prácticas de ingeniería genética - bbm1.ucm.es · ciones debemos conocer como funciona el...
TRANSCRIPT
Prácticas de Ingeniería genética
AG C TT
T
ATCG AAATCA
T
AA
AGT
T
T
GA
GACCTTG
AGA
CT
CT
5’
5’3’
3’
AG C TT
T
ATCG AAATCA
T
AA
AGT
T
T
GA
GACCTTG
AGA
CT
CT
5’
5’3’
3’
lacZα Guión de alumnos (2012-2013)
malE http://www.bbm1.ucm.es/public_html/public_html//teach/biol/web05/index.html


Pág Cuadro de tareas 0 Introducción 1 Experimentos 5
Esquemas generales 5 Esquema diario 9
Día 1 9
Día 2 10 Día 3 11 Día 4 13 Día 5 14 Protocolos 16 PCR 16 PCR comprobación de orientación 17 Transformación 18 Digestiones 19 Inducción con IPTG y Lisados 20
Electroforesis / Western Blot 21 Cuestiones 24 Apéndice 26 El método “quick change” 26 Diseño de Oligos 27
Código genético 28 Secuencias de oligos para PCRs 28 Mapas y secuencias de pMalc2x y pUCTcKm 29

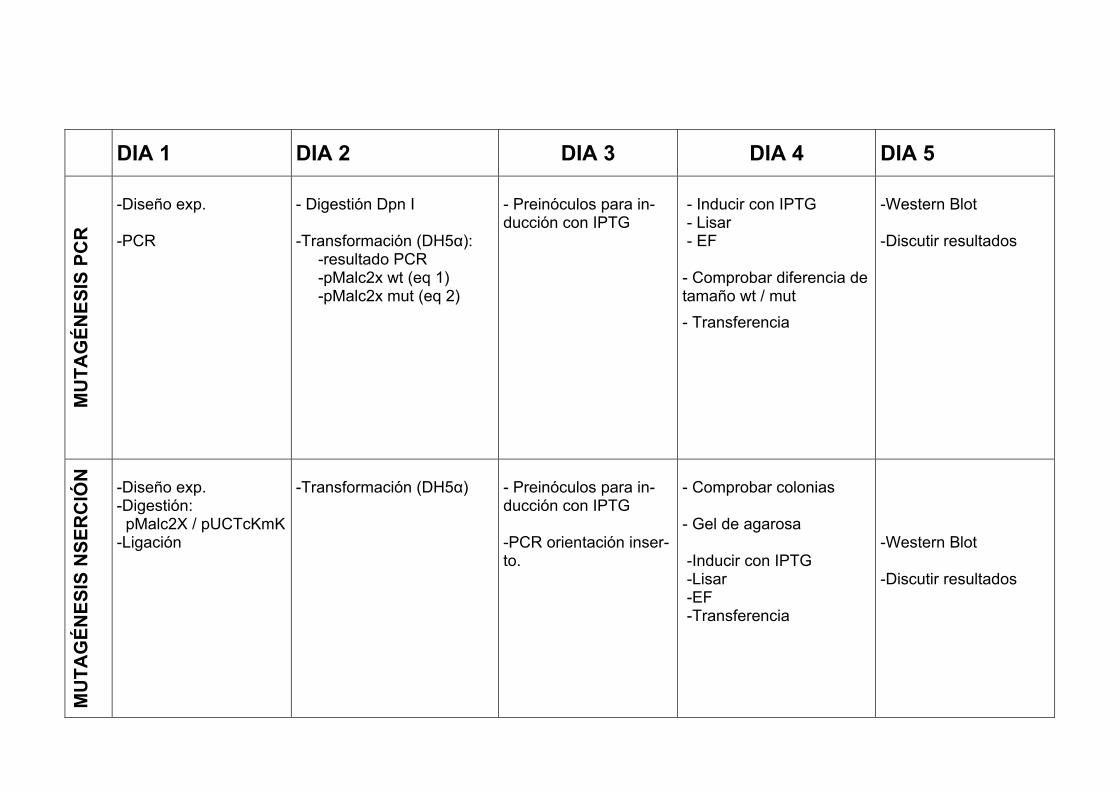
DIA 1 DIA 2 DIA 3 DIA 4 DIA 5
MU
TAG
ÉNES
IS P
CR
-Diseño exp. -PCR
- Digestión Dpn I -Transformación (DH5α): -resultado PCR -pMalc2x wt (eq 1) -pMalc2x mut (eq 2)
- Preinóculos para in-ducción con IPTG
- Inducir con IPTG - Lisar - EF - Comprobar diferencia de tamaño wt / mut - Transferencia
-Western Blot -Discutir resultados
MU
TAG
ÉNES
IS N
SER
CIÓ
N
-Diseño exp. -Digestión: pMalc2X / pUCTcKmK -Ligación
-Transformación (DH5α)
- Preinóculos para in-ducción con IPTG -PCR orientación inser-to.
- Comprobar colonias - Gel de agarosa -Inducir con IPTG -Lisar -EF -Transferencia
-Western Blot -Discutir resultados


Introducción
1
INTRODUCCIÓN El siguiente guión pretende guiaros a través de estas prácticas de Ingeniería
Genética en las que utilizaréis algunas de las técnicas más habituales de Biología Molecular. Aquí encontraréis un esquema de los experimentos a realizar y una descripción detallada de los protocolos para poder llevarlos a cabo. Además, se proporciona información complementaria en el apéndice que será de utilidad a la hora de comprender el fundamento de los experimentos. Por otra parte, hemos pre-parado una serie de cuestiones a las que deberéis responder y que nos permitirán evaluar vuestro grado de comprensión de estas prácticas.
El objetivo de las prácticas es alterar la actividad de una proteína codificada
en el plásmido pMalc2x. Para ello se realizarán mutagénesis de dicho plásmido por diferentes métodos.
Antes de diseñar las estrategias adecuadas para producir esas muta-
ciones debemos conocer como funciona el sistema donde vamos a llevarlas a cabo.
El sistema pMal/DH5α
Hoy en día se dispone de un amplio rango de bacterias modificadas genéti-camente. Muchas de esas modificaciones se han diseñado para facilitar la utiliza-ción de cada determinada estirpe en diferentes experimentos tales como: clonación de vectores, selección de esos vectores clonados o expresión eficaz de proteínas en diversas condiciones. Así, casi todas las estirpes utilizadas en el laboratorio se encuentran perfectamente caracterizadas conociéndose con exactitud las modifica-ciones que portan en su material genético. (Se puede acceder a las características de las diferentes estirpes de Escherichia coli en distintas paginas de “internet” por ejemplo: http://cgsc.biology.yale.edu)
Una de esas estirpes de E. coli es DH5α que, entre otras modificaciones que
pueden resultar interesantes, contiene la mutación lacZ∆M15. Esa mutación consiste en la eliminación de una zona del gen lacZ de manera que la β-galactosidasa que producen esas células carece de los aminoácidos 11 a 41 y no es funcional, dado que no puede interaccionar con las otras subunidades de la enzima y formar el tetrámero que es el que posee actividad enzimática. Como consecuencia, la estirpe DH5α no puede metabolizar lactosa y por tanto (lo que es más interesante desde el punto de vista de estas prácticas) no es capaz de transformar el X-Gal en el corres-pondiente derivado coloreado. En definitiva, cuando se plaquea DH5α en LB + X-Gal se obtienen colonias blancas (no coloreadas).
Aprovechando esta característica de esa y otras muchas estirpes de E. coli,
se han diseñado una serie de plásmidos que permiten α complementación, es decir, portan un fragmento del gen lacZ (también llamado lacZα) de manera que cuando una célula de la estirpe DH5α ha sido transformada con uno de esos vectores, la expresión del péptido LacZα codificado en el plásmido es capaz de restablecer la capacidad de dicha célula para metabolizar lactosa (y por tanto, X-Gal).

Introducción
2
En estas prácticas vamos a utilizar el vector pMalc2x (podréis encontrar el acceso a su secuencia completa en la página web de las prácticas)
El vector pMalc2x permite llevar a cabo α-complementación puesto que con-tiene el gen malE-lacZα. La expresión de este gen produce una proteína de fusión que resulta funcional y que lleva en su extremo N terminal la proteína de unión a maltosa (MBP) y en su extremo C terminal el péptido LacZα. Además, este gen se encuentra bajo el control del promotor Ptac, un promotor muy fuerte e inducible por IPTG. Para completar la funcionalidad del sistema, el plásmido pMalc2x lleva tam-bién el gen lac Iq que codifica el represor Lac I.
Mutagénesis
Nuestro primer objetivo será diseñar un experimento que nos permita eliminar la funcionalidad de la proteína MBP-LacZ mediante mutagénesis. En concreto, de las diferentes estrategias disponibles, a lo largo de estas prácticas emplearemos dos aproximaciones experimentales para alterar la secuencia de DNA que codifica la función de nuestra proteína:
A) Mutagénesis dirigida (que nos permitirá introducir un cambio puntual en la secuencia de MBP-LacZα)
B) Interrupción del gen mediante la introducción de una secuencia de DNA en el interior de dicho gen.

Introducción
3
A) Mutagénesis dirigida mediante PCR
1. En primer lugar deberemos determinar cual es la posición de la se-cuencia más adecuada para llevar a cabo esa mutagénesis. Para ello podéis con-sultar la secuencia del plásmido pMalc2x y también las estructuras tridimensionales de las proteínas MBP y β-galactosidasa (que se puede encontrar en el apéndice de este guión y en la página web de las prácticas: http://www.bbm1.ucm.es/teach/biol/web05/index.html).
2. A continuación habrá que diseñar los oligonucleótidos necesarios para poder llevar a cabo ese cambio mediante el método “quick change” (ver apén-dice para más información –allí podrás encontrar también una descripción de las características que deben cumplir esos oligonucleótidos).
3. Una vez seleccionado el codón a mutagenizar y obtenidos los oligonu-cleótidos ya podemos llevar a cabo la reacción de mutagénesis mediante PCR y obtener bacterias que lleven el plásmido mutante (ver protocolos).
4. Finalmente comprobaremos que efectivamente hemos conseguido la mutación esperada en nuestro plásmido comprobando que, una vez expresada, la proteína ha incorporado el cambio y que por tanto no es capaz de α-complementar.
B) Mutagénesis por inserción (knock out)
En el caso de la mutagénesis por inserción procederemos a introducir una secuencia de DNA [la de un casete que codifica un gen de resistencia a kanamicina (Km)] dentro del gen malE-lacZα con el fin de interrumpir su expresión e impedir así la traducción de una proteína capaz de α-complementar.
1. En primer lugar deberemos determinar cual es el sitio de la secuen-cia más adecuado para llevar a cabo esa inserción. Para ello podéis consultar la secuencia del plásmido pMalc2x y también la del casete de Km (que se puede en-contrar en el apéndice de este guión y en la página web de las prácticas: http://www.bbm1.ucm.es/teach/biol/web05/index.html).
2. A continuación habrá que digerir con enzimas de restricción tanto el plásmido pMalc2x como el plásmido portador del casete de Km para poder ligar el casete dentro de la región elegida de pMalc2x.
3. Una vez conseguidos los plásmidos mutantes habrá que comprobar si su introducción dentro de bacterias DH5α lleva a la obtención de células incapaces de llevar cabo α-complementación.
4. Finalmente comprobaremos que el plásmido lleva un inserto del tama-ño esperado mediante electroforesis y comprobaremos la orientación del mismo mediante PCR.

Introducción
4
A lo largo de las siguientes secciones encontraréis una descripción detallada de los pasos a seguir para llevar a cabo estos experimentos. En cualquier caso, es importante que, además del desarrollo experimental, comprendáis la base de los experimentos y el manejo de las secuencias de DNA y los programas que utilizare-mos (todo ello lo podréis encontrar tanto en el apéndice de este guión como en la página web de las prácticas: http://www.bbm1.ucm.es/public_html/public_html//teach/biol/web05/index.html
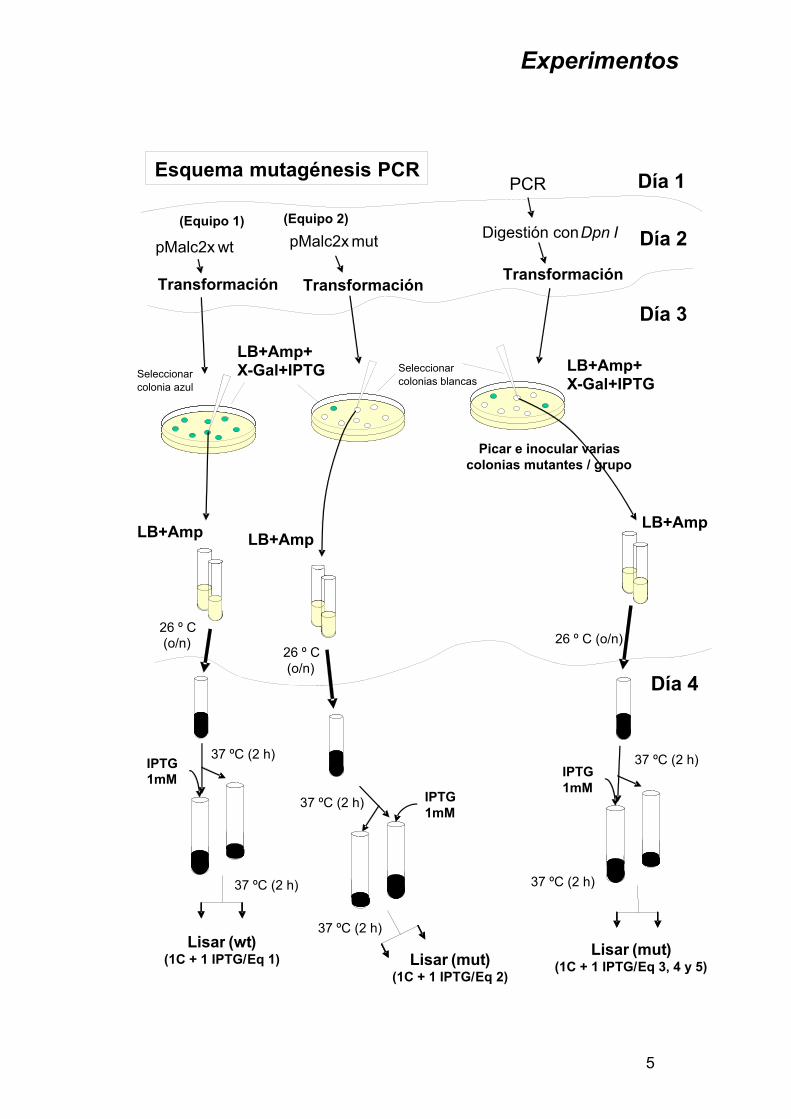
Experimentos
5
LB+Amp+X -Gal+IPTG LB+Amp+
X -Gal+IPTG
pMalc2x wt
Picar e inocular varias colonias mutantes / grupo
Transformación
(Equipo 1)
26 º C (o/n)
IPTG 1mM
37 ºC (2 h)
37 ºC (2 h)
Lisar (mut)(1C + 1 IPTG/Eq 2)
Seleccionar colonias blancas
37 ºC (2 h)
37 ºC (2 h)
Lisar (wt)(1C + 1 IPTG/ Eq 1)
Esquema mutagénesis PCR Día 1
Día 2
Día 3
26 º C (o/n) 26 º C (o/n)
Día 4
Seleccionar colonia azul
LB+Amp
pMalc2x mut
PCR
Digestión con Dpn I
Transformación
Lisar (mut)(1C + 1 IPTG/ Eq 3, 4 y 5)
Transformación
IPTG 1mM
IPTG 1mM
37 ºC (2 h)
37 ºC (2 h)
LB+Amp LB+Amp
(Equipo 2)

Experimentos
6
2 col / equipo
LB+Amp+KmXGal+IPTG
pMalc2x wt
Transformación
Día 1
Día 2
Día 3
pMalc2x
Transformación
pUCTcKm
Digerir con Pst I Digerir con Pst I
Inactivar PstI
16 ºC (o/n)
pMalc2xKm Or1Transformación
Día 4
Esquema mutagénesis por inserción
Mezclar y añadir ligasa
26 º C (o/n)
IPTG
26 º C (o/n)
IPTG
Lisados Lisados
LB+Amp+Km
LB+Amp+Km
Común con experimento mutagénesis PCR
pMalc2x Km Or2
Transformación
LB+Amp+KmXGal+IPTG
LB+Amp +Km
26 º C(o/n)
IPTG
Lisados
(Equipo 3) (Equipo 4)
Verificar orientaciónen las colonias
con inserto (PCR de colonia)

Experimentos
7
Preparar muestras y aplicar en
Día 4 (cont)
Día 5
Correr electroforesis (1 hora /100 Vol)
Preparar muestras
Lisados (Inserción)
Aplicar muestras en un gel
Gel
Cada gel debe llevar lisados de-Control (-/+ IPTG)-Control mutante PCR (+ IPTG) -Control mutante Ins Or 1(+ IPTG) -Control mutante Ins Or 2(+ IPTG) -Mutantes PCR e Ins (-/+ IPTG) -DH5α
Teñir gel y analizar resultado
Bloquear membrana
Incubar con anticuerpo 1º
Incubar con anticuerpo 2º
lavar
Transferir a membrana
Revelar y analizar resultado
Lisados(PCR)
Verificar orientaciónen las colonias
con inserto (PCR de colonia)
Verificar 3 col/grupoLos controles de
pMalc2Km Orientación 1 y
Orientación 2 comunes para todo el laboratorio
gelde agarosa

Experimentos
8

Experimentos / Día 1
9
MUTAGÉNESIS PCR / Día 1 1. Mezclar los componentes de la reacción e iniciar la PCR (ver protocolo
PCR/mutagénesis de la página 16) (una reacción por equipo) MUTAGÉNESIS POR INSERCIÓN / Día 1
1. En primer lugar se llevarán a cabo las digestiones de los plásmidos pMalc2x y pUCTcKm (ver protocolo digestiones en la página 19).
-pMalc2x con Pst I (apertura del vector) -pUCTcKm con Pst I (obtención del inserto)
Componente volumen Componente volumen pMalc2x 5 μl pUCTcKm 10 μl Tampón 10x 2 μl Tampón 10x 2 μl PstI 1 μl PstI 1 μl H2O 12 μl H2O 7 μl Volumen final 20 μl Volumen final 20 μl
2. Una vez añadidas las enzimas, las reacciones se incubarán durante 45-60 min a 37 ºC
3. Finalizada la reacción se procederá a inactivar las enzimas incubando
los tubos durante otros 20 min a 60ºC o en el termobloque 10 min.
4. Finalmente se ligará mezclando vector e inserto
Componente volumenVector (pMalc2x / Pst I) 6 μl Inserto (pUCTcKm / Pst I ) 15 μl Tampón 10 x ligasa 2,5 μl Ligasa 1,5 μl
Volumen final 25 μl
5. Se le da un pulso con la microfuga a la mezcla y se incuba a temperatu-
ra ambiente durante toda la noche.

Experimentos / Día 2
10
MUTAGÉNESIS PCR / Día 2 1. Una vez terminada la reacción de PCR, sacar los tubos del termociclador,
añadir 1μl de Dpn I a cada tubo e incubar durante 2 h a 37 ºC (ver protocolo de mutagénesis en la página 16) (una reacción por equipo)
2. Una vez finalizada la digestión con Dpn I se transformarán células DH5α
competentes (ver protocolo de transformación en la página 19). Para ello se emplearán:
- 20 μl de la reacción de PCR digerida con Dpn I (una trans-
formación por equipo)
Además, para llevar en paralelo controles de los plásmidos wt y mutado los equipos 1 y 2 de cada grupo utilizarán:
- 1 μl del plásmido control (pMalc2x) (Equipo 1) - 1 μl del plásmido mutado (pMalc2x mut) (Equipo 2)
para transformar células competentes (DH5α)
3. Por último se sembrarán las células en placas de LB+Amp+X-Gal+IPTG MUTAGÉNESIS POR INSERCIÓN / Día 2 1. Las reacciones de ligación serán empleadas para transformar células com-
petentes. Para ello cada equipo utilizará:
- 10 μl de la mezcla de ligación (una transformación por equipo)
Además, para llevar en paralelo controles de los plásmidos mutados los equipos 3 y 4 de cada grupo utilizarán:
-1 μl del plásmido mutado por inserción (pMalc2x Km Or 1) (Equipo 3) -1 μl del plásmido mutado por inserción (pMalc2x Km Or 2) (Equipo 4)
para transformar células competentes (DH5 α).
2. Finalmente las células que hayan sido transformadas con el plásmido con-
trol (equipo 1) se sembrarán en placas de LB+Amp+X-Gal+IPTG mientras que aquellas células que hayan sido transformadas con el plásmido pMalc2x Km Or 1 u Or2 (equipos 3 y 4) o con el resultado de la ligación (to-dos los equipos) se sembrarán en placas de LB+Amp+Km+X-Gal+IPTG.

Experimentos / Día 3
11
MUTAGÉNESIS PCR / Día 3 1. A partir de las colonias obtenidas tras la transformación de células DH5α
con el producto de la PCR (plásmido mutado), cada equipo tomará con un palillo 1 de esas colonias (de entre las que sean de color blanco, mu-tantes), la introducirá en un eppendorff con 50 µl de agua destilada y la resuspenderá cuidadosamente. A continuación se emplearán 20µl/tubo para inocular 2 tubos Falcon con 1,5 ml de LB + Amp (los tubos se cre-cerán a 26ºC o/n para posterior inducción con IPTG y lisado).
2. A partir de las colonias obtenidas tras la transformación con el plásmido control [pMalc2x (wt)], el equipo 1 tomará con un palillo 1 colonia que sea azul, la introducirá en un eppendorff con 50 µl de agua destilada y la resuspenderá cuidadosamente. A continuación se emplearán 20µl/tubo para inocular 2 tubos Falcon con 1,5 ml de LB + Amp (crecimiento a 26ºC o/n).
3. Por último a partir de las colonias obtenidas tras la transformación con el plásmido mutado [pMalc2x mut], el equipo 2 tomará con un palillo 1 co-lonia que sea de color blanco, la introducirá en un eppendorff con 50 µl de agua destilada y la resuspenderá cuidadosamente. A continuación se emplearán 20µl/tubo para inocular 2 tubos Falcon con 1,5 ml de LB + Amp (crecimiento a 26ºC o/n).
MUTAGÉNESIS POR INSERCIÓN / Día 3 1. A partir de las colonias obtenidas tras la transformación de células DH5α
con el producto de la ligación (plásmido mutado Km), cada equipo to-mará con un palillo 1 de esas colonias (de entre las que sean de color blanco), la introducirá en un eppendorff con 50 µl de agua destilada y la resuspenderá cuidadosamente. A continuación se emplearán 20µl/tubo para inocular 2 tubos Falcon con 1,5 ml de LB + Amp + Km (los tubos se crecerán a 26ºC o/n para posterior inducción con IPTG y lisado). .
2. Por último, a partir de las colonias obtenidas tras la transformación con el plásmido mutado [pMalc2x Km Or1 y Or2], los equipos 3 (Or1) y 4 (Or2) tomarán con un palillo 1 colonia que sea de color blanco de cada una de las orientaciones, la introducirá en un eppendorff con 50 µl de agua destilada y la resuspenderá cuidadosamente. A continuación se emple-arán 20µl/tubo para inocular 2 tubos Falcon con 1,5 ml de LB + Amp + Km (los tubos se crecerá a 26ºC o/n para posterior inducción con IPTG y lisa-do).

Experimentos / Día 3
12
3. Comprobaremos además la orientación de la inserción mediante PCR de
colonias. Para ello, emplearemos el volumen restante tras la resuspensión de las colonias seleccionadas y la inoculación en tubos Falcon realizada en los puntos 1 y 2, y seguiremos el protocolo de la página 17.
a. Para los cultivos provenientes de bacterias transformadas con el
resultado de la mutagénesis por inserción (mutantes) que sean defectivos en actividad β-galactosidasa (colonias blancas).
b. Para los cultivos provenientes de bacterias transformadas con los
controles del plásmido mutante pMalc2x Km Or1 y Or2 (mutan-tes), que también sean defectivos en actividad β-galactosidasa (colonias blancas).

Experimentos / Día 4
13
MUTAGÉNESIS PCR / Día 4 1. 2 horas antes de realizar el lisado de los cultivos seleccionados, se lle-
vará a cabo la inducción con IPTG, y se aplicarán las muestras en los ge-les de electroforesis (ver protocolos en la página 20).
2. Se hará una transferencia de los geles a una membrana de nitrocelulosa
para realizar posteriormente un Western blot de la proteína expresada.
MUTAGÉNESIS POR INSERCIÓN / Día 4
1. 2 horas antes de realizar el lisado de los cultivos seleccionados, se lle-vará a cabo la inducción con IPTG, y se aplicarán las muestras en los geles de electroforesis (ver protocolos en la página 20) utilizando:
- 2 cultivos de entre los generados a partir de células transfor-
madas con el resultado de la ligación (plásmidos mutantes)
- 2 cultivos de entre los generados a partir de células transfor-madas con el plásmido pMalc2x Km (uno con Or1 y otro con Or2)
2. Las muestras se cargarán en 1 gel de electroforesis que se correrá du-
rante 1h a 150-200 V. Transcurrido ese tiempo, se transferirá a una membrana de nitrocelulosa para posterior Western blot.
3. Con objeto de visualizar la orientación del inserto en las muestras de
DNA amplificadas por PCR de colonia en el día 3, se mezclarán 10 μl del resultado de la amplificación con 2,5 μl del tampón de carga 5x.
Se aplicarán 10 µl de cada una de las muestras en un gel de agarosa al 1,5 % y se correrán durante 45-60 min a 100 V.
Finalmente los geles se teñirán durante 10 min en una solución de Bro-muro de etidio y se analizarán los resultados.

Experimentos / Día 5
14
MUTAGÉNESIS PCR / Día 5
1. Tras el bloqueo de la membrana con leche al 5% se incubará con el anticuerpo primario (anti-MBP) (ver protocolo de Western Blot en página 23).
2. Después de los correspondientes lavados se incubará con el anti-
cuerpo secundario conjugado con peroxidasa.
3. Finalmente se obtendrá el correspondiente luminograma mediante la utilización de una solución conteniendo agua oxigenada (que permita revelar la presencia de peroxidasa) y una película fotográfica (ver protocolo).

15

Protocolos
16
MUTAGÉNESIS / PCR 1. Para llevar a cabo la mutagénesis por el método “quick change” (se descri-
be en la página 26 del guión) en la zona elegida del plásmido pMac2x se mezclarán en un tubo de PCR los siguientes componentes en el orden indi-cado:
[stock] Reactivo [final] Volúmen
- H2O destilada - 22 μl
5 x Tampón Polimerasa 1 x 10 μl
2.5 mM dNTPs 200 μM 4 μl
5 μM Oligo Mut-F 0.5 μM 5 μl
5 μM Oligo Mut-R 0.5 μM 5 μl
50 mM MgCl2 1.5 mM 1.5 μl
10 ng/μl DNA molde (pMalc2x) 10-20 ng 2 μl (1/20)
2 u/μl Polimerasa “Phusion” 1 u 0.5 μl 2. A continuación, se colocarán los tubos en los correspondientes orificios del
termociclador y se programará y ejecutará una secuencia de PCR tal y co-mo se describe en el cuadro adjunto.
3. Una vez finalizada la reacción (día 2) añadir 1 μl de la enzima Dpn I (20 uni-
dades) a cada reacción de PCR e incubar a 37 ºC durante dos horas 4. Transformar células supercompetentes E. coli de la cepa DH5α con el resul-
tado de la reacción.
98 ºC72 ºC
69 ºC
10 ºC
3’
x25hold
20’’
30’’
3’ 30”98 ºC72 ºC
69 ºC
10 ºC
3’
x25hold
20’’
30’’
3’ 30”98 ºC72 ºC
69 ºC
10 ºC
3’
x25hold
20’’
30’’
3’ 30”
3’ 98 ºC20” 98 ºC30” 69 ºC3’30” 72 ºCHold 4 ºC

Protocolos
17
COMPROBACIÓN DE ORIENTACIÓN / PCR Obtención de DNA de colonia 1. Se toma con un asa de siembra o con un palillo una colonia (o un poco de la estría seleccionada), se introduce en un eppendorff con 50 µl de agua destilada y se resuspende. 2. Se calienta la muestra a 95ºC durante 5’ en el baño de bloques. 3. Se enfría 1-2 min en hielo agitando después en el vortex 4. Centrifugar en microfuga a 14000 rpm, 5 min. 5. Finalmente se tomarán 4 μl del sobrenadante para realizar la PCR Protocolo PCR 1. Para comprobar la orientación del inserto dentro del plásmido pMalc2X se
mezclarán en un tubo de PCR los siguientes componentes en el orden indi-cado:
[stock] Reactivo [final] Volúmen
- H2O destilada - 26 μl
10 x Tampón Polimerasa 1 x 5 μl
2.5 mM dNTPs 200 μM 4 μl
5 μM Oligo MalEF 0,5 μM 5 μl
5 μM Oligo KM2 o KM2R 0,5 μM 5 μl
- Sobrenadante - 4 μl
5 u/μl Taq Polimerasa - 1 μl 2. A continuación, se colocarán los tubos en los correspondientes orificios del
termociclador y se programará y ejecutará una secuencia de PCR tal y co-mo se describe en el cuadro adjunto.
3’ 95 ºC15” 95 ºC30” 68 ºC40” 72 ºCHold 10 ºC
3’ 95 ºC15” 95 ºC30” 68 ºC40” 72 ºCHold 10 ºC
hold
95 ºC72 ºC
60 ºC
10 ºC
x28
15’’95 ºC72 ºC
68 ºC
ºC
15’’
30’’
40’’95 ºC72 ºC
ºC
3’ 15’’
hold
95 ºC72 ºC
60 ºC
10 ºC
x28
15’’95 ºC72 ºC
68 ºC
ºC
15’’
30’’
40’’95 ºC72 ºC
ºC
3’ 15’’

Protocolos
18
TRANSFORMACIÓN 1. El primer paso será descongelar tubos conteniendo alicuotas de 200 μl de
células competentes (E. Coli -DH5α) en un barreño con hielo. 2. Una vez que las células competentes estén descongeladas se añadirá 1 μl
(en el caso de una transformación utilizando los plásmidos control wt o con-trol mutantes - plásmidos intactos que no presentan nicks), 20 μl en el caso de los productos de PCR mutados) o 10 μl (en el caso de la mezcla de liga-ción) del correspondiente plásmido y se incubarán las células durante 30 min en hielo.
3. Shock térmico: A continuación se introducirán las células en un baño a 42
ºC durante exactamente 1’30” y a continuación se volverán a incubar en hielo durante 5 min.
4. Periodo de recuperación: Se añadirán 750 μl de LB a cada uno de los tubos
y se incubarán las células durante 45 min - 1 h a 37 ºC con agitación mecánica.
5. Al cabo de ese tiempo se centrifugarán las células durante 1 min a 12.000
rpm en la microfuga. 6. Tras la centrifugación se elimina el sobrenadante por decantación dejando
aproximadamente las células en unos 100-150 μl de LB. 7. Finalmente se plaquean las células en placas de LB/agar con el correspon-
diente antibiótico.

Protocolos
19
DIGESTIONES CON ER Para llevar a cabo las digestiones con endonucleasas de restricción se mez-clarán los siguientes componentes: -Aproximadamente (0.8- 1μg) (unos 8 μl) del correspondiente plásmido -1 μl de la correspondiente endonucleasa de restricción -*X μl de tampón 10 x (**) -H2O hasta completar el volumen deseado * NOTA: Para que las ER sean activas deben actuar en una disolución que contenga < 10% de glicerol. Considerando que las enzimas son suministradas en una disolución de glicerol, es necesario preparar un volumen de reacción al menos 10 veces mayor que el volumen de la enzima que se va a añadir (por ejemplo si se va a añadir 1 μl de enzima se suelen hacer las reacciones en un volumen final de 15 μl) **NOTA: La elección del tampón y la temperatura de reacción son esenciales para conseguir la correcta funcionalidad de las enzimas de restricción. Por tan-to, antes de llevar a cabo cualquier digestión, hay que elegir el tampón más adecuado para cada enzima (consultar tablas según la marca comercial). En el caso de digestiones dobles hay que comprobar que el tampón utili-zado sea compatible para ambas enzimas. En caso contrario las digestiones deben hacerse de forma secuencial.

Protocolos
20
INDUCCIÓN CON IPTG, LISADOS Y PREPARACIÓN DE MUESTRAS PARA ELECTROFORESIS
Inducción con IPTG Para la inducción con IPTG, se requiere que el cultivo se encuentre creciendo en fase exponencial. 1. Para ello se picarán colonias (utilizando una punta amarilla o un palillo esté-
ril) a partir de las placas de bacterias conteniendo el plásmido pMalc2x wt, mutante o pMalc2x Km y se inocularán 2 tubos Falcon con 1,5 ml de medio de LB+Amp o LB+Amp+Km según corresponda.
2. Los inóculos se dejarán crecer toda la noche a 26 ºC con agitación. 3. A la mañana siguiente se cambiarán a 37 ºC durante una hora (hasta que el
cultivo alcance una densidad óptica de 0.8). 4. A 1 de los tubos del cultivo se le añadirán 1.5 μl de IPTG 1M. 5. Ambos tubos se incubarán otras 2 horas a 37 ºC con agitación. Lisados 6. Después de dos horas de incubación se transferirán 200 μl de cada cultivo a
tubos eppendorf y se centrifugarán durante 1 min a 12.000 rpm. 7. Tras descartar los sobrenadantes se añadirán 100 μl de SBL (1x) Preparación de muestras para electroforesis 8. Finalmente las muestras serán calentadas durante 5 min a 95 ºC 9. Las muestras (10 μl ) serán cargadas en los geles.

Protocolos
21
ELECTROFORESIS (PAGE-SDS), TRANSFERENCIA Y WESTERN BLOT Electroforesis (PAGE-SDS)
1. Las muestras (aproximadamente 10 μl) se aplicarán en geles disconti-nuos de poliacrilamida (gel separador al 10%) (que serán preparados previamente por los técnicos de laboratorio).
2. Los geles cargados se conectarán a la fuente de electroforesis y se co-
rrerán a 150-200 v (aproximadamente 1 h) hasta que el frente de azul de bromofenol esté llegando al final del gel.
3. Los geles de electroforesis se separarán cuidadosamente de los crista-
les de montaje y se colocarán en una bandeja con tampón de transfe-rencia.
Transferencia
1. Se sumergirá el gel durante 15 min en tampón de transferencia 2. En paralelo se prepararán:
-2 almohadillas para transferencia -2 rectángulos (de tamaño algo mayor al del gel a transferir) de papel Whatman 3MM. -1 rectángulo de membrana de PVDF (de tamaño algo mayor que el gel a transferir)
y también se empaparán en tampón de transferencia durante 10-15 min.
3. A continuación se procederá al montaje del sándwich:
- Sobre la parte negra del equipo de transferencia (que corresponderá al polo negativo) se colocarán sucesivamente: una almohadilla, un rectángulo de papel Whatman, el gel de electroforesis, la membrana de nitrocelulosa, el otro rectángulo de papel Whatman y la otra almohadilla (cada paso deberá realizarse asegurando –utilizando para ello una pi-peta pasteur o un tubo de ensayo- que no quedan burbujas y que el contacto entre cada una de las capas superpuestas y la anterior es completo)
- Posteriormente se procederá al cerrado del sándwich y su colocación dentro de la cubeta de transferencia.

Protocolos
22
- A continuación, se llenará la cubeta con tampón de transferencia fresco hasta que cubra completamente las almohadillas conteniendo los ge-les.
4. Para comenzar la transferencia se conectará la cubeta de transferencia a una fuente de electroforesis y se procederá a electrotransferir los geles durante 1,5 h a 100 v (también se pede transferir o/n a 24 v)
5. Una vez terminada la transferencia se sacarán las membranas de los
sandwiches y se teñirán con rojo Ponceau durante 2 min con agitación (La tinción con rojo Ponceau permite localizar la posición de las proteí-nas en la membrana verificando que la transferencia ha tenido lugar).
6. Finalmente se desteñirán las membranas enjuagándolas con agua desti-
lada durante 5-10 min..
Almohadilla
Papel Whatman
Gel
Membrana de nitrocelulosa

Protocolos
23
Western Blot
La técnica de Western blot (WB) permite identificar de manera específica la presencia de proteínas que sean reconocidas por un anticuerpo. Para ello una vez transferidas las proteínas a una membrana de nitrocelulosa:
1. Se incubará la membrana en una cubeta con 10-15 ml de leche desna-
tada al 5 % durante 30-60 min (también se puede incubar o/n a 4º C) con el fin de bloquear la membrana e impedir uniones inespecíficas a la misma.
2. Tras eliminar la leche, se incubará la membrana dutrante 1 h con 10 ml
del anticuerpo primario anti-MBP (dilución 1:4000 + TBS-tween 0.1%- BSA 1%) a temperatura ambiente, con agitación en balancín.
3. A continuación se recogerá el anticuerpo primario y se lavará la mem-
brana con TBS-tween 0.1 % utilizando el siguiente protocolo : -2 lavados rápidos, -1x 10 min -2x 5 min
4. Después se incuba la membrana a temperatura ambiente durante 45
min - 1 h con 10-15 ml del anticuerpo secundario conjugado con peroxi-dasa “anti mouse” (dilución 1:5000 en TBS-tween 0.1%).
5. Tras eliminar el anticuerpo secundario, se lava la membrana con TBS-
tween 0.1%: -2 lavados rápidos -1x 20 min -4x 5 min
6. A continuación hay que proceder a incubar la membrana con la solución
de revelado que contiene H2O2. Para ello: -Se prepara la mezcla añadiendo 4 ml de solución A + 100 μl de H2O2. -Se incuba la membrana durante 1 min en presencia de dicha so-lución -Tras escurrir levemente la membrana se coloca dentro de un re-cipiente de plástico (una bolsa de plástico para termo-sellado o entre dos láminas de acetato para transparencias) y se coloca en una casete para autorradiografía.
7. Finalmente (en la cámara oscura) se procede a exponer la membrana a
una película fotográfica durante 1–5 min y se revela la película en la máquina reveladora.

Cuestiones
24
CUESTIONES SOBRE LAS PRÁCTICAS
1. Comparar el método de mutagénesis por PCR con el utilizado. Cuáles
son las principales diferencias entre los dos métodos?
2. ¿Qué interés puede tener la modificación mediante mutagénesis de una proteína?
3. Localiza la(s) zona(s) de la secuencia del gen más adecuada(s) para
modificar la proteína MBP-LacZα mediante mutagénesis dirigida. Explica las diferentes estrategias que se pueden utilizar para inactivar la proteí-na y razona la respuesta en función del tipo de mutación.
4. Diseña los oligonucleótidos adecuados para poder hacer la mutagénesis
dirigida en el sitio elegido en la cuestión anterior (en un sitio diferente al que se lleva a cabo en el laboratorio) por el método “quick change”.
5. Identifica las regiones a digerir en los plásmidos pMalc2x y pUCTcKm
para poder insertar el casete de Km dentro del gen malE-LacZα.
6. Predice la secuencia de la proteína que se obtendrá tras la introducción del inserto. ¿qué ocurre con la expresión del gen malE-lacZα?
7. Diseña un experimento (incluyendo la secuencia de los oligonucleótidos
necesarios) para determinar la orientación del casete de Km dentro del gen malE-LacZα. Utiliza unos oligonucleótidos diferentes a los utilizados en las prácticas.
8. ¿Cual sería la estrategia a seguir si quisieras que el gen de resistencia a
Km se introdujese dentro del marco de lectura del gen malE-LacZα?. Describe el método a utilizar incluyendo, si procede, el diseño de oligo-nucleótidos, esquematiza el proceso de forma detallada y muestra la se-cuencia de nucleótidos de la región correspondiente del plásmido así como la de aminoácidos de la proteína antes y después. Detalla cada uno de los pasos a seguir.
9. Tras la reacción en el termociclador ¿por qué es necesario digerir con la
enzima Dpn I en el caso de la mutagénesis por el método “quick chan-ge”?
10. ¿Crees que la mutación introducida es la única que puede tener lugar al
llevar a cabo la reacción en el termociclador?¿Cómo podrías comprobar-lo?
11. Haz un esquema que explique como afectan las mutaciones introduci-
das a la capacidad de metabolizar lactosa en nuestro sistema.
12. ¿Se podría comprobar la eficacia de la mutagénesis llevada a cabo en el gen malE-lacZα en cualquier cepa de E. coli?

25

Apéndice
26
El método “quick change” Existen diversos métodos para introducir mutaciones dentro de una determina-da secuencia utilizando la técnica de PCR. Nosotros aquí vamos a utilizar uno de ellos denominado “quick change”. Como su propio nombre indica, este método tiene la ventaja de que una sola reacción de PCR permite introducir la mutación dentro de las dos hebras de DNA del plásmido que se desea mutar. Podréis conocer más acerca de este método en la página http://www.stratagene.com/products/showProduct.aspx?pid=131 Este procedimiento se basa en la utilización de: dos oligonucleótidos com-plementarios y antiparalelos que lleven la mutación elegida (así, cada oligo-nucleótido hibridará con una de las dos hebras del plásmido a amplificar) y de un tipo de polimerasa como es la Phusion, la Pfu turbo (o alguna equivalente) que sea capaz de polimerizar varias kilobases con una tasa de mutación muy baja (este tipo de enzimas poseen una actividad correctora de errores). Así, se desarrollará una reacción PCR en la que se utilizará como molde un plásmido que lleve la secuencia a mutagenizar, como cebadores los oligonu-cleótidos mutagénicos diseñados específicamente y como polimerasa la Phu-sion polimerasa u otra equivalente.
Una vez desarrollada la reacción, nos aprovecharemos del hecho de que el DNA obtenido a partir de aquellas estirpes de E. Coli -como DH5α− que po-seen el sistema Dam1 se encuentra metilado en las adeninas de las secuencias GAmTC y digeriremos con la enzima Dpn I (que precisamente reconoce y digie-re exclusivamente el DNA que se encuentra metilado en GAmTC) con lo que se digerirá el DNA molde pero no el plásmido mutado que proviene de una reac-ción de PCR y por tanto no se encuentra metilado. Finalmente se transforma con el DNA mutado.

Apéndice
27
1(Para más información sobre los sistemas de metilación consultar la página: http://rebase.neb.com/rebase/rebase.html ) Diseño de oligonucleótidos (mutagénesis dirigida) El diseño de oligonucleótidos para reacciones de PCR tiene una serie de nor-mas que deben ser respetadas para conseguir que las hibridaciones que tienen que tener lugar durante la reacción ocurran de la manera adecuada. Esas nor-mas se refieren sobre todo al porcentaje de GC en la secuencia y la existencia de posibles zonas de estructura 2ª que puedan dificultar la hibridación específi-ca de los oligonuceótidos. En el caso de los cebadores para llevar a cabo la mutagénesis mediante el método “quick change” es necesario contemplar algunas normas adicionales:
1. Los dos oligonucleótidos mutagénicos deben contener la mutación de-seada e hibridar en la misma secuencia en hebras opuestas del plásmi-do.
2. Los oligos deben tener de 25 a 45 pares de bases de longitud, y la Tm de
los cebadores debe ser mayor o igual a 78ºC. Se puede utilizar la siguiente fórmula para estimar dicha tempera-tura: Tm= 81.5 + 0.41 (%GC) - 675 / (N -% de no hibridación) N= nº de nucleótidos del cebador
3. La mutación deseada debe estar en el medio del oligonucleótido con 10
o 15 bases de secuencia correcta en ambos lados 4. Los cebadores deben tener un % mínimo de GC del 40% y deben termi-
nar en C o G.
5. Los cebadores no tienen que estar fosforilados en 5´ pero deben estar purificados por FPLC o PAGE
Calculo de Tm de oligonucleótidos (PCR convencional)
En el caso de las PCR convencionales lo normal es hacer oligos de 18-20 nu-cleótidos de longitud que sean complementarios al DNA en la región corres-pondiente del mismo. En este caso como todos los nulcleótidos son comple-mentarios a región que se pretende amplificar no es necesario que los oligos sean tan largos como en el caso anterior. Se puede utilizar esta fórmula para el cálculo de Tm de estos oligos: Tm = (A+C) x 2 + (C+G) x 4

Apéndice
28
El código genético T C A G
T
TTT Phe (F) TTC " TTA Leu (L) TTG "
TCT Ser (S) TCC " TCA " TCG "
TAT Tyr (Y)TAC TAA Ter TAG Ter
TGT Cys (C)TGC TGA Ter TGG Trp (W)
C
CTT Leu (L) CTC " CTA " CTG "
CCT Pro (P) CCC " CCA " CCG "
CAT His (H)CAC " CAA Gln (Q)CAG "
CGT Arg (R)CGC " CGA " CGG "
A
ATT Ile (I) ATC " ATA " ATG Met (M)
ACT Thr (T) ACC " ACA " ACG "
AAT Asn (N)AAC " AAA Lys (K)AAG "
AGT Ser (S)AGC " AGA Arg (R)AGG "
G
GTT Val (V) GTC " GTA " GTG "
GCT Ala (A) GCC " GCA " GCG "
GAT Asp (D)GAC " GAA Glu (E)GAG "
GGT Gly (G)GGC " GGA " GGG "
Oligonucleótidos mutagénicos A continuación se encuentra la secuencia de los oligonucleótidos mutagénicos que se han seleccionado para la mutación en la proteína MBP-LacZα: Mutmal E1 (F): GATGTCCGCTTTCTGATATGCCGTGCGTACTG Mutmal E2 (R): CAGTACGCACGGCATATCAGAAAGCGGACATC Oligonucleótidos para comprobar la orientación A continuación se encuentra la secuencia de los oligonucleótidos que se han seleccionado para verificar la orientación del inserto Km en el plásmido pMalc2x: MalEF GATGAAGCCCTGAAAGACGCGC KM2 GAAGGAGAAAACTCACCGAGGC KM2R GCCTCGGTGAGTTTTCTCCTTC

Apéndice
29
PLÁSMIDOS

Apéndice
30
Nota: En las secuencias incluidas en esta sección, los aminoácidos (en código de una letra) están colocados debajo del nucleótido central del triplete codificante.

31

Apéndice
32

33

Apéndice
34

35

36
Nota: En las secuencias incluidas en esta sección, los aminoácidos (en código de una letra) están colocados debajo del nucleótido central del triplete codificante. En las secuencias de las regiones de los genes detallados, es importante comprobar el sentido de los marcos de lectura abiertos codificantes.

Apéndice
37

Apéndice
38