protocolo de ensayo clÍnico código de protocolo
TRANSCRIPT

PROTOCOLO DE ENSAYO CLÍNICO
Fármacos: Bortezomib (Velcade) y Lenalidomida
Código de Protocolo: GEM2010MAS65
Título del estudio: Estudio Fase IIb Nacional, Abierto, Multicéntrico, Randomizado,
Comparativo de tratamiento con un esquema secuencial Melfalán/Prednisona/Bortezomib
(Velcade ) (MPV) seguido de Lenalidomida/Dexametasona a bajas dosis (Rd) versus un
esquema alternante de Melfalán/Prednisona/Velcade (MPV) con
Lenalidomida/Dexametasona a bajas dosis (Rd) en pacientes con Mieloma Múltiple (MM)
sintomático de nuevo diagnóstico mayores de 65 años.
Fase de desarrollo: IIb
Número de EudraCT: 2010-018379-70
Fecha de protocolo: 30 de marzo de 2011
Versión: Tercera
Promotor: Fundación PETHEMA
Coordinadores del ensayo:
Dr. Jesús F. San Miguel Hospital Universitario de Salamanca
En colaboración con:
Dra Mª Victoria Mateos Hospital Universitario de Salamanca Dr. Joaquín Martínez Hospital 12 de Octubre. Madrid Dr. Juan José Lahuerta Hospital 12 de Octubre. Madrid Dra. Laura Rosiñol Hospital Clinic i Provincial de Barcelona Dr. Joan Bladé Hospital Clinic i Provincial de Barcelona
CONFIDENCIAL
PROPIEDAD DE FUNDACIÓN PETHEMA
NO PUEDE SER UTILIZADO, DIVULGADO O PUBLICADO SIN EL CONSENTIMIENTO DE LA FUNDACIÓN PETHEMA.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 2
Tercera versión: 30 de marzo de 2011
PÁGINA DE FIRMAS DEL PROTOCOLO
Código de Protocolo: GEM2010MAS65
Fármacos: Bortezomib (VELCADE ) + Lenalidomida
He leído este protocolo y acepto supervisar y dirigir la realización de este ensayo de acuerdo
con todas las estipulaciones del protocolo, consentimiento informado, directrices de la
Buena Práctica Clínica e ICH y de la Declaración de Helsinki.
Firma del personal de Fundación PETHEMA
Dr. J.F. San Miguel ------------------------- -----------------------
(Coordinador de ensayo) Firma Fecha
Dr. J. Díaz Mediavilla ------------------------- -----------------------
(Representante F. PETHEMA) Firma Fecha

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 3
Tercera versión: 30 de marzo de 2011
PÁGINA DE FIRMAS DEL PROTOCOLO
Código de Protocolo: GEM2010MAS65
Fármacos: Bortezomib (VELCADE ) + Lenalidomida
He leído este protocolo y acepto supervisar y dirigir la realización de este ensayo de acuerdo
con todas las estipulaciones del protocolo, consentimiento informado, directrices de la
Buena Práctica Clínica e ICH y de la Declaración de Helsinki.
Investigador Principal: __________________________ ___________________ _________________ Nombre Firma Fecha Co-Investigador: __________________________ ___________________ _________________ Nombre Firma Fecha Co-Investigador: __________________________ ___________________ _________________ Nombre Firma Fecha Co-Investigador: __________________________ ___________________ _________________ Nombre Firma Fecha

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 4
Tercera versión: 30 de marzo de 2011
LISTA DE ABREVIATURAS
Abreviatura Definición AA Acontecimiento Adverso AAG Acontecimiento Adverso Grave ALT Alanina transaminasa AST Aspartato transaminasa CEIC Comité Ético de Investigación Clinica CM Componente Monoclonal CRD Cuaderno de Recogida de Datos CTC (NCI) Criterios de toxicidad común CTD Ciclofosfamida / Talidomida / Dexametasona EBMT European Bone Marrow Transplantation ECOG Eastern Cooperative Oncology Group EMEA European Medicines Agency FDA Food and Drug Administration G-CSF Granulocite-colony stimulating factor GEM Grupo Español de Mieloma ICH International Conference on Harmonisation IF Inmunofijación IMWG International Mieloma Working Group ISS International Staging System IV Intravenoso LDH L-lactate dehydrogenase mg Miligramo ml Mililitro MM Mieloma Múltiple mm3 Milímetro cúbico mmol Milimol MO Médula ósea MP Melfalán / Prednisona MPV Melfalán / Prednisona / Velcade NCI Instituto Nacional del Cáncer NYHA New York Heart Association RAGI Reacción Adversa Grave e Inesperada RC Remisión completa Rd Lenalidomida / Dexametasona a bajas dosis RP Remisión parcial SAE Serious Adverse Event sFLC Cadenas ligeras libres en suero SG Supervivencia Global SLP Supervivencia Libre de Progresión TNT Tiempo hasta iniciar nuevo tratamiento TTP Tiempo hasta la Progresión VGPR Very Good Partial Response

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 5
Tercera versión: 30 de marzo de 2011
1. RESUMEN
1.1. Tipo de Solicitud
Ensayo clínico con un fármaco en nuevas condiciones de uso.
1.2. Identificación del promotor
Fundación PETHEMA CIF: G-81245706 Representante: Dr. Joaquín Díaz Mediavilla Servicio de Hematología Hospital Clínico San Carlos C/ Profesor Martín Lagos s/n 28040 Madrid Telf. 91- 330 33 12 Fax: 91- 330 33 11 Correo electrónico: [email protected]
1.3. Título del ensayo clínico
Estudio Fase IIb Nacional, Abierto, Multicéntrico, Randomizado, Comparativo de tratamiento con un esquema secuencial Melfalán/Prednisona/Bortezomib (Velcade ) (MPV) seguido de Lenalidomida (Revlimid ®)/Dexametasona a bajas dosis (Rd) versus un esquema alternante de Melfalán/Prednisona/Velcade (MPV) con Lenalidomida/Dexametasona a bajas dosis (Rd) en pacientes con Mieloma Múltiple (MM) sintomático de nuevo diagnóstico mayores de 65 años.
1.4. Código del Protocolo
GEM2010MAS65
1.5. Coordinadores del ensayo
Dr. Jesús F. San Miguel
Servicio de Hematología. Hospital Universitario de Salamanca
Paseo de San Vicente 58-182. Salamanca – 37007. Telf. 923-291384
España
En colaboración con:
Dra. Mª Victoria Mateos:
Servicio de Hematología. Hospital Universitario de Salamanca
Paseo de San Vicente 58-182. Salamanca – 37007. Telf. 923-291384
España

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 6
Tercera versión: 30 de marzo de 2011
Drs. Juan José Lahuerta y Joaquín Martínez
Servicio de Hematología. Hospital Universitario 12 de Octubre.
Ctra. Andalucía, km 5,5. Madrid – 28048 Telf. 91 3908525
España
Drs. Joan Bladé y Laura Rosiñol
Servicio de Hematología. Hospital Clínico y Provincial de Barcelona
Villarroel, 170. Barcelona – 08036 Telf. 93 2275428
España
1.6. Centros en los que se prevé realizar el ensayo
Ver anexo 1
1.7. Comités Éticos de Investigación Clínica
Ver anexo 2
1.8. Nombre y calificación de las personas responsa bles de la monitorización
Monitores de Ensayos Clínicos de Trial Form Support (TFS) Jefe de Proyecto de Trial Form Support (TFS) Dirección: C/ Arturo Soria 336 7º Izda. 28033 Madrid
1.9. Fármacos experimentales y control: dosis, form a farmacéutica, vía de administración y grupo terapéutico.
� Fármaco experimental: Bortezomib (Velcade) Forma farmacéutica: Viales de polvo liofilizado estéril para reconstituir. Vía de administración: Intravenosa Grupo terapéutico: Inhibidor de proteosomas. Antineoplásicos (L01XX32)
� Fármaco experimental: Lenalidomida
Forma farmacéutica: Cápsulas Vía de administración: Oral Grupo terapéutico: Inmunomodulador (L04AX04)
1.10. Fase del ensayo clínico
Fase IIb
1.11. Objetivos del ensayo clínico
Los objetivos primarios de este estudio son:

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 7
Tercera versión: 30 de marzo de 2011
� Analizar la eficacia en términos de supervivencia libre de progresión (SLP) a los 18
meses de MPV y Rd administrados en un esquema secuencial vs alternante en
pacientes con MM de nuevo diagnóstico, mayores de 65 años.
� Evaluar la seguridad y tolerabilidad de los dos esquemas de inducción propuestos,
medida en función de la incidencia de toxicidades clínica y de laboratorio.
Los objetivos secundarios de este estudio son:
� Analizar la eficacia en términos de respuesta, duración de la respuesta, tiempo
hasta la progresión (TTP), tiempo hasta iniciar nue vo tratamiento (TNT) y
supervivencia global (SG) de los dos esquemas de inducción propuestos.
� Identificar las características biológicas (incluyendo un análisis genómico) de aquellos
pacientes que fueran resistentes a uno u otro esquema o a ambos esquemas
propuestos.
1.12. Diseño
Este protocolo es un estudio nacional, multicéntrico, comparativo, abierto y randomizado
diseñado para comparar, en primer lugar, la SLP a 18 meses de dos esquemas de
quimioterapia de inducción –MPV seguido de Rd versus MPV alternando con Rd- en
pacientes con MM de nuevo diagnóstico mayores de 65 años. Este estudio está diseñado
para comparar ambos esquemas en términos de eficacia y toxicidad. En cada rama de
tratamiento se incluirán 120 pacientes y las evaluaciones y visitas programadas se
realizarán en los periodos de pre-tratamiento (periodo de selección) y tratamiento. Después
de finalizado el protocolo a los pacientes se les seguirá cada dos meses aproximadamente,
dentro de la práctica clínica habitual, para poder observar la supervivencia y respuestas a
otros tratamientos.
En el periodo pre-tratamiento se realizarán los procedimientos para la selección de los
pacientes. Después de proporcionar el consentimiento informado por escrito para participar
en el estudio se evaluará a los pacientes para establecer su elegibilidad. Los
procedimientos para la selección se realizarán, en su mayoría, en un período de 15 días
antes de la primera dosis de medicación (días –14 al –0). Los pacientes elegibles serán
incluidos en el estudio y serán randomizados en una proporción 1:1 para recibir el esquema
secuencial Melfalán/Prednisona/Velcade seguido de Lenalidomida/Dexametasona a bajas
dosis (Grupo A de tratamiento de inducción) o el esquema alternante
Melfalán/Prednisona/Velcade alternando con Lenalidomida/Dexametasona a bajas dosis
(Grupo B de tratamiento de inducción). Dentro del grupo B de tratamiento habrá una

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 8
Tercera versión: 30 de marzo de 2011
segunda randomización en una proporción 1:1 para iniciar el tratamiento con MPV o Rd
(B1: inicio con MPV y B2: inicio con Rd)
Los pacientes asignados al Grupo A de tratamiento de inducción recibirán nueve ciclos
de MPV consistentes en un ciclo de 6 semanas de duración con Velcade administrado
dos veces por semana (días 1, 4, 8, 11, 22, 25, 29 y 32) en combinación con Melfalán y
Prednisona, administrados los días 1-4 del ciclo, seguido de ocho ciclos de cuatro
semanas de duración consistentes en Velcade administrado una vez por semana (días 1,
8, 15 y 22) en combinación con Melfalán y Prednisona, administrados los días 1-4 del ciclo.
Posteriormente, los pacientes recibirán nueve ciclos adicionales de Rd de cuatro
semanas de duración con Lenalidomida los días 1-21 seguidos de 7 días de descanso
(días 22-28) más Dexametasona una vez por semana (días 1, 8, 15 y 22) seguidos de 6
días de descanso (días 23-28).
Los pacientes asignados al Grupo B de tratamiento recibirán el mismo esquema de
tratamiento que los pacientes asignados al Grupo A, pero los ciclos MPV serán alternados
con ciclos Rd. Los pacientes serán de nuevo randomizados en este grupo en una
proporción 1:1 a iniciar el tratamiento con un ciclo MPV o Rd.
Globalmente, todos los pacientes recibirán idéntico número de ciclos: nueve ciclos MPV y
nueve ciclos Rd. Los pacientes asignados al grupo A de tratamiento secuencial que
recaigan o progresen durante el tratamiento con los nueve ciclos iniciales MPV pasarán a
recibir tratamiento con Rd. Previamente se deberá informar a los coordinadores, vía fax,
para que éstos confirmen la progresión y aprueben el paso a ciclos de Rd.
Los pacientes deberán ser evaluados durante el periodo de tratamiento en el día 1 de cada
nuevo ciclo (± 4 días) y un mes después de finalizados los 18 ciclos del periodo de
tratamiento (± 1 semana). La respuesta al tratamiento se medirá según los criterios
modificados del EBMT y los del IMWG (Anexo 7) actualizados según el último panel de
consenso.
La seguridad será evaluada mediante la monitorización de todos los acontecimientos
adversos graves y de los acontecimientos adversos no graves, hematológicos y no
hematológicos, relacionados con los fármacos en estudio.
Finalizado el tratamiento, y después de realizada la visita final, se realizará un seguimiento
de los pacientes cada 2 meses.
1.13. Enfermedad en estudio
Mieloma Múltiple (MM)

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 9
Tercera versión: 30 de marzo de 2011
1.14. Variables principales de valoración
Las variables principales son:
• Variable principal de eficacia: supervivencia libre de progresión (SLP) con los dos
esquemas de tratamiento propuestos, entendiendo como SLP la duración en meses
desde la fecha de la inclusión en el ensayo hasta la fecha de la progresión de la
enfermedad o muerte debido a cualquier causa.
• Variables principales de seguridad: seguridad y tolerabilidad de los dos esquemas de
tratamiento propuestos, secuencial versus alternante.
• Las variables secundarias son:
• Variables secundarias de eficacia: eficacia, en términos de tasa de respuestas,
duración de la respuesta, TTP, TNT y SG en ambos grupos de tratamiento.
• Variables secundarias de asociación genética: características biológicas (incluyendo
análisis de perfil de expresión génica) de aquellos pacientes resistentes a uno de los
dos regímenes propuestos o ambos.
1.15. Población del estudio
Población es estudio
Pacientes ˃ 65 años con mieloma múltiple sintomático de nuevo diagnóstico, en función de
los criterios estándar (véase Anexo 6), que precisen tratamiento y que no hayan sido
tratados previamente.
Número total de pacientes
Serán incluidos 240 pacientes en total, 120 asignados a cada rama de tratamiento. La
inclusión será competitiva, por lo que no se prevé un número determinado de pacientes por
centro participante.
1.16. Duración del tratamiento
Los pacientes incluidos en el ensayo recibirán tratamiento en ambos grupos durante un
máximo de 74 semanas (38 semanas para el esquema MPV y 36 para Rd).

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 10
Tercera versión: 30 de marzo de 2011
1.17. Calendario y fecha prevista de finalización
El inicio del ensayo está previsto aproximadamente para junio de 2010. El periodo de
inclusión será aproximadamente de 19 meses, por lo que se prevé la finalización de la
inclusión en enero de 2012. Los pacientes recibirán tratamiento durante 74 semanas.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 11
Tercera versión: 30 de marzo de 2011
2. ÍNDICE PÁGINA DE FIRMAS DEL PROTOCOLO................................................................................. 2 LISTA DE ABREVIATURAS...................................................................................................... 4 1. RESUMEN..................................................................................................................... 5 1.1. Tipo de Solicitud................................................................................................................. 5 1.2. Identificación del promotor.................................................................................................. 5 1.3. Título del ensayo clínico ..................................................................................................... 5 1.4. Código del Protocolo .......................................................................................................... 5 1.5. Coordinadores del ensayo.................................................................................................. 5 1.6. Centros en los que se prevé realizar el ensayo .................................................................. 6 1.7. Comités Éticos de Investigación Clínica ............................................................................. 6 1.8. Nombre y calificación de las personas responsables de la monitorización ......................... 6 1.9. Fármacos experimentales y control: dosis, forma farmacéutica, vía de
administración y grupo terapéutico. ............................................................................... 6 1.10. Fase del ensayo clínico .................................................................................................... 6 1.11. Objetivos del ensayo clínico ............................................................................................. 6 1.12. Diseño .............................................................................................................................. 7 1.13. Enfermedad en estudio..................................................................................................... 8 1.14. Variables principales de valoración .................................................................................. 9 1.15. Población del estudio ....................................................................................................... 9 1.16. Duración del tratamiento................................................................................................... 9 1.17. Calendario y fecha prevista de finalización ..................................................................... 10 2. INFORMACIÓN GENERAL ......................................................................................... 14 2.1. Identificación del ensayo .................................................................................................. 14 2.1.1. Código de protocolo:.................................................................................................... 14 2.1.2. Título del ensayo: ........................................................................................................ 14 2.2. Tipo de ensayo clínico:..................................................................................................... 14 2.3. Descripción de los productos del ensayo:......................................................................... 14 2.4. Datos relativos al promotor:.............................................................................................. 14 2.5. Director Técnico responsable del control de las muestras: ............................................... 14 2.6. Identificación del Monitor: ................................................................................................. 15 2.7. Datos de los centros e investigadores participantes en el ensayo: ................................... 15 2.8. Duración prevista del ensayo: .......................................................................................... 15 2.9. Número de pacientes previstos: ....................................................................................... 15 2.10. Lista de tablas en el texto ............................................................................................... 15 3. INTRODUCCION Y JUSTIFICACION DEL ESTUDIO ................................................. 15 3.1. Antecedentes científicos................................................................................................... 15 3.1.1. Repaso general del Mieloma Múltiple .......................................................................... 15 3.1.2. Tratamiento actual del Mieloma Múltiple ...................................................................... 16 3.2. Justificacion del estudio y selección de las dosis.............................................................. 18 3.3. Objetivos del ensayo ........................................................................................................ 19 4. TIPO DE ENSAYO CLÍNICO Y DISEÑO ..................................................................... 19 4.1. Diseño global.................................................................................................................... 19 4.2. Plan de estudio................................................................................................................. 19 4.3. Procedimientos del estudio............................................................................................... 21 5. POBLACIÓN OBJETO DEL ESTUDIO ........................................................................ 27 6.1. Criterios de Inclusión ........................................................................................................ 27 6.2. Criterios de exclusión ....................................................................................................... 28 6.3. Número de sujetos previstos ............................................................................................ 29 6.4. Retirada de los pacientes del tratamiento o del ensayo.................................................... 29

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 12
Tercera versión: 30 de marzo de 2011
6.5. Duración del tratamiento de los pacientes ........................................................................ 30 6. DESCRIPCIÓN DEL TRATAMIENTO DEL ENSAYO .................................................. 30 7.1. Materiales del ensayo clínico............................................................................................ 30 7.2. Preparación, cuidados, y almacenamiento de las drogas del ensayo ............................... 31 7.3. Administración de las drogas de estudio y esquema de dosis .......................................... 33 7.4. Modificación de dosis y retrasos....................................................................................... 36 7.5. Envasado y etiquetado ..................................................................................................... 40 7.6. Tratamiento concomitante ................................................................................................ 40 7.7. Medicaciones permitidas y tratamientos de soporte ......................................................... 42 7.7.1. Medicaciones permitidas ............................................................................................. 42 7.7.2. Medicaciones prohibidas ............................................................................................. 42 7.8. Cumplimiento del tratamiento ........................................................................................... 42 7. DESARROLLO DEL ENSAYO Y EVALUACIÓN DE LA RESPUESTA ........................ 43 8.1. Variables principales y secundarias.................................................................................. 43 8.2. Controles a realizar .......................................................................................................... 43 8.3. Valoración de la respuesta ............................................................................................... 47 8. ACONTECIMIENTOS ADVERSOS ............................................................................. 47 9.1. Información mínima .......................................................................................................... 47 9.1.1. Acontecimientos Adversos (AA) .................................................................................... 47 9.1.2. Acontecimientos Adversos Graves (AAG) ..................................................................... 48 9.2. Calificación de un acontecimiento adverso....................................................................... 50 9.3. Procedimientos para la notificación rápida de acontecimientos adversos
graves.......................................................................................................................... 52 9.4. Notificación a las Autoridades Sanitarias y a los Comités Éticos ...................................... 53 9.5. Embarazo......................................................................................................................... 54 9.6. Tratamiento de las reclamaciones de calidad de Velcade®............................................... 55 9. ASPECTOS ÉTICOS................................................................................................... 56 10.1. Buena práctica clínica .................................................................................................... 56 10.2. Consideraciones éticas................................................................................................... 56 10.3. Información al paciente y consentimiento informado ...................................................... 57 10.4. Confidencialidad de los pacientes .................................................................................. 57 10.5. Cumplimiento del protocolo ............................................................................................ 57 10.6. Finalización prematura del estudio ................................................................................. 58 10.7. Responsabilidad y seguro .............................................................................................. 58 10. CONSIDERACIONES PRÁCTICAS............................................................................. 58 11.1. Responsabilidades de todos los participantes en el ensayo ........................................... 58 11.1.1. Investigador................................................................................................................. 58 11.1.2. Monitor ........................................................................................................................ 59 11.1.3. Promotor...................................................................................................................... 60 11.2. Auditoría......................................................................................................................... 60 11.3. Contabilidad de la medicación........................................................................................ 61 11.4. Retención de los registros .............................................................................................. 61 11.5. Publicaciones de los resultados del ensayo y uso de la información .............................. 61 11.5.1. Normativa básica del ensayo....................................................................................... 61 11.5.2. Condiciones de publicación del grupo PETHEMA ....................................................... 62 11. ANÁLISIS ESTADÍSTICO............................................................................................ 64 12.1. Estimación del tamaño de la muestra............................................................................. 64 12.2. Población para el análisis ............................................................................................... 64 12.3. Procedimientos para el manejo de los datos inexistentes, no usados y
confusos ...................................................................................................................... 65 12.4. Métodos estadísticos...................................................................................................... 65

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 13
Tercera versión: 30 de marzo de 2011
12.4.1. Análisis de eficacia ...................................................................................................... 66 12.4.2. Comparaciones basales .............................................................................................. 70 12.4.3. Análisis de seguridad................................................................................................... 70 12.5. Procedimientos para la comunicación de desviaciones del plan estadístico
original......................................................................................................................... 71 12.6. Análisis preliminar .......................................................................................................... 71 12. REFERENCIAS BIBLIOGRÁFICAS............................................................................. 72 ANEXO 1: LISTADO DE CENTROS E INVESTIGADORES PARTICIPANTES....................... 74 ANEXO 2: LISTADO DE CEIC ................................................................................................ 77 ANEXO 3: ESQUEMA DE VISITAS......................................................................................... 80 ANEXO 4: ESCALAS ECOG Y KARNOFSKY PARA EL ESTADO GENERAL........................ 81 ANEXO 5: FÓRMULAS UTILIZADAS EN ESTE PROTOCOLO.............................................. 82 ANEXO 6: CRITERIOS DIAGNÓSTICOS DE MIELOMA MÚLTIPLE...................................... 83 ANEXO 7: CRITERIOS DE RESPUESTA DE LA ENFERMEDAD .......................................... 84 ANEXO 8: CENTRALIZACION DE MUESTRAS DE MO......................................................... 88 ANEXO 9: DECLARACIÓN DE HELSINKI .............................................................................. 89 ANEXO 10: PROGRAMA DE PREVENCIÓN DE EMBARAZO (PPE) -
LENALIDOMIDA.......................................................................................................... 94

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 14
Tercera versión: 30 de marzo de 2011
2. INFORMACIÓN GENERAL
2.1. Identificación del ensayo
2.1.1. Código de protocolo:
GEM2010MAS65
2.1.2. Título del ensayo:
Estudio Fase IIb Nacional, Abierto, Multicéntrico, Randomizado, Comparativo de tratamiento con un esquema secuencial Melfalán/Prednisona/Bortezomib (Velcade) (MPV) seguido de Lenalidomida (Revlimid®)/Dexametasona a bajas dosis (Rd) versus un esquema alternante de Melfalán/Prednisona/Velcade (MPV) con Lenalidomida/Dexametasona a bajas dosis (Rd) en pacientes con Mieloma Múltiple (MM) sintomático de nuevo diagnóstico mayores de 65 años.
2.2. Tipo de ensayo clínico:
Ensayo clínico con un fármaco en nuevas condiciones de uso.
2.3. Descripción de los productos del ensayo:
- Bortezomib (Velcade ) Viales de Polvo liofilizado para reconstituir. Administración Intravenosa. Johnson&Johnson Pharmaceutical Research&Development (J&JPR&D)
- Lenalidomida Cápsulas duras de 25 mg, 15 mg, 10 mg y 5 mg Administración oral Celgene
2.4. Datos relativos al promotor:
Fundación PETHEMA CIF: G-81245706 Representante: Dr. Joaquín Díaz Mediavilla
Servicio de Hematología Hospital Clínico San Carlos C/ Profesor Martín Lagos s/n 28040 Madrid Telf. 91- 330 33 12 Fax: 91- 330 33 11 Correo electrónico: [email protected]
2.5. Director Técnico responsable del control de la s muestras:
Fisher Clinical Services UK Ltd para el Velcade

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 15
Tercera versión: 30 de marzo de 2011
Fisher Clinical Services UK Ltd para Lenalidomida.
2.6. Identificación del Monitor:
Trial Form Support (TFS) Dirección: Arturo Soria 336, 7º Izda. 28033 Madrid
2.7. Datos de los centros e investigadores particip antes en el ensayo:
Ver anexo 1
2.8. Duración prevista del ensayo:
Hasta julio de 2013
2.9. Número de pacientes previstos:
Un total de 240 pacientes. El ensayo es competitivo, no se prevé la inclusión de un número determinado de pacientes por centro.
2.10. Lista de tablas en el texto
Tabla 1: Esquemas de tratamiento MPV y Rd para los grupos A, B1 y B2 de tratamiento
Tabla 2. Esquema de tratamiento grupo A: Esquema secuencial MPV-Rd.
Tabla 3. Esquema de tratamiento grupo B1 y B2: Esquema alternante MPV-Rd
Tabla 4: Manejo de los pacientes con neuropatía sensitiva y/o dolor neuropático
relacionado con Velcade
Tabla 5.- Reducción de dosis de lenalidomida
Tabla 6.- Reducción de dosis de Dexametasona
Tabla 7: recomendaciones para el uso de profilaxis antitrombótica
3. INTRODUCCION Y JUSTIFICACION DEL ESTUDIO
3.1. Antecedentes científicos
3.1.1. Repaso general del Mieloma Múltiple
El Mieloma Múltiple (MM) es una neoplasia de células B en sus últimos estadios de
maduración (células plasmáticas). Representa la segunda neoplasia hematológica
más común después del linfoma no Hodgkin. La incidencia anual es aproximadamente
de 4 casos por 100.0001. El Mieloma Múltiple sigue siendo una enfermedad incurable.
A medida que la enfermedad avanza, la disminución de la resistencia a las

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 16
Tercera versión: 30 de marzo de 2011
infecciones, la importante destrucción del esqueleto (con dolores óseos, fracturas
patológicas e hipercalcemia), la anemia, la insuficiencia renal y, con menor frecuencia,
las complicaciones neurológicas y la hiperviscosidad son responsables de la
morbilidad y posterior mortalidad de estos pacientes2. La supervivencia de los
pacientes con MM ha mejorado significativamente en los últimos años, especialmente
para aquellos pacientes diagnosticados a partir del año 20013. Sin embargo, esta
mejoría en la supervivencia se ha observado fundamentalmente en los pacientes
jóvenes, menores de 65 años y candidatos a recibir un trasplante autólogo. Sin
embargo, en los pacientes mayores de 65 años, esta mejoría es menos clara y, por
ello, exisite una necesidad urgente de nuevos tratamientos para aumentar la
supervivencia de estos enfermos.
3.1.2. Tratamiento actual del Mieloma Múltiple
Antes de disponer de los agentes alquilantes la mediana de supervivencia de los
pacientes con mieloma múltiple (MM) era inferior a un año. Con el tratamiento
quimioterápico convencional, básicamente ciclos intermitentes de melfalán y prednisona
(MP), se logran entre un 50-60% de respuestas y la mediana de supervivencia se sitúa
entre 2 y 3 años4. Sin embargo, la introducción de nuevos fármacos en el tratamiento de
pacientes con MM no candidatos a trasplante ha conllevado importantes avances. Hay
cinco estudios randomizados en que Talidomida junto con MP ha demostrado ser
superior a MP en términos de tasa global de respuestas, tasa de remisiones completas
(RC) y supervivencia libre de evento o TTP; sin embargo, sólo en dos de ellos MPT ha
demostrado ser superior a MP en SG5-9. Talidomida y dexametasona (TalDex) fue
tambien superior a MP en respuestas en un estudio randomizado, pero esta superioridad
no se tradujo en ventaja en TTP ni SG, y esto fue especialmente evidente en el grupo de
pacientes mayores de 75 años, debido fundamentalmente a la elevada tasa de
mortalidad durante el primer año en el grupo de pacientes tratados con TalDex por
acontecimientos adversos, ya que el esquema incluía dosis altas de Talidomida y
Dexametasona10. Finalmente, el agente alquilante ciclofosfamida se ha añadido a
TalDex, a bajas dosis, y comparado con MP; el esquema CTD ha demostrado ser
superior en respuestas globales y RC, aunque no se conoce todavía los resultados en
supervivencia11. A la vista de todos estos resultados, MPT ha sido también recientemente
aprobado por la EMEA para el tratamiento de pacientes con MM no candidatos a
trasplante.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 17
Tercera versión: 30 de marzo de 2011
Con respecto a Lenalidomida, los resultados son más preliminares pero hay datos de un
estudio fase I/II en que Lenalidomida se combinó con MP con resultados prometedores
de eficacia, así como de supervivencia12. Un importante estudio randomizado realizado
en pacientes de nuevo diagnóstico en que se ha comparado Lenalidomida más
Dexametasona a altas dosis (RD) con Lenalidomida más Dexametasona a bajas dosis
(Rd) ha demostrado que tras cuatro ciclos de inducción, RD es superior en tasa de
respuestas, aunque no en SLP y SG, debido a la menor incidencia de acontecimientos
adversos observados con Rd y si se selecciona el grupo de pacientes que reciben esta
combinación, Rd, durante más de cuatro ciclos (población con mediana de edad 66 años)
se ha observado que el tratamiento continuado mejora la tasa de respuestas (91%), RCs
(21%), con menor incidencia de acontecimientos adversos que en la rama RD no
hematológicos y una supervivencia global estimada a 3 años similar a los pacientes
tratados con RD13. A la luz de los resultados del esquema Rd, por su eficacia y
tolerabilidad, se está convirtiendo oen una de las alternativas terapeúticas más atractivas
para le tratamiento del MM.
Bortezomib fue combinado con MP en un ensayo fase I/II realizado por el grupo español
de MM, demostrando que se trataba de un esquema eficaz y seguro en pacientes
mayores de 65 años14, lo que justificó la realización del ensayo VISTA, randomizado,
fase III en que MPV fue comparado con MP en 682 pacientes. MPV demostró ser
superior a MP en tasa global de respuestas (71% vs 31%), RCs (30% vs 4%), TTP
(mediana para MPV de 24 meses), TNT y SG; además, la eficacia de MPV fue
independiente de la presencia de factores pronósticos adversos, como edad avanzada
(>75 años), presencia de insuficiencia renal o alteraciones citogenéticas de mal
pronóstico. La toxicidad fue aceptable, destacando la toxicidad gastrointestinal y la
neuropatía periférica como las más frecuentes, si bien es importante señalar que la
neuropatía periférica mejoró o se resolvió completamente en más de 3/4 de los pacientes
con suspensión del tratamiento o ajustes de dosis15. Estos resultados condujeron a la
aprobación del esquema MPV por la FDA y EMEA, siendo actualmente uno de los
estándares de tratamiento del MM. Recientemente el grupo español de MM y el grupo
italiano han realizado ensayos clínicos fase III utilizando esquemas MPV modificados con
respecto al original propuesto en el VISTA (basados, en líneas generales, en el uso de
bortezomib en esquema semanal en lugar de dos veces por semana) con el doble
objetivo de mantener la eficacia y disminuir la toxicidad16;17. La tasa de RCs fue del 22% y
20% en el estudio español e italiano, respectivamente y la toxicidad disminuyó
significativamente con una incidencia de neuropatía periférica del 8% y 5%,

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 18
Tercera versión: 30 de marzo de 2011
respectivamente y una incidencia de discontinuaciones debido a toxicidad
significativamente más bajas.
3.2. Justificacion del estudio y selección de las d osis
Los resultados obtenidos de los ensayos clínicos realizados en primera línea con esquemas
basados en bortezomib y lenalidomida en pacientes con MM no candidatos a trasplante están
contribuyendo a mejorar la supervivencia de este grupo de pacientes. Los nuevos ensayos
clínicos a realizar en pacientes mayores de 65 años no candidatos a trasplante deberían ir
encaminados a optimizar las opciones de tratamiento, con un doble objetivo: mantener o
aumentar la eficacia, disminuyendo la toxicidad. En este ensayo proponemos el uso del
esquema MPV modificado tal y como se hizo en el ensayo realizado previamente por el grupo
español de MM, ya que ha demostrado ser eficaz, obteniendo tasa de RCs del 22%, pero
además añadiendo el esquema Rd que, por separado, ha demostrado también ser efectivo en
pacientes con MM de nuevo diagnóstico. El uso de estos dos esquemas, en ensayos clínicos
independientes, ha demostrado también disminuir significativamente la toxicidad. En este
sentido, el esquema MPV modificado ha demostrado ser menos tóxico con respecto al
esquema convencional MPV del VISTA y el uso de lenalidomida con dexametasona a bajas
dosis consiguió disminuir la incidencia de acontecimientos adversos de una manera
significativa con respecto al uso de lenalidomida y dexametasona a altas dosis. Por ello, para
intentar no incrementar la toxicidad de estos esquemas modificados, no proponemos la
administración simultánea de los cuatro fármacos, bortezomib y lenalidomida con un agente
alquilante y corticoide, sino administrarlos, bien de una manera secuencial o alternante.
Nuestra hipótesis es que el esquema alternante resultaría potencialmente más eficaz con
menor toxicidad acumulada ya que las células tumorales van a estar precozmente expuestas
a todos los agentes disminuyendo las posibilidades de que las células tumorales desarrollen
mecanismos de resistencia y “escape” de las mismas dando lugar a recaídas precoces,
mientras que las células hematopoyéticas normales tendrán la posibilidad de recuperarse de
la toxicidad de los diferentes fármacos, ya que su mecanismo de acción es diferente para
cada una de ellas y finalmente, la toxicidad sería menor. Además, el uso de dos esquemas
diferentes de tratamiento en el mismo paciente puede contribuir a identificar pacientes que
sean, bien sensibles a ambos regímenes, resistentes a ambos, o resistentes a uno de los dos.
Por ello, además, en este ensayo clínico se realizará una correcta caracterización de todos
los pacientes, a través de estudios genómicos, que puede contribuir en un futuro a
individualizar el tratamiento en función de las características de los pacientes así como
mejorar el conocimiento de los mecanismos de resistencia a drogas.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 19
Tercera versión: 30 de marzo de 2011
3.3. Objetivos del ensayo
Los objetivos primarios de este estudio son:
• Analizar la eficacia en términos de supervivencia libre de progresión a 18 meses de
MPV y Rd administrados en un esquema secuencial vs alternante en pacientes con MM
de nuevo diagnóstico, mayores de 65 años.
• Evaluar la seguridad y tolerabilidad de los dos esquemas de inducción propuestos,
medida en función de la incidencia de toxicidades clínica y de laboratorio.
Los objetivos secundarios de este estudio son:
• Analizar la eficacia en términos de respuesta, duración de la respuesta, tiempo hasta
progresión (TTP), tiempo hasta iniciar nuevo tratam iento (TNT) y supervivencia
global (SG) de los dos esquemas de inducción propuestos.
• Identificar las características biológicas (incluyendo un análisis genómico) de aquellos
pacientes que fueran resistentes a uno u otro esquema o a ambos esquemas
propuestos.
4. TIPO DE ENSAYO CLÍNICO Y DISEÑO
4.1. Diseño global
Este protocolo es un estudio nacional, multicéntrico, comparativo, abierto y randomizado
diseñado para comparar, en primer lugar, la supervivencia libre de progresión (SLP) a 18
meses de dos esquemas de quimioterapia de inducción –MPV seguido de Rd versus MPV
alternando con Rd - en pacientes con MM de nuevo diagnóstico mayores de 65 años. Este
estudio está diseñado para comparar ambos esquemas en términos de eficacia y toxicidad.
En cada rama de tratamiento se incluirán 120 pacientes pertenecientes a hospitales
incluidos dentro del Grupo Español de Mieloma.
4.2. Plan de estudio
Los pacientes serán evaluados en visitas a lo largo de todo el ensayo.
En el periodo Pre-tratamiento o de selección se evalúa a los pacientes para establecer su
elegibilidad, después de proporcionar el consentimiento informado por escrito para
participar en el estudio. Este período de selección tiene lugar en los 15 días anteriores a la
visita basal (primera dosis de medicación) (días –14 al –0), excepto para la serie ósea y el
aspirado de médula ósea, que podrán ser de un mes anterior a la primera dosis. Los
pacientes elegibles serán incluidos en el estudio y serán randomizados en una proporción

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 20
Tercera versión: 30 de marzo de 2011
1:1 para recibir el esquema secuencial Melfalán/Prednisona/Velcade seguido de
Lenalidomida/Dexametasona a bajas dosis (Grupo A de tratamiento de inducción) o el
esquema alternante Melfalán/Prednisona/Velcade alternando con
Lenalidomida/Dexametasona a bajas dosis (Grupo B de tratamiento de inducción). Dentro
del grupo B de tratamiento habrá una segunda randomización en una proporción 1:1 para
iniciar el tratamiento con MPV (B1) o Rd (B2).
Los pacientes asignados al Grupo A de tratamiento de inducción recibirán nueve ciclos
de MPV consistentes en un ciclo de 6 semanas de duración con Velcade administrado
dos veces por semana (días 1, 4, 8, 11, 22, 25, 29 y 32) en combinación con Melfalán y
Prednisona, administrados los días 1-4 del ciclo, seguido de ocho ciclos de cuatro
semanas de duración consistentes en Velcade administrado una vez por semana (días 1,
8, 15 y 22) en combinación con Melfalán y Prednisona, administrados los días 1-4 del ciclo.
Posteriormente, los pacientes recibirán nueve ciclos adicionales de Rd de cuatro
semanas de duración con Lenalidomida los días 1-21 seguidos de 7 días de descanso (22-
28) más Dexametasona una vez por semana (días 1, 8, 15 y 22) seguidos de 6 días de
descanso (23-28).
Los pacientes asignados al Grupo B de tratamiento recibirán el mismo esquema de
tratamiento que los pacientes asignados al Grupo A, pero los ciclos MPV serán alternados
con ciclos Rd. Los pacientes serán de nuevo randomizados en este grupo en una
proporción 1:1 a iniciar el tratamiento con un ciclo MPV o Rd.
Globalmente, todos los pacientes recibirán idéntico número de ciclos, nueve ciclos MPV y
nueve ciclos Rd.
Los pacientes deberán ser evaluados durante el periodo de tratamiento en el día 1 de cada
nuevo ciclo (± 4 días) y un mes después de finalizados los 18 ciclos del periodo de
tratamiento (± 1 semana). La respuesta al tratamiento se medirá según los criterios
modificados del EBMT y los del IMWG (Anexo 7) actualizados según el último panel de
consenso.
La seguridad será evaluada mediante la monitorización de los acontecimientos adversos
graves y de los acontecimientos adversos no graves, hematológicos y no hematológicos,
relacionados con los fármacos en estudio, de acuerdo al plan de monitorización
establecido.
Finalizado el tratamiento, y después de realizada la visita final del protocolo, se realizará un
seguimiento de los pacientes cada 2 meses.
Todos los pacientes recibirán bisfofonatos cada 4 semanas durante el periodo de
tratamiento a partir del tercer ciclo. También recibirán profilaxis antiviral para evitar

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 21
Tercera versión: 30 de marzo de 2011
reactivaciones del virus herpes zoster durante el tratamiento con bortezomib con aciclovir a
dosis de 400 mg/12 horas, profilaxis antitrombótica durante el tratamiento con lenalidomida
con aspirina, heparina de bajo peso molecular o anticoagulación oral en función de las
características personales de cada paciente y profilaxis antibiótica con levofloxacino
durante los cuatro primeros ciclos de tratamiento (Ver punto 7.6, “Medicación
Concomitante”, para la descripción de las pautas de administración).
Los pacientes que desarrollen progresión de la enfermedad confirmada durante el periodo
de Tratamiento deberán ser retirados del ensayo clínico, con la excepción de los pacientes
asignados al grupo A de tratamiento secuencial que recaigan o progresen durante el
tratamiento con los nueve ciclos iniciales MPV, que pasarán a recibir tratamiento con Rd.
Previamente se deberá informar a los coordinadores, vía fax, para que éstos confirmen la
progresión y aprueben el paso a ciclos de Rd.
Los detalles de cada visita y sus procedimientos están contenidos en el Anexo 3.
4.3. Procedimientos del estudio
Antes de iniciar el fármaco en estudio:
Advertencia sobre embarazos
Lenalidomida está relacionada estructuralmente con talidomida. Talidomida es una
sustancia activa con conocidos efectos teratogénicos en el ser humano, que causa
defectos congénitos severos y amenazantes para la vida. Lenalidomida induce, en
primates, malformaciones parecidas a las descritas con talidomida. Es esperable que
lenalidomida tenga efectos teratogénicos en humanos si se toma durante el embarazo.
Las condiciones del Programa de Prevención de Embarazo deben ser cumplidas por todos
los pacientes a menos que haya pruebas fiables de que no existe capacidad de gestación.
Criterios correspondientes a mujeres sin capacidad de gestación:
Se considera que una paciente o la pareja mujer de un paciente tiene capacidad de
gestación a menos que cumpla alguno de los siguientes criterios:
• Edad ≥ 50 años y amenorrea natural desde hace ≥ 1 año*
• Fallo ovárico prematuro confirmado por un ginecólogo especialista
• Salpingooforectomía bilateral o histerectomía previa
• Genotipo XY, síndrome Turner o agenesia uterina
* La amenorrea secundaria al tratamiento antineoplásico no descarta la capacidad de
gestación.
Procedimientos

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 22
Tercera versión: 30 de marzo de 2011
Las evaluaciones a realizar durante el estudio se resumen a continuación mediante una
referencia al período, día y ciclo del estudio. En el Anexo 3 se incluye un diagrama del
programa de procedimientos del estudio.
La práctica totalidad de las exploraciones forman parte de la rutina habitual, con la única
posible excepción, en función de los centros, de la realización de un aspirado de médula
ósea tras los nueve primeros ciclos de inducción. Esta prueba adicional nos permitirá llevar
un control más estrecho de la enfermedad y es muy importante para los estudios biológicos
relacionados con el ensayo.
Procedimientos de Selección
Cada paciente debe firmar y fechar el modelo de consentimiento informado antes de llevar a
cabo cualquier procedimiento del estudio. No obstante, son válidos para la selección todos
los procedimientos que formen parte de la rutina habitual en el diagnóstico del MM
realizados antes de firmar el consentimiento informado, siempre que se hayan realizado en
los 14 días previos al inicio del tratamiento. La serie ósea y el aspirado de médula ósea
podrán ser de hasta un mes antes de la primera dosis de medicación de ensayo. Si se tiene
un paciente candidato, a la espera del diagnóstico por falta de aspirado de médula ósea, se
enviará la muestra a los laboratorios de referencia indicándolo (ver anexo 8), siempre que se
haya firmado previamente el consentimiento informado.
Los siguientes procedimientos serán realizados durante la Visita de Selección. Todas las
evaluaciones del Periodo de Selección se realizarán antes de recibir la medicación del
estudio.
� Presentación del consentimiento informado por escrito.
� Demografía
� Historia médica, incluyendo enfermedades concomitantes, tratamiento de
soporte y condiciones médicas basales.
� Examen físico completo.
� Electrocardiograma.
� Serie ósea para documentar lesiones óseas, así como otras radiografías,
scanner o resonancia magnética que sean necesarias para documentar
plasmocitomas extramedulares.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 23
Tercera versión: 30 de marzo de 2011
� Peso y altura, para cálculo de la superficie corporal.
� Estado general mediante el índice de ECOG.
� Determinación del estadío del MM (tanto Durie y Salmon como ISS).
� Extracción de muestras de sangre para análisis: Hematología y Bioquímica.
� Extracción de muestras de sangre para cuantificación de inmunoglobulinas y
componente monoclonal.
� Orina de 24 horas para proteinuria, estudio de componente monoclonal e
inmunofijación.
� Cadenas ligeras libres en suero (sFLC): Sólo se realizarán en el momento del
diagnóstico si el paciente presenta un MM oligosecretor/no secretor o MM
Bence Jones puro.
� Extracción de sangre para proteína-C reactiva y B2 microglobulina.
� Aspirado de MO. Los estudios de MO incluirán: morfología, citometría de flujo
(con análisis del ciclo celular), biología molecular y citogenética. Estos últimos
estudios se realizarán de forma centralizada en laboratorios de referencia
(Anexo 8).
Procedimiento de Randomización para el periodo de t ratamiento
Los pacientes que cumplan todos los criterios, previamente a la realización de la visita Basal
(día 1 del ciclo 1) serán randomizados en una proporción 1:1 para recibir unos de los dos
esquemas de quimioterapia de inducción propuestos. La randomización se realizará
mediante el CRD electrónico. El estudio es abierto por lo que tanto el investigador, el
personal del centro así como el propio paciente conocerán la rama de tratamiento asignada.
Los pacientes randomizados al Grupo B de tratamiento serán de nuevo randomizados,
previamente a la visita Basal, en una proporción 1:1 para iniciar el tratamiento con el
esquema MPV o Rd. Se le asignará un número que será usado para identificar al paciente
en el CRD. En este momento se podrá iniciar el tratamiento de inducción.
Procedimientos generales del Periodo de tratamiento
Durante el periodo de tratamiento, independientemente de la rama asignada, el día 1 de
cada nuevo ciclo (± 4 días), se recogerán las siguientes muestras de laboratorio y se
realizarán las siguientes evaluaciones antes de la administración de la quimioterapia
correspondiente:

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 24
Tercera versión: 30 de marzo de 2011
� Exploración física dirigida en función de la sintomatología.
� Estado general mediante el índice de ECOG.
� Extracción de muestras de sangre para análisis: Hematología y Bioquímica.
� Extracción de muestras de sangre para cuantificación de inmunoglobulinas, y
componente monoclonal. La inmunofijación sólo es necesaria si la electroforesis es
negativa.
� Orina de 24 horas para proteinuria de Bence Jones, siendo necesaria la
inmunofijación si la electroforesis es negativa.
� Recogida de acontecimientos adversos relacionados con la medicación de estudio
y de todos los acontecimientos adversos graves.
� Documentación de la medicación concomitante y tratamientos de soporte.
� El asesoramiento acerca de las precauciones contra el embarazo y los posibles
riesgos de exposición fetal se deben realizar como mínimo cada 28 días. Durante
el asesoramiento, se debe recordar a los pacientes que no compartan el fármaco
en estudio y no donen sangre.
� En el caso de Lenalidomida, no se administrará más de un suministro de 28 días
del fármaco en estudio cada vez.
En caso de detectar Segundas Neoplasias Primarias d e tipo hematológico, se llevará a cabo la recogida de muestras biológicas y se remiti rán al centro de referencia del Hospital de Salamanca para su análisis centralizado y el posterior envío de los resultados a Celgene, y, en el caso de Segundas Neo plasias Primarias no hematológicas se llevará a cabo una recogida estric ta de todos los datos del paciente que pudieran ser de interés para una evaluación det allada.
Evaluaciones particulares pueden ser repetidas más frecuentemente si están clínicamente
indicadas para el manejo del paciente.
El día 1 del ciclo 1 no hará falta repetir la extracción de muestras de sangre para bioquímica,
cuantificación de inmunoglobulinas y componente monoclonal, ni la obtención de orina de 24
horas para proteinuria, componente monoclonal, siempre que estas pruebas se hayan
realizado para la selección, como máximo, una semana antes.
Durante el Periodo de Tratamiento, a todos aquellos pacientes en los que se objetive una
desaparición del componente monoclonal (CM), tanto en la electroforesis como en la
inmunofijación será necesario realizar, en el momento en que se detecte dicha desaparición,
los siguientes procedimientos:

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 25
Tercera versión: 30 de marzo de 2011
• Aspirado de MO, incluyendo estudios de morfología, citometría de flujo y biología
molecular.
• Cadenas ligeras libres en suero (sFLC).
Además, durante el Periodo de tratamiento se realizará un aspirado de MO, que incluirá
estudios de morfología, citometría de flujo y biología molecular en los siguientes momentos:
• Tras el ciclo 9 de tratamiento, antes de iniciar el ciclo 10.
• Al finalizar los 18 ciclos de tratamiento, en la visita final de tratamiento.
Procedimientos generales a todas las ramas al final izar el último ciclo de tratamiento
Se recogerán las siguientes muestras de laboratorio y se realizarán las siguientes
evaluaciones cuatro semanas aproximadamente después (±1 semana) de terminar el último
ciclo de tratamiento, siempre teniendo en cuenta que los efectos adversos deben ser
recogidos hasta un mes posterior a la última dosis de medicación de ensayo ,
independientemente de la rama asignada, para la evaluación global de la respuesta al final
del tratamiento:
� Examen físico en función de la sintomatología.
� Estado general mediante el índice de ECOG.
� Extracción de muestras de sangre para análisis: Hematología y Bioquímica.
� Extracción de muestras de sangre para cuantificación de inmunoglobulinas,
componente monoclonal e inmunofijación si hubiese desaparecido el componente
monoclonal.
� Orina de 24 horas para proteinuria, estudio de componente monoclonal e
inmunofijación si hubiese desaparecido el componente monoclonal.
� Cadenas ligeras libres en suero (sFLC) en el caso de pacientes con MM
oligosecretor/no secretor o si el paciente ha alcanzado RC con desaparición del
CM por electroforesis e inmunofijación.
� Serie ósea no obligada a no ser que existan datos clínicos que sugieran incremento
o aparición de nuevas lesiones óseas.
� Aspirado de MO. Los estudios de MO incluirán: morfología, citometría de flujo y
biología molecular. Estos últimos estudios se realizarán de forma centralizada en
laboratorios de referencia (Anexo 8).

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 26
Tercera versión: 30 de marzo de 2011
� En caso de MM con plasmocitomas extramedulares para documentar la respuesta,
se utilizará si es posible, la misma técnica que se empleó en el momento del
diagnóstico (scanner, resonancia magnética,...)
Procedimientos especiales de la Visita Fin de Trata miento
La visita Fin de Tratamiento, en los pacientes que hayan completado todo el Periodo de
tratamiento, coincide con la visita anteriormente indicada, y se realizarán los mismos
procedimientos.
Los pacientes que finalicen prematuramente el Periodo de Tratamiento deberán realizar esta
Visita Fin de Tratamiento aproximadamente cuatro semanas después del último ciclo de
tratamiento (±1 semana), siempre teniendo en cuenta que los efectos adversos deben ser
recogidos hasta un mes posterior a la última dosis de medicación de ensayo . Se
recogerán también las muestras de laboratorio y evaluaciones anteriormente indicadas,
siendo necesario recoger además:
� Motivo de interrupción prematura del Periodo de Tratamiento.
En caso de que el tratamiento haya sido suspendido prematuramente por toxicidad a
algunas de las medicaciones y se haya solicitado una excepción para que el paciente
continúe sin medicación más de un mes, se continuarán haciendo controles mensuales para
comprobar si se puede reintroducir la medicación. Si, finalmente, el paciente es
discontinuado sin volver a recibir medicación de ensayo se considerará como visita Final
aquella que se haya realizado aproximadamente cuatro semanas después del último ciclo
de tratamiento (±1 semana). Se recogerá también la fecha en la que se toma la decisión de
discontinuación y la razón de la misma.
Procedimientos durante el seguimiento
Fuera de protocolo, después de realizar la Visita Final del mismo, los pacientes deberán
realizar visitas de seguimiento cada 2 meses en las que se recogerán:
� Nueva línea de tratamiento, en caso que el motivo de interrupción haya sido
recaída o progresión bajo el Tratamiento propuesto.
� Fecha de la visita.
� Respuesta alcanzada con las nuevas líneas de tratamiento.
� Documentación del estado de supervivencia y la progresión.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 27
Tercera versión: 30 de marzo de 2011
5. POBLACIÓN OBJETO DEL ESTUDIO
6.1. Criterios de Inclusión
Todos los pacientes deben cumplir los siguientes criterios de inclusión:
• El paciente debe, en opinión del investigador, ser capaz de cumplir con todos los
requerimientos del ensayo clínico.
• El paciente debe firmar voluntariamente el consentimiento informado antes de la
realización de cualquier prueba del ensayo que no forme parte de la atención
habitual de los pacientes, con el conocimiento por parte del paciente de que puede
abandonar el ensayo en el momento que quiera, sin que se vea perjudicado en
ningún momento su atención posterior.
• Edad ˃ 65 años.
• El paciente debe estar diagnosticado de Mieloma Múltiple sintomático según los
criterios establecidos y no puede haber recibido previamente ningún tratamiento para
su enfermedad (ver Anexo 6). Está permitida la administración de pulsos de
esteroides por alguna urgencia que lo requiera previo a iniciar el tratamiento de
inducción o la administración de bisfofonatos.
• El paciente debe tener enfermedad medible, definida como sigue:
� Para Mieloma Múltiple secretor, la enfermedad medible es definida por la
presencia de componente monoclonal cuantificable en suero, ≥ 1g/dL o, en orina
si la excreción de cadenas ligeras es superior o igual a 200 mg/24 horas.
� Para Mieloma Múltiple oligosecretor o no secretor, nivel en suero de la cadena
ligera libre afectada ≥ 10 mg/dL (≥ 100 mg/L, con una ratio de cadenas ligeras
libres en suero anormal)
• El paciente debe tener un estado general medido mediante la escala de ECOG ≤ 2
(ver Anexo 4).
• El paciente debe tener una esperanza de vida superior a 3 meses.
• El paciente debe tener los siguientes valores de laboratorio previamente al inicio del
tratamiento de inducción correspondiente:
1. Recuento de plaquetas ≥ 50000/mm3, hemoglobina ≥ 8 g/dl y recuento absoluto
de neutrófilos ≥ 1000/mm3. Valores más bajos están permitidos si son debidos a
infiltración de la MO.
2. Calcio ≤14mg/dl o calcio sérico corregido ≤14mg/dl en pacientes con la albúmina
fuera de rango
3. Aspartato transaminasa (AST): ≤ 2.5 x el límite superior de lo normal.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 28
Tercera versión: 30 de marzo de 2011
4. Alanina transaminasa (ALT): ≤ 2.5 x el límite superior de lo normal.
5. Bilirrubina total: ≤1.5 x el límite superior de lo normal.
6. Creatinina sérica ≤ 2 mg/dl.
• Todo paciente varón deberá:
� Comprometerse a utilizar preservativos durante todo el tratamiento con el
fármaco en estudio, incluidos todos los períodos de interrupción de dosis y hasta
una semana después de finalizar el tratamiento si su pareja es una mujer con
capacidad de gestación y no utiliza métodos anticonceptivos.
� Comprometerse a no donar semen durante el tratamiento con el fármaco en
estudio y hasta una semana después de finalizar el tratamiento.
• Todos los pacientes deberán:
� Abstenerse de donar sangre mientras reciban el tratamiento con el fármaco en
estudio y durante la semana siguiente a la finalización del mismo.
� Abstenerse de compartir el fármaco en estudio con otras personas y devolver
todo el fármaco en estudio no utilizado al investigador o al farmacéutico.
6.2. Criterios de exclusión
Los pacientes que presenten alguno de los siguientes criterios de exclusión no podrán ser
incluidos en el ensayo clínico:
• Pacientes que hayan recibido previamente tratamiento para el Mieloma Múltiple, con
la excepción de pulsos de esteroides por alguna urgencia que lo requiera previo a
iniciar el tratamiento de inducción, la administración de bisfofonatos o radioterapia
bien antiálgica o debido a la presencia de plasmocitomas que la requieran por alguna
urgencia.
• Pacientes con enfermedad no medible ni por sFLC.
• Pacientes que tengan una neuropatía periférica ≥ Grado 2 dentro de los 14 días
previos a su inclusión.
• Pacientes con hipersensibilidad conocida al bortezomib, ácido bórico o manitol.
• Pacientes con hipersensibilidad conocida a la lenalidomida.
• Pacientes que hayan recibido cualquier agente en investigación en los 30 días
previos a su inclusión.
• Paciente que sea conocido portador del virus de la inmunodeficiencia humana (VIH),
antígeno de superficie del virus de la hepatitis B o infección activa por el virus de la
hepatitis C (aquellos en los que es posible detectar RNA del virus de hepatitis C).

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 29
Tercera versión: 30 de marzo de 2011
• Pacientes que hayan tenido un infarto de miocardio en los 6 meses previos a la
inclusión en el ensayo clínico o posea una clase funcional III o IV de acuerdo con
New York Heart Association (NYHA) , fallo cardiaco, angina no controlada, arritmias
ventriculares no controladas o isquemia aguda detectada electrocardiográficamente
o anormalidades del sistema de conducción.
• Pacientes que estén actualmente en otro ensayo clínico o recibiendo cualquier
agente en investigación.
• Paciente con enfermedad pulmonar infiltrativa difusa aguda y/o enfermedad
pericárdica.
• Historia previa de otras enfermedades malignas diferentes al mieloma (excepto para
carcinoma basal o escamoso de la piel, o carcinoma in situ de cérvix o mama) a
menos que el paciente se encuentre libre de enfermedad más allá de 5 años.
• Hipertensión arterial o diabetes mellitus mal controladas o cualquier otra enfermedad
orgánica grave que suponga un riesgo excesivo para el paciente o cualquier
alteración psiquiátrica que interfira con la comprensión del consentimiento informado.
• Pacientes que no sean capaces o no estén dispuestos a recibir tratamiento de
prevención antitrombótica.
6.3. Número de sujetos previstos
Un total de hasta 240 pacientes ˃ 65 años con Mieloma Múltiple sintomático de nuevo
diagnóstico y que no hayan recibido previamente tratamiento alguno para su enfermedad
serán incluidos en el ensayo. Serán incluidos 120 pacientes en cada rama de tratamiento.
El ensayo es competitivo, no se prevé la inclusión de un número determinado de pacientes
por centro.
6.4. Retirada de los pacientes del tratamiento o de l ensayo
Los pacientes serán retirados del estudio cuando cumplan alguno de los siguientes
criterios:
� Progresión confirmada de la enfermedad. Los pacientes asignados al Grupo A de
tratamiento y que recaigan o progresen durante el tratamiento con MPV pasarán a
recibir tratamiento con Rd, previa aprobación del promotor del estudio.
� Desarrollo de algún efecto adverso inaceptable.
Los pacientes serán informados que pueden abandonar el ensayo clínico en el momento
que ellos quieran sin que ello conlleve algún perjuicio para su atención médica posterior. El

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 30
Tercera versión: 30 de marzo de 2011
investigador también podrá, a su criterio, suspender el tratamiento en pacientes en que
aparezca alguna de las siguientes razones:
� Negación a recibir el tratamiento.
� Enfermedad concomitante.
� Violaciones mayores al protocolo.
� Requerimiento del paciente.
� No cumplimiento con las visitas establecidas.
� Razones administrativas.
� Cambios específicos o generales en el paciente que hace que, a criterio del
investigador, no deban recibir más medicación.
Los pacientes que abandonen el ensayo por cualquier motivo, no podrán volver a ser
incluidos.
En el momento de abandonar el ensayo, la razón principal para la retirada del ensayo debe
ser recogida y, si es posible, el paciente deberá ser reevaluado.
6.5. Duración del tratamiento de los pacientes
Los pacientes incluidos en el estudio recibirán tratamiento, en cualquiera de las ramas
asignadas, durante 74 semanas: 38 semanas con MPV y 36 con Rd.
6. DESCRIPCIÓN DEL TRATAMIENTO DEL ENSAYO
7.1. Materiales del ensayo clínico
Melfalán para administración oral viene presentado en tabletas de 2 mg.
Prednisona para administración oral viene presentada en tabletas de 30, 10, 5 y 2,5 mg
para ajustar la dosis correcta a administrar.
Velcade (Bortezomib) para inyectables es un polvo liofilizado estéril para reconstituirse y
se suministra en viales que contienen VELCADE y manitol en una proporción de 1:10. Por
ejemplo, viales que contienen 3,5 mg de VELCADE contienen 35 mg de manitol. Serán
suministrados, en nombre del promotor, por J&JPR&D a través de Fisher. Janssen-Cilag
suministrará al promotor Velcade ® de forma gratuita para la conducción del ensayo clínico.
Lenalidomida para administración oral viene presentado en cápsulas de 25 mg, 15 mg, 10

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 31
Tercera versión: 30 de marzo de 2011
mg y 5 mg. Será suministrado, en nombre del promotor, por Fisher Clinical Services UK Ltd
a través de OFSA. Celgene suministrará al promotor lenalidomida de forma gratuita para la
conducción del ensayo clínico.
Dexametasona para administración oral, presentado en ampollas de 40 mg.
Los fármacos Melfalán; Dexametasona y Prednisona no serán suministrados por el
promotor por ser fármacos de uso habitual en el tratamiento del MM y formar parte de la
terapia farmacológica de base que es obligada para el tratamiento de este tipo de
pacientes. Se utilizarán las presentaciones comerciales que cada centro utilice
habitualmente.
7.2. Preparación, cuidados, y almacenamiento de las drogas del ensayo
El Melfalán, la Prednisona y la Dexametasona se conservarán en las condiciones
habituales de mantenimiento de cada hospital.
Velcade : Los viales contienen Velcade liofilizado para inyección y debe ser almacenado a
temperatura ambiente. Los datos de estabilidad indican que el producto liofilizado es
estable durante al menos 12 meses si es almacenado bajo las condiciones recomendadas.
No obstante, se están llevando a cabo estudios de estabilidad y J&JPR&D notificará al
investigador si esta información precisase ser revisada a lo largo del ensayo.
Velcade es un anti-neoplásico citotóxico y, al igual que con otros compuestos
potencialmente tóxicos, se deberá tener precaución al manejar y preparar la solución de
Velcade. El farmacéutico deberá preparar Velcade utilizando una vitrina (campana)
biológica de flujo laminar vertical y una técnica aséptica adecuada. Se recomienda llevar
guantes y ropa protectora durante la preparación de Velcade.
Velcade debe ser administrado en viales de un único uso, conteniendo 3.5 mg de
Velcade. Cada vial de Velcade para inyección debe ser reconstituido en una campana de
flujo laminar dentro de las 8 horas anteriores a su administración con 3.5 ml de suero salino
(0.9%), de tal manera que la solución reconstituida contenga Velcade a una concentración
de 1 mg/ml. La disolución es completa en aproximadamente 10 segundos. La solución
reconstituida es clara, con un pH final de 5 a 6. Velcade reconstituido debe ser
administrado cuanto antes y en ningún caso deben transcurrir más de 8 horas desde su
reconstitución. Si la solución de Velcade entra en contacto con la piel, límpiese
inmediatamente la piel con jabón, agua y peróxido de hidrógeno diluido. Si la solución de
Velcade entra en contacto con las membranas mucosas, irrigue bien con agua. Siempre
se debe contactar con un médico después del contacto con cualquier parte del cuerpo.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 32
Tercera versión: 30 de marzo de 2011
Todos los materiales usados durante la preparación deben ser eliminados de acuerdos a
los estándares establecidos.
Lenalidomida: Las cápsulas de Lenalidomida deberán ser almacenadas bien cerradas y
protegidas de la luz, y según se indica en la etiqueta.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 33
Tercera versión: 30 de marzo de 2011
7.3. Administración de las drogas de estudio y esqu ema de dosis
Administración y esquemas de tratamiento
Los pacientes candidatos serán incluidos en el estudio bajo la supervisión del investigador
o de los sub-investigadores identificados. Si es posible, los pacientes recibirán el
tratamiento de forma ambulatoria. Todos los fármacos del ensayo serán administrados a
los pacientes bajo la prescripción del investigador o sub-investigadores identificados y
dispensados en el Servicio de Farmacia del hospital.
De acuerdo a la primera randomización tras la visita de selección, los pacientes recibirán
MPV seguido de Rd (Grupo A de tratamiento) o MPV alternando con Rd (Grupo B de
tratamiento). En el grupo B de tratamiento los pacientes serán randomizados en proporción
1:1 a iniciar el tratamiento con MPV o Rd. En todos los grupos recibirán tratamiento durante
un máximo de 74 semanas. En la Tabla 1 aparece el esquema general de tratamiento de
los ciclos MPV y Rd.
Tabla 1: Esquemas de tratamiento MPV y Rd para los grupos A, B1 y B2 de tratamiento
1er ciclo MPV de 6 semanas Días
1 2 3 4 8 11 12-21 22 25 29 32 33-42
Melfalán 9mg/m 2 ���� ���� ���� ����
Prednisona 60 mg/m 2 ���� ���� ���� ���� Velcade 1,3 mg/m 2 ���� ���� ���� ���� ���� ���� ���� ����
2, 3, 4, 5, 6, 7, 8 y 9º ciclo MPV de 4 semanas D ías
1 2 3 4 8 15 22 23-28
Melfalán 9mg/m 2 ���� ���� ���� ����
Prednisona 60 mg/m 2 ���� ���� ���� ���� Velcade 1,3 mg/m 2 ���� ���� ���� ����
Esquema Rd
Dexametasona, 40 mg/día
Periodo descanso
Periodo descanso
Periodo descanso
1 8 15 22

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 34
Tercera versión: 30 de marzo de 2011
Lenalidomida, 25 mg/día
PERIODO DE TRATAMIENTO
Grupo A de tratamiento: Melfalán/Prenisona/Velcade seguido de
Lenalidomida/dexametasona
Los pacientes asignados al Grupo A recibirán un ciclo de 6 semanas de duración de
Melfalán/Prednisona/Velcade consistente en Melfalán , 9 mg/m2 vía oral los días 1 a 4 y
Prednisona , 60 mg/m2 vía oral los días 1 a 4, en combinación con Velcade , a dosis de
1,3 mg/ m2 vía iv en bolo dos veces por semana (días 1, 4, 8, 11, 22, 25, 29 y 32), seguido
de un periodo de descanso de 10 días (días 33 al 42); y ocho ciclos de cuatro semanas de
duración de Melfalán/Prednisona/Velcade consistente en Melfalán , 9 mg/m2 vía oral los
días 1 a 4 y Prednisona , 60 mg/m2 vía oral los días 1 a 4, en combinación con Velcade , a
dosis de 1,3 mg/ m2 vía iv semanalmente (días 1, 8, 15 y 22) seguido de un periodo de
descanso de 6 días (días 23 al 28). Posteriormente recibirán nueve ciclos de 4 semanas de
duración de Lenalidomida/dexametasona consistente en Lenalidomida , 25 mg vía oral los
días 1 a 21 seguido de un periodo de descanso de 7 días (días 22 a 28), en combinación
con dexametasona , 40 mg vía oral los días 1, 8, 15 y 22, seguido de un periodo de
descanso de 6 días (días 23 a 28). En la tabla 2 se recoge un esquema del Grupo A de
tratamiento.
Melfalán y Prednisona serán dispensados para su administración vía oral. El Melfalán será
tomado en ayunas y la Prednisona será tomada por la mañana con o inmediatamente
después de la comida. La cantidad en mg será calculada en función de la superficie
corporal, que será calculada el día 1 de cada ciclo. Para la administración de Velcade, la
dosis calculada en el día 1 del ciclo será la misma a lo largo de todo el ciclo. Si un paciente
experimenta una ganancia o pérdida de peso muy notable dentro del ciclo, la dosis a
administrar será recalculada en función de la nueva superficie corporal.
La cantidad apropiada de Velcade será dispensada en una inyección y será administrada
mediante un bolo IV durante un tiempo entre 3 y 5 segundos seguido de lavado de la vía
con suero fisiológico. Los viales son de administración única. Entre dos dosis de Velcade
deberán transcurrir, como mínimo, 72 horas.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 35
Tercera versión: 30 de marzo de 2011
Lenalidomida y dexametasona serán dispensadas para su administración oral. Serán
tomados por la mañana, con o inmediatamente después de la comida. La cantidad es fija y
no deberá ajustarse por superficie corporal.
Tabla 2. Esquema de tratamiento grupo A: Esquema se cuencial MPV-Rd.
Grupo B de tratamiento: Melfalán/Prenisona/Velcade alternando con
Lenalidomida/dexametasona
Los pacientes asignados al Grupo B de tratamiento recibirán ciclos MPV alternando con Rd,
en los mismos esquemas y dosis antes indicados para el Grupo A. El primer ciclo MPV
tendrá una duración de 6 semanas, mientras que los ocho siguientes tendrán una duración
de cuatro semanas. Los ciclos Rd tendrán una duración de cuatro semanas.
Dentro de este grupo B de tratamiento, los pacientes serán de nuevo randomizados a iniciar
el tratamiento con el ciclo MPV o Rd.
En la tabla 3 aparece un esquema resumido del grupo B de tratamiento, con las dos
posibilidades: B1 y B2.
Tabla 3. Esquema de tratamiento grupo B1 y B2: Esqu ema alternante MPV-Rd
MPV Rd
1º 2 º 3º 4º 5º 6º 7º 8º 9º 1º 2º 3º 4 º 5º 6 º 7º 8º 9º
MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd
Grupo B1 de tratamiento alternante: MPV alternando con Rd
Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV Rd MPV
Grupo B2 de tratamiento alternante: Rd alternando con MPV

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 36
Tercera versión: 30 de marzo de 2011
7.4. Modificación de dosis y retrasos
PERIODO DE TRATAMIENTO
Todos los pacientes serán evaluados en el día 1 de cada nuevo ciclo (± 4 días), no siendo
necesarias las visitas intermedias, salvo cualquier situación clínica que lo requiera.
Como regla general, si no se observase recuperación de la toxicidad hematológica o no
hematológica en 4 semanas, el paciente deberá abandonar el ensayo clínico. En caso de
claro beneficio clínico del tratamiento para el paciente, se consultará con los coordinadores
del estudio la permanencia del paciente en el ensayo.
• Durante la administración de Melfalán/Prednisona/Velcade ® los pacientes serán
evaluados en el día 1 (± 4 días) de cada ciclo acerca de todas las posibles toxicidades que
hayan podido presentarse desde el ciclo anterior. Las toxicidades serán manejadas de
acuerdo a los Criterios de Toxicidad Común del NCI (CTC), versión 3.0.
http://ctep.cancer.gov/reporting/ctc.html.
� MODIFICACIONES POR TOXICIDAD HEMATOLÓGICA DE MELFAL ÁN
En el día 1 de la administración de los ciclos Melfalán/Prednisona/Velcade se requerirán
al menos 8 g/dl de Hemoglobina, 1000 granulocitos/mm3 y 50000 plaquetas/mm3 en el
hemograma para poder iniciar el ciclo. Si no los presentara se podrá retrasar el inicio del
ciclo siguiente hasta 4 semanas.
Si el día 1 de la administración de los ciclos Melfalán/Prednisona/Velcade se observa
que durante el ciclo anterior el paciente ha tenido neutropenia grado 3 (< 1000/mm3) que
se haya mantenido durante una semana o trombopenia aislada grado 4 (< 25000/mm3),
será atribuido al Melfalán administrado en el ciclo anterior y la dosis de Melfalán se
reducirá un 25%. Si la toxicidad persiste se podrá aplicar una reducción adicional del
25% (supondría un 50% de la dosis inicial).
� MODIFICACIONES POR TOXICIDAD NO HEMATOLÓGICA DE MEL FALÁN
Si el paciente experimenta cualquier toxicidad no hematológica grado ≥3 considerada
por el investigador en relación con el melfalán/prednisona deberán ser suspendidas
la/las medicación/es con las que se considere relacionada la toxicidad hasta que ésta
vuelva a ser grado 1 o niveles basales.
Si se observa insuficiencia renal (creatinina > 2 mg/dl), la dosis de melfalán en el
siguiente ciclo se reducirá al 50%.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 37
Tercera versión: 30 de marzo de 2011
� MODIFICACIONES POR TOXICIDAD HEMATOLÓGICA DE VELCAD E
Como se ha indicado previamente, los valores requeridos para la administración de
Velcade en el día 1 de cada nuevo ciclo son: hemoglobina ≥8g/dl, granulocitos >
≥1000/mm3 y plaquetas ≥50000/mm3. Si el paciente no presenta estos valores, se podrá
esperar hasta 4 semanas para recuperación de la toxicidad.
En cualquiera de los días de administración de Velcade distinto del día 1 de cada ciclo,
los valores hematológicos mínimos requeridos para su administración son: hemoglobina
≥8 g/dl, granulocitos ≥750/mm3 y plaquetas ≥30000/mm3. Cuando una dosis de Velcade
no se administra por toxicidad hematológica, esa dosis se pierde y no es recuperada.
Cuando se reanude el tratamiento, se reanudará en el día correspondiente y en el CRD
quedará reseñada su no administración por toxicidad hematológica.
Si durante el primer ciclo de tratamiento, tres o más dosis de Velcade de las ocho
programadas o, durante los siguientes ciclos, dos o más de las cuatro dosis debieran ser
suspendidas por toxicidad hematológica, la dosis de Velcade será reducida de 1.3 mg/m2
a 1.0 mg/m2 o de 1.0 mg/m2 a 0.7 mg/m2. No pudiendo incrementarse posteriormente.
� MODIFICACIONES POR TOXICIDAD NO HEMATOLOGICA DE VEL CADE
Si el paciente experimenta cualquier toxicidad no hematológica grado ≥3 consideradas
por el investigador en relación con Velcade, entonces debe ser suspendido hasta que la
toxicidad vuelva a ser grado 1 o niveles basales. Posteriormente la dosis de Velcade
deberá ser reducida un nivel.
Los pacientes que desarrollen dolor neuropático y/o neuropatía periférica sensitiva
relacionada con Velcade, serán manejados de acuerdo a la siguiente tabla:
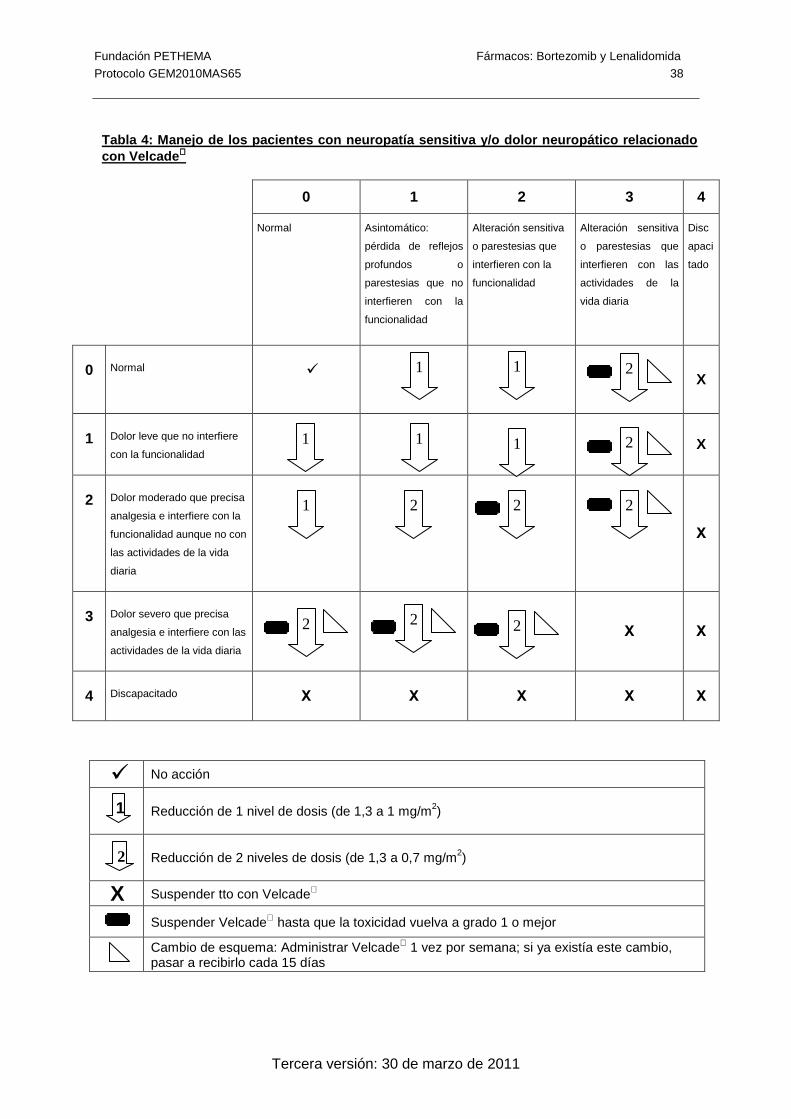
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 38
Tercera versión: 30 de marzo de 2011
Tabla 4: Manejo de los pacientes con neuropatía sen sitiva y/o dolor neuropático relacionado con Velcade
0 1 2 3 4
Normal Asintomático:
pérdida de reflejos
profundos o
parestesias que no
interfieren con la
funcionalidad
Alteración sensitiva
o parestesias que
interfieren con la
funcionalidad
Alteración sensitiva
o parestesias que
interfieren con las
actividades de la
vida diaria
Disc
apaci
tado
0 Normal � X
1 Dolor leve que no interfiere
con la funcionalidad X
2 Dolor moderado que precisa
analgesia e interfiere con la
funcionalidad aunque no con
las actividades de la vida
diaria
Х
3 Dolor severo que precisa
analgesia e interfiere con las
actividades de la vida diaria
Х Х
4 Discapacitado Х Х Х Х Х
� No acción
Reducción de 1 nivel de dosis (de 1,3 a 1 mg/m2)
Reducción de 2 niveles de dosis (de 1,3 a 0,7 mg/m2)
X Suspender tto con Velcade
Suspender Velcade hasta que la toxicidad vuelva a grado 1 o mejor
Cambio de esquema: Administrar Velcade 1 vez por semana; si ya existía este cambio, pasar a recibirlo cada 15 días
1 2
1
2
2
2
2
2
2 1
2
1
1 1
1
2

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 39
Tercera versión: 30 de marzo de 2011
• Durante la administración de Lenalidomida/dexametasona, los pacientes serán
evaluados en el día 1 de cada ciclo acerca de todas las posibles toxicidades que hayan
podido presentarse desde el ciclo anterior. Las toxicidades serán manejadas de acuerdo a
los Criterios de Toxicidad Común del NCI (CTC), versión 3.0.
� MODIFICACIONES POR TOXICIDAD HEMATOLÓGICA DE LENALI DOMIDA
En el día 1 de cada nuevo ciclo de Lenalidomida/dexametasona los valores
hematológicos mínimos requeridos para su administración son: Hemoglobina ≥ 8 g/dl,
granulocitos ≥ 1000/mm3 y plaquetas ≥ 50000/mm3. Si el paciente no presentara estos
valores el ciclo podrá ser retrasado hasta 4 semanas para que la toxicidad revierta,
siendo necesario hacer recuentos semanales. En el momento de reiniciar el tratamiento
tras la recuperación de cifras deberá aplicarse un nivel de reducción de dosis tal y como
se indican en la tabla 5; si la neutropenia fuera la única toxicidad presente que requiera
una reducción de dosis, la primera reducción de dosis será el nivel de dosis -1, seguido
por los niveles -2, -3 y -4, que aparecen en la tabla 5.
Tabla 5.- Reducción de dosis de lenalidomida
Dosis inicial 25 mg durante 21 días cada 28 días
Nivel de dosis -1 (sólo aplicable para
neutropenia aislada)
25 mg durante 21 días cada 28 días más G-CSF
Nivel de dosis -2 15 mg diarios durante 21 días cada 28 días
Nivel de dosis -3 10 mg diarios durante 21 días cada 28 días
Nivel de dosis -4 5 mg diarios durante 21 días cada 28 días
� MODIFICACIONES POR TOXICIDAD NO HEMATOLÓGICA DE LEN ALIDOMIDA
Si el paciente desarrolla cualquier toxicidad no hematológica G3-4 relacionada con
lenalidomida, ésta deberá ser suspendida hasta que la toxicidad revierta a ≤G2 y en ese
momento reiniciar aplicando un nivel de reducción de dosis, de acuerdo a la tabla 5,
teniendo en cuenta que el nivel de reducción -1 es sólo para la neutropenia.
Si la toxicidad no se resuelve en un plazo de 4 semanas será necesario contactar con el
monitor del estudio para valorar continuar en el estudio.
Los pacientes que no puedan tolerar el nivel de dosis -4 interrumpirán el estudio.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 40
Tercera versión: 30 de marzo de 2011
� MODIFICACIONES POR TOXICIDAD DE DEXAMETASONA
Si el paciente presenta cualquier toxicidad G3-4 relacionada con la dexametasona será
necesario interrumpir el tratamiento hasta que la toxicidad revierta a un grado ≤2 y
reiniciar el tratamiento modificando la dosis, de acuerdo a la tabla 6.
Tabla 6.- Reducción de dosis de Dexametasona
Dosis inicial 40 mg vía oral los días 1, 8, 15 y 22
Nivel de dosis -1 20 mg vía oral los días 1, 8, 15 y 22 de cada ciclo
Nivel de dosis -2 10 mg vía oral los días 1, 8, 15 y 22 de cada ciclo
Nivel de dosis -3 5 mg vía oral los días 1, 8, 15 y 22 de cada ciclo
Si el paciente no pudiera tolerar el nivel de reducción de dosis -3 de dexametasona,
entonces se discontinuará la dexametasona y el paciente interrumpirá la fase de
tratamiento del estudio.
� MODIFICACIONES POR TOXICIDAD DE PREDNISONA
Si el paciente presenta cualquier toxicidad G3-4 relacionada con la prednisona se podrá
disminuir al 50% la dosis de la misma. Si continúa la toxicidad, se podrá suspender la
prednisona y continuar con bortezomib y melfalán.
7.5. Envasado y etiquetado
Todos los fármacos del estudio se administrarán de forma descubierta, no se empleará
ningún método de enmascaramiento en este estudio.
El Velcade y la Lenalidomida serán suministrados a las farmacias envasados y etiquetados
según los requerimientos habituales para ensayos clínicos.
7.6. Tratamiento concomitante
Los tratamientos de soporte para el Mieloma Múltiple que estuviesen utilizándose en la
visita basal serán permitidos durante la fase de tratamiento del ensayo, siguiendo las
recomendaciones publicadas.
No está permitido ningún agente antineoplásico para el mieloma múltiple.
• Es altamente recomendable que todos los pacientes reciban bisfofonatos
mensualmente durante el periodo de tratamiento, a partir del 3er mes;
posteriormente, a criterio de cada centro.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 41
Tercera versión: 30 de marzo de 2011
• Todos los pacientes en ambos grupos de tratamiento recibirán profilaxis antibiótica
con levofloxacino, a dosis de 500 mg/ día durante los cuatro primeros ciclos de
tratamiento.
• Durante el tratamiento con bortezomib es obligado realizar profilaxis de la
reactivación de la infección por el virus Varicela Zoster. Se hará con aciclovir, a dosis
de 400 mg/12 horas mientras dure el tratatamiento. Alternativamente se podría
utilizar valaciclovir, a dosis de 500 mg tres veces al día o famciclovir, a dosis de 500
mg tres veces al día. En el esquema secuencial se administrará la profilaxis sólo
durante los nueve ciclos de VMP y en el alternante debería usarse sólo los meses en
que el paciente recibe VMP. Con Rd no es necesario.
• Durante todo el periodo de Tratamiento es altamente recomendable el uso
profiláctico de G-CSF para asegurar la administración del tratamiento en los días
correspondientes a cada ciclo y evitar demoras y discontinuaciones.
• En el grupo A de tratamiento, durante el periodo en que reciben tratamiento con
lenalidomida y dexametasona deberán recibir profilaxis antitrombótica con ácido
acetilsalicílico, heparina de bajo peso molecular o anticoagulación oral, en función de
las características de cada paciente. En el grupo B de tratamiento, durante los meses
en que reciben tratamiento con lenalidomida y dexametasona deberán también
recibir profilaxis antitrombótica (incluso durante la semana de descanso), a elegir por
el investigador y en función de los factores de riesgo que presente cada paciente. No
es necesaria la profilaxis antitrombótica durante el tratamiento con MPV, en esquema
secuencial o alternante. En la tabla 7 aparecen resumidas las recomendaciones para
el uso de profilaxis antitrombótica durante el tratamiento con lenalidomida18.
Tabla 7: recomendaciones para el uso de profilaxis antitrombótica
Profilaxis TVP/TEP
Si ≤≤≤≤1 factor de riesgo* Profilaxis con aspirina
Si≥2 factores de riesgo HBPM (a dosis profilácticas**) al menos los primeros 4-6 meses***
* Si dexametasona a dosis altas, doxorrubicina o poliquimioterapia: utilizar HBPM independientemente de la existencia o no de más factores de riesgo.
** Enoxaparina 40 mg / Deltaparina 5000 U. *** Considerar más tiempo si factores de riesgo. Si los factores de riesgo hubieran desparecido, se
podría cambiar a aspirina a dosis de 100 mg diarios. Si insuficiencia renal: Uso de anticoagulantes orales.
Suspender profilaxis de TVP/TEP si plaquetas <50.00 0/µµµµL
Durante la administración de profilaxis antitrombótica, se deberán tener en cuenta las
siguientes recomendaciones de acuerdo al recuento plaquetario:

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 42
Tercera versión: 30 de marzo de 2011
- Si las plaquetas descienden por debajo de 50000/mm3, la profilaxis antitrombótica que
se esté usando deberá ser suspendida, reinstaurándola cuando se observe recuperación
de la trombopenia.
7.7. Medicaciones permitidas y tratamientos de sopo rte
7.7.1. Medicaciones permitidas
• Factor estimulador de colonias granulocíticas, factor estimulador de colonias
granulocíticas y de macrófagos, eritropoyetinas, heparinas de bajo peso molecular,
ácido acetilsalicílico, aciclovir, transfusiones de hematíes y plaquetas son
permitidos, a criterio del investigador. Es altamente recomendable el uso de factor
estimulante de colonias granulocíticas para permitir la correcta administración del
tratamiento, sobre todo durante la administración de los ciclos MPV 2º a 8º que se
administrarán cada cuatro semanas.
• Los siguientes procedimientos son permitidos durante el ensayo:
Vertebroplastia, Kifoplastia, procedimientos ortopédicos de urgencia, radioterapia
(siempre que sea necesaria por otros motivos diferentes a la progresión de la
enfermedad).
7.7.2. Medicaciones prohibidas
Las siguientes medicaciones y tratamientos de soporte, así como procedimientos
están prohibidos durante el ensayo:
• Cualquier otro agente en investigación diferente a bortezomib y lenalidomida.
• Cualquier tratamiento anti-neoplásico contra el Mieloma Múltiple, diferentes al
tratamiento propuesto en el estudio.
7.8. Cumplimiento del tratamiento
Todos los fármacos del ensayo serán administrados a los pacientes bajo la prescripción del
investigador o sub-investigadores identificados. El farmacéutico guardará las peticiones de
los fármacos del estudio, así como los números de lote aplicados a cada paciente, peso del
paciente y superficie corporal, así como la cantidad total de producto dispensado, en ml y
mg. Cualquier discrepancia entre la dosis calculada y la dosis administrada y la razón de la
discrepancia debe ser recogida.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 43
Tercera versión: 30 de marzo de 2011
7. DESARROLLO DEL ENSAYO Y EVALUACIÓN DE LA RESPUES TA
8.1. Variables principales y secundarias
Las variables principales son:
� Variable principal de eficacia: supervivencia libre de progresión (SLP) con los dos
esquemas de tratamiento propuestos, entendiendo como SLP la duración en meses
desde la fecha de la inclusión en el ensayo hasta la fecha de la progresión de la
enfermedad o muerte debido a cualquier causa.
� Variables principales de seguridad: seguridad y tolerabilidad de los dos esquemas de
tratamiento propuestos, secuencial versus alternante.
Las variables secundarias son:
� Variables secundarias de eficacia: eficacia, en términos de tasa de respuestas,
duración de la respuesta, TTP, TNT y SG en ambos grupos de tratamiento.
� Variables secundarias de asociación genética: características biológicas (incluyendo
análisis de perfil de expresión génica) de aquellos pacientes resistentes a uno de los
dos regímenes propuestos o ambos.
8.2. Controles a realizar
CM en Suero
Serán recogidas a todos los pacientes durante la visita de Selección muestras de sangre
para cuantificación de inmunoglobulinas así como identificación y cuantificación del
componente monoclonal. Posteriormente, dichas muestras serán recogidas:
� Durante el periodo de tratamiento, en el día 1 (±4 días) de cada nuevo ciclo de
tratamiento. La inmunofijación sólo será necesaria si la electroforesis es negativa.
� Al finalizar el periodo de tratamiento, en la visita final, un mes después (±1 semana) del
último ciclo de tratamiento.
Después de la visita final se continuará evaluando el CM en suero cada 2 meses, hasta
que se objetive progresión de la enfermedad.
CM en Orina
Se tomarán muestras de orina en 24 horas para la identificación y cuantificación de
proteína M a todos los pacientes durante el período de Screening. Posteriormente, dichas
muestras serán recogidas:

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 44
Tercera versión: 30 de marzo de 2011
� Durante el periodo de tratamiento, en el día 1 de cada nuevo ciclo de tratamiento (± 4
días).
� Al finalizar el periodo de tratamiento.
Después de la visita final se continuará evaluando el CM en orina cada 2 meses, hasta que
se objetive progresión de la enfermedad.
Cadenas ligeras libres en suero
Se realizara determinación de cadenas ligeras libres en suero en la visita de selección a
todos aquellos pacientes con MM Bence Jones puro y MM oligosecretores o no secretores
para evaluar si tienen enfermedad medible por esta técnica. Posteriormente, dichas
muestras serán recogidas:
� Durante el periodo de tratamiento, en el día 1 de cada nuevo ciclo de tratamiento (± 4
días) y al finalizar el tratamiento, para aquellos pacientes en que sea ésta la técnica de
detección de la enfermedad medible. En este grupo de pacientes, se realizará
posteriormente cada dos meses durante el periodo de seguimiento.
� En el resto de pacientes con enfermedad medible por técnicas convencionales, sólo
será obligado en el momento de documentación de la RC con IF negativa.
Aspirado de MO
Un aspirado de MO deberá ser realizado para todos los pacientes durante la visita de
selección. Los estudios de MO incluirán: morfología, citometría de flujo (con análisis del
ciclo celular), citogenética y biología molecular. Estos se realizarán de forma centralizada
en laboratorios de referencia (ver Anexo 8). Posteriormente se realizará al finalizar el ciclo
9, antes del inicio del ciclo 10 y al finalizar el tratamiento, tras el ciclo 18.
Esta prueba será también obligada en el momento en que se objetive la RC con IF
negativa, y en cualquier momento del estudio, a criterio del investigador principal del
ensayo.
Análisis genético
Se llevarán a cabo estudios de hibridación in situ para detectar alteraciones cromósomicas
(deleciones, translocaciones) que, en la actualidad, se conoce que afectan al pronóstico del
mieloma múltiple y a la respuesta al tratamiento. También se anlizarán los marcadores
genéticos que surjan, en el futuro, relacionados con esta enfermedad y con la respuesta de
los pacientes al tratamiento de la misma.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 45
Tercera versión: 30 de marzo de 2011
Serie ósea y otras radiografías
Se realizará una serie ósea completa, mediante radiografías durante la visita de selección.
Posteriormente, sólo será necesario repetir la serie ósea si aparecen datos clínicos que
sugieran la aparición de nuevas lesiones óseas, o en cualquier momento, a criterio del
investigador.
A veces los pacientes tienen enfermedad progresiva que se manifiesta con síntomas de
dolor debido a las alteraciones óseas. Por lo tanto, se puede documentar la enfermedad
progresiva en estos casos por medio de series óseas u otras radiografías, dependiendo de
los síntomas que experimente el paciente. Si el diagnóstico de la enfermedad progresiva es
obvio en las investigaciones radiográficas, no es necesario repetir radiografías para
confirmación. En los casos en que los cambios sean más sutiles, es posible que sea
necesario repetir una radiografía en un período de 1 a 3 semanas.
En la visita de selección es importante documentar las localizaciones de la enfermedad
mielomatosa, especialmente en zonas extra-medulares. Es posible que esta
documentación requiera la realización de exámenes clínicos, tomografía computerizada o
evaluaciones de imágenes de resonancia magnética. En estos casos, para documentar la
respuesta, es necesario repetir la misma prueba realizada para su documentación y
evaluación.
B2-microglobulina y Proteina C Reactiva
Muestras de sangre para B2 microglobulina y Proteína C Reactiva deben ser realizadas
durante la visita de Selección y en cualquier momento del estudio, a criterio del
investigador.
Evaluaciones de laboratorio
Las muestras de sangre para los tests de laboratorio serán realizados durante la visita de
selección; durante el periodo de tratamiento se realizarán el día 1 de cada ciclo (± 4 días).
Estas evaluaciones serán repetidas a criterio del investigador si existe justificación clínica.
Serán evaluados los siguientes parámetros de laboratorio:
Hematología, hemoglobina, recuento de leucocitos con recuento diferencial y plaquetas.
Bioquímica: urea, creatinina, ácido úrico, bilirrubina, fosfatasa alcalina, LDH, AST, ALT,
sodio, cloro, calcio, potasio, glucosa, proteinograma e Igs.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 46
Tercera versión: 30 de marzo de 2011
Nota: En caso de detectar Segundas Neoplasias Primarias d e tipo hematológico, se llevará a cabo la recogida de muestras biológicas y se remitirán al centro de referencia del Hospital de Salamanca para su anális is centralizado y el posterior envío de los resultados a Celgene, y, en el caso de Segun das Neoplasias Primarias no hematológicas se llevará a cabo una recogida estric ta de todos los datos del paciente que pudieran ser de interés para una evaluación det allada.
Estado general mediante la escala ECOG
El estado general mediante la escala ECOG será determinado durante la visita de
selección, en el día 1 de cada nuevo ciclo durante el periodo de tratamiento (± 4 días), al
finalizar el tratamiento de inducción, y, luego, durante el periodo de seguimiento, cada dos
meses hasta progresión o retirada del estudio.
Electrocardiograma
Durante la visita de selección se debe realizar un electrocardiograma, y será repetido a lo
largo del tratamiento si existe alguna alteración o si existe algún dato clínico que, a criterio
del investigador, requiera la evaluación electrocardiográfica.
Historia Médica y Examen físico
La historia médica será obtenida durante la visita de selección. La historia médica incluirá
toda la información demográfica y resumen de toda su patología. En el momento de la
visita de selección se realizará un examen físico completo al enfermo durante todas las
visitas es obligado realizar un examen físico dirigido en función de la sintomatología
referida por el paciente.
Peso, talla y superficie corporal
Los pacientes deben ser medidos y tallados en el momento de la visita de selección, y
posteriormente siempre que se objetive algún cambio evidente en el peso que sugiera un
cambio significativo de la superficie corporal, para un adecuado ajuste de la medicacion. La
superficie corporal se calcula en función de un normograma (ver Anexo 5).
Terapia de soporte
Las terapias de soporte serán recogidas desde la visita de selección hasta la salida del
ensayo.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 47
Tercera versión: 30 de marzo de 2011
8.3. Valoración de la respuesta
El investigador realizará pruebas que permitirán la valoración de la respuesta a la terapia
según los criterios de respuesta del EBMT presentados en el Anexo 7 y los criterios del
Grupo de Trabajo Internacional de Mieloma (IMWG) para documentar la RC por citometría
de flujo y biología molecular, actualizados según el último panel de consenso.
Una valoración de la respuesta se realizará el día 1 (± 4 días) de cada nuevo ciclo de
tratamiento, así como durante el periodo de seguimiento, cada dos meses.
8. ACONTECIMIENTOS ADVERSOS
Los acontecimientos adversos relacionados con la medicación y todos los acontecimientos
adversos graves se registrarán en los documentos originales y el CRD del paciente desde la
firma del consentimiento informado hasta 30 días después de la última dosis de medicación.
Todos los acontecimientos adversos deberán seguirse hasta que remitan o hasta que se
determine definitivamente que se deben a una afección crónica o estable del paciente o
enfermedad intercurrente.
9.1. Información mínima
9.1.1. Acontecimientos Adversos (AA)
Un acontecimiento adverso (AA) es cualquier acontecimiento médico nocivo, no
intencionado o desfavorable que no necesariamente presenta relación causal con el
tratamiento de estudio, y que, a cualquier dosis, aparece o empeora durante el
transcurso de un estudio. Puede tratarse de una nueva enfermedad intercurrente, del
empeoramiento de una enfermedad concomitante, de una lesión o de cualquier
deterioro concomitante del estado de salud del paciente, incluyendo valores analíticos
(según se indica a continuación), independientemente de su etiología. Cualquier
condición médica que estuviera presente antes del tratamiento del estudio y que
permanezca inalterada o mejore no debe considerarse ni registrarse como AA. Si
hubiera un empeoramiento de esa condición médica, entonces sí debe considerarse
un AA.
La información sobre todos los acontecimientos adversos que el investigador
considere relacionados con la medicación de estudio, tanto si son comunicados por el
paciente, descubiertos por el investigador como resultado de un cuestionario general o
detectado en la exploración física, en exámenes de laboratorio o en otras

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 48
Tercera versión: 30 de marzo de 2011
exploraciones, se registrarán en el formulario de Acontecimientos Adversos del CRD y
se controlarán adecuadamente.
Sólo se recogerán en el CRD aquellos AA que el investigador considere relacionados
con la administración del tratamiento.
En la medida de lo posible, cada acontecimiento adverso también se describirá en
función de:
• Su duración (fechas de inicio y de finalización)
• El grado de intensidad (grado 1, 2, 3, 4 o 5)
• La gravedad del AA (grave o leve)
• Fármaco/s de estudio con el que se sospecha relación de causalidad.
• La(s) acción(es) tomada(s).
En el apartado 9.2 se muestran ejemplos del grado de intensidad, relación con la
medicación del estudio y acciones tomadas, tal y como deben cumplimentarse en los
CRDs.
9.1.2. Acontecimientos Adversos Graves (AAG)
La información sobre todos los acontecimientos adversos graves se recogerá y se registrará en el formulario de Acontecimientos Adversos Graves y en la página correspondiente del CRD. Para garantizar la seguridad del paciente, cada acontecimiento adverso grave también deberá notificarse a Trial Form Support (TFS) dentro de un plazo de 24 horas desde su conocimiento. Un acontecimiento adverso grave se define en general como un acontecimiento adverso (desfavorable) que:
1. Es mortal o amenaza la vida del paciente
2. Requiere o prolonga la hospitalización
3. Causa incapacidad / discapacidad permanente o significativa
4. Causa una anomalía congénita o un defecto de nacimiento
5. Constituye un acontecimiento médico importante.
Los acontecimientos médicos importantes se definen como aquellos acontecimientos
que pueden no amenazar la vida de forma inmediata o provocar la muerte, una
hospitalización o una discapacidad, pero que pueden poner en peligro al paciente,
requerir una intervención médica o quirúrgica para prevenir uno de los otros
desenlaces descritos arriba, o ser considerado clínicamente significativo. Se debe
utilizar el juicio médico y científico para decidir si un AA debe considerarse grave. Por
último, se considerarán AAG médicamente significativos, la trasmisión de un agente
infeccioso a través de la medicación.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 49
Tercera versión: 30 de marzo de 2011
Para cualquier AAG el investigador proporcionará información sobre su intensidad,
fechas de inicio y terminación, relación con el fármaco del estudio, acción llevada a
cabo con respecto al fármaco del estudio y resultado, mediante el Formulario de
Notificación de AAG
No se considerarán AAGs las hospitalizaciones que ocurran bajo las siguientes
circunstancias: estar programadas antes de incluirse al paciente en el estudio, ir
destinadas al tratamiento, ocurrir en una urgencia en régimen ambulatorio sin causar
ingreso (a menos que cumplan los criterios anteriores) o formar parte del tratamiento
normal o monitorización de la indicación estudiada y no estar asociadas con un
empeoramiento de la enfermedad.
El embarazo, no siendo en sí un acontecimiento adverso grave, también deberá
gestionarse expeditivamente igual que un AAG. Deberá registrarse en un formulario
específico de notificación de embarazo, notificarse de manera expeditiva desde que se
tenga conocimiento del acontecimiento y seguirse hasta el final, aunque ocurra una
interrupción voluntaria o espontánea del mismo, describiéndose los detalles del
nacimiento y la presencia o ausencia de algún defecto en el feto o alguna anomalía
congénita.
En el caso de aparición de Segundas Neoplasias Prim arias, se revisarán y notificarán
como acontecimientos adversos graves, independiente mente de que cumplan o no
criterios de gravedad y de su relación con la medic ación del ensayo o placebo, y que
hayan sucedido a los pacientes en cualquier momento del estudio, incluyendo:
a. Pacientes actualmente activos en el estudio b. Pacientes discontinuados por cualquier causa (in cluyendo fallecimiento) c. Pacientes en período de seguimiento, garantizan do que el seguimiento
de seguridad es de idéntica duración en ambos brazo s del estudio respecto a SNP, para que sean realmente comparables . Así, se registrarán todas las SNP ocurridas en los particip antes en el ensayo clínico, en ambos brazos de tratamiento, hasta 18 m eses después de la última administración de cualquiera de los fármacos en estudio.
Las instrucciones sobre el modo de cumplimentar y notificar los AAGs se presentan en
el apartado 9.3 “Instrucciones para la notificación rápida de AAGs”.
Reacción Adversa Grave e Inesperada (RAGI)
Reacción nociva y no intencionada a un medicamento en investigación,
independientemente de la dosis administrada. La naturaleza o gravedad de esta

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 50
Tercera versión: 30 de marzo de 2011
reacción no se corresponde con la información referente al producto (manual del
investigador en el caso de un medicamento en investigación no autorizado para su
comercialización, o la ficha técnica del producto en el caso de un medicamento
autorizado.)
9.2. Calificación de un acontecimiento adverso
El grado de intensidad de un acontecimiento adverso proporciona una evaluación
cualitativa del alcance o la intensidad de un acontecimiento adverso, determinada por el
investigador o comunicada por el paciente. El grado de intensidad no refleja la gravedad
clínica del acontecimiento, sino sólo el grado o alcance de la aflicción o incidencia (por
ejemplo, náusea severa, crisis leve) y tampoco refleja la relación con la medicación del
estudio.
Grado de intensidad de un acontecimiento adverso
El investigador valorará la intensidad de los AA y de los AAG. La intensidad de los
acontecimientos adversos (AA) se clasificará con arreglo a una escala del 1 al 5 según la
última versión de los criterios de toxicidad comunes para acontecimientos adversos
(CTCAE) del National Cancer Institute (NCI), que pueden consultarse por internet en la
siguiente dirección del NCI: http://ctep.cancer.gov/reporting/ctc.html
Si un acontecimiento concreto no figura en la escala de toxicidad del NCI CTCAE, se
utilizará la siguiente tabla para su clasificación:

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 51
Tercera versión: 30 de marzo de 2011
Grado Definición
1 Leve. El paciente refiere el signo, síntoma o acontecimiento, que por lo general
suele ser pasajero y no requiere tratamiento especial ni interfiere en las
actividades cotidianas habituales.
2 Moderado. Molestias que interfiere en las actividades habituales pero que
suelen mejorar con medidas terapéuticas básicas.
3 Intenso. Incapacita y no hace posible realizar actividades habituales o que
afecta significativamente al estado clínico y garantiza la intervención
terapéutica. La hospitalización puede ser necesaria o no.
4
Amenza la vida. Riesgo de muerte inminente que requiere hospitalización e
intervención clínica.
5 Muerte.
Relación de los acontecimientos adversos y aconteci mientos adversos graves con el tratamiento del estudio
La relación entre la administración de la medicación del estudio y la aparición de un
acontecimiento adverso se describe bajo una de las dos categorías siguientes, sospechada
o no sospechada por el investigador.
2= NO SOSPECHADA
La relación temporal del acontecimiento clínico con la medicación del estudio hace que la relación causal sea improbable , o bien el acontecimiento puede explicarse satisfactoriamente por otros fármacos, intervenciones terapéuticas o trastornos subyacentes.
1= SOSPECHADA La relación temporal del acontecimiento clínico con la medicación del estudio hace que la relación causal sea posible , y otros fármacos, intervenciones terapéuticas o trastornos subyacentes no proporcionan una explicación suficiente para el acontecimiento observado
Sólo es necesario registrar en el CRD aquellos AAs que el investigador considere
relacionados con la administración del tratamiento de estudio y todos los AAG,
relacionados o no.
Acciones a llevar a cabo en respuesta al acontecimi ento adverso Las medidas a tomar ante un acontecimiento adverso se describen en una escala
numérica, de 0 a 5, que cubre distintas posibilidades. Deberán seleccionarse una o más de
ellas.
0 = Ninguna 1 = Ajuste/interrupción temporal de la dosis de la medicación del estudio

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 52
Tercera versión: 30 de marzo de 2011
2 = La medicación del estudio se discontinúa permanentemente debido al acontecimiento adverso
3 = Administración de medicación concomitante 4 = Administración de tratamiento no farmacológico 5 = Hospitalización/prolongación de la hospitalización
9.3. Procedimientos para la notificación rápida de acontecimientos adversos graves.
Responsabilidad de la notificación
Cualquier acontecimiento adverso grave que se produzca desde la firma del
Consentimiento Informado y hasta transcurridos 30 días después de haber recibido la
última dosis del fármaco del estudio deberá ser notificado. El período posterior a la
interrupción del fármaco del estudio puede ampliarse en caso de que exista una sospecha
sólida de que el fármaco todavía no se ha eliminado o si se sospecha que el AAG esté
relacionado con la medicación del estudio, aunque hayan pasado esos 30 días.
Cada AAG deberá ser notificado inmediatamente por el investigador y completar su
notificación en el plazo de 24 horas desde su conocimiento, en el formulario de AAG,
incluso aunque no parezca estar relacionado con el tratamiento. La información sobre el
seguimiento de un acontecimiento adverso grave previamente comunicado también debe
notificarse en el mismo plazo. Si estuviese justificado, se enviará una alerta a los
investigadores para informar de dicho AAG a todos los investigadores que participen en
algún estudio con la misma medicación.
Todos los AAG deben ser objeto de seguimiento hasta que suceda una de las siguientes
situaciones:
- Resolución del acontecimiento.
- Estabilización del acontecimiento.
- Reestablecimiento de la situación basal del acontecimiento, si se dispone de un
valor basal.
- El acontecimiento puede atribuirse a productos distintos de la medicación del
estudio o a factores no relacionados con la realización del estudio.
- Resulta improbable obtener más información (el sujeto o el médico de atención
primaria rechazan proporcionar información adicional, pérdida para el seguimiento
después de la demostración de diligencia debida con los esfuerzos de seguimiento)
Se realizará el seguimiento de todos los acontecimientos adversos graves que no se hayan
resuelto al final del estudio, o que no se hayan resuelto con la retirada de la participación
en el estudio, hasta que se produzca cualquiera de las mencionadas situaciones.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 53
Tercera versión: 30 de marzo de 2011
La causa de la muerte de un sujeto en un estudio clínico, tanto sea el evento esperado o
no, y tanto el acontecimiento esté o no relacionado con el/los fármacos de estudio, se
considera un acontecimiento adverso grave notificable en 24h.
El investigador debe conservar una copia de toda la información relacionada con el AAG,
incluida la correspondencia con Trial Form Support (TFS) y el Comité Ético.
Procedimientos para la notificación
El investigador deberá cumplimentar un formulario de Acontecimientos Adversos Graves en
inglés, evaluar la relación con el fármaco del estudio y enviar el formulario completo por
Fax, en un plazo de 24 horas, a las oficinas de Trial Form Support (TFS) (FAX: 91 125 05
51). El original del Formulario de Acontecimientos Adversos Graves, junto con el Fax de
confirmación, deberán guardarse con los cuadernos de recogida de datos en el centro de
estudio. El monitor recogerá una copia del formulario de AAGs.
La información de seguimiento se enviará a la misma persona a la que se envió el
Formulario de Acontecimientos Adversos Graves original, incluyendo la fecha del informe
original. Se enviará un formulario de Acontecimientos Adversos Graves nuevo (indicando
que se trata de un seguimiento y la nueva fecha anotada). El seguimiento deberá describir
si el acontecimiento se ha resuelto o continúa, si fue tratado y de qué forma y si el paciente
permaneció o no en el estudio. Tanto el formulario como la hoja de confirmación del envío
deberán archivarse.
El informe inicial debe ser lo más completo posible, incluyendo detalles de la enfermedad
actual y del acontecimiento adverso grave y la evaluación de la relación causal entre el
acontecimiento adverso y el producto en investigación. La información no disponible en el
momento del informe inicial deberá ser recogida en el formulario de seguimiento del AAG.
Cualquier informe de un evento adverso debe incluir el número asignado al paciente, edad,
sexo, grado de intensidad, relación con la medicación del estudio, fecha de la
administración de la medicación del ensayo, medicaciones concomitantes y tratamiento.
El promotor proporcionará a Celgene y a Janssen-Cilag S.A. una copia de todos los
informes de AAG en el plazo de 24 horas a partir de que se tenga conocimiento del mismo,
independientemente de si éste está descrito o no en el Manual del Investigador.
9.4. Notificación a las Autoridades Sanitarias y a los Comités Éticos
El promotor comunicará a las Autoridades Sanitarias competentes y a los Comités Éticos:
- Toda la información relevante sobre acontecimientos adversos graves e inesperados
que se sospeche guarden relación con el fármaco del estudio y que sean fatales o

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 54
Tercera versión: 30 de marzo de 2011
amenacen la vida, a la mayor brevedad posible y en todo caso, en el plazo máximo de
siete días a partir del momento en que se tenga conocimiento del caso. La información de
seguimiento relevante para estos casos se notificará posteriormente en un plazo adicional
de ocho días.
- Cualquier otro acontecimiento grave e inesperado que se sospeche guarde relación con
la medicación fármaco del estudio, a la mayor brevedad posible, pero como máximo en
un plazo de quince días a partir del momento en que el investigador tenga conocimiento
del mismo
Para la evaluación de la esperabilidad se utilizarán los manuales del investigador del
Velcade y la Lenalidomida y la ficha técnica de la Prednisona, Dexametasona y Melfalán.
9.5. Embarazo
Dada la edad de los pacientes que se incluirán en este ensayo, las notificaciones de
posibles embarazos se refieren, exclusivamente, a las parejas mujeres, con capacidad de
gestación, de los pacientes hombres participantes en el mismo.
Se considerarán acontecimientos de notificación inmediata (24 horas) al promotor (en su
nombre, a Trial Form Support ) y éste a Celgene y Janssen-Cilag S.A., todos los casos de
embarazo y sospecha de embarazo de las parejas fértiles de los pacientes varones del
ensayo (incluidos los positivos de las pruebas del embarazo independientemente de la
edad o del cuadro patológico) mientras el paciente esté tomando el fármaco del estudio o
en los 28 días siguientes a la última dosis del mismo, debido a que la medicación del
estudio puede tener un efecto sobre los espermatozoides y, por tanto, sobre el feto.
Para esto, el paciente deberá comunicarle al investigador si su pareja se queda
embarazada. Se le solicitará autorización a la mujer, mediante un consentimiento escrito,
para hacer un seguimiento de los datos clínicos del embarazo y comunicarlos al promotor y
laboratorios titulares de los fármacos, que quedará archivado junto al resto de la
documentación del estudio en el centro.
Se indicará a las parejas mujeres de los varones que tomen el fármaco del estudio que
deben dirigirse al médico de inmediato en caso de quedarse embarazadas.
El embarazo se seguirá hasta el final, aunque ocurra una interrupción voluntaria o
espontánea del mismo, describiéndose los detalles del nacimiento y la presencia o
ausencia de algún defecto en el feto o alguna anomalía congénita.
El embarazo, sospecha de embarazo o resultado positivo de la prueba deberán notificarse
al promotor (en su nombre, a Trial Form Support ), quien lo comunicará a Celgene de
inmediato por vía telefónica (Tfno.: 630 56 48 73) y posteriormente (en ningún caso

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 55
Tercera versión: 30 de marzo de 2011
transcurridas más de 24 horas) a Celgene por correo electrónico (drugsafety-
[email protected]) o por fax al (fax local de Farmacovigilancia 91 422 90 95 ó 91 564 11
84 si el anterior no funciona) y a Janssen-Cilag S.A. por fax (Fax de Janssen-Cilag S.A. 91
722 85 20) mediante el impreso de notificación de embarazo.
Se derivará a la embarazada a un ginecólogo/obstetra experto en toxicidad reproductiva
para que le realice las evaluaciones necesarias y le brinde el asesoramiento oportuno.
Si el desenlace del embarazo reúne los criterios de clasificación inmediata como AAG (es
decir, se trata de un aborto natural o provocado [se documentará toda anomalía congénita
detectada en el feto abortado], bebé mortinato, muerte neonatal o anomalía congénita
[inclusive en un feto abortado]), el investigador seguirá los procedimientos de notificación
de AAG.
Se comunicará como AAG toda muerte neonatal que sobrevenga en los 28 días siguientes
al parto, independientemente de la causalidad. Asimismo, también deberá notificarse a
Celgene y a Janssen-Cilag S.A. la muerte de un lactante, una vez transcurrido dicho plazo
de 28 días, si el investigador considera que guarda relación con la exposición al fármaco
del estudio durante el embarazo, en el plazo de 24 horas a partir del momento en que el
investigador tenga conocimiento de ello. Se remitirá un impreso de notificación de AAG a
Celgene por correo electrónico a [email protected] o por fax al 91 422 90 95
(ó al 91 564 11 84 si el anterior no funciona) y por fax a Janssen-Cilag S.A. (91 722 85 20).
9.6. Tratamiento de las reclamaciones de calidad de Velcade ®
Una reclamación de calidad (product quality complaint (PQC)) se define como cualquier
sospecha de un defecto en un producto relacionada con la fabricación, etiquetado, o
empaquetado, por ejemplo, cualquier tipo de insatisfacción relativa a la identidad, calidad,
caducidad o fiabilidad de un producto incluyendo la integridad del envasado. Las
reclamaciones de calidad, pueden tener un impacto en la seguridad y eficacia del producto.
La notificación en tiempo, precisa y completa y el análisis de las reclamaciones de calidad
de los estudios clínicos es crucial para la protección de los sujetos, investigadores y
promotor y dicha notificación es obligatoria por las Autoridades Sanitarias mundiales. Las
notificaciones de calidad del fármaco Velcade® deberán notificarse, al igual que los AAG,
en 24 horas, en el formulario correspondiente de notificación (Product Quality Complaint)
por fax a Janssen Cilag S.A. Las reclamaciones de calidad debidas a problemas logísiticos
(rotura de cadena de frío, de transporte,...) o de caducidad quedan fuera de este reporte.
Números de teléfono y fax y personas de contacto
.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 56
Tercera versión: 30 de marzo de 2011
Los números de teléfono y de fax de las personas responsables de TFS para el envío de AAG son: Laura Aragón/ Pablo Rodriguez/ Victor Socias/ Begoñ a García Telf: 91 125 0550 Fax: 91 125 0551 Los números de teléfono y de fax de la persona responsable de recibir las reclamaciones de calidad de Janssen Cilag son: Dra. Concepción Suárez Telf: 91 722 81 00
LA FECHA Y LA FIRMA DEL INVESTIGADOR RESPONSABLE O DE UNO DE LOS
COLABORADORES AUTORIZADOS DE SU PERSONAL DEBE FIGUR AR EN TODOS
LOS INFORMES.
EL FORMULARIO DE PRODUCT QUALITY COMPLAINT PUEDE SER FIRMADO POR EL
FARMACEÚTICO RESPONSABLE DE LA MEDICACIÓN DE ESTUDI O EN CADA
CENTRO
9. ASPECTOS ÉTICOS
10.1. Buena práctica clínica
El estudio se desarrollará de acuerdo con la Conferencia Internacional sobre la
Armonización en relación con la buena práctica clínica y los requisitos reglamentarios
correspondientes. El investigador estará perfectamente familiarizado con el uso correcto
del fármaco del estudio según se describe en el protocolo y en el Manual del investigador.
Los documentos clínicos esenciales se conservarán para demostrar la validez del estudio y
la integridad de los datos recopilados. Los archivos principales se deben determinar al
comienzo del estudio, mantener durante el transcurso de dicho estudio y conservar de
acuerdo con las normativas que correspondan.
10.2. Consideraciones éticas
El estudio se desarrollará de acuerdo con los principios éticos de la Declaración de Helsinki
(véase Anexo 9). El CEIC examinará toda la documentación correspondiente al estudio con
el objetivo de proteger los derechos, seguridad y bienestar de los pacientes. El estudio sólo
se realizará en los centros para los que se haya obtenido la aprobación por parte del CEIC.
El investigador hará entrega al CEIC del protocolo, el Manual del investigador, el
consentimiento informado, la publicidad (si procede), la información por escrito facilitada a
los pacientes (incluyendo las tarjetas diario), las actualizaciones relacionadas con la

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 57
Tercera versión: 30 de marzo de 2011
seguridad, los informes de progreso anuales y cualquier modificación de estos
documentos.
10.3. Información al paciente y consentimiento info rmado
Una vez descrito el estudio en su totalidad, se obtendrá el consentimiento informado por
escrito del paciente o bien de su tutor o representante legal antes de hacer efectiva su
participación en el estudio. El método de obtención y documentación del consentimiento
informado y su contenido debe estar de acuerdo con la Conferencia Internacional sobre la
Armonización en relación con la buena práctica clínica y todos los requisitos reglamentarios
correspondientes.
10.4. Confidencialidad de los pacientes
Con el fin de respetar la privacidad de los pacientes, en todos los cuadernos de recogida
de datos, registros de contabilidad del fármaco del estudio, informes y comunicaciones del
estudio se identificará a los pacientes por el código de paciente asignado. El investigador
facilitará a los inspectores y posibles auditores del promotor o a las personas designadas y
a las autoridades reguladoras el acceso a los registros médicos originales de los pacientes
de modo que puedan verificar los datos reunidos en los cuadernos de recogida de datos y
auditar el proceso de recopilación de datos. Se mantendrá la confidencialidad y no se hará
pública la identidad de los pacientes en la medida que lo permitan las leyes y normativas
pertinentes.
10.5. Cumplimiento del protocolo
El investigador realizará el estudio en conformidad con el protocolo proporcionado el
promotor y una vez obtenida la aprobación o dictamen favorable del CEIC y las autoridades
reguladoras correspondientes. No se debe modificar el protocolo sin el consentimiento
tanto del investigador como del promotor. Las modificaciones del protocolo requieren la
aprobación o dictamen favorable por escrito del CEIC con anterioridad a su implementación
salvo en el caso de que la modificación resulte necesaria para eludir riesgos inmediatos
para los pacientes. El CEIC puede facilitar, si las autoridades reguladoras correspondientes
lo permiten, una revisión y aprobación o dictamen favorable a la mayor brevedad para
realizar los cambios pequeños de los estudios en curso que tengan la aprobación o
dictamen favorable del CEIC. El promotor presentará todas las modificaciones hechas al
protocolo ante las autoridades reguladoras de acuerdo con las normativas vigentes.
Cuando se requiera la desviación inmediata del protocolo para eludir riesgos inmediatos

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 58
Tercera versión: 30 de marzo de 2011
para los pacientes, el investigador se pondrá en contacto con el promotor, si las
circunstancias lo permiten, para tratar de las medidas que se ha planeado adoptar.
Cualquier desviación con respecto al protocolo se debe documentar detalladamente en el
CRD y la documentación original.
10.6. Finalización prematura del estudio
Este estudio puede ser interrumpido prematuramente si en opinión del promotor o de las
autoridades reguladoras existe una causa razonable suficiente. El investigador recibirá una
notificación por escrito en que la parte cesante documenta el motivo de la suspensión del
estudio.
Las circunstancias que justifican la suspensión del estudio incluyen, pero no se limitan a:
� Determinación de riesgos imprevistos, considerables o inaceptables para los
pacientes
� Imposibilidad de inscribir a un número aceptable de pacientes
� Insuficiente cumplimiento de los requisitos del protocolo
� Planes de modificación, suspensión o discontinuidad del desarrollo del fármaco
del estudio
10.7. Responsabilidad y seguro
El promotor ha suscrito una póliza de seguros que cubre, en sus términos y condiciones, la
responsabilidad legal por daños ocasionados a las personas participantes y derivados de
esta investigación, realizada estrictamente en conformidad tanto con el protocolo científico
como con la ley aplicable y los estándares profesionales.
10. CONSIDERACIONES PRÁCTICAS
11.1. Responsabilidades de todos los participantes en el ensayo
11.1.1. Investigador
Las obligaciones del investigador quedan especificadas en el artículo 37 del Real
Decreto 223/2004.
El investigador debe estar de acuerdo con el presente protocolo y conocer a fondo las
propiedades de los productos utilizados en el ensayo clínico.
El investigador debe entregar la hoja de información al paciente y colaborar con éste
para que pueda entender la explicación que se le proporciona en la misma. Es

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 59
Tercera versión: 30 de marzo de 2011
importante que comunique al paciente que su participación en el estudio es
completamente voluntaria y que ello no afectará a la relación paciente / médico,
además de garantizarle que todas las personas implicadas en el estudio respetarán la
confidencialidad de cualquier información relacionada con el paciente.
El Investigador principal o alguno de sus colaboradores, serán los encargados de
recoger, registrar y notificar los datos de forma correcta y garantizará la notificación
inmediata de los acontecimientos adversos graves o inesperados dentro de un plazo
de 24 horas a Trial Form Support (TFS). (Fax: 91- 125 05 51).
Es obligación del investigador preparar y mantener las historias de los pacientes
adecuadas y exactas, diseñadas para registrar todas las observaciones y otros datos
relevantes para el estudio de cada participante en el estudio. Toda la información
registrada en los CRDs para este estudio debe ser coherente con la documentación
original de los pacientes.
Todos los datos del estudio registrados en los documentos originales se transcribirán
en los CRDs. Los datos del estudio se introducirán en un sistema de procesamiento de
datos validado y seguro y se conservarán las copias de seguridad. Se documentará
cualquier modificación en los datos del estudio.
Es deber del investigador informar regularmente al Comité Ético de Investigación
Clínica de la marcha del estudio y corresponsabilizarse con el promotor de la
elaboración del informe final.
Los investigadores principales participantes en el ensayo estarán personalmente
involucrados en la decisión de una posible interrupción prematura del ensayo por
motivos de seguridad (ej.: un aumento de eventos tromboembólicos, neuropatía u otro
tipo de SAE en uno de los brazos del ensayo) o eficacia. Para cumplir este objetivo
deberán mantener un estrecho seguimiento del desarrollo del ensayo para poder
detectar precozmente posibles problemas, tanto en lo que respecta a seguridad como
a eficacia, y tomar las decisiones oportunas al respecto.
11.1.2. Monitor
El monitor se encargará de efectuar un seguimiento directo de la realización del
ensayo siguiendo las directrices de las Buenas Prácticas clínicas (GCP). Las
obligaciones del monitor quedan especificadas en el artículo 36 del Real Decreto
223/2004.
Todos los datos del estudio registrados en los documentos originales se transcribirán
en los CRDs. Los datos del estudio se introducirán en un sistema de procesamiento de

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 60
Tercera versión: 30 de marzo de 2011
datos validado y seguro y se conservarán las copias de seguridad. Se documentará
cualquier modificación en los datos del estudio.
Durante el curso del estudio, el monitor hará visitas a los centros para comprobar el
cumplimiento del protocolo, comparar los CRDs y las historias médicas de los
pacientes individuales, evaluar la contabilidad de los fármacos y garantizar que el
estudio se está desarrollando de acuerdo con los requisitos reguladores pertinentes.
Los CRDs se verificarán frente a la documentación original. La comprobación de las
historias médicas se realizará de manera que se garantice la confidencialidad del
paciente.
Los casos de omisión de datos o datos que no se puedan interpretar se tratarán con el
investigador para su resolución.
11.1.3. Promotor
Las obligaciones del promotor quedan especificadas en el artículo 35 del Real Decreto
223/2004.
El promotor del ensayo clínico es la persona física o jurídica que tiene interés en su
realización, firma las solicitudes de autorización dirigidas al CEIC y/o a la Agencia
Española del Medicamento (AEM) y se responsabiliza del mismo, incluyendo su
realización, comienzo y finalización. El promotor será el responsable de velar por el
cumplimiento de las normas legales pertinentes.
Fundación PETHEMA asume las obligaciones del promotor recogidas en el Artículo 35
del Real Decreto 223/2004, disponiendo todos los medios y colaboradores precisos
para cumplir con plenas garantías dicha responsabilidad.
11.2. Auditoría
Las autoridades reguladoras, el CEIC y el promotor o un representante designado pueden
solicitar el acceso a todos los documentos originales, cuadernos de recogida de datos del
paciente y el resto de la documentación del estudio para llevar a cabo una auditoria o
inspección en el centro. El investigador debe garantizar el acceso directo a estos
documentos y colaborar en todo momento en la realización de estas actividades. Se
atenderá en tales procedimientos a la debida protección de los datos privados de
identificación personal de acuerdo a la ley de protección de datos.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 61
Tercera versión: 30 de marzo de 2011
11.3. Contabilidad de la medicación
La contabilidad del fármaco en el centro de estudio es responsabilidad del investigador. El
investigador debe garantizar que el fármaco del estudio sea utilizado únicamente en
conformidad con este protocolo. En los casos en que sea permitido, el investigador puede
optar por transferir algunas de las responsabilidades relacionadas con la contabilidad del
fármaco a un farmacéutico o cualquier otra persona cualificada. Los registros de la
contabilidad del fármaco que indican la fecha de entrega del fármaco al centro de estudio,
el inventario del centro de estudio, la dispensación a los pacientes y la devolución o
eliminación del fármaco serán conservados por el centro de estudio. Estos registros
documentarán de la forma adecuada el hecho de que se administren a los pacientes las
dosis conforme se especifica en el protocolo. Los registros de contabilidad incluirán las
fechas, cantidades, números de lote y serie, fechas de caducidad (si procede) y números
de los pacientes.
Todos los fármacos del estudio devueltos por pacientes, no utilizados o caducados serán
devueltos a quien el promotor designe o bien destruidos en el Servicio de Farmacia del
centro si así se autoriza. Cualquiera de los procedimientos deberá quedar bien
documentado.
11.4. Retención de los registros
El investigador conservará todos los registros del estudio de acuerdo con la buena práctica
clínica de la Conferencia Internacional sobre la Harmonización y los requisitos
reglamentarios correspondientes.
11.5. Publicaciones de los resultados del ensayo y uso de la información
Los resultados de este ensayo clínico serán comunicados en Congresos científicos y
publicados en una revista científica. Los resultados finales del ensayo y un manuscrito de la
publicación será enviado a J&JPR&D y Celgene, al menos 60 días antes de su envío para
su publicación. También serán comunicados a Celgene, al menos 5 días antes de su envío,
los pósters, abstracts u otro material oral o escrito relacionado con el ensayo.
11.5.1. Normativa básica del ensayo.
El Investigador Principal y/o el HOSPITAL, se obligan a no utilizar ni transmitir a
terceros, ni a divulgar y/o publicar los resultados obtenidos en este Ensayo sin el
consentimiento previo y por escrito de la FUNDACIÓN PETHEMA, promotor único del
ensayo. En cualquier caso, deberán respetarse las siguientes condiciones:

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 62
Tercera versión: 30 de marzo de 2011
a) Los resultados del presente estudio no podrán ser publicados hasta la finalización
del ensayo o anteriormente, si se acuerda por ambas partes.
b) El promotor no citará el nombre de los investigadores sin su autorización, excepto
en el caso de referencias a trabajos ya publicados
c) El promotor permitirá la publicación de los datos obtenidos en este ensayo a
revistas de reconocido prestigio científico y su divulgación en seminarios y
conferencias dentro del ámbito profesional médico, siempre que se respete lo
establecido en el párrafo a) de este apartado y que se permita la revisión del
borrador definitivo del artículo por el promotor en un plazo mínimo de treinta días
11.5.2. Condiciones de publicación del grupo PETHEMA
En los trabajos derivados de los estudios de PETHEMA se deberán observar las
siguientes indicaciones:
a) En todos los trabajos se hará constar el nombre de los centros participantes, y
figurarán como autores los participantes en el diseño, seguimiento, análisis de los
resultados y redacción del trabajo.
b) Los responsables del protocolo serán también los encargados de comunicar los
resultados a las reuniones de PETHEMA y congresos científicos, así como
proceder a la redacción de los trabajos derivados del protocolo. En dicho
cometido los responsables del protocolo contarán, si lo estiman necesario, con la
ayuda en la redacción de los trabajos.
c) La redacción definitiva del texto del artículo podrá ser sometida a juicio y visto
bueno de algún otro miembro de PETHEMA con reconocida experiencia, para
que con sus sugerencias y modificaciones mejore la calidad del trabajo. Esta
persona podrá figurar como coautor a criterio de los responsables del protocolo.
En caso de que no figurara como coautor se deberá mencionar en el apartado de
agradecimientos.
d) La inclusión del Data Manager como coautor queda a criterio de los responsables
del protocolo. Si no figurara como coautor se debería mencionar en el apartado
de agradecimientos
e) Estadístico (si lo hubiere) dependiendo del grado de participación y del criterio de
los responsables del protocolo podrá figurar como coautor o en los
agradecimientos.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 63
Tercera versión: 30 de marzo de 2011
f) Tanto el estadístico como el Data Manager no se computarán para la
determinación de número máximo de autores según la cifra de casos incluidos
por centro.
g) Se determinará un máximo de autores por centro participante en el estudio para
su inclusión en el encabezamiento del artículo. Esta selección por razón de casos
incluidos se hará de acuerdo al criterio de casos aportados al estudio (siempre y
cuando sean evaluables y seguidos hasta la última actualización) y de acuerdo a
la norma siguiente:
- Protocolos en los que se hayan reclutado menos de doscientos
pacientes: cada centro tendrá derecho a un autor por cada cinco por
ciento de pacientes incluidos del total de reclutados para el estudio.
En cualquier caso habrá un límite máximo de quince autores para la
publicación. Esta restricción se aplicará también a las publicaciones
a congresos.
- Protocolos en los que se hayan incluido doscientos enfermos o más.
Todo centro que haya incluido diez casos tendrá derecho a un autor.
Por cada quince casos adicionales se tendrá derecho a otro autor.
En cualquier caso habrá un límite máximo de veinticinco autores para
la publicación. Esta restricción se aplicará también en las
comunicaciones a congresos.
h) Si existiera alguna limitación de autores por parte de la organización del congreso
o de la revista, el número se reducirá a partir de los últimos que tienen derecho a
firmar como tales. En caso de discrepancias prevalecerá el criterio de los
responsables del protocolo.
i) En un apéndice constarán todas las instituciones, así como las personas que
hayan participado en el protocolo.
Antes de remitir el trabajo para publicación se enviará una copia a cada uno de los
autores para que efectúen las modificaciones y modificaciones que consideren
oportunas. En caso de que no exista respuesta en el caso máximo que fije l
responsable del protocolo se considerará que se está de acuerdo con el contenido
del trabajo. Dado que puede existir un número considerado de autores y que, en
algunas ocasiones, las sugerencias pueden ser redundantes, contradictorias o no
sustanciales, la versión definitiva del trabajo queda bajo la responsabilidad de las
personas que hayan efectuado la redacción del mismo.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 64
Tercera versión: 30 de marzo de 2011
11. ANÁLISIS ESTADÍSTICO
El análisis estadístico será realizado por los coordinadores del grupo español de
Mieloma.
12.1. Estimación del tamaño de la muestra
La variable principal de eficacia del estudio es evaluar la supervivencia libre de
progresión a 18 meses en las dos ramas de tratamiento programados. La SLP a 18
meses con MPV en el estudio VISTA es del 60%, esta es la proporción mínima esperada
para la rama control (tratamiento secuencial). Cabe esperar que la adición de tratamiento
con Rd de manera secuencial o alternante aumente potencialmente la SLP a 18 meses a
un 75%. Este aumento podría producirse en ambas ramas o bien, en sólo una de ellas.
El diseño de este estudio “pick de winner”19,20 permite evaluar cuál de las dos
combinaciones (secuencial o alternante) es la que permite obtener el beneficio clínico
estimado (75% SLP a 18 meses) así como descartar también si uno de los brazos
obtiene un beneficio clínico inferior (“drop de looser”) a lo esperado (44,5% de SLP a 18
meses), ya que el esquema VMP propuesto en este ensayo es ligeramente menos
intenso que el esquema utilizado en el ensayo VISTA utilizado como referencia.
Los parámetros utilizados en los cálculos son: α = 0,05 bilateral (nivel de significación de
la prueba estadística), β = 0,20 (correspondiente a la potencia del 80%), tasa máxima
esperada de abandonos del 4%. Con un periodo de reclutamiento de 19 meses y 37,5
meses de duración del estudio en total, el tamaño muestral requerido es de 240
pacientes, incluyendo los posibles abandonos y pérdidas de seguimiento.
TTP: Alfa: 5% (2 colas) y potencia del 80%, incluyendo 4% de abandonos
HR *
(SLP a 18 m)
Tasa de
reclutamiento
Eventos
necesarios
Duración del estudio
(reclutamiento) en
meses
N para la
comparación entre
grupos
1.78
(60% vs 75% a 18 m) 12,5 pts/mes 94 37.5 (19) 240
12.2. Población para el análisis
El análisis de eficacia incluirá a todos los pacientes que se incluyan en cualquiera de los
grupos de tratamiento de estudio (análisis por intención de tratamiento), y que hayan
recibido al menos una dosis de las combinaciones del estudio MPV o Rd. Los análisis de

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 65
Tercera versión: 30 de marzo de 2011
seguridad de administración del tratamiento incluirá tan sólo a aquellos pacientes que
reciban, al menos, una dosis de los fármacos en estudio, bortezomib o lenalidomida.
12.3. Procedimientos para el manejo de los datos in existentes, no usados y confusos
Todos los datos relativos a la seguridad y eficacia disponibles se incluirán en las listas y
tablas de datos. No se realizará imputación de valores para datos no disponibles.
Los pacientes tratados con los fármacos del estudio a los que no se realiza un
seguimiento relacionado con la seguridad no serán incluidos en los análisis de seguridad
debido a que su inclusión no haría más que disminuir los porcentajes de pacientes con
acontecimientos adversos o toxicidades de laboratorio.
Los posibles datos confusos o erróneos serán examinados de acuerdo con los
procedimientos de control de datos normalizados.
12.4. Métodos estadísticos
Se realizarán tablas-resumen donde se mostrará el número de observaciones, la media,
la desviación estándar, la mediana, el mínimo y el máximo para las variables contínuas,
y el número y porcentaje por categoría cuando se trate de datos categóricos. Los datos
de tiempo hasta el evento se resumirán mediante los percentiles 25, 50 (mediana) y 75
además de los intervalos de confianza del 95% de dos lados relacionados y el porcentaje
de observaciones excluidas. Las categorías del resumen se facilitarán para los pacientes
tratados en cada una de las ramas de tratamiento, tanto de inducción como de
mantenimiento. Las pruebas de hipótesis estadísticas formales relativas a la superioridad
de alguna de las dos ramas de tratamiento se realizarán con el nivel de significación de
0,05 de dos lados.
El primer análisis de eficacia será la evaluación de la SLP a 18 meses con los dos
esquemas de tratamiento propuestos.
El segundo análisis de eficacia será la evaluación de la tasa de respuestas (con especial
atención a la tasa de RC, incluyendo las RC estrictas, RC por citometría de flujo y
biología molecular) dentro de cada grupo de tratamiento. Se facilitarán los intervalos de
confianza (límites del 95%) de los índices de respuesta. Se calculará el Riesgo Relativo
en todos los pacientes incluidos en el ensayo.
Dentro de los análisis de eficacia, se determinarán también los tiempos de Supervivencia
Global, Tiempo hasta la Progresión y tiempo hasta el inicio de un nuevo tratamiento, en

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 66
Tercera versión: 30 de marzo de 2011
cada unos de los grupos de tratamiento. También se evaluará la sensibilidad a los
tratamientos de rescate en el momento de la recaída o progresión de la enfermedad.
Se identificarán las características biológicas (incluyendo análisis de perfil de expresión
génica) de aquellos pacientes resistentes a uno de los dos regímenes propuestos, o
ambos.
12.4.1. Análisis de eficacia
Definiciones y análisis de los objetivos de eficaci a
• El primer análisis de eficacia es la evaluación del SLP a 18 meses , que será
evaluado como:
Duración en meses desde la fecha de aleatorización en el ensayo hasta la fecha de la
progresión de la enfermedad o muerte debido a cualquier causa. Los pacientes que
tengan una pérdida de seguimiento serán censurados en la fecha de la última visita.
Este análisis consistirá en un análisis de Kaplan-Meier para la supervivencia libre de
progresión. Los datos del tiempo hasta la muerte se analizarán para buscar diferencias
entre los distintos grupos de tratamiento utilizando la prueba del orden logarítmico, con
una conclusión que alguno de los esquemas de tratamiento propuestos produce una
supervivencia libre de progresión más larga si la prueba del orden logarítmico es
estadísticamente significativa al nivel de significación α = 0,05 de dos lados. Los
factores de estratificación serán usados en el análisis.
Si la progresión de la enfermedad está basada en un aumento de los niveles de
paraproteína monoclonal, entonces los criterios propuestos para valorar la respuesta
requerirían que el aumento del nivel de paraproteína se documente en dos ocasiones
consecutivas. Por tanto, si la segunda medición del nivel de paraproteína confirma
progresión de la enfermedad, se calculará la progresión para la SLP desde la
randomización hasta el momento de la primera medición del nivel de paraproteína que
demostró una recaída o progresión de la enfermedad. Si la segunda medición no
confirmara la progresión, el paciente continuará el tratamiento del estudio hasta que
se documente progresión. La SLP se censurará como la última fecha en que se supo
que el paciente no tenía progresión de la enfermedad para: 1) los pacientes cuya
enfermedad no haya progresado en el momento del análisis y 2) para los pacientes
que se hayan retirado de la fase de tratamiento del estudio antes de documentar la
progresión (incluidos aquellos que murieran por causas no relacionadas con el MM).
Los pacientes a quienes se les administre otro tratamiento contra el MM antes de

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 67
Tercera versión: 30 de marzo de 2011
objetivar la progresión o toxicidad inaceptable, su SLP y su TTP serán censurados en
el día anterior al inicio del nuevo tratamiento.
Como consecuencia de este primer análisis de eficacia se podrá analizar cual de las
dos estrategias combinatorias (secuencial y/o alternante) proporcionan mayor
beneficio clínico (“winner”) para los pacientes respecto al esquema MPV del estudio
VISTA. Cabe la posibilidad que las dos estrategias (“winners”) alcancen el punto de
mejora de la SLP estimado a 18 meses del 75% versus el 60% del VISTA. Y también
cabe la posibilidad que una de las combinaciones sea clínicamente inferior (“looser”)
44.5% versus 60% del MPV del estudio VISTA.
• El segundo análisis de eficacia será evaluar la tasa de respuestas con los
esquemas propuestos, así como la duración de la respuesta, supervivencia global,
tiempo hasta la progresión y tiempo hasta iniciar nuevo tratamiento:
Las categorías de la respuesta de la enfermedad que serán usadas para los análisis
de eficacia serán: RC estricta, RC, Muy Buena Respuesta parcial (VGPR), RP,
Enfermedad Estable y progresión de la enfermedad, añadiendo la categoría de RC por
Biología Molecular.
El análisis descriptivo de eficacia consistirá en la evaluación de la tasa de respuesta
(RC estricta, RC, VGPR, RP, Enfermedad Estable, Progresión de la enfermedad y RC
por Biología Molecular) en cada uno de los grupos de tratamiento inducción.
Supervivencia global: Duración en meses desde la fecha de la aleatorización en el
ensayo hasta la fecha de la muerte. Los pacientes que tengan una pérdida de
seguimiento, serán censurados en la fecha de la última visita. Este análisis consistirá
en un análisis de Kaplan-Meier para la supervivencia global. Los datos del tiempo
hasta la muerte se analizarán para buscar diferencias entre los distintos grupos de
tratamiento utilizando la prueba del orden logarítmico, con una conclusión que alguno
de los esquemas de tratamiento propuestos produce una supervivencia global más
larga que los otros dos si la prueba del orden logarítmico es estadísticamente
significativa al nivel de significación α = 0,05 de dos lados. Los factores de
estratificación serán usados en el análisis.
Duración de la respuesta: La duración de la respuesta en meses será calculada
desde la primera fecha de evidencia de, al menos, una respuesta parcial, hasta la
primera fecha de evidencia de progresión de la enfermedad (o recaída si el paciente
hubiese alcanzado RC). La duración de la respuesta será usando un análisis de

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 68
Tercera versión: 30 de marzo de 2011
Kaplan-Meier, ajustado con los factores de estratificación, similar al análisis de
supervivencia global y supervivencia libre de progresión.
Tiempo hasta la progresión (TTP): El tiempo transcurrido desde la aleatorización de
los pacientes hasta la primera documentación de progresión de la enfermedad. Si la
progresión de la enfermedad está basada en un aumento de los niveles de
paraproteína monoclonal, entonces los criterios propuestos para valorar la respuesta
requerirían que el aumento del nivel de paraproteína se documente en dos ocasiones
consecutivas. Por tanto, si la segunda medición del nivel de paraproteína confirma
progresión de la enfermedad, se calculará el TTP desde la randomización hasta el
momento de la primera medición del nivel de paraproteína que demostró una recaída
o progresión de la enfermedad. Si la segunda medición no confirmara la progresión, el
paciente continuará el tratamiento del estudio hasta que se documente progresión. El
TTP se censará como la última fecha en que se supo que el paciente no tenía
progresión de la enfermedad para: 1) los pacientes cuya enfermedad no haya
progresado en el momento del análisis y 2) para los pacientes que se hayan retirado
de la fase de tratamiento del estudio antes de documentar la progresión (incluidos
aquellos que murieran por causas no relacionadas con el MM). Los pacientes a
quienes se les administre otro tratamiento contra el MM antes de objetivar la
progresión o toxicidad inaceptable, su TTP será censado en el día anterior al inicio del
nuevo tratamiento.
• El tercer análisis de eficacia tratará de identificarlas características biológicas
(incluyendo análisis de perfil de expresión génica) de aquellos pacientes resistentes a
uno de los dos regímenes propuestos, o ambos.
Fuera de protocolo, en el periodo de seguimiento se evaluará también lo siguiente:
Tiempo hasta iniciar nuevo tratamiento: Duración en meses desde la fecha de
inclusión en el ensayo hasta la fecha de inicio del siguiente tratamiento o muerte de
cualquier causa, lo primer que ocurra. Para la medición de esta variable de una
manera uniforme, se deberán tener en cuenta los criterios requeridos actualizados por
el panel de consenso del IMWG para decidir iniciar una nueva linea de tratamiento y
que aparecen en el anexo 7.
• El cuarto análisis de eficacia consistirá en evaluar la sensibilidad a los
tratamientos de rescate en el momento de la recaída o progresión de la enfermedad.
Para la valoración de este análisis de eficacia se evaluarán los tratamientos de

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 69
Tercera versión: 30 de marzo de 2011
rescate administrados en el momento de la recaída o progresión de la enfermedad,
con la respuesta alcanzada valorando ésta de acuerdo a las características antes
definidas, así como la duración de la misma, y la supervivencia global.
Análisis adicionales de eficacia
Análisis descriptivos
Los análisis descriptivos de eficacia consistirán en evaluar los cambios que se han
producido desde la visita de selección en los siguientes valores de importancia en el
mieloma:
o Componente monoclonal en suero y/u orina o cadenas ligeras libres en suero
para los pacientes con MM oligo/no secretor.
o Porcentaje de células plasmáticas en aspirado de MO mediante morfología y
citometría de flujo.
o Análisis de citometría de flujo (porcentaje de células plasmáticas en fase S y
enfermedad residual).
o Análisis citogenéticos por Hibridación in situ.
o Estudios de Biología Molecular
o Medida de los plasmocitomas en tejidos blandos y hueso mediante estudios
radiológicos.
Estas variables se pueden analizar en relación con los cambios desde la visita de
selección a los intervalos de medidas sucesivos o mediante otros sistemas métricos
adecuados como el método del área bajo la curva.
Ajuste para las co-variables
Con el fin de garantizar que las conclusiones del estudio no estén influenciadas en
exceso por los factores presentes en este estudio con unos índices muy distintos entre
los distintos grupos de tratamiento, se realizarán análisis exploratorios adicionales del
tiempo hasta la progresión de la enfermedad, duración de la respuesta, la
supervivencia y la tasa de respuestas mediante la selección de un conjunto de
posibles variables de pronóstico (obtenidas con anterioridad o en la visita de
selección) como covariables de los modelos de regresión de Cox (para el tiempo
hasta la progresión, duración de la respuesta y la supervivencia) y métodos de

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 70
Tercera versión: 30 de marzo de 2011
modelación de datos categóricos como la regresión logística (para la tasa de
respuesta). Los factores incluirán:
o Alteraciones citogenéticas.
o Presencia de enfermedad extramedular.
o B2-microglobulina.
o Proteína C-reactiva.
o Tipo/estadio de la enfermedad al diagnóstico.
o Edad al diagnóstico.
o Análisis mediante citometría de flujo (porcentaje de células plasmáticas en fase
S y enfermedad residual).
Estos análisis se realizarán con la inclusión de todas la covariables en el modelo a fin
de determinar la influencia relativa de cada una en la variable de eficacia respectiva.
Las conclusiones de estos análisis ajustados contribuirán a determinar si existe
alguna razón por la cual considerar que la eficacia relativa del tratamiento del estudio
puede variar en función de las características que presentan los pacientes.
12.4.2. Comparaciones basales
Se evaluarán los 2 regímenes de tratamiento para poder establecer una comparación
descriptiva de las características demográficas y de la visita de selección. Los datos
que se van a evaluar incluirán al menos la edad, el sexo, y los componentes de la
evaluación del tipo y estadio de la enfermedad. Los posibles factores pronóstico que
se vayan a examinar como covariables serán además resumidos para estudiar las
diferencias de los grupos de tratamiento en la visita de selección. Se llevarán a cabo
análisis secundarios con ajustes de las covariables de los factores de la visita de
selección, razón por la que cualquier influencia de estos factores en el resultado de la
eficacia debe ser evidente a partir de estos análisis ajustados.
12.4.3. Análisis de seguridad
Las evaluaciones relativas a la seguridad dependerán de la incidencia, intensidad y
tipo de acontecimientos adversos así como de los cambios clínicamente significativos
de los hallazgos de la exploración física del paciente y los resultados del laboratorio
clínico. Las variables de seguridad serán incluidas en una tabla y se facilitarán para
todos los pacientes que reciban cualquier cantidad de la medicación del estudio, en

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 71
Tercera versión: 30 de marzo de 2011
dos cohortes por separado, una que incluya todos los que han completado el ensayo y
otra que incluye aquéllos que han recibido, al menos, una dosis de fármaco del estudio
aunque no lo hayan completado. Se incluirán asimismo en una tabla la exposición a
los fármacos del estudio y las razones por las que se suspende el tratamiento del
estudio. Se realizarán análisis para determinar si existen diferencias estadísticamente
significativas entre los 2 grupos de tratamiento con índices de incidencia de
acontecimientos adversos graves, índices de acontecimientos adversos derivados del
tratamiento con una clasificación por sistemas u órganos, índices de suspensión del
tratamiento debido a los acontecimientos adversos o a la toxicidad conforme a la
evaluación del laboratorio clínico y los índices de toxicidad hematológica.
Todos los acontecimientos adversos considerados por el investigador como
relacionados con la medicación de estudio y los acontecimientos adversos graves que
se produzcan con posterioridad a la administración de la primera dosis del fármaco del
estudio (bortezomib o lenalidomida) y durante 30 días después de ser administrada la
última dosis de dicho fármaco se incluirán en las listas de datos organizadas por
pacientes. Se facilitará asimismo una tabla que enumere los acontecimientos adversos
según la intensidad máxima. Los fallecimientos, los acontecimientos adversos graves y
los acontecimientos adversos que implican la suspensión del estudio se clasificarán
igualmente en otra tabla.
El cambio de la línea basal de los parámetros del laboratorio clínico será resumido
durante el estudio y la frecuencia de los valores de laboratorio anormales clínicamente
significativos se mostrará en una tabla. En estas tablas se incluirá la rama de
tratamiento asignada, así como el número de ciclo y nivel de dosis mostrando el
número de pacientes con cada grado de criterios comunes de toxicidad de la línea
basal y los cambios hasta el grado máximo de criterios comunes de toxicidad en el
ciclo. Los cambios en los parámetros de las constantes vitales se resumirán con el
tiempo de un modo similar a los parámetros de laboratorio y cualquier valor anormal se
incluirá en una tabla.
12.5. Procedimientos para la comunicación de desvia ciones del plan estadístico original
Todas las desviaciones del plan de análisis estadístico original se incluyen en el informe
final del estudio clínico.
12.6. Análisis preliminar
Un análisis preliminar será realizado anualmente desde la inclusión del primer paciente.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 72
Tercera versión: 30 de marzo de 2011
12. REFERENCIAS BIBLIOGRÁFICAS
Reference List
1. Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer
J.Clin. 2000;50:7-33.
2. Garcia-Sanz R, Mateos MV, San Miguel JF. [Multiple myeloma]. Med.Clin.(Barc.)
2007;129:104-115.
3. Kumar SK, Rajkumar SV, Dispenzieri A et al. Improved survival in multiple myeloma
and the impact of novel therapies. Blood 2008;111:2516-2520.
4. Alexanian R, Haut A, Khan AU et al. Treatment for multiple myeloma. Combination
chemotherapy with different melphalan dose regimens. JAMA 1969;208:1680-1685.
5. Palumbo A, Bringhen S, Liberati AM et al. Oral melphalan, prednisone, and
thalidomide in elderly patients with multiple myeloma: updated results of a
randomized controlled trial. Blood 2008;112:3107-3114.
6. Hulin C, Facon T, Rodon P et al. Melphalan-Prednison-Thalidomide (MP-T)
demonstrates a significant survival advantage in elderly patients > 75 years with
Multiple Myeloma compared with Melphalan-Prednisona (MP) in a randomized,
double-blind, placebo-controlled trial, IFM 01/01. Blood 2007;110:75a.
7. Facon T, Mary JY, Hulin C et al. Melphalan and prednisone plus thalidomide versus
melphalan and prednisone alone or reduced-intensity autologous stem cell
transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised
trial. Lancet 2007;370:1209-1218.
8. Wijermans P. Melphalan + Prednisone Versus Melphalan + Prednisone +
Thalidomide in Induction Therapy for Multiple Myeloma in Elderly Patients: Final
Analysis of the Dutch Cooperative Group HOVON 49 Study [abstract]. Blood
2008;112:
9. Waage AGPJGTIFP. Melphalan-Prednisone-Thalidomide to Newly Diagnosed
Patients with Multiple Myeloma: A Placebo Controlled Randomised Phase 3 Trial.
Blood 2007;110:
10. Ludwig H, Tothova E, Hajek R et al. Thalidomide-Dexamethasone versus Melphaln-
Prednisone as first line treatment in elderly patients with multiple myeloma: second
interim analysis. Haematologica 2007;92:0446a.
11. Morgan GJ, Faith D, Roger O et al. Thalidomide Combinations Improve Response
Rates; Results from the MRC IX Study. Blood 2007;110:3593a.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 73
Tercera versión: 30 de marzo de 2011
12. Palumbo A, Falco P, Corradini P et al. Melphalan, prednisone, and lenalidomide
treatment for newly diagnosed myeloma: a report from the GIMEMA--Italian Multiple
Myeloma Network. J.Clin.Oncol. 2007;25:4459-4465.
13. Rajkumar SV, Jacobus S, Callander N et al. Randomized trial of lenalidomide plus
high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone in
newly diagnosed myeloma (E4A03), a trial coordinated by the Eastern Cooperative
Oncology Group: Analysis of response, survival, and outcome wi. J.Clin.Oncol.
2008;26:8504a.
14. Mateos MV, Hernandez JM, Hernandez MT et al. Bortezomib plus melphalan and
prednisone in elderly untreated patients with multiple myeloma: results of a
multicenter phase 1/2 study. Blood 2006;108:2165-2172.
15. San Miguel JF, Schlag R, Khuageva NK et al. Bortezomib plus melphalan and
prednisone for initial treatment of multiple myeloma. N.Engl.J.Med. 2008;359:906-
917.
16. Mateos MV, Oriol A, Martinez J et al. Bortezomib (Velcade)-Melphalan-Prednisone
(VMP) Versus Velcade- Thalidomide-Prednisone (VTP) in Elderly Untreated Multiple
Myeloma Patients: Which Is the Best Partner for Velcade: An Alkylating or An
Immunomodulator Agent? Blood 2008;112:
17. Palumbo A, Bringhen S, Rossi G et al. A Prospective, Randomized, Phase III Study
of Bortezomib, Melphalan, Prednisone and Thalidomide (VMPT) Versus Bortezomib,
Melphalan and Prednisone (VMP) in Elderly Newly Diagnosed Myeloma Patients.
Blood 2008;112:
18. Palumbo A, Rajkumar SV, Dimopoulos MA et al. Prevention of thalidomide- and
lenalidomide-associated thrombosis in myeloma. Leukemia 2008;22:414-423.
19. Simon R, Wittes RE, Ellenberg SS. Randomized phase II Clinical Trials. Cancer
Treatment Reports 1985;69:1375-1381.
20. Rubinstein L, Crowley J, Ivy P, Leblanc M, Sargent D. Randomized phase II designs
Clinical cancer research 2009;15:1883-1890.
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 74
Tercera versión: 30 de marzo de 2011
ANEXO 1: LISTADO DE CENTROS E INVESTIGADORES PARTIC IPANTES
HOSPITAL INVESTIGADOR PRINCIPAL
ANDALUCÍA
Complejo Hospitalario Regional Virgen del Rocío Jesús Martín Sánchez
Hospital Nuestra Señora de Valme Eduardo Ríos Herranz
Complejo Hospital Costa del Sol María Casanova Espinosa
Hospital de Especialidades de Jerez de la Frontera José Luis Guzmán Zamudio
Hospital Puerta del Mar Francisco Javier Capote Huelva
ARAGÓN
Hospital Clínico Universitario Lozano Blesa Luis Palomera Bernal
Hospital Miguel Servet Pilar Giraldo Castellano
ISLAS BALEARES
Complejo Asistencial Son Dureta Antonia Sampol Mayol
Hospital Son Llàtzer Joan Bargay Lleoneart
CANARIAS
Hospital de Gran Canaria Dr. Negrín Alexia Suárez Cabrera
Hospital Universitario de Canarias Miguel Teodoro Hernández García
CANTABRIA
Hospital Universitario Marqués de Valdecilla Eulogio Conde García
CASTILLA LA MANCHA
Hospital Universitario Guadalajara Dunia de Miguel Llorente
Complejo Hospitalario de Toledo Luis Felipe Casado Montero
Hospital Nuestra Señora del Prado José Luis Bueno Cabrera
Hospital General de Ciudad Real Carmen Calle Primo
Hospital General Virgen de la Luz Carlos Javier Cerveró Santiago
CASTILLA LEÓN
Hospital Clínico de Salamanca Jesús San Miguel Izquierdo
Hospital General de Segovia José Mariano Hernández Martín
Hospital Virgen de la Concha Monserrat Pérez Sánchez
Hospital de León Fernando Escalante
CATALUÑA
Hospital Universitari Joan XXIII de Tarragona Lourdes Escoda Teigell
Hospital Clínic i Provincial de Barcelona Joan Bladé Creixentí
Hospital de Sabadell (Parc Taulí) Elena Rámila Herrero
Hospital Germans Trias i Pujol Albert Oriol Rocafiguera
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 75
Tercera versión: 30 de marzo de 2011
HOSPITAL INVESTIGADOR PRINCIPAL
Hospital de la Santa Creu i Sant Pau Miquel Granell
Hospital del Mar Eugenia Abella Monreal
Hospital Universitari Dr. Josep Trueta – ICO de Girona Yolanda Gonzalez Montes
Althaia (red asistencial de Manresa) Albert Altés Hernández
Hospital Durán y Reynals - ICO L´Hospitalet José Sarrá Escarrer
Hospital Vall d'Hebron Mercedes Gironella Mesa
EXTREMADURA
Complejo Hospitalario de Cáceres Carmen Cabrera Silva
GALICIA
Complejo Hospitalario Universitario de Santiago José Luis Bello López
MADRID
Hospital Universitario Fundación Alcorcón Francisco Javier Peñalver Párraga
Hospital Universitario 12 de Octubre Juan José Lahuerta Palacios
Hospital de Fuenlabrada Pilar Bravo Barahona
Hospital Universitario la Paz Raquel de Paz Arias
Hospital Universitario de la Princesa Adrián Alegre Amor
Clínica Puerta de Hierro Isabel Krsnik Castelló
Hospital Ramón y Cajal María Jesús Blanchard
Complejo Universitario de San Carlos Rafael Martínez Martínez
Centro Oncológico MD Anderson International España Rebeca Iglesia del Barrio
Hospital de Madrid, S.A.- Norte Hospital General Jaime Pérez de Oteyza
Hospital Infanta Leonor José Ángel Hernández Rivas
Hospital del Tajo Ana Paz Lafuente Gujosa
Hospital Severo Ochoa Carolina Bombín Canal
Hospital Universitario Príncipe de Asturias Juan Jose Gil Fernández
Hospital Infanta Sofía Eugenio Giménez Mesa
Hospital General Universitario Gregorio Marañón Cristina Encinas Rodríguez
MURCIA
Hospital Morales Meseguer Felipe de Arriba de la Fuente
Hospital Universitario Virgen de la Arrixaca José M. Moraleda Jiménez
NAVARRA
Clínica Universitaria de Navarra Felipe Prósper Cardoso
Complejo Hospitalario de Navarra Mª Ángeles Goñi He rranz
PAÍS VASCO
Hospital de Cruces Elena Amutio Díez
Hospital Donostia María Asunción Echeveste Gutiérrez
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 76
Tercera versión: 30 de marzo de 2011
HOSPITAL INVESTIGADOR PRINCIPAL
Hospital de Txagorritxu Ernesto Pérez Persona
COMUNIDAD VALENCIANA
Hospital Arnau de Vilanova Aurelio López Martínez
Hospital General de Castellón Inmaculada García Navarro
Hospital Clínico Universitario de Valencia Ana Isabel Teruel Casasús
Hospital Universitario Dr. Peset Aleixandre Paz Ribas García
Hospital Universitario La Fe Javier De La Rubia Comos
Hospital Francesc de Borja de Gandía María José Fernández Llavador
Consorcio Hospital General Universitario de Valencia Pedro Luís Pérez Rodríguez

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 77
Tercera versión: 30 de marzo de 2011
ANEXO 2: LISTADO DE CEIC
CEIC del Hospital de Badalona Germans Trias i Pujol (CEIC DE REFERENCIA) CEIC Autonómico de Andalucía CEIC Regional de Aragón CEIC Regional de Islas Baleares CEIC del Hospital de Gran Canaria Dr. Negrín CEIC del Hospital Universitario de Canarias CEIC de Cantabria CEIC del Hospital de León CEIC del Hospital Clínico Universitario de Salamanca CEIC del Hospital General de Segovia CEIC del Hospital Virgen de la Concha CEIC del Complejo Hospitalario de Toledo CEIC del Hospital Universitario de Guadalajara CEIC del Hospital General Virgen de la Luz CEIC del Hospital General de Ciudad Real CEIC del Hospital Nuestra Señora del Prado CEIC del Hospital Clinic I Provincial de Barcelona CEIC del ICO de Girona, Hospital Dr. Josep Trueta CEIC del Hospital Joan XXIII CEIC del Institut Municipal d.Assistència Sanitària (IMAS) CEIC de la Corporació Sanitaria Parc Taulí CEIC Hospital Universitari de Bellvitge CEIC de Fundació de G.S. del Hospital de la Santa Creu i Sant Pau

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 78
Tercera versión: 30 de marzo de 2011
CEIC del Hospital Vall D'Hebron CEIC Fundació Unió Catalana dHospitals CEIC del Complejo Hospitalario de Cáceres CEIC Regional de Galicia CEIC de la Fundación Hospital Alcorcón CEIC del Hospital 12 de Octubre CEIC del Hospital Universitario La Paz CEIC del Hospital Universitario La Princesa CEIC del Hospital Universitario Puerta de Hierro CEIC del Hospital Ramón y Cajal CEIC del Hospital Clínico San Carlos CEIC del Hospital Gregorio Marañón CEIC Hospital de Madrid CEIC del Hospital Príncipe de Asturias CEIC del Hospital Severo Ochoa CEIC del Hospital General Morales Messeguer CEIC del Hospital Universitario Virgen de la Arrixaca CEIC Regional de Navarra CEIC de Euskadi CEIC del Hospital Arnau de Vilanova CEIC del Hospital General de Castellón CEIC del Hospital Clínico Universitario de Valencia CEIC del Hospital Universitario Doctor Peset Aleixandre CEIC del Hospital Universitario La Fe

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 79
Tercera versión: 30 de marzo de 2011
CEIC del Consorcio Hospital General Universitario d e Valencia
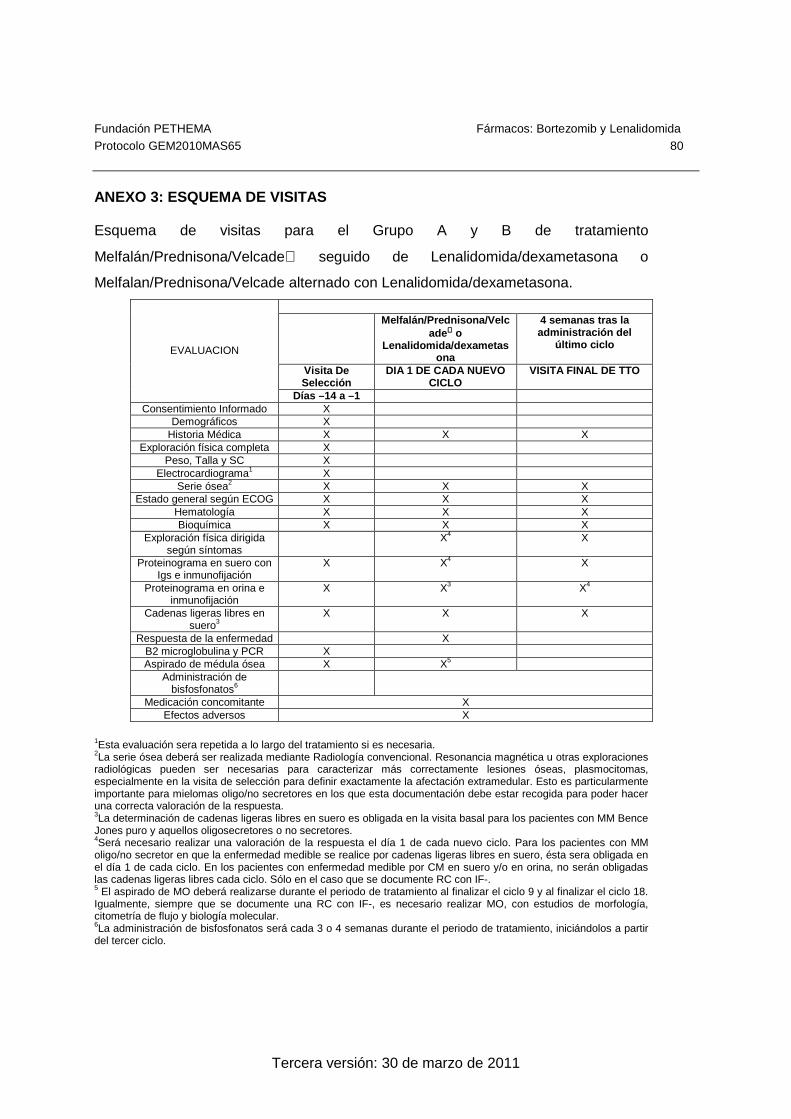
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 80
Tercera versión: 30 de marzo de 2011
ANEXO 3: ESQUEMA DE VISITAS
Esquema de visitas para el Grupo A y B de tratamiento
Melfalán/Prednisona/Velcade seguido de Lenalidomida/dexametasona o
Melfalan/Prednisona/Velcade alternado con Lenalidomida/dexametasona.
Melfalán/Prednisona/Velc
ade o Lenalidomida/dexametas
ona
4 semanas tras la administración del
último ciclo
Visita De Selección
DIA 1 DE CADA NUEVO CICLO
VISITA FINAL DE TTO
EVALUACION
Días –14 a –1 Consentimiento Informado X
Demográficos X Historia Médica X X X
Exploración física completa X Peso, Talla y SC X
Electrocardiograma1 X Serie ósea2 X X X
Estado general según ECOG X X X Hematología X X X Bioquímica X X X
Exploración física dirigida según síntomas
X4 X
Proteinograma en suero con Igs e inmunofijación
X X4 X
Proteinograma en orina e inmunofijación
X X3 X4
Cadenas ligeras libres en suero3
X X X
Respuesta de la enfermedad X B2 microglobulina y PCR X
Aspirado de médula ósea X X5 Administración de
bisfosfonatos6
Medicación concomitante X Efectos adversos X
1Esta evaluación sera repetida a lo largo del tratamiento si es necesaria. 2La serie ósea deberá ser realizada mediante Radiología convencional. Resonancia magnética u otras exploraciones radiológicas pueden ser necesarias para caracterizar más correctamente lesiones óseas, plasmocitomas, especialmente en la visita de selección para definir exactamente la afectación extramedular. Esto es particularmente importante para mielomas oligo/no secretores en los que esta documentación debe estar recogida para poder hacer una correcta valoración de la respuesta. 3La determinación de cadenas ligeras libres en suero es obligada en la visita basal para los pacientes con MM Bence Jones puro y aquellos oligosecretores o no secretores. 4Será necesario realizar una valoración de la respuesta el día 1 de cada nuevo ciclo. Para los pacientes con MM oligo/no secretor en que la enfermedad medible se realice por cadenas ligeras libres en suero, ésta sera obligada en el día 1 de cada ciclo. En los pacientes con enfermedad medible por CM en suero y/o en orina, no serán obligadas las cadenas ligeras libres cada ciclo. Sólo en el caso que se documente RC con IF-. 5 El aspirado de MO deberá realizarse durante el periodo de tratamiento al finalizar el ciclo 9 y al finalizar el ciclo 18. Igualmente, siempre que se documente una RC con IF-, es necesario realizar MO, con estudios de morfología, citometría de flujo y biología molecular. 6La administración de bisfosfonatos será cada 3 o 4 semanas durante el periodo de tratamiento, iniciándolos a partir del tercer ciclo.
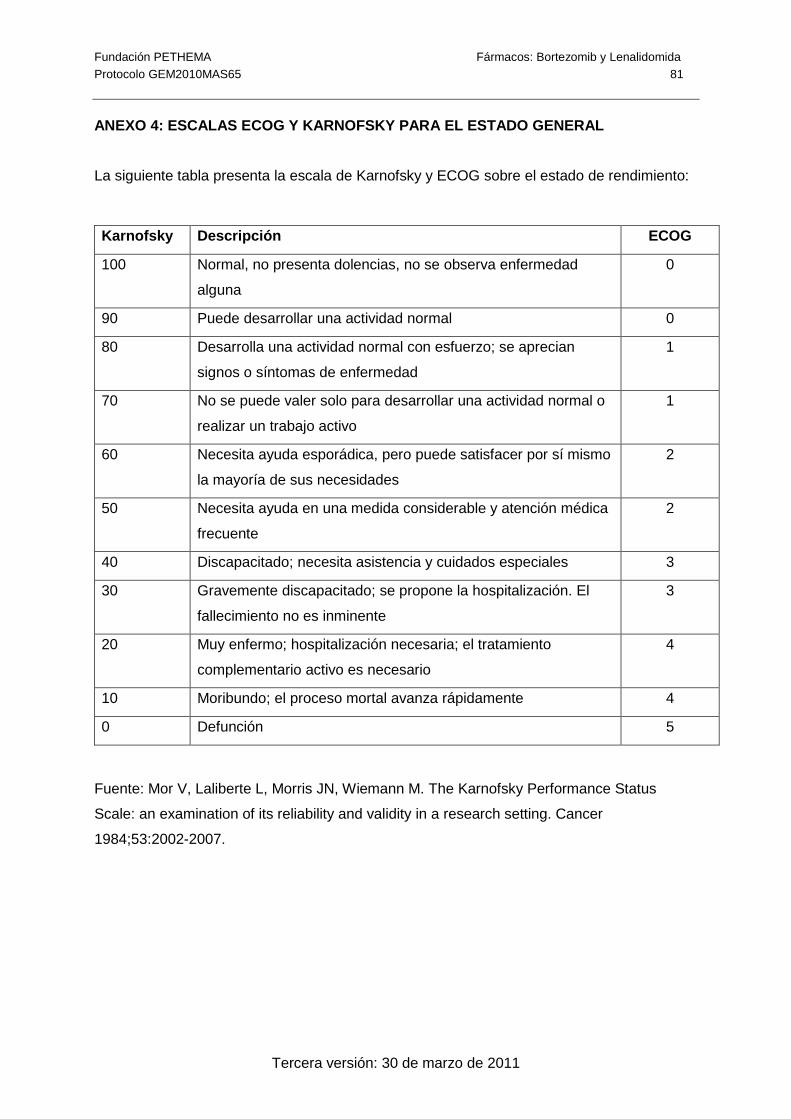
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 81
Tercera versión: 30 de marzo de 2011
ANEXO 4: ESCALAS ECOG Y KARNOFSKY PARA EL ESTADO GE NERAL
La siguiente tabla presenta la escala de Karnofsky y ECOG sobre el estado de rendimiento:
Karnofsky Descripción ECOG
100 Normal, no presenta dolencias, no se observa enfermedad
alguna
0
90 Puede desarrollar una actividad normal 0
80 Desarrolla una actividad normal con esfuerzo; se aprecian
signos o síntomas de enfermedad
1
70 No se puede valer solo para desarrollar una actividad normal o
realizar un trabajo activo
1
60 Necesita ayuda esporádica, pero puede satisfacer por sí mismo
la mayoría de sus necesidades
2
50 Necesita ayuda en una medida considerable y atención médica
frecuente
2
40 Discapacitado; necesita asistencia y cuidados especiales 3
30 Gravemente discapacitado; se propone la hospitalización. El
fallecimiento no es inminente
3
20 Muy enfermo; hospitalización necesaria; el tratamiento
complementario activo es necesario
4
10 Moribundo; el proceso mortal avanza rápidamente 4
0 Defunción 5
Fuente: Mor V, Laliberte L, Morris JN, Wiemann M. The Karnofsky Performance Status
Scale: an examination of its reliability and validity in a research setting. Cancer
1984;53:2002-2007.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 82
Tercera versión: 30 de marzo de 2011
ANEXO 5: FÓRMULAS UTILIZADAS EN ESTE PROTOCOLO
- Área de la superficie corporal:
El área de la superficie corporal se debe calcular mediante un nomograma estándar. A
continuación se muestra un ejemplo de nomograma:
ASC = 3600
)()( kgPesocmH ×
- Calcio corregido:
Se calculará mediante la siguiente fórmula y sólo en aquellos pacientes en los que, en la
visita de selección, la albúmina esté fuera de rango:
Ca corregido = Ca total – Albúmina + 4

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 83
Tercera versión: 30 de marzo de 2011
ANEXO 6: CRITERIOS DIAGNÓSTICOS DE MIELOMA MÚLTIPLE
MM SINTOMÁTICO
• CM en suero y/u orina
• Presencia de CP clonales* en médula ósea o presencia de plasmocitomas
• Sintomatología derivada de la afectación de órganos o tejidos (incluyendo
afectación ósea) por el Mieloma (ROTI (Myeloma-Related Organ or Tissue
Impairment due to the plasma cell proliferative process)).
* Si se realiza estudios de citometría de flujo, >90% de las células plasmáticas tendrán un fenotipo patológico. ** Algunos pacientes pueden no tener síntomas, pero sí deterioro de la función de algún órgano por afectación del mieloma.
Sintomatología derivada de afectación de órganos o tejidos por el Mieloma (ROTI) • *Hipercalcemia: Calcio corregido > 11,5 mg/dl • *Insuficiencia renal: Creatinina > 2 mg/dl • *Anemia: Hemoglobina 2g/dl por debajo del límite bajo de la normalidad o
Hemoglobina <10 g/dl • *Lesiones óseas: lesiones líticas u osteoporosis con fracturas que produzcan
compresión • Otros: Hiperviscosisdad, amiloidosis, infecciones recurrentes (>2 episodios en
1 año)
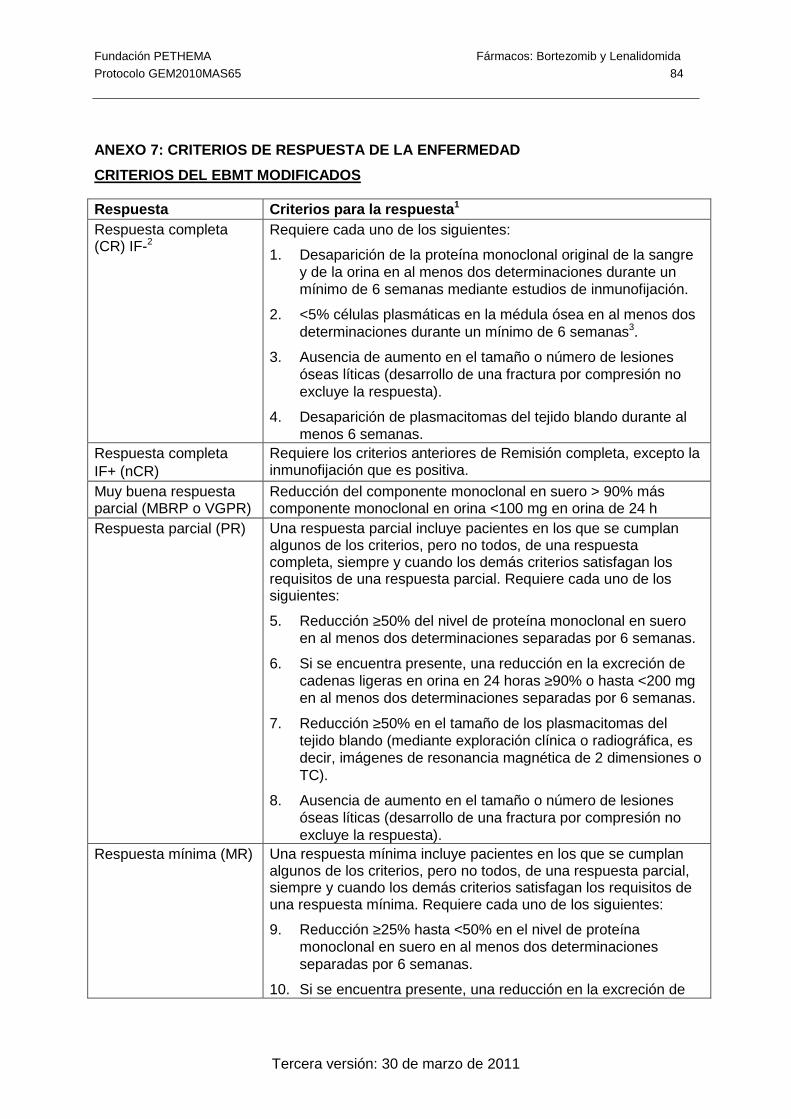
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 84
Tercera versión: 30 de marzo de 2011
ANEXO 7: CRITERIOS DE RESPUESTA DE LA ENFERMEDAD
CRITERIOS DEL EBMT MODIFICADOS Respuesta Criterios para la respuesta 1 Respuesta completa (CR) IF-2
Requiere cada uno de los siguientes:
1. Desaparición de la proteína monoclonal original de la sangre y de la orina en al menos dos determinaciones durante un mínimo de 6 semanas mediante estudios de inmunofijación.
2. <5% células plasmáticas en la médula ósea en al menos dos determinaciones durante un mínimo de 6 semanas3.
3. Ausencia de aumento en el tamaño o número de lesiones óseas líticas (desarrollo de una fractura por compresión no excluye la respuesta).
4. Desaparición de plasmacitomas del tejido blando durante al menos 6 semanas.
Respuesta completa IF+ (nCR)
Requiere los criterios anteriores de Remisión completa, excepto la inmunofijación que es positiva.
Muy buena respuesta parcial (MBRP o VGPR)
Reducción del componente monoclonal en suero > 90% más componente monoclonal en orina <100 mg en orina de 24 h
Respuesta parcial (PR) Una respuesta parcial incluye pacientes en los que se cumplan algunos de los criterios, pero no todos, de una respuesta completa, siempre y cuando los demás criterios satisfagan los requisitos de una respuesta parcial. Requiere cada uno de los siguientes:
5. Reducción ≥50% del nivel de proteína monoclonal en suero en al menos dos determinaciones separadas por 6 semanas.
6. Si se encuentra presente, una reducción en la excreción de cadenas ligeras en orina en 24 horas ≥90% o hasta <200 mg en al menos dos determinaciones separadas por 6 semanas.
7. Reducción ≥50% en el tamaño de los plasmacitomas del tejido blando (mediante exploración clínica o radiográfica, es decir, imágenes de resonancia magnética de 2 dimensiones o TC).
8. Ausencia de aumento en el tamaño o número de lesiones óseas líticas (desarrollo de una fractura por compresión no excluye la respuesta).
Respuesta mínima (MR) Una respuesta mínima incluye pacientes en los que se cumplan algunos de los criterios, pero no todos, de una respuesta parcial, siempre y cuando los demás criterios satisfagan los requisitos de una respuesta mínima. Requiere cada uno de los siguientes:
9. Reducción ≥25% hasta <50% en el nivel de proteína monoclonal en suero en al menos dos determinaciones separadas por 6 semanas.
10. Si se encuentra presente, una reducción en la excreción de
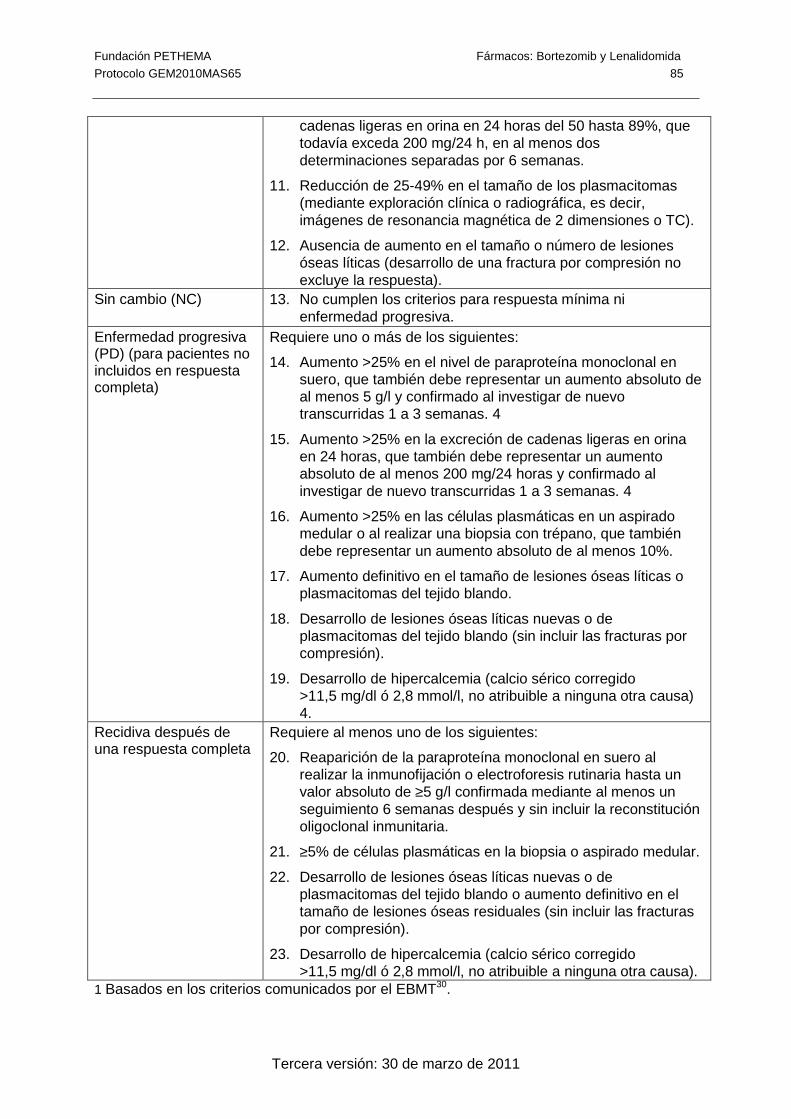
Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 85
Tercera versión: 30 de marzo de 2011
cadenas ligeras en orina en 24 horas del 50 hasta 89%, que todavía exceda 200 mg/24 h, en al menos dos determinaciones separadas por 6 semanas.
11. Reducción de 25-49% en el tamaño de los plasmacitomas (mediante exploración clínica o radiográfica, es decir, imágenes de resonancia magnética de 2 dimensiones o TC).
12. Ausencia de aumento en el tamaño o número de lesiones óseas líticas (desarrollo de una fractura por compresión no excluye la respuesta).
Sin cambio (NC) 13. No cumplen los criterios para respuesta mínima ni enfermedad progresiva.
Enfermedad progresiva (PD) (para pacientes no incluidos en respuesta completa)
Requiere uno o más de los siguientes:
14. Aumento >25% en el nivel de paraproteína monoclonal en suero, que también debe representar un aumento absoluto de al menos 5 g/l y confirmado al investigar de nuevo transcurridas 1 a 3 semanas. 4
15. Aumento >25% en la excreción de cadenas ligeras en orina en 24 horas, que también debe representar un aumento absoluto de al menos 200 mg/24 horas y confirmado al investigar de nuevo transcurridas 1 a 3 semanas. 4
16. Aumento >25% en las células plasmáticas en un aspirado medular o al realizar una biopsia con trépano, que también debe representar un aumento absoluto de al menos 10%.
17. Aumento definitivo en el tamaño de lesiones óseas líticas o plasmacitomas del tejido blando.
18. Desarrollo de lesiones óseas líticas nuevas o de plasmacitomas del tejido blando (sin incluir las fracturas por compresión).
19. Desarrollo de hipercalcemia (calcio sérico corregido >11,5 mg/dl ó 2,8 mmol/l, no atribuible a ninguna otra causa) 4.
Recidiva después de una respuesta completa
Requiere al menos uno de los siguientes:
20. Reaparición de la paraproteína monoclonal en suero al realizar la inmunofijación o electroforesis rutinaria hasta un valor absoluto de ≥5 g/l confirmada mediante al menos un seguimiento 6 semanas después y sin incluir la reconstitución oligoclonal inmunitaria.
21. ≥5% de células plasmáticas en la biopsia o aspirado medular.
22. Desarrollo de lesiones óseas líticas nuevas o de plasmacitomas del tejido blando o aumento definitivo en el tamaño de lesiones óseas residuales (sin incluir las fracturas por compresión).
23. Desarrollo de hipercalcemia (calcio sérico corregido >11,5 mg/dl ó 2,8 mmol/l, no atribuible a ninguna otra causa).
1 Basados en los criterios comunicados por el EBMT30.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 86
Tercera versión: 30 de marzo de 2011
1 Para la valoración correcta de la respuesta completa, la médula ósea deberá ser ≥20% celular y el calcio sérico deberá estar entre los límites normales.
2 Para documentar la respuesta completa es necesario tomar y evaluar una muestra de médula ósea. No se requiere la repetición de la extracción y valoración de médula ósea para confirmar la respuesta completa en pacientes con mieloma secretor que muestran una ausencia sostenida de proteína monoclonal al realizar la inmunofijación durante un mínimo de 6 semanas; sin embargo, se requiere la repetición de la extracción y valoración de médula ósea en la visita de confirmación de la respuesta para los pacientes con mieloma no secretor.
3 La necesidad de tratamiento urgente puede exigir que se repitan estas pruebas de forma prematura o que se elimine la repetición de un examen.
CRITERIOS IMWG DE RESPUESTA A LA ENFERMEDAD Adaptado con permiso de Durie et al. International uniform response criteria for multiple myeloma. Leukemia 2006; 20:1467-73 6
Categoria de respuesta Criterios de respuesta Remision Completa 1
(RC)
Requiere cada uno de los siguientes: • Inmunofijación negativa en suero y orina, y • Desaparición de todos los plasmocitomas de partes blandas, y • ≤ 5% de células plasmáticas en MO
Remisión completa estricta (RCs) 2
RC como se ha definido previamente, más: • Ratio de cadenas ligeras libres en suero normal, y • Ausencia de células plasmáticas con fenotipo patológico en MO
siendo necesario analizar un mínimo de 3000 células por citometría de flujo multiparamétrico (utilizando cuatro colores)
Muy buena Respueta Parcial(VGPR/MBRP) 1
• Componente Monoclonal en suero y orina detectable por inmunofijación, pero no por electroforesis, o
• Reducción del componente monoclonal en suero ≥ 90% más componente monoclonal en orina <100 mg en orina de 24 h
Respuesta Parcial (RP)
• Reducción del componente monoclonal en suero ≥50% y en orina de 24h > 90% o <200 mg en la orina de 24 h
• Si el componente monoclonal en suero y orina no son medibles, se requiere un descenso de ≥50% en la diferencia entre los niveles de cadenas ligeras libres afectada y no afectada en suero
• Si el componente monoclonal en suero y orina no son medibles, y tampoco las cadenas ligeras libres en suero, se requiere una reducción de ≥50% en la infiltración por células plasmáticas en MO (siempre que la infiltración basal sea ≥30%)
• Además de los criterios anteriores, si hubiera plasmocitomas de partes blandas al diagnóstico, éstos deberán haberse reducido 50% o más de su tamaño
Enfermedad Estable (EE) • No cumple criterios de RC, RCs, VGPR o PR ni progresión de la enfermedad
Enfermedad Progresiva (PD)1
Incremento de ≥25% con respecto al nivel más bajo alcanzado de uno o más de los siguientes parámetros:
• Componente monoclonal en suero (el incremento absoluto debe ser ≥ 0,5 g/dl) y/o
• Componente monoclonal en orina (el incremento absoluto debe ser ≥ 200 mg en orina de 24 horas) y/o
• Para los pacientes con enfermedad no medible en suero y orina por componente monoclonal: incremento de >25% de la diferencia entre los niveles de cadena ligera libre afectada y no

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 87
Tercera versión: 30 de marzo de 2011
Categoria de respuesta Criterios de respuesta afectada en suero (el incremento absoluto debe ser >10 mg/L)
• Incremento de ≥25% de la infiltración en MO por células plasmáticas (el incremento absoluto debe ser ≥ 10%)
• Aparición de nuevas lesiones óseas o plasmocitomas de partes blandas o aumento del tamaño de los previamente existentes.
• Aparición de hipercalcemia (calcio corregido en suero >11.5 mg/dl) que pueda ser atribuido exclusivamente al mieloma
Todos las categorías de respuesta (RC, RCs, VGPR, RP) requieren dos mediciones consecutivas realizadas en cualquier momento antes del inicio de un nuevo tratamiento; RC, RP y EE requieren también evidencia de no aparición de nuevas lesiones óseas si se realizaron radiografías. Los estudios radiológicos no son necesarios para establecer los criterios de respuesta antes indicados. Los estudios de MO no necesitan ser repetidos para confirmar una respuesta. 1Para codificar RC y VGPR de acuerdo a los actuales criterios en pacientes en que la enfermedad medible se hace solo por cadenas ligeras libres en suero: la RC en estos pacientes requiere una ratio de cadenas ligeras libres en suero normal entre 0.26-1.65 además de los criterios indicados anteriormente. La VGPR se define como un descenso >90% entre el nivel de cadena ligera libre afectada y no afectada en suero. 2La RC estricta ha sido actualizado y refleja la necesidad de citometría de flujo multiparamétrica como técnica de elección a la inmunofluorescencia o inmunohistoquímica.
Criterios para iniciar una nueva línea tratamiento:
- Recaída clínica/biológica de acuerdo a los criterios IMWG antes señalados en el apartado enfermedad progresiva, o
- Incremento significativo de la paraproteína o componente monoclonal en ausencia de datos de recaída clínica/biológica, si el componente monoclonal se duplica en dos mediciones consecutivas separadas en, al menos, dos meses; o si se produce un incremento de los valores absolutos de componente monoclonal de 1g/dl o 500 mg de proteinuria en orina de 24 horas, o de 20 mg/dl de la cadena ligera libre en suero afectada (más una ratio de cadenas ligeras libres en suero anormal) en dos mediciones consecutivas separadas en, al menos, dos meses.
Criterios adicionales de respuesta Categoria de respuesta Criterios de respuesta Respuesta Menor (RM) Sólo para pacientes con MM en recaída y refractarios
• Descenso de >25% y <49% del componente monoclonal en suero y reducción del componente monoclonal en orina de 24 horas entre el 50 y 89%, siendo éste superior a 200 mg en orina de 24 horas
• Si existiesen plasmocitomas de partes blandas, reducción del tamaño de los mismos entre un 25 y 49%
• No aumento en el número o tamaño de las lesiones óseas Remisión completa molecular
RC estricta, más: • ASO-PCR negativa, con una sensibilidad de 10-5

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 88
Tercera versión: 30 de marzo de 2011
ANEXO 8: CENTRALIZACION DE MUESTRAS DE MO
Se han establecido tres laboratorios de referencia (Hospital Clínico Universitario de Salamanca, Hospital 12 de Octubre de Madrid y Hospital La Fe de Valencia), que aún podrían incrementarse según la evolución del reclutamiento.
Distribución . Inicialmente, los Hospitales de la Comunidad de Castilla-León dirigirán sus muestras al Hospital Clínico de Salamanca mientras que los de la Comunidad de Madrid lo harán al Servicio de Hematología del Hospital 12 de Octubre y los de la Comunidad Valenciana al Hospital La Fe. El resto de los centros participantes podrán elegir según sus preferencias a cualquiera de los tres laboratorios de referencia (siempre el mismo).
Muestras . 10 ml de suero (seroteca), 8 ml de médula ósea en EDTA (CMF y DNA) y 5 ml de médula ósea en heparina-litio (citogenética).Las muestras serán enviadas a temperatura ambiente y en menos de 24 horas desde la extracción.
Identificación de las muestras . Código del paciente, códgio de protocolo, momento de la enfermedad (diagnóstico, ciclo 9, ciclo 18 u otra situación, como la documentación de la RC), hospital que lo envía y médico de contacto con teléfono.
Momento del envío. Se requieren varios sets de muestras, en el momento del diagnóstico, al finalizar los ciclos 9 y 18, si se alcanza RC por inmunofijación, o cualquier otra situación a criterio del investigador.
Transporte y contactos
Contenedores. Desde PETHEMA serán remitidos a la lista de hospitales contenedores aptos para el estudio. En cualquier caso los posibles fallos en la distribución o carencias puntuales pueden subsanarse solicitando contenedores a la secretaría de PETHEMA en Madrid, Rocío Aguirre, por medio de email: [email protected], o dejando un mensaje en el contestador del número: 915040259. En caso de solicitudes de urgencia pueden contactar en el móvil: 629853789
Horario. Se aceptarán muestras con horario de llegada de lunes a viernes, última hora de recepción 10 a.m. del viernes (lo que supone que las muestras deben extraerse de lunes a jueves ). Se ruega acordar la hora de entrega antes de las 10 de la mañana (consultar desde cada destino a la empresa de mensajería). Fuera de estos horarios se hace necesaria una llamada telefónica personal para acordar la entrega.
Transporte: Se dispone de transporte a cargo de la Fundación PETHEMA con la empresa MRW. Para solicitarlo es necesario informar a la operadora del número de abonado y del código de este proyecto.
Agencia MRW 2615 (Castilla 47. 28039- Madrid)
Teléfono: 913110051 (único para toda España)
Fundación PETHEMA (Nº de Abonado 15018)
Código de proyecto: GEM2010MAS65
Destinos:
Salamanca: Unidad de Inmunopatología Servicio de Hematología. Hospital Clínico Universitario. Pº San Vicente 58-182. 37007-Salamanca. Tfnos: 923291384 y 923291375. Fax 923294624. E-mail: [email protected]
Madrid: Unidad de Biología Molecular: Dra. Montalbán / Dr. Lahuerta / Dr. Martínez López
Servicio de Hematología. Hospital 12 de Octubre. Avda. de Córdoba s/n. 28041-Madrid Tfno: 913908000-Ext. 1027/1771; Fax 913908510. E-mail: [email protected]
Valencia: Servicio de Hematología. Hospital La Fe de Valencia. Avda Campanar 21
46009 Valencia. Telefono: 963 862 700 E-mail: [email protected]

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 89
Tercera versión: 30 de marzo de 2011
ANEXO 9: DECLARACIÓN DE HELSINKI
Principios éticos para las investigaciones médicas en seres humanos Adoptada por la
18ª Asamblea Médica Mundial, Helsinki, Finlandia, junio 1964 y enmendada por la
29ª Asamblea Médica Mundial, Tokio, Japón, octubre 1975 35ª Asamblea Médica Mundial, Venecia, Italia, octubre 1983 41ª Asamblea Médica Mundial, Hong Kong, septiembre 1989
48ª Asamblea GeneralSomerset West, Sudáfrica, octubre 1996 52ª Asamblea General, Edimburgo, Escocia, octubre 2000
Nota de Clarificación del Párrafo 29, agregada por la Asamblea General de la AMM, Washington 2002
Nota de Clarificación del Párrafo 30, agregada por la Asamblea General de la AMM, Tokio 2004
59ª Asamblea General, Seúl, Corea, octubre 2008 A. INTRODUCCION 1. La Asociación Médica Mundial (AMM) ha promulgado la Declaración de Helsinki como una propuesta de principios éticos para investigación médica en seres humanos, incluida la investigación del material humano y de información identificables. La Declaración debe ser considerada como un todo y un párrafo no debe ser aplicado sin considerar todos los otros párrafos pertinentes. 2. Aunque la Declaración está destinada principalmente a los médicos, la AMM insta a otros participantes en la investigación médica en seres humanos a adoptar estos principios. 3. El deber del médico es promover y velar por la salud de los pacientes, incluidos los que participan en investigación médica. Los conocimientos y la conciencia del médico han de subordinarse al cumplimiento de ese deber. 4. La Declaración de Ginebra de la Asociación Médica Mundial vincula al médico con la fórmula "velar solícitamente y ante todo por la salud de mi paciente”, y el Código Internacional de Etica Médica afirma que: "El médico debe considerar lo mejor para el paciente cuando preste atención médica”. 5. El progreso de la medicina se basa en la investigación que, en último término, debe incluir estudios en seres humanos. Las poblaciones que están subrepresentadas en la investigación médica deben tener un acceso apropiado a la participación en la investigación. 6. En investigación médica en seres humanos, el bienestar de la persona que participa en la investigación debe tener siempre primacía sobre todos los otros intereses. 7. El propósito principal de la investigación médica en seres humanos es comprender las causas, evolución y efectos de las enfermedades y mejorar las intervenciones preventivas, diagnósticas y terapéuticas (métodos, procedimientos y tratamientos). Incluso, las mejores intervenciones actuales deben ser evaluadas continuamente a través de la investigación para que sean seguras, eficaces, efectivas, accesibles y de calidad. 8. En la práctica de la medicina y de la investigación médica, la mayoría de las intervenciones implican algunos riesgos y costos.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 90
Tercera versión: 30 de marzo de 2011
9. La investigación médica está sujeta a normas éticas que sirven para promover el respeto a todos los seres humanos y para proteger su salud y sus derechos individuales. Algunas poblaciones sometidas a la investigación son particularmente vulnerables y necesitan protección especial. Estas incluyen a los que no pueden otorgar o rechazar el consentimiento por sí mismos y a los que pueden ser vulnerables a coerción o influencia indebida. 10. Los médicos deben considerar las normas y estándares éticos, legales y jurídicos para la investigación en seres humanos en sus propios países, al igual que las normas y estándares internacionales vigentes. No se debe permitir que un requisito ético, legal o jurídico nacional o internacional disminuya o elimine cualquiera medida de protección para las personas que participan en la investigación establecida en esta Declaración. B. PRINCIPIOS PARA TODA INVESTIGACION MÉDICA 11. En la investigación médica, es deber del médico proteger la vida, la salud, la dignidad, la integridad, el derecho a la autodeterminación, la intimidad y la confidencialidad de la información personal de las personas que participan en investigación. 12. La investigación médica en seres humanos debe conformarse con los principios científicos generalmente aceptados y debe apoyarse en un profundo conocimiento de la bibliografía científica, en otras fuentes de información pertinentes, así como en experimentos de laboratorio correctamente realizados y en animales, cuando sea oportuno. Se debe cuidar también del bienestar de los animales utilizados en los experimentos. 13. Al realizar una investigación médica, hay que prestar atención adecuada a los factores que puedan dañar el medio ambiente. 14. El proyecto y el método de todo estudio en seres humanos debe describirse claramente en un protocolo de investigación. Este debe hacer referencia siempre a las consideraciones éticas que fueran del caso y debe indicar cómo se han considerado los principios enunciados en esta Declaración. El protocolo debe incluir información sobre financiamiento, patrocinadores, afiliaciones institucionales, otros posibles conflictos de interés e incentivos para las personas del estudio y estipulaciones para tratar o compensar a las personas que han sufrido daños como consecuencia de su participación en la investigación. El protocolo debe describir los arreglos para el acceso después del ensayo a intervenciones identificadas como beneficiosas en el estudio o el acceso a otra atención o beneficios apropiados. 15. El protocolo de la investigación debe enviarse, para consideración, comentario, consejo y aprobación, a un comité de ética de investigación antes de comenzar el estudio. Este comité debe ser independiente del investigador, del patrocinador o de cualquier otro tipo de influencia indebida. El comité debe considerar las leyes y reglamentos vigentes en el país donde se realiza la investigación, como también las normas internacionales vigentes, pero no se debe permitir que éstas disminuyan o eliminen ninguna de las protecciones para las personas que participan en la investigación establecidas en esta Declaración. El comité tiene el derecho de controlar los ensayos en curso. El investigador tiene la obligación de proporcionar información del control al comité, en especial sobre todo incidente adverso grave. No se debe hacer ningún cambio en el protocolo sin la consideración y aprobación del comité. 16. La investigación médica en seres humanos debe ser llevada a cabo sólo por personas con la formación y calificaciones científicas apropiadas. La investigación en pacientes o

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 91
Tercera versión: 30 de marzo de 2011
voluntarios sanos necesita la supervisión de un médico u otro profesional de la salud competente y calificado apropiadamente. La responsabilidad de la protección de las personas que toman parte en la investigación debe recaer siempre en un médico u otro profesional de la salud y nunca en los participantes en la investigación, aunque hayan otorgado su consentimiento. 17. La investigación médica en una población o comunidad con desventajas o vulnerable sólo se justifica si la investigación responde a las necesidades y prioridades de salud de esta población o comunidad y si existen posibilidades razonables de que la población o comunidad, sobre la que la investigación se realiza, podrá beneficiarse de sus resultados. 18. Todo proyecto de investigación médica en seres humanos debe ser precedido de una cuidadosa comparación de los riesgos y los costos para las personas y las comunidades que participan en la investigación, en comparación con los beneficios previsibles para ellos y para otras personas o comunidades afectadas por la enfermedad que se investiga. 19. Todo ensayo clínico debe ser inscrito en una base de datos disponible al público antes de aceptar a la primera persona. 20. Los médicos no deben participar en estudios de investigación en seres humanos a menos de que estén seguros de que los riesgos inherentes han sido adecuadamente evaluados y de que es posible hacerles frente de manera satisfactoria. Deben suspender inmediatamente el experimento en marcha si observan que los riesgos que implican son más importantes que los beneficios esperados o si existen pruebas concluyentes de resultados positivos o beneficiosos. 21. La investigación médica en seres humanos sólo debe realizarse cuando la importancia de su objetivo es mayor que el riesgo inherente y los costos para la persona que participa en la investigación. 22. La participación de personas competentes en la investigación médica debe ser voluntaria. Aunque puede ser apropiado consultar a familiares o líderes de la comunidad, ninguna persona competente debe ser incluida en un estudio, a menos que ella acepte libremente. 23. Deben tomarse toda clase de precauciones para resguardar la intimidad de la persona que participa en la investigación y la confidencialidad de su información personal y para reducir al mínimo las consecuencias de la investigación sobre su integridad física, mental y social. 24. En la investigación médica en seres humanos competentes, cada individuo potencial debe recibir información adecuada acerca de los objetivos, métodos, fuentes de financiamiento, posibles conflictos de intereses, afiliaciones institucionales del investigador, beneficios calculados, riesgos previsibles e incomodidades derivadas del experimento y todo otro aspecto pertinente de la investigación. La persona potencial debe ser informada del derecho de participar o no en la investigación y de retirar su consentimiento en cualquier momento, sin exponerse a represalias. Se debe prestar especial atención a las necesidades específicas de información de cada individuo potencial, como también a los métodos utilizados para entregar la información. Después de asegurarse de que el individuo ha comprendido la información, el médico u otra persona calificada apropiadamente debe pedir entonces, preferiblemente por escrito, el consentimiento informado y voluntario de la

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 92
Tercera versión: 30 de marzo de 2011
persona. Si el consentimiento no se puede otorgar por escrito, el proceso para lograrlo debe ser documentado y atestiguado formalmente. 25.Para la investigación médica en que se utilice material o datos humanos identificables, el médico debe pedir normalmente el consentimiento para la recolección, análisis, almacenamiento y reutilización. Podrá haber situaciones en las que será imposible o impracticable obtener el consentimiento para dicha investigación o podría ser una amenaza para su validez. En esta situación, la investigación sólo puede ser realizada después de ser considerada y aprobada por un comité de ética de investigación. 26. Al pedir el consentimiento informado para la participación en la investigación, el médico debe poner especial cuidado cuando el individuo potencial está vinculado con él por una relación de dependencia o si consiente bajo presión. En una situación así, el consentimiento informado debe ser pedido por una persona calificada adecuadamente y que nada tenga que ver con aquella relación. 27. Cuando el individuo potencial sea incapaz, el médico debe pedir el consentimiento informado del representante legal. Estas personas no deben ser incluidas en la investigación que no tenga posibilidades de beneficio para ellas, a menos que ésta tenga como objetivo promover la salud de la población representada por el individuo potencial y esta investigación no puede realizarse en personas competentes y la investigación implica sólo un riesgo y costo mínimos. 28. Si un indivividuo potencial que participa en la investigación considerado incompetente es capaz de dar su asentimiento a participar o no en la investigación, el médico debe pedirlo, además del consentimiento del representante legal. El desacuerdo del individuo potencial debe ser respetado. 29. La investigación en individuos que no son capaces física o mentalmente de otorgar consentimiento, por ejemplo los pacientes inconscientes, se puede realizar sólo si la condición física/mental que impide otorgar el consentimiento informado es una característica necesaria de la población investigada. En estas circunstancias, el médico debe pedir el consentimiento informado al representante legal. Si dicho representante no está disponible y si no se puede retrasar la investigación, el estudio puede llevarse a cabo sin consentimiento informado, siempre que las razones específicas para incluir a individuos con una enfermedad que no les permite otorgar consentimiento informado hayan sido estipuladas en el protocolo de la investigación y el estudio haya sido aprobado por un comité de ética de investigación. El consentimiento para mantenerse en la investigación debe obtenerse a la brevedad posible del individuo o de un representante legal. 30. Los autores, directores y editores todos tienen obligaciones éticas con respecto a la publicación de los resultados de su investigación. Los autores tienen el deber de tener a la disposición del público los resultados de su investigación en seres humanos y son responsables de la integridad y exactitud de sus informes. Deben aceptar las normas éticas de entrega de información. Se deben publicar tanto los resultados negativos e inconclusos como los positivos o de lo contrario deben estar a la disposición del público.En la publicación se debe citar la fuente de financiamiento, afiliaciones institucionales y conflictos de intereses. Los informes sobre investigaciones que no se ciñan a los principios descritos en esta Declaración no deben ser aceptados para su publicación. C. PRINCIPIOS APLICABLES CUANDO LA INVESTIGACION ME DICA SE COMBINA CON LA ATENCION MEDICA

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida
Protocolo GEM2010MAS65 93
Tercera versión: 30 de marzo de 2011
31. El médico puede combinar la investigación médica con la atención médica, sólo en la medida en que tal investigación acredite un justificado valor potencial preventivo, diagnóstico o terapéutico y si el médico tiene buenas razones para creer que la participación en el estudio no afectará de manera adversa la salud de los pacientes que toman parte en la investigación. 32. Los posibles beneficios, riesgos, costos y eficacia de toda intervención nueva deben ser evaluados mediante su comparación con la mejor intervención probada existente, excepto en las siguientes circunstancias: - El uso de un placebo, o ningún tratamiento, es aceptable en estudios para los que no hay una intervención probada existente. - Cuando por razones metodológicas, científicas y apremiantes, el uso de un placebo es necesario para determinar la eficacia y la seguridad de una intervención que no implique un riesgo, efectos adversos graves o daño irreversible para los pacientes que reciben el placebo o ningún tratamiento. Se debe tener muchísimo cuidado para evitar abusar de esta opción. 33. Al final de la investigación, todos los pacientes que participan en el estudio tienen derecho a ser informados sobre sus resultados y compartir cualquier beneficio, por ejemplo, acceso a intervenciones identificadas como beneficiosas en el estudio o a otra atención apropiada o beneficios. 34. El médico debe informar cabalmente al paciente los aspectos de la atención que tienen relación con la investigación. La negativa del paciente a participar en una investigación o su decisión de retirarse nunca debe perturbar la relación médico-paciente. 35. Cuando en la atención de un enfermo las intervenciones probadas han resultado ineficaces o no existen, el médico, después de pedir consejo de experto, con el consentimiento informado del paciente, puede permitirse usar intervenciones no comprobadas, si, a su juicio, ello da alguna esperanza de salvar la vida, restituir la salud o aliviar el sufrimiento. Siempre que sea posible, tales intervenciones deben ser investigadas a fin de evaluar su seguridad y eficacia. En todos los casos, esa información nueva debe ser registrada y, cuando sea oportuno, puesta a disposición del público. Traducción realizada por el Departamento de Internacional del Consejo General de Colegios
Oficiales de Médicos. Madrid. Noviembre 2008.

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 94
Tercera versión: 30 de marzo de 2011
ANEXO 10: PROGRAMA DE PREVENCIÓN DE EMBARAZO (PPE) - LENALIDOMIDA
(Esta versión del PPE con fecha del 24/feb/09 debe aplicarse durante este Estudio. Cualquier
modificación posterior reemplazará esta versión en su totalidad)
Advertencia sobre embarazos Lenalidomida está relacionada estructuralmente con talidomida. Talidomida es una sustancia activa con conocidos efectos teratogénicos en el ser humano, que causa defectos congénitos severos y amenazantes para la vida. Lenalidomida induce, en primates, malformaciones parecidas a las descritas con talidomida. Se espera que lenalidomida tenga efectos teratogénicos en humanos si se toma durante el embarazo. Las condiciones del Programa de Prevención de Embarazo deben ser cumplidas por todos los pacientes a menos que haya pruebas fiables de que no existe capacidad de gestación. Criterios correspondientes a mujeres sin capacidad de gestación: Se considera que una paciente o la pareja mujer de un paciente tiene capacidad de gestación a menos que cumpla alguno de los siguientes criterios: • Edad ≥ 50 años y amenorrea natural desde hace ≥ 1 año* • Fallo ovárico prematuro confirmado por un ginecólogo especialista • Salpingoooforectomía bilateral o histerectomía previa • Genotipo XY, síndrome Turner o agenesia uterina. * La amenorrea secundaria al tratamiento antineoplásico no descarta la capacidad de gestación.
Criterios de inclusión de pacientes
Los pacientes deberán cumplir todos los criterios de inclusión expuestos a continuación para poder participar en el ensayo:
• Todo paciente varón deberá:
1.1. Comprometerse a utilizar preservativos durante todo el tratamiento con el fármaco en estudio, incluidos todos los períodos de interrupción de dosis y hasta una semana después de finalizar el tratamiento si su pareja es una mujer con capacidad de gestación y no utiliza métodos anticonceptivos.
1.2. Comprometerse a no donar semen durante el tratamiento con el fármaco en estudio y hasta una semana después de finalizar el tratamiento.
• Todos los pacientes deberán:
1.3. Abstenerse de donar sangre mientras reciban el tratamiento con el fármaco en estudio y durante la semana siguiente a la finalización del mismo.
1.4. Abstenerse de compartir el fármaco en estudio con otras personas y devolver todo el fármaco en estudio no utilizado al investigador o al farmacéutico.
Informes de embarazo
Los embarazos o sospechas de embarazo (incluida una prueba de embarazo positiva sin tener en
cuenta la edad ni el estado de la enfermedad) de la pareja de un paciente varón que se produce
mientras el paciente está tomando el fármaco en estudio, o en un plazo de 28 días desde la última
dosis del fármaco en estudio del paciente se consideran episodios que se deben comunicar

Fundación PETHEMA Fármacos: Bortezomib y Lenalidomida Protocolo GEM2010MAS65 95
Tercera versión: 30 de marzo de 2011
inmediatamente al Promotor y éste a Celgene. Si el paciente está tomando el fármaco en estudio,
éste debe ser interrumpido inmediatamente y el paciente debe ser instruido para que devuelva
cualquier parte no utilizada del fármaco en estudio al investigador/es. El embarazo, sospecha de
embarazo o prueba de embarazo positiva debe ser comunicada al Promotor quien informará al
Departamento de Seguridad del Medicamento de Celgen e en España en el teléfono: 630 56 48
73, Fax: 91 422 90 95, email: drugsafety-spain@celg ene.com inmediatamente por teléfono o por
fax con el Formulario de Informe de embarazo.
La mujer debe ser remitida a un tocólogo/ginecólogo con experiencia en toxicidad reproductiva para
posterior evaluación y asesoramiento.
El Investigador/es hará un seguimiento de la mujer hasta que complete el embarazo y debe informar
al Promotor y éste al Departamento de Seguridad del Medicamento de Celgen e en España en el
tel.: 630 56 48 73, Fax: 91 422 90 95, email: drugs [email protected] de los resultados del
embarazo (incluidas las notificaciones de pruebas positivas falsas) en un plazo de 24 horas de tener
conocimiento del caso como un seguimiento del informe inicial.
Si el resultado del embarazo cumple los criterios para la clasificación inmediata de AAG (esto es,
aborto espontáneo o terapéutico [cualquier malformación congénita detectada en un feto abortado se
debe documentar], mortinatalidad, muerte neonatal o anomalía congénita [incluida la de un feto
abortado]), el Investigador/es debe seguir los procedimientos para el informe de AAG (esto es,
informar al Departamento de Seguridad del Medicamento de Celgen e en España en el tel.: 630
56 48 73, Fax: 91 422 90 95, email: drugsafety-spai [email protected] por fax en el plazo de 24
horas desde que el Investigador conozca el caso).
Se debe informar de todas las muertes neonatales que se produzcan en un plazo de 28 días desde
el nacimiento, sin tener en cuenta la causa, como AAG. Además, cualquier muerte infantil después
de 28 días que el Investigador/es sospeche que está relacionada con la exposición del útero al
fármaco en estudio debe ser comunicada al Promotor que a su vez informará al Departamento de
Seguridad del Medicamento de Celgene en España en e l tel.: 630 56 48 73, Fax: 91 422 90 95,
email: [email protected] por fax en un plazo de 24 horas desde que el
Investigador/es conozca el caso.
Hombres:
Las parejas mujeres de hombres que estén tomando el fármaco en investigación serán aconsejadas
para avisar a su profesional sanitario inmediatamente si se quedan embarazadas. El hombre debe
notificar al Investigador del embarazo de su pareja y darle la información del profesional sanitario. El
investigador proporcionará entonces esta información al Promotor y a Celgene para el seguimiento
según sea necesario.