clase no.5
DESCRIPTION
gfgdTRANSCRIPT

0
ENSAYOS
CLINICOSFARMACIA CLINICA
GRUPO 1 y 3 2009 2010CBuenestado

1
� CONCEPTOCONCEPTOCONCEPTOCONCEPTO: REAL DECRETO 223/2004, 6 de febrero, por el que se
regulan los ensayos clínicos con medicamentos. (que deroga el R.D. 561/1993 � incorpora el ordenamiento jurídico de la Directiva Europea 2001/20/CE)
“Toda investigación experimental de una “Toda investigación experimental de una “Toda investigación experimental de una “Toda investigación experimental de una sustancia o medicamento, a través de su sustancia o medicamento, a través de su sustancia o medicamento, a través de su sustancia o medicamento, a través de su administración o aplicación a administración o aplicación a administración o aplicación a administración o aplicación a seres seres seres seres humanoshumanoshumanoshumanos, orientada hacia alguno de los , orientada hacia alguno de los , orientada hacia alguno de los , orientada hacia alguno de los siguientes fines:siguientes fines:siguientes fines:siguientes fines:
1. Establecer su eficacia para una indicación terapéutica, profiláctica o diagnóstica determinada.
2. Poner de manifiesto sus efectos farmacodinámicos o recoger datos referentes a su absorción, distribución, metabolismo y excreción en el organismo humano.
3. Conocer el perfil de sus reacciones adversas y establecer su seguridad.

2
1. EMEA: Agencia Europea del Medicamento
2. FDA: Food and Drug Administration
3. AEM: Agencia Española del Medicamento
ENSAYOS CLÍNICOS : Agencias ReguladorasENSAYOS CLÍNICOS : Agencias ReguladorasENSAYOS CLÍNICOS : Agencias ReguladorasENSAYOS CLÍNICOS : Agencias Reguladoras

3
Desarrollo de un fármaco nuevoDesarrollo de un fármaco nuevoDesarrollo de un fármaco nuevoDesarrollo de un fármaco nuevo
Etapa Precomercialización
Fase 0 : Evaluación preclínica o
de experimentación animal
Evaluación Clínica (en el ser humano)
Fase I: de seguridadFase II: de eficacia terapéuticaFase III: de utilidad comparada
Etapa de Poscomercialización
Fase IV: farmacovigilancia

4
Evaluación Clínica
Fase I: de seguridad� 1ª toma de contacto del fármaco con elorganismo.
� Objetivos: verificar la seguridad del fármaco en humanos, regímenes seguros de dosificación, estudios de farmacodinamia y farmacocinética.
� Realizado en: un nº reducido de voluntarios adultos sanos (consentimiento informado: por escrito).
� En hospitales: ensayos abiertos ( no controlado, ni aleatorio).

5
Fase II: de eficacia terapéutica
� Objetivos: demostrar la eficacia en el tratamiento de una enfermedad y delimitar un rango de dosis terapéuticas.
� Realizado en: un nº limitado de voluntarios que padezcan la enfermedad.

6
Fase III: de utilidad comparada� Objetivos: verificar la seguridad y eficacia del fármaco a corto y largo plazo, comparando con; otras alternativas terapéuticas o con un placebo.
� También debe establecer la incidencia de los efectos secundarios.
� Realizado en: un nº más elevado de pacientes.� Requiere condiciones determinadas : ensayos controlados, aleatorios, ciegos, cruzados multicéntricos.

7
Etapa de Poscomercialización
Fase IV: farmacovigilancia� Objetivos:
� Detectar reacciones adversas (no descritas previamente), interacciones, otros efectos secundaeios; efectos teratógenos.
� Investigar nuevas aplicaciones terapéuticas etc.
� (son reacciones descubiertas en el uso del
medicamento; usos compasivos etc (enviadas por
pacientes,farmaceuticos,medicos...etc a los
Centros de Farmacovigilancia.).
� Son estudios postcomercialización.

8
Historia del ensayo clínico.� 1747: James Lind, observó la mejora en marinos con escorbuto, al
ser tratados con cítricos.
� 1870:Joseph Lister,eficacia de la antisepsia con fenol� 1923: Fisher: introduce “aleatorizacion” o randomización
� l1931: Amberson, comprobó que la administración de sanocrisina a
pacientes con tuberculosos, empeoraba la enfermedad, en
comparación con los controles. Primer E.C. con asignación aleatoria
rudimentaria
� 1947 Tribunal del Juicio de Nüremberg: Código de Nüremberg:
investigación en humanos.
� 1948 Bradford Hill (Medical Research Council) : Primer
E.C.randomizado: Estreptomicina en la tuberculosis

9
Historia del ensayo clínico.
� 1950 B.Hill (MRC): Primer E.C. randomizado y doble-ciego.
Antihistamínicos en el catarro común
� 1964 Asamblea Médica Mundial: Código de Helsinki (DECLARACIÓN DE HELSINKI Asociación Médica Mundial (AMM)Recomendaciones para orientar a los médicos en la investigación biomédica con sujetos humano)
� 1977 FDA : Normas de Buena Práctica Clínica (BPC) en EEUU
� 1990 Comité de especialidades farmacéuticas: Normas de BPC
de la Comunidad Europea
Tres dimensiones del E.C.: metodología, ética, autenticidad“El E.C. tiene que ser científicamente correcto, éticamente aceptable,
y sus datos y procedimientos verificables.”

10
ASPECTOS LEGALES
� La Ley del Medicamento establece que los ensayos clínicos deberán realizarse en condiciones de respeto a los derechos fundamentales de la persona.
� Las bases de la filosofía ética en la investigación en humanos fueron fijadas por la Asamblea Médica Mundial en la declaración de Helsinki(1964).
� En Edimburgo se realizó una revisión en el año 2000, pero la última normativa para la realización de ensayos clínicos con medicamentos es el R.D.223/2004 de aplicación europea.

11
ASPECTOS LEGALES
� En la revisión de Edimburgo se estableció:necesidad de que la población sobre la que se realiza la investigación clínica sea beneficiaria de sus resultados.consideraciones sobre el empleo del placebo.obligación de publicar los resultados negativos de la investigación.transparencia e información sobre las fuentes de financiación.

12
ASPECTOS METODOLÓGICOS(Buena Práctica clínica)
� Los EC deben realizarse bajo planteamientos científicos y metodológicos CORRECTOS, que aseguren la validez de los resultados.
Las normas de BUENA PRACTICA CLINICA: son una serie de medidas, de carácter administrativo, que deben ser observadas para que el informe de un EC sea aceptado por las autoridades sanitarias como demostración de la SEGURIDAD y EFICACIA de un NUEVO medicamento.

13
Las normas de buena practica clínica:
� Pretenden asegurar que los datos son fiables
� Que se protegen los derechos, integridad y confidencialidad de los sujetos
� Señalan las responsabilidades de los implicados en cada una de las fases de planificación y ejecución de un EC
� Obligan a la existencia de unos procedimientos preestablecidos (PROCEDIMIENTOS NORMALIZADOS DE TRABAJO) que se apliquen de forma sistemática en la organización, dirección, recogida de datos, documentación y verificación de los EC.

14
CLASIFICACIÓN DE LOS ENSAYOS CLÍNICOS
� Se han propuesto numerosas clasificaciones de los ensayos clínicos, siendo una de las más utilizadas la recogida en el R.D. 561/1993 que clasifica estos estudios de acuerdo a una serie de factores:
1.-FINALIDAD: según el grado de desarrollo clínico del fármaco.
2.-INVESTIGADOR Y CENTROS que intervienen.3.-METODOLOGÍA Y DISEÑO:
►CERRADO/ ABIERTO►NO CONTROLADO/ CONTROLADO►ALEATORIO/ NO ALEATORIO►PARALELO/CRUZADO

15
1.-E.C. SEGÚN SU FINALIDAD
� Son los ensayos clínicos denominados:
-FASE I-FASE II-FASE III-FASE IV

16
2.- E.C. SEGÚN INVESTIGADOR Y CENTROS
� ESTUDIOS UNICÉNTRICOS↪Son aquellos estudios realizados por un solo
investigador o equipo investigador, en un centro hospitalario o en centroS dependientes o tutelados del mismo (Areas de Salud..extrahospitalario).↪↪↪↪Son rápidos y fáciles de realizar.↪↪↪↪INCONVENIENTE: que al realizarse con un
número pequeño de pacientes será muy difícil el demostrar posibles diferencias entre tratamientos.↪↪↪↪UTILIZACIÓN: valoración de terapias que no
utilizan fármacos (innovaciones en cirugía o radioterapia, estudios de nutrición>).

17
2.- E.C. SEGÚN INVESTIGADOR Y CENTROS
� ESTUDIOS MULTICÉNTRICOS.↪↪↪↪Ensayo clínico que utilizan un protocolo común y único
pero en más de un centro y, por tanto, realizado por más de un investigador.↪↪↪↪VENTAJAS:
-Permiten estudiar un número suficiente de pacientes en menor tiempo.
-Conclusiones más fiables que permiten una mejor valoración y son más representativas que si se hubiera realizado el estudio en un solo centro. Tiene mayor margen de confianza.↪↪↪↪INCONVENIENTES: -Complejidad en la planificación y la administración del
estudio.-Necesidad de una buena coordinación.-Difícil control, supervisión, recogida y manipulación de
datos.

18
3.- E.C. SEGÚN METODOLOGÍA Y DISEÑO
� Uno de los principales objetivos de un ensayo clínico es demostrar que un nuevo fármaco es más eficaz y seguro que un placebo o que otro tratamiento. Para ello se utilizan los siguientes ensayos:
►►►► CERRADO/ABIERTO:- Cerrado: Suelen ser controlados, se elabora una normativa de actuación rígida, recogida en el protocolo del ensayo, que no se puede modificar durante su desarrollo (IMPRESCINDIBLES EN ENSAYOS MULTICENTRICOS)
-Abierto: En este ensayo pueden modificarse las características del estudio durante su realización.

19
►NO CONTROLADOS /CONTROLADOS
-No controlados:↪↪↪↪Son aquellos que no se comparan con un
grupo control. ↪↪↪↪UTILIZACIÓN:Para generar experiencia en la
mejor utilización de un medicamento, para estudiar efectos secundarios, cambios bioquímicos en terapias a largo plazo, tolerancia, interacciones o eficacia de los fármacos.
3.- E.C. SEGÚN METODOLOGÍA Y DISEÑO

20
- CONTROLADOS:- ↪Son aquellos que se se se se comparacomparacomparacomparannnn estadestadestadestadíííísticamente los sticamente los sticamente los sticamente los resultados obtenidos en un resultados obtenidos en un resultados obtenidos en un resultados obtenidos en un grupo tratadogrupo tratadogrupo tratadogrupo tratado con la sustancia en con la sustancia en con la sustancia en con la sustancia en experimentaciexperimentaciexperimentaciexperimentacióóóón y otro n y otro n y otro n y otro grupo controlgrupo controlgrupo controlgrupo control: placebo o sustancia de : placebo o sustancia de : placebo o sustancia de : placebo o sustancia de referenciareferenciareferenciareferencia
- Se asignan los tratamientos al azar de forma que ni el sujeto ni el investigador puedan influir en su asignación. Dentro de los estudios controlados hay que distinguir:
a) ESTUDIOS PILOTO: son un sondeo o ensayo preliminar destinado a la puesta a punto de una modalidad para un ensayo posterior más importante.
Ayudan al investigador a decidir qué numero de pacientes; tipo de pacientes, tratamientos y observaciones son las más apropiadas.
3.- E.C. SEGÚN METODOLOGÍA Y DISEÑO

21
b) ESTUDIOS NO CIEGOS: son aquellos ensayos en los que tanto el sujeto como el investigador conocen a qué tipo de tratamiento o intervención médica ha sido asignado.
Estos estudios interesan por dos razones principales:-Son más fáciles de realizar que los otros estudios.-Los investigadores pueden sentirse más cómodos, a la
hora de tomar decisiones, si conocen la intervención.
El que el sujeto(paciente) sepa que está recibiendo un tratamiento nuevo puede ser beneficioso desde un punto de vista psicológico.Todo puede influir en la respuesta al tratamiento.
3.- E.C. SEGÚN METODOLOGÍA Y DISEÑO

22
C) ESTUDIOS SIMPLE CIEGO: son aquellos en los que el sujeto desconoce el grupo de tratamiento a que ha sido asignado. El investigador si lo conoce. En ellos se trata de neutralizar el efecto placebo.EJEMPLO: este método se utiliza frecuentemente en la valoración de fármacos para el tratamiento del cáncer.
D) ESTUDIOS DOBLE CIEGO: son aquellos estudios en los que tanto el sujeto como el investigador desconocen la asignación a los grupos de tratamiento. En ellos se trata de neutralizar el efecto placebo y la subjetividaddel observador.
VENTAJA: se reduce el riesgo de sesgo, se considera a priori como los estudios preferibles a la hora de un mejor diseño y garantía de objetividad en los datos obtenidos.
INCONVENIENTE: mayor complejidad a la hora de su preparación, ya que exigen una formulación galénica exacta en apariencia, para la muestra en estudio y el placebo;Y la necesidad de que elfarmaceutico conozca los tratamientos(muestra y placebo) y los sujetos(enfermos) a los que se le han asignado.(para informar al investigador una vez terminado el ensayo).
3.- E.C. SEGÚN METODOLOGÍA Y DISEÑO

23
3.-E. C. SEGÚN SU METODOLOGÍA Y DISEÑO.ESTUDIOS CONTROLADOS.
⇨Cada ensayo clínico requiere una consideración cuidadosa de los siguientes aspectos:
a) ÉTICA: el procedimiento doble ciego no debe representar ningún daño o riesgo indebido para el paciente.
b) VIABILIDAD: para algunos tratamientos puede ser totalmente imposible diseñar un ensayo doble ciego, bien sea por el tipo de patología en estudio o por las formas galénicas comparadas.
c) PREVENCIÓN DEL SESGO: hay que comprobar la influencia que puede tener el “sesgo” si el estudio no es ciego.

24
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.En cuanto a la asignación del tratamiento a los grupos o pacientes
� ESTUDIOS ALEATORIOS O RANDOMIZADOSSon aquellos en los que el paciente es asignado a un grupo o a otro de tratamiento siguiendo un método de randomización preestablecido. Se utilizan para probar la eficacia de innovaciones terapéuticas siempre y cuando el tipo de fármaco o patología que estamos valorando lo permita.
� ESTUDIOS NO ALEATORIOSSon aquellos en los que los pacientes son asignados a uno u otro tratamiento, bien sea siguiendo un método de asignación sistemática predeterminada o el juicio del investigador o del paciente.
Ejemplo: Fecha de nacimiento (días de nacimientos impares/pares = tratamiento nuevo/estandar)
ESTUDIOSALEATORIOS
YNO
ALEATORIOS
ESTUDIOSPARALELOS
YCRUZADOS
OTROSDISEÑOS

25
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
MÉTODOS PARA REALIZAR LA ASIGNACION ALEATORIA:
• ALEATORIZACIÓN SIMPLE:
• ALEATORIZACION ESTRATIFICADA
• ALEATORIZACIÓN DESIGUAL
ESTUDIOSALEATORIOS
YNO
ALEATORIOS
ESTUDIOSPARALELOS
YCRUZADOS
OTROSDISEÑOS

26
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
MÉTODOS PARA REALIZAR LA ASIGNACION ALEATORIA:
• ALEATORIZACIÓN SIMPLE:El investigador asigna el tratamiento a los pacientes siguiendo el orden numérico de la lista, pero desconociendo el tratamiento que corresponde a dicho número (lista preparada por alguien ajeno al estudio)
ESTUDIOSALEATORIOS
YNO
ALEATORIOS
ESTUDIOSPARALELOS
YCRUZADOS
OTROSDISEÑOS

27
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
• ALEATORIZACIÓN ESTRATIFICADA:Su principal objetivo es tratar de evitar que los grupos de tratamientos presenten diferencias importantes en las características de los pacientes. Hay que decidir de forma cuidadosa que factores de los pacientes se deben estratificar.
(Ejemplo: para ver si el tabaco influye en el efecto broncodilatador de un nuevo fármaco los pacientes se dividen en fumadores y no fumadores y dentro de cada grupo se procede a la aleatorización)
• ALEATORIZACIÓN DESIGUAL:Cuando se compara un fármaco nuevo frente a uno estándar, se tiene también interés en ganar experiencia y conseguir información adicional sobre el nuevo tratamiento, en estos casos, en lugar de incluir igual numero de pacientes en cada grupo, se incorporan más pacientes al grupo del tratamiento en experimentación siguiendo proporciones de 2:1 ó 3:2.

28
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
ENSAYOS ALEATORIOS
VENTAJAS:-reducen los posibles “sesgos” que pudiera
originar el investigador cuando tiene libertad de decisión para asignar el tratamiento.
-los datos que se obtienen corresponderán a grupos comparables en cuanto a factores relevantes.
-permiten garantizar la validez del test de significado estadístico.
DESVENTAJA:-consideraciones éticas que podría tener el
investigador (por no tener la opción de decidir que tipo de tratamiento es mas adecuado para cada paciente).

29
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
ENSAYOS NO ALEATORIOS
INCONVENIENTES:-el investigador conoce el tratamiento que
va a recibir el paciente, por lo que puede decidir si éste es incluido o no en el estudio.
-los grupos no son estrictamente comparables.
ESTUDIOSALEATORIOS
YNO
ALEATORIOS
ESTUDIOSPARALELOS
YCRUZADOS
OTROSDISEÑOS
30
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
En cuanto a la naturaleza del grupo de comparación PARALELOS CRUZADOS
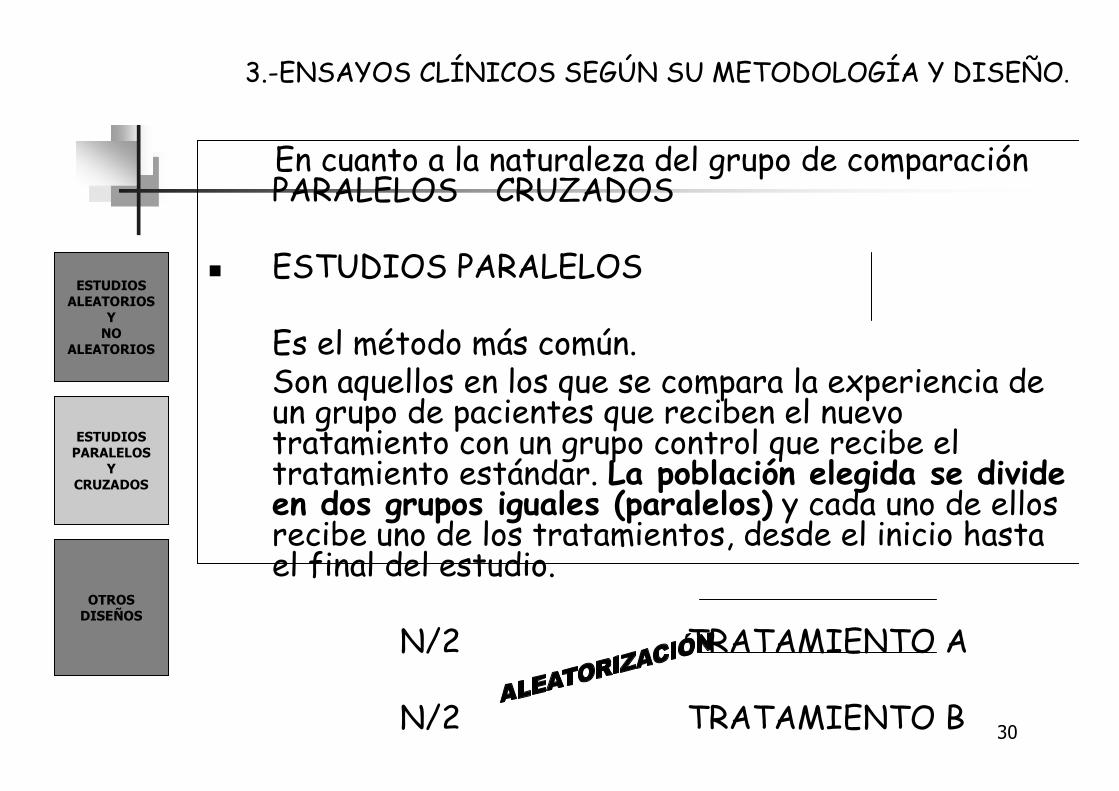
� ESTUDIOS PARALELOS
Es el método más común.Son aquellos en los que se compara la experiencia de un grupo de pacientes que reciben el nuevo tratamiento con un grupo control que recibe el tratamiento estándar. La población elegida se divide en dos grupos iguales (paralelos) y cada uno de ellos recibe uno de los tratamientos, desde el inicio hasta el final del estudio.
N/2 TRATAMIENTO A
N/2 TRATAMIENTO B
ESTUDIOSALEATORIOS
YNO
ALEATORIOS
ESTUDIOSPARALELOS
YCRUZADOS
OTROSDISEÑOS

31
� ESTUDIOS CRUZADOSEn ocasiones, la variabilidad de cada grupo o dificultad de encontrar un número de pacientes suficiente para cada grupo aconseja la realización de estudios cruzados.
En este diseño cada sujeto del estudio recibe un tratamiento en la primera fase y el otro tratamiento en una etapa posterior.
N/2 TRAT. A TRAT. BN/2 TRAT. B TRAT. A
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
ESTUDIOSALEATORIOS
YNO
ALEATORIOS
ESTUDIOSPARALELOS
YCRUZADOS
OTROSDISEÑOS

32
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
ESTUDIOS PARALELOS
VENTAJAS:-los resultados obtenidos pueden ser razonablemente
extrapolados a la población.-no se requiere un orden de llegada de los pacientes
para su inclusión en el estudio.-los grupos comparados son iguales
cuantitativamente. DESVENTAJA:
-la principal desventaja es el número de sujetos requeridos, convirtiéndolos en ensayos clínicos muy costosos en términos de tiempo, fuente de pacientes y personal involucrado en los mismos. A pesar de ello, una regla fundamental en investigación clínica es que los estudios en fase III siempre deben ser paralelos

33
ESTUDIOS CRUZADOS
VENTAJAS:-no influyen factores relevantes como
criterios de inclusión y exclusión, ya que cada paciente sirve de su propio control.
-se reduce la variabilidad (se necesita un tamaño inferior de la muestra para poder detectar diferencia estadística entre el grupo de intervención y el grupo control)
3.-ENSAYOS CLÍNICOS SEGÚN SU METODOLOGÍA Y DISEÑO.
ESTUDIOSALEATORIOS
YNO
ALEATORIOS
ESTUDIOSPARALELOS
YCRUZADOS
OTROSDISEÑOS

34
ELEMENTOS DEL ENSAYO ELEMENTOS DEL ENSAYO CLÍNICOCLÍNICO
1.- Introducción:- Equipo multidisciplinar.- Comité Ético de Investigación Clínica.
2.- Documentación técnica del ensayo clínico:
- Consentimiento informado del paciente.- Información básica.- Protocolo del ensayo clínico.- Formulario de registro de casos.
3.- Muestras para investigación clínica.

35
1.- Introducción. -- Equipo multidisciplinar.Equipo multidisciplinar.
� La investigación clínica, requiere la participación responsable e informada de todos los que intervengan en su realización.
� El intercambio de información entre participantes debe ser antes, durante y después del ensayo clínico.
� Las figuras más representativas son:- Sujeto del ensayoSujeto del ensayo.- Promotor.Promotor.- Monitor.Monitor.- Investigador principalInvestigador principal.

36
Introducción. Equipo multidisciplinar.
� Sujeto del ensayo: persona, sana o enferma, que participa en un ensayo libremente, tras otorgar su consentimiento informado.
� Promotor: persona física o jurídica, interesado en la realización del ensayo, firma la solicitud de autorización dirigida al Comité Ético de Investigación Clínica(CEIC), y al Ministerio de Sanidad y Consumo, responsabilizándose de dicho ensayo. Suelen ser las industrias farmacéuticas.
� Monitor: profesional capacitado, elegido por el promotor, para el seguimiento directo de la realización del ensayo.
� Investigador principal: dirige la realización práctica del ensayo. Junto con el promotor asume la responsabilidad del proyecto.
Es requisito indispensable, que el investigador principal sea un profesional sanitario cualificado y que todo el proceso esté bajo control médico.

37
Introducción. Equipo multidisciplinar.
PROMOTOR(monitor)
INVESTIGADORPRINCIPAL
Colaboradores preclínicos(coinvestigadores)
SERVICIOSCENTRALES SERVICIO DE
FARMACIAPERSONALSANITARIO
PACIENTES(familiares)
Comunicaciónen la
investigación clínica

38
Fase previa a la realización de los E.C.Evaluación y aprobación por:
COMITÉ ÉTICO DE INVESTIGACION COMITÉ ÉTICO DE INVESTIGACION CLINICA (CEIC)CLINICA (CEIC)

39
Comité Etico de Investigación Clínica:
� Son equipos interdisciplinarios, integrados por:
� Médicos (al menos uno será farmacólogo clínico)� Farmacéuticos del hospital� Personal de enfermería� Personas ajenas a profesiones sanitarias ( al menos uno será
jurista)
� Sus funciones se recogen en el RD. 223/2004.� Actualmente, la designación de sus componentes y su
ámbito de actuación, es competencia de las CC.AA.

40
� Encargado de revisar, y aprobar o denegar los ensayos clínicos.
� Debe estudiar y aceptar los protocolos, como paso previo, IMPRESCINDIBLE, para la tramitación administrativa de la autorización.
� Otras funciones de las que se encarga son:� Análisis relación beneficio/riesgo.�Seguro de responsabilidad civil.� Realización de auditorias ( ver si hay fraudes).� Consentimiento informado.� Información. Fiabilidad de datos.
� Y todo ello protegiendo al PACIENTE, al investigador y a la institución.
COMITÉ ETICO DE INVESTIGACION CLÍNICA

41
ELEMENTOS DEL ENSAYO ELEMENTOS DEL ENSAYO CLÍNICOCLÍNICO
1.- Introducción:- Equipo multidisciplinar.- Comité Ético de Investigación Clínica.
2.- Documentación técnica del ensayo clínico:
- Consentimiento informado del paciente.- Información básica.- Protocolo del ensayo clínico.- Formulario de registro de casos.
3.- Muestras para investigación clínica.

42
2.1.CONSENTIMIENTO INFORMADO (223/2004).Hoja de información para el posible participante.
Contendrá información referente a los siguientes aspectos del ensayo clínico:1) Objetivo 2) Metodología empleada 3) Tratamiento que puede serle administrado, haciendo referencia al placebo si procede. 4) Beneficios derivados del estudio. 5) Incomodidades y riesgos derivados del estudio (número de visitas, pruebas
complementarias a que se someterá...). 6) Posibles acontecimientos adversos. 7) Tratamientos alternativos disponibles. 8) Carácter voluntario de su participación, así como posibilidad de retirarse del estudio en
cualquier momento, sin que por ello se altere la relación médico-enfermo ni se produzca perjuicio en su tratamiento.
9) Personas que tendrán acceso a los datos del voluntario y forma en que se mantendrá la confidencialidad.
10) Modo de compensación económica y tratamiento en caso de daño o lesión por su participación en el ensayo, tal como consta en la Ley de medicamento.
11) Investigador responsable del ensayo y de informar al sujeto y contestar a sus dudas y preguntas, y modo de contactar con él en caso de urgencia.
2.- Documentación técnica del ensayo clínico.

43
Modelo de consentimiento por escrito
Título del ensayo...........................................................Yo .................................................... (Nombre y apellidos)He leído la hoja de información que se me ha entregado.He podido hacer preguntas sobre el estudio.He recibido suficiente información sobre el estudio.He hablado con..................... (Nombre del investigador).Comprendo que mi participación es voluntaria.Comprendo que puedo retirarme del estudio:
� Cuando quiera � Sin tener que dar explicaciones � Sin que esto repercuta en mis cuidados médicos
Presto libremente mi conformidad para participar en el ensayoFechaFirma del participante

44
2.- Documentación técnica del ensayo clínico.2.2- INFORMACIÓN BÁSICA.
- Resumen para el investigador clínico de las características del preparado a estudiar.
- También se conoce como “Manual del investigador”.- Las partes que lo componen:
1.- Portada: nombre del laboratorio, denominación del preparado, y fecha de la redacción del documento.
2.- Índice.3.- Introducción: razones de porque se ha
desarrollado un nuevo fármaco.

45
Documentación técnica del E.C. Información básica.
4.- Descripción del preparado: características químicas y galénicas.
5.- Datos preclínicos: toxicología, farmacocinética y farmacología.
6.- Datos clínicos: procedentes de ensayos terminados, para establecer: dosis, duración del tratamiento, resultado y efectos adversos.
7.- Otros: incompatibilidades, precauciones, contraindicaciones, etc.

46
Documentación técnica del E.C. Protocolo del E.C.
2.3.- PROTOCOLO DEL ENSAYO CLÍNICO.- La realización del ensayo, debe ajustarse al contenido del
protocolo, tal como se especifica en el Art. 66 de la ley del Medicamento.
- Desde un punto de vista general, la elaboración de un protocolo, consta de las siguientes etapas:
1. Preparación del borrador del protocolo.2. Discusión con estadística de los medios y métodos
que se van a usar.3. Revisión por la Unidad de Garantía de Calidad.4. Aprobación por el investigador clínico.5. Aprobación por el CEIC.

47
- Un diseño correcto de un ensayo, se basa en la elaboración de un protocolo bien estructurado y completo.
- Según la legislación los apartados que deben ser verificados cuando se proyecta un ensayo, son:1. Información general 9. Reacciones adversas..2. Justificación y objetivos. 10. Cuestiones prácticas.3. Consideraciones éticas generales. 11. Recogidas de datos.4. Programación y duración del ensayo. 12. Evaluación de la respuesta.5. Diseño general. 13. Tratamiento estadístico.6. Selección de los sujetos. 14. Solucionar problemas. 7. Tratamiento. 15. Resumen extenso y anexos.8. Evaluación de la eficacia. 16. Lista de referencias import.
Documentación técnica del E.C. Protocolo del E.C.

48
Documentación técnica del E.C. Formulario del registro de casos.
2.4- Formulario de registro de casos.- Es el documento, en el que se encuentran, todos los datos
referentes al sujeto en estudio, además de resultados y observaciones.
- También conocida como Hoja de Recogida de Datos.- Su contenido debe presentar:
1. Tipo de información a registrar: toda información que me garantice la seguridad y eficacia del fármaco, identificación de paciente, datos del tto, etc.
2. Forma de cumplimentar las respuestas.3.- Diseño gráfico:para una interpretación adecuada de
la información.

49
3.- Muestras para investigación clínica.
- el promotor garantiza ell medicamento en estudio, y que el producto se encuentra bajo control y en perfectas condiciones, con el fin de que dicho producto, sea bien dispensado, además del correcto uso tanto por parte de paciente como por el personal de enfermería.
- Es importante que los fármacos usados en un ensayo clínico estén dotados de una etiqueta que los identifique fácilmente, en dicha etiqueta deben quedar reflejados los siguientes datos:

50
Muestras para investigación clínica.
1.- Código del protocolo.2.- Nº de unidades y forma galénica.3.- Vía de administración.4.- Nombre y dirección de la entidad farmacéutica elaboradora.5.- Nombre del director técnico responsable.6.- Nº de lote.7.- Fecha de caducidad.8.- Condiciones especiales de conservación.9.- Inscripción: “Muestra para investigación clínica.”

51
Muestras para investigación clínica.
- El servicio de farmacia de acuerdo con la normativa vigente, se responsabiliza de la: recepción, almacenamiento, control y dispensación del medicamento en estudio.
- Además de garantizar el uso correcto de los medicamentos, proporciona apoyo logístico a los investigadores y al promotor.

52
Una vez finalizado el E.C
Presentación de los resultados:
• Introducción•Metodología•Resultados•Discusión de los resultados•Conclusiones•Métodos estadísticos utilizados•Evaluacion

53
EVALUACIÓN EFICACIA Y SEGURIDAD

54
No PublicadoPublicado
Drotrecogina alfa en sepsis
Atazanavir.
•“Es el ensayo clínico que ha justificado la aprobación del medicamento por parte de las agencias reguladoras y la base de la ficha técnica”Es la principal fuente de información sobre eficacia y seguridad
ENSAYO CLÍNICO PIVOTAL:Eficacia
Autorización sin articulo publicado
Peginterferón alfa 2b para la Hepatitis C crónica

55
Emiten informes públicos donde revisan y discuten el/
los ensayos clínicos “pivotales”
Son muy útiles para:Contrastar e interpretar dichos
ensayos clínicos.
•Acceder a datos originales en caso de ensayos no
publicados
•Ampliar la información publicada:
Contiene opinión de expertos que permite contrastar la
información.
EMEA y FDA
ENSAYO CLÍNICO PIVOTAL: Agencias Reguladoras
56
INFORMES FDA
ENSAYO CLÍNICO PIVOTAL: Agencias Reguladoras
Medical Review

57
•Evaluación internacional:Evaluación internacional:Evaluación internacional:Evaluación internacional:
- Prescrire
- UKMi: CIM del Reino Unido
•Evaluación nacional:
-Información terapéutica del SNS
-PAM (Colegio Farmacéutico)
• Evaluaciones centros autonómicos
- Fichas de evaluación de nuevos medicamentos
•Otras fuentes útiles:
- Medical Letter. (Acceso restringido)
- Micromedex (Acceso desde la SEFH)
EVALUACIONES INDEPENDIENTES
FUENTES DE EVALUACION

58
Revista Prescrire http://www.prescrire.org/
Revista independiente francesa •Pionera en la clasificación y evaluación de novedades farmacéuticas. •Los medicamentos clasificados según su aportación:
•1.Bravo.•2. Importante mejora•3.Aporta alguna ventaja•4.Utilidad eventual•5.Nada nuevo•6.Juicio reservado (Insuficiente experiencia)•7.No aceptable.
EVALUACIONES INDEPENDIENTES: Internacional
FUENTES SECUNDARIAS

59
FUENTES SECUNDARIASEVALUACIONES INDEPENDIENTES: Internacional

60
FUENTES SECUNDARIASEVALUACIÓN NACIONAL
Sistema Nacional de Salud: Información Terapéutica.
Revisiones de Nuevos Principios Activos.A*:Novedad terapéutica excepcionalA:Importante mejora terapéuticaB: Modesta mejora terapéuticaC: Nula o pequeña mejora terapéuticaD: Sin Calificación.
•CGCOF: Panorama Actual del Medicamento.
•Sin Innovación
•Innovación moderada
•Innovación importante
•Innovación excepcional

61
CADIME: Centro Andaluz de Información de Medicamentos
CEVIME: Centro Vasco de Información de Medicamentos
CONSELLERIA SANITAT GENERALITAT VALENCIANA
SERGAS: Servicio Gallego de Salud
NAVARRA
SESCAM: Servicio Salud Castilla La Mancha
Direcciones en material adjunto.
FUENTES SECUNDARIASEVALUACIONES INDEPENDIENTES
Centros Autonómicos

62
FICHAS NOVEDAD TERAPÉUTICA
FUENTES SECUNDARIAS