evaluación de la actividad antidiabética y antioxidante … y... · iii resumen en el presente...
TRANSCRIPT

UNIVERSIDAD TECNOLÓGICA DE LA MIXTECA
Evaluación de la actividad antidiabética y antioxidante in vitro de extractos
polares de Justicia spicigera y elucidación estructural de los compuestos
fenólicos mayoritarios.
TESIS PARA OBTENER EL GRADO DE:
MAESTRÍA EN CIENCIAS:
PRODUCTOS NATURALES Y ALIMENTOS
PRESENTA:
I. A. NANCY GIRÓN CRUZ
DIRECTOR DE TESIS:
DR. RAÚL SALAS CORONADO
Huajuapan de León, Oaxaca, México, Junio 2014.

II
Este trabajo se realizó en las instalaciones de los Laboratorios de Productos Naturales y
Alimentos del Instituto de Agroindustrias de la Universidad Tecnológica de la Mixteca.

III
RESUMEN
En el presente proyecto se cuantificaron fenoles totales en los extractos obtenidos a
partir de infusiones, decocciones, y asistidos por ultrasonido, denominados extractos crudos.
Esta cuantificación, así como el contenido de flavonoides totales y actividad antirradicalar
frente al radical DPPH• también se realizó en extractos obtenidos a partir de una limpieza
realizada por extracción en fase sólida (SPE, por sus siglas en inglés), estos extractos se
denominaron extractos limpios. Para los extractos limpios, los flavonoides totales se
cuantificaron a tres diferentes longitudes de onda (405, 490 y 515 nm). También se calculó el
TEC50 con la primera derivada de los datos obtenidos a partir de las cinéticas correspondientes
(tiempo vs % DPPH• consumido), que posteriormente junto con el IC50 permitieron obtener las
eficiencias antirradicalares de todos los extractos. Los extractos crudos con el mayor
contenido de fenoles totales fueron el extracto de la decocción de la hoja fresca con 767.04 ±
0.01 mg EAG/100 g y el extracto de etanol 70% con 618.62 ± 0.01 mg EAG/100 g. Mientras
que los extractos limpios con mayor contenido de fenoles totales fueron el extracto de la
decocción de la hoja fresca y el extracto de etanol 70% con valores de 687.2 ± 0.01 mg
EAG/100 g y 486.05 ± 0.01 mg EAG/100 g, respectivamente. Los valores obtenidos para
flavonoides totales variaron significativamente en función de la longitud de onda utilizada
para la lectura de la absorbancia. Consecuentemente, se realizó una correlación de Pearson de
los valores de flavonoides totales calculados a las tres longitudes de onda para cada extracto
con el contenido de fenoles totales para encontrar la mejor correlación y de esa manera
proponer la mejor longitud de onda para cuantificar los flavonoides totales. La mejor
correlación se obtuvo con los valores calculados a una longitud de onda de 490 nm. El mayor
contenido de flavonoides totales se encontró en el extracto de la decocción de hoja fresca,
668.65 ± 0.01 mg equivalentes de quercetina (EQ)/100 g, seguido del extracto de etanol al
70%, 492.15 ± 0.01 mg EQ/100 g. El extracto limpio con la mayor actividad antirradicalar
correspondió a la fracción acuosa proveniente del extracto crudo de etanol al 70%, el cual
presentó un IC50 de 1115.00 ± 3.43 g/mL, un TEC50 de 18 min y una eficiencia antirradicalar
de 1 x 10-6 kg DPPH•/(g muestra seca*min).

IV
ÍNDICE
Página
RESUMEN………………………………………………………………………………….…II
ABREVIATURAS…………………………………………………………………………….V
ÍNDICE DE APÉNDICES………………………………………………………………...…VII
ÍNDICE DE FIGURAS………………………………………………………………...……VIII
ÍNDICE DE TABLAS………………...…………………………………...………………….IX
1. INTRODUCCIÓN .................................................................................................................. 1
2. ANÁLISIS DE FUNDAMENTOS ........................................................................................ 3
2.1. Características taxonómicas del género Justicia .............................................................. 3
2.2. Características taxonómicas y distribución geográfica de J. spicigera ............................ 4
2.3. Propiedades biológicas del género Justicia ...................................................................... 4
2.4. Plantas con propiedades antidiabéticas............................................................................. 7
2.5. Propiedades de los compuestos fenólicos ......................................................................... 9
2.6. Actividad antidiabética de extractos de plantas .............................................................. 11
2.6.1. Actividad antidiabética in vitro ............................................................................... 11
2.7. Actividad antioxidante de extractos de plantas .............................................................. 13
2.8. Métodos de extracción .................................................................................................... 16
2.9. Extracción en fase sólida (SPE)...................................................................................... 17
2.10. Elucidación estructural ................................................................................................. 18
2.10.1. Espectroscopía de infrarrojo .................................................................................. 19
2.10.2. Espectrometría de masa (MS)................................................................................ 19
2.10.3. Espectroscopía de resonancia magnética nuclear (RMN) ..................................... 20

V
3. ORIGINALIDAD ................................................................................................................. 23
4. OBJETIVOS ......................................................................................................................... 24
4.1. Objetivo general ............................................................................................................. 24
4.2. Objetivos específicos ...................................................................................................... 24
5. METAS ................................................................................................................................. 25
6. METODOLOGÍA ................................................................................................................. 26
6.1. Equipos, reactivos y disolventes ..................................................................................... 27
6.2. Obtención de la muestra ................................................................................................. 27
6.3. Preparación de la muestra ............................................................................................... 28
6.4. Obtención de los extractos .............................................................................................. 28
6.5. Limpieza de los extractos por extracción en fase sólida (SPE) ...................................... 29
6.6. Cuantificación de fenoles totales en los extractos .......................................................... 29
6.7. Cuantificación de flavonoides totales en los extractos ................................................... 30
6.8. Evaluación de la actividad antirradical de los extractos ................................................. 31
6.9. Hidrólisis de los extractos............................................................................................... 32
6.10. Evaluación de la inhibición de la difusión de glucosa.................................................. 32
6.11. Evaluación de la inhibición de la actividad enzimática ................................................ 33
6.11.1. Inhibición de los extractos en presencia de α-amilasa ........................................... 33
6.11.2. Inhibición de la α-glucosidasa en presencia de los extractos. ............................... 34
6.12. Aislamiento de los compuestos fenólicos ..................................................................... 34
6.13. Caracterización de los compuestos fenólicos presentes en los extractos ..................... 35
6.14. Análisis estadístico ....................................................................................................... 36
7. RESULTADOS Y DISCUSIONES ..................................................................................... 37
7.1. Obtención de la muestra ................................................................................................. 37

VI
7.2. Obtención de extractos ................................................................................................... 38
7.3. Cuantificación de fenoles totales .................................................................................... 39
7.4. Cuantificación de flavonoides totales ............................................................................. 46
7.5. Evaluación de la actividad antioxidante ........................................................................ 51
8. REFERENCIAS ................................................................................................................... 56
9. ANEXOS .............................................................................................................................. 61
10. APÉNDICES ...................................................................................................................... 64

VII
ABREVIATURAS
A 1 Anexo 1
A 2 Anexo 2
ABTS•+ Catión radical ácido 2,2-azinobis-(3-etil-benzotiazolinal)-6-sulfónico
CID Disociación inducida por colisión
DMSO Dimetilsulfóxido
DPPH• Radical 2,2-difenil-1-picrilhidracilo
EAG Equivalentes de ácido gálico
EC Equivalentes de catequina
EQ Equivalentes de quercetina
FRAP Poder reductor del ion férrico
HPLC Cromatografía líquida de alta resolución
IC50 Concentración de extracto requerida para inhibir el 50% del DPPH•
IR Espectroscopía de infrarrojo
MS Espectrometría de masa
MS/MS Espectrometría de masa en cascada
OMS Organización mundial para la salud
ORAC Capacidad de absorción de radicales oxígeno
RMN Resonancia magnética nuclear

VIII
SPE Extracción en fase sólida
TEAC Capacidad antioxidante en equivalentes de Trolox
UV/Vis Ultravioleta/visible

IX
ÍNDICE DE ANEXOS
Página
A 1. Relación entre el mecanismo de acción de fármacos y plantas. ........................................ 61
A 2. Medicamentos orales para el tratamiento de la diabetes Tipo 2. ....................................... 62

X
ÍNDICE DE APÉNDICES
Página
AP 1. Fenoles totales en extractos crudos de las hojas de J. spicigera. .................................... 64
AP 2. Flavonoides totales en equivalentes de catequina. .......................................................... 66
AP 3. Flavonoides totales en equivalentes de quercetina. ......................................................... 68
AP 4. Curva de calibración en equivalentes de ácido gálico para la cuantificación de fenoles
totales. ........................................................................................................................................ 70
AP 5. Curva de calibración en equivalentes de (+) - catequina para la cuantificación de
flavonoides totales. .................................................................................................................... 71
AP 6. Curva de calibración en equivalentes de quercetina para la cuantificación de flavonoides
totales. ........................................................................................................................................ 73
AP 7. Curva de calibración en equivalentes de ácido gálico para DPPH•................................ 75
AP 8. IC50 y TEC50 del BHT para el ensayo del DPPH•. ............................................................ 76
AP 9. IC50 y TEC50 de la decocción de la hoja fresca de J. spicigera, fracción acuosa para
DPPH•. ....................................................................................................................................... 77
AP 10. IC50 y TEC50 de la decocción de la hoja fresca de J. spicigera, fracción MeOH/H2O
para DPPH•. ............................................................................................................................... 78
AP 11. IC50 y TEC50 de la decocción del polvo de la hoja de J. spicigera, fracción acuosa para
DPPH•. ....................................................................................................................................... 79
AP 12. IC50 y TEC50 de la decocción del polvo de la hoja de J. spicigera, fracción MeOH/H2O
para DPPH•. ............................................................................................................................... 80

XI
AP 13. IC50 y TEC50 de la infusión del polvo de la hoja de J. spicigera, fracción acuosa para
DPPH•. ....................................................................................................................................... 81
AP 14. IC50 y TEC50 de la infusión del polvo de la hoja de J. spicigera, fracción MeOH/H2O
para DPPH•. ............................................................................................................................... 82
AP 15. IC50 y TEC50 del extracto H2O por ultrasonido del polvo de la hoja de J. spicigera,
fracción acuosa para DPPH•. ..................................................................................................... 83
AP 16. IC50 y TEC50 del extracto H2O por ultrasonido del polvo de la hoja de J. spicigera,
fracción MeOH/H2O para DPPH•. ............................................................................................. 84
AP 17. IC50 y TEC50 del extracto MeOH por ultrasonido del polvo de la hoja de J. spicigera,
fracción acuosa para DPPH•. ..................................................................................................... 85
AP 18. IC50 y TEC50 del extracto MeOH por ultrasonido del polvo de la hoja de J. spicigera,
fracción MeOH/H2O para DPPH•. ............................................................................................. 86
AP 19. IC50 y TEC50 del extracto EtOH 70% por ultrasonido del polvo de J. spicigera, fracción
acuosa para DPPH•. ................................................................................................................... 87
AP 20. IC50 y TEC50 del extracto EtOH 70% por ultrasonido del polvo de J. spicigera, fracción
MeOH/H2O para DPPH•. ........................................................................................................... 88
AP 21. IC50 y TEC50 del extracto acetona 70% por ultrasonido del polvo de J. spicigera,
fracción acuosa para DPPH•. ..................................................................................................... 89
AP 22. IC50 y TEC50 del extracto de acetona 70% por ultrasonido del polvo de J. spicigera,
fracción MeOH/H2O para DPPH•. ............................................................................................. 90

XII
ÍNDICE DE FIGURAS
Página
Figura 1. Hojas de J. gendarussa1, J. pectoralis2, J. isularis3, J. adhatoda4, J. flava5 y J.
procumbes6. ................................................................................................................................. 3
Figura 2. Hojas y flores de J. spicigera. ..................................................................................... 4
Figura 3. Compuestos fenólicos. ................................................................................................ 9
Figura 4. Biosíntesis de un lignano. ......................................................................................... 10
Figura 5. Modelo de Gallagher et al. (2003), Imagen de la Tesis de Cruz-Gallegos, 2012. .... 12
Figura 6. Extracción en fase sólida........................................................................................... 18
Figura 7. Procedimiento del tratamiento de la muestra. ........................................................... 26
Figura 8. Secado de hojas de J. spicigera. ............................................................................... 37
Figura 9. Diagrama para la obtención de extractos limpio. ...................................................... 39
Figura 10. Contenido de fenoles totales en extractos crudos de J. spicigera. .......................... 40
Figura 11. Contenido de fenoles totales en hojas de J. spicgera. ............................................. 41
Figura 12. Contenido de fenoles totales de las hojas de J. spicigera y del tallo J. gendarussa.
................................................................................................................................................... 42
Figura 13. Contenido de fenoles totales en hojas de diferentes plantas. .................................. 43
Figura 14. Contenido de fenoles totales de extractos limpios de J. spicigera.......................... 44
Figura 15. Comparativo de fenoles totales de extractos crudos y extractos limpios de J.
spicigera. ................................................................................................................................... 45

XIII
Figura 16. Contenido de fenoles totales en extractos limpios de J. spicigera.......................... 46
Figura 17. Flavonoides totales de la suma de fracciones en equivalentes de quercetina de las
hojas de J. spicigera. ................................................................................................................. 47
Figura 18. Contenido de flavonoides totales en hojas de J. spicigera. .................................... 48
Figura 19. Contenido de flavonoides totales en equivalentes de quercetina. ........................... 49
Figura 20. Flavonoides totales de la suma de fracciones en equivalentes de catequina de las
hojas de J. spicigera. ................................................................................................................. 50
Figura 21. IC50 de las fracciones de los extractos de J. spicigera. ........................................... 52
Figura 22. Cinética de la fracción acuosa de la decocción de la hoja fresca de J. spicigera. .. 53
Figura 23. Cinética de la fracción acuosa del extracto de etanol al 70% de J. spicigera. ........ 53

XIV
ÍNDICE DE TABLAS
Página
Tabla 1. Actividad biológica del género Justicia. ...................................................................... 5
Tabla 2. Metabolitos secundarios aislados del género Justicia. ................................................. 6
Tabla 3. Actividad antioxidante de algunas plantas. ................................................................ 14
Tabla 4. Actividad antioxidante de J. pectoralis. ..................................................................... 15
Tabla 5. Actividad antioxidante de J. gendarussa. ................................................................... 15
Tabla 6. Bandas del espectro de IR de algunos flavonoides..................................................... 19
Tabla 7. Desplazamiento químico de protón. ........................................................................... 21
Tabla 8. Desplazamientos químicos de varios tipos de carbonos de flavonoides. ................... 22
Tabla 9. Determinación de humedad en hojas de J. spicigera. ................................................ 38
Tabla 10. Correlación de Pearson entre fenoles y flavonoides. ................................................ 48
Tabla 11. TEC50 y eficiencia antirradicalar (AE) de los extractos de J. spicigera. ................... 55

1
1. INTRODUCCIÓN
La diabetes mellitus es un trastorno metabólico que se caracteriza por la pérdida de la
homeostasis de la glucosa con alteraciones en el metabolismo de carbohidratos, lípidos y
proteínas. Esto se refleja en la secreción de insulina, la acción de la insulina o en ambos. De
acuerdo con datos de la OMS, se estima que el 3% de la población en el mundo padece
diabetes y se espera que se duplique para el año 2025 a 6.5% aproximadamente. Para la
comunidad médica continúa siendo un reto el empleo de medicamentos que no ejerzan efectos
negativos en la salud de los pacientes diabéticos (Basha et al., 2012).
La diabetes mellitus se puede clasificar en dos tipos principales, Tipo 1 o diabetes
juvenil (insulinodependiente) y Tipo 2 o diabetes de adultos (no insulinodependiente). La
diabetes Tipo 1 se presenta en la infancia, principalmente se debe a la destrucción de las
células pancreáticas β, lo cual trae como consecuencia a la deficiencia absoluta de insulina. La
diabetes Tipo 2 está más asociada con la edad adulta, en este caso se desconoce por qué se da
la resistencia a la insulina, sin embargo se asocia al impacto ambiental, los hábitos
alimentarios, obesidad, a una etapa en una enfermedad determinada, entre otros aspectos
(Hemalatha et al., 2012).
El uso de medicamentos está restringido por sus propiedades farmacocinéticas y por los
efectos secundarios adversos, A 2. Por ejemplo, la glibenclamida puede llegar a provocar
vómito, hipoglucemia, estreñimiento, hiperacidez gástrica, cefalea, entre otros. La metformina,
otro medicamento empleado para tratar la diabetes Tipo 2, puede llegar a causar diarrea, gases,
distensión abdominal, dolor muscular, etc. Por lo tanto la búsqueda de una nueva clase de
compuestos es esencial para superar problemas asociados con la diabetes (Basha et al., 2012).
De acuerdo a la OMS, aproximadamente el 80% de la población mundial,
especialmente las poblaciones rurales de los países en desarrollo, ingieren infusiones o

2
decocciones de plantas para tratar enfermedades como cáncer, diarrea, parasitarias, diabetes,
etc. (Kadir et al., 2012). Particularmente, personas con diabetes Tipo 2 utilizan plantas
medicinales para regular los niveles de glucosa en sangre, obteniendo resultados positivos. Sin
embargo, los tratamientos siguen siendo empíricos y generalmente se desconoce su
mecanismo de acción. Algunas plantas que comúnmente se utilizan para la diabetes en México
son las hojas de aguacate, el nopal, la sábila, el wereke y el muicle (Justicia spicigera). Esta
última, objeto del presente estudio, es un arbusto de hoja perenne con flores tubulares de color
naranja que crece en climas cálidos y se extiende desde México hasta América del Sur (Ortiz-
Andrade et al., 2012).
Actualmente existe un estudio reciente que está relacionado con la evaluación de la
actividad antidiabética in vivo e in vitro de J. spicigera, Ortiz-Andrade et al. (2012). En este
estudio se evaluó la actividad antidiabética in vivo de extractos etanólicos de J. spicigera. Los
resultados del estudio demostraron que esos extractos poseen una fuerte actividad
antidiabética respecto a la glibenclamida, repaglinida y saxagliptina, fármacos
hipoglucemiantes comerciales. Los autores proponen cinco posibles mecanismos por los
cuales se podría llevar a cabo la actividad antidiabética de los extractos de J. spicigera, los
extractos pueden modular la acción y/o secreción de la insulina, pueden involucrar la
participación de un sistema antioxidante, aumentar el metabolismo de la glucosa en las células
β, activar un sistema enzimático y pueden servir como un mensajero de derivados de
fosfolípidos. Cabe resaltar que, no existe mucha información en la literatura científica sobre
quiénes son los metabolitos secundarios y el mecanismo por el cual los extractos de J.
spicigera ejercen dicha actividad.
Debido a lo anterior, en el presente proyecto se evaluó la actividad antidiabética in
vitro de los extractos de J. spicigera a través de dos mecanismos, la inhibición de la difusión
de glucosa mediante el método modificado de Gallagher et al. (2003) y la inhibición
enzimática de las enzimas -amilasa y -glucosidasa. Adicionalmente se determinó la
actividad antirradical, y se cuantificaron los flavonoides y fenoles totales presentes en los
extractos. Finalmente, se caracterizaron los compuestos fenólicos mayoritarios presentes en
los extractos. Este estudio permitió cuantificar in vitro la actividad antidiabética y elucidar los
metabolitos secundarios que confieren a la planta dicha propiedad.

3
2. ANÁLISIS DE FUNDAMENTOS
En la presente sección se describirán las características taxonómicas de Justicia
spicigera y algunas propiedades biológicas tanto para esta planta como para otras del mismo
género. También se describirán los métodos utilizados para la determinación de la actividad
antidiabética y antioxidante, así como los métodos para la elucidación estructural de los
metabolitos secundarios a aislar.
2.1. Características taxonómicas del género Justicia
El Género Justicia, Figura 1, se encuentra ampliamente distribuido por todo el mundo y
se caracteriza por tener usos medicinales y algunos de estos usos se han estudiado.
Figura 1. Hojas de J. gendarussa1, J. pectoralis2, J. isularis3, J. adhatoda4, J. flava5 y J.
procumbes6.
1 2 3
4 5 6

4
2.2. Características taxonómicas y distribución geográfica de J. spicigera
Esta planta es nativa de México y puede llegar a crecer hasta en algunas regiones de
América del Sur. En algunos estados como Yucatán se conoce con el nombre de chak lool,
ts’i’its y yichkaan, y en la Huasteca Potosina se le conoce comúnmente como muicle o muite
(Ortiz-Andrade et al., 2012).
Se puede describir como una hierba erecta o trepadora perene. Sus hojas son pecioladas
con un margen de la hoja que generalmente es entero. Las brácteas y bractéolas son
generalmente visibles y superpuestas. La corola se puede observar, con un labio posterior que
es generalmente de dos lóbulos, un labio anterior que es de tres lóbulos, dos estambres, una
cápsula con cuatro semillas, y la porción basal estéril, Figura 2 (Ortiz-Andrade et al., 2012).
Figura 2. Hojas y flores de J. spicigera.
2.3. Propiedades biológicas del género Justicia
En la Tabla 1 se muestran algunas especies del género Justicia así como su actividad
biológica y en la Tabla 2 se muestran lo metabolitos secundarios presentes en el género
Justicia que poseen actividad biológica (Corrêa & Alcântara, 2012).
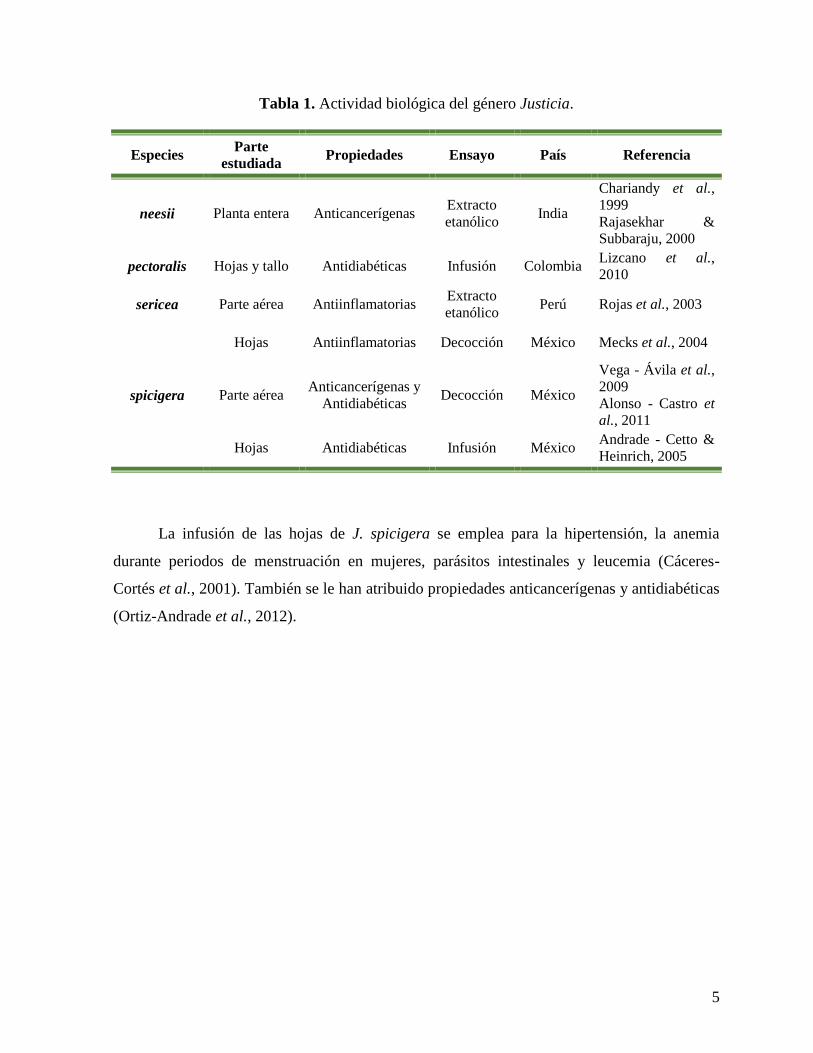
5
Tabla 1. Actividad biológica del género Justicia.
Especies Parte
estudiada Propiedades Ensayo País Referencia
neesii Planta entera Anticancerígenas Extracto
etanólico India
Chariandy et al.,
1999
Rajasekhar &
Subbaraju, 2000
pectoralis Hojas y tallo Antidiabéticas Infusión Colombia Lizcano et al.,
2010
sericea Parte aérea Antiinflamatorias Extracto
etanólico Perú Rojas et al., 2003
spicigera
Hojas Antiinflamatorias Decocción México Mecks et al., 2004
Parte aérea Anticancerígenas y
Antidiabéticas Decocción México
Vega - Ávila et al.,
2009
Alonso - Castro et
al., 2011
Hojas Antidiabéticas Infusión México Andrade - Cetto &
Heinrich, 2005
La infusión de las hojas de J. spicigera se emplea para la hipertensión, la anemia
durante periodos de menstruación en mujeres, parásitos intestinales y leucemia (Cáceres-
Cortés et al., 2001). También se le han atribuido propiedades anticancerígenas y antidiabéticas
(Ortiz-Andrade et al., 2012).
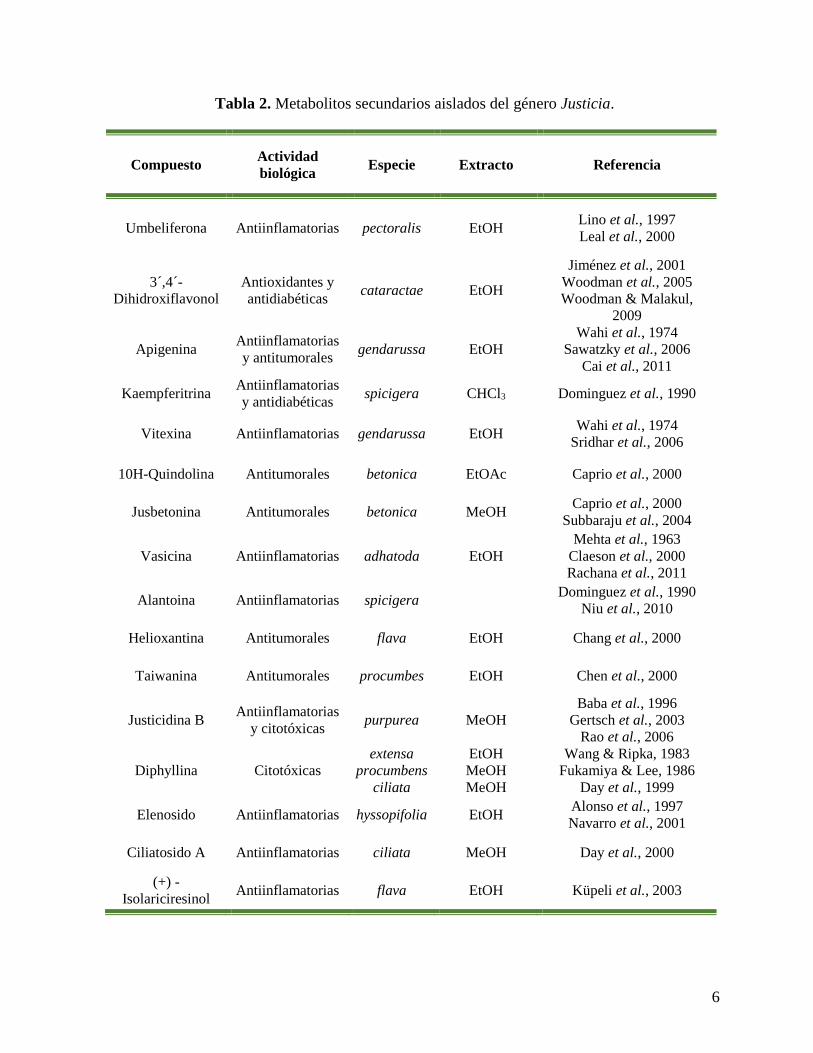
6
Tabla 2. Metabolitos secundarios aislados del género Justicia.
Compuesto Actividad
biológica Especie Extracto Referencia
Umbeliferona Antiinflamatorias pectoralis EtOH Lino et al., 1997
Leal et al., 2000
3´,4´-
Dihidroxiflavonol
Antioxidantes y
antidiabéticas cataractae EtOH
Jiménez et al., 2001
Woodman et al., 2005
Woodman & Malakul,
2009
Apigenina Antiinflamatorias
y antitumorales gendarussa EtOH
Wahi et al., 1974
Sawatzky et al., 2006
Cai et al., 2011
Kaempferitrina Antiinflamatorias
y antidiabéticas spicigera CHCl3 Dominguez et al., 1990
Vitexina Antiinflamatorias gendarussa EtOH Wahi et al., 1974
Sridhar et al., 2006
10H-Quindolina Antitumorales betonica EtOAc Caprio et al., 2000
Jusbetonina Antitumorales betonica MeOH Caprio et al., 2000
Subbaraju et al., 2004
Vasicina Antiinflamatorias adhatoda EtOH
Mehta et al., 1963
Claeson et al., 2000
Rachana et al., 2011
Alantoina Antiinflamatorias spicigera Dominguez et al., 1990
Niu et al., 2010
Helioxantina Antitumorales flava EtOH Chang et al., 2000
Taiwanina Antitumorales procumbes EtOH Chen et al., 2000
Justicidina B Antiinflamatorias
y citotóxicas purpurea MeOH
Baba et al., 1996
Gertsch et al., 2003
Rao et al., 2006
Diphyllina Citotóxicas
extensa
procumbens
ciliata
EtOH
MeOH
MeOH
Wang & Ripka, 1983
Fukamiya & Lee, 1986
Day et al., 1999
Elenosido Antiinflamatorias hyssopifolia EtOH Alonso et al., 1997
Navarro et al., 2001
Ciliatosido A Antiinflamatorias ciliata MeOH Day et al., 2000
(+) -
Isolariciresinol Antiinflamatorias flava EtOH Küpeli et al., 2003

7
2.4. Plantas con propiedades antidiabéticas
Se ha reportado la existencia de 800 plantas utilizadas para el control de la diabetes en
todo el mundo; de las cuales, se ha comprobado experimentalmente que 450 poseen
propiedades antidiabéticas. Sin embargo, el mecanismo de acción solo se ha comprobado en
109 plantas, A 1 (Kumar et al., 2011). Los mecanismos de acción antidiabética propuestos
para las plantas se enlistan a continuación, de los cuales los más estudiados son los
mecanismos 3 y 9 (Bhushan et al., 2010):
1. Estimulan la síntesis y/o liberación de insulina
2. Mimetizan a la insulina
3. Incrementan el metabolismo de la glucosa
4. Incrementan la utilización de glucosa periférica
5. Inhiben la absorción de glucosa en el tracto gastrointestinal
6. Reducen los niveles de glucosa en sangre por regeneración de células β
7. Reducen la insulinorresistencia
8. Disminuyen la gluconeogénesis y glicogénesis
9. Inhiben a la α-amilasa y α-glucosidasa
A continuación se mencionan algunos ejemplos representativos de mecanismos de
acción de plantas antidiabéticas:
Basha et al. (2012), evaluaron la actividad antidiabética in vitro de las hojas de Psidium
guajava. Los extractos metanólicos de esta planta presentan un efecto potencial de la
inhibición de la difusión de glucosa usando un tubo de diálisis. En ese estudio se muestra la
inhibición de la difusión y consecuentemente un posible efecto sobre el movimiento de la
glucosa en el tracto intestinal (Mecanismo 5).
Los extractos metanólicos de las partes aéreas de Passiflora incarnata, disminuyen los
niveles de glucosa en ratones con diabetes inducida con estreptozotocina. Los extractos
pueden estar involucrados en la liberación de insulina, aumentando la utilización de glucosa
periférica (Mecanismos 4 y 6), esto está asociado con la regeneración de los islotes de

8
Langerhans. Estos islotes contienen células β, las cuales son las encargadas de producir
insulina y se encuentran localizadas en el interior del páncreas (Gupta et al., 2012).
Ramkumar et al. (2010), evaluaron la actividad antidiabética in vitro con el uso de
enzimas α-amilasa y α-glucosidasa de los extractos etanólicos de las hojas de Gymnema
montanum. Los resultados obtenidos muestran que los extractos inhiben la actividad de estas
enzimas y por lo tanto se reducen los niveles de glucosa en sangre. Esto indica que los
extractos pueden suprimir la absorción de carbohidratos desde el intestino y con ello reducir la
hiperglicemia, mediante la inhibición enzimática (Mecanismo 9).
En un estudio en el cual se evaluó la actividad antidiabética in vivo de los extractos de
las hojas de Biophytum sensitivum, se encontró que estos extractos estimulan a las células β
del páncreas para liberar insulina en conejos machos diabéticos, ejerciendo así una actividad
hipoglucemiante. La administración del extracto B. sensitivum en ayunas a los conejos
diabéticos mostraron un aumento significativo en los niveles séricos de insulina, lo que sugiere
un modo de acción a nivel del páncreas. La respuesta hipoglicémica de esta planta es mediada
a través de la estimulación de la síntesis/liberación de insulina de las células β de los islotes de
Langerhans (Mecanismo 3) (Patel et al., 2012).
Los metabolitos secundarios presentes en plantas con propiedades antidiabéticas
pertenecen a los grupos de compuestos químicos llamados: terpenos, sapogeninas,
carotenoides, alcaloides, esteroles, taninos, polisacáridos, cumarinas, esteroides, ácidos
fenólicos, lignanos y flavonoides (Andrade-Cetto & Heinrich, 2005). Cabe mencionar que
tanto los lignanos como los flavonoides, son compuestos fenólicos que están presentes en los
extractos polares de plantas del género Justicia. Consecuentemente, a continuación se
describirán brevemente las propiedades biológicas de estos dos grupos de compuestos.

9
2.5. Propiedades de los compuestos fenólicos
Los compuestos fenólicos son metabolitos secundarios que poseen un anillo aromático
con uno o más grupos hidroxilo. Sus estructuras pueden variar de una molécula fenólica
simple a un polímero complejo de peso molecular alto. Los compuestos fenólicos se dividen
en diversas clases, basándose en el número de anillos aromáticos que contengan y a los
elementos estructurales que se encuentren unidos a esos anillos. Los principales grupos de
compuestos fenólicos son: ácidos fenólicos, taninos, estilbenos, flavonoides y lignanos, Figura
3 (Ignat et al., 2011). Los flavonoides y lignanos se han identificado en el género Justicia y se
les han atribuido una gran parte de las propiedades medicinales de esas plantas. A
continuación se describen brevemente las características estructurales de estas dos clases de
compuestos.
Figura 3. Compuestos fenólicos.
Nombre R1 R2 R3
Ácido p - hidroxibenzoico H OH H
Ácido gálico OH OH OH
Ácido protocatecuico OH OH H
Nombre R1 R2 R3 R4 R5 R6
trans - resveratrol OH H OH H OH H
Piceatannol OH H OH OH OH H
trans - astringina GlcO H OH H OH OH
Nombre R1 R2 R3 R4 R5 R6
Enterodiol CH2OH CH2OH OH H OH H
Secoisolariciresinol CH2OH CH2OH OCH3 OH OCH3 OH

10
La estructura básica de los flavonoides es un núcleo flavano que está constituido por 15
átomos de carbono en tres anillos (C6-C3-C6), nombrados como A, B, C. Las diferentes clases
de flavonoides difieren en el nivel de oxidación y saturación del anillo C, mientras que los
compuestos individuales difieren en la sustitución del anillo A y B. Las diferencias en la
estructura y sustitución influyen en la estabilidad del radical y por lo tanto en las propiedades
antioxidantes de los flavonoides (Wojdylo et al., 2007).
Los flavonoides tienen propiedades biológicas tales como, actividad antimicrobiana,
antiulcerosa, antiinflamatoria, antioxidante, antitumoral, antiespasmódica, antidiabética, entre
otras (Ghasemzadeh & Ghasemzadeh, 2011).
Particularmente, los lignanos son un grupo de compuestos comúnmente encontrados en
las plantas, están constituidos por la unión de dos moléculas de compuestos fenólicos con
cadenas insaturadas. En la Figura 4 se muestra un ejemplo de la biosíntesis de un lignano a
partir del alcohol coniferílico. Estos metabolitos secundarios poseen una gama amplia de
efectos benéficos a la salud, tales como actividad antioxidante, antiviral, antitumorigénica,
antidiabética, antiobesidad y contra enfermedades cardiovasculares (Smeds et al., 2012).
+MeO
OH
OH
Alcohol coniferílico
-H
MeO
O
OH
.O
OH
MeO
.
OH
O
OH
OMe
OH
OH
OMe
Lignano
. +H.
, + HO.
Figura 4. Biosíntesis de un lignano.

11
2.6. Actividad antidiabética de extractos de plantas
Los efectos antidiabéticos de los extractos de plantas pueden ser estudiados usando
modelos in vivo o usando una variedad de sistemas in vitro. Los sistemas in vitro juegan un
papel muy importante en la evaluación de la actividad antidiabética de los fármacos, y
proporcionan las herramientas iniciales para la selección de un gran número de candidatos
terapéuticos potenciales. Estos sistemas pueden proporcionar información útil sobre el
mecanismo de acción del agente terapéutico. Los materiales biológicos en los sistemas in vitro
pueden ser órganos enteros, tejidos, cultivos celulares, entre otros. En los sistemas in vivo, se
emplean animales vivos, los cuales son necesarios para estudiar cómo se comportan en
condiciones clínicas o fisiopatológicas (Thorat et al., 2012). Los métodos propuestos para
evaluar la actividad antidiabética de los extractos polares de J. spicigera en el presente
proyecto consistirán en modelos in vitro. Consecuentemente a continuación se describen de
manera breve.
2.6.1. Actividad antidiabética in vitro
Investigaciones recientes indican que niveles de glucosa postprandial altos en la sangre
son el factor más importante que favorece el inicio y desarrollo de la diabetes Tipo 2,
denominada hiperglicemia postprandial. Esta hiperglicemia es una absorción rápida de la
glucosa en el intestino después de ingerir alimentos, en la cual la α-amilasa y α-glucosidasa
juegan un papel importante debido a la hidrólisis del almidón y de oligosacáridos. La
inhibición de estas enzimas puede controlar la elevación de los niveles de glucosa en sangre.
Por lo tanto es importante establecer estrategias para reducir la hiperglicemia postprandial a
través de la inhibición de la actividad de estas enzimas (Dong et al., 2012).
Actualmente, los inhibidores de α-amilasa y α-glucosidasa como la acarbosa, voglibosa
y miglitol son ampliamente utilizados en pacientes con diabetes Tipo 2. Sin embargo, se ha
documentado que estos inhibidores sintéticos tienen efectos indeseables tales como diarrea y
cólicos abdominales. Hoy en día se han estudiado inhibidores de α-amilasa y α-glucosidasa
provenientes de fuentes naturales, especialmente de plantas del género Gymnema, tales como
G. sylvestre, G. inodorum y G. yunnanense (Ramkumar et al., 2010). También se ha

12
demostrado que extractos de la cáscara de Camellia sinensis tienen la capacidad de inhibir la
α-amilasa y α-glucosidasa (Wang et al., 2012). Los resultados obtenidos en este último estudio
mostraron que los responsables de la bioactividad de estos extractos son los compuestos
fenólicos.
Dentro de los métodos para evaluar la actividad antidiabética se encuentra la inhibición
de la enzima α-amilasa, este método se fundamenta en mezclar el extracto con una disolución
de la enzima. Después, esa mezcla se expone a una disolución de almidón. Posteriormente se
adiciona un reactivo colorimétrico y se miden los cambios de color con un espectrofotómetro
(Nickavar & Yousefian, 2011). De manera análoga, la inhibición de la α-glucosidasa se
determina por la adición de esta enzima al extracto a evaluar. Para iniciar la reacción es
necesario adicionar un sustrato (p-nitrofenil glucopiranósido), que al ser consumido durante la
reacción libera al p-nitrofenil. Éste último compuesto puede ser medido
espectrofotométricamente e indicar de manera indirecta el efecto del extracto sobre la
actividad enzimática (Eom et al., 2011).
Gallagher et al. (2003), evaluaron la actividad antidiabética in vitro de Agaricus
campestris, Agrimony eupatoria, Coriandrum sativum, Eucalyptus globulus, Juniperus
communis, Medicago sativa, Persea americana, Sambuus nigra, Urtica diocia y Viscum
álbum. Los extractos acuosos de estas plantas presentaron una inhibición de la difusión de
glucosa, lo cual está relacionado directamente con la reducción de la absorción de glucosa en
el tracto gastrointestinal. El método consiste en el uso de una membrana de diálisis, Figura 5.
Este método fue evaluado con 10 plantas con propiedades antidiabéticas.
Figura 5. Modelo de Gallagher et al. (2003), Imagen de la Tesis de Cruz-Gallegos, 2012.

13
En el presente estudio se utilizarán estos métodos de evaluación de la actividad
antidiabética.
2.7. Actividad antioxidante de extractos de plantas
En el cuerpo humano el desequilibrio entre especies antioxidantes y oxidantes conduce
al daño celular y problemas en la salud. Esto trae como consecuencia el desarrollo de
enfermedades degenerativas, cardiovasculares, cáncer, diabetes, entre otras. Por lo tanto es
necesario que el ser humano consuma fuentes de antioxidantes no enzimáticos, tales como,
ácido ascórbico, α-tocoferol, carotenoides, flavonoides, entre otros (Krishnaiah et al., 2011).
Las propiedades medicinales de las plantas han sido ampliamente investigadas
alrededor del mundo, debido a su actividad farmacológica y baja toxicidad. Los compuestos
bioactivos que se encuentran en las plantas han demostrado tener posibles beneficios como
actividad antioxidante, anticancerígena, antihipertensiva, antimutagénica, antidiabética, entre
otras. Los compuestos fenólicos en las plantas se componen principalmente de los ácidos
fenólicos y flavonoides (Rodríguez Vaquero et al., 2010).
Dentro de los métodos para determinar la actividad antioxidante de los extractos de las
plantas se encuentran el método del radical DPPH•, del catión radical ABTS•+, entre otros. El
método del radical 2,2-difenil-1-picrilhidracilo (DPPH•), radical que produce un color azul
intenso en solución, es uno de los métodos más utilizados para evaluar la actividad
antioxidante de extractos. Este método se basa en la capacidad del DPPH• para reaccionar con
compuestos que pueden donar un radical hidrógeno, esta reacción induce una decoloración de
la solución que puede medirse espectrofotométricamente. Otro método muy utilizado es el del
radical catión ácido 2,2-azinobis-(3-etil-benzotiazolina)-6-sulfonico (ABTS•+) que se basa en
su reducción en presencia de antioxidantes donadores de electrones. Esta reducción produce
una decoloración medible espectrofotométricamente (Krishnaiah et al., 2011).
La actividad antioxidante ha sido investigada ampliamente en diversas plantas. En la
Tabla 3, se muestran algunas plantas con los métodos más utilizados para determinar dicha
actividad (Wojdylo et al., 2007).
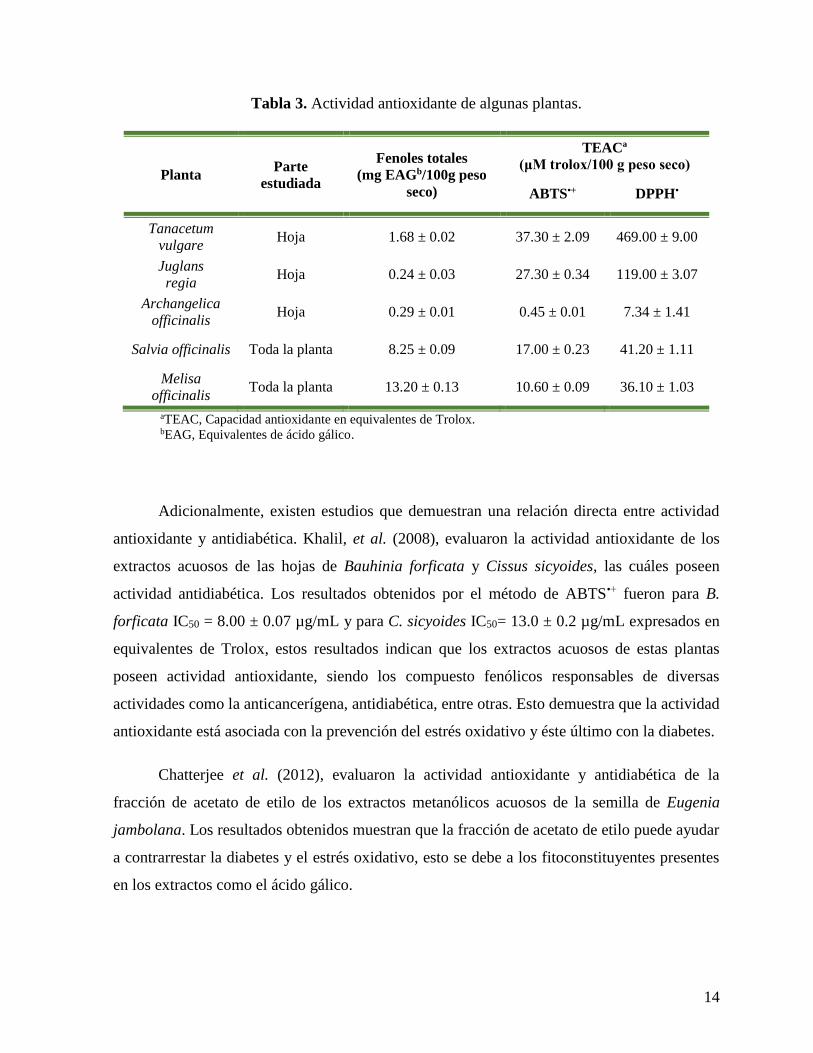
14
Tabla 3. Actividad antioxidante de algunas plantas.
Planta Parte
estudiada
Fenoles totales
(mg EAGb/100g peso
seco)
TEACª
(μM trolox/100 g peso seco)
ABTS•+ DPPH•
Tanacetum
vulgare Hoja 1.68 ± 0.02 37.30 ± 2.09 469.00 ± 9.00
Juglans
regia Hoja 0.24 ± 0.03 27.30 ± 0.34 119.00 ± 3.07
Archangelica
officinalis Hoja 0.29 ± 0.01 0.45 ± 0.01 7.34 ± 1.41
Salvia officinalis Toda la planta 8.25 ± 0.09 17.00 ± 0.23 41.20 ± 1.11
Melisa
officinalis Toda la planta 13.20 ± 0.13 10.60 ± 0.09 36.10 ± 1.03
aTEAC, Capacidad antioxidante en equivalentes de Trolox.
bEAG, Equivalentes de ácido gálico.
Adicionalmente, existen estudios que demuestran una relación directa entre actividad
antioxidante y antidiabética. Khalil, et al. (2008), evaluaron la actividad antioxidante de los
extractos acuosos de las hojas de Bauhinia forficata y Cissus sicyoides, las cuáles poseen
actividad antidiabética. Los resultados obtenidos por el método de ABTS•+ fueron para B.
forficata IC50 = 8.00 ± 0.07 µg/mL y para C. sicyoides IC50= 13.0 ± 0.2 µg/mL expresados en
equivalentes de Trolox, estos resultados indican que los extractos acuosos de estas plantas
poseen actividad antioxidante, siendo los compuesto fenólicos responsables de diversas
actividades como la anticancerígena, antidiabética, entre otras. Esto demuestra que la actividad
antioxidante está asociada con la prevención del estrés oxidativo y éste último con la diabetes.
Chatterjee et al. (2012), evaluaron la actividad antioxidante y antidiabética de la
fracción de acetato de etilo de los extractos metanólicos acuosos de la semilla de Eugenia
jambolana. Los resultados obtenidos muestran que la fracción de acetato de etilo puede ayudar
a contrarrestar la diabetes y el estrés oxidativo, esto se debe a los fitoconstituyentes presentes
en los extractos como el ácido gálico.

15
Existen algunos reportes de la evaluación de la actividad antidiabética para algunas
especies de Justicia. Lizcano et al. (2010), evaluaron la actividad antioxidante de las hojas y el
tallo Justicia pectoralis, reportando los datos que se muestran en la Tabla 4, los resultados
están expresados por g de planta fresca.
Tabla 4. Actividad antioxidante de J. pectoralis.
Parte
estudiada
Fenoles
(mg EAGb/g)
Flavonoides
(mg ECc/g)
TEACa
(μmol Trolox/g)
ORACd
(μmol Trolox/g)
Hoja 5.65 ± 0.01 0.75 ± 0.04 18.80 ± 0.70 89.70 ± 3.70
Tallo 1.18 ± 0.00 0.12 ± 0.00 5.30 ± 0.70 21.90 ± 1.00
aTEAC, Capacidad antioxidante en equivalentes de Trolox. bEAG, Equivalentes de ácido gálico. cEC, Equivalentes de catequina. dORAC, Capacidad de absorción de radicales oxígeno.
Los resultados están expresados en base fresca.
Sulaiman et al. (2011), evaluaron la actividad antioxidante del tallo de J. gendarussa
con diferentes disolventes, los resultados obtenidos se muestran en la Tabla 5.
Tabla 5. Actividad antioxidante de J. gendarussa.
Disolvente
Fenoles totales
(mg EAGa/100
g peso seco)
Flavonoides
totales
(mg EQb/100 g
peso seco)
FRAP
(mg EAGa/100 g
peso seco)
DPPH•
(mg EAGa/100 g
peso seco)
Acetona 70% 40.00 ± 0.10 120.00 ± 0.00 40.00 ± 0.00 70.00 ± 0.10
Etanol 70% 180.00 ± 1.20 210.00 ± 0.30 40.00 ± 0.00 40.00 ± 0.10
Metanol 70% 20.00 ± 0.10 110.00 ± 0.10 30.00 ± 0.00 10.00 ± 0.00
Agua destilada 10.00 ± 0.10 10.00 ± 0.00 10.00 ± 0.00 30.00 ± 0.00
aEAG, Equivalentes de ácido gálico. bEQ, Equivalentes de quercetina.
Los resultados anteriores muestran que la actividad antioxidante es dependiente de la
especie estudiada, la parte de esta planta, el disolvente y método utilizado para la extracción.
Además es importante mencionar que aunado a lo anterior también el estado de madurez de la
planta, condiciones ambientales donde crece la planta y el método de obtención de los

16
extractos también afectan significativamente la actividad antioxidante de los extractos de
plantas.
2.8. Métodos de extracción
Las técnicas de extracción para la obtención de compuestos bioactivos se pueden
clasificar en tradicionales y modernas. Dentro de las técnicas tradicionales se encuentran la
maceración, infusión, decocción y Soxhlet. En las técnicas modernas podemos encontrar a la
extracción asistida por microondas, extracción asistida por ultrasonido, extracción con fluidos
supercríticos, entre otras. La infusión y la decocción son técnicas comúnmente utilizadas, la
primera consiste en adicionar agua a punto de ebullición al material a extraer y se deja enfriar
para posteriormente filtrar, en la segunda se coloca el material a extraer con agua y se calienta
hasta alcanzar el punto de ebullición, se deja enfriar y se filtra (Brusotti et al., 2014).
Particularmente, para el presente estudio es de interés la extracción asistida por
ultrasonido. En este tipo de extracción por lo general, se trabaja en un intervalo de 20 kHz a
100 MHz. Las ondas de ultrasonido pasan a través de un medio creando compresión y
expansión. Este proceso produce un fenómeno llamado cavitación, que significa la
producción, el crecimiento y el colapso de las burbujas que se crean en el medio. El
mecanismo de extracción por ultrasonido implica principalmente dos fenómenos físicos, la
difusión a través de la pared de la célula, el crecimiento y rompimiento de la burbuja. Para que
una extracción por ultrasonido sea eficaz depende de diversos factores, tales como el
contenido de humedad de la muestra, el grado de molienda, el tamaño de las partículas, el
disolvente, la temperatura, el tiempo de exposición de la muestra a las ondas de ultrasonido
(Azmir et al., 2013).
Li et al. (2005) emplearon la extracción por ultrasonido con metanol por 30 min en las
hojas frescas, corteza fresca y corteza seca Eucommia ulmodies, ellos encontraron una
recuperación del 70% de ácido clorogénico, un compuesto fenólico, en comparación con las
técnicas de extracción convencionales.

17
2.9. Extracción en fase sólida (SPE)
Generalmente, las muestras obtenidas a partir de la extracción están constituidas de una
mezcla compleja de sustancias químicas, por lo que se torna importante un tratamiento
posterior previo al estudio de los componentes químicos, incluyendo el aislamiento y
caracterización. Para lograr esto, existen diversos pretratamientos que se pueden aplicar a una
muestra, los más comunes son: la extracción líquido-líquido y la extracción en fase sólida
(SPE). La SPE tiene las ventajas de realizarse en tiempos cortos, poder utilizar muestras
pequeñas (50 a 100 µL), y por lo tanto un consumo de disolventes reducido, entre otras. Los
cartuchos de SPE pueden estar constituidos por sorbentes en fase normal, en fase revesa, entre
otros (He et al., 2007).
Para llevar a cabo la extracción en fase sólida, se deben realizar los pasos siguientes
1. Acondicionamiento del cartucho
2. Aplicación de la muestra
3. Elución de solutos retenidos (interferencias)
4. Elución del analito de interés
5. Limpieza del cartucho
Los analitos de interés se retendrán por el sorbente y posteriormente serán eluidos
dependiendo de la afinidad que tengan con el material del sorbente y la fase móvil, Figura 6,
(Harris, 2007).

18
Figura 6. Extracción en fase sólida.
2.10. Elucidación estructural
Una vez obtenido el compuesto puro, (aproximadamente 30 mg de cada analito para la
caracterización) se obtendrán los datos espectroscópicos y espectrométricos, y mediante la
combinación de estos datos se elucidará la fórmula molecular de los flavonoides aislados.
Por la gran importancia que representan en el análisis químico moderno, se describirán
a continuación.
Analito Interferencias

19
2.10.1. Espectroscopía de infrarrojo
En la Tabla 6, se muestran las bandas del espectro de IR de flavonoides examinados,
así como también las asignaciones para los compuestos citados (Heneczkowski et al., 2001).
Tabla 6. Bandas del espectro de IR de algunos flavonoides.
Flavonoide
Flavona Fisetina Miricetina Quercetina
Grupo asignado a la
banda dada (cm-1)
Banda C=O
Vibraciones de
deformación -C-OH
1649 1619 1638 1638
1332 m 1377 1368, 1355, 1348
1313 w 1304 1333, 1326 1317, 1296
1283 w 1285 m, 1252 w 1245, 1211
1260 w 1252 w 1228, 1202
1224 m 1193 m 1186
Vibraciones de
estiramiento –C-OH
1129, 1112 m –
s 1168 s 1164 s, 1142 m
s – fuerte; m- mediano; w-débil; vw – muy débil.
, número de onda.
2.10.2. Espectrometría de masa (MS)
La espectrometría de masa es una técnica poderosa para el análisis de compuestos
fenólicos por su sensibilidad alta y por la posibilidad de combinarse con diferentes técnicas
cromatográficas. De manera independiente, esta técnica permite determinaciones cuantitativas
y cualitativas.
El detector en espectrometría de masa es crítico para la identificación de compuestos
fenólicos debido a la complejidad y diversidad estructural de estos compuestos y sus bajas
concentraciones en plantas. La sensibilidad y selectividad de la detección puede ser
incrementada usando espectrometría de masa en cascada (MS/MS), produciendo mayor
fragmentación de los precursores e iones hijos, por consiguiente proveen de información
estructural adicional para la identificación de flavonoides.

20
Los espectros de iones positivos obtenidos por Disociación Inducida por Colisión (CID,
por sus siglas inglés) se utilizan con gran frecuencia. A partir de estos se puede identificar la
pérdida de moléculas pequeñas o iones [M+H]+, tales como H2O, COCO, C2H2O, CO2CO y
CH3 se observan comúnmente y son útiles para identificar la presencia de grupos funcionales
específicos. De manera adicional, el modo de ionización negativo es bastante sensible para el
análisis de flavonoides, ya que se utiliza una energía de colisión alta para rendir una adecuada
fragmentación, proporcionando información complementaria en el espectro. En el modo de
ionización negativo varios iones (aniones radicales) tales como CH2CHOH, COCO, CO2CO, y
CH3 también son observados después de estudiar el espectro de masa.
En la identificación de glucosilflavonoides, se puede obtener información muy útil del
espectro de masa, tal como la masa molecular, la estructura de la aglicona, información acerca
de la acilación de los grupos hidroxilo de azúcares, metilación de los grupos hidroxilo de la
aglicona, número de azúcares, en algunos casos la posición de los enlaces glicosídicos. Sin
embargo, MS no provee información acerca de la estereoquímica del enlace glicosídico y no
distingue entre unidades de azúcar diasteroméricas.
2.10.3. Espectroscopía de resonancia magnética nuclear (RMN)
2.10.3.1. RMN de 1H
El espectro de RMN-1H de los flavonoides permite reconocer características
estructurales importantes. Un resumen de los δ para los tipos de protones más comúnmente
hallados en los flavonoides se presenta en la Tabla 7.

21
Tabla 7. Desplazamiento químico de protón.
Desplazamiento químico
δ (ppm) Tipos de protones
1.0 – 1.2 Metilo de ramnosa (da)
1.7 Metilos del grupo isopentenilo
3.0 – 4.8 H de azúcares
3.5 Metileno del grupo isopentenilo
3.7 – 4.1 Metoxilos aromáticos
4.1 – 4.6 Protones 2 y 3 de isoflavanonas
4.2 – 6.0 H-1 de azúcares , protón 2 de flavanonoles y flavanonas
(dd)
5.4 H-2 de flavanonoles y flavanonas (dd)
5.9 – 6.0 Metilendioxi
6.0 – 6.8 H-3, 6 y 8 de flavonas
6.8 – 8.0 H aromáticos del anillo B
7.5 – 8.0 H-2 de isoflavonas
12.0 – 14.0 H de hidroxilo 5
da, doble ancho.
dd, doble de doble.
2.10.3.2. RMN de 13C
En el espectro de RMN-13C se pueden reconocer varios tipos de carbonos, en la Tabla 8
se presentan los desplazamientos químicos para varios tipos de carbonos de flavonoides.

22
Tabla 8. Desplazamientos químicos de varios tipos de carbonos de flavonoides.
Desplazamiento químico
δ (ppm)
Tipos de carbonos
18 C-6 de ramnosa
30 C-4 de flavan-3-oles
42 – 46 C-3 de flavanonas
56 – 61 Metoxilos
60 – 80 C-OH de carbohidratos
70 – 75 C-3 de flavanonoles
80 C-2 de flavanonas
85 C-2 de flavanonoles
100 – 115 C-3 de flavonas, C-1 de carbohidratos, C-10 de
flavonas 5-hidroxiladas
115 – 128 Aromáticos con H
145 C-3 de flavonoles, C-5 de flavonas 5-hidroxiladas, C-3
y C-4 de antocianinas
150 – 165 C aromáticos hidroxilados y metoxilados, C-2 de
flavonas, C-4´oxigenado, C-9 de flavonas.
175 – 178 Carbonilo C-4 sin OH en C-5 en flavonas, C-4 de
flavonoles
182 Carbonilo C-4 con OH en C-5 de flavonas
190 – 196 Carbonilo C-4 de flavanonas
197 – 200 Carbonilo C-4 de flavanonoles
Es posible también diferenciar entre un C-glicosilflavonoide y un O-glicosilflavonoide.
En los O-glicósidos, el C-1 presenta un desplazamiento químico alrededor de 100 ppm para
los carbohidratos más comunes, mientras que en los C-glicósidos este carbono se encuentra en
alrededor de 75 ppm. Por otro lado, en los C-glicósidos el carbono de la aglicona enlazado al
carbohidrato está en alrededor de 10 ppm a frecuencias más altas respecto a su valor normal
(sin sustituyente).

23
3. ORIGINALIDAD
El objeto del presente estudio es Justicia spicigera, una planta que tiene propiedades
antidiabéticas comprobadas (Ortiz-Andrade et al., 2012). Sin embargo no existe mucha
información en la literatura científica sobre quiénes son los metabolitos secundarios y el
mecanismo por el cual los extractos de J. spicigera ejercen dicha actividad.

24
4. OBJETIVOS
4.1. Objetivo general
Determinar la actividad antioxidante y antidiabética in vitro de los extractos de Justicia
spicigera, así como relacionar esta actividad con los compuestos fenólicos presentes en los
extractos.
4.2. Objetivos específicos
1. Obtener extractos ricos en compuestos fenólicos a partir de las hojas de J.
spicigera.
2. Determinar la actividad antidiabética y antioxidante in vitro de los extractos.
3. Elucidar la estructura de los compuestos fenólicos mayoritarios presentes en los
extractos.
4. Correlacionar la actividad antioxidante y antidiabética de los extractos.

25
5. METAS
Objetivo 1.
1.1. Obtener una muestra homogénea de las hojas de J. spicigera.
1.2. Obtener un método eficiente de extracción de compuestos fenólicos de las hojas de J.
spicigera.
Objetivo 2.
2.1 Determinar la actividad antidiabética por el método de inhibición de difusión de
glucosa y mediante la inhibición de las enzimas α-amilasa y α-glucosidasa.
2.2 Determinar la actividad antioxidante mediante la reducción del DPPH•.
2.3 Cuantificar los fenoles y flavonoides totales de los extractos.
Objetivo 3.
3.1 Separar mediante métodos cromatográficos los compuestos fenólicos mayoritarios
presentes en el extracto que contenga el mayor contenido de estos compuestos.
3.2 Caracterizar los compuestos fenólicos aislados mediante técnicas espectrométricas y
espectroscópicas.
Objetivo 4.
4.1 Correlacionar la actividad antidiabética con la actividad antioxidante mediante una
correlación de Pearson.

26
6. METODOLOGÍA
Hojas de J.
spicigera
Secado
Molienda y
tamizado
Almacenamiento
-20 °C
Decocción Infusión
Ultrasonido: metanol,
acetona 70%, etanol
70%, agua
Limpieza del
extracto (SPE)
Fenoles totalesFlavonoides
totalesDPPH•
Elucidación
estructural (IR,
MS, RMN)
Actividad
antidiabética
Figura 7. Procedimiento del tratamiento de la muestra.

27
6.1. Equipos, reactivos y disolventes
El trabajo experimental se desarrolló en los laboratorios en Productos Naturales y
Alimentos del Instituto de Agroindustrias de la Universidad Tecnológica de la Mixteca. A
continuación se enlistan los equipos y reactivos utilizados en este proyecto.
Termobalanza, Balanza analítica Sartorius BL210S, molino ciclónico Foss Cyclotec
1093, Sonicador Ultrasonic Cleaner SB-3200 DTN, lector de microplacaS BioTek® ELx808,
parrillas de calentamiento CIMAREC®, vórtex, rotavapor de vacío IKA® RU10, recirculador
de agua ECO10 SEV, micropipeta, tamiz #60, balanza granataria OHAUS®, material de
vidrio.
Metanol 99.5%, etanol 99.5%, acetona 99.5%, ácido gálico monohidratado 98%, 2,2-
difenil-1-picrilhidracilo (DPPH•) <100%, (+)-catequina hidratada 96%, reactivo de Folin-
Ciocalteau 2 M, hidróxido de sodio en lentejas 99%, carbonato de sodio 99%, nitrito de sodio
97%, cloruro de aluminio 99.4%, BHT 99%.
Los reactivos y disolventes utilizados fueron grado analítico. Los disolventes y el agua,
se destilaron y degasificaron antes de ser utilizados para cada experimento.
6.2. Obtención de la muestra
La muestra de J. spicigera se colectó en el mes de agosto del 2013 en el municipio de
Asunción Cuyotepeji, ubicado en la región de la mixteca, Oaxaca. El arbusto de J. spicigera se
ubicó a 1765 m de altitud, 17º 55’ 45.12’’ N y -97º 40’ 22.44’’ O. Posteriormente se
transportó a los laboratorios de la Universidad Tecnológica de la Mixteca (UTM). La planta se
envió a la Universidad Autónoma de Chapingo (UACh) para su identificación etnobotánica.

28
6.3. Preparación de la muestra
El proceso de secado consistió en colocar las hojas de J. spicigera entre papel
periódico. El papel periódico se cambió periódicamente hasta alcanzar una remoción del 90%
de humedad presente en las hojas. Las hojas deshidratadas de J. spicigera se molieron en un
molino ciclónico. La muestra se tamizó a través de una malla número 60 y el polvo obtenido
se almacenó en frascos de color ámbar a -20 °C.
6.4. Obtención de los extractos
Se utilizaron tres métodos de extracción: Decocción, infusión y por ultrasonido, en este
último método se usaron diferentes disolventes como metanol, etanol 70%, acetona 70% y
agua desionizada. El uso de estos tres métodos tiene como finalidad determinar el efecto del
método de extracción sobre la actividad antidiabética del extracto. A continuación se describe
brevemente cada uno de los métodos de extracción utilizados.
Decocción. Se pesaron 700 mg del polvo de J. spicigera y se adicionaron 5 mL de agua
desionizada. Esta mezcla se calentó en una parrilla eléctrica con agitación magnética. Una vez
alcanzado el punto de ebullición se retiró de la fuente de calor y se dejó enfriar.
Posteriormente se filtró sobre papel Whatman No. 1. El filtrado se almacenó a -20 °C en un
frasco color ámbar hasta su análisis (Cruz-Gallegos et al., 2012).
Infusión. A 700 mg del polvo de J. spicigera se le adicionaron 5 mL de agua
desionizada en ebullición. Se agitó y se dejó en reposo por 15 min. Posteriormente se filtró
con un papel Whatman No. 1. El filtrado se almacenó a -20 °C en un frasco color ámbar hasta
su análisis (Cruz-Gallegos et al., 2012).
Extractos por ultrasonido: metanol, etanol 70%, acetona 70%, agua. Se pesaron 100
mg del polvo de J. spicigera, se colocó en un tubo eppendorf y se adicionó 1 mL de
disolvente. Posteriormente se colocó en un baño de ultrasonido a 35 °C por 15 min. Después,
el extracto se centrifugó, decantó, y se colocó en un matraz bola, este procedimiento se realizó

29
por duplicado. El disolvente se removió en un rotavapor a 40 °C a vacío. Los extractos
obtenidos se almacenaron a -20 °C hasta su análisis.
6.5. Limpieza de los extractos por extracción en fase sólida (SPE)
La etapa de limpieza de los extractos tuvo como objetivo eliminar las clorofilas, debido
a que estas absorben aproximadamente a 510 nm y por consiguiente interferían en la
determinación de flavonoides y DPPH•.
Se utilizaron cartuchos de extracción en fase sólida Supelco ENVI-18 en fase reversa.
El cartucho se acondiciono con 5 mL de H2O. Posteriormente se adicionó el extracto, en el
caso de los extractos de metanol, acetona 70% y etanol 70% se llevó a sequedad en un
rotavapor a 40 ºC y se redisolvió con 5 mL de H2O. Posteriormente se le adicionó 5 mL de
MeOH/H2O (50/50). Por último se le adicionó 5 mL de MeOH. Se obtuvieron dos eluatos por
cada extracto, el eluato acuoso y el de MeO/H2O. Todos los eluatos se almacenaron a -20 ºC
hasta su análisis.
6.6. Cuantificación de fenoles totales en los extractos
Preparación de soluciones.
EtOH acuoso al 90% (v/v). Se mezcló 90 mL de EtOH con 10 mL de agua.
Na2CO3 al 0.5% (p/v). Se pesaron 50 mg de Na2CO3, se disolvieron en agua y llevaron
hasta el afore a 10 mL.
Reactivo de Folin-Ciocalteu 0.1 M. Se midieron 0.5 mL del reactivo de Folin-Ciocalteu
2 M, se disolvieron en agua en ausencia de luz y se llevaron hasta el afore a 10 mL.
Ácido gálico a 100 ppm. Se pesó 1 mg de ácido gálico y se disolvió en EtOH al 90%
hasta un volumen de 10 mL. Los estándares de ácido gálico 5, 10, 15, 20 y 25 ppm se
prepararon a partir de la disolución de ácido gálico de 100 ppm.

30
Cuantificación por el método Folin-Ciocalteau (Julián-Loaeza et al., 2011). 40 µL de
extracto o del estándar se mezclaron con 40 µL del Reactivo de Folin-Ciocalteau 0.1 M en una
microplaca. La mezcla se dejó en reposo por 3 min a temperatura ambiente y se agitó por 15 s
a baja velocidad. Pasado este tiempo, a la mezcla anterior se le adicionaron 40 µL de Na2CO3
al 0.5%. La mezcla se dejó reposar por 30 min a 40 oC y se agitó por 1 min a velocidad media.
La absorbancia de la muestra y de los estándares se determinó a 750 nm. Al blanco de la
muestra o del estándar se le adicionaron agua en lugar del reactivo de Folin-Ciocalteu. Se
utilizó el estándar de 25 ppm como blanco de los estándares. Para la curva de calibración se
utilizó ácido gálico como estándar. Todas las determinaciones se realizaron por triplicado.
6.7. Cuantificación de flavonoides totales en los extractos
Preparación de soluciones.
NaNO2 al 1.5% (p/v). 375 mg de NaNO2 se disolvieron en agua y la mezcla se llevó a
un volumen de 25 mL.
AlCl3 al 3% (p/v). 300 mg de AlCl3•6H2O se disolvieron en agua y se llevó a un
volumen de 10 mL.
NaOH 1 M. 400 mg de NaOH, se disolvieron en agua y se llevó a un volumen de 10
mL.
(+)-Catequina 1000 ppm. 10 mg de (+)-catequina, se disolvieron en MeOH y se llevó a
un volumen de 10 mL en ausencia de luz. A partir de esta solución se prepararon estándares de
catequina de 10, 20, 30, 40 y 50 ppm.
Quercetina 1000 ppm. 10 mg de quercetina, se disolvieron en MeOH y se llevó a un
volumen de 10 mL en ausencia de luz. A partir de esta solución se prepararon estándares de
quercetina de 20, 40, 60, 80 y 100 ppm.
Cuantificación, Julián-Loaeza et al., (2011). En un vial ámbar se mezcló 1 mL del
extracto o del estándar contenido con 1 mL de NaNO2 al 1.5%. La mezcla se agitó por 5 min

31
en un vórtex a temperatura ambiente. 50 µL de esta mezcla se colocó en una microplaca.
Posteriormente, a esta mezcla se le agregaron 50 µL de AlCl3 al 3% y se agitó por 1 min.
Pasado este tiempo, se le adicionaron 50 µL de NaOH 1 M. Esta mezcla se agitó por 1 min.
Pasado este tiempo se determinó la absorbancia a 405, 490 y 515 nm. El blanco de la muestra
o del estándar contenía agua en lugar de AlCl3. Como blanco de los estándares se utilizó el
estándar de 50 ppm. Para la curva de calibración se utilizaron (+)-catequina y quercetina.
Todas las determinaciones se realizaron por triplicado.
6.8. Evaluación de la actividad antirradical de los extractos
Preparación de soluciones
Solución patrón de DPPH• al 0.1% (p/v). Se pesaron 10 mg de DPPH•, se disolvieron
en MeOH hasta un volumen de 10 mL en ausencia de luz. De esta disolución se preparó una
solución de DPPH• al 0.004%, que se resguardó de la luz y se dejó en refrigeración entre
mediciones.
Soluciones de estándares de ácido gálico 0.5, 1.0, 1.5, 2.0 y 2.5 ppm. Primero se
preparó una solución patrón de 100 ppm de ácido gálico (1 mg de ácido gálico llevado a un
volumen final de 10 mL con EtOH al 90%). A partir de esta disolución se prepararon los
estándares antes mencionados.
Soluciones de extractos. Estas soluciones se prepararon a concentraciones que
permitieron reducir al DPPH• entre el 20 y el 90%.
Cuantificación (Julián-Loaeza et al., 2011). 70 µL del extracto o del estándar se
mezclaron con 70 µL de DPPH• al 0.004% en una microplaca, y se dejó en reposo por 30 min
a temperatura ambiente en ausencia de luz y con agitación. Posteriormente, se determinó la
absorbancia a 515 nm. La mezcla de 70 µL del DPPH• al 0.004% y 70 µL de MeOH se usó
como control. El blanco del estándar se obtuvo con el estándar menos concentrado. La
actividad antirradical se expresó como el % de inhibición [((Acontrol – Aextracto)/Acontrol) *
100]. El valor IC50, que corresponde a la cantidad de muestra requerida para reducir el 50%
del radical DPPH• se reportó en gramos de muestra/kg de DPPH•.

32
% Inhibición = [ADPPH• − AEXT
ADPPH•] x 100
Cinética de eficiencia antirradical. La cinética se realizó con el extracto a la
concentración que exprese el valor de IC50. Se realizó una lectura de absorbancia en intervalos
de 30 s durante 120 min. Los valores de absorbancia ajustados se graficaron contra el tiempo
para determinar el tiempo en el cual inicie el estado estable, ese punto se denomina TEC50. Con
los valores de IC50 y TEC50 se calculó el valor de la eficiencia antirradicalar (AE) expresada
como kg de DPPH•/kg muestra seca•min.
AE =1
IC50 x TEC50
6.9. Hidrólisis de los extractos
Se sometió a una hidrólisis ácida el extracto que presentó la mayor actividad
antidiabética, para emular las condiciones del estómago y así determinar posibles cambios en
la actividad antidiabética debido a los cambios sufridos por los compuestos bioactivos durante
su paso a través del mismo. La hidrólisis consistió en hacer reaccionar 1 g del polvo de J.
spicigera con 20 mL de HCl 2 M por 30 min. Después, la mezcla de reacción se llevó a
sequedad a 50 oC utilizando un rotavapor a presión reducida (Harborne, 1998).
Al extracto hidrolizado se le determinó la actividad antidiabética y la actividad
antirradicalar, de acuerdo a los procedimientos descritos en las secciones 6.8, 6.10 y 6.11.
6.10. Evaluación de la inhibición de la difusión de glucosa
Para evaluar la difusión de glucosa se empleó el método de Gallagher et al. (2003). Se
usó un tubo de diálisis de 6 cm x 15 mm (Spectra/Por, MWCO: 2000), en el cual se colocaron
15 mL del extracto correspondiente, 2 mL de una solución de NaCl 0.15 M y de una solución
de D-glucosa 0.22 mM, el tubo se selló. Una vez sellado, el tubo de diálisis se colocó dentro
de un tubo de centrifuga de 50 mL el cual contenía 45 mL de una solución de NaCl 0.15 M. Se
realizó una medición de la concentración de glucosa cada hora, durante 24 horas, tanto en la

33
solución externa como interna. Como blanco se utilizó una solución del extracto con NaCl
0.15 M en ausencia de D-glucosa. Estas mediciones se realizaron con un biosensor.
6.11. Evaluación de la inhibición de la actividad enzimática
6.11.1. Inhibición de los extractos en presencia de α-amilasa
Solución de almidón (0.5% p/v). A 0.25 g de almidón de papa se le adicionaron 50 mL
de agua desionizada, posteriormente se agitó y ebulló por 15 min.
Solución de la enzima. La solución de la enzima se preparó mezclando 1 mg de α-
amilasa de páncreas porcino en 100 mL de buffer de fosfato de potasio 20 mM (pH 6.9) que
previamente contenía NaCl 6.7 mM.
Reactivo colorimétrico. Se preparó con la mezcla de 20 mL de ácido 3,5-
dinitrosalicílico 96 mM, 8 mL de KNaC4H4O6•4H2O 5.31 M en NaOH 2 M, y 12 mL de agua
desionizada.
Generación de la reacción. 1 mL del extracto y 1 mL de la solución de α-amilasa se
mezcló y se incubó a 25 °C por 30 min. A 1 mL de la mezcla se le adicionó 1 mL de la
solución de almidón y se incubó a 25 °C por 3 min. Posteriormente, se adicionó 1 mL del
reactivo colorimétrico y se colocó en un baño de agua a 85 °C. Después de 15 min, la mezcla
se retiró del baño y se enfrió, después se realizó una dilución con 9 mL de agua destilada y se
determinó la absorbancia a 540 nm en un espectrofotómetro. Se usó acarbosa como control
positivo. El porcentaje de inhibición de α-amilasa se calculó con la fórmula siguiente
(Nickavar & Yousefian, 2011).
Iα−amilasa =∆Acontrol − ∆Amuestra
∆Acontrol
Donde: ∆Acontrol = Aprueba − Ablanco
∆Amuestra = Aprueba − Ablanco

34
6.11.2. Inhibición de la α-glucosidasa en presencia de los extractos.
Solución de α-glucosidasa. Se utilizaron 0.2 U de α-glucosidasa por 1 mL de buffer de
fosfato (0.2 U/mL).
Solución de buffer de fosfato de potasio (pH 6.8). Se pesaron 1.36 g de K2HPO4 y 2.72
g de KH2PO4, se disolveron con agua destilada hasta aforar a 20 mL.
Solución de p-nitrofenil glucopiranósido 2.5 mM (sustrato). Se pesaron 7.53 mg del
sustrato en 10 mL del buffer de fosfato.
Solución de Na2CO3 0.2 M. Se pesaron 0.21 g de Na2CO3 en 10 mL de agua destilada.
Generación de la reacción. 8 µL del extracto en DMSO, se mezclaron con 112 µL del
buffer de fosfato y 20 µL de la solución de la enzima y se incubó a 37 oC por 15 min.
Posteriormente se le adicionaron 20 µL de la solución del sustrato y se dejó reaccionar a 37 oC
por 15 min. La reacción se terminó por la adición de 80 µL de Na2CO3. La actividad
enzimática se cuantificó por la liberación del p-nitrofenilo a 405 nm. Los datos de inhibición
enzimática se expresaron como EC50. Como control positivo se usó acarbosa. Las velocidades
inhibitorias (%) se calcularon de acuerdo a la fórmula siguiente (Li et al., 2011):
Velocidad de inhibición ((%))= (1-(Aensayo-Ablanco)
Control Aensayo-Control Ablanco) x 100 %
6.12. Aislamiento de los compuestos fenólicos
Previo a la separación por HPLC se llevó a cabo una etapa de limpieza del extracto,
mediante una extracción en fase sólida (SPE, por sus siglas en inglés) utilizando un cartucho
Supelco ENVI-18. El cartucho se acondicionó con 5 mL de H2O, posteriormente se adicionó
el extracto con mayor contenido de compuestos fenólicos al cartucho y se eluyó con 5 mL de
H2O. Posteriormente se le adicionaron 5 mL de MeOH/H2O al 50%. Por último se le

35
adicionaron al cartucho 5 mL de metanol. Los tres eluatos se almacenaron a -20 ºC hasta su
análisis. Para determinar que extracto se utilizó en la separación química, se le cuantificaron
los fenoles totales de acuerdo al procedimiento descrito en la sección 6.6. El extracto que
contenía la mayor cantidad de compuestos fenólicos se evaporó a sequedad para obtener el
extracto seco.
Una vez obtenido el extracto seco, se disolvió en acetonitrilo (Merck, grado HPLC) en
una concentración de 20 mg/mL (p/v) y se inyectó en un bucle de 500 µL de un equipo de
HPLC LC1150 de GBC.
La separación se llevó a cabo en una columna semipreparativa de fase reversa C18 (10
mm i.d. x 200 mm, 4 µm) (GENESIS, Número de catálogo 8P25960) con una fase
estacionaria de gel de sílice modificada con grupos octadecilos, a una temperatura de columna
de 25 oC. La fase móvil estuvo compuesta de acetonitrilo-agua-ácido acético (Merck, grado
HPLC), (50:50:1, v/v). Esta fase fue eluída isocráticamente a una velocidad de flujo de 0.2
mL/min, el eluato se monitoreó con un detector UV/Vis modelo LC1205 de GBC a 260 nm ya
que a esta longitud de onda absorben los compuestos fenólicos. Las distintas fracciones se
colectaron en viales de acuerdo al perfil detectado en el cromatograma.
6.13. Caracterización de los compuestos fenólicos presentes en los
extractos
La caracterización de los compuestos fenólicos puros aislados se realizó mediante el
análisis e interpretación de sus constantes físicas (punto de fusión, rotación óptica, solubilidad,
Rf, etc.) y datos espectroscópicos de UV, IR, RMN y espectrométricos (EM de baja y alta
resolución). El análisis e integración de la información proporcionada por los datos anteriores
condujeron a una hipótesis estructural; si se trata de un compuesto ya descrito, su estructura se
corroborará comparando los parámetros obtenidos con los parámetros reportados en la
literatura química. En caso de ser nuevos compuestos químicos no descritos en la literatura, se
realizará un estudio detallado por técnicas de Resonancia Magnética nuclear (RMN) en dos
dimensiones, tales como el HETCOR (correlación heteronuclear 1H-13C), NOESY
(Espectroscopía del Efecto Nuclear Overhauser en dos dimensiones) para determinar

36
interacciones a través del espacio entre 1H. Es importante señalar que todos los datos
espectroscópicos y espectrométricos se obtuvieron en el laboratorio de la Universidad del
Papaloapan a cargo del Dr. Lemuel Pérez Picasso.
6.14. Análisis estadístico
Para el análisis estadístico se realizó un ANOVA. Los valores promedio obtenidos de las
diferentes pruebas se analizaron mediante un comparativo de medias con un p<0.05, utilizando
la prueba de Duncan (Walpole et al., 1999). El objetivo de este análisis fue determinar si
existe diferencia significativa entre los valores obtenidos. También, se llevaron a cabo
correlaciones de Pearson para el contenido de fenoles totales, flavonoides totales, la actividad
antidiabética expresada como % de inhibición de difusión de glucosa e inhibición de la
actividad enzimática de la -glucosidasa y -amilasa, así como la actividad antirradical. Estas
correlaciones permitieron determinar si existe interdependencia entre las actividades
estudiadas.

37
7. RESULTADOS Y DISCUSIONES
7.1. Obtención de la muestra
La muestra ubicada a 1765 m de altitud, 17º 55’ 45.12’’ N y -97º 40’ 22.44’’ O se
colectó en el mes de agosto del 2013 en el municipio de Asunción Cuyotepeji, Oaxaca,
México. Se revisó que la muestra estuviera en buen estado, es decir, que no presentara ninguna
enfermedad o plaga. El proceso de secado se realizó con papel periódico, colocando una capa
de hojas frescas y una de papel periódico, y así sucesivamente, las muestras se almacenaron a
temperatura ambiente. Posteriormente las muestras se prensaron y se realizaron cambios de las
hojas de papel periódico diariamente por los primeros 4 días y posteriormente se cambiaron
cada 3 días hasta alcanzar una remoción del 87% de humedad, en un lapso de tiempo de 2
semanas y media.
Figura 8. Secado de hojas de J. spicigera.

38
La determinación de humedad se realizó en una termobalanza, tomándose muestras
tanto de hoja fresca como de hoja seca, las cuales se trocearon finamente. En la Tabla 9, se
muestran los resultados obtenidos para la determinación de humedad.
Tabla 9. Determinación de humedad en hojas de J. spicigera.
Como se puede observar en la Tabla 9, el contenido de humedad de la hoja fresca es de
63.87% y el porcentaje de humedad de la hoja después del proceso de secado fue de 8.30%.
Con base en estos resultados, se determinó que la remoción de humedad después del proceso
de secado fue del 87%. Una vez que se tuvo la muestra seca de procedió a la molienda y
tamizado del polvo.
La masa de la muestra fresca colectada fue de 6330.47 g, después del proceso de
secado se obtuvo una masa de 2287.20 g. La muestra seca se troceó manualmente para reducir
el tamaño de las hojas y con ello facilitar el proceso de molienda en el molino ciclónico, la
masa obtenida después del proceso de molienda fue de 2219.7 g con una rendimiento del
97.05%.
7.2. Obtención de extractos
Se obtuvieron dos tipos de extractos, los extractos crudos, obtenidos a partir de la
extracción de acuerdo al diagrama descrito en la Figura 9, y los extractos que se sometieron a
una etapa de limpieza con cartuchos de SPE, denominados extractos limpios. A los extractos
crudos se les cuantificaron fenoles totales y a los extractos limpios se les determinó fenoles
totales, flavonoides totales y actividad antioxidante. De los extractos limpios se obtuvieron dos
Réplica Masa inicial (g) Humedad hoja
fresca (%)
Humedad hoja
seca
(%)
1 0.99 62.73 7.34
2 1.00 65.14 8.46
3 1.00 63.74 9.09
Promedio 1.00 ± 0.00 63.87 ± 1.21 8.30 ± 0.89

39
eluatos uno contenido en agua y otro contenido en MeOH/H2O, en total se obtuvieron 14
eluatos, dos por cada extracto los cuales se almacenaron a -20 ºC. El eluato contenido en
MeOH se desechó debido a que en este se encuentran contenidas las clorofilas. En la Figura 9,
se muestra un esquema general de los extractos obtenidos.
Eluato
H2O
Limpieza por
SPE
Decocción
Polvo Hoja seca
Infusión
Polvo Hoja seca
H2O
MeOH
Etanol 70%
Acetona 70%
Decocción
Hoja Fresca
Ultrasonido
Polvo Hoja seca
Eluato
MeOH/H2O
Figura 9. Diagrama para la obtención de extractos limpio.
7.3. Cuantificación de fenoles totales
Se cuantificó el contenido de fenoles totales en siete diferentes extractos crudos de J.
spicigera. Como se puede observar en la Figura 10, el extracto obtenido por decocción de la
hoja fresca posee un contenido alto de fenoles totales con 767.04 ± 0.01 mg EAG/100 g en
comparación con los otros seis extractos. Así mismo, el extracto obtenido por ultrasonido con
etanol al 70% presenta un mayor contenido de fenoles totales con 618.62 ± 0.01 mg EAG/100
g en comparación con los diferentes extractos que se estudiaron a partir del polvo de J.
spicigera. Comparando entre los extractos obtenidos por decocción de la hoja fresca y del

40
polvo de las hojas se puede decir que las hojas frescas presentan un contenido alto de fenoles
totales en comparación con los deshidratados, esto se puede deber a la influencia del proceso
de secado en el cual se presentó una degradación de estos compuestos. Realizando una
comparación entre la infusión y la decocción del polvo de la hoja de acuerdo a la Figura 10, se
puede apreciar que la decocción presenta un mayor contenido de fenoles totales que en la
infusión.
Por otra parte, de los extractos obtenidos por ultrasonido el extracto etanólico al 70%,
presenta mayor contenido de fenoles totales, seguido por el extracto de acetona al 70%, el
metanólico y por último el acuoso. También dependiendo del disolvente empleado para la
extracción, el contenido de fenoles totales puede variar, esto se puede ver en los valores
reportados en la Figura 10.
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto obtenido por
decocción a partir de muestra seca. INF-MS, Extracto obtenido por infusión a partir de muestra seca.
H2O-US, Extracto acuoso obtenido por ultrasonido. MeOH-US, Extracto de metanol obtenido por
ultrasonido. EtOH-US, Extracto de etanol obtenido por ultrasonido al 70%. ACT-US, Extracto de
acetona obtenido por ultrasonido al 70%. Los datos expresados son el promedio de tres mediciones ±
desviación estándar. Todas las muestras son significativamente diferentes (p<0.05). Se usó la prueba
de Duncan y un ANOVA para el análisis estadístico.
Figura 10. Contenido de fenoles totales en extractos crudos de J. spicigera.
0
100
200
300
400
500
600
700
800
DEC-MH DEC-MS INF-MS H2O-US MeOH-US EtOH-US ACT-US
767.04 ± 0.01
509.91 ± 0.01
422.74 ± 0.01
220.72 ± 0.01
492.41 ± 0.01
618.62 ± 0.01
610.81 ± 0.01
mg E
AG
/100g
Extracto

41
Sepúlveda-Jiménez et al. (2009), cuantificaron fenoles totales a los extractos obtenidos
por maceración en agua y metanol de las flores, hojas y tallo de J. spicigera. Como se puede
observar en la Figura 11, el contenido de fenoles totales en el extracto metanólico es mayor en
comparación con el extracto acuoso en ambos casos. Esta diferencia en el contenido de fenoles
totales se puede atribuir a diferentes factores tales como: el mes de colección, la edad y el
entorno en el cual se encuentra la planta, el método de secado y al método de extracción.
Figura 11. Contenido de fenoles totales en hojas de J. spicgera.
Sulaiman et al. (2011), cuantificaron fenoles totales de la maceración del tallo de J.
gendarussa con diferentes disolventes. Comparando los resultados de Sulaiman et al., y los
valores para las hojas de J. spicigera, Figura 12, se puede ver que la hojas de J. spicigera
poseen un contenido alto de fenoles totales en comparación con el tallo de J. gendarussa. Así
mismo en ambos casos el extracto de etanol al 70%, es el mejor disolvente para extraer
compuestos fenólicos. Un factor que influye en el contenido de fenoles totales es la parte de la
planta de cual se va a realizar el análisis, esto se ve reflejado en la Figura 12, en el cual las
hojas llegan a tener un alto contenido de compuestos fenólicos en comparación con el tallo.
Otro factor importante y que de ello depende el contenido de fenoles es el tipo de extracción,
0
500
1000
1500
2000
2500
3000
3500
4000
H2O MeOH
3850 ± 0.02 3910 ± 0.02
220.72 ±0.011
492.41 ± 0.003
mg
EA
G/1
00
g
Extracto
Sepúlveda-Jiménez et al.,
(2009).
Objeto de estudio de este
proyecto.

42
para extraer los compuestos fenólicos del tallo de J. gendarussa se usó maceración y para J.
spicigera se usó ultrasonido, claramente se pude ver la ventaja de usar la extracción del
ultrasonido reflejándose en el contenido de fenoles totales.
H2O, Extracto acuoso. MeOH, Extracto de metanol. EtOH 70%, Extracto de etanol al 70%. ACT 70%,
Extracto de acetona al 70%.
Figura 12. Contenido de fenoles totales de las hojas de J. spicigera y del tallo J. gendarussa.
Kirca & Arslan (2008), cuantificaron fenoles totales a los extractos metanólicos
obtenidos de una extracción sólido-líquido de las hojas de diferentes plantas, como se puede
observar en la Figura 13, los extractos de J. spicigera poseen un alto contenido en
comparación con las diferentes plantas mostradas en la misma figura. Se puede observar que a
pesar de ser el mismo disolvente, el tipo de extracción y la planta influyen en el contenido de
fenoles totales a obtener.
0
100
200
300
400
500
600
700
H2O MeOH EtOH 70% ACT 70%
220.72 ± 0.01
492.41 ± 0.01
618.62 ± 0.01 610.81 ± 0.01
10.00 ± 0.1 20.00 ± 0.1
180 .00± 1.2
40.00 ± 0.1
mg
EA
G/1
00
g
Extracto
J. spicigera
J. gendarussa
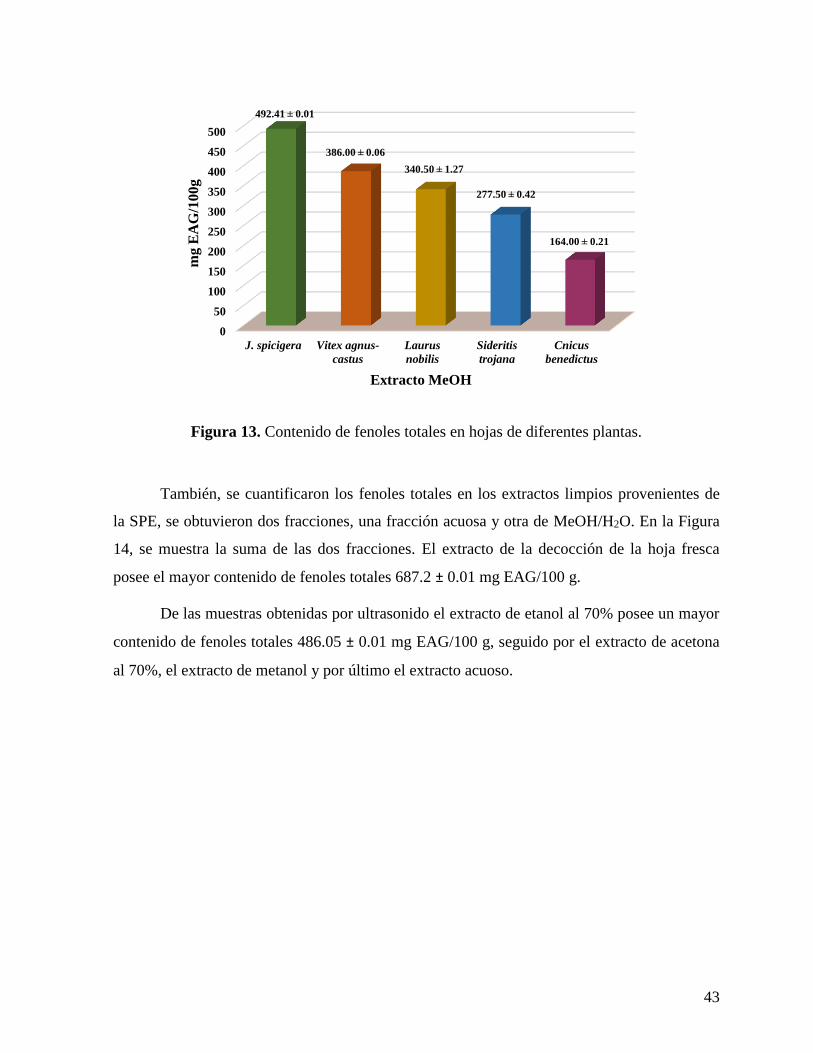
43
Figura 13. Contenido de fenoles totales en hojas de diferentes plantas.
También, se cuantificaron los fenoles totales en los extractos limpios provenientes de
la SPE, se obtuvieron dos fracciones, una fracción acuosa y otra de MeOH/H2O. En la Figura
14, se muestra la suma de las dos fracciones. El extracto de la decocción de la hoja fresca
posee el mayor contenido de fenoles totales 687.2 ± 0.01 mg EAG/100 g.
De las muestras obtenidas por ultrasonido el extracto de etanol al 70% posee un mayor
contenido de fenoles totales 486.05 ± 0.01 mg EAG/100 g, seguido por el extracto de acetona
al 70%, el extracto de metanol y por último el extracto acuoso.
0
50
100
150
200
250
300
350
400
450
500
J. spicigera Vitex agnus-
castus
Laurus
nobilis
Sideritis
trojana
Cnicus
benedictus
492.41 ± 0.01
386.00 ± 0.06
340.50 ± 1.27
277.50 ± 0.42
164.00 ± 0.21
mg
EA
G/1
00
g
Extracto MeOH

44
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto obtenido por
decocción a partir de muestra seca. INF-MS, Extracto obtenido por infusión a partir de muestra seca.
H2O-US, Extracto acuoso obtenido por ultrasonido. MeOH-US, Extracto de metanol obtenido por
ultrasonido. EtOH-US, Extracto de etanol obtenido por ultrasonido al 70%. ACT-US, Extracto de
acetona obtenido por ultrasonido al 70%. Los datos expresados son el promedio de tres mediciones ±
desviación estándar. Todas las muestras son significativamente diferentes (p<0.05). Se usó la prueba
de Duncan y un ANOVA para el análisis estadístico.
Figura 14. Contenido de fenoles totales de extractos limpios de J. spicigera.
Sadiki & Martin (2012), realizaron un estudio de los compuestos fenólicos de la miel
de maple utilizando SPE, los autores varían las concentraciones de metanol en el eluyente y
determinan que la concentración de metanol influye en la recuperación de los compuestos
fenólicos presentes en una muestra. Comparando los resultados de fenoles totales de los
extractos crudos y de los extractos limpios de las hojas de J. spicigera, Figura 15, se puede
observar una disminución en dicho contenido en los extractos limpios. Esta disminución se
puede deber a la polaridad de los compuestos presentes en las muestras, así como también a la
polaridad del eluyente, lo cual provoca que exista una disminución en de los compuestos
fenólicos, los cuales se quedan retenidos en el cartucho de SPE y con ello existe una baja
recuperación de los compuestos fenólicos.
0
100
200
300
400
500
600
700
DEC-MH DEC-MS INF-MS H2O-US MeOH-US EtOH-US ACT-US
687.2 ± 0.01
192.3 ± 0.02
232.06 ± 0.02
178.05 ± 0.01
236.05 ± 0.02
486.05 ± 0.01
372.45 ± 0.02
mg
EA
G/1
00
g
Extracto

45
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto obtenido por decocción a
partir de muestra seca. INF-MS, Extracto obtenido por infusión a partir de muestra seca. H2O-US, Extracto
acuoso obtenido por ultrasonido. MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US,
Extracto de etanol obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por ultrasonido al
70%. Los datos expresados son el promedio de tres mediciones ± desviación estándar. Todas las muestras son
significativamente diferentes (p<0.05). Se usó la prueba de Duncan y un ANOVA para el análisis estadístico.
Figura 15. Comparativo de fenoles totales de extractos crudos y extractos limpios de J.
spicigera.
Sulaiman et al. (2011), cuantificaron fenoles totales de la maceración del tallo de J.
gendarussa con diferentes disolventes. Comparando los resultados de Sulaiman et al., y los
valores para las hojas de J. spicigera, Figura 16, se puede ver que la hojas de J. spicigera
poseen un alto contenido de fenoles totales en comparación con el tallo de J. gendarussa. Así
mismo en ambos casos el extracto de etanol al 70%, es el mejor disolvente para extraer
compuestos fenólicos. Tanto los resultados obtenidos de los extractos limpios y los extractos
crudos de las hojas de J. spicigera muestran un contenido alto de compuestos fenólicos en
comparación con los valores reportados para J. gendarussa.
0
100
200
300
400
500
600
700
800
mg
EA
G/1
00
g
Extracto
Extracto crudo
Extracto limpio
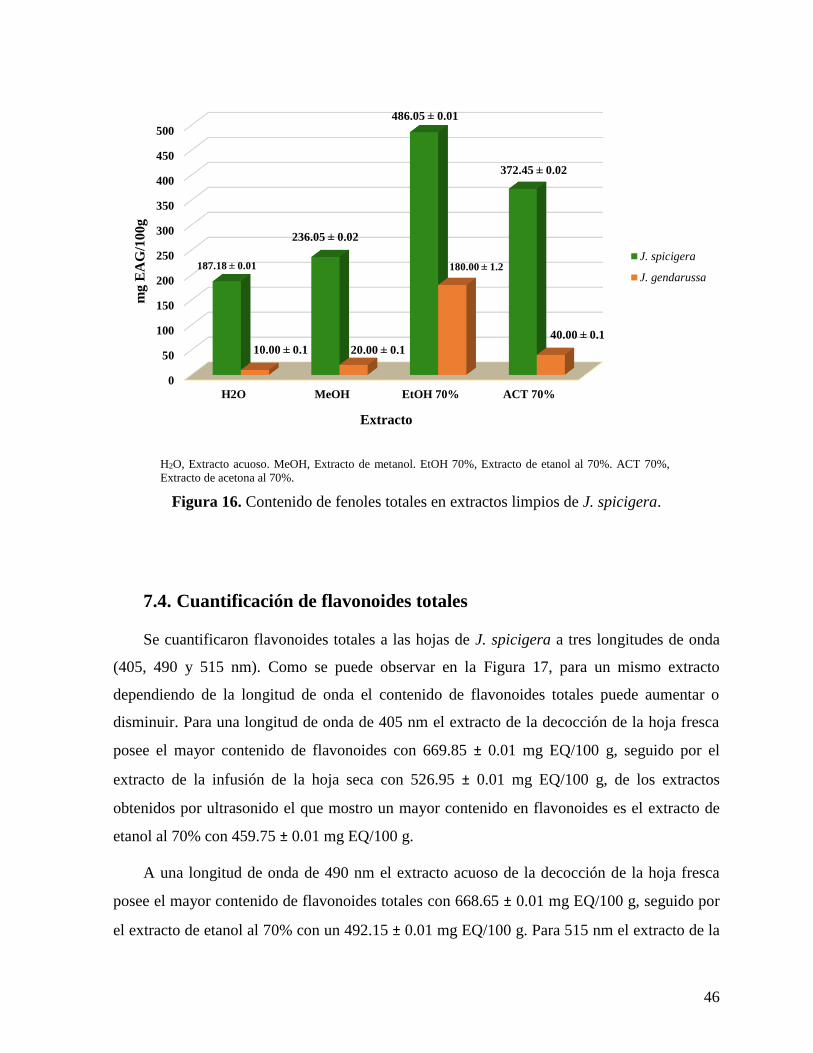
46
H2O, Extracto acuoso. MeOH, Extracto de metanol. EtOH 70%, Extracto de etanol al 70%. ACT 70%,
Extracto de acetona al 70%.
Figura 16. Contenido de fenoles totales en extractos limpios de J. spicigera.
7.4. Cuantificación de flavonoides totales
Se cuantificaron flavonoides totales a las hojas de J. spicigera a tres longitudes de onda
(405, 490 y 515 nm). Como se puede observar en la Figura 17, para un mismo extracto
dependiendo de la longitud de onda el contenido de flavonoides totales puede aumentar o
disminuir. Para una longitud de onda de 405 nm el extracto de la decocción de la hoja fresca
posee el mayor contenido de flavonoides con 669.85 ± 0.01 mg EQ/100 g, seguido por el
extracto de la infusión de la hoja seca con 526.95 ± 0.01 mg EQ/100 g, de los extractos
obtenidos por ultrasonido el que mostro un mayor contenido en flavonoides es el extracto de
etanol al 70% con 459.75 ± 0.01 mg EQ/100 g.
A una longitud de onda de 490 nm el extracto acuoso de la decocción de la hoja fresca
posee el mayor contenido de flavonoides totales con 668.65 ± 0.01 mg EQ/100 g, seguido por
el extracto de etanol al 70% con un 492.15 ± 0.01 mg EQ/100 g. Para 515 nm el extracto de la
0
50
100
150
200
250
300
350
400
450
500
H2O MeOH EtOH 70% ACT 70%
187.18 ± 0.01
236.05 ± 0.02
486.05 ± 0.01
372.45 ± 0.02
10.00 ± 0.1 20.00 ± 0.1
180.00 ± 1.2
40.00 ± 0.1
mg
EA
G/1
00
g
Extracto
J. spicigera
J. gendarussa

47
decocción de la hoja fresca posee el mayor contenido de flavonoides totales con 428.90 ±
0.0031 mg EQ/100 g, seguido por el extracto etanólico con 391.25 ± 0.01 mg EQ/100 g.
Se realizó una correlación de Pearson del contenido de fenoles totales con el contenido de
flavonoides totales para el conjunto de valores de cada longitud de onda y se encontró que
existe una correlación de 0.95 y 0.96 de fenoles con flavonoides totales a 490 nm, Tabla 10.
λ
(nm) DEC-MH DEC-MS INF-MS H2O MeOH EtOH 70% ACT 70%
• 405 669.85 ± 0.01 274.59 ± 0.01 526.95 ± 0.01 310.76 ± 0.01 326.40 ± 0.01 459.75 ± 0.01 368.80 ± 0.01
• 490 668.65 ± 0.01 275.27 ± 0.01 424.63 ± 0.01 275.10 ± 0.01 316.35 ± 0.01 492.15 ± 0.01 438.00 ± 0.01
•515 428.90 ± 0.01 154.12 ± 0.01 287.75 ± 0.01 192.27 ± 0.01 309.65 ± 0.01 391.25 ± 0.01 362.25 ± 0.01
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto obtenido por decocción a
partir de muestra seca. INF-MS, Extracto obtenido por infusión a partir de muestra seca. H2O-US, Extracto
acuoso obtenido por ultrasonido. MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US, Extracto
de etanol obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por ultrasonido al 70%. Los
datos expresados son el promedio de tres mediciones ± desviación estándar. De la fracción MeOH/H2O a 490 nm
entre los extractos H2O-US y MeOH-US no existe diferencia significativa (p<0.05). Se usó la prueba de Duncan
y un ANOVA para el análisis estadístico.
Figura 17. Flavonoides totales de la suma de fracciones en equivalentes de quercetina de las
hojas de J. spicigera.
0
100
200
300
400
500
600
700
DEC-MH DEC-MS INF-MS H2O-US MeOH-US EtOH-US ACT-US
mg
EQ
/10
0m
g
Extracto

48
Tabla 10. Correlación de Pearson entre fenoles y flavonoides.
Absorbancia
@ λ
Correlación
Fenoles/Flavonoides
nm (mg EAGa*100 g-1)/
(mg EQb*100 g-1)
(mg EAGa*100 g-1)/
(mg ECc*100 g-1)
405 0.77 0.61
490 0.95 0.96
515 0.84 0.84
aEAG, Equivalentes de ácido gálico. bEQ, Equivalentes de quercetina.
cEC, Equivalentes de catequina.
Sepúlveda-Jiménez et al. (2009), cuantificaron flavonoides totales a los extractos
macerados en agua y metanol de las flores, hojas y tallo de J. spicigera. Como se puede
observar en la Figura 18, el contenido de flavonoides totales en el extracto metanólico es
mayor en comparación con el extracto acuoso en ambos casos. Como en el caso para fenoles
totales la diferencia en dicho contenido se puede atribuir como ya se mencionó antes a el mes
de colecta, la edad y el entorno en el cual se encuentra la planta, la etapa de limpieza, el
método de secado y al método de extracción.
Figura 18. Contenido de flavonoides totales en hojas de J. spicigera.
0
100
200
300
400
500
600
700
800
900
1000
H2O MeOH
620.00 ± 0.13
980.00 ± 0.13
106.94 ± 0.003131.46 ± 0.003
mg
EC
/100g
Extracto
Sepúlveda-Jiménez et al., 2009.
Objeto de estudio para este
proyecto.

49
Sulaiman et al. (2011), cuantificaron flavonoides totales de la maceración del tallo de
J. gendarussa con diferentes disolventes en equivalentes de quercetina a 415 nm. Comparando
el contenido de flavonoides totales de J. gendarussa con J. spicigera se puede ver en la Figura
19, que todos los extractos J. spicigera poseen un mayor contenido de flavonoides totales. Así
como también en ambos casos se sigue la misma tendencia, el extracto de etanol al 70% posee
mayor contenido de flavonoides, seguido por el extracto de acetona al 70%, el extracto de
metanol y por último es extracto acuoso. Esta diferencia en el contenido de flavonoides se
debe al tipo de extracción, así como también de la parte de la planta estudiada y el disolvente
empleado.
Figura 19. Contenido de flavonoides totales en equivalentes de quercetina.
Cabe resaltar, como ya se mencionó anteriormente, que el tipo de extracción es
determinante debido a que dependiendo de la extracción el contenido de flavonoides puede
variar, como se pude observar en la Figura 20, la extracción asistida por ultrasonido es más
eficaz que la maceración, los valores reportados para J. spicigera son superiores a los
reportados para J. gendarussa.
Para una longitud de onda de 490 nm el extracto acuoso de la decocción de la hoja fresca
posee el mayor contenido de flavonoides totales con 251.15 ± 0.01 mg EC/100 g, seguido por
0
50
100
150
200
250
300
350
400
450
500
H2O MeOH EtOH 70% ACT 70%
310.76 ± 0.01 326.4 ± 0.01
459.75 ± 0.01
368.85 ± 0.01
10 ± 0.0
110 ± 0.1
210 ± 0.3
120 ± 0.0
mg E
Q/1
00g
Extracto
J. spicigera
J. gendarussa
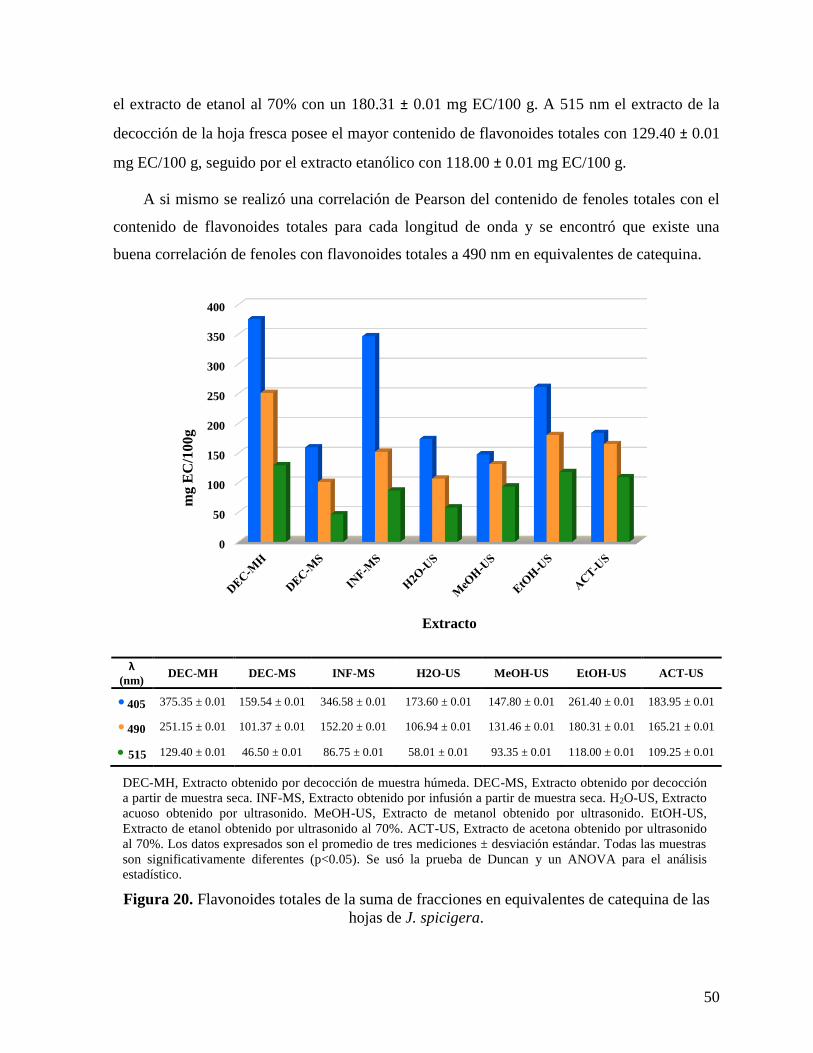
50
el extracto de etanol al 70% con un 180.31 ± 0.01 mg EC/100 g. A 515 nm el extracto de la
decocción de la hoja fresca posee el mayor contenido de flavonoides totales con 129.40 ± 0.01
mg EC/100 g, seguido por el extracto etanólico con 118.00 ± 0.01 mg EC/100 g.
A si mismo se realizó una correlación de Pearson del contenido de fenoles totales con el
contenido de flavonoides totales para cada longitud de onda y se encontró que existe una
buena correlación de fenoles con flavonoides totales a 490 nm en equivalentes de catequina.
λ
(nm) DEC-MH DEC-MS INF-MS H2O-US MeOH-US EtOH-US ACT-US
• 405 375.35 ± 0.01 159.54 ± 0.01 346.58 ± 0.01 173.60 ± 0.01 147.80 ± 0.01 261.40 ± 0.01 183.95 ± 0.01
• 490 251.15 ± 0.01 101.37 ± 0.01 152.20 ± 0.01 106.94 ± 0.01 131.46 ± 0.01 180.31 ± 0.01 165.21 ± 0.01
• 515 129.40 ± 0.01 46.50 ± 0.01 86.75 ± 0.01 58.01 ± 0.01 93.35 ± 0.01 118.00 ± 0.01 109.25 ± 0.01
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto obtenido por decocción
a partir de muestra seca. INF-MS, Extracto obtenido por infusión a partir de muestra seca. H2O-US, Extracto
acuoso obtenido por ultrasonido. MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US,
Extracto de etanol obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por ultrasonido
al 70%. Los datos expresados son el promedio de tres mediciones ± desviación estándar. Todas las muestras
son significativamente diferentes (p<0.05). Se usó la prueba de Duncan y un ANOVA para el análisis
estadístico.
Figura 20. Flavonoides totales de la suma de fracciones en equivalentes de catequina de las
hojas de J. spicigera.
0
50
100
150
200
250
300
350
400
mg E
C/1
00g
Extracto

51
7.5. Evaluación de la actividad antioxidante
Se evaluó la actividad antioxidante de los extractos frente al radical DPPH•. Como se
puede observar en la Figura 21, en general la fracción acuosa posee una mayor actividad
antioxidante en comparación de la fracción MeOH/H2O para los extractos de la hoja fresca, de
la decocción de la hoja seca, metanólico y el etanólico al 70%. La fracción acuosa del extracto
de la hoja fresca (EFD1) posee un IC50 415.00 ± 11.18 ppm, esto indica que tiene una mejor
actividad antioxidante en comparación con los otros extractos. El extracto de etanol al 70%,
fracción acuosa (ETOH1), tiene un IC50 de 1115.00 ± 3.43 ppm, este extracto posee una mejor
actividad antioxidante en comparación con los extractos obtenidos a partir del polvo
deshidratado. Sin embargo la fracción acuosa de la infusión del polvo de la hoja deshidratada
posee un IC50 de 9370.00 ± 503.59 ppm y la fracción de MeOH/H2O del extracto de metanol
presenta un IC50 de 12812.00 ± 88.36 ppm, estos valores indican que las fracciones de estos
extractos tienen una baja actividad antioxidante en comparación con los otras fracciones de los
diferentes extractos, por tal razón no se muestran en la Figura 21.
En la Figura 21, se puede observar que el tipo de disolvente influye en la actividad
antioxidante de un extracto, así como también el tipo de extracción puede afectar a la actividad
antioxidante del extracto. Comparando los valores de IC50 de la decocción de la hoja fresca
con los valores de la infusión y decocción de la muestra deshidratada se puede decir que las
fracciones de la hoja fresca poseen una mayor actividad antioxidante que las fracciones de los
extractos antes mencionados. De los extractos obtenidos por ultrasonido el extracto que
presento mayor actividad antioxidante fue el de etanol al 70%, se puede decir que el tipo de
disolvente influye en la actividad antioxidante de una fracción de un extracto.

52
EFD1 415.00 ± 11.18 DEC1 1375.00 ± 28.85 ETOH1 1115.00 ± 3.43
EFD2 1336.00 ± 22.80 DEC2 2264.00 ± 93.40 ETOH2 1528.00 ± 55.99
INF2 2567.00 ± 86.22 H2O US 3869.00 ± 113.76 ACET1 1314.00 ± 52.04
MEOH1 1195.00 ± 21.88 H2O US 1571.00 ± 70.76 ACET2 1262.00 ± 22.56
EFD1, Extracto obtenido por decocción de muestra húmeda, fracción acuosa. EFD2, Extracto obtenido por decocción de
muestra húmeda, fracción MeOH/H2O. DEC1, Extracto obtenido por decocción a partir de muestra seca, fracción acuosa.
DEC2, Extracto obtenido por decocción a partir de muestra seca, fracción MeOH/H2O. INF2, Extracto obtenido por infusión a
partir de muestra seca, fracción MeOH/H2O. H2O US1, Extracto acuoso obtenido por ultrasonido, fracción acuosa. H2O US2,
Extracto acuoso obtenido por ultrasonido, fracción MeOH/H2O. MeOH1, Extracto de metanol obtenido por ultrasonido,
fracción acuosa. EtOH1, Extracto de etanol obtenido por ultrasonido al 70%, fracción acuosa. EtOH2, Extracto de etanol
obtenido por ultrasonido al 70%, fracción MeOH/H2O. ACET1, Extracto de acetona obtenido por ultrasonido al 70%, fracción
acuosa. ACET2, Extracto de acetona obtenido por ultrasonido al 70%, fracción MeOH/H2O. Los datos expresados son el
promedio de tres mediciones ± desviación estándar. Todas las muestras son significativamente diferentes
(p<0.05). Se usó la prueba de Duncan y un ANOVA para el análisis estadístico.
Figura 21. IC50 de las fracciones de los extractos de J. spicigera.
Se llevó a cabo la cinética para cada fracción de los extractos de J. spicigera. A partir
de los datos de las curvas de cinética, se calculó la primera derivada para conocer el TEC50. El
TEC50 de la fracción acuosa del extracto de la decocción de la hoja fresca fue de 35 min, Figura
22. El TEC50 de la fracción acuosa del extracto de etanol al 70% fue de 18 min, Figura 23.
0
500
1000
1500
2000
2500
3000
3500
4000
IC5
0(p
pm
)
Extracto
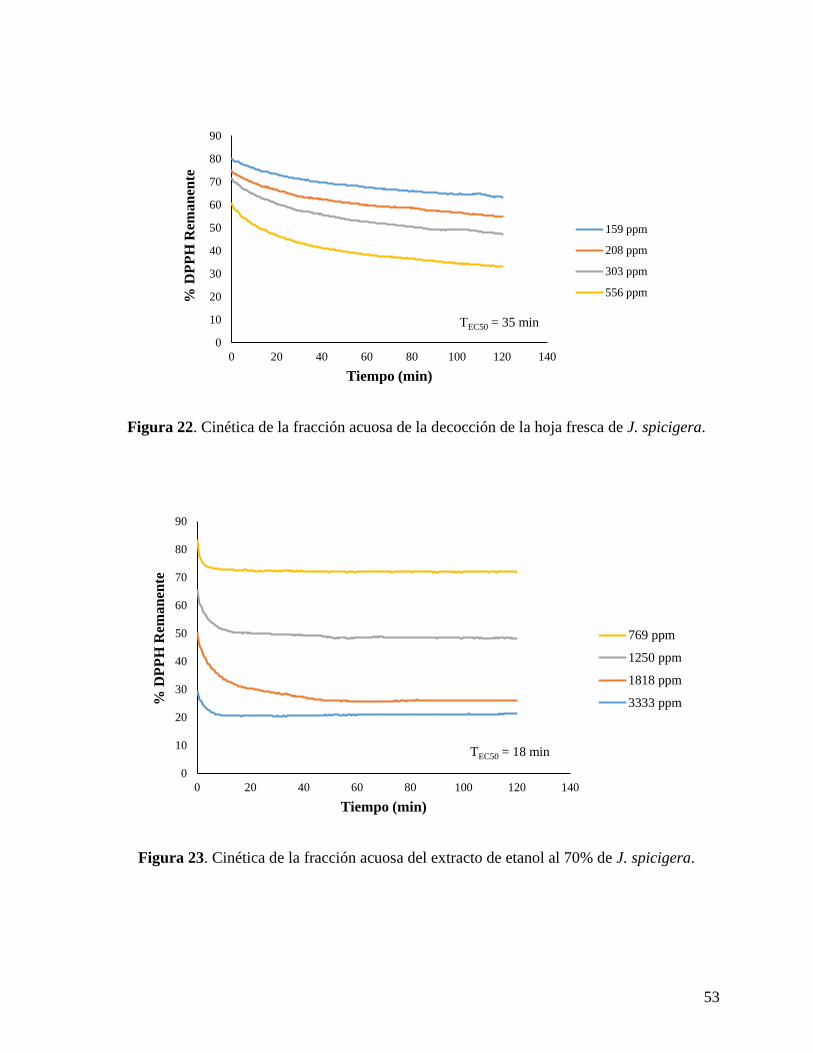
53
Figura 22. Cinética de la fracción acuosa de la decocción de la hoja fresca de J. spicigera.
Figura 23. Cinética de la fracción acuosa del extracto de etanol al 70% de J. spicigera.
0
10
20
30
40
50
60
70
80
90
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 35 min
159 ppm
208 ppm
303 ppm
556 ppm
0
10
20
30
40
50
60
70
80
90
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 18 min
769 ppm
1250 ppm
1818 ppm
3333 ppm

54
En la Tabla 11 se muestra el TEC50 para cada una de las fracciones, así como su
eficiencia antirradicalar expresada en kg de DPPH•/g muestra seca*min. Como se puede
observar en la tabla, la fracción acuosa obtenida de la SPE del extracto H2O asistido por
ultrasonido (H2O US1) presenta el menor TEC50, esto indica que el extracto reacciona
rápidamente frente al radical DPPH•. En general los extractos presentan tiempos pequeños esto
indica que reaccionan rápidamente frente a un radical, la capacidad antioxidante está
relacionada con su contribución potencial en la prevención de enfermedades de diversa
naturaleza; teniendo gran importancia su determinación en la eficiencia antirradicalar la cual
indica que tan efectivo es un extracto.
En la Tabla 11, se puede observar que a pesar de que un extracto posea un tiempo corto
no necesariamente indica que tenga una buena eficiencia antirradicalar, los extractos EFD1 y
ETOH1 muestran una mayor eficiencia antirradicalar en comparación con los otros extractos.
Sin embargo estos dos extractos reaccionan de manera diferente siendo más rápidos el extracto
ETOH1. El extracto de INF1 muestra una baja eficiencia antirradicalar así como también
reacciona de manera más lenta si se compara con las fracciones de los demás extractos. En
general se puede decir que las fracciones acuosas de los diferentes extractos son más eficientes
que las fracciones de MeOH/H2O, esto se pude atribuir a la diferente composición de
compuestos fenólicos presentes en ambas fracciones.

55
Tabla 11. TEC50 y eficiencia antirradicalar (AE) de los extractos de J. spicigera.
Extracto EC50
(kg m s/kg DPPH•)
TEC50
(min)
AE
(kg DPPH•/ kg m s *min)
EFD1 20.75 35 1.38
EFD2 66.80 22 0.68
DEC1 68.75 15 0.97
DEC2 113.20 33 0.27
INF1 468.50 46 0.05
INF2 128.35 20 0.39
H2O US1 193.45 11 0.47
H2O US2 78.55 22 0.58
MEOH1 59.75 18 0.93
MEOH2 640.60 16 0.1
ETOH1 55.75 18 1.00
ETOH2 76.40 28 0.47
ACET1 65.70 23 0.66
ACET2 63.10 28 0.57
BHT 1.42 83 0.01
EFD1, Extracto obtenido por decocción de muestra húmeda, fracción acuosa. EFD2, Extracto obtenido
por decocción de muestra húmeda, fracción MeOH/H2O. DEC1, Extracto obtenido por decocción a partir
de muestra seca, fracción acuosa. DEC2, Extracto obtenido por decocción a partir de muestra seca,
fracción MeOH/H2O. INF1, Extracto obtenido por infusión a partir de muestra seca, fracción acuosa.
INF2, Extracto obtenido por infusión a partir de muestra seca, fracción MeOH/H2O. H2O US1, Extracto
acuoso obtenido por ultrasonido, fracción acuosa. H2O US2, Extracto acuoso obtenido por ultrasonido,
fracción MeOH/H2O. MeOH1, Extracto de metanol obtenido por ultrasonido, fracción acuosa. MeOH2,
Extracto de metanol obtenido por ultrasonido, fracción MeOH/H2O. EtOH1, Extracto de etanol obtenido
por ultrasonido al 70%, fracción acuosa. EtOH2, Extracto de etanol obtenido por ultrasonido al 70%,
fracción MeOH/H2O. ACET1, Extracto de acetona obtenido por ultrasonido al 70%, fracción acuosa.
ACET2, Extracto de acetona obtenido por ultrasonido al 70%, fracción MeOH/H2O.

56
8. REFERENCIAS
Andrade-Cetto, A. & Heinrich, M. 2005. Mexican plants with hypoglycaemic effect used in
the treatment of diabetes. Journal of Ethnopharmacology, 99, 325-348.
Azmir, J.; Zaidul, I. S. M.; Rahman, M. M.; Sharif, K. M.; Mohamed, A.; Sahena, F.; Jahurul,
M. H. A.; Ghafoor, K.; Norulaini, N. A. N.; Omar, A. K. M. 2013. Techniques for
extraction of bioactive compounds from plat materials: A review. Journal of Food
Engineering, 117, 426-436.
Basha, S. K. & Kumari, V. S. 2012. In vitro antidiabetic activity of psidium guajava leaves
extracts. Asian Pacific Journal of Tropical Disease, 98,100.
Brusotti, G.; Cesari, I.; Dentamaro, A.; Caccialanza, G.; Massolini, G. 2014. Isolation and
characterization of biactive compounds from plants resources: The role of analysis in the
ethnopharmacological approach. Journal of Pharmaceutical and Biomedical Analysis, 87,
218-228.
Cáceres-Cortés, J. R.; Cantú-Garza, F. A.; Mendoza-Mata, M. T.; Chávez-González, M. A.;
Ramos- Mandujano, G.; Zambrano-Ramírez, A. R. 2001. Cytotoxic Activity of Justicia
spicigera is Inhibited by bcl-2 Proto-onocgene and Induces Apoptosis in a Cell Cycle
Dependent Fashion. Phytotherapy Research, 15, 691-697.
Chatterjee, K.; Monjur, A. K.; De, D.; Kumar, P. D.; Ghosh, D. 2012. Antidiabetic and
antioxidative activity of ethyl acetate fraction of hydromethanolic extract of seed of
Eugenia jambolana Linn through in vivo and in vitro study and its chromatographic
purification. Free Radicals and Antioxidants, 2, 21-30.
Corrêa, G. M. & Alcântara, A. F. 2012. Chemical constituents and biological activities of
species of Justicia – a review. Brazilian Journal of Pharmacognosy, 22 (1), 220-238.

57
Cruz-Gallegos, J. A.; Salas-Coronado, R.; Valadez-Blanco, R. 2012. Relación flavonoides
totales-actividad antidiabética (in vitro por difusion de glucosa) en extractos de Colubrina
elliptica. Tesis, Universidad Tecnológica de la Mixteca.
Dong, H. Q.; Li, M.; Zhu, F.; Liu, F. L.; Huang, J. B. 2012. Inhibitory potencial of trilobatin
from Lithocarpus polystachyus Rehd against α-glucosidase and α-amylase linked to type
2 diabetes. Food Chemistry, 130, 262-266.
Eom, S.H.; Lee, S. H.; Yoon, N. Y.; Jung, W. K.; Jeon, Y. J.; Kim, S. K.; Lee, M. S.; Kim, Y.
M. 2012. α-Glucosidase- and α-amylase-inhibitory activities of phlorotannins
from Eisenia bicyclis. Journal of Science of Food Agriculture, 92, 2084–2090.
Gallagher, A.; Flatt, P.; Duffy, G.; Abdel-Wahab, Y. 2003. The effects of traditional
antidiabetic plants on in vitro glucose difussion. Nutrition Research. 23, 413-424.
Ghasemzadeh, A. & Ghasemzadeh, N. 2011. Flavonoids and phenolic acids: Role and
biochemical activity in plants and human. Journal of Medicinal Plants Research, 5 (31),
6697-6703.
Gupta, R. K.; Kumar, D.; Chaudhary, A. K.; Maithani, M.; Singh, R. 2012. Antidiabetic
activity of Passiflora incarnata Linn. in streptozotocin-induced diabetes in mice. Journal
of Ethnopharmacology, 801-806.
Harborne J. B. 1998. Phytochemical methods a guide to modern techniques of plants analysis.
United Kingdom. Springer. pp. 302.
Harris, D. C. 2007. Análisis químico cuantitativo. Tercera Edición. Reverte. pp. 744.
He, C.; Long, Y.; Pan, J.; Li, K.; Liu, F. 2007. Application of moleculary imprinted polymers
to solid-phase extraction of analytes from real samples. Journal of Biochemical and
Biophysical Methods, 70, 133-150.
Hemalatha, S.; Patel, D. K.; Kumar, R.; Laloo, D. 2012. Diabetes mellitus: An overview on its
pharmacological aspects and reported medicinal plants having antidiabetic activity. Asian
Pacific Journal of Tropical Biomedicine, 411-420.
Heneczkowski, M.; Kopacz, M.; Nowak, D.; Kuzniar, A. 2001. Infrared spectrum analysis of
some flavonoids. Acta Poloniae Pharmaceutica-Drug Research, 58 (6), 415-420.

58
Ignat, I.; Volf, I.; Popa, I. V. 2011. A critical review of methods for characterisation of
polyphenolic compounds in fruits and vegetables. Food Chemistry, 126, 1821-1835.
Julián-Loaeza, A. P.; Santos-Sánchez, N. F.; Valadez-Blanco, R.; Sánchez-Guzmán, B. S.;
Salas-Coronado, R. 2011. Chemical composition, color, and antioxidant activity of three
varieties of Annona diversifolia Safford fruits. Industrial Crops and Products, 34 (2),
1262-1268.
Kadir, M. F.; Sayeed, M. S. B.; Shams, T.; Mia, M. M. K. 2012. Ethnobotanical survey of
medicinal plants used by Bangladeshi traditional health practitioners in the management
of diabetes mellitus. Journal of Ethnopharmacology, 144, 605-611.
Khalil, N. M.; Pepato, M., Brunetti, I. L. 2008. Free radical scavenging profile and
myeloperoxidase inhibition of extracts from antidiabetic plants: Bauhinia forficata and
Cissus sicyoides. Biological Research, 41, 165-171.
Kirca, A. & Arslan, E. 2008. Antioxidant capacity and total phenolic content of selected plants
from Turkey. International Journal of Food Science and Technology, 43, 2038-2046.
Krishnaiah, D.; Sarbatly, R.; Nithyanandam, R. 2011. A review of the antioxidant potentional
of medicinal plant species. Food and bioproducts processing, 89, 217-233.
Kumar, P. P. & Doble, M. 2011. Mechanism of Action of Natural Products Used in the
Treatment of Diabetes Mellitus. Chinese Journal of Integrative Medicine, 17 (8), 563-
574.
Kumar, K. S.; Vijayan, V.; Bhaskar, S.; Krishnan, K.; Shalini, V.; Helen, A. 2012. Anti-
inflammatory potential of an ethyl acetate fraction isolated from Justicia gendarussa
roots through inhibition of iNOS and COX-2 expression via NF-κB pathway. Cellular
immunology, 272(2), 283–9.
Li, H.; Chen, B.; Yao, S. 2005. Application of ultrasonic technique for extracting chlorogenic
acid from Eucommia ulmodies Oliv. (E. ulmodies). Ultrasonics Sonochemistry, 12 (4),
295–300.
Li, Y.; Li, C.; Xu, Q.; Kang, W. 2011. Antioxidant, α-glucosidase inhibitory activities in vitro
and alloxan-induced diabetic rats’ protective effect of Indigofera stachyodes Lindl. root.
Journal of Medicinal Plants Research, 5 (14), 3321-3328.

59
Lizcano, L. J.; Bakkali, F.; Ruiz-Larrea, M. B.; Ruiz-Sanz, J. I. 2010. Antioxidant activity and
polyphenol content of aqueous extracts from Colombian Amazonian plants with
medicinal use. Food Chemistry, 119, 1566-1570.
Nickavar, B. & Yousefian, N. 2011. Evaluation of α-amylase inhibitory activities of selected
antidiabétic medicinal plants. Journal für Verbraucherschutz und Lebensmittelsicherheit,
6, 191-195.
Ortiz-Andrade, R.; Cabañas-Wuan, A.; Arana-Argáez, V. E.; Alonso-Castro, A. J.; Zapata-
Bustos, R.; Salazar-Olivo, L. A.; Domínguez, F.; Chávez, M.; Carranza-Álvarez, C.;
García-Carrancá, A. 2012. Antidiabetic effects of Justicia spicigera Schltdl
(Acanthaceae). Journal of Ethnopharmacology, 143 (2), 455-462.
Patel, D. K.; Prasad, S. K.; Kumar, R.; Hemalatha, S. 2012. An overview on antidabetic
medicinal plants having insulin mimetic property. Asian Pacific Journal Tropical
Biomedicine, 230-330.
Ramkumar, K. M.; Thayumanavan, B.; Palvannan, T.; Rajaguru, P. 2010. Inhibitory effect of
Gymnema montanum leaves on α-glucosidase activity and α-amylase activity and their
relationship with polyphenolic content. Medicinal Chemistry Research, 19, 948-961.
Reddy, S. N. V. L.; Anarthe, S.; Raghavendra. 2010. In vitro Antioxidant and Antidiabetic
activity of Asystasia gangetica (Chinese Violet) Linn. (Acanthaceae). International
Journal of Research in Pharmaceutical and Biomedical Sciences, 1 (2), 72-75.
Rodríguez Vaquero, M. J.; Tomassini Serravalle, L. R.; Manca de Nadra, M. C.; Strasser de
Saad, A. M. 2010. Antioxidant capacity and antibacterial activity of phenolic compounds
from argentinean herbs infusions. Food Control, 21 (5), 779–785.
Sadiki, M.; Martin, N. 2012. Solid-pashe extraction and procedure for determination of
phenolic compounds in maple syrup. Food Analytical Methods, 6, 737-744.
Sepúlveda-Jiménez, G.; Reyna-Aquino, C.; Chaires-Martínez, L.; Bermúdez-Torres, K.;
Rodríguez-Monroy, M. 2009. Antioxidant activity and content of phenolic compounds
and flavonoids from Justicia spicigera. Journal of Biological Sciences, 9 (6), 629-632.

60
Sulaiman, S. F.; Sajak, B. A. A.; Ooi, L. K.; Supriatno, Seow, A. M. 2011. Effect of solvents
in extracting polyphenols and antioxidants of selected raw vegetables. Journal of Food
Composition and Analysis, 24, 506-515.
Smeds, I. A.; Eklund, P. C.; Willför, S. M. 2012. Content, composition, and stereochemical
characterization of lignans in berries and seeds. Food Chemistry, 134, 1991-1998.
Sowemimo, A. A.; Adio, O.; Fageyinbo, S. 2011. Anticonvulsant activity of the methanolic
extract of Justicia extensa T. Anders. Journal of ethnopharmacology, 138 (3), 697–9.
Thorat, K.; Patil, L.; Limaye, D.; Kadam, V. 2012. In vitro Models for Antidiabetic Activity
Assessment. International Journal of Research in Pharmaceutical and Biochemical
Sciences, 3 (2), 730-733.
Walpole, R. E.; Myers, R. H.; Myers, S. L. 1999. Probabilidad y estadística para ingenieros.
Prentince-Hall. pp. 752.
Wang, Y.; Huang, S.; Shao, S.; Qian, L.; Xu, P. 2012. Studies on bioactivities of tea (Camellia
sinensis L.) fruit peel extracts: Antioxidant activity and inhibitory potencial against α-
glucosidase and α-amylase in vitro. Industrial Crops and Products, 37, 520-526.
Wojdylo, A.; Oszmianski, J.; Czemerys, R. 2007. Antioxidant activity and phenolic
compounds in 32 selected herbs. Food Chemistry, 105, 940-949.
Won Lee, K.; Jun Kim, Y.; Joo Lee, H.; Yong Lee, C. 2003. Cocoa has more phenolic
phytochemicals and higher antioxidant capacity than teas and red wine. Journal of
Agriculture and Food Chemistry, 51, 7292-7295.

61
9. ANEXOS
A 1. Relación entre el mecanismo de acción de fármacos y plantas.
Fármaco Mecanismo de acción Planta
Metformina Disminuyen la gluconeogénesis y
glicogénesis.
Psidium guajava
(Basha et al., 2012)
Glipizida
Glibenclamida
Glyvurida
Reducen los niveles de glucosa en sangre
por regeneración de células β.
Estimulan la secreción de insulina de
células β.
Passiflora incarnata
(Gupta et al., 2012)
Prandin
Starlix
Reducen los niveles de glucosa en sangre
por regeneración de células β.
Estimulan la secreción de insulina de
células β.
Passiflora incarnata (Gupta
et al., 2012)
Precose
Glyset
Reducen la absorción de glucosa
intestinal.
Inhiben a la α-amilasa y α-glucosidasa.
G. sylvestre, G. inodorum,
G. yunnanense
(Ramkumar et al., 2010)
Camellia sinensis
(Wang et al., 2012)
Asystasia gangetica
(Reddy et al., 2010)
Amaranthus viridis
(Kumar et al., 2012)
Indigofera stachyodes
(Li et al., 2011)
Januvia
Inician la liberación de insulina.
Estimulan la síntesis y/o liberación de
insulina.
Passiflora incarnata
(Gupta et al., 2012)
Actos
Avandia
Mimetizan a la insulina.
Disminuyen los niveles de glucosa en
sangre.
Incrementan la utilización de la glucosa
periférica.
Passiflora incarnata
(Gupta et al., 2012)
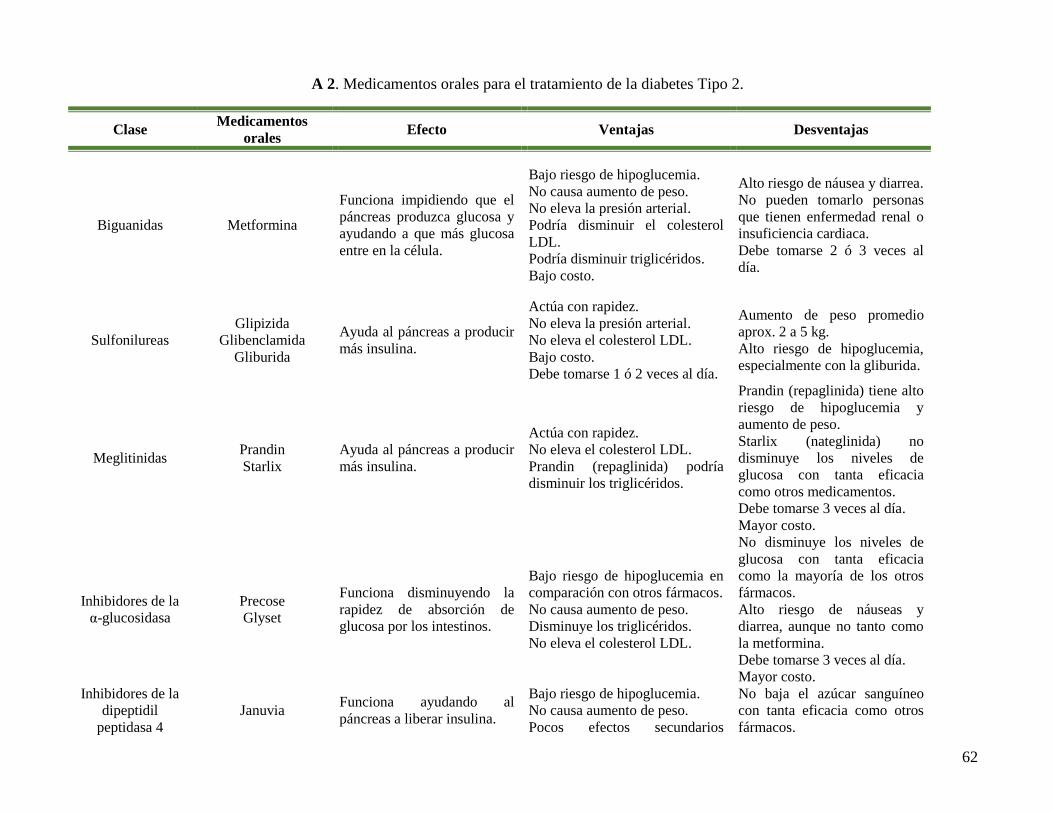
62
A 2. Medicamentos orales para el tratamiento de la diabetes Tipo 2.
Clase Medicamentos
orales Efecto Ventajas Desventajas
Biguanidas Metformina
Funciona impidiendo que el
páncreas produzca glucosa y
ayudando a que más glucosa
entre en la célula.
Bajo riesgo de hipoglucemia.
No causa aumento de peso.
No eleva la presión arterial.
Podría disminuir el colesterol
LDL.
Podría disminuir triglicéridos.
Bajo costo.
Alto riesgo de náusea y diarrea.
No pueden tomarlo personas
que tienen enfermedad renal o
insuficiencia cardiaca.
Debe tomarse 2 ó 3 veces al
día.
Sulfonilureas
Glipizida
Glibenclamida
Gliburida
Ayuda al páncreas a producir
más insulina.
Actúa con rapidez.
No eleva la presión arterial.
No eleva el colesterol LDL.
Bajo costo.
Debe tomarse 1 ó 2 veces al día.
Aumento de peso promedio
aprox. 2 a 5 kg.
Alto riesgo de hipoglucemia,
especialmente con la gliburida.
Meglitinidas Prandin
Starlix
Ayuda al páncreas a producir
más insulina.
Actúa con rapidez.
No eleva el colesterol LDL.
Prandin (repaglinida) podría
disminuir los triglicéridos.
Prandin (repaglinida) tiene alto
riesgo de hipoglucemia y
aumento de peso.
Starlix (nateglinida) no
disminuye los niveles de
glucosa con tanta eficacia
como otros medicamentos.
Debe tomarse 3 veces al día.
Mayor costo.
Inhibidores de la
α-glucosidasa
Precose
Glyset
Funciona disminuyendo la
rapidez de absorción de
glucosa por los intestinos.
Bajo riesgo de hipoglucemia en
comparación con otros fármacos.
No causa aumento de peso.
Disminuye los triglicéridos.
No eleva el colesterol LDL.
No disminuye los niveles de
glucosa con tanta eficacia
como la mayoría de los otros
fármacos.
Alto riesgo de náuseas y
diarrea, aunque no tanto como
la metformina.
Debe tomarse 3 veces al día.
Mayor costo.
Inhibidores de la
dipeptidil
peptidasa 4
Januvia Funciona ayudando al
páncreas a liberar insulina.
Bajo riesgo de hipoglucemia.
No causa aumento de peso.
Pocos efectos secundarios
No baja el azúcar sanguíneo
con tanta eficacia como otros
fármacos.

63
conocidos.
Debe tomarse 2 veces al día.
Es nuevo se sabe poco sobre
los posibles efectos
secundarios.
Es posible que funcione sólo
como un segundo
medicamento.
Mayor costo.
Tiazolidinedionas Actos
Avandia
Funciona ayudando a las
células a utilizar la glucosa.
Bajo riesgo de hipoglucemia.
Puede elevar ligeramente el
colesterol HDL.
Actos (pioglitazona) podría
disminuir los triglicéridos.
Debe tomarse 1 ó 2 veces al día.
Mayor riesgo de insuficiencia
cardiaca.
Aumento de peso promedio
aprox. 2 a 5 kg.
Mayor riesgo de edema
(retención de líquidos).
Mayor riesgo de anemia.
Eleva el colesterol LDL.
Avandia aumenta los
triglicéridos y podría aumentar
el riesgo de un ataque al
corazón.
Raro riesgo de causar
problemas del hígado.
No actúa con rapidez.
Mayor costo.
El A 2 se tomó parcialmente de Consumer Reports (Best Buy Drugs), http://www.consumerreports.org/health/resources/pdf/best-
buy-drugs/DbtesSp5Jan112008final.pdf (Accesado Febrero 2013).

64
10. APÉNDICES
AP 1. Fenoles totales en extractos crudos de las hojas de J. spicigera.
Extracto mg EAG/100g
DEC-MH 767.04 ± 0.01a
DEC-MS 509.91 ± 0.01b
INF-MS 422.74 ± 0.01c
H2O-US 220.72 ± 0.01d
MeOH-US 492.41 ± 0.01e
EtOH-US 618.62 ± 0.01f
ACT-US 610.81 ± 0.01g
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-
MS, Extracto obtenido por decocción a partir de muestra seca. INF-MS,
Extracto obtenido por infusión a partir de muestra seca. H2O-US,
Extracto acuoso obtenido por ultrasonido. MeOH-US, Extracto de
metanol obtenido por ultrasonido. EtOH-US, Extracto de etanol obtenido
por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por
ultrasonido al 70%. Los datos expresados son el promedio de tres
mediciones ± desviación estándar. Todas las muestras son
significativamente diferentes (p<0.05). Se usó la prueba de Duncan y un
ANOVA para el análisis estadístico.

65
Fenoles totales extractos limpios de las hojas de J. spicigera.
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto
obtenido por decocción a partir de muestra seca. INF-MS, Extracto obtenido por
infusión a partir de muestra seca. H2O-US, Extracto acuoso obtenido por ultrasonido.
MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US, Extracto de etanol
obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por
ultrasonido al 70%. Los datos expresados son el promedio de tres mediciones ±
desviación estándar. Todas las muestras son significativamente diferentes (p<0.05). Se
usó la prueba de Duncan y un ANOVA para el análisis estadístico.
Extracto
mg EAG/100g
Fracción
acuosa
Fracción
MeOH/H20
Suma de
fracciones
DEC-MH 114.20 ± 0.01ª 573.00 ± 0.02h 687.20 ± 0.02º
DEC-MS 83.70 ± 0.01b 108.60 ± 0.01i 192.30 ± 0.01p
INF-MS 192.09 ± 0.01c 39.97 ± 0.01j 232.06 ± 0.02q
H2O-US 132.40 ± 0.01d 45.65 ± 0.01k 178.05 ± 0.01r
MeOH-US 93.50 ± 0.01e 142.55 ± 0.01l 236.05 ± 0.01s
EtOH-US 308.00 ± 0.01f 178.05 ± 0.01m 486.05 ± 0.01t
ACT-US 153.05 ± 0.01g 219.60 ± 0.01n 372.65 ± 0.02u

66
AP 2. Flavonoides totales en equivalentes de catequina.
Extracto
λ = 405 nm λ = 490 nm λ = 515 nm
Fracción
acuosa
Fracción
MeOH/H2O
Fracción
acuosa
Fracción
MeOH/H2O
Fracción
acuosa
Fracción
MeOH/H2O
DEC-MH 141.35 ±
0.01
234.00 ±
0.01
66.45 ±
0.01a
184.70 ±
0.01
35.70 ±
0.01b
93.70 ±
0.01
DEC-MS 109.54 ±
0.01 50.00 ± 0.01
64.67 ±
0.01a 36.70 ± 0.01
29.27 ±
0.01
17.23 ±
0.01
INF-MS 315.45 ±
0.01 31.13 ± 0.01
125.29 ±
0.01 26.91 ± 0.01
74.12 ±
0.01
12.63 ±
0.01
H2O-US 74.20 ±
0.01 99.40 ± 0.01c
26.38 ±
0.01 80.56 ± 0.01d
15.66 ±
0.01
42.35 ±
0.01e
MeOH-US 88.25 ±
0.01 59.55 ± 0.01
64.10 ±
0.01a 67.36 ± 0.01
49.45 ±
0.01
43.90 ±
0.01e
EtOH-US 159.55 ±
0.01
101.85 ±
0.01c
100.45 ±
0.01 79.86 ± 0.01d
62.70 ±
0.01
55.30 ±
0.01
ACT-US 104.65 ±
0.01 79.30 ± 0.01
63.20 ±
0.01a
102.01 ±
0.01
38.60 ±
0.01b
70.65 ±
0.01
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto obtenido por decocción a partir
de muestra seca. INF-MS, Extracto obtenido por infusión a partir de muestra seca. H2O-US, Extracto acuoso
obtenido por ultrasonido. MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US, Extracto de etanol
obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por ultrasonido al 70%. Los datos
expresados son el promedio de tres mediciones ± desviación estándar. De la fracción MeOH/H2O a 405 nm los
extractos H2O-US y EtOH-US no existe diferencia significativa (p<0.05). En la fracción acuosa a 490 nm entre los
extractos DEC-MH, DEC-MS, MeOH-US y ACT-US no existe diferencia significativa (p<0.05). De la fracción
MeOH/H2O a 490 nm entre los extractos H2O-US y EtOH-US no existe diferencia significativa (p<0.05). En la
fracción acuosa a 515 nm entre los extractos DEC-MH y ACT-US no existe diferencia significativa (p<0.05). De la
fracción MeOH/H2O a 515 nm entre los extractos H2O-US y MeOH-US no existe diferencia significativa (p<0.05).
Se usó la prueba de Duncan y un ANOVA para el análisis estadístico.

67
Suma de las dos fracciones obtenidos después de la limpieza del extracto (eluato acuoso y
eluato MeOH/H2O).
Extracto λ = 405 nm λ = 490 nm λ = 515 nm
DEC-MH 375.35 ± 0.01 251.15 ± 0.01 129.40 ± 0.01
DEC-MS 159.54 ± 0.01 101.37 ± 0.01 46.50 ± 0.01
INF-MS 346.58 ± 0.01 152.20 ± 0.01 86.75 ± 0.01
H2O-US 173.60 ± 0.01 106.94 ± 0.01 58.01 ± 0.01
MeOH-US 147.80 ± 0.01 131.46 ± 0.01 93.35 ± 0.01
EtOH-US 261.40 ± 0.01 180.31 ± 0.01 118.00 ± 0.01
ACT-US 183.95 ± 0.01 165.21 ± 0.01 109.25 ± 0.01
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto
obtenido por decocción a partir de muestra seca. INF-MS, Extracto obtenido por
infusión a partir de muestra seca. H2O-US, Extracto acuoso obtenido por ultrasonido.
MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US, Extracto de etanol
obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por
ultrasonido al 70%.Los datos expresados son el promedio de tres mediciones ±
desviación estándar. Todas las muestras son significativamente diferentes (p<0.05). Se
usó la prueba de Duncan y un ANOVA para el análisis estadístico.

68
AP 3. Flavonoides totales en equivalentes de quercetina.
Extracto
λ = 405 nm λ = 490 nm λ = 515 nm
Fracción
acuosa
Fracción
MeOH/H2O
Fracción
acuosa
Fracción
MeOH/H2O
Fracción
acuosa
Fracción
MeOH/H2O
DEC-MH 242.35 ± 0.01 427.50 ±
0.01
160.65 ±
0.01
508.00 ±
0.01
118.40 ±
0.01
310.50 ±
0.01
DEC-MS 194.06 ± 0.01 80.53 ± 0.01 165.56 ±
0.01
109.71 ±
0.01
96.98 ±
0.01 57.14 ± 0.01
INF-MS 468.58 ± 0.01 58.37 ± 0.01 350.16 ±
0.01
74.47 ±
0.01
245.89 ±
0.01 41.86 ± 0.01
H2O-US 117.66 ± 0.01 193.10 ±
0.01a
63.60 ±
0.01
211.50 ±
0.01b
51.92 ±
0.01
140.35 ±
0.01
MeOH-US 180.05 ± 0.01 146.35 ±
0.01
152.35 ±
0.01
164.00 ±
0.01
164.05 ±
0.01
145.60 ±
0.01
EtOH-US 263.75 ± 0.01 196.00 ±
0.01a
283.15 ±
0.01
209.00 ±
0.01b
207.90 ±
0.01
183.35 ±
0.01
ACT-US 199.25 ± 0.01 169.55 ±
0.01
149.00 ±
0.01
289.00 ±
0.01
128.05 ±
0.01
234.20 ±
0.01
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto obtenido por decocción a partir
de muestra seca. INF-MS, Extracto obtenido por infusión a partir de muestra seca. H2O-US, Extracto acuoso
obtenido por ultrasonido. MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US, Extracto de etanol
obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por ultrasonido al 70%.Los datos
expresados son el promedio de tres mediciones ± desviación estándar. De la fracción MeOH/H2O a 405 nm los
extractos H2O-US y EtOH-US no existe diferencia significativa (p<0.05). De la fracción MeOH/H2O a 490 nm entre
los extractos H2O-US y EtOH-US no existe diferencia significativa (p<0.05). Se usó la prueba de Duncan y un
ANOVA para el análisis estadístico.

69
Suma de las dos fracciones obtenidos después de la limpieza del extracto (eluato acuoso y
eluato MeOH/H2O).
Extracto λ = 405 nm λ = 490 nm λ = 515 nm
DEC-MH 669.85 ± 0.01 668.65 ± 0.01 428.90 ± 0.01
DEC-MS 274.59 ± 0.01 275.27 ± 0.01a 154.12 ± 0.01
INF-MS 526.95 ± 0.01 424.63 ± 0.01 287.75 ± 0.01
H2O-US 310.76 ± 0.01 275.10 ± 0.01a 192.27 ± 0.01
MeOH-US 326.40 ± 0.01 316.35 ± 0.01 309.65 ± 0.01
EtOH-US 459.75 ± 0.01 492.15 ± 0.01 391.25 ± 0.01
ACT-US 368.80 ± 0.01 438.00 ± 0.01 362.25 ± 0.01
DEC-MH, Extracto obtenido por decocción de muestra húmeda. DEC-MS, Extracto
obtenido por decocción a partir de muestra seca. INF-MS, Extracto obtenido por
infusión a partir de muestra seca. H2O-US, Extracto acuoso obtenido por ultrasonido.
MeOH-US, Extracto de metanol obtenido por ultrasonido. EtOH-US, Extracto de etanol
obtenido por ultrasonido al 70%. ACT-US, Extracto de acetona obtenido por
ultrasonido al 70%.Los datos expresados son el promedio de tres mediciones ±
desviación estándar. De la fracción MeOH/H2O a 490 nm entre los extractos H2O-US y
MeOH-US no existe diferencia significativa (p<0.05). Se usó la prueba de Duncan y un
ANOVA para el análisis estadístico.

70
AP 4. Curva de calibración en equivalentes de ácido gálico para la cuantificación de fenoles totales.
y = 0.0152x - 0.0009
R² = 0.9956
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.45
0 5 10 15 20 25 30
Ab
s
ppm
y = 0.0124x + 0.0011
R² = 0.9963
0.000
0.050
0.100
0.150
0.200
0.250
0.300
0.350
0 5 10 15 20 25 30
Ab
s
ppm

71
AP 5. Curva de calibración en equivalentes de (+) - catequina para la cuantificación de flavonoides
totales.
Curva a una longitud de onda de 405 nm.
Curva a una longitud de 490 nm.
y = 0.0054x - 0.013
R² = 0.9813
0
0.05
0.1
0.15
0.2
0.25
0.3
0 10 20 30 40 50 60
Ab
s
ppm
y = 0.0072x - 0.025
R² = 0.9848
0.000
0.050
0.100
0.150
0.200
0.250
0.300
0.350
0.400
0 10 20 30 40 50 60
Ab
s
ppm

72
Curva a una longitud de onda de 515 nm.
y = 0.0063x
R² = 0.9824
0.000
0.050
0.100
0.150
0.200
0.250
0.300
0.350
0.400
0 10 20 30 40 50 60
Ab
s
ppm

73
AP 6. Curva de calibración en equivalentes de quercetina para la cuantificación de flavonoides totales.
Curva a una longitud de onda de 405 nm.
Curva a una longitud de onda de 490 nm.
y = 0.0046x - 0.0833
R² = 0.9962
0.000
0.050
0.100
0.150
0.200
0.250
0.300
0.350
0.400
0.450
0 20 40 60 80 100 120
Ab
s
ppm
y = 0.002x + 0.0064
R² = 0.9737
0.000
0.050
0.100
0.150
0.200
0.250
0 20 40 60 80 100 120
Ab
s
ppm

74
Curva a una longitud de onda de 515 nm.
y = 0.0019x
R² = 0.9935
0.000
0.020
0.040
0.060
0.080
0.100
0.120
0.140
0.160
0.180
0.200
0 20 40 60 80 100 120
Ab
s
ppm

75
AP 7. Curva de calibración en equivalentes de ácido gálico para DPPH•.
y = 27.012x + 2.381
R² = 0.9983
IC50 = 1.76
0
10
20
30
40
50
60
0 0.5 1 1.5 2 2.5
% I
nh
ibic
ión
ppm

76
AP 8. IC50 y TEC50 del BHT para el ensayo del DPPH•.
y = -0.0152x2 + 2.0072x + 5.228
R² = 0.9968
IC50 = 28.43 ± 0.104
0
10
20
30
40
50
60
70
80
0 10 20 30 40 50 60
% I
nh
ibib
icio
n
ppm
0
20
40
60
80
100
120
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 83 min
10 ppm
28.43 ppm
40 ppm

77
AP 9. IC50 y TEC50 de la decocción de la hoja fresca de J. spicigera, fracción acuosa para DPPH•.
y = 21.544ln(x) - 79.875
R² = 0.9942
IC50 = 415 ppm ± 11.175
0
10
20
30
40
50
60
0 100 200 300 400 500 600
% I
nh
ibic
ión
ppm
0
10
20
30
40
50
60
70
80
90
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 35 min
159 ppm
208 ppm
303 ppm
556 ppm

78
AP 10. IC50 y TEC50 de la decocción de la hoja fresca de J. spicigera, fracción MeOH/H2O para DPPH•.
y = -2E-05x2 + 0.0683x - 5.5432
R² = 1
IC50 = 1336 ± 22.799
0
10
20
30
40
50
60
70
80
90
0 1000 2000 3000 4000 5000 6000
% I
nh
ibic
ión
ppm
0
10
20
30
40
50
60
70
80
90
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 22 min
625 ppm
909 ppm
1667 ppm
5000 ppm

79
AP 11. IC50 y TEC50 de la decocción del polvo de la hoja de J. spicigera, fracción acuosa para DPPH•.
y = -2E-06x2 + 0.0295x + 13.366
R² = 0.9995
IC50 = 1375 ± 28.846
0
10
20
30
40
50
60
70
80
0 500 1000 1500 2000 2500 3000
% I
nh
ibic
ión
ppm
0
10
20
30
40
50
60
70
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 15 min
959 ppm
1207 ppm
1657 ppm
2500 ppm

80
AP 12. IC50 y TEC50 de la decocción del polvo de la hoja de J. spicigera, fracción MeOH/H2O para
DPPH•.
y = 7E-06x2 - 0.0049x + 25.238
R² = 1
IC50 = 2264 ± 93.404
0
10
20
30
40
50
60
70
0 500 1000 1500 2000 2500 3000
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100 120 140
% D
PP
H R
ema
nen
te
Tiempo (min)
TEC50 = 33 min
1522 ppm
1784 ppm
1944 ppm
2692 ppm

81
AP 13. IC50 y TEC50 de la infusión del polvo de la hoja de J. spicigera, fracción acuosa para DPPH•.
y = -1E-07x2 + 0.0056x + 6.3131
R² = 0.9996
IC50 = 9370 ± 503.596
0
10
20
30
40
50
60
0 2000 4000 6000 8000 10000 12000 14000
% I
nh
ibic
ión
ppm
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 46 min
2500 ppm
3889 ppm
8750 ppm
12727 ppm

82
AP 14. IC50 y TEC50 de la infusión del polvo de la hoja de J. spicigera, fracción MeOH/H2O para
DPPH•.
y = 3E-06x2 + 0.0123x - 1.2167
R² = 0.9996
IC50 = 2567 ± 86.215
0
10
20
30
40
50
60
0 500 1000 1500 2000 2500 3000
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 20 min
1522 ppm
1944 ppm
2295 ppm
2692 ppm

83
AP 15. IC50 y TEC50 del extracto H2O por ultrasonido del polvo de la hoja de J. spicigera, fracción
acuosa para DPPH•.
y = -2E-06x2 + 0.0248x - 16.016
R² = 0.9997
IC50 = 3869 ± 113.757
0
10
20
30
40
50
60
70
80
90
0 2000 4000 6000 8000 10000
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
ppm
TEC50 = 11 min
8333 ppm
4545 ppm
3125 ppm
1923 ppm

84
AP 16. IC50 y TEC50 del extracto H2O por ultrasonido del polvo de la hoja de J. spicigera, fracción
MeOH/H2O para DPPH•.
y = -9E-06x2 + 0.0515x - 8.6826
R² = 1
IC50 = 1571 ± 70.760
0
10
20
30
40
50
60
70
80
0 2000 4000 6000 8000 10000 12000
% I
nh
ibic
ión
ppm
0
10
20
30
40
50
60
70
80
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 22 min
1250 ppm
1818 ppm
3333 ppm
10000 ppm

85
AP 17. IC50 y TEC50 del extracto MeOH por ultrasonido del polvo de la hoja de J. spicigera, fracción
acuosa para DPPH•.
y = -3E-05x2 + 0.1315x - 64.311
R² = 1
IC50 = 1195 ± 21.882
0
10
20
30
40
50
60
70
80
90
0 500 1000 1500 2000 2500 3000 3500
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 18 min
769 ppm
1818 ppm
1250 ppm
3333 ppm

86
AP 18. IC50 y TEC50 del extracto MeOH por ultrasonido del polvo de la hoja de J. spicigera, fracción
MeOH/H2O para DPPH•.
y = -5E-07x2 + 0.0185x - 104.95
R² = 1
IC50 = 12812 ± 88.356
0
10
20
30
40
50
60
70
0 2000 4000 6000 8000 10000 12000 14000 16000 18000
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
ppm
TEC50 = 16 min
2500 ppm
10000 ppm
13333 ppm
16000 ppm

87
AP 19. IC50 y TEC50 del extracto EtOH 70% por ultrasonido del polvo de J. spicigera, fracción acuosa
para DPPH•.
y = -1E-05x2 + 0.0774x - 23.865
R² = 0.9995
IC50 = 1115 ± 3.427
0
10
20
30
40
50
60
70
80
90
0 500 1000 1500 2000 2500 3000 3500
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
90
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 18 min
769 ppm
1250 ppm
1818 ppm
3333 ppm

88
AP 20. IC50 y TEC50 del extracto EtOH 70% por ultrasonido del polvo de J. spicigera, fracción
MeOH/H2O para DPPH•.
y = -2E-06x2 + 0.0335x + 3.5498
R² = 0.9919
IC50 = 1528 ± 55.998
0
10
20
30
40
50
60
70
80
0 500 1000 1500 2000 2500 3000
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 28 min
556 ppm
769 ppm
1667 ppm
2500 ppm

89
AP 21. IC50 y TEC50 del extracto acetona 70% por ultrasonido del polvo de J. spicigera, fracción acuosa
para DPPH•.
y = -7E-06x2 + 0.0464x + 1.1262
R² = 0.9986
IC50 = 1314 ± 52.038
0
10
20
30
40
50
60
70
80
90
0 500 1000 1500 2000 2500 3000 3500
% I
nh
ibic
ón
ppm
0
10
20
30
40
50
60
70
80
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 23 min
769 ppm
1250 ppm
1818 ppm
3333 ppm
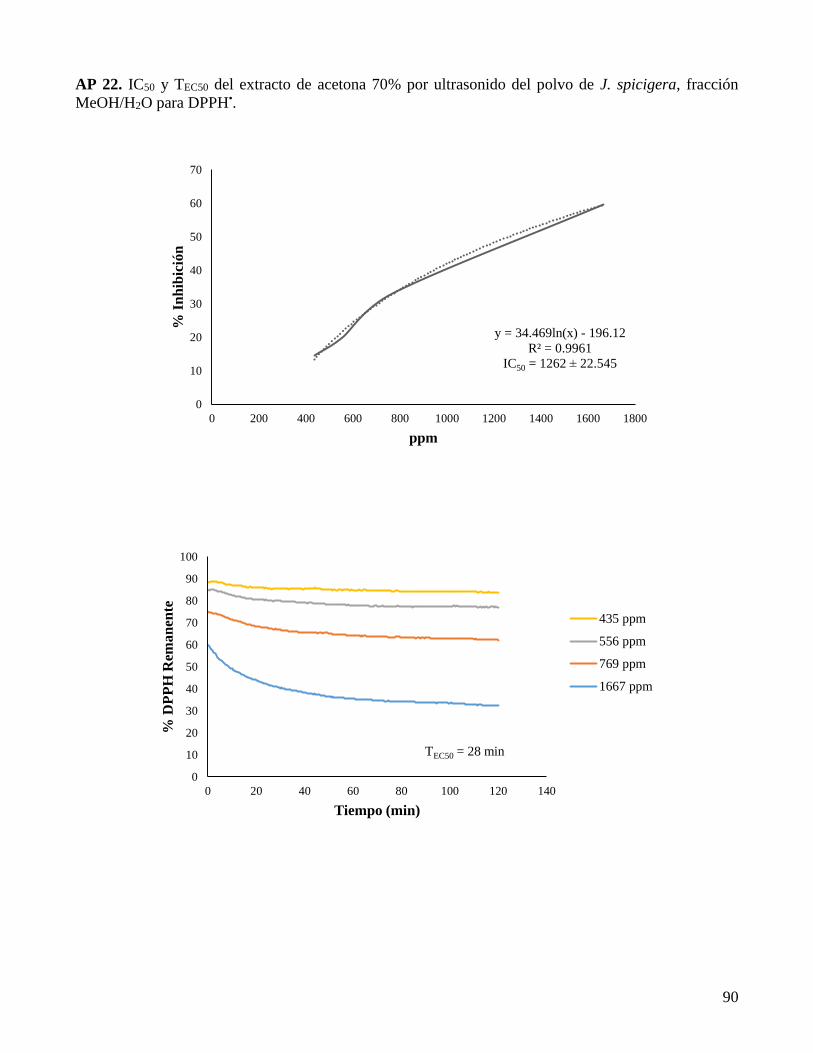
90
AP 22. IC50 y TEC50 del extracto de acetona 70% por ultrasonido del polvo de J. spicigera, fracción
MeOH/H2O para DPPH•.
y = 34.469ln(x) - 196.12
R² = 0.9961
IC50 = 1262 ± 22.545
0
10
20
30
40
50
60
70
0 200 400 600 800 1000 1200 1400 1600 1800
% I
nh
ibic
ión
ppm
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100 120 140
% D
PP
H R
eman
ente
Tiempo (min)
TEC50 = 28 min
435 ppm
556 ppm
769 ppm
1667 ppm