universidad central del ecuador facultad de … · a los chicos de la familia de nanoestructuras...
TRANSCRIPT

UNIVERSIDAD CENTRAL DEL ECUADOR
FACULTAD DE CIENCIAS QUÍMICAS
CARRERA DE QUÍMICA FARMACÉUTICA
TITULO DE TESIS
“Diseño y elaboración de micropartículas cargadas con Diclofenaco mediante la técnica de
doble emulsificación”
AUTORA: Geovanna Catalina Valenzuela Molineros
Tesis para optar por el Título Profesional de
QUÍMICO FARMACÉUTICO
TUTOR: Msc. Pablo Mauricio Bonilla Valladares
Quito, Septiembre 2015

ii
Geovanna Catalina Valenzuela Molineros (2015).
Diseño y elaboración de micropartículas cargadas con
Diclofenaco mediante la técnica de doble
emulsificación. Trabajo de investigación para optar por
el título de Químico Farmacéutico. Carrera de Química
Farmacéutica. Quito: UCE. 115 p.

iii
DEDICATORIA
A mis padres
Porque así como hoy, mis logros han sido el resultado de su sacrificio, apoyo y amor
incondicional.
Les amo

iv
AGRADECIMIENTO
Mi primer agradecimiento va dirigido hacia Dios, por la manera en que ha guiado mi vida,
poniéndome situaciones y personas en el camino que me han dado la mano y me han ayudado a
avanzar para la culminación de este objetivo.
A mis padres por todo el amor, por su sacrificio, por cuidar de mis bebes como si fueran suyos y
permitirme terminar este trabajo.
Al Msc. Pablo Bonilla director de esta tesis quien sin conocerme confío en mí, me tendió su mano
y supo acompañarme durante todo este proceso, quien a más de compartirme sus conocimientos
me alentaba día a día para no caer en este largo camino.
A la Msc. Verito Taco y al BF. Jorge Moncayo por su apoyo y paciencia.
A Diego Lucero por su visión crítica de muchos aspectos, por compartir conmigo sus
conocimientos y apoyarme con el manejo de los equipos.
A mis queridos maestros quienes han sido parte tanto de mi formación profesional como personal,
a la Dra Liliana Naranjo, Dra Ximena Chiriboga, (†) Dr. Patricio Miño y BF. Miguel de la Cadena,
por estar pendientes no solo del avance de este trabajo sino también por sus consejos y amistad.
A ti esposo mío por haberme acompañado en este camino con paciencia, disfrutando de mis logros
y siendo mi soporte cuando las cosas se ponían difíciles y junto contigo a mis compañeros Matias
y Esteban.
A ustedes Kari y Cami por el amor y la compañía, por su preocupación y la ayuda que me han
dado.
A ti abuelito por estar pendiente de mi desarrollo profesional y personal, y por tu amor.
A los ayudantes de los laboratorios por los que esta tesis ha cruzado, por facilitarme los materiales
que he necesitado. A los chicos de la familia de Nanoestructuras por hacer que los días en el
laboratorio sean toda una aventura.
A “Los de siempre” por que estuvieron y siguen estando presentes en mi vida. Magui, Geomi,
Pablo gracias por haber estado junto a mí en momentos cruciales de mi vida y por demostrarme
tanto cariño.
A todos mis amigos por permitirme tener en mi memoria momentos que me recuerdan lo mucho
que disfrute de esta etapa de mi vida.

v
UNIVERSIDAD CENTRAL DEL ECUADOR
FACULTAD DE CIENCIAS QUÍMICAS
CARRERA DE QUÍMICA
Yo, GEOVANNA CATALINA VALENZUELA MOLINEROS, declaro conocer y aceptar la
disposición del Artículo 214, numerales 3 y 8 del Estatuto de la Universidad Central del Ecuador
que señala: “El patrimonio de la Universidad Central está constituido por lo siguiente:
Las publicaciones, memorias, obras de arte, tesis, investigaciones científicas y
tecnológicas, y lo que a futuro produjere.
Los beneficios provenientes de inversiones, investigación, prestación, cursos, seminarios,
consultorías y venta de servicios, patentes, marcas y otros conceptos, desarrollados por
las diferentes unidades académicas de la Universidad”
Geovanna Valenzuela
1715950554

vi
UNIVERSIDAD CENTRAL DEL ECUADOR
FACULTAD DE CIENCIAS QUÍMICAS
CARRERA DE QUÍMICA Y FARMACIA
Por la presente, dejo constancia que he leído la tesis presentada por la Señorita Valenzuela
Molineros Geovanna Catalina, para optar por el título profesional cuyo tema es; “Diseño y
elaboración de micropartículas cargadas con Diclofenaco mediante la técnica de doble
emulsificación” la misma que reúne los requerimientos, y los méritos suficientes para ser
sometida a evaluación por el tribunal calificador
En la ciudad de Quito, a los 07 días del mes de agosto del 2015
Msc. Pablo Mauricio Bonilla Valladares
C.C.: 1709888240

vii

viii
LUGAR EN DONDE SE REALIZÓ LA INVESTIGACIÓN
La presente investigación se realizó en la Facultad de Ciencias Químicas de la Universidad
Central del Ecuador, en los laboratorios de: Coloideoquímica, Nanoestructuras, Tecnología
Farmacéutica, y Productos Naturales.

ix
CONTENIDO
RESUMEN .................................................................................................................................... 1
ABSTRACT .................................................................................................................................. 2
INTRODUCCION ........................................................................................................................ 3
CAPÍTULO I ................................................................................................................................. 4
PLANTEAMIENTO DEL PROBLEMA. ..................................................................................... 4
FORMULACIÓN DEL PROBLEMA .......................................................................................... 5
HIPÓTESIS ................................................................................................................................... 5
OBJETIVOS DE LA INVESTIGACION ..................................................................................... 6
IMPORTANCIA Y JUSTIFICACIÓN ......................................................................................... 7
CAPÍTULO II ............................................................................................................................... 8
MARCO TEÓRICO ...................................................................................................................... 8
ANTECEDENTES ........................................................................................................................ 8
FUNDAMENTO TEÓRICO....................................................................................................... 10
2.1 SISTEMAS DE LIBERACION MODIFICADA (SLM) ............................................ 10
2.1.1 CLASIFICACIÓN DE LOS SISTEMAS DE LIBERACIÓN MODIFICADA 10
2.1.2 SISTEMAS TRANSPORTADORES DE FARMACOS .................................... 12
2.1.3 MICROCÁPSULAS ........................................................................................... 14
2.1.4 MICROENCAPSULACIÓN ..................................................................................... 15
2.1.5 METODO DE DOBLE EMULSIFICACIÓN EVAPORACION-EXTRACCION
DEL SOLVENTE ............................................................................................................... 20
2.1.5.1 EMULSIONES.................................................................................................... 21
2.1.6 TECNICA DE DOBLE EMULSIFICACION, EVAPORACION –
EXTRACCION DEL SOLVENTE ..................................................................................... 28
2.1.7 RUTAS DE ADMINISTRACIÓN DE LAS MICROPARTICULAS ................ 34
2.1.8 MATERIAS PRIMAS UTILIZADAS EN LA INVESTIGACIÓN .................. 34
2.1.10 LIBERACION DEL PRINCIPIO ACTIVO DESDE LA MICROPARTICULA 45
2.1.11 DESVENTAJAS DE LOS SISTEMAS DE LIBERACION MODIFICADA .... 53
MARCO LEGAL ........................................................................................................................ 54

x
2.2.1 REGLAMENTO DE BUENAS PRÁCTICAS DE MANUFACTURA (B.P.M.)
PARA LA INDUSTRIA FARMACÉUTICA ..................................................................... 54
2.2.2 NORMA TECNICA ECUATORIANA (NTE INEN-ISO/TR 12885) .............. 54
CAPÍTULO III ............................................................................................................................ 55
METODOLOGÍA ....................................................................................................................... 55
TIPO DE INVESTIGACIÓN ...................................................................................................... 55
POBLACION Y MUESTRA .................................................................................................. 55
DISEÑO EXPERIMENTAL................................................................................................... 55
DIAGRAMA DE FLUJO METODOLÓGICO ...................................................................... 56
3.1.1 VARIABLES ...................................................................................................... 57
3.1.2 INSTRUMENTOS ANALITICOS ..................................................................... 57
3.1.2.1 MATERIALES.................................................................................................... 57
3.1.2.2 EQUIPOS ............................................................................................................ 57
3.1.2.3 REACTIVOS ...................................................................................................... 58
3.1.3 MÉTODOS.......................................................................................................... 58
CAPITULO IV ............................................................................................................................ 64
4.1 RESULTADOS Y DISCUCIONES ................................................................................ 64
4.1.1 DISEÑO DE LA EMULSION ................................................................................... 64
4.1.1 EXTRACCION DE LAS MICROPARTICULAS POLIMERICAS .................. 72
4.1.2 DETERMINACIÓN DEL TAMAÑO DE LAS PARTÍCULAS ........................ 73
4.1.3 CONTENIDO DE PRINCIPIO ACTIVO ........................................................... 74
4.1.4 RENDIMIENTO DE LA PRODUCCION .......................................................... 75
4.1.5 EFICACIA DE LA ENCAPSULACION ............................................................ 75
4.1.6 PERFILES DE LIBERACIÓN DEL PRINCIPIO ACTIVO EN LAS
MICROPARTICULAS. ...................................................................................................... 76
4.1.7 PERFILES DE LIBERACIÓN DEL PRINCIPIO ACTIVO DESDE LA
EMULSION ........................................................................................................................ 77
4.1.8 PERFILES DE LIBERACIÓN DEL PRINCIPIO ACTIVO DESDE EL
EMULGEL (VOLTAREN®) ............................................................................................... 79

xi
4.1.9 MORFOLOGIA DE LAS MICROPARTICULAS OBTENIDAS EN LA
EMULSION ........................................................................................................................ 82
CAPITULO V ............................................................................................................................. 84
CONCLUSIONES Y RECOMENDACIONES .......................................................................... 84
5.1 CONCLUSIONES ...................................................................................................... 84
5.2 RECOMENDACIONES ............................................................................................. 85
BIBLIOGRAFÍA ......................................................................................................................... 87
ANEXOS..................................................................................................................................... 92

xii
LISTA DE TABLAS
Tabla 2. 1 Propiedades de algunos solventes utilizados en la elaboración de micropartículas. ________ 29 Tabla 2. 2 Efecto de los factores que influencian en las características de las micropartículas ________ 33 Tabla 2. 3 Propiedades del Limoneno _____________________________________________________ 35 Tabla 2. 4 Propiedades Limoneno Estandar. Vs Limoneno muestra _____________________________ 35 Tabla 2. 5 Propiedades de los surfactantes utilizados en la experimentación ______________________ 38 Tabla 3. 1 Cantidad de agente surfactante probado en la formulación __________________________ 59
Tabla 4. 1 Adición del agente surfactante. _________________________________________________ 64
Tabla 4. 2 Adición de la gelatina. _______________________________________________________ 66
Tabla 4. 3 Adición del PVA en solución. ___________________________________________________ 68
Tabla 4. 4 Incremento en la concentración de alcohol polivinílico. _____________________________ 70
Tabla 4. 5 Porcentajes de los componentes utilizados en la formulación _________________________ 71
Tabla 4. 6 Datos y contenido de principio activo, determinación espectrofotométrica. ______________ 74
Tabla 7. 1 Curva de calibración para la determinación del contenido de principio activo. ____________ 94

xiii
LISTA DE FIGURAS
Figura 2. 1 Perfiles de concentración plasmática ____________________________________________ 11 Figura 2. 2 Clasificación de los sistemas transportadores de fármacos. __________________________ 12 Figura 2. 3 Representación de una microcápsula ____________________________________________ 14 Figura 2. 4 Diagrama del proceso de microencapsulación por coacervación. ______________________ 18 Figura 2. 5 Clasificación de las emulsiones , ________________________________________________ 22 Figura 2. 6 Emulsiones dobles vistas al microscopio. _________________________________________ 22 Figura 2. 7 Proceso de coalescencia en una emulsión. ________________________________________ 24 Figura 2. 8 Representación del movimiento browniano _______________________________________ 24 Figura 2. 9 Descripción del proceso de floculación ___________________________________________ 25 Figura 2. 10 Pasos generales para la formación de microcápsulas por el método de emulsificación
evaporación-extracción del solvente mediante una emulsión w/o/w ___________________________ 28 Figura 2. 11 Estructura del limoneno _____________________________________________________ 35 Figura 2. 12 Alcohol polivinílico __________________________________________________________ 37 Figura 2. 13 Estructura molecular del Tween ® 80. __________________________________________ 38 Figura 2. 14 Estructura molecular del Span® 80. ____________________________________________ 38 Figura 2. 15 Estructura del Diclofenaco de sodio ____________________________________________ 39 Figura 2. 16 Estructura del diclofenaco dietilamonio (DDEA) __________________________________ 40 Figura 2. 17 Tamaño de partícula analizado con el programa ImageJ ___________________________ 43 Figura 2. 18 Lectura del tamaño de partícula en el equipo DLS. ________________________________ 43 Figura 2. 19 Esquema de la columna de flujo continuo. _______________________________________ 50 Figura 2. 20 Curvas de concentración plasmática ___________________________________________ 52 Figura 4. 1 Formulaciones A y B respectivamente vistas al microscopio. (100x). ___________________ 65
Figura 4. 2 Formulaciones C y D respectivamente vistas al microscopio. (100x). ___________________ 65 Figura 4. 3 Formulaciones A, B, C y D (izquierda a derecha) ___________________________________ 65 Figura 4. 4 Formulación D1 y D2 vista al microscopio. (100x). _________________________________ 66 Figura 4. 5 Formulación D3 y D4 vista al microscopio. (100x). _________________________________ 67 Figura 4. 6 Formulaciones D1, D2, D3 y D4. ________________________________________________ 67 Figura 4. 7 Formulaciones con polivinilalcohol ______________________________________________ 68 Figura 4. 8 Formulación D1-1 y D1-2 ______________________________________________________ 69 Figura 4. 9 Formulaciones a pH 6.8 y _____________________________________________________ 69 Figura 4. 10 Formulación D1-5. __________________________________________________________ 70 Figura 4. 11 Partículas obtenidas. ________________________________________________________ 72 Figura 4. 12 Espectro Ultravioleta obtenido de la prueba de liberación del _______________________ 76 Figura 4. 13 Comparación de los perfiles de liberación de las emulsiones ________________________ 77 Figura 4. 14 Linealización para la cinética de orden cero ______________________________________ 77 Figura 4. 15 Linealización para la cinética de primer orden ____________________________________ 78 Figura 4. 16 Linealización para la cinética de segundo orden __________________________________ 78 Figura 4. 17 Linealización para la cinética de Higuchi ________________________________________ 78 Figura 4. 18 Curva de liberación del diclofenaco dietilamonio (promedio)________________________ 79 Figura 4. 19 Linealización para la cinética de orden cero ______________________________________ 79 Figura 4. 20 Linealización para la cinética de primer orden ____________________________________ 80 Figura 4. 21 Linealización para la cinética de segundo orden __________________________________ 80 Figura 4. 22 Linealización para la cinética de Higuchi ________________________________________ 80 Figura 4. 23 Comparación de la cinética de liberación entre la emulsión vs Voltaren _______________ 81 Figura 4. 24 Morfología determinada mediante microscopia de fuerza atómica. __________________ 82 Figura 4. 25 Fotografía obtenida mediante Microscopia de fuerza atómica en 3D (Fase) ____________ 83

xiv
Figura 7. 1 Procedimiento de elaboración de la emulsión múltiple ______________________________ 92 Figura 7. 2 Extracción de las partículas ____________________________________________________ 92 Figura 7. 3 Determinación del tamaño de partícula __________________________________________ 93 Figura 7. 4 Proceso De liberación del principio activo ________________________________________ 93 Figura 7. 5 Morfología de las partículas durante el proceso de secado, determinación por microscopia
óptica (60X). _________________________________________________________________________ 94 Figura 7. 6 Gráfico de la curva de calibración de Diclofenaco sódico ____________________________ 95 Figura 7. 7 Gráfico de la curva de calibración de Diclofenaco sódico ____________________________ 96 Figura 7. 8 Barrido espectral de los componentes del sistema _________________________________ 97 Figura 7. 9 Cromatograma de la liberación desde la emulsión _________________________________ 97 Figura 7. 10 Cromatograma de la liberación desde Voltaren® emulgel __________________________ 98

xv
LISTA DE ECUACIONES
Ecuación 2. 1 Relación entre la energía libre y el área superficial de las gotas _____________________ 21 Ecuación 2. 2 Contenido de principio activo en la micropartícula _______________________________ 44 Ecuación 2. 3 Rendimiento de la producción. _______________________________________________ 44 Ecuación 2. 4 Porcentaje de principio activo encapsulado ____________________________________ 44 Ecuación 2. 5 Cinética de orden cero ______________________________________________________ 47 Ecuación 2. 6 Cinética de primer orden ____________________________________________________ 47 Ecuación 2. 7 Cinética de segundo orden __________________________________________________ 47 Ecuación 2. 8 Ecuación de Higuchi________________________________________________________ 48 Ecuación 2. 9 Porcentaje de liberación del fármaco __________________________________________ 48
Ecuación 4. 1 Ecuación de Grubbs ________________________________________________________ 75

xvi
GLOSARIO
Equivalentes farmacéuticos
Son productos que contienen cantidades idénticas del mismo principio activo, por ejemplo, la
misma sal o éster de la especie terapéutica, en la misma forma farmacéutica, pero que no
necesariamente contienen los mismos ingredientes inactivos, y que reúnen idénticos estándares
de identidad, potencia, calidad y pureza y, cuando es aplicable, uniformidad de contenido, tiempo
de desintegración y/o velocidad de disolución.
Doble emulsificación
Proceso mediante el cual se obtiene una emulsión simple dentro de otra emulsión.
Nanoemulsión
Son emulsiones cuyo tamaño de gota está en la escala manométrica (20-200 nm) y es debido a su
tamaño que aparecen transparentes a simple vista y posee buena estabilidad ante la sedimentación
y formación de crema.
Microscopio de fuerza atómica o AFM por sus siglas en inglés
Equipo utilizado para el análisis de superficies a escala atómica mediante las fuerzas de repulsión
electrónica entre la superficie y la punta de un microscopio de sonda que se desplazan por encima
de la muestra.
DLS (Dinamyc Light Scattering)
Técnica físico-química empleada para la determinación de la distribución de tamaños de
partículas en suspensión mediante la lectura de las fluctuaciones en la intensidad en función del
tiempo.
Partículas con menor tamaño debido al movimiento browniano se mueven rápidamente con lo
que se acelera la variación de intensidad de dispersión, lo que no sucede con partículas más
grandes que llevan a variaciones más lentas.

xvii
LISTA DE ANEXOS
Anexo 7. 1 Procedimiento utilizado en la técnica de doble emulsificación-extracción del solvente
__________________________________________________________________________ 92
Anexo 7. 2 Curva de calibración para la cuantificación de diclofenaco sódico _____________ 94
Anexo 7. 3 Curva de calibración para la determinación de Diclofenaco dietilamonio _______ 95
Anexo 7. 4 Barridos espectrales realizados a los componentes de la matriz de las
micropartículas elaboradas con diclofenaco sódico _________________________________ 97
Anexo 7. 5 Cromatogramas ____________________________________________________ 97
Anexo 7. 6 Calculo del BHL requerido para la emulsión _______________________________ 98

1
RESUMEN
Se ha diseñado una metodología para la elaboración de micropartículas poliméricas siguiendo la
técnica de doble emulsificación (w/o/w). La plantilla para la obtención de partículas esféricas es
una emulsión preparada con limoneno como sustituto a los solventes orgánicos utilizados de
forma común en esta técnica, una solución de gelatina-diclofenaco como fase acuosa interna y
una solución de polivinilalcohol como fase acuosa externa.
En una primera etapa se diseñó y preparó una emulsión con diclofenaco de sodio para la
formación de partículas sólidas y otra con diclofenaco dietilamonio como vehículos del fármaco.
En una segunda etapa se realizaron los ensayos de caracterización y la determinación de la cinética
de liberación de los vehículos. Las micropartículas sólidas con un 28.08 % de principio activo
encapsulado, 27.81% de eficacia de la encapsulación y 67.84% de rendimiento de producción no
permitió los resultados esperados en cuanto a la liberación, mientras que desde la emulsión se
liberó el 91.82% de diclofenaco a las 72 horas comparado con el 98% liberado a las 24 horas
desde la forma comercial concluyendo la obtención de un sistema de liberación prolongada.
PALABRAS CLAVE: MICROPARTICULAS, DOBLE EMULSIFICACIÓN, SISTEMAS DE
LIBERACION MODIFICADA, PERFILES DE LIBERACION.

2
ABSTRACT
This document presents the design of a methodology for the elaboration of polymeric micro
particles following the double emulsification technique (w/o/w). The emulsion has been used as
a template to obtain spherical particles, prepared with limonene as a substitute for organic solvents
used in common form of this technique, a solution of gelatin-diclofenac as an internal aqueous
phase and a polyvinyl alcohol solution as an external aqueous phase.
In the first stage it was designed and prepared an emulsion containing sodium diclofenac and
other with diethyl ammonium diclofenac such as drug vehicle
In a second stage assays characterization and determination of the kinetics of release of the drug
were performed. The solid micro particles with 28.08 % of drug encapsulated, 27.81 % of
encapsulation efficiency and 67.84 % production yield did not allow the desired results in terms
of release, while allowing the emulsion release diclofenac 91.82 % at 72 hours compared to 98%
released after 24 hours from completing commercially obtaining a prolonged release system.
Keywords: MICRO PARTICLES, DOUBLE EMULSIFICATION, MODIFIED LIBERATION
SYSTEMS, LIBERATION PROFILES

3
INTRODUCCION
En el campo de la tecnología se ha llegado a obtener sistemas terapéuticos que liberan uno o más
principios activos de forma continua bajo una pauta preestablecida, en un sitio del organismo y
bajo un periodo de tiempo determinado, estos sistemas han sido muy aplicables a la práctica
clínica de manera que ahora existen las formas farmacéuticas convencionales y las nuevas formas
farmacéuticas o de dosificación.
Las nuevas formas farmacéuticas incluyen los Sistemas de Liberación Modificada (SLM) y los
Sistemas de Vectorización que dirigen el fármaco a un órgano diana, pudiendo haber también la
asociación de estos dos mecanismos. (Suñé Negre, 2003)
En un contexto más profundo se puede diseñar sistemas inteligentes, de manera que reconozcan
y se adhieran a un sitio específico del cuerpo en donde se requiere el efecto terapéutico, que
permitan la liberación dirigida y localizada de los agentes médicos en estas regiones únicas, para
evitar la distribución no selectiva del fármaco que es la responsable de que el principio activo no
llegue en las cantidades adecuadas a la zona en donde debe actuar, además de que por ello se
producen las reacciones adversas. De esta manera se limitan los efectos secundarios, permitiendo
tener un producto con adecuadas características de biodisponibilidad, estabilidad, toxicidad, y
elegancia farmacéutica. (Fery & Weinkamer, 2007)
La microencapsulación es un proceso mediante el cual ciertas sustancias químicas, sustancias
biológicamente activas y otro tipo de sustancias, son introducidas en una matriz de biopolímeros
con el objetivo de impedir su pérdida, protegerlos de la reacción con otras sustancias del
ambiente, impedir que sufran reacciones de oxidación debido a la luz o la presencia de oxígeno
de este modo se conservan las características fisicoquímicas y biológicas de estas sustancias. La
cubierta de las partículas además de proteger al principio activo, permite la liberación de este con
un perfil de cesión determinado, y enmascara olores y sabores desagradables. (Castañeta, 2011)
La investigación básica farmacéutica se centró en la búsqueda de nuevas moléculas con
potencialidad terapéutica más selectiva y más duradera, con menos efectos secundarios tóxicos o
indeseables para el paciente, lo que podría interpretarse como la búsqueda de compuestos
terapéuticamente útiles, más seguros y confiables. En la actualidad sin abandonar este aspecto los
esfuerzos de la industria farmacéutica están dirigidos a la búsqueda de nuevas formas de
presentación con tres objetivos claros:
i. Obtener una máxima economía o aprovechamiento del fármaco administrado, para
disminuir en lo posible la dosificación diaria.
ii. Disminuir o eliminar el riego de toxicidad y de efectos secundarios no deseados.
iii. Disminuir el número de tomas por día de manera que el paciente se sujete al tratamiento
al sentir la comodidad del sistema posológico.

4
CAPÍTULO I
PLANTEAMIENTO DEL PROBLEMA.
Hoy en día en el mercado existe una amplia variedad de fármacos destinados a diagnosticar,
prevenir, aliviar y curar un sinnúmero de dolencias que aquejan a la población; sin embargo la
industria farmacéutica continúa en la búsqueda de nuevas moléculas así como formas de
administración de estas, conseguir acciones terapéuticas más selectivas, duraderas y con menor
intensidad o cantidad de efectos colaterales no deseables.
Las formas convencionales de administración de fármacos son terapéuticamente eficaces, pero
para que éstas alcancen y mantengan concentraciones de fármaco dentro del margen terapéutico
es necesaria la administración de estos sistemas varias dosis al día debido a que los fármacos se
distribuyen en el organismo de acuerdo a sus propiedades como solubilidad, coeficiente de
partición y carga lo cual conlleva a una considerable fluctuación de los niveles plasmáticos y un
riesgo de incumplimiento por parte del paciente.
El deseo de facilitar una adecuada terapia al paciente hace necesaria la investigación y desarrollo
de nuevas vehículos así como métodos de manufactura de fármacos; utilizando compuestos que
permitan una liberación controlada del principio activo y sean más selectivos con el sitio de
acción. Dentro de los portadores coloidales utilizados con este objetivo están los liposomas,
nanopartículas y micropartículas (Ramos, Gómez, Fernández, & Núñez, 2000).
Uno de los procedimientos utilizados actualmente es la microencapsulación, que exhibe algunas
ventajas sobre sistemas habituales de entrega de fármacos debido a que:
i. El paciente se siente complacido, pues con una sola dosificación se logra un efecto
terapéutico prolongado.
ii. El sistema proporciona el grado de control necesario para lograr un orden de liberación
del principio activo cercano a cero.
iii. La velocidad y duración de la liberación del principio activo in vivo pueden ser
determinadas mediante la selección del tamaño de las partículas.
iv. Estabilidad física, química y microbiológica.
v. La reducción de las concentraciones sistémicas de la droga, lo que promueve la
concentración local en el órgano blanco, de manera que se obtiene la máxima actividad
farmacológica con mínimos efectos adversos sistémicos.
vi. Protección del principio activo frente a posibles inactivadores en el medio biológico antes
de alcanzar el lugar de acción.
vii. Fácil fabricación con buena reproducibilidad. (Ramos, Gómez, Fernández, & Núñez,
2000)

5
Igualmente las formas farmacéuticas de liberación modificada permiten disponer de un
medicamento prácticamente nuevo, lo que le permite ser patentable, para esto la forma
farmacéutica debe cumplir con algunos requisitos como son:
Tener estudios comparativos que demuestren ventaja en eficacia y seguridad.
Demostrar que el medicamento sea sustancialmente mejor que el innovador.
Por lo mencionado, en el presente estudio se propone la elaboración de micropartículas
poliméricas cargadas con Diclofenaco como fármaco modelo a través del método de doble
emulsificación.
FORMULACIÓN DEL PROBLEMA
En el país, el desarrollo de técnicas para la elaboración de Sistemas de Liberación Modificada es
muy limitado, por lo que el presente estudio delineará los procesos farmacéuticos para la
preparación de micropartículas poliméricas cargadas con Diclofenaco utilizando como plantilla
una doble emulsión w/o/w.
HIPÓTESIS
PRIMERA ETAPA:
H0: No es posible elaborar micropartículas poliméricas cargadas con diclofenaco como fármaco
modelo a partir de la técnica de doble emulsificación.
H1: Si es posible elaborar micropartículas poliméricas cargadas con diclofenaco como fármaco
modelo a partir de la técnica de doble emulsificación.
SEGUNDA ETAPA:
H0: La formación de micropartículas poliméricas como vehículo de principios activos no incide
en la cinética de liberación del diclofenaco.
H1: La formación de micropartículas poliméricas como vehículo de principios activos incide en
la cinética de liberación del diclofenaco.

6
OBJETIVOS DE LA INVESTIGACION
1.1 OBJETIVO GENERAL.
Diseñar y elaborar micropartículas poliméricas cargadas con Diclofenaco utilizando como matriz
una emulsión obtenida mediante la técnica de doble emulsificación.
1.1.1 OBJETIVOS ESPECÍFICOS.
Obtener las micropartículas poliméricas cargadas con principio activo.
Caracterizar las micropartículas obtenidas mediante determinación de tamaño de
partícula, contenido de principio activo, rendimiento de la producción y eficacia de
encapsulación.
Evaluar la cinética de liberación del principio activo desde las micropartículas.
Determinar la existencia de un sistema de liberación modificada.

7
IMPORTANCIA Y JUSTIFICACIÓN
El uso de los Sistemas de Liberación Modificada (SLM) permite evitar fluctuaciones de la
concentración plasmática del fármaco en el organismo, esto posibilita un control de la aparición
de efectos adversos, y favorece el cumplimiento de la terapia por parte del paciente, además que
se puede hacer uso de estos sistemas para mejorar las propiedades de solubilidad y absorción de
moléculas nuevas como de las ya conocidas. (Sanchez, Carreño, & Castelletto, 2001)
Los sistemas que controlan la liberación de fármacos, entre los que destacan las micropartículas,
han tenido un gran desarrollo en los últimos años, sin embargo en el Ecuador no existen estudios
que permitan ayudar al avance de estas técnicas por lo que su aplicación en terapéutica se ha visto
muy limitada, mientras que en otros países ya existe estudios que han revelado la importancia de
las microcápsulas al ser administradas intravenosamente, estos estudios destacan que partículas
mayores de 7 µm se depositarán en el lecho capilar de los pulmones con una alta eficiencia y este
hecho es el principal atractivo para el tratamiento de cáncer pulmonar con drogas antineoplásicas;
partículas con diámetro menor a los 3 µm serán removidas del torrente circulatorio por células
del sistema fagocítico mononuclear, lo que ofrece una oportunidad para el tratamiento de
infecciones intracelulares; también se ha experimentado la administración de microcápsulas por
vía oral e intramuscular mediante un vehículo oleoso para direccionar agentes antitumorales hacia
el sistema linfático. (Pena, Martinez, & Nacucchio, 1990)
En los últimos años la industria farmacéutica ha crecido en el mercado a un ritmo muy acelerado
sin embargo, los altos costos que demanda el desarrollo de nuevas moléculas, el incremento de
los requerimientos de las autoridades farmacéuticas para nuevas entidades moleculares y el
vencimiento de las patentes que convierten en genéricos drogas establecidas en el mercado, han
marcado una limitante para el desarrollo de estos sistemas.
No obstante el desarrollo de los nuevos sistemas de liberación modificada se advierte como una
de las oportunidades de desarrollo, permite reformular moléculas terapéuticas ya existentes, con
lo que se lograra incrementar el retorno de la inversión de Investigación y Desarrollo, extender la
patente y proteger a la molécula frente a genéricos, además que establecerá la diferencia frente a
la competencia.
Por lo expuesto anteriormente resulta importante desarrollar una técnica que permita obtener
micropartículas de estructura polimérica cargadas con Diclofenaco como fármaco modelo,
elaborados por el método de doble emulsificación que permitan la liberación modificada del
fármaco.

8
CAPÍTULO II
MARCO TEÓRICO
ANTECEDENTES
La Tecnología Farmacéutica nos ha permitido en el transcurso del tiempo, desarrollar
presentaciones farmacéuticas que sirvan como vehículos para la administración de fármacos en
el ser humano, y en la búsqueda de estos nacen los sistemas de liberación modificada (SLM),
entre ellas las micropartículas.
Los procesos de microencapsulación fueron desarrollados entre los años 1930 y 1940 por la
National Cash Register para la aplicación comercial de un tinte a partir de gelatina como agente
encapsulante mediante un proceso de coacervación.
En 1940 se publica la patente de Israel Lipowski en la misma que se describe un método para
obtener pellets recubiertos, que brindaban una liberación lenta y constante del principio activo,
este es de los primeros registros sobre el uso de sistemas de liberación modificada aplicados a la
industria farmacéutica. (Baena, Aragón , Sandoval, Rosas, & Ponce , 2006).
Históricamente, la microencapsulación fue introducida de manera comercial en 1954 como medio
de hacer copias múltiples sin el uso del papel carbón.
Las primeras investigaciones en el área farmacéutica fueron realizadas en la Universidad de
Wisconsin (Estados Unidos) y datan de los años 50. (Yáñez Fernandez & et. al, 2002). Desde
entonces, se ha adquirido experiencia en el área, controlando parámetros en el proceso como ruta
de preparación, polímeros utilizados, cantidad de fármaco incorporado, estabilidad del fármaco,
estabilidad del sistema de liberación, entre otras; lo que ha posibilitado la existencia de
numerosos mecanismos para la obtención de estos productos, y la disponibilidad de
medicamentos a través de ellos conseguidos. (Ramirez Rigo, 2010).
Valdés Oscar (2005) en su tesis “Diseño de soportes combinados a base de polímeros naturales y
sintéticos para su evaluación en cultivos celulares y la liberación de principios activos”, propone
la formación de microcápsulas por el método de Coacervación o Separación de fases, utilizando
como polímeros Alginato de sodio y poli(Q9/HEMA) en una concentración 2:1.5, con una
velocidad optima de agitación de 300rpm; el producto con un tamaño promedio de 50±1µm,
mostró un porcentaje de encapsulación del 25%, el porcentaje de liberación del principio activo
fue de 7%; concluyendo que se debe optimizar el proceso de absorción para garantizar una
liberación sostenida más duradera y eficiente. (Valdés Lizama, Diseño de soportes combinados a

9
base de polímeros naturales y sintéticos para su evaluación en cultivos celulares y la liberación
de principios activos, 2005)
En el artículo “Elaboración y Caracterización de microcápsulas gastrorresistentes de Diclofenaco
obtenidas por Gelificación Iónica”, realizado por la Universidad Autónoma de Nicaragua (2008),
se describe la elaboración de microcápsulas por el método de Gelificación Iónica, con este proceso
se obtuvo microcápsulas con un diámetro máximo de 2.5mm; el rendimiento de la producción de
76.14%; la eficacia del método de encapsulación de 89.56% y el contenido de principio activo de
81.04% (Universidad Autónoma de Nicaragua, 2008).
En el VI Congreso SEFIG y 3° jornadas de Tecnología Farmacéutica se expuso un estudio sobre
la Influencia que ejerce el emulgente en la elaboración de microcápsulas por el método de
polimerización interfacial. Los autores concluyen que se obtienen emulsiones estables de tipo
O/A utilizando menor cantidad de agente surfactante (HBL=12); las microcápsulas obtenidas
vistas al microscopio se presentaron aisladas, de forma esférica y superficie regular, con un
tamaño promedio de 55µm, la eficacia del procedimiento de microencapsulación fue superior al
98%. (Rivas Esteban, Gil Alegre, & Torres Suárez, 2009)

10
FUNDAMENTO TEÓRICO
2.1 SISTEMAS DE LIBERACION MODIFICADA (SLM)
Sistemas de Liberación Modificada son aquellas especialidades farmacéuticas a las que se les ha
modificado el lugar o la velocidad de liberación del principio activo en relación a las formas de
farmacéuticas de liberación convencional, administradas por la misma ruta. (Jover Botella & Et.
al, 2004)
La Farmacopea de los Estados Unidos (USP 30, 2007) describe a las Formas de Liberación
Modificada como: “aquellas en las cuales se eligen las características de la liberación en el curso
del tiempo y/o en la localización para lograr objetivos terapéuticos o de conveniencia que no
ofrecen las formas farmacéuticas convencionales”.
La liberación modificada de fármacos en el tracto digestivo implica un suministro de fármaco en
el organismo mediante una forma farmacéutica que actúa como un dispositivo con un perfil de
cesión determinado, generado como consecuencia de un mecanismo conocido (Universidad de
Salamanca - Fundación Grunenthal, 2008), el cual es provocado por el retardo en el proceso de
desintegración de la forma farmacéutica sólida, lo que permite la modulación de la liberación de
principios activos, de modo de conseguir la programación de la velocidad de cesión con el fin de
reducir el ritmo de administración en aquellos fármacos de corta vida media de eliminación
2.1.1 CLASIFICACIÓN DE LOS SISTEMAS DE LIBERACIÓN MODIFICADA
De manera general los SLM se clasifican en base a dos mecanismos de liberación del fármaco.
i) Sistemas que liberan el principio activo durante un periodo prolongado de tiempo de
acuerdo con una cinética predecible, con el fin mantener la concentración en la zona
terapéutica por un periodo de tiempo prolongado.
ii) Sistemas diseñados para modificar la velocidad de tránsito de la forma farmacéutica
a lo largo del tracto digestivo y/o liberar el principio activo en un área específica para
obtener un efecto local o sistémico.
De estas características surgen los diferentes tipos de liberación modificada, los que incluyen
liberación prolongada, controlada, retardada, sostenida.
En la Figura 2.1 se observa la diferencia en las curvas de concentración plasmática para los
distintos sistemas. A pertenece a la de una forma farmacéutica convencional, B corresponde a un
sistema de liberación retardada, C muestra la curva de concentración plasmática para un
medicamento de liberación repetida, D corresponde a un sistema de liberación prolongada y E
para el tipo de liberación sostenida.

11
1
Formas farmacéuticas de liberación retardada
Están situadas en este grupo aquellas formas farmacéuticas cuyo principio activo es liberado en
el organismo después de un tiempo de latencia; presentan una cubierta entérica que permite la
cesión del fármaco en un lugar específico del tracto digestivo.
Como ejemplo tenemos los AINEs, omeprazol. (Mateu López & Herrera LLópiz, 2007)
Formas farmacéuticas de liberación sostenida
Estas formas liberan inicialmente una cantidad de fármaco de manera que se obtiene la respuesta
farmacológica esperada rápidamente y se mantiene la liberación al tracto gastrointestinal en una
cantidad adecuada y constante de manera que la velocidad de absorción se iguale a la velocidad
de eliminación, esto en un periodo de 10 a 24 horas, con lo que se reduce la pulsación en el
plasma al ser comparado con los sistemas de dosificación unitaria. En términos cinéticos la
velocidad de liberación es de orden cero.
Como ejemplo de estas formas de liberación tenemos los Comprimidos Osmóticos, estos sistemas
permiten la entrega de fármacos desde un núcleo, que se halla cubierto por una membrana
semipermeable, provista de un orificio por donde ocurre la liberación.
La liberación del principio activo involucra la entrada del disolvente al núcleo a través de la
membrana y posterior salida de la solución (fármaco + disolvente) a través del orificio de la
membrana. (Baena, Aragón , Sandoval, Rosas, & Ponce , 2006)
El principio activo se libera a una velocidad constante, debido a que el flujo de agua a través de
la membrana, cuyo tamaño de poro determina la velocidad, también es constante.
Formas farmacéuticas de liberación prolongada
Se refiere a aquellas formas en donde el principio activo se libera inicialmente en una cantidad
suficiente como para producir el efecto terapéutico, después se sigue liberando lentamente en un
periodo que dura al menos de 6-8 horas, sin que la liberación sea constante; describen una curva
Posibilidad de efectos secundarios
Concentración eficaz
Débil o nula respuesta farmacológica
Figura 2. 1 Perfiles de concentración plasmática para los distintos sistemas de liberación del principio activo. Suñe Negre, 2003.

12
de concentración plasmática amplia que varía dentro de la zona terapéutica. Ejemplo:
Comprimidos de liberación prolongada de oxicodona. (Gómez Vega & et al., 2007)
Formas farmacéuticas de liberación repetida
Estas formas farmacéuticas permiten la liberación inicial de principio activo, pasado un tiempo
vuelven a liberar otra dosis, la liberación ocurre de manera periódica por lo que el perfil de
liberación plasmática es igual al de la liberación repetida de las formas farmacéuticas
convencionales, es decir que presenta valores máximos y mínimos de concentración.
Normalmente las formas farmacéuticas que permiten este patrón son aquellas dotadas de un doble
núcleo. (Arias, 1999)
2.1.2 SISTEMAS TRANSPORTADORES DE FARMACOS
Los sistemas de transporte para fármacos han sido ampliamente estudiados durante los últimos
años como una forma de mejorar la biodistribución de los principios activos.
Al estar el principio activo dentro de un transportador la distribución de este en el organismo ya
no va a depender de sus propiedades físico-químicas y estructura molecular, sino de las
características que presente el sistema transportador.
Los sistemas transportadores logran proteger al principio activo de la inactivación por condiciones
externas, además que pueden ser utilizados para lograr terapias dirigidas con lo que se logra mayor
efectividad con disminución de efectos secundarios.
Figura 2. 2 Clasificación de los sistemas transportadores de fármacos. Elaborado por: Valenzuela G.
La Figura 2.2 muestra una clasificación de los sistemas transportadores de fármacos de los cuales
las micro y nanoemulsiones así como las micro y nanopartículas han sido desarrolladas por ser el
propósito de la investigación.
Sistemas transportadores de farmacos
Sistemas Moleculares
Ciclodextrinas Dendrímeros
Sistemas Coloidales
Micro y Nano emulsiones
Liposomas
Micro y nano partículas

13
2.1.2.1 Sistemas coloidales
a. Micro y Nano-emulsiones
El término micro-emulsión inicialmente introducido por Schulman et al. fue utilizado para
describir dispersiones transparentes termodinámicamente estables de aceite y agua estabilizadas
por una molécula anfifílica.
La diferencia entre nano-emulsiones y micro-emulsiones radica en que las primeras son muy
inestables desde el punto de vista termodinámico lo que genera una separación de fases, las
microemulsiones una vez que las condiciones son adecuadas se forman de manera espontánea y
su formación depende del tipo de surfactante y su estructura.
Algunos autores consideran que el término microemulsión es erróneo, además de que corresponde
a un sistema termodinámicamente estable y que no tiene interfase ni gotas; En vez de esto las
dispersiones de dos fases líquido-líquido con tamaño de gota inferior a 0,5 µm son llamadas
emulsiones ultrafinas o emulsiones submicrometricas.
Las nano-emulsiones exhiben las propiedades típicas de todas las emulsiones pero con algunos
detalles específicos. Su tamaño de gota sub – micrométrico las hace translúcidas dependiendo de
la distribución de tamaño de gota y del contenido de fase dispersa.
Las nano-emulsiones no son sistemas particularmente viscosos cuando poseen poca proporción
de fase dispersa. Se conoce que la viscosidad de las nano-emulsiones aumenta a medida que el
tamaño de gota disminuye, así, y contrariamente a las macroemulsiones, las nano-emulsiones
podrían ser viscosas a un porcentaje de fase dispersa entre 40-50% especialmente el tamaño de
gota es uniforme, en cuyo caso frecuentemente se las llama “emulsiones – gel”. (C. & P. , 2005)
Debido a su pequeño tamaño de gota, las nano-emulsiones tienen mayor área interfacial y por lo
tanto se requiere una mayor cantidad de surfactante para estabilizarlas. (Forgiarini, Marquez, &
Salager, 2006)
b. Micropartículas y Nanopartículas
Son partículas poliméricas esféricas, contienen en su estructura el material activo. El tamaño para
las micropartículas se sitúa en el rango de los micrómetros a milímetros y para las nanopartículas
es de nanómetros.
Existen dos tipos de partículas que según su estructura los podemos clasificar como:
a. Partículas de Sistema reservorio o capsular: Son sistemas vesiculares donde el material
activo se encuentra incluido en el núcleo que puede ser de naturaleza líquida o sólida y
se halla envuelto por una fina película del material de recubrimiento, se les denomina
también microcápsulas.
b. Partículas de sistema matricial: el material activo se encuentra altamente disperso en la
matriz polimérica, se los conoce como microesferas.

14
Los equivalentes morfológicos a las microcápsulas y microesferas son las nanocápsulas y
nanoesferas, teniendo estas tamaños menores a 1µm. (Sáez, 2004)
Las microcápsulas y nanocápsulas, presentan características en su composición que le permite
modificar la liberación del principio activo, ya sea en tiempo o segmento del organismo.
2.1.3 MICROCÁPSULAS
Las microcápsulas son el producto del proceso por el cual partículas individuales o gotas de un
material activo (Núcleo) se rodean por una cubierta para producir capsulas en el rango de
micrómetros a milímetros.
Núcleo: Está conformado por partículas sólidas o por líquido en forma de finas gotas que
constituyen el principio activo, además se puede incorporar otros compuestos como
estabilizadores, antioxidantes, antisépticos, diluyentes u otras sustancias con el propósito de
modificar la cesión.
Cubierta: Es una cáscara dura soluble o suave que contiene al principio activo y excipientes. La
selección del material de recubrimiento se realiza de acuerdo a la función que se quiere dar a la
microcápsula. Normalmente se forma de gelatina, pero también puede ser almidón u otras
sustancias poliméricas.
Figura 2. 3 Representación de una microcápsula polimérica el núcleo formado por las sustancias activas.
Fuente: elmundo.es
Además de la protección el material de recubrimiento debe dar a la microcápsula propiedades de
flexibilidad, fuerza o resistencia, permeabilidad y facilidad de aplicación para lo cual puede
emplearse junto con otras sustancias como plastificantes.
Es de tener en cuenta que cambios mínimos en el recubrimiento puede influir de gran manera en
la liberación del principio activo contenido en el núcleo. (Técnicas de microencapsulación, 2013)
A diferencia de las formulaciones convencionales las microcápsulas presentan una vida media
más larga, lo que permite una menor dosificación e incrementa el cumplimiento por parte del

15
paciente. Estos sistemas pueden ser administrados en dosis única o como multidosis. (Jelvehgari
& Montazam, 2012)
2.1.4 MICROENCAPSULACIÓN
La microencapsulación es el proceso físico que se utiliza para empacar en miniatura sustancias
activas. Esta técnica permite envolver cantidades pequeñas de gases, líquidos o sólidos dentro de
una pared que funciona como escudo del ingrediente activo, la cual le protege de agentes que
causan su deterioro y usualmente pueden controlar su liberación.
2.1.4.1 Métodos de manufactura.
Existen descritos una amplia variedad de métodos empleados para la elaboración de
microcápsulas; como visión general se puede decir que existen procesos basados exclusivamente
en fenómenos físicos, otros procesos usan reacciones químicas de polimerización para producir
la pared de la cápsula, y otros combinan los métodos físicos y químicos. (Vilstrup, 2004)
Procesos físicos de microencapsulación:
a. Recubrimiento en bombo.
b. Secado por atomización (Spray drying).
c. Enfriamiento tras atomización (Spray chilling).
d. Recubrimiento en lecho fluido.
e. Disco giratorio con orificios múltiples.
Procesos químicos de microencapsulación:
a. Polimerización interfacial.
b. Inclusión molecular
Procesos físico-químicos de microencapsulación:
a. Coacervación
b. Polímero-polímero incompatible.
c. Gelificación iónica.
d. Atrapamiento por liposomas
e. Proceso de inyección sumergido. (Parra Huertas, 2010)
f. Co-cristalización.
g. Emulsificación Evaporación-Extracción del solvente

16
Una vez que las cápsulas se han formado, es necesario someterías a un procedimiento de
consolidación o endurecimiento, los métodos pueden incluir reacciones químicas, evaporación de
solvente o enfriamiento (Técnicas de microencapsulación, 2013).
La técnica a utilizar es seleccionada en función de las propiedades del núcleo (solubilidad, estado
sólido o líquido, estabilidad frente a los solventes y temperatura, compatibilidad química con el
material de recubrimiento), del agente utilizado como material de recubrimiento y las posibles
interacciones con el núcleo, además del equipamiento disponible (recipientes termostatizados
dotados de sistemas de agitación para las técnicas basadas en formación de emulsiones, mientras
que para los métodos mecánicos se precisa de equipamiento específico). Asimismo se debe
convenir el tamaño de la microcápsula, la manera como se une la película al núcleo, la forma de
cesión del principio activo, las características de producción y la economía del proceso antes de
elegir el método.
Procesos físicos de microencapsulación
a. Recubrimiento en bombo.
Es una de las técnicas más antiguas, empleada para el recubrimiento de comprimidos, se utiliza
el mismo tratamiento para la formación de cobertura de la microcápsula pero la variación radica
que el tamaño de partícula a recubrir va de 100 a 600 µm.
Este proceso permite obtener micropartículas de acción sostenida o es utilizado para la protección
de sustancias de la acción de la luz o la humedad.
La elaboración del núcleo puede hacerse utilizando partículas inertes como gránulos de azúcar y
sobre estos depositar en forma de capas el material activo o mediante la preparación de granulados
que contenga el principio activo.
Una vez preparados los núcleos, estos se colocan en el bombo y son recubiertos con capas de
material de recubrimiento alternando capa y aire frío o caliente con el objeto de evaporar el
solvente.
b. Secado por atomización
Consiste de manera general, en atomizar el material que se encuentra en estado líquido, ya sea
como disolución o como dispersión, en forma de finas gotas sobre una corriente de gas calentado.
Cuando las pequeñas gotas del líquido se ponen en contacto con el gas a mayor temperatura, se
produce una rápida evaporación del disolvente, formándose una fina película del material de
recubrimiento que se encuentra disuelto en él. ( Lozano Berna, 2009)
El tamaño de las micropartículas que se obtienen por este procedimiento varía entre 5 y 600 µm.

17
La estructura de la cubierta casi siempre es porosa, por lo que es recomendable cuando se procesan
materiales por este método, emplear una baja proporción del activo y que la cubierta ocupe un
porcentaje importante de la microcápsula total para asegurar una adecuada protección.
c. Enfriamiento tras atomización (spray chilling)
La microencapsulación por este procedimiento se realiza, en general, suspendiendo el principio
activo en el material de recubrimiento fundido. El líquido caliente se atomiza dentro de una
cámara fría empleando un equipo de secado por atomización, que se opera inyectando aire frío.
El material se recolecta en el fondo del aparato en forma de polvo y consiste en partículas más o
menos esféricas, cada una de las cuales contiene el ingrediente activo suspendido en una matriz
del agente de recubrimiento.
d. Recubrimiento en lecho fluido
En este procedimiento las micropartículas se forman al suspender las pequeñas partículas que
forman el material activo en un lecho de aire, al mismo tiempo que se dispersa sobre ellas, en
forma de fina lluvia, una disolución del material de recubrimiento. La película se forma por
evaporación del disolvente el cual a su vez, es separado por el aire o el gas que abandona el
sistema. Cuando las partículas alcanzan un cierto grado de recubrimiento, sobrepasan el peso que
soporta la columna de aire y caen sobre una malla, deslizándose en el plano inclinado hacia la
zona de salida, de donde pueden retirarse.
La microencapsulación por recubrimiento en lecho fluido se aplica ampliamente como tecnología
farmacéutica para producir micropartículas de acción sostenida, para mejorar las características
de flujo de las partículas y para el recubrimiento de numerosas sustancias en tecnología de
alimentos y otras industrias relacionadas ( Lozano Berna, 2009)
e. Disco giratorio con orificios múltiples
Este procedimiento aprovecha la fuerza centrífuga para proyectar el material activo contra la
película del material que formará la cubierta de la microcápsula.
Al chocar las partículas con la película de recubrimiento se produce un englobamiento del
material activo y cuando las fuerzas centrífugas de la masa del material activo y del material de
recubrimiento sobrepasan la fuerza de cohesión de la película, se forman pequeñas cápsulas que
se proyectan hacia fuera.
Procesos químicos de microencapsulación
a. Polimerización interfacial
Este proceso se basa en la formación de un polímero en la interfase por interacción de dos solutos
que se encuentran en dos fases líquidas.

18
El procedimiento descrito por Chang consiste en emulsionar la sustancia que se va a recubrir y la
hexanodiamina, disueltos en agua, con ayuda de Span® 85 al 1%, en un solvente orgánico
constituido por cloroformo y ciclohexano en proporción 1 a 4. Una vez producida la emulsión de
las características apropiadas, se adiciona, agitando, una solución de cloruro de sebacilo en el
mismo solvente orgánico. Al producirse la polimerización en la interfase, la membrana se deposita
sobre los glóbulos de la emulsión recubriéndolas. (Técnicas de microencapsulación, 2013)
b. Inclusión molecular
La microencapsulación por inclusión molecular se da por la capacidad de algunos materiales
como las ciclodextrinas, de alojar a moléculas hidrofóbicas en la cavidad interna que se forma
gracias a su estructura.
Este método no ha sido incursionado con fuerza en el ámbito industrial debido al alto costo y baja
eficiencia de encapsulación. (Gordillo Galeano, 2013)
Procesos físico-químicos de microencapsulación
a. Coacervación
La coacervación son técnicas de microencapsulación basadas en la inducción de la desolvatación
parcial del polímero, quedando éste disponible para depositarse en forma de gotículas de
coacervado alrededor de las partículas de principio activo.
Justificación del fenómeno:
Cuando en un sistema coloidal se varía el pH o se adiciona otras sustancias como una sal o un
no-solvente, se puede disminuir la solubilidad de la macromolécula, provocando su separación en
una nueva fase, de manera que en el sistema exista dos zonas, una rica en sustancia coloidal y la
otra que contiene una baja proporción de la macromolécula; en la fase rica en coloide, la
macromolécula puede quedar constituyendo una fase en forma de pequeñas gotas. (Ramos Picos,
Gómez Carril, & Fernández Mena, 2001)
Figura 2. 4 Diagrama del proceso de microencapsulación por coacervación. Elaborado por: Valenzuela G.
Preparación de un sistema: vehículo
liquido + material de recubrimiento (fase continua), material
que va a encapsularse (fase dispersa).
Modificación de las características del
medio para producir la
separación de fases.
Depósito del líquido coloidal en forma de una membrana continua sobre el
material que constituye el núcleo
Endurecimiento de la cubierta
depositada sobre el material
microencapsulado

19
b. Polímero- polímero incompatible
Se basa en inducir la separación de fases añadiendo un polímero “incompatible” con el polímero
formador de cubierta. Es incompatible el polímero que presenta una mayor solubilidad en el
disolvente que el propio polímero de recubrimiento, no teniendo, en cambio, afinidad por el
material que se va a encapsular. Por lo tanto, a medida que se añade el polímero incompatible, se
produce la desolvatación del de recubrimiento, que se separa y deposita alrededor de las partículas
suspendidas en el medio. (Berna Lozano, 2009)
c. Gelificación Iónica
La Gelificación Iónica es la técnica que permite la formación de partículas poliméricas usando un
agente iónico de entrecruzamiento. Se basa en la formación de complejos debido a la atracción
de cargas opuestas; las atracciones electrostáticas crean interacciones físicas, lo que permite la
transformación de un líquido en gel.
d. Atrapamiento en liposomas.
Los liposomas son vesículas esféricas que constan de doble capa de fosfolípidos, teniendo dos
partes una hidrosoluble y una liposoluble.
Los liposomas se forman cuando películas de fosfolípidos son dispersadas en un medio acuoso.
Al igual que las membranas naturales, los liposomas son selectivamente permeables a iones.
Estructuralmente existen tres tipos de liposomas: multilamelar, vesículas de un compartimiento y
macrovesículas.
La sonicación permite la formación de un solo compartimiento de vesículas, mientras que las
macrovesículas son formadas por inyección de soluciones de lípido en un buffer de fosfatos.
Los liposomas pueden ser obtenidos con cargas positivas por la adición de aminas o con cargas
negativas por la adición de fosfatidil serina o diacetil fosfato.
Los compuestos hidrofílicos que se desea sean atrapados son disueltos en agua y mezclados con
una película lípídica para formar liposomas, mientras que los materiales hidrofóbicos son
embebidos en una delgada película de lípido.
La liberación del principio activo se realiza por difusión a través de la bicapa, por destrucción de
la vesícula, por medio de una concentración crítica de iones calcio o por un cambio de pH. (Yáñez
Fernández, 2005)
e. Procesos de inyección sumergida
Varios procesos de microencapsulación por métodos químicos utilizan la fuerza centrífuga o
boquillas de dos fluidos sumergidas para formar las micropartículas. En un proceso, una copa
perforada con una serie de los agujeros contiendo la fase acuosa, se sumerge en un baño del aceite.
La fase del agua de esta emulsión es una disolución concentrada de un polímero soluble en agua.

20
Se rota la copa, que está sumergida en el aceite, de tal modo que en la fase del aceite se forma una
corriente de gotitas de una emulsión aceite/agua. Controlando la temperatura del baño del aceite,
la fase externa de las gotitas de la emulsión se secan y gelifican para crear capsulas de aceite-
cargados del gel que pueden ser aisladas y ser secados. Cuando están aisladas las cápsulas
consisten en un número de gotitas pequeñas del material activo dispersadas a través de una matriz
del material de la pared. Este proceso fue desarrollado en 1942 para producir las cápsulas que
mejoraron la estabilidad de oxidación de vitaminas y de aceites de los pescados.
f. Co-cristalización
Es un proceso de microencapsulación donde dos ingredientes son incorporados en un
conglomerado poroso de microcristales de sacarosa. El paso inicial para la encapsulación es la
concentración de jarabes de sacarosa hasta supersaturación, posteriormente se adiciona el
material central y se agita constantemente para inducir la nucleación y la aglomeración. (Astolfi
et al., 2005).
La cápsula obtenida es granular, de fácil manejo y con buenas características de flujo.
La co-cristalización es una alternativa flexible y económica por ser un procedimiento
relativamente simple; numerosos productos pueden ser encapsulados como jugos de frutas,
aceites esenciales, saborizantes, aromatizantes, entre otros. (Sandoval Aldana, Rodriguez , &
Ayala, 2004)
g. Método de emulsificación evaporación-extracción del solvente.
Esta técnica de manera general podría describirse como la formación de partículas esféricas que
contienen el principio activo usando como plantilla una emulsión, mediante un agente
dispersante.La deposición del polímero alrededor de las partículas de fármaco se produce por el
particionamiento del disolvente del polímero de la fase dispersa a la fase continua, seguido de la
remoción del solvente del polímero mediante la evaporación-extracción del mismo, lo que permite
el depósito del polímero sobre las micropartículas.
2.1.5 METODO DE DOBLE EMULSIFICACIÓN EVAPORACION-EXTRACCION
DEL SOLVENTE
Para concebir de mejor manera esta y otras técnicas de microencapsulación que requieren de la
formación previa de una emulsión que sirva como plantilla o molde de las partículas, es preciso
definir que es una emulsión y las características que se necesita para su formación.

21
2.1.5.1 Emulsiones.
Una emulsión es una dispersión coloidal de un líquido en otro inmiscible con él, que se produce
por agitación y se destruye después de un periodo de tiempo, este efecto se da debido a que las
fuerzas de cohesión de cada líquido son superiores a las fuerzas de adhesión existentes entre
ambos líquidos. Las fuerzas de cohesión de cada líquido se manifiesta como la energía o tensión
interfacial en la superficie de separación de ambos líquidos. La estabilización de las fases se
produce por la incorporación de un agente surfactante que impide la coalescencia, y por la
aportación de energía mecánica con la ayuda de calor.
En el proceso de emulsificación se forman pequeñas partículas dispersas cuyo tamaño varía entre
0.1 µm y 10 µ, pero algunas veces pueden llegar a ser tan pequeñas llegando al tamaño
nanométrico. (Aranberri, 2006)
2.1.5.2 Teoría de las emulsiones
Desde el punto de vista termodinámico las emulsiones son sistemas inestables, esto se debe a
que en la formación de los pequeños glóbulos durante la emulsificación, el área superficial se
incrementa con relación al área de la superficie inicial del líquido (∆A) aunque el volumen del
líquido no haya sufrido alteración, lo que produce un incremento de la energía libre de Gibbs
(∆G) y este incremento es lo suficientemente grande para hacer que las gotitas formadas en el
proceso de emulsificación muestren tendencia a unirse.
La relación del área superficial con la energía libre superficial viene dada por la siguiente
ecuación:
∆G = γ . ∆A
Ecuación 2. 1 Relación entre la energía libre y el área superficial de las gotas
Donde:
∆G= representa el incremento de la energía libre de Gibbs
γ= representa la tensión superficial.
∆A= representa el incremento de áreas producto de la formación de los glóbulos en el proceso
de emulsificación.
La energía libre superficial necesaria para formar la emulsión disminuye al utilizar los agentes
emulsionantes, los que debido a su estructura se ubican en la interface líquido- líquido. Los
agentes emulsificantes no solo son importantes porque disminuyen la energía necesaria para la
formación de la emulsión sino que también por su naturaleza química envuelven las gotitas
dispersas con una fuerte y elástica película mono o multimolecular. (Aranberri, 2006)

22
2.1.5.3 Clasificación de las emulsiones
Las emulsiones se clasifican de acuerdo a las características de la fase dispersa, las características
del surfactante utilizado o según la consistencia que presenten.
Figura 2. 5 Clasificación de las emulsiones por la fase dispersa, tipo de surfactante y consistencia
Emulsiones dobles: Se forman estableciendo una matriz (w/o/w) que se elabora en dos
etapas: en la primera se emulsiona agua en aceite y en la segunda se reemulsiona la
emulsión anterior en una solución acuosa con emulsificantes-estabilizantes, dejando
gotas pequeñas encerradas otras más grandes.
Figura 2. 6 Emulsiones dobles vistas al microscopio.
Fuente: Binks , 2007
Clasificación de las emulsiones
Por la fase dispersa
Emulsión directa (o/w)
Emulsión inversa (w/o)
Emulsiones dobles
(w/o/w)
Por el tipo de surfactante
Emulsión aniónica
Emulsión aniónica
Emulsión no iónica
Por la consistencia
Emulsión fluida o leche
Emulsión consistente o
crema

23
2.1.5.4 Métodos para determinar el tipo de emulsión
a. Método del colorante: Se espolvorea una pequeña cantidad de uno de los colorantes
solubles en agua (azul de metileno o azul brillante) sobre la superficie de la emulsión, si
el agua forma la fase externa (emulsión o/w), el colorante se disolverá y se difundirá
uniformemente en el agua, por el contrario si la emulsión es de tipo w/o, el colorante se
distribuirá en grupos sobre la superficie de la emulsión.
b. Dilución de la emulsión: Se diluye la emulsión con agua, si el agua se mezcla con
facilidad la emulsión es de tipo o/w.
c. Método de electrodos: Se conectan dos electrodos a una fuente de energía eléctrica y se
sumergen en la emulsión, si la fase externa es agua, la corriente eléctrica pasará a través
de la emulsión, pero si la fase oleosa constituye la fase continua la corriente eléctrica no
pasará a través de la emulsión.
Mediante la determinación de la conductividad se puede también determinar si hay
inversión de fases al producirse un cambio brusco en valores de conductividad debido a
que las emulsiones o/w tienen agua como fase continua y son altamente conductoras,
mientras que las emulsiones w/o al tener aceite como fase continua son poco conductoras.
(Aranberri, 2006)
2.1.5.5 Estabilidad de las emulsiones
La estabilidad de una emulsión se expresa como la resistencia a la coalescencia de la fase interna
y de la formación de crema.
a. Sedimentación y formación de crema
La formación de crema se trata de un proceso causado por la acción de la gravedad. En el caso de
las emulsiones o/w la densidad de la fase dispersa en menor que la fase continua, la velocidad de
sedimentación será negativa, por lo que se forma la crema ascendente; si la fase dispersa es más
densa que la fase continua se dará lugar a la formación de crema descendente; la velocidad de
este proceso aumenta conforme aumenta la diferencia de densidades entre las dos fases.

24
b. Coalescencia y destrucción de la emulsión
Este es un proceso irreversible, no se puede volver a tener una emulsión por simple mezcla debido
a que la cubierta de agente emulsionante que rodea a los glóbulos ha sido destruida.
Figura 2. 7 Proceso de coalescencia en una emulsión. Fuente: Química Nova
c. Movimiento browniano
Es el movimiento aleatorio que se observa en partículas microscópicas suspendidas en un medio
fluido, este movimiento ocasiona choques entre las partículas lo que podría originar que las
partículas se unan y aumenten su tamaño con posterior floculación y desestabilización del sistema.
Figura 2. 8 Representación del movimiento browniano de las partículas.
Fuente: www.educación.unComo.com
d. Floculación
La floculación es la adhesión de las gotas sin fusionarse y sin que exista variación en la
distribución del tamaño de la gota. El proceso de la floculación está controlado por un
equilibrio global entre las fuerzas de atracción electrostáticas de van der Waals, y repulsivas
de tipo estéricas y de hidratación.

25
Figura 2. 9 Descripción del proceso de floculación Fuente: www.weschile.com
e. Maduración de Ostwald
La maduración de Ostwald es el proceso por el cual las gotas grandes crecen a expensas de las
más pequeñas a causa del transporte de masa de la fase dispersa, este efecto está limitado para
gotas pequeñas y no tiene efecto sobre las macroemulsiones.
La diferencia en solubilidad produce un gradiente de concentración entre los alrededores de la
pequeña gota a los alrededores de las gotas grandes y esto da origen a la transferencia de masa
por difusión. Como consecuencia, las pequeñas gotas tienden a desinflarse dentro de las grandes.
Teóricamente el proceso no se detendrá hasta que la separación de fases ocurra, pero en la práctica
el proceso se ralentiza con el crecimiento de las gotas.
Como este proceso es dependiente de la solubilidad de la fase dispersa en la fase continua, la
velocidad del proceso de maduración podría reducirse utilizando aceites menos solubles en agua.
La maduración de Otswald puede reducirse a concentraciones de surfactante entre 5-10% ya que
el surfactante forma fases de cristal líquido que envuelven las gotas y evitan la transferencia de
masa. (Forgiarini, Marquez, & Salager, 2006)
2.1.5.6 Factores que se influyen en la estabilidad de las emulsiones
Tamaño de las partículas
El tamaño de las partículas es un factor determinante de la coalescencia, no siempre las partículas
pequeñas proveen de estabilidad a la emulsión, cuando existe partículas de diverso tamaño las
partículas más pequeñas son englobadas entre las más grandes dando lugar a las fuerzas de
cohesión que producen la coalescencia.
Viscosidad
El incremento en la viscosidad puede generar aumento en la estabilidad debido a que esta
propiedad retarda la floculación y la coalescencia.

26
Efecto del volumen entre la fase acuosa y oleosa
Hace algún tiempo se estableció una regla empírica que señala que la proporción 50:50 permite
emulsiones más estables.
Punto crítico
Se le conoce como la concentración máxima de la fase interna por encima de la cual el emulgente
no puede producir una emulsión estable del tipo deseado; en una emulsión de tipo o/w no se puede
incorporar más del 74% de la fase oleosa ya que la mayoría de las veces los glóbulos de aceite
coalescen.
Carga eléctrica
No existe un acuerdo de tipo general pero cuando se tiene emulsión de tipo o/w, las partículas
oleosas que de forma general tienen carga negativa, al estar rodeadas por una película de
emulgente de tipo no iónico, casi no presentan efecto electrocinético, manteniendo la estabilidad
del sistema.
Efecto del surfactante
Posiblemente este sea el factor que mayor influencia tienen sobre la estabilidad de la emulsión
debido a que el emulgente o la mezcla de estos permite un descenso en la tensión interfacial,
produce pequeños glóbulos uniformes y permite la formación de una película que protege a estos
de la reagrupación.
Las emulsiones w/o/w requieren al menos dos surfactantes, uno lipófilo y otro hidrófilo que
ayuden a estabilizar la emulsión. (Kosegarten & Jiménez, 2012)
2.1.5.7 Surfactantes
Los agentes surfactantes conocidos también como agentes emulsionantes son sustancias que
presentan un carácter anfifílico debido a que en su estructura existe una porción lipofílica y una
porción hidrofílica. El grupo hidrófilo ejerce un efecto solubilizante y tiende a llevar a la molécula
a disolución completa, por el contrario el grupo hidrófobo debido a su insolubilidad tiende a
contrarrestar la tendencia del otro. Sí se logra el equilibrio adecuado entre los dos grupos se ve
que la sustancia no se disuelve por completo, ni queda sin disolver del todo, concentrándose en la
interfase con sus moléculas orientadas de tal forma que los grupos hidrófilos se orientan hacia la
fase acuosa, mientras que los hidrófobos hacia la no acuosa o a la fase vapor, con esto se reduce
la tensión superficial de la fase continua, se evita la coalescencia y aglomeración de gotas y por
tanto se estabiliza la emulsión.
Clasificación de los surfactantes.
Se clasifican en base al poder de disociación en presencia de un electrolito.

27
a. Iónicos: Dentro de estos se encuentran los aniónicos, catiónicos y anfóteros.
Surfactantes aniónicos: Están constituidos por una cadena alquílica lineal o
ramificada que va de 10 a 14 átomos de carbono, y en su extremo polar de la molécula
presentan un anión.
Generalmente tienen un HBL alto por su característica hidrofílica.
Surfactantes catiónicos: Son aquellos que en solución acuosa se carga positivamente
la molécula, generalmente son compuestos de amonio cuaternario.
Surfactantes anfóteros: actúan dependiendo del medio en que se encuentren, en medio
básico son aniónicos y en medio ácido son catiónicos.
b. No iónicos: Son a aquellas sustancias que no presentan carga. Se solubilizan mediante un
efecto de grupos solubilizantes débiles hidrófilos como los enlaces de tipo éter o grupos
hidroxilos en su molécula. Ejemplo: Nonil fenol etoxilado o el nonanol etoxilado, Laurato
de sorbitán. Tween, Span (Li, Rouaud, & Poncelet, 2008)
El tipo de emulsión que se tiende a formar depende del balance entre las propiedades hidrófilas e
hidrófobas del agente emulsificante. Generalmente se suele cumplir la regla de Bancroft, regla
empírica que afirma que la fase continua de una emulsión es aquella la cual solubiliza al agente
emulsificante; es decir que la emulsión a formar depende más del surfactante empleado que de
las proporciones relativas de las fases oleosas y acuosas o de la metodología de preparación de la
emulsión.
La naturaleza anfipática de los agentes surfactantes puede ser expresado en términos de una escala
empírica que comúnmente se denomina HBL, que es la relación entre los grupos hidrófilos y
lipófilos de la molécula.
De acuerdo a su balance hidrofílico/lipofílico a los agentes emulsificantes se los puede reclutar
en tres grupos:
a. Surfactantes con BHL entre 3 y 9: Son compuestos lipofílicos, en este grupo se
incluyen los monoacilgliceroles y sus derivados acetilados, etoxilados, y lactilados de
los monoglicéridos, así como el estearato de propilenglicol.
b. Surfactantes con BHL entre 8 y 12: Son compuestos moderadamente hidrofílicos,
corresponde al éster diacetilado del ácido tartárico y los monoacilgliceroles
succinilados.
c. Surfactantes con BHL entre 12 y 20: Son compuestos hidrofílicos entre los que
están los polisorbatos, el estearoil-2-lactilato y los esteres del poliglicerol. (Bello
Gutiérrez, 2000)

28
2.1.6 TECNICA DE DOBLE EMULSIFICACION, EVAPORACION –
EXTRACCION DEL SOLVENTE
Existen diferentes técnicas a usar en la microencapsulación por emulsificación, la elección del
método depende de las características de hidrofilicidad o lipofilicidad del principio activo a
encapsular. Para principios activos insolubles o poco solubles en agua el método de la
emulsificación aceite en agua (o/w) es comúnmente utilizada; mientras que para principios activos
hidrofílicos el método ideal para la formación de la partícula es la doble emulsificación (w/o/w).
La metodología general utilizada para la elaboración de micropartículas utilizando la técnica de
doble emulsificación de basa en encontrar un material formador de pared, tal como un polímero,
el cual es disuelto o disperso en un disolvente adecuado, el que además de solubilizar al polímero
debe ser químicamente inerte con respecto a los demás componentes de la formulación.
El principio activo a encapsular se dispersa o disuelve luego en el disolvente que contiene el
material que forma la pared.
La solución formada por el principio activo-polímero y solvente se añade a un medio de proceso
que será la fase continua de la emulsión para formar microgotitas. Este medio de proceso en el
caso de las emulsiones w/o/w es en generalmente un disolvente orgánico y aceites cuando el agua
se usa para disolver el material de pared y/o principio activo.
La fase continua se agita a medida que se adiciona la que será la fase dispersa constituida por el
disolvente, el material formador de pared y el principio activo.
Después de formada la primera emulsión, esta se adiciona sobre una cantidad adecuada de fase
acuosa la que sería la fase externa en la emulsión múltiple.
El proceso de emulsificación se da con la adición agentes surfactantes para permitir la formación
de una emulsión estable y prevenir la aglomeración. La concentración del agente surfactante
normalmente va desde el 0.1% al 20%. (Oficina Española de patentes y marcas, 1996)
Figura 2. 10 Pasos generales para la formación de microcápsulas por el método de emulsificación evaporación-extracción del solvente mediante una emulsión w/o/w
Elaborado por: Valenzuela G.
Emulsificación de la fase acuosa en la fase organica (w/o)
La emulsion anterior se reemulsifica en una
solución de PVA (w/o/w)
Extracción del solvente de la fase dispersa por la fase continua, seguido de
la evaporación del solvente
Recuperación y secado de las
microparticulas
Emulsificación w/o/w Extracción del solvente
Obtención de partículas sólidas

29
La elección de la técnica que podría traer un incremento en la eficiencia de encapsulación
dependerá de la hidrofilicidad o hidrofobicidad de la droga.
En las emulsiones aceite en agua, se agregan polímeros hidrofílico naturales o sintéticos junto con
los agentes surfactantes. Estas sustancias se acumulan en la interfase y aumentan la viscosidad de
la fase acuosa, por lo cual disminuye la velocidad de formación de los agregados. (Kosegarten &
Jiménez, 2012)
FACTORES QUE AFECTAN EN LA ELABORACIÓN DE LAS MICROPARTICULAS
POR ESTA TECNICA
2.1.6.1 Los solventes utilizados
Un disolvente adecuado para ser utilizado con la técnica de microencapsulación por doble
emulsificación evaporación-extracción del solvente debe cumplir con los siguientes criterios:
Debe ser poco soluble en la fase continua.
Los solventes a ser eliminados por evaporación deben tener el punto de ebullición
más bajo que el del medio de suspensión.
No debe ser tóxico (Li, Rouaud, & Poncelet, 2008)
La Tabla 2.1 muestra algunas de las características de los solventes comúnmente utilizados en la
técnica de doble emulsificación evaporación-extracción del solvente para microencapsulación.
Tabla 2. 1 Propiedades de algunos solventes utilizados en la elaboración de micropartículas.
SOLVENTE PUNTO DE
EBULLICIÓN
(°C)
SOLUBILIDAD
EN AGUA (g/L)
20°C
CARACTERÍSTICAS
Cloroformo 61 8 Baja solubilidad en agua, mayor toxicidad
que el diclorometano
Diclorometano 39.7 20 Solvente de la mayoría de polímeros Alta
toxicidad, baja temperatura de ebullición,
casi inmiscible en agua.
Acetato de
etilo 77 90 Baja toxicidad, parcialmente soluble en agua
Formiato de
etilo 54 105 Baja toxicidad, parcialmente soluble en agua
Por: (Li, Rouaud, & Poncelet, 2008)
El acetato de etilo presenta características menos tóxicas que el diclorometano, pero al ser
parcialmente miscible en agua no permite la formación de las microesferas durante la
emulsificación, además al extraer el solvente de forma repentina se forman aglomerados de
polímero debido al rápido intercambio del solvente. (Ramos Picos, Gómez Carril, & Fernández
Mena, 2001)

30
Para superar los inconvenientes que produce la miscibilidad en agua de los solventes se ha
propuesto tres técnicas que se detalla a continuación.
La solución acuosa se pre-saturada con disolvente
a. La fase dispersa se emulsiona primero en una pequeña cantidad de
solución acuosa, después de la formación de gotas de esta emulsión
se vierte en una gran cantidad de solución acuosa
b. La fase dispersa se emulsiona en una pequeña cantidad de solución acuosa, la solución se
agita y se evapora el disolvente dando lugar a la solidificación de las microesferas. (Li,
Rouaud, & Poncelet, 2008)
Las técnicas antes mencionadas facilitan la elaboración de microcápsulas con acetato de etilo, sin
embargo al comparar la morfología se observa microcápsulas de forma esférica y regular cuando
se utiliza diclorometano.
Al usar solventes miscibles con el agua o parcialmente miscibles también se ve disminuida la
eficiencia de encapsulación debido a que al estar una porción del solvente en la fase continua, se
facilita la disolución del p.a. en esta fase, habiendo disminución del p.a. aprovechable para la
elaboración de la microcápsula.
El aceite mineral como fase apolar dispersante es de uso común para la microencapsulación de
componentes activos hidrofílicos. (Ramos Picos, Gómez Carril, & Fernández Mena, 2001)
2.1.6.2 Uso de co-solventes
Los co-solventes son sustancias que ayudan a solubilizar los principios activos que no se logran
solubilizar de manera total con los solventes.
Se ha visto útil la mezcla de solventes como diclorometano-metanol, etanol o propilenglicol para
alcanzar disolución del principio activo, otros compuestos necesitan mezclas de acetonitrilo-
etanol y opcionalmente agua, o solamente acetona o una mezcla de metanol y etanol para su
solubilización; el uso de las mezclas también permite una rápida eliminación del solvente así
como rápida precipitación del polímero.
La acetona como co-solvente de compuestos halogenados ha permitido disminuir el tamaño de
partícula, produciendo el mismo efecto la mezcla de acetona y metanol, este efecto se explica
porque la acetona es miscible con el agua y algunos solventes orgánicos por lo que al ser añadida
se produce un incremento de la solubilidad en agua de los solventes orgánicos, facilitando así la
extracción del solvente por la fase externa. (Jelvehgari & Montazam, 2012) (Li, Rouaud, &
Poncelet, 2008)
2.1.6.3 Efecto de los generadores de porosidad
Llamados también porosígeno o porógeno, son sustancias que incrementan la porosidad de la
microcápsula con ello aumenta la velocidad de liberación del principio activo.

31
Sustancias utilizadas con esta finalidad son n-hexano, gel de dextrano reticulado, cloruro de sodio,
lactosa, heparina de sodio, polietilenglicol, co-polímeros de óxido de polietileno/óxido de
propileno y sus mezclas.
2.1.6.4 Efecto de la temperatura
La temperatura no es crítica pero debe estar en un intervalo que prevenga la ebullición del
solvente, gelificación del polímero o al principio activo o material formador de pared degradarse.
2.1.6.5 Efecto de la viscosidad
El incremento de la viscosidad de la fase dispersa mejora la eficiencia de encapsulación, así como
provoca el aumento en el tamaño de las micropartículas; esto se logra por adición de un polímero
o incremento en el peso molecular de este, así como por variación del solvente utilizado, aunque
en producción este es lo que menos se toma en cuenta ya que lo primordial es proveerse de un
solvente con alta velocidad de evaporación. (Li, Rouaud, & Poncelet, 2008)
2.1.6.6 Efecto de la concentración de polímero
El incremento en la concentración de polímero da a lugar micropartículas con superficie más lisa,
y por otro lado se incrementa la eficiencia de encapsulación.
La superficie formada por el polímero se vuelve más gruesa, esto conlleva una disminución en la
velocidad de liberación del fármaco.
Sin embargo el control en la concentración del polímero es crítica ya que si bien los polímeros de
alto peso molecular ayudan a mejorar la estabilidad de la emulsión, también pueden provocar la
agregación de las gotas mediante el mecanismo de depleción por floculación cuando la
concentración es muy pequeña. (Aranberri, 2006)
2.1.6.7 Efecto de la cantidad de principio activo
El equilibrio de fuerzas dispersivas y polares (balance hidrofílico- lipofílico) de las grasas y
surfactantes, así como la presencia de ciertos grupos funcionales específicos del principio activo
que se desea encapsular, van a determinar el comportamiento del fármaco al momento de ser
atrapado por la matriz. Dichas fuerzas determinan la solubilidad, el coeficiente de reparto, la
adsorción del fármaco sobre la superficie de las partículas y/o la tendencia a la cristalización de
los materiales de la matriz.
En consecuencia determinan la eficiencia de carga en la matriz y la estabilidad física de las
partículas.
De manera general de se considera que el incremento de la cantidad de principio activo mejora la
eficiencia de encapsulación. (Garzón S., Vázquez R., & Villafuerte R., 2009)

32
2.1.6.8 Los emulsificantes en la microencapsulación
Antes de elegir el tipo de surfactante y su concentración, es importante saber la polaridad de las
dos fases no miscibles, el tamaño deseado de microesferas y la demanda de la esfericidad de las
microesferas para que la liberación del principio activo sea lo más constante.
Los agentes emulsificantes forman una fina capa alrededor de las gotículas de aceite, polímero o
principio activo, evitando la coalescencia principal problema en el proceso de emulsificación,
cuando se está eliminando el solvente el emulsificante continúa manteniendo las gotículas de
aceite en su configuración esférica y las previene de la agregación hasta que el solvente es
eliminado completamente y las microesferas son endurecidas como partículas discretas.
Las propiedades físico-químicas, las propiedades estructurales y la concentración del
emulsificante influyen en las características de las microesferas. Generalmente se dice que para
un emulsificante dado, el incremento en la concentración disminuye la tensión superficial de la
fase continua, lo que se traduce en la disminución en el tamaño de los micropartículas. Sin
embargo, han aparecido concentraciones limitantes, por encima de las cuales el emulsificante no
aumenta su efecto. Esto se debe probablemente a que se haya alcanzado la concentración óptima
de empaquetamiento de la emulsión lo que se conoce como concentración micelar crítica.
La concentración requerida y la efectividad de cada emulsificante es diferente, y el mejor
emulsificante para una aplicación en particular es determinado experimentalmente. (Ramos Picos,
Gómez Carril, & Fernández Mena, 2001) (Li, Rouaud, & Poncelet, 2008)
2.1.6.9 Uso de antiespumantes
Cuando en el proceso de elaboración de micropartículas se va a utilizar altas velocidades de
agitación es adecuado añadir a la formulación un antiespumante a la fase acuosa que evite la
formación de burbujas que podrían afectar la formación de micropartículas.
Existe antiespumantes con o sin silicona los que van a ayudar a disipar las burbujas con aire de la
formulación.
Una alternativa es reemplazar el agente surfactante por un polímero biodegradable anfifílico, con
esto se evita el posible efecto del surfactante sobre el microgránulo. (Li, Rouaud, & Poncelet,
2008)
2.1.6.10 Materiales de recubrimiento
Los materiales de recubrimiento utilizados en la elaboración de micropartículas son de tres tipos:
a. Grasas: Cera carnauba, alcohol estearílico, ácido esteárico.
b. Proteínas: Gelatina, Albúmina.
c. Polímeros: Naturales, sintéticos, semisintéticos, biodegradables.

33
Los polímeros utilizados para la elaboración de los sistemas de liberación modificada deben
cumplir con características de biodegradabilidad y biocompatibilidad, es decir que deben ser
químicamente inertes, no deben provocar una respuesta inflamatoria o tóxica, con una estructura
física adecuada, fácilmente degradados en el organismo formando compuestos inertes que son
después metabolizados o excretados, y debe tener un periodo de vida útil aceptable. (Jelvehgari
& Montazam, 2012)
Otro aspecto importante para la elección del polímero es la velocidad con la que se desea liberar
el principio activo; cuando un polímero no cumple con las características de liberación requeridas,
es posible sintetizar a partir de otros dos, un nuevo polímero con mejores características de
permeabilidad, a este nuevo se le denomina co-polímero. Como ejemplo de puede citar el co-
polímero (PEG/PLA), sintetizado a partir de polietilenglicol y homopolímeros de ácido láctico.
Los segmentos de hidrofílicos de polietilenglicol en la estructura incrementando la difusión del
agua a través del polímero con lo que se logra incrementar la velocidad de liberación. (Li, Rouaud,
& Poncelet, 2008)
Tabla 2. 2 Efecto de los factores que influencian en las características de las micropartículas
IMPACTO EN LAS PROPIEDADES DE LAS MICROESFERAS
FACTORES
(Incremento)
Tamaño Morfología de la
superficie
Encapsulación de la droga
con
dic
ion
es d
e o
per
aci
ón
Viscosidad de la fase
dispersa
Diámetro alto Superficie suave Incremento de la eficiencia,
liberación lenta del fármaco
Proporción entre la fase
dispersa y la fase
continua
Disminuye el
diámetro o no
influencia
No influye Incremento de la eficiencia.
Cantidad de droga en la
fase dispersa
No influye Superficie porosa
e irregular
Baja eficiencia cuando la
cantidad de fármaco es alta
por la formación de poros
Concentración del
surfactante
Menor diámetro No influye No influye
Concentración del
porosígeno
Menor diámetro Superficie gruesa
con grandes poros
No influye
Velocidad de agitación Menor diámetro,
menor tamaño de
distribución.
No influye No influye

34
IMPACTO EN LAS PROPIEDADES DE LAS MICROESFERAS co
nd
icio
nes
de
op
era
ció
n
Incremento de
temperatura
Diámetro alto Superficie gruesa Disminución de la eficiencia
Presión reducida en
comparación con la
presión atmosférica
Diámetro pequeño o
no influencia
Superficie suave Aumento o disminución de la
eficiencia de encapsulación
(según diferentes autores),
liberación lenta del fármaco
Nota: Modificado de (Li, Rouaud, & Poncelet, 2008)
2.1.6.11 DESVENTAJAS DE LA TECNICA
La técnica de doble emulsificación evaporación-extracción del solvente no es muy recomendable
debido a que existe una pérdida importante de principio activo durante el proceso debido a que la
emulsificación debe realizarse en una fase acuosa y al ser el principio activo hidrofílico se filtrará
rápidamente desde la solución polimérica hacia el entorno acuoso. (Jelvehgari & Montazam,
2012)
2.1.7 RUTAS DE ADMINISTRACIÓN DE LAS MICROPARTICULAS
Los sistemas empleados en la liberación modificada de fármacos permiten la administración el
principio activo por cualquiera de las vías tradicionales siendo la oral, transdérmica y parenteral
subcutánea las que mayor éxito terapéutico han tenido.
2.1.8 MATERIAS PRIMAS UTILIZADAS EN LA INVESTIGACIÓN
Para la presente investigación se seleccionó como fase externa de la que será la primera emulsión
(w/o) el limoneno, debido a la reducida toxicidad en comparación a los solventes orgánicos que
de manera habitual se utiliza en el desarrollo de la técnica de microencapsulación por doble
emulsificación evaporación-extracción del solvente.
Como polímero formador de pared se seleccionó la gelatina ya que al ser un material
biodegradable, fácil de adquirir, y de bajo costo ha sido utilizado en muchos otros trabajos.
Los agentes surfactantes con los que puso a prueba la estabilidad de la emulsión fueron el Span®
80 Tween® 80.

35
Como fase externa de la emulsión w/o/w se utilizó una solución de alcohol polivinílico (PVA).
2.1.8.1 Limoneno
El limoneno es una sustancia natural que se extrae del aceite de las cáscaras de los cítricos, en
nuestro caso se extrajo del aceite esencial de naranja. Pertenece al grupo de los terpenos, en
concreto a de los limonoides.
Es un líquido incoloro a temperatura ambiente, es insoluble en agua, inflamable a temperaturas
superiores a 48°C.
Tabla 2. 3 Propiedades del Limoneno
Estructura del limoneno
Figura 2. 11 Estructura del limoneno con sus respectivos isómeros.
Fuente: www.quimicaorgánica.net
El limoneno utilizado para esta investigación fue extraído del aceite esencial de naranja y presenta
las siguientes propiedades.
Tabla 2. 4 Propiedades Limoneno Estandar. Vs Limoneno muestra pH Índice de refracción
Limoneno estándar 2.6 1.471
Limoneno muestra 3.8 1.471
PROPIEDADES DEL LIMONENO
Nombre IUPAC 1-Methyl-4-(1-methylethenyl)-cyclohexene
Fórmula molecular C10H16
Densidad 0.842 g/mL
Masa molar 136.23 g/mol
Punto de fusión -74,35 °C, (199 K)
Punto de ebullición 176 °C, (449 K)

36
Aplicaciones del limoneno
En los últimos años el limoneno ha adquirido una singular importancia debido a su demanda
como disolvente biodegradable. En la industria farmacéutica y alimentaria es utilizado como
componente aromático y para dar sabor.
Recientes estudios parecen apuntar a que el limoneno tiene efectos anticancerígenos. Incrementa
los niveles de enzimas hepáticas implicadas en la detoxificación de carcinógenos. La glutatión S-
transferasa (GST) es un sistema que elimina carcinógenos. El limoneno parece promover el
sistema GST del hígado y los intestinos atenuando el efecto dañino de los carcinógenos.
Estudios en animales demuestran que limoneno en la dieta reduce el crecimiento tumoral en
mamíferos.
2.1.8.2 Agua tipo I
El agua utilizada es el “AGUA TIPO I” obtenida con el equipo purificador de agua Mili-Q.
Las características del agua que este equipo nos proporciona en parámetros de resistividad
es de 18.2mΩ, la purificación que se da es mediante ósmosis inversa, filtración por filtros
de carbono, resinas de intercambio iónico, fotooxidación con luz ultravioleta a doble
longitud de onda (254 nm y 320 nm) y finalmente es filtrada por membrana de 0.22 µm
de tamaño de poro, lo que permite tener características adecuadas para evitar partículas
externas que interfieran en la determinación del tamaño de partícula, componentes
químicos o microorganismos tengan una acción negativa en la formulación.
2.1.8.3 Polímeros
Gelatina
La gelatina es una proteína natural, biodegradable obtenida por hidrólisis del colágeno. La
gelatina es una mezcla heterogénea de hebras de glicina, prolina, y residuos de hidroxiprolina. En
vivo se degradan a estos aminoácidos
El proceso de formación de la micropartículas con gelatina involucra la adición de otro no
solvente miscible en agua para la gelatina o la adición de otro polímero más soluble en agua, a la
solución de gelatina que se encuentra por encima de los 40°C.
Las partículas de gelatina se aíslan y endurecen por reticulación química con glutaraldehido.
Alternativamente, estas partículas se pueden preparar usando una simple emulsión o/w o w/o/ w
método de microemulsión. Nanopartículas de gelatina se han utilizado para entregar el paclitaxel,
metotrexato, doxorubicina, ADN, oligonucleótidos de doble cadena, y los genes. PEGilación de
las partículas aumenta significativamente su tiempo de circulación en el torrente sanguíneo y
aumenta su absorción en las células por endocitosis. Nanopartículas de gelatina modificada por
anticuerpos se han utilizado para la absorción selectiva de los linfocitos.

37
Alcohol polivinílico
El alcohol polivinílico o polivinilalcohol, PVA, es un polímero hidrofílico, no toxico,
biocompatible, con buenas propiedades mecánicas, y muy estable durante largos periodos en
diferentes condiciones de temperatura y pH. Su fórmula química es (C2H4O)n.
El alcohol polivinílico es soluble en agua fría, pero su disolución es lenta mientras que puede
disolverse rápidamente en agua caliente y puede normalmente disolverse a más de 90°C.
Las disoluciones acuosas no son particularmente estables, especialmente si hay presentes trazas
de ácido o base. Las disoluciones pueden sufrir una compleja serie de reacciones de gelación
reversibles e irreversibles. Por ejemplo, puede producirse entre-cruzamiento en los enlaces éter,
lo que resulta en un aumento de viscosidad por la formación de productos insolubles.
El alcohol polivinílico (PVA) es un polímero anfifílico, utilizado como estabilizante de las
emulsiones; el incremento en la viscosidad de la fase continua acuosa que este polímero produce
el movimiento de las gotas dispersas y por tanto la probabilidad de que las gotas se acerquen y
se fusionen
El alcohol polivinílico es utilizado para estabilizar las micelas de la emulsión. Este compuesto
presenta una temperatura de transición vítrea (Tg) a los 85°C, que es la temperatura a la cual se
produce la transición del estado vítreo al estado gomoso.
Figura 2. 12 Alcohol polivinílico A) Estructura química del PVA. (Tecnología de los plásticos. 2012) B) Polímero al estado sólido. (Por Valenzuela G.)
2.1.8.4 Agentes surfactantes
Tween ® 80
El Tween® 80, cuya fórmula molecular es C64H124O26 es un surfactante no iónico hidrofílico con
un amplio e intenso poder emulgente y suspensor, permite formar emulsiones de tipo o/w, estables
y de textura fina, poco afectables por altas concentraciones de electrolitos o por cambios ligeros
de pH.
El Tween® 80 además posee acción protectora y emoliente, En formulaciones orales y
parenterales se lo usa como agente humectante.
Es bien tolerado y no es irritante para la piel y mucosas.
B
)
A
)

38
Figura 2. 13 Estructura molecular del Tween ® 80. Por Sigma Aldrich®
Span ® 80
El Span® 80 de fórmula molecular es C24H44O6, es utilizado en preparaciones farmacéuticas y
alimenticias. Con frecuencia es utilizado en cosmética para solubilizar aceites esenciales dentro
de productos de base acuosa, por su estructura permite formar emulsiones del tipo w/o.
Figura 2. 14 Estructura molecular del Span® 80. Por Sigma Aldrich®
Propiedades de los surfactantes utilizados en la formulación
Tabla 2. 5 Propiedades de los surfactantes utilizados en la experimentación Nombre Comercial Span ® 80 Tween ® 80
Surfactante Monooleato de sorbitán Monooleato de sorbitan
polietoxilado
HLB 4.3 15
Estado
(25°C)
Líquido, claro, viscoso, de
color café oscuro Líquido
Peso molecular (g/mol) 428 1310
Densidad relativa
(25°C) 0,986 g/mL 1,06 g/mL
Fuente: (Bullón , 2007)

39
2.1.8.5 Diclofenaco
El diclofenaco es un antiinflamatorio y analgésico no esteroideo, del grupo de los derivados
arilacéticos.
Sus efectos farmacológicos se deben a la inhibición de las isoenzimas de la ciclooxigenasa, COX-
1 y COX-2, bloqueando la conversión del ácido araquidónico a prostaglandina G2, siendo mayor
su inhibición sobre COX-2.
Diclofenaco de sodio
Estructura del Diclofenaco de sodio
Figura 2. 15 Estructura del Diclofenaco de sodio Fuente: Biblioteca digital UNAL, 2012
Propiedades del Diclofenaco sódico
PROPIEDADES
Nombre IUPAC [2[(2,6 diclorofenill) amino] fenil] acetato de sodio.
Fórmula condensada: C14H10Cl2NNaO2
Masa molar 318.1g/mol.
Descripción Polvo de color blanco opaco, inodoro
Punto de Fusión 283° - 285° C
Solubilidad en Agua (pH 5,2) 9mg/mL
Solubilidad en acetona 4,45mg/g
Por:(Martinez-Pla et al. 2005)
Vía de administración y dosis
La administración diaria de 75 mg a 100 mg de Diclofenaco produce efectos analgésicos y
antiinflamatorios significativos en el tratamiento de diversas condiciones reumáticas

40
Farmacocinética
a. Absorción
Se caracteriza por tener un tiempo de vida media biológico corto de 1,2 a 2 horas, después de su
administración por vía oral; es absorbido casi completamente en el tracto gastrointestinal pero sus
biodisponibilidad es solamente del 50-60% debido a un gran efecto de primer paso, posiblemente
por el citocromo intestinal P450 3A4.
b. Biotransformación
El diclofenaco sódico sufre un significativo metabolismo de primer paso, indican los autores, y
sólo un 60% de la droga alcanza la circulación sanguínea sin modificaciones luego de la
administración oral.
c. Eliminación
Se elimina principalmente por metabolismo hepático y posterior excreción urinaria de los
conjugados de sus metabolitos. En humanos, el principal metabolito es el 4-hidroxi-diclofenaco,
que posee una actividad antiinflamatoria despreciable en comparación con la droga madre.
De acuerdo a su comportamiento en cuanto a la solubilidad y permeabilidad, se encuentra en la
categoría II del Sistema de Clasificación Biofarmacéutica (Chuasuwan et al. 2009).
d. Contraindicaciones
No se recomienda utilizar diclofenaco en niños menores de 18 meses, advierten, y sólo en casos
especiales en mujeres embarazadas o en período de lactancia.
Diclofenaco dietilamina
Estructura del diclofenaco dietilamonio
Figura 2. 16 Estructura del diclofenaco dietilamonio (DDEA) Fuente: Mulgund et al. 2009

41
Indicaciones
El diclofenaco está indicado como antiinflamatorio y analgésico en procesos osteoarticulares
como artritis reumatoide, lumbalgias, poliartritis. Es utilizado como analgésico y antitérmico en
reumatismos que han producido tumefacción inflamatoria.
Vía de administración y dosis
Vía de administración: tópica.
Adultos: aplicar una fina capa del producto en la zona dolorida de 3 a 4 veces al día mediante un
ligero masaje para facilitar la penetración.
La cantidad a aplicar depende del tamaño de la zona dolorosa. Por ejemplo, 2 a 4 gramos de
diclofenaco.
La duración del tratamiento depende de la indicación y de la respuesta obtenida.
No aplicar más de 7 días seguidos.
Farmacocinética
a. Absorción
La cantidad de diclofenaco absorbido a través de la piel es proporcional al tiempo de contacto y
el área de la piel cubierta por diclofenaco, y depende de la dosis tópica total y de la hidratación
de la piel.
b. Distribución
Tras la administración tópica de diclofenaco tópico en las articulaciones de la mano y la rodilla,
se puede medir el diclofenaco en plasma, tejido sinovial, y líquido sinovial. Las concentraciones
plasmáticas máximas de diclofenaco tras la administración tópica de diclofenaco son unas 100
veces más bajas que tras la administración oral de diclofenaco comprimidos. El 99,7% de
diclofenaco se une a las proteínas plasmáticas, principalmente a albúmina (99,4%).
c. Biotransformación
La Biotransformación de diclofenaco supone por una parte glucuronidación de la molécula
intacta, pero principalmente hidroxilación simple y múltiple que genera diversos metabolitos
fenólicos, la mayoría de los cuales se convierten en glucurónidos conjugados. Dos de estos
metabolitos fenólicos son biológicamente activos, aunque en una proporción mucho menor que
diclofenaco.
d. Eliminación
El aclaramiento sistémico total de diclofenaco del plasma es 263 +/- 56 mL/min. El diclofenaco
y sus metabolitos se excretan principalmente por la orina.

42
Contraindicaciones
No utilizar diclofenaco dietilamonio sobre quemaduras solares.
No administrar a pacientes que hayan presentado reacciones alérgicas (rinitis, asma, prurito,
angiodema, urticaria, shock u otras), provocadas por ácido acetilsalicílico u otros AINES debido
a la posibilidad de hipersensibilidad cruzada.
No utilizar durante el último trimestre del embarazo
2.1.9 CARACTERIZACIÓN
2.1.9.1 Estabilidad de la emulsión
Las emulsiones al formar gotas sirven de plantilla para formación de las partículas poliméricas,
por lo que es necesario direccionarse a obtener una emulsión con las mejores características de
estabilidad, que nos permitan obtener partículas lo más esféricas, uniformes y con los
componentes adecuados para mantener la estructura aun después de eliminado el solvente.
La estabilidad en una emulsión a más de la observación macroscópica y microscópica, puede de
forma indirecta determinarse mediante dispersión de luz dinámica (DLS), esta es una técnica
óptica utilizada para estimar la distribución de los tamaños de partícula y si estos varian en el
transcurso del tiempo es un indicativo de inestabilidad
La determinación se hace al valorar la luz dispersada por las partículas cuando son iluminadas
con una luz monocromática.
2.1.9.2 Caracterización de las micropartículas
Las micropartículas deben ser caracterizadas y controladas con ensayos que aseguren su calidad
y homogeneidad, así como su comportamiento en la liberación del principio activo.
Los ensayos característicos que se suelen realizar a las micropartículas son:
a) Tamaño de partícula.
b) Contenido de principio activo.
c) Rendimiento de la producción de partículas.
d) Eficacia de la encapsulación.
a. Tamaño De Partícula
La uniformidad en la dosificación mejora con la uniformidad en el tamaño de partícula.
La microscopia óptica además de permitirnos monitorizar de forma constante las características
de estabilidad de la emulsión, nos permite determinar valores de los tamaños de las partículas

43
mediante programas informáticos como el ImageJ, que permite el procesamiento de imagen
digital de dominio público.
La lectura se realizó a 25 ºC empleando un láser con un ángulo de dispersión de 173º. Se
realizaron cinco mediciones por muestra.
Figura 2. 17 Tamaño de partícula analizado con el programa ImageJ Izquierda: Vista microscópica. Derecha Histograma
El DLS también permite determinar el índice de polidispersión. Este índice provee información
sobre la muestra: valores cercanos a 0 indican que la muestra es monodispersa así como valores
cercanos a la unidad indican que la muestra presenta una amplia variedad de tamaños.
Otro parámetro determinado mediante esta técnica es el potencial zeta es una medida de la
estabilidad de una partícula e indica el potencial que se requiere para penetrar la capa de iones
circundante en la partícula para desestabilizarla. Por lo tanto, el potencial zeta es la potencia
electrostática que existe entre la separación de las capas que rodean a la partícula. Puede ser un
parámetro indicativo de aglomeración.
Figura 2. 18 Lectura del tamaño de partícula en el equipo DLS.
A. Posicionamiento de la muestra B. Software para el análisis
A B
05
10152025
3,0
7,2
11
,5
15
,7
19
,9 y…

44
b. Contenido de principio activo
El contenido de principio activo o capacidad de encapsulación hace referencia a la cantidad de
principio activo encapsulado en la microcápsulas. Se calcula de la siguiente manera:
𝐶𝑜𝑛𝑡𝑒𝑛𝑖𝑑𝑜 𝑝. 𝑎. = 𝐶𝑎𝑛𝑡𝑖𝑑𝑎𝑑 𝑑𝑒 𝑝𝑟𝑖𝑛𝑐𝑖𝑝𝑖𝑜 𝑎𝑐𝑡𝑖𝑣𝑜 𝑒𝑛𝑐𝑎𝑝𝑠𝑢𝑙𝑎𝑑𝑜
𝑃𝑒𝑠𝑜 𝑓𝑖𝑛𝑎𝑙 𝑑𝑒 𝑙𝑎 𝑚𝑖𝑐𝑟𝑜𝑝𝑎𝑟𝑡í𝑐𝑢𝑙𝑎𝑥100
Ecuación 2. 2 Contenido de principio activo en la micropartícula
c. Rendimiento de producción de partículas
El rendimiento de producción refleja el porcentaje de microcápsulas obtenidas con respecto a la
cantidad total de material (material activo + polímero) empleado. Se trata de un control muy
importante desde el punto de vista económico, teniendo en cuenta el elevado costo de la mayoría
de los polímeros y materiales activos utilizados; por lo que es necesario recuperar en forma de
microcápsulas la mayor cantidad posible del material de partida.
La siguiente ecuación nos permite calcular la cantidad de microcápsulas obtenidas en relación a
lo esperado en términos de porcentaje.
%𝑅𝑒𝑛𝑑. 𝑃𝑟𝑜𝑑𝑢𝑐𝑐𝑖ó𝑛 =𝐶𝑎𝑛𝑡𝑖𝑑𝑎𝑑 𝑑𝑒 𝑚𝑖𝑐𝑟𝑜𝑝𝑎𝑟𝑡í𝑐𝑢𝑙𝑎𝑠 𝑜𝑏𝑡𝑒𝑛𝑖𝑑𝑎𝑠
𝐶𝑎𝑛𝑡𝑖𝑑𝑎𝑑 𝑑𝑒 𝑚𝑖𝑐𝑟𝑜𝑝𝑎𝑟𝑡í𝑐𝑢𝑙𝑎𝑠 𝑒𝑠𝑝𝑒𝑟𝑎𝑑𝑎𝑠𝑥100
Ecuación 2. 3 Rendimiento de la producción.
d. Eficacia de la encapsulación
Para cuantificar la cantidad de principio activo encapsulado en las micropartículas se disuelve
previamente el polímero formador de cubierta en un disolvente adecuado o se extrae el material
activo utilizando un disolvente en el cual el compuesto activo es soluble y el polímero insoluble.
Se calcula a partir de la siguiente ecuación:
% 𝐸𝑛𝑐𝑎𝑝𝑠𝑢𝑙𝑎𝑑𝑜 = 𝐶𝑎𝑛𝑡𝑖𝑑𝑎𝑑 𝑑𝑒 𝑝𝑟𝑖𝑛𝑐𝑖𝑝𝑖𝑜 𝑎𝑐𝑡𝑖𝑣𝑜 𝑒𝑛𝑐𝑎𝑝𝑠𝑢𝑙𝑎𝑑𝑜
𝐶𝑎𝑛𝑡𝑖𝑑𝑎𝑑 𝑡𝑒ó𝑟𝑖𝑐𝑎 𝑑𝑒 𝑝𝑟𝑖𝑛𝑐𝑖𝑝𝑖𝑜 𝑎𝑐𝑡𝑖𝑣𝑜𝑥100
Ecuación 2. 4 Porcentaje de principio activo encapsulado
Lo óptimo dentro del proceso de microencapsulación es que tanto el contenido de principio activo
como la eficacia de encapsulación sean lo más elevados posibles. Es decir, que se haya
incorporado la mayor cantidad posible de principio activo dentro de la; además, desde el punto
de vista económico conviene que todo o prácticamente todo el material activo utilizado en el
proceso sea encapsulado. (Jelvehgari & Montazam, 2012)

45
2.1.10 LIBERACION DEL PRINCIPIO ACTIVO DESDE LA MICROPARTICULA
2.1.10.1 Liberación del principio activo en sistemas poliméricos.
La velocidad de liberación de los principios activos puede ser modificada mediante el uso de
polímeros hidrófilos en la formulación, estos consiguen hincharse en un medio acuoso pero sin
disolverse, generando una velocidad de liberación prácticamente constante.
La velocidad de liberación del principio activo se ve regulada por mecanismos como la difusión,
el hinchamiento y la erosión, además de las restricciones de transferencia que se presenten en la
interfase polímero/líquido. (Andreeta, 2003)
El proceso de liberación de principios activos en sistemas hidrofílicos es descrito en cuatro pasos
que se muestran a continuación:
a. La penetración del líquido del medio de disolución o del tracto gastrointestinal en la
forma farmacéutica, junto con la disolución simultánea de una cantidad pequeña de
fármaco que se encuentra en la superficie.
b. Hinchamiento del polímero hidrófilo por adsorción de agua y formación de una barrera
gelificada
c. Penetración de los líquidos circundantes en la profundidad de la forma farmacéutica por
difusión a través de la capa de gel y disolución del fármaco
d. Liberación del fármaco disuelto a través de la barrera gelificada.
Los sistemas poliméricos controlan la liberación del p.a. mediante mecanismos físicos y
mecanismos químicos.
Mecanismos físicos de liberación.
a. Sistemas controlados por difusión: La liberación se da a través de la estructura
molecular del compuesto o por macro/micro poros o por ambos. Estos sistemas pueden
presentarse de la siguiente manera:
Sistemas Depósito: Caracterizados porque el principio activo se encuentra en el
núcleo de la forma farmacéutica, rodeado de una fina membrana, homogénea y no
porosa de polímero, el cual pueden o no hincharse.
En estos sistemas el transporte del p.a. al exterior se da por disolución de este en la
interfase p.a./polímero y posterior difusión.
Sistemas Matriciales: Son sistemas monolíticos, en donde el principio activo está
distribuido uniformemente en un soporte de polímero. El fármaco puede estar
disuelto o disperso en la matriz polimérica.

46
En las matrices lipídicas el principio activo se cede paulatinamente al medio a medida
que los líquidos del tracto gastrointestinal erosionan lentamente la masa de la forma
farmacéutica; en estos sistemas la velocidad de liberación del p.a. puede ser regulada
añadiendo sustancias hidrofílicas a la formulación, estas sustancias permiten la formación
de canales, es así que la liberación del p.a. será controlado por erosión de la matriz y
difusión a través de los poros.
b. Sistemas controlados por el disolvente: Pueden ser sistemas de depósito o matriciales
en donde la salida del principio activo está controlada por la penetración del disolvente,
sea por osmosis o hinchamiento.
Sistemas controlados por hinchamiento: Son sistemas monolíticos en los que el activo
esta disuelto o disperso en un soporte polimérico hidrofílico, pudiendo ser
entrecruzado o no. El soporte se hincha al contacto con el agua, siendo dependiente
del balance hidrofílico/lipofílico, así como del grado de entrecruzamiento del
polímero.
Sistemas Osmóticos: Son sistemas de depósito, cuya membrana permite el paso del
agua mas no del fármaco. Presentan un orificio en la membrana por el cual se libera
el fármaco cuando se incrementa la presión osmótica. (Infante Blanco & et. al, s.f.)
Cabe resaltar que también existen mecanismos químicos que regulan la liberación, en este caso
el sistema libera el principio activo en respuesta a una reacción química.
Como conclusión se obtiene que no existe en términos generales una regla para la liberación de
la droga, esta dependerá de la composición de la partícula polimérica y los factores que a esta
afecten, factores que deben ser tomados en cuenta para evitar riesgos de ineficacia terapéutica.
(Andreeta, 2003)
2.1.10.2 Etapas de liberación del fármaco
Para que el material activo sea liberado desde la microcápsula es necesario que esta se someta a
condiciones particulares como cambios en la humedad, pH, incremento de presión o fuerza que
se ejerza sobre las microcápsulas o una combinación de todos estos factores externos; por otro
lado la liberación del fármaco encapsulado puede presentarse porque el principio activo difunde
a través de los canales y poros que se forman tras la degradación de la superficie de la matriz
polimérica.
Es así que los factores que influyen en el perfil de liberación incluyen factores externos que se
aplican sobre la microcápsula y los factores internos como las propiedades de la matriz
polimérica, el fármaco utilizado, la estructura de la micropartícula, la técnica de encapsulación y
las condiciones experimentales.

47
Las sustancias hidrofílicas encapsuladas en microcápsulas o microesferas poliméricas son
comúnmente liberadas siguiendo un patrón de tres pasos.
a. Fase de liberación explosiva del fármaco que está determinado por la cantidad de fármaco
en la superficie, los canales y poros de las microesferas
b. Fase de liberación lenta, en donde poco o nada se libera el fármaco
c. Fase de liberación rápida del principio activo debido a la erosión de las partículas.
(Jelvehgari & Montazam, 2012)
2.1.10.3 Cinética de liberación de principios activos
Al determinar la cinética de liberación de un principio activo se mide la cantidad de fármaco que
se va liberando desde una matriz o vehículo farmacéutico a través del tiempo.
La velocidad de absorción, es decir del número de moléculas de fármaco que se absorben por
unidad de tiempo depende de la constante de absorción (K) y de la cantidad de moléculas que se
encuentren en solución en el lugar de absorción; en los sistemas de liberación inmediata todas las
moléculas de fármaco se encuentran expuestas para su absorción, la que disminuye de manera
exponencial con el transcurso del tiempo, a esto se le conoce como proceso de orden uno (1),
mientras que en los sistemas de liberación modificada el proceso es de orden cero puesto que el
número de moléculas a absorber está limitado a la velocidad de liberación de la estructura que es
bastante constante, el número de moléculas no disminuye con el tiempo debido a que las
moléculas absorbidas son repuestas desde el depósito, aquí la velocidad de absorción permanece
constante durante todo el proceso. (Baños Díez & Farré Albaladejo, 2002). Esta dependencia
puede ser representada por la siguiente ecuación:
[𝐴] = [𝐴]0 − 𝐾𝑡
Ecuación 2. 5 Cinética de orden cero
Los sistemas de liberación inmediata pueden ser representados por una cinética de primer o
segundo orden y las ecuaciones que definen dichas cinéticas son:
𝑙𝑛[𝐴] = 𝑙𝑛[𝐴]0 − 𝐾𝑡
Ecuación 2. 6 Cinética de primer orden
1
[𝐴] =
1
[𝐴]0+ 𝐾𝑡
Ecuación 2. 7 Cinética de segundo orden

48
Donde:
[𝐴]: Representa la concentración del analito.
[𝐴]0: Representa la concentración inicial del analito.
K: Representa la constante cinética.
𝑡: Representa los diferentes tiempos a los que se toman las muestras.
Para matrices más complejas existe un modelo matemático propuesto por Higuchi en 1963, el
cual describe el proceso de liberación de fármacos que cumple con la ley de Fick ya que toma en
cuenta parámetros como el coeficiente de difusión, el área superficial del sistema de liberación
controlada, la concentración del principio activo en la matriz y su solubilidad. El modelo de
Higuchi se describe con la siguiente ecuación:
𝑀𝑡
𝑀∞= 𝑘 √𝑡
2
Ecuación 2. 8 Ecuación de Higuchi
Donde:
𝑀𝑡: Representa la cantidad absoluta de fármaco liberado en un tiempo t.
𝑀∞: Representa la cantidad de fármaco liberado en un tiempo infinito, correspondiente a la
cantidad total incorporada en el sistema a t = 0
𝑘: Representa la constante de velocidad de liberación que incorpora características estructurales
y geométricas del sistema de liberación.
t: representa el tiempo transcurrido desde el inicio de la liberación.
La relación 𝑀𝑡
𝑀∞⁄ puede ser expresada como el porcentaje de liberación de fármaco “Q”,
entonces:
𝑄 = 𝑘 √𝑡2
Ecuación 2. 9 Porcentaje de liberación del fármaco
Las condiciones que se deben tener en cuenta para que se cumpla este modelo son las siguientes:
La concentración inicial de droga en el sistema es mucho mayor que la solubilidad de la
droga en el mismo (condición de estado pseudo estacionario-condición sink)
El análisis matemático se basa en una difusión unidimensional (de esta manera
despreciamos efectos de contorno.

49
El tamaño de partícula de la droga suspendida en el sistema es tal, que el diámetro de la
misma no es significativo frente al espesor del sistema.
Es despreciable el hinchamiento y disolución del polímero.
La difusividad de la droga es constante.
En todo el proceso de liberación se mantiene la condición Sink. (Andreeta, 2003)
2.1.10.4 Métodos de disolución.
Existen algunos métodos oficiales recomendados por la Farmacopea de los Estados Unidos para
determinar la velocidad de liberación desde las formas farmacéuticas, siendo el método de cesta
y paleta los más empleados.
Método de cesta (Aparato 1)
Método de paleta (Aparato 2)
Método de cilindro de doble acción (Aparato 3)
Método de sistema celular de flujo continuo (Aparato 4)
La cinética de liberación del principio activo ha sido determinada con el aparato 2 y una
modificación al Método de sistema celular de flujo continuo.
a. Método de paleta, Aparato 2
El método de paleta consiste en vasos cilíndricos de vidrio o plástico y de fondo es esférico. Los
vasos van inmersos en un baño de agua termostatizado, de modo que el medio receptor que se
encuentra en su interior permanezca a 37°C ± 0,5°C.
El medio de disolución o medio receptor es el especificado en la monografía de cada producto.
La agitación se da mediante una paleta de diámetro entre 74 y 75 mm, recubierta por un polímero
fluorocarbonado. Una vez accionado el sistema de agitación, se deja caer la forma farmacéutica
al fondo del vaso deslizándola por las paredes, evitando la formación de burbujas en su superficie.
Las alicuotas para la determinacion de la velocidad de liberación del principio activo se toman en
tiempos determinados, reponiendo con un volumen igual el medio de disolucion que debe estar a
la misma temperatura.
b. Método de sistema celular de flujo continuo, Aparato 4
Es un método basado en la columna de flujo continuo descrito por Langenbucher.
El aparato consiste en un depósito y una bomba para el medio de disolución, una celda de flujo y
un baño de agua para mantener la temperatura del medio de disolución a 37°C ± 0.5°C.
La bomba impulsa el medio de disolución a través de la celda de flujo desde la parte inferior a la
superficie. La bomba tiene un intervalo de operación de 240 mL a 900 mL por hora y las

50
velocidades de flujo estándares son 4mL, 8mL o 16mL por minuto. La bomba debe suministrar
un flujo constante.
La celda de flujo está montada verticalmente con un sistema de filtro que impide que se escapen
partículas no disueltas. La base cónica de la celda esta generalmente llena de pequeñas perlas de
vidrio de aproximadamente 1mm de diámetro y una de esas perlas de aproximadamente 5 mm la
que está ubicada en el ápice para proteger al tubo de entrada de fluido.
El líquido, después de pasar por la columna se recolecta para su análisis.
Figura 2. 19 Esquema de la columna de flujo continuo. Aparato 4. Métodos oficiales USP
Medio de disolución
El medio de disolución utilizado debe simular en lo posible el ambiente gastrointestinal, lo que
permitirá tener un acercamiento a la cinética del fármaco en medio fisiológico.
Volumen: Por lo general es de 500, 900 o 1000 mL.
Agitación
Cuando el método utilizado es de cesta la velocidad común es de 50 a 100 rpm, y con el método
de paleta es de 50 a 75 rpm. (FDA U.S. Food and Drug Administration, 1997)
En esta investigación el ensayo de Perfiles de Liberación se realizó haciendo una adaptación
del Aparato 2 y el Aparato 4. Se utilizó un equipo de disolución con vasos para 150 mL de medio
receptor. La solución receptora es una Buffer de fosfatos pH 6,8 ± 0.2.
El principio activo contenido en el vehículo se colocó en la membrana de celulosa para
diálisis, desde donde difundió al medio receptor. El sistema se mantuvo en recirculación
con la ayuda de la bomba de flujo continuo.

51
Para la determinación del diclofenaco dietilamonio se extrajo la muestra para ser leída
por HPLC siendo necesario hacer reposición de medio con un volumen exactamente igual
al tomado después de cada extracción.
La solubilidad del diclofenaco dietilamina en la solución buffer de fosfatos pH 6.8 es
cerca de 2.7mg/mL, el 20 % de este valor equivale a 0.54mg/mL que es la concentración
máxima de diclofenaco dietilamonio que la solución buffer puede contener para que el
proceso de liberación no se vea afectado por la concentración.
2.1.10.5 Condiciones SINK
Uno de los requerimientos para la aplicación del modelo de Higuchi es que el proceso de
liberación se mantenga bajo condiciones SINK.
Esto es importante porque cuando una sustancia se disuelve en un medio de disolución, las
partículas disueltas generan una disminución del gradiente de concentración, con lo que la
velocidad de disolución va disminuyendo con el tiempo.
Con el fin de conseguir estas condiciones, en los ensayos se utiliza volúmenes grandes de medio
receptor o bien, la cantidad del activo no en una concentración mayor al 20% de su solubilidad
máxima.
2.1.10.6 Factores que influencian la velocidad de disolución
La velocidad con que migra la droga desde el interior de la microcápsula hacia el medio
circundante está determinada absolutamente por la cubierta polimérica, su espesor y solubilidad
en el medio de disolución. El recubrimiento polimérico actúa como una membrana
semipermeable y permite la creación de un gradiente de presión osmótica entre el interior y el
exterior de la microcápsula, lo que origina que pequeñas cantidades de fármaco salgan a través
de los pequeños poros de la capa.
Se ha visto también que existe un efecto de descarga inicial debido a la presencia de fármaco
adherido en la superficie de la microcápsula, el mismo que es liberado de manera instantánea al
entrar en contacto con el medio de disolución. (Jelvehgari & Montazam, 2012)
Normalmente para evaluar la velocidad de disolución de varios productos se utiliza parámetros
como la Eficiencia de Disolución y el Factor de Diferencia, sin embargo al tratar de SLM como
nuevas formas farmacéuticas los estudios de farmacología clínica deben realizarse para la
determinación de los parámetros farmacocinéticos.

52
2.1.10.7 Perfiles de liberación del principio activo.
La velocidad de absorción, es decir del número de moléculas de fármaco que se absorben por
unidad de tiempo depende de la constante de absorción (K) y de la cantidad de moléculas que se
encuentren en solución en el lugar de absorción; en los sistemas de liberación inmediata todas las
moléculas de fármaco se encuentran expuestas para su absorción, y su absorción disminuyen de
manera exponencial con el transcurso del tiempo, a esto se le conoce como proceso de orden 1,
mientras que en los sistemas de liberación modificada el proceso es de orden cero puesto que el
número de moléculas a absorber está limitado a la velocidad de liberación de la estructura que es
bastante constante, el número de moléculas no disminuye con el tiempo debido a que las
moléculas absorbidas son repuestas desde el depósito, aquí la velocidad de absorción permanece
constante durante todo el proceso. (Baños Díez & Farré Albaladejo, 2002)
Figura 2. 20 Curvas de concentración plasmática para
diferentes velocidades de absorción
En la gráfica se muestra las curvas de concentración plasmática de diferentes formulaciones de
un fármaco administrado por vía oral, en donde se aprecia que el área bajo la curva de a, b y c es
similar, sin embargo en a el efecto es rápido e intenso, en b la intensidad disminuye y en c no
llega a haber concentración eficaz debido a que la absorción es muy lenta aunque completa; la
curva de d nos indica que aunque la absorción no es completa, es rápida de manera que el efecto
se da rápidamente, pero menos duradero en comparación con a. (Baños Díez & Farré Albaladejo,
2002).
Factores a tomar en cuenta
El coeficiente de variación para los las muestras tomadas a tiempos tempranos no deberá ser
mayor al 20% y en otros puntos de muestra no deberá ser mayor al 10%. (FDA U.S. Food and
Drug Administration, 1997)

53
2.1.10.8 Control De Disolventes Orgánicos Residuales
La determinación de solventes orgánicos residuales en las micropartículas es un control muy
importante que se realiza cuando se utiliza solventes orgánicos de elevada toxicidad como fase
oleosa para elaboración de la emulsión; en este caso el limoneno extraído del aceite esencial de
naranja presenta baja toxicidad y más bien presenta propiedades terapéuticas a concentraciones
adecuadas por lo que no se ve necesaria esta prueba.
2.1.11 DESVENTAJAS DE LOS SISTEMAS DE LIBERACION MODIFICADA
A pesar del potencial que se observa en estas formas de administración de fármacos, es posible
manifestar algunas desventajas que pudieran presentarse.
a. Riesgo de acumulación e intoxicación si la velocidad de eliminación es lenta, si existe
dificultad para eliminar el fármaco
b. Falta de regularidad de la respuesta terapéutica de acuerdo con la velocidad de
vaciamiento gástrico;
c. Variaciones en el esquema de liberación cuando la forma farmacéutica se rompe,
mastica o tritura, con problemas de sobredosificación. (Costa Castro, 2004)

54
MARCO LEGAL
2.2.1 REGLAMENTO DE BUENAS PRÁCTICAS DE MANUFACTURA (B.P.M.)
PARA LA INDUSTRIA FARMACÉUTICA
(N° 4640 19 de Julio del 1994 Registro Oficial N° 486)
Título I
Capítulo V: De la higiene
Art 30.- A fin de garantizar la seguridad del producto, del personal y la del consumidor, el
personal que trabaja en una empresa farmacéutica debe cumplir con normas escritas de limpieza
e higiene.
Art 31.- Al personal de planta deberá proveérsele de uniformes adecuados a las operaciones a
realizar a fin de protegerlo de la contaminación con los productos o insumos que manipulan y
además preservar el producto de la contaminación proveniente de la ropa de calle.
2.2.2 NORMA TECNICA ECUATORIANA (NTE INEN-ISO/TR 12885)
Nanotecnologías. prácticas de seguridad y salud en lugares de trabajo relacionados con las
nanotecnologías (iso/tr 12885:2008, idt)
Al estar este trabajo de investigación relacionado con los fundamentos y técnicas de
nanotecnología, se toma en consideración esta norma que describe directrices para el desarrollo
responsable de esta tecnología: Caracterización del peligro, Evaluación de la exposición a nano
materiales, Evaluación de riesgos en los lugares de trabajo.

55
CAPÍTULO III
METODOLOGÍA
TIPO DE INVESTIGACIÓN
La investigación es de tipo experimental cuantitativa porque se basa en la recolección de datos
numéricos obtenidos de las características de los micropartículas.
POBLACION Y MUESTRA
De acuerdo al tipo de investigación realizado, la población se considera como las infinitas
formulaciones que permitan obtener micropartículas y como la muestra las formulaciones
obtenidas mediante experimentación en este trabajo.
DISEÑO EXPERIMENTAL
PORCENTAJE DE ´PRINCIPIO ACTIVO
LIBERADO
Tiempo de
liberación
Repetición 1 Repetición 2 Repetición 3 Promedio
T1
T2
T3
T4
T5
Tn

56
Surfactantes
Gelatina
Polivinilalcohol
pH
Tamaño de partícula
Contenido de principio activo
Rendimiento de la producción
Eficacia de encapsulación
DIAGRAMA DE FLUJO METODOLÓGICO
Evaluación de proporciones adecuadas de los componentes para la formación de la
emulsión
Micropartículas sólidas
Obtención de las partículas por procesos de congelamiento y liofilizaciones sucesivas
Perfiles de liberación del principio activo
Lavado y secado de las partículas
Adición del principio activo
Micropartículas en la emulsión
Perfiles de liberación del principio activo desde la emulsión
Diseño y elaboración de micropartículas poliméricas cargadas con Diclofenaco dietilamina
mediante la técnica de doble emulsificación
Liberación adecuada
Caracterización de las partículas
1 2
FIN
NO
FIN
SI

57
3.1.1 VARIABLES
Primera etapa: Diseño y elaboración de la emulsión
Se realizó el análisis de estabilidad tanto macroscópica como microscópica de la emulsión en un
tiempo determinado, descartando las formulaciones que no cumplan con la característica
mencionada.
Segunda etapa: Perfiles de disolución
VARIABLES INDEPENDIENTES: Tiempo
VARIABLES DEPENDIENTES: Cantidad de principio activo liberado
3.1.2 INSTRUMENTOS ANALITICOS
3.1.2.1 MATERIALES
Vasos de precipitación
Espátula
Tubos de ensayo
Agitadores magnéticos
Papel aluminio
Balones aforados de 10 mL, 25 mL, 50 mL, 100 mL, 1000 mL.
Micropipetas
Pipetas Pasteur de vidrio y plásticas.
Frascos de vidrio
Celda de vidrio de cuatro caras transparentes
Celdas de cuarzo
Bolsas de diálisis – Cellu Sep® T3 tamaño de poro 20µm.
Pinzas para bolsa de díalisis
Filtros Millipore Millex-GV 0,22 μm.
Jeringuillas
Viales de vidrio con tapa perforada para HPLC
3.1.2.2 EQUIPOS
Balanza analítica de precisión - Mettler Toledo ML204 .
Potenciómetro -WTW inoLab pH 720
Equipo mezclador/emulsificador - Silverson L4R

58
Cronómetro digital
Estufa con regulación de temperatura
Equipo de Dispersión de luz dinámica (DLS) - Horiba Scientific SZ-100
Cromatógrafo HPLC – DIONEX Ultimate 3000
Liofilizador – LABCONCO Freeze dry system/Freezone® 4,5
Rotavapor – BUCCHI R-210V
Homogeneizador Ultra Turrax - IKA® T10
Equipo de disolución – COPLEY DIS 6000
Bomba de flujo continuo - Thermo Scientific
3.1.2.3 REACTIVOS
Limoneno extraído del aceite esencial de naranja (VER ANEXO 1)
Diclofenaco de sódio
Diclofenaco Dietilamonio
Tween 80® - Sigma Aldrich®
Span 80® - Sigma Aldrich®
Gelatina (grado alimenticio)
Polivinilalcohol (AIRVOL® 325) – AIR PRODUCTS AND CHEMICALS, INC.
Fosfato dibásico de sodio (p.a.)
Fosfato monobásico de potasio (p.a.)
Cloruro de Sodio (p.a.)
Agua purificada Tipo I
Acetonitrilo grado HPLC
Metanol grado HPLC
3.1.3 MÉTODOS
3.1.3.1 Diseño de la emulsión
Debido a que el principio activo es soluble en agua se opta por realizar una emulsión del tipo
w/o/w.
La relación agua-aceite es 30:70 respectivamente para todas las pruebas, basado en la técnica
“Elaboración de micropartículas poliméricas utilizando la gelatina como material encapsulante”.
(Oficina española de patentes y marcas, 2008)
La agitación en las pruebas para la formación de la emulsión w/o se realiza en el
mezclador/emulsificador Silverson L4R durante 30 minutos a 3000 rpm .
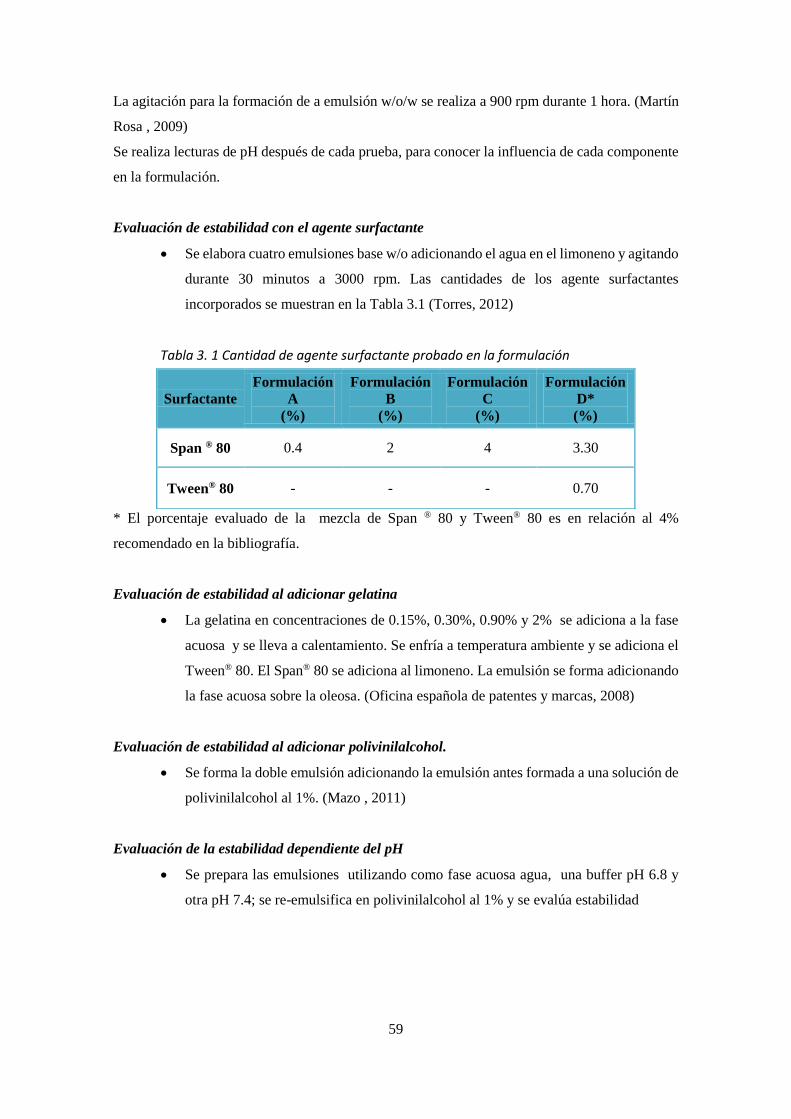
59
La agitación para la formación de a emulsión w/o/w se realiza a 900 rpm durante 1 hora. (Martín
Rosa , 2009)
Se realiza lecturas de pH después de cada prueba, para conocer la influencia de cada componente
en la formulación.
Evaluación de estabilidad con el agente surfactante
Se elabora cuatro emulsiones base w/o adicionando el agua en el limoneno y agitando
durante 30 minutos a 3000 rpm. Las cantidades de los agente surfactantes
incorporados se muestran en la Tabla 3.1 (Torres, 2012)
Tabla 3. 1 Cantidad de agente surfactante probado en la formulación
* El porcentaje evaluado de la mezcla de Span ® 80 y Tween® 80 es en relación al 4%
recomendado en la bibliografía.
Evaluación de estabilidad al adicionar gelatina
La gelatina en concentraciones de 0.15%, 0.30%, 0.90% y 2% se adiciona a la fase
acuosa y se lleva a calentamiento. Se enfría a temperatura ambiente y se adiciona el
Tween® 80. El Span® 80 se adiciona al limoneno. La emulsión se forma adicionando
la fase acuosa sobre la oleosa. (Oficina española de patentes y marcas, 2008)
Evaluación de estabilidad al adicionar polivinilalcohol.
Se forma la doble emulsión adicionando la emulsión antes formada a una solución de
polivinilalcohol al 1%. (Mazo , 2011)
Evaluación de la estabilidad dependiente del pH
Se prepara las emulsiones utilizando como fase acuosa agua, una buffer pH 6.8 y
otra pH 7.4; se re-emulsifica en polivinilalcohol al 1% y se evalúa estabilidad
Surfactante
Formulación
A
(%)
Formulación
B
(%)
Formulación
C
(%)
Formulación
D*
(%)
Span ® 80 0.4 2 4 3.30
Tween® 80 - - - 0.70

60
Evaluación de la estabilidad con incremento de la cantidad y concentración de la solución de
polivinilalcohol.
Se prepara la emulsión base y re-emulsifica en una solución de polivinilalcohol 1.5%,
la relación de fases es de 0.5, se prueba en agua y en buffer pH 7.4. (Mazo , 2011)
Evaluación de la estabilidad con adición del principio activo.
Se incorpora el principio activo en la emulsión con mejores características de
estabilidad. La cantidad de principio activo se determina en base a la solubilidad que
presenta este y a la proporción adecuada de vehículo-principio activo para su
administración.
3.1.3.2 Extracción de las micropartículas poliméricas
La emulsión luego de pasar por un proceso de liofilización de 6 horas se somete a
congelamiento a -20°C.
Se repetirte el proceso liofilización-congelamiento 4 veces más retirando el aceite de
la superficie del hielo cuando sea posible.
La muestra se somete a centrifugaciones sucesivas con lo que se extrae los residuos
de la fase oleosa.
Se elimina el agua mediante secado en estufa a 30°C durante 48 horas.
El residuo se lava con agua tipo I y se repite el secado a las mismas condiciones
Se pesa el polvo obtenido. (Fundación Mapfre, 2007)
3.1.3.3 Caracterización de las micropartículas
Tamaño de partícula
El tamaño de partícula fue determinado en la muestra tras los procesos de
congelamiento y liofilización mediante técnica microscópica óptica.
Contenido de principio activo
a. Preparación de la muestra
Se pesó 20.1 mg de muestra seca, y se colocó en un balón de 25 mL junto con 10 mL
de Metanol y 10 mL de agua destilada. La muestra se sometió a baño ultrasónico

61
durante 30 minutos, luego a calentamiento en baño maría durante otros 30 minutos, se
enfrió y llevó a aforo.
Se tomó 5 mL y se llevó a 25 mL con agua destilada.
La muestra fue leída espectrofotométricamente a 276 nm, luego de pasar a través de
un filtro 0.22 μm. Adaptado de Jelvehgari & Montazam (2012)
b. Preparación de la curva de calibración
Se pesó 24.9 mg de principio activo y se llevó a 100 mL con 25 mL de Metanol; A
partir de esta solución se prepararon soluciones de 62.25 ppm, 31.125 ppm, 12.45
ppm, 5 ppm y 0.5 ppm las que fueron leídas espectrofotométricamente a 276 nm.
Con los datos obtenidos realizo el cálculo para la determinación del Rendimiento de la
producción y Eficacia de la encapsulación.
3.1.3.4 Perfiles de liberación del principio activo
Determinación de la solubilidad del principio activo
Para conocer las condiciones SINK del experimento se debe determinar la solubilidad
del principio activo en el medio de disolución.
Colocar un peso exacto del principio activo en un vaso de precipitación,
Adicionar el solvente a 25 °C y en cantidades exactas con una pipeta semiautomática.
Tras cada adición agitar levemente.
La concentración del fármaco solubilizado se conoce tras la adición del solvente hasta
la formación de una solución clara y transparente.
Liberación del principio activo (diclofenaco de sodio) desde las partículas poliméricas.
La liberación del principio activo se realiza en una solución buffer pH 6.8, para lo cual
se debe pesar 0.19g de KH2PO4, 2.38g de Na2HPO4, y 8g de NaCl. Disolver
todas las sales con agua Tipo I y aforar a 1 Litro.
Ajustar el pH a 6.8 ± 0.2 con ácido fosfórico
Colocar 100 mL del medio de disolución en los vasos del equipo de disolución
y estabilizar a 37 °C ± 0.5.
Acoplar la bomba de recirculación al espectrofotómetro y equipo de disolución.
La longitud de onda a la que se realiza la lectura es de 276 nm.

62
La toma y lectura de la muestra es automática. No se necesita hacer reposición
de medio
Encender los equipos y colocar inmediatamente en el medio de disolución
aproximadamente 910 mg de muestra seca, según cálculos realizados para
mantener las condiciones Sink.
Liberación del principio activo (diclofenaco dietilamonio) desde las partículas en emulsión,
lectura mediante HPLC
Preparar el medio de disolución buffer fosfatos pH 6.8, colocar 100 mL en los vasos
del equipo de disolución y llevar a 37°C ± 0.5
Cortar aproximadamente 5cm de membrana de celulosa para diálisis, doblar un
extremo de la membrana y sujetarlo con una pinza. Se toma el peso de la membrana
con la pinza.
Colocar dentro de la membrana la cantidad exacta de emulsión desde donde se va a
liberar el principio activo según los cálculos realizados para mantener las condiciones
Sink. Se debe trabajar con cuidado de no producir derrames.
Doblar el extremo libre de la membrana y sujetarlo con la pinza.
Acoplar la bomba de recirculación al equipo de disolucón de manera que haya
recirculación en el medio mediante las mangueras.
Iniciar los equipos y colocar las membranas de diálisis que contienen la emulsión en
el cada vaso de manera que queden totalmente introducidas y no tengan contacto con
las paletas del equipo de disolución.
Inmediatamente de iniciado el ensayo se tomar una alícuota de 1.5 mL desde cada
vaso en viales para HPLC con una pipeta semiautomática, esta muestra cataloga como
muestra a tiempo cero.
La tomas de las alícuotas se deben realizar siempre desde el mismo punto y utilizando
una punta para cada vaso de manera de evitar transferencia de muestra desde un vaso
a otro.
Tomar 1.5 mL de muestra en los viales para HPLC según los tiempos establecidos
hasta las 72 horas, y reponer el volumen retirado de cada vaso con el medio de
reposición después de cada toma de muestra.
Leer la concentración del principio activo que ha sido liberada desde las partículas en
emulsión en el equipo HPLC.
Liberación del principio activo (diclofenaco dietilamonio) desde el emulgel VOLTAREN®
mediante lectura en HPLC

63
Repetir el procedimiento de liberación descrito anteriormente. El peso de la muestra
que se pone en la membrana de diálisis es exactamente alrededor de 6g y el volumen
del medio de disolución a colocar en cada vaso del equipo de disolución es de 100 mL.
El tiempo que dura la prueba de liberación es 24 horas.
Las determinaciones se deben realizar por triplicado, con un vaso adicional que mantenga el
medio de disolución a la temperatura establecida y sirva para reposición.
Elaboración de la curva de calibración para HPLC
Pesar exactamente alrededor de 80 mg de diclofenaco dietilamonio estándar y llevar
a 100 mL con agua Tipo I
A partir de esta solución preparar estándares de 80 ppm, 40 ppm, 20 ppm, 10 ppm y 1
ppm.
Condiciones para la lectura en HPLC
Fase móvil: La fase móvil es la mezcla Acetonitrilo - Agua en proporciones 70:30, pH
3.
Flujo: 1.2 mL/min
Temperatura: 25° C
Longitud de onda: 276 nm
Columna: ACCLAIM ® Polar Advantage, C16, 2.1 x 150 mm. (Método desarrollado
y validado por Diego Lucero)

64
CAPITULO IV
4.1 RESULTADOS Y DISCUSIÓN DE RESULTADOS
4.1.1 DISEÑO DE LA EMULSION
Debido a que no existen estudios previos de los parámetros bajo los que se ha realizado la presente
investigación, el estudio se ha realizado mediante prueba-error.
1. Evaluación de estabilidad con el agente surfactante
Tabla 4. 1 Adición del agente surfactante. Componentes de la formulación w/o con sus respectivas proporciones
El porcentaje evaluado de la mezcla de Span® 80 y Tween® 80 es en relación al 4% de la
formulación según lo recomendado en la bibliografía.
La figuras 4.1 y 4.2 permiten apreciar como mejora la homogeneidad de la emulsión conforme se
incrementa la cantidad de Span® y aún más con el incremento del BHL con la adición del Tween®
80 en la formulación.
FORMULACION (%)
A B C D
Fase acuosa Agua 29.70 29.70 29.70 29.00
Fase oleosa Limoneno 69.90 68.30 66.30 67.00
Surfactante
Span® 80
0.4 2 4 3.30
Tween® 80 - - - 0.70
pH 3.5 3.4 3.1 3.6
Aspecto Crema
descendente
Crema
descendente
Crema
descendente Homogéneo
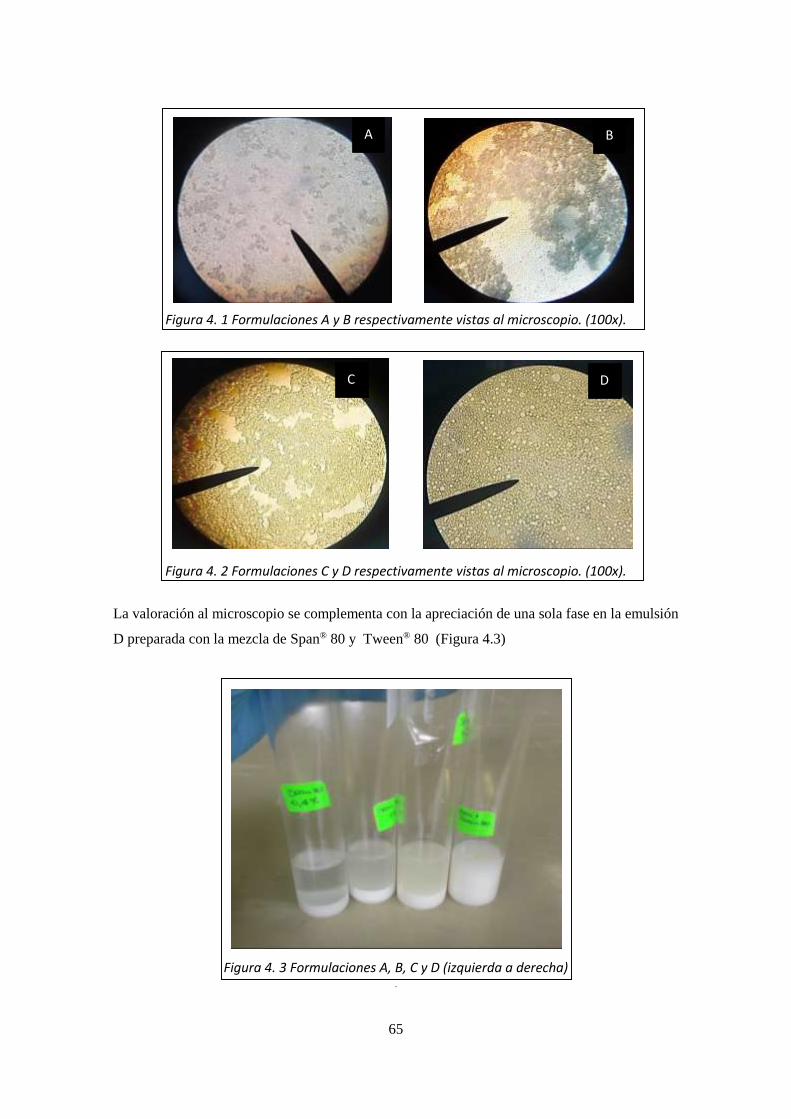
65
Figura 4. 1 Formulaciones A y B respectivamente vistas al microscopio. (100x).
Figura 4. 2 Formulaciones C y D respectivamente vistas al microscopio. (100x).
La valoración al microscopio se complementa con la apreciación de una sola fase en la emulsión
D preparada con la mezcla de Span® 80 y Tween® 80 (Figura 4.3)
Figura 4. 3 Formulaciones A, B, C y D (izquierda a derecha) .
A
) B
)
C
AD
A

66
2. Evaluación de estabilidad al adicionar gelatina
Tabla 4. 2 Adición de la gelatina. Componentes de la formulación w/o con sus respectivas proporciones.
Las figuras 4.4, 4.5 y 4.6, muestran un incremento en la polidispersión con respecto a la emulsión
D, cuando se adiciona gelatina a la formulación; además el incremento en la concentración de
gelatina favorece la desestabilización de la emulsión.
Figura 4. 4 Formulación D1 y D2 vista al microscopio. (100x).
FORMULACIÓN, (%)
D1 D2 D3 D4
Fase acuosa Agua 28.85 28.70 28.1 27.00
Gelatina 0.15 0.30 0.90 2.00
Fase oleosa Limoneno 67.00 67.00 67.00 67.00
Surfactante
Span® 80 3.30 3.30 3.30 3.30
Tween® 80 0.70 0.70 0.70 0.70
pH 3.5 3.6 3.6 3.7
Aspecto Homogéneo Crema
descendente
Crema
descendente
Crema
descendente
D1 D2

67
Figura 4. 5 Formulación D3 y D4 vista al microscopio. (100x).
La figura 4.6 es una vista macroscópica de las emulsiones preparadas luego de las 24 horas, con
lo que se comprueba la desestabilización que produce el incremento en la concentración de
gelatina.
Figura 4. 6 Formulaciones D1, D2, D3 y D4. Vista macroscópica
D3 D4
D1 D2 D3 D4

68
3. Evaluación de estabilidad al adicionar Polivinilalcohol.
Tabla 4. 3 Adición del PVA en solución. Componentes de la formulación w/o/w con sus respectivas proporciones.
Se prueban dos formulaciones con un contenido de polivinilalcohol del 50% y 70% con respecto
a la formulación D, las dos emulsiones son inestables ya que se observa separación de fases de
manera inmediata, además al microscopio no se observa formación de partículas homogéneas.
Figura 4. 7 Formulaciones con polivinilalcohol D1-1 50%, D1-2 70%, vista al microscopio (100x)
FORMULACIÓN w/o/w, (%)
D1-1, D1-2
Fase acuosa 1 Agua 14.42 8.66
Gelatina 0.08 0.04
Fase oleosa Limoneno 33.50 20.10
Surfactante
Span® 80 1.65 0.99
Tween® 80 0.35 0.21
Fase acuosa 2 Solución PVA 1% 50.00 70.00
pH 3.5 3.5
Aspecto Crema
ascendente
Crema
ascendente
D1-1 D1-2

69
Figura 4. 8 Formulación D1-1 y D1-2 respectivamente (derecha a izquierda)
Las figuras 4.7 y 4.8 nos permiten identificar que pese a la inestabilidad de la emulsión bajo estas
dos condiciones, se aprecia mejor formación de las gotas en la emulsión preparada con 70% de la
solución de PVA al 1%
La imagen microscópica de la emulsión D1-2 (Figura 4.7) nos deja apreciar la formación de
partículas pequeñas al interior de las grandes, las mismas que presentan movimiento browniano.
4. Evaluación de la estabilidad dependiente del pH
Debido a los valores bajos de pH que se presentan, se opta por realizar la misma formulación a
pH 6.8 y 7.4, lo que se logra preparando las fases acuosas con soluciones buffer a los pH
requeridos. Los porcentajes utilizados para la preparación de la emulsión son los descritos en la
Tabla 4.3
Figura 4. 9 Formulaciones a pH 6.8 y pH 7.4 (Izquierda a derecha)

70
La figura 4.9 muestra las dos fases que no logran incorporarse cuando el pH de la solución
asciende. No se logra observar la muestra al microscopio óptico.
5. Evaluación de la estabilidad con incremento de y concentración de la solución de
polivinilalcohol.
Tabla 4. 4 Incremento en la concentración de alcohol polivinílico. Componentes de la formulación w/o/w con sus respectivas proporciones
Aspecto de la emulsión
Figura 4. 10 Formulación D1-5. A. Emulsión estable.
FORMULACIÓN w/o/w, D1-5
COMPONENTES (%)
Fase acuosa 1 Agua 14.97
Gelatina 0.08
Fase oleosa Limoneno 35.09
Surfactante
Span® 80 1.68
Tween® 80 0.39
Fase acuosa 2 Solución PVA 1.5% 47.79
pH 4.8
Aspecto Emulsión estable
A B

71
B. Vista al microscopio de la formulación D1-6 (100x)
6. Evaluación de la estabilidad con adición del principio activo.
Después de evaluar las mejores condiciones de formulación para la elaboración de las
micropartículas poliméricas, se adiciona el principio activo en la cantidad adecuada para cumplir
con las condiciones SINK en el momento de la liberación.
Tabla 4. 5 Porcentajes de los componentes utilizados en la formulación
DISCUSION
La emulsión preliminar agua- limoneno presenta mejores características cuando se incorpora a
la formulación la mezcla de surfactantes Span® 80-Tween® 80, esto debido a que el BHL
requerido es de 6.28 (Ver Anexo 1.6) mayor al que proporciona solo el Span® 80 que es de 4.3.
Debido a que esta emulsión presenta uniformidad tanto visual como microscópica, se procede a
incorporar la gelatina en esta formulación pudiendo observar en el análisis visual que mientras
menor cantidad de gelatina se incorpora en la formulación mayor es la estabilidad de esta. Sin
embargo la polidispersión se ve afectada con relación a la emulsión preliminar.
La emulsión D1 con solo 0.15% de gelatina es seleccionada para adicionar la solución al 1% de
polivinilalcohol en concentraciones del 50% y 70%. Las emulsiones formadas bajo estas
condiciones son inestables y al microscopio se observa maduración de Otswald, también es
posible apreciar algunas gotas pequeñas en el interior de otras más grandes por la formación de
la doble emulsión, estas gotas pequeñas presentan movimiento browniano.
El pH en las emulsiones elaboradas varía de 3.5 a 3.7 por lo que se consideró evaluar el efecto de
este al preparar las emulsiones utilizando como fase acuosa una solución Buffer pH 7.4, con lo
que se logró determinar que este sistema es susceptible del pH debido a que no existe formación
de emulsión a estas condiciones.
Componente Peso, (g) Porcentaje respecto al peso
total, (%)
Limoneno 114.25 34.29
Span 80 5.39 1.62
Agua 48.00 14.40
Gelatina 0.25 0.08
Tween 80 1.25 0.37
Diclofenaco 12.57 3.77
PVA (solución al 1,5%) 151.50 45.47

72
La investigación gira en torno al mejoramiento de la estabilidad de la emulsión mediante la
adición de la solución de polivinilalcohol al 1.5% y a condiciones propias de pH, según el análisis
de los resultados obtenidos en las emulsiones D1-1 y D1-2 y el estudio bibliográfico (Mazo ,
2011). La emulsión obtenida bajo estos parámetros presenta buena estabilidad visual y al
microscopio se observa gotas más esféricas y mejor formadas, el pH es de 4.8.
Se determinó el tipo de emulsión mediante la técnica de dilución en agua sin observar separación
de las fases, por tanto la fase externa de la emulsión es de naturaleza hidrofílica como lo que se
espera en una emulsión w/o/w.
El tamaño presenta alta dispersión según los histogramas realizados para el análisis de tamaño de
partícula.
Al adicionar el Diclofenaco sódico en la formulación D1-5, el valor del pH de la formulación no
se ve influenciado ni tampoco la estabilidad.
4.1.1 EXTRACCION DE LAS MICROPARTICULAS POLIMERICAS
Figura 4. 11 Partículas obtenidas. A) Polvo obtenido después del proceso de secado.
B) Vista microscópica del polvo resuspendido en agua.
No se logra obtener un polvo fino, después del proceso de lavado y secado la muestra se aglomera,
sin embargo al resuspender en agua las partículas se observan aisladas como muestra la figura
4.11 B).
Las partículas obtenidas presentan una alta polidispersión y aunque se observan partículas
aisladas, existen aglomerados de polímero también.
DISCUSION
Según se va eliminando el solvente en la etapa de liofilización se forma una masa gelatinosa la
que al pasar por el proceso de secado forma un sólido bastante compacto, cuya formación puede
A) B)

73
deberse al colapso de las partículas por el rápido intercambio del solvente. (Ramos Picos, Gómez
Carril, & Fernández Mena, 2001)
La vista microscópica nos indica que además de las partículas hay aglomerados de polímero, los
que no pueden ser eliminados con los lavados que se realizan por lo que no se logra obtener polvo
fino.
4.1.2 DETERMINACIÓN DEL TAMAÑO DE LAS PARTÍCULAS
Se determinó el tamaño de partícula en solución mediante técnica microscópica, para lo que se
tomó un lote de 100 partículas y se midió su diámetro con el programa ImageJ.
Repetición 1 (R1)
Repetición 2 (R2)
05
10152025
30
Fre
cue
nci
a
Tamaño (μm)
Tamaño de partícula (R2)
0
5
10
15
20
25
Fre
cuen
cia
Tamaño (μm)
Tamaño de partícula (R1)

74
Repetición 3 (R3)
DISCUSION
El límite superior de detección del equipo DLS es 8µm (HORIBA Scientific, 2003), mientras que
las partículas obtenidas tienen valores mayores a este máximo, por lo tanto se realiza la lectura
del tamaño de partícula en microscopio óptico con el programa informático ImageJ.
Las partículas pese a ser polidispersas se encuentran en el rango de los micrómetros, el tamaño
mayor encontrado es de 27.4 µm. Sin embargo partículas con tamaños menores a 3 µm. no
pudieron ser determinadas por las limitaciones del método.
La dispersión en partículas elaboradas con materiales poliméricos es un parámetro difícil de
controlar debido a la estructura misma del polímero y la falta de conocimiento de la
estequiometria del proceso, a diferencia de las partículas obtenidas por síntesis tales como los
dendrímeros y las ciclodextrinas.
4.1.3 CONTENIDO DE PRINCIPIO ACTIVO
El contenido de principio activo se ha calculado con la ecuación 2.7
Tabla 4. 6 Datos y contenido de principio activo, determinación espectrofotométrica.
Muestra
Peso
muestra
(mg)
Absorbancia
de la
muestra
Conc.
(ppm)
Factor
De
Dilusión
Principio
Activo en
muestra
(mg)
Contenido
de p.a.
(%)
R1 17.4 0.6541 19.69 0.25 4.92 28.27
R2 19.2 0.3557 10.71 0.50 5.35 27,87
R3 19.0 0.3547 10.68 0.50 5.34 28.11
0
10
20
30
40
Fre
cuen
cia
Tamaño (μm)
Tamaño de partícula (R3)

75
Promedio 28.08
Desv. St. 0.20
Análisis de contraste Grubbs para rechazo de datos anómalos
𝐺 = 𝑉𝑎𝑙𝑜𝑟 𝑎𝑛ó𝑚𝑎𝑙𝑜 − �̅�
𝜎
Ecuación 4. 1 Ecuación de Grubbs
𝐺 = 27.87−28.08
0.20= 1.05
G critico (P=0.05) es 1.155
G calculado (1.05) < G critico (1.155); no se rechaza el valor sospechoso.
4.1.4 RENDIMIENTO DE LA PRODUCCION
En base a la ecuación 2.8 se calcula el rendimiento de la producción
La cantidad de solidos obtenidos al final del proceso es de: 12.38 g
%𝑅𝑒𝑛𝑑. 𝑃𝑟𝑜𝑑𝑢𝑐𝑐𝑖ó𝑛 =12.38 𝑔
18.2475 𝑔𝑥100
%𝑹𝒆𝒏𝒅. 𝑷𝒓𝒐𝒅𝒖𝒄𝒄𝒊ó𝒏 = 𝟔𝟕. 𝟖𝟒 %
4.1.5 EFICACIA DE LA ENCAPSULACION
A partir de la ecuación 2.9 se obtienen los valores de eficiencia de encapsulación
%𝐸𝑛𝑐𝑎𝑝𝑠𝑢𝑙𝑎𝑑𝑜 = 3.48 𝑔
12.5125 𝑔∗ 100
%𝑬𝒏𝒄𝒂𝒑𝒔𝒖𝒍𝒂𝒅𝒐 = 𝟐𝟕. 𝟖𝟏%
DISCUSION
Tanto el contenido de principio activo como la eficacia de la encapsulación son bajos debido a
los procesos de lavado continuo.
El Diclofenaco de sodio que se determinó en las micropartículas fue de 28.08%., sin llegar a
establecer si este se encontraba formando parte de una microcápsula o microesfera.
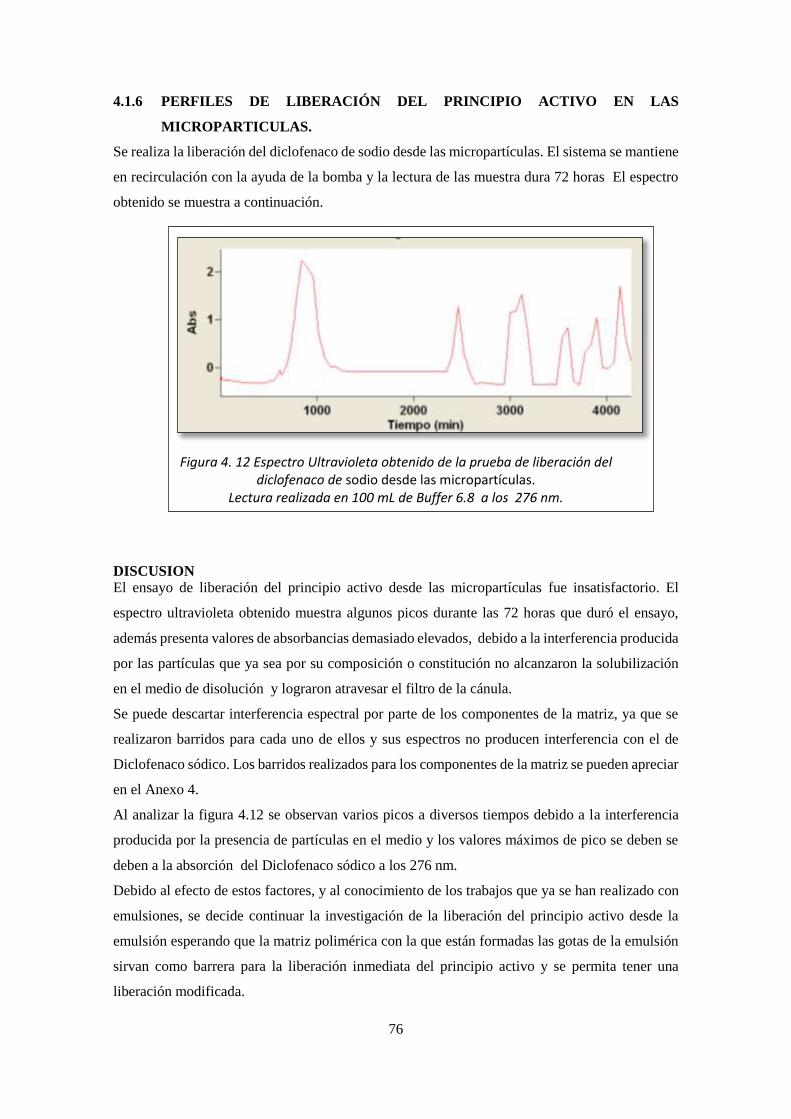
76
4.1.6 PERFILES DE LIBERACIÓN DEL PRINCIPIO ACTIVO EN LAS
MICROPARTICULAS.
Se realiza la liberación del diclofenaco de sodio desde las micropartículas. El sistema se mantiene
en recirculación con la ayuda de la bomba y la lectura de las muestra dura 72 horas El espectro
obtenido se muestra a continuación.
Figura 4. 12 Espectro Ultravioleta obtenido de la prueba de liberación del diclofenaco de sodio desde las micropartículas.
Lectura realizada en 100 mL de Buffer 6.8 a los 276 nm.
DISCUSION
El ensayo de liberación del principio activo desde las micropartículas fue insatisfactorio. El
espectro ultravioleta obtenido muestra algunos picos durante las 72 horas que duró el ensayo,
además presenta valores de absorbancias demasiado elevados, debido a la interferencia producida
por las partículas que ya sea por su composición o constitución no alcanzaron la solubilización
en el medio de disolución y lograron atravesar el filtro de la cánula.
Se puede descartar interferencia espectral por parte de los componentes de la matriz, ya que se
realizaron barridos para cada uno de ellos y sus espectros no producen interferencia con el de
Diclofenaco sódico. Los barridos realizados para los componentes de la matriz se pueden apreciar
en el Anexo 4.
Al analizar la figura 4.12 se observan varios picos a diversos tiempos debido a la interferencia
producida por la presencia de partículas en el medio y los valores máximos de pico se deben se
deben a la absorción del Diclofenaco sódico a los 276 nm.
Debido al efecto de estos factores, y al conocimiento de los trabajos que ya se han realizado con
emulsiones, se decide continuar la investigación de la liberación del principio activo desde la
emulsión esperando que la matriz polimérica con la que están formadas las gotas de la emulsión
sirvan como barrera para la liberación inmediata del principio activo y se permita tener una
liberación modificada.

77
4.1.7 PERFILES DE LIBERACIÓN DEL PRINCIPIO ACTIVO DESDE LA
EMULSION
De acuerdo a los valores declarados mediante técnica de cromatografía líquida de la liberación
del diclofenaco dietilamonio a los diferentes intervalos de tiempo se obtiene las siguientes curvas
de liberación.
Figura 4. 13 Comparación de los perfiles de liberación de las emulsiones
Gráficas para la determinación de la cinética de liberación en la emulsión
Figura 4. 14 Linealización para la cinética de orden cero
0102030405060708090
100
0 20 40 60 80Pri
nci
pio
act
ivo
lib
erad
o (
%)
Tiempo (horas)
CINETICA DE LIBERACION DEL DICLOFENACO DIETILAMONIO
Promedio
Emul 1
Emul 2
Emul 3
y = 1,2938x + 20,193R² = 0,7622
0
20
40
60
80
100
120
0 2 0 4 0 6 0 8 0
% L
IBER
AD
O
TIEMPO DE LIBERACION (HORAS)
CINETICA DE ORDEN CERO

78
Figura 4. 15 Linealización para la cinética de primer orden
Figura 4. 16 Linealización para la cinética de segundo orden
Figura 4. 17 Linealización para la cinética de Higuchi
y = 0,0378x - 1,9344R² = 0,4017
-5
-4
-3
-2
-1
0
1
0 20 40 60 80
LN (
%
LIB
ERA
DO
)
TIEMPO DE LIBERACION (HORAS)
CINÉTICA DE PRIMER ORDEN
y = -0,2621x + 12,535R² = 0,0683
-20
0
20
40
60
80
100
0 2 0 4 0 6 0 8 0
% L
IBER
AD
O -
1
TIEMPO DE LIBERACION (HORAS)
CINETICA DE SEGUNDO ORDEN
y = 11,092x + 8,5617R² = 0,911
0
20
40
60
80
100
120
0 2 4 6 8 10
% L
IBER
AD
O
RAIZ CUADRADA DEL TIEMPO (HORAS)
CINETICA DE HIGUCHI

79
4.1.8 PERFILES DE LIBERACIÓN DEL PRINCIPIO ACTIVO DESDE EL
EMULGEL (VOLTAREN®)
Grafica de la liberación del diclofenaco dietilamonio en función del tiempo. Determinación
en el emulgel.
Figura 4. 18 Curva de liberación del diclofenaco dietilamonio (promedio)
Determinación del orden de liberación del principio activo desde el emulgel
Figura 4. 19 Linealización para la cinética de orden cero
0
20
40
60
80
100
0 5 10 15 20 25
% l
iber
ado
tiempo (horas)
LIBERACION DEL PRINCIPIO ACTIVO EN Voltaren® (promedio)
y = 4,1557x + 19,016R² = 0,6974
0
20
40
60
80
100
0 5 10 15 20 25
% L
IBE
RA
DO
TIEMPO (HORAS)
CINÉTICA DE CERO ORDEN

80
Figura 4. 20 Linealización para la cinética de primer orden
Figura 4. 21 Linealización para la cinética de segundo orden
Figura 4. 22 Linealización para la cinética de Higuchi
y = 0,1817x + 1,7797R² = 0,2224
0
1
2
3
4
5
0 5 10 15 20 25LN
(%
LIB
ER
AD
O)
TIEMPO (HORAS)
CINETICA DE PRIMER ORDEN
y = -0,0836x + 1,1253R² = 0,0743
-1
0
1
2
3
4
5
6
0 5 10 15 20 25
TIEMPO (HORAS)
CINETICA DE SEGUNDO
ORDEN
1/%
LIB
ER
AD
O
y = 24,767x - 3,086R² = 0,9021
0
20
40
60
80
100
0,00 1,00 2,00 3,00 4,00 5,00 6,00
% L
IBE
RA
DO
RAIZ CUADRADA DEL TIEMPO (HORAS)
CINÉTICA DE HIGUCHI

81
Comparación de la cinética de liberación de la emulsión vs Voltaren ®
Figura 4. 23 Comparación de la cinética de liberación entre la emulsión vs Voltaren
DISCUSIONES
De la manera esperada, el sistema polimérico presente en la emulsión permite tener un tipo de
liberación hasta las 72 horas.
La cinética de liberación que sigue la emulsión desarrollada corresponde la definida por Higuchi,
ya que el R2 es el más aproximado a la linealidad, con respecto a las cinéticas de cero, primer y
segundo orden que también fueron evaluadas. (Ver figuras 4.14-4.17)
En cuanto a la liberación del Diclofenaco dietilamonio desde la forma farmacéutica comercial
Voltaren® emulgel, se observa que esta también sigue el modelo matemático de Higuchi debido
a que el R2 para esta cinética es el más próximo a la unidad. (Ver figura 4.22)
Comparado con la emulsión desarrollada la liberación para esta forma farmacéutica es más rápida
obteniéndose casi el 100% de liberación a las 24 horas. (Ver figura 4.23)
La emulsión desarrollada muestra un tipo de liberación prolongada de acuerdo a lo descrito en la
Figura 2.1. Esto puede deberse a que el Diclofenaco dietilamina se encuentra menos disponible
ya sea por efectos estéricos que proporcionan las grandes cadenas poliméricas, o por la difusión
a través de una doble barrera generada por la formación de la doble emulsion w/o/w lo que se
comprueba al analizar la figura 4.13 en el cual existen dos cambios de pendientes en la curva de
liberación, la primera debido a una rápida difusión del fármaco desde la fase acuosa externa hacia
la solución receptora y la otra más lenta de la fase acuosa interna hacia la fase orgánica externa
(w/o), de ahí a la acuosa externa y luego a la solución receptora.
La polaridad también es un factor que influye en la liberación ya que al formar una doble
emulsión, el principio activo que es de carácter hidrofílico y por la técnica utilizada debería
0
20
40
60
80
100
0 2 0 4 0 6 0 8 0
% D
E LI
BER
AC
ION
TIEMPO (HORAS)
COMPARACION DE LA C INETICA DE L IBERACION EMULSION VS VOLTAREN ®
Emulsion promedio Emulgel ppromedio

82
mantenerse en el núcleo, tiene que atravesar una fase hidrofóbica hasta llegar a la fase hidrofílica
externa y de ahí difundir hasta el medio receptor.
Aunque la técnica utilizada no permitió una correcta liberación del principio activo desde las
partículas sólidas, al estar dentro de una emulsión se permite la liberación del principio activo de
forma prolongada obteniendo así un nuevo sistema de liberación modificada y de esta manera
podemos aceptar la hipótesis nula planteada en esta investigación.
4.1.9 MORFOLOGIA DE LAS MICROPARTICULAS OBTENIDAS EN LA
EMULSION
La morfología de las partículas es esférica lo que se puede comprobar con la fotografía tomada
mediante el equipo AFM (Microscopio de Fuerza Atómica)
Figura 4. 24 Morfología determinada mediante microscopia de fuerza atómica. Imagen en 3D (Topografía). Por Diego Lucero
Para la determinación, la emulsión fue filtrada con filtros de 0.22 μm y colocada en un
cubreobjetos, se dejó secar al ambiente tapada para evitar contaminación con partículas externas.
Luego se observó al microscopio.
En la imagen se visualiza cráteres formados de la ruptura de las gotas poliméricas cuando se secó
la emulsión.

83
Figura 4. 25 Fotografía obtenida mediante Microscopia de fuerza atómica en 3D (Fase) Izquierda: Superficie de la muestra. Derecha: Base de la muestra.
Por: Diego Lucero
La figura 4.25 muestra la Imagen de Fase determinada por microscopia de fuerza atómica. En
esta imagen se puede apreciar la existencia de partículas esféricas que colapsaron al secarse la
muestra, la presencia de tres colores indica que existen tres tipos de materiales con propiedades
de adhesión y viscoelasticidad diferente.

84
CAPITULO V
CONCLUSIONES Y RECOMENDACIONES
5.1 CONCLUSIONES
En la elaboración de la emulsión se consiguió obtener un sistema estable constituido por
agua como fase acuosa interna, la que contiene el principio activo y la gelatina como
material formador de pared, limoneno como fase oleosa y una solución de
polivinilalcohol como fase acuosa externa. Los surfactantes utilizados fue una mezcla de
de Span® 80 y Tween® 80 en proporciones de 1.62% y 0.37% respectivamente
Los tamaños de partícula obtenidos bajo las condiciones de: pH 4.8, concentración de
fase oleosa 34.29%, concentración de fase acuosa 14.40%, mezcla de surfactantes Span®
y Tween®, agitación de la emulsión w/o a 3000 rpm y 900 rpm para la emulsión w/o/w
van desde 3µm hasta las 27.4µm, lo que indica un índice alto de polidispersión.
Las partículas al estado sólido no permitieron la determinación de la liberación del
principio activo, sin embargo al realizar el ensayo de contenido de principio activo
sometiendo las partículas a molienda, calentamiento y baño ultrasónico, se obtuvo un
28.08% de principio activo encapsulado. La eficacia de encapsulación fue del 27.81% y
el rendimiento de la producción fue de 67.84%.
La cinética de liberación del principio activo desde la emulsión obedece al modelo
propuesto por Higuchi donde la liberación del principio activo se debe principalmente a
fenómenos de difusión; debido a que la matriz se expande muy poco por encontrarse ya
en un medio acuoso, la difusión del principio activo depende del fluido que ingresa al
sistema, en otros términos la velocidad de liberación depende del gradiente de
concentración. (Andreeta, 2003)
La emulsión diseñada permitió obtener la liberación prolongada del principio activo,
alcanzando el 90.60% de liberación a las 72 horas, mientras que el sistema de la muestra
comercial Voltaren® emulgel liberó el 98% del principio activo hasta las 24 horas.

85
5.2 RECOMENDACIONES
El método de emulsificación extracción del solvente obtiene bajas eficiencias de
encapsulación con las moléculas activas solubles en agua, debido a la repartición del
principio activo entre la fase acuosa interna y la fase acuosa externa por lo que se puede
corregir la migración del principio activo saturando la fase oleosa externa con el solvente
orgánico utilizado, siempre y cuando el sistema lo permita.
Se puede probar la adición de las sustancias generadoras de porosidad descritas en el
fundamento teórico para incrementar la porosidad de las partículas sólidas y facilitar así
la liberación del principio activo desde el núcleo de la partícula.
Se debe probar con otras técnicas de extracción del principio activo, ya sea por
evaporación en rotavapor o utilizando solventes, teniendo en cuenta que si el solvente se
extrae de forma repentina se producen aglomerados debido al rápido intercambio del
solvente.
Existen parámetros que podrían mejorar la eficiencia de encapsulación como son:
incremento en la viscosidad de la emulsión w/o, disminución de los volúmenes de las
fases, disminución de la duración del proceso de precipitación de las microesferas los
que podrían ser evaluados en la formulación para reducir la pérdida de sustancia activa.
El volumen de solventes utilizados para la obtención de micropartículas por esta técnica
es muy alto, por lo que se debe desarrollar sistemas de tratamientos de residuos para
evitar problemas relacionados con la seguridad de la operación y los problemas
ambientales.
El trabajo se desarrolla tomando las medidas adecuadas de seguridad para la muestra. La
materias primas deben ser filtradas previamente a la preparación de la emulsión, colocarse
guantes para evitar impregnar grasa en las membranas de celulosa.
Cuando se trabaja con partículas de tamaño tan pequeño es importante mantener el control
de las partículas presentes en el área de trabajo, ya que cualquier impureza puede variar
de manera significativa los resultados de la experimentación.
La evaluación de la estabilidad en la emulsión se ha realizado de forma visual, por lo que
se recomienda realizar ensayos de estabilidad mediante dispersión de luz en función del
tiempo.
Después de tomar en cuenta estas consideraciones y mejorar el sistema, se recomienda
continuar con ensayos de seguridad de la forma farmacéutica, ya que al ser un nuevo
sistema de liberación prolongada debe analizarse su comportamiento en organismos
vivos.

86

87
BIBLIOGRAFÍA
Andreeta, H. (2003). Fármacos de Acción Prolongada: Mecanismos de Liberación. Obtenido
de Lat. Am. J. Pharm:
http://www.latamjpharm.org/trabajos/22/4/LAJOP_22_4_4_1_IA69LWUQMI.pdf
Andueza, I. (2012). Desarrollo de un Nanosistema para la Liberación deFármacos Intravítreo.
Obtenido de http://saber.ucv.ve/jspui/handle/123456789
Aranberri, I. (2006). Elaboracion y caracterización de emulsiones estabiliazadas por polímeros
y agentes tensoactivos. Obtenido de revista Iberoamericana de Polímeros :
http://www.ehu.es/reviberpol/pdf/AGO06/aranberri.pdf
Arias, T. (1999). Glosario de medicamentos: desarrollo, evaluación y uso.
Baena, Y., Aragón , M., Sandoval, P., Rosas, J., & Ponce , L. (Diciembre de 2006). Revista
Colombiana de Ciencias Químico - Farmacéuticas. Obtenido de Sistemas osmóticos de
administración oral: http://www.scielo.org.co/scielo.php?pid=S0034-
74182006000200004&script=sci_arttext
Baños Díez, J.-E., & Farré Albaladejo, M. (2002). Principios de Farmacología Clínica. Bases
científicas de la utilización de medicamentos. Barcelona, España: Masson.
Bello Gutiérrez, J. (2000). Ciencia Bromatológica. Principios generales de los alimentos.
Madrid, España: Díaz de santos.
Berna Lozano, M. (Marzo de 2009). Obtención de microencapsulados funcionales de zumo de
Opuntia stricta mediante secado por atomización. Proyecto fin de carrera. Cartagena.
Bravo Osuna, I., & Herrero Vanrell, R. (2007). Potencial de dendrímeros como vehículos de
fármacos en Oftalmología. Arch Soc Esp Oftalmol [online]. Recuperado el 03 de 06 de
2014, de http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0365-
66912007000200002
Bullón , J. (2007). Nano-emulsification of trigliceride oils for prenteral use by mean of a low-
energy emulsification metod. Obtenido de
http://www.scielo.org.ve/pdf/rtfiuz/v30nespecial/art047.pdf
C., S., & P. , I. (2005). Nano-emulsions. Current opinion in Colloid and Interface Science.
ELSEVIER, 102-110.
Castañeta, H. (2011). Microencapsulación, un método para la conservacion de propiedades
fisicoquímicas y biologicas de sustancias quimicas. Obtenido de
http://www.academia.edu/9206518/REVISTA_BOLIVIANA_DE_QU%C3%8DMICA
_MICROENCAPSULACION_UN_METODO_PARA_LA_CONSERVACION_DE_P
ROPIEDADES_FISICOQUIMICAS_Y_BIOLOGICAS_DE_SUSTANCIAS_QUIMIC
AS
Constitución de la República del Ecuador. (2008).

88
Costa Castro, E. (2004). Características y propiedades de los sistemas matriciales. (L. S.
Científica, Entrevistador) Santiago, Chile: Acta Farmacéutica Bonaerense.
ECHEVERRI, C., VALLEJO, C., & LONDONO, M. (2009). Síntesis y Caracterización de
hidrogeles de alcohol polivinílico por la tecnica de congelamiento/descongelamiento
para aplicaciones médicas. Recuperado el 22 de 09 de 2014, de
Rev.EIA.Esc.INg.Antioq [online]:
<htpp://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S1794-
12372009000200005&Ing=en&nrm=iso>
FDA U.S. Food and Drug Administration. (Agosto de 1997).
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm
200707.htm. Recuperado el 12 de 03 de 2013
Fery , A., & Weinkamer, R. (30 de Noviembre de 2007). Mechanical properties of micro-and
nanocapsules: Single capsule measurements,. Polymer- Elsevier, 48(25), 7221-7235.
Forgiarini, A., Marquez, L., & Salager, J. (2006). Nanoemulsiones . Obtenido de Cuaderno
FIRP S237-A, modulo de enseñnza en fenómenos interfaciales:
http://www.firp.ula.ve/archivos/cuadernos/S237A.pdf
Fundación Mapfre. (2007). Microencapsulación de factores neutrofílicos: Aplicación al
tratamiento de la enfermedad de parkinson. MAPFRE Medicina, 18(4).
Garzón S., M., Vázquez R., M., & Villafuerte R., L. (2009). Efecto de los componentes de la
formulación en las propiedades de las nanopartículas lipídicas sólidas. Recuperado el
13 de Noviembre de 2014, de Revista Mexicana de Ciencias Farmacéuticas :
http://www.redalyc.org/pdf/579/57912942005.pdf
Gómez Vega , & et al. (2007). Oxicodona en el dolor crónico no oncológico. Obtenido de
Revista de la Sociedad Española del Dolor:
http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1134-80462007000200006200
Gordillo Galeano, A. (2013). Preparación y Caracterización de Microparticulas sólidad
lipídicas cargadas con ac eite esencial de Cidrón (Aloysia tryphylla) mediante
emulsificación por ultrasonido. Recuperado el 2014, de Tesis presentada como requisito
parcial para optar al título de:Magister en Ciencias Farmacéuticas:
http://www.bdigital.unal.edu.co/10787/1/192598.2013.pdf
Jelvehgari, M., & Montazam, S. (Octubre de 2012). Comparison of Microencapsulation by
Emulsion-Solvent Extraction/ Evaporation Technique Using Derivatives Cellulose and
Acrylate-Methacrylate Copolymer as Carriers. Jundishapur Journal of Natural
Pharmaceutical Products [online], 7(4), 144-152.
Jover Botella , A., & Et. al. (2004). Manual del auxiliar de Farmacia, Módulo II. España: MAD
S.L.

89
Kosegarten, C., & Jiménez, M. (2012). Factores principales que intervienen en la estabilidad
de una emulsión doble. Obtenido de http://web.udlap.mx/tsia/files/2013/12/TSIA-
62Kosegarten-Conde-et-al-2012.pdf
Li, M., Rouaud, O., & Poncelet, D. (2008). Microencapsulation by solvent evaporation: State of
the art for process engineering approaches. ELSEVIER, INternational Journal of
Pharmaceutics, 363, 26-39.
Lopretti, M., & et al. (2007). Microencapsulación de compuestos de actividad biológica.
Obtenido de
http://www.google.com.ec/url?sa=t&rct=j&q=&esrc=s&source=web&cd=3&ved=0CD
YQFjAC&url=http%3A%2F%2Fojs.latu.org.uy%2Findex.php%2FINNOTEC%2Farticl
e%2Fdownload%2F22%2F18&ei=CJsWU7eyFqbQ0QH6-
IGYDw&usg=AFQjCNFFMS_vHCG3DjaDVHdPVwLC-
4n9Jw&sig2=h34VbCC97aU7W90QuzC
Lucinda-Silva, R., & Evangelista, R. (2005). Studies on the Formation of Complex Coacervates
between Chitosan and Alginate During Microparticles Preparation. Acta Fram.
Bonaerence, 24(3), 366-70.
Martín Rosa , M. (2009). Empleo de microparticulas para uso como vacunas y la liberacion de
moléculas biologicamente activas. Obtenido de
http://www.google.com/patents/WO2009063116A1?cl=es
Mateu López , L., & Herrera LLópiz, A. (2007). Revista Cubana de Farmacia . Obtenido de
http://www.bvs.sld.cu/revistas/far/vol41_1_07/far13107.htm
Mazo , P. (2011). Síntesis de Microcápsulas de Poliurea a partir de Aminas Renovables,
Mediante Doble Emulsificación. Obtenido de
http://www.scielo.br/pdf/po/v21n2/AOP_0703.pdf
Muriel , C., Santos Juan, & Sánchez Montero, F. (2013). Master del Dolor. Obtenido de
http://www.catedradeldolor.com/PDFs/Cursos/Tema%206.pdf
Navarro, G., Cabral , P., Malanga, A., & Savio, E. (2008). Diseño de liposomas para el
transporte de Diclofenac Sódico. (F. Martínez R., Ed.) Revista Colombiana de Ciencias
Químico-Farmacéuticas, 37(2).
Oficina Española de patentes y marcas. (1996). Procedimiento de encapsulación. Obtenido de
http://www.espatentes.com/pdf/2084698_t3.pdf
Oficina española de patentes y marcas. (2008). Método para preparar micropartículas que
tienen un peso molecular polimérico seleccionado. Obtenido de
www.oepm.es/pdf/ES/0000/000/02/30/50/ES-2305071_T3.pdf
Oficina Española de Patentes y Marcas. (s.f.). Procedimiento de encapsulación . Obtenido de
http://www.espatentes.com/pdf/2084698_t3.pdf

90
Organización Panamericana de la Salud. (2008). Conceptos Básicos. Obtenido de
new.paho.org/paho-usaid/documents/events/ciess08/bd-be_conceptos_basicos-ops-
nelly_marin.pps
Paniagua Del Cid, M. (2003). Evaluación fisicoquímica de diclofenaco sodico inyectable e
ibuprofeno tabletas, adquiridos por contrato abierto en el hospital Roosevelt.
Parra Huertas, R. (2010). Revisión: Microencapsulación de alimentos. Obtenido de
http://www.scielo.org.co/pdf/rfnam/v63n2/a20v63n01.pdf
Pimentel Figueira, L. (2007). Nanotecnología farmacêutica aplicada ao tratamento da malária.
Recuperado el 03 de 06 de 2014, de Rev. Bras. Cienc. Farm.[online]:
http://www.scielo.br/scielo.php?pid=S1516-93322007000400003&script=sci_arttext
Ramirez Rigo, M. V. (21 de Septiembre de 2010). Los Sistemas de Liberación Modificada de
Fármacos. Bahía Blanca, CONICET. Argentina.
Ramos Picos, D., Gómez Carril, M., & Fernández Mena, D. (2001). Métodos de obtención de
microesferas biodegradables. Revista cubana de farmacia [online], 35(2), 126-135.
Rivas Esteban, I., Gil Alegre, M., & Torres Suárez, A. (2009). Influencia del emulgente en las
características de microcápsulas obtenidas por polimerización interfacial. VI Congreso
SEFIG y 3° jornadas TF, (págs. 225,226,227,228). Madrid.
Rosas, J. (2007). Microesferas de PLGA: Un sistema para la liberación controlada de
moléculas con actividad inmunogénica . Obtenido de
www.revistas.unal.edu.co/index.php/rccquifa/article/view/1574/2225
Sáez, V. (2004). Liberación controlada de fármacos. Micropartículas. Obtenido de Revista
Iberoamericana de Polímeros: http://www.ehu.es/reviberpol/pdf/JUL04/Saez.pdf
Sanchez, V., Carreño, P., & Castelletto, M. (2001). Liberación y Permeación de Diclofenaco
Sódico desde matrices hidrofílicas. Acta farmacéutica bonaerense, 20(2),
101,102,103,104.
Santos Ramos, B., & Guerrero Aznar, M. (1994). Administración de medicamentos: Teoría y
Práctica. Madrid: Díaz de Santos, S.A.
Suñé Negre, J. (2003). Nuevas aportaciones galénicas a las formas de administración.
Obtenido de http://www.ub.edu/legmh/capitols/sunyenegre.pdf
Torres, C. (2012). Diseño y desarrollo de una crema repelente a partir del aceite escencial de
la especie Bursera graveolens. Obtenido de
dspace.utpl.edu.ec/bitstream/123456789/7760/1/1019279.pdf
Universidad Autónoma de Nicaragua. (2008). Elaboración y Caracterización de microcápsulas
gastrorresistentes de Diclofenaco obtenidas por Gelificación Iónica. Universitas, 1(2),
27,28,29,30.
Valdés Lizama, O. (Septiembre de 2005). Diseño de soportes combinados a base de polímeros
naturales y sintéticos para su evaluación en cultivos celulares y la liberación de

91
principios activos. Tesis en Opción del Título de Master en Ciencia y Tecnología de
Materiales, Universidad de la Habana. La Habana.
Yáñez Fernández. (2005). Aplicaciones Biotecnológicas de la Microencapsulación. Obtenido de
http://www.alimentariaonline.com/apadmin/img/upload/MA005_microencapsula_WSF.

92
ANEXOS
Anexo 7. 1 Procedimiento utilizado en la técnica de doble emulsificación-extracción del solvente
Preparación de la emulsión
Figura 7. 1 Procedimiento de elaboración de la emulsión múltiple A) Solubilización de los componentes de la emulsión
B) Elaboración de la emulsión por agitación en el equipo mezclador/emulsificador – Silverson C) Emulsión obtenida
Extracción de las partículas
Figura 7. 2 Extracción de las partículas A) Proceso de congelamiento y liofilización
B) Lavados, eliminación de los residuos por centrifugación C) Muestra seca
A) B) C)
A) B)
A) B)
A)
C) A) B)

93
Determinación del tamaño de partícula, contenido y liberación de principio activo
Figura 7. 3 Determinación del tamaño de partícula A) Prueba en el equipo DLS
B) Determinación del tamaño mediante el programa imageJ
Figura 7. 4 Proceso De liberación del principio activo A) Membrana de diálisis con la emulsión
B) Equipo de disolución y bomba de recirculación
A)
B)
A) B)
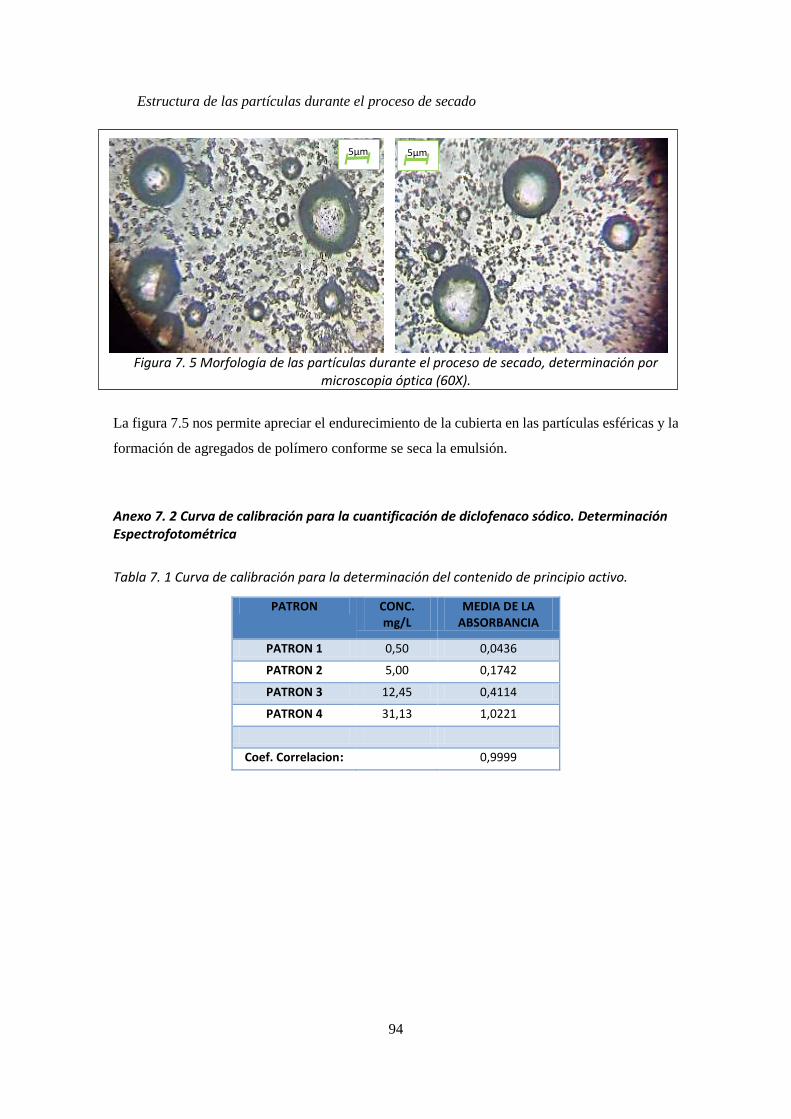
94
Estructura de las partículas durante el proceso de secado
Figura 7. 5 Morfología de las partículas durante el proceso de secado, determinación por microscopia óptica (60X).
La figura 7.5 nos permite apreciar el endurecimiento de la cubierta en las partículas esféricas y la
formación de agregados de polímero conforme se seca la emulsión.
Anexo 7. 2 Curva de calibración para la cuantificación de diclofenaco sódico. Determinación Espectrofotométrica
Tabla 7. 1 Curva de calibración para la determinación del contenido de principio activo.
PATRON CONC. mg/L
MEDIA DE LA ABSORBANCIA
PATRON 1 0,50 0,0436
PATRON 2 5,00 0,1742
PATRON 3 12,45 0,4114
PATRON 4 31,13 1,0221
Coef. Correlacion: 0,9999
5µm 5µm

95
Figura 7. 6 Gráfico de la curva de calibración de Diclofenaco sódico
Anexo 7. 3 Curva de calibración para la determinación de Diclofenaco dietilamonio por HPLC
Área (mUA*min)
REPETICIÓN 1 REPETICIÓN 2 REPETICIÓN 3
0,1349 0,1376 0,1379
0,2986 0,2912 0,2888
0,7405 0,7357 0,7344
1,5574 1,4943 1,4914
3,7513 3,6371 3,6451
y = 0,0321x + 0,0187R² = 0,9997
0
0,2
0,4
0,6
0,8
1
1,2
0,0 5,0 10,0 15,0 20,0 25,0 30,0 35,0
AB
SOR
BA
NC
IA
Concentración
CURVA DE CALIBRACION DICLOFENACO SODICO
CONCENTRACION DE LOS ESTANDARES
(ppm)
St. 1 St. 2 St. 3 St. 4 St. 5
1,43 2,86 7,15 14,3 34,32

96
Figura 7. 7 Gráfico de la curva de calibración de Diclofenaco sódico
Coeficiente de Correlación Promedio 0,99998
Pendiente (Ks) 0,107
Corte con el eje 0,022
Determinación del límite de detección
𝐿𝑂𝐷 =3𝑆𝐷𝑏
𝐾𝑠
AREA DE LOS BLANCOS EN LAS EMULSIONES (mUA*min)
Blanco Emul. 1 0,0004
Blanco Emul. 2 0,0011
Blanco Emul. 3 0,0006
SDb 0,0004
𝐿𝑂𝐷 =3 ∗ 0,0004
0,107
𝑳𝑶𝑫 = 0,010
y = 0,1077x - 0,0221R² = 0,9995
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
0 10 20 30 40
Are
a (m
UA
)
Concentración (ppm)
CURVA DE CALIBRACION DICLOFENACODIETILAMONIO
DDEA Lineal (DDEA)

97
Anexo 7. 4 Barridos espectrales realizados a los componentes de la matriz de las micropartículas elaboradas con diclofenaco sódico
Figura 7. 8 Barrido espectral de los componentes del sistema A Diclofenaco de sodio, B Gelatina, C Alcohol polivinílico
Anexo 7. 5 Cromatograma de una de las muestras obtenida de la prueba liberación de diclofenaco dietilamonio desde la emulsión
Figura 7. 9 Cromatograma de la liberación desde la emulsión Pico 1 Diclofenaco dietilamonio (DDEA)
0,0 1,3 2,5 3,8 5,0 6,3 7,5 8,8 10,0
-1,00
0,00
1,00
2,00
3,00
4,00
5,00
6,00
7,00
8,00DDEA_20150512_EMULSION_1 #30 [modified by dslb] UV_VIS_2mAU
min
1 - DDEA - 0,723
WVL:276 nm
A
B
C

98
Figura 7. 10 Cromatograma de la liberación desde Voltaren® emulgel Pico 1 Diclofenaco dietilamonio (DDEA)
Anexo 7. 6 Calculo del BHL requerido para la emulsión
𝐻𝐵𝐿𝑅 = 𝑋1 𝐻𝐵𝐿1 + 𝑋2 𝐻𝐵𝐿2
𝐻𝐵𝐿𝑅 =1.62
1.994.3 +
0.37
1.9915
𝑯𝑩𝑳𝑹 = 𝟔. 𝟐𝟖
0,0 1,3 2,5 3,8 5,0 6,3 7,5 8,8 10,0
-1,00
0,00
1,00
2,00
3,00
4,00
5,00DDEA_20150513_EMULGEL_2 #3 DDEA_emulsionBA3 UV_VIS_2mAU
min
1 - DDEA - 0,710
WVL:276 nm