genetica-cromosomopatias dra martinez
TRANSCRIPT

Genética - cromosomopatías.

Genética
Ciencia de la diversidad y variabilidad.
Genética Humana
Rama de la biología que estudia las diferencias entre los individuos, determinadas por las diferencias del material genético.

DOGMA CENTRAL DE LA BIOLOGIA MOLECULAR
ACIDO DESOXIRRIBONUCLEICO
Almacena
Replica
Expresa
mRNA – mensajero
rRNA - ribosomal
tRNA - transferencia
ACIDO RIBONUCLEICO


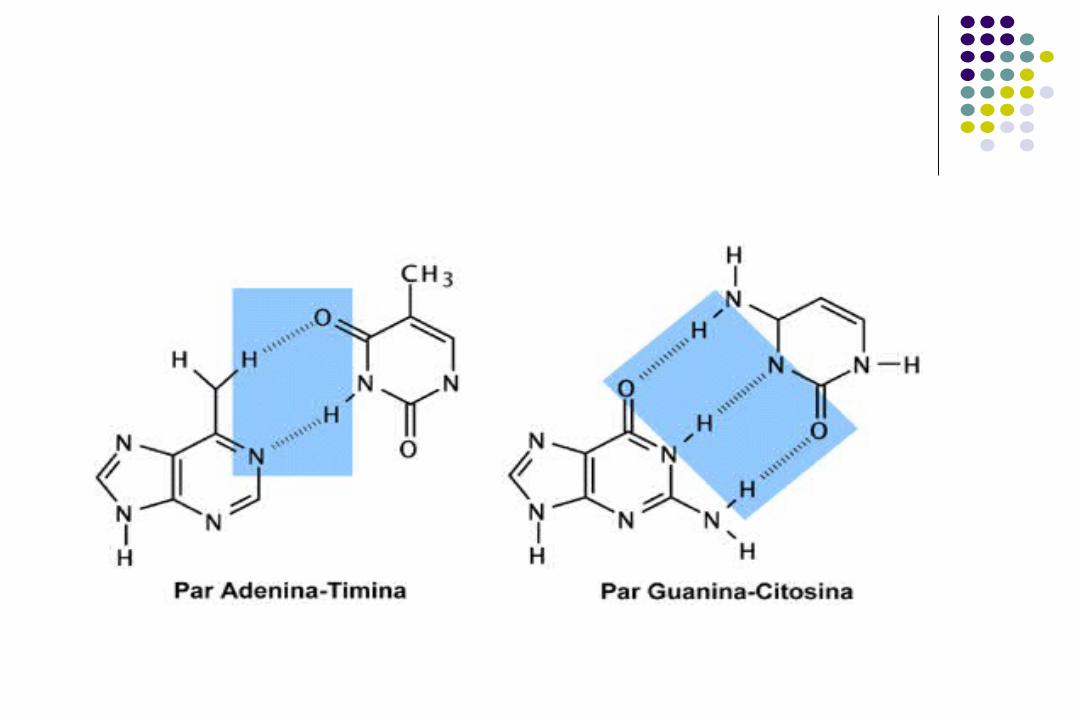
Estructura de DNA.
Extremo 5’ de la cadena: grupo fosfato. Extremo 3’ de la cadena: grupo hidroxilo. Las bases complementarias se unen por Puentes
de Hidrógeno
(2 entre A y T) (3 entre G y C) Las bases adyacentes se unen por enlaces 5’3’
fosfodiester.

Strachan &Read,
Cadena de polinucleótidos
Enlaces fosfodiester
Polaridad de la molécula

Clasificación de las aberraciones cromosómicas
Alteraciones en el número.
Alteraciones en la estructura.

Euploidías - Repetición del número haploide.
Triploidía 3n
Tetraploidía 4n
Pentaploidía 5n
Poliploidías
Númericas
Rearreglos Cromosómicos Constitucionales
Triploidía

Aneuploidías - Alteraciones en el número diploide.
Trisomía
Monosomía
Doble Trisomía
Tetrasomía

Alteraciones en la estructura.
Duplicación - Secuencia igual de genes aparece doble en un mismo cromosoma.
Inversión - Segmento cromosómico gira 180˚ sobre si mismo y se coloca en forma invertida.
- Paracéntrica
- Pericéntrica

Translocación - Intercambio de segmentos entre cromosomas.
- Recíproca
- Robertsoniana
Cromosoma en Anillo - Se presenta un rompimiento simultáneo en el brazo corto y largo, y los extremos se unen.
Isocromosoma - División transversal del cromosoma.

Cromosoma Dicéntrico - Tiene dos centrómeros y puede resultar de una translocación de cromosomas no homólogos o de una translocación de las dos cromátides del mismo cromosoma.

Rearreglos Cromosómicos Constitucionales
Mecanismo de formación de un Isocromosoma

Rearreglos Cromosómicos Constitucionales
Cromosoma dicéntrico por translocación de cromátides

Rearreglos Cromosómicos Constitucionales
Heredados de un portador ó de novo en los gametos que resultan en el cigoto.
Se detecta en 0.6% de los recién nacidos.
25% de todos los abortos.
50-60% de abortos durante el 1er. Trimestre.
La mayoría causados por aneuploidías.

Rearreglos Cromosómicos Constitucionales
Rearreglos balanceados presentes en cerca de 1 en 500 individuos.
No balanceados presentes en el 3% de todas las anormalidades cromosómicas.
Primeras anormalidades no balanceadas descritas antes de los metódos convencionales de bandeo.

Rearreglos Cromosómicos Constitucionales
Tipos
Rearreglos Intercromosómicos.
Involucran dos cromosomas distintos y pueden ocurrir en cromosomas no homólogos.
Translocaciones Robertsonianas.
Translocaciones Recíprocas.

Rearreglos Cromosómicos Constitucionales
Translocaciones Robertsonianas. Intercambios completos de brazos largos entre cromosomas acrocéntricos.
Cr. 13, 14, 15, 21 y 22.
Rearreglo cromosómico recurrente más frecuente. Frecuencia de 1 en 1000 individuos.
rob(13q14q) y rob(14q21q) constituyen cerca del 85% de todas las translocaciones robertsonianas.

Rearreglos Cromosómicos Constitucionales
Translocaciones Recíprocas. Resulta de una sola ruptura en cada uno de los dos cromosomas involucrados.
Frecuencia de 1 en 625 individuos en población general.
La t(11;22)(q23;q11.2) es común.
Todos los cromosomas han sido reportados.

Rearreglos Cromosómicos Constitucionales
Rearreglos Intracomosómicos. Aberraciones que involucran un solo cromosoma.
Deleciones intersticiales y terminales. Duplicaciones intersticiales. Cromosomas marcadores. Inversiones. Isocromosomas. Inserciones.
Pueden involucrar un solo homólogo ó a los dos.

Rearreglos Cromosómicos Constitucionales

Rearreglos Cromosómicos Constitucionales
• Inserción

Rearreglos Cromosómicos Constitucionales
Formación de un Anillo Cromosómico

Rearreglos Cromosómicos Constitucionales
Deleciones y Duplicaciones intersticiales son resultado de intercambios en brazo del cromosoma conservando el telómero.
Cromosomas Marcadores. Anormalidades estructurales pequeñas de origen no identificable.
Más comunes. Cr. X, 15 y 22.

Rearreglos Cromosómicos Constitucionales
Inversiones. Paracéntrica y pericéntrica.
Inv(9)(p11q12). Presente en 2% de la población.
Isocromosomas. Imágenes en espejo ya sea de brazos cortos ó largos.
Más frecuentemente observados en Cr. X y acrocéntricos.
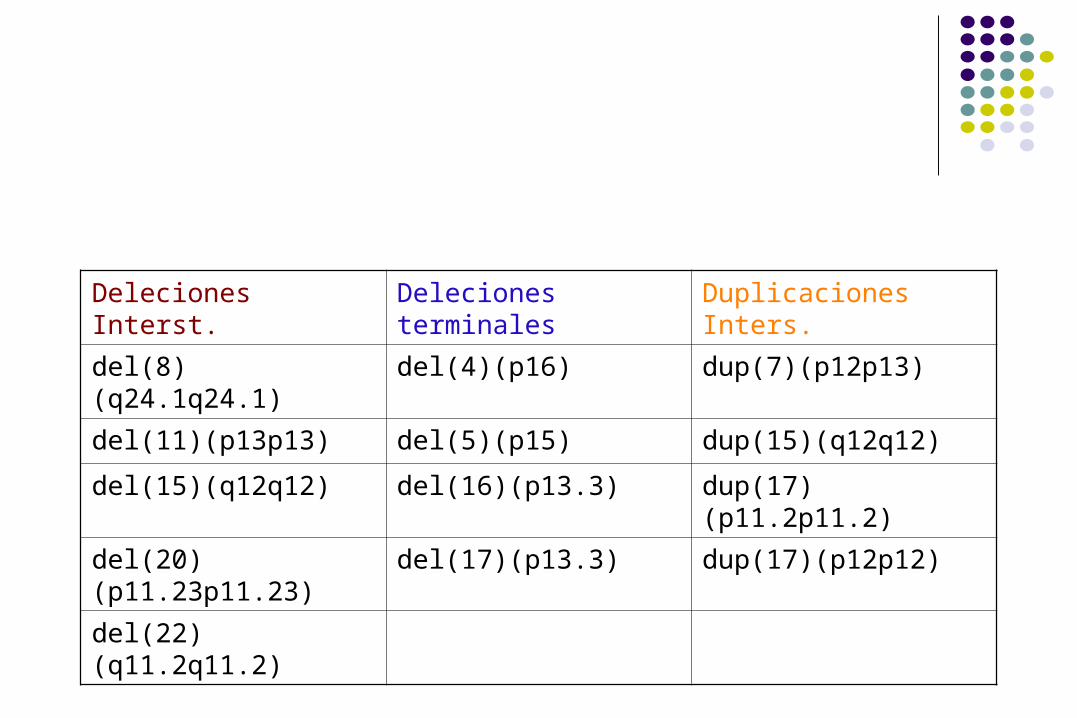
Deleciones Interst. Deleciones terminales
Duplicaciones Inters.
del(8)(q24.1q24.1) del(4)(p16) dup(7)(p12p13)
del(11)(p13p13) del(5)(p15) dup(15)(q12q12)
del(15)(q12q12) del(16)(p13.3) dup(17)(p11.2p11.2)
del(20)(p11.23p11.23)
del(17)(p13.3) dup(17)(p12p12)
del(22)(q11.2q11.2)
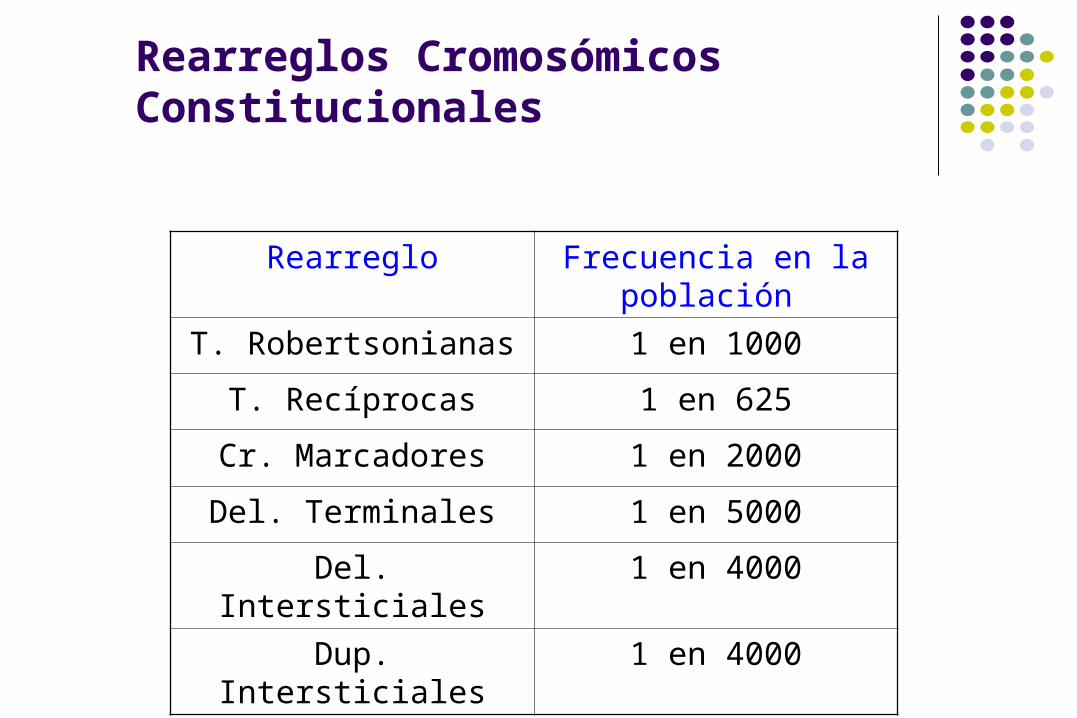
Rearreglos Cromosómicos Constitucionales
Rearreglo Frecuencia en la población
T. Robertsonianas 1 en 1000
T. Recíprocas 1 en 625
Cr. Marcadores 1 en 2000
Del. Terminales 1 en 5000
Del. Intersticiales 1 en 4000
Dup. Intersticiales 1 en 4000

Rearreglos Cromosómicos Constitucionales
Metódos de Estudio.
Cariotipo
FISH
SKY
Convencional 400-500 bandas.
Alta Resolución 800-1000 bandas.
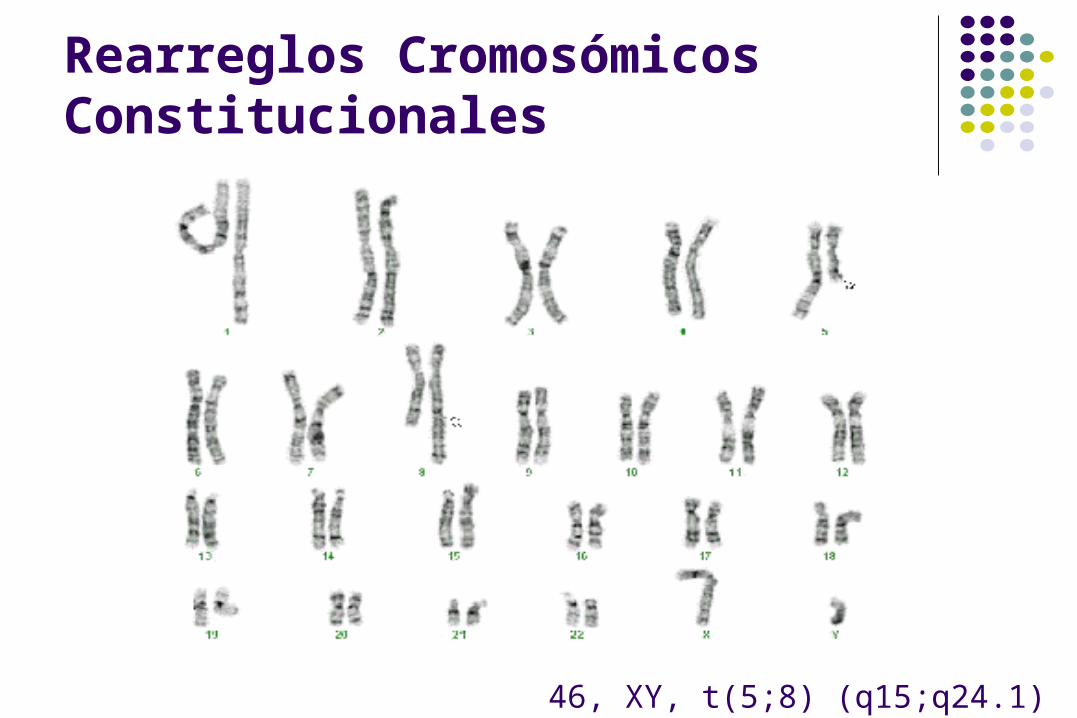
Rearreglos Cromosómicos Constitucionales
46, XY, t(5;8) (q15;q24.1)

Rearreglos Cromosómicos Constitucionales
Formación del Cuadrivalente en paquiteno de la meiosis para la segregación en una translocación balanceada.
Cr. X Cr. 9 der (X) der (9)
Cuadrivalente en
Paquiteno
Alterna Adyacente 1 Adyacente 2

der 9,X
X normal
9 normal
der X,9
FISH
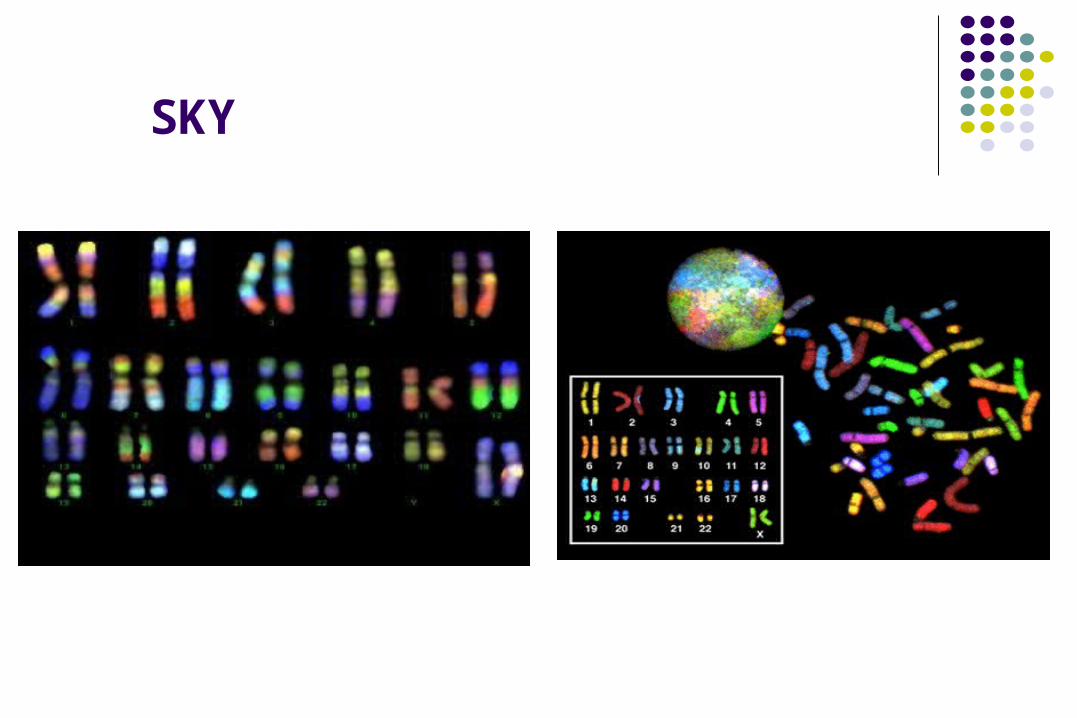
SKY
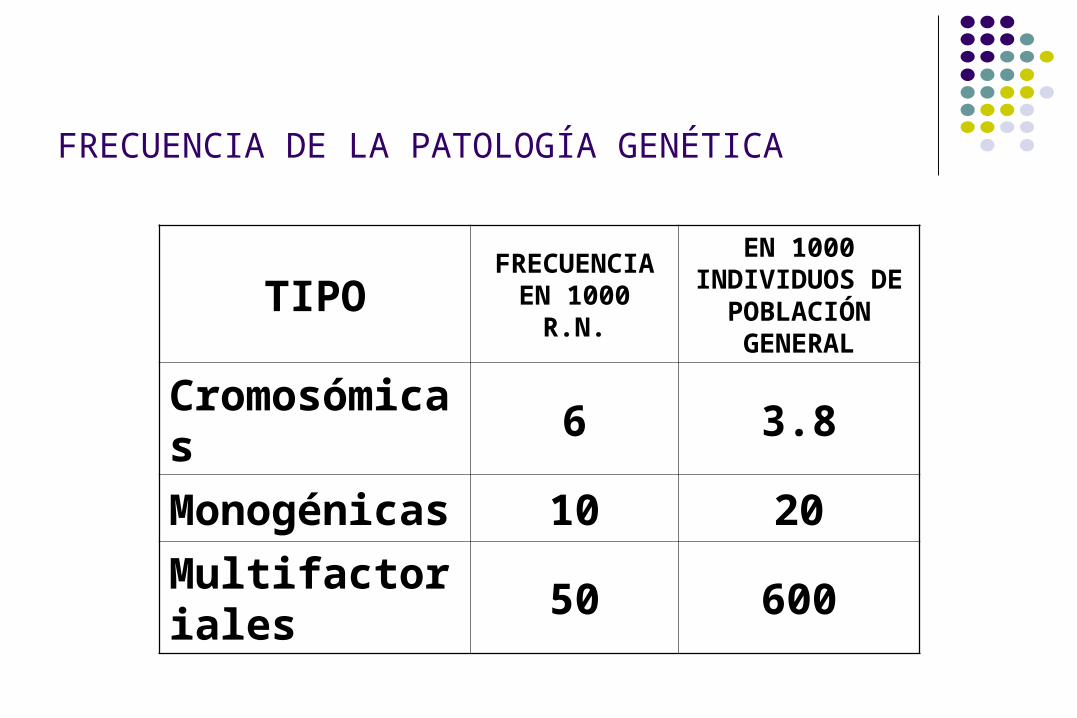
FRECUENCIA DE LA PATOLOGÍA GENÉTICA
TIPO FRECUENCIA EN 1000 R.N.
EN 1000 INDIVIDUOS DE POBLACIÓN
GENERAL
Cromosómicas 6 3.8
Monogénicas 10 20
Multifactoriales 50 600
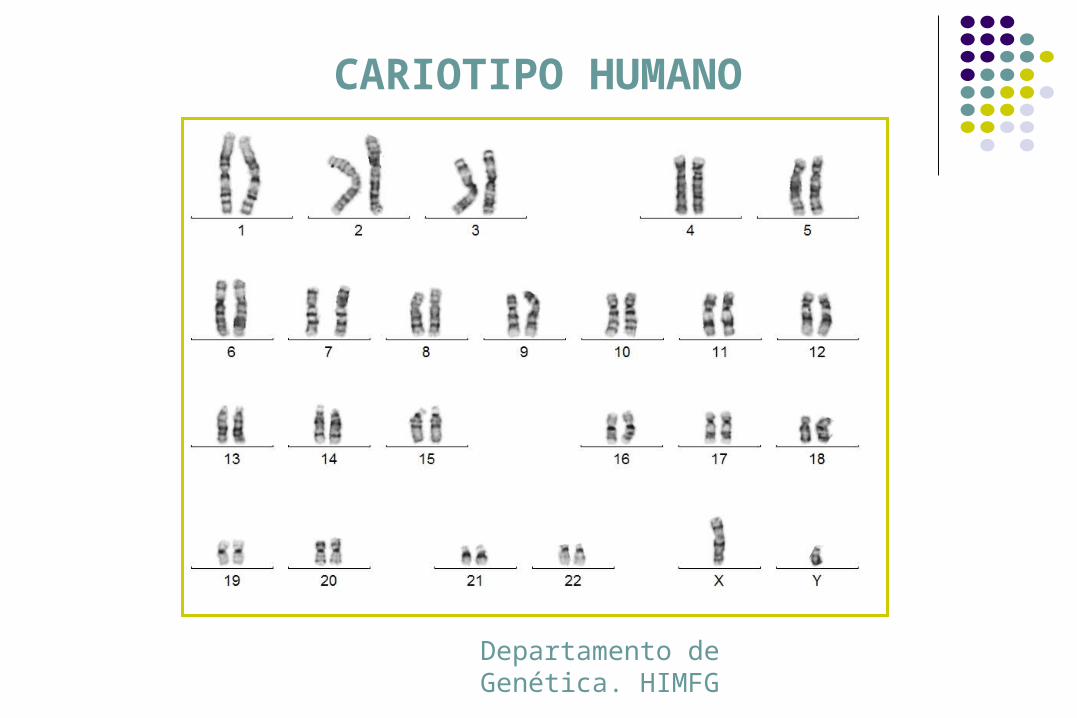
CARIOTIPO HUMANO
Departamento de Genética. HIMFG
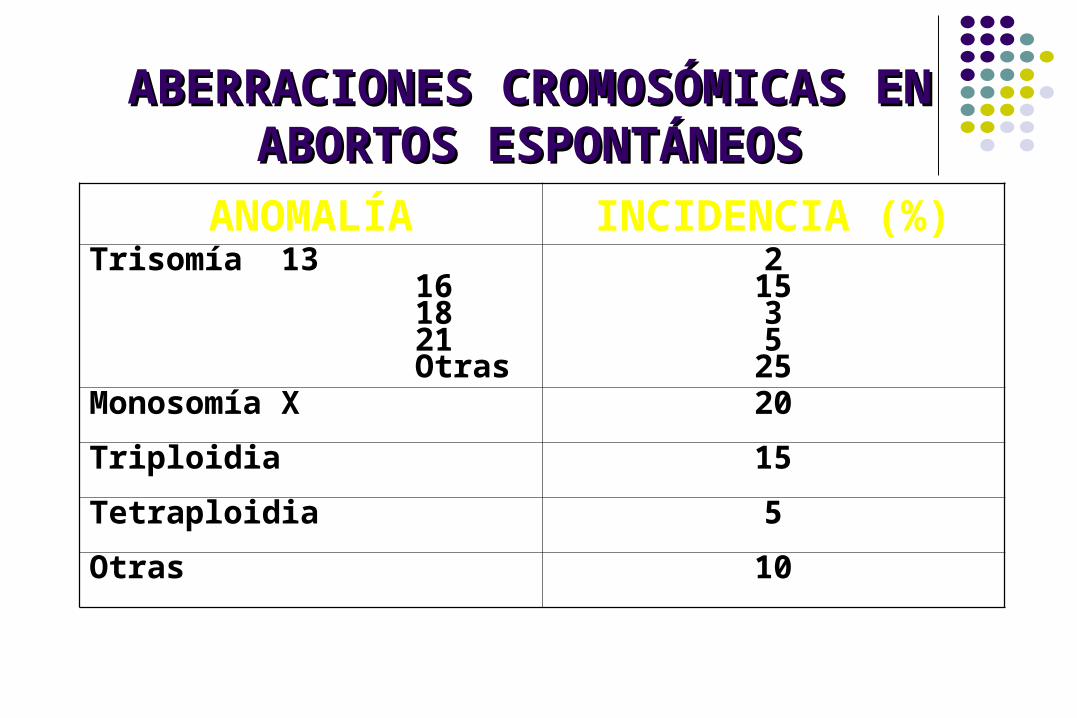
ANOMALÍA INCIDENCIA (%)Trisomía 13 16 18 21 Otras
2153525
Monosomía X 20
Triploidia 15
Tetraploidia 5
Otras 10
ABERRACIONES CROMOSÓMICAS EN ABERRACIONES CROMOSÓMICAS EN ABORTOS ESPONTÁNEOSABORTOS ESPONTÁNEOS

ABERRACIONES CROMOSÓMICAS EN RECIÉN NACIDOSABERRACIONES CROMOSÓMICAS EN RECIÉN NACIDOSAUTOSOMAS
Trisomía 21 1: 700 Trisomía 18 1: 6,000 Trisomía 13 1: 12,000 Rearreglos desbalanceados 1: 17,000 Rearreglos balanceados Translocaciones robertsonianas 1: 1,000 Translocaciones recíprocas 1: 11,000

ABERRACIONES CROMOSÓMICAS EN RECIÉN ABERRACIONES CROMOSÓMICAS EN RECIÉN NACIDOSNACIDOS
CROMOSOMAS SEXUALES
47,XXY 1: 1000 nacimientos de varones
47,XYY 1: 1000 nacimientos de varones
45,X 1: 2500 nacimientos de mujeres
47,XXX 1: 1000 nacimientos de mujeres
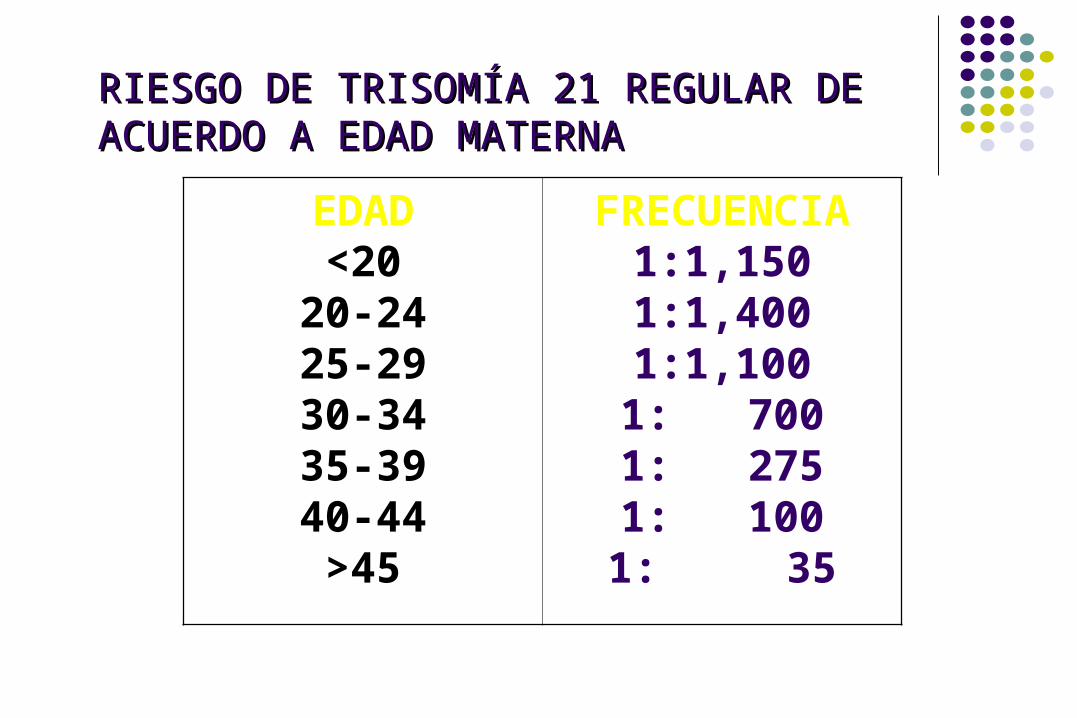
RIESGO DE TRISOMÍA 21 REGULAR DE ACUERDO A RIESGO DE TRISOMÍA 21 REGULAR DE ACUERDO A EDAD MATERNAEDAD MATERNA
EDAD<20
20-2425-2930-3435-3940-44>45
FRECUENCIA1:1,1501:1,4001:1,1001: 7001: 2751: 1001: 35

SÍNDROME DE DOWNSÍNDROME DE DOWNGENERALIDADES
65 a 80% de las gestaciones con trisomía 21
son abortos espontáneos.
Frecuencia en R.N.: 1/600
Aneuploidia más común compatible con la vida

SÍNDROME DE DOWNSÍNDROME DE DOWN
GENERALIDADES Peso y talla bajos pre y postnatal IQ: 30-50 Causas de mortalidad Infancia: cardiopatía congénita,
infecciones respiratorias, leucemia Adulto: Alzheimer, infecciones

SÍNDROME DE DOWN
CARACTERÍSTICAS CRANEOFACIALES

SÍNDROME DE DOWNSÍNDROME DE DOWN CARACTERISTICAS CLINICAS OJOS
Fisuras palpebrales hacia arribaEpicanto interno
Manchas de Brushfield Estrabismo Nistagmos PABELLONES AURICULARES
Malformados y pequeñosAusencia de lóbulos

SÍNDROME DE DOWNSÍNDROME DE DOWN CARACTERÍSTICAS CLÍNICAS
CAVIDAD ORALBoca abiertaComísuras labiales hacia abajoProtrusión lingual
Macroglosia Surcos linguales Paladar angosto Malposición dentaria

SÍNDROME DE DOWNSÍNDROME DE DOWN
CARACTERÍSTICAS CLÍNICAS
CUELLOCorto y ancho
TORAXPectum excavatumPectum carinatumXifosis dorsolumbar

SÍNDROME DE DOWNSÍNDROME DE DOWN
CARACTERÍSTICAS CLÍNICAS
ABDOMEN Diastasis de rectos Hernia umbilical GENITALES Pene pequeño Criptorquidia Escroto pequeño

SÍNDROME DE DOWNSÍNDROME DE DOWN
CARACTERÍSTICAS CLÍNICAS MANOS
Anchas y pequeñas
Braquidactilia
Pliegue palmar único
5to dedo pequeño
Pliegue de flexión
Clinodactilia de 5o. dedo

SÍNDROME DE DOWNSÍNDROME DE DOWN
CARACTERÍSTICAS CLÍNICAS
PIES
Diastasis entre 1ro y 2do ortejos.
Surco plantar.

SÍNDROME DE DOWNSÍNDROME DE DOWN
Los varones son infértiles mientras que las mujeres son fértiles

SÍNDROME DE DOWNSÍNDROME DE DOWN
MALFORMACIONES ASOCIADASCardiopatía congénita: 45% Canal atrio-ventricular 45% CIV 35% CIA 8% PCA 7% Tetralogía de Fallot 4%

SÍNDROME DE DOWNSÍNDROME DE DOWN
MALFORMACIONES ASOCIADASGastrointestinales: 10 – 18%
Atresia duodenal 2 -3% Hirschsprung 2% Onfalocele, bandas duodenales, páncreas
anular, atresia de ileo o yeyuno, microcolon, alteraciones de recto y ano.

SÍNDROME DE DOWNSÍNDROME DE DOWN
PROBLEMAS MÉDICOS ASOCIADOS Infecciones recurrentes Leucemia aguda linfoblástica (15 a 20
veces más frecuente) Hipotiroidismo (1 : 10) Hipoacusia conductiva o
neurosensorial Inestabilidad atlanto-axoidea Alzheimer

SÍNDROME DE DOWN
Puente nasal deprimidoHipoplasia medio facial

SÍNDROME DE DOWNSÍNDROME DE DOWNFRECUENCIA DE CONSTITUCIÓN CROMOSÓMICAFRECUENCIA DE CONSTITUCIÓN CROMOSÓMICA
CARIOTIPO FRECUENCIA EN %
Trisomía 21 regular
Translocación
Mosaicismo
95
3
2

CARIOTIPO POR TRISOMIA 21 REGULAR
47,XY +21

MEIOSIS NORMAL
MEIOSIS I
23 DUPLICADOS
MEIOSIS II
23 SENCILLOS
2n
n

MECANISMOS DE FORMACIÓN ABERRACIONES NÚMERICAS
No disyunción meiosis II
No disyunción meiosis I
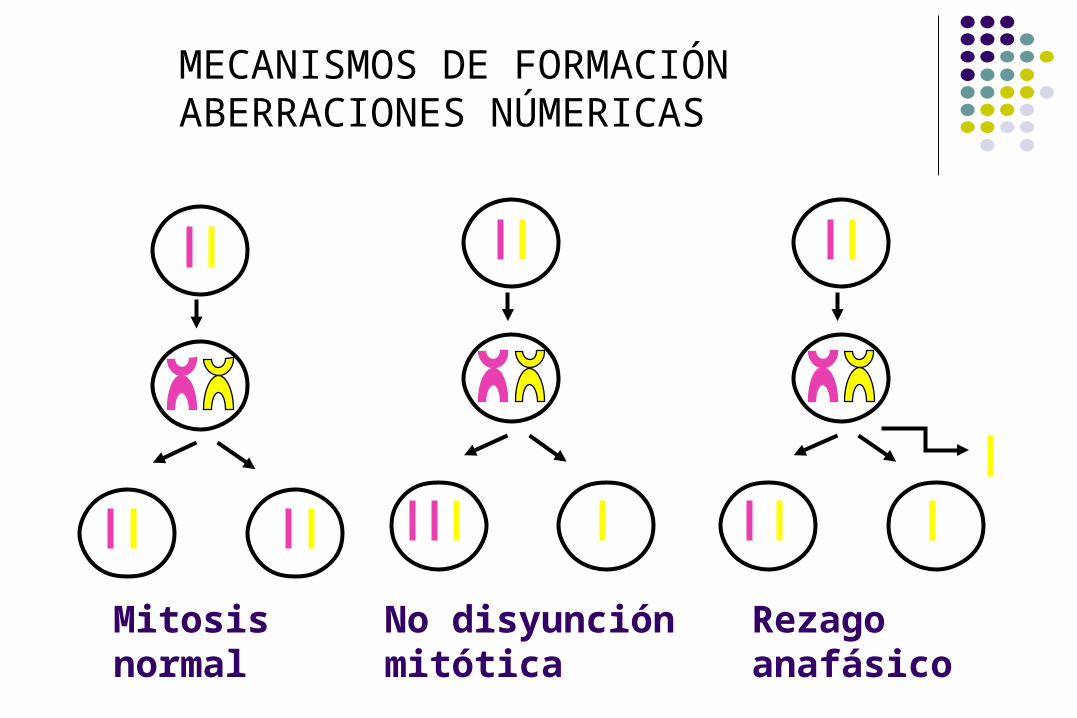
MECANISMOS DE FORMACIÓN ABERRACIONES NÚMERICAS
Rezago anafásicoNo disyunción mitóticaMitosis normal

SÍNDROME DE DOWNSÍNDROME DE DOWNETIOLOGÍA ETIOLOGÍA
FRECUENCIA EN %
No disyunción materna 95
Meiosis I 75
Meiosis II 25
RIESGO DE RECURRENCIA 1%

CARIOTIPO CON TRISOMIA 21 POR t (14;21)
n
x
ia

CARIOTIPO DE PORTADOR DE t(14;21)
aa

TRISOMÍA 18 (EDWARDS)TRISOMÍA 18 (EDWARDS)GENERALIDADES
Frecuencia: 1/6,000 R. N. Representa la segunda alteración
cromosómica de autosomas después de Down
Vida media: 48 días Fallecimientos ¿cardiopatía, apnea
central?30% en el primer mes50% en los primeros 6 meses90% en el primer año

Retraso en el crecimiento 96% Retraso severo del desarrollo psicomotor
96% Hipertonia Dolicocefalia 93% Occipucio prominente 91% Micrognatia 96% Pabellones auriculares bajos y malformados 88%
TRISOMÍA 18 (EDWARDS)TRISOMÍA 18 (EDWARDS)MANIFESTACIONES CLÍNICASMANIFESTACIONES CLÍNICAS
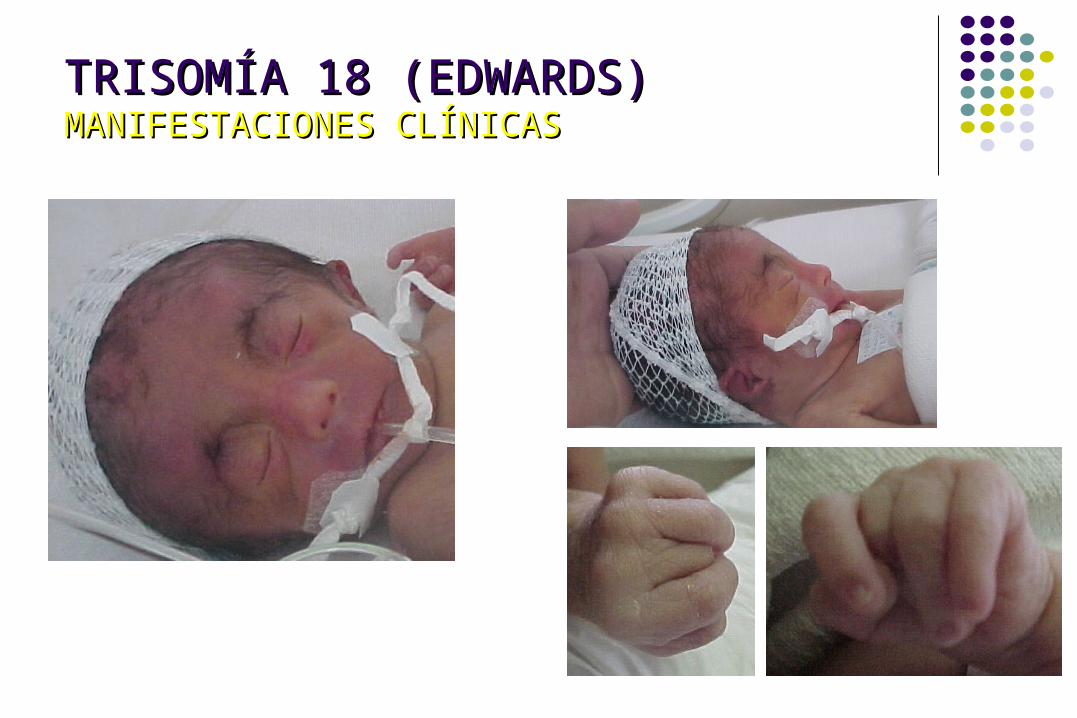
TRISOMÍA 18 (EDWARDS)TRISOMÍA 18 (EDWARDS)MANIFESTACIONES CLÍNICASMANIFESTACIONES CLÍNICAS


Esternón corto Teletelia 90% Hernia umbilical o
inguinal Criptorquidia 100% Clitoromegalia 89% Pelvis pequeña
TRISOMÍA 18 (EDWARDS)TRISOMÍA 18 (EDWARDS)MANIFESTACIONES CLÍNICASMANIFESTACIONES CLÍNICAS

Sobreposición de dedos 89% Uñas hipoplásicas 100% Dermatoglifos: arcos 96% Pie equinovaro Calcáneo prominente Pies en mecedora Dorsiflexión del primer ortejo Sindactilia de 2do y 3ro
TRISOMÍA 18 (EDWARDS)TRISOMÍA 18 (EDWARDS)MANIFESTACIONES CLÍNICASMANIFESTACIONES CLÍNICAS

TRISOMÍA 18 (EDWARDS)TRISOMÍA 18 (EDWARDS) MALFORMACIONES ASOCIADAS Cardiopatía congénita: 85% Enfermedad polivalvular CIV PCA Foramen oval permeable Urogenitales: 30%
Riñones poliquísticosEn herradura

TRISOMÍA 18 (EDWARDS)TRISOMÍA 18 (EDWARDS) ETIOLOGÍA
CARIOTIPO FRECUENCIA EN %
Trisomía 18
regular
Meiosis II el 50%
Translocación
Mosaicismo
94
1
5
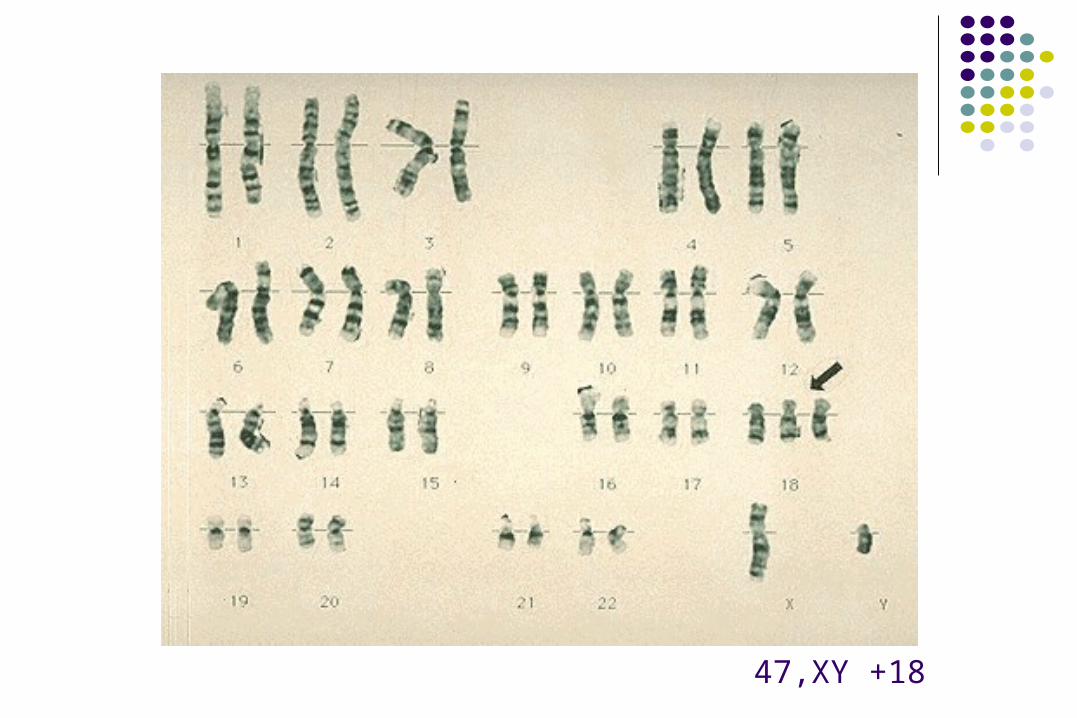
47,XY +18

TRISOMÍA 13 (PATAU)TRISOMÍA 13 (PATAU)
Frecuencia: 1/12,000 R. N. Representa el 1% de los abortos
espontáneos Vida media: 130 días Fallecimientos
45% en el primer mes70% en los primeros 6 meses86% en el primer año
GENERALIDADES

TRISOMÍA 13 (PATAU)TRISOMÍA 13 (PATAU)
Retraso del crecimiento Microcefalia Holoprosencefalia Episodios de apnea y
convulsiones Retraso severo del desarrollo
psicomotor
MANIFESTACIONES CLÍNICAS
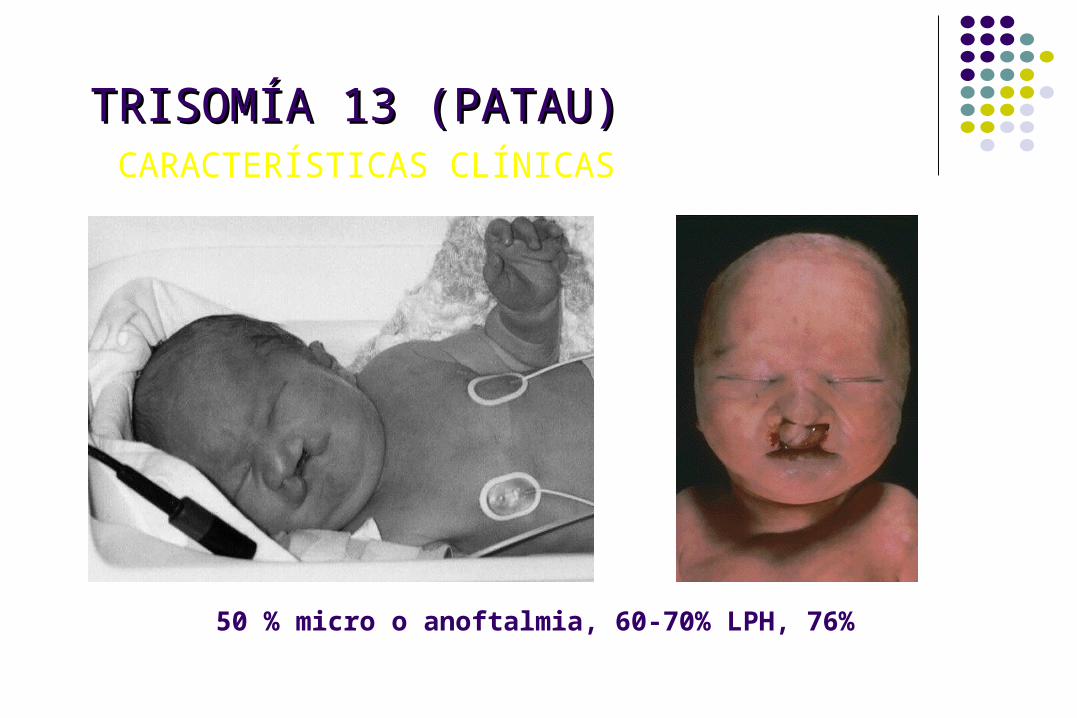
TRISOMÍA 13 (PATAU)TRISOMÍA 13 (PATAU) CARACTERÍSTICAS CLÍNICAS
50 % micro o anoftalmia, 60-70% LPH, 76%
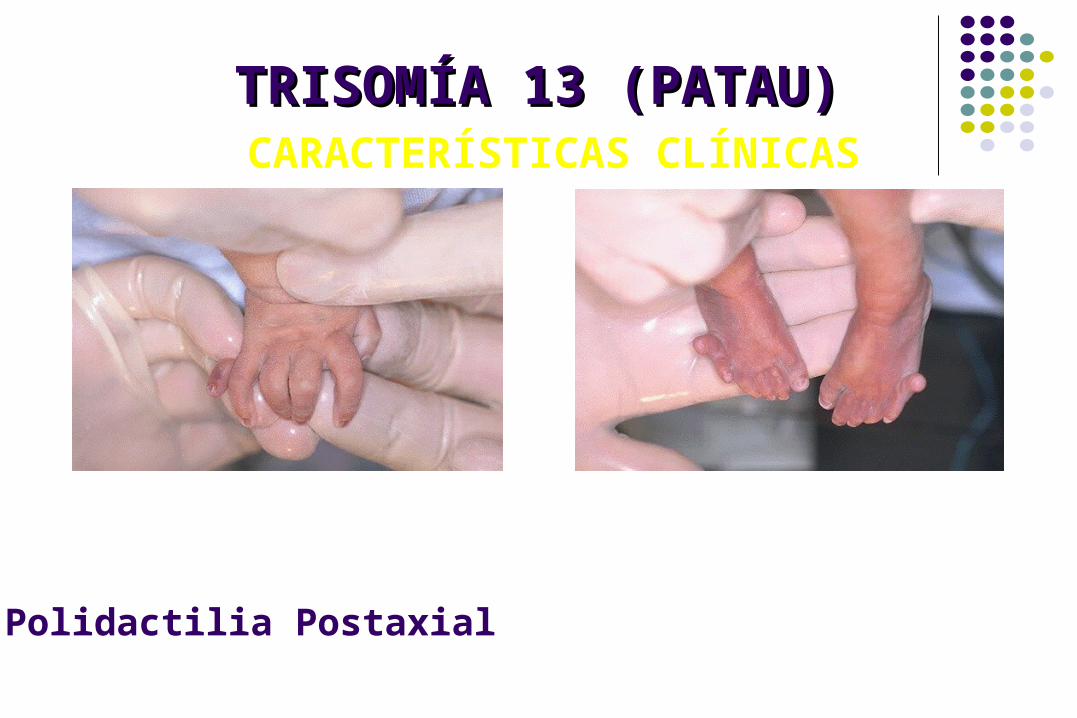
TRISOMÍA 13 (PATAU)TRISOMÍA 13 (PATAU) CARACTERÍSTICAS CLÍNICAS
Polidactilia Postaxial

TRISOMÍA 13 (PATAU)TRISOMÍA 13 (PATAU) MALFORMACIONES ASOCIADAS Cardiopatía congénita: 90%
Persistencia del conducto arterioso CIV CIA Urogenitales 30-60%
Riñones poliquísticosHiperlobulacíones renalesHidronefrosisCriptorquidiaUtero bicorne

TRISOMÍA 13 (PATAU)TRISOMÍA 13 (PATAU) ETIOLOGÍA
CARIOTIPO FRECUENCIA EN %
Trisomía 13
regular
Translocación
Mosaicismo
90
5-10
1
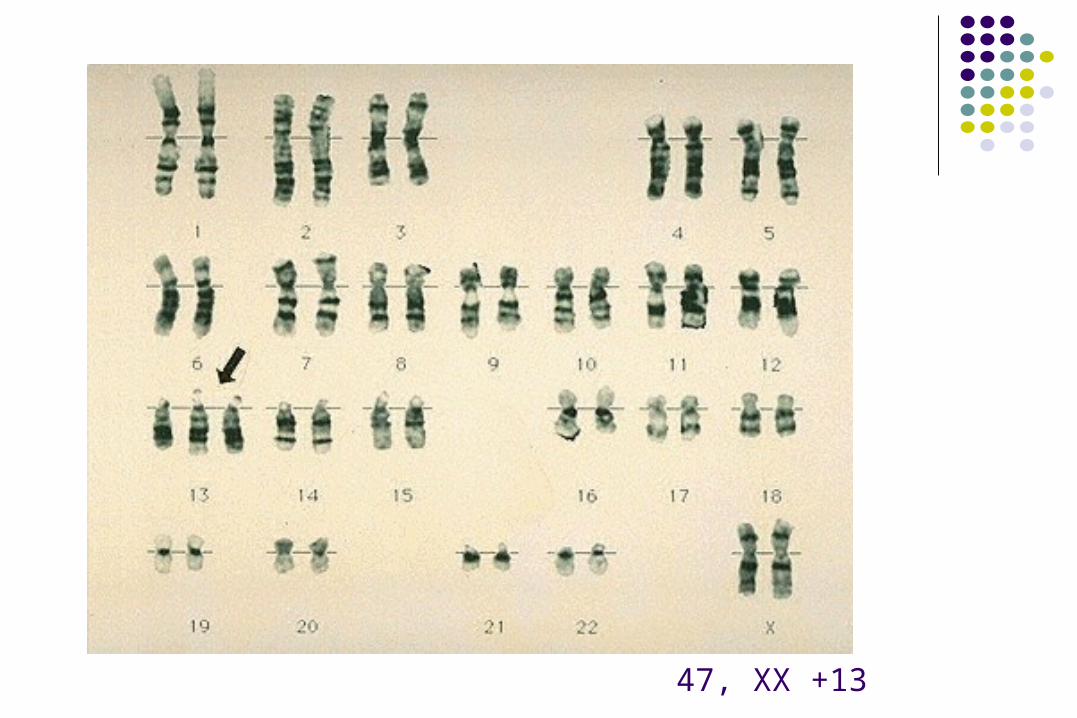
47, XX +13

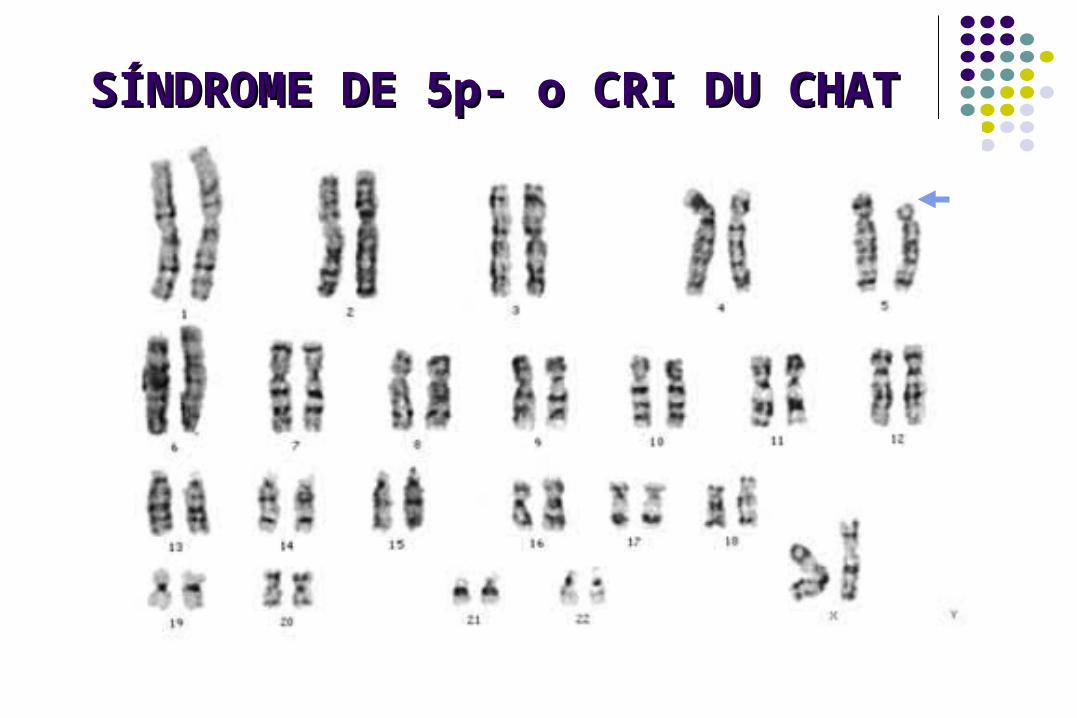
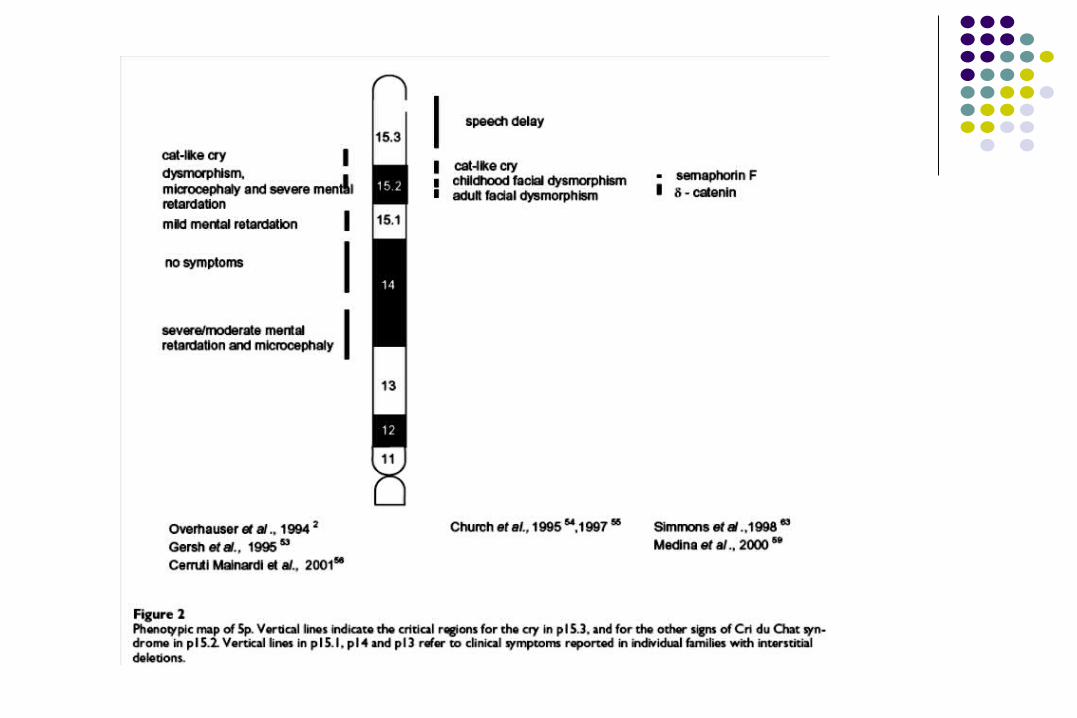
SÍNDROME DE 5p- o CRI DU SÍNDROME DE 5p- o CRI DU CHATCHAT
GENERALIDADES
Frecuencia: 1/50,000 R. N.
Deleción 5p14 a p15
Deleción de novo 85%, heredada 15%
Retraso severo del crecimiento y mental
Llanto débil de origen central no laríngeo
Cardiopatías en el 30 a 50%

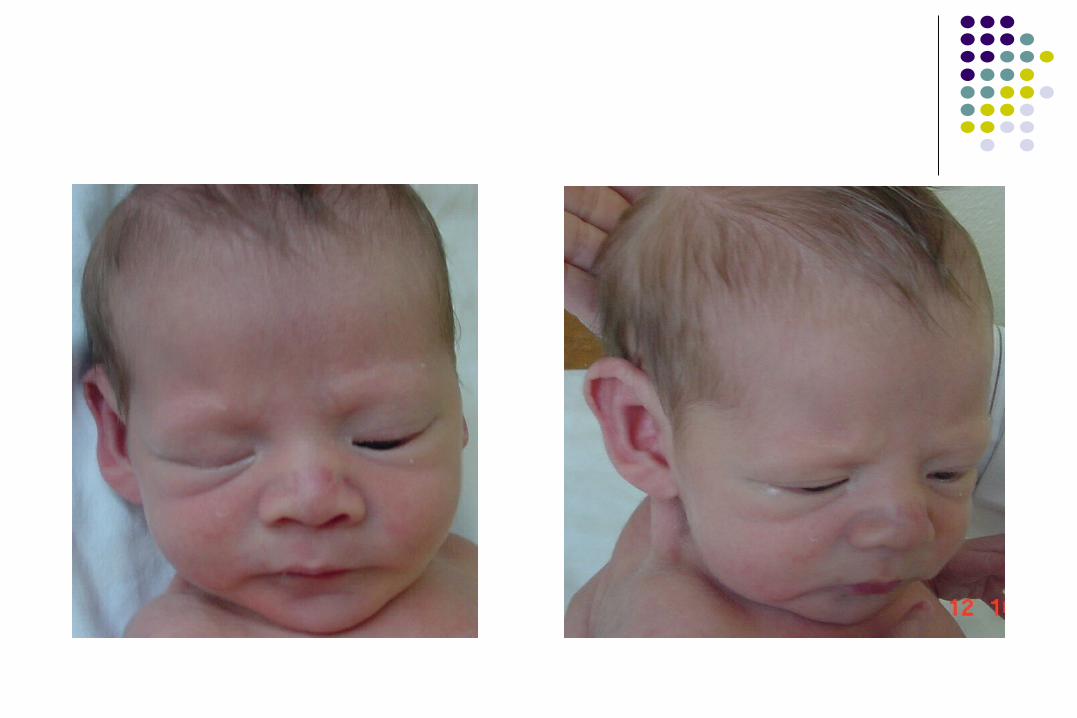
SÍNDROME DE 5p- o CRI DU SÍNDROME DE 5p- o CRI DU CHATCHAT
• Microcefalia
• Epicanto interno
• Telecanto
• Facies redonda

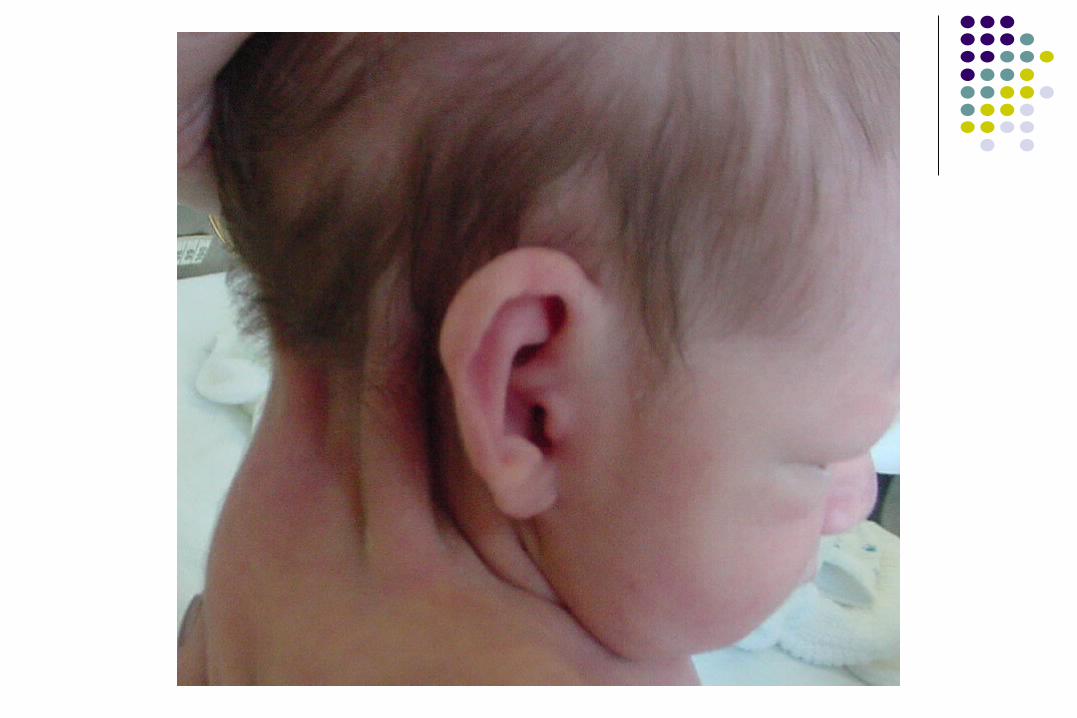
SÍNDROME DE 5p- o CRI DU SÍNDROME DE 5p- o CRI DU CHATCHAT
• Pabellones auriculares con rotación posterior
• Apéndices preauriculares 20 %
• Puente nasal ancho
• Raíz nasal prominente

SÍNDROME DE 5p- o CRI DU SÍNDROME DE 5p- o CRI DU CHATCHAT

SÍNDROME DE 5p- o CRI DU CHATSÍNDROME DE 5p- o CRI DU CHAT

SÍNDROME DE 4p- o WOLF-SÍNDROME DE 4p- o WOLF-HIRSCHHORNHIRSCHHORN
GENERALIDADES
Frecuencia: 1/50,000 R. N. Región crítica 4p16, pero la mayoría 1/3
o 2/3 de p deletado Deleción de novo 85%, heredada 15% 35% fallecen en los primeros dos años
de vida Retraso severo del crecimiento y
mental con crisis convulsivas.

Datos clínicos prácticamente
constantes:
Microcefalia leve
Frente amplia
Puente nasal ancho con glabela
prominente
Naríz hacia abajo
Hipertelorismo
Epicanto interno
SÍNDROME DE 4p- o WOLF-HIRSCHHORNSÍNDROME DE 4p- o WOLF-HIRSCHHORN

SÍNDROME DE 4p- o WOLF-HIRSCHHORNSÍNDROME DE 4p- o WOLF-HIRSCHHORN
En el 50% ptosis, fisuras palpebrales hacia abajo, estrabismo divergente

Coloboma de iris 35% Conducto auditivo
externo estrecho Apéndices preauriculares Filtrum corto y profundo Comisuras bucales hacia
abajo Micrognatia 50%
SÍNDROME DE 4p- o WOLF-HIRSCHHORNSÍNDROME DE 4p- o WOLF-HIRSCHHORN

Labio hendido 10% Paladar hendido 40% Cardiopatías 50% : CIA o CIV Criptorquidia e
hipospadias Alteraciones renales
SÍNDROME DE 4p- o WOLF-HIRSCHHORNSÍNDROME DE 4p- o WOLF-HIRSCHHORNMALFORMACIONES ASOCIADASMALFORMACIONES ASOCIADAS

Sx. Wolf-Hirschhorn

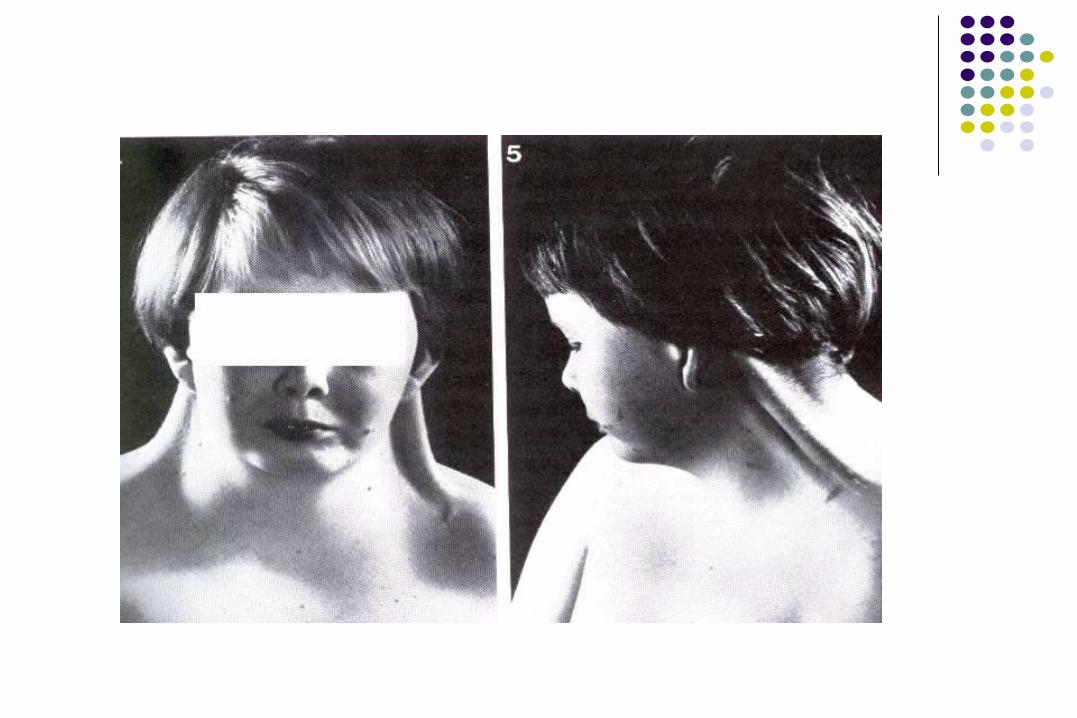
SINDROME DE TURNER
99% de los embriones 45,X se abortan. 10% de los abortos. 1 en 2,500 niñas. Causado por monosomía completa o parcial del
cromosoma X. 2/3 de los casos son origen materno.

Gen SHOX (Xp22)
Alteraciones esqueléticas
Talla baja Acortamiento de 4to metacarpiano Cubitus valgus Paladar alto y arqueado Cuello corto

CARACTERISTICAS CLINICAS
Talla Baja (100%) Cubitus valgus Uñas hipoplasicas Acortamiento de 4to metacarpiano Mal oclusión y apiñonamiento de piezas dentarias Cuello corto y ancho Cuello alado (pterygium coli) con baja inserción de
cabello: resolución del higroma quistico de la vida fetal.

Riñones en herradura o ectópicos Hipertensión Sordera conductiva o neurosensorial Hipotiroidismo autoinmune Diabetes mellitus (5%) Artritis reumatoide

Craneofacial
Fisuras palpebrales hacia abajo. Pliegue epicantico. Ocasional ptosis palpebral o estrabismo. Hipoplasía medio facial. Puente nasal ancho. Paladar alto y arqueado. Micrognatia. Pabellones auriculares rotados.

Tórax en escudo Aumento en la distancia entre los pezones Cardiopatías congénitas 30%
Coartación de la aorta Válvula aorta bicúspide Estenosis aórtica

ANOMALÍAS VASCULARES
Telangiectasias intestinales múltiples. Hemangiomas. Linfangiectasias. Linfaedema periférico en el recién nacido en
dorso de manos y pies. Estasis de Liquido linfático debido a hipoplasía
de vasos linfáticos: Extensión mecánica y alrededor de los tejidos.
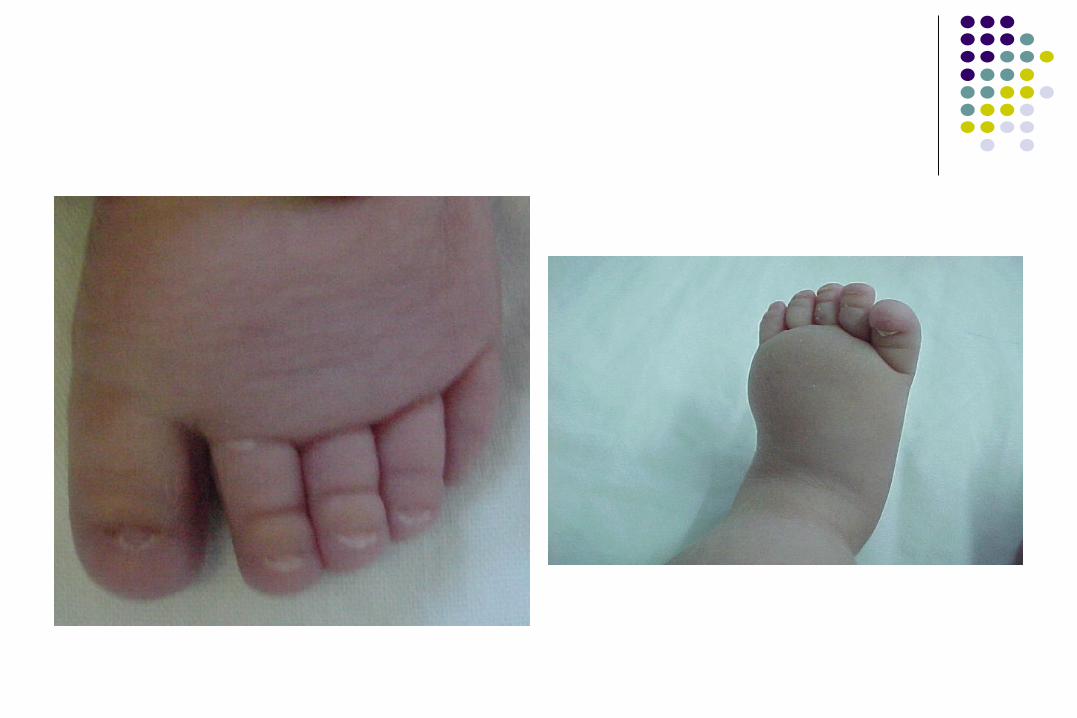

DISGENESIA GONADAL
Los ovocitos entran en acelerada degeneración durante meiosis I, perdiéndolos todos en la infancia.
Infantilismo sexual Amenorrea primaria Menstruación espontánea solo en el 5% Falla ovárica progresiva Esterilidad


45,X

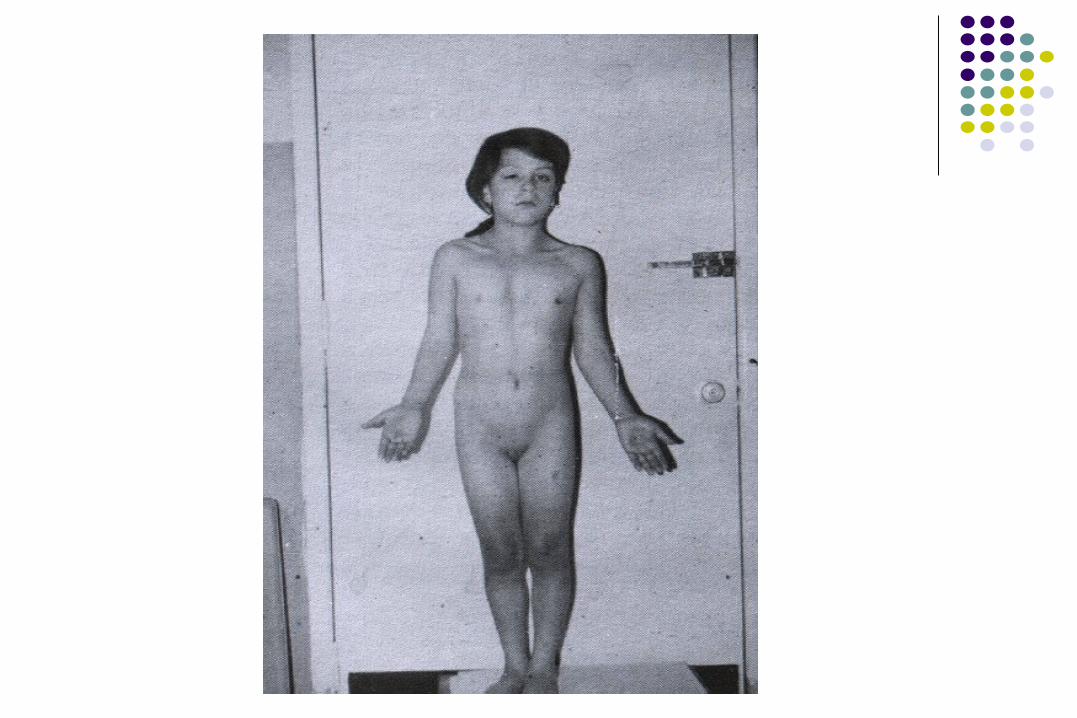
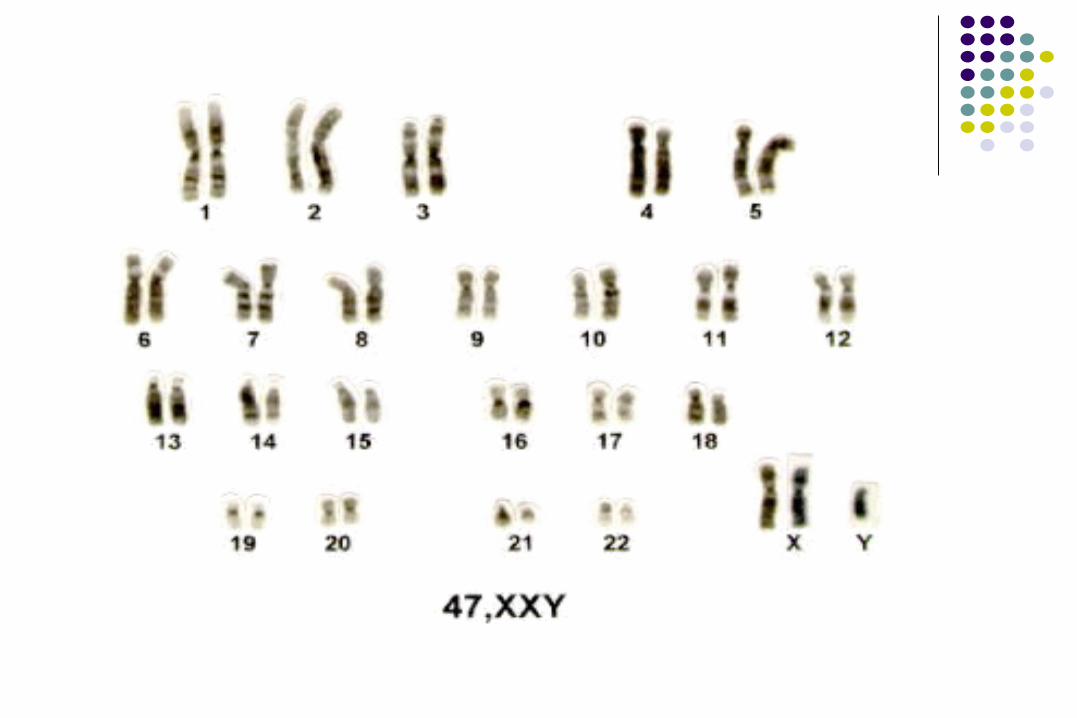
Síndrome de Klinefelter
47, XXY

Síndrome de Klinefelter o disgenesia de túbulos seminíferos.
Incidencia de 1 en 1,000 RN vivos varones. Hipogonadismo hipogonadotrofico. Falla testicular primaria con infertilidad e
hipogonandrogenismo. 60% de los pacientes se deben a no disyunción
materna (meiosis I 75%).

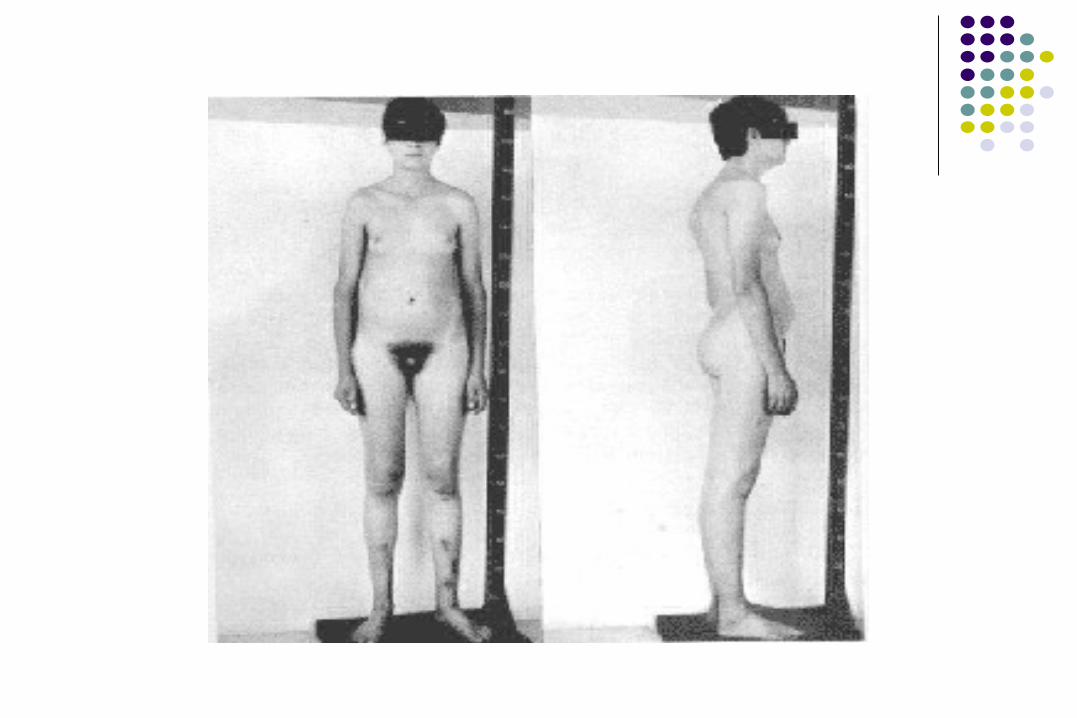
Cuadro clínico
Azoospermia 99-100%Testículos pequeños 99-100%Ginecomastia 55-75%Vello púbico escaso 30-60%Vello facial escaso 60-80%

Testosterona ↓ 65-85%Longitud peneana ↓ 10-25%Habitus eunocoide >80%Problemas sociales y
de aprendizaje 70%

Falla testicular primaria LH, FH, testosterona en sujetos prepuberes es
normal En individuos de 12-14 se incrementan las
gonadotrofinas y la testosterona permanece en limites inferiores normales

Posterior a la pubertad:
Hialinización y fibrosis de tubulos seminiferos Ausencia de células germinales, agregación de
células de Leydig por la hiperestimulación de la LH.

Prolapso de la válvula mitral 55% Varices 20-40% Leve incremento en enfermedades auto
inmunes LUPUS Enfermedad tiroidea Artritis reumatoide Diabetes mellitus


VARIANTES KLINEFELTERVARIANTES KLINEFELTER48, XXXY
Etiología: No disyunción en meiosis I y II
paterna o materna
Fenotipo muy similar a los varones 47, XXY
excepto que puede existir retraso mental
Generalmente son tranquilos y pasivos

VARIANTES KLINEFELTERVARIANTES KLINEFELTER
49, XXXXY Etiología: No disyunción postcigótica de
un cigoto XXY, origen materno de los X’s
Retraso mental con un IQ entre 20 y 60
Facies tosca
Hipogenitalismo

49, XXXXY
ANOMALÍAS ESQUELÉTICAS
Limitación movimientos de codos Sinostosis radiocubital Clinodactilia del 5to. dedo Coxa valga Coxa vara Xifosis o escoliosis Pie plano

47, XYY47, XYY
Frecuencia: 1 / 1000 varones Etiología: No disyunción en meiosis II paterna No presentan un fenotipo característico Crecimiento acelerado en infancia temprana Talla alta (180 a 186 cm.) Estado gonadal y fertilidad normal

47, XYY47, XYY
Inteligencia: IQ varía de 80 a 140
Los datos con relación a personalidad
son controversiales: conducta antisocial
o impulsiva, temperamento fuerte

48,XYYY y 49,XYYYY48,XYYY y 49,XYYYY
Frecuencia muy baja
Retraso mental
Retraso en desarrollo somático
Hipogonadismo
Sinostosis radiocubital