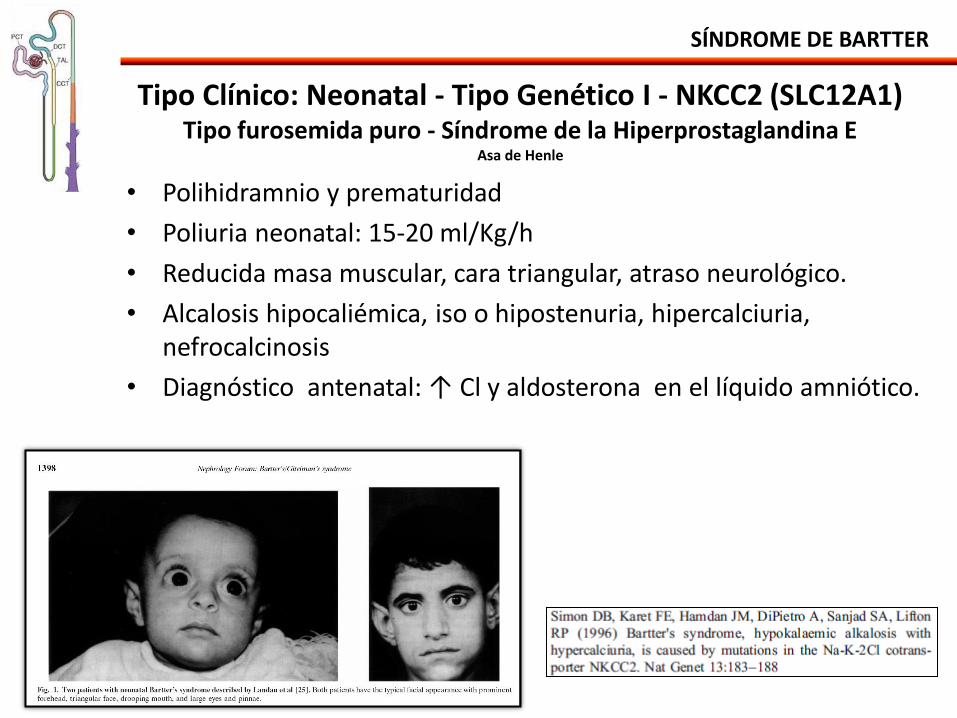
Tipo Clínico: Neonatal - Tipo Genético I - NKCC2 (SLC12A1) Tipo furosemida puro - Síndrome de la Hiperprostaglandina E
Asa de Henle
• Polihidramnio y prematuridad
• Poliuria neonatal: 15-20 ml/Kg/h
• Reducida masa muscular, cara triangular, atraso neurológico.
• Alcalosis hipocaliémica, iso o hipostenuria, hipercalciuria, nefrocalcinosis
• Diagnóstico antenatal: ↑ Cl y aldosterona en el líquido amniótico.
SÍNDROME DE BARTTER
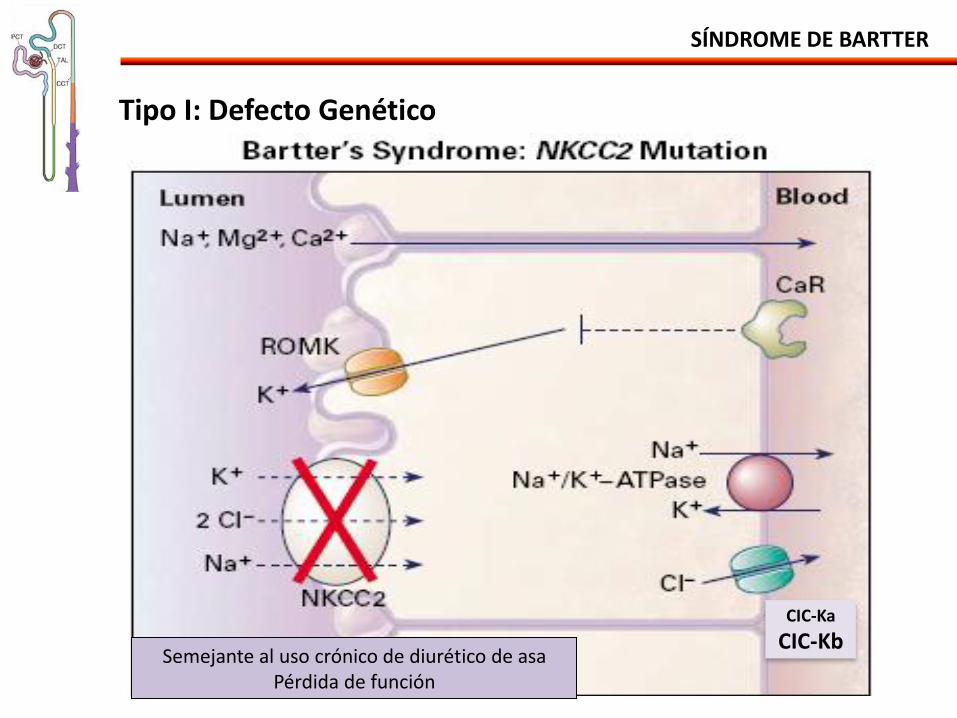
Tipo I: Defecto Genético
Semejante al uso crónico de diurético de asa Pérdida de función
CIC-Ka
CIC-Kb
SÍNDROME DE BARTTER
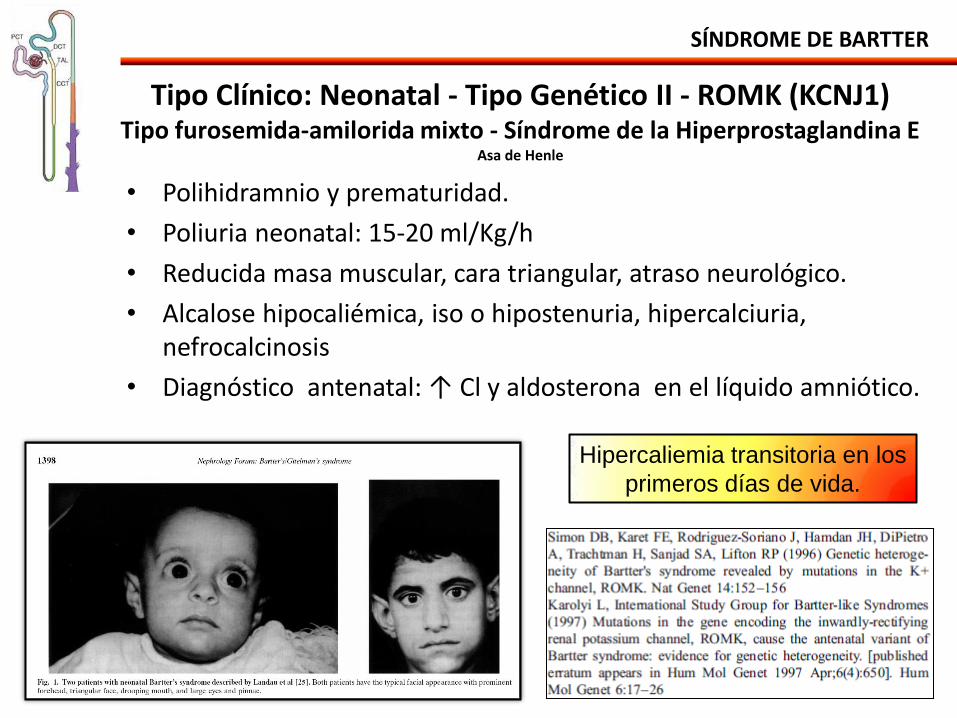
Tipo Clínico: Neonatal - Tipo Genético II - ROMK (KCNJ1) Tipo furosemida-amilorida mixto - Síndrome de la Hiperprostaglandina E
Asa de Henle
Hipercaliemia transitoria en los
primeros días de vida.
• Polihidramnio y prematuridad.
• Poliuria neonatal: 15-20 ml/Kg/h
• Reducida masa muscular, cara triangular, atraso neurológico.
• Alcalose hipocaliémica, iso o hipostenuria, hipercalciuria, nefrocalcinosis
• Diagnóstico antenatal: ↑ Cl y aldosterona en el líquido amniótico.
SÍNDROME DE BARTTER
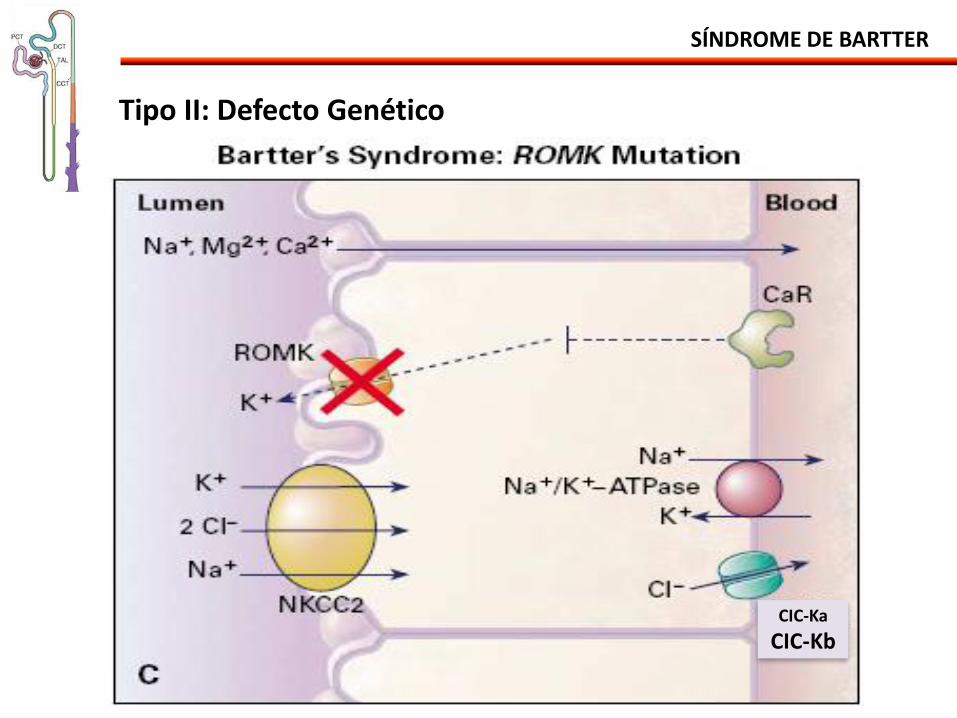
Tipo II: Defecto Genético
CIC-Ka
CIC-Kb

SÍNDROME DE BARTTER
• Historia de polihidramnio y prematuridad, menos grave • Sinales clínicos mas tardios (infancia) • Poliuria, polidipsia, vómitos, desidratación, dificultad de defecar • Retardo del crescimiento y mental • Fraqueza muscular, calambres • Capacidad de concentración preservada, nefrocalcinosis es rara • Magnesio sérico geralmente normal • DD entre SB III y SG puede ser difícil
Tipo Clínico: Clásica - Tipo Genético III - CIC-Kb (CLCNKB) Tipo tiazida-furosemida mixto
Túbulo contorneado distal
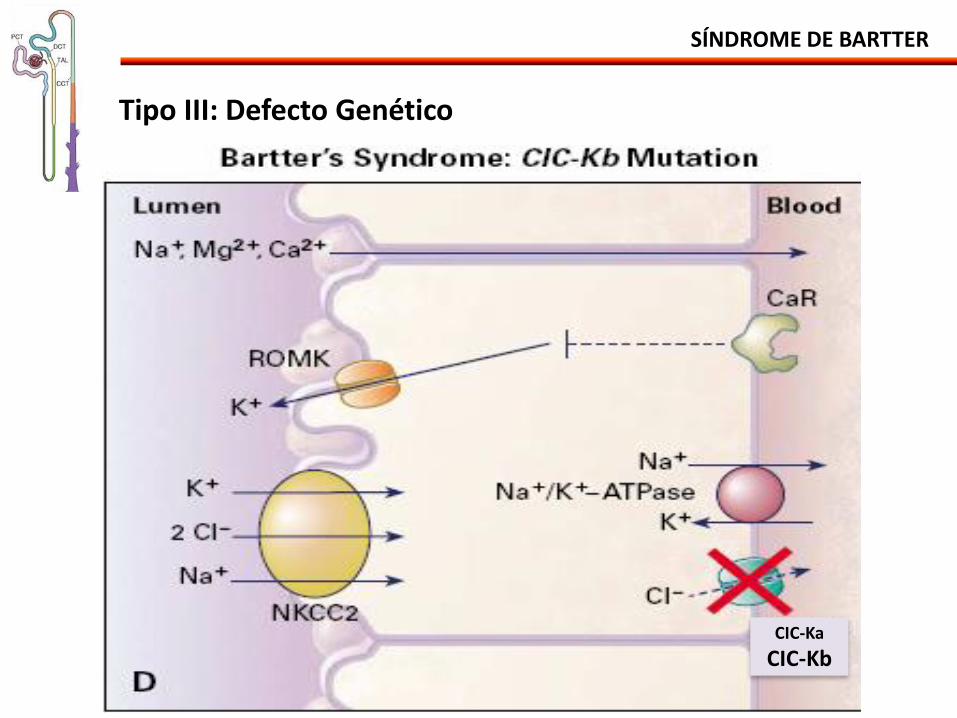
SÍNDROME DE BARTTER
Tipo III: Defecto Genético
CIC-Ka
CIC-Kb

SÍNDROME DE BARTTER
• Mutación del gene BSND proteina bartina - Subunidad β esencial para el transporte del cloro de los canales de Cl
• Células de la rama gruesa de la asa de Henle (CIC-Kb) • Células oscuras secretoras de K en el oído interno (CIC-Ka e CIC-Kb)
• Grave, pérdida de la función renal, sin nefrocalcinosis y la hipercalciuria es transitoria.
Tipo Clínico: Neonatal con sordera - Tipo Genético IV - Bartina (BSND) Tipo furosemida-tiazida mixto - Síndrome da Hiperprostaglandina E, con surdera
Asa de Henle y Túbulo contorneado distal
1. Birkenhäger R et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 2001;29:310–4.
2. Kitanaka S et al. A compound heterozygous mutation in the BSND gene detected in Bartter syndrome type IV. Pediatr Nephrol 2006;21(2):190-3.
3. Zaffanello M et al. Type IV Bartter syndrome: report of two new cases. Pediatr Nephrol. 2006;21(6):766-70.
4. García-Nieto V et al. Mutation G47R in the BSND gene causes Bartter syndrome with deafness in two Spanish families. Pediatr Nephrol 2006;21(5):643-8.
5. Janssen AG et al. Disease-causing dysfunctions of barttin in Bartter syndrome type IV. J Am Soc Nephrol 2009;20(1):145-53.
6. de Pablos AL et al.Severe manifestation of Bartter syndrome Type IV caused by a novel insertion mutation in the BSND gene. Clin Nephrol 2014;81(5):363-8.
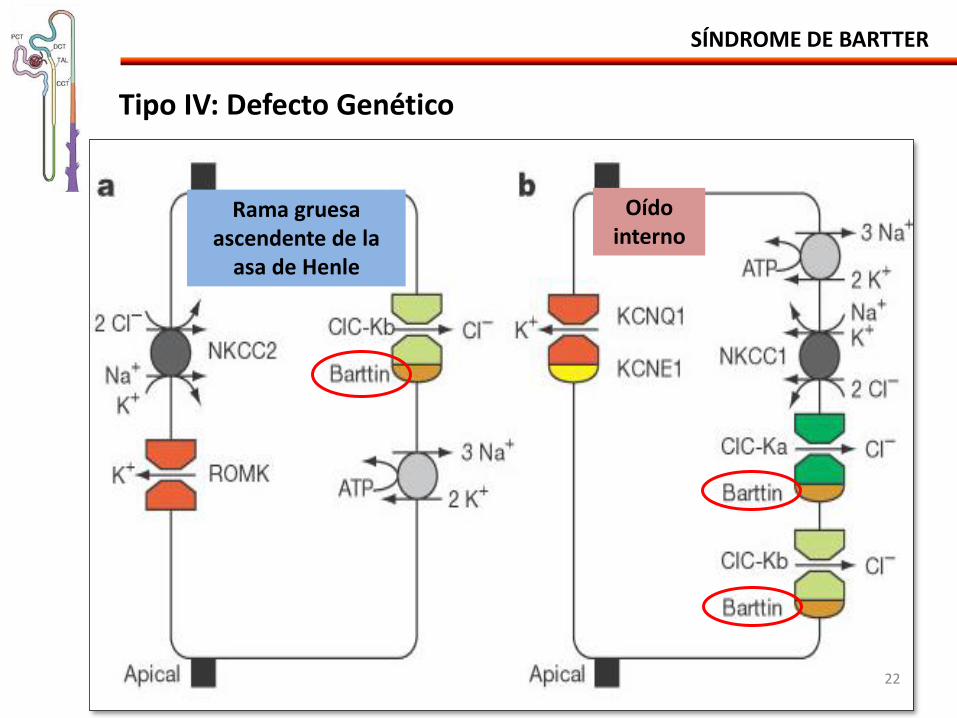
SÍNDROME DE BARTTER
Tipo IV: Defecto Genético
Rama gruesa ascendente de la
asa de Henle
Oído interno
22

SÍNDROME DE BARTTER
• Alteración digénica.
• Mutación de los genes CLCNKA y CLCNKB CIC-Ka y CIC-Kb
- Grande deleción en el CLCNKA y un punto de mutación en el CLCNKB.
• Células de la rama gruesa de la asa de Henle (CIC-Kb). • Células oscuras secretoras de K en el oído interno (CIC-Ka e CIC-Kb).
Tipo Clínico: Neonatal con surdera- Tipo Genético IVb CCl-Ka y CCl-Kb (CLCNKB y CLCNKA)
Tipo furosemida-tiazida mixto - Síndrome de la Hiperprostaglandina E, con sordera
Asa de Henle e Túbulo contorneado distal
1. Schalingmann et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 2004;350:1314–9.
2. Krämer BK et al. Mechanisms of Disease: the kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat Clin Pract Nephrol 2008;4(1):38-46.
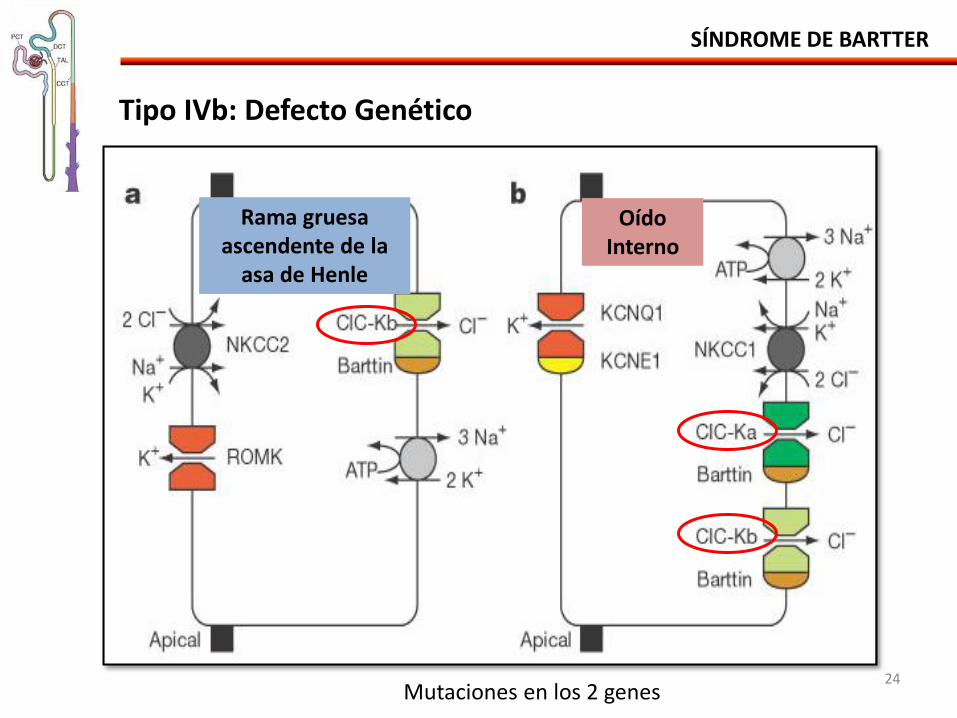
SÍNDROME DE BARTTER
Tipo IVb: Defecto Genético
Rama gruesa ascendente de la
asa de Henle
Oído Interno
Mutaciones en los 2 genes 24

SÍNDROME DE BARTTER
Tipo Clínico: Clásico - Tipo Genético V - CaSR (gene CASR) Asa de Henle
• Hipocalcemia Autosómica Dominante o Hipoparatireoidismo Autosómico Dominante
• Mutaciones en el gene CaSR CaSR: receptor sensible de calcio. - Con ganancia de función - Autosómica dominante
• Caracterizada por sintomas mas leves y mas tardios (infancia y adolescencia).
• Ha pérdida leve del Na, Cl, K y Ca. Pérdida acentuada de Mg (hipermagnesuria).
• Puede haber ↑ moderado de la aldosterona y renina.
1. Wang WH et al. Cytochrome P-450 metabolites mediate extracellular Ca(2+)-induced inhibition of apical K+ channels in the TAL. Am J Physiol 1996; 271:C103.
2. Hebert SC. Extracellular calcium-sensing receptor: implications for calcium and magnesium handling in the kidney. Kidney Int 1996; 50:2129.
3. Watanabe S et al. Association between activating mutations of calcium-sensing receptor and Bartter's syndrome. Lancet 2002;360:692-4.
4. Gunn IR & Gaffney D. Clinical and laboratory features of calcium-sensing receptor disorders: a systematic review. Ann Clin Biochem 2004;41:441-58
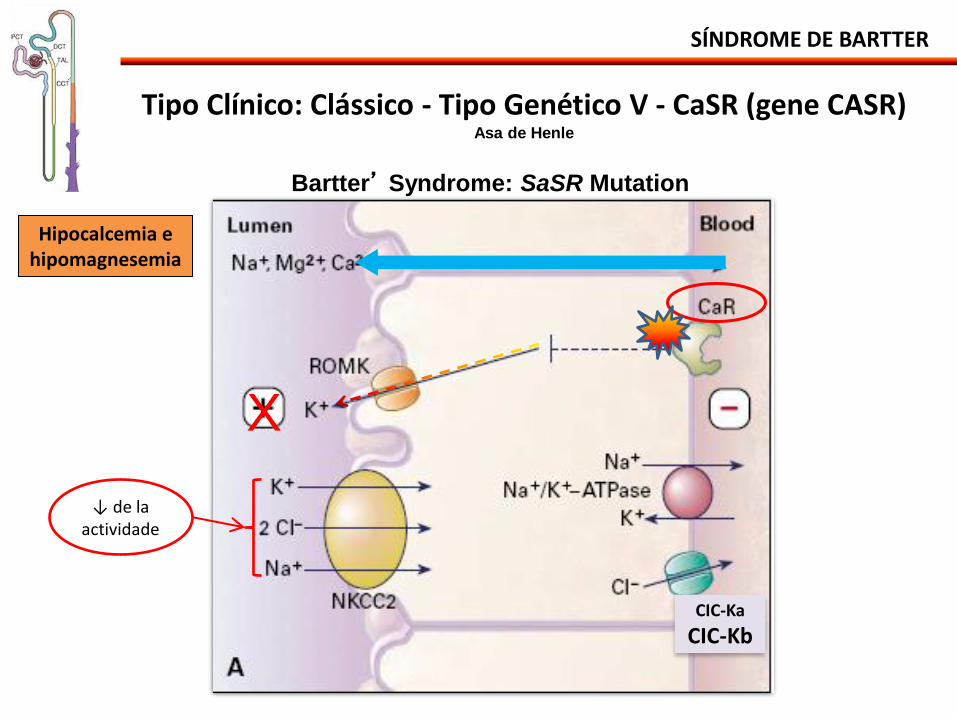
SÍNDROME DE BARTTER
CIC-Ka
CIC-Kb
Tipo Clínico: Clássico - Tipo Genético V - CaSR (gene CASR) Asa de Henle
Bartter’ Syndrome: SaSR Mutation
↓ de la actividade
Hipocalcemia e hipomagnesemia
X

SÍNDROME DE BARTTER
Tratamiento
• El defecto no puede ser corregido.
• Orientaciones sobre la enfermedad y sobre el tratamiento.
• Evitar ejercícios exagerados.
• Objetivo: minimizar los efectos del exceso de PGs y aldosterona.
• AINES: reduzir la síntesis de PGs (Indometacina, ibuprofeno, cetoprofeno, rofecoxib ??). Algunos casos del tipo IV no responden bien.
• Diuréticos ahorradores de K: espironolactona, amilorida.
• Reposición de K: KCl.
• IECA: puede agravar hipotensión.
1. Vaisbich MH et al. Bartter syndrome: benefits and side effects of long-term treatment. Pediatr Nephrol 2004; 19:858-63.
2. Vinci JM et al. The kallikrein-kinin system in Bartter's syndrome and its response to prostaglandin synthetase inhibition. J Clin Invest 1978; 61:1671-82.
3. Griffing GT et al. Amiloride in Bartter's syndrome. Clin Pharmacol Ther 1982; 31:713-8. 4. Hené RJ et al. Correction of hypokalemia in Bartter's syndrome by enalapril. Am J Kidney Dis 1987; 9:200-5. 5. Morales JM et al. Long-term enalapril therapy in Bartter's syndrome. Nephron 1988; 48:327.

Síndrome de Gitelman Mutaciones del Co-transportador Na/Cl - NCCT - SLC12A3
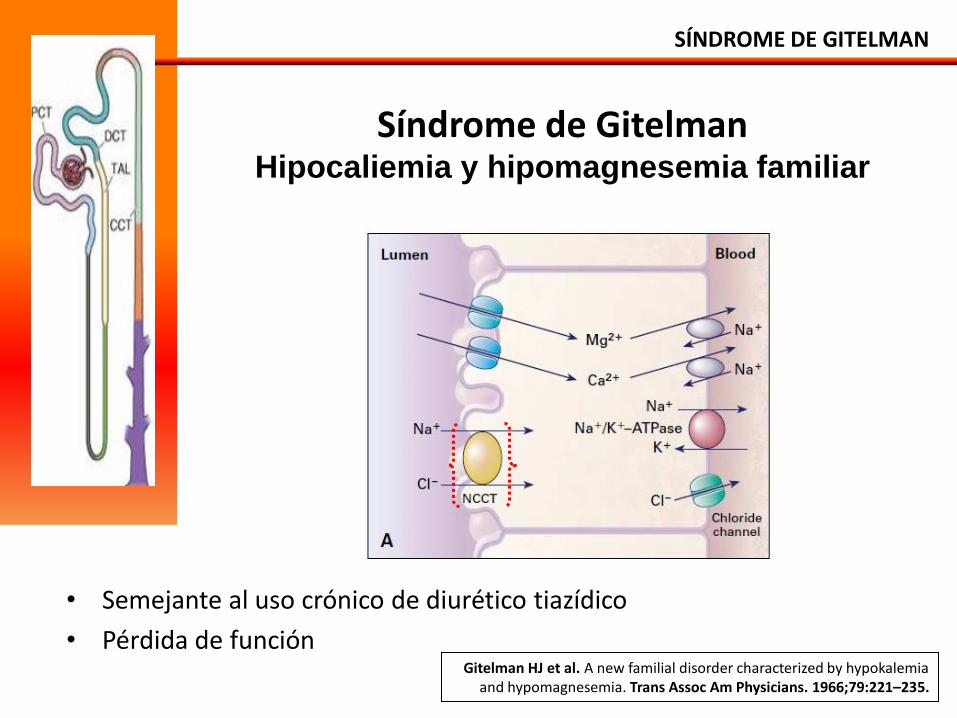
Síndrome de Gitelman Hipocaliemia y hipomagnesemia familiar
• Semejante al uso crónico de diurético tiazídico
• Pérdida de función
SÍNDROME DE GITELMAN
Gitelman HJ et al. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235.

SÍNDROME DE GITELMAN
• Mutación del gene SLC12A3 NCCT (> 140 mutaciones descriptas)
- Codifica el co-transportador Na-Cl tiazída sensible, en el túbulo distal.
• Diagnóstico: niños, adolescentes o adultos (condrocalcinosis).
• Hipocaliemia y hipomagnesemia: calambres, tetania, fraqueza muscular.
• Hipocalciuria, alcalosis metabólica.
• Normo o hipotensión.
• Poliuria leve y nocturia.
Tipo Clínico: Clásica - Tipo Genético - NCCT - (SLC12A3) Tipo tiazida puro - Túbulo contorneado distal
1. Simon DB et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 1996; 12:24-30.
2. Monnens L et al. Gitelman syndrome comes of age. Nephrol Dial Transplant 1998; 13:1617-9. 3. Bettinelli A et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J
Pediatr 1992; 120:38-43. 4. de Jong JC et al. Functional expression of mutations in the human NaCl cotransporter: evidence for impaired routing mechanisms in Gitelman's syndrome. J Am
Soc Nephrol 2002; 13:1442-8. 5. Vargas-Poussou R et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol 2011; 22:693-703. 6. Jeck N et al. Salt handling in the distal nephron: lessons learned from inherited human disorders. Am J Physiol Regul Integr Comp Physiol 2005;288:R782–R795. 7. Cruz DN et al. Gitelman"s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int 2001;59:710–7. 8. Riveira-Munoz E et al. Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol
2007;18:1271–83. 9. de Jong et al. Effects of chemical chaperones on partially retarded NaCl cotransporter mutants associated with Gitelman's syndrome in a mouse cortical collecting
duct cell line. Nephrol Dial Transplant. 2004;19:1029–32.
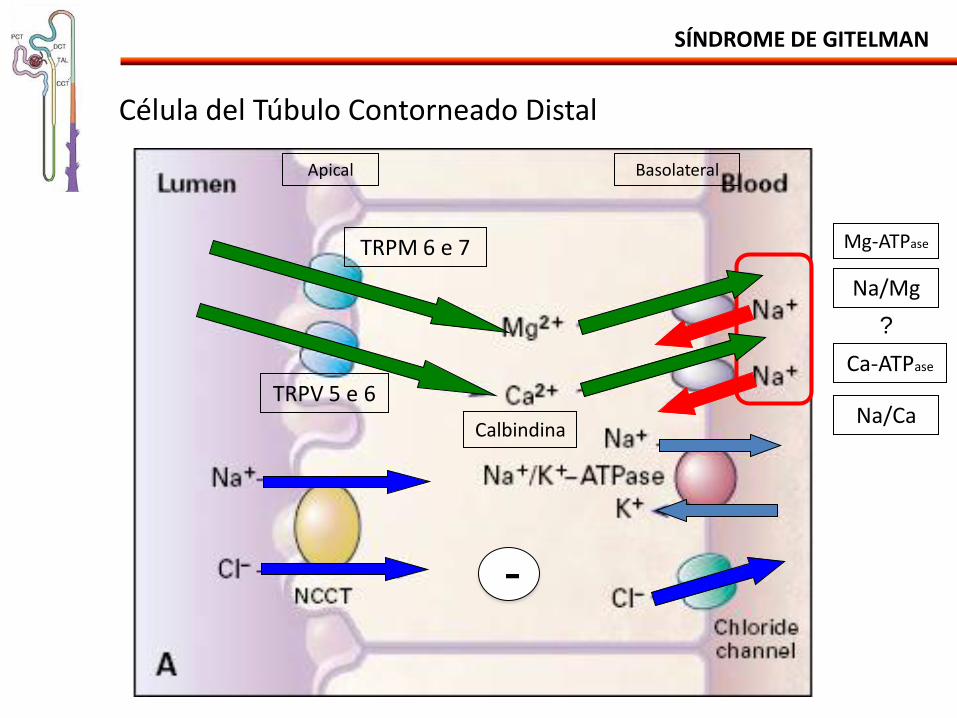
SÍNDROME DE GITELMAN
TRPM 6 e 7
TRPV 5 e 6
Célula del Túbulo Contorneado Distal
Ca-ATPase
Na/Mg
Mg-ATPase
Na/Ca Calbindina
Apical Basolateral
-
?
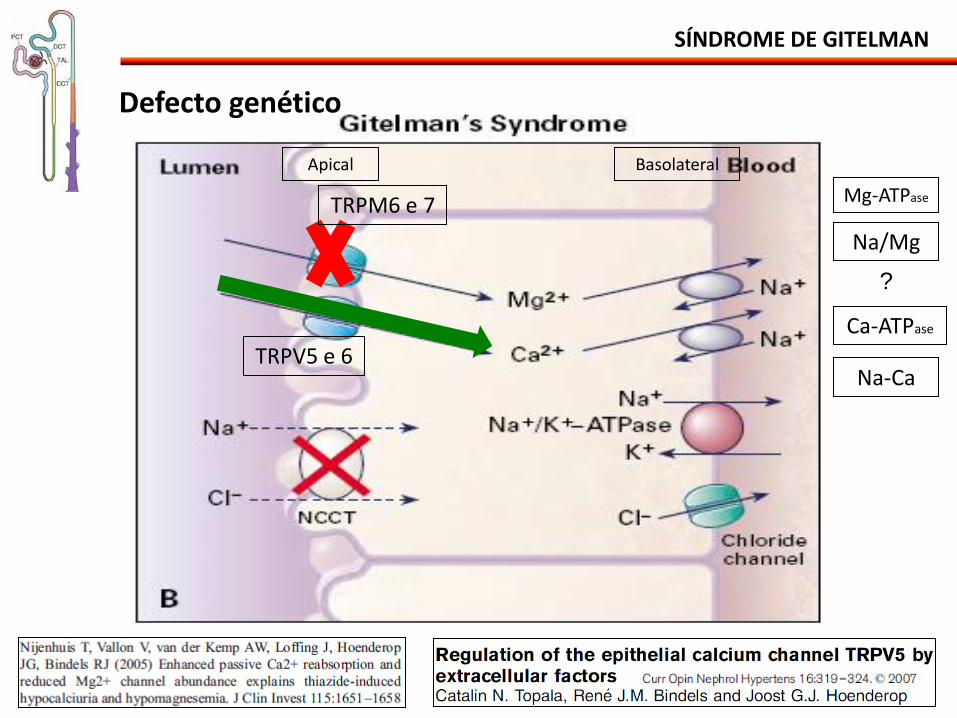
SÍNDROME DE GITELMAN
Defecto genético
TRPM6 e 7
TRPV5 e 6
Na/Mg
Mg-ATPase
?
Apical Basolateral
Ca-ATPase
Na-Ca

SÍNDROME DE GITELMAN
Fisiopatología:
• Clínica menos grave que S. Bartter.
• Reducción de la reabsorción del NaCl hipovolemia leve SRAA.
• Hiperaldosteronismo + aporte del Na al Túbulo Colector:
- Alcalosis metabólica + hipocaliemia.
• Estímulo a la reabsorción del Ca: hipocalciuria.
• Reducción de la cantidad de los canales epiteliales de Mg: hipomagnesemia.
• Síntese de PG normal.
-6.

SÍNDROME DE GITELMAN
Tratamiento:
• Dieta rica en sodio y potasio.
• Diuréticos ahorradores de K: espironolactona, amilorida.
• Reposición de K: KCl.
• Reposición de Mg: (MgCl).
• AINES: sin valor (no ha exceso de PGs).
1. Peters M et al (2002) Clinical presentation of genetically defined patients with hypokalemic salt-losing
tubulopathies. Am J Med 112:183–90. 2. Knoers NV, Levtchenko EN (2008) Gitelman syndrome. Orphanet J Rare Dis 3:22-6. 3. Liaw LC et al(1999) Dose related growth response to indometacin in Gitelman syndrome. Arch Dis Child
81:508–10.

Pseudohipoaldosteronismo Tipo I
Mutaciones en el canal epitelial de Na – ENaC - Subunidades α (SCNN1A), β (SCNN1B) o γ (SCNN1G) ; y em el receptor de mineralocorticoide - MR
(NR3C2)
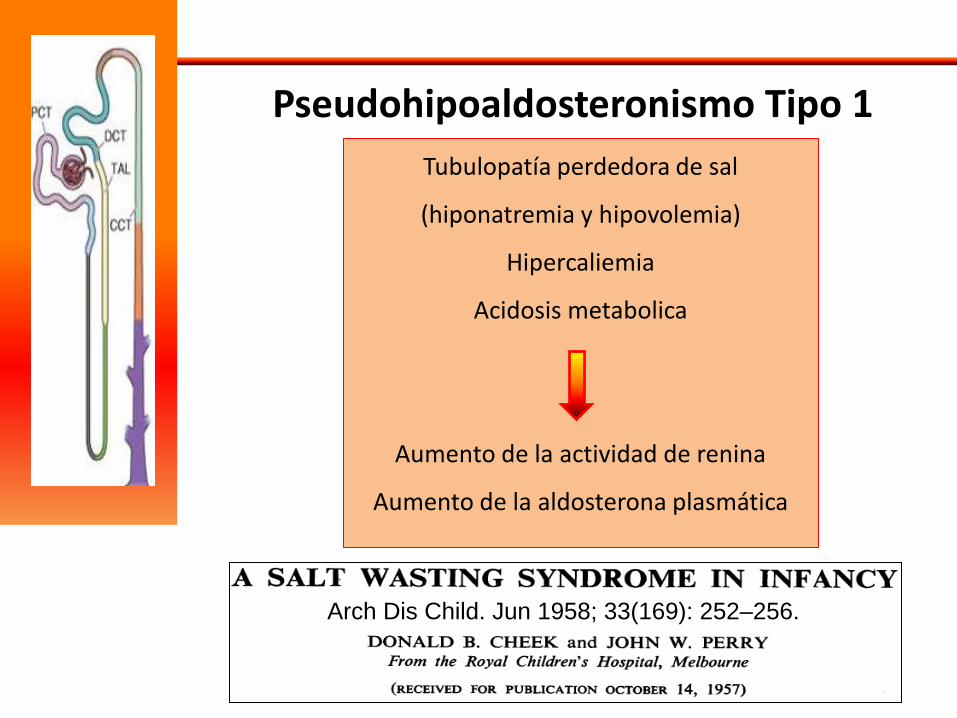
Pseudohipoaldosteronismo Tipo 1
Arch Dis Child. Jun 1958; 33(169): 252–256.
Tubulopatía perdedora de sal
(hiponatremia y hipovolemia)
Hipercaliemia
Acidosis metabolica
Aumento de la actividad de renina
Aumento de la aldosterona plasmática
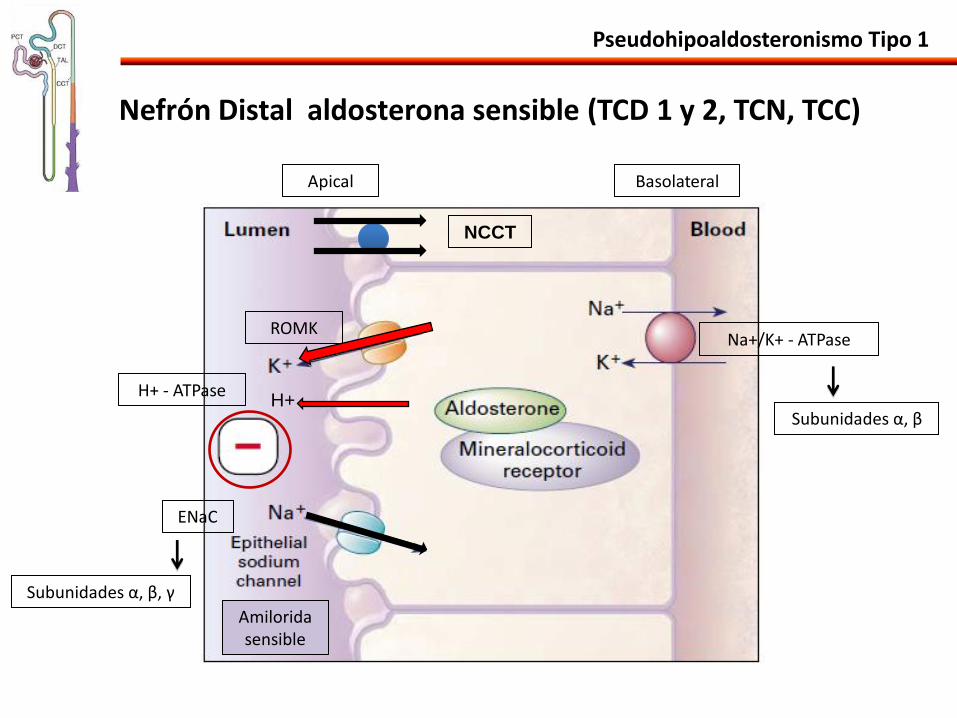
Pseudohipoaldosteronismo Tipo 1
Nefrón Distal aldosterona sensible (TCD 1 y 2, TCN, TCC)
Na+/K+ - ATPase
Apical Basolateral
ROMK
ENaC
H+ H+ - ATPase
NCCT
Subunidades α, β
Subunidades α, β, γ
Amilorida sensible
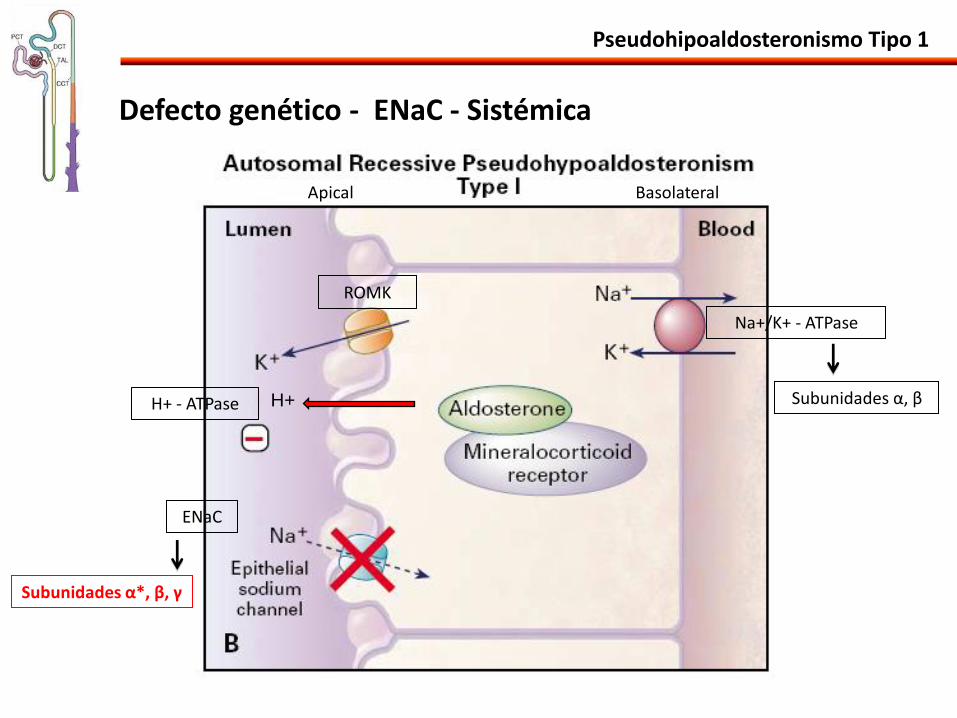
Pseudohipoaldosteronismo Tipo 1
Defecto genético - ENaC - Sistémica
Na+/K+ - ATPase
ROMK
ENaC
H+ H+ - ATPase
Apical Basolateral
Subunidades α, β
Subunidades α*, β, γ

Pseudohipoaldosteronismo Tipo 1
• Mutaciones em el ENaC: α*, β ou γ pérdida de la actividad.
• Resistencia a la acción estimulatoria de la aldosterona.
• Afecta otros órganos (expresión del ENaC):
Pulmones incapacidad de reabsorver líquido alveolar en el periodo neonatal y infecciones de repetición.
Aumento del NaCl en la saliva y en el sudor rash miliária like.
Baja ganancia de peso y de estatura.
• Forma mas grave:
Alta mortalidad neonatal, sintomas precoces desidratación y hospitalizaciones.
No mejora con el tiempo.
Forma Sistémica - Autosómica Recesiva
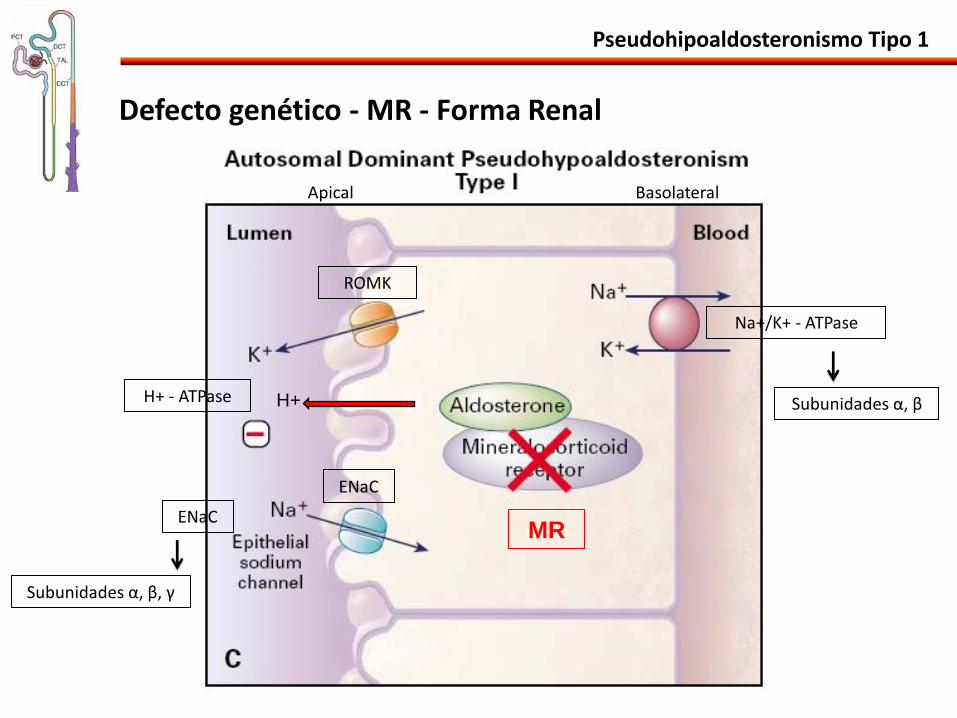
Defecto genético - MR - Forma Renal
Pseudohipoaldosteronismo Tipo 1
Na+/K+ - ATPase
ROMK
ENaC
H+ H+ - ATPase
Apical Basolateral
ENaC
Subunidades α, β
Subunidades α, β, γ
MR

Pseudohipoaldosteronismo Tipo 1 - Autosómica Dominante
Pseudohipoaldosteronismo Tipo 1
• Mutaciones en el MR: (NR3C2) pérdida de la actividad.
• > 50 mutaciones identificadas.
• Sin involucramiento de otros órganos.
• Cuadro mas suave.
• Mejora con el tiempo.

Tratamiento
Pseudohipoaldosteronismo Tipo 1
• Dieta rica en NaCl:
Previne hipovolemia.
Aumenta excreción del K+ ↑ de la cantidad de Na en el TC.
• Suplementación de sal:
AD ≈ 3-20 mEq/kg/día NaCl
AR ≈ 20-50 mEq/kg/día NaCl
• Fludrocortisona en altas dosis (1-2mg/día):
Potente efecto mineralocorticoide.
• Carbenoxolona:
Antagoniza la conversión del cortisol en cortisona.
Cortisol acción en el receptor mineralocorticoide. Arai K et al. Physiological and molecular aspects of mineralocorticoid receptor action in
pseudohypoaldosteronism: a responsiveness test and therapy. J Clin Endocrinol Metab 1994; 79:1019-23.
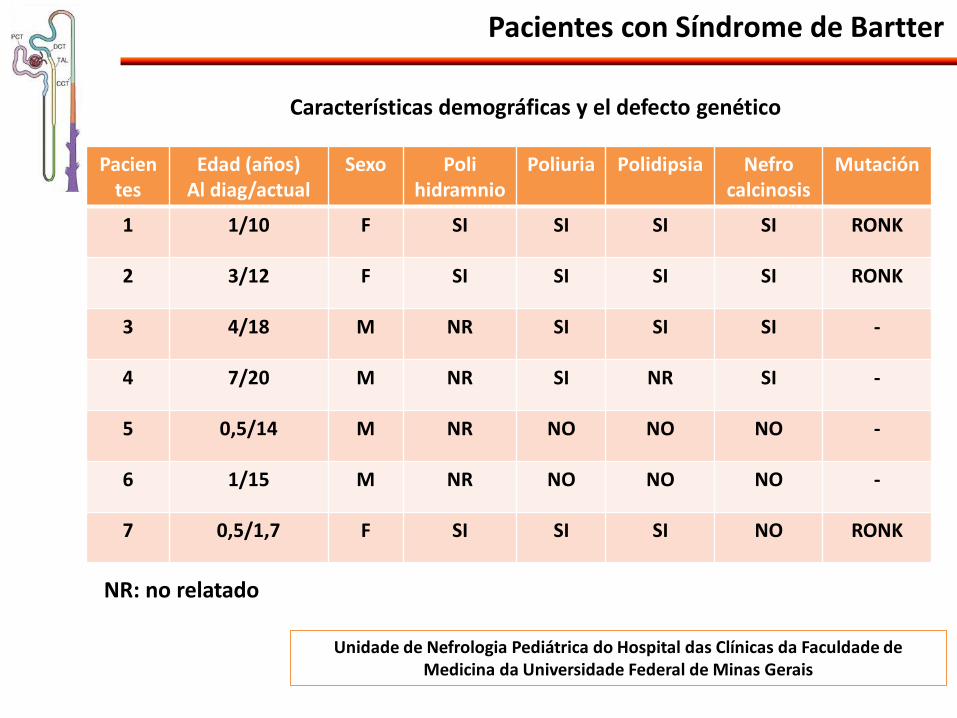
Pacientes con Síndrome de Bartter
Pacientes
Edad (años) Al diag/actual
Sexo Poli hidramnio
Poliuria Polidipsia Nefro calcinosis
Mutación
1 1/10 F SI SI SI SI RONK
2 3/12 F SI SI SI SI RONK
3 4/18 M NR SI SI SI -
4 7/20 M NR SI NR SI -
5 0,5/14 M NR NO NO NO -
6 1/15 M NR NO NO NO -
7 0,5/1,7 F SI SI SI NO RONK
NR: no relatado
Características demográficas y el defecto genético
Unidade de Nefrologia Pediátrica do Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Minas Gerais

Pac Urea Creat Na K Cl Mg Ca Bic
1 19 / 37 0,3 / 0,5 137 / 138 2,9 / 3,9 99 / 99 2,1 / 2,24 1,23 / 1,21 33 / 29
2 32 / 30 0,3 / 0,5 139 / 139 4,1 / 4,3 100 / 102 2,0 / 1,9 1,27 / 1,24 26 / 26
3 21 / 38 0,5 / 0,9 138 / 134 2,8 / 3,6 97 / 95 1,5 / 1,7 9,1 / 9,7 30,2 / 24,3
4 16 / 28 0,2 / 0,6 132 / 134 1,0 / 3,0 76 / 93 2,3 / 1,8 1,03 / 9,7 31 / 25
5 42 / 29 0,4 / 0,6 131 / 144 2,8 / 3,4 92 / 99 2,2 / 1,9 10,8 / 9,2 29 / 27,7
6 59 / 46 0,6 / 0,6 137 / 136 2,0 / 3,8 105 / 95 2,1 / 1,9 9,6 / 9,5 26,5 / 28,5
7 18 / 28 0,27 / 0,3 127 / 141 2,2 /3,9 87 / 89 2,0 / 2,3 1,0 / 1,2 33,5 /25,1
Niveles de los electrolitos y bicarbonato en el sangre al diagnóstico/y actual
Pacientes con Síndrome de Bartter
Pac Calcio (mg/kg/24h) Magnesio (mg/24h)
1 8,9 / 6,0 32 / 22
2 8 / 3,8 79 / 58
3 6,5 / 1,4 54 / 45,9
4 7,9/3,1 32 / 123
5 6,9 / 2,6 34 / 198
6 6,5 / 1,5 79 / -
7 7,0 / 3,4 33 / 52
Niveles del calcio y magnesio en la orina 24h al diagnóstico/y actual
Unidade de Nefrologia
Pediátrica do Hospital das Clínicas da
Faculdade de Medicina da Universidade
Federal de Minas Gerais

Paciente con Síndrome de Gitelman
Paciente Edad (años) Al diag/actual
Sexo Poli hidramnio
Poliuria Polidipsia Nefro calcinosis
Mutación
12/24 M NR SI SI NO -
NR: no relatado
Característica demográfica y el defecto genético
Niveles de los electrolitos y bicarbonato en el sangre al diagnóstico/y actual
Urea Creat Na K Cl Mg Ca Bic
20 / 38 0,5 / 0,8 137 / 138 1,9 / 3,3 98 / 99 1,5 / 2,0 1,2 / 1,2 33 / 29
Calcio (mg/kg/24h) Magnesio (mg/24h)
0,9 / 2,0 111 / 122
Niveles del calcio y magnesio en la orina 24h al diagnóstico/y actual
Paciente con Pseudohipoaldosteronismo Tipo I
Paciente
Edad (años) Al diag/actual
Sexo Poliuria Polidipsia Nefro calcinosis
Mutación
0,25/5,5 M SI SI NO -
Niveles de los electrolitos y bicarbonato en el sangre al diagnóstico/y actual
Urea Creat Na K Ca Bic Aldosterona
19 / 33 0,3 / 0,4 118 / 138 5,7 / 4,2 1,2 / 1,3 18 / 23 43,2 / 31
Característica demográfica y el defecto genético Con el permiso

Gracias! Obrigada!