talasemias dra. silvia varas [email protected] fundamentos técnicas moleculares
TRANSCRIPT

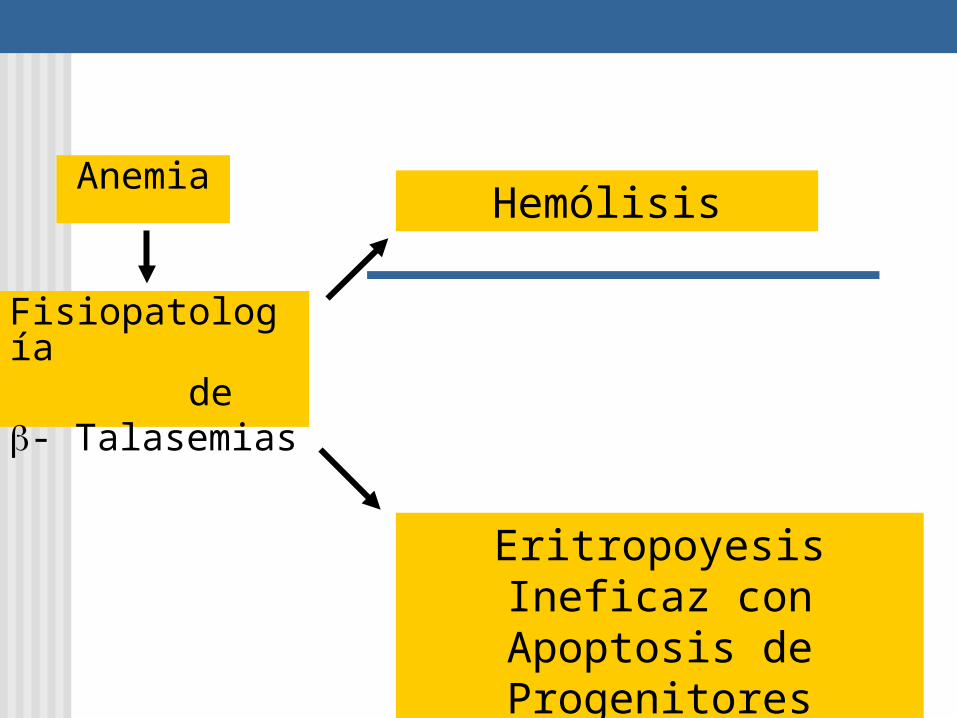
Fisiopatología de - Talasemias
Hemólisis
Eritropoyesis Ineficaz con Apoptosis de
Progenitores
Anemia

Hemólisis:
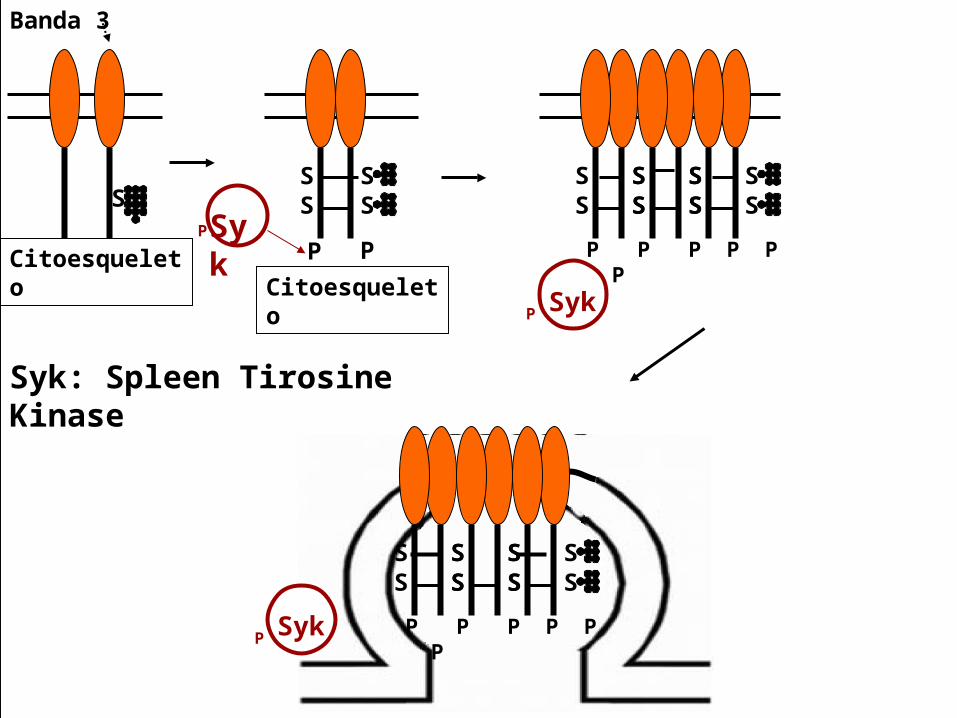
Formación de hemicromos Los hemicromos se unen a banda 3, catalizan la
oxidación de Banda 3. Entrecruzamiento oxidativo con Banda 3. Inmediata asociación con Syk(Spleen Tyrosine
Kinase), fosforilación de treoninas de Banda 3. La fosforilación marca la disminución de afinidad
con la anquirina y liberación del citoesqueleto (espectrina/actina).
Aumenta la movilidad lateral en la bicapa (clustering) de Banda 3.
Formación de agregados de Banda 3. Progresiva vesiculación. Perdida de la membrana plasmática y LISIS
celular

Citoesqueleto
SP
SS
SS
Citoesqueleto
Syk P P
SS
SSSS
SSSS
SS
P P P P P P
SykP
SS
SSSS
SSSS
SS
P P P P P PSykP
Syk: Spleen Tirosine Kinase
Banda 3
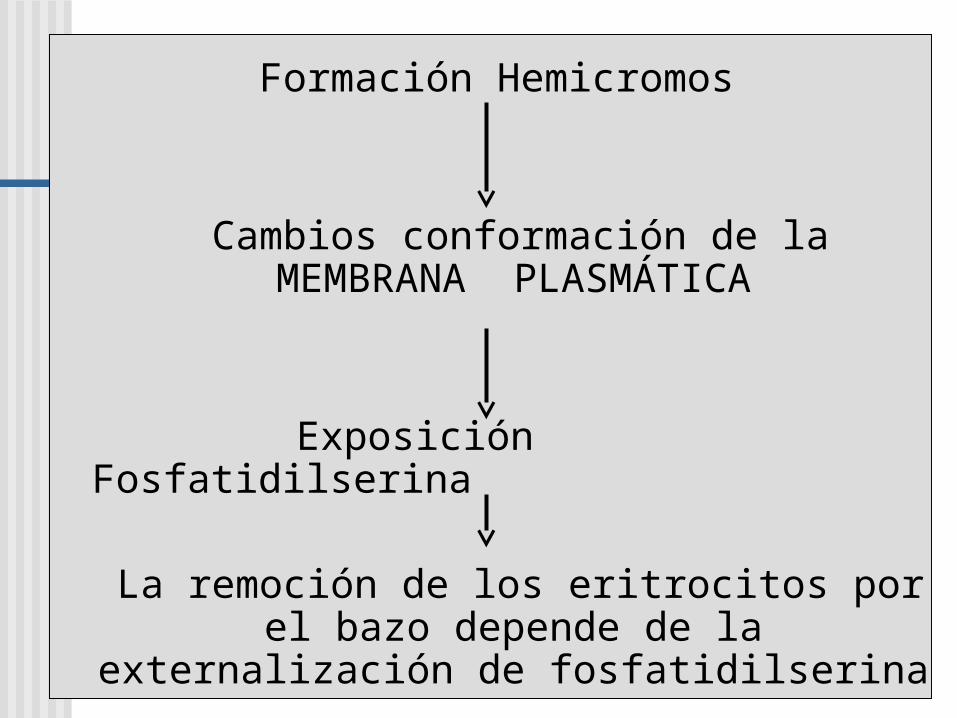
Formación Hemicromos
Cambios conformación de la MEMBRANA PLASMÁTICA
Exposición Fosfatidilserina
La remoción de los eritrocitos por el bazo depende de la externalización de
fosfatidilserina
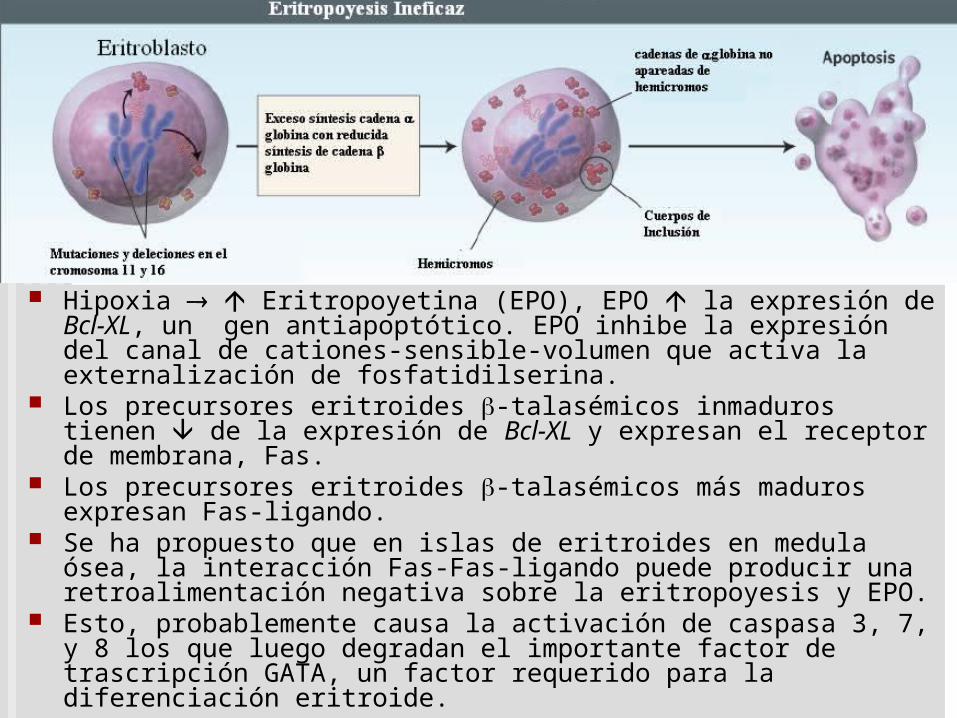
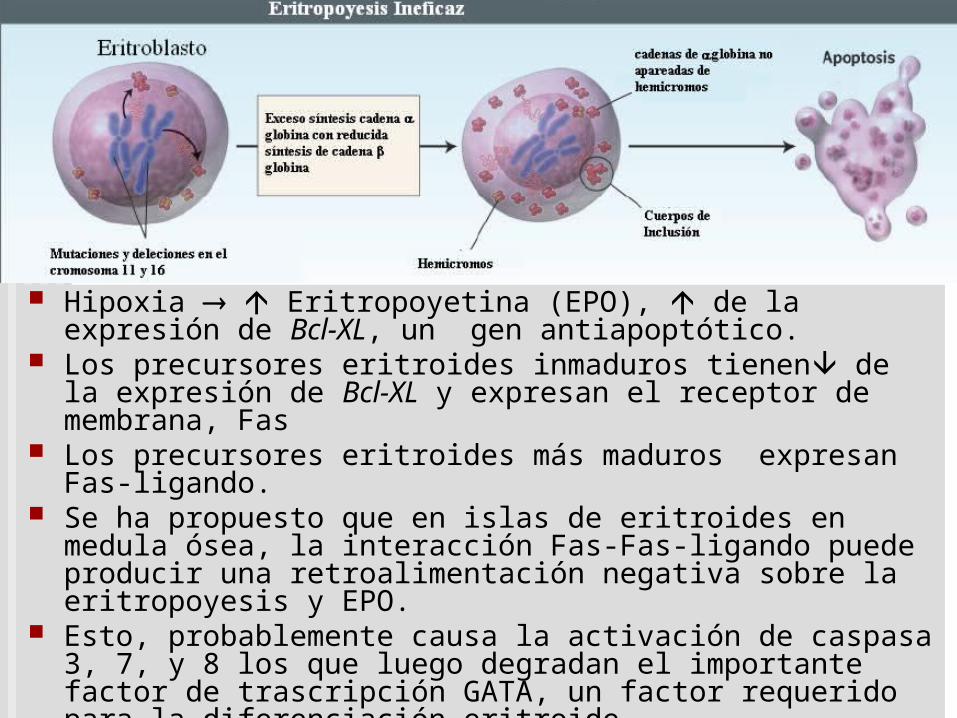
Apoptosis de precursores
Hipoxia Eritropoyetina (EPO), EPO la expresión de Bcl-XL, un gen antiapoptótico. EPO inhibe la expresión del canal de cationes-sensible-volumen que activa la externalización de fosfatidilserina.
Los precursores eritroides -talasémicos inmaduros tienen de la expresión de Bcl-XL y expresan el receptor de membrana, Fas.
Los precursores eritroides -talasémicos más maduros expresan Fas-ligando.
Se ha propuesto que en islas de eritroides en medula ósea, la interacción Fas-Fas-ligando puede producir una retroalimentación negativa sobre la eritropoyesis y EPO.
Esto, probablemente causa la activación de caspasa 3, 7, y 8 los que luego degradan el importante factor de trascripción GATA, un factor requerido para la diferenciación eritroide.
Apoptosis de precursores
Hipoxia Eritropoyetina (EPO), de la expresión de Bcl-XL, un gen antiapoptótico.
Los precursores eritroides inmaduros tienen de la expresión de Bcl-XL y expresan el receptor de membrana, Fas
Los precursores eritroides más maduros expresan Fas-ligando.
Se ha propuesto que en islas de eritroides en medula ósea, la interacción Fas-Fas-ligando puede producir una retroalimentación negativa sobre la eritropoyesis y EPO.
Esto, probablemente causa la activación de caspasa 3, 7, y 8 los que luego degradan el importante factor de trascripción GATA, un factor requerido para la diferenciación eritroide.

Biología molecular
Talasemias

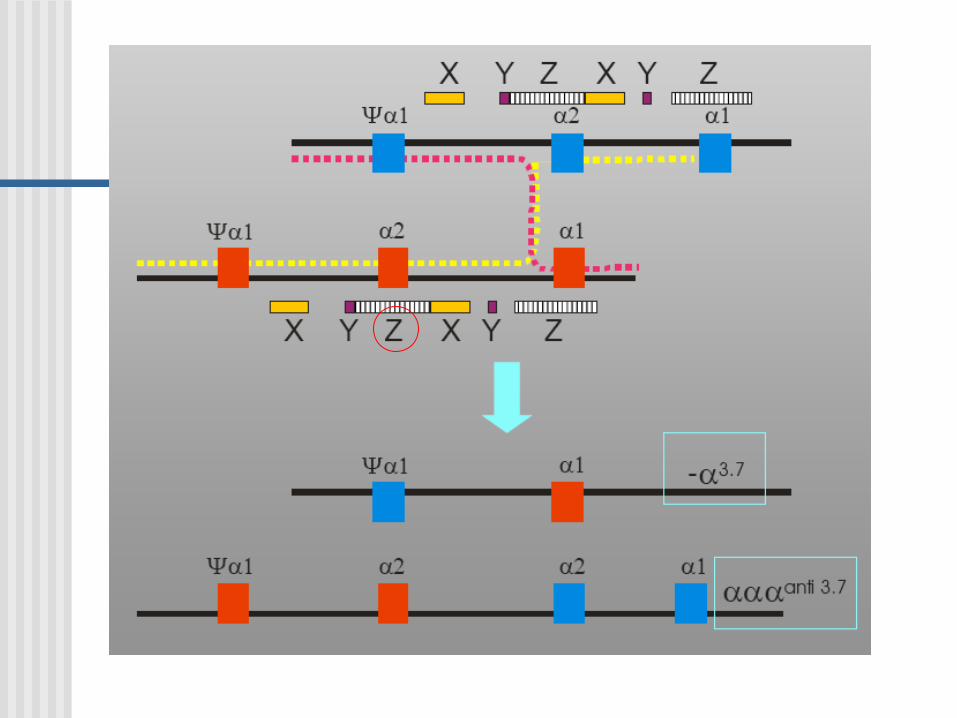
Estructura molecular de los genes globina duplicados
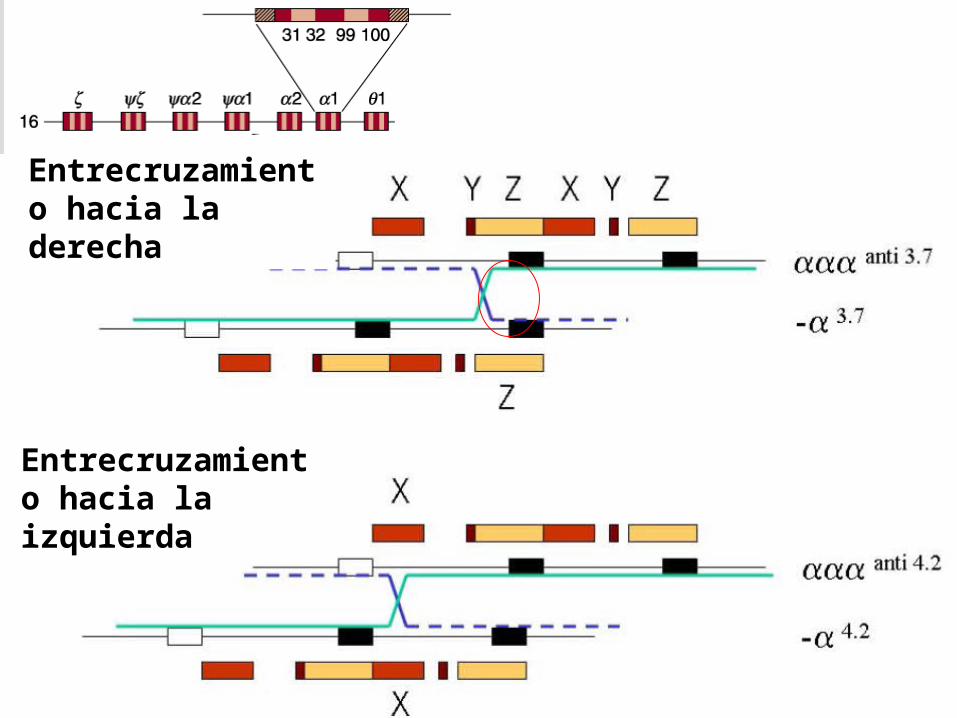
Entrecruzamiento hacia la derecha
Entrecruzamiento hacia la izquierda


Clasificación Relación Fenotipo/ Mutación:
• Beta +: La mutación no impide que el gen sintetice algo de globinas beta.
• Beta 0: La mutación impide totalmente la síntesis de globinas beta, bien porque no se produce RNAm o bien porque se produzca pero no sea traducible.
-talasemia
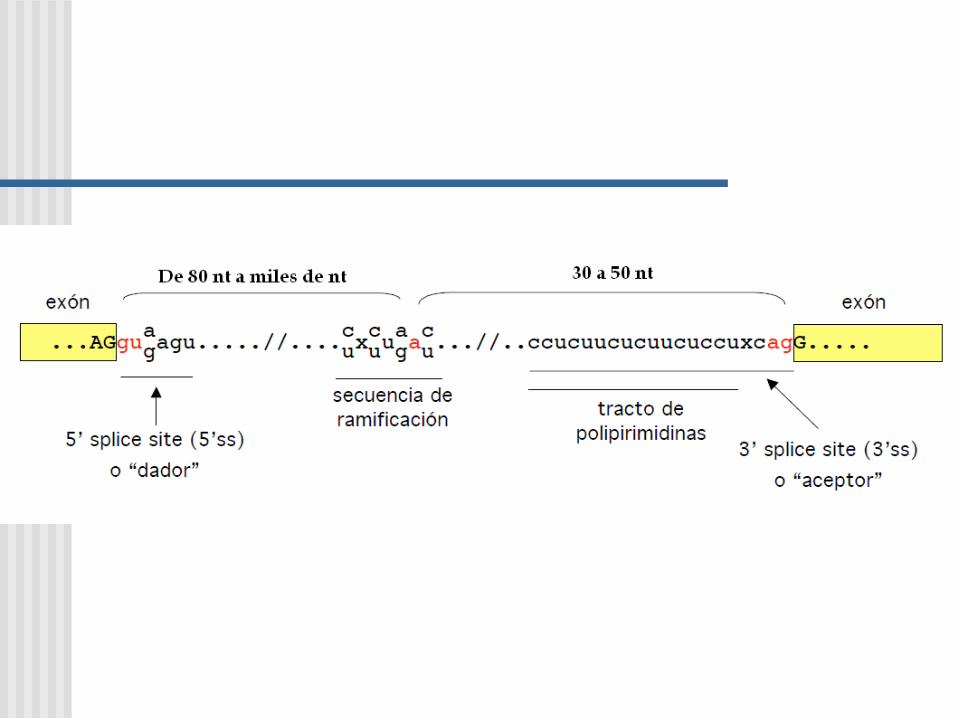

Consiste de estructuras exon e intrones. Se refieren al ADN codificante y no codificante, respectivamente, del gen.
INTRONES
Los genes de las globinas:
•Inicia con un sitio dador consenso (G100U100A62A68G84U63..)
•Tiene un punto de ramificación cercano al sitio 3’ del intron (no es un sitio consenso muy conservado UACUAAC)
•Finaliza con un sitio aceptor consenso (12Py..NC65A100G100)
AGUACUAAC
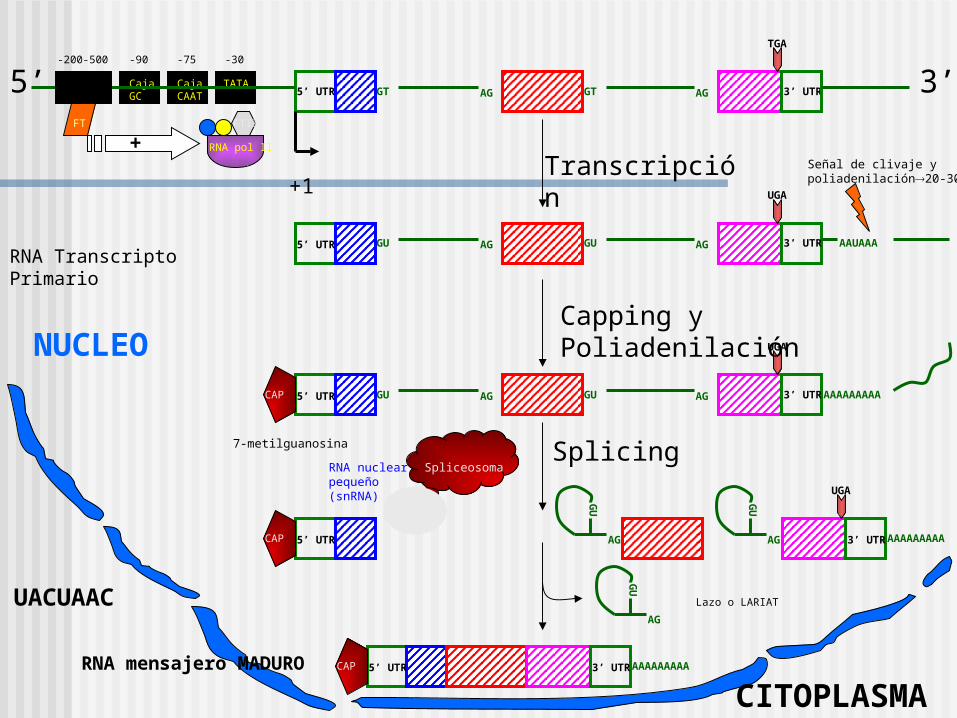
EnhancerCaja GC
-90
Caja CAAT
-75 -30
TATA5’ UTR
+1
3’ UTRGT AG GT AG
TGA
5’ 3’FTII
RNA pol II
-200-500
FT
+Transcripción
RNA Transcripto PrimarioGU GU5’ UTR AG AG 3’ UTR AAUAAA
UGA
Señal de clivaje y poliadenilación20-30nt
Capping y Poliadenilación
GU GU
UGA
AG AG 3’ UTR5’ UTRCAP
7-metilguanosina
AAAAAAAAA
SplicingSpliceosoma
5’ UTRCAP
GU
AG
UGA
AAAAAAAAA
GU
AG
GU
AG
Lazo o LARIAT
5’ UTRCAP AAAAAAAAA
3’ UTR
3’ UTRRNA mensajero MADURO
NUCLEO
CITOPLASMA
RNA nuclear pequeño (snRNA)
UACUAAC


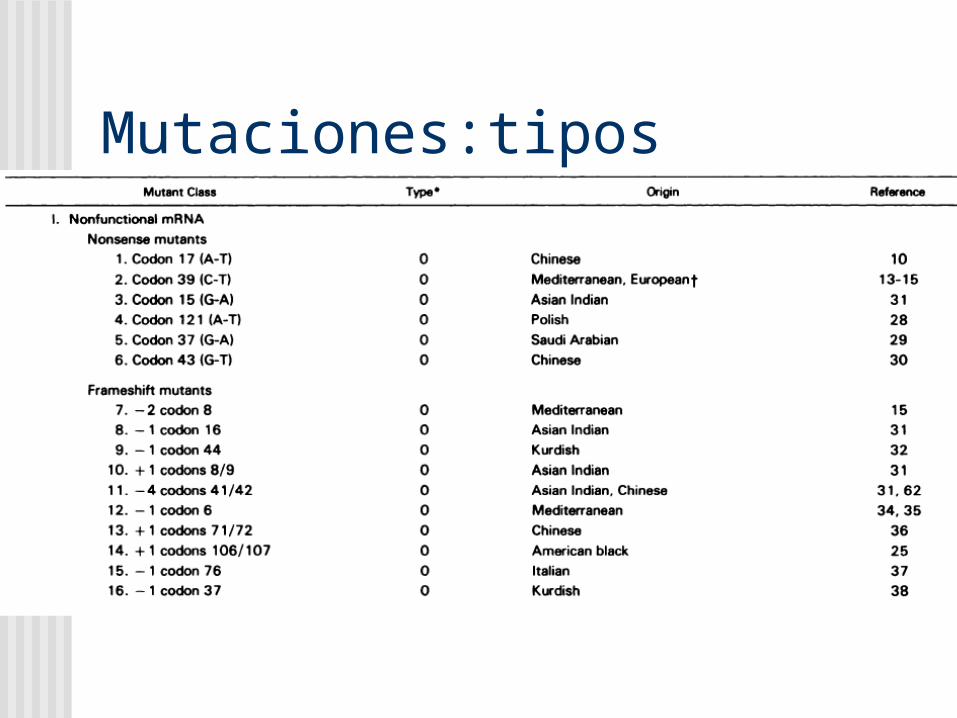
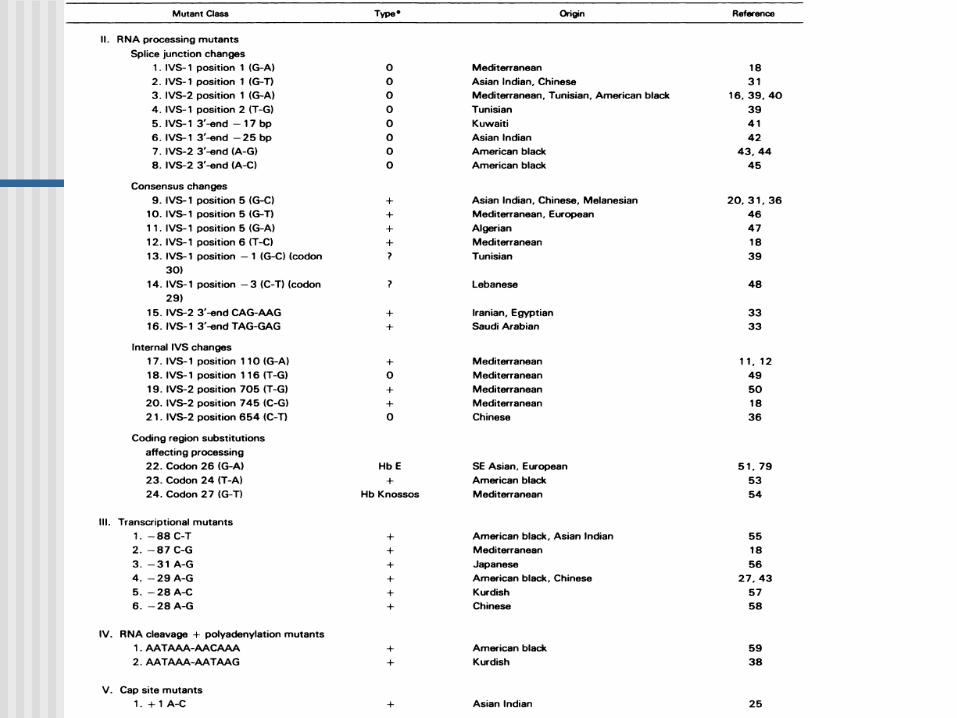
Mutaciones:tipos



PCR+ ER
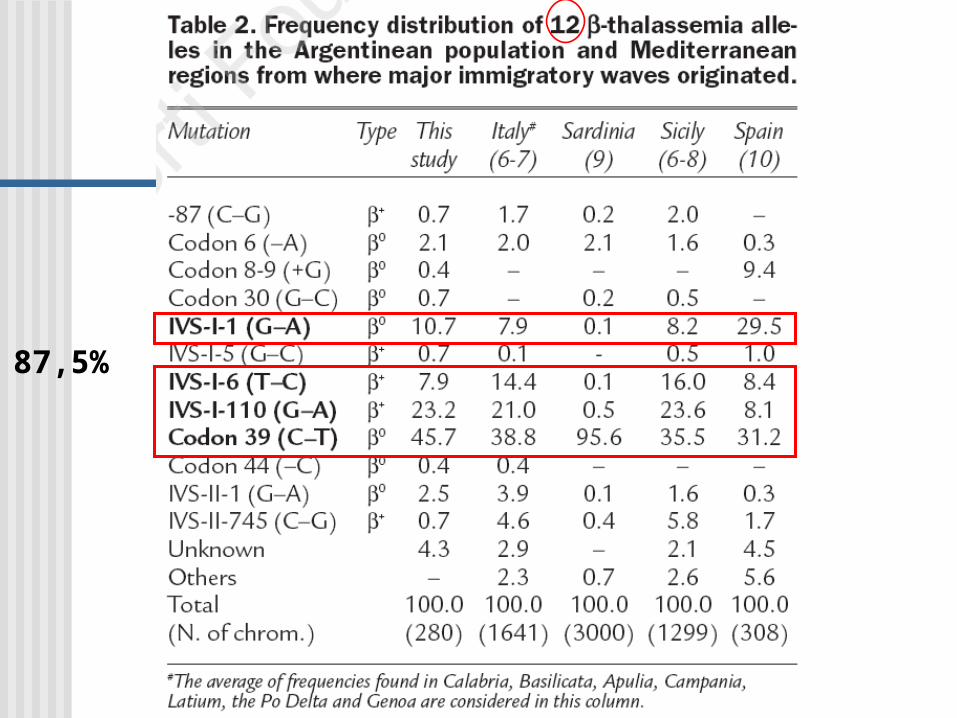
87,5%
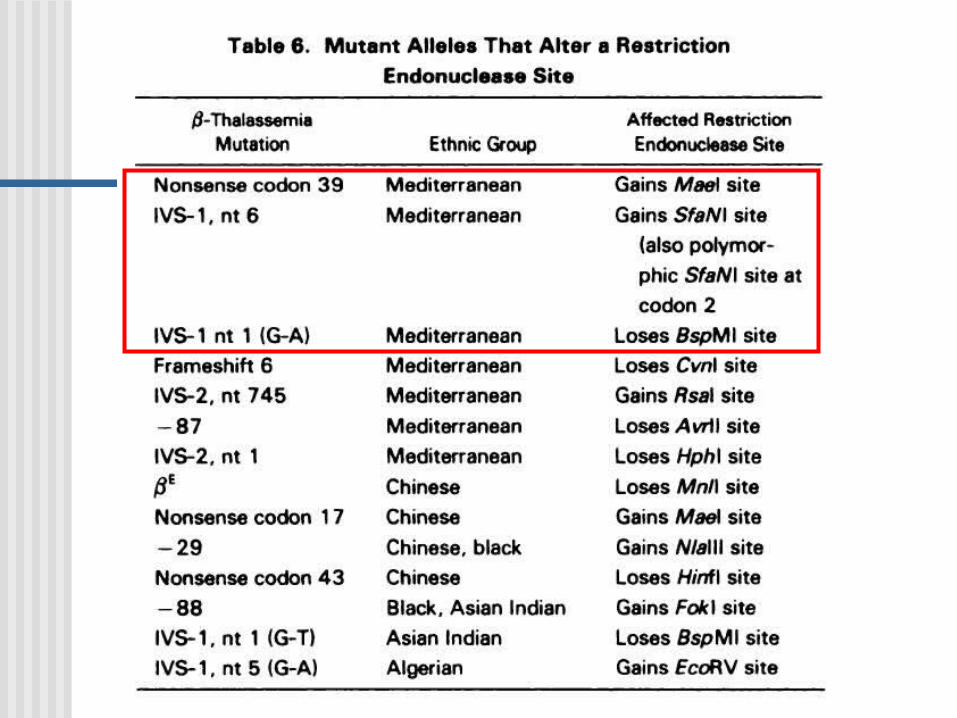


Estrategias: Se produce una perdida o ganancia
sitio de corte ER: PCR+ ER (algunos la refieren como RFLP-PCR).
No hay cambio: Análisis con Dot Blot + Sondas ASO (Oligonucleótidos Alelo Especificas), ARMS

1. Extracción de ADN
2. Amplificación con un par de primers específicos a la región flanqueante al sitio de restricción polimorfico
3. Digestión con la Enzima de Restricción apropiada
4. Separación de los fragmentos por electroforesis, teñido con Gel Red
5. Análisis de los resultados
PCR + Análisis con Endonucleasa de Restricción (RFLP-PCR)
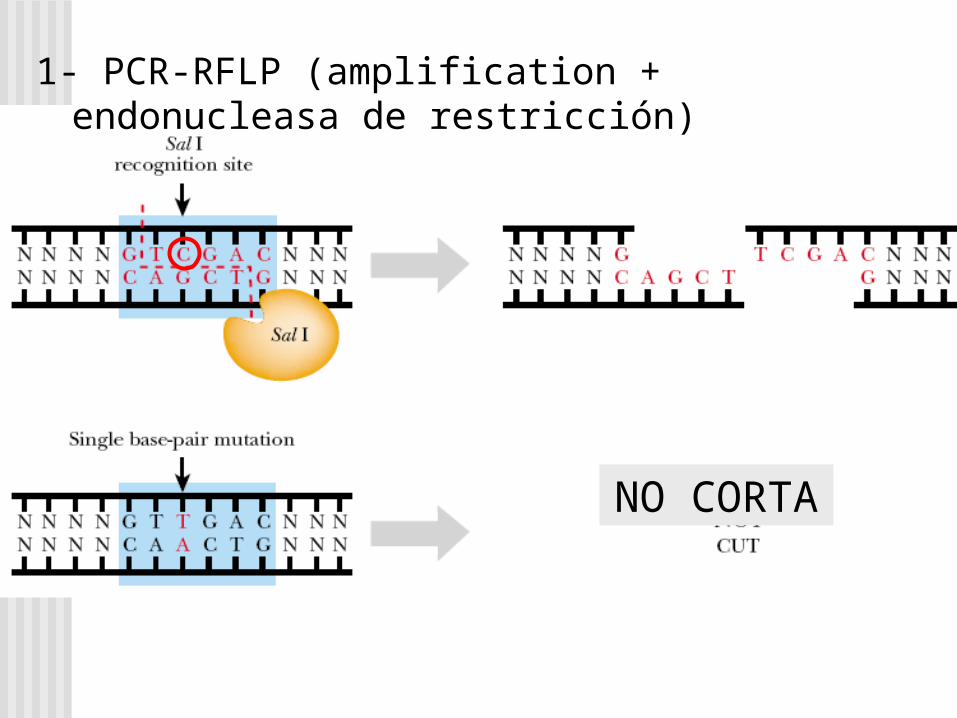
1- PCR-RFLP (amplification + endonucleasa de restricción)
NO CORTA
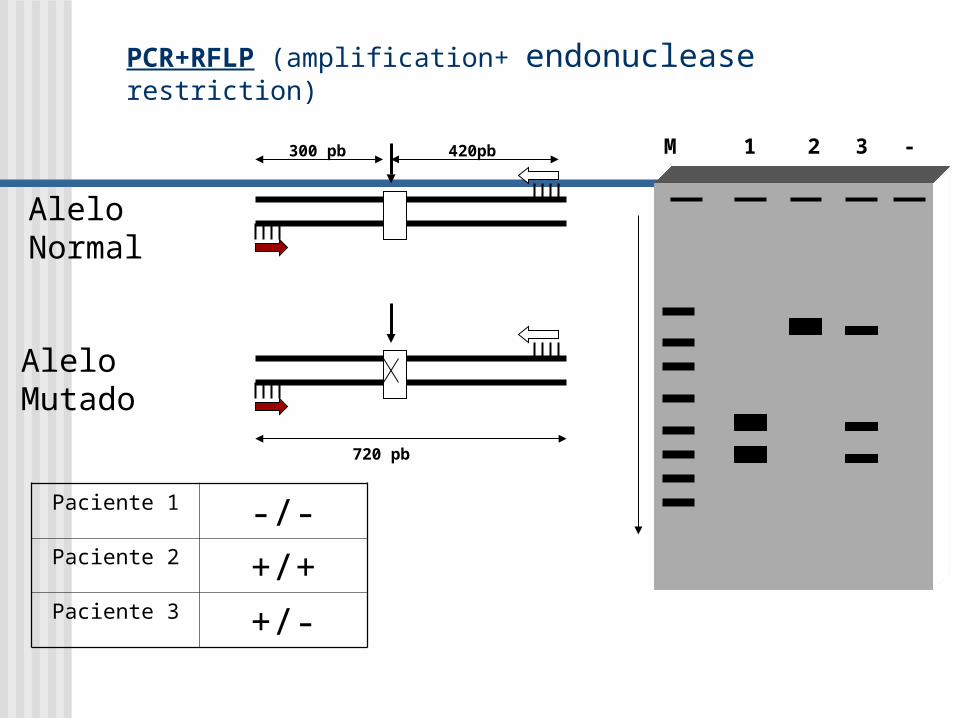
PCR+RFLP (amplification+ endonuclease restriction)
Alelo Normal
Alelo Mutado
420pb300 pb
720 pb
M 1 2 3 -
Paciente 1 -/-Paciente 2 +/+Paciente 3 +/-

726 pb
WT
WT

Otro ejemplo:Hb S: La mutación de Hb S en el
codon 6, destruye el sitio de reconocimiento de la enzima Dde I.

Se usa para la detección de Mutaciones puntuales (que no afectan sitios de cortes de una Enzima de restricción)
Análisis con Dot Blot + Sondas ASO (Oligonucleótidos Alelo Especificas)

Equipo de DOT BLOT

Armado:
1. Tapa inferior
2. Membrana selladora
3. Membrana de Nitrocelulosa
4. Tapa superior
5. Ajuste de los tornillos

-Volumen Total PCR: 100 l
Para sembrar un punto del dot blot:
Producto de PCR: 10 l
EDTA 100 mM 50 l
NaOH 2N 40 l
Agua 100 l
Teniendo en cuenta cuántos sondas vamos a probar y cuántas muestras se van a analizar, hacer los cálculos necesarios para TODOS los dot blots, multiplicando los volúmenes anteriores por el número de sondas + 1. En este ejemplo, se usaron 7 sondas
x 8.
Producto de PCR: 80 l
EDTA 100 mM 400 l
NaOH 2N 320 l
Agua 800 l
X 8
Preparación de los productos a transferir:
1- Tomar tantos tubos eppendorf grandes como muestras.
7
2- Preparar las mezclas calculadas antes muestras listas para transferir a membranas.
Se utilizan membranas
de NITROCELULOSA
Cortar un rectángulo de 12 x 8 cm trabajando siempre con guantes.
Hidratar 30 minutos con agua destilada y luego equilibrar durante 5 minutos en buffer SSPE 20X, pH 7,4.
Preparación de la membrana:

HYBRI-DOT MANIFOLD, BRL (Gibco)
+
membrana
Colocar la plancha inferior del aparato, conectar a la bomba de vacío. Identificar un extremo de la membrana con un corte.
Utilizando guantes, colocar la membrana sobre la plancha inferior.
Siembra de las Muestras- Dot Blot:

•Colocar la plancha superior y ajustar perfectamente los tornillos en forma enfrentada. Encender la bomba de vacío y verificar que ésta elimina el líquido remanente del lavado de la membrana.
• En cada punto de siembra colocar sucesivamente 200 ml de dilución de cada producto de amplificación y 200 ml de SSPE 20X
Finalizada la siembra, y luego que la bomba eliminó todo el líquido, colocar la membrana entre dos papeles Whatman y sellarlos con una cinta. Secar durante una hora a 80° C en estufa (o por cross – linker)
1 2 3 4 5 6 7 8

El marcado se puede realizar:
• con radiactivo
• con métodos no radiactivos.
En el primer caso, se marca la sonda con P32. Hay dos métodos
para hacerlo:
1. en uno de ellos, la sonda queda marcada en varios sitios a lo largo de la misma, para ello se utiliza el fragmento Klenow de la DNA polimerasa. --------------------- * * * * *
2. en el segundo, sólo se marca el extremo de la sonda y para ello se usa una kinasa. ------------------*
El primer método, para ello se colocan 250 ng de ADN de la sonda en un tubo Eppendorf (contenido en 20 l) y luego, trabajando detrás de una pantalla de acrílico y con doble guante, se adiciona 2 l de cada dNTP, el dNTP marcado (32P- dATP), random primers y la enzima Klenow. Se incuba dos horas a 25° C y luego se detiene la reacción. La sonda marcada se purifica por columnas de Sephadex y se mide la actividad en un contador.

•En el caso de métodos no radiactivos, suelen comprarse las sondas ya marcadas, generalmente con una molécula de BIOTINA incorporada en uno de sus extremos.
•De esta forma ya tenemos el ADN fijado a la membrana y las sondas marcadas. Podemos comenzar con las hibridaciones.
•Con el objeto de poder analizar cada muestra con cada una de las sondas, se debe cortar la membrana en tantas tiras como sondas haya.
•RECORDAR QUE TODO ESTO SE DEBE REALIZAR CON GUANTES PARA NO CONTAMINAR LAS MUESTRAS

•Se deben colocar cada una de las tiras de la membrana en recipientes individuales, donde se van adicionando las distintas soluciones.
•Se adiciona a las membranas el medio de pre hibridación que contiene SDS y DNA de esperma de salmón previamente desnaturalizado en baño de agua hirviendo durante cinco minutos. Esto es para BLOQUEAR la membrana, es decir, para evitar las reacciones inespecíficas.
•Se tapan los recipientes para evitar evaporación, y se los incuba con agitación, durante dos horas a 37º C.
•Para evitar confusiones, rotular cada recipiente con el nombre correspondiente a la sonda que se va a utilizar en él.
1° Paso: HIBRIDIZACION:

Una vez que tenemos los dot blot revelados, identificamos cada una de las muestras sembradas y observamos el resultado correspondiente a cada sonda.
1 2 3 4 5 6 7 8
7: control negativo de la PCR
8: control negativo de hibridación (gen de tiroglobulina)
1: portador de la mutación 1 –1
2: portador de la mutación 1 –1
3: portador de la mutación 1 - 110
4: portador de la mutación 39
5: portador de la mutación 39
6: normal


Detección de Mutaciones por el Sistema Refractario de
Amplificación de MutaciónARMS
(Detection of Mutations by the Amplification Refractory Mutation System)

Para el screnning de una mutación particular se necesita :
El primers ARMS Mutante o Normal El primer común Par primers control: Los primers control que amplifica un segmento de ADN a alguna distancia del sitio de mutación y que no interfiera con la amplificación del fragmento de ADN producida por el primers del ARMS y el primer común.
Línea 1: Madre: DNA de un heterocigoto para la mutación IVS-1nt 5 con un primer ARMS mutante.
Línea 2: Padre: DNA de un heterocigoto para la mutación IVS-1nt 5 con un primer ARMS mutante.
Línea 3: DNA de un individuo normal con un primer ARMS mutante.Línea 4: DNA Fetal con un primer ARMS normal.Línea 5: DNA Fetal con un primer ARMS mutante.El feto es homocigoto para la mutación IVS-1nt 5 del gen de –globina.
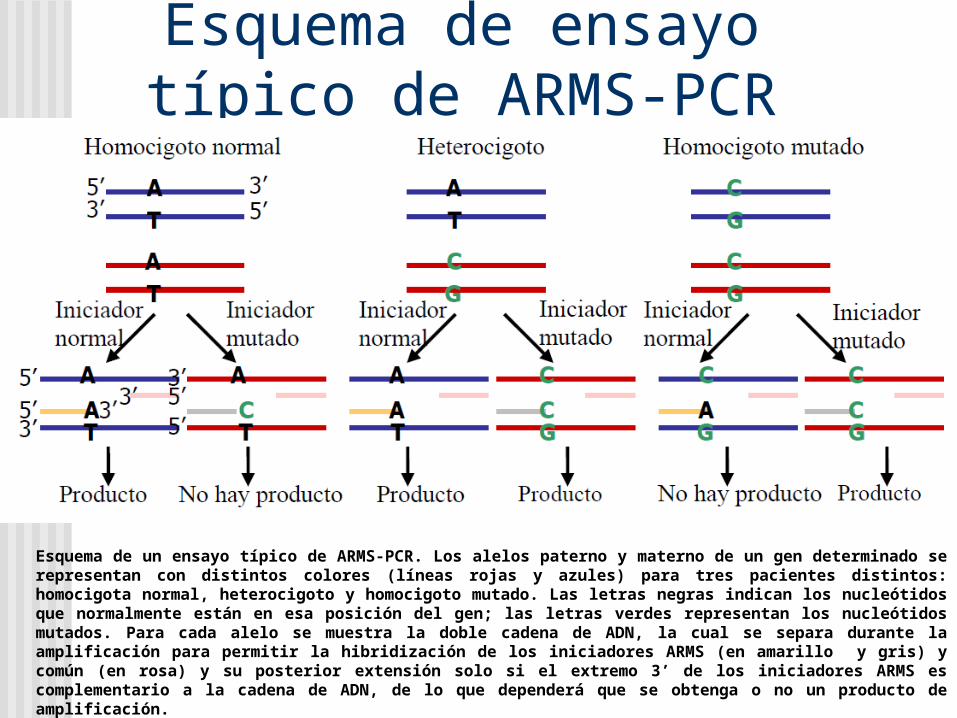
Esquema de ensayo típico de ARMS-PCR
Esquema de un ensayo típico de ARMS-PCR. Los alelos paterno y materno de un gen determinado se representan con distintos colores (líneas rojas y azules) para tres pacientes distintos: homocigota normal, heterocigoto y homocigoto mutado. Las letras negras indican los nucleótidos que normalmente están en esa posición del gen; las letras verdes representan los nucleótidos mutados. Para cada alelo se muestra la doble cadena de ADN, la cual se separa durante la amplificación para permitir la hibridización de los iniciadores ARMS (en amarillo y gris) y común (en rosa) y su posterior extensión solo si el extremo 3’ de los iniciadores ARMS es complementario a la cadena de ADN, de lo que dependerá que se obtenga o no un producto de amplificación.
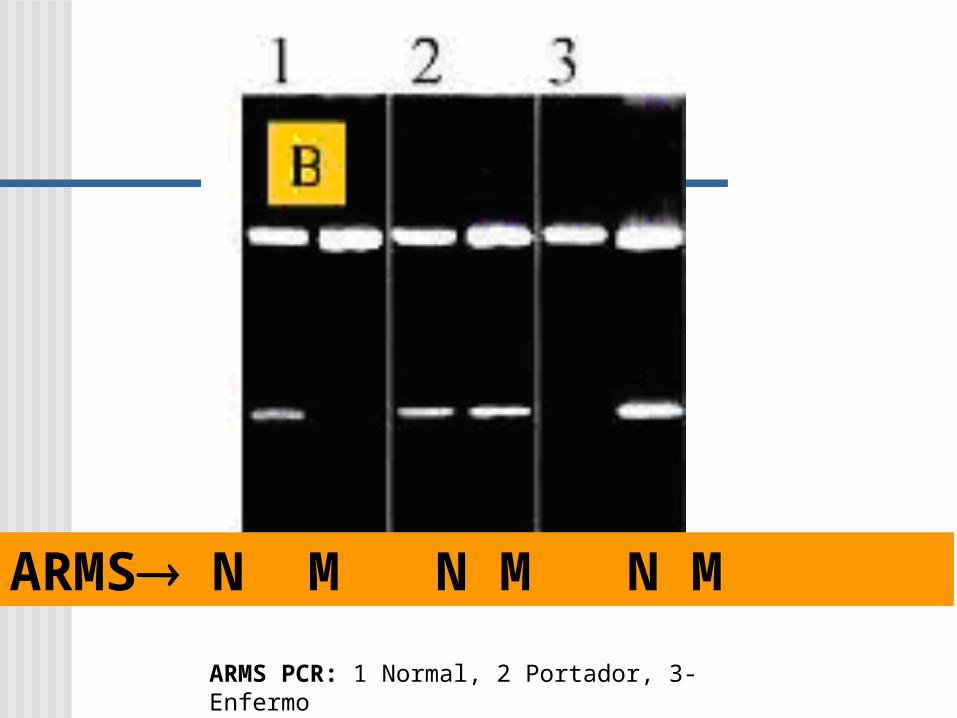
ARMS PCR: 1 Normal, 2 Portador, 3- Enfermo
ARMS N M N M N M


Alfa Talasemias
MUTACIONES
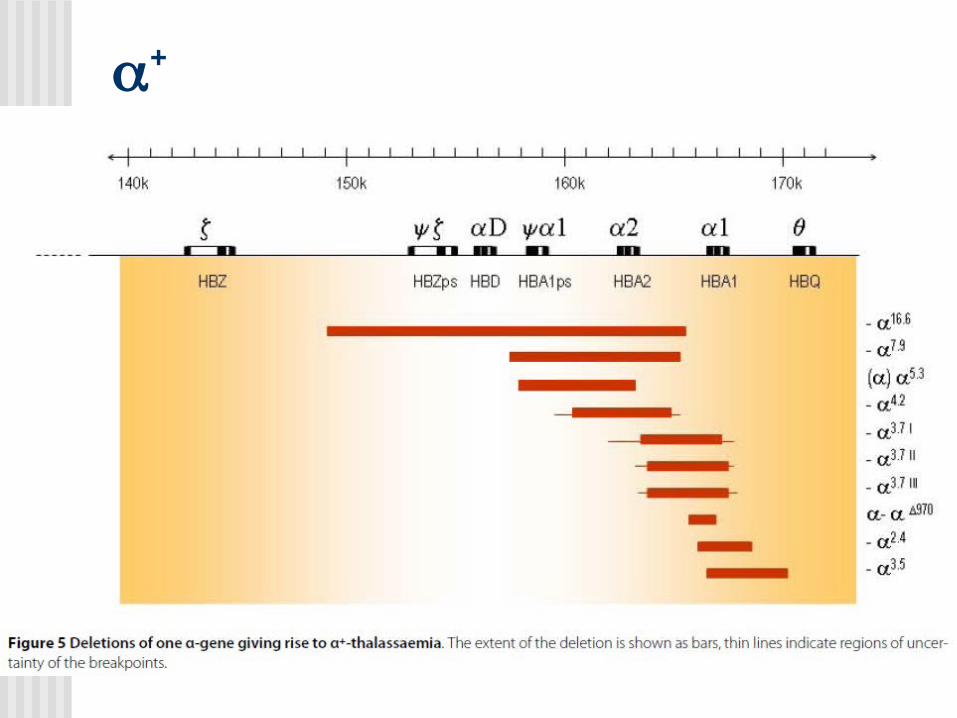
+
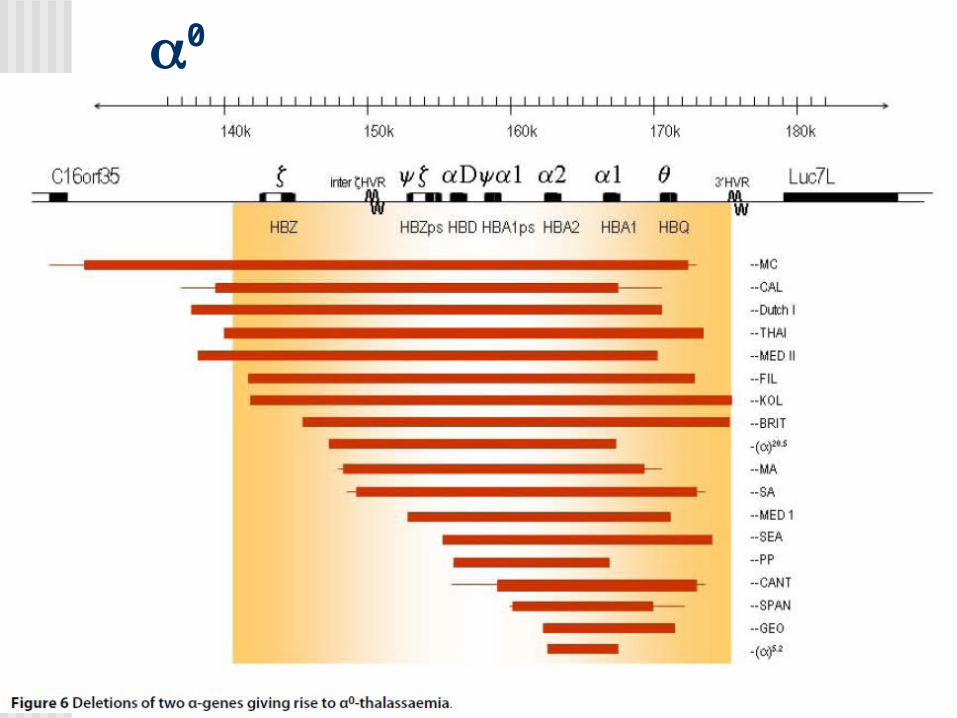
0

Southern Blot y análisis de la secuencia de ADN
Gap-PCR Otro método es el MLPA (Amplificación
de Sondas dependiente de Ligamiento Múltiple
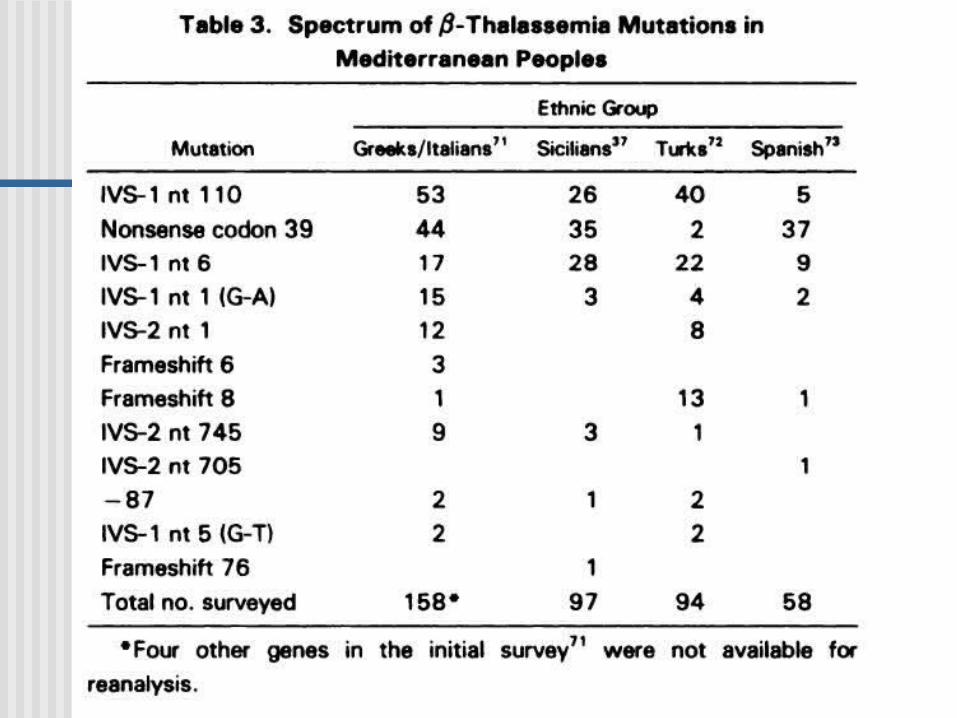
- Talasemias:
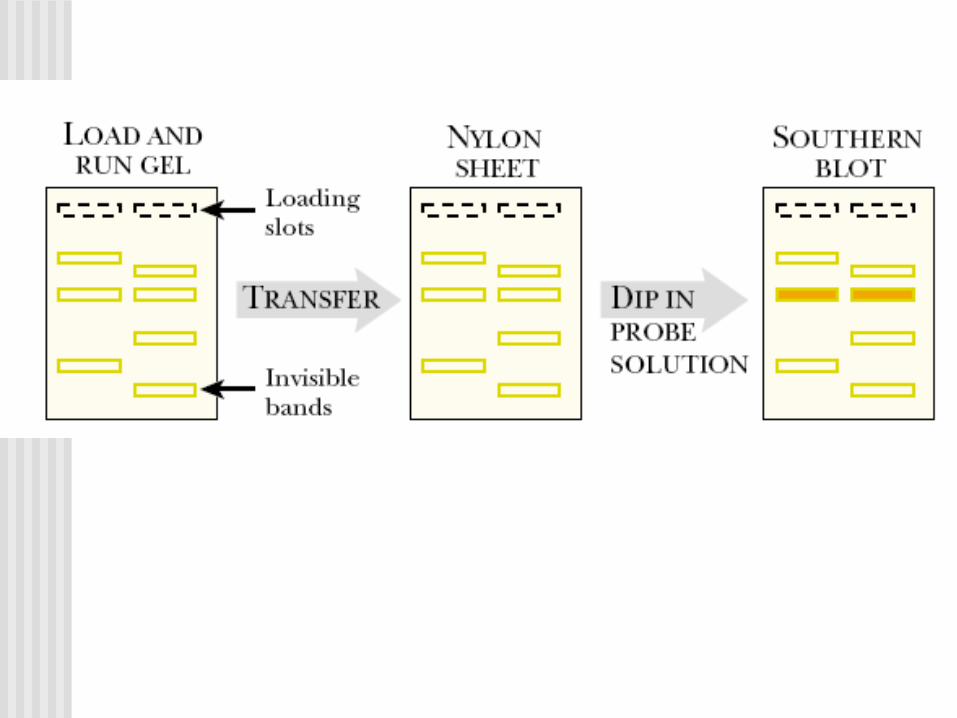
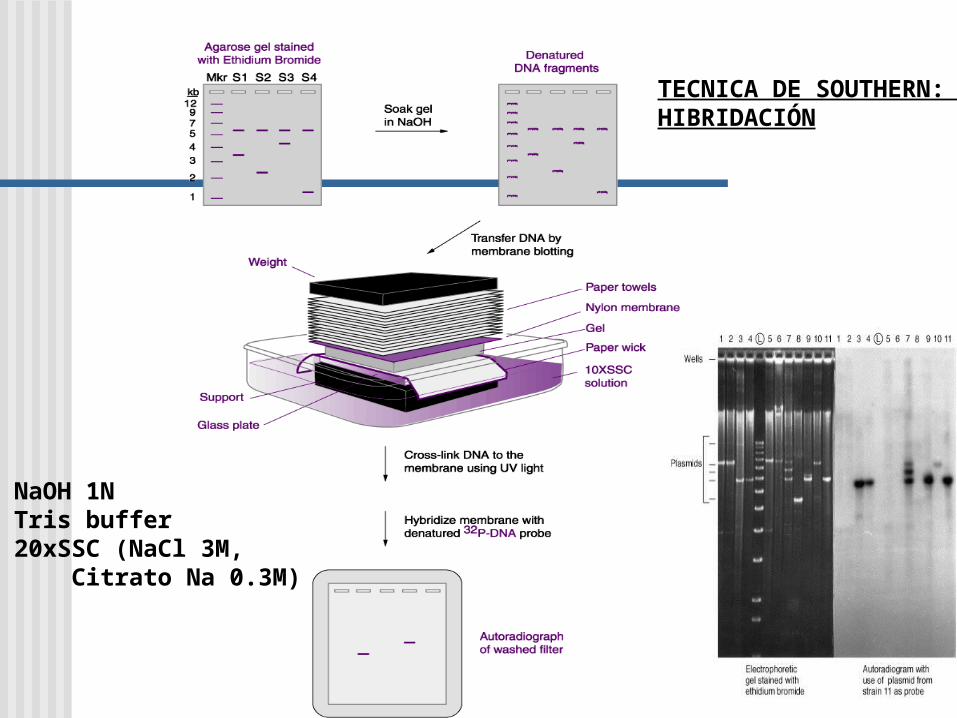
TECNICA DE SOUTHERN: HIBRIDACIÓN
1. NaOH 1N2. Tris buffer3. 20xSSC (NaCl 3M, Citrato Na 0.3M)

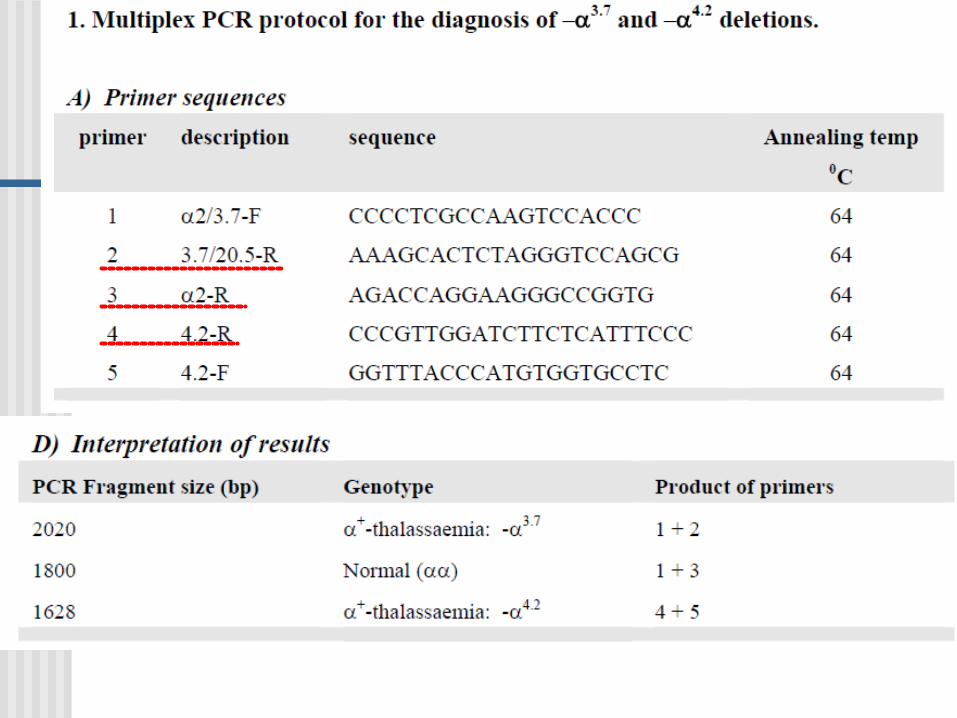
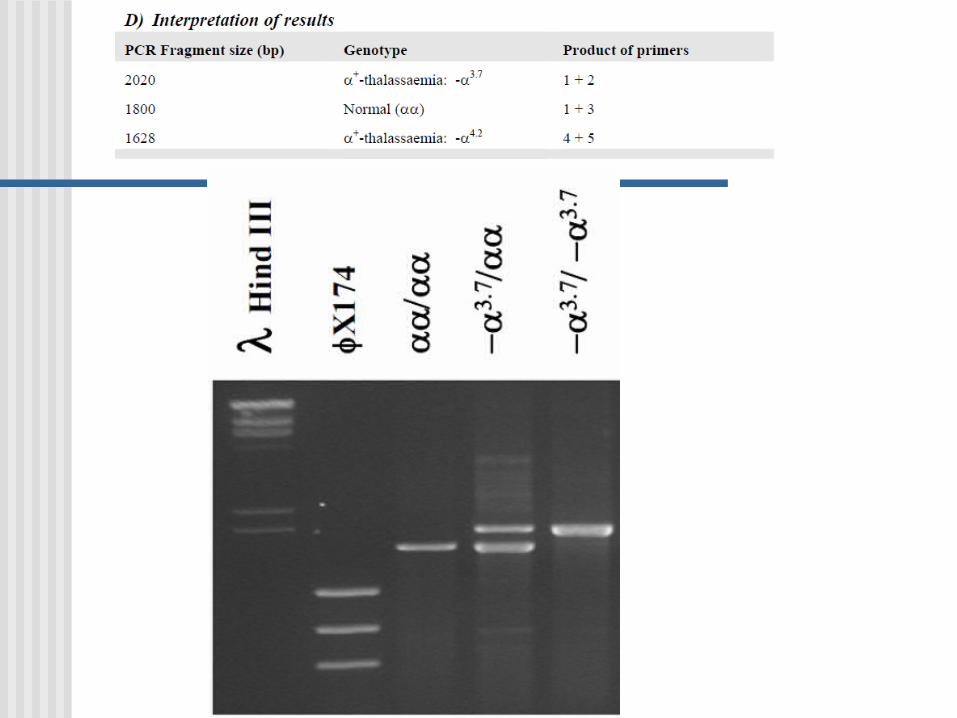
PCR Multiplex
Es una variante de PCR en donde dos o más loci son simultáneamente amplificados en la misma reacción.

Cuidados especiales:1-Los primers de 18-24 pb deben tener un contenido de GC 35-60% de esa forma tiene una T anneling de 55-58ºC o mayor.2-Distribuir los amplicones de distintos tamaños.3-Diseñar primers con características similares: sin estructura secundaria ni interacciones entre ellos.4-Se debe calcular el Tm (no muy diferentes entre primers) y analizar las interacciones primer-primer).
PCR Multiplex

Cuidados especiales II:
5- Se deberá optimizar: Tº extensión, Tiempo de extensión, Tiempo y temperatura de anneling, número de ciclos, cantidad de primers, concentración de dNTP y MgCl2, uso de adyuvantes (glicerol, DMSO, BSA), etc.
6- Para testear posible secuencias repetidas se puede usar banco de datos del National Center for Biotechnology Information (NCBI) usando Basic Local Alignment Search Tool (BLAST)
PCR Multiplex

Gap- PCR
Variante de una PCR-multiplex
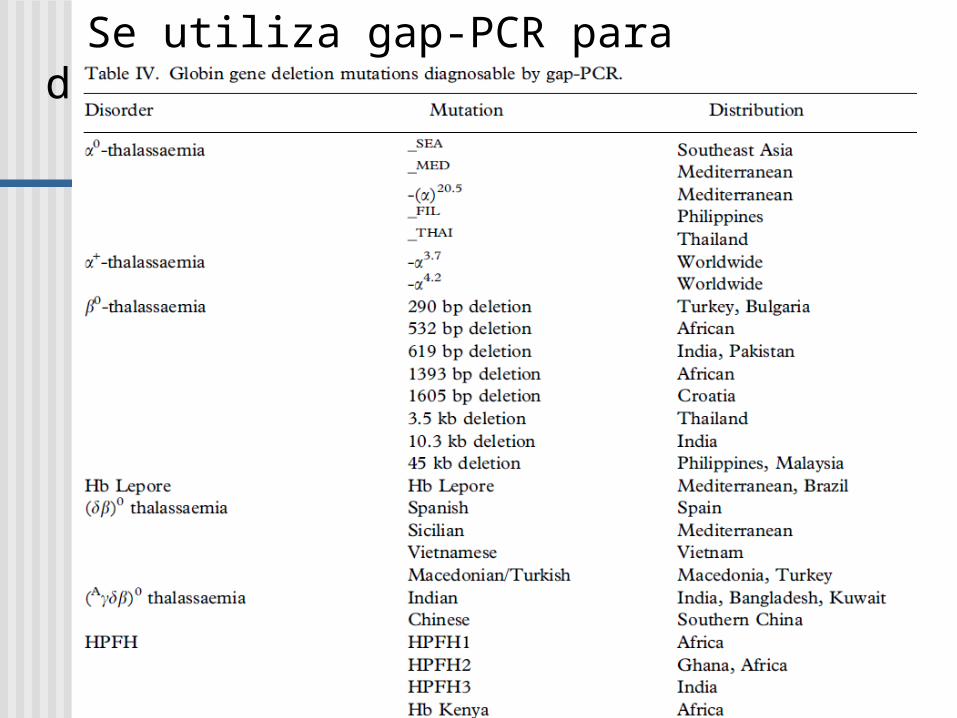
Se utiliza gap-PCR para diagnóstico de:

GAP-PCR Una gran deleción reune a los primers AC y se
produce la amplificación
Usada en la identificación de deleciones en el gen de - globina.
A
B C
C
Normal: A and B da un producto; A y C están demasiados apartados no hay producto PCR
Deleción: A y C ahora dan un producto, el cual es diferente en tamaño al producto A y B.
A

- Talasemias:
Amplificación de Sondas
dependiente de Ligamiento
Múltiple (MLPA)

Amplificación de Sondas dependiente de Ligamiento Múltiple
• Uso Primario:– Detección múltiple de cambios pequeños números de copias de
secuencias ADN/RNA, por ejemplo deleciones/duplicación de exones de un gen
• Fue descrito por primera vez en 2002: Schouten JP et al. Nucleic Acids Res.
• Mas de 750 artículos publicados con diferentes usos de MLPA• Mas de 1.000.000 reaccciones de MLPA son llevadas a cado cada
año• Se aplica a estudios en– genética humana– citogenética– investigación en cáncer

Instrumentos
Un termociclador y un sistema de electroforesis tipo de secuencia son requeridos para MLPA
Los resultados son obtenidos en 24 hs. Mas de 96 Ms pueden ser procesados en un experimento
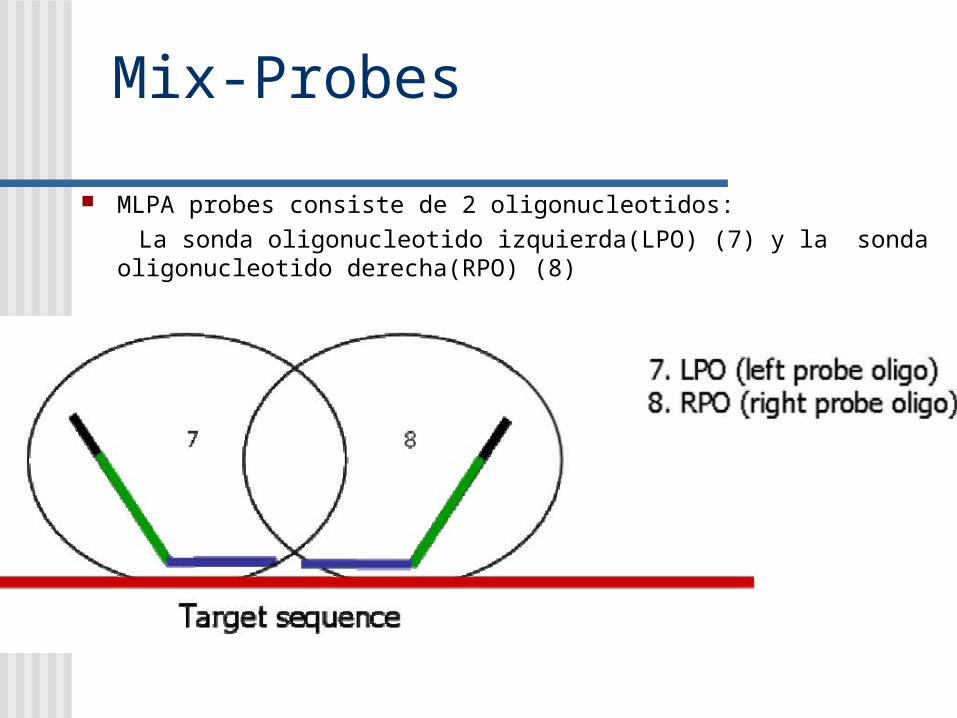
Mix-Probes
MLPA probes consiste de 2 oligonucleotidos: La sonda oligonucleotido izquierda(LPO) (7) y la sonda oligonucleotido
derecha(RPO) (8)

Cada oligo a su vez contiene 2 partes: El LPO (OI) contiene en su extremo 5’ el F y en su extremo 3’La secuencia de la sonda de hibridización izquierda (LHS). El RPO (OD) contiene en su extremo 5’ la sonda de
hibridización derecha (RHS) y el primer R en su extremo 3’
Secuencia relleno (de longitud variable)


Protocolo típico de MLPA

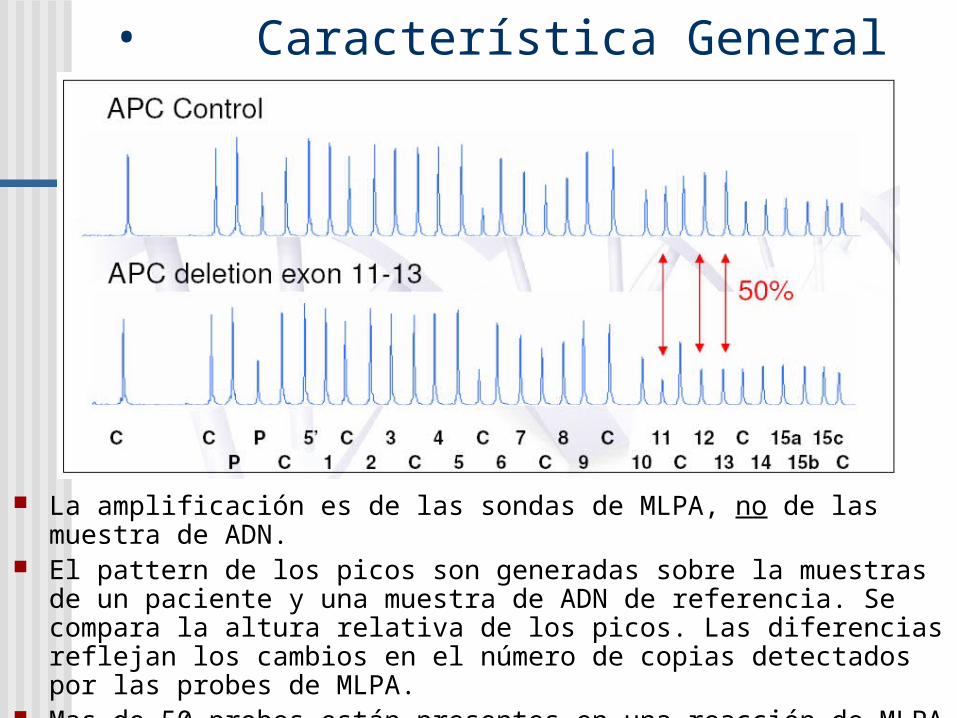
• Característica General
La amplificación es de las sondas de MLPA, no de las muestra de ADN.
El pattern de los picos son generadas sobre la muestras de un paciente y una muestra de ADN de referencia. Se compara la altura relativa de los picos. Las diferencias reflejan los cambios en el número de copias detectados por las probes de MLPA.
Mas de 50 probes están presentes en una reacción de MLPA
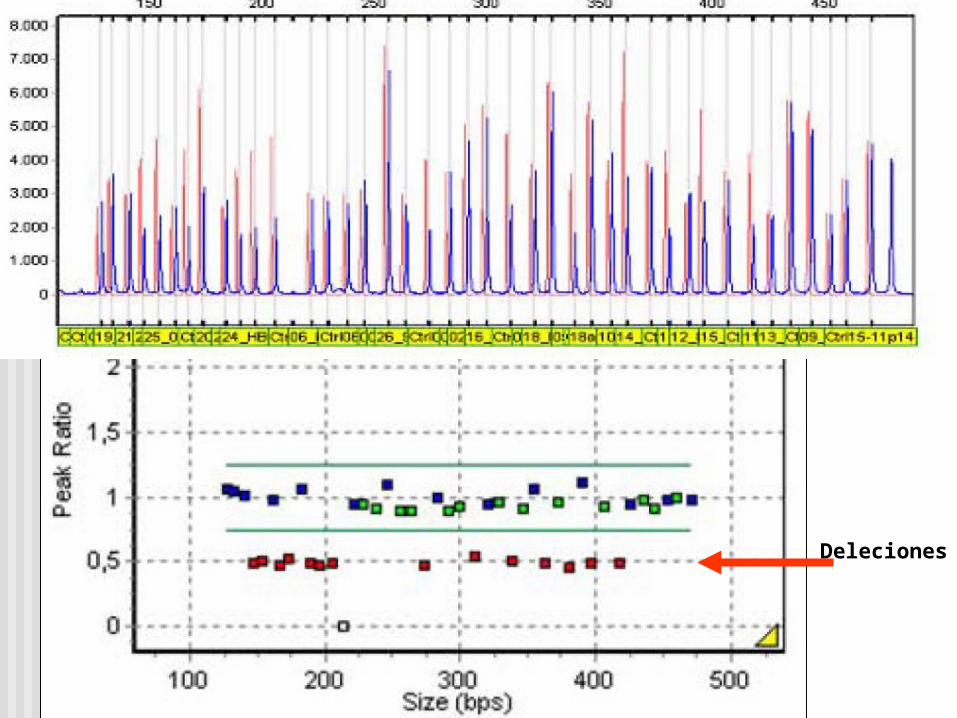
Deleciones

MLP versus otras técnicas Secuenciamiento, DHPLC, SSCE y otras técnicas
para la detección de mutaciones no pueden detectar cambios en el numero de copias de exones completos
FISH no puede detectar pequeñas mutaciones Southern Blot son extremadamente laboriosos PCR en tiempo real es menos sensible a
pequeños cambios en el numero de copias y no es una técnica de amplificación múltiple

VENTAJAS de MLPA:
Detección de número de copias de 40-50 secuencias de ADN genómicos en una simple reacción, basada en PCR
Requiere únicamente 20 ng de ADN humano (3.000 células/0,5 ml de fluido amniótico).
Discrimina secuencias que difieren en SOLO un nucleótido.
MLPA puede ser usado sobre Ms de ADN parcialmente degradadas tales como ADN extraído de tacos de tejidos parafinados.

PROBLEMAS de MLPA:
MLPA es mas sensible a las impurezas en la muestras que una PCR ordinaria.
Disminución de la señal de una sonda puede ser debido a un SNP/polimorfis_ mo raro. Deleciones detectadas por una única sonda requiere confirmación independiente





Bibliografía general: LEHNINGER ALBERT L., COX MICHAEL M., NELSON DAVID L. “Principios de Bioquímica”. Editorial OMEGA 4º Edición. 2006. Diapositivas en Power Point (formato ppt) (Manual para docentes)
LEHNINGER ALBERT L., COX MICHAEL M., NELSON DAVID L. “Principios de Bioquímica” Web: http://bcs.whfreeman.com/lehninger/ The Journal of Biological Chemistry: Classic Articles Web: www.jbc.org
VOET DONALD, VOET JUDITH G. “Bioquímica” Editorial MÉDICA PANAMERICANA.
3º Edición, en Español. 2006
WATSON, BAKER, BELL, GANN, LEVINE, LOSICK “Biología Molecular del Gen”Editorial MÉDICA PANAMERICANA. 5º Edición, en Español. 2006
STRYER LUBERT, BERG JEREMY M., TYMOCZKO JOHN L.”Bioquímica”Editorial REVERTE. Edición 5º Edición, en Español. 2003
MATHEWS CHRISTOPHER K., AHERN KEVIN G., VAN HOLDE K. E.”Bioquímica” Editorial PEARSON EDUCACION. 3º Edición en Español. 2003
Diapositivas (formato ppt), Problemas (Manual para docentes) y Casos Clínicos de Aplicación
VOET DONALD, VOET JUDITH G. and PRATT CHARLOTTE W. “Fundamentals of Biochemistry”Second Edition.Copyright © 2006 by John Wiley & Sons, IncWeb: www. medicapanamericana.com/voet/

Bibliografía especifica: Alvarez SM, Varas SM, Meloni AM, Giménez AI y
Giménez MS. 2000. Incidencia de la beta -talasemia en San Luis, Argentina. Acta Bioquímica Clínica Latinoamericana: 35 (1) 75-82
Alvarez, Silvina M. (1999). Trabajo Final de Tesina en Biología Molecular ‘’ Rastreo de pacientes - talasémicos: estudio bioquímico y molecular. UNSL. Biblioteca Central.
Weatherall DJ y col. Chapter 93: The hemoglobinopathies. Book II. The metabolic and molecular basis of inherited disease. Scriver CR, Beaudet AL, Sly WS & Valle D. 7º Edition. 1995. New York Mc Graw-Hill.
Fundación Argentina de Talasemia "FUNDATAL" http://www.fundatal.org.ar Ronald J A Trent. Diagnosis of the Haemoglobinopathies. Clin. Biochem. Rev. Vol 27 February 2006

Bibliografía especifica: Charles R. Scriver, Arthur L. Beaudet, William
S. Sly and David Valle: THE METABOLIC AND MOLECULAR BASES OF INHERITED DISEASE. Volume I,II and III. Seventh Edition.Mc Graw-Hill Editors
Trabajos publicados en revistas de la especialidad.
BLOG: http://qbpatologica.wordpress.com/