estudio de prevalencia de la enfermedad de fabry dialisis.pdf · comunicación del protocolo ......
TRANSCRIPT

Estudio de prevalencia de la
ENFERMEDAD DE FABRY en pacientes en diálisis
CONFIDENCIAL: Este documento contiene información confidencial que no debe ser divulgada o publicada a otras personas distintas de los investigadores clínicos, miembros de los Comités de Ética de Investigación Clínica de los centros participantes y Autoridades Sanitarias Españolas.


• 3 •
ÍNDICE
1. Resumen .........................................................................................................................................................5
2. Información general ..................................................................................................................................62.1. Identificación del estudio ..................................................................................................................6
3. Justificación, hipótesis y objetivos......................................................................................................63.1. Introduccion ...........................................................................................................................................63.2. Objetivos del estudio ..........................................................................................................................8
4. Diseño del estudio .....................................................................................................................................84.1. Tipo de estudio .....................................................................................................................................84.2. Investigadores ........................................................................................................................................84.3. Selección de la muestra ....................................................................................................................84.4. Tamaño de la muestra .......................................................................................................................84.5. Periodos de inclusión y seguimiento ............................................................................................8
5. Población del estudio ...............................................................................................................................85.1. Criterios de inclusión y exclusión ...................................................................................................9
6. Desarrollo del estudio ..............................................................................................................................96.1. Variables del estudio ...........................................................................................................................96.2. Descripción de las visitas del estudio ...........................................................................................9
7. Aspectos éticos .........................................................................................................................................107.1. Disposiciones legales vigentes .....................................................................................................107.2. Información del paciente ................................................................................................................107.3. Confidencialidad de los datos del paciente .............................................................................107.4. Comunicación del protocolo .........................................................................................................11
8. Consideraciones prácticas ...................................................................................................................118.1. Monitorización ....................................................................................................................................118.2. Recogida y archivo de la documentación del estudio ........................................................11
9. Consideraciones estadísticas y plan de análisis .......................................................................119.1. Gestión de datos ...............................................................................................................................119.2. Análisis estadístico ............................................................................................................................11
10. Bibliografía ..................................................................................................................................................12
11. Anexos...........................................................................................................................................................1411.1. Anexo 1: Instrucciones de recogida de muestras ..............................................................1411.2. Anexo 2: Consentimientos informados ..................................................................................1611.3. Anexo 3: Cuaderno de recogida de datos ............................................................................25

• 5 •
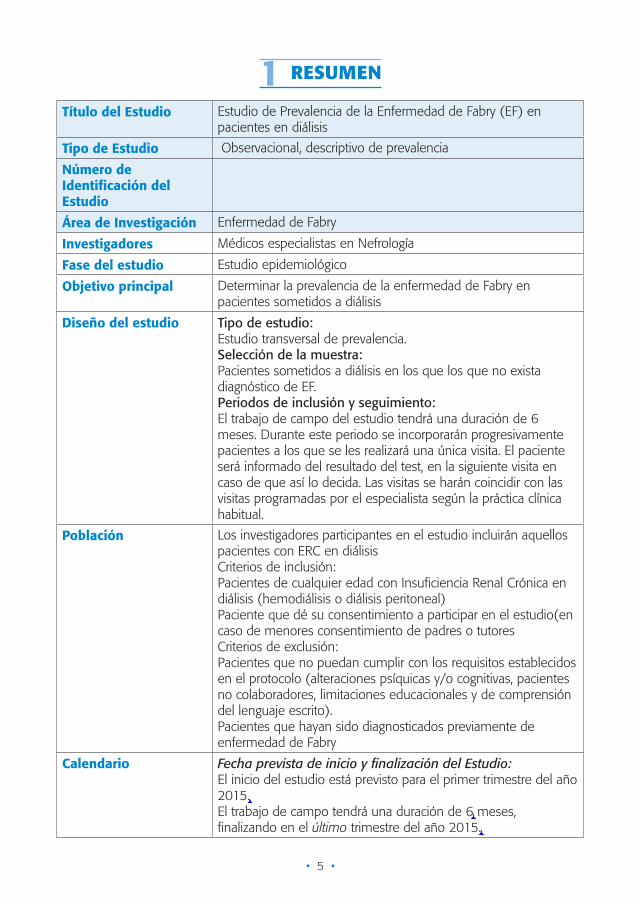
1 RESUMEN
Título del Estudio Estudio de Prevalencia de la Enfermedad de Fabry (EF) en pacientes en diálisis
Tipo de Estudio Observacional, descriptivo de prevalencia
Número de Identificación del Estudio
Área de Investigación Enfermedad de Fabry
Investigadores Médicos especialistas en Nefrología
Fase del estudio Estudio epidemiológico
Objetivo principal Determinar la prevalencia de la enfermedad de Fabry en pacientes sometidos a diálisis
Diseño del estudio Tipo de estudio:Estudio transversal de prevalencia.Selección de la muestra:Pacientes sometidos a diálisis en los que los que no exista diagnóstico de EF.Periodos de inclusión y seguimiento:El trabajo de campo del estudio tendrá una duración de 6 meses. Durante este periodo se incorporarán progresivamente pacientes a los que se les realizará una única visita. El paciente será informado del resultado del test, en la siguiente visita en caso de que así lo decida. Las visitas se harán coincidir con las visitas programadas por el especialista según la práctica clínica habitual.
Población Los investigadores participantes en el estudio incluirán aquellos pacientes con ERC en diálisisCriterios de inclusión:Pacientes de cualquier edad con Insuficiencia Renal Crónica en diálisis (hemodiálisis o diálisis peritoneal)Paciente que dé su consentimiento a participar en el estudio(en caso de menores consentimiento de padres o tutoresCriterios de exclusión:Pacientes que no puedan cumplir con los requisitos establecidos en el protocolo (alteraciones psíquicas y/o cognitivas, pacientes no colaboradores, limitaciones educacionales y de comprensión del lenguaje escrito).Pacientes que hayan sido diagnosticados previamente de enfermedad de Fabry
Calendario Fecha prevista de inicio y finalización del Estudio:El inicio del estudio está previsto para el primer trimestre del año 2015.El trabajo de campo tendrá una duración de 6 meses, finalizando en el último trimestre del año 2015.

• 6 •
2 INFORMACIÓN GENERAL
2.1. Identificación del estudio
2.1.1. Título del estudioEstudio de Prevalencia de la Enfermedad de Fabry en pacientes en diálisis
2.1.2. Tipo de estudioEstudio observacional, descriptivo de prevalencia
3 JUSTIFICACIÓN, HIPÓTESIS Y OBJETIVOS
3.1. IntroducciónLa enfermedad de Fabry es una enfermedad lisosomal que se caracteriza por el déficit de la
enzima α-galactosidasa A (α-GAL A) lo que origina depósito de glicoesfingolípidos principalmen-te globotriaosilceramida (Gb3), en el endotelio vascular y otros tejidos. Es una enfermedad pro-gresiva que causa manifestaciones a nivel renal, cardiaco, neurológico, cutáneo, ocular y de otros sistemas. La forma clásica o severa de la enfermedad es originada por la ausencia de actividad de la enzima (< 1%), mientras que defectos parciales (1-30%) producen formas incompletas con afectación predominantemente cardiaca y/o renal(1,2).
Se considera una enfermedad rara, que afecta a 1 de entre 40.000 a 238.000 varones(2-4). Sin embargo en los estudios realizados mediante cribado neonatal se ha observado una inciden-cia mayor de entre 1/ 3.100 a 1/4.100 nacidos vivos para las formas de comienzo tardío y de 1 cada 37.000 para los fenotipos clásico(5,6). Obviamente la frecuencia es mayor en la poblaciones de alto riesgo como en la afectada por Enfermedad renal crónica (ERC) que requiere hemodiá-lisis o entre los afectados de hipertrofia ventricular izquierda (HVI) o ictus(7).
La primera causa de muerte en pacientes con EF se debe a fallo renal y ocurre muy tempra-namente, en la cuarta y quinta década de la vida, particularmente en los varones(8,9).
La proteinuria es un signo temprano de la afectación renal de la EF está presente en el mo-mento del diagnóstico de la EF en el 44-54% de los varones y en el 33-41% de las mujeres(10) y constituye la manifestación más frecuente de la enfermedad(11,12), de hecho es un factor de riesgo independiente del grado y afectación renal en pacientes con o sin tratamiento y determina el éxito del mismo(13-16).
La nefropatía de la EF puede tener distintos grados de severidad, pero el porcentaje de pro-gresión a ERC es muy similar al de patologías como la diabetes, lo que sugiere que los pacientes no tratados pueden evolucionar ERC estadio 5 a edad temprana (en torno a los 50 años)(17,18).
Se ha determinado un amplio rango de manifestaciones histológicas en la EF(19).Todas las células renales están afectadas en mayor o menor medida, incluso en aquellos
pacientes con filtrados glomerulares normales y mínima proteinuria(20). Es característica la va-cuolización de los podocitos y de las células epiteliales con expansión mesangial y progresiva glomeruloesclerosis(9,20-22), de hecho la infiltración de los podocitos juega un papel importante en la aparición de la proteinuria.
Los pacientes con EF y que tienen la función renal ya afectada tienen un pronóstico más desfavorable que aquellos que no la tienen(23).
• 7 •
El tratamiento específico de la EF es la terapia de reemplazo enzimático (TES) que sustituye la enzima deficitaria que caracteriza la enfermedad, además se complementa con terapias dirigi-das al tratamiento de los distintos órganos afectados.
Distintos estudios sugieren que el tratamiento precoz con la TES permite estabilizar la progre-sión de la enfermedad renal y que este es más efectivo cuando la afectación renal está menos avanzada(24-26).
La mayoría de los estudios de cribado realizados para determinar la EF en pacientes con nefropatía se han llevado a cabo en poblaciones sometidas a hemodiálisis, algunos incluyeron también pacientes en diálisis peritoneal.
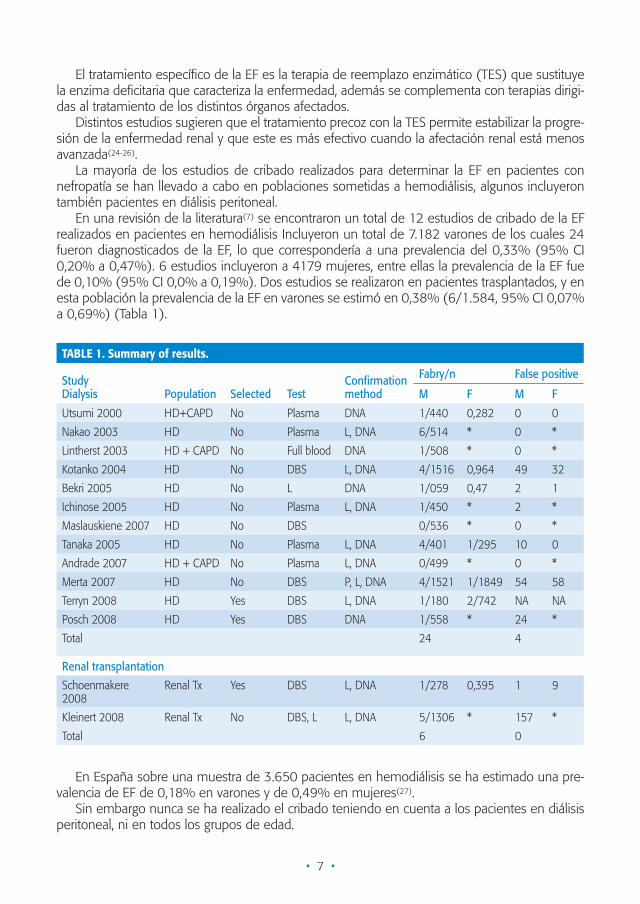
En una revisión de la literatura(7) se encontraron un total de 12 estudios de cribado de la EF realizados en pacientes en hemodiálisis Incluyeron un total de 7.182 varones de los cuales 24 fueron diagnosticados de la EF, lo que correspondería a una prevalencia del 0,33% (95% CI 0,20% a 0,47%). 6 estudios incluyeron a 4179 mujeres, entre ellas la prevalencia de la EF fue de 0,10% (95% CI 0,0% a 0,19%). Dos estudios se realizaron en pacientes trasplantados, y en esta población la prevalencia de la EF en varones se estimó en 0,38% (6/1.584, 95% CI 0,07% a 0,69%) (Tabla 1).
En España sobre una muestra de 3.650 pacientes en hemodiálisis se ha estimado una pre-valencia de EF de 0,18% en varones y de 0,49% en mujeres(27).
Sin embargo nunca se ha realizado el cribado teniendo en cuenta a los pacientes en diálisis peritoneal, ni en todos los grupos de edad.
TABLE 1. Summary of results.
StudyDialysis Population Selected Test
Confirmation method
Fabry/n False positive
M F M F
Utsumi 2000 HD+CAPD No Plasma DNA 1/440 0,282 0 0
Nakao 2003 HD No Plasma L, DNA 6/514 * 0 *
Lintherst 2003 HD + CAPD No Full blood DNA 1/508 * 0 *
Kotanko 2004 HD No DBS L, DNA 4/1516 0,964 49 32
Bekri 2005 HD No L DNA 1/059 0,47 2 1
Ichinose 2005 HD No Plasma L, DNA 1/450 * 2 *
Maslauskiene 2007 HD No DBS 0/536 * 0 *
Tanaka 2005 HD No Plasma L, DNA 4/401 1/295 10 0
Andrade 2007 HD + CAPD No Plasma L, DNA 0/499 * 0 *
Merta 2007 HD No DBS P, L, DNA 4/1521 1/1849 54 58
Terryn 2008 HD Yes DBS L, DNA 1/180 2/742 NA NA
Posch 2008 HD Yes DBS DNA 1/558 * 24 *
Total 24 4
Renal transplantation
Schoenmakere 2008
Renal Tx Yes DBS L, DNA 1/278 0,395 1 9
Kleinert 2008 Renal Tx No DBS, L L, DNA 5/1306 * 157 *
Total 6 0

• 8 •
Por tanto nos planteamos la realización de un cribado de la EF en la población en diálisis tanto hemodiálisis como en diálisis peritoneal, que permita la detección de los pacientes con Enfermedad de Fabry lo que permitiría evitar otras complicaciones secundarias y potencialmente mortales de la enfermedad además de poder realizar un diagnóstico precoz de sus familiares en riesgo.
3.2. Objetivos del estudio
3.2.1. Objetivo principalDeterminar la prevalencia de enfermedad de Fabry entre los pacientes en diálisis.
3.2.2. Objetivos secundarios• Analizar la relación entre la EF y el cuadro de base.• Valorar el tiempo de demora entre inicio de síntomas y diagnóstico de la EF.
4 DISEÑO DEL ESTUDIO
4.1. Tipo de estudioEstudio observacional, transversal de prevalencia
4.2. InvestigadoresNefrólogos que se adscriban al proyecto
4.3. Selección de la muestraSe incluirán aquellos pacientes en diálisis y que cumplan con los criterios de inclusión.
4.4. Tamaño de la muestraDado la baja prevalencia de la EF, se incluirán todos los pacientes que cumplan con los cri-
terios de evaluación durante el periodo de recogida y que den su consentimiento a participar en el estudio.
4.5. Periodos de inclusión y seguimientoEl trabajo de campo del estudio tendrá una duración de 12 meses. Durante este periodo se
incorporarán progresivamente pacientes que serán evaluados en una única visita. La visita se hará coincidir con las visitas programadas por el especialista según la práctica clínica habitual. El paciente será informado del resultado en la siguiente visita programada por su especialista en caso de que así lo decida.
5 POBLACIÓN DEL ESTUDIO
• Pacientes que acuden a unidades de diálisis y/o consultas de nefrología y que cumplan los criterios descritos a continuación. Los pacientes con EF ya diagnosticada serán inclui-dos para la estimación de la prevalencia.

• 9 •
5.1. Criterios de inclusión y exclusión
5.1.1. Criterios de inclusiónLos investigadores participantes en el estudio incluirán aquellos pacientes en diálisis.1. Pacientes de cualquier edad con insuficiencia renal Crónica en diálisis (hemodiálisis o
diálisis peritoneal).2. Paciente que dé su consentimiento a participar en el estudio(en caso de menores con-
sentimiento de padres o tutores.
5.1.2. Criterios de exclusión• Pacientes que no puedan cumplir con los requisitos establecidos en el protocolo (altera-
ciones psíquicas y/o cognitivas, pacientes no colaboradores, limitaciones educacionales y de comprensión del lenguaje escrito).
• Pacientes que hayan sido diagnosticados previamente de enfermedad de Fabry. • Pacientes que tengan realizada biopsia renal previa que haya descartado razonablemente
la enfermedad.
6 DESARROLLO DEL ESTUDIO
6.1. Variables del estudioVariables clínicas:• Variablessociodemográficas: en la visita basal se recogerá la edad, el género, la existen-
cia de antecedentes familiares de la patología de estudio y el tipo de parentesco. • Eventosclínicosdelaenfermedad: Se registrarán las complicaciones o afectaciones de
órganos o sistemas que presente el paciente• Diagnóstico: se anotará, en la visita basal, el diagnóstico de presunción, las fechas en que
la que el paciente comenzó con su patología, en la comenzó la diálisis y en la que acudió a consulta por primera vez.
• Enfermedadescrónicasconcomitantes: se registrarán aquellas enfermedades crónicas concomitantes no relacionadas con la EF que el paciente padezca.
6.2. Descripción de las visitas del estudioTodas las variables se recogerán en un cuaderno de recogida de datos (CRD) Se estima que el trabajo de campo es de 12 meses, a lo largo de los cuales se podrán ir
incluyendo nuevos pacientes. Durante estos 12 meses se realizarán a cada paciente las visitas programadas por el inves-
tigador para el seguimiento habitual de la enfermedad no interfiriendo en la práctica clínica habitual.
Los pacientes serán entrevistados por el médico que atiende habitualmente al paciente ase-gurando la confidencialidad y el rigor de la investigación.
A continuación se detallan las visitas del estudio:
Visita 1-BasalEn esta visita se explicará el objetivo principal del estudio y se pedirá el Consentimiento
Informado por escrito al paciente para participar en el estudio. En caso de ser menor de edad, el consentimiento del representante legal se pedirá además del asentimiento del paciente en

• 10 •
caso de ser mayor de 14 años. Posteriormente se comprobará que el paciente cumple con los criterios de evaluación.
Se recogerá en el CRD (Anexo 3): la fecha de la visita,y las siguientes variables:• Variables sociodemográficos: edad, género, antecedentes familiares y tipo de parentesco• Diagnóstico actual y fecha• Fecha de inicio de síntomas • Eventos clínicos de la enfermedad • Sintomatología del paciente• Enfermedades crónicas concomitantes• Test en gota de sangre seca y fecha del mismo
– DETERMINACIÓN ACTIVIDAD ENZIMÁTICA α-Galactosidasa A en gota de sangre seca DBS · Varones: se determinara el valor de actividad enzimática, en caso de resultado por
debajo del límite marxcado por el laboratorio de referencia, se procederá a estudio genético del gen GLA.
· Mujeres: independientemente del resultado de actividad enzimática, TODAS las mujeres pasarán a estudio genético del den GLA.
7 ASPECTOS ÉTICOS
7.1. Disposiciones legales vigentesEl estudio se llevará a cabo de acuerdo con los requerimientos éticos de las declaraciones de
Helsinki, para la investigación con seres humanos, las Guías de Buena Práctica Epidemiológicas de la ICH (International Conference of Harmonization) y la normativa existente en España.
Se trata de un estudio observacional de base poblacional. No existe intervención farmaco-lógica. De este modo, el presente estudio no es relevante para la evaluación de aspectos de seguridad de medicamentos. No se desarrollará ninguna evaluación de seguridad sobre pro-ductos farmacológicos (estudios post-autorización), ni se desarrollará ningún informe periódico de seguridad. Puesto que no se investiga ningún fármaco, no se necesita la aprobación de las autoridades competentes en temas de seguridad o eficacia de medicamentos.
7.2. Información del pacienteAntes de la inclusión de un paciente en el estudio, a través de la Hoja de Información al
Paciente y antes de conceder su consentimiento para participar en el mismo, se le informará al paciente acerca de los objetivos del estudio, la metodología a seguir durante el mismo, las molestias y riesgos, los posibles beneficios y la confidencialidad.
Los pacientes deberán ser informados sobre su derecho a poder retirarse del estudio en cualquier momento.
Se proporciona en el Anexo 2 un ejemplar de la Hoja de Información al Paciente y un ejem-plar del Consentimiento Informado del paciente
7.3. Confidencialidad de los datos del pacienteTodos los pacientes del estudio serán identificados mediante un código del paciente. El
investigador guardará un listado de identificación de los pacientes, que incluirá el número del paciente, el nombre y la dirección completa de todos los pacientes. Los datos serán tratados con absoluta confidencialidad según la Ley Orgánica de protección de datos de carácter personal.

• 11 •
7.4. Comunicación del protocoloEl estudio se ha comunicado al Comité Ético de Investigación Clínica (CEIC) del Hospital
Clinic de Barcelona que ha dado su aprobación.
8 CONSIDERACIONES PRÁCTICAS
8.1. MonitorizaciónLos Investigadores principales monitorizarán y supervisarán el estudio personalmente. Al ini-
cio del estudio se realizará una sesión con todos los investigadores con la finalidad de mostrar el funcionamiento del cuaderno de recogida de datos (CRD), así como de aclarar posibles dudas en el protocolo del estudio.
8.2. Recogida y archivo de la documentación del estudioEl investigador dispondrá, como material en papel del estudio, de un ejemplar del protocolo
y del CRD, así como copias del Consentimiento Informado del paciente. La recogida de datos se realizará mediante un Cuaderno de Recogida de Datos breve y fácil
de cumplimentar por el investigador.Estos datos pasarán automáticamente a una base de datos que será revisada por el equipo
investigador.El investigador será el responsable de mantener un archivo del estudio que contendrá los
datos de identificación del paciente, el protocolo y una copia de los CRD de cada paciente.
9 CONSIDERACIONES ESTADÍSTICAS Y PLAN DE ANÁLISIS
9.1. Gestión de datosLos datos cumplimentados por un investigador, tan solo serán visibles por él mismo, por
la persona encargada de la monitorización durante el trabajo de campo y por la persona que llevará a cabo el análisis de los datos. Todas las partes se comprometerán a guardar la máxima confidencialidad.
9.2. Análisis estadístico Se realizará análisis de datos en distintas fases del periodo de recogida de datos. Se realiza-
rá un análisis descriptivo del estado de la inclusión de los pacientes de forma trimestral, y a la finalización del mismo
9.2.1. Población para el análisis Cada uno de los análisis de datos se realizará con aquellos pacientes que hayan sido inclui-
dos en el estudio y que cumplan con los criterios de evaluación para el estudio.
9.2.2. Análisis Se realizará una descripción de las características sociodemográficas, el tiempo con diagnós-
tico y los eventos clínicos padecidos por la población de estudio durante el periodo de segui-miento. Para la descripción de variables continuas se utilizará la media y la desviación estándar

• 12 •
y para la descripción de variables categóricas el número y porcentaje de pacientes por categoría de respuesta. La descripción de las características de los pacientes se realizará para el total de la muestra de estudio
Para el análisis de los datos se utilizará el programa estadístico SPSS. En todas las pruebas estadísticas realizadas con las variables de resultados se utilizará un
nivel de significación estadística de 0,05. Se utilizarán técnicas estadísticas preliminares a la realización de las pruebas descritas en los apartados anteriores para asegurar el cumplimiento de los supuestos estadísticos. En el caso de que no se cumplan los supuestos establecidos se utilizarán pruebas equivalentes que no presenten dichas limitaciones, como por ejemplo prue-bas no paramétricas.
10 BIBLIOGRAFÍA
1. Meikle PJ, Hopwood JJ, Clague AE, et al. Prevalence of lysosomal strorage disordes. JAMA. 1999; 281: 249-54.
2. Desnick RJ, Brady R, Barraguer J, et al. Fabry disease, an under-recognized multisystemic disorder: Ex-pert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Inter Med. 2003; 138: 338-46.
3. Poorthuis BJ, Wevers RA, Keijer WJ, et al. The frequency of lysosomal storage diseases in the Nether-lands. Hum Genet. 1999; 105: 151-6.
4. Poupetová H, Ledvinová J, Berná L, Dvoráková L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis. 2010; 33: 387-96.
5. Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet. 2012; 379: 335-41.
6. Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by new-born screening. Am J Hum Genet. 2006; 79: 31-40.
7. Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations:a systematic review. J Med Genet. 2010; 47: 217-22.
8. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and manage-mentof multi-organ systeminvolvement. Genet Med. 2006; 8: 539-48.
9. Sessa A, Meroni M, Battini G, et al. Renal pathological changes in Fabry disease. J Inherit Metab Dis. 2001; 24(Suppl 2): 66-70.
10. Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 pa-tients in the Fabry OutcomeSurvey. Eur J Clin Invest. 2004; 34: 236-42.
11. Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ in-volvement: lessons from the Fabry Registry. Mol Genet Metab. 2008; 93: 112-28.
12. Tondel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008, 51: 767-76.
13. Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007; 18: 1547-57.
14. Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO. Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant. 2006; 21: 345-54.
15. Wanner C, Oliveira JP, Ortiz A, et al. Prognostic indicators of renal diseaseprogression in adults with Fabry disease: natural history data from the Fabry Registry. Clin J Am Soc Nephrol. 2010, 5: 2220-8.

• 13 •
16. Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a rand-omized trial. Ann Intern Med. 2007; 146: 77-86.
17. Branton MH, Schiffmann R, Sabnis SG, et al. Natural history of Fabry renal disease: influence of al-pha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002; 81: 122-38.
18. Schiffmann R, Warnock DG, Banikazemi M, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009; 24: 2102-11.
19. Faraggiana T, Churg J, Grishman E, et al. Light- and electron-microscopic histochemistry of Fabry’s dis-ease. Am J Pathol. 1981; 103: 247-62.
20. Gubler MC, Lenoir G, Grunfeld JP, Ulmann A, Droz D, Habib R. Early renal changes in hemizygous and heterozygous patients with Fabry’s disease. Kidney Int. 1978, 13: 223-35.
21. Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002; 13(Suppl 2): S134-8.
22. Fischer EG, Moore MJ, Lager DJ. Fabry disease: a morphologic study of 11 cases. Mod Pathol. 2006; 19: 1295-301.
23. Waldek S, Feriozzi S. Fabry nephropathy: a review - how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014; 15: 72.=
24. Breunig F, Weidemann F, Strotmann J, Knoll A, Wanner C. Clinical benefit of enzyme replacement ther-apy in Fabry disease. Kidney Int. 2006; 69: 1216-21.
25. Warnock DG, Ortiz A, Mauer M, et al. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant. 2012, 27: 1042-9.
26. Feriozzi S, Torras J, Cybulla M, Nicholls K, Sunder-Plassmann G, West M; FOS Investigators. The effec-tiveness of long-term agalsidase alfa therapy in the treatment of Fabry nephropathy. Clin J Am Soc Nephrol. 2012; 7: 60-9.
27. Herrera J, Miranda CS. Prevalence of Fabry’s disease within hemodialysis patients in Spain. Clin Neph-rol. 2014; 81: 112-20.
28. Terryn W, Cochat P, Froissart R, et al. Fabry nephropathy: indications for screening and guidance for di-agnosis and treatment by the European Renal Best Practice. Nephrol Dial Transplant. 2013; 28: 505-17.

• 14 •
11 ANEXOS
11.1. Anexo 1: Instrucciones de recogida de muestras

• 15 •

• 16 •
14.2. Anexo 2: Consentimientos informados
HOJA DE INFORMACIÓN AL PACIENTE MAYOR DE 18 AÑOS Y AUTORIZACIÓN para la participación en el estudio sobre:
EstudiodePrevalenciadelaEnfermedaddeFabry en pacientes en diálisis
Objetivos y antecedentes del estudioLos síntomas que UD presenta en este momento podrían ser compatibles con la Enferme-
dad de Fabry. La Enfermedad de Fabry se asocia con eventos renales.Esta enfermedad es muy poco frecuente y hereditaria. El origen está en un fallo en la des-
trucción de determinadas sustancias del organismo que se acumulan en diferentes órganos (el corazón, el riñón, el tubo digestivo o los nervios periféricos) y pueden dañarlos. A veces es detectada en edades tempranas pero lo habitual es que pase desapercibida hasta la aparición de una complicación. Dado que por su rareza (1 de cada 117.000 nacidos vivos) no se realizan pruebas para su diagnóstico de forma habitual, por ello se está promoviendo este estudio de detección en personas con problemas renales a fin de poder determinar de forma más exacta la prevalencia de esta enfermedad.
La investigación se desarrollará con el máximo respeto de los derechos individuales, según los postulados aceptados internacionalmente por las Naciones Unidas y la Comisión Europea Por ello, todas las investigaciones que se lleven a cabo contarán con la aprobación y supervisión de los diferentes Comités de Ética quienes velarán por el cumplimiento de los postulados ante-riormente citados.
Beneficios y compensacionesLa participación en el estudio no conlleva ningún beneficio económico para el sujeto partici-
pante en el estudio y no le supone coste alguno. Así mismo, le informamos que la inclusión en este estudio no conlleva riesgo alguno para
el sujeto participante. En el caso de que se le diagnosticara una enfermedad de Fabry, usted se beneficiaria de la posibilidad de un tratamiento precoz y eficaz para su enfermedad. Sin embar-go, le informamos que si así lo desea, tiene derecho a rechazar el conocer los resultados de las pruebas.
ProcedimientoPara poder realizar el estudio se necesita extraer una gota de sangre que impregne el papel
de una tarjeta, especialmente preparada para el estudio de la enfermedad de Fabry. En todos los pacientes, la extracción se hará coincidir con otra que precise para su tratamiento. La tarjeta será enviada al Laboratorio Centogene de Alemania
Con la muestra, se procederá a la determinación del déficit enzimático/análisis genético de la enfermedad sospechada por su médico.
ConfidencialidadLas bases de datos de trabajo no contendrán datos personales que permitan la identificación
de los participantes en el estudio. El nombre del sujeto participante se vinculará a un código for-mado por dos números, el primero corresponderá al Hospital y el segundo al sujeto participante.
La información será manejada de forma anónima por el investigador, de forma que el par-ticipante solamente podrá ser identificado por su médico, debiendo dirigirse al mismo para el

• 17 •
ejercicio de los derechos de acceso, rectificación, cancelación y oposición a sus datos de carácter personal.
Bajo ciertas circunstancias, el personal responsable de garantizar que la investigación se está haciendo adecuadamente podría revisar sus datos médicos (p.ej. datos recogidos en su historia médica). Los citados a continuación podrán tener acceso a estos datos: investigadores y coor-dinadores del estudio, autoridades reguladoras españolas, comités éticos. A todos ellos se les requiere asimismo mantener su identidad oculta y revisarán sus datos bajo secreto profesional.
Toda la información obtenida de los sujetos participantes será confidencial, cumpliendo en todo momento la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carác-ter Personal y el Real Decreto 1720/2007, de 21 de diciembre, que la desarrolla.
El uso que se haga de la información obtenida será confidencial. Por lo tanto, su identidad será siempre preservada. Igualmente los datos obtenidos solo podrán ser publicados de forma anónima y agregada, es decir, en forma de porcentajes o datos numéricos sin identificación del participante, y nunca de manera individual.
Las muestras de sangre serán destruidas tras la finalización del estudio.Usted tiene derecho a no ser informado de los resultados de los análisis realizados. Cuando
esta información sea necesaria para evitar un grave perjuicio, bien para usted o bien para sus familiares biológicos, se podrá informar a los afectados o a su representante legalmente autori-zado. En todo caso, la comunicación se limitará exclusivamente a los datos necesarios para estas finalidades.
Derecho a rehusar o a abandonarLa participación en este proyecto es enteramente voluntaria, pudiendo abandonar el estudio
cuando quiera, sin tener que dar explicación alguna y sin que esto repercuta en sus cuidados médicos. Por otra parte, si en algún momento desea retirar sus muestras del laboratorio, podrá hacerlo, salvo que dichas muestras hayan sido previamente disociadas y por lo tanto no sea posible la identificación del sujeto participante.
Para ello deberá ponerse en contacto con el Dr./Dra. ………………………………………… del Hospital ………………………………………….……………………………………………… en el teléfono de contacto ……………………………….

• 18 •
Firma del paciente Firma del médico
He leído y entiendo este consentimiento informa-do. Mis dudas han sido aclaradas. Voluntariamente consiento participar.
Apellidos: _________________________________
__________________________________________(mayúsculas)
Nombre: _________________________________
__________________________________________ (mayúsculas)
Fecha: ____ / ____ / ________DD/MM/AAAAA
Firma:
Reconozco que he explicado personalmente la na-turaleza, objetivos, duración, riesgos y beneficios potenciales del estudio al paciente y que él/ella ha otorgado hoy su consentimiento de forma libre y de carácter revocable.
Apellidos: _________________________________
__________________________________________(mayúsculas)
Nombre: _________________________________
__________________________________________ (mayúsculas)
Fecha: ____ / ____ / ________DD/MM/AAAAA
Firma:
CONSENTIMIENTO INFORMADO
He tenido suficiente tiempo para evaluar y comentar con mi médico mi inclusión, y he recibido una respuesta a todas mis preguntas. También he leído y acepto las condiciones que se explican en esta infor-mación. Recibiré una copia de esta hoja y el original se conservará junto con mi historia clínica para indicar mi participación en este estudio. Entiendo que mi participación en el estudio es voluntaria y que puedo negarme a participar o retirar mi consentimiento en cualquier momento.
La participación en el estudio dará lugar a un tratamiento de datos de salud. Usted autoriza expresamente que dichos datos se mantengan en el fichero, en tanto no solicite su cancelación y que sean tratados para la finalidad de la investigación objeto del proyecto. Usted podrá acceder, rectificar, cancelar, oponerse al trata-miento de los datos dirigiéndose al DDr.…………………………………………………………………………
Los datos recogidos en el estudio se conservarán de forma estrictamente confidencial. Acepto que el equipo médico, el personal designado por él así como los representantes tanto de las autoridades sanitarias como de los comités éticos, que están sujetos a secreto profesional tengan acceso a estos datos y a los documentos fuente solamente con objetivos relacionados con el estudio.
Consiento y autorizo que mi muestra de sangre sea enviada al Laboratorio Centogene en Alemania y sólo podrá ser utilizada para la determinación enzimática/ genética de la Enfermedad de Fabry, objeto del presente estudio, de forma que se pedirá permiso para cualquier otro tipo de análisis complementario en el caso de que fueran necesarios.
Consiento que las muestras puedan ir etiquetadas con mi código de participación en el estudio y que solamente mi médico pueda asociar dicho código con mis datos de carácter personal y así poder conocer mi identidad.
Deseo r, no deseo r (marque con una X su opción) que se me comuniquen los resultados de los análisis y de las pruebas a través del Dr.…………………………………………………………………………

• 19 •
HOJA DE INFORMACIÓN PARA PADRES O TUTORES DEL PACIENTE Y AUTORIZACIÓN para la participación en el estudio sobre:
EstudiodePrevalenciadelaEnfermedaddeFabry en pacientes en diálisis
Objetivos y antecedentes del estudioLos síntomas que su hijo/tutorando presenta en este momento podrían ser compatibles con
la Enfermedad de Fabry. La Enfermedad de Fabry se asocia con eventos renales.Esta enfermedad es muy poco frecuente y hereditaria. El origen está en un fallo en la des-
trucción de determinadas sustancias del organismo que se acumulan en diferentes órganos (el corazón, el riñón, el tubo digestivo o los nervios periféricos) y pueden dañarlos. A veces es detectada en edades tempranas pero lo habitual es que pase desapercibida hasta la aparición de una complicación. Dado que por su rareza (1 de cada 117.000 nacidos vivos) no se realizan pruebas para su diagnóstico de forma habitual, por ello se está promoviendo este estudio de detección en personas con problemas renales a fin de poder determinar de forma más exacta la prevalencia de esta enfermedad.
La investigación se desarrollará con el máximo respeto de los derechos individuales, según los postulados aceptados internacionalmente por las Naciones Unidas y la Comisión Europea (Acta de Helsinki de 2008 y Convenio de Oviedo de 1997). Por ello, todas las investigaciones que se lleven a cabo contarán con la aprobación y supervisión de los diferentes Comités de Ética quienes velarán por el cumplimiento de los postulados anteriormente citados.
Beneficios y compensacionesLa participación en el estudio no conlleva ningún beneficio económico para el sujeto partici-
pante en el estudio y no le supone coste alguno. Así mismo, le informamos que la inclusión en este estudio no conlleva riesgo alguno para el
sujeto participante. En el caso de que se le diagnosticara una enfermedad de Fabry, el paciente se beneficiaria de la posibilidad de un tratamiento precoz y eficaz para su enfermedad. Sin em-bargo, le informamos que si así lo desea, tiene derecho a rechazar el conocer los resultados de las pruebas.
ProcedimientoPara poder realizar el estudio, se necesita extraer una gota de sangre que impregne el papel
de una tarjeta, especialmente preparada para el estudio de la enfermedad de Fabry. En todos los pacientes, la extracción se hará coincidir con otra que precise para su tratamiento. La tarjeta será enviada al Laboratorio Centogene de Alemania
Con la muestra, se procederá a la determinación del déficit enzimático/análisis genético de la enfermedad sospechada por su médico.
Le informamos que a su hijo/a/tutorando, si tuviera entre 14 y 18 años, se le va a entregar una Hoja de Información y Consentimiento Informado adaptados a su capacidad de entendi-miento y deberá firmarlos él/ella dando su asentimiento a participar en el estudio.
ConfidencialidadLas bases de datos de trabajo no contendrán datos personales que permitan la identificación
de los participantes en el estudio. El nombre del sujeto participante se vinculará a un código for-mado por dos números, el primero corresponderá al Hospital y el segundo al sujeto participante.

• 20 •
La información será manejada de forma anónima por el investigador, de forma que el par-ticipante solamente podrá ser identificado por su médico, debiendo dirigirse al mismo para el ejercicio de los derechos de acceso, rectificación, cancelación y oposición a sus datos de carácter personal.
Bajo ciertas circunstancias, el personal responsable de garantizar que la investigación se está haciendo adecuadamente podría revisar sus datos médicos (p.ej. datos recogidos en su historia médica). Los citados a continuación podrán tener acceso a estos datos: investigadores y coor-dinadores del estudio, autoridades reguladoras españolas, comités éticos. A todos ellos se les requiere asimismo mantener su identidad oculta y revisarán sus datos bajo secreto profesional.
Toda la información obtenida de los sujetos participantes será confidencial, cumpliendo en todo momento la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carác-ter Personal y el Real Decreto 1720/2007, de 21 de diciembre, que la desarrolla.
El uso que se haga de la información obtenida será confidencial. Por lo tanto, su identidad será siempre preservada. Igualmente los datos obtenidos solo podrán ser publicados de forma anónima y agregada, es decir, en forma de porcentajes o datos numéricos sin identificación del participante, y nunca de manera individual.
Las muestras de sangre serán destruidas tras la finalización del estudio o, en su caso, trans-currido el periodo máximo de almacenamiento, siendo éste de 30 años.
Usted tiene derecho a no ser informado de los resultados de los análisis realizados a su hi-jo/a/tutorando. Cuando esta información sea necesaria para evitar un grave perjuicio, bien para su hijo/a/tutorando, usted o bien para cualquiera de sus familiares biológicos, se podrá informar a los afectados o a su representante legalmente autorizado. En todo caso, la comunicación se limitará exclusivamente a los datos necesarios para estas finalidades.
Derecho a rehusar o a abandonarLa participación en este proyecto es enteramente voluntaria, pudiendo abandonar su hijo/a/
tutorando el estudio cuando quiera, sin tener que dar explicación alguna y sin que esto repercuta en sus cuidados médicos. Por otra parte, si en algún momento desea retirar las muestras de su hijo/a/tutorando del laboratorio, podrá hacerlo, salvo que dichas muestras hayan sido previa-mente disociadas y por lo tanto no sea posible la identificación del sujeto participante.
Para ello deberá ponerse en contacto con el Dr./Dra. ………………………………………… del Hospital ………………………………………….……………………………………………… en el teléfono de contacto ……………………………….

• 21 •
CONSENTIMIENTO INFORMADO
He tenido suficiente tiempo para evaluar y comentar con mi médico la inclusión de mi hijo/a/tutorando, y he recibido una respuesta a todas mis preguntas. También he leído y acepto las condiciones que se explican en esta información. Recibiré una copia de esta hoja y el original se conservará junto con la historia clínica de mi hijo/a para indicar su participación en este estudio. Entiendo que la participación en el estudio de mi hijo/a es voluntaria y que puedo negarme a su participación o retirar mi consentimiento en cualquier momento.
La participación en el estudio dará lugar a un tratamiento de datos de salud. Usted autoriza expresa-mente que dichos datos se mantengan en el fichero, en tanto no solicite su cancelación y que sean tratados para la finalidad de la investigación objeto del proyecto. Usted podrá acceder, rectificar, cancelar, oponerse al tratamiento de los datos dirigiéndose al Dr…………………………………………………………………
Los datos recogidos en el estudio se conservarán de forma estrictamente confidencial. Acepto que el equipo médico, el personal designado por el promotor así como los representantes tanto de las autoridades sanitarias como de los comités éticos, que están sujetos a secreto profesional tengan acceso a estos datos y a los documentos fuente solamente con objetivos relacionados con el estudio.
Consiento y autorizo que la muestra de sangre de mi hijo/a sea enviada al Laboratorio Centogene en Alemania y sólo podrá ser utilizada para la determinación enzimática (análisis genético) de la Enfermedad de Fabry objeto del presente estudio, de forma que se pedirá permiso para cualquier otro tipo de análisis complementario en el caso de que fueran necesarios.
Consiento que las muestras puedan ir etiquetadas con un código de participación en el estudio y que solamente mi médico pueda asociar dicho código con los datos de carácter personal de mi hijo/a y así poder conocer su identidad.
Deseo r, no deseo r (marque con una X su opción) que se me comuniquen los resultados de los análisis y de las pruebas realizadas a mi hijo/a a través del Dr………………………………………………
Firma del paciente Firma del médico
He leído y entiendo este consentimiento informa-do. Mis dudas han sido aclaradas. Voluntariamente consiento la participación de mi hijo/a.
Apellidos: _________________________________
__________________________________________(mayúsculas)
Nombre: _________________________________
__________________________________________ (mayúsculas)
Fecha: ____ / ____ / ________DD/MM/AAAAA
Firma:
Reconozco que he explicado personalmente la na-turaleza, objetivos, duración, riesgos y beneficios potenciales del estudio a los padres o tutor y que él/ella ha otorgado hoy su consentimiento de for-ma libre y de carácter revocable.
Apellidos: _________________________________
__________________________________________(mayúsculas)
Nombre: _________________________________
__________________________________________ (mayúsculas)
Fecha: ____ / ____ / ________DD/MM/AAAAA
Firma:

• 22 •
HOJA DE INFORMACIÓN AL PACIENTE ENTRE 14 Y 18 AÑOS DE EDAD Y AUTORIZACIÓN para la participación en el estudio sobre:
EstudiodePrevalenciadelaEnfermedaddeFabry en pacientes en diálisis
¿Por qué se realiza este estudio?Los síntomas que presentas en este momento podrían ser compatibles con la Enfermedad
de Fabry. La Enfermedad de Fabry se asocia con alteraciones renales.Esta enfermedad es muy poco frecuente y hereditaria. El origen está en un fallo en la des-
trucción de determinadas sustancias del organismo que se acumulan en diferentes órganos (el corazón, el riñón, el tubo digestivo o los nervios periféricos) y pueden dañarlos. A veces es detectada en edades tempranas pero lo habitual es que pase desapercibida hasta la aparición de una complicación. Dado que por su rareza (1 de cada 117.000 nacidos vivos) no se realizan pruebas para su diagnóstico de forma habitual, por ello se está promoviendo este estudio de detección en personas renales a fin de poder determinar de forma más exacta la prevalencia de esta enfermedad.
La investigación se desarrollará con el máximo respeto de los derechos individuales, según los postulados aceptados internacionalmente por las Naciones Unidas y la Comisión Europea (Acta de Helsinki de 2008 y Convenio de Oviedo de 1997). Por ello, todas las investigaciones que se lleven a cabo contarán con la aprobación y supervisión de los diferentes Comités de Ética quienes velarán por el cumplimiento de los postulados anteriormente citados.
¿Qué beneficios y recompensa obtendría?La participación en el estudio no conlleva ningún beneficio económico para ti en el estudio
y no supone coste alguno. Así mismo, te informamos que la inclusión en este estudio no conlleva riesgo alguno para ti.
En el caso de que se te diagnosticara una enfermedad de Fabry, te beneficiarias de la posibilidad de un tratamiento precoz y eficaz para tu enfermedad. Sin embargo, te informamos que si así lo deseas, tienes derecho a rechazar el conocer los resultados de las pruebas.
¿En qué consiste el estudio?Para poder realizar el estudio, se necesita extraer una gota de sangre que impregne el papel
de una tarjeta, especialmente preparada para el estudio de la enfermedad de Fabry. En todos los pacientes, la extracción se hará coincidir con otra que precise para su tratamiento. La tarjeta será enviada al Laboratorio Centogene de Alemania
Con la muestra, se procederá a la determinación del déficit enzimático/ análisis genético de la enfermedad sospechada por tu médico.
¿Qué derechos tengo en este estudio?Toda la información obtenida sobre ti será confidencial, cumpliendo en todo momento la Ley
Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal y el Real Decreto 1720/2007, de 21 de diciembre, que la desarrolla.
Las bases de datos de trabajo no contendrán datos personales que permitan tu identificación en el estudio. Tu nombre se vinculará a un código formado por 2 números, el primero corres-ponderá al Hospital y el segundo a ti.

• 23 •
La información será manejada de forma anónima por el investigador, de forma que sola-mente podrás ser identificado por tu médico, debiendo dirigirte al mismo para el ejercicio de los derechos de acceso, rectificación, cancelación y oposición a sus datos de carácter personal.
En algunos casos, el personal responsable de garantizar que la investigación se está haciendo adecuadamente podría revisar tus datos médicos (p.ej. datos recogidos en tu historia médica).
Los citados a continuación podrán tener acceso a estos datos: investigadores y coordinado-res del estudio, autoridades reguladoras españolas, comités éticos. A todos ellos se les requiere asimismo mantener su identidad oculta y revisarán sus datos bajo secreto profesional El uso que se haga de la información obtenida será confidencial. Por lo tanto, tu identidad será siempre preservada. Igualmente los datos obtenidos solo podrán ser publicados de forma anónima y agregada, es decir, en forma de porcentajes o datos numéricos sin identificación del participante, y nunca de manera individual.
Las muestras de sangre serán destruidas tras la finalización del estudio o, en su caso, trans-currido el periodo máximo de almacenamiento, siendo éste de 30 años.
Tienes derecho a no ser informado de los resultados de los análisis realizados. Cuando esta información sea necesaria para evitar un grave perjuicio, bien para ti o bien para tus familiares biológicos, se podrá informar a los afectados o a su representante legalmente autorizado. En todo caso, la comunicación se limitará exclusivamente a los datos necesarios para estas finali-dades.
¿Qué ocurriría si no quiero participar o quiero retirarme del estudio?La participación en este proyecto es enteramente voluntaria, pudiendo abandonar el estudio
cuando tú quieras, sin tener que dar explicación alguna y sin que esto repercuta en tus cuidados médicos.
Por otra parte, si en algún momento deseas retirar tus muestras del laboratorio, podrás hacerlo por medio de tus padres o tutor, salvo que dichas muestras hayan sido previamente disociadas y por lo tanto no sea posible tu identificación. Para ello, tus padres o tutor deberán ponerse en contacto con el Dr./Dra………………………………… del Hospital…………………………………………. en el teléfono de contacto……………………………….

• 24 •
Firma del paciente Firma del médico
He leído y entiendo este asentimiento. Mis dudas han sido aclaradas. Voluntariamente consiento participar.
Apellidos: _________________________________
__________________________________________(mayúsculas)
Nombre: _________________________________
__________________________________________ (mayúsculas)
Fecha: ____ / ____ / ________DD/MM/AAAAA
Firma:
Reconozco que he explicado personalmente la na-turaleza, objetivos, duración, riesgos y beneficios potenciales del estudio al paciente y que él/ella ha otorgado hoy su asentimiento de forma libre y de carácter revocable.
Apellidos: _________________________________
__________________________________________(mayúsculas)
Nombre: _________________________________
__________________________________________ (mayúsculas)
Fecha: ____ / ____ / ________DD/MM/AAAAA
Firma:
ASENTIMIENTO DEL PARTICIPANTE
He tenido suficiente tiempo para evaluar y comentar con mi médico mi inclusión, y he recibido una respuesta a todas mis preguntas. También he leído y acepto las condiciones que se explican en esta infor-mación. Recibiré una copia de esta hoja y el original se conservará junto con mi historia clínica para indicar mi participación en este estudio. Entiendo que mi participación en el estudio es voluntaria y que puedo negarme a participar o retirar mi consentimiento en cualquier momento.
La participación en el estudio dará lugar a un tratamiento de datos de salud. Autorizo expresamente que dichos datos se mantengan en el fichero, en tanto no solicite su cancelación y que sean tratados para la finalidad de la investigación objeto del proyecto. Podrás acceder, rectificar, cancelar, oponerse al tratamiento de los datos dirigiéndote al Dr……………….…………………………………….
Los datos recogidos en el estudio se conservarán de forma estrictamente confidencial. Acepto que el equipo médico, el personal designado por el promotor así como los representantes tanto de las autoridades sanitarias como de los comités éticos, que están sujetos a secreto profesional tengan acceso a estos datos y a los documentos fuente (por ejemplo, mi historia clínica) solamente con objetivos relacionados con el estudio.
Consiento y autorizo que mi muestra de sangre sea enviada al Laboratorio Centogene de Alemania y solo podrá ser utilizada para la determinación enzimática/análisis genético de la Enfermedad de Fabry objeto del presente estudio, de forma que se pedirá permiso para cualquier otro tipo de análisis complementario en el caso de que fueran necesarios.
Consiento que las muestras puedan ir etiquetadas con mi código de participación en el estudio y que solamente mi médico pueda asociar dicho código con mis datos de carácter personal y así poder conocer mi identidad.
• 25 •
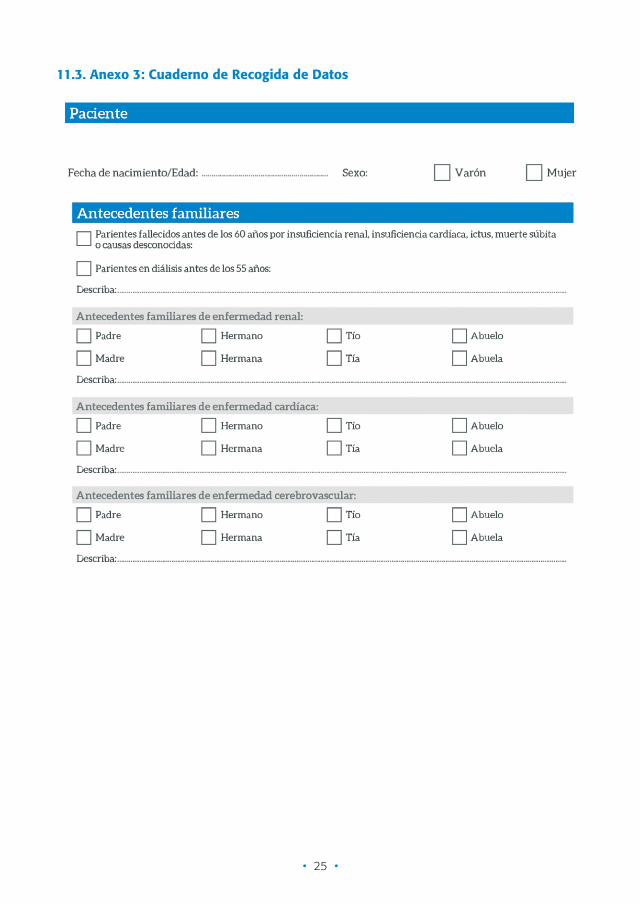
11.3. Anexo 3: Cuaderno de Recogida de Datos

• 26 •
Fecha (dd/mm/aaaa) Resultado enzimatico Resultado genético
Test DBSResultado
*
Lyso Gb3
*especificar mutaciones y variantes en el gen GLA.

