investigación y desarrollo farmacéutico -...
TRANSCRIPT

• Entre 10 y 17 años para llegar al mercado• Probabilidad de éxito menor al 10%
Investigación y desarrollo farmacéutico
Target Discovery Lead Discovery Lead Optimisation ADMET Desarrollo Registro
2-3 años 0.5-1 año 1-3 años 1-2 años 5-6 años 1-2 años
•modelos de la enf•Identificación de blancos
• desarrollodiseño de fármacos
• química farmacéutica• diseño orientado defármacos
• biodisponibilidad• exposición sistémica
• Fase I• Fase II• Fase III • USA
• Europa• Japón• Resto del mundo
Pre-clínica ($300 millones US) Clínica ($500m)
Lipinski, C. J. Pharmacol. Tox. Meth. 44,235-249 (2000)

• La farmacocinética es el estudio del tiempo de residencia de un fármaco dentro del cuerpo. Incluye los procesos de Absorción, Distribución, Metabolismo y Excreción (ADME).
Hígado
Metabolismo
Metabolismo
Biodisponibilidad
Pared intestinalDosis
Heces Tejidos
Farmacocinética

BIODISPONIBILIDAD Y BIOEQUIVALENCIA
Como control de calidad para asegurar la intercambiabilidad de los productos farmacéuticos
Dra Inés Fuentes Noriega.
Facultad de Química UNAM
Lab. 112 Biofarmacia.

HistoriaBiodisponibilidadFactores que afectan la biodisponibilidadBiodisponibilidad absoluta y relativaParámetros importantes, Cmax, tmax y ABCBioequivalenciaRegulación sanitaria, NOM 177Sistema de clasificación biofarmacéutica
Contenido

Historia
� 1960, ineficiencia del dicumarol en un grupo de pacientes. Se modificó la formulación de la tableta para que se pudiera romper fácilmente.
� 1963, prednisona, se cambió por otra marca y no tuvo la eficacia esperada.

Historia
� 1968-1969, se cambió el sulfato de calcio por lactosa en productos de fenitoína, esto provocómayor incidencia de efectos secundarios por existir mayor absorción con lactosa.
� 1978 fenitoína, se dan a conocer formulaciones de liberación rápida y sostenida (no intercambiar fenitoína por fenitoína sódica).

Historia
� 1975, intoxicación por digoxina en casos de hospitales en Israel, el problema fue el cambio de formulación que realizaron para que el proceso de disolución fuera más rápido.

� ¿Como garantizar la equivalencia terapéutica de los medicamentos con el mismo principio activo, misma dosis y misma forma farmacéutica?
� La respuesta farmacológica esta generalmente relacionada con la concentración del fármaco en el sitio receptor.

� ¿Es posible medir rápida y directamente la concentración del fármaco en el sitio de acción?
� Hípótesis: Existe una relación entre la concentración del fármaco en sangre y su efecto terapéutico

Biodisponibilidad
� Término farmacocinético
� Describe el grado y la velocidad a la cual una sustancia o molécula terapéutica es liberada desde una forma farmacéutica hasta la circulación sanguínea general.

Factores que afectan la biodisponibilidad
� Propiedades fisicoquímicas del fármaco
� Tamaño de partícula
� Estructura cristalina
� Grado de hidratación del cristal
� Coeficcciente de partición
� Pka
� Tipo de sal o ester

Factores que afectan la biodisponibilidad
� Variables de formulación y manufactura
� Cantidad de desintegrante
� Cantidad de lubricante
� Recubrimientos especiales
� Naturaleza de diluentes
� Fuerza de compresión
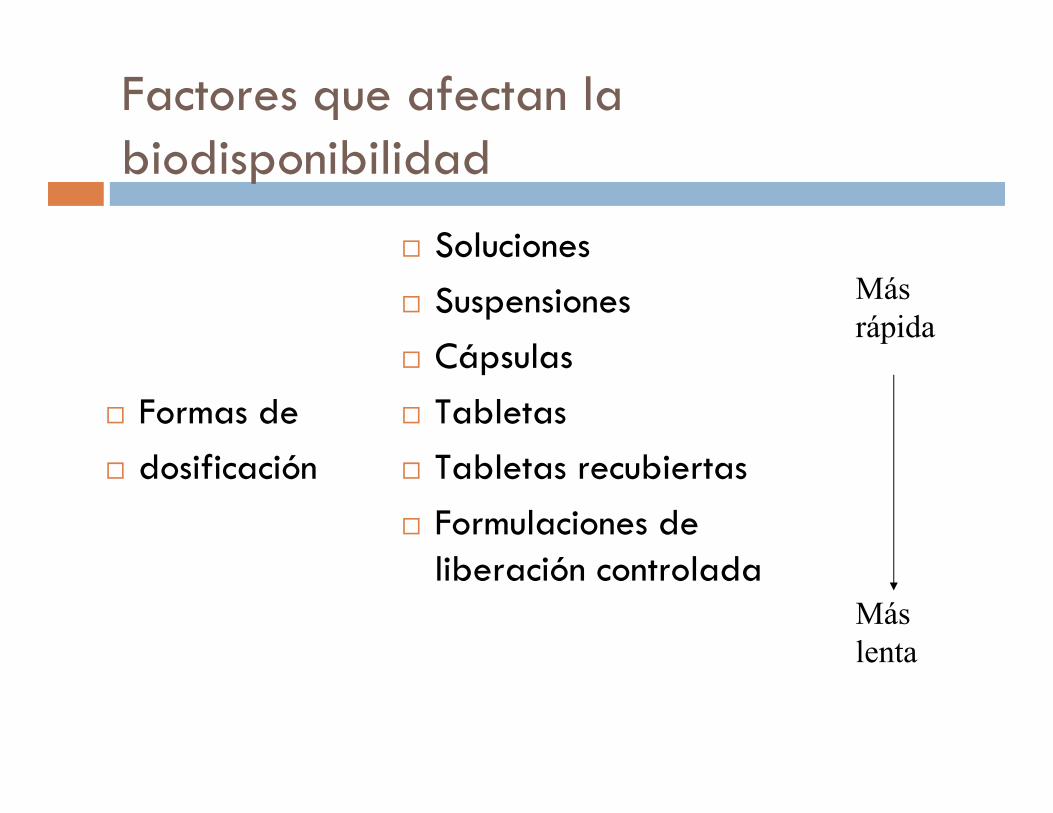
Factores que afectan la biodisponibilidad
� Formas de
� dosificación
� Soluciones
� Suspensiones
� Cápsulas
� Tabletas
� Tabletas recubiertas
� Formulaciones de liberación controlada
Más
rápida
Más
lenta

Factores que afectan la biodisponibilidad
� Factores fisiológicos
� Variaciones en el poder de absorción a lo largo del tracto GI
� Variaciones en el pH del fluído GI
� Velocidad de vaciamiento gástrico
� Motilidad intestinal� Perfusión del tracto GI� Metabolismo presistémico y
de primer paso� Edad, sexo y peso� Estados de enfermedad
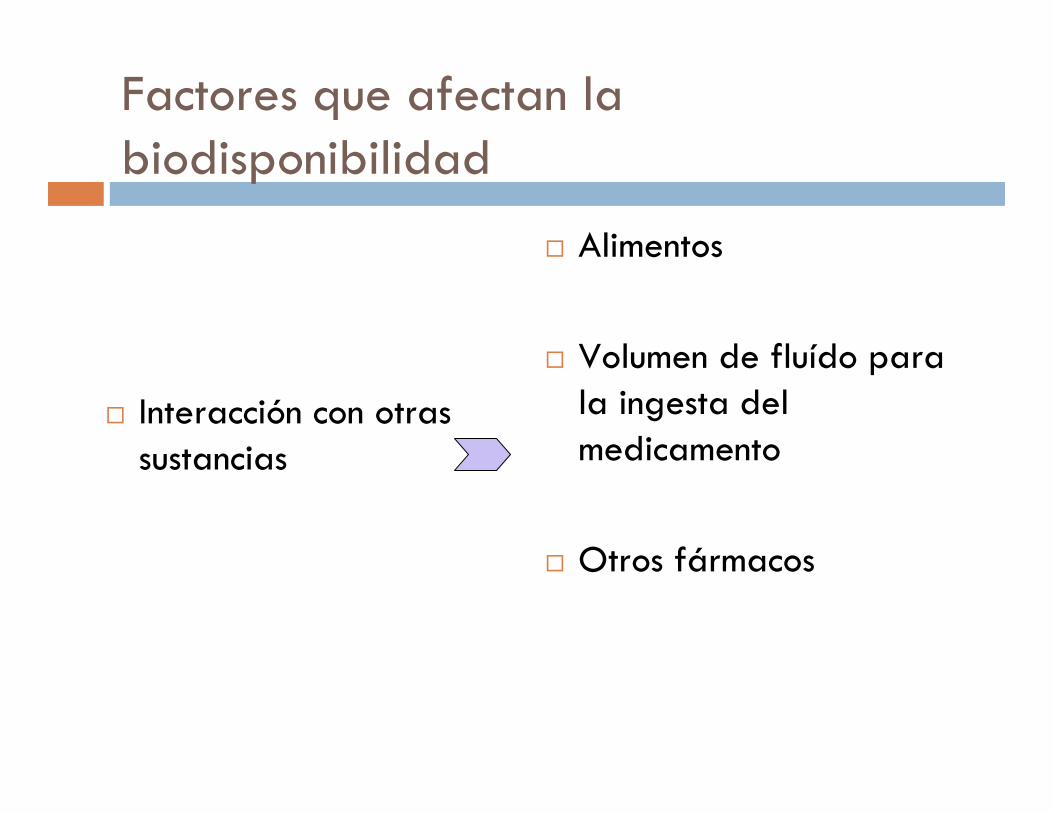
Factores que afectan la biodisponibilidad
� Interacción con otras sustancias
� Alimentos
� Volumen de fluído para la ingesta del medicamento
� Otros fármacos

Transporte en la membrana gastrointestinal

Tránsito gastrointestinal
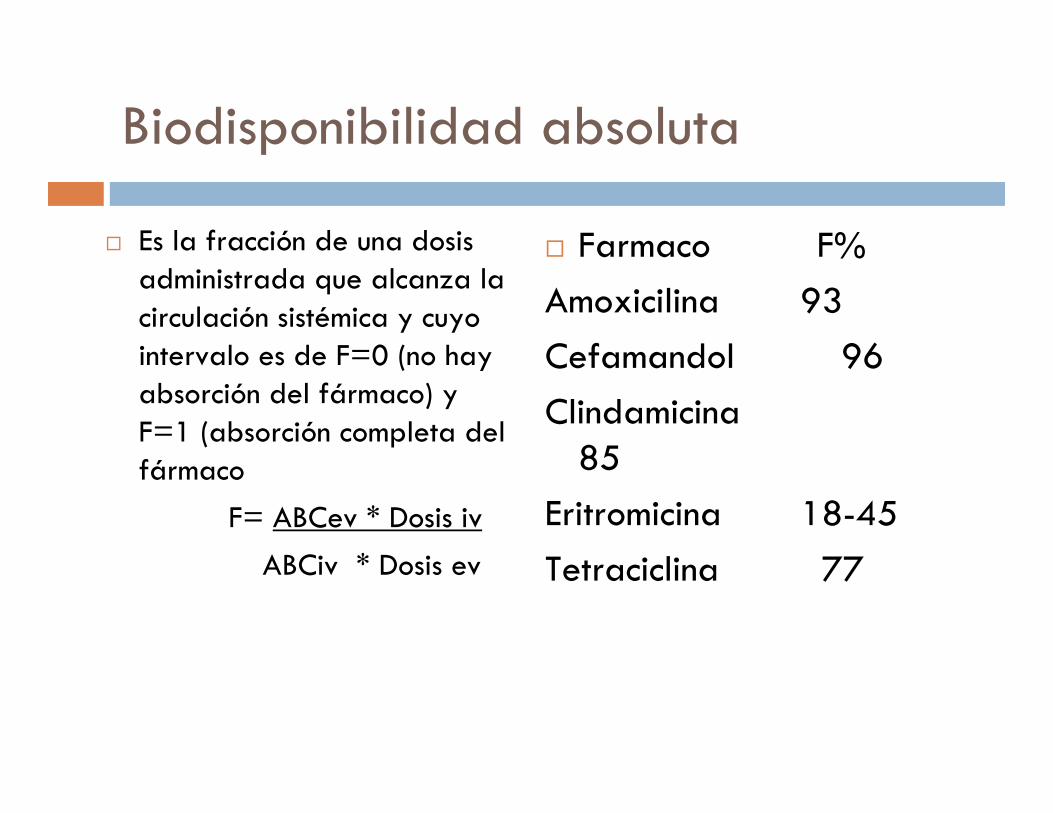
Biodisponibilidad absoluta
� Es la fracción de una dosis administrada que alcanza la circulación sistémica y cuyo intervalo es de F=0 (no hay absorción del fármaco) y F=1 (absorción completa del fármaco
F= ABCev * Dosis iv
ABCiv * Dosis ev
� Farmaco F%
Amoxicilina 93
Cefamandol 96
Clindamicina85
Eritromicina 18-45
Tetraciclina 77
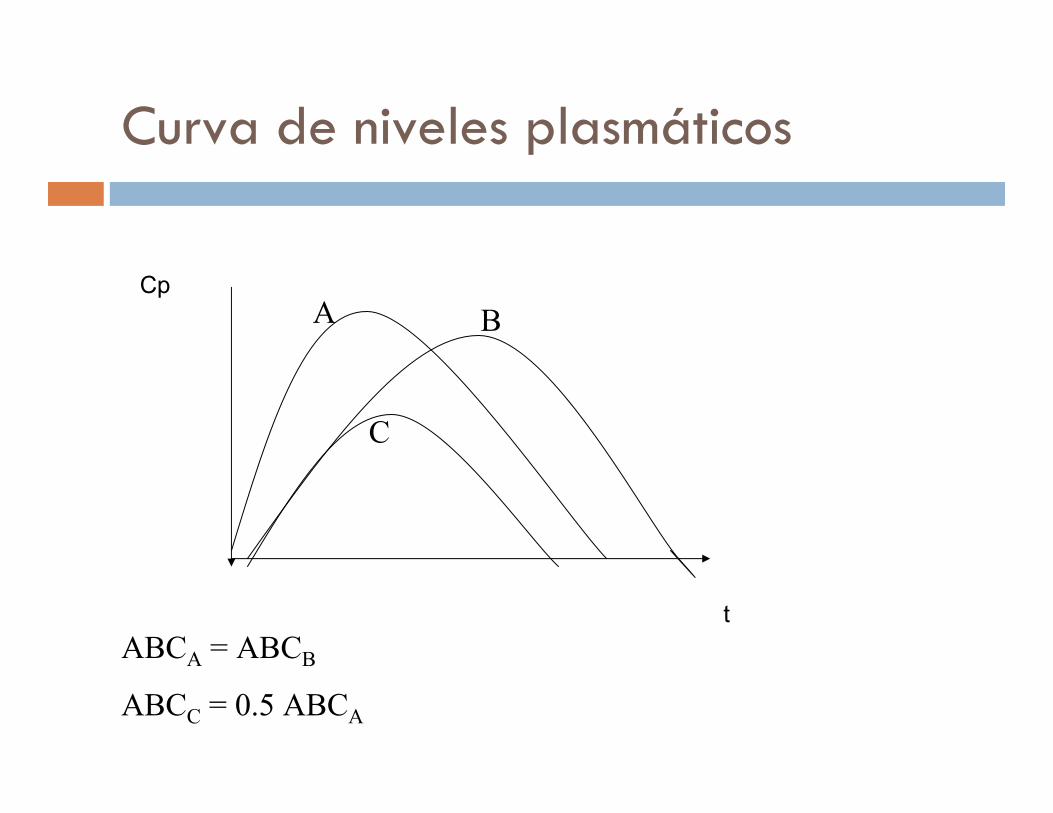
Curva de niveles plasmáticos
ABCA = ABCB
ABCC = 0.5 ABCA
A B
C
Cp
t

Biodisponibilidad absoluta
� Con datos urinarios:
� F = Du∞ po / dosis oral
Du ∞ iv / dosis iv
Sólo cuando el fármaco se libera inalterado en un 85% en orina
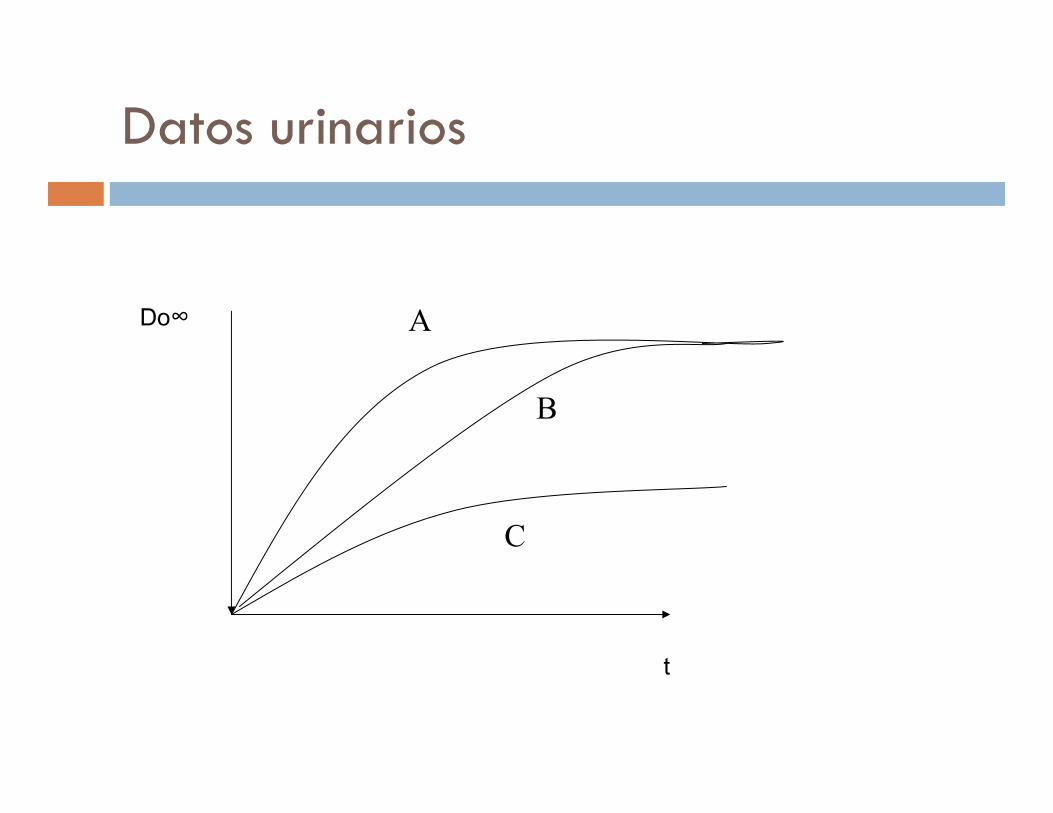
Datos urinarios
A
B
C
t
Do∞

Biodisponibilidad relativa
� Es la biodisponibilidad de un fármaco en un producto farmacéutico comparado con otra forma de dosificación o producto del mismo fármaco dado en la misma dosis
F = ABCev1
ABC ev2

Factores que afectan la biodisponibilidad
� Absorción incompleta
� Metabolismo
� Flujo sanguíneo

Parámetros
� Miden la velocidad� Cmax y T max
� Miden la extensión
� ABC o-∞ y ABC0-t
Se puede analizar por momentos estadísticos

Parámetros de biodisponibilidad cuando hay cambios en velocidad y extensión.
Extensión del fármaco, disminución de biodisponibilidad
Velocidad del fármaco, disminución de biodisponibilidad
Parámetro Cambio Parámetro Cambio
Datos
plasmáticos
Tmax
Cmax
ABC
Mismo
Disminuye
disminuye
Tmax
Cmax
ABC
Aumenta
Disminuye
misma
Datos urinarios
T∞
(dDu/dt)max
Du∞
Mismo
Disminuye
disminuye
T∞
(dDu/dt)max
Du∞
Aumenta
Disminuye
misma

Bioequivalencia
� Ausencia de una diferencia significativa en la velocidad y el grado al cual el ingrediente activo o molécula activa, en equivalentes farmacéuticos o alternativas farmacéuticas, llega a ser disponible en el sitio de acción cuando es administrado a la misma dosis y bajo condiciones similares ( sitio diseñado apropiadamente)

Bioequivalencia
� Si dos o mas productos son bioequivalentes entre sí, deberían ser terapéuticamente equivalentes y pueden ser intercambiados por los profesionales del sector Salud

Política del gobierno
� Uso de medicamentos genéricos con el fin de abaratar el costo y que la población tenga acceso

NOM 177-SSA1-1998
� Norma oficial mexicana que establece las pruebas y procedimientos para demostrar que un medicamento es intercambiable
� Requisitos a que deben sujetarse los terceros autorizados que realicen las pruebas
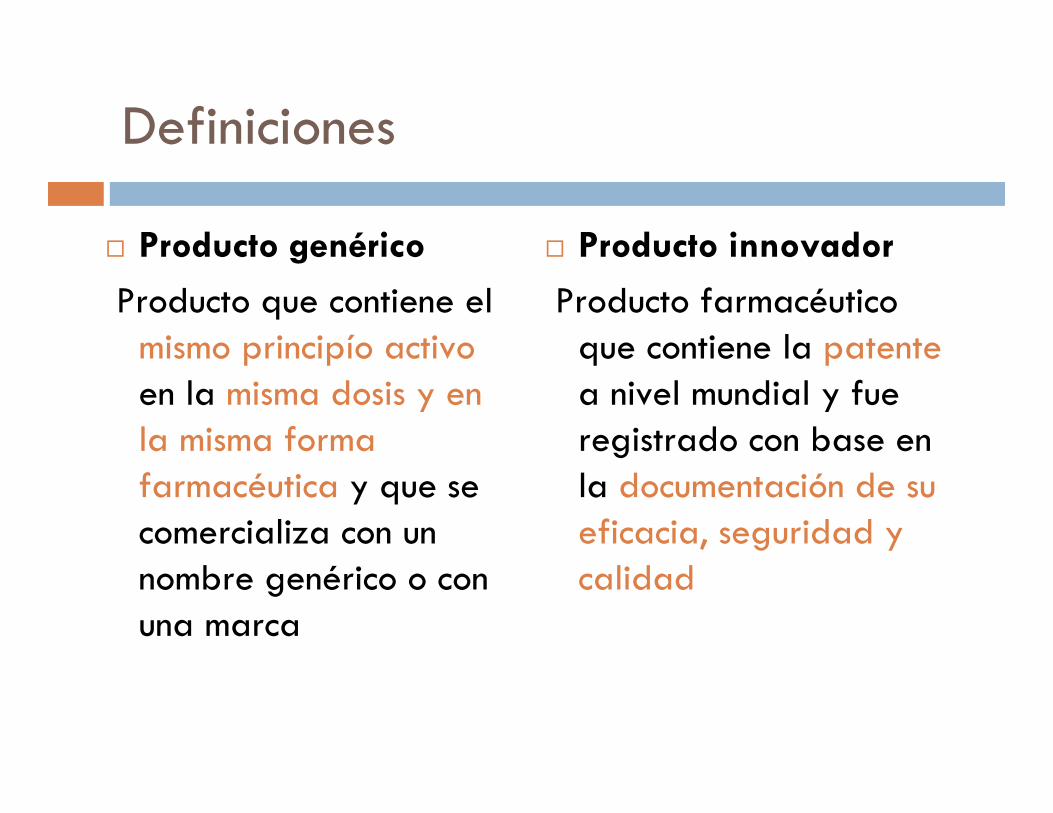
Definiciones
� Producto genérico
Producto que contiene el mismo principío activoen la misma dosis y en la misma forma farmacéutica y que se comercializa con un nombre genérico o con una marca
� Producto innovador
Producto farmacéutico que contiene la patentea nivel mundial y fue registrado con base en la documentación de su eficacia, seguridad y calidad

Definiciones
� Medicamento de referencia
El indicado por la SSA, que cuenta con el registro de dicha dependencia, se encuentra disponible comercialmente y es selleccionado conforme a los siguientes criterios:
-Medicamento innovador y en caso de no existir cualquiera de los siguientes:
Producto cuya bioequivalencia esta determinada
Producto que cuente con el registro mas antiguo ante la autoridad sanitaria y que haya demostrado su eficacia y seguridad
Producto cuya correlación in vitro-in vivo esta establecida
CBF

BCS, Sistema de Clasificación Biofarmacéutica
“Las diferencias de la disolución in vivo en el tracto gastrointestinal son la razón principal para observar diferencias en la biodisponibilidad de dos productos de liberación inmediata (LI) conteniendo el mismo fármaco”

BCS
� Un fármaco, de acuerdo a BCS, se puede clasificar en:� Alta o baja solubilidad
� Alta o baja permeabilidad

Altamente soluble
� La dosis mas alta se solubiliza en 250 mL o menos en agua en un pH de 1-8.� (250mL: Fármaco administrado con 8 onzas de fluído)

Altamente permeable
� La extensión de la absorción en humanos se establece como no mas del 90% de una dosis administrada en una comparación a una dosis iv. F≥0.9

Determinación solubilidad
� Objetivo: Determinar la solubilidad al equilibrio de un fármaco bajo condiciones fisiológicas aproximadas:� Determinar perfiles de pH-solubilidad en un intervalo de pH 1-8.� Preferiblemente 8 o mas condiicones de pH� no se deben usar Sols. Amortiguadores que reaccionen con el fármaco
El tipo de solubilidad determinada calculando el volumen de un medio acuoso es suficiente para disolver la dosis más alta
*Altamente soluble: cuando la dosis mas alta es soluble en 250 mLo menos de un medio acuoso en un intervalo de pH de 1-8.

BCS
� Basada en la solubilidad del fármaco y permeabilidad:� Caso 1: Alta solubilidad- alta permeabilidad
� Caso 2 :Baja solubilidad-alta permeabilidad
� Caso3: Alta solubilidad-baja permeabilidad
� Caso4: Baja solubilidad-baja permeabilidad

Disuelve rápidamente
� Condiciones de USP: Cuando se disuelve no menos del 85% de la cantidad de la etiqueta en 30 min.
� Aparato 1 a 100 rpm o aparato 2 a 50 rpm en un vol. De 900 mL o menos en cada uno de los siguientes medios:
� Medio ácido Ej: HCl 0.1N, fluído gástrico sin enzimas (USP).
� Sol. Amortiguadora pH 4.5
� Sol. Amortiguadora pH 6.8 o fluído intestinal sin enzimas.

BCS, caso 1
� Disolución, no velocidad limitante.
� Difusión pasiva, no velocidad limitante
� No se puede medir una velocidad limitante in-vitro
� El significado de correlación in vitro in vivo no es posible.

Caso 2
� La disolución puede ser velocidad limitante
� Difusión pasiva no es velocidad limitante
� Puede medirse una velocidad limitante in vitro
� Es significativo el IVIVC.

Caso 3
� La disolución No es velocidad limitante
� La difusión pasiva puede ser la velocidad limitante
� No puede medirse la velocidad limitante in vitro
� Significado IVIVC, probabilidad limitada.

Caso 4
� RETO, fármaco para administración por vía oral
� Grandes probabilidades de que el proyecto no se realice.

� Solubilización, proceso por el cual una sustancia pura se disuelve
� Disolución, proceso por el cual una sustancia impura se disuelve.(fármaco con excipientes).


Introducción
Historia
� 1978 - La FDA publica “Guías para la prueba de disolución”(“Guidelines for disolución testing”).
� 1990 - Se incorporan a la USP XXII aparatos 3, 4, y 5 para la disolución de parches transdérmicos
� 1995 - La USP XXIII incorpora 2 nuevos aparatos para productos de liberación prolongada y renumera los 7 aparatos
� 2000 - La FDA publica varias Guías relacionadas con disolución (http://www.fda.gov/cder/guidance.htm)

Prueba de Disolución
� Es una prueba físicoquimica utilizada para evaluar la calidad de un producto farmacéutico.
� Se usa para evaluar el control de calidad de los diferentes lotes de producción.
� Las especificaciones de disolución permiten la liberación de nuevos lotes dentro del mercado de venta.

Factores que Influyen en la Disolución de Formas Farmacéuticas Sólidas
� Características físicas de las formas farmacéuticas sólidas
� Capacidad de humectación de la forma farmacéutica
� Capacidad de penetración en el medio de disolución
� Proceso de hinchazón
� Desintegración
� Disgregación
� Propiedades físico-químicas del fármaco

Importancia de la Prueba de Disolución ...cont.
� Provee los datos para facilitar la aprobación inicial y los cambios referentes al escalamiento y post-aprobación del producto.
� Permite a las entidades regulatorias tomar la decisión de aprobar cambios menores en la formulación y procesos de fabricación .
� Es un requisito regulatorio en las pruebas de evaluación de formas farmacéuticas sólidas.

Clasificación biofarmacéutica
� Permeabilidad: Grado de absorción en humanos ómétodos de permeabilidad intestinal� Alta, grado de absorción mayor al 85% en humanos

Bioexención
� Los estudios de biodisponibilidad in vivo y/o los de bioequivalencia pueden ser exentos en determinados productos genéricos administrados por vía oral.
� Ventajas, simplificación y reducción del tiempo requerido para la aprobación de un producto.

Bioexención
� Un producto debe presentar características de muy rápida o rápida disolución in vitro, depende de las características del ingrediente activo
� Clasificación clase 1� Muy rápida disolución: se disuelve el 85% O MÁS EN 15 MINUTOS
� Rápida disolución: se disuelve el 85% O MÁS EN 30 MINUTOS.
� Aparato I a 100 rpm� Aparato II a 50 rpm� Volumen 900 mL.