nombre del producto yondelis formas … · observaron aumentos de la cpk (de grado 3-4) asociados...
TRANSCRIPT

NOMBRE DEL PRODUCTO YONDELIS® Trabectedina para inyección (1 mg) FORMAS FARMACÉUTICAS Y CONCENTRACIONES Polvo para concentrado de solución para infusión. El producto farmacéutico YONDELIS® se comercializa en forma de polvo estéril liofilizado, de color blanco o blanquecino. Cada frasco contiene 1 mg de trabectedina. 1 ml de solución reconstituida contiene 0,05 mg de trabectedina (véase la sección “Instrucciones de uso y manipulación”). Consulte los excipientes en la Lista de excipientes. INFORMACIÓN CLÍNICA Indicaciones YONDELIS® combinado con hidrocloruro de doxorubicina liposomal pegilada (PLD) está indicado para el tratamiento de pacientes con cáncer de ovario recurrente. YONDELIS® está indicado para el tratamiento de pacientes con sarcoma avanzado de los tejidos blandos tras un tratamiento fallido con antraciclinas e ifosfamida, o que no son elegibles para el tratamiento con estos agentes. Los datos de eficacia se basan principalmente en pacientes con liposarcoma y leiomiosarcoma. Posología y forma de administración YONDELIS® se debe administrar bajo la supervisión de un médico experimentado en el uso de la quimioterapia. Su uso se debe restringir al personal especializado en la administración de agentes citotóxicos. La dosis inicial recomendada para el tratamiento del sarcoma de tejidos blandos es de 1,5 mg/m2 de superficie corporal, administrados en infusión intravenosa durante 24 horas, con un intervalo de tres semanas entre ciclos. Para tratar el cáncer de ovario, YONDELIS® se usa en combinación con PLD cada tres semanas. Se administra una dosis de 1,1 mg/m2 de YONDELIS® en infusión intravenosa de 3 horas, después de 30 mg/m2 de PDL en infusión intravenosa de 90 minutos. Para consultar las instrucciones de administración de PLD, ver el prospecto del fabricante local. Se recomienda especialmente administrar mediante un catéter central (véase las secciones “Advertencias y precauciones” e “Instrucciones de uso y manipulación”). Todos los pacientes deben recibir medicación previa con corticosteroides, como 20 mg IV de dexametasona 30 minutos antes de cada infusión de YONDELIS®, no solo como profilaxis antiemética, sino también porque parece tener efectos protectores sobre el hígado. Se puede administrar medicación antiemética adicional según necesidad (véase la sección “Interacciones”). Para permitir un tratamiento con YONDELIS®, se requieren los siguientes criterios: • Recuento absoluto de neutrófilos (RAN) ≥ 1500/mm3 • Recuento de plaquetas ≥ 100.000/mm3 • Hemoglobina ≥ 9 g/dl • Bilirrubina ≤ límite normal superior (LNS) • Fosfatasa alcalina de origen no óseo ≤ 2,5 × LNS (considerar las isoenzimas hepáticas 5
nucleotidasa o GGT para distinguir si el aumento puede ser de origen óseo) • Albumina ≥ 25 g/l • Alanina aminotransferasa (ALT) y aspartato aminotransferasa (AST) ≤ 2,5 x LNS

• Aclaramiento de creatinina ≥ 30 ml/min; terapia de combinación para el cáncer de ovario: creatinina sérica ≤ 1,5 mg/dl (≤ 132,6 mmol/l) o aclaramiento de creatinina ≥ 60 ml/min
• Creatina fosfoquinasa (CPK) ≤ 2,5 x LNS Estos mismos criterios se deben cumplir antes de comenzar los siguientes ciclos. De otro modo, se debe posponer el tratamiento un máximo de 3 semanas, hasta que se cumplan los criterios. Si estos efectos adversos persisten por más de 3 semanas, se considerará la interrupción del tratamiento. Además, se deben monitorizar los parámetros hematológicos y bioquímicos [fosfatasa alcalina, bilirrubina, CPK y aminotransferasas (AST y ALT)] semanalmente, durante los dos primeros ciclos de tratamiento y al menos una vez entre tratamientos, durante los ciclos siguientes. Se debe administrar la misma dosis en todos los ciclos, siempre que no se observen toxicidades de grado 3-4 y que los pacientes cumplan con los criterios para el re-tratamiento. Ajustes de la dosis durante el tratamiento Antes del re-tratamiento, los pacientes deben cumplir con los criterios basales antes definidos. Si se presenta cualquiera de los siguientes eventos en cualquier momento entre un ciclo y otro, se debe reducir la dosis de YONDELIS® a 1,2 mg/m2 en los siguientes ciclos de monoterapia, o a 0,9 mg/m2 en terapia combinada. • Neutropenia < 500/mm3 durante más de 5 días o neutropenia acompañada de fiebre o
infección • Trombocitopenia < 25.000/mm3 • Aumento de la bilirrubina > LNS • Fosfatasa alcalina de origen no óseo > 2,5 × LNS • Aumento de las aminotransferasas (AST o ALT) > 2,5 × LNS que no se recupera para el
día 21; terapia combinada para cáncer de ovario con AST o ALT > 5 × LNS que no se recupera para el día 21. La dosis de PLD se debe reducir también a 25 mg/m2.
• Cualquier otra reacción adversa de grado 3 o 4 (como náuseas, vómitos, fatiga)
Una vez que se ha reducido la dosis debido a los efectos adversos, se recomienda no volver a aumentarla en los siguientes ciclos. Si reaparece cualquiera de estos efectos adversos en ciclos subsiguientes, en un paciente que mostraba efectos clínicos beneficiosos, se puede volver a reducir la dosis de YONDELIS® a 1 mg/m2 en monoterapia o a 0,75 mg/m2 en terapia combinada con PLD. Si resulta necesario reducir aún más la dosis, se considerará la interrupción del tratamiento. Se pueden administrar factores estimulantes de la formación de colonias para los efectos adversos, en ciclos subsiguientes, según las prácticas locales estándar. Para realizar ajustes adicionales en la dosis de PLD, ver la información del prospecto del fabricante local. Para consultar las instrucciones de reconstitución y dilución del medicamento, antes de su administración, véase la sección “Instrucciones de uso y manipulación”. Poblaciones especiales Pacientes pediátricos En un estudio de fase 2 que investiga la actividad de YONDELIS a una infusión de 1,5 mg/m2 durante 24 horas, una vez cada 3 semanas, en 42 pacientes pediátricos con sarcomas (osteosarcoma, STS, sarcoma de Ewing y rabdomiosarcoma), el perfil de seguridad fue coherente con el de los adultos. No obstante, dado que no se observó eficacia alguna, YONDELIS no debe utilizarse en pacientes pediátricos con sarcomas pediátricos.

Pacientes ancianos En función de un análisis de seguridad integrado de ensayos clínicos de un solo agente en varios tipos de tumores, no se observaron diferencias relevantes en el perfil de seguridad o la eficacia en la población de pacientes ancianos, al comparar con pacientes de <65 años de edad. De manera coherente con este análisis, no se observaron diferencias en la seguridad y eficacia en la población de pacientes ancianos en un estudio de fase 3 (ET743-SAR-3007). En un estudio en pacientes con cáncer de ovario, no se observó ninguna diferencia en la seguridad en la población de pacientes ancianos. En este estudio, un análisis multivariado de supervivencia sin avance de la enfermedad, demostró que la edad mayor de 65 años no afectaba los resultados. Los resultados de un análisis farmacocinético poblacional indican que la edad no influye en el aclaramiento plasmático y el volumen de distribución de la trabectedina. Por lo tanto, no se recomienda ajustar la dosis basándose únicamente en la edad. Pacientes con función hepática reducida Los pacientes con insuficiencia hepática corren mayor riesgo de sufrir efectos adversos. No se puede recomendar una dosis inicial para estos pacientes, porque no se han realizado estudios adecuados sobre el uso de trabectedina en pacientes con función hepática disminuida. Sin embargo, se aconseja tener especial precaución y puede que sea necesario ajustar la dosis para estos pacientes, dado que puede aumentar la exposición sistémica y el riesgo de toxicidad hepática. No se debe tratar con YONDELIS® a los pacientes que presentan bilirrubina elevada en el momento de iniciar un ciclo (véase la sección “Advertencias y precauciones”). Pacientes con función renal reducida No se han realizado estudios en pacientes con insuficiencia renal (aclaramiento de creatinina < 30 ml/min); terapia combinada para cáncer de ovario < 60 ml/min) y por lo tanto, no se debe utilizar YONDELIS® en esta población (véase la sección “Advertencias y precauciones”). No es de esperar que la farmacocinética de trabectedina se vea influenciada por una insuficiencia renal leve o moderada (véase la sección “Propiedades farmacocinéticas”). CONTRAINDICACIONES YONDELIS® no se debe administrar a madres en período de lactancia (véase la sección “Embarazo, lactancia y fertilidad”). YONDELIS® no se debe administrar a pacientes con hipersensibilidad conocida a cualquiera de sus componentes. YONDELIS® no se debe administrar a pacientes con una infección activa grave o no controlada. ADVERTENCIAS Y PRECAUCIONES Insuficiencia hepática Para comenzar un tratamiento con YONDELIS®, los pacientes deben cumplir criterios específicos en cuanto a los parámetros de función hepática. Dado que la exposición sistémica a la trabectedina puede aumentar debido a una insuficiencia hepática y, por lo tanto, también puede aumentar el riesgo de hepatotoxicidad, se debe vigilar estrechamente a los pacientes con enfermedades hepáticas relevantes y ajustar la dosis si fuera necesario. Los pacientes con bilirrubina elevada en el momento de iniciar un ciclo no deben tratarse con trabectedina (véase la sección “Posología y forma de administración”). Insuficiencia renal Se debe controlar el aclaramiento de creatinina antes y durante el tratamiento. La trabectedina no se debe usar como agente único en pacientes con aclaramiento de creatinina < 30 ml/min, ni en combinación con PLD en pacientes con aclaramiento de creatinina < 60 ml/min (véase la sección “Posología y forma de administración”). Mielosupresión

Las anormalidades hematológicas de laboratorio de grado 3-4 (neutropenia, leucopenia, trombocitopenia y anemia) se informaron como muy comunes (>10%) en los estudios clínicos de fase 2 y 3 de pacientes con sarcoma de tejidos blandos y de cáncer de ovarios tratados con YONDELIS. Entre los pacientes con sarcoma de tejidos blandos con disminución en los recuentos de neutrófilos de grado 3-4, el primer valor mediano de severidad de grado 3 se observó en el Día 12, seguido de una recuperación al grado 2 para el Día 24. Las anormalidades en los recuentos de neutrófilos no fueron acumulativas, incluso en pacientes que recibieron un tratamiento prolongado con trabectedina (≥ 6 ciclos). Entre los pacientes con cáncer de ovario con disminución en los recuentos de neutrófilos de grado 3-4, se observaron niveles mínimos de neutrófilos después de una mediana de 15 días, que se recuperaron en una semana. Se debe realizar un recuento completo de células sanguíneas, que incluya recuento diferencial y recuento de plaquetas, en el período basal, semanalmente durante los dos primeros ciclos y una vez entre ciclo y ciclo a partir de ahí (véase la sección “Posología y forma de administración”). YONDELIS® no debe administrarse a pacientes con recuento basal de neutrófilos menor a 1500 células/mm3, recuento plaquetario menor a 100.000 células/mm3 o de hemoglobina < 9 g/dl. Si se observa neutropenia (RAN < 500 células/mm3) por más de 5 días o neutropenia asociada con fiebre o infección, o trombocitopenia (recuentos de plaquetas < 25.000/μl) se recomienda reducir la dosis (véase la sección “Posología y forma de administración”). Deberán administrarse factores estimulantes de asistencia sintomática/de la formación de colonias, de ser necesario, conforme a los lineamientos institucionales. Náuseas y vómitos Con frecuencia se ha informado la ocurrencia de náuseas y vómitos de grado 3 o 4. Se debe medicar a todos los pacientes con corticosteroides tales como la dexametasona. Se puede administrar medicación antiemética adicional según necesidad (véase la sección “Posología y forma de administración e interacciones”). Rabdomiólisis y aumento grave de la CPK (> 5 x LNS) En estudios clínicos de fase 2 y 3 de pacientes tratados contra el sarcoma de tejidos blandos, se observaron aumentos de la CPK (de grado 3-4) asociados con insuficiencia renal, rabdomiólisis y otras toxicidades relacionadas con lo muscular tales como miositis, debilidad muscular o dolor muscular en el 3,5% de los pacientes (n = 25). De ellos, tres pacientes presentaron resultados mortales; uno a causa de rabdomiólisis y dos a causa de insuficiencia renal. Se informó rabdomiólisis en cuatro pacientes en total (0,6%). Ocasionalmente, se ha observado la ocurrencia de rabdomiólisis y aumento grave de la CPK en el 2% de los pacientes tratados con YONDELIS en combinación con PLD, generalmente asociados con mielotoxicidad, anomalías graves en las pruebas de función hepática o insuficiencia renal. Por lo tanto, se debe vigilar atentamente la CPK y respetar estrictamente las pautas de tratamiento durante la fase del tratamiento y antes del retratamiento. No se debe utilizar trabectedina en pacientes con CPK > 2,5 × LNS (véase la sección “Posología y forma de administración”). Si se presenta una rabdomiólisis, se deben establecer en seguida medidas de apoyo como hidratación parenteral, alcalinización urinaria y diálisis, según sea necesario. El tratamiento con YONDELIS debe interrumpirse hasta que el paciente se recupere por completo. Se actuará con precaución si se administran medicamentos asociados con rabdomiólisis (por ej, estatinas) simultáneamente con trabectedina, ya que esto puede aumentar el riesgo de rabdomiólisis. Anomalías en las pruebas de función hepática (PFH) Se han comunicado aumentos agudos y reversibles en los niveles de AST y ALT en pacientes tratados con YONDELIS® en monoterapia o en combinación con PLD. El aumento de las transaminasas de grado 3 o 4 fue muy frecuente; el aumento de grado 4 fue frecuente. La

mediana de tiempo hasta el aumento de ALT o AST hasta el grado 3 o 4 fue de 8 días. Los niveles elevados cayeron por debajo del grado 3 o 4 en aproximadamente 8 días. Los aumentos en las transaminasas no fueron acumulativos y disminuyeron en magnitud e incidencia en los ciclos subsiguientes. Para los pacientes que tengan niveles aumentados de AST, ALT o fosfatasa alcalina entre ciclos puede ser necesario reducir la dosis (véase la sección “Posología y forma de administración”). YONDELIS no se debe utilizar en pacientes que presenten bilirrubina elevada al momento de iniciar un ciclo. Antes de la siguiente dosis deberán revisarse los aumentos en la bilirrubina que sucedieran desde la dosis previa en busca de la causa y considerarse una reducción de la dosis (véase “Dosis y administración”). Reacciones en el sitio de inyección Se recomienda especialmente utilizar un acceso venoso central (véase la sección “Posología y forma de administración”). Los pacientes pueden presentar reacciones graves en el sitio de inyección cuando se administra trabectedina mediante un catéter venoso periférico. Se han informado algunos casos de extravasación de la trabectedina con la consiguiente necrosis tisular, que requirió debridamiento. No hay ningún antídoto específico para la extravasación de trabectedina. La extravasación se debe manejar según las prácticas locales. Reacciones alérgicas Durante la experiencia posterior a la comercialización, se han informado casos raros de reacciones de hipersensibilidad, rara vez con desenlace mortal, en conexión con la administración de trabectedina, sola o en combinación con doxorubicina liposomal pegilada (PLD) (véase las secciones “Contraindicaciones” y “Reacciones adversas”). INTERACCIONES FARMACOLÓGICAS Se requiere una estrecha monitorización de las toxicidades en pacientes tratados con trabectedina en combinación con inhibidores potentes de CYP3A4 (por ejemplo, ketoconazol por vía oral, itraconazol, posaconazol, voriconazol, claritromicina, indinavir, lopinavir, ritonavir, boceprevir, nelfinavir, saquinavir, telaprevir, nefazodona, conivaptan) y tales combinaciones deben evitarse si es posible. Además, aprepitant y fluconazol sistémico deberán utilizarse con precaución durante el tratamiento con YONDELIS. Si tales combinaciones son necesarias, en caso de toxicidades deberían aplicarse los ajustes de dosis apropiados (véase la sección “Posología y forma de administración”). Se deben tomar precauciones al administrar productos que causan hepatotoxicidad, de forma concomitante con trabectedina, porque esto puede aumentar el riesgo de hepatotoxicidad. Se debe evitar el uso concomitante de trabectedina y alcohol. Hombres y mujeres en edad fértil Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento y hasta 3 meses después de finalizado el mismo. Los hombres fértiles deben utilizar métodos anticonceptivos efectivos durante el tratamiento y hasta 5 meses después de finalizado el mismo (véase la sección “Embarazo, lactancia y fertilidad”). En caso de embarazo se debe informar inmediatamente al médico a cargo del tratamiento. Advertencias y precauciones de PLD Ver la información del prospecto del fabricante de PLD para consultar las advertencias y precauciones sobre este producto. INTERACCIONES Efectos de otras sustancias sobre la trabectedina Un análisis poblacional, basado en el muestreo disperso de datos de un estudio de fase 3, demostró que el aclaramiento plasmático de la trabectedina disminuía en aproximadamente un

31% en 86 pacientes a los que se coadministró PLD 30 mg/m2 en comparación con 745 pacientes que participaron en 14 estudios en los que recibieron trabectedina sola. Los datos de otro estudio, de fase 1, en el cual se obtuvieron los perfiles farmacocinéticos de trabectedina de 16 pacientes que recibieron entre 0,9 y 1,3 mg/m2 de trabectedina combinada con 30 mg/m2 de PLD, mostraron un aclaramiento plasmático de trabectedina similar (diferencia media del 16%) al de la misma dosis de trabectedina administrada como agente único. Los resultados del análisis de farmacocinética poblacional (n=831 sujetos) indicaron que el aclaramiento plasmático de la trabectedina fue un 19% mayor en los pacientes que recibieron administración concomitante de dexametasona que en los que no la recibieron. Dado que la trabectedina se metaboliza principalmente mediante CYP3A4, es probable que el aclaramiento de la trabectedina disminuya en aquellos pacientes a los que se les coadministran medicamentos capaces de inhibir la actividad de esta isoenzima. Del mismo modo, la administración concomitante de trabectedina con potentes inductores de CYP3A4 puede aumentar el aclaramiento metabólico de trabectedina. Dos estudios de fase 1 de interacción fármaco-fármaco han confirmado tendencias al aumento y disminución de la exposición a la trabectedina cuando se la administra junto con ketoconazol y rifampina, respectivamente. En un estudio de interacciones fármaco-fármaco (n=8) con ketoconazol, un potente inhibidor de la CYP3A4, la exposición sistémica de trabectedina se incrementó en aproximadamente un 21% (Cmáx) y 66% (AUCúlt) cuando trabectedina se administra concomitantemente con ketoconazol (dosis total diaria de 400 mg). Se requiere una estrecha monitorización de las toxicidades en pacientes tratados con trabectedina en combinación con inhibidores potentes de CYP3A4 (por ejemplo, ketoconazol por vía oral, itraconazol, posaconazol, voriconazol, claritromicina, indinavir, lopinavir, ritonavir, boceprevir, nelfinavir, saquinavir, telaprevir, nefazodona, conivaptan) y tales combinaciones deben evitarse si es posible. Además, aprepitant y fluconazol sistémico deberán utilizarse con precaución durante el tratamiento con YONDELIS. Si tales combinaciones son necesarias, en caso de toxicidades deberían aplicarse los ajustes de dosis apropiados (véase la sección “Posología y forma de administración”). En un estudio de interacciones fármaco-fármaco (n = 8) con rifampicina, un potente inductor de la CYP3A4, la exposición sistémica de trabectedina se redujo en aproximadamente un 22% (Cmáx) y 31% (AUCúlt), cuando trabectedina se administró concomitantemente con rifampicina (dosis total diaria de 600 mg). Por lo tanto, el uso concomitante de trabectedina con inductores potentes de la CYP3A4 (por ejemplo, rifampicina, fenobarbital, hierba de San Juan) se debe evitar si es posible. Los estudios preclínicos in vitro han demostrado que la trabectedina es un sustrato de múltiples transportadores de eflujo, entre ellos, la P-gp, MRP2 y potencialmente MRP3 y MRP4, pero no BCRP. La administración concomitante de inhibidores de la P-gp, como la ciclosporina y el verapamil, pueden alterar la distribución de la trabectedina. No se ha establecido la relevancia clínica de esta interacción, por ej., en cuanto a toxicidad sobre el SNC, y se debe actuar con precaución al administrar inhibidores de la P-gp junto con trabectedina. El potencial de otros compuestos para desplazar a la trabectedina de su unión a proteínas plasmáticas se considera muy limitado, sobre la base de datos in vitro. Impacto de la trabectedina sobre medicamentos coadministrados In vitro, la trabectedina no induce ni inhibe las principales enzimas del citocromo P450. Un análisis poblacional basado en el muestreo disperso de datos de un estudio de fase 3 demostró que la farmacocinética plasmática de 30 mg/m2 de PLD es similar cuando se administra de manera concomitante con 1,1 mg/m2 de trabectedina (86 pacientes) y cuando se administra sola (80 pacientes).

Los niveles en plasma de trabectedina no unida y el total máximo alcanzados en pacientes con STS son de alrededor de 0,05 y 1,8 nM respectivamente, y en pacientes con cáncer de ovarios recurrente son de alrededor de 0,26 y 10,3 nM, respectivamente, y solamente están presentes durante la infusión, es decir, durante el día 1 de un ciclo de tratamiento de 3 semanas. En este nivel bajo de concentración, no se espera que la trabectedina produzca una inhibición clínicamente importante de los transportadores. En vista de los niveles extremadamente bajos de trabectedina plasmática relativos a los niveles fisiológicos de proteínas plasmáticas, se considera que el potencial de la trabectedina para a desplazar otros compuestos de su unión a proteínas plasmáticas es muy poco probable. EMBARAZO, LACTANCIA Y FERTILIDAD Embarazo No existen datos suficientes sobre la exposición durante el embarazo. Sin embargo, según su mecanismo de acción conocido, la trabectedina puede causar defectos congénitos graves, si se administra durante el embarazo. La trabectedina traspasó la placenta cuando se la administró a ratas embarazadas. No se recomienda la administración de trabectedina durante el embarazo. Si una paciente quedara embarazada durante el tratamiento, se debe informar a la paciente del riesgo potencial para el feto (véase la sección “Datos no clínicos”). Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento y hasta 3 meses después de finalizado el mismo. Los hombres fértiles deben utilizar métodos anticonceptivos efectivos durante el tratamiento y hasta 5 meses después de finalizado el mismo (véase las secciones “Advertencias y precauciones” y “Datos no clínicos”). En caso de embarazo se debe informar inmediatamente al médico a cargo del tratamiento. Si se produce un embarazo durante el tratamiento, hay que considerar buscar asesoramiento genético. Lactancia Se desconoce si la trabectedina se excreta en la leche humana. La excreción de trabectedina en la leche no se ha estudiado en animales. La lactancia materna está contraindicada durante el tratamiento y durante los 3 meses posteriores (véase la sección “Contraindicaciones”). Fertilidad La trabectedina puede tener efectos genotóxicos. Debido a la posibilidad de esterilidad irreversible causada por el tratamiento con YONDELIS®, se aconseja buscar asesoramiento sobre la conservación de ovocitos o esperma. También se recomienda buscar asesoramiento genético a los pacientes que deseen tener niños después de la terapia. EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS No se han estudiado los efectos sobre la capacidad para conducir y utilizar máquinas. Sin embargo, se ha comunicado astenia y fatiga en pacientes que recibían trabectedina. Aquellos pacientes que experimenten alguno de estos eventos durante el tratamiento, no deben operar máquinas. REACCIONES ADVERSAS En esta sección, se presentan las reacciones adversas. Las reacciones adversas son acontecimientos adversos que se consideran razonablemente asociados con el consumo de la trabectedina con base en la evaluación completa de la información disponible sobre acontecimientos adversos. No se puede determinar con fiabilidad una relación causal con la trabectedina en casos individuales. Además, dado que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos
de un medicamento no pueden compararse directamente con las tasas de los ensayos clínicos de otro medicamento y pueden no reflejar los índices observados en la práctica clínica. YONDELIS® en monoterapia en sarcoma de tejidos blandos avanzado En estudios clínicos de fase 2 y 3 de pacientes con sarcoma de tejidos blandos que reciben YONDELIS en la dosis recomendada (n = 717), se informaron reacciones adversas de grado de severidad 3 o 4 en el 54% de los pacientes, con el 13% clasificado como grave. Las reacciones adversas más comunes (≥20%) de cualquier grado de severidad fueron náuseas, fatiga, vómitos, diarrea, estreñimiento, disminución del apetito, disnea, dolor de cabeza, aumento de AST/ALT, neutropenia, anemia, leucopenia, trombocitopenia, aumento de la creatinina en sangre, aumento de la fosfatasa alcalina en sangre y disminución de albúmina en sangre. Se han producido reacciones adversas mortales en el 2,1% de los pacientes. Estas a menudo fueron el resultado de una combinación de eventos, incluidos mielosupresión, neutropenia febril (algunas con sepsis), disfunción hepática, insuficiencia renal o multiorgánica y rabdomiólisis.
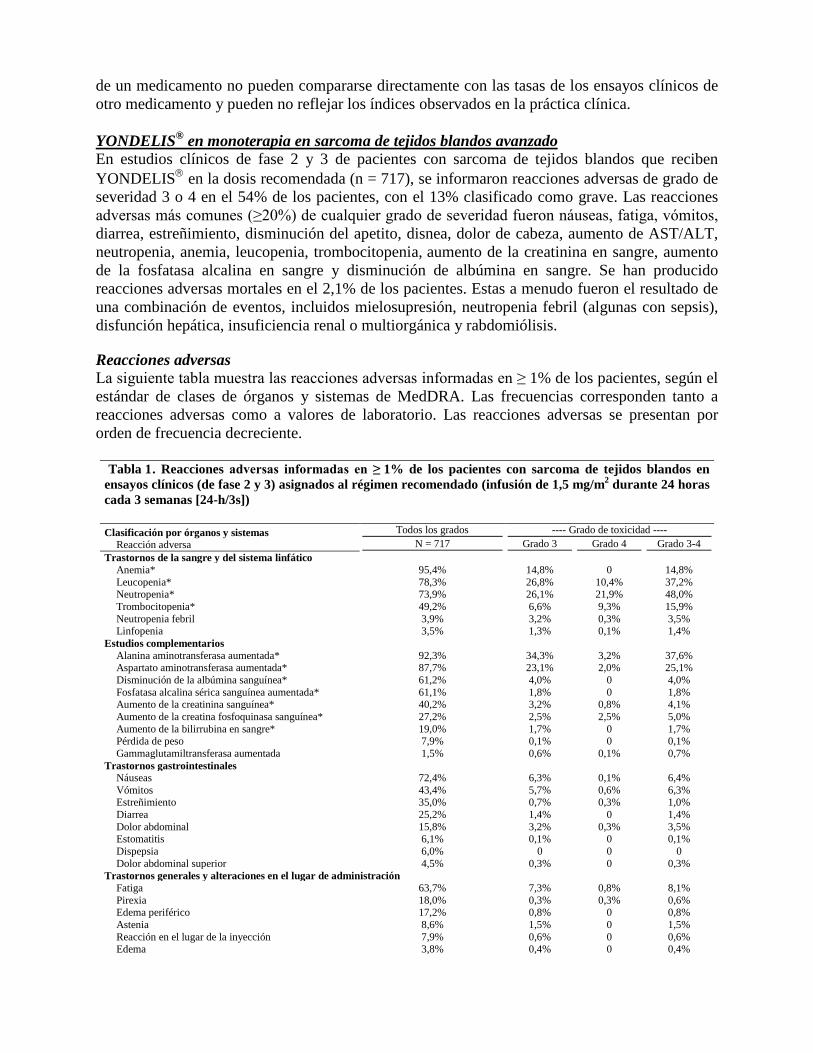
Reacciones adversas La siguiente tabla muestra las reacciones adversas informadas en ≥ 1% de los pacientes, según el estándar de clases de órganos y sistemas de MedDRA. Las frecuencias corresponden tanto a reacciones adversas como a valores de laboratorio. Las reacciones adversas se presentan por orden de frecuencia decreciente.
Tabla 1 . Reacciones adversas informadas en ≥ 1% de los pacientes con sarcoma de tejidos blandos en ensayos clínicos (de fase 2 y 3) asignados al régimen recomendado (infusión de 1,5 mg/m2 durante 24 horas cada 3 semanas [24-h/3s])
Clasificación por órganos y sistemas Reacción adversa
Todos los grados ---- Grado de toxicidad ---- N = 717 Grado 3 Grado 4 Grado 3-4
Trastornos de la sangre y del sistema linfático Anemia* 95,4% 14,8% 0 14,8% Leucopenia* 78,3% 26,8% 10,4% 37,2% Neutropenia* 73,9% 26,1% 21,9% 48,0% Trombocitopenia* 49,2% 6,6% 9,3% 15,9% Neutropenia febril 3,9% 3,2% 0,3% 3,5% Linfopenia 3,5% 1,3% 0,1% 1,4%
Estudios complementarios Alanina aminotransferasa aumentada* 92,3% 34,3% 3,2% 37,6% Aspartato aminotransferasa aumentada* 87,7% 23,1% 2,0% 25,1% Disminución de la albúmina sanguínea* 61,2% 4,0% 0 4,0% Fosfatasa alcalina sérica sanguínea aumentada* 61,1% 1,8% 0 1,8% Aumento de la creatinina sanguínea* 40,2% 3,2% 0,8% 4,1% Aumento de la creatina fosfoquinasa sanguínea* 27,2% 2,5% 2,5% 5,0% Aumento de la bilirrubina en sangre* 19,0% 1,7% 0 1,7% Pérdida de peso 7,9% 0,1% 0 0,1% Gammaglutamiltransferasa aumentada 1,5% 0,6% 0,1% 0,7%
Trastornos gastrointestinales Náuseas 72,4% 6,3% 0,1% 6,4% Vómitos 43,4% 5,7% 0,6% 6,3% Estreñimiento 35,0% 0,7% 0,3% 1,0% Diarrea 25,2% 1,4% 0 1,4% Dolor abdominal 15,8% 3,2% 0,3% 3,5% Estomatitis 6,1% 0,1% 0 0,1% Dispepsia 6,0% 0 0 0 Dolor abdominal superior 4,5% 0,3% 0 0,3%
Trastornos generales y alteraciones en el lugar de administración Fatiga 63,7% 7,3% 0,8% 8,1% Pirexia 18,0% 0,3% 0,3% 0,6% Edema periférico 17,2% 0,8% 0 0,8% Astenia 8,6% 1,5% 0 1,5% Reacción en el lugar de la inyección 7,9% 0,6% 0 0,6% Edema 3,8% 0,4% 0 0,4%
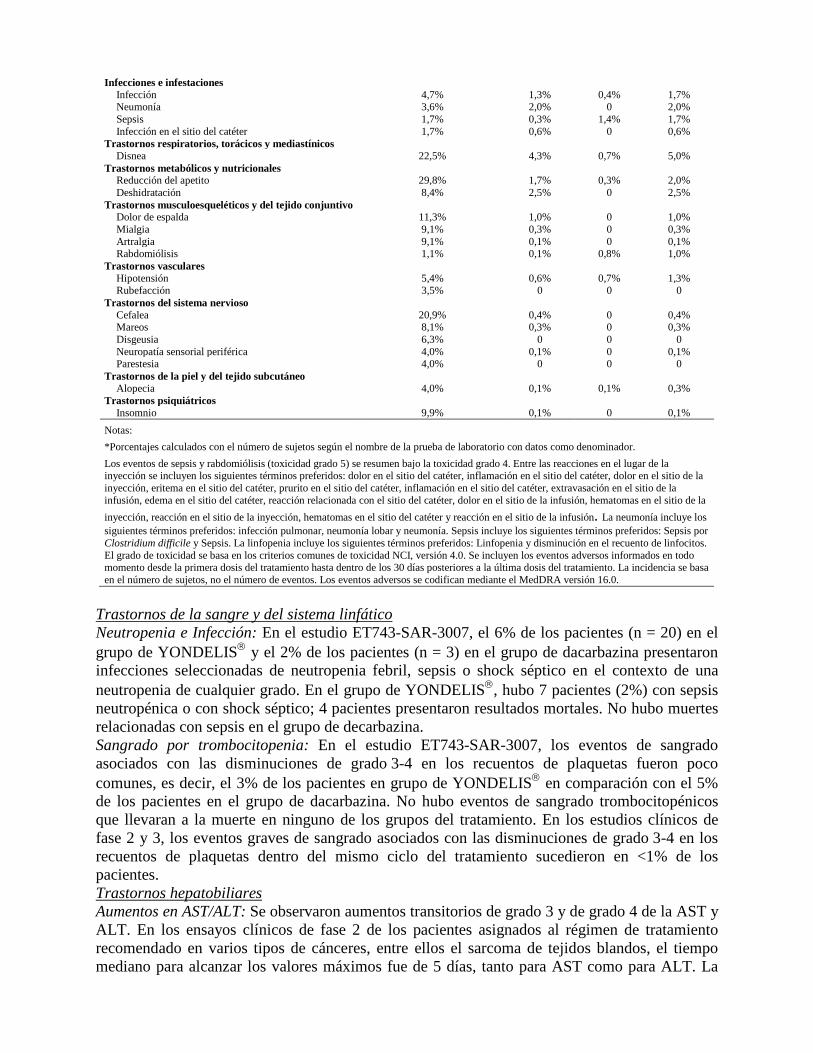
Infecciones e infestaciones Infección 4,7% 1,3% 0,4% 1,7% Neumonía 3,6% 2,0% 0 2,0% Sepsis 1,7% 0,3% 1,4% 1,7% Infección en el sitio del catéter 1,7% 0,6% 0 0,6%
Trastornos respiratorios, torácicos y mediastínicos Disnea 22,5% 4,3% 0,7% 5,0%
Trastornos metabólicos y nutricionales Reducción del apetito 29,8% 1,7% 0,3% 2,0% Deshidratación 8,4% 2,5% 0 2,5%
Trastornos musculoesqueléticos y del tejido conjuntivo Dolor de espalda 11,3% 1,0% 0 1,0% Mialgia 9,1% 0,3% 0 0,3% Artralgia 9,1% 0,1% 0 0,1% Rabdomiólisis 1,1% 0,1% 0,8% 1,0%
Trastornos vasculares Hipotensión 5,4% 0,6% 0,7% 1,3% Rubefacción 3,5% 0 0 0
Trastornos del sistema nervioso Cefalea 20,9% 0,4% 0 0,4% Mareos 8,1% 0,3% 0 0,3% Disgeusia 6,3% 0 0 0 Neuropatía sensorial periférica 4,0% 0,1% 0 0,1% Parestesia 4,0% 0 0 0
Trastornos de la piel y del tejido subcutáneo Alopecia 4,0% 0,1% 0,1% 0,3%
Trastornos psiquiátricos Insomnio 9,9% 0,1% 0 0,1%
Notas: *Porcentajes calculados con el número de sujetos según el nombre de la prueba de laboratorio con datos como denominador. Los eventos de sepsis y rabdomiólisis (toxicidad grado 5) se resumen bajo la toxicidad grado 4. Entre las reacciones en el lugar de la inyección se incluyen los siguientes términos preferidos: dolor en el sitio del catéter, inflamación en el sitio del catéter, dolor en el sitio de la inyección, eritema en el sitio del catéter, prurito en el sitio del catéter, inflamación en el sitio del catéter, extravasación en el sitio de la infusión, edema en el sitio del catéter, reacción relacionada con el sitio del catéter, dolor en el sitio de la infusión, hematomas en el sitio de la inyección, reacción en el sitio de la inyección, hematomas en el sitio del catéter y reacción en el sitio de la infusión. La neumonía incluye los siguientes términos preferidos: infección pulmonar, neumonía lobar y neumonía. Sepsis incluye los siguientes términos preferidos: Sepsis por Clostridium difficile y Sepsis. La linfopenia incluye los siguientes términos preferidos: Linfopenia y disminución en el recuento de linfocitos. El grado de toxicidad se basa en los criterios comunes de toxicidad NCI, versión 4.0. Se incluyen los eventos adversos informados en todo momento desde la primera dosis del tratamiento hasta dentro de los 30 días posteriores a la última dosis del tratamiento. La incidencia se basa en el número de sujetos, no el número de eventos. Los eventos adversos se codifican mediante el MedDRA versión 16.0.
Trastornos de la sangre y del sistema linfático Neutropenia e Infección: En el estudio ET743-SAR-3007, el 6% de los pacientes (n = 20) en el grupo de YONDELIS y el 2% de los pacientes (n = 3) en el grupo de dacarbazina presentaron infecciones seleccionadas de neutropenia febril, sepsis o shock séptico en el contexto de una neutropenia de cualquier grado. En el grupo de YONDELIS, hubo 7 pacientes (2%) con sepsis neutropénica o con shock séptico; 4 pacientes presentaron resultados mortales. No hubo muertes relacionadas con sepsis en el grupo de decarbazina. Sangrado por trombocitopenia: En el estudio ET743-SAR-3007, los eventos de sangrado asociados con las disminuciones de grado 3-4 en los recuentos de plaquetas fueron poco comunes, es decir, el 3% de los pacientes en grupo de YONDELIS en comparación con el 5% de los pacientes en el grupo de dacarbazina. No hubo eventos de sangrado trombocitopénicos que llevaran a la muerte en ninguno de los grupos del tratamiento. En los estudios clínicos de fase 2 y 3, los eventos graves de sangrado asociados con las disminuciones de grado 3-4 en los recuentos de plaquetas dentro del mismo ciclo del tratamiento sucedieron en <1% de los pacientes. Trastornos hepatobiliares Aumentos en AST/ALT: Se observaron aumentos transitorios de grado 3 y de grado 4 de la AST y ALT. En los ensayos clínicos de fase 2 de los pacientes asignados al régimen de tratamiento recomendado en varios tipos de cánceres, entre ellos el sarcoma de tejidos blandos, el tiempo mediano para alcanzar los valores máximos fue de 5 días, tanto para AST como para ALT. La

mayoría de los valores se había reducido a grado 1 o se habían resuelto, para los días 14-15 y menos del 2% de los ciclos tuvieron tiempos de recuperación mayores de 25 días. Los aumentos en ALT y AST no siguieron un patrón acumulativo, sino que su gravedad tendió a disminuir con el tiempo. En el estudio ET743-SAR-3007, la incidencia de anormalidades de laboratorio en ALT y AST de grado 3-4 fueron superiores en el grupo YONDELIS en comparación con el grupo de dacarbazina (29% y 16% en comparación con el 0,7% y el 1,3% respectivamente). Los aumentos en la transaminasa se manejaron mediante la reducción o retraso de las dosis de YONDELIS (véase la sección “Advertencias y precauciones”). Hiperbilirrubinemia: La bilirrubina alcanza un máximo aproximadamente una semana después del inicio y se resuelve aproximadamente dos semanas después del inicio. Lesión hepática grave: En el estudio ET743-SAR-3007, el 30% de los pacientes en el grupo de YONDELIS presentó un evento grado 3 y el 2% presentó eventos de grado 4 relacionados con lesiones hepáticas, que fueron principalmente anormalidades de laboratorio en las pruebas de función hepática (PFH). Fue rara la presencia de lesiones hepáticas severas inducidas por el medicamento (AST/ALT>3 × LNS, bilirrubina total ≥ 2 × LNS, ALP <2 × ULN antes de e incluido el día de la primera aparición de la elevación de la bilirrubina total ≥ 2 × ULN); con dos pacientes en el grupo de YONDELIS, ninguno de los cuales avanzó a insuficiencia hepática. No hubo muertes a causa de lesiones hepáticas en este estudio. En los estudios clínicos de fase 2 y 3, las manifestaciones de lesiones hepáticas severas fueron poco comunes, con una incidencia del 1%. Los signos y síntomas individuales incluyeron ictericia, hepatomegalia y dolor hepático. La mortalidad en presencia de daño hepático ocurrió en menos del 1% de los pacientes. Rabdomiólisis y aumentos de la CPK En el estudio ET743-SAR-3007, se observaron elevaciones de la CPK de cualquier grado en el 26% de los pacientes tratados con YONDELIS en comparación con el 7% en el grupo de dacarbazina. En el grupo de YONDELIS 18 pacientes (5%) y un paciente (< 1%) en el grupo de dacarbazina informaron aumentos en los niveles de la CPK de grado 3-4. De los 18 pacientes en el grupo de YONDELIS, cinco pacientes presentaron debilidad muscular, miositis o dolor musculoesquelético y cuatro pacientes presentaron rabdomiólisis. La rabdomiólisis fue mortal para un paciente. No hubo muertes por causa de la rabdomiólisis asociada con una CPK elevada (grado 3-4) en el grupo de decarbazina. Otras reacciones adversas Insuficiencia hepática: Se han comunicado casos raros de insuficiencia hepática (incluidos casos con resultados mortales) en pacientes con graves afecciones médicas subyacentes, tratados con trabectedina. Algunos factores de riesgo potenciales que pueden haber fomentado el incremento de la toxicidad de la trabectedina en dichos casos fueron la gestión de dosis no coherente con las pautas recomendadas, la potencial interacciones con CYP3A4 debido a múltiples substratos de CYP3A4 competidores o inhibidores de CYP3A4, o la falta de profilaxis con dexametasona. YONDELIS® en combinación con PLD para el tratamiento del cáncer de ovario avanzado El siguiente perfil de seguridad de YONDELIS® está basado en la evaluación de un estudio clínico de fase 3, OVA 301 de 663 pacientes con cáncer de ovario avanzado recurrente, que recibieron ya sea PLD (30 mg/m2) seguido de YONDELIS® (1,1 mg/m2) cada 3 semanas o PLD solo (50 mg/m2) cada 4 semanas. La combinación de YONDELIS® con PLD se administró a 333 pacientes de este estudio. En el grupo de combinación, la mediana del número de ciclos administrados fue de 6,0 ciclos (rango: 1 a 26) durante una mediana de 19 semanas. En el grupo de PLD solo, la mediana del número de ciclos administrados fue de 5,0 ciclos (rango: 1 a 22) durante una mediana de 20 semanas. La mayoría de las reacciones adversas se manejaron con reducciones o retrasos en la dosis (véase la sección “Posología y forma de administración”).
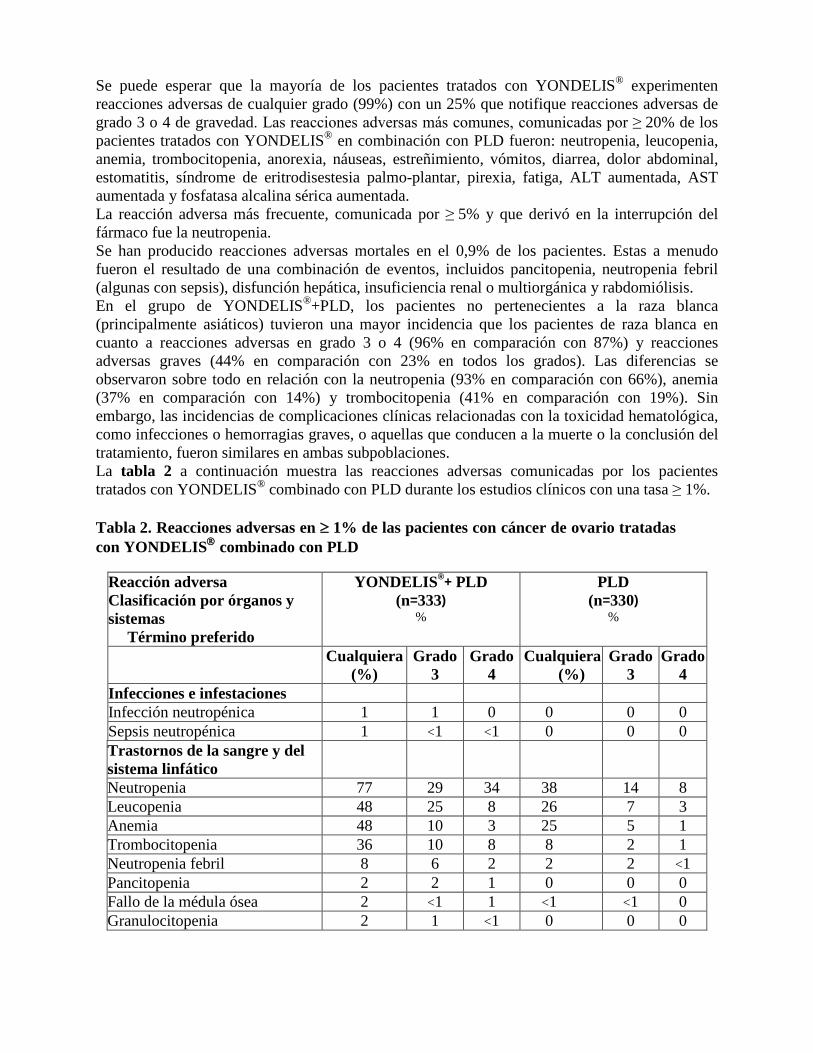
Se puede esperar que la mayoría de los pacientes tratados con YONDELIS® experimenten reacciones adversas de cualquier grado (99%) con un 25% que notifique reacciones adversas de grado 3 o 4 de gravedad. Las reacciones adversas más comunes, comunicadas por ≥ 20% de los pacientes tratados con YONDELIS® en combinación con PLD fueron: neutropenia, leucopenia, anemia, trombocitopenia, anorexia, náuseas, estreñimiento, vómitos, diarrea, dolor abdominal, estomatitis, síndrome de eritrodisestesia palmo-plantar, pirexia, fatiga, ALT aumentada, AST aumentada y fosfatasa alcalina sérica aumentada. La reacción adversa más frecuente, comunicada por ≥ 5% y que derivó en la interrupción del fármaco fue la neutropenia. Se han producido reacciones adversas mortales en el 0,9% de los pacientes. Estas a menudo fueron el resultado de una combinación de eventos, incluidos pancitopenia, neutropenia febril (algunas con sepsis), disfunción hepática, insuficiencia renal o multiorgánica y rabdomiólisis. En el grupo de YONDELIS®+PLD, los pacientes no pertenecientes a la raza blanca (principalmente asiáticos) tuvieron una mayor incidencia que los pacientes de raza blanca en cuanto a reacciones adversas en grado 3 o 4 (96% en comparación con 87%) y reacciones adversas graves (44% en comparación con 23% en todos los grados). Las diferencias se observaron sobre todo en relación con la neutropenia (93% en comparación con 66%), anemia (37% en comparación con 14%) y trombocitopenia (41% en comparación con 19%). Sin embargo, las incidencias de complicaciones clínicas relacionadas con la toxicidad hematológica, como infecciones o hemorragias graves, o aquellas que conducen a la muerte o la conclusión del tratamiento, fueron similares en ambas subpoblaciones. La tabla 2 a continuación muestra las reacciones adversas comunicadas por los pacientes tratados con YONDELIS® combinado con PLD durante los estudios clínicos con una tasa ≥ 1%. Tabla 2. Reacciones adversas en ≥ 1% de las pacientes con cáncer de ovario tratadas con YONDELIS combinado con PLD
Reacción adversa Clasificación por órganos y sistemas
Término preferido
YONDELIS®+ PLD (n=333)
%
PLD (n=330)
%
Cualquiera (%)
Grado 3
Grado 4
Cualquiera (%)
Grado 3
Grado 4
Infecciones e infestaciones Infección neutropénica 1 1 0 0 0 0 Sepsis neutropénica 1 <1 <1 0 0 0 Trastornos de la sangre y del sistema linfático
Neutropenia 77 29 34 38 14 8 Leucopenia 48 25 8 26 7 3 Anemia 48 10 3 25 5 1 Trombocitopenia 36 10 8 8 2 1 Neutropenia febril 8 6 2 2 2 <1 Pancitopenia 2 2 1 0 0 0 Fallo de la médula ósea 2 <1 1 <1 <1 0 Granulocitopenia 2 1 <1 0 0 0
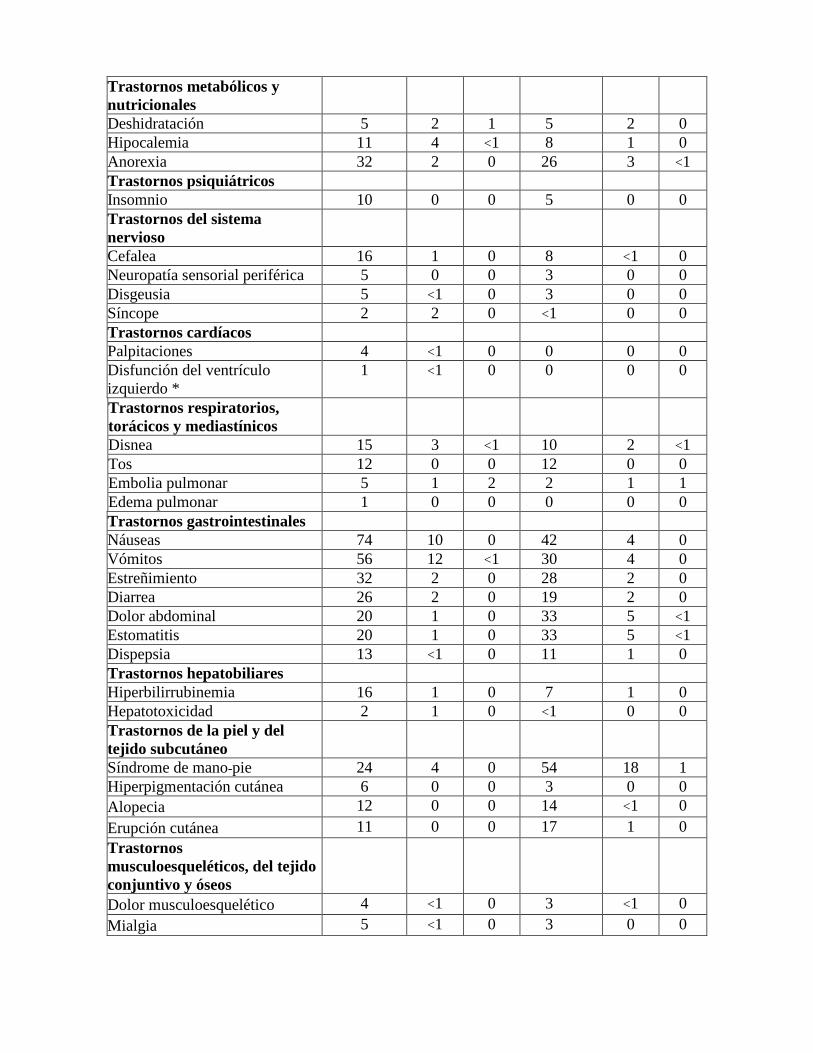
Trastornos metabólicos y nutricionales
Deshidratación 5 2 1 5 2 0 Hipocalemia 11 4 <1 8 1 0 Anorexia 32 2 0 26 3 <1 Trastornos psiquiátricos Insomnio 10 0 0 5 0 0 Trastornos del sistema nervioso
Cefalea 16 1 0 8 <1 0 Neuropatía sensorial periférica 5 0 0 3 0 0 Disgeusia 5 <1 0 3 0 0 Síncope 2 2 0 <1 0 0 Trastornos cardíacos Palpitaciones 4 <1 0 0 0 0 Disfunción del ventrículo izquierdo *
1 <1 0 0 0 0
Trastornos respiratorios, torácicos y mediastínicos
Disnea 15 3 <1 10 2 <1 Tos 12 0 0 12 0 0 Embolia pulmonar 5 1 2 2 1 1 Edema pulmonar 1 0 0 0 0 0 Trastornos gastrointestinales Náuseas 74 10 0 42 4 0 Vómitos 56 12 <1 30 4 0 Estreñimiento 32 2 0 28 2 0 Diarrea 26 2 0 19 2 0 Dolor abdominal 20 1 0 33 5 <1 Estomatitis 20 1 0 33 5 <1 Dispepsia 13 <1 0 11 1 0 Trastornos hepatobiliares Hiperbilirrubinemia 16 1 0 7 1 0 Hepatotoxicidad 2 1 0 <1 0 0 Trastornos de la piel y del tejido subcutáneo
Síndrome de mano-pie 24 4 0 54 18 1 Hiperpigmentación cutánea 6 0 0 3 0 0 Alopecia 12 0 0 14 <1 0 Erupción cutánea 11 0 0 17 1 0 Trastornos musculoesqueléticos, del tejido conjuntivo y óseos
Dolor musculoesquelético 4 <1 0 3 <1 0 Mialgia 5 <1 0 3 0 0
Trastornos renales y urinarios Fallo renal agudo 2 1 <1 1 1 0 Trastornos generales y alteraciones en el lugar de administración
Pirexia 20 1 0 13 1 0 Fatiga 46 8 <1 36 5 <1 Astenia 17 2 0 12 2 0 Inflamación de la mucosa 12 2 0 19 6 0 Edema periférico 9 1 0 8 0 <1 Edema 3 <1 0 1 0 0 Dolor en el sitio del catéter 3 0 0 0 0 0 Eritema en el sitio del catéter 2 0 0 0 0 0 Inflamación en el sitio del catéter 2 0 0 1 0 0 Estudios complementarios Alanina aminotransferasa aumentada
55 29 2 9 1 0
Aspartato aminotransferasa aumentada
40 6 1 10 1 <1
Fosfatasa alcalina sérica aumentada
23 1 0 8 1 0
Creatina fosfoquinasa sérica aumentada
7 1 1 3 0 0
Creatinina sérica aumentada 6 <1 <1 6 <1 0 Gamma-glutamiltransferasa aumentada
4 2 0 2 0 0
Bilirrubina conjugada aumentada 1 0 0 0 0 0 * Todos los pacientes que comunicaron disfunción ventricular izquierda mejoraron tras la interrupción del tratamiento. Rabdomiólisis Se observó la siguiente reacción adversa clínicamente significativa en menos del 1% de los pacientes tratados con YONDELIS® en combinación con PLD: rabdomiólisis (YONDELIS® + DLP ≤ 1% (grado 3: 0%, grado 4: ≤ 1%), y PLD solo 0%). Reacciones alérgicas Durante los ensayos clínicos, se comunicó hipersensibilidad en el 2% de los pacientes tratados con trabectedina sola o en combinación con doxorubicina liposomal pegilada (PLD), y la mayoría de dichos casos fue de grado 1 o 2 de gravedad. Durante la experiencia posterior a la comercialización, se han informado casos raros de reacciones de hipersensibilidad, rara vez con desenlace mortal, en conexión con la administración de trabectedina, sola o en combinación con doxorubicina liposomal pegilada (PLD) (véase las secciones “Contraindicaciones” y “Advertencias y precauciones”). Extravasación y necrosis tisular Durante los estudios clínicos y la vigilancia posterior a la comercialización, se han informado algunos casos de extravasación de trabectedina con subsiguiente necrosis tisular, lo que requirió desbridamiento (véase la sección “Advertencias y precauciones”).

Shock séptico Los casos de shock séptico, algunos de los cuales fueron mortales, se han comunicado con poca frecuencia en los estudios clínicos y en la experiencia posterior a la comercialización, en los pacientes tratados tanto con monoterapia como con terapia combinada. SOBREDOSIS Los datos sobre los efectos de la sobredosis de trabectedina son limitados. Los principales efectos adversos son gastrointestinales, de supresión de la médula ósea y hepáticos. Actualmente no existe un antídoto específico para la trabectedina. En caso de sobredosis, se debe monitorizar estrechamente a los pacientes e instituir las medidas de asistencia sintomática necesarias. PROPIEDADES FARMACOLÓGICAS Propiedades farmacodinámicas Grupo farmacoterapéutico: Agente antineoplásico, código ATC: L01CX01. Mecanismo de acción La trabectedina se une al surco menor del ADN, curvando la hélice hacia el surco mayor. Esta unión al ADN dispara una cascada de eventos que afectan a varios factores de transcripción, proteínas de unión al ADN y vías de reparación del ADN, todo lo cual perturba el ciclo celular. Se ha demostrado que la trabectedina ejerce una actividad antiproliferativa in vitro e in vivo frente a un rango de líneas celulares tumorales humanas y tumores experimentales, incluidos cánceres tales como el sarcoma, cáncer de mama, cáncer pulmonar de células no pequeñas, cáncer de ovario y melanoma. Se ha demostrado en modelos xenográficos in vitro e in vivo que YONDELIS® tiene efectos aditivos y sinérgicos cuando se lo combina con doxorubicina. Electrocardiograma Los efectos de la trabectedina sobre el intervalo QT/QTc se evaluaron en un estudio simple ciego, controlado con placebo y de diseño secuencial, con pacientes que presentaban tumores sólidos avanzados, localizados o metastásicos, que recibieron ≤ 3 líneas previas de quimioterapia. En este estudio, 75 pacientes recibieron placebo (solución salina) y trabectedina (1,3 mg/m2) en forma de infusión IV de 3 horas, los días 1 y 2, respectivamente. En este estudio, ningún paciente presentó un QTc de más de 500 ms o un aumento correspondiente en el tiempo del QTc con respecto al nivel basal de más de 60 ms en ningún momento. Una dosis terapéutica de trabectedina no prolonga el intervalo QTc. Recuento absoluto de neutrófilos (RAN) Se observó una asociación entre la evolución clínica del RAN durante el tratamiento y las concentraciones plasmáticas de trabectedina, lo cual indica que una exposición más alta a la trabectedina puede aumentar la incidencia y la severidad de neutropenia (véase la sección “Advertencias y precauciones”). Alanina aminotransferasa (ALT) La elevación de la ALT en suero durante la administración de trabectedina es transitoria y no acumulativa tras los ciclos posteriores del tratamiento con trabectedina. La magnitud de la elevación de ALT en suero transitoria tras la administración de la trabectedina depende de la dosis, pero no de la duración de la infusión. La elevación de la ALT en suero no parece estar asociada con la exposición a la trabectedina en la dosis recomendada. Se ha demostrado un efecto de protección hepática de la dexametasona cuando se trata con trabectedina a sujetos con cáncer. Bilirrubina total

Se ha observado una asociación entre la incidencia de la bilirrubina total de grado de toxicidad ≥ 2 y la AUC y Cmáx de trabectedina en plasma (véase la sección “Advertencias y precauciones”). Eficacia clínica Monoterapia La eficacia y seguridad clínica de trabectedina para el tratamiento de pacientes con leiomiosarcoma o liposarcoma (L-sarcoma) se demostró en el estudio ET743-SAR-3007, un estudio controlado aleatorizado de fase 3 de trabectedina en comparación con decarbazina en pacientes con L-sarcoma avanzado. Los pacientes en el estudio ya habían sido tratados en cualquier orden con al menos un régimen que contenía antraciclina e ifosfamida, o una antraciclina que contenía el régimen y un régimen adicional de quimioterapia citotóxica. En el punto de corte de los datos clínicos, se aleatorizó a 345 pacientes para el grupo de trabectedina (1,5 mg/m2 una vez cada 3 semanas [q3s 24h]) y a 173 pacientes para el grupo de dacarbazina (1 g/m2 una vez cada 3 semanas). La edad mediana de los pacientes fue de 56 años (rango de 17 a 81), el 30% era de sexo masculino; el 76% caucásico, el 12% afroamericano y el 4% asiático. Los factores de pronóstico importantes estuvieron bien equilibrados entre los pacientes; el 73% tuvo leiomiosarcomas, el 27% liposarcomas y todos los pacientes obtuvieron un puntaje ECOG de ≤ 1. La mayoría de los pacientes en ambos grupos del tratamiento recibió 2 rondas previas de quimioterapia y el tiempo mediano desde la última progresión de la enfermedad fue menos de 1 mes (0,85 mes). Los pacientes en los grupos de trabectedina y de decarbazina recibieron una mediana de 4 y 2 ciclos, respectivamente. El criterio de valoración de la eficacia primario del estudio fue la supervivencia general (SG), con un análisis final que se llevaría a cabo al momento de los 376 eventos de muerte observados. Los criterios de valoración secundarios importantes según la evaluación del investigador de la respuesta de la enfermedad fueron la supervivencia libre de progresión (PFS), la tasa de respuesta objetiva (ORR), la duración de la respuesta (DOR) y el tiempo hacia la progresión (TTP). Los criterios de respuesta a la enfermedad se evaluaron con los lineamientos de los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST, por su sigla en inglés). En el análisis intermedio (n = 518), fallecieron 189 pacientes (126 [37%] en el grupo de tratamiento con trabectedina y 63 [36%] en el grupo de tratamiento de dacarbazina). La SG mediana fue de 12,39 meses (IC del 95%: [11,17-14,55] meses) para el grupo de tratamiento con trabectedina y 12,91 meses (IC del 95%: [7,79-16,43] meses) para el grupo de tratamiento con dacarbazina. La HR fue de 0,872 meses (IC del 95%: [0,644-1,181]; p = 0,3741), lo cual representa una reducción del 13% en el riesgo de muerte para los pacientes en el grupo de tratamiento de trabectedina. En la tabla a continuación, se presentan otros resultados de eficacia del estudio ET743-SAR-3007 al momento del análisis intermedio. Los resultados de la PFS se auditaron por medio de una revisión retrospectiva por radiólogos independientes. Tabla 3. Resultados de eficacia del estudio ET743-SAR-3007
Criterio de valoración de la eficacia Trabectedina Dacarbazina
N: Pacientes IDT 345 173 Supervivencia libre de progresióna Eventos n (%) 217 (63%) 112 (65%) Mediana [IC del 95%] (meses) 4,21 [2,99- 4,83] 1,54 [1,48- 2,60] HR [IC del 95%]b 0,550 [0,436-0,696] valor pc < 0,0001 Tiempo hasta la progresión Eventos n (%) 205 (59%) 112 (65%) Mediana [IC del 95%] (meses) 4,24 [3,22-4,93] 1,[1,48-2,60]
HR [IC del 95%]b 0,522 [0,412-0,661] valor pc < 0,0001 Tasa de respuesta objetiva (ORR; CR+PR) Tasa de respuesta objetiva n (%) 34 (10%) 12 (7%) ORR [IC del 95% ]e [6,9-13,5] [3,6-11,8] O [IC del 95%]d 1,467 [0,717-3,197] valor pd 0,3269 Duración de la respuesta (CR+PR) n: respuesta-paciente evaluable 34 12 Medianac [IC del 95%] (meses) 6,47 [3,58-7,62] 4,17 [2,14- NE] HR [IC del 95%]b 0,471 [0,168-1,318] valor p 0,1415 Tasa de beneficio clínico (CBR) CBR, n (%) 118 (34%) 32 (19%) CBR [IC del 95%]e [29,2-39,5] [13,0-25,1] O [IC del 95%]d 2,291 [1,446-3,692] valor pd 0,0002 Duración de la enfermedad estable n (%): pacientes con la mejor respuesta de enfermedad estable
177 (51%) 60 (35%)
Eventos, n (%) 90 (51%) 35 (58%) Mediana [IC del 95%], meses 6,01 [4,86- 6,97] 4,17 [2,89-5,49] HR [IC del 95%]b 0,449 [0,300-0,673] valor pc 0,0001 a Según la evaluación del investigador. b El cociente de riesgo se estima mediante el uso del modelo de riesgo proporcional COX con un grupo del tratamiento como la
única covariante; una tasa de riesgo < 1 indica una ventaja para la trabectedina c El valor p se basa en la prueba de rango logarítmico no estratificada. d Con base en la prueba exacta de Fisher. e La ORR IC del 95% se basa en el IC exacto de Fisher; CR = Respuesta completa; CRu = Respuesta completa no confirmada; PR = Respuesta parcial; IC = Intervalo de confianza, HR = Cociente de riesgo; IDT = Intención de tratar, OR = Cociente de probabilidades; CBR = Tasa de beneficio clínico. Nota: La tasa de respuesta objetiva (ORR) se define como la proporción de sujetos que alcanzaron una CR o PR como mejor respuesta. Beneficio clínico se define como un sujeto cuya mejor respuesta general es CR, PR o SD > = 18s.
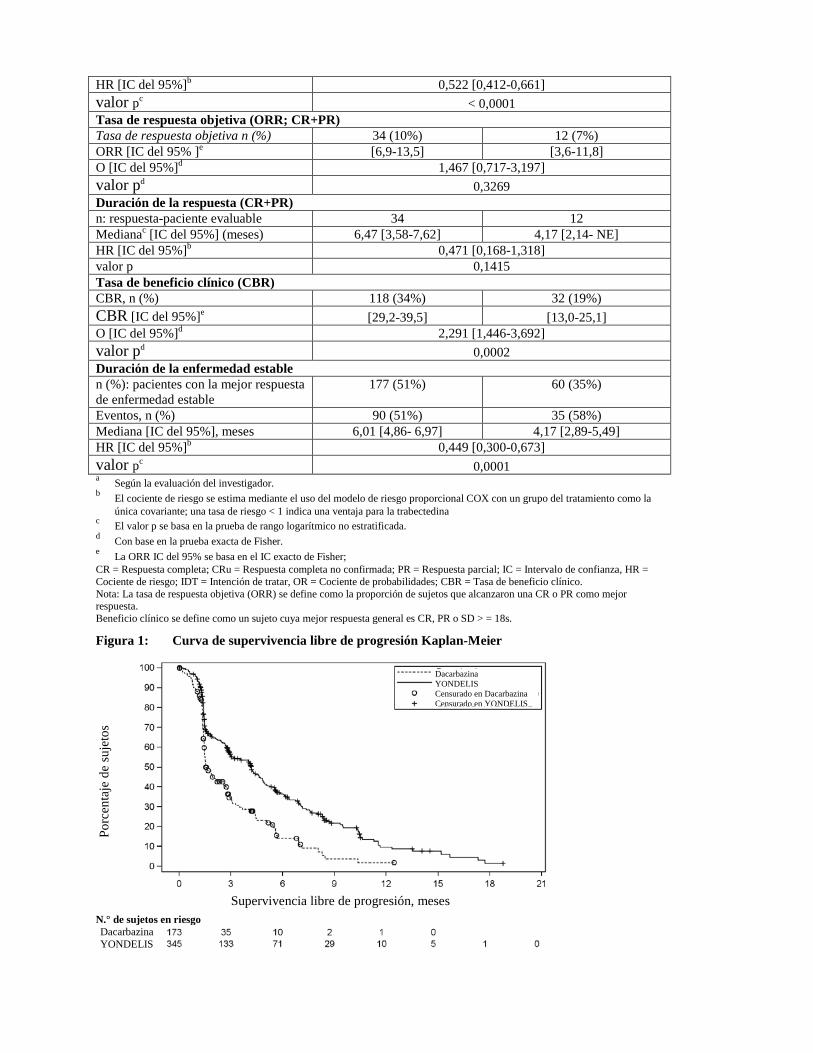
Figura 1: Curva de supervivencia libre de progresión Kaplan-Meier
Porc
enta
je d
e su
jeto
s
Supervivencia libre de progresión, meses N.° de sujetos en riesgo Dacarbazina YONDELIS
Dacarbazina YONDELIS Censurado en Dacarbazina Censurado en YONDELIS

En el estudio ET743-SAR-3007, se utilizaron los puntajes del Inventario de Síntomas M. D. Anderson (MDASI, por su sigla en inglés) para evaluar la carga de los síntomas percibida por el paciente (severidad del síntoma e interferencia del síntoma en relación con la función física y mental) y para determinar el efecto del tratamiento sobre el cambio o la estabilidad del síntoma. Los puntajes MDASI demostraron niveles bajos de los síntomas en el punto basal tanto en el grupo de trabectedina como en el de dacarbazina; esto se mantuvo durante la terapia sin diferencia significativa alguna entre los grupos del tratamiento. El estudio aleatorizado ET743-STS-201 evaluó la eficacia y seguridad de la trabectedina en pacientes con liposarcoma o leiomiosarcoma avanzados, localizados o metastásicos, cuya enfermedad había avanzado o recurrido tras un tratamiento con, al menos, antraciclinas e ifosfamida. En este estudio, se administró trabectedina ya sea en dosis de 1,5 mg/m2, en infusión intravenosa de 24 horas cada 3 semanas, o en dosis de 0,58 mg/m2 semanales, en infusión intravenosa de 3 horas durante 3 semanas de un ciclo de 4 semanas. No se determinó previamente un límite para el número de ciclos administrado. El tratamiento continuó mientras se observaron beneficios clínicos. No se observaron efectos adversos acumulativos en los pacientes tratados con múltiples ciclos. El análisis final especificado en el protocolo del tiempo hasta la progresión (THP) evidenció un 26,6% de reducción del riesgo relativo de progresión en los pacientes tratados con 24h/3s (cociente de riesgo = 0,734; IC [0,554-0,974]). La mediana de valores de THP fue de 3,7 meses (IC: [2,1-5,4] m) en el grupo de 24h/3s y 2,3 meses (IC: [2,0-3,5] m) en el grupo de 3h/s (p=0,0302). No se detectaron diferencias significativas en la sobrevida general (SG). La mediana de SG con el régimen 24h/3s fue de 13,9 meses (IC: [12,5 -18,6] y el 60,3% de los pacientes estaban vivos a 1 año (IC: [52,0- 68,5] %). Se encuentran disponibles más datos de eficacia, de 3 ensayos de fase 2 de un solo grupo con poblaciones similares, tratadas con el mismo régimen. En estos ensayos se evaluó un total de 100 pacientes con liposarcoma y leiomiosarcoma y 83 pacientes con otros tipos de sarcoma. Los resultados de un programa de acceso ampliado para pacientes con STS muestran que, entre los 903 pacientes evaluados para SG, la mediana de tiempo de supervivencia fue de 11,9 meses (IC del 95%: [11,2-13,8]). La mediana de supervivencia por tipo de histología tumoral fue de 16,2 meses (IC del 95%: [14,1-19,5]) en pacientes con leiomiosarcomas y liposarcomas, y 8,4 meses (IC del 95%: [7,1-10,7]) en pacientes con otros tipos de sarcomas. La sobrevida mediana para pacientes con liposarcoma fue de 18,1 meses (IC95%: [15,0-26,4]) y para pacientes con leiomiosarcoma fue de 16,2 meses (IC del 95%: [11,7-24,3]). Terapia combinada La eficacia y seguridad de YONDELIS® combinado con PLD en pacientes con cáncer de ovario recurrente se demostró en un estudio abierto de fase 3 multicéntrico, aleatorizado y con control activo. Este estudio incluyó 672 pacientes aleatorizados para recibir ya sea YONDELIS® (1,1 mg/m2 i.v. durante 3 horas) administrado después de PLD® (30 mg/m2 i.v durante 90 min) cada 3 semanas o PLD (50 mg/m2 i.v. durante 90 min) cada 4 semanas. La mediana de edad de los pacientes de este estudio fue de 57 años (rango 26 - 87); un 78% eran caucásicos, un 20% asiáticos y un 2% de raza negra/otras razas. Las características basales demográficas y patológicas se muestran a continuación en la tabla 4:
Tabla 4. Resumen de las características basales demográficas y patológicas de los pacientes
YONDELIS + PLD n = 337
PLD n = 335
Mediana de edad (rango) 56 (26; 82) 58 (27; 87)

YONDELIS + PLD n = 337
PLD n = 335
Estado funcional ECOG basal (%)
0 68 57 1 29 39 2 3 3 Sensibilidad al platino (%) Sensible al platino 65 63 Resistente al platino 35 37 Terapia anterior con taxano (%)
80 81
Intervalo libre de platino (ILP)*
n (%) n (%)
< 6 118 (35) 124 (37) ≥6-12 123 (37) 90 (27) ≥12 95 (28) 121 (36)
*PFI: desde la terminación del último tratamiento con platino hasta el tiempo de progresión. Los beneficios clínicos de YONDELIS® + PLD se observan en la supervivencia libre de progresión (SLP) y en la tasa de respuesta objetiva; la supervivencia muestra una tendencia a favor del grupo de terapia combinada al momento del análisis final. El criterio de valoración principal, la supervivencia libre de progresión (SLP), fue significativamente mayor en los pacientes tratados con YONDELIS® combinado con PLD® que en los tratados con PLD solo (mediana de SLP: 7,3 en comparación con 5,8 meses, respectivamente). El tratamiento con YONDELIS® + PLD redujo en un 21% el riesgo de avance de la enfermedad, en comparación con PLD solo [CR = 0,79; IC del 95% [0,65-0,96], p = 0,0190]. El análisis final de supervivencia se llevó a cabo basado en los 522 eventos con una mediana de seguimiento de 47,4 meses. Los resultados de eficacia se resumen en la tabla 5.
Tabla 5. Eficacia de YONDELIS combinado con PLD en el tratamiento de pacientes con cáncer de ovario (Estudio OVA-301)
YONDELIS+PLD PLD valor p
Revisión por un radiólogo independiente
n=328 n=317
0, 0190a Mediana de SLP [IC del 95%] meses*
7,3 [5,9-7,9] 5,8 [5,5-7,1]
Cociente de riesgo [IC del 95%]
0,79 [0,65-0,96]
Población ITT n=337 n=335 Tasa de respuesta objetiva (%)
27,6 18,8 0,0080b

Cociente de probabilidades**
1,646 (1,144-2,367)
Duración mediana de la respuesta (meses)*
7,9 (7,4; 9,2) 7,7 (6,5; 9,0)
0,8203a Cociente de riesgo [IC del 95%]
0,95 [0,62-1,46]
Revisión por un oncólogo independiente
n=336 n=335
0,0008a Mediana de SLP [IC del 95%] meses*
7,4 [6,4-9,2] 5,6 [4,2-6,8]
Cociente de riesgo [IC del 95%]
0,72 [0,60-0,88]
Tasa de respuesta objetiva (%)
30,4 19,1 0,0009b
Cociente de probabilidades** 1,846 (1,290-2,641)
Supervivencia general (SG) (Análisis final n = 522 eventos) Todos aleatorizados n=337 n=335 Mediana de SG [IC del 95%] (meses)
22,2 [19,3-25,0] 18,9 [17,1-21,5] 0,0835
Cociente de riesgoa 0,86 (0,72-1,02) Supervivencia general en población sensible al platino (Análisis final n = 316
eventos) n=218 n=212 Mediana de SG [IC del 95%] (meses)
27 [24,1-31,4] 24,1 [20,9-25,9] 0,1056
Cociente de riesgoa 0,83 (0,67-1,04) Supervivencia general en población resistente al platino (Análisis final n = 206
eventos) n=119 n=123 Mediana de SG [IC del 95%] (meses)
14,2 [11,1-16,8] 12,4 [10,6-14,8] 0,5452
Cociente de riesgoa 0,92 (0,70-1,21)
*Con base en las estimaciones de Kaplan Meier; un cociente de riesgo < 1 indica una ventaja para YONDELIS+PLD. **Cociente de probabilidades > 1 indica una ventaja para (YONDELIS+PLD) calculada según Cochran-Mantel-Haenszel. a prueba de rango logarítmico b prueba exacta de Fisher Según una revisión hecha por un oncólogo independiente, los pacientes que tuvieron intervalos libres de platino (ILP) de menos de 6 meses, tuvieron SLP similares entre los dos grupos, ambos con una mediana de SLP de 3,7 meses [CR = 0,89 (IC del 95%: [0,67-1,20])]. La tasa de respuesta objetiva fue del 14,3% en el grupo de YONDELIS®+PLD en comparación con 11,4% en el grupo de monoterapia con PLD. Para los pacientes con ILP > 6 meses, la mediana de SLP

fue de 9,7 meses en el grupo de YONDELIS®+PLD en comparación con 7,2 meses en el grupo de monoterapia con PLD [CR = 0,66 (IC del 95% [0,51-0,85]), valor de p = 0,0010]. La tasa de respuesta objetiva fue del 39,2% en el grupo de YONDELIS®+PLD en comparación con 23,6% en el grupo de monoterapia con PLD. En el análisis final, el efecto del tratamiento sobre la supervivencia general fue más pronunciado en la combinación YONDELIS®+PLD en comparación con PLD solo en pacientes con ILP ≥ 6 meses (población sensible al platino: 27,0 en comparación con 24,1 meses, CR = 0,83, IC: [0,67-1,04] que aquellos con ILP < 6 meses (población resistente al platino: 14,2 en comparación con 12,4 meses, CR = 0,92, IC: [0, 70-1,21]. En los análisis multivariados incluido ILP, el efecto del tratamiento fue estadísticamente significativo a favor de la combinación de YONDELIS®+PLD en oposición al PLD solo (SLP, p = 0,0157; SG, p = 0,0285). Propiedades farmacocinéticas La exposición sistémica tras la administración intravenosa en forma de infusión a velocidad constante es proporcional a la dosis, con dosis de hasta 1,8 mg/m2 inclusive. El perfil farmacocinético de la trabectedina es consistente con un modelo de eliminación de múltiples compartimentos, con una vida media terminal en plasma de 175 horas. Las concentraciones de trabectedina en plasma no se acumulan cuando se la administra cada 3 semanas. Distribución La trabectedina tiene un gran volumen de distribución (más de 5000 l), indicativo de una extensa distribución en los tejidos periféricos. La trabectedina se une extensivamente a la alfa-1 glicoproteína ácida y a la albúmina plasmática. La fracción libre (no unida) plasmática media es de 2,23% y 2,72% a concentraciones plasmáticas totales de 10 ng/ml y 100 ng/ml, respectivamente. Los estudios preclínicos in vitro han demostrado que trabectedina es un sustrato de transportadores múltiples de eflujo que incluye la P-gp, MRP2 y potencialmente MRP3 y MRP4, pero no BCRP. Los modelos preclínicos indican que la P-gp, MRP2 y MRP3 están involucradas en el eflujo hepático de los metabolitos de trabectedina y tienen una función importante y parcialmente redundante en la protección contra la toxicidad (hepática) mediada por trabectedina. Metabolismo La trabectedina se metaboliza en alto grado. La isoenzima 3A4 es la principal isoenzima del citocromo P450 responsable del metabolismo oxidativo de la trabectedina en concentraciones clínicamente relevantes. No se puede descartar que otras enzimas del P450 contribuyan al metabolismo de la trabectedina. No se ha observado una glucuronidación apreciable de la trabectedina. Eliminación La media (DE) de la radiación total recuperada fue del 58% (17%) y del 5,8% (1,73%) en heces (24 días) y orina (10 días), respectivamente, tras administrar una dosis de trabectedina marcada a 8 pacientes con cáncer. La cantidad de medicamento inalterado que se excreta en heces y orina es insignificante (< 1% de la dosis). El aclaramiento corporal de la trabectedina es de aproximadamente 35 l/h. Este valor representa aproximadamente la mitad de la tasa de flujo sanguíneo hepático humano. Por lo tanto, la proporción de extracción de la trabectedina se puede considerar moderada. La variabilidad estimada entre pacientes de la población, en cuanto al aclaramiento plasmático de la trabectedina, es del 49%, y la variabilidad dentro de un mismo paciente es del 28%. Poblaciones especiales

Un análisis farmacocinético poblacional indica que el aclaramiento plasmático de la trabectedina no está influenciado por el peso total del cuerpo (rango: de 36 a 148 kg), la superficie corporal (rango: de 0,9 a 2,8 m2), la edad (rango: de 19 a 83 años) o el sexo. Función renal disminuida No se ha observado una influencia relevante de la función renal, medida como aclaramiento de creatinina, sobre la farmacocinética de la trabectedina dentro del rango de valores (≥ 30,3 ml/min) de los pacientes incluidos en los estudios clínicos. No hay datos disponibles en pacientes con un aclaramiento de creatinina menor a 30,3 ml/min. La escasa recuperación (< 9% en todos los pacientes estudiados) de radioactividad total en orina, tras una única dosis de trabectedina marcada con 14C sugiere que la insuficiencia renal tendría poca influencia en la eliminación de la trabectedina o sus metabolitos. Función hepática disminuida El aclaramiento de la trabectedina, puede disminuir en pacientes con insuficiencia hepática, lo cual resulta en mayores concentraciones de trabectedina en plasma. Es indispensable vigilar atentamente la toxicidad cuando se administra trabectedina a pacientes con la función hepática disminuida. Pediatría Se ha estudiado la farmacocinética de la trabectedina en niños (rango etario: de 1 a 18 años) con tumores sólidos refractarios. En general, los valores de los parámetros se encontraban dentro del rango observado previamente en adultos. Sin embargo, no se pueden derivar conclusiones definitivas dado el tamaño relativamente pequeño de la muestra de pacientes pediátricos en cada grupo de dosis (véase la sección “Posología y forma de administración”). Ancianos Los resultados de análisis farmacocinéticos poblacionales indican que la edad no influye en el aclaramiento plasmático y el volumen de distribución de la trabectedina. Por lo tanto, no se recomienda el ajuste de la dosis inicial de trabectedina debido a cambios potenciales relacionados con la edad en sus propiedades farmacocinética (véase la sección “Posología y forma de administración”). Raza/origen étnico Un análisis farmacocinético poblacional de un número limitado de sujetos mostró que no se espera que la raza y el origen étnico tengan efectos clínicamente relevantes sobre la farmacocinética de la trabectedina. DATOS NO CLÍNICOS Farmacología/toxicología Los datos preclínicos indican que la trabectedina tiene efectos limitados sobre los sistemas cardiovascular, respiratorio y nervioso central, con exposiciones por debajo del rango terapéutico clínico, en cuanto al AUC. Los efectos de la trabectedina sobre la función cardiovascular y respiratoria se han investigado in vivo (en monos macacos anestesiados). Se seleccionó un régimen de 1 hora de infusión para lograr niveles plasmáticos máximos (valores Cmáx) en el rango de aquellos observados en la clínica. Se alcanzaron niveles de trabectedina plasmática de 10,6 ± 5,4 ng/ml (Cmáx), similares a los que se alcanzan tras la administración de 1,1 mg/m2 en infusión de 3 horas (Cmáx de 7,9±2,0 ng/ml). Los principales efectos adversos de la trabectedina identificados fueron la mielosupresión y la hepatotoxicidad. Los hallazgos observados incluyeron toxicidad hematopoyética (leucopenia severa, anemia y agotamiento linfoide y de la médula ósea) y aumentos en las pruebas de función

hepática, degeneración hepatocelular, necrosis epitelial intestinal y reacciones graves en el sitio de inyección. En ratones, ratas, conejos y monos, se observó con regularidad inflamación local dependiente de la dosis en el sitio de inyección, tras la inyección i.v., en especial después de varios ciclos. En estudios de toxicidad con dosis repetidas, realizados en monos macacos, se observó tromboflebitis grave con inflamación perivascular extensa y fibrosis, generalmente con necrosis pronunciada, que también afectaba a los tejidos de alrededor, después del cuarto ciclo, lo cual condujo al sacrificio prematuro o la muerte de algunos animales. Estos efectos adversos se observaron al administrar trabectedina a animales de menos de 3 kg. Se observó mortalidad con 0,42 mg/m2 y más (véase la sección “Posología y forma de administración”; “Poblaciones especiales; Pacientes pediátricos”). En estudios de toxicidad con ciclos repetidos realizados en monos, se observaron efectos adversos renales. Estos hallazgos fueron secundarios a una intolerancia local grave en el sitio de administración (en la punta del catéter) con daño severo de los tejidos de alrededor (por ej., los riñones) y por lo tanto, no pueden atribuirse con certeza a la trabectedina. Sin embargo, se debe actuar con precaución al interpretar los hallazgos renales, ya que no se pueden descartar los efectos adversos del tratamiento. La trabectedina es genotóxica tanto in vitro como in vivo. No se han llevado a cabo estudios de carcinogenicidad a largo plazo. No se han realizado estudios de fertilidad con trabectedina pero se han observado algunos cambios histopatológicos en las gónadas en estudios de toxicidad de dosis repetidas. Considerando la naturaleza del compuesto (citotóxico y mutagénico), es probable que afecte a la capacidad reproductiva. DATOS FARMACÉUTICOS Lista de excipientes Sacarosa, potasio dihidrógeno fosfato, ácido fosfórico (para ajustar el pH), hidróxido de potasio (para ajustar el pH). Incompatibilidades YONDELIS® no se debe mezclar ni diluir con medicamentos excepto aquellos mencionados en la sección “Instrucciones de uso y manipulación”. Período de validez Observe la fecha de caducidad en el embalaje exterior. Después de la reconstitución, la estabilidad química y física se ha demostrado durante 30 horas hasta una temperatura de 25° C. Después de la dilución, se ha comprobado la estabilidad química y física durante 30 horas hasta una temperatura de 25° C. El tiempo total de espera entre la reconstitución inicial y el final del tratamiento no debe ser superior a 30 horas. Desde el punto de vista microbiológico, la solución reconstituida se debe diluir y utilizar inmediatamente. Si no se diluye el producto reconstituido y utiliza inmediatamente, las condiciones durante su uso y los tiempos de almacenamiento anteriores a su uso son responsabilidad del usuario. No se deberían superar las 24 horas de 2° a 8 º C, a menos que la reconstitución se haya realizado bajo condiciones controladas y asépticas validadas. Condiciones de almacenamiento Observe las precauciones de conservación en el embalaje exterior. Para consultar las condiciones de almacenamiento del medicamento reconstituido y diluido, véase la sección “Período de validez”.

Manténgase fuera del alcance de los niños. Naturaleza y contenido del envase YONDELIS® se comercializa en un frasco de vidrio incoloro de tipo 1 con un tapón de goma de butilo, cubierto por un precinto de aluminio. Cada frasco contiene 1 mg de trabectedina. Cada caja contiene un frasco. Instrucciones de uso y manipulación Preparación para infusión intravenosa La reconstitución de YONDELIS® y la dilución de la solución reconstituida se deben realizar en condiciones asépticas, de acuerdo con los procedimientos de seguridad recomendados para manipular compuestos citotóxicos. Instrucciones para la reconstitución Cada frasco con 1 mg de trabectedina se reconstituye con 20 ml de agua esterilizada para inyecciones. Se usa una jeringa para inyectar agua esterilizada para inyecciones en el frasco. Se agita el frasco hasta la disolución completa. La solución resultante debe ser límpida y entre incolora y amarillo-parda, básicamente sin partículas visibles. Esta solución reconstituida contiene 0,05 mg/ml de trabectedina. Requiere más dilución y es para un único uso. Instrucciones para la dilución La solución reconstituida se debe diluir con una solución para infusión de cloruro sódico 9 mg/ml (0,9%), o una solución para infusión de glucosa 50 mg/ml (5%). El volumen necesario se calcula de la siguiente forma: Volumen (ml) = ASC (m2) × dosis individual (mg/m2)
0,05 mg/ml ASC = Área de superficie corporal Se debe retirar la cantidad necesaria de solución del frasco y agregarla a una bolsa de infusión que contenga 500 ml de solución salina normal 0,9% para infusión o solución de dextrosa 5% para infusión, si se va a administrar mediante un catéter venoso central. Si el acceso venoso central no es posible y hace falta usar un catéter venoso periférico, la solución reconstituida se debe diluir aún más, en una bolsa de infusión que contenga ≥ 1000 ml de solución salina normal, 0,9% para infusión o solución de dextrosa 5% para infusión. Tras la administración de la infusión de PLD, se debe enjuagar bien el catéter intravenoso con dextrosa 5% en agua (D5W) antes de administrar YONDELIS®. La PLD no se debe mezclar con solución salina. Los productos farmacéuticos parenterales se deben inspeccionar visualmente en busca de partículas o coloración, antes de su administración, siempre que la solución y el envase lo permitan. Después de la reconstitución y dilución, se ha demostrado la estabilidad química y física del producto durante 30 horas hasta una temperatura de 25° C. La solución reconstituida debe diluirse y usarse inmediatamente. El tiempo total entre la reconstitución inicial y el final del tratamiento no debe superar las 30 horas. Instrucciones para la manipulación YONDELIS® es un medicamento citotóxico anticancerígeno y, al igual que con otros compuestos potencialmente tóxicos, se debe tener precaución durante su manipulación. Se deben seguir los procedimientos para la adecuada manipulación y eliminación de medicamentos

citotóxicos. YONDELIS® se debe manipular y eliminar igual que otros medicamentos anticancerígenos. El contacto accidental con la piel, los ojos o las mucosas, se debe tratar con abundantes cantidades de agua. No se han observado incompatibilidades entre YONDELIS® y frascos de vidrio tipo 1, bolsas y catéteres de cloruro de polivinilo (PVC) y polietileno (PE), bolsas de mezcla de PE y polipropileno, depósitos de poliisopreno y sistemas de acceso vascular implantables de titanio o resina plástica. Instrucciones para la eliminación Cualquier producto no utilizado o material de desecho debe eliminarse de acuerdo con las normas locales. En Paraguay: Ante la eventualidad de una sobredosificación recurrir al Centro Nacional de Toxicología del Centro de Emergencias Médicas del M.S.P. y B.S. – Av. Gral. Máximo Santos entre Herminio Giménez y Teodoro S. Mongelós – Teléfonos: (021)220-418; (021)204-800(interno 1011). En Uruguay: Ante sobredosis o intoxicación comunicarse con el Centro de Información y Asesoramiento Toxicológico (CIAT): Tel: 1722. FABRICADO POR Véase el embalaje exterior. BOLIVIA: Importado y Distribuido por: SCHMIDTS PHARMA S.R.L., Av. Mariscal Santa Cruz esquina Yanacocha, Ed. Hansa 6° piso, La Paz, Bolivia. Venta bajo receta médica. Reg. Far. Gabriela Ayala M. Registro Sanitario N°: II-39023/2015 PARAGUAY: Importado y distribuido por VICENTE SCAVONE & CIA., Pastora Céspedes y Don Vicente Scavone, San Lorenzo, Paraguay. Director Técnico: Q.F. María Belén Vega. Reg. Prof. Nº: 5051. Venta Autorizada por el M.S.P. y B.S. Venta bajo receta. Registro Sanitario Nº: 16505-02-EF URUGUAY: Representante: JOHNSON & JOHNSON DE URUGUAY S.A, Av. Italia 7519 piso 3, Montevideo, Uruguay. Tel.: 2901517. Reg. Imp. 651. Ley 15443. D.T.Q.F. Andrea Bonatti. Venta bajo receta profesional. Registro MSP N°: 42450 Centro de atención al cliente: Por correo electrónico (Bolivia, Paraguay y Uruguay): [email protected] Por teléfono: Bolivia: 800-100-990. Paraguay: 00980-0521-0040. Uruguay: 000-405-296638. FECHA DE REVISIÓN DEL TEXTO 27 de febrero de 2015, basada en CCDS del 26 de febrero de 2015