nociones de farmacocinÉticas30f9b4ac704b1040.jimcontent.com/download/version... · hasselbach, la...
TRANSCRIPT

NOCIONES DE FARMACOCINÉTICA
Q.F. NIDIA JACKELINE HERNÁNDEZ ZAMBRANO

DESARROLLO DE FÁRMACOS
FÁRMACO EN EL MERCADO
ENSAYOS CLÍNICOS
Baja biodisponibilidad Escasa distribución intracelular Inactivación elevada
Farmacocinética desfavorable
El éxito de un tratamiento farmacológico depende en gran medida del régimen posológico utilizado, pero la selección del mismo se ve dificultada por las variaciones o diferencias interindividuales en el perfil, tanto farmacocinético como farmacodinámico, del medicamento.
evaluación

FARMACOCINETICA
• Es la rama de la Farmacología que estudia el paso de las drogas a través del organismo en función del tiempo y la dosis.
Es el estudio de la evolución temporal de las concentraciones y cantidades de fármaco y sus metabolitos en los diferentes fluidos, tejidos del organismo, así como el estudio de la evolución de la respuesta farmacológica y la construcción de modelos adecuados para interpretar los datos obtenidos

Es importante tener en cuenta que la cantidad del fármaco que hay en el organismo no permanece estática, sino que varía con el tiempo. Por tanto el curso temporal de la cantidad de fármaco depende de la influencia conjunta de los procesos de absorción, distribución, metabolismo y excreción.

PROCESOS FARMACOCINÉTICOS BÁSICOS
Concentración mínima eficaz (CME): representa el umbral a partir del cual comienza a observarse un efecto Concentración mínima tóxica (CMT): es la concentración a partir de la cual se producen efectos tóxicos Entre ambos puntos se establece el índice terapéutico (IT) que corresponde a las concentraciones entre las cuales el fármaco tiene un efecto sobre el organismo. Respecto al efecto se observa un período de latencia (PL) que representa el tiempo que transcurre desde la administración del fármaco hasta que éste comienza a actuar El área bajo la curva (AUC): representa la cantidad de fármaco que ha habido en la sangre durante un periodo de tiempo (se relaciona con la biodisponibilidad.
T 1/2: tiempo necesario para que la concentración plasmática de un fármaco presente en un organismo se reduzca al 50% Se necesitan aprox. 5 t ½ para eliminar 97% de la dosis inicial. Si un farm. Tiene t ½ de 1h, se eliminará en aprox. 5h Sirve para determinar la frecuencia con que se debe administrar un fármaco

La absorción: proceso por el cual el fármaco entra en el organismo.
La distribución: con lo que se logra que el fármaco llegue primero del lugar de absorción a la circulación sistémica y desde ella a los tejidos. Esto se logra atravesando diversas membranas para alcanzar la circulación sistémica y para alcanzar de esta al liquido intersticial, llegando al interior de las células e incluso a estructuras intracelulares.
La eliminación: El fármaco puede ser eliminado por metabolismo (principalmente hepático) o por excreción del fármaco inalterado en la orina, bilis u otro. En algunos casos el metabolismo puede producir metabolitos activos.
La concentración que alcanza un fármaco en el lugar de acción depende de:
PROCESOS FARMACOCINÉTICOS BÁSICOS

Paso de las moléculas del medicamento desde su punto de administración a la sangre.
ABSORCIÓN
La gran mayoría de fármacos son moléculas pequeñas, con PM menor de 1000 Da y atraviesan las membranas por difusión, en su estado sin carga.
El pH del entorno influye en el proceso de absorción. Si se considera el pH fisiológico (7.4) y que los fármacos son ácidos o bases débiles o anfóteros, la absorción no será constante, pues es la forma no ioizada la que difunde a través de las membranas.
El grado de ionización se determina con la ecuación de Henderson-Hasselbach, la cual relaciona el pKa del fármaco con el pH del medio
Según esta ecuación, un fármaco podrá existir en la forma ionizada cuando esté expuesto a entornos químicos de pH opuestos al suyo.

Propiedades y variables que influyen en la absorción
Propiedades químicas Naturaleza química Peso molecular Solubilidad solubilidad
lipídica del medicamento
Coeficiente de partición
Variables fisiológicas Motilidad GI pH en el lugar de absorción Área de la superficie
absorbente Flujo sanguíneo Eliminación presistémica Ingesta con o sin alimentos Vía de administración

Representa el % de la dosis del medicamento administrada que alcanza la circulación sistémica en forma inalterada.
La vía parenteral es la única que garantiza que la dosis de medicamento llegue en su totalidad al sitio de acción
MECANISMOS DE TRANSPORTE:
BIODISPONIBILIDAD
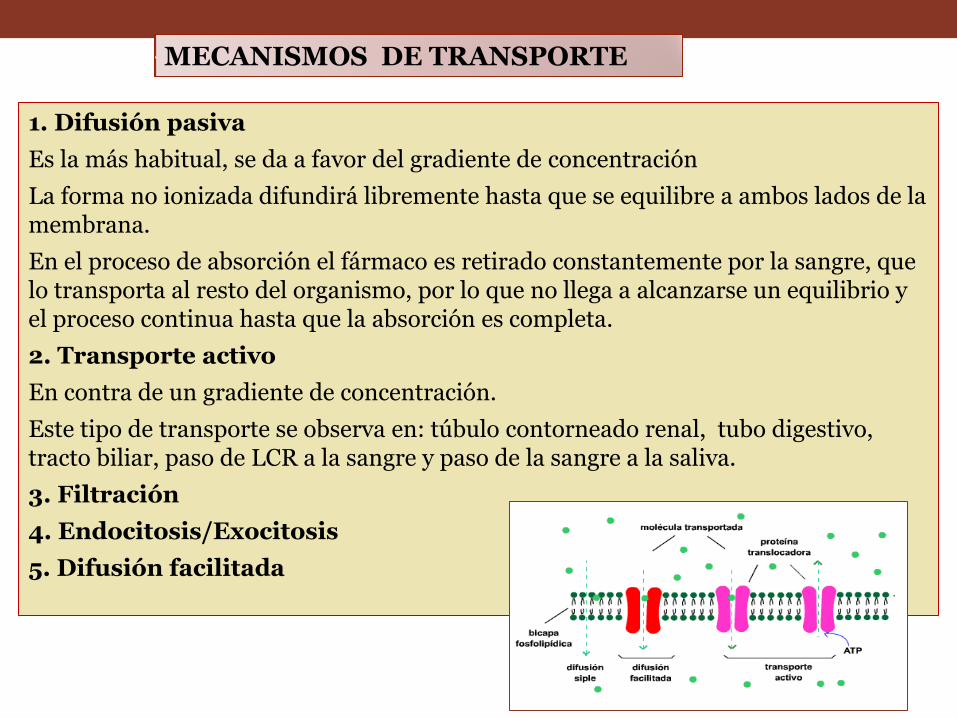
1. Difusión pasiva
Es la más habitual, se da a favor del gradiente de concentración
La forma no ionizada difundirá libremente hasta que se equilibre a ambos lados de la membrana.
En el proceso de absorción el fármaco es retirado constantemente por la sangre, que lo transporta al resto del organismo, por lo que no llega a alcanzarse un equilibrio y el proceso continua hasta que la absorción es completa.
2. Transporte activo
En contra de un gradiente de concentración.
Este tipo de transporte se observa en: túbulo contorneado renal, tubo digestivo, tracto biliar, paso de LCR a la sangre y paso de la sangre a la saliva.
3. Filtración
4. Endocitosis/Exocitosis
5. Difusión facilitada
MECANISMOS DE TRANSPORTE

Liposolubilidad e hidrosolubilidad
Forma farmacéutica
Biodisponibilidad oral de soluciones
Biodisponibilidad oral de comprimidos.
pH gastrointestinal
Superficie de la mucosa GI
Gran superficie de absorción
La mayoría de los fármacos se absorben en intestino delgado.
Riego sanguíneo
Vaciamiento gástrico
Es retardado por ingesta de: comida grasa, bebida ácida, anticolinérgicos
Biotransformación intestinal y hepática.
Enzimas digestivas
Enzimas hepáticas
Factores que afectan la biodisponibilidad (enteral)

DISTRIBUCIÓN
Proceso mediante el cual el fármaco se incorpora desde la circulación sanguínea a los diferentes órganos y tejidos corporales
Los proceso de distribución son, en consecuencia, procesos cinéticos en los que se realiza una transferencia, en general reversible, del fármaco entre distintos compartimientos corporales
VOLUMEN DE DISTRIBUCIÓN
Concepto introducido para establecer un parámetro matemático que relaciona la cantidad del fármaco en el organismo con la concentración plasmática.
Qt = cantidad del fármaco en el organismo
Ct = concentración a un tiempo determinado Vd= Qt/Ct
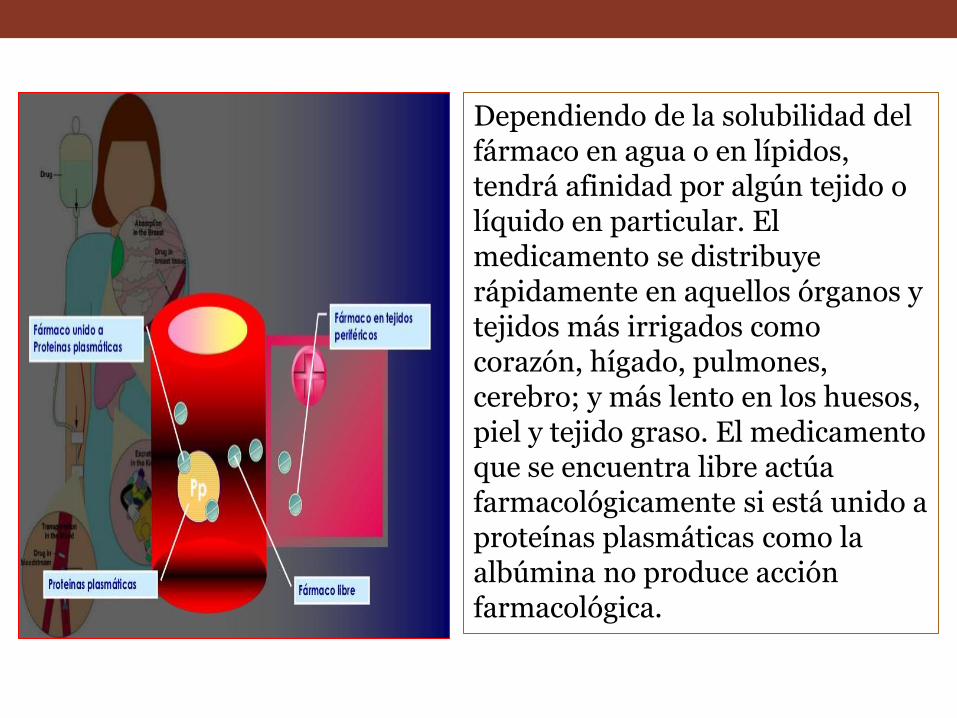
Dependiendo de la solubilidad del fármaco en agua o en lípidos, tendrá afinidad por algún tejido o líquido en particular. El medicamento se distribuye rápidamente en aquellos órganos y tejidos más irrigados como corazón, hígado, pulmones, cerebro; y más lento en los huesos, piel y tejido graso. El medicamento que se encuentra libre actúa farmacológicamente si está unido a proteínas plasmáticas como la albúmina no produce acción farmacológica.
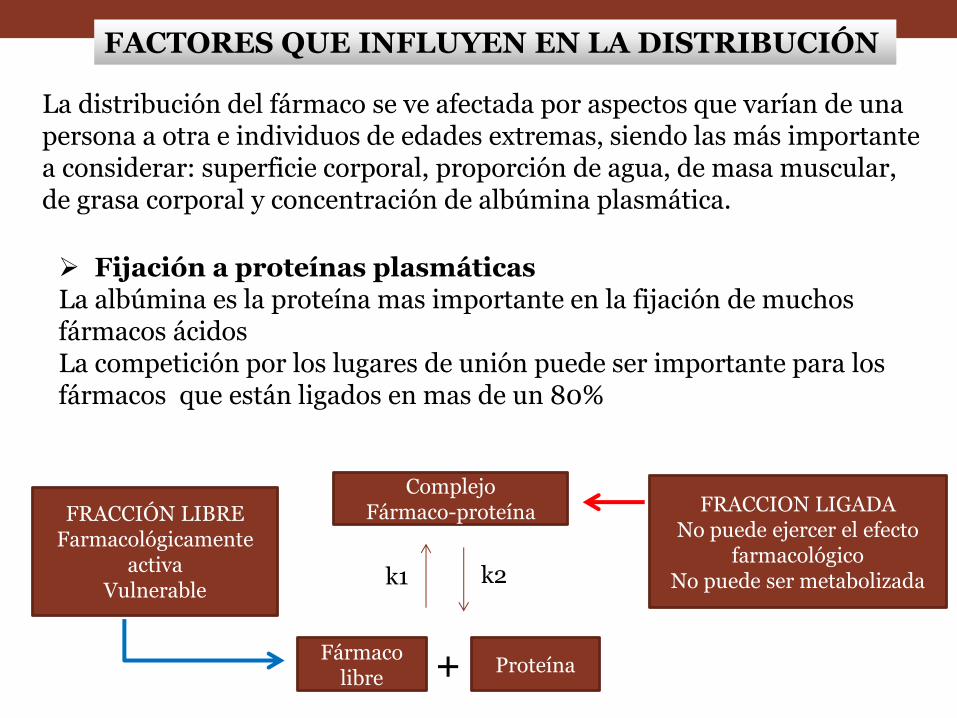
La distribución del fármaco se ve afectada por aspectos que varían de una persona a otra e individuos de edades extremas, siendo las más importante a considerar: superficie corporal, proporción de agua, de masa muscular, de grasa corporal y concentración de albúmina plasmática.
FACTORES QUE INFLUYEN EN LA DISTRIBUCIÓN
Fijación a proteínas plasmáticas La albúmina es la proteína mas importante en la fijación de muchos fármacos ácidos La competición por los lugares de unión puede ser importante para los fármacos que están ligados en mas de un 80%
FRACCIÓN LIBRE Farmacológicamente
activa Vulnerable
Complejo Fármaco-proteína
Proteína Fármaco
libre
FRACCION LIGADA No puede ejercer el efecto
farmacológico No puede ser metabolizada
+
k1 k2

P. PLASMATICAS
ALBUMINA: se unen fármacos con pH neutro o ácidos débiles (penicilina)
Glucoproteína α1: se unen fármacos con pH alcalino (eritromicina)
Lipoproteínas: se unen fármacos muy lipofílicos (pesticidas).
Los fármacos con gran afinidad por lipoides llegan primero a Encéfalo y se depositan en panículo adiposo. El tejido adiposo almacena cantidad de fármaco liposoluble y se convierte en sitio de almacenamiento

• Insuficiencia renal
• Enfermedades hepáticas
• Quemaduras severas
• Embarazo
• Edad
• FLUJO SANGUINEO: Órganos menos irrigados: piel, hueso, tejido adiposo, músculo en reposo.
Órganos más irrigados: Cerebro, corazón, hígado, riñón
• SALIDA DEL FÁRMACO DEL
INTERIOR VASCULAR: El fármaco disuelto en sangre se distribuye por todo el organismo, saliendo del compartimento vascular a favor de un gradiente mediante diferentes mecanismos
FACTORES QUE AFECTAN LA FIJACIÓN A PROTEÍNAS
FACTORES QUE INFLUYEN EN LA DISRIBUCIÓN

TRANSFERENCIA DE FÁRMACOS A TRAVÉS DE BARRERAS FISIOLÓGICAS
BARRERA HEMATOENCEFÁLICA
Localizada entre plasma sanguíneo de los vasos cerebrales y el espacio extracelular del encéfalo
BARRERA HEMATOOCULAR
El epitelio de los procesos ciliares es una barrera difícil de atravesar, por ello la mayoría de fármacos no alcanzan sus niveles terapéuticos
BARRERA SNGRE LÍQUIDO
CEFALORAQUIDEO
Localizada a nivel de plexos coroideos e impide el paso de ciertas drogas al LCR, son similares a los capilares intracraneales cubiertos con células responsables de la diferencia de absorción
BARRERA PLACENTARIA
Drogas administradas a la madre pueden ejercer efectos en el feto. Es importante tener en cuenta la administración de fármacos en periodo de organogénesis
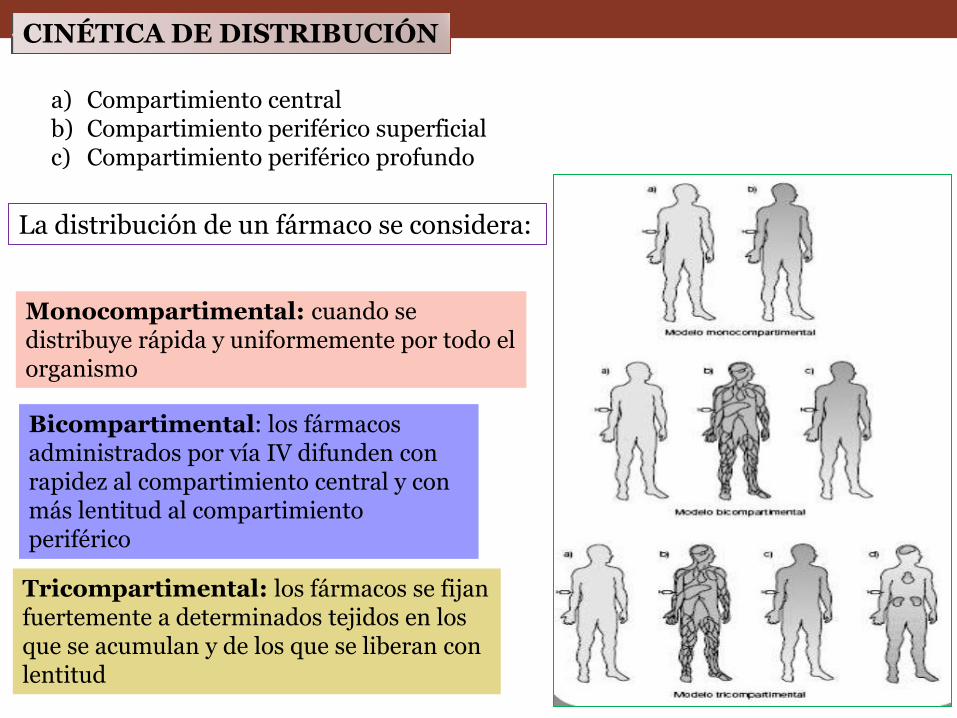
CINÉTICA DE DISTRIBUCIÓN
a) Compartimiento central b) Compartimiento periférico superficial c) Compartimiento periférico profundo
La distribución de un fármaco se considera:
Monocompartimental: cuando se distribuye rápida y uniformemente por todo el organismo
Bicompartimental: los fármacos administrados por vía IV difunden con rapidez al compartimiento central y con más lentitud al compartimiento periférico
Tricompartimental: los fármacos se fijan fuertemente a determinados tejidos en los que se acumulan y de los que se liberan con lentitud

También se denomina biotransformación debido a que implica la transformación biológica de un fármaco en un metabolito inactivo, un compuesto más soluble o un metabolito más potente.
Son los cambios con los cuales los xenobióticos (fármacos) se convierten en sustancias más ionizadas, más polares, menos difusibles y más fácilmente eliminables que la sustancia original. Las reacciones de biotransformación corren a cargo del sistema microsomal, mitocondrias, enzimas solubles, lisosomas y la microflora.
FARMACO ACTIVO
METABOLITO TOXICO
METABOLITO INACTIVO
METABOLITO ACTIVO
Resultado de la biotransformación
metabolismo

El hígado es el órgano principal; en el interior de sus hepatocitos tiene una gran batería de enzimas que son capaces de llevar a cabo reacciones químicas de oxidación, reducción, hidrólisis y de conjugación. Los metabolitos que se producen, son fácilmente eliminados a través de las vías comunes de eliminación como bilis, heces y orina. El metabolismo prolongado de un fármaco genera su acumulación y por consiguiente efectos o respuestas derivadas de la acción prolongada.
LUGARES DE BIOTRANSFORMACIÓN
Estomago
Intestino
Receptores celulares
Sangre
Riñón
SNC, placenta, pulmones
Hígado

Los medicamentos hidrosolubles se metabolizan a nivel hepático lo que varia de acuerdo a la edad; se sabe que la masa hepática se reduce con los años en relación a la superficie total del cuerpo, como así mismo la irrigación y las enzimas hepáticas necesarias para la oxidación de los fármacos. En los niños menores, recién nacidos y lactantes, por inmadurez, el metabolismo se puede ver afectado
Existe una minoría de fármacos que no sufre transformación alguna y son excretados sin modificar

EFECTO DEL PRIMER PASO
Metabolismo del fármaco antes de ingresar a la circulación sistémica
Pared intestinal (CYP3A4)
Sangre portal
Hígado
Reducción de la biodisponibilidad
FARMACO METABOLITO INACTIVO
Liposoluble No polar Activo
+/- polar +/- inactivo
Polar hidrosoluble
Sist. Enz microsomal hepático
FASE I FASE II
Las reacciones del metabolismo tienen lugar en dos fases: Reacciones de fase I Reacciones de fase II

Consisten en reacciones de oxidación, reducción e hidrólisis.
Producen en general un aumento en la polaridad de la molécula y determina:
FASE I (funcionalización)
Adquisición de carga eléctrica
Menor liposolubilidad
Menor ingreso a los tejidos
Menor reabsorción renal
Excreción mas rápida
Inactivación Conversión de un producto inactivo
en otro activo, en cuyo caso el producto original se denomina profármaco
Conversión de un producto activo en otro también activo, cuya actividad puede ser similar o distinta de la del fármaco original
Conversión de un producto activo en otro activo, pero cuya actividad resulta toxica

Oxidación …. Son catalizados por el sistema enzimático denominado citocromo P450
Oxidación alifática
Hidroxilación aromática
N-desalquilacion
O-desalquilación
S-desalquilación
Epoxidación
Desaminación oxidativa
Formación de sulfoxidos
Desulfuración
N-oxidación y N-hidroxilación
Oxidación …mecanismos no microsómicos
Oxidación de alcohol y aldehidos
Oxidación de purinas
Desaminación oxidativa (monoaminooxidasa y diaminooxidasa)
Reducción…..ocurren en la fracción microsómica
Azorreduccion y nitrorreducción
Hidrólisis….se producen en el plasma y en diversos tejidos
Hidrolisis de esteres y amidas
Hidrólisis de enlaces peptídicos, de epoxidos

Citocromo P450

El metabolito procedente de la fase I se acopla a un sustrato endógeno, como el ácido glucorónido, el acido acético y el ácido sulfúrico.
Aumenta el tamaño de la molécula, con la cual casi siempre se inactiva el fármaco y se facilita se excreción.
Glucoronidación
Acetilación
Formación de acido mercaptúrico
Conjugación con sulfato
N, O y S-metilación
Transulfuración
FASE II (reacciones de conjugación)


FISIOLÓGICOS
Edad: mayor susceptibilidad de actividad farmacológica o toxica en personas muy jóvenes y de edad avanzada.
A partir de los 25 años el flujo sanguíneo hepático va disminuyendo
GENÉTICOS Y ÉTNICOS
Variaciones metabólicas de una raza a otra y de una especie a otra.
Los factores genéticos influyen en las concentraciones enzimáticas.
Por lo general los defectos se trasmiten como rasgos autosómico recesivo.
FACTORES QUE MODIFICAN EL METABOLISMO DE FARMACOS
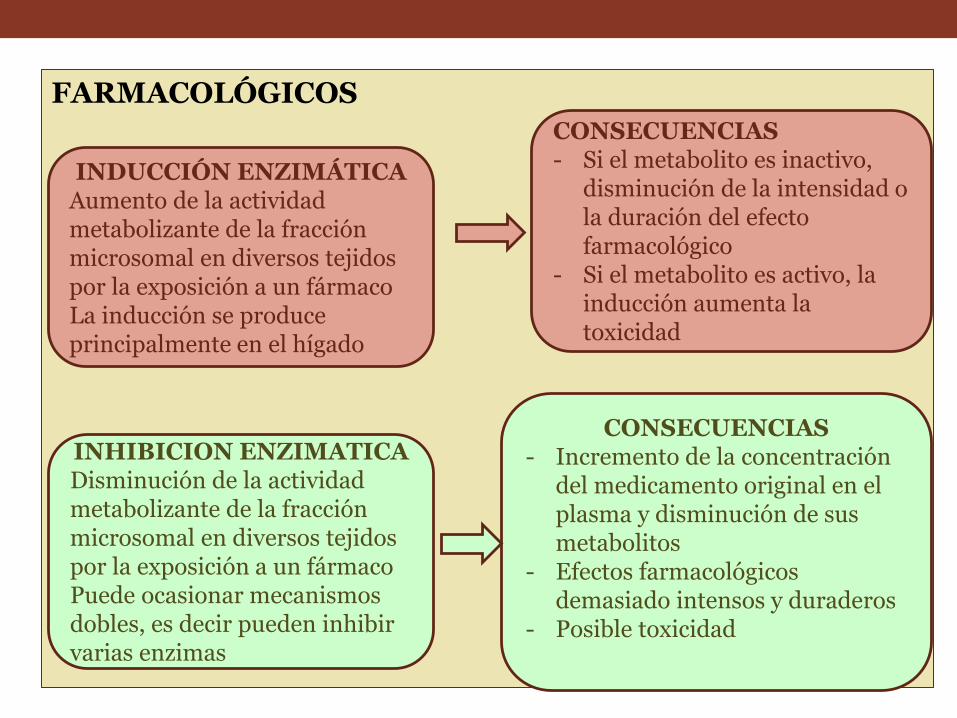
FARMACOLÓGICOS
INDUCCIÓN ENZIMÁTICA Aumento de la actividad metabolizante de la fracción microsomal en diversos tejidos por la exposición a un fármaco La inducción se produce principalmente en el hígado
CONSECUENCIAS - Incremento de la concentración
del medicamento original en el plasma y disminución de sus metabolitos
- Efectos farmacológicos demasiado intensos y duraderos
- Posible toxicidad
CONSECUENCIAS - Si el metabolito es inactivo,
disminución de la intensidad o la duración del efecto farmacológico
- Si el metabolito es activo, la inducción aumenta la toxicidad
INHIBICION ENZIMATICA Disminución de la actividad metabolizante de la fracción microsomal en diversos tejidos por la exposición a un fármaco Puede ocasionar mecanismos dobles, es decir pueden inhibir varias enzimas

PATOLÓGICOS
Hepatitis alcohólica
Cirrosis alcohólica activa o inactiva
Hepatitis activa o crónica
Cirrosis biliar
Hepatitis viral o fármaco-inducido
Enfermedades cardiacas
Ca de pulmón
Envenenamiento por metales pesados
Disfunción tiroidea.
AMBIENTALES Y DIETA
Los fumadores metabolizan algunos fármacos con mayor rapidez que los no fumadores
Verduras crucíferas (lechuga, nabos, rábanos) aumentan reacciones de oxidación y de glucoronidacion.
Jugo de toronja, inhibe la CYP3A4.

Paso de un fármaco del medio interno al medio externo
EXCRECIÓN RENAL
Es la más importante para la eliminación de fármacos.
Con abundante irrigación, recibe el 25% del gasto cardiaco.
Es relevante cuando se eliminan de forma preferente por esta vía, en forma inalterada o como metabolitos activos
Por el contrario, es poco importante en los fármacos que se eliminan principalmente por metabolismo, aun cuando una parte sustancial de sus metabolitos inactivos se eliminen por el riñón
Implica:
Filtración glomerular
Secreción tubular
Reabsorción tubular
EXCRECIÓN
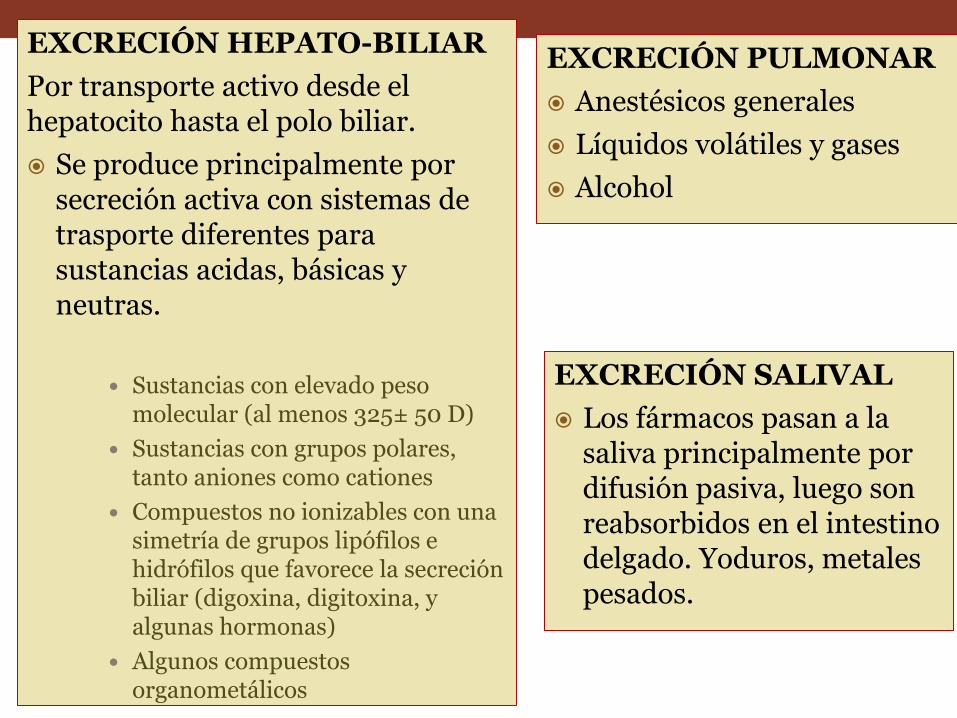
EXCRECIÓN HEPATO-BILIAR
Por transporte activo desde el hepatocito hasta el polo biliar.
Se produce principalmente por secreción activa con sistemas de trasporte diferentes para sustancias acidas, básicas y neutras.
Sustancias con elevado peso molecular (al menos 325± 50 D)
Sustancias con grupos polares, tanto aniones como cationes
Compuestos no ionizables con una simetría de grupos lipófilos e hidrófilos que favorece la secreción biliar (digoxina, digitoxina, y algunas hormonas)
Algunos compuestos organometálicos
EXCRECIÓN PULMONAR
Anestésicos generales
Líquidos volátiles y gases
Alcohol
EXCRECIÓN SALIVAL
Los fármacos pasan a la saliva principalmente por difusión pasiva, luego son reabsorbidos en el intestino delgado. Yoduros, metales pesados.

EXCRECIÓN GASTRICA
Las bases débiles pasan del plasma al jugo gástrico
EXCRECIÓN INTESTINAL
Algunos fármacos pueden pasar de la sangre a la luz intestinal, por difusión pasiva, eliminándose por las heces.
EXCRECIÓN CUTANEA
A través de la piel y el sudor
Se eliminan sustancias como el alcohol, antipirina.
EXCRECIÓN LACRIMAL
A través de las lágrimas y luego pasan por el conducto lagrimal pasan a las fosas nasales.
EXCRECIÓN POR LA GLANDULA MAMARIA
Algunos fármacos (liposolubles o bases débiles) llegan a la secreción láctea que tiene un pH más ácido que el plasma.
En el lactante pueden originar reacciones idiosincrásicas y toxicas.

FARMACOCINÉTICA……..