papayas de exportación y criollas con resistencia a …glifos.concyt.gob.gt/digital/fodecyt/fodecyt...
TRANSCRIPT

Papayas de Exportación y Criollas con Resistencia a Papaya Ringspot Virus
(PRSV)
Reporte Final del Proyecto # 9 FONACYT
Consejo Nacional de Ciencia y Tecnología CONCYT
UNIVERSIDAD DEL VALLE DE GUATEMALA Dr. Guillermo E. Sánchez Licda. Margarita Palmieri
Licda. Laura Vergara Luis Lopez
UNIVERSITY OF GEORGIA Dr. Wayne Parrott Dr. Michael Deom
Guatemala, 15 de Octubre de 1999

INDICE
1 . RESUMEN DEL PROYECTO .................................................................................................................. 2
11 . ANTECEDENTES ....................................................................................................................................... 2
111 . OBJETIVOS ................................................................................................................................................ 4
IV . MATERIALES Y METODOS .............................................................................................................. 5
A . Sección de Trabajos con el virus PRSV ............................................................................................... 5 1 . Evaluación de la presencia de papaya ringspot virus (PRSV) en campos de papaya en
.............................................................................................................................................. Guatemala 5 2 . Purificación del virus y evaluación del virus purificado ............................................................... 7 3 . Selección. aislamiento y secuenciación del gen de la cáside de PRSV ......................................... 9 4 . Tranformación de A . tumefaciens e introducción del fragmento de gen a la planta ............. 11 . . . . 5 . Confirmacion de la transformacion ................................................................................................ 13
B . Cultivo de tejidos de papaya para generación de embriones somáticos y regeneración de plantas in vitro .............................................................................................................................. 14
1 . Embriogénesis somática .................................................................................................................. 14 2 . Regeneración de plántulas de papaya a partir de embriones somáticos ................................. 15
A . Sección de Trabajos con el virus PRSV ............................................................................................. 17 1 . Evaluación de la presencia de papaya ringspot virus (PRSV) en campos de papaya en
Guatemala ............................................................................................................................................ 17 2 . Purificación del virus y evaluación del virus purificado ............................................................. 22 3 . Selección. aislamiento y secuenciación del gen ............................................................................ 22 4 . Tranformación de bacteriana e introducción del fragmento de gen a la planta ................... 23 . . 5 . Confirmación de la transformacion ............................................................................................... 27
B . Cultivo de tejidos de papaya para generación de embriones somáticos y regeneración de plantas in vitro .............................................................................................................................. 29
1 . Embriogénesis somática .................................................................................................................. 29 2 . Regeneración de plántulas de papaya a partir de embriones somáticos .................................. 33
VI . CONCLUSIONES Y RECOMENDACIONES ................................................................................ 37
VI1 . IMPACTO DEL PROYECTO ........................................................................................................... 38
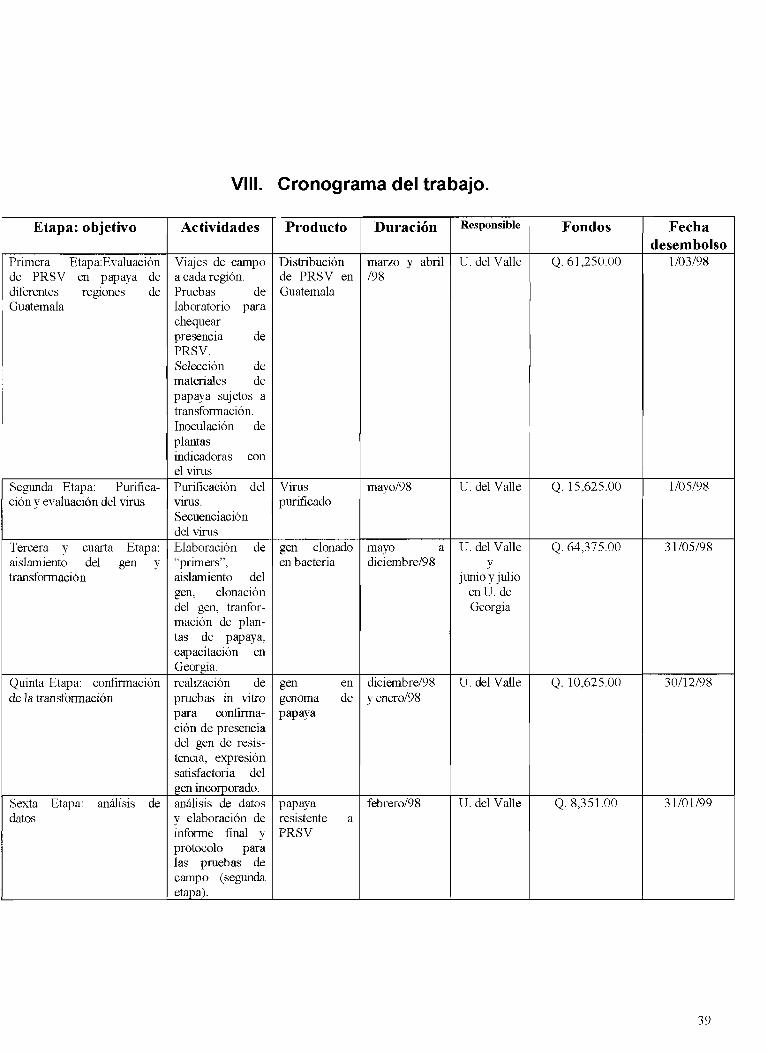
VI11 . CRONOGRAMA DEL TRABAJO .................................................................................................... 39
IX . BIBLIOGRAFIA ................................................................................................................................... 40
.................................................... X . EXPECTATIVAS SOBRE PUBLlCACIONES O PATENTES 43
.......................................................................................................................................... XI . APENDICES 44

PAPAYAS (Carica papaya L.) DE EXPORTACION Y CRIOLLAS CON RESISTENCIA A PAPAYA RINGSPOT VIRUS (PRSV).
Investigadores principales: Dr. Guillermo Sánchez y Liwla. Margarita Palrnieri (Universidad del Valle de Guatemala)
Investigadores asociados: Dr. Waqne Parrott (Universidad de Creorga) Dr. Carl Michael Deom (Universidad de Georgia)
Palabras clave: transghiw; imprimador, plásmido, tranformación, capside, auxina, explantes; embriogénesis, cultivo de tejidos, in vitro, virus, papaya.
l . Resumen del proyecto.
La producción de papaya de buena calidad en Guatemala y otras latitudes se ve amenazada por enfermedades virales que afectan al cultivo, siendo la principal la mancha anular causada por el papaya ringspot virus (PRSV). No se ha encontrado resistencia natural a este virus por lo que la investigación se ha volcado a la incorporación de genes de resistencia a PRSV, a plantas de papaya mediante transformación genética. Se ha reportado en varios trabajos recientemente, que plantas transgénicas expresando el gen de la cápside del virus muestran niveles altos de resistencia a la infección de ese virus o cepas de ese virus. Este fenómeno es conocido como resistencia de la cápside (Grumet, 1994).
El objetivo de este trabajo fue la generación de plantas transgénicas de papaya (tipo solo (Hawaiana) y criollas) resistentes cepas nativas del virus PRSV. Para el efecto se hizo inicialmente una evaluación de las diferentes cepas de PRSV existentes en los campos de papayas para poder seleccionar el genoma vira1 a purificar. Seguidamente se purificó el virus para identificar y secuenciar el gen de interés siendo éste el de la cápside viral. El gen de la cápside se insertó al vector (plásmido resistente a antibióticos) utilizado para su inserción en el genoma de plantas de papaya. Una vez llevada a cabo la transformación, se seleccionaron las células transgénicas,verifícándose la transformación mediante el uso de medios de cultivo con antibióticos los antibióticos mefoxin e higromicincr Corno resultado se obtuvieron embriones somáticos transformados tanto de papaya criolla como hawaiana.
II. Antecedentes.
La papaya es una de las frutas más ampliamente cultivadas en regiones tropicales y subtropicales por su sabor y su riqueza nutricional en vitaminas A y C, además de ser fuente de papaína, enzima digestiva de mucha utilidad en la industria. Su producción está muy restringida en diversas áreas del mundo por la mancha anular causada por el papaya ringspot virus (PRSV). Algunos síntomas típicos inducidos por este virus incluyen manchas en forma de anillo en el fruto y enanismo de las plantas, pudiendo conducir a

una reducción drástica en la producción. Asimismo dificulta la aceptación del producto para exportación.
Hasta el momento, la papaya en Guatemala no es un producto en el mercado internacional por diversas razones entre los que destacan la falta de tecnología de producción adecuada, las enfermedades, uso de semillas criollas no seleccionadas, etc. La papaya sin embargo se considera con gran potencial para ser un rubro muy alto en las exportaciones guatemaltecas, ya que estudios en Estados Unidos indican que la papaya tiene una demanda creciente de 15 años (Sullivan, comunicación personal). Además, Guatemala posee el clima y extensiones grandes con las condiciones apropiadas para su cultivo. El cultivo de papaya para consumo interno es de alta rentabilidad y de gran aceptación forma fiesca y procesada.
Sin embargo, para Guatemala, la mayor restricción para la exportación de papaya al exterior ha sido la infestación por la mosca del mediterráneo; este producto puede ser cultivado con éxito y de hecho se está sembrando en el área de Petén, la cual se espera sea reconocida por los Estados Unidos como zona libre de mosca del mediterráneo en un futuro próximo. Además existen en la actualidad tratamientos térmicos aplicados previamente a la exportación (similares a los utilizados con el mango) y que son aceptados por las regulaciones de exportación en diferentes países.
El PRSV es un potivirus transmitido por áfidos (Hom0ptera:aphididae) de manera no persistente. Un campo de papaya joven puede quedar totalmente infectado en el lapso de 3 a 4 meses. Esfuerzos para el control del vector con insecticidas de todo tipo no han sido efectivos, aunque se ha obtenido escaso éxito con el uso de aceites, ya que éstos interfieren ccn la adquisición del virus por parte del vector, al introducir éste su estilete en los tejidos vegetales (Clough, G., et al, 1995). Para el control del PRSV también se han hecho esfuerzos incluyendo protección cruzada pero no han dado los resultados esperados (Ploetz, R. et al., 1994). La situación anterior se agrava con la falta de resistencia natural (Fitch, M. et al., 1992), por lo que la incorporación de resistencia a este virus a las plantas de papaya representa la opción más viable para el control de la mancha anular. Esta resistencia surte efecto ya que el virus debe liberarse de la cápside antes de poder replicar su ARN, proceso indispensable para que la enfermedad pueda proseguir su curso. La producción en planta de proteína de cápside viral evita el ensamblaje de nuevas partículas virales. Debido a esto se ha mostrado que plantas con el gen de las proteínas de la cápside incorporado, están protegidas contra la infección de ese virus (Clough, G., et al., 1995).
La incorporación de resistencia de un virus a una planta, es una práctica que tiene ventajas sobre la obtención de resistencia del hospedero mismo. La ventaja de los genes virales es que pueden ser identificados, aislados y clonados fácilmente debido a los genomas pequeños de los virus. En cambio, en el hospedero esto se dificulta tanto por el tamaño del genoma como por las grandes cantidades de ADN repetitivo que puede tener un organismo. Este tipo de resistencia se deriva del concepto clásico de protección cruzada. Se hipotetiza que la presencia de algún componente viral (cápside) es responsable de conferir protección, interfiriendo con el anclaje, entrada y con la falta de

formación de cápside del virus. También puede ser debido a competencia de factores limitantes o interferencia con la replicación (Grumet, 1994).
Se ha reportado en varios trabajos recientemente que plantas transgénicas expresando el gen de la cápside del virus muestra niveles altos de resistencia a la infección de ese virus o cepas de ese virus. Este fenómeno de resistencia conferida por la expresión de proteína vira1 de la envoltura proteica es conocido como resistencia de la cápside. Se pueden utilizar dos metodologías para lograr introducir esta resistencia a las plantas, mediante una bacteria Agvobactevium tumefaciens (Yang, J . S., et al., 1996) y mediante una "pistola" a presión (Fitch, m. M. M. et al., 1992). En este proyecto se ha empleado la primera metodología ya que es tecnología de más fácil acceso y aplicabilidad en Guatemala.
Hasta el momento, casi todos los estudios de resistencia mediada por la cápside han sido realizados en países desarrollados, dejando a los países en desarrollo dependientes de esta tecnología. Esto tiene como consecuencia que la selección de cultivos y técnicas de transformación genética son las que son de importancia a ellos o bien se ajustan a sus condiciones e intereses particulares. Como ejemplo de lo anterior, un grupo de la Universidad de Cornell (Fitch M. M. M. et al, 1990; Namba, S., et. a1.,1992; Tricoli, D. et al., 1995; Fuchs, M. and D.Gonsalves, 1995) ha estado trabajando en este aspecto y con papaya hawaiana, reportando que han utilizado el gen de una cepa hawaiana, consiguiendo resistencia únicamente a esa cepa. Al exponer las papayas transgénicas a cepas virales de otras regiones, éstas son susceptibles a ser infectadas, perdiendo toda resistencia. Por esto es fundamental clonar el gen del virus guatemalteco para obtener papayas transformadas resistentes a las cepas guatemaltecas.
Un aspecto importante es que este estudio intenta abarcar no sólo germoplasma de papayas hawaianas de exportación (tipo "solo ") sino involucrar germoplasma de papaya criolla para incrementar y mejorar el mercado interno así como para conservar el patrimonio natural y ayudar al pequeño agricultor. Es importante hacer notar que la comercialización de papayas transgénicas ya ha sido aprobada por el congreso de los Estados Unidos, por lo que no existen medidas legales o cuarentenarias que dificulten su exportación y mercadeo.
III. Objetivos.
General: Introducir el gen de resistencia a la cápside del virus de la mancha anular a papayas criollas y de exportación guatemaltecas.
Específicos:
1- Detectar e identificar el virus de la mancha anular en cultivos de papaya criolla y tipo solo en Guatemala 2- Purificar cepas nativas del PRSV-p, aislar el gen codificante de la envoltura proteica (cápside) y clonar este gen.

3- Transformar 2 variedades seleccionadas de papaya, 1 criolla y la variedad hawaiana "Sunrise" utilizando para ello la técnica molecular de transformación mediada por Agrobacterium tumefaciens y embriogénesis somática de explantes de papaya.
4- Corroborar la transformación genética de los embriones somáticos de papaya utilizando los antibióticos mefoxin e higromicina.
IV. Materiales y métodos.
La hipótesis de trabajo fue que el gen de la cápside de cepas nativas del PRSV sería introducido exitosamente a tejido vegetal in vztro de 2 variedades de papaya.
Para comprobar la hipótesis anterior, este trabajo se dividió en dos aspectos importantes, llevados a cabo en diferentes laboratorios pero coordinados con los resultados que se iban obteniendo en ambos. Por esta razón, tanto la metodología como los resultados se han dividido en dos secciones con lo cual la información resultará mas clara para el lector. En la sección "A", se describe todo lo relativo a la colecta de muestras de papaya infectadas de PRSV, la detección del virus, su aislamiento, amplificación y purificación, así como la obtención del gen de la cápside del mismo, su inserción a los vectores necesarios, su introducción a Agrobacteviunz tumefaciens y finalmente la transformación de los embriones de papaya. En la sección "Bu se describe la obtención de plántulas de papaya a partir de semilla madura tanto de papaya criolla como hawaiana a través de técnicas in vitro.
A. Seccidn de trabajos con el virus PRSV.
1. Evaluación de la presencia de papaya n'ngspot virus (PRSV) en campos de papaya en Guatemala.
Duración de esta etapa: esta etapa se llevó a cabo durante todo el período que duró el estudio pero fue más intenso el muestre0 del 27 de abril de 1998 a junio de 1998. Lugar: Laboratorio de Virología de la Universidad del Valle de Guatemala.
Se llevaron a cabo giras para la recolecta de muestras de papaya visitándose diversas plantaciones en las distintas regiones de Guatemala a excepción de Petén. Se colectaron muestras de papaya procedentes de: región central (Guatemala, Villa Nueva), región sur (Retalhuleu, Suchitepéquez, Escuintla), oriente (Zacapa, Jalapa, El Progreso, Jutiapa, Santa Rosa), occidente (Chimaltenango) y norte (Alta Verapaz, Izabal). En cada región se visitaron plantaciones de papaya hawaiana y10 papaya criolla. Cuando no existían sembradíos comerciales de papaya se procedió a colectar muestras de plantas localizadas a lo largo de la carretera, en casas particulares o huertos familiares . En el caso de plantaciones comerciales, se determinó tomar muestras de hoja sintomaticas de

aproximadamente 5% del total de plantas presentes. El muestre0 se dirigió principalmente a plantas que presentaron diferentes síntomas de virosis (mano de mono, moteado, mosaico, clorosis, etc.). La giras de campo incluyeron visitas de colecta a parcelas experimentales de papaya hawaiana, conducidas por la Asociación Gremial de Exportadores de Productos No Tradicionales (AGEXPRONT), en las cuales se evaluaban las variedades sunrise, whaimanalo, sunset y kapoho. En estas parcelas se colectó material de todas las variedades presentes. La toma de muestras incluyó la colecta de hojas asintomáticas, con el objeto de determinar si es posible la detección temprana de PRSV. Se colectaron también, como control, muestras de papayas aparentemente sanas, alejadas de las papayas infectadas.
Se seleccionaron de 3 a 5 hojas jóvenes (no extendidas en su totalidad, situados a 3-5 nudos por debajo del ápice) de las plantas. Para evitar contaminación cruzada las hojas se introdujeron a bolsas plásticas enrollada en la mano, evitando así el contacto directo de la piel con el tejido vegetal. En caso de hojas difíciles de remover con la mano, se utilizó una cuchilla previamente descontaminada con hipoclorito de sodio al 2.5%, paso que se llevó a cabo entre planta y planta. Las bolsas fueron debidamente rotuladas e identificadas con información pertinente, incluyendo ubicación de la finca o sembradío, fecha de colecta, tipo de papaya, síntoma predominante. En la hoja de registro se anotaron todos los demás datos de importancia, entre ellos: sintomatología, altura, coordenadas, nombre del productor, datos de edad de la plantación, productos que usan para control de plagas, insectos presentes. Las muestras se almacenaron en una hielera, enfriada con hielo a base de anticongelante, para evitar que las muestras se humedecieran excesivamente.
Al llegar al laboratorio, el tejido colectado se almacenó en un cuarto frío (4 grados centígrados) y fueron procesadas de la siguiente manera:
a- Se anotó la sintomatología presentada por cada muestra: a- mosaico b- moteado o manchas, c- deformación foliar, d- clorosis y10 e- anillos en el fruto o manchas en el tallo. Se tomó fotografias de esta sintomatología cuando la presentaban las plantas y también de cualquier otro síntoma distinto a los mencionados anteriormente.
b- Las hojas fueron "picadas" con cuchillas a retazos entre 5 y 10 mm2,envueltas en papel secante y codificadas.
c- Las muestras fueron introducidas a bolsas plásticas con cierre hermético y conteniendo silica gel para deshidratarla lo más rápido posible, a cuatro grados centígrados
d- Diariamente se revisaron las muestras para cambiar la silica gel para mantener las muestras secas, esto se hizo hasta que la sílica gel no absorbió más humedad
Una vez secas, las muestras se almacenaron a 4 "C, en bolsas plásticas con cierre hermético.
Para determinar si las muestras se encontraban infectadas con PRSV, se sometieron a pruebas de ELISA (ensayo inmunosorbente de enzima ligada), utilizando en cada placa controles positivos y negativos para PRSV. Para esto se utilizó un protocolo modificado de la empresa AGDIA que se adjunta en el Apéndice 1. Las modificaciones

consistieron básicamente en el tiempo de detención de la reacción enzimática ya que no se detuvo con NaOH al tiempo sugerido por AGDIA, sino que se tomaron lecturas a la media hora, a la hora y a las dos horas de iniciada la reacción. Si la reacción era muy lenta, las placas se incubaron a 4 Celsius y en completa oscuridad hasta realizar la nueva lectura, siempre observando que la placa no tuviera "fondo" (reacción inespecífica) en los controles negativos y solución amortiguadora. Este procedimiento permitió esclarecer el estado de muestras con lecturas iniciales inciertas y ayudó a esclarecer si algunas muestras que presentaban resultados dudosos se debían considerar positivas o negativas. Otra modificación fue el uso de leche descremada al 2% en casos en que la placa presentara mucho fondo, esto ayudó a diferenciar muestras positivas de negativas también. Se compararon los resultados de ELISA con la sintomatología para determinar qué síntomas tienen más relación con la presencia del virus.
Para transportar las muestras a la Universidad de Georgia, se seleccionó tejido infectado por PRSVp y exhibiendo uno o más de los síntomas siguientes: a- mosaico b- moteado o manchas, c- deformación foliar, d- clorosis y/o e- anillos en el fruto o manchas en el tallo. Estas muestras se almacenaron con desecante (silica gel) a 4 "C hasta el momento de la extracción y purificación del ARN viral, llevado a cabo en los laboratorios del Dr. Wayne Parrott del Departamento de Agronomía y Dr. Michael Deom, del Departamento de Patología Vegetal de la Universidad de Georgia (Athens, Georgia). Previo al viaje, se hizo una gira a la costa sur, específicamente al parcelamiento "Los Angeles" en el Puerto San José, para colectar tejido fresco de papaya y llevarlo a Georgia en frío, después de haber hecho la prueba de ELISA. Esto aseguraría la presencia del virus en alta concentración en las muestras deshidratadas o en las frescas.
2. Purificación del virus y evaluación del virus purificado.
Duración: aproximadamente un mes y medio. Lugar: Laboratorio de Dr. Wayne Parrott, Departamento de Cultivos, Suelos y Meteorología, y Laboratorio de Dr. Michael Deom, Departamento de Patología Vegetal, Universidad de Georgia.
Muestras seleccionadas de papaya provenientes del sur, oriente y norte de Guatemala, se llevaron a Georgia,extrayéndoseles el ácido ribonucleico (ARN)con el kit de Qiagen para extracción de ARN de plantas (ver Apéndice 2). Se trabajó el mismo método tanto con muestras frescas como deshidratadas. Se evaluó la cantidad de ARN obtenido mediante espectofotometría, calculándose la concentración final de ARN. Las muestras de papaya hawaiana y criolla que exhibieron las concentraciones más altas de ARN fueron utilizadas para continuar con los pasos necesarios para aislar el gen. Para todo el trabajo con ARN se utilizó agua libre de ARNasas. Todas las muestras de ARN fueron concentradas y purificadas utilizando la metodología que aparece en el Apéndice 3. El "pellet" o depósito de ARN se reconstituyó con 20ul de agua libre de ARNasas y se midió su concentración final.
Para poder amplificar el gen se utilizó la técnica de reacción de polimerasa en cadena (PCR). Para esto se hizo necesario diseñar imprimadores (primers) que indicaban

la sección del ARN a amplificarse. Se procedió a elaborarlos de la siguiente manera: Primeramente se buscó en la literatura (en el banco de genes de tres regiones del mundo) secuencias reportadas de la cápside del virus de la mancha anular de la papaya (PRSV-p), incluyendo la secuencia de la proteína de la cápside del virus de Hawaii reportado por Quemada et al. (1 990). Seguidamente se buscó en la información de las secuencias de la cápside, un fragmento que fuera diferente en la mayoría de las secuencias pero que incluyera alguna secuencia de nucleótidos similares. Esto se hizo con el objeto de poder establecer diferencias entre la cápside del virus guatemalteco y las cápsides reportadas en la literatura. Las regiones conservadas en la cápside se hacen necesarias para poder utilizar los mismos imprimadores para todas las muestras, de manera que éstos tuvieran áreas precisas en donde unirse antes del proceso de amplificación. El tamaño final de los imprimadores diseñados fue entre 18 y 25 bases, largo adecuado según la literatura. A los imprimadores también se les añadió la secuencia de las enzimas de restricción presentes en el vector al cual se iba a introducir el fragmento para ser secuenciado. De esta manera, a la hebra 5' - 3' se le añadió la secuencia de la enzima Xhol y a la hebra de sentido negativo (3'-5') la secuencia de la enzima Sal l. La secuencia de los imprimadores usados aparece a continuación:
a- Imprimador 5'-3 ':
PRVSXHO 5' -- CCG CTC GAC ATA CGC ATG TGT TTC ATC AGT C -- 3' Número de bases: 3 1 incluyendo la secuencia para Xho 1
b- Imprimador del extremo 3 '- 5 ' :
PRV3S 5' -- GCG TCG ACT TAG TTG CGC ATA CCC AGG AG -- 3' Número de bases: 29 incluyendo la secuencia para Sal 1
Se mandó a hacer una cantidad mínima, 40 nanomoles, para poder evaluar su efectividad.
Ya que los imprimadores se diseñan para ser utilizados específicamente con ADN, el ARN (único ácido nucleico presente en PRSV) debe ser transcritos a cADN. Para esto se utilizó la técnica llamada RT-PCR (Reverse Transcriptase-PCR, por sus siglas en inglés) cuyo protocolo aparece en el Apéndice 4. Para todos los PCRs llevados a cabo se utilizó como enzima amplificadora a PJir en lugar de la Taq polimerasa pues se ha visto que comete menos errores que la Taq a la hora de copiar y amplificar el fragmento. Los productos del PCR se visualizaron con una electroforesis horizontal utilizando geles de agarosa al 1%, utilizando como marcador de peso, lamda cortado con Hind m. El fragmento a amplificar posee un peso de 862 pares de bases. El gel se coloreó con bromuro de etidio (10 p1 en 200 m1 de agua). Se visualizó con luz ultravioleta y se tomó una fotografia del gel.
Después de amplificado el fragmento se extrajo o recuperó el ADN utilizando un kit de Qiagen denominado QIAquick Gel Extraction kit, cuyo protocolo se presenta en el Apéndice 5. Este método consistió en la extracción de ADN por medio de geles de agarosa al 0.8%. Para esto se utilizan geles grandes y pozos con capacidad de 100 p1 de las muestras amplificadas. Para llevarlo a cabo, las muestras se colocaron en el gel

mezclando 3 partes de ADN (aprox. 5 pg), 1 parte de buffer No. 2 1X (Tris HC110 mM, MgC12 mM, NaCl 50 mM, DTT ImM, pH 7.9 a 25 grados centígrados), y 2 partes del colorante de montaje (1X). El marcador de peso, se diluyó también en 1 p1 de buffer No. 2, 6 p1 de agua y 2 y1 de colorante de montaje. La electroforesis se corrió a 80 mv, hasta que la banda más rápida del colorante llegó al borde opuesto del gel. El gel se coloreó con bromuro de etidio, evitándose el uso de luz ultravioleta ya que causa mutaciones en el ADN que se va a utilizar. Una vez identificados y cortados los trozos de gel conteniendo el ADN, se comó una porción (-0.5~1) del ADN en un minigel de agarosa al 1% para calcular la concentración del fragmento. Para esto se utilizaron diferentes concentraciones del marcador lamda cortado con Hind 111 (1 pg, 0.5 pg y 0.25 pg de ADN). A partir del minigel, se calcularon las concentraciones comparando la de la muestra con las del marcador de peso. Con esto, el fragmento o gen estuvo listo para ser introducido al primer vector (PCR-ScriptAmp SK+) previo a su secuenciación, llevada a cabo en un laboratorio especializado de la Universidad de Georgia. Este trabajo se llevó a cabo con fragmentos de cápside purificados de muestras del sur y del oriente de Guatemala, tanto de papaya hawaiana como de criolla.
3. Selección, aislamiento y secuenciación del gen de la cápside de PRSV-p.
Duración: aproximadamente medio mes. Lugar: Departamento de Cultivos, Suelos y Meteorología, y Departamento de Patología Vegetal, Universidad de Georgia.
Sabiendo la concentración del ADN existente (gen de la cápside) en la solución extraída del gel (ver paso anterior) y con el objeto de conocer la secuencia de nucleótidos del gen de las cápsides de las cepas guatemaltecas de PRSV-p, se procedió a clonar el fragmento en el primer vector, el plásmido PCR-Script Amp SK(+) de 2,961 pares de bases. Este plásmido tiene la característica de tener un gen lacz para la beta galactosidasa, lo que permite que las colonias bacteriales (E. coli) transformadas (conteniendo el plásmido con el gen incorporado) adquieran una tonalidad blanca y las no transformadas permanezcan de color azul. Además este vector posee en la región disponible para clonar fragmentos, una variedad de secuencias que permitan sea cortado por diferentes enzimas. Para nuestro caso inicialmente se utilizaron B o I y Sal I (Apéndice 6), sin embargo, al observar que Sal I cortaba el fragmento en dos segmentos, se evaluaron otras enzimas que supuestamente no cortaban el fragmento. Las enzimas seleccionadas y utilizadas finalmente fueron Not I y Eco RV, por lo que se tuvo la necesidad de enviar a hacer otro imprimador para la dirección 5'-3 que tuviera terminaciones para Not I. Un imprimador con la secuencia de EcoRV no fue necesaria porque ya estaba incluída en la secuencia del plásmido. Estas enzimas son importantes porque con ellas se puede cortar el fragmento deseado y trasladarlo a otros vectores (Apéndice 6).
Para poder introducir y ligar el fragmento o gen al vector (plásmido) Script Amp SK(+), se calculó la cantidad de inserto que se necesitaba para llevar a cabo el primer paso de la

inserción ( la ligación) al vector. Para esto se utilizó la fórmula: ng de producto PCR= [(número de bp de PCR) x (10 ng del vector pPCR Script para clonaje)]/2961 bp del vector pPCR Script para clonaje; donde bp indica pares de bases.
Una vez calculada la cantidad de inserto, se preparó la reacción de ligación añadiendo los componentes siguientes, en orden, a un tubo de 0.5 ml:
a- 1 p1 del vector de clonaje pPCR Script Amp SK(+) (1OngIpl) b- 1 pl de buffer de reacción para PCR-Script 10X c- 0.5 plof 1OmMrATP d- 2-4 pl del producto de PCR o 4 p1 del inserto control de PCR e- 1 p1 dela enzima de restricción SrfI (5UIpl) f- 1 pl de la T4 DNA ligasa (4UIpl) g- Agua destilada hasta completar un volumen final de 1 Opl
Se mezcló la reacción suavemente y se incubó por una hora a temperatura ambiente seguido por diez minutos a 65 grados centígrados. Seguidamente el tubo de reacción se colocó en hielo hasta que se utilizó para la transformación con células ultracompetentes (en este caso se utilizaron células Epicurianas Coli XL 10 Gold Kan).
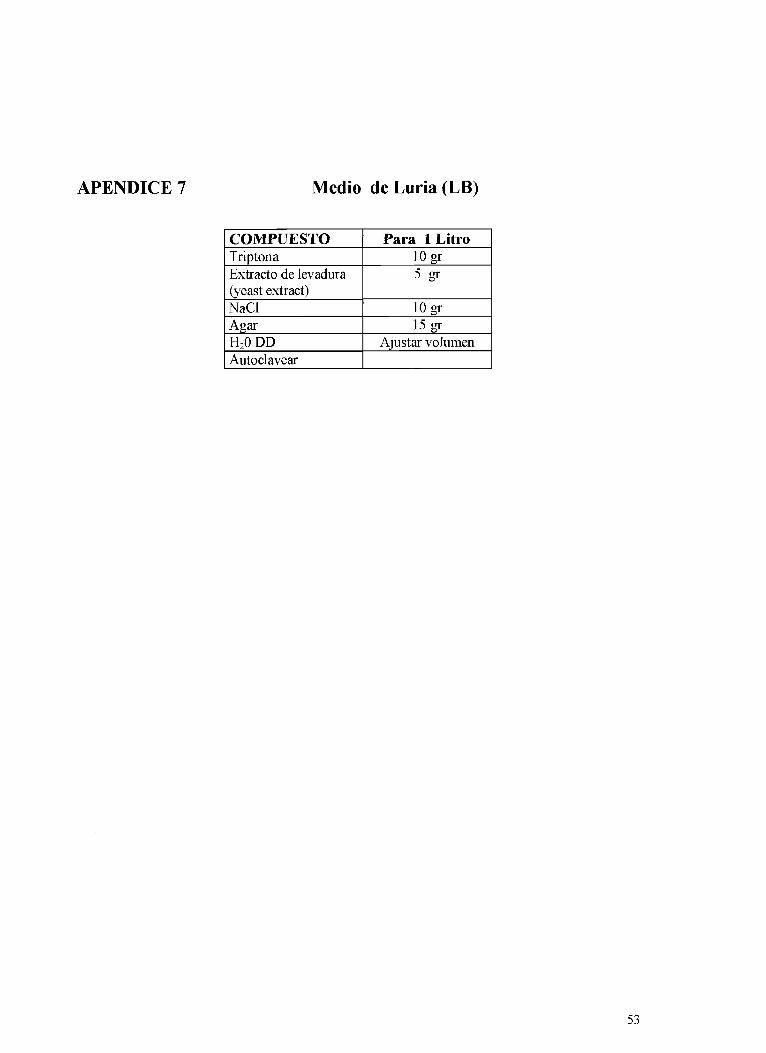
La transformación de E. coli se inició con el descongelamiento de las células, agregándose 15 pl del ligado a 100 p1 de células. Se mezclaron suavemente rotando el tubo e incubándose 30 minutos en hielo. Seguidamente se les produjo el golpe de calor "heat shock" poniendo el tubo 45 segundos en agua a 42 grados centígrados sin moverlo. Luego se transfirieron nuevamente a hielo por dos minutos y se añadió 0.9 m1 de Caldo de Luria a temperatura ambiente sin antibióticos. Seguidamente se incubaron en un agitador a 37 grados centígrados y a 225 rpm durante una hora. Mientras tanto se prepararon las placas con medio Luria (LB, Luria broih, Apéndice 7) y 200 pg de ampicilina. Se utilizaron dos placas a las cuales se les agregó 16 p1 de X gal(50mglml) y 33 p1 de 100 mM de IPTG. Se dispersó por toda la placa y se dejó reposar 45 minutos antes de que la incubación de la muestra finalizara. Los reactivos Xgal e IPTG sirven para diferenciar colonias transformadas de las no transformadas, esto se debe a la presencia del gen LacZ (promotor lac) en el plásmido el cual permite la fusión de la proteína expresada con el producto de la beta galactosidasa. Si la colonia se torna azul, la bacteria no está transformada mientras que las transformadas permanecen de color blanco. Al finalizar la hora de incubación de las células, se rayó una de las placas conteniendo LB con 100 pl de la solución, la cual se incubó a 37 grados centígrados durante toda la noche.
Al día siguiente se seleccionaron las colonias blancas (transformadas), evitando cualquier contaminación con bacterias azules. Se hizo una placa de LB-ampicilina conteniendo X-gal y IPTG para verificar el fenotipo Lac-. Se sembraron en medio líquido Luria con 200 pg de ampicilina y se dejaron incubando durante la noche. Se tomaron 500 m1 de este caldo con bacterias y se almacenó con 500 p1 de glicerol a menos ochenta grados centígrados como stock. El resto se usó para hacer minipreparaciones y recuperar el ADN del plásmido con el gen insertado ( Apéndice 8). Una vez aislado el ADN, se utilizaron enzimas de restricción, en este caso Xho I y Sal I para cortar el

inserción ( la ligación) al vector. Para esto se utilizó la fórmula: ng de producto PCR= [(número de bp de PCR) x (10 ng del vector pPCR Script para clonaje)]/2961 bp del vector pPCR Script para clonaje; donde bp indica pares de bases.
Una vez calculada la cantidad de inserto, se preparó la reacción de ligación añadiendo los componentes siguientes, en orden, a un tubo de 0.5 ml:
a- 1 pl del vector de clonaje pPCR Script Amp SK(+) (IOngIpl) b- 1 p1 de buffer de reacción para PCR-Script 10X c- 0.5 plof 10 mMrATP d- 2-4 pl del producto de PCR o 4 pl del inserto control de PCR e- 1 pl dela enzima de restricción SrfI (5UIpl) f- 1 pl de la T4 DNA ligasa (4UIpl) g- Agua destilada hasta completar un volumen final de 10p1
Se mezcló la reacción suavemente y se incubó por una hora a temperatura ambiente seguido por diez minutos a 65 grados centígrados. Seguidamente el tubo de reacción se colocó en hielo hasta que se utilizó para la transformación con células ultracompetentes (en este caso se utilizaron células Epicurianas Coli XL 10 Gold Kan).
La transformación de E. coli se inició con el descongelamiento de las células, agregandose 15 p1 del ligado a 100 p1 de células. Se mezclaron suavemente rotando el tubo e incubándose 30 minutos en hielo. Seguidamente se les produjo el golpe de calor "heat shock" poniendo el tubo 45 segundos en agua a 42 grados centígrados sin moverlo. Luego se transfirieron nuevamente a hielo por dos minutos y se añadió 0.9 m1 de Caldo de Luria a temperatura ambiente sin antibióticos. Seguidamente se incubaron en un agitador a 37 grados centígrados y a 225 rpm durante una hora. Mientras tanto se prepararon las placas con medio Luria (LB, Luria broth, Apéndice 7) y 200 pg de ampicilina. Se utilizaron dos placas a las cuales se les agregó 16 pl de X gal(50mglml) y 33 p1 de 100 mM de PTG. Se dispersó por toda la placa y se dejó reposar 45 minutos antes de que la incubación de la muestra finalizara. Los reactivos Xgal e IPTG sirven para diferenciar colonias transformadas de las no transformadas, esto se debe a la presencia del gen LacZ (promotor lac) en el plásmido el cual permite la fusión de la proteína expresada con el producto de la beta galactosidasa. Si la colonia se torna azul, la bacteria no está transformada mientras que las transformadas permanecen de color blanco. Al finalizar la hora de incubación de las células, se rayó una de las placas conteniendo LB con 100 pl de la solución, la cual se incubó a 37 grados centígrados durante toda la noche.
Al día siguiente se seleccionaron las colonias blancas (transformadas), evitando cualquier contaminación con bacterias azules. Se hizo una placa de LB-ampicilina conteniendo X-gal y P T G para verificar el fenotipo Lac-. Se sembraron en medio líquido Luria con 200 pg de ampicilina y se dejaron incubando durante la noche. Se tomaron 500 m1 de este caldo con bacterias y se almacenó con 500 pl de glicerol a menos ochenta grados centígrados como stock. El resto se usó para hacer minipreparaciones y recuperar el ADN del plásmido con el gen insertado ( Apéndice 8). Una vez aislado el ADN, se utilizaron enzimas de restricción, en este caso Xho I y Sal I para cortar el

fragmento de cápside del plásmido y comprobar su presencia por medio de geles de agarosa al 0.8%. El marcador de peso utilizado en el gel, en este caso lamda cortado con Hind 111, se preparó en tres concentraciones (0.25 pg, 0.5 pg y 1.0 pg) para poder comparar y estimar la concentración que tenía el fragmento de aproximadamente 862 bp.
Se mandaron a secuenciar minipreparaciones de papayas hawaiana y criolla de las regiones sur, oriente y nororiente. Al recibirlas de vuelta se compararon dichas secuencias entre sí y con las del PRSV de Hawaii.
4. Tranformación de Agrobacteríum tumefaciens e introducción del fragmento de gen a la planta.
Duración: 1 mes en la Universidad de Georgia 8 meses en Universidad del Valle de Guatemala
En esta etapa se hicieron varias transformaciones y varios clonajes. En total se llevaron a cabo 3 clonaciones y cuatro transformaciones.
Una vez identificado y aislado el fragmento de cápside, el siguiente paso fue la inserción del fragmento al vector pAPCH V, con el objeto de proporcionar resistencia a higromicina, Este vector, de 6823 pares de bases posee también resistencia a ampicilina, aunque no posee el gen para lac-z, por lo cual las bacterias no se seleccionan por color. Una vez preparado el gen y sabiendo su concentración, se procedió a descongelar el vector que estaba almacenado (-80 C) en minipreparaciones de células de E. coli DH 5 alpha. El ADN del pAPCH V se purificó según protocolo que aparece en el Apéndice 8 y se cuantificó utilizando un fluorímetro. Previo a insertar el gen de la cápside al pAPCH V anibos ADN fueron cortados con NotI y con EcoRK Se hicieron geles de agarosa al 0.8% para medir las concentraciones de ambos previo a la inserción del gen al plásmido.
La ligación de ambos ADN se hizo utilizando la ligasa T4 a una relación entre vector y gen de 8:1, dejándolos en contacto por un período de 4 hr a 37 "C. El producto ligado (vector pPACH V transformado) se puso en hielo y se procedió a la transformación de células de E. coli DH 5 alpha siguiendo el procedimiento previamente descrito.
El proceso de incorporación de ADN del pAPCH V a células alpha DH5 de E. coli (transformación), se hizo colocando 1 pl de ADN (255 nglpl) por 100 pl de células competentes en hielo por treinta minutos. Se les aplicó un choque de calor (42 "C por dos minutos) e inmediatamente después se colocaron en hielo y se añadió caldo de Luria hasta completar 1 ml. La solución se agitó por 45 minutos (37 "C) y se sembraron en placas con Luria y 200 pg de ampicilina las cuales se incubaron a 37 C durante la noche. A la mañana siguiente se tomaron cinco colonias que se sembraron en medio de Luria líquido con ampicilina, incubándose durante la noche agitando a 37 grados centígrados. Al día siguiente se tomaron 500 pl de cultivo para almacenarlo con 500 pl de glicerol a menos ochenta grados centígrados y el resto se usó para hacer minipreparaciones. Se hicieron maxipreparaciones tanto del vector como del gen para que se pudiera obtener

concentraciones altas de ambos y poder así proceder a la transformación de ambos (ver Apéndice 9).
Para verificar que la inserción del gen (al plásmido) y la introducción del plásmido a E. coli se llevó a cabo adecuadamente, se procedió a hacer minipreparaciones de todas las colonias obtenidas, incluyéndose también colonias no transformadas como control. Se cortaron todas con Not I y EcoRV y se corrieron en geles de agarosa al 0.8%. De las colonias transformadas (banda del gen presente en el gel de agarosa) se tomo un stock, el cual se almacenó con glicerol (-80 C) para tomar de allí el gen y ser introducido al plásmido pPzP-201B, vector a ser utilizado en A. tumefaciens para la transformación del tejido de papaya.
Este tercer vector, denominado pPzP-20 1 B, es un vector binario para Agvobacterium, es pequeño, versátil, estable y completamente secuenciado. Tienen dos bordes, ambos necesarios junto con el sitio Bom del pBR322 para movilización del gen desde Escherichia coli a Agvobacterizrm tumefaciens. Posee tambien dos genes, el ColEl y pVS1, necesarios para replicación en E. coli y A. tumefaciens, respectivamente. Incluye además marcadores como resistencia a estreptomicina. Los bordes mencionados anteriormente son importantes para hacer la transferencia del gen del plásmido al núcleo del tejido vegetal (papaya).
El plásmido pPzP-201B fue descongelado y preparado como se mencionó con anterioridad para el plásimido pPACH V. Se transformaron bacterias E. coli alpha DH 5. Se hicieron minipreparaciones y se cortó y linearizó el plásmido con la enzima BAMH 1. Se ligó y se transformó según protocolos anteriores, sólo que se seleccionaron las bacterias transformadas utilizando agar Luria con kanamicina 50. Se picaron unas cinco o seis colonias y se hicieron stocks y minipreparaciones.
Por último, se procedió a transformar Agvobacterizrm tumefaciens, insertándole el vector modificado. Para esto se sembraron células de A. tumefaciens, cepa LBA 4404, en agar YEP con kanamicina 50 (Apéndice 10) y se incubaron a 28°C durante 2 días. De las colonias resultantes se seleccionaron cinco colonias y se sembraron individualmente en tubos de 3 ml. con caldo YEP y kanamicina 50. Se incubaron durante la noche a 28 grados centígrados y con agitación de 250 rpm. Se tomaron 2.5 m1 de uno de los cultivos y se añadió a los 50 m1 de YEP líquido con kanamicina 50, se incubó de nuevo a 28 grados centígrados con agitación de 150 rpm. El resto se guardó como stock con glicerol. Cuando la densidad óptica del cultivo alcanzó 0.80 unidades de absorbancia (Ua), se centrifugaron las células a 5000 rpm por 5 minutos a cuatro grados centígrados. Se descartó el sobrenadante. Se enfriaron en hielo los cultivos, tubos eppendorf de 1.5 m1 y la solución de cloruro de calcio (Ca C12) 20 mM. Se resuspendieron las células en 1 m1 de cloruro de calcio 20 mM frío. Finalmente se hicieron alícuotas de éstas en tubos eppendorf de 1.5 ml. Se colocaron 100 p1 en cada tubo y se congelaron inmediatamente en nitrógeno líquido. Se almacenaron a - 70 grados centígrados.
Para la transformación de células de A. tumefaciens, se añadieron 10 p1 del plásmido aislado a una concentración de 1 pg a 100 p1 de células LBA 4404 congeladas. Se

incubaron en baño de María a 37 grados centígrados por cinco minutos. Se descongelaron completamente y se dejó que la bacteria expresara los genes de resistencia a estreptomicina. Para esto se añadió un m1 de YEP con kanamicina 50 al tubo, se incubó por 28 grados centígrados por cuatro horas con una agitación suave. Se centrifugó por 30 segundos, se descartó el sobrenadante y se resuspendieron las células en 100 p1 de YEP. Se esparció en una placa de agar YEP con kanamicina 50 y se incubaron a 28 grados por más o menos 2 días. Se picaron 5 colonias y se crecieron en 5 m1 de YEP con kanamicina 50 con agitación. Luego se analizaron los plásmidos por electroforesis, se hicieron minipreparaciones y los stocks respectivos.
Una vez se tiene el Agrobacterium tumefaciens transformado, se deben tener listos los embriones de la planta, en este caso de papaya. Estos embriones se vinieron preparando paralelamente con todo lo descrito anteriormente. La descripción de esta parte, obtención y preparación de embriones in vitro, se describe en la sección B.
El Agrobacterium tumefaciens se descongeló y se sembró en caldo de YEP de estreptomicina a una concentración de 2.5 mglml. Se hizo una alícuota de 3 m1 de este caldo y se incubó a 28 grados centígrados por 4-8 horas. Se tomó 2.5 m1 de este cultivo y se agregó a 50 m1 de YEP-con estreptomicina a la concentración anterior. Se agitó a 150 rpm por una noche a 28 grados centígrados. El resto del caldo se almacenó con glicerol a menos 80 grados centígrados como stock. Al día siguiente se centrifugaron las bacterias a 3000 rpm, a temperatura ambiente y se descartó el sobrenadante. El sedimento se diluyó en 10 m1 de MSD20 líquido a pH 5.8 (Apéndice 11). Los embriones de papaya se pusieron en contacto con la suspensión bacterial, aproximadamente 100 embriones por los 10 m1 de A. tumejacierzs en MSD20, por un periodo de cinco minutos a 28°C. Luego se lavaron dos veces con el mismo medio, separando los embriones de la bacteria mediante centrifugación de 5 minutos a 3000 rpm. Se secaron los embriones con toallas estériles tratando de eliminar al Agrobacterium tumqfacieizs de los embriones. Los embriones se sembraron en agar MSD20 pH 5.8 (Apéndice 11) en placas de Petri por dos días y finalmente se trasladaron a MSD20 pH 7.0 (Apéndice 11) con una concentración de higromicina de 10mgI m1 y con mefoxin 100mg/ml.
5. Confirmación de la transformación. Duración: 2 meses Lugar: Universidad del Valle.
Durante esta etapa se procedió a seleccionar los embriones. Esto fue posible mediante la siembra éstos en embriones en medio Murashige and Skoog con 20 mgllt de ácido 2,4-diclorofenoxiacético 2-4D (MSD20, pH 7) con diferentes concentraciones de higromicina y mefoxin 500 para evitar la contaminación. Se usaron concentraciones de higromicina de 10 mglml, 20mglml y de 30 mglml. Esta última concentración es muy alta ya que se espera que los embriones estarán transformados si crecen a una concentración de 20 mglml y mas aún si crecen a 30 mglml. Se sembraron embriones no transformados a estas mismas concentraciones de higromicina como controles. Cualquier embrión no transformado se muere con concentraciones más bajas de 10 mglml, pero

para eliminar cualquier posibilidad de mezcla, se decidió trabajar con concentraciones mayores pero tolerables para el embrión. Siempre se añadió mefoxín 500 al medio para evitar la contaminación con otros organismos. Todos los embriones que se desarrollaron a estas concentraciones de higromicina se trasladarán al medio con carbón activado para estimular la diferenciación, lo cual se detalla en la sección de cultivo de tejidos.
B. Cultivo de tejidos de papaya para generación de embriones somáticos y regeneracidn de plantas in vitro.
1. Embriogénesis somática
Duración: 3 meses, Laboratorios de Drs Parrott y Deom, Deplo. de Agronomía y Fitopatología de launiversidad de Georgia. Laboratorio de Virología, Universidad del Valle de Guatemala (3 meses).
Los trabajos en esta sección se iniciaron con una búsqueda de metodologías adecuadas para el cultivo de papaya in vitro. Se encontraron metodologías basadas en semilla inmadura como explante, pero no con semilla madura por lo que se tuvieron que adaptar técnicas para la obtención de embriones a partir de semilla madura.
Inicialmente se trató de germinar in vitro la semilla madura inalterada, con el objeto de tomar de la plántula resultante, distintos tipos de tejido como explante. Estos explantes de tallo, hoja y raíz se utilizarían para estimular la producción de embriones somáticos (embriogénesis somática) seguido de organogénesis. Como primer paso previo a sembrar in vitro la semilla madura inalterada, se Ilevó a cabo la descontaminación de la misma, procedimiento llevado a cabo tanto en Georgia como en Guatemala. En Georgia, el proceso de descontaminación consistió en dejar las semillas una hora en agua corriente, tres minutos en etanol al 70%, 20 minutos en cloro al 2% con 0.01% de tween 20 con agitación de 150 rpm, según lo reportado por Yang y Ye, 1992. En la campana de flujo laminar, se hicieron tres cambios con agua estéril y luego se establecieron las semillas en medio Murashige and Skoog (MSO) pH 5.8, sembrándose de 10 a 15 semillas inalteradas por caja petri. Se dejaron incubando a 27 "C con luz difusa, hasta que se visualizara el rompimiento de la capa externa de la semilla. La germinación in vitro de semilla sexual también se evaluó con otros medios incluyendo: ?4 MSO con 5.5 pM de 2-4D, MSO con vitamina de los medios B5, MSDl O pH 5.8 y pH 7, MSD20 pH 5.8 y pH 7, MSO sin sulfato de amonio, MSO con 0.5 BA y O. 1 NAA (Apéndice 11). Estas placas se pusieron en las mismas condiciones que las anteriores.
La germinación de semilla sexual inalterada fue muy pobre (menor del 3%) bajo condiciones in vitro, por lo que decidió utilizar distintas porciones del embrión sexual. Para esto se seccionó el embrión sexual en tres tipos de explantes, siendo éstos 1) radícula, 2) cotiledones y 3) radícula + una porción cotiledonar. Estas partes de la semilla se sembraron en un medio con el regulador 2-4 D a dos concentraciones (10 pg/ml y 20 pg/ml) y dos pHs diferentes (5.8 y 7.0) (Apéndice 11). Se sembraron aproximadamente 20 a 25 explantes de cada tipo en los medios descritos arriba. Se dejaron incubando a 27°C con luz difusa.

Ya en Guatemala se corrieron pruebas evaluando distintos tiempos de descontaminación superficial de semilla en etanol y cloro (Tabla I), ya que las pruebas realizadas en los laboratorios de la Universidad de Georgia exhibieron índices de contaminación superiores al 30%. Seguidamente a la descontaminación superficial, la semilla se disectó, utilizándose como explante único la radícula conteniendo parte de los cotiledones del embrión sexual de la semilla. En pruebas anteriores se había observado que sembrando una porción del embrión sexual en lugar de la semilla completa aceleraba el proceso de diferenciación y crecimiento in vitro.
Los explantes que lograron desarrollar masas embrionarias (tejido calloso en el que se generan embriones somáticos) se trasladaron a medio MSD20 pH7 para que lo embriones somáticas desarrollaran mejor. Se dejaron allí hasta que alcanzaron el tamaño adecuado, apariencia cristalina con varios embriones en un pie y sin callo. Se trasladaron de medio cada vez que se contaminaban o desarrollaban mucho callo. Estos embriones al tener apariencia de flor fueron utilizados para la transformación, como se describió en la sección respectiva.
Tabla 1. Tratamientos utilizados en las pruebas de descontaminación superficial de semilla de papaya criolla y hawaiana previo a su establecimiento in vitro.
Tratamiento
2. Regeneración de plántulas de papaya a partir de embriones somáticos Duración: 12 meses, Georgia y Guatemala
6
7
8
9
Con el objeto de estandarizar las técnicas de regeneración y micropropagación in vitro previo a su aplicación a embriones transgénicos, se llevaron a cabo pruebas de
1
1
1
1
5
1 o
10
1 O
60
20
3 O
60
3
3
3
3

regeneración de plántulas in vitro de papaya a partir de embriones somáticos no transformados. Los embriones de prueba que mostraron apariencia de flor, sin callo y cristalinos, se trasladaron a un medio con carbón activado (MSMóAC, apéndice 11). para incitar diferenciación del tejido vegetal (organogénesis). Cuando los embriones mostraban indicios de diferenciación de tejidos, procedió a secarlos (hardening), colocándolos durante unos días en placas de Petri con una capa muy delgada de MSO. Cuando estos embriones diferenciados adquirieron una apariencia amarillenta, se trasladaron a medio de maltosa (MSM6, apéndice 11) Aquí se dejaron de dos a cuatro semanas observar la formación de tallo y hojas. Seguidamente se cambiaron a % MSO para que formaran raíces. Las plántulas que no formaron raíces se transfirieron a MSO suplementado con ácido indolbutírico (IBA) a una concentración de 2pgíml para estimular el crecimiento de éstas. Al formarse las raíces, se trasladaron las plantitas a !h MSO (Apéndice 11) y se dejaron allí hasta que la mismas adquirieran el tamaño adecuado para su traslado al invernadero.
Como colaboración del IPMICRSP con el proyecto, se envió a un estudiante de agronomía a un intercambio de verano a la Universidad de Purdue. Se aprovechó aquí para hacer pruebas de cultivo de tejidos con papaya. Se trabajó particularmente en la germinación, enraizamiento y propagación de explantes (yemas axilares) provenientes de plantas in vitro. Para esto se esta utilizando medio MS con 0.1 uM de NAA y 0.8 uM de 6 BA y MS mas IBA y riboflavina.

V. Resultados y Discusión.
Los resultados de este proyecto se presentarán de acuerdo a las etapas que se mencionaron en la metodología. Aún cuando se incluyen algunos resultados cuantitativos, la naturaleza de este proyecto presenta logros qualitativos, producto de los resultados de laboratorio. Esto se debe a que en la ciencia de ingeniería genética el logro más importante consiste en obtener por lo menos un ejemplar (en nuestro caso papaya) transformado y con las características deseadas.
A. Sección de trabajos con el virus PRSV.
1. Evaluación de la presencia de papaya ringspot virus (PRSV) en campos de papaya en Guatemala.
En el siguiente mapa (Figura 1) se señalan las principales regiones en las que se cultiva papaya en Guatemala, la cuales fueron incluídas en la colecta de muestras, con excepción del Petén.
Figura 1. Regiones de Guatemala incluídas en el muestre0 de papaya dirigido a detectar la presencia del virus (PRSV) de la mancha anular de la papaya.
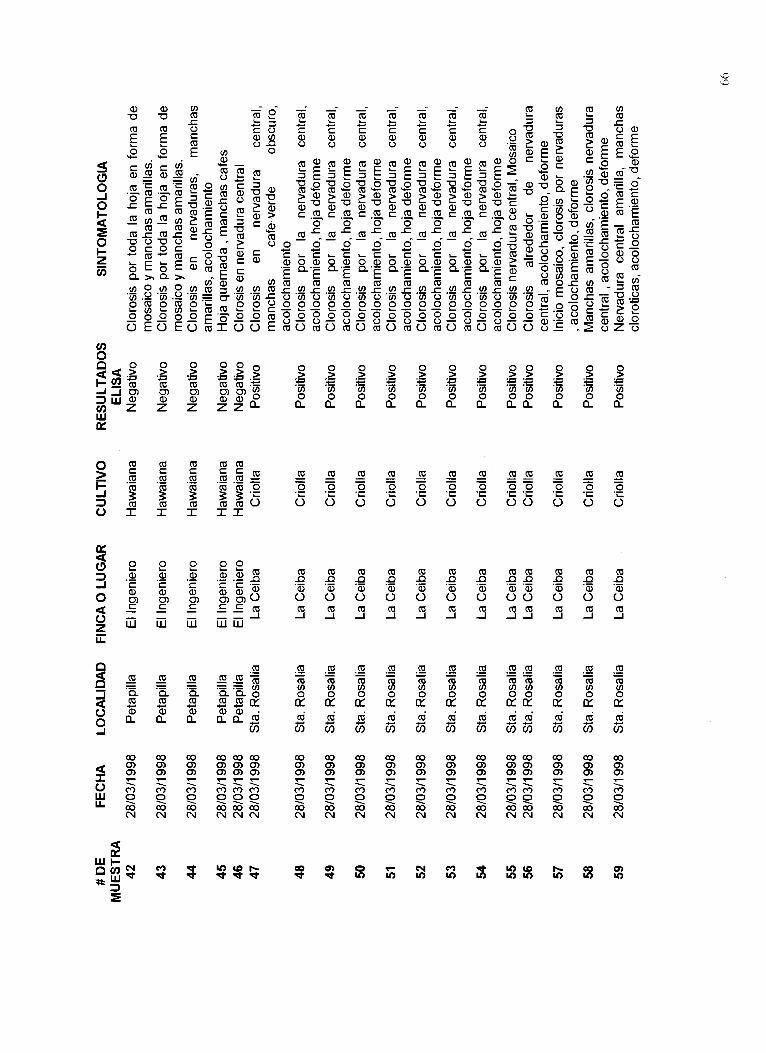
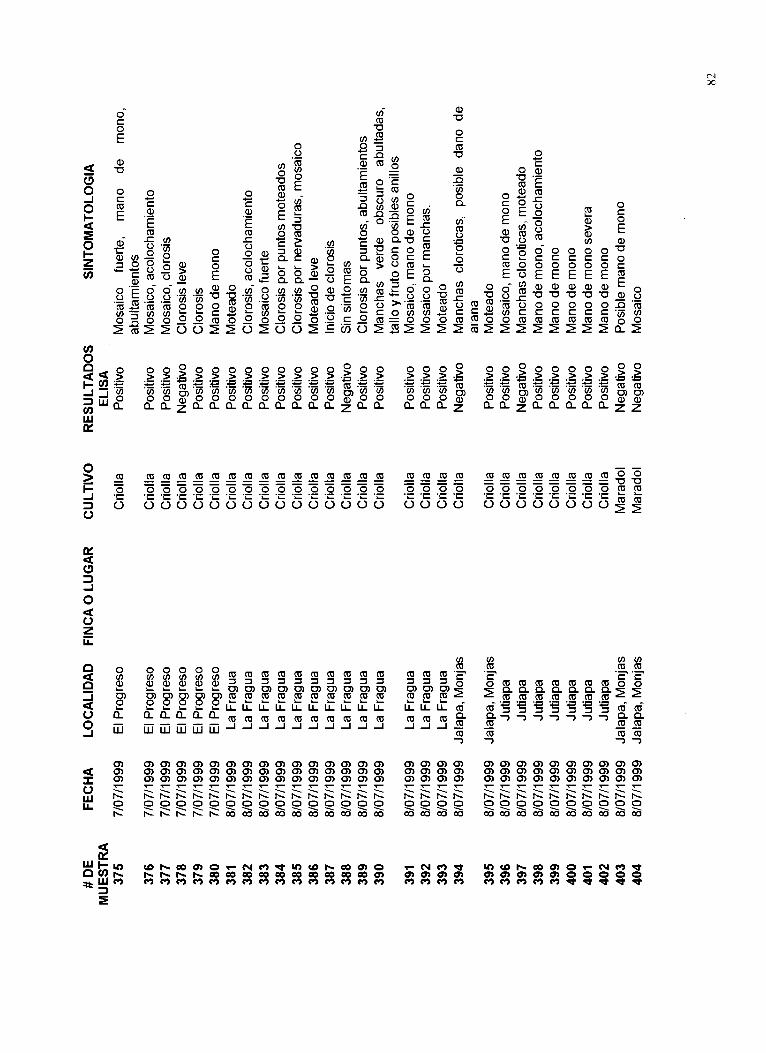
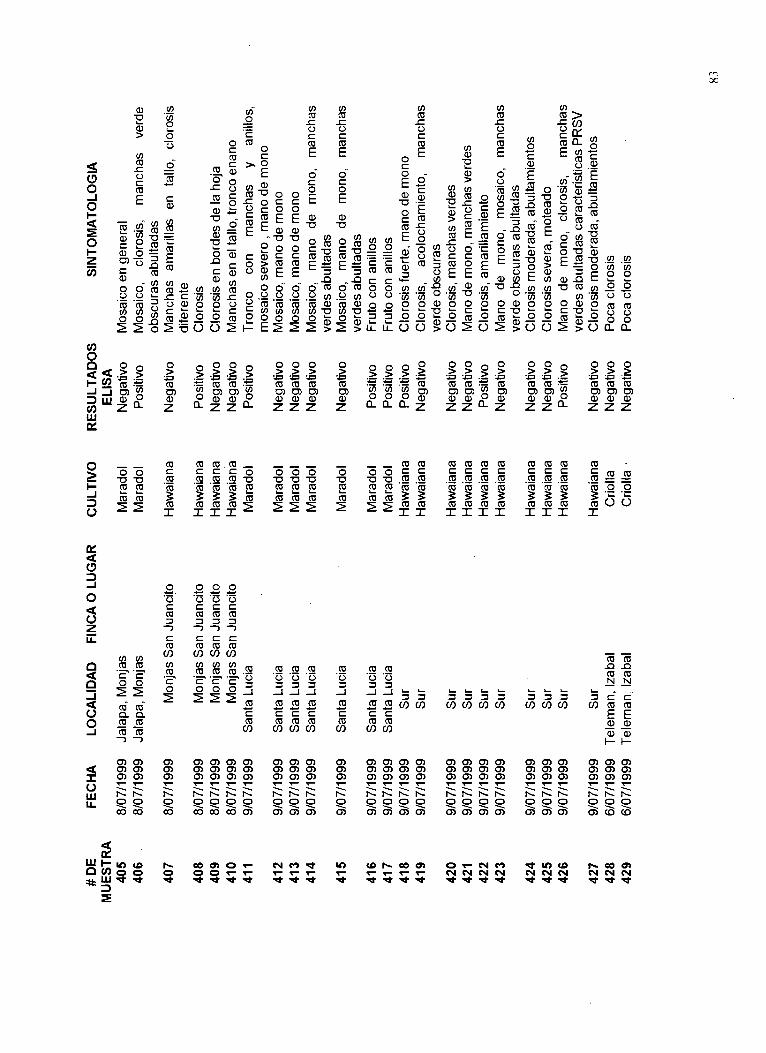
Un listado de las muestras recolectadas, el lugar de procedencia, las fechas de colecta, síntomas y resultados de la presencia del virus PRSV se muestra en el Apéndice 12.

En la Figura 2 se muestra un campo de papaya hawaiana con fines comerciales. La tecnología empleada en estos campos es significativamente mas intensiva en comparación al cultivo de papaya criolla, ya que se incluye riego por goteo, cobertura plástica, distanciamientos de siembra adecuados al porte de las variedades y planes de manejo distintos al manejo tradicional.
Figura 2. Recolecta de hojas en campo experimental de papaya hawaiana tipo "solo".
Los síntomas más frecuentemente encontrados en las plantaciones de papaya fueron clorosis, mosaico, acolochamiento, deformidades de la hoja o fi-uto, enanismo, manchas cloróticas o verdes oscuro en fi-uto, tallo u hoja, anillos en el fruto,y una combinación de mosaico clorótico y deformidad comúnmente llamada "mano de mico" (Figura 3).
a. Anillos en fruto de papaya criolla
b. Deformidad de la hoja

d. Mosaico en hojas de papaya a c. Mancha grasienta del tallo
e. Mano de mico f. Enanismo n Figura 3. Síntomas típicos asociados a mancha anular en plantas de papaya
ubicadas en Guatemala.
Durante la colecta de muestras a través de las distintas regiones de Guatemala puede ver que este virus predomina en la zona sur y en la zona oriental del país principalmente y que aparece tanto en papaya criolla como en hawaiana tipo 'solo'. En la visita a la zona oriental (Figura 4C y 4D) se incluyeron plantaciones ubicadas en Jalapa (Monjas), Jutiapa (Asunción Mita) y Zacapa (Finca Nueva, La Ceiba y La Fragua). Se visitó también una plantación de papaya hawaiana en "El Ingeniero", Chiquimula. Como se observa en la Figura 4 (C y D) en el oriente del país se detectó un 25% de muestras PRSV positivas en papaya criolla y 34% en papaya Hawaiana.
En la costa sur de Guatemala (Figura 4 E y F) la mancha anular se detectó tanto en papaya criolla como hawaiana, con 48% y 40% de muestras positivas, respectivamente. En la zona norte del país se detectó el virus en proporciones similares
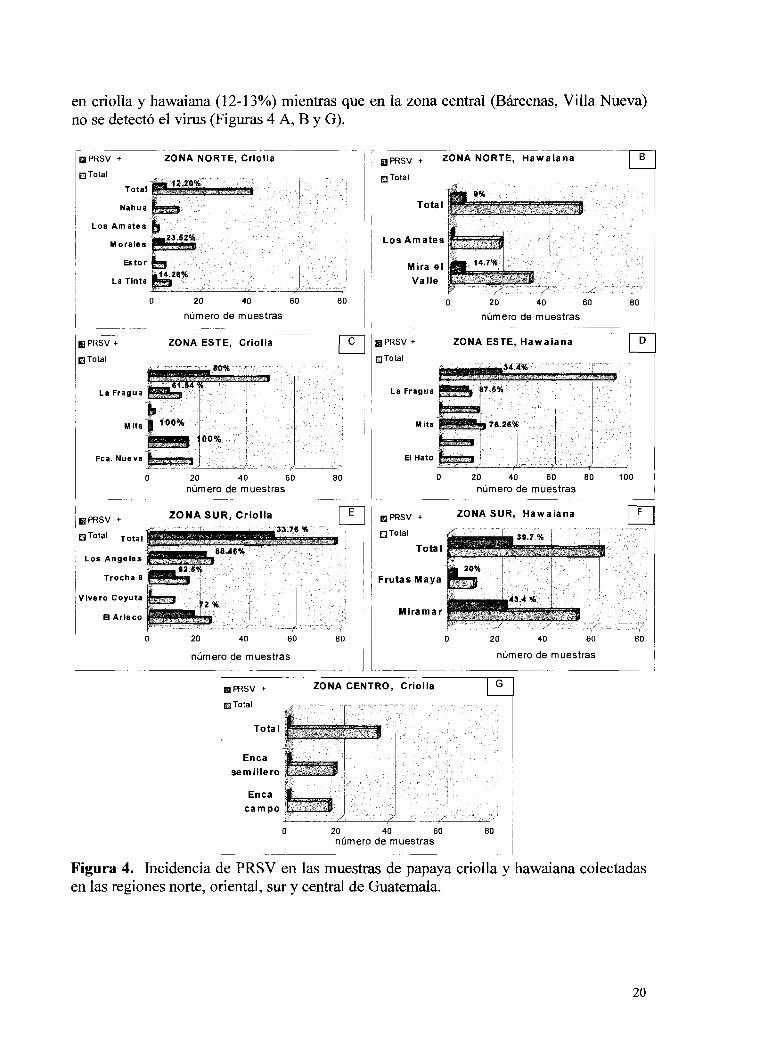
en criolla y hawaiana (1 2- 13%) mientras que en la zona central (Bárcenas, Villa Nueva) no se detectó el virus (Figuras 4 A, B y G).
-- -- -- -- 1 61 PRSV + ZONA NORTE, criol la
Total
Los
I Total 1
Nahua 1 Am ates
Mora les
Estor
La Tlnta
i - --
1 número de muestras 1 - -
ZONA ESTE, ~ r i o ñ a
R Total
La
Fca
Fragua
Mita
. Nueva
--
m~~~~ + ZONA NORTE, Hawaiana
[il Total
Total
Los Amates
Mira e l Val le
O 20 40 60 80 1 número de muestras I
L --
-- - --
1 M PRSV + ZONA ESTE, Hawaiana ID(
o L- .~
20 40 60 O 20 40 60 80 100 1 número de muestras número de muestras
~ ~ ~ . - -. p . ~ ~ . -
' g PRSV + ZONA SUR, Hawaiana
BTota I Total
Los Angeles
j Trocha 8 1
V i v e r o Coyuta
U Arisco
O 2 O 40 60
número de muestras número de muestras
1 Total
1 Total
Enca semillero
l
campo
O 20 40 60 80 número de muestras
--
Figura 4. Incidencia de PRSV en las muestras de papaya criolla y hawaiana colectadas en las regiones norte, oriental, sur y central de Guatemala.

La incidencia de PRSV de acuerdo al tipo de papaya (criolla o hawaiana) se presenta en la Figura 5. En dicha figura se puede observar que tanto la papaya criolla como hawaiana presentan niveles similares de incidencia general, con 40% y 33% de plantas positivas respectivamente.
- - -- -- - -- - - - - - -
l Incidencia de PRVp PRV-p en Papaya Criolla
-----
Incidencia de PRVp en Papaya Hawaiana
Figura 5. Incidencia general de mancha anular en muestras de papaya criolla y hawaiana recolectadas en Guatemala.
Los resultados presentados en la figura anterior parecen sugerir que la papaya criolla y hawaiana presentan una disposición similar a ser infectadas por el virus de la mancha anular.
A nivel de cultivo, se determinó que un 37% de las muestras recolectadas se encontraban infectadas por PRSV (Figura 6). Estos resultados ofrecen información valiosa acerca de la importancia de la mancha anular en Guatemala ya que muestran que la incidencia es alta en nuestro país.
Presencia de PRV-p en Guatemala
Figura 6. Porcentaje de mancha anular (PRSV) detectado en muestras de papaya colectadas en Guatemala.

2. Purificación del virus y evaluación del virus purificado.
Los resultados de la amplificación, purificación y estimación de la concentración del virus PRSV extraído de las papayas infectadas, tanto de criolla como de hawaiana, se resumen en el gel siguiente:
Marcador lamda cortado con Hind 111 a
concentraciones de lug, 0.5 ug y 0.25 ug
respectivamente
PRV-p (peso 862 bp)
Figura 7. Gel de Agarosa mostrando el fragmento de gen de cápside de PRSV-p de 862 pares de base (bp).
Es importante hacer notar que el fragmento purificado mediante geles de agarosa no se pueden fotografiar porque se daña el fragmento al someterlo a luz ultravioleta por un período largo o relativamente largo. Por lo tanto solamente se cortó el fragmento y se extrajo el ADN con el kit de Qiagen. La concentración de ADN obtenida fue de 0.2320 yglyl para el gen de PRSV.
3. Selección y aislamiento del gen.
El vector pPCR-Script Arnp SK(+), resultado de la inserción del fragmento o gen de PRSV tuvo la arquitectura mostrada en la Figura 8 (esta es la forma como se enviaron los fragmentos a Ser secuenciados):

Figura 8. Arquitectura del vector pPCR-Script Amp SIC(+), utilizado para determinar la secuencia de nucleótidos en el ADN del fragmento de gen de la cápside del virus PRSV.
Como se observa en la Figura 9, la homología entre la cepa control de PRSV proveniente de Hawaii (% de la secuencia de bases que es idéntica entre fragmentos) y las cepas PRSV Guatemaltecas es del 95% en tres de ellas, específicamente la G-92 (cepa del oriente de Guatemala, papaya hawaiana), G-61 (cepa del oriente, papaya criolla) y la G-326 (cepa del sur del país, papaya criolla). En contraste a éstas, la cepa G-352 (cepa del sur, papaya hawaiana) mostró una homología del 91%, aunque debe mencionarse que la longitud del fragmento de ésta última era de 550 bases, en comparación a 865 de las primeras mencionadas.
G-352 G-326 G81 G-92 Hawaii
Cepa PRSV
Figura 9. Comparación (% de homología) entre la secuencia de bases en los fragmentos de gen de la cápside de las cepas Guatemaltecas (G) y la cepa control proveniente de Hawaii. Las cepas G-352 y 326 fueron purificadas de papaya hawaiana y criolla del sur de Guatemala, respectivamente, mientras que las cepas G-61 (criolla) y G-92 (hawaiana) fueron obtenidas de cepas del oriente del país.
Por razones de espacio, las secuencias completas de los fragmentos enviados a secuenciar se muestran en el Apéndice 13. Es importante hacer notar que secuencias del

virus purificado de la zona norte no se tienen pues el virus se encontró en muy baja concentración en los pocos casos que presentaron infección. Se trató de aislar de nuevo de otras muestras de papaya de la misma zona pero no se encontraron más casos.
De estos resultados se puede notar que las cepas de Guatemala que provienen de papaya hawaiana y de papaya criolla son altamente similares y al compararlas con el virus de Hawaii, la diferencia más notoria se presenta a nivel de las bases 75 y 82, en donde se presenta una deleción de 6 bases en las cepas de Guatemala. Sin embargo el efecto que esta diferencia, equivalente a 2 aminoácidos en la cápside proteica, pueda tener en la estructura de la capa proteica se desconoce y no se sabe si esto puede ser motivo suficiente para que el virus rompa la resistencia de las papayas transgénicas resistentes al virus hawaiano, generadas por investigadores de las Universidades de Hawaii y Cornell (Fitch, et al, 1992 ). Por otro lado, esta diferencia en la secuencia de bases de los fragmentos Guatemalteco y Hawai podría ser suficiente para inutilizar la resistencia exhibida por la plantas trangénicas hawaianas, tornándolas susceptibles al virus guatemalteco. La inefectividad de la resistencia transgénica en papaya a cepas de PRSV provenientes de otras localidades del mundo ha sido comprobada (Fitch, et al, 1995) y es una de las razones por las cuales la transformación de papayas utilizando fragmentos de cepas virales locales se hace sumamente necesaria.
4. Tranformación bacteriana e introducción del gen a la planta.
En esta etapa se trasladó el fragmento o gen de PRSV del plásmido pPCR Script (SK+) al plásmido pAPCH V el cual, como se dijo con anterioridad, daría la resistencia a la higromicina. Para realizar esto se hace necesario conocer con certeza las concentraciones de tanto del gen como del vector. Para esto se corrieron geles de agarosa con los cuales se calculó la concentración de ambos, tal y como se muestra en la Figura 10
cortado con HindIII, 1 ug,
PAPCH V (6823 bp)
PRV- p -L
Figura 10. Detección por medio de gel de agarosa del plásmido PAPCH V y el gen de la cápside de PRSV-potyvirus de 862 pares de bases (bp).

Después de ligar el plásmido pAPCH-V al fragmento, se obtuvo como resultado el plásmido cuya arquitectura se muestra en la Figura 1 1.
Figura 11. Arquitectura del plásmido pAPCH V con el gen de la cápside PRSV y el gen de resistencia a higromicina.
Como se muestra en la Figura 12, la presencia del plásmido pAPCH V en las colonias de Agrobacterum tumesfaciens transformadas se verificó por medio de geles de agarosa. Esta prueba es de gran importancia ya que este plásmido no posee un gen lacZ
Marcador lamda/HindIII
pAPCH V con gen
1 pAPCH V sin gen I Figura 12. Gel de agarosa mostrando la presencia del plásmido pAPCH V y del fragmento de gen de cápside del PRSV-potivirus.
para seleccionar las colonias transformadas de las no transformadas, por lo que se utilizó el antibiótico (higromicina) como criterio de selección. Sin embargo dado que al cortar el vector con las enzimas de restricción podría darse el caso de no localizar el fragmento de gen de cápside, se hizo necesario cortar el plásmido con las enzimas NotI y EcoRV.

La tercera transformación se hizo con el vector pZP201B. Para esto se tuvo que tener tanto el vector nuevo como el fragmento con altas concentraciones. Se siguió el mismo procedimiento que con pAPCHV sólo que se usó como enzima de restricción a BAMH 1 para linearizar el plásmido. Este vector agregó al gen y a la resistencia a higromicina, la resistencia a kanamicina, los bordes para el traslado del gen al genoma de la célula vegetal y otras estructuras el ori y bom que servirán para la inserción de este plásmido binario a Agrobacterium tumefaciens. El plásmido con las modificaciones insertadas se muestra en la Figura 13.
(ori, bom)
Resistencia lappomicha
Figura 13. Arquitectura de plásmido pPZP-201B con el gen de la cápside insertado.
Las colonias de Agrobacterium tumefaciens obtenidas en medio YEP con kanamicina 50 eran de color blanco. La kanamicina sirvió para seleccionar las bacterias transformadas, ya que las no transformadas no sobreviven en este medio debido a la presencia del antibiótico.
Una vez transformado el Agrobacterium, se procedió a ponerlo en contacto (sembrado y multiplicado en medio líquido) con los embriones somáticos diferenciados como se observa en la Figura 14.

Embriones listos para transformar
Figura 14. Embriones somáticos de papaya listos para ser transformados con el gen de la cápside del virus de la mancha anular de la papaya (PRSV).
Se pusieron en contacto 50 estructuras embrionarias (Figura 13) por 10 ml. de Agrobacterium tumefaciens incubado durante una noche. La turbidez estuvo alrededor de (OD600) = 0.80. Se dejaron en contacto por 5 minutos y luego se lavó con medio MSD20 pH 5.8 líquido dos veces.
5. Confirmación de la transformación.
A los dos días de sembrados en MSD20 pH 5.8, los embriones transformados se trasladaron a MSD20 pH 7.0 con mefoxin 100 y con higromicina a dos concentraciones diferentes, 10 pglml, 20pglml. Como se observa en la Tabla 2, la papaya criolla mostró porcentajes mayores de recuperación de embriones transformados con 36% y 31% de recuperación para embriones lavados y no lavados respectivamente. En contraste, los embriones somáticos de papaya hawaiana exhibieron una aparente capacidad menor de soportar el proceso de transformación ya que se recuperó el 28% y 27% de embriones con lavado y sin lavado post-transformación, respectivamente. Experimentos similares llevados a cabo en otras regiones del mundo, utilizando A. tumefaciens como vector para transformación de papaya reportan porcentajes de recuperación similares; Yang, et al, (1996) reportaron una recuperación entre el 20 y 40% de los tejidos sujetos a transformación
Tabla 2. Número de masas embrionarias de papaya putativamente transformadas y sobrevivientes despues de 8 semanas en medio MSD20 sólido conteniendo higromicina.
Número de Sobrevivientes Papaya Número Inicial de con lavado' sin lavado
Masas Embrionarias H,,' H2, % total H,, H,, % total
Criolla 292 38 15 36 33 12 31
Hawaiana 150 13 8 28 12 8 27 1 Con lavado se refiere a que seguidamente al proceso de transformación y antes a la centrifugación (3,000 rpm por 2 minutos), la mitad de los embriones de papaya criolla y hawaiana fueron lavados en medio

líquido MSD20 pH 5.8, tres veces por tres minutos. Sin lavado significa que el 50% restante de los embriones sujetos a transformación no fueron lavados sino solamente secados en papel absorbente estkril. ' H , ~ y HZ0 se refiere a medio MSD20 conteniendo 10 yg/ml y 20 yg/ml de higromicina, respectivamente.
Una vez desarrollados los embriones, se trasladarán a medio de cultivo conteniendo maltosa y carbón activado para promover la diferenciación del tejido vegetal. Estas plantitas diferencias serán el material a utilizarse en la segunda parte del proyecto que será la diferenciación de las plantitas transformadas, su traslado a invernadero y al campo, para todas las pruebas finales de resistencia al PRSV-p y fenotipo del producto obtenido.
En la Figura 15 se muestra la secuencia de cambios observados en la apariencia de los embriones sometidos al proceso de transformación mediada por A. tumefaciens. Como se observa en la Figura15.A las masas embrionarias constan de múltiples embriones y cada uno de ellos puede dar lugar a una planta transformada. En la Figura 15 .B se observa una masa embrionaria luego haber sido sometida a la transformación. Se observa que la masa embrionaria ha tomado un tinte amarillento y que los embriones ya no son definidos. Los embriones no transformados se toman necróticos y mueren (15.C) en el medio conteniendo higromicina, mientras que los embriones transformados vuelven a diferenciarse de la masa y prosiguen su crecimiento (1 5.D). Como puede apreciarse en la Figura 15.E. algunas masas embrionarias pueden incluir embriones vivos (transformados) y no transformados (muertos).
transformar en crecimieento
tranofnrmadn
C. Embriones muertos, sin actividad D. Embrión transformado de papaya hawaiana

1 ~ m b n o n e s muertos ]
-
1 E. Masa ernbrionaria transformada (criolla) 1
Embriones regenerados = transformados
Figura 15. Comportamiento observado en los embriones somáticos de papaya sometidos a transformación genética con Agrobacterium tumefaciens.
Estos embriones se dejan por un total de 8 semanas en MSD20 con higromicina, posterior a lo cual se trasladarán a medio con carbón activado para promover su diferenciación. Es importante mencionar que bajo la presencia del medio conteniendo antibiótico, los embriones de papaya criolla se han desarrollado más rapidamente que los de papaya hawaiana tanto previo a la transformación como posterior a la misma.
B. Cultivo de tejidos de papaya para generación de embriones somáticos y regeneración de plantas in vitro.
1. Embriogénesis somática
La semilla madura de papaya fue descontaminada de diferentes maneras ya que con el tratamiento de descontaminación sugerido por los colaboradores de la Universidad de Georgia se detectaron pérdidas por contaminación superiores al 30%. Por lo tanto, y con el objeto de mejorar la eficiencia del proceso de descontaminación del explante, se procedió a variar los tiempos de permanencia de las semillas de papaya en etanol y cloro al 2%. El proceso se evaluó para cada tipo de papaya ya que se observó que los materiales de papaya no responden de la misma manera al proceso de descontaminación. Los resultados de estos tratamientos se observan en la Tabla 3.
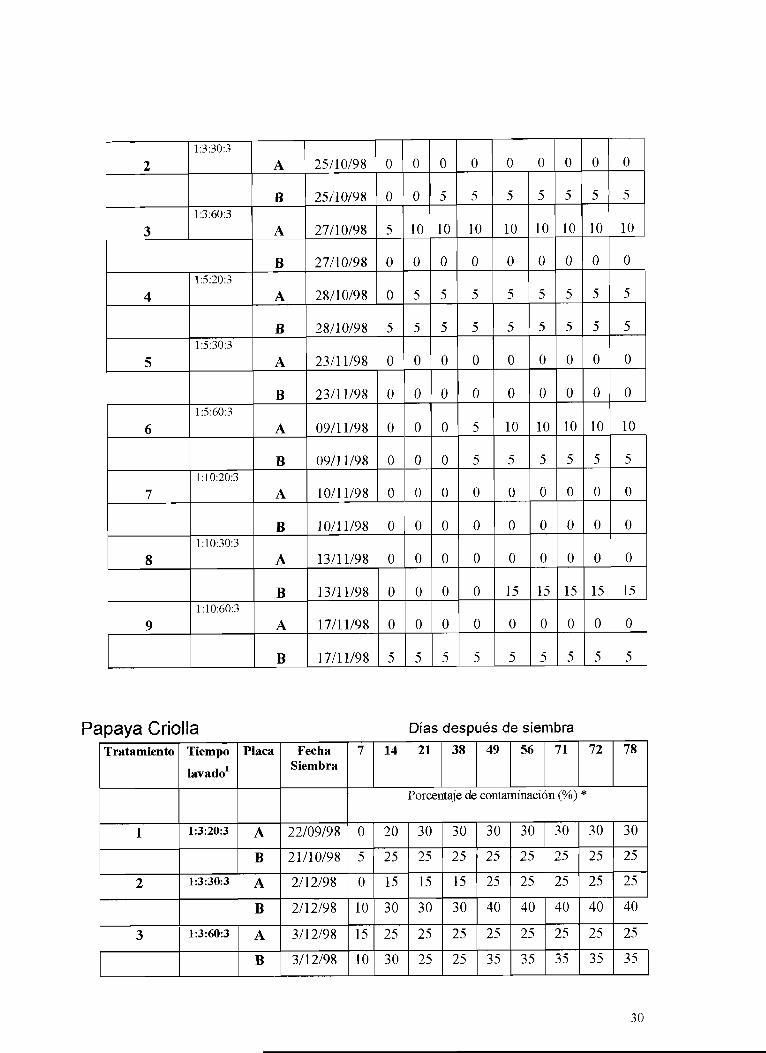
Tabla 3. Porcentajes de contaminación en pruebas de esterilización superficial de semilla de papaya criolla y hawaiana
Pa~ava Hawaiana Días Deswues de Siembra a
1
Tratamiento 78
~ a v a d o ' ---- 1:3:20:3
105 Tiempo
A
B
Fecha de Placa
Siembra Porcentaje de contaminación (%)
22/08/98
21/10/98 O O O 5 5 10 15 15 20
14 21 38 49 56 71 72