por ricardo mÁcarron larumbe - eprints.ucm.es · 12.5. determinación de la masa molecular 37...
TRANSCRIPT

AltI¿
UNIVERSIDAD COMPLUTENSE DE MADRID
FACULTAD DE CIENCIAS BIOLOGICAS
DEPARTAMENTO DE BIOQUíMICA Y BIOLOGIA MOLECULAR 1
*53Q957Q334*
UNIVERSIDAD COMPLUTENSE
PURIFICACION Y CARACTERIZACION DE
ENDOGLUCANASA III DE TrichodennareeseiQM9414
TESIS DOCTORAL
por
RICARDO MÁCARRON LARUMBE
Directores:
Dra. CarmenAcebalSarabia Dra. MU Pilar CastillónBorreguero
MADRID, JU DE 1992

A mis padres,a mis hernianosy a Esther
El presente trabajo ha sido realizado en el Departamentode Bioquímica y BiologíaMolecular 1 de las Facultadesde Ciencias Biológicasy de CienciasQuímicas de la UniversidadComplutensede Madrid bajo la dirección de las Dras. CarmenAcebaly M’~ Pilar Castillón, aquienesagradezcosuapoyoincondicionaly suvaliosaamistad
Gran parte de los resultadosaquí apuestosse han obtenido en el Laboratorium voorBiochemiede la Universidadde Gante (Bélgica) bajo la tutela del Dr Marc Claeyssens,cuyaentusiastacolaboraciónha sidofundamentalpara la conclusiónde estaTesi&
Tambiéndeseoapresarmi agradecimientoa lassiguientespersonas:
- a los Ores. Ma Isabelde la Mata,Juan ManuelDomínguezy Pilar Estrada,compañerosde laboratorio,por suinestimableayudaa lo largo de tantosañosysu estímuloconstante;a Angelespor haberhechomuyllevaderoel agotadortrabajo conel HPLC.
- al Dr JoséG. GavilanesFranco, directordel Departamento,y a los demáscompañerosyprofesoresdel Departamentopor su cordialidady buenadisposiciónpara echarmeuna
manocuandoha hechofalta
- al Dr Jos VanBeeumen(Lab. Microbiologie, Universidadde Gante)por haberrealizadoel análisisdepéptidoscon enormeinterésy dedicación.
- a los Dres. Ge5ranPetterssonyJenyStdhlbergdel BiomedicalCenterdela UniversidaddeUppsala <Suecia)por sugran ayudaen el inicio de estetrabajo.
- al Dr. Bernard Henrissat (CNRS, Grenoble)por cedermeamablementelos estudiosdehomología.
- al Dr Eric Messens(¡ab. Genetika,Universidadde Gante)porhabermefacilitado el asodel detector “¡‘hoto DiodeAn-ay”.
- al Dr. Peter Tomme, a Madeleine Obreno y a Jules Venneulen <Lab. Biochemie,Universidadde Gante) por su colaboración; a ellos, a Marc y EditAr, y a los demáscompañerosde Gante (Wilson,Marco, Bern4 Roberto, etc)por haberconseguidoconsucariño, quemesintiera comoen casa.

INDICE
INTRODUCCION 1
1. APROVECHAMIENTO INDUSTRIAL DE LA CELULOSA 2
1.1. Naturalezade los residuoslignocelulósicos 3
1.2. Hidrólisis enzimáticade celulosa 4
2. MICROORGANISMOSCELULOLITICOS 5
3. CELULASAS DE Trichodermareesei 6
3.1. Especificidadde sustrato 8
3.2. Mecanismode la hidrólisis enzimáticade la celulosa 9
3.3. Regulaciónde la síntesisde celulasas 11
3.4. Relaciónestructura-funciónen celulasas 12
3.5. Manipulacióngenéticade T. reeseiy de sus celulasas 16
4. ESTUDIOS PREVIOS SOBREENDOGLUCANASA III DE T reesei 17
5. OBJETIVOS DEL PRESENTETRABAJO 18
MATERIALES Y METODOS 20
1. MATERIALES 21
2. FERMENTACION DEL HONGO 21.
2.1. Mantenimientode la cepa 21
2.2. Medio de producciónde celulasas 22
2.3. Inóculo 22
2.4. Crecimientodel hongo 22
3. PURIFICACION DE BO III 23
3.1. Precipitacióncon sulfato amónico 23
3.2. Cromatografíaen DEAE-Sepharosea pH 7.0 23
3.3. Cromatografíaen Ultrogel Aca-44 25
3.4. Cromatografíaen DEAE-Sepharosea pH 6.5 25

4. DETERMINACIONES ANALITICAS 25
4.1. Cuantificaciónde prote(na 25
4.2. Valoraciónde azúcaresreductores 25
4.3. Valoraciónde glucosa 26
4.4. Separacióny análisisde celooligosacáridosy derivadosporHPLC 26
5. ENSAYOS ENZIMATICOS. ANALISIS DE DATOS 27
5.1. Actividad y adsorciónsobrecelulosamicrocristalina(Avicel) 27
5.2. Actividad sobrecarboximetilcelulosa(CMC) 27
5.3. Actividad sobresustratoscromofóricos:CNP(Glc)3y MeUmb(Glc), 28
5.4. Actividad sobrecelooligosacáridos 28
5.5. Actividad sobremananos 28
5.6. Actividad sobrexilano 29
5.7. Actividad 8-glucosidasa 29
6. TECNICAS ELECTROFORETICAS 29
6.1. Electroforesisen gelesde poliacrilamidaen presenciade SDS 29
6.2. Isoelectroenfoqueanalítico 30
6.3. Curva de titulación 30
6.4. Localizaciónde actividadesenzimáticasen los geles 30
7. CARACTERIZACION ESTRUC1’URAL DE EG III 30
7.1. Determinaciónde la masamolecular 30
7.2. Determinacióndel punto isoeléctrico 31
7.3. Análisis de aminoácidos 31
7.4. Centrifugaciónanalítica 31
7.5. Rotura con bromurode cianógeno 31
7.6. Determinaciónde grupossultbidrio libres 32
8. EFECTO DEL pH Y LA TEMPERATURA SOBRELA CINETICA ENZIMATICA 32
9. MODIFICACION QUIMI~A DE EG III 33
9.1. Modificación con dietilpirocarbonato(DEPC) 33
9.2. Modificación con tetranitrometano(TNM) 33
9.3. Modificación concarbodiimidas(EDC y EAC) 33
9.4. Modificación con N-etoxicarbonil-2-etoxi-1,2-dihidroquinoleína(EEDQ) 33
9.5. Modificación con reactivoK de Woodward(RKW) 34
9.6. Estudiosde protección 34

10. MODIEICACION DE TRIPTOFANOS CON N-BROMOSUCCINIMIDA (NBS) 34
10.1. Reaccióncon NBS 34
10.2. Cálculo del númerode Trp oxidados 35
10.3. Preparaciónde formas de EO III con distinto grado de
oxidación (W10, y W20,) 35
10.4. Estudiosde termoestabiidad 35
10.5. Espectrosde dicroísmo circular 35
11. MARCAJE DE AFINIDAD DE EG III CON 4’,5’-EPOXIPENTIL-
B-CELOBIOSIDO (EPOS) 36
11.1. Reacciónde inactivacicin de ECli III con epoxialquil B-celobiósidos 36
1 1.2. Preparaciónde EPO5-EGIII 36
12. ANALISIS DE PEPTIDOS 36
12.1. Digestión contripsina 36
12.2. Digestióncon proteasaV8 36
12.3. Separaciónde péptidospor HPLC 36
12.4. Secuenciaciónamino-terminal 37
12.5. Determinaciónde la masamolecular 37
RESULTADOS 38
1. PURIFICACION E IDENTIFICACION DE EG III 39
1.1. Purificaciónde EG III 39
1.2. Propiedadesmolecularesde EG III 42
1.3. Efectode pH, temperaturay fuerza iónica en la estabilidady actividad
de ECli III 44
2. ESPECIFICIDAD Y MODO DE ACCION DE ECli III 44
2.1. Actividad sobresustratoscromofóricos 44
2.1.1. Estudiosde partición con metanol 46
2.2. Actividad sobrecelooligosacáridos 48
2.3. Actividad sobresustratoscelulósicos 49
2.4. Actividad sobremananos 50

3. IDENTIFICACION DE RESIDUOSESENCIALES DE EO III 52
3.1. Efecto del pH y la temperaturaen la cinéticade una reaccióncatalizada
por ECli 111 52
3.2. Modificación de histidinascon dietilpirocarbonato(DEPC) 53
3.3. Modificación de tirosinascon tetranitrometano(TNM) 53
3.4. Modificación de triptófanoscon N-bromosuccinimida(NES) 54
3.4.1. Extensióny especificidadde la modificación 55
3.4.2. Proteccióncon ligandos 56
3.4.3. Efecto de la modificación en la actividadcatalíticay en la
capacidadde adsorciónacelulosa 57
3.4.4. Efecto de la modificación en la termoestabijidady estructura
secundaria 59
3.4.5. Identificación de un residuode Trp esencial
en el dominio de unión a celulosa 60
3.4.6. Identificación de un residuode Trp esencial
en el dominio catalítico 64
3.5. Modificación degruposcarboxilo con agentesquímicos
específicos 66
3.6. Modificación de un carboxioesencialcon el marcadorde afinidad
4’,5’-epoxipentil-B-celobiósido(EPOS) 67
3.6.1. Inactivaciónde EO III por epoxialquil B-celobió-
sidos.Efectode la longitud del brazoaglicónico 68
3.6.2. Especificidadde la inactivacién 68
3.6.3. Cinéticade la inactivaciónpor EPOS 69
3.6.4. Dependenciacon el pH de la inactivaciónpor EPOS 71
3.6.5. Identificación del grupo carboxioesencialunido a EPOS 71
APENDICE. Análisis cinéticode la hidrólisis enzimáticadesustratoscondos
víasalternativasde ataque. 74

DISCUSION
1. PURIFICACION Y PROPIEDADESMOLECULARES DE EG III
2. ESPECIFICIDADY MODO DE ACCION DE EG III
2.1. Modelo de organizaciónen subsitiosdel centroactivo
22 Mecanismocinético
2 3 Actividad sobremananos
3. RESIDUOSDE TRIPTOFANO ESENCIALES
4. IDENTIFICACION DE DOS GRUPOSCARBOXILO ESENCIALES:
MECANISMO QUíMICO DE EG III
4.1. Estudiosde pH: Glu2l8
4.2. Marcaje de afinidad: Glu329
4.3. Mecanismoquímico de EG III
CONCLUSIONES
76
77
78
79
80
80
81
85
85
85
86
89
BIBLIOGRAFíA 91

ABREVIATURAS
AvicelCBHCMCCNBrCNPCNPGIcCNP(Glc)2CNP(Glc),DEAEDEPCDTEDTNEDUCEACEDCEEDQEGEG III W,0,~EPO3EPO4EPOSEPO5-EGIIGuHCIHCA
HPLCIAAmLBOMESMeUmbMeUmbGlcMeUmb(Olc)2MeUmb(Glc)3NBSPDMS
ProteasaVSRKWSDSTNM
celulosamicrocristalinapara cromatografíaen capafinacelobiohidrolasa,1,4-I3-D-glucancelobiohidrolasa(EC 3.2.1.91)carboximetilcelulosabromurode cianógeno2-cloro-4-nitrofenol2’-cloro-4’nitrofenil-I3-D-glucósido2’-cloro-4’nitrofenil-13-celobiósido2’-cloro-4’nitrofenil-13-celotriósidodietilaminoetiodietilpirocarbonato1,4-ditioeritritolácido 5,5’-ditiobis-(2-nitrobenzoico)dominio de unión a celulosa1-etil-3-(4-azonio-4,4-dimetilpentil).carbodiimida1-etil-3-(-dimetilaminopropil)-carbodiimidaN-etoxicarbonil-2-etoxi-1 ,2-dihidroquinoleínaendoglucanasa,l,4-13-D-glucanglucanohidrolasa(EC 3.2.1.4)endoglucanasaIII con n Trp oxidadosporNBS2’,3’-epoxipropil-13-celobiósido3’,4’-epoxibutil-B-celobiósido4’,5’-epoxipentil-13-celobiósidoendoglucanasaIII unida apentil-l3-celobiósidocloruro de guanidinioanálisisde secuenciasproteicaspor gruposhidrofóbicos (“hydrophobic
clusteranalysis)cromatografíalíquida de alta resolución
iodoacetamidagomade algarroba(Ceratoniasiliqua), galactomanano(“locust beangum’)ácido 2-N-morfolin-etanosuífónico4-metilumbeliferona4’-metilumbeliferil-B-D-glucósido4’-metilumbeliferil-B-celobiósido4’-metilumbeliferij-l3-celotriósidoN-bromosuccinimidaespectroscopiade masasde desorciónde plasma(‘plasmadesorption
massspectrometry’)endoproteinasaOlu-C (EC3.4.21.19)de StaphylococcusaureuscepaVSreactivoK de Woodward,N-etil-5-feniisoxazolio-3’-sulfonatododedilsulfatosádicotetranitrometano

INTRODUCCION

INTRODUCCION 2
La celulosa es el compuestoorgánico más abundanteen la Naturaleza.Su potencialenergéticoha sido aprovechadodesde tiempos remotos por numerososorganismos;masrecientementeha atraído la atenciónde los biotecnólogos,que estudianlos medios parautilizarla como fuentede combustiblesy otros productosquímicos de interésindustrial.
La investigaciónen estecampocomenzóen los años50, cuandoel equipo de los Drs.Reesey Mandels,del Ejército de los EstadosUnidos, evaluó la capacidadcelulolítica de másde 14 000 microorganismos.La finalidad de su trabajo era determinarlas causasde ladegradaciónde los materialescelulósicos(uniformes,cordajes,tiendas)observadadurantelaSegundaGuerra Mundial, en las unidadesmilitares desplegadasen el Pacífico.
A partir de la crisis del petróleo de los años70, el estudiode la hidrólisis de la celulosapasó a ser una línea de investigaciónprioritaria en muchospaíses, habida cuentade suspotencialidadescomofuentede energíaalternativa,renovabley limpia. Prontosehizo evidente,que la viabilidad económicade estaalternativaenergéticaestabaseriamentelimitada por lafalta de conocimientosbásicossobrelos complejosmecanismosbiológicos queoperanen lacelulolisis. El interésde los gobiernosen la financiaciónde esta línea de investigacióndecrecióen losaños80; sin embargo,elempeñode microbiólogos,bioquímicosy genéticosporresolverlas incógnitasplanteadasen esteatractivocampode investigaciónha permitidola continuidaddel proyecto.
Teniendosiempreenel horizontela idea original de aprovecharla celulosacomo fuentede energía,otrasaplicacionesindustrialessehanderivadodelconocimientoadquiridoalo largode estos años. Actualmente las enzimas celuloliticas se emplean en la elaboracióndedetergentesparalavarropa, y en la clarificacióndevinos y cervezas(Pokornyy col., 1990); eldominio de unión a celulosa presenteen muchas celulasasconstituye una interesanteherramientabiotecnológica,quepuedefacilitar lapurificación o la inmovilizacióndecualquierproteínapor fusión a nivel génico (Ongy col., 1989); la enormecapacidadde producciónde
celulasasde algunosmutantesde microorganismoscelulolíticospuedeseraprovechadaparalasecrecióneficientede proteínasheterólogas(Teeri y col., 1990).
1. APROVECHAMIENTO INDUSTRIAL DE LA CELULOSA
La producciónanualde celulosapor fotosíntesisseestimaen 4x10’0 Tm (Coughlan,1985).De esta cantidadsólo un 2% es aprovechadapor el hombre mediantecombustiónu otrosprocesosindustriales(Vegay col., 1983);el restosedesechaen forma deresiduos,o escapaalcontrol humano.Cerca de un 20% de esta biomasano utilizada (unas8x109 Tm, con uncontenidoenergéticopróximo a 3x10’8 J), podría recuperarsecomo materia prima para laproducciónde combustibles,alimentosu otrosproductosquímicosde interés(Detroy y Julian,1982). Para este fin, es esencial la degradaciónde la celulosa hasta glucosa, pero lascaracterísticasestructuralesde este polisacáridoy su asociacióncon hemicelulosay lignina
dificultan enormementesu hidrólisis.

INTRODUCCION 3
1.1. Naturaleza de los residuos lignocelulósicos
La celulosa forma partede las paredesde las célulasvegetalesen íntima asociaciónconlignina y hemicelulosas,constituyendoen conjuntola denominadalignocelulosa.La proporciónde estospolímerosvaría segúnel origen del residuolignocelulósicoconsiderado.
La celulosaestáformadaporcadenaslinealesdeD-glucosaunidasporenlacesglucosidicos13(1-.4). El grado de polimerización,esdecir, el númerode restosde glucosaqueforman unamoléculade celulosa,puedeoscilar entre50 y 15 000 (Gilberty Tsao, 1983). Lasmoléculasdeglucosa se encuentranen conformación de silla, giradas 1800 cada una con respectoa lasiguiente.Por ello, desdeun punto de vista estereoquímica,es la celobiosala unidad queserepite (Fig. 1).
GLUCOSA
Figura 1. Estructura molecular de la celulosa.
Las cadenasdecelulosaseunenmediantepuentesde hidrógeno,formandomicrofibrillas,en las que las moléculasde celulosasedisponenen ordenaciónantiparalela(Ltitzen y col.,1983). A su vez, variasmicrofibrillas se agrupanen fibrillas, cuyaagregaciónda origen a lasfibras de celulosa.Esta estructurase mantienegraciasa un elevadonúmero de puentesdehidrógenoy de interaccionesde van derWaals, queproporcionanen conjuntouna unión muyfuerte, haciendoa la fibra de celulosa insolubley muy resistenteal ataquequímico.
Aunqueno seconoceexactamentela estructurafibrilar de la celulosa,todoslos modelospropuestoscoinciden en señalar la existencia de regiones cristalinas, con alto grado deordenación,quesealternanconregionesamorfas,menosordenadas(Cowlingy Kirk, 1976; Fany col., 1987). La proporción en que aparecenvaría según el origen de la celulosa y eltratamientoa quese someta.
La hemicelulosaestaconstituidapor varios heteropolisacáridosde composicióndistintaen cadaplanta. Los másabundantessonxiianos, mananosy galactanos.Se trata de polímeroscortosy en generalramificados,incapacesde agregar,y por tanto,susceptiblesde hincharseydispersarsefácilmenteen agua.Su funciónprincipal en la paredvegetalesla deunir lacelulosay la lignina. (Fan y col., 1987).
La lignina es un polímero muy complejo y de elevadopeso molecular. Se forma pordeshidrogenaciónenzimática de alcoholes derivados del fenilpropano, seguida por unapolimerizaciónno controlada,lo quehace quela lignina no tengaunaestructuradefinida, nisiquieraen una misma especievegetal (Barcelóy col., 1987).
El papel de la lignina es cementarlos polisacáridosde la paredvegetaly actuarcomobarrerade la degradaciónmicrobianadel materiallignocelulósico(Blanch y Wilkes, 1983). Laíntima unión de la lignina alarmazónvegetales un impedimentoparala hidrólisisde celulosa
II
CELOB IOSA

INTRODUCCION 4
de la que sólo se consiguesepararmediantetratamientosenérgicos.Su propia hidrólis¡srequiereseverascondicionesdebidoa su complejaestructuraquímica.
1.2. Hidrólisis enzimíticade celulosa
La hidrólisisde la celulosahastaglucosapuederealizarseenzimáticao químicamente.La hidrólisis enzlináticaha sido investigadaintensamenteen los últimos 20 años.Hasta elmomento no se ha instalado ninguna planta industrial para hidrolizar celulosa por víaenzimática;no obstante,existenplantaspiloto en Japón(Fany col., 1987)y Francia(Pourquiéy col., 1988),y la tecnologíadesarrolladaestámuy cercanaal umbralde viabilidad comercial.
La hidrólisis química implica generalmenteel uso de ácidos. En 1913 se instaló enCarolinadel Sur(EEUU)unaplantaparahidrolizarporestavía desechosprocedentesde unaserrería.La fábricasecerróa raíz de la caídade los preciosdel azúcar.EnAlemania,durantela SegundaGuerra Mundial, seempleóla hidrólisis ácidaparala obtenciónde azúcar(Fanycol., 1987).
La sacarificación de residuos lignocelulósicos por vía enzimática requiere unpretratamientopara aumentarla susceptibilidadde los mismos a la hidrólisis. Sin unpretratamientoadecuado,la biodegradaciónde la lignocelulosa es lenta y con un bajorendimientoen azúcaresfermentables(Dale, 1987).La hidrólisis química esventajosaen esteaspectoya queno esnecesariopretratarla materiaprima. Sin embargo,la hidrólisisenzimáticapresentavarias ventajas sobre la química: las celulasas,enzimas que llevan a cabo ladegradacióndela celulosa,no formanproductossecundarios;alfinal delprocesono serequiereunaneutralizacióndelproducto;los costessereducenporquelas temperaturasde trabajosonsuaves,no hay necesidaddeemplearmaterialesresistentesa la corrosión,y el rendimientoenglucosaesmayor (Fany col., 1987).
El productode la hidrólisis, la D-glucosa,tienemúltiplesposibilidadesde uso:- Refinada,puededestinarsea la industriaalimentaria.- Se puedetransformarpor isomerizaciónen fructosa,que esun potenteedulcorante.- Puedesersustratopara procesosfernientativosque rinden:
• etanol• biomasamicrobiana• biopolímeros• productosquímicosy fármacostalescomo antibióticos,L-aminoácidos,ácido
cítrico, acetona,butanol,etc.
En la Figura 2 seesquematizael procesoglobal de sacarificaciónde lignocelulosaparaobtenerproductosde interésindustrial. Los gastosde producciónde las enzimascelulolíticassuponenuna importantepartida de los costesdel proceso de sacarificaciónde residuoslignocelulósicos.Avanzaren el conocimientode las celulasas,de su biosíntesis,propiedadesymecanismode acción, seleccionarbuenosproductoresy buenastécnicasde producción,haabaratadoestabiotecnologíahastahacerlacasicompetitivacon otrastecnologíasempleadasenla produccióndeazúcares(fundamentalmentela hidrólisisdealmidones).Paraquelleguea seraplicableindustrialmentedebecontinuarel esfuerzoinvestigadorque acabarápordesvelarlasincógnitasquerodeana estecomplejoprocesobiológico: ¿cuálesson los mecanismoscelularesde regulaciónde la biosíntesisde celulasas?,¿quépapeldesempeñacadatipo de celulasaenla degradaciónde celulosa?,¿cuál es la causa del sinergismo encontradoentre algunascelulasas?,¿existealgún factor, todavía no identificado, quedisgregelas fibrillas de celulosaparahacerlamássusceptibleal ataqueenzimático?.

INTRODUCCION 5
MATERIALESLIGNOCELULOSICOS
FermentaciónMICROORGANISMO
CELULOUTICO
*Pretratamiento
tCELU LOSA
n CELU LASAS
rHIDROLISISENZIMATICA
GLUCOSA
Hemicelulosa
Hidrólisisenzimática
$Pentosas
Lignina
Compuestosfenálicos
FERMENTACION
ETANOL, BIOMASA MICROBIANA, FARMACOS, ETC.
Figura2. Esquemadel procesode sacarificaciónde residuoslignocelulósicos.Elaprovechamientode hemicelulosa y lignina incidiría favorablementeen elbalanceeconómicoglobal.
2. MICROORGANISMOS CELULOLITICOS
La degradaciónnaturalde la celulosaesllevada a cabopornumerososmicroorganismos.Entreéstoshay bacteriasy hongos,aerobiosy anaerobios,mesófijosy termófios.En la Tabla1 serecogenlos microorganismoscelulolíticos mejor caracterizados.
Todos los organismoscapacesde degradarcelulosacristalinasecretanun conjunto decelulasascon diferentesespecificidadesy modo de acción,que actúan sinérgicamenteparahidrolizar la celulosa (Béguin, 1990). Dicho conjuntose ha venido denominandosistemaocomplejo celulasa. Esta última denominaciónes confusa ya que sólo en ciertas bacteriasanaerobiaslas enzimasceluloliticasse agrupanen complejosmultienzimáticosextracelulares;el casomásconocidoeselde Clostridumthermocellumquesecretaun complejocelulolitico con14-18 polipéptidosdistintos, formando una estructuramuy establedenominadacelulosoma(Lamedy Bayer, 1988).

INTRODUCCION
Tabla 1. Microorganismos celulolíticos más estudiados (Robson y Chambliss,1989; Coughlan y Ljungdahl, 1988).
BACTERIAS HONGOS
Acetivibrio celulo/yticus . Aspergillus spp.Bacillus spp. . Fusarium solan¡Gel/u/amonas spp. . Neocallimastix frontalisClostridium spp. . Penicillium spp.Erwínia chrysanthemi Schizophyllum communeMiorobispora bispara . Sporotrichum pulverulentumRuminococcus albus . Talaromyces emersonhiStreptomyces spp. . Tríchoderma spp.Thermonospora spp.
Uno de los microorganismoscelulolíticosmásextensamenteestudiadosha sido el hongoaerobioy mesófiloTrichodennareesei(e.g.Kubiceky col., 1990a).Se aisló en 1944 en la isla deBougainville(Nueva Guinea),dentrode una prospecciónrealizadaporel cuerpocientífico delEjército de los EstadosUnidos, que estudiabala alarmantedegradaciónde los tejidos dealgodón (tiendas,vestimenta,cordajes)en las unidadesdesplegadasen el Pacífico Sur.Inicialmenteseclasificó como T. viride. E. Simmonsconsideróque se tratabade una nuevaespecie,y la bautizóen 1977 con su denominaciónactual,en honorde Elwyn T. Reese,pioneroenel estudiode las celulasasjunto con su colaboradoraMary Mandeis; ambosseleccionaroneste hongo, entre 14 000 microorganismos,por su extraordinariacapacidadcelulolitica(Simmons,1977; Reesey Mandels, 1984).
T. reeseisecretael sistemaenzímáticocelulasaen cantidadesimportantes,siendocapazde hidrotizartotalmentecelulosacristalina;susenzimassonestablesdurantevariosdíasa 450Cy pH 5. De la estirpeoriginal “QM 6W’ se han derivadopor mutacionesal azarmediantedistintastécnicas(Uy, nitrosoguanidina,aceleradorlineal, dietil sulfato, radiación ) másde50 cepassuperproductoras(EI-Gogaryy col., 1990).La estirpeestudiadaen nuestrolaboratorio(Acebaly col., 1985) y empleadaen la realizacióndel presentetrabajoesla QM 9414.Data de1971 y ha sido una de las más intensamenteinvestigadaspor bioquímicosy genéticos,a pesarde que actualmentesedisponede cepasmejoresproductorasde celulasas(Pourquiéy col.,1988).
3. CELULASAS DE Trichodenna
Las celulasasde origen fúngico se han agrupadotradicionalmenteen tres categorías:celobiohidrolasas(CBH; exoglucanasas;EC 3.2.1.91),queatacanlas moléculasde celulosaporsuextremono reductor,liberandosecuencialmentesubunidadesde celobiosa;endoglucanasas(EG; EC 3.2.1.4), que rompen al azar enlaces internos de las moléculasde celulosa; II-
6

.7INTRODUCCION
glucosidasas (EC 3.2.1.21),quehidrolizancelobiosay celodextrinasde bajopesomolecular.Ensentidoestricto sólo las EGs y CBHs seconsiderancelulasas;las ¡3-glucosidasasparticipanindirectamenteen la degradaciónde celulosaal hidrolizarcelobiosaqueinhibe a las CBHs.
La composicióndel sistemacelulasade T. reeseiha sido estudiadapordiferentesgruposde investigación,con resultadosgeneralmentecontradictoriosen cuantoal númerode enzimasy sus propiedadesmoleculares(M, pI) (Griztali y Brown, 1979; Shoemakery col., 1983;Bhikhabhaiy col., 1984; Schtilein, 1988). La confusión en estepunto se ha debido a variascausas:
- existenciarealde variosgenes;hastael momentosehandonadoenE. colí y secuenciadolos genesdedos CBHs,CBH 1 (Teeriy col., 1983)y CBH II (Cheny col., 1987),y dedosEGs,EG 1 (Penttiláy col., 1986) y EG III (Saloheimoy col., 1988); otraEG, denominada“EG debajopesomolecular” (EGLMW) ha sido caracterizaday pareceno tenerrelacióncon ningunade las dos secuenciadas(Ulker y Sprey,1990).
- modificacionespost-traduccionalesde los productosgénicos, tales como proteolisis(St~hlbergy col., 1988),glicosilación o desamidación.Se han descritovariasisoenzimasparacadauna de las enzimasmencionadas,que aparecentardíamenteen los caldosde cultivo delhongo (Messnery col., 1988; Biely, 1990).
- dificultadesen la purificacióny caracterizaciónde las diferentescelulasasdebidoa lasimilitud de susmasasmoleculares(en el intervalode 50 a 60 kDa)y de suspuntosisoeléctricos(de 3.5 a 6.5) (EG LMW esla únicacon propiedadespeculiares)(Biely, 1990),a su naturalezaglicoproteica (Bhikhabhaiy col., 1984), a la utilización tardía de sustratosque permiten ladetecciónespecíficade cadaactividadcelulolítica (Claeyssens,1988), y a la capacidaddeagregarde algunasde ellas (Spreyy Lambert, 1983; Domínguezy col., 1992).
La composicióndel sistemacelulasade T reeselha quedadoestablecidacomo serecoge
en la Tabla II, despuésdel CongresoInternacionalTricel 89 (Kubiceky col., 1990a).
Tabla 11. Composicióndel sistemacetulasade Trichodermareesei (Biely, 1990;Ulker y Sprey, 1990).
ENZIMA Ma píb % ch~(koa) <% (p/p)
)
Celobiohidrolasas
CBH ¡ 62 (58) 3.4-3.9 6
CBH II 59 (50) 4.4-6.5 18
Endoalucanasas
EG 1 51<46) 3.3-4.4 10EG III 50 <42) 3.8-6.7 15EG LMW 25 7.5 0
A Los valores entreparéntesisrepresentanla masa deducidaa partir de la
secuenciade la proteínab Intervalo de puntosisoeléctricosde las distintasisoenzimas.
Porcentajede carbohidratos.

INTRODIJCCION 8
3.1. Especificidad de sustrato
La clasificaciónde las celulasascomo endoglucanasaso celobiohidrolasasseha basadoclásicamenteen su capacidadpara hidrolizar sustratoscelulósicosde distinta naturaleza;lasenzimasactivassobrecelulosassustituidassolubles,carboximetilcelulosa(CMC), e inactivassobrecelulosacristalina,Avicel, seconsiderabanEGs,y CBHs aquellascapacesde hidrolizarAvicel pero no CMC. La simple división en dos subgruposde las enzimascelulolíticasesinsuficienteparaexplicarfenómenoscomoelsinergismoentrealgunoscomponentesdelsistema,o la razóndeserdevariosgenesparacadatipo de enzima.Sehacenecesaria,en consecuencia,unadiferenciaciónmásrigurosade lascelulasasen funcióndesu especificidaddesustrato;éstaha sido posiblegraciasal empleo de sustratosde bajamasamolecular:celodextrinas(Pereiray col., 1988), y derivados cromofóricos de celooligosacáridos(Claeyssens, 1988). Lasespecificidadesfrentea metilumbeliferil B-glicósidosdelas principalescelulasasdeT reeseiserecogenen la Figura 3.
GEN! EG 1
~EJ= ½~EJp1 1
EnEJe mEJe1 ti
EJEJEJe Encime1 1
ci ci ci ci e En EJ En EJ OIII It
En ci ci ci EJ e EJ EJ En ci -~
III 1 ¶GENIl EG III
Encime EJEJEJe1 t
r~ EJ EJ EJ p
¶ EJEJEJEJO_____________________________________________________________ fEJ EJ EJ EJ En e r~ ci EJ EJ ci e
ji ItFigura 3. Principales puntos de ataque de las celulasasde T. reesei a 4-metilunibeliferil-B-glicósidosderivados de celooligosacáridosy lactosa. Los productos se analizaron por HPLC. Símbolos:
residuos de I~-galactopiranosilo; , residuos de 8-glucopiranosilo; ,grupos4-metilumbeliferilo.(van Tilbeurgh y col., 1988).
Los sustratoscromofóricosson,además,degranutilidad en la detecciónespecíficade lascelulasasen gelesde isoelectroenfoqueo deelectroforesisen condicionesno desnaturalizantes(van Tilbeurgh y Claeyssens,1985), así como en el estudiodel mecanismocinéticode estasenzimas, imposiblecon los sustratospoliméricosclásicospor las dificultadesque entrañasuensayo,y la no linearidadde las curvas de dilución enzimáticacorrespondientes(Sharrock,1988).
La multiplicidad de celulasasha sido explicadapor algunosautoresteniendoen cuentala rotaciónde 1800 quepresenta,en la moléculade celulosa,cadaresiduode glucosarespectodel siguiente. Cada enlace glucosídico es estéricamentedistinguible del siguiente, y, enconsecuencia,podría haber dos tipos de EG y dos tipos de CBH, con distintaestereoespecificidad(Wood y col., 1988).Esta teoría no esválida, al menosen el casode Treesei (Sinnot, 1990).

INTRODUCCION 9
La capacidad de algunas celulasas para degradar sustratos no celulósicoses un datollamativo; EG 1 poseeactividadxi]anasa(Biely y col, 1991), EO 1 y EG III son activassobremanano(Fig. 12, pág. 40) y ambasCBHshidrolizan 13-glucanos,quecontienenenlacesB-( l-.3)y l3-(í-.4), máseficazmentequecelulosa(Enariy Niku-Paavola,1987).Hayqueteneren cuentaqueel sustratonaturaldel hongoes lignocelulosay no celulosapura. En los caldosde cultivose encuentranademásde celulasas,xiianasas(Biely, 1990) y mananasas(Eig. 12, pág. 40).Esclarecerel mecanismode acción de una mezclaenzimáticatan complejasobreun sustratoigualmentecomplejoparece,hoy porhoy, lejano;no obstante,esel únicocaminoparadilucidarinequívocamentela función biológica decadacomponente.
3.2. Mecanismo de la hidrólisisenzimáticade celulosa
El primermodeloparaexplicarel mecanismode la hidrólisis enzimáticade celulosafuepropuestopor Reeseen 1950 ; segúnestemodelo, la celulosaseríaactivadaporun factor dehinchamiento o activador, enzima no hidrolítica C1; a continuación actuaría una enzimahidrolítica C~; la celobiosay otros celooligosacáridosproducidos,seríanhidrolizados hastaglucosapor 13-glucosidasa(Fig. 4) (Reesey col., 1950).
13-glucosidasaCELULOSA —CELULOSA ACTIVADA —CELOBIOSA GLUCOSA
Figura 4. Mecanismo de la hidrólisis enzimAtica de celulosa según el modelo de Reese (Reese y col., 1950>.
Pocosañosdespués,Gilligany Reesedescribieronporprimeravezel sinergismoentrelasenzimascelulolíticasen la digestión de la celulosa(Gilligan y Reese,1954); la hidrólisis desustratosinsolublespor una combinacióndeEGs y CBHs esmuchomásefectivaque lo quecabría esperarde la sumade suscapacidadesh¡drolíticas individuales(Mandelsy Reese,1964;Henrissaty col., 1985). Las basesmolecularesdel sinergismono estánaclaradas;el grado desínergismodependede la composiciónde las mezclasenzimáticasempleadas(Henrissaty col.,1985).Tambiénseha observadosinergismoentreCBH 1 y CBH II (Henrissaty col., 1985), yen algunoscasosausenciade sinergismoentreCBH 1 y EG 1 (Niku-Paavolay col., 1986).CBH1 pareceactuarcomo una endoglucanasaen estassituaciones,o cuandohidroliza B-glucanos(Henrissaty col., 1985).
El análisisde la adsorciónde las celulasasa las fibras de celulosaesfundamentalen elestudiocinéticode la hidrólisis, ya quees un prerrequisitoparaque tengalugar la reacción(Fan y col. 1987). La capacidadde adsorciónvaria de una enzimaa otra (Woodwardy col.1988; Kyriacou y col., 1989),y tambiénes distinta la afinidadde cadacelulasapor las distintasregionespresentesen el sustrato (Nieves y col., 1991). La adsorcióndebejugar un papelimportanteen los fenómenosde sinergismo.
Probablemente,en la degradaciónde celulosaparticipantambiénotrasenzimascomo lacelobiosa-oxidasa,detectadaen caldos de cultivo de otros hongos,que oxida celobiosaycelodextrinasa suscorrespondientesácidosaldónicos.Una función deestaenzimapuedeserla oxidación del extremoreductorformado tras la acciónde una EG, evitandoque el enlacepudieraformarsedenuevoportransglicosilación.La celobiono-lactonano sólo seha encontradoen caldos de cultivo de T. reesei, sino que ademásesun potentey específicoinductorde lasíntesisde celulasas(Iyayi y col., 1989). La formación de fibras cortas de celulosa en los

INTRODUCCIONlo
primerosestadiosde la celulolisisesdependientede oxígeno(Vahen, 1982).
Ningunodelos modelospropuestos(Montenecourty Eveleigh,1979;Enariy Niku-Paavo¡a,1987; Coughlany Ljungdahl, 1988; Wood y col., 1988) explica todos los datosexperimentalesde quesedisponeactualmente,y pareceprobableque cadatipo de sustratosea hidrolizadodeun modo distinto, por unacombinaciónparticular del arsenalde enzimasde quedisponeelhongo (Knowlesy col., 1988). Tampocosepuededescartarla existenciade algunaenzimaofactorno enzimático(C1 en el modelode Reese),cuyadetecciónno hayasido aún posibleporla carenciade un método adecuadoparaello (Wood, 1985).
Activación ~ OxidaciónRegionescristalinas
yRegionesamodas
B
AEndoglucaflasa (EG)
ffl
Celobiohidrolasa (CBH)
— xQl t
EG/CBH
U _
1:1
B-glucos¡dasa
1 1Formación defibras cortas
Segmentación Disgregación
Sinergismo SinergismoEndo/Exo CALI l/CBH II
Celodextrinas + Celobiosa —* Celobiono. 24 Acido celobiónicolactona
n
GLUCOSA
311
Glucosa -% Gí~~no. —~-~ Mido glucér,icolactona
Figura 5. Modelos del mecanismo de hidrólisis enzimática de celulosa. A) Modelo de Montenecourt(Montenecourt y Eveleigh, 1979); B) Modelo de Coughlan (Coughlan y Ljungdahl, 1988, modificado).(1), celobiosa oxidasa/deshidrogenasa; (2), lactonasa; (3), 8-glucosidasa; (4), endoglucanasa; (5),celobiohidrolasa; (6), glucosa oxidasa.
El modelo de Montenecourt (Fig. SA) (Montenecourty Eveleigh, 1979), se aceptageneralmentecomo una simplificaciónválida del mecanismode hidrólisis enzimáticade lacelulosa (Béguin, 1990); las endoglucanasasefectuaríanel ataqueinicial por ias regionesamorfasdel sustrato,creandoextremoslibressobrelos queactuaríanlas celobiohidrolasas.Laacción sinérgica de EGs y CBHs se repetiría hastasolubilizar totalmentela celulosa; laB-glucosidasaaceleraríala reacciónal hidrolizar hastaglucosa,las celodextrinasy celobiosaliberadaspor las celulasas.
Sin embargo,otrosmodeloscomoel deEnari(Enariy Niku-Paavola,1987),postulanqueel ataqueinicial a las fibras de celulosacorre a cargo de las CBHs. El mecanismoreal dehidrólisis debeparecersemás al esquemapropuestopor Coughlan(Fig. SB) (CoughlanyLjungdahl, 1988),quecontemplala participaciónde otrosfactores,y haceespecialhincapiéenla activacióninicial de la celulosa(volvemosde nuevoa la hipótesisde Reese)poruna o más

INTRODUCCION 11
vías alternativas.Estudiosde microscopiaelectrónicahan demostradola capacidadde CBI-1 1paradisgregarlas fibras de celulosa(Chanzyy col., 1983).
3.3. Regulaciónde la síntesisde celulasas
T. reeseisecretacelulasascuandohaycelulosaen el mediodecultivo, siguiendoel principiodeeconomíacelular;pero,¿cómoun sustratoinsolublepuedeinducir la síntesisde proteínas?.Más de treintaañosdespuésde que fueraplanteadaestacuestión(Mandelsy Reese,1960),tenemosun conocimientotan sólo aproximado de los mecanismosde inducción (Kubicek y col.,1990b). En la Figura 6 seesquematizanlos mecanismosque regulanla síntesisde celulasas.
e
Figura 6. Modelo de la regulación de la síntesis de celulasas en 2? reesel. (1) cejulasas constitutivas;(2) 13-glucosidasa unida a membrana; (3) 13-disacárido permeasa; CBL, l,S-celobionolactona.(EI-Gogary y col., 1990, modificado).
La existenciadenivelesbasalesde celulasasconstitutivashasidodescritaporvariosgrupos(EI-Gogaryy col., 1989; Messnery Kubicek, 1991); también ha quedadoestablecidosu papelfundamentalen la índuccíónde la síntesisdecelulasas(EI-Gogaryy col., 1989).Seha postuladola necesidadde un contactofísico entrela celulosay la paredmiceliar para quese inicie lahidrólisis del polímero (Bindery Ohose,1978);pareceprobablequelascelulasasdetectadasenlas paredesdel conidio (CBH 1 y, especialmenteCBH II) actúencomosensoresparadetectarla presenciade celulosaen el medioy dispararel mecanismoinductivo (Messnery col., 1991).
Los oligómeros producidos por el ataque inicial de las celulasasconstitutivassonhidrolizadospor 13-glucosidasa.Existe una ¡3-glucosidasaconstitutiva, ligada a la membranaplasmática,y esencialparala inducciónde la síntesisdecelulasas(Kubicek, 1987).La actividadtransglicosilasadeestaenzimaprovocala formaciónde soforosa(13-(1-.2)-glucobiosa),inductorde la síntesis de celulasas(Mandels y col., 1962). La soforosatambién se puede formarextracelularmentepor transgJicosilacióncatalizadapor otrascelulasascomo EG 1 (Claeyssens
.1
e e.C B-glucosidasaRepresión por catabolito .7
Glucosa

INTRODUCCION 12
y col., 1990a). La presenciade inductoresadicionalesha sido propuestapor varios autores,debidoa quela soforosano inducela síntesisdetodo el sistemacelulasa(Sternbergy Mandels,1979; Messner y col., 1988). Ni celobiosa, ni celooligosacáridosde mayor grado depolimerizaciónparecentenercapacidadinductora (Kubicek y col., 1990b). Sin embargo, la1,5-celobionolactonase ha reveladocomo un potenteinductor de la formación de celulasas(Iyayi y col., 1989); la celobionolactonaprocedeprobablementede la acción conjuntade unacelulosaoxidasay de una CBH.
La entradaen la célula de los inductoresformadosen el medioextracelularesposibleatravésde una 13-disacáridopermeasalocalizadaen la pareddel hongo (Fritschery col., 1990).Se desconocela naturalezade los receptoresque transducenel efecto inductor. Recientesestudios(Messnery Kubicek, 1991)demuestranque la regulaciónde la síntesisde celulasastiene lugar sobrela transcripcióny no sobre la traducción como se había propuestoconanterioridad(Nísizaway col., 1972).Dichosestudiosdescartan,porotra parte,quela represiónpor catabolito sea un mecanismoreguladorde la síntesis de celulasas,como se aceptabatradicionalmente(Kubicek y col., 1990b).
3.4. Relaciónestructura-funciónen celulasas
Las principalescelulasasde T. reesei (CBH 1, CBH II, EG 1 y ECli III) compartenunaarquitecturacomún: el centroactivo se localiza en un dominio o núcleo catalítico; uno de losextremosde la proteínaconsisteen un dominio de unión a celulosa;ambosdominiosseunenmedianteuna bisagraglicosilada(Fig. 7) (Cíaeyssensy Tomme, 1990).Más de 60 celulasasyxilanasasde diverso origen han sido donadasy secuenciadas.Muchasde estas13-glicanasaspresentanuna organizaciónsimilar a la descritaparalas celulasasde T. reesei (Gilkes y col.,1991).
Figura 7. Modelo de estructura tridimensional de una enzima celulotitica de 71 reesel (Knowles y col.,1987). Se señala el punto de ataque proteolitico por papaína en CBH 1 y CBH IT (Claeyssens yTommne, 1990).
Los dominioso núcleoscatalíticoscomprendenla mayorpartedecadacelulasade T.reesel (unos 300 aminoácidos).Análisis porproteolisislimitada de CBH 1 y CBH II (Tommey col., 1988),y el estudiodel núcleocatalíticodeEGIII aisladodel caldode cultivo del hongo(StAhlbergy col., 1988),permitierondemostrarquelos núcleoscatalíticospuedenactuarcomoentidadesindependientesconservandointactasu capacidadparahidrolizar sustratossolubles;
Centro activo

INTRODUCCION 13
disminuyedrásticamente,en cambio,suactividad sobrecelulosanativa,de lo quesededucequeel núcleocontieneel centroactivo de la enzima,pero no el sitio de unión a celulosa.EIanálisispor gruposhidrofóbicos(HCA, Gaboriaudy col., 1987)de lassecuenciasconocidasdecelulasas(cerca de 50) ha permitido clasificar en seis familias sus núcleoscatalíticos,a pesarde laaparentediversidadentrelas celulasasde distinto origen o incluso procedentesde un mismoorganismo(en T. reeseisólo CBH 1 y EQ 1 exhibenun alto gradode homología)(Henrissatycol., 1989; Gilkes y col., 1991).En estaclasificaciónEClI III quedaincluida en la familia A, lamásnumerosa;CEN II estáencuadradaen la familia B; CBH 1 y EG 1 pertenecenambasa lafamilia C (Tabla III).
Tabla III. Clasificaciónen familias de las celulasasen funciónde la homologíadeterminadapor HCA de sus núcleos catalíticos (Henrissaty col.,1989). Elanálisisde secuenciaspublicadascon posterioridada estetrabajoha supuestolaincorporaciónde nuevosmiembrosa cadafamilia (Gilkes y col., 1991).
Familia Código Enzima Organismo
A A ¡ FO Bacillus sutúhis£0 C CIastridi¡~nz thcr,nocelkn,
A £0 III lYichader,,zaA
4 £6 E Closiridiun, Ihermocel/un,
A5 EG B Claslridiun, ¡hermocúlun,A6 EG Badilus sp. sirain 1139A,, A8 £0 Bocilus sp. strain N-4
(genespNKI andpNK2)A9 EG Z Erwinia chrysanthenii
A,0 FO 1 Schizophy//uni comniune
B B, CHI] II Trichoderina reeselFO A Ce/luíamonasfinaFO Serepwmyces sp. (KSM-9)
C C, CHI-II Phonerochae~e chrvsosporiu.n
CHI] 1 Trichodermo reese,FO 1 Trichoderma
D D, £0 Ce//u jotuonas udc
FO A Clos¿ridium mher,nocel/um
E E, FO O Closzridiuni thermocelluniE, £0 Escudomonas fluoresceos
E E, GB li Ce/luíamonasfinilE, Xyn Z Closíridiun, thermoceiium
Xyn Crypwcoccus alhidu9
El alineamientodesecuenciasen cadafamilia revelala existenciadealgunosaminoácidosácidoso básicosconservadosquedebenseresencialesbien parala actividadenzimática,bienparamantenerla estructuratridimensionalde laproteína(Henrissaty Mornon, 1990).Estudiosde modificación química y mutagénesisdirigida permiten determinarla función de dichosaminoácidos.Por el momentoseha identificadoun carboxioesencialen CEH 1, implicadoprobablementeen la unión del sustratoo en la estabilizacióndel complejo enzima-sustrato

INTRODUCCION 14
(Tommey Claeyssens,1989; Mitsuishi y col., 1990). En EG 1, el carboxilo homólogoparecetener una implicación más directa en el fenómenocatalítico (Mitsuishi y col., 1990). Lasglicosidasas,comomuchashidrolasas,actúanporcatálisisácido-basegeneral;enla granmayoríade las glicosidasascuyo mecanismocatalíticoesconocido(lisozima,B-galactosidasa,amilasa)al menosun grupocarboxioparticipadirectamenteen la catálisis(Sinnot, 1990).
Estudiosde resonanciamagnéticanuclearde la reacciónde hidrólisisdesustratosdebajopesomolecularcatalizadapor celulasasde distintas familias han reforzadola validez de laclasificaciónantescomentada;lashomologíasdetectadasentremiembrosdeuna mismafamiliaresponden a similitudes en la topología del centro activo, y en consecuenciasuestereoespecifidades la misma. Las celulasasde las familia A (EG III), C (EG 1 y CBH 1) yF actúancon retenciónde la configuracióndel carbonoanomérico,esdecir, los productosson13-glicósidoscomolos sustratos,mientrasque las encuadradasen las familias E (CBH II) y E,invierten la misma (Knowlesy col., 1988;Claeyssensy col., 1990b; Barrasy col., 1992; Gebblery col., 1992).
Los núcleoscatalíticosdeCBH 1 y CBH II han sido cristalizados(Bergsforsy col., 1989),y la estructuratridimensionaldel segundoha podidoserdeterminadapor difracción de rayosX (Fig. 8) (Rouvineny col., 1990);consisteen un barril a/lA formadopor 7 láminas13 paralelas,conectadaspor a-hélices;dosde las horquillasqueunen láminas¡3 y a-hélices,seextiendenamodo de túnel para facilitar la unión del sustrato.El sitio activo está constituido por los
extremosC-terminal de las láminas13, al igual que en otrasenzimasde estructuraparecida(triosa fosfato isomerasa).Los aminoácidosesencialespara la catálisis parecen ser dosaspárticos.Ambos estánsituadosen el mismo lado del sitio activo, como cabía esperarde laestereoquímicade la reacciónenzimática.
Figura 8. Representaciónesquemática de la estructuratridimensional del núcleo catalítico de CBH11 (Tecri y col., 1990).

INTRODUCCION ‘5
La functón dela bisagraglicosilada(Fig. 7, pág. 12) es la de conectarel núcleocatalíticoy el dominio de unión a celulosa. Se trata de un nexo flexible lo que ha impedido lacristalizaciónde celulasasintactas.Es una región rica en prolinasy aminoácidoshidroxilados.Estos últimos están fuertementeglicosilados, y las prolinas intercaladaspuedenayudar alempaquetamientode las cadenasde azúcares,como ocurre en otrasglicoproteinas(Allen,1983); la función de la glicosilación ha sido objeto de muchasconjeturas,pero estudiosconcelulasasdonadas en E. coil (no glicosiladas en consecuencia)o hiperglicosiladasenSaccharomycescerevisiaehan desechadocualquierpapelen la activiad enzimática(Knowles ycol., 1990).La 0-glicosilaciénesnecesariaparala secreciónde celulasasen T reesei (Kubiceky col., 1990b);su presenciaademásprotegedelataqueproteolítico (Claeyssensy Tomme,1990).Las bisagrassonregionesbienconservadasen las cuatrocelulasasde secuenciaconocidade Treesei;se denominanbloquesB, y en el casode CBH II apareceduplicada(Fig. 9).
Los dominiosde uniónacelulosa(DUC) deCBH 1, CBH II, EG 1 y EGIII de T. reesei,secorrespondencon los denominadosbloquesA, regionesbienconservadasen todasellas,perosituadasen el extremoN-terminal de la proteínaen unasy en el C-terminalen otras(Fig. 9).Contienenunos30 aminoácidos,en contrastecon los casi 100 de los DUC de origenbacteriano(Gilkes y col., 1991). Entre los aminoácidosconservadosen los DUC de 71 reesei,hay cuatrocisteinasqueformandospuentesdisulfuro, dosglutaminasy cuatroresiduosaromáticos;estosúltimos son fundamentalespara la unión a celulosa(Tomme, 1991). El DUC de CBH 1 sepreparóporsíntesisquímica,y suestructurafue determinadapor resonanciamagnéticanuclearbidimensional;tiene forma de cuña,con unacarahidrofóbica y otra hidrofílica en la que trestirosinassedisponena modo de tren de aterrizajepara interaccionarcon la superficie de lacelulosa(Kraulisy col., 1989).La posibilidadde queel DUC, ademásdeunirseacelulosa,tengacapacidadparasolubilizarlahaciendounalabordezapapreviaa la hidrólisisha sido postuladaporel grupode Knowles (Knowlesy col., 1988).En el casodel DUC de Cellulomonafimi talsupuestoha sido confirmadoporestudiosde microscopiaelectrónica(Din y col., 1991),y muyprobablementeel de 71 reeseiposeala misma capacidadde disgregación.
Centro activoIntrón 8 A
* ¡ II
CaRI NR2 . .. GOOH
*CERn D mr
*EGIII
(*) ____________
EG 1 _____________________ _____________________
Figura 9. Representación esquemática de las celulasas de T. reesei. El dominio de unión a celulosa(bloque A) conecta mediante una bisagra (bloque B) con el dominio catalítico, donde se localiza elcentro activo (). (Knowles y col., 1988).
Comolos núcleoscatalíticos,los DUC dediferentesmicroorganismoshansidoclasificadosen familiasdeacuerdocon su gradode homología.Pero,al contrariode lo quesucedíacon losprimeros,dentrodelas celulasasde un microorganismosólo seencuentranDUC deuna mismafamilia. Estehechopareceindicar quelos DUC se unierona los núcleoscatalíticos en unaetapa tardía de la evolución de las celulasas (Béguin, 1990). Las celulasas proceden

INTRODUCCION 16
probablementede un númerolimitado de secuenciasprogenítoras,quehanevolucionadoporfusión y/o translocaciónde dominios; los DUC guardanrelacióncon secuenciasconservadasen otrasproteínasque interaccionancon polisacáridoscomo lectinas,amilasasy quitinasas(Gilkes y col., 1991).
Apartede sus implicacionesen la hidrólisis de celulosa,los DUC seperfilan como unainteresanteherramientabiotecnológica.La fusión a una proteína de interés por ingenieríagenéticapuedefacilitar su purificacióne inmovilización (Fig. 10); la viabilidad deesta ideahasido ya probadacon unalA-glucosidasadeAgrobacteriu,nsp.y fosfatasaalcalinadeE. coli (Ongy col., 1989).
oLA con~:Z:t0r~~to
K ~zv ~
i Adsorción a celulosa
Celulosa
Inmovilización con celulosa de le
un reactor enzimát¡coproteino híbrida para su empleo enDesorción de la proteína
h,brida con agua Sustralo de la enzima
4 proleolifica
del DUC4 Eliminaciónon Celulosa
Proleina purificada Producto de lareacción eruirnática
Figura 10. Esquema de las posibles aplicaciones de los dominios de unión a celulosa (DUC) en la
purificación e inmovilización de proteínas. La proteína de interés se fusiona a un DUC ( ) poringeniería genética. La proteína híbrida tiene un sitio específico de rolura proteolítica ( ) (Ongy col., 1989).
3.5. Manipulacióngenéticade T. reeseiy de sus celulasas
La viabilidad industrialde la hidrólisis enzimáticadecelulosano dependesimplementedela producción a gran escala de celulasas,sino que requiere,además,profundizar en lainvestigaciónbásicadel proceso,hastadilucidar cómo actúanlas celulasas.Así, se podrándiseñarmezclasde enzimasadecuadasparacadatipo desustrato.La ingenieríagenéticaaporta

INTRODUCCION 17
las herramientasadecuadasparatrabajaren estadirección;las celulasas“a la carta”sepuedenprepararsiguiendo dos estrategias:mezclar celulasaspuras producidaspor separado,odesarrollarnuevosmicroorganismoscapacesde producir la mezclade interés(Teeri y col.,1990).Ya se han obtenidocepasde 71 reeselcon niveles alteradosde algunade suscelulasas(Mántylá y col., 1989).
Entrelas desventajasque presentanlas celulasasde 71 reesel respectoa ciertascelulasasde otros microorganismoshay que señalarsu termostabiidadmoderada,su baja actividadespecíficasobrecelulosanativa, y su bajo contenidoen 13-glucosidasa(Klyosov, 1988). Esteúltimo punto sepuedesubsanarpor la vía antescomentada.La mejora de las propiedadescatalíticasy de la estabilidadtérmicadebeemprendersepor ingenieríade proteínas,cuyodesarrollorequiereun conocimientoexhaustivode los aminoácidosesencialesdecadaenzima.
Algunascepasde 71 reeselseencuentranentrelos microorganismoscon mayorcapacidadde secreción(hasta40 g/l) de unasólaproteína,CBH 1, de cuyo gen existe unasola copiaenel genomadel hongo (EI-Gogaryy col., 1990); resultamuy prometedorala posibilidad de usarel promotorde dicho genpara mejorar la producciónde hormonas,enzimasy proteínas engeneral con aplicacionesbiotecnológicas(industriales, clínicas, alimentarias, etc). Por elmomentoelúnico intentode producciónheterólogaen 71 reesel,por la vía indicada,ha llevadoa la obtenciónde célulastransformadasquesecretanrelativamentealtosnivelesde quimosina,correctamenteprocesaday enzimáticamenteactiva (Teeriy col., 1990).
4. ESTUDIOS PREVIOS SOBRE ENDOGLUCANASA III DE T. reesei
Entrelos componentesmayoritariosdel sistemacelulasade 71 reesel, EG III ha sido elmenosestudiado;su correspondientegen (egl3) y su cDNA fuerondonadosy secuenciados(lasecuenciade EG III se muestra en la Figura 49 (pág. 84), y ademásse realizó unacaracterizaciónparcial de la enzimapurificada a homogeneidad(Saloheimoy col., 1988). Setrata de una glicoproteina ácida; sus principales propiedadesfísico-químicasasí como sucomposiciónaminoacídicaserecogenen la Tabla IV.
En EG III, como en CBH II, el dominio de unión a celulosa(bloqueA) y la bisagraO-glicosilada(bloque B) se encuentranen el extremo N-terminal de la proteína. El dominiocatalíticoha sido aisladoen caldosde cultivo del hongo,productode la degradaciónnaturaldelaproteína;el puntode ataqueproteolíticoestáentreThr-61 y Ser-62en el bloqueE. El núcleoresultantetiene una M = 38 kDa, un pI = 4.4, y un contenidoen carbohidratosde un 4 %.
En la secuenciadeEG III existeun único punto de posibleN-glicosilación,Asn-Phe-Thr,en laposición 103-105 (St~hlbergy col., 1988).
Se ha descritola existenciade una isoenzimade EG III denominada“A 1:2”, con igualmasapero distinto pl que el productogénico original, y que apareceprobablementepordesamidaciónde éste(StAhlbergycol., 1988).
EGIII ha sido encuadradaen la familia A de celulasas(Tabla III), quecomprendehastael presente27 celulasas, la mayoría de origen bacteriano (Gilkes y col., 1991). Unaendoglucanasadel hongoSchizophyllumcomniuneguardauna estrechahomologíacon EG III(Saloheimoy col., 1988).Ambasforman la subfamilia A5 (Béguin, 1990).

INTRODUCCION IR
En cuantoa suspropiedadescatalíticas,sólo se handeterminadolos patronesde roturade algunossustratoscromofóricos(Fig. 3, pág. 8); EG III esla única celulasade 71 reesei quehidroliza el enlaceheterosídicodel 4-metilumbeliferilcelotriósido(Saloheimoy col., 1988). ECliIII, como todaslas enzimasde la familia A analizadashastala fecha,actúacon retenciónde laconfiguracióndel carbonoanoméricodel sustrato(Gebblery col., 1992).
Tabla IV. Composición de aminoácidos y principales característicasde EG III(Saloheimoy col., 1988).
Masa molecular calculada a partir de la composición
de aminoácidos y el contenido de carbohidratos
5. OBJETIVOSDEL PRESENTETRABAJO
El conocimiento profundo de las característicasestructuralesy catalíticas de cadacomponentedelsistemacelulasa,ha sido señaladocomo un requisitoimprescindibleparallegara dilucidar el mecanismode la hidrólisisenzimáticade celulosa,y porende,conseguirqueesteprocesoseaviable a nivel industrial (Béguin, 1990).
En estesentido,nuestrolaboratorioemprendióhacealgunosañosel estudiodelsistemacelulasade TrichodemiareeselQM 9414. Caracterizadaexhaustivamentela endoglucanasa1
Aa
ArgLysHisAspGluAsn
GinTrpTyrPheCysMalSerThrGlyPro
AlaValIleLeu
Total
1065
196
302511141312
4
42
44
431928
22
2123
397
2.51.51.34.81.57.66.32.83.5
3.33.01.0
10.611.110.84.87.15.55.35.8
Contenido en Carbohidratos = 15 % (p/p)
M (SOS-PAGE) = 48 lOa
M (teárioa)a = 49.8 koa
pl = 5.5
= 77000 M1•cm1
a
100

INTRODUCCION 19
(Domínguez,1991)y la lA-glucosidasa(de la Mata, 1992),consideramosinteresanteabordarlacaracterizaciónde endoglucanasaIII; setrata del componentedel sistemamenosestudiado,probablementepor dificultadesen su purificación.
El trabajo seha desarrolladode acuerdocon el siguienteesquema:
1. Purificacióny caracterizaciónestructuralde endoglucanasaIII.
- Desarrollodel métodode purificación.- Determinaciónde los parámetrosfísico-químicos(M, pI, titulación, parámetros
hidrodinámicos).- Estudiodel efectodepH, temperaturay fuerzaiónica sobrela estabilidady la
actividadde la enzima.
2. Especificidadde sustrato.Modo de acción.
- Actividad sobresustratoscromofóricos.Estudiosde partición con metanol.- Actividad sobrecelooligosacáridos.- Actividad sobresustratoscelulósicos.- Actividad sobremananos.
3. Identificación de aminoácidosesenciales.
- Estudiosdel efectodel pH y la temperaturasobrelos parámetroscinéticosde lareacciónenzimática.Determinaciónde valoresdepK degruposionizables.
- Estudiosde modificación químicacon agentesespecíficos.- Identificaciónde residuosesencialesde triptófano,susceptiblesde oxidaciónpor
N-bromosuccinimida.- Identificación de un carboxioesencialpormarcajede afinidad.

MATERIALES Y METODOS

MATERIALES Y METODOS 21
1. MATERIALES
El microorganismoempleadoen la presenteinvestigación, Trichodemzareesel cepaQM9414, fue suministradoporMycologicalServices(EEUU).La pajade trigoprocedíade unafinca de la provincia de Madrid.
Soportescroniatográficos:DEAE-SepharoseCL-ÓB y Sephadex0-25 son productosdePharmacia(Suecia); Ultrogel AcA 44 era de LKB (Suecia), actualmenteabsorbidaporPharmacia,que comercializaun gel con similarespropiedades:SephacrylSIOO HR.
Sustratos:4’-nitrofenil-J3-D-glucopiranósido,CMC (viscosidadmedia,gradodesustitución0.8),celobiosa,O-glucosay xilano seobtuvierondeFluka (Suiza),Avicel de Merck(Alemania),y LBG de Sigma(EEUU). Los siguientessustratosfueronproporcionadosgentilmentepor elDr. M. Claeyssens(Universidadde Gante,Bélgica),quien los preparódeacuerdocon métodosdescritosen las referenciasadjuntas:celooligosacáridos(van Tilbeurgh y col., 1982), metil-lA-glicósidos(Wolfrom y Hag, 1964), 2’-cloro-4’-nitrofenil-13-glicósidosy 4’-metilumbeliferil-13-glicósidos(Claeyssens,1988),exceptoMeUmbprocedentedeSigma(EEUU) y MeUmb(Glc)3de Lambda(Austria).
Proteínas: los patronesde masamoleculareran de Pharmacia(Suecia)y los de punto
isoeléctricode Biorad (EEUU). Albúmina de suerobovino y proteasaV8 se obtuvierondeSigma(EEUU) y tripsina de Serva (Alemania).El núcleo catalíticodeFO III, purificado deacuerdocon el métododescritoporSaloheirnoy col. (1988),fueamablementecedidoporel Dr.O. Pettersson(Universidadde Upsala,Suecia).
Reactivosespecíficospara modificación química: DTNB, TNM, DEPC, EDC, EAC yRKW procedíande Sigma(EEUU),FF00 de Fluka (Suiza),y N?BS deAldrich (Bélgica); esteúltimo se recristalizódos vecesa partir de solución acuosaantesde su uso. EPO3y EPO4fuerongentilmentedonadosporel Dr. P. B. H0j (UniversidadLaTrobe,Bundoora,Australia),y EPO5 por el Dr. M. Claeyssens(Universidad de Gante, Bélgica); estos productossesintetizaronsiguiendoel métodode H0j, Rodríguez,Stick y Stone(140] y col, 1989).
Todos los demásreactivosy disolventesempleadoseran de grado analítico, o superior
en casosque así lo requerían(productosparaHPLC).
2. FERMENTACION DEL HONGO
2.1. Mantenimientode la cepa
La cepaseha mantenidoa cortoy medioplazomedianteresiembrasen placasPetriconmedio agar-patata(Haynesy col., 1955), incubadasen estufade cultivo a 28
0C durante7 a 10días (hastaque se observa abundanteesporulación)y conservadasen neveraa 40C. Lasresiembrassehan efectuadocada30 días. El cultivo seha mantenidoa largopíazo medianteInmersión en parafina.

MATERIALES Y METODOS 22
2.2. Medio de producciónde celulasas
Las condicionesóptimas de fermentaciónde 71 reesel 0M94 14 para la producción decelulasas, utilizando paja de trigo como fuente de carbono, fueron determinadasconanterioridaden nuestrolaboratorio(Acebaly col., 1986).El medioempleadotienela siguientecomposición:
KH2PO4 8.00 g(NH4)2S04 1.40 gUrea 030 gTween 80 2.00 gPeptona 1.00 gDisolución de oligoelementos~” 1.00 ml
CaCI . 0.79 gMgSO47H2O 0.30 gAgua destiladahasta1000 mí; el pH se ajustaa 5.5.
<>La disoluciónde oligoelementoscontiene:
FeSO4 7H,O 4.57 gZnSO4- 7H20 2.82 gCoCí2 -61420 4.33 gHCI 5.00 mlAgua destiladahasta500 ml.
Comofuentedecarbonoseha empleadopaja detrigo molida y tamizada,con un tamañode partícula inferior a 0.177mm. La concentraciónde paja en el mediode cultivo era 25 g/l.
La fermentaciónserealiza en matracesErlenmeyerde 250 ml. En cadauno de ellosseintroducen100 ml de mediodecultivo y 2.5 g depaja.A continuaciónseesteriizaen autoclave(1 kg/cm
2, 1200C, 30 mm).
23. Inóculo
A un cultivo de resiembraen placa Petri se añadenunos10 ml de soluciónsalina(9 g/lde NaCí) estéril. La placa se agita ligeramentecon objeto de obteneruna suspensiónde
esporas;éstase transvasaaun matrazErlenmeyerconunos15 ml de soluciónsalinaestéril. Laconcentracióndeesporassedeterminaporrecuentoen unacámarade Neubauer;la inoculacióndel medio de cultivo selleva a cabointroduciendoen cadamatrazun volumen de suspensiónquecontenga— 4 . 10~ esporas.
2.4. Crecimientodel bongo
Los matracesinoculadosse introducen en una incubadora(New Brunswick, EEUU)termostatizaday provistade agitaciónrotatoria.La fermentacióntienelugara 330Cdurantelasprimeras50 h, y a 290C hastael final del proceso(duración total, 7 días); la agitación se

MATERIALES Y METODOS 23
mantieneconstantea 200 rpm. El crecimientodelhongoseobservapor apariciónde un micelioen forma de cinta, adheridoa las paredesdel matraz.
Finalizadala fermentaciónseañade1 ml deazidasódica(20 gIl) porcadalitro de medio,y secentrifugaa 1 500 g durante15 mm, con el fin de eliminarlos restosde pajay el miceliodel hongo. El sobrenadanteconstituyeel denominado“caldo crudo”.
3. PURIFICACION DE EGIII
La purificación de FO III a partir del caldo crudo seha realizadode acuerdocon elprocedimientoesquematizadoen la Figura 11.
3.1. Precipitaciónconsulfatoamónico
A un volumendeterminadode caldo crudo (normalmente500 mi) se añadelentamentesulfatoamónicofinamentepulverizadohastaalcanzarun 20 % (p/v) de saturación.Despuésde30 mm de agitaciónen frío (— 40C) la mezclasecentrifugaa 12 000 g durante20 mm. Alsobrenadanteobtenidose le añadesulfato amónicoparallegar a un 60 % de saturación,y tras30 mm de agitaciónen frío secentrifugaen las condicionesantesdescritas,desechándoseelsobrenadante.
El sedimentoresultantese resuspendeen el mínimo volumen posiblede aguadestilada(unos30 mI) y seprocedea sudesaladomediantecromatografíaen unacolumnadeSephadex0-25Medium(5.5x20cm) equilibradaen agua;serecogenfraccionesde7.5 mi, y sejuntanparasuliofilización las quecontienenproteína(apreciablea simplevista por su íntima asociaciónconpigmentospresentesen el mediode cultivo), queesexcluidade la columna.La elución de
la sal sedetectacon cloruro bárico,ya queel sulfato báricoesinsolubley forma un precipitadoblanco.
3.2. Cromatografíaen DEAE-Sepharosea pH 7.0
El residuosecoprocedentedela precipitaciónsalinaseresuspendeen un volumenmínimo(10 mí) de tampónfosfato sódico5 mM, pH 7.0 con azida sódica(0.2 gil); trascentrifugación(3 000 g, 10 mm) se aplica en una columna de DEAE-SepharoseCL-ÓB de 2.6x42 cm,equilibradaen el mismo tampón. La elución se lleva a cabo sucesivamentecon 500 ml deltampóndeequilibrado,250mIdeun gradientelinealde NaCí(0-500mM) en el mismotampón,y 250 ml de tampóncon NaGI 500 mM. El flujo se mantieneconstantea 60 ml/fi con unabombaperistáltica(Pharmacia,Suecia).Se recogenfraccionesde6 mí, en lasquesedeterminanactividadescelulolíticasy hemicelulolíticas.La concentraciónde proteínase estimapor lecturade la absorbanciaa 280 nm.
Las fraccionescorrespondientesal segundopico de actividadCMCasase reúnen,y seconcentran,primeropor ultrafiltración (unidad202de Amicon, EEUU, con membranaPM-lode Diaflo, EEUU) y finalmentepor evaporaciónavacio (Univap de Uniscience,Reino Unido).

MATERIALES Y METODOS 24
CALDO CRUDO
PRECIPITACION CON <NH4)2S04(20 % saturación)
Centrifugación
Sobrenadante
Sedimento
PRECIPITACION CON (NH4)2S04(60 % saturación)
Centrifugación
Sobrenadante
Redisolución en agua
Desalado <Sephadex 0-25)
Liofilización
Residuo seco
CROMATOGRAFíA DE INTERCAMBIO ANIONICOen DEAE-Sepharose a pH 7.0
2~ pico de actividad CMCasa
Ultrafiltración
Concentración
CROMATOGRAFíA DE PENETRABILIDADen Ultrogel AcA-44
Pico de actividad CMCasa
1ler pico de actividad CMCasa
1CROMATOGRAFíA DE INTERCAMBIO ANIONICO
en DEAE-Sepharose a pH 6.5
Desalado (Sephadex 0-25)
Liofilización
EG III “pura”
Sedimento
Figura 11. Esquemadel procedimientode purificación de FO III.

MATERIALES Y METODOS 25
3.3. Cromatografíaen Ultrogel Aca-44
El concentrado(3 mí) de FO III procedentede la cromatografíaanteriorseaplica enuna columnade Ultrogel AcA-44 (1.6x95 cm) equilibradaen imidazol-HCI 8mM, pH 6.5 conazidasódica (0.2g/l). La elución se lleva a cabocon el mismotampón,a un flujo de 10 ml/h.Se recogenfraccionesde 2 ml en las que sedeterminaactividadsobreCMC y absorbanciaa280 nm.
3.4. Cromatografíaen DEAE-Sepharosea pH 6.5
El pico de actividadCMCasade la cromatografíade penetrabilidadse cargaen unacolumnadeDEAE-SepharoseCI-6B equilibradaen imidazol-HCI8mM, pH6.5con azidasódica(0.2 g/l). La columnase lava con 50 ml deestetampón,antesdeaplicarun gradientelineal deNaCí (200 mí, 0-300mM). El flujo semantienea 30 ml/h, y serecogenfraccionesde 3 ml enlas que sedeterminaactividadsobreCMC y absorbanciaa 280 nm.
El primerpico de actividadCMCasa,correspondientea EO III, seconcentrahastaunos10 ml por evaporacióna vacio, y sedesalaen columnasde Sephadex0-25 equilibradasyeluidascon agua destilada(columnasPD-i0, Pharmacia,Suecia).La proteína así purificada,y libre de sales,se liofiliza.
4. DETERMINACIONES ANALITICAS
4.1. Cuantificaciónde proteína
La concentraciónde proteína en los distintospasosdel procesode purificación sehadeterminadoporel métododeLowry (Lowry y col., 1951),empleandoalbúminadesuerobovinocomopatrón.En elcaldo crudoy el precipitadoconsulfato amónico,antesde la valoración,seprecipitacon ácido tricloroacéticoal 10 % (p/v) (de la Mata, 1992) con el fin de eliminar loscompuestosfenólicosprocedentesde lignina queinterferiríanen la determinacióndeproteínaal reducir el reactivode Folin.
La concentración de FO III, una vez purificada, se ha determinado porespectrofotometríateniendoen cuentael valor del coeficientede extinción molar a 280 nm(77 000M’ - cm’) descritopreviamente(Saloheimoycol., 1988).En lacuantificacióndel núcleocatalítico se ha empleadoun valor de e2~ = 62 700 calculadoa partir de su composicióndeaminoácidos(Stáhlbergycol., 1988)yconsiderandoe,~Trp = 5500M
4 -cm’y c280Tyr = 1100
cm’.
4.2. Valoraciónde azúcaresreductores
Se ha seguidoel métododel ácido3,5-dinitrosalicílico(Miller, 1959; Domínguez,1991)con glucosacomo patrón. En las muestrasen que erade esperaruna baja concentracióndeazúcaresse hanañadido100ggde glucosaantesdela adicióndelreactivo(Woody Bhat, 1988)y la lectura de absorbanciaseha realizadoa 540 nm (en lugarde a 640 nm), con el objeto deaumentarla sensibilidad.

MATERIALES Y METODOS 2<
4.3. Valoración de glucosa
La valoraciónespecíficadeD-glucosaseha llevadoa caboporel métodode la EJ-glucosaoxidasa-peroxidasa,utilizando un preparadocomercial(BoehringerMannheim,Alemania), ysiguiendolas instruccionesdel fabricante.
4.4. Separacióny análisisde celooligosacáridosy derivadospor HPLC
Los productos de la degradaciónenzimática de celooligosacáridosy sus derivadosmetilados,sehanseparadoenun sistemaHPLC (Waters,EEUU) conunacolumnaRsilPolyol(0.46x25cm) de Biorad (EEUU). La elución se lleva a cabocon acetonitrio:agua(60:40) encondicionesisocráticas(flujo de 1 mí/min) y losazúcaresse detectanpor cambiosen el índicede refraccióndel eluyente(detectorR401 de Waters,EFUU). El productocorrespondienteacadapico en los cromatogramasseidentificaporsu tiempode retencióncaracterístico(Tablay); la cuantificaciónse realizapor interpolaciónde la altura en unacurva patrón específicapara cadaproducto.
Tabla V. Tiemposde retenciónde los distintos celoligosacáridos((Oic)~) y susderivados1-metilados(Me-(Glc)~)enlas condicionescromatográficasdescritas.
Azúcar t~ (mm) Azúcar tr (mm)
Ole 4.4 Me-Olc 3.8(Glc)2 5.0 Me-(Olc)2 4.2(Glc)3 5.6 Me-(Olc)3 4.7(Olc)4 6.3 Me-(Glc)4 5.3(Olc)5 7.2
Los derivadoscromofóricosde celotriosaMeUmb(Glc)3y CNP(Glc)3y los productosdesu hidrólisiscatalizadaporEGIII sehananalizadoporHPLCenun sistemaBeckman(EEUU)computerizado(SystemGoid), conuna columnaSpherisorbN112 (0.46x25cm) suministradaporTecnokroma(España).La elución se realiza en isocrático en el caso de CNP(Olc)3 (50 %acetonitrilo, 50 % fosfatosódico 1 mM, pH 7.3; flujo 0.3 mí/mm), y en gradienteen el casodeMeUmb(Glc)3(100 % acetonitrioa 70 % acetonitrilo,30 % agua,en 3 mm, con un flujo de
Tabla VI. Tiempos de retención de MeUmb(Glc)3 y CNP(Olc)3 y de sus
productosde degradaciónen las condicionescromatográficasdescritas.
Producto t~ (mm) Producto t~ (mm)
CNP 6.81 MeUmb 3.31CNPOIc 7.12 MeUmbGlc 3.70CNP(Olc)2 7.49 MeUmb(Glc)2 4.28CNP(Olc)3 8.90 MeUmb(Olc)3 4.68

MATERIALES Y METODOS 27
2 mí/mm). Los derivadoscromofóricossedetectanporabsorbanciaa 313 nm (detectorSystemGoId 166 de Beckman,EEUU).El productocorrespondientea cadapico en los cromatogramasse identifica por su tiempo de retencióncaracterístico(TablaVI); la cuantificaciónse realizapor interpolacióndel áreaen unacurva patrón específicapara cadaproducto.
5. ENSAYOS ENZIMATICOS. ANALISIS DE DATOS
A lo largodel procesode purificaciónde FO III la actividadenzimáticaseha expresadoen unidadesinternacionales(UI), queequivalenamicromolesdeproducto(azúcaresreductoresen la mayoría de los sustratosensayados)liberadospor minuto de incubación.
5.1. Actividad y adsorciónsobrecelulosamicrocristalina(Avicel)
En la determinaciónde la actividadAvicelasa de eluidos cromatográficos,a 1 ml demuestraseañade1 mIdeAvicel (20 g/l) en tampóncitrato sódico0.1 M, pH 4.8. La incubaciónserealiza a 5~C durante1 h; la reacciónsedetienepor inmersiónen bañode hielo; la mezclade ensayose centrifuga (5 000 g, 10 mm) para eliminar el sustrato no hidrolizado, y en elsobrenadantese valoranazúcaresreductores(apartado4.2, pág. 25).
Los productosde la hidrólisis de Avicel (10 g/l) catalizadaporFO III (8 gM) en tampónacetatosódico0.1 M, pH 5.0 a 370C,sehananalizadoporHPLC (apartado4.4, pág.26). Antesde su inyección en el cromatógrafo,las muestras(140 pI) extraídasa distintos tiemposdeincubaciónse concentranpor evaporaciónavacío hasta20 pl.
La adsorcióna Avicel de muestrasde EO III se ha determinadocomo se describeacontinuación:100 pl de FO III (0.6 pM) seañadena un volumenigual de Avicel (20 gIl), y lamezcla se incubaa 40C (para evitar la hidrólisis del sustrato)durante30 mm, con agitacióncontinua;transcurridoestetiempo secentrifuga(5 000g, 10 mm) y la enzimano adsorbidasecuantificaporvaloraciónde la actividadsobreCNP(Olc)
3del sobrenadante.
5.2. Actividad sobrecarboximetilcelulosa(CMC)
La actividad CMCasase ha determinadoa 500C,en tampóncitrato sódíco 0.1 M, pH 4.8
con CMC (10 gIl) y unadilución adecuadade la muestraenzimáticaa valorar(volumentotal,2 mí). Tras 10 mm de incubaciónla reacciónse detienepor inmersión en baño de hielo, y sedetermina la concentraciónde azúcaresreductoresliberados(apartado4.2, pág. 25).
Parala determinaciónde las constantescinéticas,EO III (60 nM) seincubaa 300C o a500C con variasconcentracionesde CMC (entre2 y 20 g/l) en tampónacetatosódico0.1 M,PH 5.0. Despuésde 2, 4, 6, 8 y 10 mm se toman alícuotas en las que se determinalaconcentraciónde azúcaresreductores(apartado4.2, pág. 25). Los valoresde K,,, y kat se hancalculadoporajuste,medianteun programainformáticode regresiónlineal (Cornish-Bowden,1976), de los datosde velocidadinicial paracadaconcentraciónde sustratoa la ecuacióndeHanes-Woolf:
S/v = Km/(k~t ‘[E]0) + S/(kat - [E]0) (1)
donde5 es la concentraciónde sustrato,y es la velocidadinicial, y [E]0 es la concentraciónínicial de enzima.

MATERIALES Y METODOS 28
5.3. Actividad sobresustratoscromofóricos:CNP(GIc)3y MeUmb(Glc)3
La hidrólisis de estossustratoscatalizadaporFO III tiene lugarpordosvíasalternativassegúnel modo de unión al centro activo de la enzima; el correspondienteanálisiscinético,basadoen los datosexperimentalesobtenidos,sedesarrollamásadelante(Apéndice,pág. 74).La vía de hidrólisis que conducea la liberación del cromóforo (CNP o MeUmb) resultaadecuadapara evaluarla actividadde FO III.
La liberaciónde CNP (pI< = 5.5) a partirde CNP(Olc)3sehadeterminadoen continuo,por espectrofotometría,a valoresdepH ligeramenteácidos(Claeyssens,1988); los ensayosserealizancon CNP(Glc), 0.5 mM y FO III 0.5-2 pM, en tampón Mcllvaine (0.1 M citratosódico/fosfatosódico) pH 5.6 (volumen total, 200 pl). Esta mezclase incuba a 25
0C, y laabsorbanciaa 405 nm seregistracada20 s (espectrofotómetroDU-8 de Beckman,EEUU).
La formaciónde MeUmb a partir de MeUmb(Olc),seha cuantificadopor fluorimetria(Claeyssens,1988); la mezcla de ensayo,con una concentración100 pM de sustratoy 25 a100 nM de enzima,en tampónacetatosódico 0.1 M, pH 5.0, se incuba a 250C; a 1, 2, 3, 4 y5 mm setomanalícuotasde 100 pl a lasqueseadiciona900 pl deglicocola0.2 M, pH 10.2paradetenerla reaccióny permitir la ionización de MeUmb (pK, = 7.2). La concentracióndeMeUmb se determinamidiendola intensidadde fluorescencia~ = 360 nm, Ae,,, = 455 nm)
e interpolandoen una rectapatrónquedebeactualizarsecadavezqueserealizauna tandademedidas.El fluorímetro empleadoha sido un MPF-44 E de Perkin-Elmer(EEUU).
El análisisde las dos vías alternativasde ataqueenzimáticose haefectúadopor HPLC(apartado4.4, pág. 26); concentracionesvariables de sustrato(MeUmb(Olc)
3 25-200 pM;CNP(Olc),50-500pM) seincubana 25
0C con FO III 50 nM, en tampónpiridina/ácidoacético10 mM, pH 5.0; a distintostiempos (5, 10, 15, 20 y 25 min) setomanalícuotasde 50 pl y seadicionana 50 pl deacetonitrio,inyectándoseen el cromatógrafo20 pl de estasolución. Losvaloresdelas constantescinéticasteóricassedeterminanporajustedelosdatosexperimentalesa las ecuaciones(10) y (11) (pág. 74), medianteun programade regresiónno lineal (BMDP-AR EMDPStatisticalSoftware,EEUU). Los valoresde las constantesaparentessedeterminansustituyendolos valoresteóricosen las ecuaciones(12), (13) y (14) (pág. 74).
Fnlos estudiosdeparticióncon ¡netanolsesiguenlos protocolosdescritos,perola mezclade ensayocontieneademásmetanol(u otro alcohol)en la concentracióndeseada(normalmente2.5 M).
5.4. Actividad sobrecelooligosacáridos
Los productos de la hidrólisis de celoligosacáridos(10 mM) catalizadapor FO III(200 nM) a 370Cen tampónpiridinaíácidoacético10 mM, pH 5.0, sehananalizadoporHPLC(apartado4.4, pág. 26). La liberación de EJ-glucosaa partir de celotriosay celobiosase hadeterminadocon mayor precisión(apartado4.3, pág. 26) permitiendola evaluaciónde losparámetroscinéticos correspondientespor ajuste a la ecuación(1) como se ha descritoanteriormente(apartado5.2, pág. 27).
5.5. Actividad sobre¡nananos
El sustratoempleadomásfrecuentementeha sido LBG, galactomanano,que sepreparadel siguientemodo: unasuspensiónde LBO (20 gil) en tampónacetatosódico0.1 M, pH 5.0

MATERIAI.FS Y METODOS 29
semantieneen agitacióndurante24 h, a temperaturaambiente,y seguidamentese centrifuga(5 000g, 20 mm) paraeliminar el sustratono solubiizado.La concentraciónrealde LBO es,en consecuencia,menor de la nominal.
En la determinaciónde la actividadmananasade eluidos cromatográficos,a 100 pl demuestraseañade1 ml de LBO (10 gIl) en tampónacetatosódico0.1 M, PH 5.0. La incubaciónse realiza a 500C durante1 h; la reacciónse detienepor inmersión en baño de hielo y sevaloranazúcaresreductores(apartado4.2, pág. 25).
Parala determinaciónde las constantescinéticas,FO III (600nM) seincuba a 500C convarias concentracionesde LBO (entre2 y 18 gIl) en tampón acetatosódico 0.1 M, pH 5.0.Despuésde 5, 10, 15, 20 y 25 min setoman alícuotasen las quesedeterminala concentraciónde azúcaresreductores(apartado4.2, pág. 25). Los valoresde K~ y k~
1 sehan calculadoporajustede los datosde velocidad inicial paracadaconcentraciónde sustratoa la ecuación(1)(apartado5.2, pág. 27).
Los productos de la hidrólisis de LBO (galactomanano)y glucomananano(20 gIl)catalizadapor FO 111 (8 pM) en tampónacetatosódico0.1 M, pH 5.0 a 37
0C,sehananalizadopor HPLC (apartado4.4, pág. 26). Antes de su inyecciónen el cromatógrafo,las muestras(140 pl) extraídasa distintos tiemposde incubaciónseconcentranhasta20 pl, por evaporaciónavacto.
5.6. Actividad sobrexilano
Enla determinacióndela actividadxilanasadeeluidoscromatográficos,a 1 ml demuestraseañade1 ml de xilano (40 g/l) en tampón citrato sódico 0.1 M, pH 6.0. La incubaciónserealizaa370Cdurante20 mm; la reacciónsedetieneporinmersiónen bañodehielo. La mezclade ensayose centrifuga(5 000 g, 10 mm) para eliminar el sustratono hidrolizado, y en elsobrenadantesevaloranazúcaresreductores(apartado4.2, pág. 25).
5.7. Actividad 6-glucosidasa
En la determinaciónde la actividad13-glucosidasade eluidos cromatográficos,la mezclade ensayo(2 mí) contiene4-nitrofenil 13-D-glucopiranósido5 mM en tampón citrato sódico0.1 M, pH 4.8 y unadilución adecuadade la muestraavalorar.La incubaciónserealizaa 500Cdurante10 mm; la reacciónsedetienepor inmersión en baño de hielo, y seadicionaNaOH0.1 M (3 mí) paradeterminarla concentraciónde4-nitrofenolpor lecturade la absorbanciaa405 nm.
6. TECNICAS ELECTROFORETICAS
6.1. Electrol’oresis en gelesde poliacrilamida en presenciade SDS
Se ha realizadoen geles al 10 % (p/v) de acrilamida,segúnel método descrito porLaemmli (1970) y modificadopor Chappmanny col. (1976).El equipoempleadoha sido unsistemavertical de placas (20x20 cm) SE 600 de Hoeffer (EEUU). Las proteínas se hanvisualizadopor tinción con azul Coomassie.

MATERIALES Y METODOS 30
6.2. Isoelectroenl’oqueanalítico
Se ha llevadoa cabocongelesde poliacrilamidapreparadoscon anfolitosdepH 3.5 a 9.5,Ampholine PAO, productocomercialde Pharmacia(Suecia),siguiendolas instruccionesdelfabricante.El equipoempleadoha sido un Multiphor de LKB (Suecia).Las proteínasse hanvisualizadopor tinción con azul Coomass¡e.
6.3. Curva de titulación
Se ha realizado en un equipo Phast System de Pharmacia(Suecia) con geles deisoelectroenfoquedepH 3 a 9, preparadosporPharmaciaparaestesistema.El procedimientoexperimentalempleadoha sido el propuestoporel fabricante:un gel de isoelectroenfoquesesometea un campoeléctrico paraestablecerun gradientede pH; seguidamenteseaplica laproteína en una línea perpendicularal gradientecreado,y que pasapor el centrodel gel;finalmentese lleva a cabounaelectroforesisaplicandoun campoeléctricoentrelos extremosdel gel paralelosa la línea de aplicación.Lasproteínassehanvisualizadopor tinción conazulCoomassieo con nitrato de plata.
6,4. Localizaciónde actividadesenzimáticasen los gelesde isoelectroenfoque
En los gelesde titulación con el pH antesde la detecciónde actividadesenzimáticasselava el gel en tampónacetatosódico0.1 M, pH 5.0 durante5 mm.
La actividad sobre MeUmb(Glc)1 se ha localizado siguiendoel método descrito porSaloheimoy col. (1988): finalizadoel isoelectroenfoquese rocía el gel con unasolución 1 mMde MeUmb(Olc)3 en tampón acetatosódico0.1 M, pH 5.0; la liberaciónde MeUmb se hacepatenteiluminandoel gel con una lámparaUy.
La actividad sobreLBG se ha localizado medianteincubacióndel gel a 500C, durante
25 mm con una réplica de agar(20 gIl) con LBO (4 gIl) en acetatosódico 0.1 M, pH 5.0; acontinuaciónla réplica se lava sucesivamentecon etanol al 50 % (y/y) y NaCí 1 M, y sesumergeen unasoluciónde rojo Congo(2 gIl); transcurridos45 mm selavadosvecescon NaCí
1 M, apareciendoentoncesbandasclarasdondeel sustratoha sido hidrolizado.
7. CARACTERIZACION ESTRUCTURAL DE EG III
7.1. Determinaciónde la masamolecular
Se ha realizadopor medio de electroforesisen SDS, así como por cromatografíadepenetrabilidaden UltrogelAcA-44. En el primercasolasproteínasempleadascomo patrónhansido: a-lactoalbúminade bovino (14.4kDa), inhibidor detripsina de soja(20.1 kDa), anhidrasacarbónicade eritrocito de bovino (30 kDa), ovoalbúmina(43 kDa),albúminadesuerobovino(67 kDa) y fosforilasab de músculode conejo (94 kDa). En el segundosehan empleadolosmismospatronesexceptoa-lactoalbúmina,sustituidapor citocromoc de corazónde caballo(12.3kfla). En elcálculodelvolumendeexclusióndela columna(y
0 = 65.9 mí) seha utilizado
azul de dextrano,y en la determinacióndel volumen total (‘
1k = 184.4 mí), 4-nitrofenol. Elcoeficientede partición K~ sedefine como: (Ve¡uáón - V
0)/(V1 - y).

MATERIALES Y METODOS 31
7.2. Determinación del punto isoeléctrico
Seha llevado a cabopor comparaciónde la posiciónen la que seestabiizaEO III conla de patronesde pl conocido: ficocianina (azul), 4.65; B-lactoglobulinaE, 5.10; anhidrasacarbónicabovina, 6.00; anhidrasacarbónicahumana,6.50; mioglobinaequina(marrón), 7.00;mioglobina de ballena(marrón),8.05; a-quimotripsina,8.80; y citocromoc (rojo), 9.60.
7.3. Análisis de aminoácidos
La composición de aminoácidos se ha determinado con un analizador automático DurrumD-500,despuésde hidrólisis ácidadelas proteínasen HCl 6 M con fenol2 g/l, a 1 100C durante24 h.
7.4. Centrifugación analítica
Los paramétroshidrodinámicos de FO III se han determinado empleando unaultracentrífugaanalítica MSE Centriscan75 equipadacon un sistemaóptico de schlieren.Dossolucionesde EG III de distinta concentración(0.6 y 1.2 mg/mí) en acetato sódico 0.2 M,pH 5.0 se introdujeronen sendasceldilias de 400 pi de capacidad;la centrifugaciónse llevó acabo a 38 500 rpm (4 034 rad/s),a 200C, durante3 h, registrándosegráficosde schlierencadahora.El valor del coeficientede sedimentacións se ha calculadoa partir de la representaciónde la posicióndel máximo en los gráficos,r, a cadatiempo, t, de acuerdoconla función:
£ = <1/c*9)~ d(ln r)/dt (2)
donde w es la velocidad angular.
El valor de s~ se ha obtenido introduciendofactorescorrectoresparala viscosidady ladensidad(Tanford, 1961). La razón friccionalf/f, se ha evaluadoutilizando las ecuacionesdeSvedberg(3) y Stokes(4):
II = sNvf/<1 - v~p) (3)
y
considerandolos valoresde masamolecular,M (49.8 kDa), y volumen especificoparcial, y
(0.702ml/g) previamentedescritos(StAhlbergy col., 1988)
7.5. Rotura con bromuro de cianógeno
EO III (1 mg) se disolvió en 200 pi de Tris-HCI 0.1 M, pH 8.8 con OuHCI 6 M. Tras
reducir (5 ¡sí de OTE0.5 M, atmósferade N2, 15 h) y carboximetilar(9 pl de IAAm 0.5 M,
30 min) la proteína, se eliminaron las salesy secambió el disolventea ácido acéticoal 50 %(y/y) en una columnade Sephadex0-25 (PD 10 de Pharmacia,Suecia); a continuaciónlamuestrase liofilizó y se resuspendióen 50 pl de ácido fórmico al 70 % (y/y). A estasoluciónse añadieron10 pl de CNBr 0.5 M, y la mezcla se incubó a temperaturaambientey enoscuridad;transcurridas15 h se secó en atmósferade N~ y se resuspendióen tampón de
muestrasdeelectroforesis(SDS),aplicándoseenun geldepoliacrilamidaconunaconcentracióndel 18 % (p/v) de acrilamida.

MATERIALES.Y METODOS 32
716. Determinación de grupos sulihidrilo libres
La determinaciónde cisteinaslibres se realizó con el reactivo de Fllman (DTNB) deacuerdocon el métododescritopor Habeeb(1972).
8. EFECTODEL pH Y LA TEMPERATURA SOBRE LA CINETICA ENZIMATICA
Seha estudiadoel efectodelpH y la temperaturaen la reaccióndeliberaciónde MeUmbapartir de MeUmb(Olc)3catalizadapor FO III. Las cinéticas se han realizado de acuerdoconla metodologíadescritaen el apartado5.3 (pág. 28), fijando la concentraciónde enzimaen50 nM, y variandoel pH de la mezclade ensayoentre4.0 y 6.3 (intervalo de estabilidadde laenzima), y la temperaturaentre 30 y 55
0C (límite superior de termoestabilidad).En lostamponesempleadosen esteestudio(50 mM acetatosódico/SOmM fosfato sódicoajustadosal pH deseadocon NaOH o HCI) se ha mantenidoconstantela fuerza iónica en 0.2 M,mediantela adiciónde cantidadesapropiadasde NaCí,calculadasconla ayudade un programainformático (Ellis y Morrison, 1982). La constantede especificidad(k~,
1/K~,) de la reacciónacada pH y temperaturase ha determinadopor ajustede los datosde velocidadinicial a laecuación(16) (pág. 74), medianteun programade regresiónlineal.
Los valores de pICa a cadatemperaturase han calculadopor ajuste de los datosdeconstantede especificidada la ecuación (5), mediante un programa BMDP-AR(BMDPStatisticalSoftware,EEUU) de regresiónno lineal:
= c/(1 + Ka/[H~1) (5)
dondec es el valor máximo teórico de k~t/Km, Ka es la constantede disociaciónde un grupode la enzimaque estáprotonadoen la forma activa de la misma,y [Hl] es la concentraciónmolar de protones.
La entalpíade ionización ( H10) se ha determinadopor ajustede los datosde pKa adistintastemperaturasa la ecuación(6):
pK, =zNH0,,/(2.3‘R ~T) + b (6)
dondeR es la constantede los gases,T la temperaturaabsolutay b unaconstante.

MATERIALES Y METODOS -33
9. MODIFICACION QUIMICA DE EG III
9.1. Modificación con dietilpirocarbonato (DEPC)
FO III 4 pM se incubé a temperatura ambienteen distintos tampones (MES 50 mM, pH6.5; fosfato sódico 100 mM, pH 6.0) con DEPC 0.5-2.5 mM (la solución madre de DEPC era50 mM en etanolpuro); a distintostiempos,hasta30 mm, setomaronalícuotasde 10 pl a lasqueseadicionaron50 pl deimidazol2 mM en tampónMdllvaine pH 5.6. La actividadresidualsobre CNP(Olc)3 (liberación de CM’) se determinédespuésde añadir 100 pl de CNP(Olc)31 mM en tampón Mcllvaine pH 5.6, y secomparécon la de un controlincubadosin DEPC.
La concentraciónefectivade la partidadeDEPCempleadasedeterminópor el métododescritopor Mijes (1977).
92. Modificación con tetranitrometano(TNM)
EG III 4 pM se incubéa temperaturaambienteen tampónTris-UCI 50 mM, pH 8.3, conTNM 5 mM (soluciónmadrede TNM, 80 mM en etanolpuro); a distintostiempos,hasta2 fi,setomaronalícuotasde 10 pl a las que seadicionaron50 pl de cisteina 1.25 mM en tampónMcílvaine pH 5.6. La actividad residualsobreCNP(Olc)3 (liberación de CNP) se determinódespuésdeañadir 100 pl de CNP(Glc)3 1 mM en tampón Mcllvaine pH 5.6, y secomparéconla de controlesincubadossin TNM.
9.3. Modificación con carbodilmidas <EDC y EAC)
FOIII 4 pMse incubé a temperatura ambiente en distintos tampones (MES 50 mM, pH
5.1 ó 5.6; fosfato sódico 50 mM, pH 3.1 6 4.5; piridina-UCI 100 mM, pH 4.0) conconcentraciones de carbodiimida(EAC o EDC)de hasta10 mM; adistintostiempos,hasta3 h,se tomaron alícuotas de 10 pl a las que se adicionaron 50 pl de tampónMcílvaine pH 5.6. Laactividad residual sobre CNP(Olc)3 (liberación de CNP) sedeterminódespuésdeañadir100 plde CNP(Olc)3 1 mMen tampón Mcllvaine pH 5.6, y secomparéconla de controlesincubadossm carbodiimida.
Paracomprobarsi la falta de inactivaciónen estascondicionessedebía a la reversiónpor aguade la modificacióncovalentede un grupocarboxioesencial(Hoarey Koshland,1967),la incubacióntambiénseefectuéen presenciade esteretílico de glicina (4 mM), nucleófiloalternativo capazde hacerirreversiblela supuestamodificación.
9.4. Modificación con N-etoxicarbonil-2-etoxi-1,2-dibidroquinoleina (EEDQ)
FO III 4 pM seincubéa temperaturaambienteen distintostampones(MES 50 mM, pH5.1 ó 5.6; fosfato sódico 50 mM, pH 3.1 6 4.5; piridina-HCI 100 mM, pH 4.0) conconcentracionesde EEDQdehasta10 mM; a distintostiempos,hasta3 h, se tomaronalícuotasde 10 pl alas queseadicionaron50 pl de tampónMcllvaine pH 5.6. La actividadresidualsobreCNP(Olc)3 (liberación de CNP) sedeterminédespuésde añadir 100 pl de CNP(Olc)3 1 mMen tampónMcllvaine pH 5.6, y secomparécon la de controlesincubadossin EEDQ.

MATERIALES Y METODOS 34
9.5. Modificación con reactivo 1< de Woodward (RKW)
FO III 4 pM se incubé a temperaturaambienteen distintostampones(MES 50 mM, pH4.5, 5.1, 5.6 é 6.4; fosfato sódico50 mM, pH 3.1 ó pH 6.0) con concentracionesde RKW dehasta 10 mM; a distintos tiempos, hasta3 h, se tomaronalícuotasde 10 pl a las que seadicionaron 50 pl de tampón Mcllvaine pH 5.6. La actividad residualsobre CNP(Olc)3(liberaciónde CNP) sedeterminédespuésde añadir100 pl de CNP(Glc)3 1 mM en tampónMcllvaine pH 5.6, y secomparécon la decontrolesincubadossin RKW.
Paradeterminarel númerodecarboxiosmodificadoscuandosealcanzala inactivacióntotal dela proteínaseprocedióa incubarFO III 10 pM con RKW 100 mM en tampónfosfatosódico0.1 M pH 6.0. El pH semantuvoconstantea 6.0 medianteadición continuade NaOH0.5 M (la hidrólisis espontáneadel reactivo K acidifica la mezcla de reacción) con unamícrojeringuilla montadaen un nonius, de forma que cada giro de la rueda del noniusprovocabala adición de 1 pl de NaOH al medio de reacción(1 mí), en el cual se habíaintroducido el electrodode un pUmetroparapoderseguir la evolucióndel pH. Transcurridos30 mm la enzimaestabatotalmenteinactivada,y entoncesse separódel excesode reactivomediantedospasoscromatográficossucesivosen Sephadex0-25(columnaPD 10 de Pharmacia,Suecia)equilibraday eluidacon tampónTris-HCI 50 mM, PH 8.3.A continuaciónseregistraronlos espectrosUV (360 a 240 nm) de la muestrade FO III modificada,y de unamuestracontroltratadadel mismo modo pero en ausenciade RKW. El númerode carboxiosmodificadossedeterminóconsiderandoel coeficientede extinciónmolar a340 nm (7 000 M’ - cm’) (ShinayBrewer, 1985)delfenol cromofóricoqueseincorporaal grupocarboxiocomoconsecuenciadela reacciónde modificación (Pétra, 1971).
9.6. Estudios de protección
En aquelloscasosen queseobservóinactivaciónde la enzima,serealizaronexperimentosde proteccióndel centro activo de FO III, preincubando5 mm la solución enzimáticaconcelobiosa250 mM antesde la adicióndel modificador.Simultáneamentese trataronmuestrascontrol de la enzimasin celobiosa.El procedimientoexperimentalempleadoen estosestudiosespor lo demásigual al especificadoparacadauno de los reactivosensayados.
10. MODIFICACION DE TRIPTOFANOSCON N-BROMOSUCCINIMIDA (NES)
10.1. Reaccióncon NES
La oxidación de residuosde Trp de FO III con NBS se ha llevado a cabo siguiendoesencialmenteel métododescritoporSpandey Witkop (1967).Unasoluciónde EOIII 9.1 pMen acetatosédico50 mM, pH 5.0 sepipeteaen doscubetasde cuarzode 1 cm depasoóptico(1 ml en cadauna), colocandouna en la célula de referencia y la otra en la de muestra, de unespectrofotómetrode doble haz (Uvikon 810 de Kontron,Suiza);la temperaturasemantienea 15
0C con un baño de aguatermostatizado.Se registrael espectrodediferenciaentre 330 y230 nm, y se tomanalícuotasde 20 pl de cadacubeta;a continuaciónseañaden20 pl de NES0.15 mM (en aguadestilada)a la solución “muestra”, y se agita rápidamente;en la solución“referencia” se añaden20 pl de aguaen lugardel reactivo. Despuésde 2 mm se registrael

MATERIALES Y METODOS 35
espectrode diferencia,se extraenalícuotasde 20 pl y se diluyen con L-triptófano 0.1 mM entampónacetatosódico50 mM, pH 5.0 (volumennecesarioparaquela concentraciónde enzimasea 1.2 pM) paraeliminarcualquierNESremanente.La adicióndel oxidante(agua)sesucedepor esteprocedimientohastaquela soluciónde proteínamodificadaseenturbia.Finalmentese determinanlas actividadessobreMeUmb(Olc)3 y CNP(Olc)3,así como la capacidaddeadsorcióna Avicel, de las alícuotasde enzimatomadas.
FI mismo métodose sigueparala modificación del núcleocatalíticode EO III, o en losexperimentosde proteccióncon ligandos.
102.Cálculo del número de Trp oxidados
FI número de Trp oxidados (n~0~) en cada estadio de modificación m seha calculado deacuerdo con la fórmula empírica propuesta por Spandey Witkop (1967):
n~0~ =—AA2~4.31/(5500’[E]m) (7)
donde A2~ es la diferencia de absorción a 280 nm entre la proteína modificada y el control(solución “referencia’), 1.31 es un factor de corrección empírico, 5 500 es el coeficientedeextinción molar del Trp a 280 nm, y [E],,, es la concentración de proteína tras m ciclos demodificación cada ciclo supone una dilución 98011000 de la solución precedentey enconsecuencta[E],,, = [E]0 .(980/1000)m.
10.3. Preparaciónde fornasde EG III con distinto gradode oxidación (W10~ y ~
Paraprofundizaren el estudiode la modificación de FO III con NBS ha sido necesarioprepararcantidadesmayoresde enzimacon distintosgradosde oxidación.FO III W10~y W~se obtienenpor adición de 10 pl de NIBS 1.5 y 3 mM, respectivamente,a 90 pl de FO III100 pM en tampón acetatosódico50 mM, pH 5.0. Análogamentesepreparala forma W10~ delnúcleocatalítico de FO III a partir de éste.
10.4. Estudiosde termoestabilidad
Alícuotas de 150 pl de soluciones de FO III intacta, W10,, yW2~ (concentración3.4 gM,en tampón acetato sódico 50 mM, pH 5.0) se incubaron a distintas temperaturas durante30 mm; el tratamiento térmico se parapor inmersiónen bañode hielo. La actividadresidualse determiné con CNP(Olc)3 como sustrato, y se comparé con la de muestras no incubadas.
10.5. Espectrosde dicroísmocircular
Los espectrosde dicroísmocircular (250-200nm) seregistraronpor triplicado paracada
muestra(concentraciónde proteína 3.4 pM, tampónacetatosédico50 mM, pH 5.0) con undicrógrafo Jobin Yvon Mark III (Francia)dotadode una lámpara de xenon de 250 W. Lavelocidad de barrido fue de 0.5 nm/s, la sensibilidad de 5 ‘lot las muestrassedispusieronenuna cubetade cuarzode 0.1 cm de pasoóptico.
Los valoresde elipticídadseexpresancomo elipticidadmolarpor residuo,que secalculateniendoen cuentaquela masamolecularmediapor residuodeEO III es 106.2Da, segúnsededucede sucomposiciónde aminoácidos.FI cálculode los valoresde elipticídad,así como elde estructurasecundaria se han realizado mediante el programa informático CDPROTdesarrolladopor Menéndez-Ariasy col. (1988).

MATERIALES Y METODOS 36
11. MARCAJE DE AFINIDAD DE EG III CON 4’,5’-EPOXIPENTIL--I3-CELOBIOSIDO(EPOS)
11.1. Reacciónde inactivación de EG III con epoxialquil8-celobiósidos
Los experimentosde inactivaciónde FO III con epoxialquil ¡3-celobiósidosen general,ycon EPOS en particular, se han llevado a cabo de acuerdocon el siguiente protocoloexperimental(los detallesconcretosde cadaexperienciaseespecificanen Resultados):FO III0.8 pM en tampón Mcílvaine pH 4.6 se incuba a 370C con inhibidor; a distintostiempossetoman alícuotasde 10 pl de la mezclade reaccióny se diluyen en 0.5 ml de MeUmb(Olc)
3100 pM en acetato sódico 0.1 M, pH 5.0 paradeterminarla actividad residual.
112. Preparación de EPOS-EGIII
La identificación del grupo carboxilo de la proteína al que se une EPOS, ha requerido la
preparación de grandescantidadesde FO III con FF05 covalentementeunido, quese hanobtenidopor incubaciónde 1 ml de enzima20 pM en tampónMcílvaine pH 4.6 con EPOS7 mM, a 37
0C hastainactivartotalmenteFO III (dos días). La preparaciónde EPOS-FOIIIsediluye con agua(hasta3.5 mí) y seconcentrapor ultrafiltración (Centricon 10 de Amicon,EEUU); este lavado serepite otra vez para eliminar el tampóny el excesode epóxido.Enparalelo,se preparancontrolessin inhibidor.
12. ANALISIS DE PEPTIDOS
12.1. Digestión con tripsina
Las muestras de FO III (300 pl con una concentración10 pM, en tampónbicarbonatoamónico 0.4 M, pH 7.8 con urea 8 M) se reducen (DTE 1.5 mM, 10 mm a 500C) ycarboximetilan(LXAm 3 mM, 15 mm a 370C). Despuésde diluir (1/4) con agua,se añaden45 ¡sg de tripsina.La mezclase incubadurante10 h a 370C,y el digerido resultantese inyectadirectamenteen el sistemaHPLC (apartado12.3).
122. l)igestión con proteasaVS
Las muestrasde FO III (300 pl con unaconcentración10 pM, en tampónbicarbonatoamónico 50 mM, pH 7.8 con GuHCI 6 M) se reducen (DTF 1.5 mM, 10 mm a 500C) y sediluyen (1/4) con tampón sin GuHCI. Seguidamente se añaden30 ¡sg de proteasaV8 y seincubadurante10 h a 370C. Los péptidosresultantesse carboximetilan(IAAm 2 mM, 15 mma 370C) antesde su inyecciónen el sistemaHPLC (apartado12.3).
12.3. Separaciónde péptidos por HPLC
Los péptidosderivadosde la proteolisis de FO III y sus derivadosoxidados (NBS) omarcadoscon EPOS,sehanseparadoporHPLC en fasereversaen un sistemaWaters(EEUU)

MATERIALES Y METODOS 37
con unacolumna C4 modelo 214 TP-54 (0.46x25cm) de Vydac (EEUU). La eluciénserealizacon un gradiente lineal del 5 al 70 % de acetonitrio en agua, con ácido trifluoroacético al 1 %(y/y) (concentración constante), a un flujo de 1 mí/mm. La detección de péptidos se lleva acabo con un Photo Diode Array (Waters, EEUU), almacenandoen una memoria electrónica,cada 5 s, espectrosde absorción(190-320nm) del eluido. Los péptidosseleccionadospara suposterior análisis se concentran por evaporación a vacío hasta unos 20 pl, y se conservana-20
0C.
12.4. Secuenciaciónamino-terminal
La determinaciónde la secuenciaamino-terminalde los péptidosseleccionadossehallevado a cabocon un secuenciadorautomáticode fasegaseosa477A (Applied Biosystems,EEUU), acoplado a un analizador de P’I’H-aminoácidos 120A (Applied Biosystems,EEUU).El procedimiento empleadoha sido el recomendadopor el fabricante. Las muestrassecarganen filtro de fibra de vidrio (GFC de Whatman, EEUU) tratado con ácido trifluoroacético puro,y se recubren con una solución acuosade polibreno (3 mg).
12.5. Determinación de la masamolecular
La masa molecular de los péptidos seleccionadosse ha determinado por espectrometriade masassiguiendo la técnica denominada de desorción de plasma (“plasma desorption massespectrometry”,PDMS),prolijamentedescritapor Macfarlane(1990). El aparatoempleadohasido un Analizador de Biopolímeros NO ION 20 de Applied Biosystems(EEUU), y elprocedimientoexperimentalel descritopor el fabricante.Los espectrosse registraronpara1 millón decuentasde fisión de la fuente(californiJ2).
El errormáximoen la determinaciónde la masamoleculardepéptidosporestemétodo,esdel 0.2 %. FI métodoesválido parapéptidosde hasta50 kDa.

RESULTADOS

RESULTADOS 39
1. PURIFICACION E IDENTIFICACION DE EG III
1.1. Purificación de EG Hl
La caracterizaciónde cualquierproteína requierela disponibilidadde muestrasde lamismacon un gradodepurezaóptimo (normalmente> 95 %). En el casode lascelulasasesterequisito cobra especial importancia, debido a la gran cantidadde enzimascon similaresaptitudescatalíticaspresentesen el materialde partida,y al sinergismoobservadoentreellas.
La purificaciónde FO III a partir de caldosde cultivo de TrichodermareeseiQM 9414seha llevadoa cabosiguiendoel esquemade la Figura 11 (pág. 24). La evaluacióncuantitativadel procesode purificaciónserecogeen la Tabla VII.
TablaVII. Purificación de endoglucanasaIII
Actividad CMCasa
Fracción VolLinen Proteína Actividad Actividad Rendimiento Factor detotat total específica (actividad) purificación
<mt> <mg) ([JI) (UI/m9) (% total>
Caldo crudo 500 430 11695 27.2 100 1.0
Precipitadocon (NH
4)2504 10 341 7395 21.7 63 0.8
OEAF-SepliarosepH 7.0 86 5.7 1634 287 14 10.6
Ultrogel AcA-44 31 3.5 1200 343 10 12.6
DEAE-SepharosepH 6.5 29 2.5 866 346 7 12.7
El primerpasocromatográficosobreDEAE-SepharoseCL-6B a pH 7.0 permitesepararlas distintascelulasasy hemicelulasaspresentesen el caldode cultivo del hongo (Fig. 12A).FO III eluye isocráticamente,aunquees retenidaparcialmenteen la columna. El análisiselectroforéticodel pico de actividadCMCasacorrespondientea FO III revela la presenciadevarioscontaminantesde distinta masa molecular (Hg. 13), por lo que el siguientepaso depurificación es una cromatografíade penetrabilidadsobre Ultrogel AcA-44 (Fig. 12B). Lapreparaciónresultantemuestraunaúnica bandade proteínaen electroforesisen presenciadeSDS (Fig. 13); sin embargo,cuandoseanalizapor isoelectroenfoqueserevela la existenciadedos isocomponentes(Fig. 14A), ambos activos sobre el sustratocromoférico específicoMeUmb(Olc)3(Fig. 14B). El componentemásbásico(pI = 5.1) se identificó con FO III y elmásácido (pI = 4.6) con la isoenzima“A 1:2”, previamentedescritas(Saloheimoy col., 1988),mediantecomparaciónde suscaracterísticasestructurales(M, pí, composiciónde aminoácidos,mapaspeptídicos tras rotura con CNBr) con muestrasprocedentesdel laboratoriodel Dr.Pettersson(Universidadde Upsala,Suecia).Posteriormentela secuenciaciónde fragmentoscorroboréla identidadde FOIII, purificadaa partir del caldodecultivo de 71 reeselcrecidoconpaja de trigo, como productodel gen egl3 (Saloheimoy col., 1988). EG III se separade su

40RESULTADOS
isoenzimaA 1:2 en cromatografíade intercambioaniónicosobreDEAE-SepharoseCL-6B apH 6.5 (Fig. 12C).
La muestrade FO III resultante(factor de purificación — 13, rendimiento deI 7 %respectoa la actividad CMCasatotal del caldo de cultivo) parecehomogéneaen los análisiselectroforétícos(Fig. 13) y por isoelectroenfoque(Fig. 14A); la curva de titulación (Fig. 16 yFig. 23, pág. 51) y los experimentosde velocidadde sedimentación(ultracentrifugación),enlos quesedetectaun únicopico simétrico(no mostrado)demuestranigualmenteel alto gradode purificación alcanzado.
30
o E
20- ~ ~
D oo -“o, o ~o~o,~oo — o o
o?10 _ o’
o
0.50
<.5
0.25 07
0.00
Ndmero de froccion
looE=
50 0
oo
25 ~
<-5
o
o 0
60~~~
E40 =
o20<3
oo
0.4
0.25o
7
0.0
mero de frocci¿n Nñrnero de tracción
Figura 12. Purificación de FO 111. (A), perfil de elución de la cromatografía de intercambio aniónico
en DEAE-Sepharose CL-6H a pH 7.0 (tampón fosfalo sódico 5 mM). (II), perfil dc elución de la
cromatografíade penetrabilidaden Ultrogel AcÁ. 44. (C), perfil de elución de la cromatografía deintercambioaniónico en DEAH-SepharoseCL-6B a pH 6.5 (tampónimidazol-HCI 8 mM). Losdetallesexperimentalesse describenen Materialesy Métodos(pág. 23 y sgtes.).Símbolos: (—),
A2~; (s-..-..),actividad 13-glucosidasa;(á.-.e3,xilanasa;(e---.), CMCasa;(v..--v), mananasa;(o-... o), avicelasa;( ), INací].
3
2
4
0 20 40 60 80 100 120 140 160
2
oo,
o0 20 40 60 80 100 0 10 20 30 40 50 60


RESULTADOS 42
12. Propiedadesmolecularesde EG III
Duranteel procesode purificación e identificación de FO Hl se han determinadosusprincipalesparámetrosmoleculares:
a) Masamolecular: por electroforesisen gel de poliacrilamida en presenciade SDS seha obtenidoun valorde 48 kDa,mientrasqueporcromatografíadepenetrabilidaden UltrogelAcA 44 el valor calculadoes50 kDa. En amboscasosla determinaciónse ha efectuadoconproteínaspatrónde M conocida(Fig. 15).
5.0
o4.5
4.0
5.0
cro
4.5
4.0
0.1 0.3 0.5 0.7 0.9 0.1 0.3 0.5 0.7 0.9
KCV
Figura 15. Determinaciónde la masamolecularde HO III. (A), por electroforesisen presenciadeSDS.(B), porcromatografíadepenetrabilidadenUltrogelAcA 44. La flechaindica,respectivamente,el valor de ES y el de Kav de la enzima.
b) Punto isoeléctrico: se ha determinadopor isoelectroenfoqueanalítico (Fig. 14A)utilizando patronesde pI conocido.El valor obtenido,5.1, difiere del descritoporSaloheimoy col. (1988), 5.6; estadiscrepanciano obedecea diferenciasrealesentrela proteínaestudiadaennuestrolaboratorioy el productodel genegl3 (Saloheimoy col., 1988),sinoaunacorreccióndel valor de pl de EG 111, cuyascausasse detallanmásadelante(pág. 77, Discusión).
c) Titulación: FO III exhibe una baja carganetaa valoresde pH por encimade su pI(Fig. 16).
d) Parámetroshidrodinámicos:por ultracentrifugaciónanalíticasehan determinadolosvaloresdel coeficientede sedimentación,~020w = 3.66 5, y de la razónfriccional, f/f
0 = 1.48.Esteúltimo correspondea una molécula muy asimétrica;por analogíacon otrascelulasasde71. reesel(Claeyssensy Tomme,1990) sepuedeasimilar a un elipsoideprolato,quetendríaunarelación axial de aproximadamente8.
A
II
B
IR


44RISSULTA DOS
descritas,un aumentode absorbanciade 0.08 unidades(0.16 siseconsideraquesiendopar elnúmerode Cys de la proteína,se encontraríanreducidasde dos en dos).
Sepuedeconcluir que las 12 Cys presentesen FO III seencuentranformando6 puentesdisulfuro. La existenciade cistinasen FO III esevidentepor el cambiodemigraciónobservadoen electroforesisen SDS cuandola muestrano se trata con DTE (Fig. 13, pág. 41).
La similitud entrelascaracterísticasmolecularesdeterminadasy las recogidasen la escasaliteraturasobreFO III (Saloheimoy col., 1988; StAhlbergy col., 1988) quedapatenteen laTabla VIII.
1.3. Efecto de pH, temperaturay fuerzajónicaen laestabilidady actividadde EG III
FO III es estableen el intervalo de pH 3.0-8.0 a 300C, pero sólo entrepH 4.0 y 6.3 a550C (Fig. 17A). A pH 5.0 la enzimaseinactiva a temperaturaspor encimade 600C, aunqueretieneun 90 % de actividadcuandoseincubaa 650C durante30 mm (Fig. 17B). A 500C y pH5.0 FO III es establedurante10 mm en presenciade concentracionesde NaCí hasta0.25 M).
El pH óptimo parala actividadestáen el amplio margende 4.0 a 5.0 paracasi todoslossustratosensayados(Hg. 17D). Sólo la actividad CMCasa presentaun máximo a un valor
concretoen eseintervalo,a pH 4.8. CuandoseempleaMeUmb(Glc)3comosustratola actividad
esmáximaa 700C (Fig. 17E,) y, por otra parte,decaeprogresivamenteal aumentarla fuerza
iónica del tampón (Hg. 1W).
2. ESPECIFICIDAD Y MODO DE ACCION DE EG III
El conocimientodela especificidady mododeacciónde cadaenzimadel sistemacelulasade 71. reeseies imprescindiblepara avanzaren la comprensióndel mecanismode la hidrólisisenzimáticade celulosa,así como paradiseñarmezclasadecuadaspara el tratamientodecadatipo particularde materiallignocelulósico(Teeri y col., 1990).
2.1. Actividad sobresustratoscromofóricos
La capacidadde FO III parahidrolizar MeUmb(Olc)3liberandoel fluróforo MeUmbes
única entrelas celulasasde 71. reesel(Fíg. 3, pág. 8) (vanTilbeurgh y col., 1988). Estesustratoproporcionaun método de detecciónde FO III muy sensible.Alternativamente,cuandolascondicionesde ensayolo permiten,sepuedevalorarporcolorimetríay en continuola liberacióndel cromóforo CNP (pKa = 5.5) a partir del sustratoCNP(Glc)3 (Claeyssens,1988).
El análisispor HPLC de los productosde hidrólisis ha reveladola existenciade otropunto de ataqueenzimáticoen cada uno de los sustratosmencionados(Fig. 18). El estudiocinéticode estasreaccionesseha basadoen el Esquema1 (Apéndice,pág. 74). En resumen,laformulación desarrolladapermitecalcularlos parámetroscinéticos(aparentesy teóricos) decada vía de hidrólisis siemprequese determinela apariciónde los dos posiblesproductoscromofóricos(CNP y CNPOIc, o MeUmb y MeUmbOle), mientrasquela medidade un soloproducto(normalmenteMeUmb o CNP) limita los cálculosa los parámetrosaparentesde esavía y asu constantede especificidadteórica (los términos“aparente”y “teórico” se definenen
el Apéndice,pág. 74).

RESUlTADOS 45
100
o
•0
o
80
80
40
20
O
pR pH
100
-U
o
o
80 -
60
40
20
O
Temperatura(C)
1 00
30 40 50 60 70 80
Temperatura (C)
80 -
Uo~0
o
60 -
40
20
0’o0 1 2 3 4
[Nací] (M)
1 2
[Nací] (M)
Figura 17. Estabilidad y actividad de Ecl 111 frente a pH, temperatura y fuerza iónica. (A),estabilidad frente al pH a 300C, 5 mm (Q- o) ya 550C,5 mm (e-—.). (8), estabilidadfrentea latemperaturaapH 5.0, 30 mm. (C), estabilidadfrentea la fuerzaiónica apH 5.0, 50~C. (D), variacióncon el pH dc la actividad sobreCMC (o—-o), celotriosa(A—A), MeUmb(Glc)
3 (.—.),
CNP(Glc)3 (v—.v).(E), variación con la temperaturade la actividad sobreMeUmb(Glc)3. (E),
variacióncon la fuerzajónica de la actividadsobreMcUmb(Glc)3.
loo
80
60
40
~3
o~0
ti
20
o2 4 6 8 10 3 4 5 6 7
~0
o-U
o4
1 00
80
60
40
20
o
100<
30 40 50 60 70
80 -
-UU
-p
o4
60
40 o20 -
o
F
3

RESULTA DOS 46
Las constantescinéticasparala hidrólisis de CNP(Glc)3y MeUmb(Olc)3 se incluyen enla Figura 18. El principal producto de la reacción enzimática es CNP(Glc) para CNP(Olc)3 comosustrato, y MeUmbpara MeUmb(Olc)3. Estadiferenciaen el patrónde rotura se debea lasdistintas afinidades de los restos cromofóricosy de glucosade estossustratos,por los subsitiosde unión de la enzima como reflejan los valoresde K,11.
Algunos datos concernientes a la hidrólisis catalizada por FO III de 4’-metilumbeliferilderivadosde celotetraosay celopentaosaseincluyenenla Figura 18. La inhibición porproducto(los celooligosacáridosliberadosson buenossustratosde la enzima) impide ahondaren elestudiocinéticode estossustratos.
(1> 85 lIS 071 69
97 1.5
tUi) t(2) (2) 286 521 055 53
(1) 6 51 0-12 4.5
33 05
(21 36 152 024 9
<1) nd ¡id. 0-38 <¡4. <¡4.
3.5
tú) t<2)
0.6
Figura 18. Hidrólisis de sustratoscromofóricoscatalizadaporEG 111. Los parámetroscinéticos(pH50, 25
0C) se determinaroncomose describeen Materialesy Métodos(pág. 28). Símbolos: ( O ),CNP; ( ), MeUmb; ( ~ ), residuode glucosa.Lasflechasindicanlos enlaceshidrolizados(lasde trazodiscontinuo señalanpuntossecundariosde hidrólisis).() Los valoresdc k~
1 y Km de estareacciónson aproximadosya queen estecasono se cumplelapremisade quela glicosílacióndela enzimaseael pasolimitante de la reacc,on.
2.1.1.Estudiosde partición con metanol
FO III escapazde transferirrestosglicosidicos no sólo a agua,sino tambiéna nucleófiiosalternativostalescomo alcoholesde bajo pesomolecular,como muchasotrasglicosidasasqueactúancon retenciónde la configuraciónanomérica(Sinnot, 1990).El etanol,y sobretodo elmetanol,son nucleófilosmásefectivosque el agua,y en consecuenciaaceleranreaccionesenlas cualesla desglicosilaciónesel pasolimitante (Fersht, 1985).
En la Figura 19A se puedeobservarel incrementoen la liberaciónde CNP a partir deCNP(Olc)3provocadopor la adición de alcoholes;sin embargo,la hidrólisis de MeUmb(Olc)3
no varíapor la presenciade metanolhasta2.5 M y disminuyeprogresivamenteal aumentarla
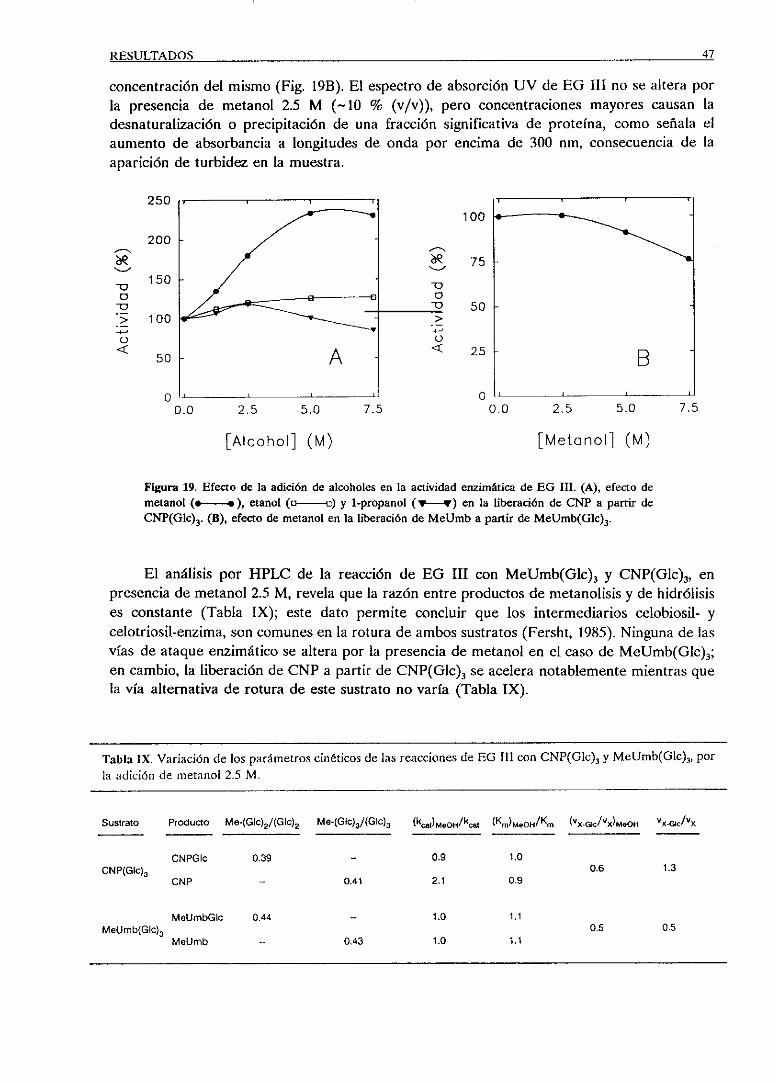
RESULTADOS 47
concentracióndel mismo (Fig. 19B). El espectrode absorciónUV deEO III no sealteraporla presenciade metanol 2.5 M (—10 % (y/y)), pero concentracionesmayorescausan ladesnaturalizacióno precipitaciónde una fracción significativa de proteína, como señalaelaumentode absorbanciaa longitudesde onda por encima de 300 nm, consecuenciade laapariciónde turbidezen la muestra.
100
‘o
ro
-o
(54
75
50
25
o0.00.0 2.5 5.0 7.5
EAIcohol] (M)
2.5
2
E
5.0 7.5
[Metonol] (M)
Figura 19. Efecto de la adición de alcoholesen la actividad enzimática de Ecl III. (A), efecto demetanol (.—.), etanol (o—c) y 1-propanol (v—v) en la liberación de CNP a partir deCNP(Glc)3. (B), efectode metanolen la liberaciónde MeUmb a partir de MeUmb(Glc).
El análisis por HPLC de la reacciónde EO III con MeUmb(Olc)3 y CNP(C}lc)3, enpresenciade metanol2.5 M, revela quela razónentreproductosde metanolisisy de hidrólisises constante(Tabla IX); este dato permite concluir que los intermediarioscelobiosil- ycelotriosil-enzima,soncomunesen la roturade ambossustratos(Fersht,1985).Ningunade lasvías deataqueenzimáticosealtera por la presenciade metanolen el casode MeUmb(Olc)3;en cambio, la liberaciónde CNP a partirde CNP(Olc)3seaceleranotablementemientrasquela vía alternativade rotura deestesustratono varía (Tabla IX).
TablaIX. Variación de los parámetroscinéticosdelas reaccionesde FC III con CNP(Clcty Meumb(Olct,porla adición de metanol2.5 M.
Sustrato Producto Me.<GIc)2/<GIc)2
CNPGIc 0.39
Me~(Glc)a/(GIc)3
—
(k~¡)M.oH/k~
0.9
(Km)M,OH/Km
1.0
<Vxck/Vx)M,o~ V,<~/Vx
cNP(oIc>acNP — 0.41 2.1 0.9
0.6 1.3
MeUn,bGlc 0.44 — 1.0 1.1MeUmb(Glc)3
MeUmb — 0.43 1.0 1.10.5 0.5
250
200
‘o
~13o-D
-4-,
(~2
4
150
100
50
0

RESULTADOS 48
Deestoshechosse infiere queel pasolimitante de la reacciónes la desglicosilaciónde FO111 en la rotura del enlace heterosídicode CNP(Olc)3, y en todos los demáscasos, laglicosilación de la enzima. En la Figura 20 se esquematizanlos experimentosde particióncomentadoscon el fin de hacermáscomprensibleal lector la interpretaciónde los mismos.
E + (GIc>2
1 Isa(ES)1 -Ns~¿<EIGlch) ~ 1 = Glicosilación
E+SE + (GIc)3 2 = Desglicosilacián
1 l-1~O<ES)2 —~-----(E-<Glc)3) -~
E + Me-(Glc)3
Figura 20. Esquema de los experimentosde partición de FO III con los sustratoscromofóricosCNP<Glc)3 y MeUmb(C;lc)3. X es CNP cuando 5 es CNP(Glc)3, y MeUmh cuando 5 esMeUmb(Glc>3.
22. Actividad sobreceloaligosacáridos
Los patronesde hidrólisis de celodextrinascatalizadaporFO III han sido determinadosporHPLC, y seilustranen la Figura 21. Debidoa la bajasensibilidaddel métodode detecciónempleado(refractómetro),las concentracionesiniciales de sustrato han sido forzosamente=10mM sin dudamuy superioresa los valoresde Km correspondientes;en consecuencia,losvalores de k~< mostradosno son más que velocidadesde desaparicióndel sustratoa unaconcentración10 mM delmismo,y debenconsiderarsemeramenteorientativos.Sólo en el casode celotriosay celobiosala determinaciónde glucosase ha realizadopor un método mássensible (glucosa oxidasa/peroxidasa),y los parámetroscinéticos se han calculado con laprecisiónadecuada.
La adición de metanol(2.5 M) al medio de reacciónha hecho posiblela determinaciónde ciertospuntos deataqueenzimáticomediantela detecciónde los metil-celooligosacáridosformados.Así sehan establecidopatronesde rotura,de otro modoambiguos;por ejemploconcelopentaosacomo sustrato se forman celobiosay celotriosa, pero ¿cuál de los productoscontieneel extremo reductor del sustrato?.El equivoco se resuelveal añadir metanol ydetectarsemetil-celotriosaperono metil-celobiosa,lo cual indica que la celobiosaprocededelextremo reductorde la celopentaosa,liberándosecomo primer productode la reacción.Esprecisamentecelopentaosael único sustratode los ensayadoscuya hidrólisis se acelera(unasdosveces) en presenciade metanol. La falta de sensibilidaddel métodode detección,antes
comentada,ha impedidoprofundizaren el estudiodeestehecho.
FO III no parecetener actividad transglicosilasa,habida cuenta de que no se handetectadoproductosdemayor masamolecularqueel sustratoconsiderado,inclusodespuésdeprolongadaincubaciónde la enzimacon altasconcentracionesde celooligosacáridos.
La celobiosaha resultadoserun mal sustratodeFO III (Fig. 21); la posibilidad de queestaactividadsedebieraacontaminaciónpor ¡3-glucosidasaen la preparacióndeFO III hasidodescartadaal no observarseinhibición en presenciade ¿-gluconolactona,potentey específico

49
RESULTADOS
inhibidor de 13-glucosidasa.La celobiosa se ha empleadocomo inhibidor competitivo enreaccionescon otros sustratosde EO III, y no se hanobservadoalteracionesen los patronesde hidrólisis de ninguno de ellos. Con celobiosa50 mM la inhibición de la actividad sobreMeUmb(GlCt 100 gM o CNP(Olc)3500 gM esde un 10 y un 50 %, respectivamente.Por otraparte,concentracionesde glucosade hasta200 mM no inhiben la actividadde FO III.
-1 1(miW1> (mM) (mirí mM
LZIyI~ 2 20 0.1
15 1 15 0.4
[Z1I}—{ZIIITjZI}T~ 800 n.d. n.d. 1.8
z4m—L3ÚmH~200 n.d. od.
LI] 11113 11] Me 500 n.d. n.d. 0.9
Figura 21. Hidrólisis de celoeligosacáridoscatalizadapor HG III. Los parámetros cinéticos(pH 5.0,370G) se determinaroncomose describeen Materialesy Métodos(pág. 28). Símbolos:( Wrestosde glucosa;( ~ ), restode glucosaconel carbonoanoméricolibre; (Me), grupometilo. Lasflechas indican los enlaceshidrolizados(las de trazo discontinuo señalanpuntos secundariosde
hidrólisis).() velocidadde desapariciónde sustratoparauna concentracióninicial 10 mM.
2.3. Actividad sobresustratoscelulósicos
Comocorrespondea unaendoglucanasatípica, EG III hidroliza eficazmentederivadossolublesde celulosacomo CMC (Km = 5 mg/mí,k~
8 = 36 &‘, a pH 5.0 y 300C)provocandouna
drásticadisminuciónde laviscosidadde la mezclade ensayo,concomitanteconla liberacióndeextremosreductores.No obstante,CMC no esun sustratoadecuadoparaestudioscinéticosyaquela curva de actividadfrentea concentraciónde enzimano eslineal (Fig. 22A). Estehechosedebea la complejidaddel sustrato,mezclaheterogéneade polimeroscon distintosgradosdecarboximetilacióny tamaño,en el que hay enlacescon distinto grado de accesibilidadparalaenzima.
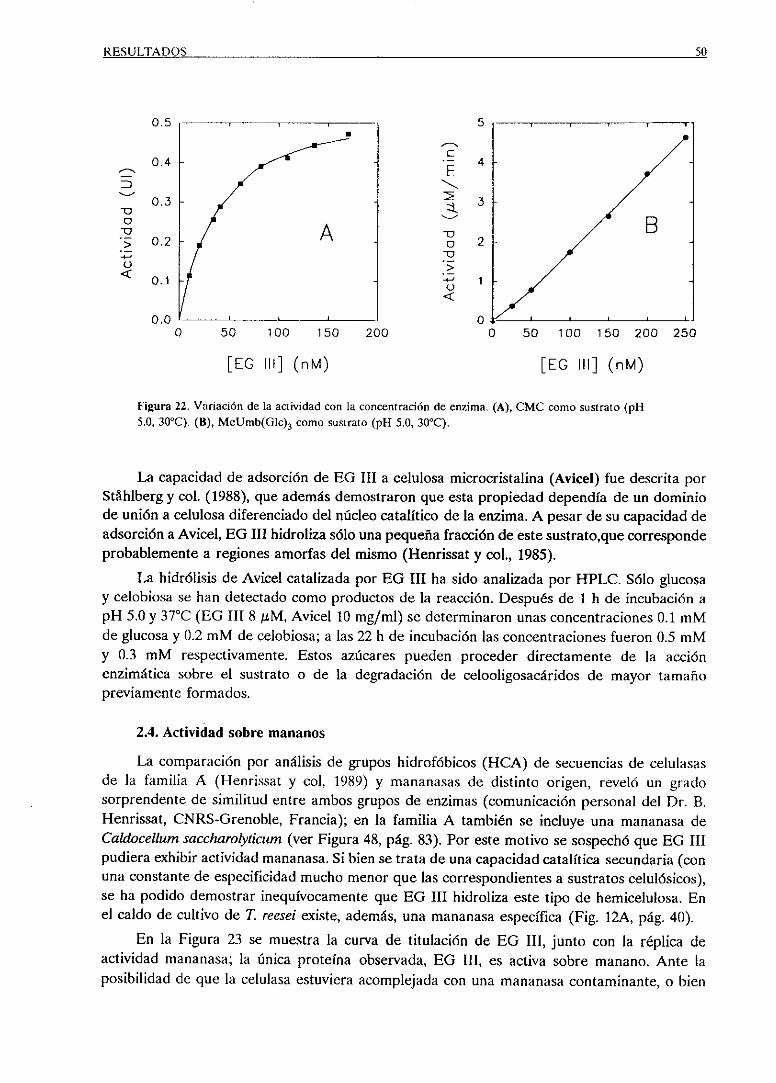
RESULTADOS St)
0 50 100 150 200
0.5 5
cN
G0.4
EEj‘o
0.3-oo ‘o
-o -o0.2 o 2
-oo
4
0.0 0
[Fc III] (nM) fEO iii] (nM)
Figura 22. Variaciónde la actividadcon la concentraciónde enzima.(A), CMC comosustrato(pH5.0, 300C). (IS), MeUmb(Glc)
3comosustrato(pH 5.0, 300C).
La capacidadde adsorcióndeEO III acelulosamicrocristalina(Avicel) fue descritaporStAhlbergy col. (1988),que ademásdemostraronqueestapropiedaddependíadeun dominiode unión a celulosadiferenciadodelnúcleocatalíticodela enzima.A pesarde su capacidaddeadsorciónaAvicel, EG III hidroliza sólo unapequeñafracciónde estesustrato,quecorrespondeprobablementea regionesamorfasdel mismo (Henrissaty col., 1985).
La hidrólisis de Avicel catalizadapor FO III ha sido analizadapor HPLC. Sólo glucosay celobiosase handetectadocomo productosde la reacción.Despuésde 1 h de incubaciónapH 5.0y 370C(EG III 8 gM, Avicel 10 mg/mí) se determinaronunasconcentraciones0.1 mMde glucosay 0.2 mM de celobiosa;a las 22 h de incubaciónlas concentracionesfueron0.5 mMy 0.3 mM respectivamente.Estos azúcarespueden proceder directamentede la acciónenzimáticasobre el sustrato o de la degradaciónde celooligosacáridosde mayor tamañopreviamenteformados.
2.4. Actividad sobremananos
La comparaciónpor análisisde gruposhidrofóbicos (HCA) de secuenciasde celulasasde la familia A (Henrissat y col, 1989) y mananasasde distinto origen, reveló un gradosorprendentede similitud entreambosgruposde enzimas(comunicaciónpersonaldel Dr. B.Henrissat,CNRS-Orenoble,Francia); en la familia A tambiénse incluye una mananasadeCaldocellumsaccharolyticum(ver Figura48, pág. 83). Porestemotivo sesospechóque FO IIIpudieraexhibir actividadmananasa.Si biensetratade unacapacidadcatalíticasecundaria(conunaconstantede especificidadmuchomenorquelas correspondientesa sustratoscelulósicos),seha podido demostrarinequívocamenteque FO III hidrolizaestetipo de hemicelulosa.Enel caldo de cultivo de T reeseiexiste,además,una mananasaespecífica(Fig. 12A, pág. 40).
En la Figura 23 se muestrala curva de titulación de EG III, junto con la réplica deactividad mananasa;la única proteína observada,FO III, es activa sobremanano.Ante laposibilidadde que la celulasaestuvieraacomplejadacon unamananasacontaminante,o bien
0 50 100 150 200 250


52RESULTADOS
Producto Sustrato
Galactomanano Manano
Manosa 0.7 mM 0.6 mM Tabla XII. Productosde la hidrólisís deManobiosa 0.8 mM 1.0 mM mananos(20 g/l) catalizadapor FO IIIManotriosa 1.3 mM 0.6 mM (8 gM), analizadospor HPLC, despuésManotetreosa 0.4 mM — de 3 h de incubacióna 370Cy pH 5.0.Manopentaosa 0.2 mM 0.2 mM
La notablediscrepanciaentrelos valoresde Km y K~ (inhibición del sustratoCNP(Olc)3)
quese observanen la Tabla XI, tanto paraLBG como paraCMC, sepuedeexplicar por lasdiferentescondicionesdepH y temperaturaen las quesehandeterminadoambosparámetros;ademásdebetenerseen cuentaqueel significado a nivel molecularde FC,,, paraun sustratosolubleclásicocomo CNP(Olc)3 es necesariamentedistinto del que correspondea sustratospoliméricosy heterogéneoscomo son CMC y LBO.
3. IDENTIFICACION DE RESIDUOS ESENCIALES DE EG III
La identificaciónde los residuosfundamentalesparala unión del sustratoy/o la actividadcatalíticadeunaenzimaes imprescindibleparadilucidar el mecanismoquímico de su acción,
y abreelcaminoasu hipotéticamejorapor la incipientey prometedoravía de la ingenieríadeproteínas.
31. Efecto del pH y la temperaturaen la cinética de unareaccióncatalizadapor EG III
Seha estudiadola dependenciacon el pH de la constantede especificidadde la reacciónde hidrólisis de Meumb(Olc)3catalizadapor FO III. De las dos vías posiblesde ataqueenzimático(Fig. 18, pág.46) seha seguidola queconducea la liberaciónde MeUmb,productoquepermiteuna valoraciónsencillay precisamediantefluorimetría. El estudioseha limitadoal intervalodepH de 4.0 a6.3, en el quela enzimaesestableinclusoa altastemperaturas(Fig.17A, pág. 45). Previamenteseanalizópor HPLC la posibilidad de que el patrónde hidrólisisdel sustratodependieradel PH; no se observaroncambios en la relación vMeumb(dlc)/vMetJmb
dentro del intervalo de pH 3.5-6.5.
A todaslas temperaturasestudiadas(entre30 y 550C), la curva de pH del logaritmo de
la constantede especificidadmuestraunaforma similar, con unacaída enla región másbásicadel intervalodepH considerado,cuya pendientees-1 (Fig. 24A). Esto indica que la reacciónenzimáticadependedel estadode ionización de un residuo de la proteína,que debeestarprotonadoparaquetengalugar la catálisis.El valor estimadoparael P1~ade dichoresiduoes5.46 ± 0.03 a 550C. La variacióndeestevalor con la temperaturaha permitidodeterminarlaentalpíade ionización correspondienteque ha resultadoser -15 9 + 2.6 .1/mol (Fig. 24B),apuntandoa un grupocarboxiocomoresponsabledeestepK~ (Cleland, 1977).

53RESULTADOS
—0.7 6.0
y~- ~~ —1.1
‘o
0 5.0
—1.5
4.5 5.0 5~5 6.0 1/T (OK1 xlO)
pH
FIgura 24. Efecto del pH y la temperatura en la constante de especificidad de la hidrólisis deMeUmb(Glc)
3 catalizada por EO III. (A), variación con el pH del log (k,~~/K,,,) a 55~C. (fl),dependenciade la temperaturadel valor de PK8 calculadoa partir de los perfilesde pH de log
3.2. Modificación de histidinas con dietilpirocarbonato(DEPC)
Los estudiosde homologíaentrecelulasasde la familia A (Henrissaty col., 1989)señalanla existenciade dos residuosde histidina conservados,que en la secuenciade FO IIIcorrespondena His173eHis28S.La posibleimplicaciónderesiduosdehistidinaenla actividaddela enzimaha sido estudiadacon DEPC,reactivoespecíficodel anillo imidazol (Miles, 1977).No se ha observadoinactivaciónde FO III despuésde incubacióncon excesode DEPC,endistintostampones(MES 50 mM, pH 6.5; fosfatosódico100mM, pH 6.0).Un resultadosimilarseobtuvo para EO 1 de £ commune(Clarkey Yaguchi, 1985), celulasamásestrechamente“emparentada”con EO III de entrelas de secuenciaconocida(Béguin, 1990).No obstante,laausenciade inactivación por DEPC no excluye la posibilidad de que una o las dos Hisconservadasen la familiaA sea(n)esencial(es)en FOIII; porcausasindeterminadaspodría(n)ser inaccesible(s)al reactivo.
3.3. Modificación de tirosinascon tetranitronietano(TNM)
La existenciade residuosaromáticos,especialmenteTrp y Tyr, que participanen la uniónde azúcareses frecuenteen proteínascapacesde interaccionarcon carbohidratos(Quiocho,1986).El estudiode la modificacióncon TNM deCBH 1 de T. reeselapuntaa la participaciónen la catálisisde un residuode Tyr (Tomme, 1991).
La incubacióndeFO III con excesodeTNM (pH 8.3, temperaturaambiente)provocaunalenta inactivación de la enzima (Fig. 25). La presenciade un ligando (celobiosa) enconcentracionessaturantesno previenela pérdidade actividad, aunquese desconocesi laenzimaescapazde unir celobiosaa valorestanbásicosde pH. En consecuencia,la modificacióncon TNM no permiteadelantarningunaconclusiónsobreel papelde los residuosde tirosinaen FO III.
A
3.05 3.15 3.25

54RESUlTADOS
100
‘o
-oo
-o
-4-,
O4
75
50
25
o
Tiempo (mm)
Figura 25. Inactivación de Ecl III por TNM (Tris-HO 50 mM, pH 8.3 a temperatura ambiente
([TNbI]/[EG 1111 = 1000). La incubaciónserealizó en ausenciade ligandos(e—--.), o en presenciade celobiosa 100 mM (o—>).
a
Md
GOLch’-
w NES
o
Eox
Md
CH, - CH
1 CH.-
6
4
2—
o220
Longitud de onda (nm)
Figura 26. Reacción de oxidación deTrp por NBS (Lundblad y Noyes, 1984).
Figura 21. Espectrosde absorciónde indol y oxindol (SpandeyWitkop, 1967).
3.4. Modificaciónde triptófanoscon N-bro,nosuccininiida(NES)
La oxidación de triptófano por NBS (Fig. 26) es unareacciónmuy rápida quese puedecuantificar fácilmente por espectroscopia,merced a la diferencia entre los espectrosdeabsorcióndel indol y el oxindol (Fig. 27) (Spandey Witkop, 1967). Este reactivo ha sidoprofusamenteempleadoen la modificación de proteínas, con el fin de estudiarla funciónbiológica de residuosde triptófano (Lundblady Noyes, 1984).
o 30 60 90
3—wetUoxindol
3—metilindol
250 280 310

RESULTADOS 55
A continuaciónsedetallael estudiode modificacióndeFO III porNBS,queha permitidoidentificar dos Trp esencialescon distinta localización,uno en el dominio de unión a celulosay el otro en el centro activo de la enzima.
3.4.1. Extensióny especificidadde la modificación
La adición sucesivadepequeñasalícuotasde NES conducea la progresivaoxidaciónderesiduosde Trp, como semuestraen la Figura28: la absorbanciadisminuyea 280 nm, y a lavezaumentaa 250 nm, indicandola formaciónde oxindolalanina(Spandey Witkop, 1967).
La oxidación de Trp eslineal con respectoa la cantidadde reactivoañadido(Fig. 29A);despuésde modificar3 residuosde Trp, trasla adiciónde4.5 molesNBS/molEOIII, la adiciónde másNBShacequeseenturbiela solucióndeproteína.Un fenómenosimilarseha observadocon frecuenciaen otrasproteínascomo por ejemplo: a-quimotripsina(Spandey col., 1966),aglutinina de germende trigo (Privat y col., 1976) o lisozima (Oreeny Witkop, 1964).
0.10
0.05
ooccl-oo(ti-n4
—0.00
—0.05
—0.10
Longitud de onda (nm)
Figura 28. Espectrosde diferenciade EG III inducidospor la oxidacióncon NBS.Los espectrosseLomaron 1 mm despuésde cadaadicióndel reactivo(20u1, 0.15 mM en agua)a la soluciónde HGIII (1 mí, concentracióninicial 9.1 gM; en acetatosódico50 mM, pU 5.0) de la cubetade muestra.A la soluciónde enzimacontenidaen la cubetade referenciase le añadíaaguaen lugar de NBS.Antesde cada adiciónde NBS (agua)se recogíanalícuotasde 20 u1 paraensayosde actividad.Latemperaturase mantuvo a l50C.
240 260 280 300

RESULTADOS 56
La reacciónde NBS con proteínasen las condicionesempleadasse limita normalmentea la oxidación de residuosde Trp, pero cabela posibilidadde queestemodificadoroxide lascadenas laterales de otros residuos: Tyr, Cys, Met o His, o bien, que rompa enlaces peptídicosen los que intervengan Trp, Tyr o His (Spandey Witkop, 1967; Lundblady Noyes, 1984).
En el presenteestudiola especificidaddel ataquedel NBS por los residuosde Trp estáfuerade todadudaporqueel excesode reactivorequeridoesmuy bajo, 1.5 molesNBS pormolde Trp oxidado, igual que el encontradoparala oxidación del estermetílico de N-acetil-L-triptófano,queseempleacomocompuestomodelo, y uno de los másbajosentrelos descritosenla bibliografía paradistintasproteínas(Lundblady Noyes,1984).Datosadicionalesa favordela especificidaddela reacciónson: el mantenimientoa lo largode la modificación,del puntoisosbésticoa 264 nm (Fig. 28) queindica que no hay oxidaciónde TS¡r (Ohnishi y col., 1980),y la ausenciade Cys libres en EO III (pág. 43).
La integridad de la estructuraprimaria de la enzima despuésde la modificación secomprobópor electroforesisen SDS; muestrasde FO III con 1 (W~0~), 2 (W~,~) ó 3 Trpoxidados(W~~) migraroncomo unasolabandaa igual distanciaque la proteína nativa.
3.42. Proteccióncon ligandos
Cuandola reaccióndeFO III conNBS se lleva a caboen presenciade celooligosacáridosen concentracionessaturantes,la modificaciónde Trp requieremayorescantidadesdereactivo.Por ejemplo,la adiciónal medio de reacciónde metil-B-celotetraósido10 mM aumentala tasade consumode NBS de 1.5 moles/moldeTrp oxidado(control) a 2.9 moles/mol(Fig. 29). Elmismoefectoseobservacon celotriosa10 mM o celobiosa100 mM, mientrasque la presenciade glucosa100mM no alterael cursode la modificación,comoeradeesperarporqueno esunligando de la enzima.
3o
~, 2o
otZ 1 Figura 29. Correlacióndel númerode Trp
oxidados por molécula de EG III y la
conceniraciónde NBS. El númerode Trpoxidados se calcula partir de la
3 disminuciónde absorbanciaa280 nm comoo se describeen Materialesy Métodos (pág.
2 35). La modificación sc llevó a cabo: (A),en ausenciadeligandoso (B), en presencia-o de metil-B-celoterraósido10 mM.0S
o0 2 4 6 8 10
[NBS]/[EC ¡iij

RESULTADOS 57
Ensayospreliminarescon el estermetílico de N-acetil-L-triptófano mostraronque losazúcaresempleadosen los estudios de protección no interferían en la reaccióndel anilloindólico con NBS.
La prevenciónparcial de la modificación por celooligosacáridossugiereque los Trpoxidadospor NESestánlocalizadosen, o cerca,del sitiode unión decualquieradelos dominiosde EO III (catalítico y de unión a celulosa).
3.43. Efecto de la modificación en la actividadcatalítica y en la capacidadde adsorcióna celulosa
La modificaciónde FO III con NIBS provocaprimero una disminuciónde la capacidadde adsorcióna celulosa(coincidentecon la oxidaciónde un primerresiduodeTrp) (Fig. 30A),y seguidamenteafectaa la actividadcatalíticacon un efectodispar sobrela hidrólisis de dossustratos(Fig. 30B).
loo
c‘oo o,
oo —op-v o,<ci
75
50
25
o200
-c
o
o
150
loo
50
o0 1 2 3 0 1 2
ox
Figura 30. Efecto dc la oxidación de Trp en la actividad y adsorción a celulosa de EG III. Enmuestras con distinto grado de modificación se han determinado las siguientes propiedades: (A),adsorcióna Avicel de EG III; (II), actividad de EG ¡II sobre CNP(Glc)3 (A—~&) y MeUmb(Glc)3
(e—s); (C), actividaddel núcleocatalítico de ECl III sobreCNP(Glc)3(á—¿) y Meumb(Glc)3
(~).
Teniendoen cuentaque la capacidadde adsorcióna celulosadependedel dominio deunión a celulosa(DUC) de EO III (StAhlbergy col., 1988),elprimer residuode Trp oxidadopareceestarsituadoen dicho dominio a juzgarpor la disminuciónde la cantidadde enzimaadsorbidaaAvicel concomitanteconlamodificacióndeesteresiduo(Fig. 30A). Paracorroborarestahipótesisseha modificadoel núcleodeEGIII (carentedeDUC); despuésde oxidar2 Trp
N de W de Wox

RESULTADOS 58
la soluciónproteicasetornabaturbia(fenómenoobservadoen FO III trasoxidaciónde 3 Trp),y los cambiosen la actividad hidrolítica observadasen EO III cuandoseoxida un segundoresiduo de Trp eranevidentesen el núcleocon la oxidaciónde un primer Trp (Fig. 30C). Seconfirma, en consecuencia,la localizaciónen el DUC del primer residuooxidadopor NBS enla enzimano truncada.La capacidadde adsorcióna Avicel se reducepero no se elimina aresultasde la conversiónde esteTrp a oxindolalanina(Fig. 31). La oxidaciónulterior de másresiduosde Trp no afectaa la adsorción(Fig. 30A).
loo‘o
a~0 75.fl1.-
o-~ 50a
uE 25NGw
o0.0
[Avicel] (% p/v)
F¡gura 31. Efecto de la oxidación de! primer Trp en la capacidad de adsorción a celulosa. HG IIIintacta (e—), W10~ HG III (y—v), y el núcleo de HG 111 (.—u), en una concentración0.6 ¡¿M,
seincubaron a 40<2 con diferentesconcentracionesdeAvicel. Una vezalcanzadoelequilibrio (30mm)se determinó la fracción de enzima no adsorbida.
La oxidaciónde un segundoresiduodeTrp afectasensiblementeala actividadcatalíticade FO III (Fig. 30B). El patrón de hidrólisis de CNP(Glc)3 y MeUmb(Olc)3 cambiadrástícamenteen la proteínamodificada,siendoopuestoel efectoen cadauno de los sustratos(Tabla XIII). La actividadsobrecelotriosa,celotetraosay celopentaosase reduceun 65 %aproximadamente(a una concentración10 mM de sustrato),sin variaciónde los patronesderotura,como consecuenciade la oxidacióndeestesegundoTrp. Los parámetroscinéticosparala hidrólisis de CMC por FO III intacta y W20~ semuestranasimismo en la Tabla XIII; lamodificación afectaa la I<~,, perono a la k~. Todoslos cambiosobservadosapuntana que elresiduo de Trp consideradoparticipa en la unión del sustrato.
Las muestrasmodificadasen presenciade ligandos mostraronlos mismoscambiosenadsorcióny actividadcon respectoalgrado deoxidación de Trp, quehansidocomentadosparala proteína modificadaen ausenciade ligandos.
El tercerresiduodeTrp accesible al NBSantes de la desnaturalización de la proteína estambiénprotegidode la modificaciónpor celooligosacáridos(Fig. 29, pág. 56); sin embargo,su oxidaciónno produceningún efectoobservableen las propiedadesenzimáticasde FO III.Desconocemossi esteresiduotienealgunaimplicaciónfuncional,difícil de estudiardespuésdelgrancambioprovocadoporla oxidacióndel “segundo”residuodeTrp, o si estálocalizadocerca,o en el centroactivo, sin participaren la unión del sustrato;tambiénpodría no tenerrelaciónalgunacon el centroactivo, e interaccionarinespecificamentecon los ligandosempleados.
0.5 1.0 1.5 2.0

RESULTADOS
TablaXIII. Efecto de la oxidacióndelsegundoTrp en las propiedadescatalíticasdeFO III. LosparámetroscinéticosKm y k~1 correspondena la hidrólisisde CMC (30
0C,pH 5.0) y las relacionesde velocidada la rotura de CNP(Olc)
3 y MeUmb(Olc)3 (250C,
pH 5.0).
Enzima
Nativa
Km 1(mg-mr)
0.48 ±0.02
(5 )
36.3 ±0.7
((mI~mg -& V~p~/V~p4p ~
75.6 1.3 0.5
0.78 ±0.06 35.1 ±1.6 45.0 0.3 0.8
3.4.4. Efecto de la modificación en la termoestabilidady estructurasecundaria
La estabilidadtérmicadeFO III W20~ es ligeramentemenorquela de la proteínanativao W10~ (Fig. 32). En cualquiercaso, despuésde incubacióna 60
0Cdurante30 mm, la muestraW~ retieneun 95 % de actividad.
100
‘o
-o
o
-4-,
aKV
80
60
40
20
o45 55 65 75
Temperatura “O
Figura 32. Termoestabilidad de ECl 111 intacta y modificada con NBS. Muestras (3.4 uM) de FOIII intacta (e—e),W
1,, (w—v), y W20~ (s—u) se incubarona distintas temperaturas a pH 5.0(acetato sádico 50 mM) durante 30 mm. La actividad residual se determinó a 25
0C con CNP(Glc)3
como sustrato.
Los espectrosde dicroísmo circular en el ultravioleta lejano de FO III nativa y condistintosgradosde oxidación (W10~y W20~) semuestranen la Figura33. La semejanzaentrelostresespectrosindicaquela modificación no afectaa la estructurasecundariadela proteína.Laposibilidadde queEO III sufra un cambio conformacionala consecuenciade la oxidación de
dos de susresiduosde Trp parecedescartablea la vista de los resultadoscomentadosen este
59

RESULTADOS 60
apartado.Porotra parte,el espectrode dicroísmocircular deEO III indica un alto contenidode hélice a y, sobretodo, estructura¡3, como ocurre en otrascelulasas:FO 1 de T reesel(Domínguez, 1991), CBH II de T. reesel (Rouvineny col., 1990), y FO D de Clostridiumthermocellum(Juy y col., 1992).
0
—2000
oE
Eo
U)
‘o
o
—4000
—6000
—6000
200 210 220 230 240
Longitud de onda (nm)
Figura33. Espectrosde dicroísmo circularde HG III intacta (—), W10~ (————), y W20~ (Las muestrasestabanen acetato sódico50 mM, pH 5.0.
3.4.5. Identificaciónde un residuode Trp esencialen el dominio de unión a celulosa
Muestrasde FO III intacta, W10~, y W20~, así como del núcleo de EO III intactoy W10~fueron digeridascon tripsina, y los hidrolizadosresultantesse analizaronpor HPLC en fasereversaempleandocomo detectorun PhotodiodeArray (Waters),quepermitedeterminardemodo continuo espectrosdel eluido de la columna. En la Figura 34 se han superpuestoloscromatogramasa 214 nmcorrespondientesa EOIII no oxidaday W20~. Seseñalanlas zonasdelmapapeptídicoen las cualesseapreciandiferenciasentrela enzimano modificaday oxidadacon NBS.
-. 4.

RESULTADOS61
o<3crono(ti-o
Tiempo (mm)
Figura 34. Comparación de los mapas trípticos de ECl III no modificada y W20,. Semuestra el perfilde absorbanciaa 214 nm correspondienteal control, sobre el que seseñalanen trazo discontinuo lasdiferencias observadasen el perfil correspondiente a ECl III W20~. En la región 1” se observan
cambioscorrespondientesala oxidacióndel primerTrp (también observadosen W~0~), mientras quela oxidación del segundoTrp se hace notar en la región ‘2’.
La oxidacióndel primerresiduodeTrp, esencialparala adsorcióna celulosa,conduceala disminución dedospicos (“a” y “b”) en la región“1” (Fig. 35A y B). La comparaciónde losperfilesdeelución de FO III no modificaday W10~, y del núcleodeEG III no modificado(Fig.35C) permitededucirque un péptidoprocedentedel DUC, presenteen los picos a0 y b0 deldigeridode FO III nativa, no apareceen a1 y b1 del digeridode FO III W10~ ni en a’0 y b’0 delnúcleo no modificado; en el primer caso a consecuenciadel tratamientocon N?BS, y en elsegundoporque el núcleo carece de DUC. En la misma posición coeluye al menos uncontaminanteen cadapico, como demuestrael análisisespectralilustradoen la Figura 35 (DyE).
20 25 30 35 40 45

RESULTADOS 62
B
20 25
Tiempo (mm)
0.10
0.05
0.00
Longitud de onda (nm) Longitud de onda (nm)
Figura 35. Comparación de los mapaspeptídicos en la región “1” de: (A), ECl III no modificada;(fl), ECl III W10~; (<2),núcleocatalítico no modificado. Análisis espectralde lospicos: (D), “a”, y <E),b”, procedentes del digerido de ECl III no modificada (línea continua) y de ECl III W10~ (línea
dtsconttnua).
El análisisdel pico b0 por PDMS (Fig. 36) señalala presenciade variospéptidos,siendoel mayoritariouno de masa835.0Da y el segundomásabundante,otro de masa1155.5 Da.Estosresultados,combinadoscon los de secuenciaciónde b0 y b1 (Tabla XIV) han permitido
0.2
A <2
o ________
0.02 ‘~
o(~2a:o
o(o-o
KV o0.05
214 nm
250 nm
o-
0.1 5
2520
o
280
o‘-5ae,-ooo,-o
4
0.10
0.05
E
0.00
200 220 240 260 280 300 200 220 240 260 280 300

RESUI .TADOS 63
identificar el componenteprincipal de b0 como el péptido Olyó-Trpl3 (M teórica:834.7 Da)procedentedelDUC,y unodeloscontaminantescomoun productodela autolisisde la tripsina(5er126-Lysl36,M teórica 1153.3 Da). Los datosobtenidosen el análisisde “a” sonconfusosporquese trata de unamezcla complejade péptidos;es posible, además,queel péptido delDUC presenteen a0 puedaestarbloqueadopor piroglutámico, correspondienteal extremoN-terminal deFO III (Saloheimoy col., 1988).
5000
-4000
‘oo,ao
oo
3000
2000
1000
o
Figura 36. Espectro de PDMS del pico b0, procedentede la digestión tríprica delcontrol (ECl III nomodificada). Los númerosindicanla masaen Da correspondientea las señalesdetectadas.
TablaXIV. Datosobtenidosdel análisisdel pico “b”. La secuenciadeFO III (Saloheimoy col.,1988) semuestraen la Figura49 (pág. 84); la dela tripsina bovinafue determinadapor Walshy Neurath (1964). (xxx indica que no se ha detectado en esa posición el aminoácidoesperado,de acuerdoconla secuenciadeducida).
Pico M por PDMS, Da
835.0 <1155.5, 706.4)
Secuencia N-terminal
GIy-GIn-xxx-Gly-GIy-IIe-
Péptido identificado (M teórica, Da)
Gly6-Trpl3 de EG III (834.7>Ser-xxx-Gly-Thr-Ser-Tyr- Serl26-Lysl36 de tripsina (1153.3>
b1 1154.9 (706.2) Ser-xxx-GIy-Thr.Ser- Serl26-Lysl3S de tripaina (1153.3)Gly-Asn-Leu-Thr-Leu- No identificado
La secuenciadel péptido del DUC identificado, 01y6-Trp13, indica que la roturaproteolítica es de tipo quimotríptico (entre un aminoácidoaromáticoy otro cualquiera)y notríptico (entreLys o Arg y cualquieraminoácido).La preparaciónde tripsina utilizada(Ref. n~37260 de Serva, Alemania), contiene—0.5 % de quimotripsina(catálogodel proveedor),y laelevada proporción tripsina:EO III empleada en la digestión(1:10) ha supuestola adición decantidadesde quimotripsinasuficientescomoparaobtenerfragmentosquimotrípticostrasun
1503 2000
M/Z

RESULTADOS 64
largo tiempo de incubación(10 h) a 37”C. Condicionesmássuaves(1:25, 1:104 h) no dieronresultadossatisfactorios,y la quimotripsinapura,tambiénensayadaparala digestióndeFO III(1:25, 1:100), produjo gran cantidadde pequeñospéptidos(observadapor electroforesisenSDS), de complicadoanálisis.
La ausencia del péptido Olyó-Trpl3 en los digeridos de EO III tratadacon NBS puedeser debida a la oxidación delTrp5 o biendelTrpl3. La quimotripsina parece incapaz de romperun enlace peptidico en el que la oxindolalanina aporte el grupo carboxilo. Esta suposiciónsebasa en el hecho de que en el el mapa “tríptico” de FO III Wiox no se observan nuevos picos(respecto al mapa del control) que contengan oxindol. Si la proteasa no rompiera tras el Trp,,5,el péptido generado,01n1-Trpl3, incluiría la Thr3, residuo muy probablementeglicosilado(Saloheimoy col., 1988); análogamentesi N?BS oxidarael Trp 13, y no hubieserotura trasesteresiduo,seformaría un péptido (01y6-Pro3O)en el que dos residuos,5er26 y Thr27, parecenestarglicosilados(Saloheimoy col., 1988). Tanto en un caso como en otro los péptidosglicosijadosproducidoseluirian en el volumen de exclusióncon restosdeproteasay FO III nohidrolizados,haciendoimposiblesu identificación.En cambio,si la proteasareconocieracomodianaun enlace peptídicoen el queel grupo carboxilo fuera aportadopor oxindolalanina,elpéptido 01y6-Trpl3 apareceríaen el mapa tríptico de FO III W1,», con igual tiempo deretenciónque en el mapadel control si el Trp5 fuera el modificadopor NES, o ligeramenteantessilo fuerael Trp 13.
Los DUCde las distintas celulasas de 7? reeseipresentanun alto grado de simiiítud~poseencuatroresiduosaromáticos(cuyaimportanciaen la interacciónproteína-polisacáridoyaha sido comentadaen la pág. 53) conservadosen todos ellos (ver Fig. 47, pág. 82). Enconsecuencia, es de esperarquela función de dichosresiduosy su localizaciónespacialen elDUCsea semejante. La estructuratridimensionaldel DUC de CBH 1 ha sido determinadaporresonanciamagnéticanuclearbidimensional(Kraulis y col., 1989);la ‘T»466,equivalentealTrp5en FO III, se sitúa en un plano hidrofflico expuestaal solvente,mientras que la Tyr474homólogaal Trpl3 deFO III forma partedel corazónhidrofóbicodel DUC. Por lo tanto, elTrp5 debeserel susceptiblede oxidaciónpor NES.
3.4.6. Identificaciónde un residuode Trp esencialen el dominio catalítico
La comparaciónde los mapaspeptídicosde EO III no modificaday EO III W20~ revelaprofundoscambiosen la región “2” (Fig. 37). Hay picosquedisminuyen(“c”, “d”) y picosqueaparecencomo consecuenciade la oxidacióndel segundoTrp: “e”, “f’, “g” y “h”; estosúltimosposeenun máximode absorbanciaa 250 nm, característicodel oxindol (Fig. 27, pág. 54) y noabsorbena 280 nm (máximo de absorbanciadel anulo de indol). El espectrodel péptido f2, elmásabundantede entrelos que aparecende novo, refleja su contenidoen oxindol (Fig. 38).
Los mismoscambiosseencuentranal compararlos mapaspeptídicosdel núcleode FO IIIno modificadoy W10,~.


RESULTADOS 66
0.015
o 0.010oa:ono
-o< 0.005
0.000
220
Longitud de onda (nm)
Figura38. Espectrode absorcióndel péptidof2 procedentede la hidrólisis tríptica de ECl III W20~.
En la tabla XV serecogenlos datosobtenidosen el análisisde algunosde los péptidosseñalados.Todoslos resultadosapuntaninequívocamenteal residuoTrp255 como el segundoen seroxidadoporNBS. La importanciafuncionaldeestegruposituadoen el Centroactivo deEO III escoherentecon su alto gradode conservaciónen las distintascelulasasde la familiaA (Fig. 48, pág. 83).
Tabla XV. Datos másrelevantesde los péptidosrelacionadoscon la oxidación del segundoTrp. La secuenciade FO III semuestraen la Figura 49 (pág. 84).
Pico
c0
M por PDMS, Da Secuencia N-terminal Péptido identificado (M teórica, Da)
1464.3<2407.3) No determinada 11e248-Phe263 de ECl III <1462.6)
No determinada I¡e-Ser-Leu-Pro-GIy-Asn-Asp-Trp-GIn- 11e248-?de ECl III
1478.7 <1639.6) lle-Ser-Leu-Pro-Gly-Asn-Asp-Trp0~-Gln- 11e248-Phe26S de ES III W2~ (1478.6)
3.5. Modificación de grupos carboxilo con agentesquímicos específicos
En todaslas glicosidasasestudiadashastala fechaseha demostradola participacióndeal menos un grupo carboxio en el fenómenocatalítico. En el caso de las que retienenlaconfiguración del carbono anomérico, la catálisistienelugarconel concursode un grupoácidoque facilita la salida del primer producto o aglicón, y de un carboxilato que actúa comonucleófilo queestabilizael intermediarioglicosil-enzima,normalmentemediantela formación
de un enlacecovalentecon el jón carboxonio(Sinnot, 1990). Recientementeseha demostradoqueFO III pertenecea estaclasede glicosidasas(Gebblery col., 1992); los estudiosde pH
250 280

RESUlTADOS 67
(pág. 52) señalan la existenciade un carboxilo esencialque debe estarprotonado.Con el fin
de identificar este residuoy el nucleófilo que presumiblementeposeeFO III en su centroactivo, se ha procedidoa la modificación de la enzimacon agentesquímicos específicosdegruposcarboxio.
En la Tabla XVI se recogenlos resultadosde los intentosde inhibición realizadosconcarbodiimidas(l-etil-3-(-dimetilaminopropil)-carbodiimida,EDC,yyodurode1-etil-3-(4-azonio-4,4-dimetilpentil)-carbodiimida,EAC), con N-etil-5-feniisoxazolio-3’-sulfonato(reactivoK deWoodward, RKW), y con N-etoxicarbonil-2-etoxi-1,2-dihidroquinoleína(EEDQ). Unicamenteel reactivo K de Woodward es capaz de inactivar EO III, pero en unas condicionesextremas.A pesar de que la celobiosaprotegede la inactivaciónpor estereactivo, lo cual indica queungrupomodificadopudieraestaren el centroactivo de la enzima,no seha profundizadoen elestudiode estareacciónde modificación debido a queel excesode reactivonecesarioparaobservarinhibición invalidacualquierconjeturaacercade la selectividady especificidadde lamodificación,y a que al ser 12 el númerode carboxilosmodificadosen la enzimainactiva, es
ínvíable la identificación de un hipotético residuo esencialentreellos.
Tabla XVI. Estudiosde modificación de FO III con reactivosespecíficosdegruposcarboxilo.
Modificador
EOC/EAC
Tampón Excesode reactivon % Inhibición~’ Observaciones
Fosfato pH 3.1 2500 0 En todos los casos sePiridina pH 4.0 2500 0 han hecho ensayos conMES pH 5.1 2500 0 y sin ester etflico deMES pH 5.6 500 0 glicina.
FEDO Fosfato pH 3.1 2500 0Piridina pH 4.0 2500 0MES pH 5.1 2500 0MES pH 5.6 2500 0
RKW Fosfato pH 3.1 2500 25Fosfato pH 6.0 2500 40MES pH 4.5 5000 30MES pH 5.1 5000 45MES pH 5.6 5000 60MES pH 6.4 5000 50
O [reactivo]/[EG lii]b después de 1 h de incubación a temperatura ambiente
3.6. Modificación de un carboxilo esencial0-celobiósido(EPO5)
con el marcador de afinidad 4’,5’-epoxlpentll-
El empleode marcadoresde afinidad,estoes,dirigidos específicamenteal centroactivodeunaenzima,suponeunavaliosaherramientaparala identificaciónde aminoácidosesenciales(Eyzaguirre, 1987). En muchosestudiossobrela función de gruposcarboxioha sido de granutilidad el acoplamientode un sustratoo análogoa un epóxido(Lundblady Noyes, 1984). Enel casode las glicosidasas,estaaproximaciónha permitido identificar carboxilosesencialesenlisozima de clara de huevo de ganso (Eshdaty col., 1973). lacZ B-galactosidasade E. coli
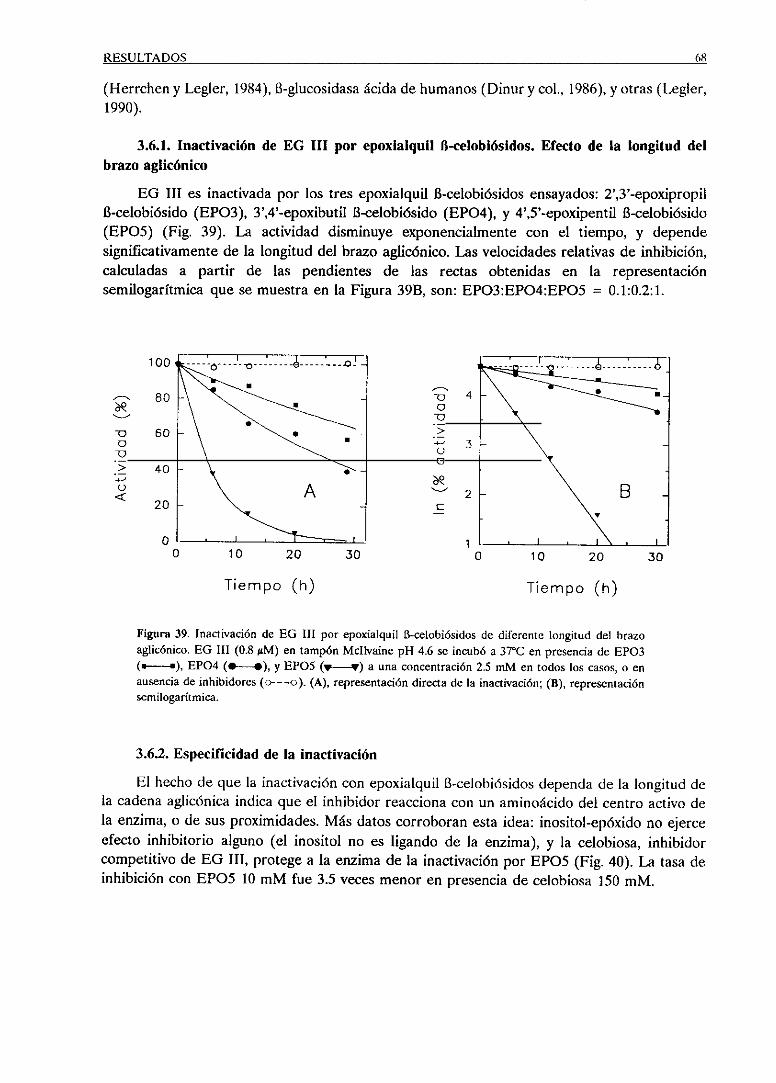
RESULTADOS 68
(Herrchen y Legler, 1984),13-glucosidasaácidade humanos(Dinury col., 1986),y otras(Legler,1990).
3.6.1. Inactivación de EG III por epoxialquil Il-celobiósidos. Efecto de la longitud delbrazo aglicónico
FO III es inactivadapor los tres epoxialquil JA-celobiósidosensayados:2’,3’-epoxipropil13-celobiósido (EPO3),3’,4’-epoxibutil 13-celobiósido(EPO4),y 4’,5’-epoxipentil ¡3-celobiósido(EPOS) (Fig. 39). La actividad disminuye exponencialmente con el tiempo, y dependesignificativamentede la longitud del brazo agilcónico.Las velocidadesrelativasde inhibición,calculadas a partir de las pendientes de las rectas obtenidas en la representaciónsemiogaritmicaque semuestraen la Figura 39B, son: EPO3:EPO4:EPO5= 0.1:0.2:1.
100
rs
‘o
ro-U
o4
80
80
40
20
o
-Uo
-o
Uo
‘o
a:
4
:5
2
Figura39. Inactivación de ECl III por epoxialquil 13-celobiósidosde diferentelongitud del brazo
aglicónico.ECl III (0.8 gM) en tampónMcllvaine pH 4.6 se incubó a 370C en presenciade EPO3(.—), EPO4(e—), y EPOS(v—q’) a una concentración2.5 mM en todoslos casos,o enausenciade inhibidores(o-— —o). (A), representacióndirectade la inactivación;(8), representación
semilogarirmica.
3.6.2. Especificidad de la inactivación
El hechode que la inactivacióncon epoxialquil 13-celobiósidosdependade la Jongitud de
la cadenaaglicónícaindica que el inhibidor reaccionacon un aminoácidodel centroactivo dela enzima,o de susproximidades.Más datoscorroboranestaidea: inositol-epóxidono ejerceefecto inhibitorio alguno (el inositol no es ligando de la enzima), y la celobiosa, inhibidorcompetitivode EG III, protegea la enzimade la inactivaciónporEPOS(Fig. 40). La tasadeinhibición con EPOS 10 mM fue 3.5 vecesmenor en presenciade celobiosa150 mM.
0 10 20 30
Tiempo (h)
0 10 20 30
Tiempo (h)

RESULTADOS 69
100
.—~ 80
‘o
r 60or.? 40-4-.
o4
20
o
Tiempo (mio)
F¡gura40. Efectoprotectordela celobiosasobrela inhibiciónporEPOS.ECl III (0.8 gM) entampónMcllvaine pR 4.6 se incubóa 370C con EPOS(10 mM) en ausencia(—.) o presencia(c—o) de celobiosa150 mM. (..--~-a), control sin inhibidor.
3.6.3. Cinética de la inactivación por EPO5
Siendo EPOSel inhibidor más efectivo de los ensayados,seha escogidoparaprofundizaren el estudio de la modificaciónespecíficadel centroactivo de FO III.
La representaciónsemiogarítmicade la actividadresidualfrenteal tiempoparadiferentesconcentracionesde inhibidor se muestranen la Figura 4tA; de las pendientesdc las rectasobtenidassecalculanlasconstantesdeprimerordende la inactivación(k
01,.), cuyadependencia
de la concentración de FF05 ha resultado ser hiperbólica (Fig. 4 iB). Esta cinética de saturaciónes la esperada para un marcador de afinidad, que se une a la enzima formando un complejodisociable enzima-inhibidor como paso previo a la inactivaciónirreversible,segúnse indicaenla ecuación (8):
E+l~El —.-E-I (8)
dondeE ‘1 es un complejo reversibleentre la enzima y el inhibidor, y E-I es la enzimainactivadapor unión covalenteal inhibidor (Eyzaguirre, 1987). Cuandola concentracióndeinhibidoresmuy superiora la deenzima,comoesel caso,y asumiendoquek2 esmuchomenorque k.1, se cumplela ecuación(9):
kapp = (k2 ‘[l])/([I] + Kd) (9)
donde sedetermina,como ya seha indicado, a partir de la ecuaciónIn (E(t)/E0) = -
k8pp ‘t, siendoE(t) la actividadresiduala tiempo t, yE0 la actividadenzimáticainicial; Kd es la
0 50 120 180 240 300

RESULTADOS 70
constantede disociación del complejo E ‘1 (Kitz y Wilson, 1962). Del ajuste de los datos
representadosen la Figura41B a la ecuación(9) sehan obtenidolos siguientesvaloresparaKd
y k2:
30 mM
= 35 1O~ min’
El orden de la reacciónde inactivaciónseha determinado,de acuerdocon Levy y col.(1963), a partir de la representación de log (k~,~) frente a log ([EPO5]) (Fig. 41C). La pendientede la recta obtenida es 0.8, indicando que la inactivación depende de la unión covalente de unamolécula de inhibidor por molécula de enzima.
‘1
o
4
3
2
-U-,
o-o
oEJ
a:
15
10
c
aaEJ
-st
5
o
rlo
c
aao-z
‘o
o
1.2
0.9
0.6
0.3
0.3 0.5 0.7 0.9 1.1 1.3
oq ([EPOS] mM)
Figura 41. Cinética de la inactivaciónde ECl III por EPOS.(A), la enzima(0.8 uM) se incubó(pH4.6, 37
0C) con EPOS2 mM (.—u), 5 mM (v—v), 7.5 mM (—s), 10 mM (o—o), 15 mM
(y—y), y 20 mM (A—t4, o sin inhibidor (o—c). (U), variaciónde ~ con la concentracióndeEPO5; la líneacontinuase ha deducidoa partir de los datosexperimentales( • ), por ajustea laecuación(9). (C), determinacióndel ordenaparentede la reacciónde inactivacién;la recta se hadibujado a partir del ajustepor regresiónlineal de los datosexperimentales( • ).
0 5 10 15 20
[EPOS] mM
0 50 100 150 200
Tiempo (mm)

RESULTADOS 71
3.6.4. Dependenciacon el pH de la inactivación por EPOS
En la Figura 42 se observala similitud entre la variación con el pH de la actividaddeEO III sobrecelotriosa,y la inactivaciónde la enzimaporel marcadorde afinidad.
100 100
80 ..-.. 80
‘ors~ 60 r 60-
‘o oaa 4oo 40
o20 20
0 03 4 5 6 7
pH pH
Figura42. (A), Dependenciacon el pH de la velocidadde inactivaciónde HG III por EPO5. Laenzima(0.8 uM) se incubó con EPOS5 mM aM0C, entampones(Mcllvaine) de distinto pH. Laactividadresidual se determinóa intervalosde tiempo apropiados,y las correspondientesk
0~, secalcularondela representaciónsemilogarítmicadeactividadresidualfrenteal tiempo. (B), variaciónconel pH de la actividaddeECl Hl sobrecelotriosa.
3.63. Identificación del grupo carboxilo esencialunido a EPOS
Para la identificación del grupo carboxilo unido a EPOSse ha procedido a la digestión demuestrasde FO III control y EPOS-FOIII, con proteasaV8, en condicionesde roturaespecíficapor enlacesOlu-X (tampónbicarbonatoamónico50 mM, pH 7.8). Esta proteasapuederompertambiénenlacesAsp-X en otrascondiciones(fosfato sódicoo potásico50 mM,pH 7.8) (Drapeau,1977). Si el marcadorseha unido a un residuode Olu, es de esperarquela enzima proteolitica sea incapazde romper tras eseresiduo provocandola desapariciónteórica de dos picos, y la aparición de uno nuevo, compendiode los dos anteriores.Lacomparaciónde los mapaspeptídicosobtenidos(Fig. 43) confirma la desapariciónde dospicos(“A’ y “B”) peroen elcromatogramacorrespondientea EPOS-FOIII no seobservaningún piconuevo. La desapariciónde E se apreciamejor en los mapastopográficosde la Figura 44. Ladisminuciónde “C” en el mapade EPO5-EOIII no es repetitiva;en experienciasparalelasnose han observado cambios en ese pico. En la Tabla XVII se muestran los datos correspondientesal análisisde los péptidos“A” y “E”.
/js
¡ 1 L
34567

GIS
0.10-
0.05
o
0.15
o-lo
0.05
o
Tiempo
Figura43. Comparacióndelos perfilesdeelución(A214)de los hidrolizadosde HG [II no modificada(control) y EPO5-EGIII, obtenidospor digestiónconproteasaV8.
(mm)
Tabla XVII. Datos de masa ymuestra en la Figura 49, pág.
secuenciade los péptidos84).
“A” y “B”. La secuencia de EO III se
Pico
Aconti
M por PDMS, Da Secuencia N-terminal Péptido identificado <M teórica, Da>
3207.3 Cys-Thr-Thr-Asn-Asn-Ile- 0ys302-Glu329 de EG III (3207.4)
Bcontroi No determinado Asp-GIy-AIa-Phe-Ser-Pro-Leu- Asp3CS-? de EG III
RESULTADOS 72
U,
uGo-nO<19-n
30 40 50 60


RESULTADOS .74
APENDICE. Análisis cinético de la hidrólisis enziniática de sustratos con dos víasalternativas de ataque.
Los parámetroscinéticosparadossustratosde EO III, MeUmb(Olc)3y CNP(Olc)3 hansido determinadosasumiendoel siguienteesquema:
(E ‘S)1-’———(E ‘(Otc)2) ~ E + (Olc)2X-Olc H20
E+S Esquema1k+r
(E ‘S)2—’——’~(E ‘(Olc)3) ‘—-~--- E + (Glc)3X 1420
dondeX esCNP cuando5 es CNP(Olc)3,y MeUmb cuando5 es MeUmb(Olc)3; E esEO III;y ku (1 = 1, 2 o 3, y] = 1 o 2) son las constantesde primer orden para cada paso ¡ y vía deataque enzimático j.
Este mecanismo es el más simple que explica todos los datos experimentales obtenidos,pero la naturalezadelos intermediariospropuestosesdesconocida,y la existenciade máspasosno es descartable. Si la glicosilación es el paso limitante de la reacción (k431 > k+2~), y se alcanzael estado estacionario para (E ‘~)~ y (E ‘5)2, se deduce que las velocidadesde formacióndeX-Olc, y1, y de Y, y2, son:
= kati’ [E]0’ [S]/(Kmi + [5] ‘(1 + K~1/K¿) (10)
y= k~ ‘[E]0 ‘[S]/(K,,.~, + [5] ~(1+ K~/Kmi)) (11)
donde esk~2~,[E]0es la concentracióninicial de enzima,Kmi = (&21 + k~1)/k+11,y K,,~ =
(k~~ + k.1j/k~12. Los experimentosde partición con metanol(pág. 46) demuestranquesóloen el caso de la reacción de formación de CNPa partir de CNP(Olc)3 la desglicosilaciónes elpaso limitante de la reacción, y por tanto la aproximación no es válida.
Los parámetros aparentesde la reacción, es decir, los que se podrían determinar a partir
de los datos experimentales si se consideraracada vía alternativa como una reacciónindependiente que cumpliera el modelo clásico de Michaelis-Menten,estánrelacionadosconlos parámetros teóricospor las siguientes ecuaciones:
(kcati)app = kcati/(1 + (12)
(kcaa)app = k~0/(1 + K¡njKmi) (13)
(Kmi)app = (K~X,~ = K~1 •K,¡.,j(Kmi + (14)
Operandoadecuadamentesededucequela relaciónentrelasvelocidadesde formacióndelos dosproductoscromofóricosalternativos(v1/v2) esconstante,y por lo tanto independientede la concentraciónde sustrato:

RESULTADOS
= (katt)app/(kc~a)app = k~11 ~ ‘Kmi)
La linearizaciónde las ecuaciones(10) o (11) permitecalculark~íj/Kmj:
[511v1= Kmj/(kcati ‘[E]~) + [S]/((kcaíj)app[E]0)
-75
(15)
(16)

DISCUSION

DISCUSION .77
1. PURIFICACION Y PROPIEDADES MOLECULARES DE EG III
Numerososprocedimientosde purificaciónpara las celulasasde Tricizodermareeseihansido desarrolladosen distintos laboratorios,haciendouso de combinacionesmás o menoscomplejasde técnicasde separaciónconvencionales(cromatografíaen todas sus vertientes,electroforesis,isoelectroenfoque)(Bhikhabhaiy col., 1984; van Tilbeurghy col., 1984; EnariyNiku-Paavola, 1987; Biely y Markovic, 1988; Schúlein, 1988, por citar algunos ejemplossobresalientes).Frente a la mayor o menorcomplejidad de estosprocedimientos,el pasocromatográfico en DEAE-Sepharosea pH 7 diseñadoen nuestro laboratorio para elfraccionamíentoinicial del materialdepartida,suponeuna forma rápiday sencilladeseparareficazmentelas principalescelulasasy hemicelulasasdel hongo(Fig. 12, pág.40).
Sin embargo,sólo se había descritohasta el presenteun método especifico para lapurificaciónde EO III (Saloheimoy col., 1988).A falta de datoscuantitativosreferentesadichométodo, no podemos estableceruna comparacióncon el procedimientoaquí expuesto.Encualquier caso, la preparaciónfinal de EO III empleadaen este estudio es altamentehomogéneacomo demuestranlos análisisde purezarealizados(Figs. 13 y 14, pág. 41).
En un trabajoprevio sehabíademostradoque la composicióndel sistemacelulasade 71reeseles independientede la fuentede carbonoempleadaen el cultivo del hongo(Domínguez,1991); no obstante,las condicionesde cultivo y la edaddel caldo determinanla aparicióndeisocomponentespor modificacionespost-traduccionalestales como agregación,glicosilación,desamidacióno proteolisis; así, en este trabajo no se ha detectado la presencia del núcleocatalítico de EO III, aislado en otras condiciones(lactosacomo fuente de carbono) porStáhlbergy col. (1988).En cambio,sí seha detectadoy purificado una isoenzimade EO III,denominadaA 1:2 (StAhlbergy col., 1988); ambasproteínasposeenigual masay forma (segúnse determina por electroforesis y cromatografía de penetrabilidad, respectivamente),composiciónde aminoácidos(determinadadespuésde hidrólisisácida,queno permiteconocerlaproporciónde Asn y Asp, o Gín y Olu, sino únicamentela sumade ácidoy amida paracadapar)y propiedadescatalíticas,diferenciándoseporsupI, másácidoen el casodeA 1:2. Parecemuy probablequeA 1:2 seauna forma desamidadadeEO III, y dado que la desamidacién esun mecanismode degradacióndeproteínasrelativamentepoco estudiado(Creighton,1985),seríainteresantedeterminarlas causasde apariciónde A 1:2 en el caldode cultivo de 71. reesei.
El comportamientodeEOIII en las doscromatografíasde intercambioiónico en DEAE-Sepharoseincluidas en el procesode purificación resultasorprendentea primera vista: a pH7 (fosfato sádico 5 mM) la enzimaesparcialmenteretenida,mientrasquea pH 6.5 (imidazol’1-ICI 8 mM) la retención es total. Esta aparentecontradicciónse explica por la diferentenaturalezaiónica de los tamponesempleadosquehace queen el primer casola cantidaddecargas negativas (que compiten con la proteína por las cargas positivas del lechocromatográfico) sea mayor que en el segundo, y, por otra parte, por la débil carga neta queexhibe la proteína a valores de PH neutros (por encima de su pI) como indica su curva detitulación (Fig. 16, pág. 43). A consecuenciade estapropiedad,la determinacióncorrectadelpI de EO III implica largostiemposde electroenfoqueparapermitir a laproteínamigrar hastael pH en que su cargase hacenula. En consecuencia,el valor depl ha sido reevaluadocomo5.1 frente al valor de 5.6 publicadopor Saloheimoy col. (1988), en un artículo dondesemostrabaun gel de isoelectroenfoqueenel queEG III aparecíacomounabandadifusay ancha

DISCUSION 78
debido, probablemente,a un tiempo insuficiente de electroenfoque,que condujo a unaimprecisadeterminacióndel pI. La posibilidadde que la discrepanciaen el valor de pI fueraachacablea diferenciasrealesentreambasproteínasquedódescartadaalobservarquemuestrasde EG III procedentesdel laboratoriodel Dr. Pettersson(Universidadde Upsala,Suecia),coautorde la citada publicación,y las preparadasen nuestrolaboratorio,exhibían el mismocomportamientoen isoelectroenfoque.
Cuandoseemprendióelestudiode EGIII existíaciertacontroversiaen cuantoalnúmerode endoglucanasassecretadaspor 71 reesei, y debidoa la similitud en masamoleculary pi de
las enzimasdescritasen distintos laboratoriosresultabacomplicadala identificación de laendoglucanasapurificada con alguna de las caracterizadashasta entonces.Los estudiosrealizadosen estadirecciónsellevaron acaboen el laboratoriodel Dr. Petterssonen Upsala,y demostraroninequívocamentela identidad de la endoglucanasapurificada en nuestrolaboratorioconel productodel genegl3 (Saloheimoy col., 1988)(pág.39; Tabla VIII, pág.43).
Las 12 Cys presentesen EO III se encuentranformando6 puentesdisulfuro. La niísmasituaciónse da en otrascelulasasde 71 reesei:EO 1 (Penttilá y col., 1986) y CBH1 (Bhikhabhaiy Pettersson,1984),y en EO 1 de Schyzophyllumcommune(Clarkey Yaguchi, 1985).En CBHII existendos Gys libres y dos puentesdisulfuro fundamentalesen el mantenimientode laestructuratridimensionaldel centro activo, que sedisponea modo de túnel para acogeralsustrato(Rouvinen y col., 1990).
2. ESPECIFICIDAD Y MODO DE ACCION DE EG III
EOIII hidroliza eficazmentecelulosassustituidassolubles,celooligosacáridosy derivadosde éstos. El estudio de los patrones de rotura de sustratos de bajo peso molecular proporcionainformación sobre la estructuradel centro activo de la enzima. El empleo de derivadoscromofóricos de celotriosa, MeUmb(Olc)3 y CNP(Olc)3, resulta muy adecuado para diferenciarEO III de otrascelulasasde 71 reesei, así como para medir la actividadenzimáticaen áquellosestudios que requieran precisión y reproducibiidad. En el caso concreto de la modificációnquímica de la enzima estos sustratossehan reveladocomoauténticassondasdel centroactivo(estudios de la modificación por NBS). Por el contrario,la carboxinietilcelulosa(CMC), quese ha utilizado tradicionalmente como sustrato de endoglucanasas, presenta seriosinconvenientesya que setrata de una mezclaheterogéneade polímerosde distinta longitud,con varias poblacionesde enlacesgiucosídicos en cuantoa su grado de accesibilidada lasenzimas(Sharrock,1988); su naturalezaiónica hacequeel pH, la fuerzaiónica o la temperaturainfluyan enormementeen la viscosidadde sussoluciones,quesuponeen sí misma un problemapráctico de manejo (Sharrock, 1988); por último, los métodos colorimétricosempleadosen ladeterminación de azúcares reductores liberados por la acción enzimática no son
estequiométricos,ya que la reaccióndel grupoaldehídoreductoresdistinta dependiendodelresiduo al cual estéunido (Aminoff y col., 1970). Así, cuandose ha ensayadoCMG comosustrato de EO 111 se ha observado que la curva de actividadfrentea pH no coincidecon la deotros sustratos (Fig. 17D, pág. 45), y que la variación de la actividad con la concentracióndeenzima dista mucho de ser lineal (Fig. 22, pág. 50). No obstante, CMCresulta útil en aquellasvaloraciones de la actividad enzimática que no requierengranprecisión,como porejemploenla detección de endoglucanasa en eluidos cromatográficos.
-I

DISCUSION .79
2.1. Modelo de organización en subsitios del centro activo
En las enzimasquedegradanpolisacáridos,como en otrasque actúansobrepotímeros,el centroactivo estácompuestoporvarios subsitioscadauno de los cualesescapazde alojarun residuoglicosídico. El ejemploarquetípicoesel de la lisozimade clara de huevode ganso,en la queexisten6 subsitiosde unión (Blakey ccl., 1967;Johnsony col., 1988).Recientementeseha determinadola estructuratridimensionalde doscelulasas,una endoglucanasa,EGD deCtostridium thermocellum(Juy y col., 1992), y una celobiohidrolasa,CBH II de 71 reesel
(Rouvineny col., 1990).El centroactivo estáconstituidopor 6 subsitiosen la primeray por 4en la segunda.La diferenciaen el modo de acción de ambasenzimas se explica por laarquitecturade la hendiduradondesealoja el sustratoquepresentalos dosextremosabiertosen la endoglucanasa,mientrasqueen la celobiohidrolasauno de ellosestácerrado,impidiendoel pasode celulosa.
En EG III los patronesde roturadesustratosde bajo pesomolecular(celooligosacáridosno sustituidosy metilados,Fig. 21, pág. 49,y derivadoscromofóricos,Fig. 18,pág. 46) permitenaventurarun modeloparaelcentroactivo, queestaríaformadopor cincosubsitios(ABCDE),localizándoselos aminoácidoscatalíticosentrelos subsitiosC y D (Fig. 45).
ynEk~ñD~n vn
A B C O E
m
nETJHEHZ½i ‘~ 1~H~O
(LJ—crJ-—w—Me) (MOOH)
vn vn vn m vn ¡
vn vn m#Ek~DFrm vn.4 E O O E
vn vn Eflpjf ~ h vn vn
EE—EJ H0
(MeOH)
(EbfXkMe)
vn vn n m vn vn
Figura 45. Representaciónesquemáticadel modelopropuestode organizacióndel Centroactivo deHG III. Las dos vías alternativasde hidrólisis de MeUmb(Glc)3 se muestrancomoejemplo. Laposibilidadde transferenciade restosglucosilo a metanoltambiénse señala.Las hendidurasen elbloque, cada unacon unaletrade la A a la E, indican los subsitiosde unión; las flechas ( ~indican gruposcatalíticosde la enzima.Otrossímbolos: ( W ), resto glucosilo;( ), grupoMeUmb; ‘Me’, grupometilo.
El extremo reductordel sustratopodría interaccionarcon el subsitioO o con el E; sitomamosla hidrólisis deMeUmb(Olc)3comoejemplo,en elprimercasoseobtendríanMeUmby celotriosacomo productos,mientrasque en el segundolos productosresultantesseríanMeUmbGlcy celobiosa.De igual forma sepodríanexplicarel resto de los patronesde rotura

DISCUSION SO
observados.Se postula la existenciade cinco subsitios, y no más, porque la inhibición porcelobiosaes competitivay no cambiael patrón de rotura de CNP(Olc)3 o MeUmb(Olc)3,indicando queno se puedenunir a la vez cualquierade estossustratosy el disacárido.
En su calidad de endoglucanasa,el centroactivo de EO III no ofrece,probablemente,ningún impedimentoal pasode celulosapor cualquierade sus extremos.
2.2. Mecanismocinético
Deacuerdocon los datosobtenidosen los estudiosdeparticióncon metanol,la reacciónde hidrólisis de oligosacáridos(y por extensión,de cualquiersustrato)catalizadapor EO IIIsigueun mecanismosecuencialordenadoUNI-BI, deacuerdocon la terminologíade Cleland(1977):
E + (Olc)~ —<E .(Glct-.-——-.cr~j——~. E ‘(Olct~—~—-—-—.’-E + (Olct~k (Olc)~ 1420
En el complejodisociableenzima-sustrato,E ‘(Olc)0, la rotura deun enlaceglicosídico delsustrato da lugar a un primer producto, (Olc)~, denominadoaglicón (Sinnott, 1990), quecomprendeel extremo reductor del sustrato,y a un complejo glicosil-enzima, E (Olc)~~.probablementede naturalezacovalente(Sinnott, 1990); el ataquenucleofílico de una moléculade agua(o alternativamentede metanol)restaurala moléculade enzimay rinde un segundoproducto,(Olc)0~ (o Me-(Olc)0~cuandoseadicionametanol).
La capacidaddeEOIII paratransferirrestosglicosio a metanoly otrosalcoholesimplicanecesariamentequela enzimaretienela configuracióndel carbonoanoméricodel sustratoenel segundoproductode la reacción,medianteunadobleinversiónde la misma(Sinnott,1990).Estudiosde RMN hanconfirmado recientementeestapropiedadde EO III (Gebblery col.,1992). Sin embargo,y en contrastecon EO 1 de 71. reesei (Claeyssensy col., 1990a), no se ha
detectadoactividad transglicosidasaen EO III.
El pasolimitante de la reacción,deducidotambiénde los experimentosde particiónconmetanol,es la glicosilaciónde la enzimaen todoslos casosestudiados,exceptoen la liberaciónde CNP a partir de CNP(Olc)3 en la que pasaa serlo la desglicosilación.La razón de estadiferenciadebeencontrarseen el bajo valor de P
1<a del CNP (5.5), que lo configuracomo el
mejor grupo salientede entre los considerados(MeUmb, pl(3 = 7.2; grupos hidroxilo de
azúcares,pK, > 14) (Fersht, 1985). Un comportamientosimilar se observaen otrasenzimascomo, por ejemplo,B’galactosidasa(Sinnott y Viratelle, 1973).
2,23. Actividad sobremananos
La actividad mananasade EO III indica que la especificidadde la enzima no es tanestrictacomosesuponía;EO1 seconsiderauna endoglucanasano específicaporsu capacidadparahidrolizarxilanosy derivadosdelactosa,y EOIII seteníaporespecífica,porno seractivasobreestossustratosno celulósicos(Biely, 1990). No obstante,teniendoen cuentala similitudentreglucosay manosa(sólo sediferencianen la orientaciónde un grupohidroxilo), no esdeextrañarque EO III sea activa sobremananossiendo su principal función la de hidrolizarcelulosa.El papel fisiológico deestaactividadhemicelulasano estáclaro, habidacuentade laexistenciade una mananasaespecíficaen el caldo de cultivo de 71 reesei.


DISCUSION 82
OBE 1 Pc 423 TvpqWGQCGGIGYtGSTtCASPyTChVLNPYYSQCYCHE 1 Tr 405 TQSHYGQCGGIGYSGPTvCASGTTCqYLNPYYSQCL **
CEE II Tr 3 *CSSVWGQCGGqnWSGPTCCASGSTCvYSNDYYSQCL
EG 1 Tr 462 TQTHWGQCGGIGYSGCKtCTSGTTCqYSNDYYSQCL **
EG III Tr 1 *qQTVWGQCGGIGWSGPTnCAPGSACStLNPYYaQCI
Figura 47. Comparaciónde secuenciascorrespondientesal dominio de unión de las celulasasdeTñehodennat-eesei(Tr> y de CBH 1 de Phanerochaetech~ysospo,-iwn(Pc). En letrasmayúsculasynegrita se indican los residuosconservados,en mayúsculaslos parcialmenteconservados,y enminúsculaslos no conservados; indica extremoN-terminaly extremoC-terminal(Gilkesy col.,1991).
La oxidación del Trp2SS, localizado en el núcleo catalítico, afecta sensiblemente4 laactividadde EO III y los estudiosrealizadosindican que setrata de un residuoesencial-parala unión del sustratoal centro activo de la enzima.Deacuerdocon el modelode organizaciónen subsitiospostulado(Fig. 45, pág. ‘79), el Trp25S debeestarsituadoen el subsitioD, ya quees la únicaposibilidad que permite explicar todos los efectosobservadosen la hidrólisis dedistintossustratoscuandose modifica dicho residuo.El patrónde rotura de MelJmb(Olc)3oCNP(Olc)3cambiadrásticamente,y en distintosentidoparacadasustrato(TablaXIII, pág.59),indicando que el Trp255 debeinteraccionarcon el grupo cromofórico correspondiente;ladistinta naturalezaquímicay volumen de estosgruposhaceque la MeUmb sea preferidoalCNP en la enzimanativa,y viceversaen EGIII W20~. Hastaaquípodríamoslocalizarel Trp255en D o E (ver Fig. 45, pág. 79), pero el hecho de que la oxidación de este residuo no cambieel patróndehidrólisisdecelooligosacáridosno sustituidos(sereducenen la mismaproporciónlas velocidadesde todaslas vías de ataqueenzimático)lleva a la conclusiónantesexpuesta,porquesólo en el subsitioO el Trp255 puedeinteraccionarcon un restoglicosídicodelsustratosea cual sea el modo de unión (productivo) considerado;si estuvieraen el subsitio E, lamodificación por NES no afectaríaa los modos de unión en los que el extremo reductorinteraccionaracon O, y en consecuenciasealteraríael patrónde rotura.
Según la nomenclatura propuesta por Karpeiskii (1976) para la designación deaminoácidosesenciales,Trp255 es un aminoácido de contacto, es decir, que participadirectamenteen la estabilizacióndel sustratoen el centro activo de la enzima. EstudiosdealineamientodesecuenciasmuestranqueesteresiduodeTrp estábienconservadoen distintascelulasasde la familia A (Fig. 48).

83DISCUSION
EC III Tr 111 n~qhfvned0I4Tl Fj4PVgwqytvnnnLcntdstsi..’--’SkYd~I ---‘‘‘‘VQGCLSLCAVCIV’Ol yarwngEa C Ci 33 dietiaeaGFDII~A$.PFdypitesddnvgeykedgt -‘‘‘‘‘ syidr LEWCKKYtN-Ct.VLOM apgyrf
EC A Ci 85 dieLvadkGlNV91~4PlatdItyawsqgiyppstdtSyflflpfiL8~LflSYCL1flfmLENFKRVClI<VIL’D petdnqEG H Ci 362 yfddfkaaGYKN~lPVrwdnh¶tmrtypytidkfif Id’’’’’’ rveqv’’---”VtJWSLSRCFVTII’N dd’wikCAlI Sc 118 dfaniasqCFNl.~lPlgywofqttdddpyvSgIqCSy’’’’~ Ldq IGIJARNNSLKVWV’DL aagsqn
AMAN A Cs 79 CMNS\i4JVLsngyrwtkipo...--’’’-’’’’’SCVGflI’’’’’’’ ISLSRSLCFKAIILEV ttgyge
EG III Tr 182 giigqggptnaq””-’”’ FTSL$kOLASKYAS--.-OSRVIJFGIMNH’hdvnintwaa”””EC C Ci 101 qdfktstkfedpnqqk FVD44Fl.AKRYINe’’’REHlAFELLN~tvepdstrwn--
EG E Ci 163 ghnyptwynttiteeí FtK4.4MVAERYlCNd’’-DTllGFDLlCN~j’htfltgtfltf kaqsafwdds¡tpnEG 14 Ci 427 edyngnier’-----’’’’’’ Fa l~OlAERFKNk---SEULLFElMN~’fgnitdeq idCAlI Sc 183 gfdr.sgtrdsykftedsntavttnvtnyitkkysaeeytDTvlGlELl NM~t9pvtárdkm”””-’--” -“‘ kn
AMAN A Cs 129 dgaacstaq--”””’-’’ aveybIkeiksvtdgn”-EDFVllNlGN~’ygnrd1yqfl”’----’’”””-’ 14V
EG III Tr 230 ‘tvqevvtairnaga’’tsOFlSLPG’nc~qsagafisdg aaatsqvt’npdgstt’’-nLl
Ea C Ci 151 klmLeyilcairei’’’dstMWLVlGC’ n4+spdet knta-diddd’’’’-ylV
EC A Ci 229 nwkrvaeetotaitevhpnVLlFVEGveij4kdgiwddetfdtsPwt9nndYY9flww99fltr9VkdYPifltgkY~SqLV
Ea N Ci 472 ckTr,sriLkiirkt---nptRJVllGC-944’IsyntL vnik-ipddp yLl
LEN Sc 246 dytapayeyLrnn’iksdqvlllsoAfq44iywddf mten-dgywg-’-’’VT
AMAN A Cs 174 ndtknaíkatrda”-gfkHTINVOA’pr~qd- ‘ -‘ wsntmrdna’qsimeadplrntV
EG III Ti- 285 Fb4jKYLdsdnsgthaecttnnidgafsptatwtrq rwlrq
EG C Ci 195 Vw44FYNpfffthqkahw---’-.’’’.’’’ sesamaynr’’’’ tvkypgqyegieefvkmpkysfn.netnntktnlce
EG E Ci 307 VSP~YGpivyeqdI.ifkgdfitfindeqakrityeqcl4rdnwayimeegiSP’-’-’-’-””----’””-”””
EG 5 Ci 516 GTFII<YYopvefthkwr9t’ ‘wgtqechidtvvrvfdfvksw’’ - - sdrrntp
CAH Sc 292 ID~~HYovfas~LersfdehikvacewgtgvL~shwt
AMAN A Cs 225 FS lj4lycvynteslcve yiqs-”- fvdlcgtp”””’----’---”---’’’-””
FC III Ti- 325 ‘‘..‘‘‘‘‘‘‘...‘‘--‘AlL§CCGNvqsciqcticqqiqvLnqnsdvytg-’y~swgagsfdstyvttetPtss9n
FC C Ci 256 ltrkdtkpaiefrekkkckt.VC4E¡CVlAiadtesrikwhedyistteey’’diggavwnykkin dfeiynedr’EG A Ci 358 LLL1~E$JCGMTegghpttdLntkytrcmrdfitenlcykthhtfwcinidsadtggtftrde
EC H Ci 561 VVFc1E>AVMAyadrtsrvkwydfisdaater~~gfacSvwdngVfgStdfl~ckMiyflrdtLEN Sc 332 CI~EjAaaLtdctkwLnsvsfgarYd9swvn9dqtssYi9scarv~ddiaYl.iSdCrkeflt
AMAN A Cs 252 LVI &JaHoEtdgdpdeeaivryakqvkigL f----swswcgnssyvgyt’”’’
Ea III Ti- 383 s---wtdtstvssctark 397
FC c Ci 329 lcpvsqetvni’-’’Larr 342
FC A Ci 418 gtpfpggrdtkwndnkyd 435
FC 14 Ci 618 rtfdteitn tfnp 631
Cali Sc 390 rcyveaqtdafemrggwi 407
AMAN A <Ss 298 ----~nvnnwdpnnptpw 311
Figura48. Alineamientode secuenciasdeldominio catalíticode las celulasasdela familia A. En mayúsculaseseñalanlos segmentosen los que se han localizadopor HCA (Gaborjaudy col.,1987)grupos de aminoácidoshidrofóbicos
topológicamenteconservadosen las celulasasde la familia A (Henrissaty col., 1989); en negritase muestranlosaminoácidosconservadosquepuedenparticiparen la unión del sustratoy/o en la catálisis.Claves:HG III Ti-, HG IIIdeTrichodermareesel(Saloheimoy col., 1988);HG C <Sí, HG C deClosírid¡wn ihennoceUwn(Schwarzy col., 1988);EGE Ci, HG E deC. ihennoceliwn(Grépinety col., 1986);EG H Ci, HG H deC. íhennocdlwn(Yagiley col., 1990);CBHSc, CBH de Sacchaminycescerevisiae(VázquezdeAldana y col., 1991); EMAN A <Ss, 13-mananasaA deCaldocellwnsaccharo¡yiicwn(Luthi y col., 1991).

DISCUSION 84
CI,, Cm mr Val ffrplCty Cín Cys Cly Cly Ile Cly Trp Ser Clv Pro Thr Asn Cys Ala Pro Cly Ser Ala Lys Ser Thr10 20
ouc NúcleoTyr Tyr Ala Cl,, Cys Ile Pro Gly Ala Thr Thr líe Thr Thr Ser Thr Arg Pro Pro Ser CIy Pro Thr rhr
40 ~o AThr Thr Arg Ala mr Ser Thr Ser Ser Ser Thr Pro Pro Thr Ser Ser Gly Val Arg Phe Ata Uy Val Asn Ile
60 70
Piie Asp Phe Cly Cys Thr Thr Asp Uy Thr Cys Val Thr Ser Lys Val Tyr Pro90
Pro100
Leu Lys Asn Phe Thr Ciy Ser Asn
Pro Asp CIV Ile Gly SIn Met Sin lIla Phe
120
Val Asn Glu Asp Cly Met Thr Ile Plie 4j
130lev Pro Val Cly Trp
SIn Tyr Leu Val Asn Asn Asn Leu SLy Cly As,, [cii Asp Ser
140Tu r
150
Ser Ile Ser Lys Tyr Asp SIn Leu Val
[cii Ser Leu Uy Ala Tyr Cys
Thr
190
líe
170
Asn Ata Cl~ Phe Thr Ser Leu Trp Ser
VaL Asp líe II~s Asn Tyr Ala Arg Trp
Cl,,200
As,,180
Leu Ala Ser Lys Tyr Ala Ser Cl~ Ser
Cly CLy Ile Ile Gly Cm Cly Cly Pro
Arg
210Val Trp Phe Gly líe Met
Asnf~3Pro lIla220
Asp Val Asn Ile Asn mr Trp Ala Ala
Thr ser SIn Phe Ile Ser [cia Pro250
Cly Asn Asp~Jtln
Thr230
Val 6l~ Chi Val Val Thr Ala líe Arg
Ser Ate Sly Ala
260
Asn Ata Cly Ala240
Leu
270
Ser Cín Val Thr As,, Pro Asp flly Ser
Tlir His Ala300
Thr280
Mu Cys Thr Ilir Asn Asn líe Asp Cly
Thr Asn Leu Ile Phe Asp Val lila Lys
Ata
310
Tyr290
Phe Ser Pro t.eu Ata Thr Trp Leu Arg
Leu Asp SerAsp Asn Ser Cly
Cín
320As,, Asn Arg Un
Ala Ile Leu Thr~j~j}Thr330
Sly Uy Cly As,, Val Cl~ Ser Cys Ile Gl~ Asp Met Cys Cm Gl~ Ile SIn Tyr Leu
340
Asn Ser Asp Val Tyr Leu Cly Tyr Val360
Cly Trp Cly Ala Cly Ser Phe Asp Ser Thr
370Tyr Val Leu Thr Síu Thr Pro Thr
Cly Asn Ser Trp Thr Asp Tlir Ser Leu Val390
Ser Ser Lys Leu Ala Arg [ya397
Figura49. SecuenciadeEG III de Trichoderma reesel(Saloheimoy col., 1988).Los aminoácidosconservadosen lascelulasasde la familia A seseñalanen rojo;se enmarcanlos residuosesencialesidentificadosen el presentetrabajo. Laflecha ( ~ ) indica el punto de rotura en la bisagra,que da lugar al núcleocatalíticoaisladodel caldode cultivo por Stáhlbergy col. (1988).
Leu Asn Pro30
Asn Tyr
110
Ala CIy80
SIn Cly Cys
160
Phe líe Ser Asp Cly Ser Ala Ala Ata
Ser Ser380
Asn Sin
350

DISCUSION 85
4. IDENTIFICACION DE DOSGRUPOSCARBOXILOESENCIALES: MECANISMO
QUíMICO DE £0 III
4.1. Estudios de pH: GIu218
Los estudiosdel efecto del pH y la temperaturasobrela hidrólisis de MeUmb(Olc)3catalizadapor EO III revelanla existenciade un grupocarboxioesencial(p
1C0 = 5.5) en el
centroactivo de la enzima.Como el residuo01u35 de lisozima,estegrupoestáprotonadoenla formaactivade la enzima,y su función debeserfacilitar la salidadel primerproductodelareacción(aglicón) por catálisisácidageneral(Sinnott, 1990).Este residuode FO III podríacorrespondera01u218, incluido en la secuenciaAsn-Glu-Proconservadaen todaslas celulasasdela familia A (Béguin, 1990)(Fig. 48).Experimentosde mutagénesisdirigida hancorroboradola importanciade esteglutámico,comodonadorpotencialdeprotones,en tresendoglucanasaspertenecientesa Ja familia A: FO de Bacílluspolirnixa, EO de fi. subtills (Baird y col., 1990),yFO de Erwinia cIu~ysanthemi(Py y col., 1991).
4.2. Marcaje de afinidad: Glu329
La rápida inactivación de EO III a valores de pH por debajo de 4 (Fig. 17A, pág. 45) halimitado al intervalo de pH de 4.0-6.3 el estudio del efecto del pH y la temperaturasobre la
reacciónenzimática,por lo queno ha podido ser detectadapor estemétodo la presumibleexistenciade un carboxilatoesencialen el centroactivo de EO III (pág. 66). Estegrupo, sinembargo,ha sido identificado como 01u329 por marcajede afinidad con el epoxipentil-
- celobiósido (EPOS). La inactivaciónpor epoxialquil-celobiósidosocurre por modificaciónespecíficade un aminoácidodel centroactivo como sugierenlos siguienteshechos:
- la inactivacióndependede la longitud del brazo aglicónicodel inhibidor (Fig. 39, pág.68); EPOSesel máspotenteinhibidor de los ensayados,probablementeporquesu brazoaglicónicoesel que mejor reproduceel tamañode un restoglicosídico en el subsitioC,permitiendo así una correcta orientaciónde la funciónepóxido(quemimetizaa un enlaceglicosídico)respectoa los aminoácidoscatalíticos(Fig. 50). Resultadosparecidoshansidodescritos para celulasas de Oxysporusspp., Aspergí/tusníger y A. wentii (Legler y Bause,1973), y para EO 1 de Schyzophyltumcornmune(Clarke, 1988; H0j y col., 1989). Sinembargo,otra endoglucanasade la familia A, EO de Bacíllus subeitís,es inactivada másrápidamente por EPO4que por EPOS (H0j y col., 1989).
- el inositol-epóxido carece de poder inhibitorio; el inositol no es ligando de la enzima.
- la celobiosa, ligando de EO III, protege de la inactivación por EPOS(Fig. 40, pág. 69).
- la variación con el pH de la velocidad de inactivación es similar a la de la actividad de
la enzimasobrecelotriosau otros sustratos(Fig. 42, pág. 71).
- la estequiometría de la reacciónde inactivación,quedependede la unión covalentedeuna moléculade inhibidor por moléculade enzima(Fig. 41C, pág. 70).
Comoseobservaen el mecanismode inactivaciónpropuesto(Fig. 50), la reaccióncon elepóxidoimplica protonacióndesdeun lado del anilio, y ataquenucleofílico por un carboxilatodesdeel otro lado; en consecuencia,sólo las glicosidasasque operan con retención de laconfiguraciónanoméricapuedensufrir inactivaciónporepóxidos(Legler, 1990).La inhibiciónde FO III por epoxialquil-celobiósidosconfirma una vez másque estacelulasaactúapor unmecanismo de doble desplazamiento.

D¡ SCUSI ON 86
y8
-tio
o4>o
n ji nrtn ji
A BCD E
k.tj ¡yH
e,o- -ori m rl r~i ji ji
I
Figura 50. Mecanismo de
inactivación de FO III por EPOS
(H0j y col., 1989, modificado).Símbolos como en la Figura 45,
pág. 79.
BHo,cI-i.
o~.. ~e—ji n n ji
El carboxilato al cual se une covalentementeel EPOS ha sido identificado medianteanálisisde péptidoscomo Glu329. Esteresiduoestáflanqueadopordos residuosdeThr, en lasecuenciaThr-Glu-Thr, bien conservadaen las celulasasde la familia A (Fig. 48). Su funcióncatalítica sería estabilizar el catión carboxoniopresumiblementeformado tras la salida delaglicón, bien por interacción electrostática, bien mediante la formación de un intermediariocovaJente (Sinnott, 1990); se trata de un grupo equivalente al Asp52 de lisozima, o al 01u461
de B-galactosidasa,que son también susceptibles de esterificación por un epóxido unido a unanálogo de sustrato (Eshdat y col., 1973; Herrcheny Legler, 1984).
4.3. Mecanismo químico de EG III
En resumen, los datos experimentalesexpuestospermitenaventurarque EG III actúamediante un mecanismo similar al de la lisozima (caso paradigmáticode ¡3-glicosidasaqueretiene la configuración anomérica).El 01u218aceleraríaporcatálisis ácidageneralla roturadel enlace glicosídico situado entre los subsitios C y D, facilitando la salidadel aglicón; el ión
carboxonioresultanteseríaestabilizadopor el 01u329bienpor interacciónelectrostática,bienmediante la formación de un intermediario covalente; una molécula de agua protonaría

DISCUSION 87
entoncesel 01u218, rindiendoun ión hidroxilo que atacaríael restoglicosio para liberar elsegundoproductode la reacción,quedandola moléculade enzimapreparadapara iniciar unnuevociclo catalítico.
;Z //////9CIu 218
c
R’O -~.OH ,
HOM—t-~on\(B)
ROH R—OH
7%oSooGQo RO OH0 H
A ~ H
HO o B
OH • OH A”
oo O o
Ac c
Ro :48=7OH HO o
HO
000$4
c
Figura 51. Modelo propuestopara el mecanismoquímico de HG 111. Se muestranlos dos tiposposiblesdeintermediariosglicosil-enzima:(A), par jónico carboxonio-carboxilato,comoenla lisozima(PhIlips, 1966), y (II), ester glicosil-enzima, como en la 8-galactosidasa(Sinnott, 1987). Elintermediadorealpuedeserun hibrido entreambasformas.En lasdosvías, los estadosdetransición
(no ilustrados> tanto de la glicosilación (iCT paso) como de la desglicosilación(2~ paso) sonsemejantesal ión oxocarboniorepresentadoen A (Sinnott, 1987).
La naturalezadel intermediarioglicosil-enzimano ha sido determinada.En el casode lalisozima se consideraba tradicionalmente que el Asp52 estabilizabael catión carboxonioporinteracción electrostática (Phiiiips, 1966; Fersht, 1985). En una revisión recientesobre el

DISCUSION SR
mecanismocatalíticodeglícosidasas,Sinnottponeen dudaestaidea,argumentandoquela faltade sustratosde lisozima en los que la etapalimitante de la reacciónsea la desglicosilaciónhaimpedido recabarinformaciónmecanisticasobreesteparticular.En otrasB-glicosidasasqueactúande forma similar (8-galactosidasasde Escherichiacoil) sí seha demostradoqueel ión
carboxonio esterifica el carboxilato esencial de la enzima (01u461) dando lugar a unintermediariocovalente(Sinnott, 1990).
Haceyacasitreintaañossesugirió quetodaslasglicosidasaspudierancompartirel mismomecanismocatalítico que la lisozima(Banksy Vernon, 1963).En el casode las celulasasestaidea se ha mantenido hasta hace pocos años (Knowles y col., 1987),perohoy día sabemosqueno existe un modo de acción común a todas las celulasas:aquellasque actúaninvirtiendolaconfiguración anomérica lo hacen por un mecanismo de desplazamiento simple, netamentedistinto al de dobledesplazamientode la lisozima. No obstante,entre las glicosidasasde unmismo tipo si se encuentran características similaresen la estrategiacatalítica. Así, en estetrabajo se ha demostrado que EG III, una endoglucanasade la familia A de celulasas(queactúan vía doble desplazamientodel sustrato),pareceseguir un mecanismosimilar al de lalisozima. Experimentos de mutagénesisdirigida, intercambioisotópicoy determinaciónde laestructura tridimensional por difracción de rayosX deberán confirmar esta hipótesis.

CONCLUSIONES

CONCLUSIONES 9<)
1. Se ha desarrolladoun métodopara la purificación de endoglucanasa111 a partir decaldosde cultivo de TrichodermareeselQM9414. La enzimase ha identificado inequívocamente
como el producto del genegl3 (Saloheimoy col., 1988).
2. EO III presentauna débil carganegativaa valoresde pH próximosa la neutralidad;su pI ha sido reevaluado como 5.1.
3. EG III esactiva sobrecelulosay derivados,y tambiénsobremananos.Los sustratoscromofóricosMeUmb(Olc)3y CNP(Olc)3 son muy adecuadospara el estudiocinético de laenzima,no así CMC, tradicionalmenteempleadacomosustratode endoglucanasas.
4. La hidrólisis de JA-glicósidoscatalizadapor EG III transcurrecon retención de laconfiguracióndelcarbonoanoméricodelsustrato,medianteun mecanismosecuencialordenadoUni-Bi, con el concursode un intermediarioglicosil-enzima.El primer productode la reacciónesel fragmentodelsustratoquecontieneel extremoreductor,esdecir,el restode glucosacuyocarbonoanoméricoestálibre (o el restoequivalenteen el casode derivados).
5. Se ha propuestoun modelo de organizacióndel centroactivo deEG III, en el quelaenzima seria capaz de alojar hasta 5 residuos de glucosa, en 5 subsitios ABCDE, estando
situados los grupos catalíticos entre C y D.
6. El Trp5, localizadoenel dominio de unión a celulosade EGIII juegaun papelesencialen la adsorción de la enzima al polisacárido. El 1rp255 participa directamenteen laestabilizacióndel sustratoen el centroactivo de la enzima.
7. EG III poseeun grupocarboxilo esencial(PKa = 5.5,zúxH.~,,, = -15.9 J/mol) que debeestarprotonadoparaque la enzimaseaactiva. Esteresiduo seha identificadocomo 01u218por estudiosde homología con otras celulasas relacionadas estructuralmente con EG III.
8. El marcadorde afinidad4’,5’-epoxipentil-13-celobiósidoseunecovalentementeal 01u329inactivandoEG III. Estegrupocarboxio es esencialpara la actividadcatalítica en su formaionizada,y estabilizael intermediarioglicosil-enzima.
9. EG III actúamedianteun mecanismodedobledesplazamiento,similaralde la lisozima.El 01u218 aceleraríapor catálisisácidageneralla rotura del enlaceglicosídico situadoentrelos subsitios C y D, facilitando la salida del agJicón; el ión carboxonio resultanteseríaestabilizadopor el Glu329 bien por interacciónelectrostática,bienmediantela formación deun intermediariocovalente;una moléculade aguaprotonaria entoncesel Glu218, rindiendoun ión hidroxilo queatacaríael restoglicosio paraliberarel segundoproductode la reacción,quedandola moléculade enzimapreparadaparainiciar un nuevo ciclo catalítico.

BIBLIOGRAFIA

BIBLIOGRAFíA 92
AbsarN., Yamasaki,N. y Funatsu,G. (1986) Idenuificalionof mhe íryptophanresiduelocatedal the saccharidebinding
site of CastorBeanhemagglutinin ¡¡gr Bid. Client. SO: 3071-3076
Alíen, A. (1983)“Mucus: a protectivesecretionof complexity TrendsBiod,em.8:169-173
Acebal,C., Castillón,M. P., Estrada,P., Mata, 1., Costa,E., Aguado,3., Romero,Uy Lacaba,F. (1985) Cellulaseproductionby Tñchodennarasel on wheatstraw” Biod:em.Saz.13: 453-455
Acebal,C., Castillón,M. P., Estrada,P., Mata, 1., Costa,E.,Aguado,S., Romero, O. y Jiménez,F. (1986) Enhancedcellulaseproduction from Trichodennareesel 0M9414 on physically treated wheat straw” Appl. Miembiol.Biotechnol. 24: 218-223
Aninoff, U., Binkiey, W. W., Schaffer,R. y Mowiy, R. W (1970)en ‘The carbohydrates”(Pigman,W., llorton, O. yHerps,A., eda) Vol. 2B, pp. 739-807,AcadeznicPress,NuevaYork, USA
Baird, 5. 0., Hefford,M. A., Johnson,0. A., Sung,W. L., Yaguchi, M y Seli~’, y. L. (1990)“The Glu residueiii theconservedAsn-Glu-Prosequenceof two highly divergentendo-B-1,4-glucanasesis essentialfor enzymaticactivity”Biochen,. Biophys. Res. Conunun. 169: 1035-1039
Barceló,J., Nicolás, G., Sabaler,E. y Sánchez,R. (1987). “Fisiología Vegetal” 4 cd. Ed. Pirámide,Madrid
Barras,E., Bortoli-German,1., Bauzan,M., Rouvier, 3., Gey, C., Heyraud,A. y Henrissat,B. (1992)“Stereochemistryof the hydrolysisreactioncatalyzedby endoglucanaseZ from Erwinia chíysaníemiFEBSLeir 300: 145-148
Béguin, P. (1990) ‘Molecular biology of cellulosedegradationAnnu. Res’. Microbiol. 44; 219-248
Bergfors, T., Rouvinen, J., Lehtovaara,P., Caldeníey,X., Tomme, P., Claeyssens,M., Pettersson,G., Teeri, T. T.,Knowles, J. y Jones,T. A. (1989) Crystallizationof the core protein of cellobiohydrolase11 from Táchodennaivesel J. Md. Rio! 209: 167-169
Bhikhabhai,R. y Peltersson,G. (1984> The disulphide bridges in a cellobiohydrolaseand an endoglucanasefromTáchodennaíeesei” Biochenz.J. 222: 729-736
Bhikhabhai, R., Johansson, O. y Peítersson, 0. (1984) “Isolation of cellulolytic enzymes from TñchodennareeselQM9414’ 4? Appl. B¡od,em.6: 336-345
Biely, P. y Markovic, 0. (1988) “Resolution of g]ycanasesof Tñchodenna reesel with respect to cellulose andxylandegradation” Biotechnof. AppLBiodicin. 10: 99-106
Biely, P. (19%) “Artificial substratesfor cellulolytic glycanasesandIheir usefor thedifferentiation of Tridiodennaenzymes”en TñchodertnaCellulases.Biochensisny,Geneñes,Physio¡ogyondApplicañon(Kubicek,C. P., Eveleigh,O. E., Esterbauer,H., Steiner,W. y Kubicek-Pranz,E. M., eds.) Pp. 30-46, Ihe Royal Society of Chemisty,Cambridge,Reino Unido
Biely, P., Vrsanska,M. y Claeyssens,M. (1991> The endo-1,4-13-glucanase ¡ from Trichodennai-eesei.Action on 13-1,4-oligomersandpoly¡nersderivedfrom D-glucoseandD-xylose” Lar J. Biod,cm. 200: 157-163
Binder, A. y Ohose,T. K. (1978)“Adsorption of celluloseby Trichodemiaviáde” Bioíechno!Bioeng.20: 187-199
Blake, C. C. F., Johnson,L. N., Mair, O. A., North, A. C. T., Phillips, O. C. y Samia,y. R. (1967)“Crystallographicstudiesof Ihe activity of henegg-whitelysozyme )‘mc. R. Sos.Lotid. [Rio!] BIÓZ: 378-388
Blanch, H. W. y Wilke, C. R. (1983)“Sugarsandchemicalsfrom cellulose”Rey. Chein.Lng 1: 71-119
Claeyssens,M. (1988) The useof chromophoricsubstratesand specific assaysin the study of structure-activityrelationshipsof cellulolyíic enzyrnes’enBioche,nisízyandGeneíicscfCellaloseDegradation(Aubert,J.-P.,Béguin,P. y Millet, J., eds.)Pp. 393-397,Acadcmic Press,Londres
Claeyssens,M. y Tomme,P. (1990)“Structure-functionrelaíionshipsof cellulolytic proleinsfrom Tñchodenna iresel en
TáchodemiaCellulases.Biod¡ernist’y, Gene¡ics, Physio/ogy andApplicotion (Kubicek, C. P., Eveleigh, 0. E.,Esterbauer,H., Steiner,W. y Kubicek-Pranz,E. M., eds.)Pp. 1-11,TheRoyal Societyof Chemistzy,Cambridge,
Reino Unido
Claeyssens,M., vanTilbeurgh,H., Kamerling, i. P., Berg, 3., Vrsanka,M. y Hiel>’, P. (1990a)“Studiesof thecellulolyticsyslem of Ihe filamentousfungus Táchodennairesei QM 9414. Substratespecificity and transfer activily ofendoghucanase1 B¡ochen:. 4? 270: 251-256
Claeyssens,M, Tomme, 1’., Brewer,C. E. y Hehre,E. J. (1990b) “Stereochemicalcourseof hydrolysisandhydrationreactionscatalysedby cellobiohydrolases1 andII from Táchodemiai-eesei’ FEBSLea. 263, 89-92.
Clarke, A. .1. y Svensson,B. (1984) Identificalion of an essentialtzyptophanylresiduein the priniary structureof
ghucoamylase 02 from Aspe’~¡llusníger’ Cartsbei-g. Res. <Somatan.49: 559-566

BIBLIOGRAFíA 93
Clarke, A. J. y Yaguchi, M. (1985) The role of carboxyl groups in the funccion of endo-13-1,4-glucanasefromSchyzophyllwncommune”Fur. J. Biochen,.149: 233-238
Clarke,A. J. (1987) Essentialtrypíophanresiduesin thefunclion of cellulasefrom Schyzophyllum conununeBiochí,n.Bioplíys. Acta 912: 424-431
Clarke,A. J. (1988) Active-site-directedinactivalion of Schyzophyllumcommunecellulaseby 4’,5’-epoxypentyl-4-D-QI-D-glucopyranosyl)-B-D-glucopyranoside”Biodícín. CefI Rio! 66: 871-879
Cleland, W. W. (1977)“Determining thechemicalmechanismsof enzyme-catalyzadreactionsby kinetic studies ¡¡dv.Enzymof. RetaL ¡¡reas Md. Bid. 4S: 273-387
Cornish-Bowden,A. (1976>“PrincipIes of enzymekinetics” Ed. Butterworths,Londres,UK
Coughlan,M. P. (1985) The propertiesof fungal and bacterialcellulaseswith cominent on their productionandapplication’ Riotech. Gener.Eng. Reviews3: 39-109
Coughlan,M. P. y Ljungdahl,L. G. (1988)“Comparativebiochemisir>’of fungalandbacterialcellulolytic enzymesystems”en Biochemisny¿md Gentilesof Ce/fu/oseDegradation (Aubert, J.-P., Béguin, P. y Millet, J., eds.) PP. 11-30,AcademicPress,Londres
Cowling, E. E. y Kirk, T. 1<. (1976) “Propertiesof cellulosematerials”Riorechnol.BioengSymp.6: 95-123
Creighton,T. E. (1984)“Proieins,W. H. FreenianandCo., NuevaYork, USA
Chanzy,U., Renrissat,E., Vuong, R. y Schúlein,M. (1983) ‘The actionof 1,4-B-D-glucancellobiohydrolaseon Va/foniocellulosamicrocrystals”FEBSLeir. 15.?: 113-118
Chapman,G. E., Hartman,P. 6. y Bradbury,E. M. (1976) Studieson the role andmodeof operationof Me ver>’-lysine-rich histoneHl in eukariotechromatin.The isolationof the globularandnon-globularregionsof the Hlhistonemolecule Ecu: 4? Iliochera. 61: 69-75
Chen, C. M, Griztali, M. y Stafford, O. W. (1987) “Nucleotide sequenceand deduced primaiy structure ofcellobiohydrolaseII of Tñehodennareesel Bio/Technof.5: 274-278
Dale, E. E. (1987)“Lignocelluloseconversionandthe futureof fermentationbiotechnology TrrndsBiorechnol. 5:287-291
de la Mata,1. (1992)“8-glucosidasade TñchodennareeselOM 9414.TesisDoctoral,Facultadde CienciasBiológicas,UniversidadComplutensede Madrid
Detroy,R. W. y Julian,G. (1982)“Biomass conversion:fermentationchemicalsandfuels” CRCCM. Rey.Micmbiot 10:
203-228
Din, N., Gilkes,N. R., Tekant,E.,Miller Jr., R. C., Warren,R.A. J. & Kilbum, 0. 6. (1991) Non-hydrolyticdisruptionof celluloselibresby the hinding domain of a bacterialcellulase” ¡lio/Fech. 9:1096-1099
Dinur, 1., Osiecki, K. M., Legler, 6., Gatt, 5., Desnick,R. J. y Grabowski,6. A. (1986) ‘Human acid 8—glucosidase:
isolation andamino acid sequenceof a peptideconrainingthe catlytic site” Proc. Nial? Acad. Sci. 83:1660-1664
Domínguez,J.M. (1991) Aislamiento,purificacióny caracterizacióndeendoglucanasaIdeTilehodennareeseiQM 9414TesisDoctoral,FacultaddeCienciasQuímicas,UniversidadComplutensede Madrid
Domínguez,1. M., Pettersson,G., Acebal,C., Jiménez,J., Macarrón, R., de la Mata, 1. & Castillón, M. 1>. (1992)
Sponraneousaggregationof endoglucanase1 from TrichodennareeseiQM9414 RiotechnotAppl. Biochem.,enprensa
Drapeau,6. It. (1977) Cleavageat glutamic acid with Staphylococcalprotease”Mci/más EnzyínoL 47: 189-191
EI-Gogar>’,S., Leite, A., Crivellaro, O., Eveleigh,O. E. y EI-Dorri H. (1989) Mechanismby which cellulosetriggers
CBH 1 geneexpressionin Tñchodennareesel Proc. Nial. Acad.ScL (iSA 86: 61384141
EI-Gogary,5., Leite,A., Crivellaro,O., EI-Dorri, U-y Eveleigh,D. E(1990> “Trlchodenna,reseicellulase-from mutantsto induction” en TáchodemiaCe/fu/ases.Biochernisuy,Generies, Physiologyand¡¡ppficaáon (Kubicek, C. P.,Eveleigh,O. E., Esterbauer,H., Steiner,W. y Kubicek-Pranz,E. M., eds.> PP. 200-211,The Royal Society of
Chemistzy,Cambridge,Reino Unido
Ellis, K. J. y Morrison, J. F. (1982) Buffers of constantionic strengthfor studying pH.dependentprocessesMeíh.Enzymot87: 405-426
Enari,T.-M. y Niku-Paavola,M.-L. (1987)“Enzymatichydrolysisof cellulose:is thecurrenttlieory of themechanismofhydrolysisvalid? CRC<Si-it. Rey.¡liotechnof. 5: 67-87
Eshdat,Y., McKelvy, J.y Sharon,N. (1973)“Identification of asparticacid 52 as thepoint of attachmentof an affinitylabel in heneggwhitelysozyme 1. Bid. CIten,. 248: 5892-5898

BIBLIOGRAFíA 94
Eyzaguirre(1987) Chemicalmodificationof enzymes.Active sitestudies BIlis Horword Ltd., Chichcster,Reino Unido.
Fan,L. T., Gharpuray,M. M.y Lee, Y.-H. (1987) Cellulosehydrolysis”. Biotechnolo~’Monographsvol. 3. Springer-Verlag, Berlín-Heidelberg
Fersht,A. (1985) Enzymestructureandmechanism,2 ed. W. H. FreemanandCo. Nueva York, USA
Fritscher,C., Messner,R. y Kubicek, C. P. (1990) Cellobiosemetabolismandcellobiohydrolase¡ by TrichodennareeselExp. Mycol. 14: 405-415
Gaboriaud,C., Biossery,Y., Benchetrit, T. y Mornon J. P. (1987) ‘Hydrophobic clusteranalysis:an efficient way to
compareandanalyseamino acidsequences”FF135 Len. 224: 149-155
Gebbler,J., Gilkes,N. Claeyssens,M., Wilson, B. Bóguin, P., Wakarchuk,W. W., Kilburn, 0. 6., Miller, R. C., WarrenA. J.y Whiters,8. G. (1992) Stereospecif¡chydrolysiscatalysedby related13-1,4glucanasesandB-1,4 xylanases”£ Biol. Chan., en prensa
Oilbert, 1. G. y Tsao G. T. (1983> “Interaction betweensolid substrateandcellulaseenzymesja cellulosehydrolysis’
¡¡¿mu. Rep.Ferin. Process.6: 323-358
Gilkes, N. R., Henrissat,E., Kilburn, D. O., Miller, R. C. Ir y Warren, R A. J. (1991) “flomains in microbial 13-1,4-glycanases:sequenceconservation,funetionandenzymefamilies” Microbio! Rey.SS: 303-315
Gilligan, W. y Reese,E. T. (1954) Evidencefor multuple componentsin microbial cellulases Ca,,. 1 Microbiol. 1:90-107
OreenN. M. y Witkop, E. (1964) Oxidation studiesof índolesandthetertiar>’ structureof proteins’ Tmns.N. Y ¡¡cad.Sci. 26: 659-669
Grépinet O. y Béguin, E. (1986)“Sequenceof thecellulasegeneof Cfosrddiwn¿hennocellumcodingfor endoglucanaseE” Nuc! AcidsRes. 14: 1791-1799
Griztali, M. y Brown, R.D. (1979)“The cellulosesystem of Trie/todenita.Relationshipsbetweenpurifiedextracellularenzymesfrom inducedor cellulose-growncelís ¡¡dv. Client. Ser. 181: 237-260
Habeeb,A. F. S. A. (1972)“Reactionof proteinsulfhydril groupswith Ellman’sreagent”Mali ods Enzymo!25:457-463
Haynes W. C., Wickerham, L. J. y Hesseltine,C. W. (1955) Maintenanceof cultures of industríall>’ importantmiaoorganisms”¡¡pp! Miembiol 3: 361-368
Henrissat,E., Driguez, H. Viet C. y Schillein, M. (1985)“Synergism of cellulasesfrom Táchodemiareesei iii thedegradationof cellulose Rio/Tech.3: 722-726
Henrissat,B.,Claeyssens,M.Tomme, P., Lemesle,L.yMomonJ.-P.(1989) Cellulasefamiliesrevealedbyhydrophobicclusteranalysis Gene81: 83-95
Henrissat B.y Mornon, J.-P. (1990) Comparisonof Tñc/wdenna cellulases with otherB-glycanasesen TñchodennaCe/fufases.Biochentisuy, Genedcs,Physiologyaítd¡¡pplicaíion (Kubicek, C. P., Eveleigh, 0. E., Esterbauer,H.
Steiner W.y Kubicek-Pranz,E. M. eds.)Pp. 11-29,The Royal Society of Chemistzy,Cambridge,ReinoUnido
Herrchen, M. y Legler, 0. (1984) “Identification of an essentialcarboxylategroupar the active site of laeZ 3-galactosidasefrom Eseheáchiacali Fur. 1 Biodte,n. 138: 527-531
Hoare,0. 0. y KoshlandJr.,0. E. (1967) Methodfor the quantitativemodificationandestimationof carboxylicacidgroups in proteins” 1 Rial. Client. 242: 2447-2453
H0j, P. E. Rodríguez,E. E. Stick, R. Y. y Sione,E. A. (1989) Differencesin activesite structurein a famil>’ of 13-glucanendohydrolasesdeducedfrom ihekineíicsof inacrivalion by epoxyalkyl13-oligoglucosides1 Rio! Client.
264: 4939-4947
lwama, M., lnokuchi N., Okazaki,Y., Takahashi,T., Yoshimoto,A. e Irie M. (1986) Oxidation of glucoamylasefromaRhizopussp. with N-bromosuccinimideClient. Pitan,,. Buf! 34: 1355-1361
lyayi, C. E., Bruchmann,E-E. y Kubicek C. P. (1989) Induction of cellulaseformation in Trichodennareesel bycellobiono-1,5-lacton¡¡‘di. Microbio! 151: 326-330
Johnson,L. N., Cheetham,1. Mctaughlin, E. J. Acharya, K. R., Basford 0. y Phillips, D. C. (1988) Protein-oligosaccharideinteractions: lysozyme, phosphoylase,amylases”<Suri-. Top. Microbio! Im,nunol. ¡39: 81-134
Juy, M., Amit, A. 6., Alzan P. M., Poljak R. 1., Claeyssens,M., Béguin, E. y Aubert J.-P.(1992) “Three-dimensionalstrucrureof athermosíablebacterialcellulase Nainre, en prensa
Karpeiskii M. Y. (1976)“Active site of enzynies:stereochemismryand dynamics Mo! Rio!. 10: 1197-1210

BIBLIOGRAFíA 95
Keskar,S.S. Srinivasan,M. C. y Oeshpande,y. V. (1989> Chemicalmodificationof axylanasefrom a thermotolerantS¡rep¿omyces.Evidencefor essentiaitryptophanandwstein residuesat the active site BlocAscm. ¿ 261:
49-55
Kitz, R. y Wilson, 1. B. (1962) Esiersof methanesulfonicacid asirreversibleinhibitors of acetylcholinesterase1. fbi.Clie,n. 237: 3245-3249
Klyosov,A-A. (1988) Cellulasesof thethird generation enRbochemisnyandGeneñcsofCe/tu/oseDegradation(Aubert,J.-P., Béguin P. y Millet J. eds.)Pp. 87-99 AcademicPress,Londres
Knowles, .1., Lehtovaara, P. y Teeri, T. T. (1987) Cellulasefamilies andtheir genes” Trends Bioteclino!. 5: 255-261
Knowles, 1., Teeri,T.T., Lehtovaara,P., Pentihl,M. y Saloheimo,M. (1988) Theuseof genetechnologrto investigatefangal cellulolytic erizymes” en Riochen:isuyaud Genolesof Ce//u/oseDegradañon (Aubert, J.-P.,Béguin, P. yMillet 1., e&.) Pp. 153-169 Academic Press,Londres
Knowles,3. 1<.C. Lehtovaara,P. Murra>’, M. y Sinnott,M .L. (1988)“Stereochemicalcoarseof actionof thecellobiosidehydrolases¡ andTI of TñchodennaIrescí 4? Cliem. Saz. Chemm. Commun. 1988: 1401-1402
Kraulis, P. 1., Clore, O. M., Nilges, M. Jones T. A. Pettersson,O., Knowles, J y Groneabora,A. M (1989)“Determinationof thethree-dimensionalsolution structureof the C-terniinaldomain of cellobiohydrolase1 fromTrichodenna reesel. A study using NMR and hybrid distance geometry-dynamicalsimulated annealing”B¡ochentisr.y28: 7241-7257
Kubicek, C. P. (1987) ¡nvolvementof a conidial endoglucanaseanda plasma-membrane-bound8-glucosidaseintheinduction of thecellulasesynlhesisby cellulosein T,iciodenna~resetJ. Gen. Microbbol. 133: 1481-1487
Kubicek,C. P., Eveleigh D. E. Esrerbauer,H., SteinerW. y Kubicek-Pranz,E. M., eds.(1990a)TñchodennaCellulases.
Biochernistry, Genetics,PhysiologyandApplication,TheRoyal Societyof Chemistiy,Cambridge,ReinoUnido
Kubicek, C. P., Messner,R. Fritscher,C. Strauss,5. y Kubicek,-PranzE. M. (1990b) ‘Regulator>’aspeasof formarionandsecretionof cellulasesby Tdciode,,nareesel en Tñciode,n,aCe/fu/ases.Bioclie¡nisúy, Genelles, Physio/ogvandilpplicaubo~t(Kubicek, C. P., Eveleigh,D. E. Esterbauer,H., Steiner,W. y Kubicek-Pranz,E. M., eds.)PP.81-102 TheRoyal Society of Chemistty,Cambridge,Reino Unido
Kyriacou,A. Neufeld, R. J.y McKenzie,C. R. (1989) Reversibilityandcompetition in theadsorptionof Tricliodennareeselcellulasecomponents”Bio¿edsno!Bioeng.33: 631437
Laemmli, U. K. (1970) Cleavageof structuralproteinsduring the assemblyof theheadof bacteriophageT4” Na¡we227: 680-685
Lamed,It. y BayerEA. (1988) Thecellulosomeconcept:exocellular/extracellularenzymereactorcentersfor effxcientbindingandcellulolysis enBiocl:entisuy¿mdGeneñcsof Ce//u/oseDegradation(Aubert,J.-P.,Béguin,P. y Millet,J., eds.)Pp. 101-116,AcademicPress,Londres
Legler, G. y Bause,E. (1973) “Epoxyalkyl oligo-(1-.4)-13-0-glucosidesas active-site-directedinhibitors of cellulasesCoitoliydr Res. 28: 45-52
Legler,0. (1990) Glycosidehydrolases:mechanisticinformationfrom studieswith reversibleandirreversibleinhibitors”Adv. Cwboliydr. Chen,.Biocien;. 48: 319-384
Lev>’, H. A., Leber, P. 0. y Ryan E. M. (1963> Inactivarionof myosin by 2,4-dinitrophenolandprotectionby adenosinetriphosphateandoyher phosphatecompounds4? Rio! Client. 238, 3654-3659
Lowry, O. 1-1., Rosebrough,N. 3., Farr, A. L. y Randalí, It. T. (1951> Protein measurementwirh Ihe Folin phenolreagení”Y Rio! <Slie,,t. 193: 256-265
Lundhlad,R. L. y Nuyes,C. M. (1984)‘Chemical reagenísfor proreinmodification CRC Press,Inc.,Boca Raton,USA
Ldthi, E., Jasmat,N. B., Grayling, R. A., Love, D. It. y Bergquist, P. L. (1991) Cíoning, sequence,anaíysisandexpressionin Escieñehlaco/i of agenecoding br a13-mannanasefrom the extremelythermophiíicbacteriumCa/doce//un,saccharofy¿icunt”¡¡ppf. Environ. Microbiof. 57: 694-700
Lútzen, N. W., Nielsen,M. H., Oxenhoelí K. M., Schulein,M. y Stenbebjerg-Olesen,B. (1983) ‘Cellulasesandtheirapplicationin Éheconversionof lignocelluloseto fermentablesugarsPhd. Ti-urs. R. Soc. i.ond. £300: 283-291
Macfarlane,It. 0. (1990)“PrincipiesofCaliforniurn-252plasmadesorptionmassspearometiyappliedLo proteinanalysisMerhodsEnzynto!193: 263-280
Mandels,M. y Reese,E. T. (1966) Inducrionof cellulase in fungi by cellobiose”4? Bacteñot79: 816-826
Mandels, M., Parrish,F. W. y Reese E. T. (1962) Sophoroseas an inducerof cellulasein Tñchodennavirlde” 4?BuceRo!83: 400-408

BIBLIOGRAFíA 96
Mandels,M. y Reese,E. T. (1964) ‘Fungal cellulasesandthe microbial decompositionof cellulosie libres” Dey. ¡ml.Mñcobio! 5: 5-20
Mtintyltl, A. Harkki, A., Pcnttilá, M., Muttilainen 5., Buhler R., Vanra, T., Knowles, J. y Nevalainen,H. (1989)
“Consiructionof Tñciodennareeselstrainswith newcwllulaseproliles EMBO-¡¡fko Wo.*sliop “Mo/ecufarbbo/ogyoffl/antentousfungi,Bspoo, Finlandia Julio de 1989 p. 40.
Mathew,C.D. y BalasubramaniamK. (1986) Chemicalmodilicationof «-galactosidasefrom coconut Phy;oehent.25:2439-2443
Menéndez-AriasL. Gómez-Gutiérrez,J., Garcia-Ferrández,lvi. García-Tejedor,A. y MorAn, F. (1988)“A BASICmicrocomputerprogramto calculatethesecondar>’structureofproteins,from their circulardichroismspectrum”CABIOS 4: 479482
Messner, R., Gruber, F. y Kubicek C. P. (1988> “Differential regulation of synthesisof multiple forms of specificendoglucanases by Tñc/u4ennareesel 0M9414” £ Bacteñol. 170: 3689-3693
Messner, It., Kubicek-Pranz, E. M. Gsur,A. y Kubicek, C. P. (1991) “Cellobiohydrolase ¡Jis the niain conidial-bound
cellulase in Tñchodennareeseland other Táchodemiastrains” ib-ch. Microbio!. 155: 601-606
Messner, It. y Kubicek, C. P. (1991)“Carbon sourcecontrol of Cellobiohydrolase1 and 11 formation by Tácliodennareesel App! Enviran. Microbio! 57: 630-635
Miles, E. W. (1977) ‘Modificarion of histidyl residuesin proteinsby diethylpyrocarbonate,Meíh, Enzymol.,47: 431-442
Miller, G. L. (1959)“Use of dinitrosalicylic acidreagentfor determinationof reducingsugars ¡¡nal. CAsen,.31: 426428
Mitsuishi, Y., Nitisinprasers,5., Saloheimo,M. Biese, 1., Reinikanien,T., Claeyssens,M. Keránen,5., Knowíes, J. K.C. y Teeri, T. T. (1990) Site-directedmutagenesisof the putativecatalyric residuesof Tñchodennareeselcellobiohydrolase1 andendoglucanase1’ FEASLea. 275: 135-138
Montenecourt,B. 8. y Eveleigh,0. E. (1979) Producrionandcharacterizarionof high yielding cellulasemutantsfrom7ñchodemtareesel’ T¡¡PP¡ Ana. MeetingProc. 1979: 101-108
Nieves, R. A., BIlis, R. P., Todd, R. .1., Johnson,T. 1. A., Grohmann,K. y Himmel, M. E. (1991) “Visualization ofTácliodennareeselcellobiohydrolase1 andendoglucanase 1 on aspencelluloseby using monoclonalantibod>’-colloidal gold conjugates”App! Envis-o,t.Microbbol. 57: 3163-3170
Niku-Paavola,M.-L., Lappalainen,A. Enari, T.-M. y Numnii, M. (1986) “Tñchodennareesel cellobiohydrolase II.
Purilication by immunoadsorptionandhydrolytic properties”Bioteclino!. Appf. Biochem.8: 449-458
Nisizawa,T., Suzuki, H. y Nisizawa,K. (1972) “Cataboliterepressionof cellulaseformation in Tácliodennavii-ide” £Biochent. 71: 999-1007
Ohnishi, M. Kawagishi,T., Abc T. e Hiromi, K. (1980) Stopped-flowstudies on the chemicalmodificationwith N-bromosuccinimide of model compounds of tryptophanresidues’4? Bbociein.87: 273-279
Ong, E. Greenwood, J. M., Gilkes, N. R. Kilbum, D. G. Miller Ir, R. C. y Warren, It. A. J. (1989) The cellulose-binding domainsof cellulases:tools for biotechnology TrendsBiotecinol. 7: 239-243
Ozaki, K. e 1ro, 8. (1991) Purification andpropertiesof an acid endo-1,4-8-glucanasefrom Bac1!lus sp. KSM-330” YGen. Microbio! /37:41-48
Penrtilli, M. Lehtovaara,P., Nevalainen,H. Bhikhabhai, It. y Knowles J.(1986> Homologybetweencellulasegenesof Tñciodennareesel: completenucleotidesequenceof the endoglucanase1 gene”Gene45: 253-263
Pereira A. N. Mobedshashi,M. y Ladisch,M. It. (1988) Prepararionof ccllodextrins” MetAs. Enzyntol.160: 26-37
Pétra,P. E. (1971) Modification of carboxyígroupsin Bovine CarboxypeptidaseA. (1). Inactivarionof the cnzymeby
N-ethyl-5-phenyl-isoxazolium-3-sulfonate(Woodward’sreageníK Bloc/temis»>’ 10: 3163-3170
Phillips, D. C. (1966> The rhrec dimensionalstruclureof an enzymemolecule ScLAm. 215: 78-90
Pokorny,M., Zupancic,5., Steiner,Th. y Kreiner, W. (1996)‘Productionanddown-streamprocessingof cellulaseson
a pilot scale” en TñchodemtaCeflufases.Biocheníisuy,Geneñcs,Physio/o~arid ¡¡pp/icañon (Kubicek, C. P.Eveleigh, 0. E. Esterbauer,H., Steiner, W. y Kubicek-Pranz,E. M., eds.) Pp. 168-184,The Itoyal Socia>’ ofChemisrr->’,Cambridge,Reino Unido
Pourquié,J., Warzywoda,M., Chevron,F., Théry,M. Lonchamp,0. y Vandecasteele3. P. <1988) “Scale up of celluloseproductionand utilization en Bioc/ie¡nis¡ry and Ge,teticsof Ce/fu/oseDegradañon (Aubert, J.-R, Béguin, P. yMiller, J., eds.) PP. 71-86,AcademicPress,Londres
Privat, 3-1>., Loran, It. Bouchard,1>., Sharon,N. y Monsigny, M. (1976) Chemical modification of the tryprophanresiduesof wheat-germagglurinin’ Eur Y Bioc/tem.68: 563-572

131RLIOC~RAFIA 97
Py E. Eortoli-Gertnan, 1., Haiech,J. Chippaux M. y Barras E. (1991) “Cellulase EGZ of Erwinia c/uysanthe~ni:structuralorganizationandimportanceof His98 andGlu133 residuesfor catalysis Protein Eng 4: 325-333
Ouiocho, E. A. (1986) Carbolrydrate-bindingproteins:rertiarystructuresand protein-sugarinteractions Annu. Rey.Biochent. 55:287-315.
Reese,E. T., Siu, It. O. H. y Levinson 1-4. S. (1950) Degradationof soluble cellulosederivatives”J. Bacteñol. 59:485-497
Iteese,E. T. y Mandels,M. (1984)“Rolling with tIte times:productionandapplicationsof Ti-ichodemia¿-cese!cellulase¡¡nn. Repoi-ts.Ferin. Pmcessses7: 1-20
Robson,L. M. y Chanbliss,O. M. (1989)“Cellulasesof bacterialorigin” EnzymeMicrob. Teclino! 11: 626-644
Roavinen,1., Bergfors, T., Teeri, T. T., Knowles, 1. K. C. y Jones,T. A. (1990) ‘Three-diinensional structureofcellobiohydrolase11 from Tñchodennareesel” Science248: 380-386
Saloheimo,M. Lehtovaara,P. Penttilá,M. Teeri,T. T. Stáhlberg,1., Johansson,O., Pettersson, O., Claeyssens, M.Tomme,P.y Knowles,1. K. C. (1988>“BO ITT, a newendoglucanas.efrom Tñchodemiareesel:tIte characterization
of both, geneandenzyme”Gene63: 11-21
Schúlein M. (1988) “Cellulasesof Tñchodennareesel Mcdi. Enzyntof.160: 234242
Schwarz,W. FI., Schimming,5., Rticknagel,K. P., Burgschwaiger5., Kreil, O. y Staudenbauer,W. L. (1988) Nucleotidesequenceof tIte ce/C geneencodingendoglucanaseC of Cfostñdiwnthennocelfum”Gene63: 23-30
Sharrock,K. It. (1988)“Cellulaseassaymethods:a review £ Biochent.Biophys. Mali. 17: 81-106
Shina V. y Brewer, 1. M. (1985> A spectrophotometricmethod of quantirarionof carboxyl group modification ofprotein usingWoodwardsReagentK Anal?Biochem. 151: 327-333
Shoemaker,5. E., Wart, K., Tsitovsky G. y Cox R. (1983) Characterizationandpropertiesof cellulasespuriliedfromTñchodennareeseistrain L27” Bio/Teclinol? 1: 687-690
Simmons,E. 0. (1977)“Classilicationof somecellulase-producingTñcliodemiaspecies”Secondln¡ema¿ionafMycofogica/Congess.Absinicis,s’ol? 2 University of SouthCalifornia,Tampa p. 618
Sinnott,M. L. y Viratelle,O. M. (1973)“11w effect of methanolanddioxanon theratesof the B-galactosidase-catalysedhydrolisesof sorneft-D-galactopyranosides:rate-limiting degalactosylation”Biochem.J. .133: 81-87
Sinnotí, M. L. (1987)“Olycosyl grouptransfer” enEnzymeMechanisms(Page,M. 1. y Williarns, A. eds.)Pp. 259-295.Ihe Itoyal Society,Londres
Sinnott, M. L. (1990)“Catalytic mechanismsof enzyrnicglycosyl transfer Client. Rey. 90: 1171-1202
Spande,1. E. Oreen,N. M. y Witkop, E. (1966) TIte reactiviry towardN-bromosuccinimideof tryptophanin enzymes,zymogensandinhibited enzynies’Biocientisuy5:1926-1933
Spande,T.E.>’ Wiíkop, E. (1967)“Determinarionof tIte tryptophancontentofproteinswithN-bromosuccinimideMe;h.
Enzyntot 11: 498-506
Sprey,E. y Lambert, C. (1983)“Titration curvesof cellulasesfrom Tñchodermazeesel:demonstrationof a cellulase-
xyíanase-13-glucosidase-containingcomplex FEMSMicrobio! Uit. 18: 217-222
Stfthlberg 1., Johansson,O. y l’ettersson,0. (1988> “A binding-site-defxcient,catalytically active, coreprotein ofendoglucanase111 from theculturefiltrate of Tñchodennateesel’Eur. .1 Biochem.173: 179-183
Sternberg, D. y Mandels,M. (1979) ‘Induction of cellulolytic enzymesiii Tñcliodennazeeselby sophorose£ Bacteño?139: 761-769
Tanford C. (1961) “Physical chemisrry of macromolecules PP. 317-366,John Wiley & Sons NuevaYork USA
leen T. 1., Salovuori, 1. y Knowles, 1. (1983) The molecular cloning of the major cellulasegenefrom Tñchodernta
reesel” Bio/Fecinofogy1: 691-699
leen,T. T., IonesA., Krauíis,P., Rouvinen,1. Penttihl, M., Harkki, A., Nevalainen,FI., Vanhanen,5., Saloheimo,M.y Knowles,1. K. C. (1990)“EngineeningTácbodennaandits cellulasesen Tñc/wdei-maCe//u/ases.Biodemist¿’y,
Geneñcs,P/sysbofogyandApp/icadon (Kubicek, C. P. Eveleigh, D. E. EsterbauerH., Steiner,W. y Kubicek-Pranz,E. M. eds.)Pp. 156-167,TIte Itoyal Society of Chemistry,Cambridge,Iteino Unido
Tomme,P., vanTilbeurgh FI. Petrersson,O. Vandekerchove,J., Knowles,1., leen,T. y Claeyssens,M. (1988) ‘Studiesof tIte celluíolysic system from Tñciodennareesel.Analysisof tIte domainfunclion in two celíobiohydrolasesbylimited proteolysis Eur. J. Bbochent.170: 575-581
Tomme,P.yClaeyssens,M. (1989)‘Identilicationof a functionallyimportanícarboxylgroupjacellobiohydrolaselfromTñciodennareesel FEBSLenas243: 239-243

98BIBLIOGRAFíA
Tomme, 1>. (1991> Cellulasen uit Táchoderníareesel en CIos¡ñdium diennocellwn: domeinstructuur chemischemodilicaties en mutagenese” Tesis Doctoral, Rijksuniversiteit Gent, Bélgica.
Olker, A. y Sprey, B. (1990) “Characterizationof anunglycosylaredlow molecularweight 14~l3~glucan-gIucanOhydro1aseof Tñcliodennareesei” FEMSMicrobio! Uit. 69: 215-220
Vahen, M. (1982) Oxidation as apart of degradation of crystalline cellulose by Tñchodennareesel £ ¡¡pp! Bioclien,.4: 356-363
van Tilbeurgh, FI., Claeyssens, M. y de Bruyne, C. K. (1982)The use of 4-methylumbelliferyl and other chromophoric
glycosides in the study of cellulolytic enzymes” FEBSUit. 149: 152-156
van Tilbeurgh, H., Bhikhabhai, R., Pettersson L. O. y Claeyssens, M. (1984) “Separation of endo- and exo-type cellulases
using a new affinity chromatography method” FEBS Uit 169: 215-216
van Tilbeurgh, FI. y Claeyssens, M. (1985) “Detection and differentiation of celiulase components using low molecular
mass fluorogenic substrates” FEBSLeir. 187: 283-288
van Tilbeurgh, H., Loontiens, E. O., De Bruyne, C. K. y Claeyssens, M. (1988) “Flurogenic and chromogenic glycosides
as substrates and ligands of carbohydrases MetAs.Enzymo! 160: 45-58
Vázquez de Aldana, C. R., Correa, J., San Segundo P., Bueno, A. Nebreda A. R., Méndez, E y del Rey, F. (1991>
Gene97: 173-182
Vega, 1. M., Castillo E. y Cárdenas,J. (1983) La bioconversiónde la biomasa Ed. Pirámide,Madrid
Ver-non, C. A. y Banks, E. (1963) ‘The enzymichydrolysisof glycosides’Bbochent..1. 86: 7p
Walsh K. A. y Neurarh,H. (1964) Typsinogenandchymot¡ypsinogenas homolognusproteins” ¡‘mc. NaO. Acad.Sci.USA 52: 884-889
Wolfrom, M. L. y Haq.S. (1964) ‘A polymer-homo~ogousseriesQf methyi 13-D-glycosidesfr-orn cellulose” T¡¡PP! 47:183- 185
Wood, 1. M. (1985) ‘Propertiesof cellulolytic systems Bbochein.Soc. Trons. 13: 407-410
Wood, T. M., McCrae,5. 1., Wilson C. A., Bhat, K. M. y Gow, L. A. (1988)“Aerobic andanaerobicfungal celiulases,with special referneeto their modeof attackon csystallinecellttlose” enBiocliemist’yami Geneticsof Ce//u/oseDegrodation (Aubert, J.-P., Béguin, P. y Millet, 1., eds.) Pp. 31-52,AcademicPress,Londres
Wood,T. M. y Bhat, K. M. (1988)“Methods for measuningcellulaseactivities” MetAs. Enzymof. 160: 87-111
Yagúe,E., Béguin,P. y Aubert,J.-P. (1990)“Nucleotidesequenceanddeletionanalysisof thecellulaseencodinggenecelH of Cfostñdiu,ntftennocellun,” Gene89; 61-67