análisis filogenético - docencia.unt.edu.ar · una de las diferencias entre las teorías...
TRANSCRIPT

A B C D E F
Terminal (especies)
Rama
Nodo interno (representa el ancestro común más reciente de D y E)
Grupos (clados) presentes en el árbol
DEBCDEFBCDEFABCDEF
Partes de un árbol filogenético
Las relaciones filogenéticas se pueden representar como una árbol filogenético
Una de las diferencias entre las teorías evolutivas de Lamarck y Darwin esta relacionada al origen de las especies.
Mientras que Lamarck planteaba que el origen de las mismas era por generación espontánea, Darwin introduce el concepto de
“comunidad de descendencia”. Planteaba que a partir de una especie podían formarse otras nuevas especies, existiendo de esta
manera una relación de parentesco entre las mismas.
La inferencia filogenética intenta establecer tales relaciones de parentesco entre especies.
Análisis Filogenético

Árbol filogenético
HOMBRE
PERRO
CARACOL
HELECHO
Permite establecer definiciones del tipo:
- “El hombre esta más relacionado con el perro que con el caracol”
- “El grupo formado por el hombre y el caracol comparten un ancestro común más reciente con el caracol que con el helecho”
- El hombre y el perro presentan pelos debido a que tal característica fue heredada de un ancestro”
- El linaje que conduce al helecho divergió antes que el linaje del caracol divergiera de aquel que conduce al hombre y el perro.
NO permite establecer definiciones del tipo:
- “El hombre es más evolucionado que el helecho”
- “El hombre y el perro descienden del caracol”
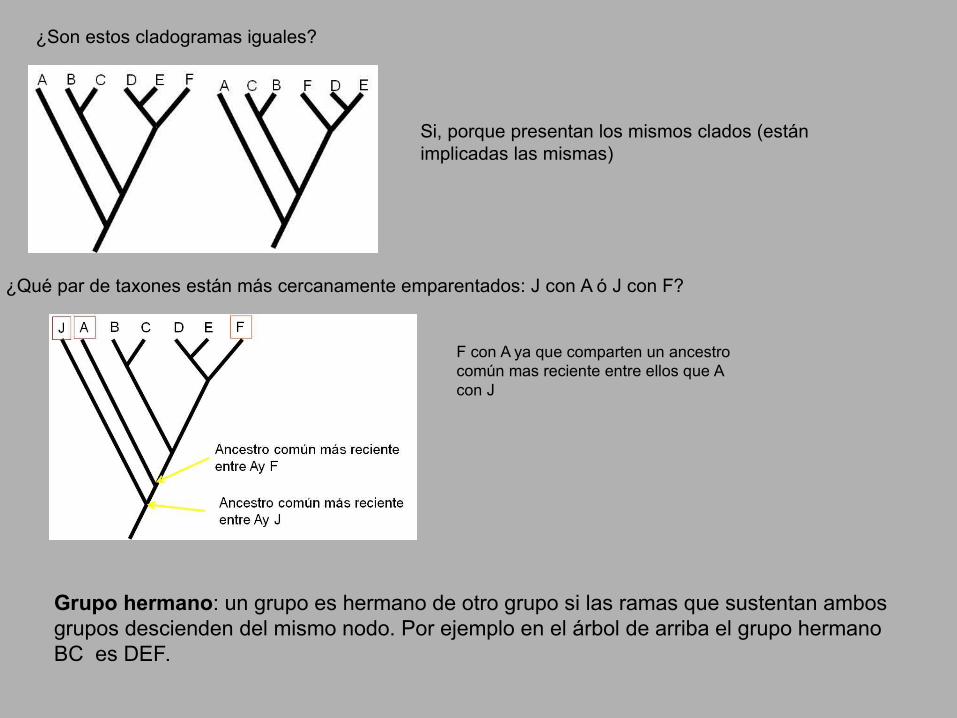
¿Son estos cladogramas iguales?
Si, porque presentan los mismos clados (están implicadas las mismas)
¿Qué par de taxones están más cercanamente emparentados: J con A ó J con F?
F con A ya que comparten un ancestro común mas reciente entre ellos que A con J
Grupo hermano: un grupo es hermano de otro grupo si las ramas que sustentan ambos grupos descienden del mismo nodo. Por ejemplo en el árbol de arriba el grupo hermano BC es DEF.

EVIDENCIA
MÉTODO
HIPÓTESIS PREFERIDA
ANALISIS FILOGENETICO
¿Cual es la evidencia puedo utilizar para inferir las relaciones filogenéticas?
Las características de las especies que pueden ser definidas en los organismos estudiados. Como requisito tales estructuras deben ser heredables (estén definidos por el genotipo del individuo y no sea variación debida al ambiente)
Caracteres Morfológicos
EVIDENCIA
Caracteres de comportamiento
Caracteres moleculares (secuencias de ADN)

Presencia /ausencia de tejidos de conducción en plantas
Presencia /ausencia de flores
Presencia /ausencia de columna vertebral
Presencia /ausencia de estructuras
Presencia /ausencia de cilias
Caracteres Morfológicos
Forma
Variación en las estructuras
Color Número
Insectos: 6 patas

Caracteres de comportamiento
Cuidado parental AlimentaciónSociabilidad
Caracteres moleculares (Secuencias de ADN)
Alineamiento de secuencias
Cada posición en la secuencia de ADN representa un carácter y cada base representa los distintos estados.

Caracter: atributo de un organismo
Estados del caracter: diferentes variantes que puede tomar el caracter
Ejemplos:
Carácter: Color de la alas Estados: Amarillo, rojo
Carácter: Forma cuerpo. Estados: largo , corto
Carácter: alas Estados: presente-ausente
Posición nucleotídica (base) 245 en el gen rbcL Estados: Adenina (A) , Guanina (G), Timina (T), Citocina (C)
Matriz de datos para análisis filogenéticos
Permite sistematizar las observaciones para poder inferir relaciones filogenéticas .
Carácter 0 : estrella: Estados: Presente (1) Ausente (0) Carácter 1: línea: Estados: recta (0) curva (1) Carácter 2: color Estados: blanco (0) amarillo (1) rojo (2) Carácter 3: luna Estados: Presente (1) Ausente (0)
Carácter 4: banda azul Estados: Presente (1) Ausente (0) Carácter 5: rombo Estados: Presente (1) Ausente (0)
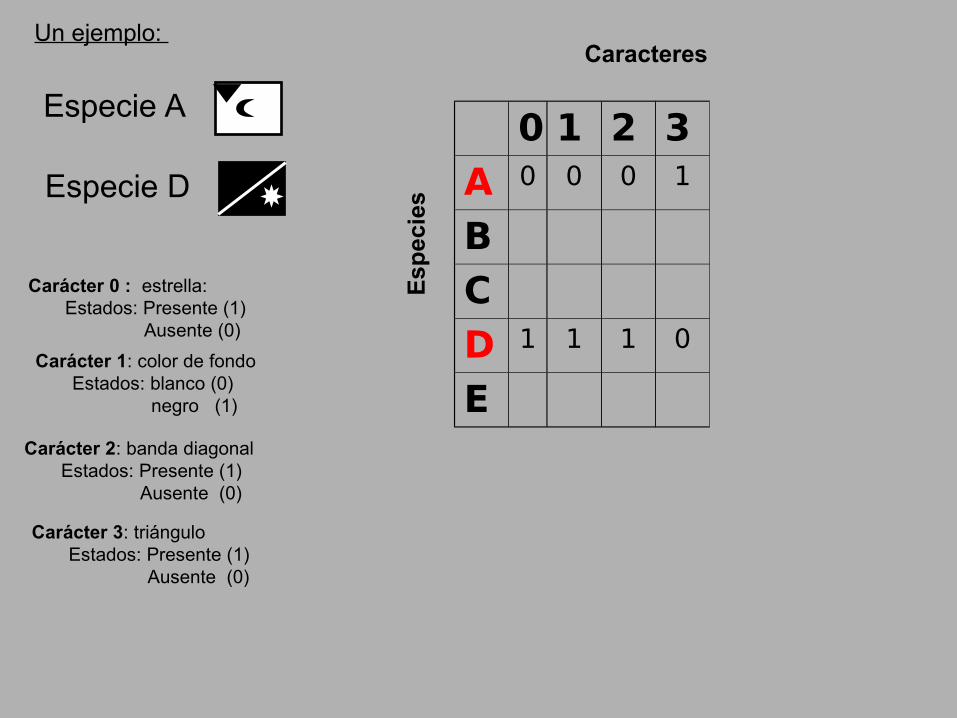
Un ejemplo:
Especie A
Especie D
0 1 2 3
A 0 0 0 1
B
C
D 1 1 1 0
EE
spec
ies
Caracteres
Carácter 0 : estrella: Estados: Presente (1) Ausente (0)
Carácter 1: color de fondo Estados: blanco (0) negro (1)
Carácter 2: banda diagonal Estados: Presente (1) Ausente (0)
Carácter 3: triángulo Estados: Presente (1) Ausente (0)

EVIDENCIA
MÉTODO
HIPÓTESIS PREFERIDA
ANALISIS FILOGENETICO
Métodos
Existe una amplia variedad de métodos y criterios para la elección de las hipótesis filogenéticas. Para analizar datos moleculares, los métodos basados en modelos probabilísticos de evolución molecular (métodos de máxima verosimilitud y métodos bayesianos; ver Felsenstein, 2004) son usados más comúnmente que parsimonia. Bajo ciertos presupuestos evolutivos, esos métodos producirían mejores resultados que el método de parsimonia; si bien existe algún disenso acerca de la aplicabilidad universal de esos presupuestos evolutivos, es cierto que los presupuestos tienen bastante asidero en nociones bien establecidas en el caso de las secuencias moleculares, y que las circunstancias ideales en que los métodos probabilísticos funcionarían correctamente son relativamente bien conocidas. En el caso de caracteres morfológicos, la mayoría de las justificaciones que pueden invocarse para los datos de secuencias moleculares carecen de validez, y en consecuencia el criterio más utilizado para al análisis de datos morfológicos es el de máxima parsimonia. El principio de parsimonia en su versión moderna está basado en los desarrollos conceptuales y metodológicos de Steve Farris - un científico estadounidense que actualmente reside en Suecia- durante la década del 70 y principios de la década del 80 del siglo pasado (Farris, 1970; Farris, 1983). A partir de estos trabajos se han seguido publicando desarrollos metodológicos basados en el principio de parsimonia,
Parsimonia
Métodos basados en Modelos Probabilísticos

El criterio de parsimonia, como se entiende hoy en día, prefiere aquella hipótesis filogenética que implique la menor cantidad de “pasos” o diferencias (potencialmente con diferente peso) a lo largo de las ramas del árbol, con los estados ancestrales reconstruídos de manera de minimizar estas diferencias. Cado “paso” en el árbol equivale a un cambio de estado (posibles variantes que puede tomar un carácter). Al minimizar el número de pasos lo que uno está haciendo es maximizar las instancias de similitud de los organismos incluidos en la filogenia que pueden ser explicadas por ancestralidad común (Fig. 2). Es decir, a la pregunta de “por qué estas dos especies A y B comparten el estado del carácter dado”, podemos responder “porque lo han heredado de un ancestro en común” sólo cuando todos los ancestros en el camino más corto entre A y B hayan sido reconstruidos con el mismo estado que A y B. En el caso contrario (es decir, cuando los ancestros intermedios entre A y B tienen una condición distinta para el carácter), nuestra única explicación posible sería … “no sé” (es decir, un paralelismo, convergencia, o reversión, que no son explicados de forma directa por una filogenia; la filogenia sólo permite explicar mediante ancestralidad común aquellas sintancias de similitud, no las de diferencias). La reconstrucción que minimiza la cantidad de casos en que soy incapaz de proveer una explicación en términos de ancestralidad común a una semejanza observada, es la misma que minimiza el número de pasos evolutivos, y por lo tanto la clasificación que minimiza la cantidad de diferencias es la que provee la mejor explicación (en términos de ancestralidad común) de las semejanzas observadas en el grupo en estudio.
Parsimonia

0 1 0 0 0 0 1
00
0
0
0
0
0 0 0 1 1 1 0
11
1
0
0
0
1 0 1 2 2 2 0
22
2
1
1
1
Reversión ConvergenciaParalelismo
Dijimos que elegiamos la hipotesis que puede explicar por ancestralidad comun la mayor cantidad de similitudes. Ahora que ocurre con las similitudes no explicadas por ancestralidad comun? Las llamamos HOMOPLASIAS
Que haya homoplasia en los datos quiere decir que existe evidencia contradictoria con respecto a los agrupamientos. Es el criterio de parsimonia el que define entonces cual va a ser el agrupamiento preferido en función de maximice la similitud explicable por ancestralidad común. Cuando uno hace esto tambien esta minimizando las homoplasias. La hopótesis filogenética óptima no es aquella que no tiene homoplasia sino la que minimiza la homoplasia.Todas las matrices filogeneticas reales presentan algún nivel de homoplasia.

- Desde un punto de vista práctico elegir el árbol bajo el criterio de parsimonia se resumen en calcular la cantidad de pasos (cambios de estado) de cada uno de los caracteres y sumarlos para todos los caracteres para calcular el largo del árbol. Aquel árbol con menor cantidad de pasos es el árbol (ó los árboles) óptimo.
- Dado un árbol se calcula el numero de pasos para cada carácter
- Se suman los pasos de todos los caracteres. Esa suma será el largo del árbol
-De la misma manera se calcula el largo para otros árboles, siendo el árbol elegido (la hipótesis preferida) aquella que tenga el mínimo numero de pasos. Esta será la hipótesis que permitirá explicar por ancestralidad común la mayor cantidad de similitudes.

Matriz filogenética c1 c2 C3 Sp. A 1 1 1 Sp. B 1 1 0Sp. C 0 0 0Sp. D 0 0 1Sp. E 0 0 0C1= carácter 1C2= carácter 2C3= carácter 3
Árbol 1 Árbol 2
Carácter 3
A B C D E A D C B E
1 0 0 1 0
0
0
00 2 PASOS
1 1 0 0 0
1
00
01 PASO
Carácter 2
Carácter 1
1 1 0 0 0
1
00
01 PASO
1 0 0 1 0
1
00
02 PASOS
1 0 0 1 0
1
00
02 PASOS
1 1 0 0 0
1
0
00 1 PASO
Largo total 1+1+2= 4 1+1+2=5
Según el criterio de parsimonia el árbol 1 es preferible sobre el árbol 2 porque tiene menor cantidad de pasos.

Búsquedas filogenéticas
Como hemos visto para elegir la hipótesis filogenetica bajo el criterio de parsimonia hay que obtener el árbol (o los árboles) que presenten la menor cantidad de pasos. Si la matríz es pequeña tanto en numero de caracteres como en numero de terminales (especies), uno podría calcular a mano el número de pasos de cada uno de lo
Cuando en número de especies es muy pequeño, se pueden evaluar cada una de las hipótesis. Sin embargo cuando el numero de especies aumenta la cantidad de árboles a evaluar puede ser enorme y hace imposible la evaluación de todas las hipótesis.

Tipos de caracteres
- Según el tipo de variaciónCaracteres discretos: la variación se representa en un numero finito de variantes (estados) Caracteres continuos: la variación se representa como valores de una variable continua.
Dicotomías y politomíasNodo dicotómico: nodo a partir del cual surgen 2 ramas descendientes.
Nodo politómico (o politomía): nodo a partir del cual surgen 3 o más ramas descendientes. Una politomía en un árbol filogenético indica que la información presente en los caracteres no permite determinar las relaciones entre las ramas descendientes.
A C B D E F G
Nodo politómico
Nodo dicotómico
Según el número de estados: Caracteres binarios: presentan dos posibles estados. Caracteres multiestado: más de dos estados
- Según los grados relativos de similitud.
Caracteres desordenados: Cuando no hay grados diferentes de similitud entre estados. Caracteres ordenados: Cuando existen grados de similitud diferentes entre los estados, es decir algunos estados se parecen más entre ellos.

Parsimonia
Métodos basados en Modelos Probabilísticos
Distancia
Máxima Verosimilitud
Análisis Bayesiano
Máxima Verosimilitud: elige el árbol que (dado el modelo de evolución) haga más probables mis observaciones (los estados de carácter para cada especie incluida en el análisis. Requiere de un modelo explícito de evolución. El método NO elige el árbol más probable
Al elegir un árbol bajo máxima verosimilitud también hay que hacer búsquedas filogenéticas como en parsimonia pero solamente el criterio de elección no es parsimonia sino máxima verosimilitud
Métodos Filogenéticos

¿Para que puede resultar relevante los estudios filogenéticos?
Establecer Clasificaciones Biológicas
Clasificación biológica: consiste en el ordenamiento de la diversidad biológica en grupos. Permite sistematizar el conocimiento y transmitir información biológica
EpidemiologíaEstudios evolutivos
EVIDENCIA
MÉTODO
HIPÓTESIS PREFERIDA
ANALISIS FILOGENETICO

Grupo Monofilético: Es un grupo que incluyen a todos los terminales descendientes de un nodo en la filogenia de referencia
Grupo Parafilético: Es un grupo que incluye a muchos pero no todos los terminales descendientes de un nodo de la filogenia de referencia.
Grupo Polifilético: Es un grupo que incluye terminales ubicados en posiciones lejanas del árbol.
La sistemática filogenética plantea que los grupos de las clasificaciones deben corresponderse con grupos presentes en la filogenia. Los grupos que pueden formar parte de clasificaciones son los GRUPOS MONOFILETICOS: grupos formados por TODOS los terminales descendientes de un nodo.
Las clasificaciones biológicas son JERÁRQUICAS (grupos dentro de grupos)

Clasificación hecha por Juan Pérez
Familia “A” integrada por spA, spB y spC:
Familia “B” integrada por spD, spE, spF, spG
Clasificación hecha por Agapito Gómez
Familia A integrada por spA, spG
Familia “B” integrada por spB, spD, spE, spF, spG
Filogenia del grupo
La clasificación de Juan Pérez está de acuerdo a los principios de la sistemática filogenética porque los grupos definidos (en este caso familias) son grupo monofiléticos en la filogenia de referencia del grupo.
La clasificación de Agapito Gómez No está de acuerdo a los principios de la sistemática filogenética porque los grupos definidos (en este caso familias) son grupos polifiléticos (Familia A) y parafiléticos (Familia B), es decir ninguno de los grupos son monofiléticos.
Ejemplo

Características de mamíferos
Glandulas mamarias
Mandíbula formada por el hueso dentario
Presencia de pelo
El análisis filogenético no solo sirve para definir que grupos son monofiléticos, sino que también permite diagnosticar los grupos de una clasificación mediante sus sinampomorfías. Se llaman SINAPOMORFÍAS a los caracteres que son compartidos por los integrantes de un grupo monofiletico y que son derivados en el nodo ancestral del grupo en cuestión.

Perro
Zorro
Gato
León
Pantera
Canguro
Trucha
Lagartija
Camarón
1
1
1
1
1
1
0
0
0= Ausencia de pelo
1 =Presencia de pelos
1
11
11
0
0
0
0
La presencia de pelos (estado 1) es una SINAPOMORFIA del grupo Mamiferos porque es un estado compartido y derivado (cambio en la rama que conduce al ancestro de Mamiferos
La ausencia de pelos es un estado de carácter compartido por el grupo “Lagartija-Trucha-Camarón”. Sin embargo como en esta filogenia es un estado primitivo, NO es una sinapomorfía de estas especies sino una SINPLESIOMORFÏA

Perro
Zorro
Gato
León
Pantera
Canguro
Trucha
Lagartija
Camarón
1
1
1
1
1
1
1
0
0= Ausencia de c. vertebral
1 =Presencia de c. vertebral
1
11
11
1
1
1
0
La presencia de columna vertebral NO es una sinapomorfía del grupo Mamíferos porque aunque es un estado compartido NO es un estado derivado (No cambia en la rama que conduce al ancestro de Mamíferos). Por esto la presencia de columna vertebral no esta asociada a la diagnosis del grupo. De hecho esta asociada a la diagnosis de otro grupo: el de vertebrados.

Estudios evolutivos
A B C D E F G H I J F
Tipo de alimentación:0= herbívoro1= carnívoro
1 0 0 1 0 0 1 0 1 1 0
0
0 0
0
0
0
0
0 0
1
La carnivoría se habría desarrollado independientemente 4 veces a lo largo de la evolución del grupo estudiado
El ancestro del grupo habría sido herbívoro
Cuando los estados entre dos nodos conectados por una rama son diferentes se infiere que ha habido un CAMBIO EVOLUTIVO. A la hora de calcular el largo del árbol a estos cambios los denominamos PASOS.
Es posible utilizar el principio de parsimonia para estudiar la evolución de caracteres sobre una filogenia. Los estados que se asignan a los nodos internos son aquellos óptimos bajo el criterio de parsimonia.