modelaciÓn de la calidad del agua · en un periodo mayor o menor, podría verse reflejada en la...
TRANSCRIPT

MODELACIÓN DE LA CALIDAD DEL AGUA
UNIVERSIDAD POLITÉCNICA DE VALENCIA
DEPARTAMENTO DE INGENIERÍA HIDRÁULICA Y MEDIO AMBIENTE
ESCUELA TÉCNICA SUPERIOR DE INGENIEROS DE CAMINOS, CANALES Y
PUERTOS
Miguel Martín Monerris. Departamento de Ingeniería Hidráulica y Medio Ambiente.
Universidad Politécnica de Valencia.
Paula Marzal Doménech. Departamento de Ingeniería Química. Universitat de València.

A la memoria del Profesor Doctor D. José Manuel Benet Granell

Introducción 1
1. INTRODUCCIÓN.
1.1 Contaminación de las aguas.
La contaminación del agua debe entenderse como un concepto relativo, asociado a las
características físicas, químicas y biológicas que impiden o dificultan su uso según las aplicaciones
a las que vaya destinada. En la naturaleza, el agua en estado líquido no se encuentra pura, sino que
va incorporando diversas sustancias desde su caída a la superficie de la tierra como agua de lluvia
hasta que llega al mar, a lo largo del ciclo del agua. Durante este recorrido, el agua va disolviendo
gases y compuestos minerales, y va incorporando partículas en suspensión de naturaleza inorgánica
y orgánica, etc. Finalmente, el agua es utilizada por el hombre para el suministro municipal y para
usos agrícolas, industriales y recreativos, impurificándose de forma considerable. Por ello, en
sentido estricto, el agua que se encuentra a disposición del ser humano está contaminada, y será su
aptitud o inaptitud para un uso determinado lo que determinará su calificación de contaminada para
ese uso.
No obstante, no debe entenderse el término "uso" en su sentido estrictamente utilitarista
para el ser humano (bebida, baño, riego, aplicación industrial, etc.), sino que el concepto debe ser
más amplio haciendo referencia, por ejemplo, al mantenimiento de las condiciones naturales de
forma que no se perturbe el funcionamiento de los ecosistemas.
El origen de la contaminación de las aguas hay que buscarlo en procesos naturales pero,
sobre todo, en la actividad humana. La contaminación inducida por el hombre es un fenómeno
antiguo si bien, la industrialización y el crecimiento demográfico y urbanístico han agravado
considerablemente el problema en muchas regiones. A pesar de su origen común, se puede
establecer una distinción entre los principales problemas de contaminación en aguas superficiales y
en aguas subterráneas.
1.1.1 Principales problemas en aguas subterráneas.
Los principales problemas de contaminación de las aguas subterráneas consisten en la
presencia de especies químicas de naturaleza orgánica o inorgánica disueltas en el agua y en la
acumulación de dichos compuestos sobre la matriz sólida de los acuíferos. El origen de la

Modelos de Calidad de Aguas 2
contaminación de las aguas subterráneas se encuentra en algunos procesos naturales
(mineralización por lixiviación, concentración por evapotranspiración) y especialmente, en la
actividad humana. Es, en gran parte, consecuencia del uso exhaustivo de productos químicos en los
países desarrollados, con la consiguiente descarga, intencionada o accidental, de sustancias
contaminantes. Paralelamente, la contaminación de las aguas superficiales puede provocar una
transferencia de agentes contaminantes hacia las aguas subterráneas, ya sea por los procesos de
infiltración natural o por los de infiltración forzada por la extracción de aguas subterráneas.
De acuerdo con Ward y col. (1985), las fuentes de contaminación de origen humano se
pueden agrupar en cuatro categorías:
• Operaciones de eliminación de residuos que utilizan el subsuelo como receptor de
agentes contaminantes: fosas sépticas, vertederos de residuos peligrosos, estanques y
lagunajes de evaporación de residuos industriales, instalaciones de tratamiento de
residuos sobre el terreno, sistemas de deposición en profundidad...
• Actividades industriales y comerciales en las que se manejan grandes cantidades de
sustancias químicas que, accidentalmente, pueden ser descargadas por fugas y derrames
durante su transporte, almacenamiento y utilización.
• Uso extensivo de productos químicos para la agricultura (fertilizantes, herbicidas,
pesticidas).
• Recarga artificial, directa o indirecta, de los acuíferos con aguas contaminadas.
Otro problema asociado a la contaminación de las aguas subterráneas se encuentra en la
dificultad para localizar las fuentes de contaminación. Ello se debe, en primer lugar, a que la
contaminación tiene lugar a nivel subterráneo y no se detecta hasta que alcanza algún pozo de
extracción que, además, puede estar situado a gran distancia de la zona de entrada de
contaminantes al acuífero. Por otro lado, la mayoría de los contaminantes más frecuentemente
encontrados en las aguas subterráneas son incoloros, inodoros y están en niveles bajos de
concentración, no apareciendo signos alarmantes de polución (color, olor, presencia de peces
muertos, etc.) como ocurre en muchas ocasiones en las aguas superficiales.
Hasta alcanzar el acuífero, los compuestos depositados sobre la superficie del terreno
percolan a través de distintas zonas dónde se dan unas condiciones físico-químicas y biológicas
específicas que pueden originar una transformación de los contaminantes (procesos de oxidación

Introducción 3
reducción, fijación microbiana, adsorción, formación de complejos con la materia orgánica
insoluble, etc.). Estas transformaciones suelen traducirse en una atenuación o disminución de la
concentración de las especies contaminantes. Una vez en el acuífero, los contaminantes se
desplazarán con el agua subterránea formando, en condiciones ideales, una pluma elíptica de
contaminación. Los fenómenos de dispersión provocan la extensión de la pluma contaminante no
solo en la dirección del flujo sino también en las direcciones transversales, originando un
ensanchamiento de la misma. La atenuación de la contaminación en el acuífero se debe,
fundamentalmente, a fenómenos de dilución, precipitación, adsorción, volatilización, filtración
mecánica y neutralización (Pye y Kelly, 1984). Si bien dicha atenuación se considera favorable
desde el punto de vista de la calidad del agua hay que tener en cuenta que un acuífero es un sistema
dinámico constituido tanto por el agua como por el propio suelo, y por lo tanto, la retención de
contaminantes en la matriz del suelo constituye, en si misma, una polución directa del acuífero, que
en un periodo mayor o menor, podría verse reflejada en la calidad del agua.
En cuanto a las especies contaminantes, desde el punto de vista de la salud pública la
atención se centra, fundamentalmente, en los compuestos orgánicos y en un reducido grupo de
sustancias inorgánicas. Entre los compuestos orgánicos los más preocupantes son aquellos
potencialmente cancerígenos, mutagénicos o simplemente tóxicos en pequeña concentración. La
lista es muy extensa, destacando, por la frecuencia de su utilización, los disolventes orgánicos
(benceno, tolueno, metanol, heptano, trihalometanos, tetracloruro de carbono, etc.) y los pesticidas,
herbicidas y fungicidas (hidrocarburos clorados, organofosfatos, carbamatos y derivados de la
urea). La atenuación de los compuestos orgánicos puede tener lugar por adsorción o por
degradación biológica. Actualmente se estudian también los problemas asociados al
desplazamiento en el subsuelo de hidrocarburos líquidos en fase no acuosa (NAPL, non-aqueous
phase liquids).
Entre las sustancias inorgánicas, los metales pesados constituyen el principal foco de
atención. La presencia de estas sustancias en las aguas subterráneas viene condicionada por su
movilidad en el sistema sólido/solución que a su vez depende de múltiples factores como son el
pH, la temperatura, el potencial redox, la posibilidad de adsorción, su solubilidad y la composición
de la solución acuosa.
Mención aparte merecen los problemas de salinización de acuíferos por la sobreexplotación
de los mismos y por fenómenos de intrusión salina en acuíferos costeros, así como la

Modelos de Calidad de Aguas 4
contaminación por nitrógeno de las aguas subterráneas. La importancia del nitrógeno como
contaminante se debe, por una parte, a que es un componente mayoritario de los fertilizantes y de
los residuos humanos y animales, y por otra, a que suele presentarse en formas solubles y
difícilmente adsorbibles. Así, se pueden citar como principales fuentes de nitrógeno en las aguas
subterráneas la infiltración de aguas residuales urbanas, de agua de riego procedente de campos
fertilizados con abonos nitrogenados, de aguas residuales procedentes de concentraciones
ganaderas estabuladas y de lixiviados de vertederos de residuos sólidos, así como la fijación del
nitrógeno atmosférico. Bajo la perspectiva de la calidad del agua, los compuestos nitrogenados más
importantes son el nitrógeno orgánico, amoníaco, nitrito, nitrato, urea y nitrógeno gaseoso. Dado
que la forma más móvil del nitrógeno en el agua es el ión nitrato, el término contaminación por
nitratos se utiliza a menudo para englobar el más genérico de contaminación por sustancias
nitrogenadas. Los riesgos potenciales de la presencia de nitrato en el agua de bebida para la salud
humana incluyen metahemoglobinemia, cáncer y malformaciones en recién nacidos. Los problemas
para la salud pública pueden ser especialmente serios en regiones en las que confluyen dos
factores: el desarrollo de una gran actividad agrícola y un uso importante del agua subterránea para
el abastecimiento de poblaciones. La situación se agrava si además existe un uso sistemático para
riego de agua bombeada del acuífero y de agua residual sin depurar.
1.1.2 Principales problemas en aguas superficiales.
A continuación se realiza un breve resumen, a modo de introducción, de los principales
problemas relacionados con la contaminación en aguas superficiales. Algunos de ellos serán objeto
de un análisis más detallado durante el desarrollo de los temas de estos apuntes.
Salinización. Se produce cuando la tasa de deposición de las sales que transportan las aguas de
riego supera la velocidad de arrastre de la sal por el curso del agua. Es un problema que se puede
plantear debido a la reutilización de aguas residuales parcialmente tratadas, puesto que estas aguas
han sufrido un pequeño incremento de sales disueltas durante su uso doméstico o industrial. Es más
habitual que se produzca debido al riego con aguas de acuíferos sobreexplotados, con altas
concentraciones de sales disueltas.
Acidificación de lagos. En lagos de baja alcalinidad, la lluvia ácida originada por la reacción del
vapor de agua con óxidos de nitrógeno y azufre provoca la acidificación de los lagos. También
puede tener lugar por deposición seca en las proximidades de los focos de emisión de los
contaminantes atmosféricos citados. El fenómeno se da en países industrializados y su corrección

Introducción 5
requiere el control y supresión de las emisiones a la atmósfera de óxidos de nitrógeno y azufre.
Reducción de los niveles de oxígeno disuelto en el agua. Diversos factores afectan de forma
negativa a los niveles de oxígeno disuelto de un sistema natural: la temperatura, la turbidez, la
materia orgánica en descomposición, la proliferación de algas, etc.
Eutrofización. La ruptura del equilibrio de un ecosistema de base planctónica por la variación de
las condiciones naturales de temperatura, luminosidad o disponibilidad de nutrientes puede
ocasionar una proliferación exagerada de algas. Dicha proliferación puede dar lugar a problemas
puntuales como la disminución del oxígeno disuelto durante la noche, o más generales si la
descomposición de las algas muertas agota el oxígeno. El aumento de la turbidez, el incremento del
pH, la generación de sustancias tóxicas por parte de algunos microorganismos, entre otras, son
consecuencias importantes de los procesos de eutrofización. La solución de los problemas de
eutrofización pasa, fundamentalmente, por la limitación de la entrada de nutrientes principales
(nitrógeno y fósforo) y la eliminación de los presentes en el sedimento.
Disminución de la biodiversidad. Relacionado con el problema de la eutrofización, aunque no de
forma exclusiva, cuando un sistema llega a un estado de hipereutrofización tan solo unas pocas
especies pueden vivir, y las que lo hacen proliferan de una forma notable.
Degradación y sustitución de especies. La contaminación de las aguas puede provocar la
desaparición de especies y la aparición de aquellas que se adaptan mejor a las nuevas condiciones.
Así por ejemplo, el vertido de agua caliente aumenta la temperatura de las aguas naturales
haciéndolas inhabitables para las especies que hasta ese momento allí existían, o el aumento de los
sólidos suspendidos en el agua provoca la disminución de la luminosidad del fondo marino. Este
último factor es, en parte, responsable de la degradación de las praderas de Posidonia Oceánica en
el Mediterráneo cuyo espacio natural está siendo ocupado por otros tipos de algas (Caulerpa
Prolifera).
Adaptación de las especies. La adaptación de las especies a la nueva situación puede también
provocar efectos altamente indeseables. Por ejemplo, la pérdida de transparencia del agua del mar
en zonas costeras hace desplazarse a cierto tipo de algas desde su profundidad normal a otras
inferiores. De este modo quedan más expuestas a la acción del oleaje y pueden ser arrancadas de
forma masiva, acumulándose en las playas. Su descomposición puede generar problemas de anoxia

Modelos de Calidad de Aguas 6
en las aguas costeras.
Contaminación de sedimentos. Los sedimentos de ríos, lagos y estuarios de zonas industrializadas
y/o densamente pobladas suelen presentar altas concentraciones de metales pesados, compuestos
orgánicos tóxicos, nutrientes y materia orgánica. En estos casos hay que tener especial cuidado
puesto que cualquier acción no controlada podría movilizar los compuestos tóxicos retenidos en los
sedimentos. En particular, el dragado de este tipo de sedimentos implica, además, la necesidad de
disponerlos en lugares adecuados, puesto que no pueden ser utilizados para el allanamiento de
terrenos agrícolas o zonas urbanas.
Bioacumulación de compuestos tóxicos. Numerosos compuestos orgánicos tóxicos y metales
pesados son ingeridos por seres vivos a través de la cadena alimenticia, acumulándose en los
tejidos grasos y musculares. estos compuestos debilitan las defensas de los organismos y provocan
numerosas enfermedades. Como último eslabón, pueden alcanzar al ser humano produciendo
lesiones y hasta la muerte.
Transmisión de enfermedades. Las aguas contaminadas por residuos de origen animal son
vehículo de agentes infecciosos (protozoos, bacterias y virus) que pueden provocar un gran número
de enfermedades en los seres vivos (conjuntivitis, poliomielitis, hepatitis, tifus, salmonelosis,
cólera, etc.). Este tipo de enfermedades de transmisión hídrica son todavía muy frecuentes en los
países del Tercer Mundo debido a la falta de condiciones de salubridad del agua. La reutilización
de aguas residuales tratadas para riego debe tener en cuenta la posible presencia de estos agentes
infecciosos. El vertido de aguas residuales a través de emisarios submarinos también puede ser otro
foco potencial de enfermedades.
Impactos puntuales. El vertido de un contaminante tóxico provocado de forma accidental o
intencionada puede provocar la degradación de amplias zonas. Ejemplos muy recientes de ello son
los vertidos de las balsas de residuos mineros de Aznalcóllar (entorno de Doñana, España) y Baia
Mare (cuenca del Danubio, Rumania). Otros ejemplos clásicos son los vertidos de hidrocarburos
debido a accidentes de petroleros o los producidos por la limpieza inadecuada de los buques.
Las consecuencias de un vertido a un río pueden ser no sólamente directas, relacionadas con
los efectos inmediatos de las sustancias vertidas sobre el medio, sino indirectas, relacionadas con
las actividades de descontaminación que no se pueden llevar a cabo. Un ejemplo clasico de esto
último se dió en el año 1986 cuando la empresa Sandoz sufrió un accidente que produjo el vertido

Introducción 7
de 30000 kg de compuestos tóxicos (pesticidas organoclorados y organofosforados) en el río Rhin.
Las estaciones de tratamiento de aguas que estaban depurando las aguas del río hubieron de cerrar
el paso de las aguas a su interior con el fin de proteger los sistemas biológicos de tratamiento. Con
ello, a la acción directa del contaminante había que añadir la de todos aquellos que no eran
eliminados en las estaciones de tratamiento.
1.2 Gestión de la calidad de las aguas.
Con el fin de preservar la salud pública y de limitar en lo posible los impactos ambientales
provocados por la contaminación de las aguas se establecen normativas para mantener la calidad en
el medio natural. Las normativas se encaminan en dos sentidos:
• Fijación de unas normas de emisión (límites de vertido).
• Establecimiento de unos objetivos de calidad.
La fijación de límites de vertido establece la cantidad máxima de contaminante que puede
ser vertido suponiendo que éste no producirá efectos indeseables sobre el medio natural. En la
fijación de los límites no se tiene en cuenta la evolución del contaminante (su acumulación,
transformación, etc.) ni su impacto en el medio una vez haya sido vertido.
El establecimiento de objetivos de calidad ha de realizarse teniendo en cuenta tanto la
cantidad de contaminante vertido como su comportamiento posterior en el medio. Precisamente
para alcanzar unos objetivos de calidad concretos, es necesario poder determinar cuál será la
evolución de un contaminante vertido en un sistema, es decir, cuál será la evolución de su
concentración con el tiempo, y cómo puede interferir con el resto de sustancias presentes en el
medio. En este sentido, se hace necesario utilizar modelos matemáticos de simulación que permitan
predecir la evolución a largo plazo del sistema en su conjunto ante una acción determinada. Estos
modelos se conocen genéricamente como "modelos de calidad" o "modelos de transporte de
contaminantes".
La validez de la predicción vendrá determinada por la capacidad del modelo propuesto de
reflejar el comportamiento del sistema. Por ello, es necesario, por una parte, conocer todos los
fenómenos que afectan al comportamiento de las distintas sustancias presentes en el sistema y, por
otra, desarrollar un conjunto de ecuaciones que describan estos fenómenos.

Modelos de Calidad de Aguas 8
El comportamiento de cualquier sustancia en el medio está determinada fundamentalmente
por dos factores:
• Su reactividad.
• La velocidad de su transporte físico en el medio.
La reactividad de una sustancia comprende el conjunto de transformaciones físico-químicas
y biológicas que afectan a la sustancia. Son de especial importancia las reacciones de degradación
y los procesos que provocan una transferencia de solutos entre las fases acuosa y sólidas o acuosa y
gaseosa del medio (precipitación, disolución, adsorción, desorción, sedimentación de materia
suspendida). Si un contaminante no experimenta procesos reactivos en el medio se dice que es un
contaminante conservativo, y su evolución se deberá exclusivamente a su desplazamiento con la
masa global de agua.
La velocidad de transporte físico en el medio vendrá determinada, fundamentalmente, por el
movimiento del agua. Para conocer las características del flujo del agua en el medio se realizan
mediciones o se utilizan los denominados "modelos de flujo" o modelos hidrodinámicos, que
proporcionan el campo de velocidades en el sistema y su variación temporal.
La aplicación de un modelo de calidad para evaluar el impacto que puede producir
cualquier compuesto en un medio requiere:
• Fijar un volumen de control claramente definido. Este volumen de control será el sistema
en estudio o una parte de él. Puede ser tan pequeño como una finísima lámina transversal
de agua de un río o tan grande como un océano.
• Conocer de la forma más exhaustiva posible de las entradas y salidas a través de las
fronteras del volumen de control.
• Conocer las características del transporte en el interior del volumen de control y a través
de sus fronteras. La capacidad de mezcla en el interior del sistema debe ser conocida, ya
sea por mediciones o por estimaciones basadas en la hidrodinámica del sistema.
• Conocer las cinéticas de reacción (físicas, químicas y biológicas) y las constantes de
velocidad en el interior del volumen de control.
La aplicación concreta de un modelo de calidad en un sistema natural dependerá, por lo

Introducción 9
tanto, del tipo de contaminante y del tipo de sistema natural objeto del estudio.
Los resultados de la simulación permitirán conocer, para cada contaminante:
• Distribución espacial y temporal de la concentración esperada en el medio natural como
consecuencia de una serie concreta de vertidos.
• Fracción del vertido original que desaparece por los procesos de transformación y
transporte.
• Persistencia en el medio: tiempo requerido para la limpieza del sistema si el vertido
cesara.

Modelos de Calidad de Aguas 10

Balances de Materia en Sistemas Naturales 11
2. BALANCES DE MATERIA EN SISTEMAS NATURALES.
2.1 Enunciado general.
El núcleo de cualquier modelo de calidad de aguas son los balances de materia. Los
balances se aplican a cada una de las sustancias objeto de interés. Pueden ser contaminantes
químicos, oxígeno disuelto, microorganismos, etc., según el tipo de problema en estudio. Dichas
sustancias constituyen los "componentes del modelo".
La concentración de cualquier sustancia en un punto cualquiera de un sistema natural ha de
cumplir la ecuación fundamental de conservación de la materia. Así, la ecuación diferencial que
representa la variación de concentración de un contaminante al cabo de un tiempo, que se
encuentra disuelto en un elemento diferencial de volumen es:
( ) ( ) ( )∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
Ct x
u Cy
u Cz
u Cx
E Cx y
E Cy z
E Cz
Sx y x x y z= − − − +
+
+
+ K Ec.( 1)
donde:
C: concentración del contaminante (mg/L o mol/L)
t: tiempo (s)
ux, uy, uz: componentes del vector velocidad real del agua; velocidad longitudinal, transversal y
vertical (m/s).
Ex, Ey, Ez: componentes del tensor de dispersión en el seno del fluido; coeficientes de dispersión
longitudinal, transversal y vertical (m2/s).
SK: fuente o sumidero del contaminante, por unidad de volumen de fluido y de tiempo (mg/(L s) ó
mol/(L s)).
Esta ecuación muestra que en un sistema completamente mezclado,
Acumulación = Entradas – Salidas ± Generación
A continuación se describen los distintos términos de la Ec.( 1).
Acumulación. El primer miembro de la Ec.(1) representa la acumulación de materia en el elemento

Modelos de Calidad de Aguas 12
diferencial de volumen, es decir, la variación con el tiempo de la cantidad de materia presente. La
acumulación viene expresada como la suma de las entradas menos las salidas del elemento
diferencial de volumen originadas por transporte convectivo-dispersivo (convección-dispersión),
más un término genérico de fuente/sumidero.
Transporte por convección. Los términos en velocidad del segundo miembro de la Ec.(1)
representan el transporte por convección. El transporte convectivo es el que se produce debido al
movimiento de contaminante conjuntamente con el fluido. El término que aparece en la ecuación
representa la diferencia entre las entradas y salidas del elemento diferencial de volumen por este
concepto. La evaluación del transporte convectivo requiere la determinación del campo de
velocidades del agua en el sistema natural. Para ello existen diversas soluciones, desde la simple
descripción empírica de las velocidades a partir de datos de campo, hasta el uso de complejos
modelos hidrodinámicos tridimensionales que resuelven simultáneamente las ecuaciones de
conservación de la cantidad de movimiento, continuidad, hidrostáticas y de estado en las tres
direcciones del espacio. El problema a estudiar determinará el método de evaluación de las
velocidades. Así, por ejemplo, en estudios donde sea importante la evolución a largo plazo o en
estado estacionario únicamente será necesario disponer de valores medios de las velocidades.
En los apartados dedicados a los distintos tipos de sistemas naturales se hará una breve
introducción a la forma de evaluar el término convectivo en cada uno de ellos. No obstante, los
fenómenos que inciden principalmente en el transporte por convección son los siguientes:
• Diferencias de nivel: El primer mecanismo de transporte de agua en un medio natural es por la
acción de la gravedad.
• Viento. El viento ejerce un esfuerzo cortante sobre la superficie del agua en los sistemas
superficiales, impulsándola en su dirección. Dicha tensión se transfiere mediante rozamiento a
las capas inferiores.
• Mareas. Las mareas son elevaciones periódicas del nivel del agua en uno de los límites del
sistema. Generan diferencias de nivel que se traducen en el movimiento de las masas de agua.
• Rozamiento con el fondo. El rozamiento de la capa de agua con el fondo produce un efecto de
frenado de la masa de agua. Dicho efecto dependerá de la naturaleza del fondo (materiales que
lo componen, existencia de vegetación, etc.).
• Gradientes de densidad. Los movimientos verticales de las masas de agua están inducidos, en
gran parte, por gradientes de densidad que dependen de la temperatura y de la salinidad del

Balances de Materia en Sistemas Naturales 13
agua.
En algunos libros de texto, al transporte convectivo se le denomina transporte advectivo. A
nuestros efectos, son sinónimos.
Transporte por dispersión. El término dispersión hace referencia a los procesos de transporte
que sufren las sustancias disueltas y/o particuladas y que no están directamente relacionados con el
desplazamiento de las masas de agua. Hay tres procesos que contribuyen a dicho transporte:
• Difusión molecular. La difusión molecular es el desplazamiento aleatorio de las moléculas de
solutos entre las moléculas de agua debido al movimiento browniano de las partículas. En la
práctica se observa una homogenización de las partículas en la solución de manera que el efecto
es como si las moléculas se “desplazaran” desde las zonas de mayor concentración a las de
menor, por lo que siempre que exista un gradiente de concentración, se dará este fenómeno. Es
un proceso extraordinariamente lento que no suele ser importante a efectos prácticos en el
estudio de sistemas naturales.
• Difusión turbulenta. La difusión turbulenta hace referencia al transporte de sustancias disueltas
y pequeñas partículas causada por turbulencias a pequeña (micro) escala. Se trata de un
transporte convectivo a microescala producido por fluctuaciones turbulentas de la velocidad
instantánea. Dichas fluctuaciones están producidas por los esfuerzos cortantes que se dan en el
interior de las masas de agua. Su magnitud es varias veces superior a la difusión molecular.
Puede darse en las tres direcciones del espacio, pero suele ser anisotrópica.
• Dispersión. La interacción de la difusión turbulenta con un perfil definido de la velocidad en la
columna de agua provoca un mayor grado de mezcla. Este fenómeno se conoce como
dispersión. La dispersión está provocada por las desviaciones de la velocidad puntual del fluido
respecto a la velocidad media. Los gradientes de velocidad del agua se originan por esfuerzos
cortantes en las fronteras del sistema (rozamiento con el fondo y paredes laterales, acción
cortante del viento en la interfase aire-agua, etc.) y por la morfología de canales y poros, por la
sinuosidad de un río o por la estratificación térmica. Cuando debido a la difusión turbulenta una
parte del fluido conteniendo sustancias disueltas cambia de posición, se encuentra en una zona
con una velocidad distinta, mayor o menor, que la que tenía. La porción de fluido desplazada y
las sustancias contenidas en ella se difunden, dependiendo de la concentración en las fronteras.

Modelos de Calidad de Aguas 14
El efecto de la dispersión se representa matemáticamente mediante una expresión análoga a
la Segunda ley de Fick de la difusión:
∂∂
∂∂x
E Cxi
ijj
Ec.( 2)
en la que Eij es el tensor de dispersión. Haciendo coincidir los ejes coordenados con los ejes
principales del tensor de dispersión, sólo quedan tres componentes no nulas de éste, las
correspondientes a Exx, Eyy, Ezz, o bien Ex, Ey y Ez, que son los tres términos de dispersión que
aparecen en la ecuación general del balance de materia (Ec.1). Si el flujo es laminar, el coeficiente
de dispersión es el coeficiente de difusión molecular. Para flujo turbulento, el coeficiente de
dispersión engloba la difusión molecular, turbulenta y dispersión. La evaluación de estos
coeficientes no es tarea sencilla. Además, el valor de los coeficientes de dispersión puede venir
afectado por el tipo de modelo matemático utilizado, por lo que es importante realizar una
cuidadosa calibración y verificación de dichos coeficientes a partir de datos de campo.
Término de fuente/sumidero. El último término del segundo miembro de la Ec.(1) representa
todas las posibles entradas y/o salidas de la sustancia en estudio no asociadas al transporte
convectivo-dispersivo que puedan producirse por unidad de tiempo y de volumen de fluido. Los
términos de entrada simple son los producidos por el vertido directo de una determinada cantidad
de contaminante. Esta entrada puede tener lugar por adición de una cantidad de agua en la que el
contaminante se encuentra disuelto (Q⋅Ce) o bien por adición directa de una cierta cantidad del
mismo (M). Los términos de salida simple son los debidos a la extracción de una determinada
cantidad de agua en la que el contaminante se encuentra disuelto (Q⋅Cs). En estos términos de
entrada/salida debe incluirse además los procesos de transporte en sedimentos. Los sedimentos
acumulados en el seno de ríos, lagos o estuarios reflejan la historia de los distintos procesos de
contaminación que haya sufrido ese sistema natural. Los sedimentos pueden actuar como fuentes o
sumideros de sustancias disueltas y/o particuladas. El problema más habitual es el sedimento como
resultado del transporte y la deposición de materia orgánica. Dicha materia puede tener un origen
externo al sistema, por ejemplo, ser resultado de un vertido continuo de aguas residuales con una
elevada carga de materia orgánica. También puede producirse la sedimentación de materia orgánica
de origen interno: la muerte de la materia viva que se ha desarrollado en el interior del sistema.

Balances de Materia en Sistemas Naturales 15
Si el término de fuente/sumidero agrupa sólo entradas y salidas simples, el contaminante
considerado es conservativo y su estudio puede realizarse simplemente en base a la resolución de la
Ec.(1).
En el caso de contaminantes no conservativos, además de los términos de entrada y salida
simple es necesario incluir los términos de generación y/o desaparición de contaminante por todos
los procesos físicos, químicos y biológicos. En este término se deberán incluir las ecuaciones
cinéticas o de equilibrio que describen dichos procesos. Aparecerán, en las ecuaciones, la
concentración del contaminante cuyo transporte se estudia y la de cualquier otra especie con la que
tenga algún tipo de interacción física, química o biológica. Si la situación es esta última, la única
forma de plantear totalmente el problema de la evolución del contaminante será la de considerar
simultáneamente las ecuaciones de transporte de todas las especies que interaccionan entre sí
mediante fenómenos físicos, químicos o biológicos, y las ecuaciones que representan dichos
fenómenos. La formulación del problema vendrá dada, por lo tanto, por un sistema de ecuaciones
diferenciales como la 9Ec.(1), una para cada sustancia que se considere, en las que el
término de fuente/sumidero será una función de la concentración del resto de especies.
La resolución de la Ecuación General del Balance de Materia en tres dimensiones es muy
compleja por lo que, en la práctica, se realizan simplificaciones con el fin de reducir el número de
dimensiones del problema. Dichas simplificaciones se realizan en base a un correcto conocimiento
de la hidrodinámica del sistema que permitirá describir el volumen de agua objeto de estudio con
dimensiones desde cero hasta tres.
La descripción de los sistemas naturales que se va a realizar a continuación estará basada,
por lo tanto, en las dimensiones necesarias para una completa representación del flujo en el
sistema. Dicha descripción determinará la discretización espacial del sistema en estudio.
2.2 Transporte convectivo y dispersivo en sistemas naturales.
2.2.1 Aguas subterráneas.

Modelos de Calidad de Aguas 16
2.2.1.1 Definición y tipos de acuíferos
Un acuífero es un estrato o formación geológica que permite la circulación de agua por sus
poros o grietas de modo que el hombre pueda aprovecharla. De esta definición, el aspecto más
destacable y lo que distingue a un acuífero de otras formaciones es la posibilidad de transmitir y
proporcionar agua en cantidades considerables. Las características principales que determinan la
capacidad de una formación geológica para llegar a constituir un buen acuífero son la porosidad y
la permeabilidad.
Un medio poroso está formado por un agregado de partículas entre las cuales existen
espacios vacíos que pueden ser ocupados por un fluido. La porosidad total se define como el
cociente entre el volumen vacío o volumen de poros y el volumen total. No obstante, se pueden
distinguir distintos tipos de poros dentro del material. Así, existe una red de poros interconectados
a través de los cuales el agua puede circular libremente y otros poros cerrados en los que el fluido
queda confinado. También existen poros semicerrados en los que la circulación del agua, si bien es
posible, se produce con dificultad y lentitud. Desde el punto de vista de la Hidrología Subterránea,
el parámetro de interés es la porosidad eficaz o efectiva, cociente entre el volumen de poros por los
que el agua puede circular y el volumen total.
La permeabilidad se define como el caudal de agua que pasa por una sección unidad del
acuífero bajo un gradiente hidráulico también unidad, a una temperatura dada. Los factores que
determinan la permeabilidad son, fundamentalmente, el tamaño de los poros y la viscosidad y
densidad del fluido.
La porosidad y la permeabilidad de una formación geológica vienen determinadas, en gran
medida, por sus características litológicas. Por lo tanto, la composición de las rocas, sus
condiciones de formación y las modificaciones estructurales posteriores influirán
considerablemente en la capacidad de almacenamiento y en la circulación del agua a través de la
formación. Así, los acuíferos formados por depósitos no consolidados de materiales sueltos (arenas,
gravas, mezclas de ambas, etc.) suelen proporcionar caudales de agua importantes si se explotan
adecuadamente. Las rocas sedimentarias consolidadas presentan, en general, menor porosidad. La
más importante en el campo de las aguas subterráneas es la caliza. Las propiedades de la caliza
varían considerablemente según el ambiente sedimentario existente durante su formación y las
modificaciones posteriores de la permeabilidad por disolución del carbonato (formación de
"karst"). Otros tipos de rocas consolidadas como los conglomerados y las areniscas únicamente

Balances de Materia en Sistemas Naturales 17
pueden ser explotados como acuíferos si presentan una ausencia parcial del cemento que las
cohesiona. Asimismo, la posibilidad de que las rocas volcánicas constituyan un buen acuífero
depende de aspectos como el tipo de erupción que las originó, el grado de alteración, edad, etc. Por
último, entre la rocas ígneas y metamórficas (granitos, dioritas, gabros, pizarras y esquistos) sólo se
pueden formar buenos acuíferos en las zonas superficiales meteorizadas o en las regiones muy
fracturadas por fallas y diaclasas. En cualquier caso, constituyen los peores acuíferos en lo que
respecta a rendimiento en caudal.
Los acuíferos se pueden clasificar en función de la presión hidrostática en:
• Acuíferos libres. Aquellos en los que existe una superficie libre de agua en contacto directo con
el aire y por tanto, a presión atmosférica.
• Acuíferos cautivos o confinados. Aquellos en los que el agua está sometida a una cierta presión
superior a la atmosférica. En ellos, el agua ocupa todos los poros de la formación geológica,
saturándola totalmente. Un caso particular lo constituyen los acuíferos semiconfinados. Se trata
de acuíferos confinados en los que el muro (parte inferior) y/o el techo (parte superior) no es
totalmente impermeable, permitiendo una filtración vertical lenta de agua procedente de un
acuífero o masa de agua situada por encima o por debajo del mismo.
En el estudio del transporte de contaminantes en aguas subterráneas se distingue entre
transporte en la zona no saturada y en la zona saturada.
La zona no saturada de un sistema acuífero es aquella capa del suelo que separa la
superficie del terreno del nivel piezométrico del acuífero. El transporte de contaminantes en esta
zona se caracteriza por estar ligado a la velocidad de percolación del fluido desde la superficie. El
transporte es, por lo tanto, unidimensional en la dirección descendente. Así, la modelización de la
zona no saturada se suele realizar en base a una discretización espacial unidimensional en la
dirección vertical.
La zona saturada es aquella en la que el flujo de agua a través de la matriz sólida del terreno
es permanente y está bien establecido. La zona saturada del acuífero suele ser considerada como un
sistema bidimensional en las direcciones x-y. No obstante, la región en la que se simula el
transporte de contaminantes se define en las tres direcciones del espacio estableciendo el espesor
del acuífero, que puede ser variable a lo largo del mismo. En este tipo de representación se admite

Modelos de Calidad de Aguas 18
la uniformidad de la concentración de las distintas especies en la dirección z, es decir, se considera
el sistema perfectamente mezclado en la dirección vertical.
2.2.1.2 Flujo del agua en un medio poroso.
La ley que regula el movimiento del agua subterránea en condiciones de flujo laminar es la
ley de Darcy. Esta ley establece la relación entre la velocidad del agua en un medio poroso y el
gradiente hidráulico. El gradiente hidráulico entre dos puntos es el cociente entre la diferencia de
niveles piezométricos (diferencia de carga) entre ambos puntos y la distancia recorrida por el agua.
La expresión de la ley de Darcy es:
u k dhdsD = − Ec.( 3)
donde:
uD: velocidad de Darcy (m/s ó m/día).
k: permeabilidad (m/s ó m/día).
h: nivel piezométrico (m).
s: distancia (m).
En la formulación original de la ley de Darcy, la velocidad viene definida como el cociente
entre el caudal que circula por una sección determinada y el área de dicha sección. En un medio
poroso, esta velocidad, conocida como velocidad de Darcy, velocidad de flujo o flujo específico, es
una velocidad ficticia puesto que en su definición se considera que el agua fluye por toda la
sección. Sin embargo, el agua no ocupa toda la sección y se mueve a velocidad variable según el
tamaño y orientación de los poros. Considerando un volumen medio suficientemente grande se
puede definir una velocidad media, denominada velocidad de filtración, velocidad intergranular o
velocidad real, cuya relación con la velocidad de Darcy viene dada por:
u uD =θ Ec.( 4)
siendo:
θ: porosidad eficaz
u: velocidad real (m/s ó m/día)
Teniendo en cuenta esta última relación, la expresión general de la ley de Darcy para un

Balances de Materia en Sistemas Naturales 19
medio anisótropo (el coeficiente de permeabilidad varía con la orientación) será:
uk h
xi ji
ij
j
= − =θ
∂∂
, , ,1 2 3 Ec.( 5)
donde:
ui: componentes del vector velocidad real del agua (m/s).
kij: componentes del tensor de permeabilidad (m/s).
xj: coordenada en la dirección j.
La resolución de la Ec.( 5) a partir de datos de permeabilidad y niveles piezométricos
proporcionará el campo de velocidades del agua en el sistema en estudio.
2.2.1.3 Dispersión en acuíferos.
El transporte dispersivo es de gran importancia en la propagación de frentes de
contaminación en acuíferos. La dispersión provoca la extensión de la contaminación en un
volumen de acuífero superior al predicho mediante el análisis del campo de velocidades del agua.
Por lo tanto, la dispersión determina la llegada de los contaminantes a los puntos de extracción y
descarga del acuífero antes de lo calculado a partir de la velocidad media del agua.
Si la dirección del vector velocidad coincide con el eje x, los coeficientes de dispersión
pueden ser calculados según:
Ex = Dm + εL [v]
Ey = Dm + εT1 [v]
Ez = Dm + εT2 [v]
Ec.( 6)
donde [v] es el módulo del vector velocidad del fluido. El parámetro Dm es la difusividad
molecular. Tiene el mismo valor en cualquier dirección y depende del fluido y del soluto de que se
trate, siendo su orden de magnitud de 10-4 m2/día. El parámetro εL se denomina dispersividad
longitudinal (en la dirección paralela al vector velocidad), y los parámetros εT1 y εT2 son las
dispersividades transversales. El valor de las dispersividades está relacionado con la fricción del
fluido con las fronteras sólidas que lo contienen.

Modelos de Calidad de Aguas 20
En el caso de las aguas subterráneas, donde la velocidad del agua es baja, el valor de la
dispersividad longitudinal está relacionado, fundamentalmente, con las características del material
del acuífero, siendo valores típicos 5 10-4 m para arenas muy homogéneas, 1 m en el caso de gravas
y hasta 100 m para rocas fisuradas. Las dispersividades transversales suelen ser entre 5 y 10 veces
menores que la longitudinal.
2.2.1.4 Transporte de contaminantes en medios porosos.
Si bien la ecuación general que representa el transporte de contaminantes es el balance de
materia dado por la Ec.(1), en el caso de las aguas subterráneas se suelen utilizar expresiones
equivalentes que tienen en cuenta, por un lado la porosidad del medio y por otro, la presencia de
fases sólidas susceptibles de intercambiar materia con la solución acuosa. Así, planteando el
balance en el sistema sólido-solución en su conjunto, se obtiene:
( ) ( ) ( )∂ θ∂
∂∂
∂∂
∂∂
∂∂
θ∂∂
∂∂
θ∂∂
∂∂
θ∂∂
( )Tt x
q Cy
q Cz
q Cx
E Cx y
E Cy z
E Cz
Sx y x x y z= − − − +
+
+
+ K
Ec.( 7)
donde T representa la concentración total de la sustancia considerada en el sistema y qx, qy, qz, son
las componentes del vector velocidad aparente del agua. En esta ecuación, el término de
fuente/sumidero contempla exclusivamente entradas y salidas simples.
O bien, planteando el balance de materia en el subsistema solución:
( ) ( ) ( )∂∂
ρθ
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
∂∂t
C Sx
u Cy
u Cz
u Cx
E Cx y
E Cy z
E Cz
Sx y x x y z( )+ = − − − +
+
+
+ K
Ec.( 8)
donde el término
∂∂
ρθt
S
Ec.( 9)
cuantifica la cantidad de soluto que es eliminada de la solución acuosa por unidad de tiempo por
transferencia hacia las fases sólidas.

Balances de Materia en Sistemas Naturales 21
2.2.2 Aguas superficiales.
2.2.2.1 Ríos.
La modelación del transporte de contaminantes en ríos requiere el conocimiento de
parámetros hidrológicos (caudal, velocidad, coeficientes de dispersión) y geométricos
(profundidad, anchura, sección transversal, pendiente).
La variación del caudal con el tiempo viene determinada por el régimen hidrológico anual:
nival, pluvio-nival, pluvial. La actividad humana también ha contribuido en gran manera a la
modificación de los caudales en los ríos mediante su regulación por medio de embalses, su
canalización, uso para riego, etc.
Los ríos son sistemas clásicos de una dimensión. La descripción de los ríos se realiza
admitiendo que en la sección transversal la concentración de sustancias es uniforme y además no
existe mezcla a lo largo del eje del río. Estas hipótesis se cumplen en ríos poco turbulentos y a
partir de cierta distancia del punto de vertido. Asimismo se consideran perfectamente mezclados en
la dirección vertical, ya que el tiempo necesario para la mezcla vertical es muy pequeño comparado
con el necesario para la mezcla lateral. Este tipo de comportamiento corresponde a un flujo de
pistón. Si se considera que la sección del río permanece constante en el tramo en estudio se puede
llegar a obtener una solución analítica para la concentración de contaminante en función del
tiempo.
En aquellos estudios en los que no se pueda utilizar la solución analítica por ser el sistema
muy complejo, presentar diversos vertidos, caudales variables con el tiempo, etc., es necesario
recurrir a soluciones numéricas.
Conceptualmente se suelen representar mediante una malla o red de trabajo lineal de
segmentos y elementos de volumen conectados. Se considera que el proceso de convección
transporta una sustancia horizontalmente debido al movimiento de la porción de agua que contiene
a dicha sustancia. En general, se utilizan dos aproximaciones para tratar la convección en ríos:
• Calibrar las propiedades del flujo en el río mediante la medición de caudales y secciones de
paso de cada segmento del río para un intervalo determinado de caudales. A partir de estos

Modelos de Calidad de Aguas 22
datos se desarrolla una función en serie en cada sección de paso para interpolar o extrapolar
valores correspondientes a otros caudales. Esta aproximación resulta especialmente
interesante en aquellos sistemas de características hidráulicas complejas y cuando la finalidad
del estudio sea el estado estacionario.
• Determinar las velocidades y caudales en cada segmento del río mediante la resolución
simultánea de la ecuación de conservación de la cantidad de movimiento y de la de
continuidad. Esta aproximación es más rigurosa y se presenta mayor capacidad predictiva
dado que los datos de caudales únicamente son necesarios para la calibración del modelo y su
posterior verificación. Esta solución es más segura y apropiada para simulaciones de la
calidad del agua en tiempo real.
La determinación de la velocidad media en el río a partir de los datos de los caudales no es
inmediata puesto que se requiere el conocimiento de la profundidad. Se hace necesario establecer
alguna relación entre velocidad, profundidad y anchura del río con el caudal. Unas expresiones
comúnmente empleadas son las siguientes:
bQaU=
dQcH= Ec.( 10)
fQeB =
en donde:
U: velocidad media de la corriente (m/s).
H: profundidad (m).
B: anchura (m).
Los coeficientes a, b, c, d, e, y f son constantes empíricas que deben ser determinadas para
cada caso en estudio. Estas constantes no son completamente independientes, y se cumple, por
ejemplo, que b + d + f es igual a 1 y que el producto a c e es igual a 1. Los valores de b oscilan
entre 0.3 y 0.7. Los de d entre 0.1 y 0.6; y los de f entre 0.05 y 0.25. Un canal rectangular tiene un
valor de b igual a 0.4, un valor de d igual a 0.6 y un valor de f igual a, lógicamente, 0.0.
Otra situación se plantea cuando el río es muy ancho y no se puede representar mediante un
sistema de una dimensión. En estos casos se sigue considerando que existe homogeneidad en la

Balances de Materia en Sistemas Naturales 23
dirección vertical y se utilizan aproximaciones bidimensionales considerando, por consiguiente, la
dispersión lateral.
Dispersión en ríos. En un sistema ideal de flujo de pistón la dispersión longitudinal es nula, es
decir, no existe mezcla hacia delante ni hacia atrás. Se ha comprobado que en realidad sí que existe
dicha mezcla y su estudio ha sido objeto de numerosas investigaciones. Así, existen numerosas
expresiones para la determinación del coeficiente de dispersión longitudinal (EL). Una de las
primeras fue propuesta por Elder (1959):
*
L uH933.5E = Ec.( 11)
siendo H la profundidad (m) y u* la velocidad friccional, calculable como:
SHgu*= Ec.( 12)
en donde:
g: aceleración gravitacional (m/s2).
S: pendiente del canal (m/m).
La expresión dada por la Ec.( 11) no considera las variaciones laterales de la corriente, por
lo que subestima los valores obtenidos en sistemas naturales. No obstante, la mayoría de las
expresiones propuestas por otros autores mantienen el esquema propuesto por Elder aunque
introducen parámetros como la velocidad de la corriente y la anchura del río:
*
22
L uHBU011.0E =
Ec.( 13)
Resulta interesante estimar si el término de dispersión puede ser importante o no en un
sistema determinado. En sistemas en los que el contaminante se vierte de forma continua y las
condiciones son de estado estacionario se puede utilizar la siguiente expresión:
k Eu
L2 0 04≤ . Ec.( 14)
donde k es la velocidad de degradación de cualquier compuesto considerado. Si se cumple esta

Modelos de Calidad de Aguas 24
expresión el perfil de concentración no se verá afectado en más de un 10% si la dispersión es
ignorada. Si el gradiente de concentración es lo suficientemente pequeño, también lo será el
transporte dispersivo.
Thomann (1973) estudió la importancia de la dispersión longitudinal en ríos afectados por
vertidos variables con el tiempo y, consecuentemente, en los que se producían gradientes de
concentración. Las conclusiones del estudio indican que en ríos grandes la dispersión era siempre
importante, mientras que en ríos pequeños, la dispersión podía ser importante cuando los vertidos
variaban en periodos de 7 días o menos.
La dispersión lateral en ríos debe ser considerada cuando se utilizan modelos
bidimensionales. Fischer (1979) propone la siguiente expresión para evaluar el coeficiente de
dispersión lateral:
*
lat uH6.0E = Ec.( 15)
La longitud necesaria para alcanzar una mezcla lateral completa puede calcularse como:
latm E
BuL2
4.0= Ec.( 16)
en el caso de descargas efectuadas en el borde del río. Si el vertido se efectúa en la zona central, la
expresión es:
latm E
BuL2
1.0= Ec.( 17)
Estas expresiones pueden ser muy útiles cuando se desee saber a qué distancia de un punto
de vertido se producirá la mezcla completa del vertido con el río. Cuando se realiza el muestreo de
las aguas de un cauce aguas abajo de un vertido, es fundamental elegir puntos representativos.

Balances de Materia en Sistemas Naturales 25
La aplicación de un análisis dimensional a las magnitudes longitud y concentración que intervienenen los procesos de transporte da lugar a la aparición del número de Peclet, término adimensionalque relaciona el transporte convectivo con el difusivo/dispersivo:
dispersivotransportedelvelocidadconvectivotransportedelvelocidad
EuLPe == . Si el número de Peclet es alto (>10), el
transporte es predominantemente convectivo, mientras que si es bajo (< 0.1) la mezcla longitudinales importante y el sistema tiende a la mezcla completa. Este análisis inicial permite estimarrápidamente qué proceso es el predominante en el río y simular en consecuencia.
2.2.2.2 Lagos.
El transporte de contaminantes y la forma en que es modelado depende en gran medida de
la forma y tamaño del lago. Con respecto a esta segunda característica es necesario introducir dos
parámetros fundamentales: el tiempo de residencia y la profundidad. El tiempo de residencia es la
relación que existe entre el caudal que entra al lago y su volumen. A efectos de analizar el
comportamiento respecto a este parámetro se distingue entre aquellos lagos cuyo tiempo de
residencia es inferior a un año y aquellos en los que es mayor. Con respecto a la profundidad, se
considera que son someros aquellos cuya profundidad es inferior a 7 metros y profundos si es
superior a dicha cifra. Aunque esta regla puede ser más o menos adecuada, sería mucho más
conveniente el obtener mediciones de alguna variable ambiental que nos permitiera deducir el
grado de mezcla vertical: si este grado de mezcla es alto, significa que el lago es lo suficientemente
somero como para considerarlo completamente mezclado en vertical. Una variable fácil de medir y
muy significativa es la temperatura del agua.
Lagos poco profundos. Los lagos poco profundos suelen ser considerados como sistemas de
dimensión cero. La suposición básica que se realiza es la de considerar que el lago se encuentra
perfectamente mezclado, gracias a las turbulencias que provocan agentes externos como el viento.
A efectos prácticos no es necesario obtener el campo de velocidades del agua puesto que el
sistema consta de un único elemento. Se considera, por lo tanto, como un Reactor Contínuo de
Tanque Agitado. Los cambios en el nivel de agua del lago vienen determinados por la diferencia
entre la entrada y la salida de agua. El modelo matemático es el más sencillo que se puede plantear
y se puede obtener fácilmente una solución analítica para casos concretos.

Modelos de Calidad de Aguas 26
Lagos profundos. En estos sistemas no se puede admitir la suposición de mezcla completa puesto
que probablemente, durante ciertas épocas del año existirán gradientes de densidad del agua que
dificulten la transferencia de materia mediante los procesos de convección y dispersión.
Los lagos con tiempos de residencia elevados y estratificación en la dirección vertical se
suelen definir como sistemas unidimensionales. Se representan mediante un conjunto de capas
horizontales en las cuales la velocidad es nula, existiendo ésta únicamente en la dirección vertical.
La entrada de afluentes al lago y la salida se realiza en las capas de la misma densidad. Teniendo
en cuenta que el agua es un fluido incompresible, el agua que entra en un nivel determinado genera
flujos convectivos en la dirección vertical en todos los elementos situados por encima del nivel de
entrada. Las capas inferiores, de densidad mayor, no se ven afectadas.
Los lagos profundos se pueden representar también mediante un sistema bidimensional en
profundidad, como se verá en el apartado dedicado a los embalses. Únicamente señalar aquí que la
mezcla vertical está fundamentalmente producida por la acción del viento sobre la superficie del
lago. Las turbulencias generadas se transmiten hacia las capas más profundas mediante la acción de
esfuerzos cortantes.
Dispersión vertical en lagos. En aquellos sistemas que pueden ser representados mediante una
dimensión en vertical, se puede evaluar el coeficiente de dispersión vertical mediante la siguiente
expresión (Baca y Arnett, 1976):
E a a V dv wz d= + −
1 24 6. / Ec.( 18)
donde:
Ev: coeficiente de dispersión vertical (m2/s).
z: profundidad (m).
Vw: velocidad del viento (m/s).
d: profundidad de la termoclina (m).
a1, a2 constantes empíricas (m2/s y m, respectivamente).
Dispersión horizontal en lagos. En líneas generales, la dispersión horizontal en lagos, embalses,
zonas costeras, etc. es varios órdenes de magnitud mayor que la dispersión vertical. Los valores de
EH en lagos son de 1 a 100 m2/s. A diferencia de como ocurría en los sistemas de flujo en canal
abierto (ríos), la dispersión en sistemas abiertos como lagos o aguas oceánicas no puede ser

Balances de Materia en Sistemas Naturales 27
relacionada con las características medias del flujo (u). En estos casos, las turbulencias tiene su
origen en una serie de fenómenos complejos como el viento, el oleaje, la ruptura de la circulación a
larga escala producida en la zona de rompientes, etc. Por ello, la mejor aproximación para los
valores de EH se obtiene relacionándola con la escala de longitud (L) del proceso. La expresión más
conocida es la llamada ley de 4/3:
E A LH D= 4 3/ Ec.( 19)
donde AD es el parámetro de disipación (igual a 0.005 si EH se expresa en cm2/s).
Existen varias aproximaciones para estimar el valor de L:
• Para descargas al mar a través de emisarios submarinos el valor de L está basado en la longitud
del difusor.
• En estuarios o zonas costeras la estimación de L se basa en la distancia de la carrera de la marea.
• En sistemas representados mediante redes bidimensionales, el valor de L puede ser la longitud
horizontal de la celda.
Okubo y Osmidov (1970) ajustaron valores de coeficientes de dispersión horizontal de
lagos y océanos obteniendo las siguientes expresiones:
EH = 2 10-3 L4/3
EH = 104
EH = 10-3 L4/3
para L < 105 cm
para 105 < L < 5 105 cm
para L > 5 105 cm
Ec.( 20)
estando EH en cm2/s y L en cm.
2.2.2.3 Embalses.
Un embalse puede ser considerado como un sistema bidimensional en profundidad si los
gradientes de densidad en la dirección vertical son importantes. Esto es, si se considera que es muy
estrecho comparado con su profundidad, se puede admitir que no existe gradiente de concentración
en la dirección transversal (en esa dirección está totalmente mezclado) con lo que el problema se
reduce a las direcciones longitudinal y vertical. Con el fin de simular correctamente los efectos de
los gradientes de densidad en profundidad en la hidrodinámica y el transporte de masa en el

Modelos de Calidad de Aguas 28
sistema, debe resolverse simultáneamente las ecuaciones de movimiento y de transporte.
Asimismo, en estos sistemas se debe tener en cuenta la transferencia de cantidad de movimiento
debido a gradientes de velocidad (mediante el coeficiente de viscosidad vertical turbulenta) y la
transferencia de materia debida a los gradientes de concentración (mediante el coeficiente de
difusividad vertical turbulenta). Ambos fenómenos están directamente relacionados con el grado de
mezcla y la estratificación en la columna de agua. En este caso, la mezcla vertical se produce,
además de por la acción del viento sobre la superficie del embalse, por el sistema de desagüe del
mismo.
2.2.2.4 Estuarios.
Los estuarios son zonas naturales donde se produce la conexión de un río con el mar. La
importancia de su estudio radica en el hecho de que numerosos asentamientos humanos se han
realizado en ellos, con el consiguiente impacto ambiental que ello ha provocado. En la
hidrodinámica de estos sistemas es muy importante el efecto de las mareas. Con respecto a la
calidad del agua, en los estuarios se distinguen dos zonas:
• Zona de intrusión salina. Aquella en la que se aprecia un marcado gradiente de salinidad
longitudinal.
• Zona de aguas dulces. Su calidad depende de los aportes del río u otras fuentes.
La localización de la separación entre ambas zonas varía a lo largo del ciclo de la marea y
con el caudal de entrada de agua dulce. La importancia de las mareas reside en el hecho de que el
caudal, la velocidad del agua y la profundidad en cada punto evolucionan con el tiempo. El
predominio de la convección o de la dispersión en los estuarios depende de la escala temporal
utilizada, relacionada, lógicamente, con el tipo de problema que se desee abordar. En problemas a
corto plazo, tales como el estudio del vertido repentino de sustancias altamente reactivas, el
fenómeno predominante es el transporte por convección. Sobre una escala temporal mayor se
observa que las corrientes generan movimientos cíclicos, por lo que predomina la dispersión. En
estuarios se denomina flujo neto a la relación que existe entre el caudal de entrada y el de salida.
En la mayoría de los casos coincide con el caudal del río que accede al estuario.
Otra característica importante de los estuarios es la existencia de una zona muerta: allí
donde la energía del río y de la marea se anulan mutuamente. Es una zona donde se acumulan
sólidos sedimentables.

Balances de Materia en Sistemas Naturales 29
Si el estuario no es excesivamente ancho, la forma más sencilla de representarlo es
mediante un sistema de una sola dimensión. La principal diferencia con respecto a un río es que en
el segmento de conexión con el mar existe una condición límite o de frontera, concretamente, el
nivel de la marea. Si el estuario es ancho y no se puede asimilar a un sistema lineal, la
aproximación más sencilla es la de un sistema pseudo bidimensional, también denominado de
dimensión 11/2. En ambos casos, el campo de velocidades se determina mediante la resolución de
las ecuaciones de conservación de la cantidad de movimiento y de continuidad en una dimensión.
La discretización espacial del estuario en celdas bidimensionales conectadas mediante "canales"
permite generar un flujo bidimensional pese a que la resolución de la ecuación sea en una única
dimensión. Esta aproximación es válida para sistemas relativamente pequeños en los que las
aceleraciones perpendiculares al flujo (como la de Coriolis) no tengan excesiva importancia. Este
tipo de modelo es muy flexible puesto que permite tratar sistemas con numerosas entradas de
caudal tanto en estado estacionario como transitorio, permite variar con facilidad las condiciones
en las fronteras y admite geometrías complejas.
El siguiente nivel de aproximación a un estuario poco profundo consiste en la
representación de un sistema bidimensional en superficie, admitiendo mezcla completa en la
dirección vertical. También puede ser tratado como un sistema bidimensional en profundidad
(como los embalses). En este caso, la mezcla vertical viene determinada por la acción del viento y
de las mareas.
Dispersión vertical en estuarios. El coeficiente de dispersión vertical en estuarios, lagos o
embalses profundos se puede estimar mediante las ecuaciones propuestas por Munk y Anderson
(1948):
( )EE
R R g zuz
v
voi i= + =
−1 3 3332
2.
ρ ∂ρ∂
∂∂
Ec.( 21)
siendo:
ρ: densidad (kg/m3).
u: velocidad media en el punto z sobre el fondo (m/s).
g: aceleración de la gravedad (m/s2).
Evo: Coeficiente de dispersión en medio no estratificado (m/s2).

Modelos de Calidad de Aguas 30
El número de Richardson, Ri, refleja la importancia de la variación de la densidad y de la
velocidad del agua en función de la profundidad: cuando el agua del mar, más salina y a menudo a
menor temperatura que la del río, penetra en el estuario, lo hace por la zona inferior del mismo,
estableciéndose una estratificación vertical. Esto hace que el coeficiente de dispersión vertical se
modifique y que, incluso, lo haga el coeficiente de dispersión longitudinal.
Dispersión longitudinal en estuarios. Si el estuario es considerado como un sistema
unidimensional únicamente será necesario considerar un coeficiente de dispersión que contemple la
dispersión efectiva. La magnitud de dicho coeficiente dependerá de los siguientes factores:
• La escala de tiempo sobre la que se realiza la simulación. La escala de tiempo determina el
intervalo en el que se promedian las cantidades que varían con el tiempo. Para escalas de tiempo
pequeñas, las ecuaciones que describen la hidrodinámica del sistema y las interacciones entre las
sustancias se pueden resolver con un mayor grado de detalle. En este caso serán necesarios unos
coeficientes de dispersión menores que, por ejemplo, si se promediara sobre un ciclo de marea.
• La localización en el interior del estuario. El coeficiente de dispersión longitudinal está referido
a un promedio espacial sobre el área de la sección transversal al flujo. Si los valores de la
velocidad o de la concentración en un instante dado a través de una sección difieren
notablemente de los valores promedio, variará el valor del coeficiente de dispersión. Estas
desviaciones suelen ser importantes en las bocas de los estuarios debido a los gradientes de
densidad que se establecen entre el agua dulce y salada.
Para la determinación de los coeficientes de dispersión en la zona de intrusión salina apenas
se han desarrollado ecuaciones y se recurre a la medición en campo de los coeficientes en base a la
concentración de una sustancia conservativa. En la zona de agua dulce se ha desarrollado un mayor
número de expresiones, aunque también en estos casos se puede recurrir al uso de trazadores para
la estimación "in situ" del coeficiente de dispersión.
Dispersión horizontal en estuarios. Cuando se representa el estuario como un sistema
bidimensional en superficie, los coeficientes de dispersión longitudinal y lateral se pueden
relacionar con las velocidades en las direcciones x e y. Las expresiones más ampliamente utilizadas
relacionan la dispersión de una sustancia con la velocidad friccional u* y la profundidad del agua,

Balances de Materia en Sistemas Naturales 31
según:
EL = 5.9 u* h
EH = 0.2 u* h
Coeficiente de dispersión en la dirección del flujo (m/s2)
Coeficiente de dispersión en la dirección normal al flujo (m/s2) Ec.( 22)
2.2.2.5 Sistemas naturales tridimensionales.
La aproximación más completa a un sistema natural es mediante su representación en tres
dimensiones. Cualquiera de los sistemas anteriores es susceptible de ser representado por un
modelo tridimensional. La cuestión está en decidir si el esfuerzo que supone la aplicación de un
modelo tridimensional compensa los resultados que se pretende obtener desde el punto de vista de
la calidad del agua. Los principales problemas que plantean este tipo de modelos son:
• La dificultad para especificar la transferencia interna turbulenta de cantidad de movimiento y de
materia. Los parámetros de los modelos han de ser calibrados a partir de una ingente cantidad de
datos de campo.
• El alto coste económico que supone la obtención de dichos datos.
• El alto coste computacional de estos modelos. Aunque este factor va siendo cada vez menos
importante debido al rápido desarrollo de los computadores, en simulaciones de la calidad del
agua a largo plazo puede ser un inconveniente puesto que restringirá el número de alternativas
que se pueden simular.
2.3 Contaminantes no conservativos.
En este apartado se exponen los principales contaminantes no conservativos de interés en
los estudios de calidad en aguas subterráneas y superficiales. Los contaminantes no conservativos
son aquellos que sufren una transformación en otras sustancias por procesos durante su
permanencia en el medio natural. Se trata, en este punto, de obtener las ecuaciones que describen
dichos fenómenos degradativos y su interrelación con otras sustancias presentes en el sistema, a fin
de representar matemáticamente la parte correspondiente del término de fuente o sumidero de la
ecuación del balance de materia.

Modelos de Calidad de Aguas 32
2.3.1 Aguas subterráneas.
La mayor parte de los estudios de calidad en aguas subterráneas se centran en la modelación
del transporte de especies químicas de naturaleza inorgánica u orgánica, susceptibles de
experimentar interacciones de naturaleza fundamentalmente físico-química, ya que la actividad
biológica en los sistemas saturados y en profundidad es escasa. No obstante, en la bibliografía
aparecen algunos trabajos recientes sobre regeneración de acuíferos contaminados utilizando
microorganismos.
Las interacciones físico-químicas engloban toda una serie de reacciones en disolución
(reacciones ácido-base, de oxidación-reducción y de formación de complejos en solución) así como
reacciones heterogéneas de precipitación-disolución y de adsorción-desorción. Desde la
perspectiva de la calidad del agua, son de especial importancia los procesos de precipitación y
adsorción ya que provocan una retención de los contaminantes sobre la matriz sólida inmóvil del
acuífero, disminuyendo el impacto de los mismos sobre la calidad del agua. Dicha retención
origina un retraso en el transporte de los contaminantes que la experimentan, que debe ser
adecuadamente evaluado para la correcta predicción de las velocidades de transporte.
A la hora de plantear un modelo de calidad en aguas subterráneas hay que considerar que en
los sistemas naturales se producen simultáneamente procesos de adsorción, desorción, disolución,
precipitación, oxidación, reducción y formación de complejos, existiendo una influencia mutua
entre ellos. Por lo tanto, un modelo de calidad que represente el comportamiento de un sistema
natural deberá incorporar simultáneamente todas estas interacciones.
Por otro lado, es importante tener en cuenta la velocidad de las reacciones. Se distingue
entre reacciones "suficientemente rápidas" y reacciones "insuficientemente rápidas" y/o
irreversibles. Bajo el término "suficientemente rápidas" se incluyen todas aquellas reacciones
reversibles que tienen lugar a gran velocidad comparada con la velocidad de cualquier otro proceso
que ocasione variaciones en la concentración de los solutos presentes en el sistema. En estas
condiciones se puede admitir la suposición de equilibrio químico local. Esta suposición implica que
en cada punto del sistema se alcanza el equilibrio químico y, por tanto, la transformación de los
solutos se produce en su máxima extensión hasta el límite marcado por las relaciones
termodinámicas. La inclusión de una reacción en la categoría de "suficientemente rápidas" no viene
fijada únicamente por la propia velocidad de la reacción, sino que depende fuertemente de las
condiciones del entorno en que se lleva a cabo dicha transformación, en especial, de la velocidad

Balances de Materia en Sistemas Naturales 33
del agua en el sistema.
Para el tratamiento de las reacciones "insuficientemente rápidas" es necesario considerar la
cinética del proceso, ya sea la cinética química o, en el caso de las reacciones heterogéneas, la
cinética de la transferencia de materia que limite la velocidad global del proceso.
2.3.1.1 Modelación de las interacciones físico-químicas.
El primer paso para la modelación de las interacciones físico-químicas en un sistema de
aguas subterráneas es la definición del sistema químico. El sistema químico se puede representar
mediante un conjunto de componentes y especies. Las especies se definen como cualquier entidad
química que se considere en el problema. Sobre esta base, se establece un conjunto de
componentes tal que todas las especies puedan ser representadas como el producto de una reacción
entre componentes, y ningún componente pueda ser representado como el producto de una reacción
entre los demás componentes. Así pues, los componentes constituyen el conjunto de variables
independientes a partir del cual se pueden definir todas las especies. Los componentes pueden
ser átomos, grupos de átomos neutros, iones e incluso el electrón. Las condiciones que se exigen a
los componentes son:
• La masa total de un componente no puede modificarse por reacción química. por lo tanto, en un
sistema cerrado, la masa total de los componentes permanece constante, mientras que en un
sistema abierto, la variación de dicha masa total se debe, exclusivamente, al transporte de masa
asociado al movimiento del fluido y a las entradas y salidas de corrientes que contienen formas
solubles del componente en cuestión.
• La elección de conjunto de componentes debe realizarse respetando la regla de las fases. Así,
por ejemplo, en un sistema en el que se considere la reacción:
Ca CO CaCOac ac ac( ) ( ) ( )2
32
3+ −+ ↔ Ec.( 23)
se podría tomar como componentes las combinaciones Ca2+/CO32-; CaCO3/Ca2+ ó CaCO3/CO3
2-,
pero no el conjunto Ca/C/O. Se puede observar cómo un problema puede ser representado por más
de un conjunto de componentes pero una vez elegido éste, cada especie tiene una definición única.
Definidos así los componentes del sistema, el modelo de calidad de aguas subterráneas
constará de una ecuación de transporte (balance diferencial de materia, Ec.( 7)) para cada

Modelos de Calidad de Aguas 34
componente junto con el conjunto de ecuaciones que describen las interacciones físico-químicas y
que se describen en el punto siguiente.
Cabe recordar que si bien en la bibliografía se pueden encontrar modelos de calidad
unicomponente, se trata de modelos simplificados que únicamente tienen en cuenta el transporte de
un componente y la adsorción como única reacción. Estos modelos al no considerar el conjunto de
reacciones que tiene lugar en el sistema presentan una aplicabilidad muy limitada.
2.3.1.1.1 Equilibrio químico.
Relaciones de equilibrio.
El equilibrio químico de las reacciones en disolución y de precipitación-disolución viene
caracterizado por las leyes de acción de masas y los balances de materia de los componentes. La
ley de acción de masas establece la constancia del producto de las actividades1 de las especies
iónicas participantes en la reacción elevadas a sus coeficientes estequiométricos. Se puede expresar
como:
C K x l Nl l ka
k
N
slk
c
= ==
∏1
1 2, ,..., Ec.( 24)
donde:
Cl: Concentración de la especie l (Kmol/m3).
Kl: Constante de formación de la especie l.
xk: Concentración del componente k (Kmol/m3).
alk: Coeficiente estequiométrico del componente k en la especie l.
Nc: Número total de componentes en el sistema.
Ns: Número de especies en el sistema.
La ley de acción de masas dada por la Ec.( 24) se ha expresado en función de las
concentraciones molares en lugar de las actividades químicas. Para ello, la constante de formación
Kl, es una constante corregida teniendo en cuenta los coeficientes de actividad:
1 Actividad de una sustancia se define como el producto de su concentración molar por el coeficiente de actividad γ.

Balances de Materia en Sistemas Naturales 35
K K l Nll
ka
k
N
slk
c
= ==
∏γγ
1
1 2, ,..., Ec.( 25)
siendo:
K: Constante termodinámica de equilibrio.
γl:: Coeficiente de actividad de la especie l.
γk: Coeficiente de actividad del componente k.
Los coeficientes de actividad pueden ser calculados en función de la fuerza iónica mediante
expresiones del tipo de la ecuación de Davis modificada:
log . ./
/γ i iz II
I= −+
−
0 50886
10 32
1 2
1 2 Ec.( 26)
donde I representa la fuerza iónica y zi la carga de la especie iónica i.
La fuerza iónica de una disolución se define como:
( )...zmzmzm21iónicafuerza 2
33222
211 +++= Ec.( 27)
donde m1, m2, m3, son las concentraciones molares de los iones en disolución y z1, z2, z3 son sus
cargas.
El balance de materia de un componente establece que su concentración total en el sistema
es igual a la suma de las concentraciones de las especies en las que participa dicho componente:
T a Cl
k Nk lk l
N
c
s
==
=∑1
1 2, ,..., Ec.( 28)
donde Tk representa la concentración total (analítica) del componente k en todo el sistema
(Kmol/m3).
Los dos conjuntos de ecuaciones algebraicas Ec.( 24) y Ec.( 28) definen matemáticamente
el sistema desde el punto de vista del equilibrio químico y su resolución iterativa permite, a partir

Modelos de Calidad de Aguas 36
de una estimación inicial de las concentraciones de los componentes, determinar la concentración
de todas las especies consideradas en el equilibrio.
La concentración total en disolución de cada componente, es decir, aquella susceptible de
ser "transportada" vendrá dada por la suma de las concentraciones de las especies solubles en las
que participa:
C a Cl
k Nk lk l
N
c
s
==
=∑1
1 2'
, ,..., Ec.( 29)
siendo Ns' el número de especies solubles en el sistema.
Las ecuaciones Ec.( 24) y Ec.( 28) representan directamente el equilibrio de formación de
complejos en disolución. También representan el equilibrio de oxidación-reducción sin más que
considerar las correspondientes semirreacciones y definir el electrón como un componente más del
sistema con concentración total igual a cero. En el caso de las reacciones de precipitación-
disolución, la presencia de una especie precipitada impone una restricción adicional al sistema, ya
que dichas especies presentan de actividad química fija igual a la unidad. El problema se puede
resolver disminuyendo los grados de libertad del sistema representando el sistema por un conjunto
de componentes alternativo en el que se sustituye uno de los componentes por la especie
precipitada. Dado que este nuevo componente tiene actividad fija, se elimina del cálculo del
equilibrio.
Procesos de Adsorción-Desorción.
Como ya se ha indicado, los procesos de adsorción-desorción son de gran importancia en el
transporte de solutos en los sistemas de aguas subterráneas. Los efectos derivados de estos
procesos son complejos debido, fundamentalmente, a la gran diversidad de especies químicas y de
fases e interfases implicadas. La distribución de la materia entre las fases viene determinada por
múltiples factores entre los que cabe destacar la naturaleza del soluto y de la fase adsorbente así
como la composición y las condiciones específicas locales (pH, temperatura, fuerza iónica...) de la
solución acuosa desde la que tiene lugar la adsorción.
Así pues, la modelación del transporte de contaminantes en aguas subterráneas requiere la
caracterización y cuantificación de los procesos de adsorción-desorción, y su expresión mediante

Balances de Materia en Sistemas Naturales 37
un modelo matemático que pueda ser implementado en el conjunto de ecuaciones que describen el
comportamiento del sistema.
Estos procesos son el resultado de la conjunción de diferentes tipos de fuerzas atractivas y
repulsivas entre las moléculas de adsorbato, de solvente y de adsorbente. En general, ocurren como
consecuencia del carácter liofóbico del soluto respecto al solvente, debido a una afinidad elevada
del soluto por el adsorbente, o bien, por una acción combinada de ambos efectos. La adsorción es
un fenómeno superficial. La acumulación de adsorbato sobre el adsorbente se encuentra restringida
a la superficie o interfase entre la solución y el adsorbente. Dependiendo de la naturaleza de las
fuerzas atractivas predominantes entre el soluto y el adsorbente se distinguen tres tipos de
adsorción: física, electrostática y química.
Las fuerzas atractivas asociadas a los procesos de adsorción física están relacionadas,
fundamentalmente, con interacciones entre los momentos dipolares, permanentes o inducidos, de
las moléculas de adsorbato y de adsorbente. Se trata de interacciones de corto alcance (fuerzas de
Van der Waals, fuerzas de dispersión de London). La magnitud de las energías implicadas en los
procesos de fisisorción es del orden de 20 kJ/mol. En estos casos, la molécula adsorbida no queda
fija en un lugar específico de la superficie, sino más bien queda libre para deslazarse a lo largo de
la interfase. La especie adsorbida puede formar varias capas superpuestas sobre la superficie del
adsorbente. La adsorción física es generalmente reversible.
En la adsorción electrostática, las fuerzas atractivas entre el soluto y el adsorbente son de
naturaleza electrostática. La adsorción se produce por interacciones electrostáticas específicas entre
cargas localizadas. Las fuerzas atractivas son de mayor intensidad y alcance que las responsables
de la adsorción física. Este tipo de adsorción se denomina a menudo adsorción por intercambio.
En la adsorción química, los enlaces formados entre las moléculas de soluto y los grupos
químicos específicos de la superficie del adsorbente presentan todas las características de
verdaderos enlaces químicos. Las energías de adsorción son elevadas, del orden de las de un enlace
químico (100-300 kJ/mol). Normalmente, la especie adsorbida forma una capa de espesor
monomolecular sobre la superficie del adsorbente y las moléculas adsorbidas no pueden
desplazarse de unos centros activos a otros.
La mayor parte de los fenómenos de adsorción son combinaciones de las tres formas

Modelos de Calidad de Aguas 38
anteriores de adsorción, resultando difícil, en muchos casos, establecer claramente el mecanismo de
la adsorción.
Para el tratamiento de los fenómenos de adsorción se utilizan, básicamente, dos tipos de
modelos de equilibrio de adsorción. El primer tipo (modelos fenomenológicos) se han desarrollado
a partir de la observación experimental y la aplicación de las teorías termodinámicas y cinéticas. En
este grupo se pueden citar, como más representativos, las clásicas isotermas de adsorción. El
segundo tipo de modelos se basan en la descripción del mecanismo del proceso y en la teorización
de las implicaciones energéticas de dicho mecanismo. Destacan en esta categoría los modelos de
formación de complejos superficiales para la adsorción de especies iónicas y polares sobre
adsorbentes con cargas superficiales.
Isotermas de adsorción.
La mayoría de los modelos utilizados para caracterizar el equilibrio de adsorción relacionan
la cantidad de soluto adsorbida por unidad de peso de adsorbente sólido, qe, con la concentración
de soluto que permanece en la solución en el equilibrio, Ce. Estas relaciones a una temperatura
dada, se conocen como isotermas de adsorción.
El modelo de equilibrio de adsorción más sencillo es la isoterma lineal, que describe la
acumulación de soluto sobre el adsorbente como directamente proporcional a la concentración de
soluto en la solución:
q K Ce D= e Ec.( 30)
donde la constante de proporcionalidad, KD, se denomina coeficiente de distribución o de partición.
En ciertos casos, las isotermas lineales describen adecuadamente el equilibrio de adsorción,
fundamentalmente para concentraciones de soluto muy pequeñas y adsorbentes con baja capacidad
de adsorción. Este modelo supone una gran simplificación desde el punto de vista matemático, pero
su extrapolación a condiciones distintas a aquellas para las que fue calibrado puede conducir con
facilidad a resultados erróneos.
El modelo de Langmuir corresponde a una isoterma de adsorción no lineal. Fue
desarrollado para sistemas en los que la adsorción da lugar a la deposición de una única capa de

Balances de Materia en Sistemas Naturales 39
moléculas de soluto sobre la superficie del adsorbente (adsorción en monocapa). Se basa en las
siguientes suposiciones:
• La energía de adsorción de cada molécula es la misma y es independiente del grado de
cobertura de la superficie.
• La superficie es homogénea.
• La adsorción ocurre únicamente sobre centros activos localizados, las moléculas
adsorbidas permanecen inmóviles y no hay interacción entre ellas.
La ecuación general del modelo de Langmuir es:
q Q bCbCe
e
e
=+
0
1 Ec.( 31)
donde Q0 representa la cantidad de soluto adsorbida por unidad de peso de adsorbente para una
monocapa completa y b es un coeficiente relacionado con la entalpía de adsorción.
El modelo de Freundlich corresponde a una isoterma no lineal con dos parámetros
ajustables. Aunque su origen es empírico, puede deducirse de forma teórica admitiendo una
disminución logarítmica de la entalpía de adsorción con el recubrimiento. La forma general de la
isoterma de Freundlich es:
q K Ce Fn= 1/e Ec.( 32)
siendo:
KF: coeficiente de Freundlich, relacionado con la capacidad de adsorción.
n: constante empírica relacionada con la intensidad de la adsorción. Los valores de n
correspondientes a los distintos tipos de adsorción son: n= 1 lineal; n>1 adsorción favorable; n<1
adsorción no favorable.
Las isotermas de adsorción se han utilizado ampliamente para caracterizar el equilibrio de
adsorción de compuestos orgánicos e inorgánicos sobre adsorbentes naturales y sintéticos, debido
fundamentalmente, a su capacidad para representar razonablemente el comportamiento de sistemas
sencillos en condiciones concretas y a la simplicidad de su formulación matemática. Sin embargo,
en sistemas complejos como son los sistemas naturales, en los que se coexisten varios adsorbatos

Modelos de Calidad de Aguas 40
(adsorción multicomponente) y fases adsorbentes, y se producen simultáneamente diversos
fenómenos que pueden modificar las condiciones del entorno, los modelos anteriores pueden
presentar importantes limitaciones. De hecho, estos modelos no son capaces, en líneas generales,
de reproducir el equilibrio de adsorción en un amplio intervalo de condiciones sin una recalibración
de los parámetros, entendiendo por tales condiciones no sólo las referentes al intervalo de
concentraciones de adsorbato, sino también las relativas a la composición de la disolución
(presencia de ligandos, adsorción competitiva, fuerza iónica y pH).
En particular, la dificultad de estos modelos para considerar la influencia del pH en los
procesos de adsorción limita su aplicación, en especial, para la adsorción de especies iónicas y
polares. La adsorción de especies iónicas presenta una marcada dependencia del pH de la
disolución. Experimentalmente se comprueba que la adsorción de las especies catiónicas pasa
desde prácticamente cero hasta el 100% de adsorción al aumentar el pH en un estrecho intervalo
(2-3 unidades de pH). El efecto sobre la adsorción de aniones es análogo si bien en este caso, la
adsorción aumenta al disminuir el pH. Por otro lado, el propio proceso de adsorción de especies
iónicas afecta al pH de la disolución acuosa. En general, la adsorción de cationes viene
acompañada de una transferencia de protones hacia la disolución, originando una disminución del
pH, mientras que la adsorción de aniones provoca una transferencia de protones al adsorbente, con
el consiguiente aumento del pH de la disolución.
Modelos de formación de complejos superficiales.
Estos modelos de equilibrio de adsorción se han venido desarrollando a partir de los años
70 sobre la base de una descripción del mecanismo del proceso a fin de poder predecirlo en un
amplio intervalo de condiciones sin necesidad de una recalibración de los parámetros del modelo.
estos modelos describen la adsorción de especies iónicas sobre adsorbentes con cargas superficiales
variables que resultan de reacciones de protonación y desprotonación de grupos funcionales
presentes en la superficie del adsorbente. Son, por lo tanto, fuertemente dependientes del pH de la
solución acuosa. Estas cargas pueden aparecer en la sílice y en los óxidos de hierro, aluminio y
manganeso, constituyentes habituales de los materiales geológicos, así como en la materia orgánica
presente en el suelo. De ahí el interés de estos modelos para la predicción de la adsorción en
sistemas naturales. Los grupos funcionales responsables de la presencia de cargas variables son el
grupo hidroxilo en los óxidos y minerales arcillosos y los grupos carboxilo y fenol en la materia
orgánica.

Balances de Materia en Sistemas Naturales 41
La presencia de cargas en la superficie del adsorbente genera una distribución de carga en la
interfase sólido-solución. Para mantener la electroneutralidad del sistema, las cargas superficiales
deben ser contrabalanceadas en la disolución, generándose una doble capa eléctrica en las
proximidades de la interfase. Dicha doble capa está constituida por los centros cargados de la
superficie del adsorbente y por un exceso equivalente de iones de carga opuesta (contraiones) de la
fase acuosa que se acumulan en el agua próxima a la superficie de la partícula. Los contraiones son
atraídos electrostáticamente a la región interfacial estableciéndose un gradiente de concentración
de los mismos. Se genera así un potencial que limita la difusión libre de los iones hacia posiciones
alejadas de la superficie. Los procesos contrapuestos de atracción electrostática y contradifusión
extienden la carga en una región difusa en la que el exceso de concentración de contraiones es
máximo en la zona adyacente a la superficie de la partícula, disminuyendo gradualmente con la
distancia a la interfase sólido-solución. Esta distribución se muestra esquemáticamente en la Figura
1.
+
+
+
+
+
+
++
+
-
--
--
-+
+
--
-
--
-
-
-
-
SuperficieCapa fija
Capa difusaDisolución
Figura 1. Representación esquemática de la Doble Capa Eléctrica
En los modelos de formación de complejos superficiales se considera que los iones
adsorbibles se desplazan a través del campo eléctrico interfacial y reaccionan con los grupos
activos superficiales del adsorbente. Dichas reacciones son, básicamente, reacciones de cambio de
ligando (protón, cationes, aniones) con los grupos activos del adsorbente, que en la mayoría de los
absorbentes son grupos hidroxilo. Por ello, se denota como SOH al adsorbente, donde S representa
la superficie del mismo y OH el grupo activo.
Las reacciones consideradas en los modelos de formación de complejos superficiales son:
• Reacciones ácido-base superficiales

Modelos de Calidad de Aguas 42
SOH SOH H2+ +↔ + Ec.( 33)
SOH SO H↔ +− + Ec.( 34)
• Reacciones de adsorción de cationes
SOH M SOM H+ ↔ ++ + Ec.( 35)
• Reacciones de adsorción de aniones
SOH A H SOH A+ + ↔− +2 Ec.( 36)
El tratamiento de estas reacciones superficiales es análogo al de las reacciones de formación
de complejos en disolución. Así, el equilibrio de adsorción se caracteriza mediante una ley de
acción de masas para cada reacción superficial pero incluyendo un término exponencial que es, en
definitiva, un factor de corrección electrostático. Este factor contempla el efecto de la variación de
la carga superficial sobre las reacciones de formación de complejos superficiales. Estas reacciones
se pueden incluir en el esquema general de cálculo del equilibrio químico definido por las Ec.( 24)
y Ec.( 28) admitiendo que se puede asimilar el término de corrección electrostática a un
componente más del sistema y establecer un balance del "componente electrostático", equivalente
al balance de materia definido para los componentes puramente químicos. El balance de
componente electrostático es un balance de densidad de carga de manera que la densidad de carga
originada por las especies superficiales sea igual a la calculada aplicando la relación carga-
potencial característica del modelo de complejación superficial.
Sobre la base de este esquema general se han desarrollado diferentes modelos de formación
de complejos superficiales. Las diferencias entre los distintos modelos se encuentran en la
estructura asignada a la región interfacial, que determina la localización de los complejos
superficiales y la variación del potencial electrostático en la capa difusa. Así, en los primeros
trabajos (modelos de Capa Difusa (Stumm, 1970) y de Capacitancia Constante (Schindler y
Gamsjaer, 1972) se admite una estructura simple de dos capas.
Posteriormente se han ido incluyendo configuraciones más complejas de la región
interfacial mediante la introducción de distintos planos de adsorción para los diferentes adsorbatos.
Uno de los modelos de mayor aceptación es el modelo de Triple Capa (Triple Layer Model, TLM)
de Yates y col. (1974). En este modelo se distinguen tres planos (o, β y d) dentro de la doble capa

Balances de Materia en Sistemas Naturales 43
eléctrica presente en la interfase sólido-solución, como se muestra en la Figura 2. La capa fija de la
doble capa eléctrica "clásica" se divide en dos, que junto con la capa difusa constituyen las tres
capas que dan nombre al modelo. Cada uno de los planos se caracteriza por un potencial
electrostático (Ψ) y una densidad de carga (σ). C1 y C2 son las capacitancias interna y externa
respectivamente y εj las constantes dieléctricas.
Las distintas especies presentes en la disolución se adsorben en un plano o en otro
dependiendo, básicamente, de su afinidad por la superficie. En general, los cationes con menor
constante de hidrólisis y mayor carga son más fuertemente adsorbidos y experimentan reacciones
en la esfera interna para formar enlaces de coordinación con los centros activos de la superficie.
Los cationes adsorbidos en menor grado, como son los alcalinotérreos, forman complejos en la
esfera externa. Los aniones de bases débiles forman pares iónicos en la esfera externa, mientras que
los de bases fuertes experimentan reacciones de intercambio de ligando con los grupos hidroxilo de
la superficie del adsorbente.
ψο −
ψd −
ψβ −
σο σdσβ
C1, ε1 C2, ε2
εdisolución
Capa difusa
Figura 2. Diagrama del modelo de la Doble Capa Eléctrica
utilizado en el Modelo de Triple Capa.
El término exponencial de la ley de acción de masas de las especies superficiales adquiere
distintas expresiones en función de la localización y de la carga de los complejos superficiales
formados. Así, para las reacciones ácido base superficiales, consideradas como de formación de
complejos en el plano o:
[ ][ ][ ]SOH H SOH K
SOH
SOH HFRT
o+ ↔ =
+ ++
+22 exp Ψ Ec.( 37)

Modelos de Calidad de Aguas 44
[ ][ ][ ]
SOH SO H KSO H
SOHFRT
o↔ + = −
− +− +
exp Ψ Ec.( 38)
siendo F el número de Faraday.
Para electrolitos que se adsorben sobre el plano β:
[ ][ ][ ][ ]
( )SOH Na SO Na H K
SO Na H
SOH Na
FRTo+ ↔ − + =
−−
−
+ − + +− + +
+exp
Ψ Ψβ Ec.( 39)
[ ][ ][ ][ ]
( )SOH NO H SOH NO K
SOH NO
SOH NO H
FRTo+ + ↔ − =
− −
− + + −+ −
− +3 2 32 3
3
expΨ Ψβ
Ec.( 40)
Para especies que se pueden adsorber sobre ambos planos:
[ ][ ][ ][ ]SOH Pb SOPb H KSOPb H
SOH PbFRT
o+ ↔ + =
+ + ++ +
+2
2 exp Ψ Ec.( 41)
[ ][ ][ ][ ]
( )SOH Pb SO Pb H K
SO Pb H
SOH Pb
FRT
o+ ↔ − + =−
−−
+ − + +− + +
+2 2
2
2
2exp
Ψ Ψβ
Ec.( 42)
Veamos con un ejemplo cuál es el conjunto de ecuaciones que define el problema de
equilibrio químico en un sistema en el que tengan lugar las reacciones dadas por las Ec.( 37) a Ec.(
41). Dicho sistema se puede definir mediante el siguiente conjunto de componentes:
SOH, H+, Na+, NO3-, Pb2+, exp(-FΨo/RT), exp(-FΨβ/RT), exp(-FΨd/RT)
Las densidades de carga sobre los planos de adsorción vienen dadas por la diferencia entre
la carga positiva aportada por las especies catiónicas y la carga negativa aportada por las especies
superficiales aniónicas, y se calculan mediante los balances de componentes electrostáticos,
totalmente análogos a los balances de los componentes químicos dados por la Ec.( 28). Para el
ejemplo considerado,

Balances de Materia en Sistemas Naturales 45
[ ] [ ] [ ] [ ] [( )σo B SOH SOH NO SOPb SO SO Na= + − + − − −+ + − + − −2 2 3 ]+
])
Ec.( 43)
[ ] [(σo B SO Na SOH NO= − − −− + + −2 3 Ec.( 44)
donde B es el factor de conversión de concentración molar a densidad de carga (C/m2):
B Fsac
= Ec.( 45)
siendo s la superficie específica del adsorbente (m2/g) y ac la concentración de adsorbente (kg/m3).
La condición de electroneutralidad del sistema impone la restricción:
σ σ σβo d+ + = 0 Ec.( 46)
La densidad de carga en la capa difusa se puede calcular mediante la ecuación de Gouy-
Chapman:
( )σ εεd odRTI F
RT= −
82
12 senh Ψ Ec.( 47)
donde I representa la fuerza iónica.
En el modelo de Triple Capa, la relación entre la densidad de carga y el potencial
electrostático viene dada por:
[ ]σ βo oC= −1 Ψ Ψ Ec.( 48)
[ ]σ βd dC= −2 Ψ Ψ Ec.( 49)
Por último, se establece un balance de centros activos del adsorbente:
T N saSOH ads c= 103 Ec.( 50)
siendo:
TSOH: concentración total de centros activos (Kmol-centros/m3).

Modelos de Calidad de Aguas 46
Nads: densidad de centros activos del adsorbente (Kmol-centros/m2).
La densidad de centros activos y la superficie específica son características del adsorbente
que se determinan experimentalmente.
En todo momento, la suma de centros activos ocupados y libres será igual a la
concentración total de centros activos:
[ ] [ ] [ ] [ ] [ ] [T SOH SOH SO SO Na SOH NO SOPbSOH = + + + − + − ++ − − + + −2 2 ]+
3 Ec.( 51)
Las ecuaciones anteriores definen matemáticamente el problema de equilibrio químico.
Conocidos Nads, s, ac, C1, C2 y la concentración analítica total de los componentes químicos, las
incógnitas para este sistema son:
• La concentración en equilibrio de 10 especies: [SOH], [H+], [Na+], [NO3-], [Pb2+],
[SOH2+], [SO-], [SO--Na+], [SOH2
+-NO3-], [SOPb+]
• Ψo, Ψβ, y Ψd
• σo, σβ, y σd
y se dispone de un número igual de relaciones independientes:
• leyes de acción de masas (Ec.( 37) a Ec.( 41))
• balances de materia de los componentes químicos (H+, Na+, NO3-, Pb2+)
• balance de centros activos (Ec.( 51))
• relaciones electrostáticas (Ec.( 43), Ec.( 44), Ec.( 46) a Ec.( 49))
En la bibliografía (Kent y col., 1986, Dzombak y Morel, 1990) puede encontrase la
metodología para la determinación de los parámetros del modelo.
Programas para el cálculo del equilibrio químico.
Existen diversos programas para el cálculo del equilibrio químico en sistemas naturales.
Uno de los más utilizados, sobre todo en los problemas relacionados con aguas subterráneas, es el
programa MINTEQA2 de la USEPA (US Environmental Protection Agency). Este programa es la
versión actualizada de los anteriores MICROQL y MINEQL. Es básicamente un programa

Balances de Materia en Sistemas Naturales 47
FORTRAN para el cálculo de la concentración en el equilibrio de las especies químicas presentes
en un sistema hidrogeoquímico. Dispone de una amplia base de datos termodinámica que incluye
las reacciones de formación de más de 20000 especies químicas. Dichas especies comprenden
complejos en disolución, pares redox y multitud de especies sólidas, precipitados y formas
alotrópicas de minerales. Para cada especie, se recogen los datos de constante y estequiometría de
la reacción de formación así como parámetros para el cálculo de la variación de la constante con la
temperatura. Admite también el tratamiento de los procesos de adsorción mediante las isotermas de
adsorción y mediante modelos de complejación superficial (Capacitancia constante, Capa difusa,
Triple Capa).
A partir de la definición del sistema en base a un conjunto de componentes como se ha
descrito en apartados anteriores, el programa determina las especies químicas a considerar en el
problema, y resuelve, en un cálculo iterativo, las ecuaciones características del equilibrio químico
(Ec.( 24) y Ec.( 28)), proporcionando la concentración en el equilibrio de las especies disueltas,
adsorbidas y precipitadas.
Por su gran capacidad, este programa ha sido implementado, con ligeras modificaciones,
como módulo para la determinación de las interacciones físico-químicas, en diversos modelos de
transporte de contaminantes en aguas subterráneas entre los que se puede citar los programas
Hydrogeochem (Yeh y Tripathi, 1991) y Aquitrach (Marzal, 1992; Marzal y col., 1994).
2.3.1.1.2 Procesos controlados por la cinética.
En muchos sistemas naturales, los tiempos requeridos para alcanzar la condición de
equilibrio termodinámico pueden ser del mismo orden, e incluso superiores, a los tiempos
asociados a los cambios en la concentración generados por el proceso de transporte. En estas
condiciones, la velocidad con que el sistema se aproxima al equilibrio, es decir, la cinética de las
interacciones físico-químicas, puede influir significativamente en el proceso de transporte y en la
distribución de las especies contaminantes entre las fases presentes en el sistema.
las situaciones de "no-equilibrio" se atribuyen a varios factores. En el caso de las reacciones
homogéneas, es la propia cinética de las reacciones químicas la que determina que las
concentraciones de las especies químicas difieran de las correspondientes al equilibrio. En las

Modelos de Calidad de Aguas 48
reacciones heterogéneas, además de por la cinética química, la ausencia de equilibrio puede
resultar de la existencia de limitaciones difusionales a la transferencia de materia entre las fases.
Si el proceso de transporte está controlado por la cinética será necesario considerar, junto a
la ecuación general de transporte (balance de materia), la ecuación diferencial que describe la
cinética.
Cinética química.
En general, para una reacción reversible homogénea:
aA bB pP qQ+ + ↔ + +... ... Ec.( 52)
la velocidad neta de la reacción vendrá dada por la diferencia entre las velocidades de la reacción
directa e inversa:
[ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ]r r r K A B P Q K A B P Qd i d iA B P Q A B P Q= − = −α α α α β β β β.... ... .... ... Ec.( 53)
donde Kd y Ki representan las constantes de velocidad directa e inversa respectivamente y los
exponentes αA. αB, y βA, βB, ..., son los órdenes de reacción directa e inversa respecto a las
especies A, B, ....La cinética de las reacciones irreversibles se expresa de forma análoga,
considerando exclusivamente el término de velocidad de reacción directa. La variación de la
concentración de soluto debida a la reacción será:
rt
Ck
k ν∂
∂−= Ec.( 54)
donde νK es el coeficiente estequiométrico del componente k (negativo para reactantes).

Balances de Materia en Sistemas Naturales 49
Las constantes de velocidad de las reacciones químicas son términos cuyo significado físico no es,en ocasiones, evidente. Está claro que representan la velocidad de la reacción, pero no siempre suvalor nos dice “algo” de una manera rápida, lo cual en ocasiones puede ser muy necesario.
Tomemos por ejemplo el caso de una reacción irreversible de orden cero con respecto a losreactantes. Esto significa que αA es igual a cero y por lo tanto la variación de la concentración del
reactante con respecto al tiempo es: dKtkC
−=∂
∂
3
. Las unidades de la constante de velocidad son
g/m3d, por lo que si su valor fuera 0.2 g/m d podríamos afirmar sin dificultad que 0.2 gramos dereactante se transforman cada día por cada metro cúbico de volumen.
Ahora bien, supongamos que se trata de una reacción irreversible de orden 1. En este caso
kCdKtkC
−=∂
∂. Este tipo de transformación va a ser muy común en gran parte de los procesos
naturales que se van a estudiar. Las unidades de la constante de velocidad son de día-1. ¿Quésignifica, por ejemplo, que la constante de velocidad de la reacción tenga un valor de 0.1 día-1?. Sepuede interpretar, sin cometer un error excesivo, que el 10% de la concentración al inicio de cadadía se transforma durante ese día. Esta aproximación se basa en el hecho de que las funciones e-x y1-x tienen valores próximos para x pequeños. Cuanto menor sea x, más acertada será lainterpretación física de Kd. Por ello, si Kd tiene un valor de 6 día-1, no se puede decir que cada día setransformará el 600% del que había al inicio del día, pero sí se puede decir, con un error de un 4%aproximadamente, que cada hora se descompondrá el 25%, o que, de modo mucho más preciso,cada minuto se transformará el 0.42%.
Aunque las reacciones de adsorción son heterogéneas, se puede establecer una analogía con
las reacciones homogéneas a fin de establecer una expresión para la velocidad de reacción. La
estequiometría de la reacción de una molécula de adsorbato A y un centro activo del adsorbente S
se puede representar como:
A S A S+ ↔ − Ec.( 55)
donde A-S representa el complejo adsorbido. En una primera aproximación, admitiendo unos
órdenes de reacción iguales a los coeficientes estequiométricos, la velocidad neta de adsorción, es
decir, la velocidad de desaparición de soluto de la disolución debido al proceso de adsorción será:
r r r K C C K Cd i d A S i A= − = − −S Ec.( 56)

Modelos de Calidad de Aguas 50
En el caso de que el número de centros activos sea suficientemente superior al número de
moléculas de soluto en la solución se puede admitir CS constante, obteniéndose así una ley de
velocidad para la reacción directa de primer orden:
r r r K C K Cd i d A i A= − = − −'
S
S
Ec.( 57)
No obstante, algunos autores proponen ecuaciones de velocidad para el proceso de
adsorción más complejas que consideran la heterogeneidad del adsorbente, por ejemplo:
• Modelos con dos tipos de centros activos en los que la rápida adsorción sobre un tipo de centro
viene seguida por una reacción más lenta sobre el segundo tipo de centro:
r K C K C K Cd A d A i A= + − −1 2' ' Ec.( 58)
• Modelos con una distribución continua de reactividades de los centros activos:
( )( )
d CC
dtB B C
C
A S
A S e A S
A S e
−
− −
−
= −
1 2exp
Ec.( 59)
siendo (CA-S)e la concentración de complejo adsorbido en el equilibrio.
• Ecuaciones de velocidad no lineales:
r K C K Cd An
i A S= − −' Ec.( 60)
Cinética de transferencia de materia entre fases.
El tratamiento riguroso de los sistemas con reacciones heterogéneas controladas por la
cinética de la transferencia de materia entre las fases requiere la descripción cuantitativa del
movimiento de las especies hacia y desde la interfase en que tiene lugar la reacción heterogénea.
Para ello es necesario especificar la geometría de la interfase y el perfil de velocidades del agua en
las proximidades de dicha interfase. En la práctica, es difícil disponer de estos datos por lo que se
recurre a aproximaciones semiempíricas.

Balances de Materia en Sistemas Naturales 51
En general se admite que las reacciones de adsorción en medios poroso tienen lugar
mediante una serie de pasos consecutivos que incluyen una o varias etapas de difusión de solutos y
una o varias etapas de reacción química propiamente dicha. Un ejemplo de este tipo de serie sería:
Difusión en la película → Difusión intrapartícula → Etapas químicas
La difusión en la película corresponde a la transferencia de adsorbato desde el seno de la
fase líquida a través de la película líquida estanca inmediatamente adyacente a la superficie exterior
del adsorbente (difusión externa). La difusión intrapartícula, difusión interna, comprende la
transferencia de soluto desde el exterior del adsorbente hasta los centros superficiales internos por
los procesos de difusión en el poro y difusión superficial. Las etapas químicas corresponden a la
adsorción sobre los centros activos y su velocidad se define mediante las ecuaciones cinéticas
expuestas en el apartado anterior.
El estudio de la cinética de estos procesos se simplifica notablemente teniendo en cuenta
que la velocidad global del proceso viene determinada, en general, por una sola de las etapas
anteriores. Así, la formulación matemática del problema se realiza expresando la cinética global del
proceso mediante la ecuación que describe la cinética de la etapa más lenta.
La importancia relativa de los procesos de difusión externa e interna en adsorbentes
granulares depende, básicamente, del tamaño y porosidad del grano y del grado de saturación del
adsorbente. A mayor tamaño de grano, mayor es la distancia que debe recorrer el adsorbato dentro
de la partícula para alcanzar los centros activos de la superficie interna del adsorbente, y por lo
tanto, mayor es la importancia relativa de la difusión interna. Análogamente, cuanto mayor es el
grado de saturación de la superficie externa, más profundamente debe difundirse el adsorbato para
encontrar centros activos libres, incrementándose la contribución de la difusión interna a la
velocidad global del proceso.
La difusión molecular a través de una película estanca se describe a menudo mediante la
ecuación empírica:
( ) (q K C CA film f Af As= − ) Ec.( 61)
donde:

Modelos de Calidad de Aguas 52
(qA)film: densidad de flujo de soluto A a través del film (mol/m2 s).
Kf: coeficiente de transferencia de materia en la película (m/s).
CAf: concentración de A en el lado de la solución de la película (mol/m3).
CAs: concentración de A en el lado de la superficie del sólido de la película (mol/m3).
En la Figura 3 se muestra una representación esquemática del perfil de concentraciones en
la película. Las concentraciones interfaciales corresponden a la concentración de adsorbato en el
seno de la solución acuosa (Caf) y la concentración en equilibrio con la especie adsorbida (CAs).
Admitiendo que el soluto es adsorbido instantáneamente una vez alcanzada la superficie externa
del adsorbente, la variación con el tiempo de la concentración de especie adsorbida vendrá dada,
directamente, por el flujo de adsorbato a través de la película:
( )∂∂
Ct
K a C CA Sf Af A
− = − s Ec.( 62)
siendo a la superficie interfacial por unidad de volumen (m-1).
Disolución
PelículaAdsorbente
CKf
CKs
CA-S
Figura 3 Perfil de concentración en procesos controlados por la difusión externa
Para el tratamiento de la difusión intrapartícula se han propuesto modelos basados en la
aplicación de la ley de Fick asumiendo una difusividad efectiva y una geometría esférica de ka
partícula. Sin embargo, se puede utilizar una expresión simplificada análoga a la utilizada para la
difusión en la película, esto es, proporcional a una fuerza impulsora lineal:
( ) ( )( )q K C CA p p A S e A S= −− − Ec.( 63)
donde:

Balances de Materia en Sistemas Naturales 53
(qA)p: densidad de flujo intrapartícula de soluto A (mol/m2 s).
Kf: coeficiente de transferencia de materia intrapartícula (m/s).
(CA-S)e: concentración de especie adsorbida en equilibrio con la concentración de adsorbato en la
disolución (mol/m3).
CA-S: concentración promedio de especie adsorbida (mol/m3).
Con esta expresión, la velocidad de adsorción resulta proporcional a la diferencia entre el
grado de saturación en la superficie exterior de la partícula y el grado de saturación promedio
dentro de la partícula. El perfil de concentraciones en este caso se muestra en la Figura 4. La
ecuación cinética será:
( )( )∂∂
Ct
K a C CA Sp A S e A S
−− −= − Ec.( 64)
DisoluciónPelícula
Adsorbente
CK CA-S
(CA-S)e
Figura 4. Perfil de concentración en procesos controlados por la difusión interna

Modelos de Calidad de Aguas 54
2.3.2 Aguas superficiales.
2.3.2.1 Alcalinidad y pH.
La alcalinidad es una medida de la capacidad para neutralizar ácidos. Los principales
contribuyentes de la alcalinidad son los iones bicarbonato, HCO3-, carbonato, CO3
2-, y oxhidrilo,
OH-. Los conceptos de pH y alcalinidad se han introducido de una forma reciente en el campo de la
simulación de la calidad de las aguas. Ello ha sido debido principalmente a la degradación a la que
han sido sometidos los sistemas naturales debido a la lluvia ácida. Se emplea la alcalinidad como
variable de estado a partir de la cual se puede estimar el pH.
Las relaciones de equilibrio entre el dióxido de carbono CO2, y los aniones bicarbonato
HCO3- y carbonato CO3
2- son de gran importancia en ríos, lagos y estuarios. Su capacidad
"tamponadora" permite corregir los aportes de ácidos o bases al sistema. Un cambio del pH puede
tener efectos indeseables como aumentar la proporción de amonio no ionizado o variar la
concentración de metales disponibles en disolución.
Si la alcalinidad es baja, el CO2 puede ser el nutriente (es elemento fundamental para la
fotosíntesis) que limite el crecimiento del fitoplancton, si la disponibilidad de los otros nutrientes
es elevada. El intercambio con la atmósfera es la principal fuente de CO2.
La forma biológicamente más importante es el CO2, puesto que es el que sufre los procesos
de reaireación superficial, respiración, fotosíntesis, etc. Por otro lado, se combina con el agua para
formar ácido carbónico, un ácido débil que se disocia en iones bicarbonato y carbonato. Dado que
estas reacciones de disociación son muy rápidas comparadas con las otras reacciones biológicas y
químicas, se considera que el carbono inorgánico disuelto es la suma de dióxido de carbono,
bicarbonatos y carbonatos.
Las reacciones de equilibrio se expresan como sigue:

Balances de Materia en Sistemas Naturales 55
k OH + H _ OHk H + CO _ HCO
k H + HCO _ COHOCH _ OH + CO
w-+
2
2+-
3-3
1+-
332
3222
Ec.( 65)
siendo las constantes de equilibrio función de la temperatura:
C)º (25 10 4.3 = ]COH[
]H[ ]HCO[ = k 7-
32
+-3
1 Ec.( 66)
C)º (25 10 5.61 = ]HCO[
]H[ ]CO[ = k 11--3
+-23
2 Ec.( 67)
C)º(25 10 = ]OH[ ]H[ = k -14-+w Ec.( 68)
la alcalinidad se calcula mediante la siguiente expresión:
] H [ - ] OH [ + ] CO [ 2+ ] HCO [ = alcal +--23
-3 Ec.( 69)
y el carbono inorgánico total, CIT:
] CO [ + ] HCO [ + ]COH[ = CIT -23
-332 Ec.( 70)
Las ecuaciones (65)-(70) pueden resolverse para obtener los valores de las concentraciones
de H2CO3, HCO3-, CO3
2-, OH-, H+.
Una vez calculado el carbono inorgánico total y la alcalinidad, se puede determinar el valor
de [H+], y luego la concentración de CO2
]H[k k +
]H[k + 1
[CIT] = CO2+
21+1
2 Ec.( 71)
Otros procesos que también pueden afectar a la alcalinidad en sistemas acuáticos:

Modelos de Calidad de Aguas 56
- La adición de ácidos y la nitrificación reducen la alcalinidad. Si las aguas vertidas tienen
una fuerte componente residual, la oxidación del amonio a nitratos introduce en el sistema
gran cantidad de H+.
- La toma de nitratos por las algas aumenta la alcalinidad.
Por otro lado, la cantidad de CO2 en el medio natural está modificada por el intercambio
con la atmósfera (un proceso idéntico a la reaireación superficial), por reacciones de
disolución/precipitación en forma de CaCO3 y por los procesos de respiración y fotosíntesis. Estos
procesos son, en general, más lentos que las reacciones puramente químicas presentes en el
equilibrio del carbonato. Una vez definidos, es preciso introducirlos en un esquema matemático
que tenga en cuenta el equilibrio del carbonato. La clave del proceso reside en considerar que las
reacciones de equilibrio son mucho más rápidas que los aportes atmosféricos o los aportes y
consumos provocados por la respiración y la fotosíntesis. Si el cálculo se realiza mediante una
escala temporal del orden de días, se puede considerar que se ha alcanzado el equilibrio local con
respecto al ciclo del carbonato y el cálculo del carbono inorgánico total sólo depende de las
interacciones bióticas y atmosféricas.
2.3.2.2 Oxígeno disuelto y materia orgánica.
El oxígeno ha sido considerado tradicionalmente, y lo es todavía, como el principal
indicador de la salud de un sistema natural. En este apartado se van a estudiar los procesos
naturales más importantes a través de los cuales la concentración de oxígeno disuelto puede
aumentar o disminuir.
Previamente, se definen a continuación una serie de conceptos de interés, que se irán
desarrollando con posterioridad:
- Concentración de saturación de oxígeno disuelto en agua: es la máxima concentración de
oxígeno que puede permanecer disuelto en el agua para unas determinadas condiciones de
temperatura y presión.
- Déficit de oxígeno: es la diferencia entre la concentración de saturación y la concentración
real del oxígeno disuelto en el sistema natural.

Balances de Materia en Sistemas Naturales 57
- Demanda biológica de oxígeno límite: oxígeno total consumido por los microorganismos
para degradar cierta cantidad de materia orgánica.
La concentración de saturación, que se representará como Cs o como [O2]sat se puede
calcular por medio de la ley de Henry, la cual establece la relación que existe entre la
concentración de un gas disuelto en un líquido y la presión parcial de dicho gas en la atmósfera en
contacto con el líquido cuando se alcanza el equilibrio. Ambas magnitudes, concentración y
presión parcial, son directamente proporcionales a través de una constante: la constante de Henry,
He.
p = cHe Ec.( 72)
donde:
He : constante de Henry (atm m3/mol).
c : concentración de gas en el agua (mol/m3).
p : presión parcial del gas en la atmósfera (atm).
Los factores que influyen en el valor de la concentración de saturación de oxígeno en agua
son, por orden de importancia, temperatura del agua, sólidos disueltos (salinidad) y presión
atmosférica y presión parcial del vapor de agua.
Existen en la bibliografía diversas expresiones empíricas que relacionan la temperatura y la
salinidad del agua con la concentración de saturación de oxígeno disuelto. Una de las más
aceptadas es la siguiente (APHA, 1992):
[ ]
+−−−
−+−+−=
−2a
3
a
12
4a
11
3a
10
2a
7
a
5
sat2
T101407.2
T100754.1107674.1S
T10621949.8
T10243800.1
T10642308.6
T10575701.134411.139Oln
Ec.( 73)
siendo:
Ta : temperatura absoluta (K).
S : salinidad ().
[O2]sat: concentración de oxígeno disuelto (mg/L).
y estando la salinidad relacionada con la concentración de cloruros (mg/m3) de la siguiente manera:

Modelos de Calidad de Aguas 58
[ ]Cl80655.1S = Ec.( 74)
La disminución de la concentración de saturación con la altitud es debida a la disminución
de la presión atmosférica y, por lo tanto, de la presión parcial de oxígeno en el aire. El cálculo de la
concentración se puede efectuar considerando que la presión atmosférica desciende un 1% por cada
82 metros que se asciende. Esta aproximación da lugar a la siguiente expresión:
[ ] [ ] 82000,2,2
z
satzsat eOO−
= Ec.( 75)
en donde la altitud, z, viene expresada en metros.
Por otro lado, Zison (1978) ha desarrollado una expresión lineal:
[ ] [ ] [ zOO satzsat 1148.010,2,2 −= ] Ec.( 76)
en donde z viene expresada en kilómetros.
Por último, la humedad relativa también, aunque en pequeña medida, puede modificar la
concentración de saturación, dado que modifica las presiones parciales de los gases en la
atmósfera.

Balances de Materia en Sistemas Naturales 59
Temas de Ampliación. Procesos de Transferencia Gas/Líquido. Teoría de la doble capa. Existen diversas teorías que desarrollan diversos modelos matemáticos postuladas con el fin de representar el fenómeno de la transferencia de un gas entre un líquido en el que está disuelto y la atmósfera que lo rodea. Una de ellas, y probablemente la más aceptada, es la teoría de Withman-Lewis, más comúnmente conocida como la teoría de la doble capa. El movimiento de las moléculas de gas se efectúa en cuatro zonas:
- Una zona de gas libre, en la cual las moléculas de gas se mueven de manera aleatoria, con la energía correspondiente a la de su temperatura. La presión parcial del gas es constante, P.
- Una fina película de gas en las proximidades de la interfase con el líquido. En esta zona, el movimiento de las moléculas está regulado por el gradiente de presiones parciales del gas entre la interfase y la zona de gas libre.
- Una fina película de líquido en las proximidades de la interfase con el gas. El paso de moléculas de gas entre la interfase y la zona de líquido libre viene determinado por el gradiente de concentraciones entre ambos puntos.
- Una zona de líquido en donde las moléculas de gas disuelto se mueven con el líquido. La concentración del gas es constante, C.
Se asume que las zonas 1 y 4 están completamente mezcladas y que la máxima resistencia al paso de moléculas de gas entre el líquido y la atmósfera se da en las zonas 2 y 3. En ellas la transferencia de materia se produce mediante difusión molecular (Ley de Fick), por lo que el flujo de materia entre el seno del líquido y la interfase vendría dado por:
)( CiCLkLJ −= E igualmente entre la atmósfera y la interfase:
)( iPPaTR
GkGJ −=
en donde kL y kG son los coeficientes de transferencia de materia en la fase líquida y en la
gaseosa, respectivamente. Se considera que en la interfase, en equilibrio, se cumple la Ley de Henry, por lo que: iCHiP =E igualmente cuando se alcanza el equlibrio, los flujos de materia se igualan:
)()( CiCLkiPPaTR
GkLJGJAJ −=−===
Un problema importante es que no es posible determinar los valores de las concentraciones ni presiones parciales de gas en la interfase. Por ello, la expresión del flujo global se reescribe introduciendo dos nuevas variables: C*: concentración del gas en equilibrio con P.
Pi Ci
C
P
P*: presión parcial del gas en equilibrio con C. Ambas obtenibles a partir de la ley de Henry.
Así, )*()*( CCLKPPaTR
GKAJ −=−=
en donde KG y KL son los coeficientes globales de transferencia de materia. En el caso de que el gas sea poco soluble en el líquido (por ejemplo, el oxígeno) y/o que la resistencia en la fase gaseosa sea despreciable, la etapa limitante estará en la zona 3 y por lo tanto el flujo se puede expresar como:
)()*( CHP
LKCCLKAJ −=−=

Modelos de Calidad de Aguas 60
Las principales fuentes de oxígeno en un sistema natural son las siguientes:
• Aportes desde afluentes. Se trata del oxígeno disuelto que entra en el sistema a través de
diversos aportes. Se obtiene a partir de mediciones in situ de oxígeno disuelto y del caudal.
• Reaireación superficial. Cuando la concentración de oxígeno disuelto en el agua es inferior
a la de saturación, se produce una transferencia de oxígeno desde la atmósfera hacia el seno
del agua. En el caso, poco habitual pero posible, de que la concentración de oxígeno
disuelto en el agua sea superior a la de saturación, el flujo de oxígeno es desde el agua hacia
la atmósfera.
• Fotosíntesis. La síntesis de materia orgánica a partir de sales minerales y absorción de
energía por parte de algas y plantas verdes genera oxígeno en el sistema.
Como principales consumos de oxígeno en un sistema natural se pueden considerar los
siguientes:
• Degradación de la materia orgánica carbonosa. Los microorganismos aerobios heterótrofos
que metabolizan la materia orgánica presente en el medio consumen oxígeno.
• Nitrificación. La transformación biológica por medio de bacterias autótrofas del nitrógeno
amoniacal en nitratos da lugar a un consumo importante de oxígeno disuelto.
• Respiración de algas, plantas y animales.
• Oxidaciones químicas naturales.
• Demanda desde los sedimentos. La degradación de la materia orgánica presente en los
sedimentos (en ocasiones mucho mayor que la existente en la columna de agua) hace
disminuir la concentración de oxígeno en el agua intersticial de los mismos, promoviendo la
transferencia de oxígeno desde la columna de agua hacia los mismos. A esta transferencia
es a lo que se denomina demanda de oxígeno de los sedimentos (DOS).
A continuación se va a realizar una aproximación a los distintos procesos de consumo de
oxígeno.
2.3.2.2.1 Materia orgánica carbonosa.

Balances de Materia en Sistemas Naturales 61
Cuando se realiza un ensayo cuya finalidad es determinar la biodegradabilidad de la materia
orgánica de una agua residual, se suele observar que el consumo de oxígeno disuelto debido a la
degradación de la materia orgánica carbonosa se ajusta bastante bien a una cinética de primer
orden, como se puede comprobar en la Figura 5, según la expresión:
o bien
001
10
0
0
0
0001
1
1
1
==−=
=
===−=
−
−
−
tDBOC)e(DBOCDBOC(t)
LL(t)
tLLe L L(t)L 'ktdLd
t'kL
tk
t'k
Ec.( 77)
en donde:
L : Demanda Biológica de Oxígeno Carbonosa no ejercida (o materia orgánica carbonosa que
queda por consumir) (mg O2/L).
L0 : Demanda Biológica de Oxígeno Carbonosa Límite (o materia orgánica carbonosa total en el
instante inicial) (mg O2/L).
k1' : constante de velocidad de degradación de la materia orgánica o de desoxigenación (base e, s-1).
k1: constante de velocidad de degradación de la materia orgánica o de desoxigenación (base 10, s-
1).
t : tiempo (s).
DBOC: Demanda Biológica de Oxígeno Carbonosa ejercida (u oxígeno consumido en la
degradación biológica de la materia orgánica carbonosa) (mg O2/L).
A partir de las expresiones anteriores se puede determinar el valor de la Demanda Biológica
de Oxígeno Carbonosa Total en función de la medida para el tiempo de 5 días, llamada DBO5, que
suele ser lo habitual.

Modelos de Calidad de Aguas 62
E n s a y o la b o r a t o r io D B O
y = 4 4 1 , 8 e - 0 , 2 2 1 2 x
R 2 = 0 , 9 8 0 1
0
5 0
1 0 0
1 5 0
2 0 0
2 5 0
3 0 0
3 5 0
4 0 0
4 5 0
0 5 1 0 1 5 2 0 2 5
D ía s
D B O e je r c id a ( m g / L )
0
5 0
1 0 0
1 5 0
2 0 0
2 5 0
3 0 0
3 5 0
4 0 0
4 5 0
D B O r e s id u a l L
( m g / L )
Figura 5.- Curva experimental de la evolución de la DBO ejercida con el tiempo y curva calculada de la DBO residual
con el modelo exponencial obtenido.
Este modelo no diferencia entre materia orgánica disuelta y particulada y asume que la
degradación sigue una cinética de 1er orden.
Entre los diversos factores que afectan a la eliminación de materia orgánica, se pueden
destacar los siguientes:
• Temperatura. El aumento de la temperatura aumenta la actividad metabólica de los
microorganismos, favoreciendo la eliminación de materia orgánica y aumentando el
consumo de oxígeno disuelto. Esto es así hasta cierta temperatura a partir de la cual, según
el tipo de organismo, su actividad comienza a disminuir. Lo más habitual es reflejar la
dependencia de la constante con la temperatura mediante una expresión tal y como:
)20()20('1)('
1−= T
oxikTk θ , siendo un valor de θoxi habitual el de 1.047.

Balances de Materia en Sistemas Naturales 63
• Turbulencias del agua. Un aumento en la agitación de las aguas mejora la mezcla entre
distintas zonas del fluido, evitando la aparición de gradientes de concentración de materia
orgánica.
• Fracciones disuelta y particulada. La proporción de materia orgánica disuelta frente a la
particulada será determinante en el conocimiento de la cantidad de materia orgánica que
será descompuesta en la columna de agua y los sedimentos.
• Velocidad de sedimentación. Cuanto mayor sea esta velocidad de sedimentación, menor
será la cantidad de materia orgánica en la columna de agua y por lo tanto menor será la
demanda total de oxígeno.
• Tipo de materia orgánica. Lógicamente, la materia orgánica más biodegradable tendrá
valores de k1’ mayores, mientras que la más refractaria tendrá valores menores. Por
ejemplo, el agua residual urbana bruta suele tener un valor de k1’ entorno a 0.35 d-1,
mientras que una vez tratada el valor se sitúa en 0.2 d-1.
2.3.2.2.2 Nitrificación.
La nitrificación es un proceso natural mediante el cual los compuestos amoniacales
presentes en el medio son transformados en nitritos y posteriormente en nitratos. Se trata de un
proceso biológico según el cual, bacterias del género nitrosomonas obtienen energía del proceso de
oxidación del NH4+ a nitritos según:
NH4
++3.5 O2 NO2- + 2 H++H20
Mientras que bacterias del género nitrobacter hacen lo propio con los nitritos:
NO2
- + 1/2 O2 NO3-
El resultado final del proceso supone que por cada gramo de nitrógeno oxidable presente se
consumen 4.57 gramos O2 (3. 12 g en la primera fase y 1.14 g en la segunda).

Modelos de Calidad de Aguas 64
Una aproximación matemática sencilla a consiste en definir una variable DBON (mg O2/L),
similar a la DBOC, como la demanda biológica de oxígeno nitrosa. Esto es, el oxígeno que
consumiría una determinada cantidad de nitrógeno oxidable. Este nitrógeno se determina
analíticamente como Nitrógeno Kjeldhal Total (NKT) y representa la suma de nitrógeno orgánico y
nitrógeno amoniacal. La relación entre ambos parámetros es: DBON = 4.57 NKT
Los resultados experimentales muestran que la velocidad de transformación de la materia
nitrosa oxidable es proporcional a la que queda por oxidar, hecho muy similar al propuesto para la
DBOC:
Ec.( 78)
001
0
0
00
0
==−=
==
=−=
−
−
tparaDBONsiendo)e(DBONDBON(t)
tparaNNsiendo
e N N(t) N 'ktdNd
t'kL
t'kN
N
N
en donde:
N : Demanda Biológica de Oxígeno Nitrosa no ejercida (o materia nitrosa oxidable que queda por
consumir) (mg O2/L).
N0 : Demanda Biológica de Oxígeno Nitrosa Límite (o materia nitrosa oxidable total en el instante
inicial) (mg O2/L).
DBON: Demanda Biológica de Oxígeno Nitrosa ejercida (u oxígeno consumido en la oxidación
biológica de la materia nitrosa) (mg O2/L).
kN : constante de nitrificación (s-1).
t : tiempo (s).
El uso de este modelo es muy atractivo puesto que es muy sencillo y conceptualmente
similar al modelo de DBOC, muy empleado. El modelo asume que en el instante inicial todo el
nitrógeno está en forma oxidable (nitrógeno amoniacal), o bien que el nitrógeno orgánico presente
es mineralizado muy rápidamente a éste. Ambos casos pueden no ser siempre ciertos. La realidad
es que existirá cierto “retraso” en la aparición de la demanda de oxígeno nitrosa cuando se
produzca un vertido compuesto de una mezcla de nitrógeno orgánico y amoniacal.
Con el fin de representar de forma más adecuada el consumo de oxígeno, se hace necesaria
la introducción de más variables: nitrógeno orgánico (NO), amonio (NH4+), nitritos (NO2
-) y

Balances de Materia en Sistemas Naturales 65
nitratos (NO3-). Las ecuaciones correspondientes a las transformaciones entre estas especies son
ahora las siguientes:
][NO'ktd
][NOd
][NO'k][NH'ktd
][NOd
][NH'k[NO]'ktd
][NHd
[NO]'ktd
[NO]d
3nitri23
2nitri24nitri12
4nitri1min4
min
−−
−+−
+
=
−=
−=+
−=
Ec.( 79)
En donde:
[NO] : Concentración de nitrógeno orgánico (mg N/L).
kmin: constante de velocidad de mineralización (s-1).
[NH4+]: Concentración de nitrógeno amoniacal (mg N/L).
Knitri1' : constante de velocidad de nitrificación (1) (s-1).
[NO2-]: Concentración de nitritos (mg N/L).
[NO3-]: Concentración de nitratos (mg N/L).
knitri2' : constante de velocidad de nitrificación (2) (s-1).
t : tiempo (s).
Y el consumo de oxígeno viene dado por:
[ ]][NOk1.14]NH[k3.43
tdOd
2'nitri24
'nitri1
2 −+ −−= Ec.( 80)
En la Figura 6 se observa la evolución de la concentración de las distintas especies de nitrógeno a
partir de una cantidad inicial de nitrógeno orgánico de 16 mg N/L.

Modelos de Calidad de Aguas 66
Procesos de Nitrificación
02468
1012141618
0 5 10 15 20 25 30Días
(mg N/L)
Amonio N. Org. Nitritos Nitratos
Figura 6.- Curvas de evolución temporal de las distintas especies de nitrógeno en un proceso de nitrificación.
En la tabla siguiente se muestran algunos valores de las constantes de velocidad
relacionadas con las formas nitrogenadas.
Constante de
velocidad
Intervalo de valores (d-1) Valor medio Factor de corrección
con la temperatura, θ
Valor medio
kmin 0.1-0.4 0.25 1.02-1.09 1.055
Knitri1' 0.1-0.5 0.35 1.055-1.103 1.085
knitri2' 0.5-2.0 1.25 1.047-1.069 1.059
2.3.2.2.3 Fracciones disueltas y particuladas.
Conocer el estado físico de la materia orgánica, esto es, su fraccionamiento en una parte
disuelta y otra particulada es de suma importancia a la hora de establecer las demandas de oxígeno
en la columna de agua. La materia orgánica particulada sedimentará con una velocidad
determinada vs, que dependerá de numerosos factores (densidad de las partículas, forma, velocidad
de la corriente, etc.). Por ello, la presencia de materia orgánica sedimentable hará disminuir la
demanda directa en la columna de agua, aumentando la indirecta desde el sedimento. Dado que en
el sedimento, como se verá en el apartado siguiente, la degradación de la materia orgánica puede
seguir vías que no implican el consumo de oxígeno, el resultado global es un menor consumo de
oxígeno en la columna de agua.

Balances de Materia en Sistemas Naturales 67
Una aproximación a esta situación se puede establecer mediante la división de la variable
anterior materia orgánica carbonosa residual L, en otras dos: Lp y Ld. La materia orgánica disuelta
tan sólo sufriría procesos de biodegradación, mientras que la particulada, además de éste, también
sedimentaría. Los términos cinéticos son los siguientes:
P0PS1
P0PPSP1P
0
D0D1
D0DD1D
LLt)k'(keL (t)LL k -L'ktd
Ld0t
LLt'keL (t)L L'ktd
Ld
=+−=−=
=
=−=−=
Ec.( 81)
en donde:
LD : Demanda Biológica de Oxígeno Carbonosa Disuelta no ejercida (o materia orgánica carbonosa
disuelta que queda por consumir) (mg O2/L).
Lp : Demanda Biológica de Oxígeno Carbonosa Particulada no ejercida (o materia orgánica
carbonosa particulada que queda por consumir) (mg O2/L).
L0D , L0P : Demanda Biológica de Oxígeno Carbonosa Límite (o materia orgánica carbonosa
disuelta y particulada en el instante inicial) (mg O2/L).
k1' : constante de velocidad de degradación de la materia orgánica carbonosa o de desoxigenación
(s-1).
ks : constante de velocidad de sedimentación de la materia orgánica carbonosa particulada (s-1).
t : tiempo (s).
Esta aproximación asume que la materia orgánica particulada, mientras está suspendida en
el agua, también sufre procesos de biodegradación por parte de los microorganismos (bien por
ingesta directa o por una rápida hidrólisis y posterior biotransformación).
Unos modelos similares podrían establecerse para la demanda nitrosa de oxígeno, siendo
además la constante de velocidad de sedimentación igual a la de la materia orgánica carbonosa
(puesto que lo que sedimenta son partículas formadas por carbono, nitrógeno, fósforo, etc.).
2.3.2.2.4 Demanda de oxígeno desde los sedimentos.

Modelos de Calidad de Aguas 68
La primera aproximación al concepto de Demanda de Oxígeno desde el Sedimento (DOS)
es la que considera la demanda con un valor constante. Aunque sumamente útil en algunos casos,
este método no aporta nada sobre el cambio que se produce con el tiempo en la composición de los
sedimentos. Tampoco refleja el posible funcionamiento aerobio/anaerobio de los mismos. En
función de la concentración de materia orgánica particulada y de su velocidad de sedimentación se
puede estimar valores de la DOS muy variables, entre 0.05 y 65.00 g O2/m2 d.
Algunos autores han propuesto expresiones empíricas con el fin de determinar la demanda
béntica de oxígeno. Así, por ejemplo, Fair, Moore y Thomas proponen:
2/10 1601
16050314.0 aT tWWCYDOS+
+= Ec.( 82)
en donde:
DOS: Demanda de Oxígeno desde los Sedimentos (g de O2/m2 d).
Y0 : DBO5 del depósito béntico (g de O2/kg SV).
CT : Factor de corrección de la temperatura ( )( ))20(5
)(5
11
k
TkT
ee−
−
−
−=C
W: Tasa diaria de deposición de sólidos volátiles (kg SV/d).
ta : número de días en que se depositan sedimentos.
Otra expresión que relaciona la DOS con la presencia de materia orgánica en el sedimento
es la obtenida por Gardiner et al. (1984) en el lago Michigan:
DQODQODOS
+=
5.15666.7 Ec.( 83)
en donde:
DOS (g de O2/m2 d).
DQO (mg de O2/m3).
La demanda de oxígeno desde los sedimentos representa un caso típico de fuente/sumidero
(en este caso, propiamente sumidero) de naturaleza difusa.
Una aproximación más rigurosa al funcionamiento de los sedimentos divide a éstos en dos
capas: una aerobia y otra anaerobia. En ambas zonas el carbono y el nitrógeno se descomponen

Balances de Materia en Sistemas Naturales 69
produciendo una demanda de oxígeno desde el sedimento. En condiciones anaerobias la materia
orgánica origina metano:
CH 21 + CO
21 OCH 422 → Ec.( 84)
el cual es difundido hacia la zona aerobia en donde puede ser oxidado originando una demanda de
oxígeno desde el sedimento.
OH + CO 21 O + CH
21 2224 → Ec.( 85)
Si el contenido en materia orgánica es muy alto el metano producido puede escapar en
forma de burbujas, por lo que esta pérdida de carbono no se reflejará en un aumento de la demanda
de oxígeno.
Otro posible efecto importante es la mineralización del nitrógeno orgánico a amonio en el
sedimento. Este amonio puede pasar a la capa superior aerobia en donde puede transformarse en
nitratos con el consiguiente consumo de oxígeno. Si los nitratos fueran el producto final, el
consumo de oxígeno sería de 4.57 g O2/g N oxidado. Ahora bien, si la concentración de oxígeno es
muy baja, el proceso puede seguir hacia la formación de N2, gaseoso (desnitrificación), provocando
un menor consumo de oxígeno.
2.3.2.2.5 Fotosíntesis y respiración.
Tanto la fotosíntesis como la respiración son dos factores que pueden tener un impacto
importante en los recursos de oxígeno del sistema natural. Desde una perspectiva estacional, la
fotosíntesis tiende a predominar durante la época de crecimiento mientras que la respiración y
descomposición lo es en la época de no crecimiento. A corto plazo, en sistemas con alta
productividad biológica, las variaciones de oxígeno disuelto entre el día y la noche pueden ser muy
elevadas.
Las formas de determinar las cantidades de oxígeno disuelto implicadas en estos dos
procesos son diversas, pero se dividen en dos grupos:
- Métodos directos en el medio natural.
- Métodos indirectos a partir de mediciones de oxígeno disuelto o estimaciones de la
biomasa.
Uno de los métodos directos consiste en situar unas botellas con agua del medio en el seno
del mismo. Una de ellas es transparente, y la otra oscura. Por consiguiente, en la primera se dan los

Modelos de Calidad de Aguas 70
procesos de fotosíntesis y respiración, mientras que en la segunda sólo respiración. Se mide
inicialmente la concentración de oxígeno disuelto en cada botella y se sitúa en el medio.
Transcurrido cierto tiempo se sacan y se vuelve a medir el oxígeno disuelto. La diferencia entre
ambas permite calcular la actividad fotosintética.
Valores aproximados del aporte de oxígeno por unidad de carbono celular mediante la
fotosíntesis de algas se situan entre 1.6 y 2.67 mg O2/mg C. Igualmente, para consumos por
respiración, los valores típicos están entre 1.87 y 2.0 mg O2/mg C.
2.3.2.2.6 Reaireación superficial.
Como ya se vió en el apartado dedicado al mecanismo de intercambio de un gas a través de
una interfase líquido-gas, siempre que la concentración de oxígeno disuelto sea inferior a la de
saturación, se producirá una transferencia de este gas desde la atmósfera hacia el seno del agua. La
velocidad de ganancia de oxígeno disuelto por parte del agua es directamente proporcional al
déficit de oxígeno:
[ ] [ ] [ ]( )2222 ' OOk =
t dO d
sat − Ec.( 86)
De manera que cuanto mayor sea el déficit, mayor será la velocidad de recuperación de
oxígeno disuelto.
Entre los factores que afectan a la reaireación superficial se pueden mencionar los
siguientes:
• Velocidad de la corriente. Cuanto mayor sea la velocidad del agua en el sistema natural,
mayor será su capacidad de reaireación.
• Profundidad del lecho. A mayor profundidad, menor será la concentración de oxígeno
transferido a la columna de agua.
• Velocidad del viento. Cuanto mayor sea la velocidad del viento, mayor también será la
reaireación. Factor importante en lagos.
• Existencia de pequeñas presas, azudes, etc. Producen una pequeña reaireación extra.
• Formación de capa de hielo. Impide la transferencia de oxígeno a través de la interfase aire-
agua.

Balances de Materia en Sistemas Naturales 71
• Temperatura del agua. A mayor temperatura también aumenta el coeficiente de reaireación
superficial.
• Partículas suspendidas.
• Surfactantes. Impiden la transferencia de oxígeno a través de la interfase aire-agua.
Existen en la bibliografía numerosas expresiones para determinar el valor de la constante de
velocidad de reaireación k’2, a partir de la velocidad del agua en el río (v) y su calado (H). En la
tabla siguiente se muestran las más habitualmente empeladas junto con el intervalo de velocidades
y calados de aplicación.
Calado (m) 0.30 - 9.14 0.61 - 3.35 0.12 - 0.73 Velocidad (m/s)
O’Connor-Dobbins
Churchill
Owens-Gibbs
0.15 - 0.49
Hv. = k .
.
851
670'2 345
0.55 - 1.52
671
970'2 095 .
.
Hv. = k
0.03 - 0.55
Hv. = k .
.
51
50'2 933
En donde:
k’2 : constante de velocidad de reaireación a 20ºC (d-1).
v : velocidad de la corriente (m/s).
H : profundidad del lecho (m).
Las expresiones anteriores son directamente aplicables a ríos, en los cuales la constante de
velocidad viene afectada fundamentalmente por la velocidad y la profundidad. La reaireación
superficial en lagos está afectada por mecanismos distintos, siendo la velocidad del viento el factor
principal a tener en cuenta. Una de las expresiones propuestas para estos casos es la de Broecker
(1978):
H
U864.0 = 'k w2 Ec.( 87)
En donde Uw es la velocidad del viento (m/s) medida a 10 m sobre la superficie.
De los factores que más afectan a la constante de velocidad de reaireación superficial, sin duda
la temperatura del agua es el más importante. De igual manera que para otras constantes de

Modelos de Calidad de Aguas 72
velocidad, Lo más habitual es reflejar la dependencia de la constante con la temperatura mediante
una expresión como:
)20()20('2)('
2−= T
reakTk θ , siendo un valor de θrea habitual el de 1.026.
2.3.2.2.7 Resumen
Los términos cinéticos resultantes de todas las interacciones descritas en los apartados
anteriores son los siguientes:
Para la materia orgánica:
( )L
HfV
]NO[ X k -
-] NH[ X k +XLk a[fito] D = tL = S
MODsMO-3DENIT
20 - Tdnitdnit
+4NIT
20) - (TnitrinitriMO
ToxiOCMO
−−
−∂∂ −
186.2
' )20(1
θ
θθ
Ec.( 88)
donde:
L : concentración de materia orgánica (mg O2/L).
aOC : relación oxígeno/carbono en la respiración del fitoplancton (mg O2/mg C).
k’1 : constante de degradación de la materia orgánica (día-1).
θoxi : coeficiente de corrección de la temperatura para la degradación de la materia orgánica (s.d.).
fMOD : fracción disuelta de L.
2.86 : factor de equivalencia mg O2/mg NO3-
VsMO : velocidad de sedimentación de la materia orgánica particulada (m/día).
XMO : factor de influencia de la concentración de oxígeno disuelto en la degradación de la materia
orgánica.
[ ] kO][O
= XMO2
2MO +
Ec.( 89)
kMO : constante de semisaturación para la limitación del oxígeno en el proceso de degradación de la
materia orgánica (mg O2/L).
Para la concentración de oxígeno disuelto:

Balances de Materia en Sistemas Naturales 73
[ ] [ ]( )
[ ]H
DOS[fito] k- XLkNHXk
OOk FPa[fito]G = t
][O = S
20 - TresprespMO
ToxiNIT
Tnitrinitri
satT
reaNHNCkO
−−−
−−+
−+
∂∂
−+−
−+
θθθ
θ
1232'
1464
'11448
1232
)20(14
)20(
22)20(
22
42
Ec.(90
)
donde:
[O2]sat : concentración de oxígeno disuelto en saturación (mg O2/L).
k’2 : constante de velocidad de reaireación (día-1).
θ rea : coeficiente de corrección de la temperatura para la reaireación (s.d.).
DOS: demanda de oxígeno por parte del sedimento (mg O2/m2 día).
2.3.2.3 Interacciones fitoplancton-nutrientes.
En este apartado se va a realizar una introducción a la modelación del proceso de
eutrofización de las aguas naturales. Para ello se va a llevar a cabo la descripción de las relaciones
entre cada una de las especies que intervienen en la aparición de este problema. Todos los procesos
de transformación que se pueden dar entre las sustancias consideradas forman parte del término
fuente/sumidero de la ecuación general del balance de masa.
En la Figura 7 se muestran las interrelaciones consideradas entre nutrientes, fitoplancton,
material carbonoso y oxígeno disuelto y que constituyen un modelo de eutrofización.

Modelos de Calidad de Aguas 74
Figura 7. Esquema de los procesos incluidos en los modelos de eutrofización.
Las variables que se consideran en el modelo anterior son las siguientes:
En la columna de agua:
- Fitoplancton, medido como pigmento fitoplanctónico clorofila a y transformado en
unidades de mg C/L mediante una relación entre la clorofila a y el carbono celular.
- Fósforo orgánico disuelto (POD), fósforo orgánico particulado (POP), fósforo soluble
reactivo (PSR) y fósforo inorgánico particulado (PIP).Unidades mg P/L.
- Nitrógeno orgánico disuelto (NOD), nitrógeno orgánico particulado (NOP), amonio
(NH4+) y nitratos (NO3
-). Unidades mg N/L.
- Oxígeno disuelto (OD). Unidades mg O2/L.
- Materia orgánica, medida como Demanda Química de Oxígeno (MO). Unidades mg O2/L.
En el agua intersticial del sedimento:
- Fósforo soluble reactivo (PSR) y fósforo total (PT). Unidades mg P/L.
- Amoniaco (NH4+) y nitratos (NO3
-). Unidades mg N/L.
En la matriz sólida del sedimento:
- Fitoplancton, medido como pigmento fitoplanctónico clorofila a.

Balances de Materia en Sistemas Naturales 75
- Fósforo total, fósforo rápidamente intercambiable (PSR), fósforo adsorbido en arcillas (P-
Fe), fósforo asociado a compuestos de calcio (P-Ca) y fósforo orgánico (PO). Unidades mg
P/L.
- Nitrógeno orgánico (NO), amoniaco (NH4+). Unidades mg N/L.
- Oxígeno disuelto (OD). Unidades mg O2/L.
Evidentemente, en función de la problemática concreta del problema que se desee abordar,
el esquema anterior y el número de variables necesarias podrá ser disminuido, simplificándose todo
el proceso. A continuación se analizan las principales transformaciones físicas, químicas y
biológicas de los distintos componentes que participan en el modelo de eutrofización: fitoplancton,
fósforo, nitrógeno, oxígeno disuelto y materia orgánica.
2.3.2.3.1 Fitoplancton.
El estudio de la evolución de la población de fitoplancton es un componente importante de
los modelos de calidad de aguas. Algunos ejemplos de ello son los siguientes:
- La eliminación de nutrientes de un sistema natural es debida principalmente a su consumo por
parte de las algas.
- En el proceso de reciclaje de nutrientes intervienen los procesos de respiración y muerte del
fitoplancton.
- La variación de oxígeno disuelto durante el día que produce el fitoplancton puede ser muy
importante, puesto que durante las horas de luz aporta oxígeno mediante la fotosíntesis y
durante la noche lo consume.
- Las variaciones estacionales de oxígeno disuelto están también estrechamente ligadas a la
evolución del fitoplancton. Se pueden originar periodos de anoxia en zonas profundas de
sistemas naturales debido a la sedimentación del fitoplancton muerto y su descomposición.
- Las algas pueden modificar el pH del agua debido al consumo de CO2 durante la fotosíntesis y
su generación durante la respiración.
- Es el principal productor primario en numerosos sistemas naturales, por lo que juega un papel
fundamental en la cadena trófica.
- En sistemas hipereutrofizados es un factor importante en el incremento de la turbidez del agua.
- Proliferaciones excesivas ("blooms") de algas pueden dificultar los usos recreacionales del
agua.

Modelos de Calidad de Aguas 76
- Pueden presentar problemas de olor y sabor en sistemas de abastecimiento de agua y
atascamiento de filtros en sistemas de tratamiento de aguas.
- Pueden ocasionar problemas de toxicidad en las aguas.
Es por todo ello que la cinética del fitoplancton asume un papel central en los problemas de
eutrofización.
El término fuente/sumidero Sk de la ecuación general del balance de masas (ec.1, pág. 8)
que corresponde al fitoplancton representa la diferencia entre la velocidad de crecimiento del
fitoplancton y sus velocidades de muerte y sedimentación en el volumen de control considerado:
[fito] )k - D -(G = S sfitok Ec.( 91)
en donde:
Sk : término de fuente/sumidero (mg C/L día).
[fito] : concentración de fitoplancton (mg C/L).
G : constante de velocidad de crecimiento del fitoplancton (día-1).
D : constante de velocidad de muerte y respiración (día-1).
ksfito : constante de velocidad de sedimentación (día-1).
Las formas principales que se han llevado a cabo hasta este momento para abordar el
estudio del fitoplancton son dos:
- Considerando el crecimiento individualizado de los diferentes grupos de fitoplancton, con
sus correspondientes factores medioambientales.
- Englobando todo el fitoplancton en un único grupo funcional y considerando el total de
biomasa.
La primera aproximación se emplea cuando se desea conocer los problemas de
eutrofización asociados a variaciones estacionales de las especies predominantes. Son simulaciones
a largo plazo (meses o años). Es necesario especificar las diferencias entre los requerimientos de
nutrientes de los diferentes grupos, sus velocidades de crecimiento, composición celular,
velocidades de absorción de nutrientes, etc. Debido a la compleja tarea que supone recabar toda
esta información, este tipo de aproximación sólo se emplea si se desea conocer sucesiones
estacionales de los grupos de fitoplancton o problemas específicos como blooms de algas.

Balances de Materia en Sistemas Naturales 77
El tratar a todo el fitoplancton como un agregado y así emplear una única variable para su
definición simplifica el tratamiento del problema. Este método es particularmente útil en ríos en
donde la escala temporal del problema es menor y los problemas están relacionados con el oxígeno
disuelto, turbidez y nutrientes.
La cuestión siguiente que hay que resolver es cómo se representa la cantidad de biomasa del
sistema. Para ello existen varias posibilidades:
- Como número de células por unidad de volumen.
- Como biomasa en forma de peso seco.
- Como carbono por unidad de volumen.
- Como pigmentos fotosintéticos por unidad de volumen: clorofila a, por ejemplo.
La primera forma de medir la población de fitoplancton es apropiada cuando se trabaja con
especies simples con las que es relativamente sencillo realizar un recuento celular en laboratorio.
En sistemas naturales relativamente complejos puede presentar los siguientes inconvenientes:
• No es sencillo distinguir entre células viables (biológicamente activas) y aquellas que
no lo son.
• Las especies que tienden a formar colonias dan lugar a confusiones puesto que el
recuento no distingue células individuales y, además, los tamaños de las colonias son
variables.
• Es un método lento, difícil y económicamente costoso.
• Su fiabilidad depende del método de muestreo y submuestreo.
El peso seco de biomasa puede ser calculado empleando los volúmenes característicos de
cada especie identificada. Un problema importante es que el volumen suele variar en función de la
disponibilidad de nutrientes. En la práctica presenta numerosas dificultades que no la hacen útil
para su empleo como variable en los modelos de calidad de aguas.
La medida del carbono orgánico total de una muestra proporcionaría el carbono asociado a
la biomasa algal más el procedente de otras fuentes. Por sí solo no sería un buen indicador del
fitoplancton presente.

Modelos de Calidad de Aguas 78
El valor de la clorofila a puede ser empleado como una variable de agregación de todas los
grupos de fitoplancton. Este parámetro presenta las siguientes ventajas:
• Su medida es directa a partir de una muestra de agua por métodos espectrofotométricos
(fluorimetría).
• Integra todas las células, edades y estados fisiológicos.
• Proporciona células viables.
• Es un parámetro de la calidad del agua comúnmente aceptado y existe abundante
información sobre el mismo.
El principal inconveniente que plantea su empleo es que no diferencia entre los diversos
grupos de fitoplancton.
De lo anteriormente expuesto se deduce que ninguno de los parámetros citados es
enteramente satisfactorio como variable de agregación que responda de la población de
fitoplancton existente. Sin embargo, a efectos prácticos se suele emplear el valor de la clorofila a,
dado que, de este modo, la relación entre las ecuaciones de balance de materia para los nutrientes y
para el fitoplancton es más sencilla. La variable número de células se emplea únicamente en
aquellos problemas cuyo objeto de estudio es la dinámica del fitoplancton.
Una vez realizada la elección de la clorofila a como variable que representará la biomasa
fitoplanctónica, el último paso para completar esta conexión es establecer una relación entre la
clorofila a de las células y el carbono de las mismas. De esta forma se simularía la cantidad de
carbono presente en el sistema, el cual podría ser fácilmente transformado a clorofila a. Procesos
como la sedimentación de la materia orgánica algal y su demanda de oxígeno desde los sedimentos
son fácilmente tratados de este modo.
Los valores habituales de la relación entre el carbono y la clorofila a están entre 20 y 100
mg C/mg Cl a. El valor menor es habitual en zonas poco iluminadas, turbias, en las cuales tiende a
predominar un fitoplancton con mayor contenidos en pigmentos fotosintéticos con el fin de
aprovechar la luz. El valor mayor tiende a darse en sistemas oligotróficos, abundantemente
iluminados, en donde se necesita menos clorofila por unidad de carbono.
Composición celular.

Balances de Materia en Sistemas Naturales 79
Se asume habitualmente que las proporciones entre carbono, nitrógeno y fósforo en el
interior de la célula y, por consiguiente, sus relaciones estequiométricas son constantes. Esto no es
del todo cierto, puesto que estas relaciones varían con la especie, el tamaño de la célula, su estado
fisiológico, presencia de nutrientes, condiciones de luz y temperatura, etc.
En algunos trabajos se han empleado relaciones estequiométicas variables con el objetivo
de, por ejemplo, evaluar los efectos de la acumulación de nutrientes en la células de fitoplancton.
En ausencia de estudios concretos sobre las especies de fitoplancton que existan en nuestro
sistema, se pueden emplear valores obtenidos a partir de la bibliografía. Algunos valores típicos de
estas relaciones se muestran en las siguientes tablas:
Porcentaje de Biomasa en peso seco
Tipo de Alga C N P Cl a
Fitoplancton Total 40 - 50 8 - 9 1.5 5 - 10
Diatomeas 40 7.2 1.0 20 - 24
Algas Verdes 40 7.2 1.0
Cianofíceas 40 7.2 1.0 1 - 3
Dinoflagelados 37 - 47 3.3 - 5.0 0.6 - 1.1
Flagelados 40 7.2 1.0
Crisofíceas 35 - 45 7.8 - 9.0 1.2 - 3.0
Algas bénticas 40 7.2 1.0
Relaciones Nutrientes / Carbono
Tipo de Alga N/C P/C Si/C
Modelos de Calidad de Aguas 80
Fitoplancton Total 0.17 - 0.25 0.025
Diatomeas 0.18 0.024 0.6
Relaciones Nutrientes / Clorofila a
Tipo de Alga C/Cl a N/Cl a P/Cl a Si/Cla
Fitoplancton Total 50 - 100 7 - 15 0.5 -
1.0
Diatomeas 100 10 -
15
0.5 -
1.0
40 -
50
Algas verdes 25 - 100
Cianofíceas 14 - 67
Dinoflagelados 275 19.3
Crecimiento.
El crecimiento de la población de fitoplancton en el medio natural es una compleja función
de la radiación solar, la temperatura y el balance entre la disponibilidad de los nutrientes y su
requerimiento por parte del fitoplancton.
El término de crecimiento G viene dado por la siguiente expresión:
G G G C)º(20 G =G NITmáx Ec.( 92)
en donde:
Gmáx (20 ºC) : velocidad de crecimiento máximo a 20 ºC (dia-1).
GT : factor de influencia de la temperatura (adim.).
GI : factor de atenuación lumínica (función de T, I, f, D y Ke, adim.).
GN : factor de limitación por nutrientes (función de NID, PID, adim.).
siendo:
T : Temperatura del agua (ºC).
I : Radiación solar incidente (langley/día = cal/cm2 día).
f : Fracción de luz diaria (adim.).
D : Profundidad de la columna de agua.

Balances de Materia en Sistemas Naturales 81
Ke : Coeficiente de extinción lumínica (m-1).
PID : Fósforo inorgánico disuelto biodisponible (mg P/L).
NID : Nitrógeno inorgánico disuelto bioasimilable (mg N/L).
Hay que hacer notar que el efecto neto de la temperatura en la cinética del fitoplancton es
difícil de establecer. Por un lado, como se verá en el apartado siguiente, tiene un efecto directo en
la velocidad de crecimiento. Por otra parte, tiene un efecto indirecto en la intensidad lumínica de
saturación del fitoplancton.
Algunos valores de Gmáx para distintas especies de fitoplancton se muestran a continuación:
Tipo de Alga Velocidad de crecimiento máximo (día-1)
Fitoplancton Total 1.3 - 2.5 (20 ºC)
Diatomeas 2.0 - 2.1 (20 ºC)
Algas Verdes 1.4 (20 ºC)
Cianofíceas 0.7 - 1.0 (20 - 25 ºC)
Dinoflagelados 0.2 - 0.28 (20 ºC)
Flagelados 1.2 (20 ºC)
Crisofíceas 1.5 (Topt)
Cocolitofóridos 1.75 - 2.16 (25 ºC)
Algas bénticas 0.5 -1.5 (Topt)
Factor de temperatura
El término GT de la expresión anterior representa el efecto de la variación de la temperatura
en la velocidad máxima de crecimiento algal bajo condiciones óptimas de luz y nutrientes. El valor
de la velocidad de crecimiento máximo depende de la especie de fitoplancton considerada y de la
temperatura de referencia: 20 ºC, la temperatura óptima o cualquier otra.
Las funciones de temperatura que se emplean, aunque tremendamente variadas, suelen
clasificarse en alguno de estos tres tipos:
- Relación lineal entre la velocidad de crecimiento y la temperatura.

Modelos de Calidad de Aguas 82
- Curva exponencial de la velocidad de crecimiento con la temperatura.
- Curva de respuesta óptima, mediante la cual el crecimiento aumenta hasta una temperatura
óptima a partir de la cual disminuye.
La forma más simple de relación lineal tiene la forma:
( ) bTa TT
TTTT1
TTTT =G
minref
min
minrefminref
minT +=
−−−=
−− Ec.( 93)
en donde:
- Tmin es la temperatura para la cual la velocidad de crecimiento es cero.
- Tref es la temperatura de referencia a la cual corresponde el valor de Gmáx.
Si Tmin se elige igual a cero, la expresión anterior queda como:
refT
TTG = Ec.( 94)
En algunas ocasiones se emplea un conjunto de funciones lineales, con diferentes
pendientes, para distintos tramos de temperaturas. En algunos de estos casos se considera que la
velocidad de crecimiento aumenta de una forma lineal con la temperatura hasta que se alcanza un
máximo. Dicho valor máximo de la temperatura de crecimiento se mantiene aunque aumente la
temperatura.
La función de ajuste exponencial más empleada está basada en la ecuación de Arrhenius-
Van't Hoff:
−
=
1T2T10
1
210
KKQ
Ec.( 95)
en donde:
K1 : velocidad de reacción a la temperatura T1.
K2 : velocidad de reacción a la temperatura T2 .
Q10 : relación que existe entre las velocidades de reacción a incrementos de la temperatura de 10
ºC.
Q K = K )10
T-T(1012
12
Ec.( 96)
o bien,

Balances de Materia en Sistemas Naturales 83
f(T) )T(K ==Q )T(K = K(T)
ref
)10T - T(
10refref
Ec.( 97)
en donde f(T) es la función de ajuste de la temperatura, la cual se suele expresar de una forma más
simplificada como:
θ )T - (T)T - (T )101(
10Trefref =Q = G Ec.( 98)
siendo θ el coeficiente de ajuste de la temperatura cuyo valor suele oscilar entre 1.01 y 1.2. Un
valor de 1.072 correspondería a una duplicación de la velocidad de crecimiento por cada
incremento de 10 ºC de la temperatura. Lo más habitual es fijar la temperatura de referencia a 20
ºC. Con esta consideración se obtiene la expresión más comúnmente conocida para el valor de GT:
θ 20) - (TT = G Ec.( 99)
Generalmente se considera que el valor de θ permanece constante para todo el rango de
temperatura. En algunos casos se ha tomado este valor variable para distintos rangos de
temperatura. Valores típicos se sitúan alrededor de 1.066.
A partir de datos de crecimiento de especies individuales de fitoplancton en función de la
temperatura se pueden deducir ecuaciones que representen curvas de crecimiento óptimo. En estos
casos la velocidad de crecimiento aumenta hasta alcanzar un máximo a una temperatura
determinada, a partir de la cual vuelve a disminuir. La temperatura de referencia es en este caso la
temperatura a la que se registra el crecimiento máximo y la función de ajuste GT se normaliza para
que su valor sea igual a 1 a esa temperatura.
Un ejemplo de este tipo de crecimiento es el siguiente:
T - TT - T
2.3 - = Goptx
opt2
T exp Ec.( 100)
siendo:
Topt : Temperatura óptima a la cual el crecimiento del fitoplancton es máximo (ºC)
Tx : Tmin para T < Topt
Tmáx para T > Topt
Tmin : Límite mínimo de la temperatura para la cual el crecimiento del fitoplancton es nulo (ºC).
Tmáx : Límite máximo de la temperatura para la cual el crecimiento se detiene (ºC).

Modelos de Calidad de Aguas 84
Este tipo de curvas óptimas son más adecuadas que las lineales o exponenciales cuando se
trata de estudiar especies individuales o grupos funcionales, dado que el crecimiento se ralentiza
notablemente, llegando a detenerse, a temperaturas elevadas. Si la finalidad del estudio es analizar
la sucesión de distintos grupos de algas (diatomeas, algas verdes, cianofíceas) en el sistema natural
en función de la temperatura, el empleo de este tipo de formulaciones es el más correcto.
Limitación por nutrientes.
El crecimiento del fitoplancton está limitado por la disponibilidad de nutrientes. Los
nutrientes principales son carbono, nitrógeno, fósforo y sílice. Los micronutrientes tales como
hierro, manganeso, azufre, etc. no suelen ser considerados como limitantes para el crecimiento
algal.
El valor del término de crecimiento viene determinado por las concentraciones externas (en
la columna de agua) de nitrógeno inorgánico disuelto (amonio más nitratos) y las de fósforo
inorgánico disuelto. El carbono inorgánico (CO2) muy raramente es limitante puesto que es un gas
bastante soluble en agua y la difusión desde la atmósfera suele ser suficiente para cubrir las
necesidades del fitoplancton. En el caso de que se incluyan las diatomeas como grupo
independiente, también hay que incluir la concentración de sílice inorgánica disuelta. Se asume
que la composición en nutrientes de las células permanece constante, es decir, no se produce
acumulación de nutrientes en las mismas.
Se definen los términos de limitación nutricional GN para cada nutriente de la siguiente
manera:
CO + kCO = G
SID+ kSID = G
PID + kPID = G
NID + kNID = G
2mC
2CO
mSiSID
mPPID
mNNID
Ec.( 101)
siendo:
NID : Concentración de nitrógeno inorgánico disuelto (amonio+nitratos+nitritos, mg N/L).
PID : Concentración de fósforo inorgánico disuelto ( fósforo soluble reactivo, mg P/L).

Balances de Materia en Sistemas Naturales 85
SID : Concentración de sílice inorgánica disuelta (ortosilicatos, mg Si/L).
CO2 : Concentración de dióxido de carbono (mg C/L).
kmN : constante de semisaturación para el nitrógeno (mg N/L).
kmP : constante de semisaturación para el fósforo (mg P/L).
kmSi : constante de semisaturación para el silicio (mg Si/L).
kmC : constante de semisaturación para el carbono (mg C/L).
Los valores de las constantes de semisaturación son aquellas concentraciones de nutrientes
para las cuales la velocidad de crecimiento es la mitad del valor máximo. La representación gráfica
de las expresiones anteriores son hipérbolas en las cuales la velocidad de crecimiento es lineal
aproximadamente para concentraciones muy bajas de nutrientes pero aumenta gradualmente hasta
un máximo para elevadas concentraciones de los mismos (crecimiento de saturación). En este
punto, el nutriente ya no tiene efectos limitantes, por lo que un incremento de los nutrientes ya no
afectará a la velocidad de crecimiento.
Existen varias formulaciones que dan cuenta de la limitación nutricional GN, (Barnwell,
1985):
1) Formulación multiplicativa: el valor de Gn es el producto siguiente:
G G G G = G COSIDPIDNIDN 2 Ec.( 102)
Esta aproximación da una excesiva importancia al efecto conjunto de la escasez de
varios nutrientes y dicho efecto también dependerá del número de nutrientes considerados
en el estudio.
2) Formulación de mínimos:
)G ,G ,G ,G ( = G COSIDPIDNIDN 2Min Ec.( 103)
En esta formulación, el factor limitante más severo es el que limita por sí solo el
crecimiento.
3) Formulación de media armónica. Combina los inversos de cada factor limitante:
G1 +
G1 +
G1 +
G1
n = G
COSIDPIDNID
N
2
Ec.( 104)

Modelos de Calidad de Aguas 86
en donde n es el número de factores limitantes (en este caso, cuatro).
Esta formulación está basada en la analogía eléctrica de una serie de resistencias.
Incluye, pues, cierto grado de interacción entre los nutrientes limitantes. No es una
formulación tan severa como la multiplicativa. Es bastante similar a la de mínimos en un
amplio número de casos.
4) Formulación de media aritmética:
nG + G + G + G
= GCOSIDPIDNID
N2 Ec.( 105)
Como la anterior, considera el efecto combinado de diversos factores limitantes. No
es una aproximación muy correcta puesto que no restringe lo suficientemente el crecimiento
en algunos casos. Por ejemplo, si el fósforo biodisponible fuera nulo pero existieran los
demás nutrientes, el valor de Gn sería mayor de cero.
Algunos valores típicos de las constantes de semisaturación se muestran en la tabla
siguiente:
Constantes de Semisaturación Km... (mg/L)
Tipo de Alga Carbono Nitrógeno Fósforo Sílice
Fitoplancton Total 0.025 0.0005-0.03
Diatomeas 0.03 0.015-0.03 0.02 0.03
Algas Verdes 0.03 0.03-0.035 0.004
Cianofíceas 0.03 0 0.01-0.02
Dinoflagelados 0.005
Flagelados 0.03 0.08 0.012
Crisofíceas 0.03 0.015 0.02
Algas bénticas 0.03-0.1 0.05-0.1 0.004-0.008
El número de los factores limitantes a considerar depende de las especies de algas a estudiar
y de las especiales condiciones del sistema natural. Por ejemplo, el tener en cuenta la sílice sólo
será importante si la presencia de diatomeas puede ser relevante. Si el grupo predominante son
cianofíceas fijadoras de nitrógeno atmosférico no tiene sentido plantear una limitación nutricional

Balances de Materia en Sistemas Naturales 87
por parte del nitrógeno inorgánico disuelto. El carbono no suele ser limitante puesto que se
considera que existe siempre un exceso de CO2 en el sistema natural (como suele suceder). En la
mayoría de los lagos sólo se considera el estudio del fósforo por ser éste el nutriente claramente
limitante siempre. Lo mismo suele suceder en muchos estuarios con el nitrógeno.
El tratamiento de nutrientes que, como el nitrógeno, pueden presentarse en distintas formas
también puede ser diverso. En algunos casos no se distingue entre amonio, nitritos y nitratos y se
engloba todo en una única variable. En otros no se tiene en cuenta los nitritos por ser una especie
inestable que, en la mayoría de los casos pasa rápidamente a nitratos. En el caso de que se
consideren amonio y nitratos, es bastante habitual introducir un factor de preferencia hacia el
consumo de amonio frente al de nitratos por parte del fitoplancton.
Limitación lumínica.
El fitoplancton necesita la radiación solar para realizar la función fotosintética. La máxima
productividad se da, en condiciones de temperatura y nutrientes constantes, para una determinada
intensidad lumínica denominada intensidad óptima. El término de limitación lumínica al
crecimiento del fitoplancton se determina mediante la combinación de dos términos:
- Una relación que describe la atenuación lumínica de la luz con la profundidad.
- Una relación que define el efecto de la luz resultante en el crecimiento del fitoplancton.
La atenuación de la luz con la profundidad viene dada por la conocida expresión de
Lambert-Beer, mediante la cual se puede determinar la intensidad luminosa a una profundidad de la
columna de agua determinada:
e I = I z K -0z e Ec.( 106)
donde:
z : profundidad (m).
Iz : intensidad luminosa a una profundidad z por debajo de la superficie (langleys/día).
I0 : Intensidad luminosa en la superficie (langleys/día).
Ke : coeficiente de extinción lumínica (m-1).

Modelos de Calidad de Aguas 88
La intensidad luminosa en superficie es una función de la localización geográfica, época del
año, hora del día, condiciones meteorológicas y posibles sombras producidas por vegetación o
accidentes topográficos. El valor empleado corresponde únicamente al rango de luz visible, el cual
es aproximadamente un 50% de la radiación solar incidente. Casi toda la radiación no visible queda
atenuada en el primer metro de la columna de agua.
Un factor importante a tener en cuenta es el de la nubosidad. La modificación de la
intensidad luminosa incidente debido a este factor se puede representar matemáticamente con la
siguiente expresión (Kremer, 1978):
)071.01(0 C I = I 0máx − Ec.( 107)
en donde C tiene un valor entre 0 y 10 y representa la mayor o menor fracción del cielo cubierto
por las nubes.
El coeficiente de extinción lumínica depende del material suspendido presente en la
columna de agua. En casos sencillos, para períodos de tiempo cortos o en zonas donde la turbidez
no varíe en gran manera, el valor de Ke se puede considerar constante. En otros casos se emplea la
siguiente expresión para su estimación:
][M a + K = K iin
1=iee 0 ∑ Ec.( 108)
donde:
Keo : coeficiente de extinción por el agua pura (m-1).
[M]i : concentración de cualquier especie i que contribuya a la extinción lumínica (mg/L).
ai : coeficiente de extinción lumínica específico (L/mg m).
El empleo de la ecuación (108) permite incluir el efecto dinámico de la auto-atenuación
lumínica por parte del fitoplancton. De esta forma, el propio fitoplancton limita su crecimiento al
aumentar el valor de Ke.
En la práctica, la estimación del coeficiente de extinción se lleva a cabo a través de la
medida de la transparencia del agua. La transparencia se mide mediante la profundidad a la que se
deja de ver el disco de Secchi y por lo tanto se mide en unidades de longitud. Cuanto menor sea esa
longitud, mayor será las concentración de las sustancias que contribuyen a la extinción lumínica,
por lo que existe una relación inversamente proporcional entre el valor de Ke y la lectura del disco
de Secchi (Margalef, 1983). La relación tiene la forma:

Balances de Materia en Sistemas Naturales 89
Secchidisco prof.Cte = K e Ec.( 109)
La constante es un valor a determinar para el sistema natural objeto de estudio.
El segundo componente de la limitación lumínica es propiamente el valor de GL. Este
término se define como la relación entre los niveles de luminosidad ambiental y la velocidad de
crecimiento del fitoplancton. En un medio natural, la intensidad luminosa a la que está expuesto el
fitoplancton no es siempre la óptima. Así, pueden producirse fenómenos de fotoinhibición en la
superficie por un exceso de radiación, y en profundidad por un defecto de la misma. Existen dos
formulaciones básicas para este término:
- Relación de saturación lumínica. La velocidad de crecimiento aumenta de una forma lineal
con la luz a bajas intensidades pero va aumentando progresivamente hasta alcanzar un valor
máximo en el valor óptimo de intensidad luminosa, a partir del cual ya no aumenta más.
- Relación de fotoinhibición. Por debajo del valor óptimo se comporta como la anterior,
pero por encima se produce una disminución de la velocidad de crecimiento debido a
efectos de fotoinhibición.
Las curvas de saturación se describen habitualmente mediante una relación del tipo
Michaelis-Menten (similar a las descritas para la limitación nutricional):
I + K I = G
LL Ec.( 110)
en donde:
GL : Función de limitación lumínica para el crecimiento del fitoplancton.
I : Intensidad luminosa (ly/día).
KL : Constante de semisaturación que fija el nivel de intensidad luminosa para el cual la velocidad
de crecimiento es la mitad del valor máximo (ly/día).
La curva de fotoinhibición empleada más comúnmente en modelos de calidad de aguas es la
relación de Steele:
e II = G
II - 1
sL s
Ec.( 111)
donde Is es la intensidad luminosa de saturación.

Modelos de Calidad de Aguas 90
En algunas situaciones el valor de Is puede variar en función de la época del año, siendo
máximo en verano y mínimo en invierno.
Las algas adaptadas a bajos niveles de luminosidad tienen una respuesta más rápida a
cambios en la intensidad luminosa que las que están adaptadas a intensidades de luz altas. Las algas
se adaptan a los cambios de luz variando el contenido de clorofila a de sus células y las algas
adaptadas a bajas intensidades lumínicas tienen más clorofila.
En un sistema natural hay que integrar, para el espesor de cada capa de agua, las
expresiones anteriores puesto que la luz varía con la profundidad. Por otro lado, hay que introducir
un nuevo factor que dé cuenta de la variación diaria de luz (día/noche). Existen dos posibilidades
para esto:
• Integrar el valor de GL para las 24 horas del día introduciendo un factor multiplicador
denominado fotoperiodo. Este factor representa la fracción del día durante el cual hay luz.
Esta aproximación es útil en simulaciones en estado estacionario o en aquellas simulaciones
dinámicas cuyos intervalos de tiempo sean elevados (horas).
• Calcular dinámicamente el valor de I0 si el intervalo de tiempo empleado es de pocos
minutos. De esta forma se simularían los días y las noches y los efectos diarios de la
fotosíntesis.
En los modelos de eutrofización habituales es necesario obtener el valor de GI medio diario
en toda la columna de agua. La formulación de saturación quedaría de la siguiente manera para una
capa de agua de espesor H:
e I + KI + K
H Kf = G H K -0L
0L
eL
eln Ec.( 112)
en donde:
f : fotoperiodo (fracción del día).
H : profundidad del agua (m).
I0 : intensidad luminosa media en superficie durante las horas de luz.
O bien de la siguiente para una capa intermedia de agua:

Balances de Materia en Sistemas Naturales 91
e I + Ke I + K
)z - z( Kf = G
z K -0L
z K-0L
12eL
2e
1eln Ec.( 113)
donde:
z1 : profundidad de la parte superior de la capa de agua (m).
z2 : profundidad de la parte inferior de la capa de agua (m).
La formulación de fotoinhibición tiene la siguientes expresión:
)II (- - H) K (-
II - f
H Ke = G
s
oe
s
o
eI
expexpexp Ec.( 114)
en donde:
Ke : coeficiente de extinción lumínica (m-1).
H : espesor de la capa de agua (m).
Io : intensidad luminosa media diaria incidente sobre la superficie (langleys/día).
Is : intensidad luminosa de saturación (langleys/día).
f : fracción del día con luz solar (s.d.).
Esta expresión refleja el hecho de que existe una intensidad lumínica óptima para la cual el
factor de luminosidad es máximo, decreciendo para cualquier otro valor de la intensidad.
El valor de GI es un valor medio temporal y espacial que únicamente depende de la época
del año a través de los valores de f (tabulados) y de Io (tabulados). Es válido para simulaciones de
largos períodos de tiempo con intervalos de cómputo muy grandes. Este planteamiento no permite
analizar las variaciones que se producen del día a la noche. En sistemas naturales
hipereutrofizados, las variaciones noche/día de la intensidad luminosa pueden provocar variaciones
importantes en la concentración del oxígeno disuelto en el agua. Para tener en cuenta esta variación
en el estudio de la evolución del oxígeno disuelto se ha empleado una expresión similar a la
ecuación (113) pero que incluye la variación de la radiación solar incidente con la hora del día.
Dicha ecuación es la siguiente:
)II (- exp - H) K (- exp
II - exp
H Ke = G
s
0e
s
o
eI Ec.( 115)
siendo el valor de Io una función de la hora del día.
Con el fin de obtener una función que represente la variación diaria de Io, se definen dos
variables auxiliares, T1 y P1:

Modelos de Calidad de Aguas 92
2f - 1 = P
)86400
T( de entera parte - 86400
T = T
1
RR1
Ec.( 116)

Balances de Materia en Sistemas Naturales 93
en donde TR es el tiempo real transcurrido de la simulación (s). Si T1 es menor que P1 o si T1 es
mayor que (P1 + f) entonces significa que es de noche, con lo cual Io es igual a cero. En caso
contrario, se ajusta la intensidad luminosa en superficie mediante una función sinusoidal como la
siguiente:
))P - 0.5
1( )P - T( ( sen3.5 (tab)I = I1
11oo π Ec.( 117)
en donde Io (tab) es el valor de la intensidad luminosa media tabulada para el mes correspondiente.
Muerte y respiración.
El término D de la ecuación que define el término Sk para el fitoplancton representa la
desaparición de fitoplancton por los procesos de respiración y excrección, muerte por
parasitización, consumo por parte del zooplancton, etc. La expresión general para este término es:
Z(t)k + k + (T)k = D GDR Ec.( 118)
en donde:
kR(T): constante de velocidad de respiración endógena (día-1).
kD: constante de velocidad de desaparición no debida a depredadores: parasitización (infección de
las células de fitoplancton por otros microorganismos, materiales tóxicos, tales como cloro
residual, etc.) (día-1).
kG: constante de velocidad específica de consumo de fitoplancton por parte del zooplancton (L/mg
C día).
Z(t): concentración de zooplancton (mg C/L).
La respiración endógena representa los procesos mediante el cual el fitoplancton consume
su masa celular propia y su muerte natural, debida a factores internos. La constante de velocidad de
respiración endógena depende de la temperatura y se expresa mediante una ecuación de la forma:
θ 20) - (T RR C)(20k = (T)k º Ec.( 119)
en donde kR(20 ºC) es la constante de velocidad de respiración endógena a 20 ºC (día-1).
La parasitización representa la muerte por infección de las células por otros
microorganismos, compuestos tóxicos, situaciones de estrés producidas por deficiencia de
nutrientes o condiciones ambientales severas, etc. Es un término que da cuenta de todas aquellas

Modelos de Calidad de Aguas 94
pérdidas de biomasa no reflejadas en los otros dos procesos. Se puede expresar mediante una
ecuación similar a la de la respiración o mediante un valor constante.
El consumo por parte del zooplancton puede representarse de varios modos dependiendo
de si las poblaciones de depredadores no son simuladas en el modelo o, si, por el contrario lo son.
Si los depredadores no son explícitamente simulados, la dinámica presa-depredador no
puede ser considerada, por lo que se asume que la pérdida de fitoplancton por depredación es un
término constante, o bien depende de la población de fitoplancton, o de una población de
zooplancton definida por el usuario. Valores habituales de kG oscilan entre 0.1 y 1.5 (L/mg C día).
Valores para este término de mortalidad para el fitoplancton total son de 0.02 día-1, o de 0.03 día-1
para las diatomeas.
Si el zooplancton es simulado se puede obtener una representación bastante buena de la
dinámica presa-depredador en el sistema natural: el fitoplancton es consumido a una velocidad que
depende de la concentración de zooplancton, cuya población también depende de la concentración
de fitoplancton. Sin embargo, la simulación del zooplancton se suele realizar para ajustar mejor la
evolución de la población de fitoplancton antes que por él mismo. El término cinético mediante el
cual se puede estimar la variación de la concentración de zooplancton es muy similar al del
fitoplancton: crecimiento (dependiendo de temperatura y nutrientes, pero no de la luz), respiración,
y muerte. En este caso no existe sedimentación. La incorporación del zooplancton a los modelos de
calidad de aguas requiere, lógicamente, mucha más información para calibrar el modelo.
Sedimentación
La sedimentación del fitoplancton en aguas naturales depende de numerosos factores como
gradientes de densidad, turbulencias verticales, estado fisiológico del fitoplancton, etc. En
numerosos sistemas naturales es un mecanismo muy importante de eliminación de fitoplancton
vivo. La sedimentación del fitoplancton se ve notablemente disminuida en lagos poco profundos,
en donde la situación es de mezcla completa debido a las turbulencias provocadas por agentes
externos.

Balances de Materia en Sistemas Naturales 95
La velocidad de sedimentación del fitoplancton se puede estimar a partir de la ley de Stokes
ligeramente modificada puesto que las células no son partículas esféricas, para las cuales fue
desarrollada dicha ley:
Ec.( 120)
donde:
V : velocidad de sedimentación (m/día). sfito
g : aceleración de la gravedad (m/día ). 2
R : radio equivalente (basado en una esfera de volumen equivalente, m).
ρ : densidad de la célula (g/m ). p3
ρ : densidad del agua (g/m ). w3
ν : viscosidad cinemática (m /día). 2
F : factor de forma. s
El factor de forma tiene un valor mayor que 1 y representa todos los factores que reducen la
velocidad de sedimentación por debajo de la correspondiente a una partícula esférica. Por ejemplo,
se han empleado valores de 1.3 para diatomeas pequeñas y de 2 para diatomeas grandes. En la
práctica, esta expresión no se suele emplear muy a menudo: el reunir a todo el fitoplancton en un
único conjunto o en varios grupos funcionales impide una definición adecuada del radio
equivalente, el factor de forma o la densidad de la célula. Tampoco la ley de Stokes tiene en cuenta
las turbulencias o la velocidad del agua que tiende a mantener en suspensión las algas o a
resuspender parte de aquellas depositadas.
Lo más habitual es que el valor de V sea un parámetro de calibración y se especifique
como un coeficiente más del modelo. El valor de k viene dado por la relación entre V
F
) - ( R g
92 = V
s
wp2
sfito ν
ρρ
sfito
sfito y la
profundidad de la capa de agua: sfito
HV = k
sfitosfito Ec.( 121)
siendo:
ksfito : constante de velocidad de pérdida efectiva de algas (día-1).
Vsfito : velocidad neta de deposición de fitoplancton (m/día).
H: profundidad de la columna de agua (m).

Modelos de Calidad de Aguas 96
En la tabla siguiente se muestran algunos valores para el coeficiente Vsfito
Tipo de Alga Velocidad de sedimentación
(m/día)
Fitoplancton Total 0.05 - 0.5
Diatomeas 0.05 - 0.4
Algas Verdes 0.05 - 0.19
Cianofíceas 0.05 - 0.15
Dinoflagelados 8.0
Flagelados 0.5
Crisofíceas 0.5
Cocolitofóridos 0.25 - 13.6
El conjunto de expresiones desarrolladas en este apartado conducen a la expresión final
empleada en el modelo de eutrofización para el término fuente/sumidero para el fitoplancton, Skfito:
[ ] ( ) [ ]fitoH
V kkCkGGGCG
tfito
= Ssfito
GDT
RRNITmáxkfito
−−−−=
∂∂ − )20()º20(º20 θ
Ec.( 122)
2.3.2.3.2 Nutrientes.
Los nutrientes son esenciales para el desarrollo de las distintas formas de vida en los
ecosistemas acuáticos. Los nutrientes principales son carbono, nitrógeno, fósforo y sílice.
También son necesarias muy pequeñas cantidades de una serie de sustancias, que se
califican como micronutrientes: hierro, manganeso, azufre, cinc, cobre, cobalto y molibdeno. Estas
sustancias no suelen ser consideradas en este tipo de estudios puesto que siempre suele existir la
suficiente cantidad de las mismas en el entorno natural para el desarrollo de la vida.

Balances de Materia en Sistemas Naturales 97
De entre las razones por las que el estudio de los nutrientes reviste gran importancia se
pueden destacar las siguientes:
- La disponibilidad de nutrientes suele ser el factor principal que controla los "blooms" de
algas.
- El crecimiento de la biomasa fitoplanctónica está limitado por el fósforo o el nitrógeno,
salvo en el caso de las diatomeas cuyo nutriente limitante suele ser el silicio.
- Algunas algas cianofíceas pueden fijar nitrógeno atmosférico, con lo que introducen
nitrógeno en el sistema natural, que de otra forma no lo tendría.
- El carbono suele estar disponible en exceso, aunque en ocasiones puede también ser
limitante.
Además, el nitrógeno tiene un interés especial, debido principalmente a:
- El paso de amonio a nitratos consume oxígeno, siendo en ocasiones un componente
importante del balance de oxígeno disuelto.
- Si el pH del agua es elevado se pueden dar altas concentraciones de amonio no ionizado
que son tóxicas para peces y otros organismos.
En los ecosistemas acuáticos los nutrientes se pueden presentar de las siguientes formas:
- Inorgánicos disueltos.
- Orgánicos disueltos.
- Orgánicos particulados (detritos).
- Fijados en los sedimentos.
- Formando parte de los seres vivos (algas, plantas, zooplancton, peces, organismos
bénticos).
Únicamente las formas inorgánicas son asimilables por las algas. Éstas son:
- CO2 disuelto.
- Amonio, nitritos y nitratos.
- Ortofosfatos.
- Sílice disuelta.

Modelos de Calidad de Aguas 98
Cada nutriente sufre un contínuo reciclaje entre las distintas formas en que se puede
presentar. Por ejemplo, los nutrientes inorgánicos disueltos son extraídos del sistema por el
consumo del fitoplancton para su crecimiento. Estos nutrientes van moviéndose a lo largo de la
cadena trófica y distribuyéndose entre otros seres vivos. Parte de ellos van siendo excretados como
materia orgánica particulada que se hidroliza a formas solubles y mineraliza a formas inorgánicas.
Parte de la materia orgánica particulada sedimenta y contribuye de esta forma al balance de
nutrientes en el sedimento. Parte de esta materia orgánica en el sedimento es descompuesta aerobia
o anaeróbicamente, liberando nuevos nutrientes hacia la columna de agua.
Los procesos que afectan a cada uno de los nutrientes se clasifican según la forma en la que
éstos se presentan:
- Disueltos inorgánicos
- Consumo por parte del fitoplancton.
- Excrección.
- Transformaciones químicas.
- Hidrólisis de compuestos orgánicos disueltos.
- Descomposición de detritos.
- Descomposición de sedimentos y transferencia.
- Entradas externas.
- Disueltos orgánicos
- Excrección.
- Hidrólisis.
- Descomposición de detritos.
- Descomposición de sedimentos y transferencia.
- Entradas externas.
- Particulados orgánicos
- Excrecciones particuladas.
- Mortalidad del plancton.
- Descomposición.
- Sedimentación.
- Consumo del zooplancton.

Balances de Materia en Sistemas Naturales 99
- Entradas externas.
- Nutrientes en sedimentos
- Sedimentación de detritos.
- Sedimentación algal.
- Descomposición de sedimentos y transferencia.
Casi todas las transformaciones siguen cinéticas de primer orden de la forma:
S = C k(T) = tC
k∂∂ Ec.( 123)
en donde:
k (T) : constante de reacción de primer orden (día-1).
Dado que el estudio exhaustivo de todas las interrelaciones entre las distintas formas de los
nutrientes implicaría un alto grado de complejidad, se suelen realizar numerosas aproximaciones
con el fin de reducir el problema a una magnitud manejable. Algunas de ellas son:
- No considerar al carbono como nutriente limitante en ningún caso. Se supone que existe
un exceso de CO2 en el sistema.
- No considerar el ciclo de la sílice si no se va a tener en cuenta las diatomeas.
- No simular la dinámica de nutrientes en el sedimento de una forma explícita (el usuario
especifica los flujos).
- No considerar las formas orgánicas disueltas. Se simula el paso directo de formas
orgánicas particuladas a formas inorgánicas disueltas.
- No considerar la fijación de nitrógeno atmosférico por cianofíceas.
Un factor importante a tener en cuenta es la temperatura. Al igual que en el caso de la
materia orgánica, reaireación, fitoplancton, la temperatura afecta de una manera importante a la
evolución de las reacciones implicadas. La forma normal de introducir esta influencia es mediante
la modificación del valor de la constante cinética k empleando la relación exponencial de
Arrhenius-van't Hoff:
θ 20) - (T C)º(20k = (T)k Ec.( 124)

Modelos de Calidad de Aguas 100
Cinética del Nitrógeno.
El término cinético Sk para las distintas formas del nitrógeno incluye los siguientes
procesos principales:
- Amonificación: mineralización del nitrógeno orgánico disuelto por parte de microorganismos y
transformación a nitrógeno amoniacal.
- Nitrificación. Oxidación de amonio a nitritos y de nitritos a nitratos. Puede suceder en una etapa o
en dos. Está producido por bacterias autótrofas (nitrosomonas) que emplean estas reacciones como
fuente de energía:
H 2 + OH + NO _ O 23 + NH +
2-22
+4 Ec.( 125)
y bacterias del género nitrobacter:
NO _ O 21 + NO -
32-2 Ec.( 126)
La velocidad de crecimiento de las bacterias autótrofas es sensiblemente inferior a la de las
heterótrofas. Además, la conversión de nitritos a nitratos es mucho más rápida que la de amonio a
nitratos.
El proceso de nitrificación consume oxígeno, por lo que es un factor importante en el
balance de O2 del sistema. El consumo total de oxígeno es de 4.57 gramos de O2 por gramo de N
oxidado. En realidad, como algo de nitrógeno pasa a formar parte del tejido celular, la demanda de
oxígeno real se aproxima a 4.2 g O2/g N.
- Desnitrificación: reducción del nitrato a N2 en condiciones anóxicas. También se genera N2O,
pero dado que no suele tener mucha importancia en la calidad del agua no se suele considerar. Si
las condiciones del medio son anóxicas puede ser un mecanismo de eliminación del nitrógeno
importante, fundamentalmente en los sedimentos.
-Consumo por parte del fitoplancton: Amonio y/o nitratos son consumidos y acumulados en forma
orgánica por el fitoplancton. Usualmente, el amonio es consumido preferentemente. Se suele
emplear una expresión como la siguiente para representar dicho consumo preferencial:

Balances de Materia en Sistemas Naturales 101
])NO[+k( ])NH[+]NO([k] NH[
+ ])NO[+k( ])NH[+k(
]NO[] NH[ = FP -
3mN+4
-3
mN+4
-3mN
+4mN
-3
+4
]NH[ +4
Ec.( 127)
en donde:
[NH4+] : concentración de amonio (mg N/L).
[NO3-] : concentración de nitratos (mg N/L).
kmN : constante de semisaturación para el nitrógeno (mg N/L).
Si la concentración de nitratos es cero, el valor del Factor de Preferencia, FP es igual a 1,
lo que lógicamente significa que el fitoplancton consume únicamente amonio para su crecimiento.
Si la concentración de amonio es cero, el valor de FP también lo es, por lo que el fitoplancton
consumirá el nitrato disponible.
- Fijación de nitrógeno atmosférico: reducción de N2 a compuestos amoniacales por las
cianobacterias. Puede ser una fuente de nitrógeno importante en el sistema. En cualquier caso, el
consumo de nitrógeno inorgánico disuelto existente es prioritario sobre la fijación de nitrógeno.
- Partición entre amonio y amoniaco no ionizado. Ciertas formas de amonio no ionizado son
tóxicas para numerosas formas de vida, incluso a concentraciones bajas. Se considera que el
amonio no ionizado está en equilibrio con el amonio y los iones hidróxido:
OH 1) - (n + OH + NH_ OH n:NH_ OH n + (g)NH 2-+
42323 Ec.( 128)
Esta reacción es bastante rápida y está controlada por la temperatura y el pH.
En la Figura 8 se muestra un esquema de los procesos incluidos en el ciclo del nitrógeno.

Modelos de Calidad de Aguas 102
Figura 8. Esquema de las relaciones entre las diversas formas de nitrógeno.
Nitrógeno orgánico [NO].
El nitrógeno orgánico aumenta en la columna de agua a partir del fitoplancton muerto que
se descompone y disminuye por su mineralización y por la sedimentación del nitrógeno orgánico
particulado. Se admite que parte del fitoplancton muerto no se transforma en nitrógeno orgánico,
sino que se mineraliza directamente a amonio, siendo su disponibilidad por parte del fitoplancton
casi inmediata. La fracción que pasa a nitrógeno orgánico se representa mediante el término fNO.
Por otra parte, la mineralización del nitrógeno orgánico depende, al igual que sucede con el
fósforo, de la cantidad de fitoplancton presente.
La expresión cinética SkNO es la siguiente:
[NO] H
)f - 1 ( V -
-[NO] X k - f a[fito] D = t
[NO] = S
NODsNO
NRC20) - (T
minNminNNONCkNO θ∂
∂
Ec.( 129)
donde:
[NO] : concentración de nitrógeno orgánico (mg N/L).

Balances de Materia en Sistemas Naturales 103
aNC : relación nitrógeno/carbono en el fitoplancton (mg N/mg C).
fNO : fracción de fitoplancton muerto que da lugar a nitrógeno orgánico (s.d.).
kminN: constante de mineralización del nitrógeno orgánico (día-1).
θminN: factor de corrección de temperatura para la mineralización del nitrógeno orgánico (s.d.).
XNRC: factor de limitación a la mineralización del nitrógeno orgánico por parte del fitoplancton
(mg C/L):
[fito] + k[fito] = X
mNCNRC Ec.( 130)
kmNC: constante de semisaturación para la limitación de la mineralización del nitrógeno orgánico
por parte del fitoplancton (mg C/L).
VsNO : velocidad de sedimentación del nitrógeno orgánico particulado (m/día).
fNOD : fracción de nitrógeno orgánico disuelto (s.d.).
Nitrógeno amoniacal [NH4+]
El contenido en nitrógeno amoniacal en la columna de agua aumenta debido a la
mineralización del nitrógeno orgánico y a la fracción del fitoplancton muerto cuyo nitrógeno se
transforma a amonio (1-fNO). El amonio disminuye debido a su consumo como nutriente por parte
del fitoplancton y por el proceso de nitrificación. El consumo está afectado por la cantidad relativa
de amonio y nitratos, existiendo una preferencia de consumo hacia el primero (ec. 126). En el
proceso de nitrificación la concentración de oxígeno disuelto actúa como limitante, de forma que a
concentraciones superiores a 2 mg/L la reacción no se ve afectada, pero conforme va disminuyendo
la concentración de oxígeno disuelto, la velocidad de la reacción también lo hace. Este efecto se
introduce mediante una función del tipo:
[OD] + k[OD] = X
NITNIT Ec.( 131)
siendo kNIT la constante de semisaturación para la limitación del oxígeno a la nitrificación (mg O2
/L).
Debido a la importante actividad fotosintética que se registra en las horas diurnas en el lago,
el pH del agua presenta valores muy elevados. En esta situación, el equilibrio entre las formas
ionizadas (NH4+) y no ionizadas (NH3) se desplaza hacia la generación de esta última. Este efecto
es indeseable puesto que disminuye la cantidad de nitrógeno biodisponible y además, la forma NH3

Modelos de Calidad de Aguas 104
es tóxica. Para calcular la cantidad de amonio en forma de NH3 se emplea la siguiente serie de
expresiones (Barnwell, 1985):
T2729.92 + 0.09018 = pK
10 = RR + 1
X 14000 =] NH[
h
) pH - pK (
3
h
Ec.( 132)
donde:
[NH3] : concentración de amoniaco (mg N /L).
14000: factor de conversión (mg N / mol N).
X : concentración molar de nitrógeno amoniacal, NH3 + NH4+, (mol N/L).
pKh : constante de hidrólisis, función de la temperatura.
T : temperatura absoluta, en K.
La cantidad de nitrógeno amoniacal en forma biodisponible será, pues:
R + 1R X 14000 =
R + 1X 14000 - X 14000 = ]NH[ +
4 Ec.( 133)
Como sustancia disuelta, el nitrógeno amoniacal se desplaza entre la columna de agua y el
agua intersticial del sedimento, provocando un flujo cuya dirección dependerá de su concentración
en ambos medios. Este mecanismo se analizará en el apartado dedicado a los sedimentos.
Teniendo en cuenta las consideraciones anteriores y algunas de las expresiones
desarrolladas para el nitrógeno orgánico, la expresión final empleada en el modelo de eutrofización
para el término fuente/sumidero para el amonio, SkNH4, es el siguiente:
]NH[ X θk - a [fito] FPG -
- [NO] X θ k + )f - (1 a [fito] D = t
]NH[ = S
+4NIT
20) - (TnitrinitriNCNH
NRC20) - (T
minNminNNONC
+4
kNH
+4
+4 ∂
∂
Ec.( 134)
donde:
knitri: constante de nitrificación del amonio (día-1).
θnitri: factor de corrección de temperatura para la nitrificación del amonio (s.d.).

Balances de Materia en Sistemas Naturales 105
Nitratos
Los nitratos proceden de la nitrificación del amonio en condiciones aerobias. Su
disminución en la columna de agua es debida a su consumo como nutriente por parte del
fitoplancton y al proceso de desnitrificación. La desnitrificación está limitada por la presencia de
oxígeno disuelto en el agua. La forma de expresar esta dependencia es mediante una función del
tipo:
[OD] + kk
= XNO
NODENIT
3
3 Ec.( 135)
en donde kNO3 es la constante de Michaelis para la desnitrificación (mg O2/L). Cuando la
concentración de oxígeno disuelto es muy elevada, el valor de XDENIT es muy pequeño, por lo que
la desnitrificación apenas tiene lugar. La introducción del factor de preferencia de consumo de
NH4+ por parte del fitoplancton, FPNH4, provoca que el consumo de nitratos sólo se produzca
cuando la concentración del primero sea lo suficientemente baja. Como sustancia disuelta, los
nitratos se desplazan entre la columna de agua y el agua intersticial del sedimento, provocando un
flujo cuya dirección dependerá de su concentración en ambos medios. Este mecanismo se analizará
en el apartado dedicado a los sedimentos.
Teniendo en cuenta las consideraciones anteriores y algunas de las expresiones
desarrolladas para el nitrógeno orgánico, la expresión final empleada en el modelo de eutrofización
para el término fuente/sumidero para los nitratos, SkNO3, es la siguiente:
]NO[ X k -
-] NH[ X k + a[fito] )FP - (1G - = t
]NO[ = S
-3DENIT
20 - Tdnitdnit
+4NIT
20) - (TnitrinitriNCNH
-3
kNO +4
-3
θ
θ∂
∂
Ec.( 136)
donde:
[NO3-] : concentración de nitratos (mg N/L).
kdnit : constante de desnitrificación (día-1).
θdnit : factor de corrección de temperatura para la desnitrificación (s.d.).

Modelos de Calidad de Aguas 106
Cinética del Fósforo
La principal característica del fósforo como elemento natural es su escasez en la corteza
terrestre. Las diferencias fundamentales con el nitrógeno son:
- El nitrógeno tiene una fase gaseosa.
- Las formas inorgánicas de nitrógeno no se fijan en los sólidos de manera tan fuerte como
las de fósforo.
- La desnitrificación supone un mecanismo de eliminación que no tiene el fósforo.
El término cinético Sk para las distintas formas del fósforo incluye los siguientes procesos
principales:
- Mineralización del fósforo orgánico.
- Consumo por parte del fitoplancton.
- Adsorción a sólidos suspendidos y sedimentación.
Figura 9. Esquema de las relaciones entre las distintas formas de fósforo.

Balances de Materia en Sistemas Naturales 107
Para definir el ciclo del fósforo se emplean dos variables: fósforo orgánico y fósforo
inorgánico.
Fósforo orgánico.
La descomposición de fitoplancton muerto introduce fósforo en el sistema. Se asume que
parte del fósforo fitoplanctónico permanece como fósforo orgánico, mientras que otra parte se
transforma directamente a fósforo inorgánico biodisponible. Se introduce el término fPO como la
fracción del fósforo fitoplanctónico que aparece como fósforo orgánico. El fósforo orgánico
desaparece de la columna de agua por mineralización a fósforo inorgánico y por la sedimentación
del fósforo orgánico presente en forma particulada (1-fPOD).
La mineralización del fósforo orgánico está afectada por la concentración de fitoplancton
presente en la columna de agua (Ambrose, 1985). Esta influencia queda reflejada mediante un
factor de limitación representado por la siguiente función:
[fito] + K[fito] = X
mPCPRC Ec.( 137)
siendo kmPC la constante de semisaturación para la limitación del fitoplancton a la mineralización
del fósforo fitoplanctónico (mg C/L). Cuando la concentración de fitoplancton es mucho mayor que
la constante de semisaturación, el valor de XPRC tiende a 1, mientras que para valores de
fitoplancton próximos a kmPc la mineralización es mucho menor.
El término fuente/sumidero para el fósforo orgánico, SkPO, es el siguiente:
[PO] H
)f - 1 ( V -
- [PO] X k - f a [fito] D = t
[PO] = S
PODsPO
PRC20) - (T
minPminPPOPCkPO θ∂
∂
Ec.( 138)
siendo:
[PO] : concentración de fósforo orgánico (mg P/L).
[fito] : concentración de fitoplancton (mg C/L).
D : constante de velocidad de muerte del fitoplancton (día-1).
aPC : relación fósforo/ carbono en el fitoplancton (mg P/mg C).
fPO : fracción de fitoplancton muerto que origina fósforo orgánico (s.d.).
kminP: constante de mineralización del fósforo orgánico (día-1).

Modelos de Calidad de Aguas 108
θminP: factor de corrección de temperatura para la mineralización del fósforo orgánico (s.d.).
VsPO : velocidad de sedimentación del fósforo orgánico particulado (m/día).
fPOD : fracción de fósforo orgánico disuelto (s.d.).
H: espesor de la columna de agua (m).
Fósforo inorgánico
El fósforo inorgánico aparece por mineralización del fósforo orgánico y por
descomposición del fitoplancton muerto. Su disminución en la columna de agua es debida
fundamentalmente al consumo del mismo por parte del fitoplancton para su crecimiento y a la
sedimentación de sólidos (arcillas) que llevan adsorbidos parte del fósforo soluble. La fracción de
fósforo soluble que está asociada a arcillas depende de la concentración de sólidos suspendidos en
el agua y del coeficiente de partición entre la forma disuelta y la adsorbida. Dicho coeficiente de
partición refleja el equilibrio local entre la concentración de fósforo inorgánico en el agua y la
concentración de fósforo adsorbida sobre los sólidos del agua. Habitualmente, los coeficientes de
partición para el fósforo oscilan entre 1000 y 16000 mg P/kg SS. En algunos modelos de calidad de
aguas, la fracción de fósforo soluble que está asociada a arcillas se considera constante, por lo que,
a falta de otros datos, se suele dar un valor fijo a la fracción disuelta de fósforo inorgánico en el
agua (Ambrose, 1985).
El intercambio desde o hacia el sedimento es una fuente o sumidero importante de fósforo
soluble reactivo en la columna de agua. Lo habitual en sistemas naturales hipereutrofizados es que
el sedimento sea un contribuyente neto de fósforo biodisponible a la columna de agua y que
acumule fósforo soluble asociado a arcillas, que en algún momento pasarán al agua intersticial y
desde ahí a la columna de agua.
Teniendo en cuenta las consideraciones anteriores y algunas de las expresiones
desarrolladas para el fósforo orgánico, la expresión final empleada en el modelo de eutrofización
para el término fuente/sumidero para el fósforo inorgánico, SkPI, es el siguiente:
[PI] H
)f - (1 V - a[fito] G -
-[PO] X k + )f - (1 a[fito] D = t
[PI] = S
PIDsPIPC
PRC20) - (T
minPminPPOPCkPI θ∂
∂
Ec.( 139)
en donde:
[PI] : concentración de fósforo inorgánico (mg P/L).

Balances de Materia en Sistemas Naturales 109
VsPI : velocidad de sedimentación del fósforo inorgánico particulado (m/día).
fPID : fracción del fósforo inorgánico disuelto.
2.3.2.4 Sustancias tóxicas.
Los contaminantes convencionales provienen de actividades naturales y su acción sobre el
entorno consiste básicamente en alterar el ciclo de producción /descomposición de materia
orgánica.
Por el contrario, los compuestos tóxicos no aparecen de forma natural e interfieren
notablemente en los procesos naturales. Su detección y eliminación va encaminada hacia una
protección de la salud y en ningún caso por motivos estéticos, como sí sucede con los
contaminantes convencionales. Su estudio es tremendamente complejo debido a su elevado número
y a que no son especies simples. Una característica general es que su impacto en la naturaleza está
relacionado con la fracción de los mismos asociada a sólidos suspendidos.
2.3.2.4.1 Compuestos químicos orgánicos.
Los principales compuestos químicos orgánicos de los cuales se va a hablar a continuación
se pueden clasificar en los siguientes grupos:
Pesticidas Carbofuranos (carbamatos), aldrín, dieldrín,
DDT (clorados), Parathion (órgano-
fosfatados).
Origen: Aplicaciones de directas a cultivos, escorrentías de jardines y zonas urbanas,
descargas en aguas de origen industrial.
Impacto ambiental: Los pesticidas son rápidamente asimilables por la fauna acuática,
liposolubles (solubles en tejidos grasos), concentrándose a lo largo de la cadena trófica. Son
muy persistentes en el suelo y en los sedimentos.

Modelos de Calidad de Aguas 110
PCB's Bifenilos policlorados: Aroclor 128.
Origen: Transformadores eléctricos, pinturas, plásticos, insecticidas.
Impacto ambiental: Características similares a los hidrocarburos clorados.
Hidrocarburos Halogenados Alifáticos Trihalometanos: cloroformo.
Origen: Extintores de fuego, refrigerantes, disolventes de grasas y aceites, procesos de
lavado en seco. Cloración de aguas de abastecimiento y residuales.
Impacto ambiental: Los HAH ocasionan daños en el sistema nervioso central e hígado. No
son muy persistentes.
Éteres Halogenados 2-Cloroetil-vinil-éter.
Origen: Disolventes de polímeros.
Impacto ambiental: Son potentes agentes carcinógenos. Su toxicidad y evolución en medios
acuáticos todavía no está bien conocida. Extremadamente volátiles.
Hidrocarburos aromáticos monocíclicos 2,4-Dimetilfenol, Pentaclorofenol.
Origen: Fabricación de otros productos químicos, explosivos, pigmentos, colorantes...
Impacto ambiental: Depresor del sistema nervioso central. Puede afectar al hígado y
riñones.
Ftalatos-ésteres Bis(2-etilhexil)ftalato.
Origen: Producción de cloruro de polivinilo y termoplásticos.
Impacto ambiental: A bajas concentraciones tiene propiedades mutagénicas y teratogénicas.
Los invertebrados acuáticos son especialmente sensibles a sus efectos tóxicos. Es
persistente.

Balances de Materia en Sistemas Naturales 111
Hidrocarburos aromáticos policíclicos Antraceno, Benzo(a)pireno.
Origen : En labores de teñido, como intermediario en la producción de otros compuestos,
pesticidas, aceites, combustibles líquidos.
Impacto ambiental: Es carcinógeno en animales y se ha relacionado indirectamente con
cáncer en los seres humanos. Es biodegradable y poco persistente.
Nitrosaminas y otros Bencidina, Dimetil nitrosamina.
Origen : Producción de compuestos orgánicos y caucho.
Impacto ambiental: Ensayos de laboratorio han mostrado que son algunos de los más
potentes agentes carcinógenos.
Todos ellos han sido introducidos en la naturaleza por el ser humano y en muchos casos no
existen mecanismos de biodegradación de los mismos o son muy lentos.
Las principales transformaciones que sufren estos compuestos en los sistemas naturales son
las siguientes:
- Biotransformación.
- Hidrólisis.
- Oxidación.
- Fotólisis.
- Volatilización.
- Adsorción.
- Bioacumulación (Bioconcentración).
En la Figura 10 se muestra un esquema con estos procesos:

Modelos de Calidad de Aguas 112
Figura 10. Especiación, transporte y procesos de transformación en medios acuáticos (adaptado de Mills et
al., 1985).
Básicamente, los procesos de transformación que sufren los compuestos que se van a tratar
a partir de este momento son dos: reacciones de equilibrio y reacciones cinéticas. Mediante las
reacciones de equilibrio se determinan las fracciones disueltas y particuladas de las distintas
sustancias que sean objeto de estudio. Se obtendrán valores de fD, que permitirán estimar la
desaparición del material particulado vía sedimentación. Las reacciones cinéticas son en su
mayoría de primer orden. El valor global de Sk para un compuesto determinado vendrá dado por:
C k - = S ik Σ Ec.( 140)
en donde ki representa las constantes cinéticas de primer orden de todas las reacciones que afectan
al compuesto de concentración C.

Balances de Materia en Sistemas Naturales 113
Transformaciones biológicas.
Este concepto hace referencia a las transformaciones que sufren los compuestos orgánicos
por mediación de microorganismos. A menudo es el principal mecanismo de desaparición natural
de estas sustancias del medio. Se pueden dar bajo condiciones aerobias o anaerobias; por bacterias,
algas u hongos; mediante diversos mecanismos (desalquilación, deshalogenación, etc.).
Biodegradación: Reacciones cuyos productos finales son, eventualmente, CO2 y H2O.
Mineralización: Reacciones que siempre finalizan en CO2 y H2O.
Se reconocen habitualmente dos tipos de biodegradación:
- Metabolismo de crecimiento. Los compuestos orgánicos tóxicos se emplean directamente
como fuente de alimento para las bacterias.
- Co-metabolismo. El Co-metabolismo hace referencia a la transformación de un sustrato
que no puede ser usado como fuente de carbono o energía pero puede ser degradado en
presencia de otros sustratos (ej. DDT).
Este proceso se desarrolla en dos fases:
- Fase de aclimatación. Los tiempos de adaptación de los microorganismos a las
sustancias que entran en el sistema pueden variar entre 2 y 20 días, según sugieren
diversos autores (Mills, 1985). En medios donde la exposición a los compuestos
químicos es crónica este tiempo puede ser nulo. En medios donde la concentración de
degradantes sea baja, el tiempo de adaptación puede ser mayor. Para los casos en los
que la biodegradación esté limitada por la población microbiana, la adaptación es más
rápida para poblaciones iniciales altas de microorganismos, y más lenta para bajas
poblaciones iniciales.
- Fase de crecimiento. Una vez alcanzada la aclimatación, la biodegradación sigue una
cinética de primer orden (Michaelis-Menten).

Modelos de Calidad de Aguas 114
La utilización de un sustrato secundario es el empleo de éste a bajas concentraciones,
menores de las necesarias para el crecimiento, en presencia de uno o más sustratos primarios que
proporcionan carbono y energía.
En el caso del cometabolismo, rara vez es necesario un proceso de aclimatación, pero por
contra, las velocidades de la reacción son lentas comparado con el metabolismo de crecimiento.
El valor de Skb viene dado por la siguiente expresión:
C) + k( YX = k
C k - = S
M
máxb
bkb
µ
Ec.( 141)
en donde:
µmáx: máxima velocidad de crecimiento del cultivo.
X : concentración de biomasa.
Y : coeficiente de rendimiento (células producidas/tóxico eliminado).
kM : constante de semisaturación.
Los factores que afectan a la biotransformación son:
• Temperatura. Los efectos son similares a los que producen en la eliminación de DBO o
amonio.
• Nutrientes. Son necesarios para el crecimiento. Importante en medios oligotróficos.
• Oxígeno disuelto. En condiciones anóxicas numerosos compuestos orgánicos son
degradados más lentamente.
• Velocidad de aclimatación.
• Densidad de población o Concentración de biomasa. Es necesaria la presencia del
suficiente número de individuos para que la degradación progrese de una forma
eficiente.
Algunos valores de la constante de velocidad de biotransformación son:
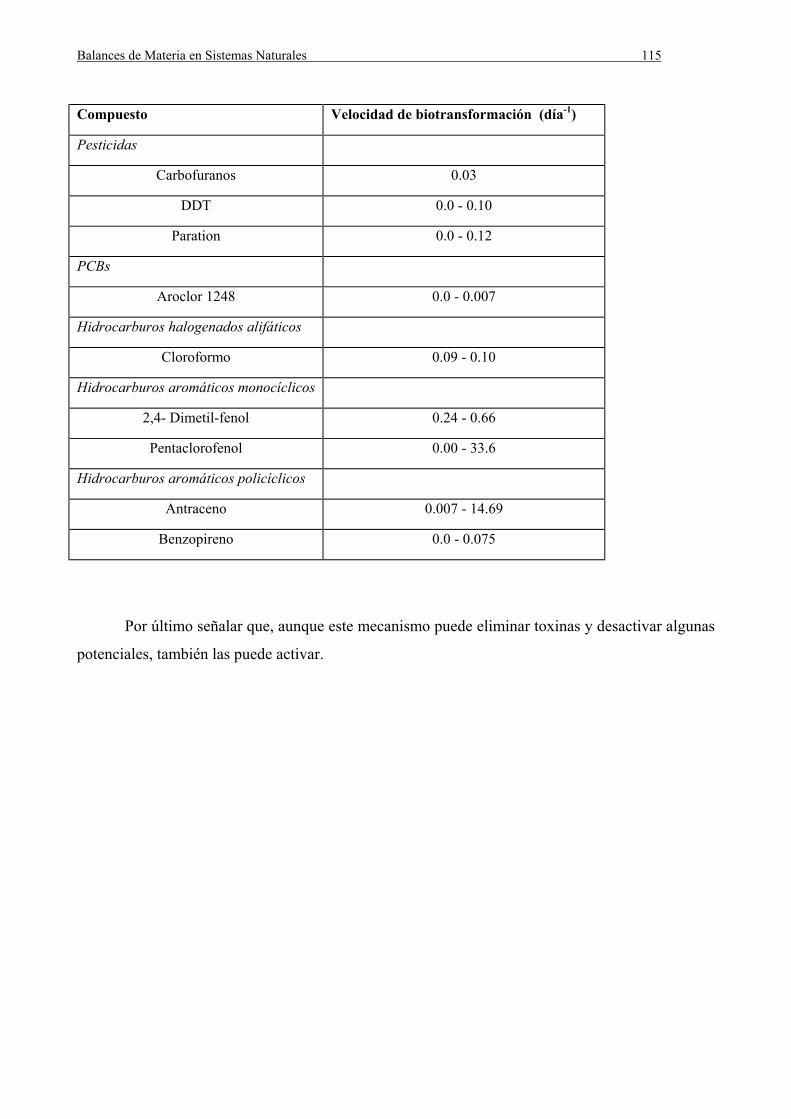
Balances de Materia en Sistemas Naturales 115
Compuesto Velocidad de biotransformación (día-1)
Pesticidas
Carbofuranos 0.03
DDT 0.0 - 0.10
Paration 0.0 - 0.12
PCBs
Aroclor 1248 0.0 - 0.007
Hidrocarburos halogenados alifáticos
Cloroformo 0.09 - 0.10
Hidrocarburos aromáticos monocíclicos
2,4- Dimetil-fenol 0.24 - 0.66
Pentaclorofenol 0.00 - 33.6
Hidrocarburos aromáticos policíclicos
Antraceno 0.007 - 14.69
Benzopireno 0.0 - 0.075
Por último señalar que, aunque este mecanismo puede eliminar toxinas y desactivar algunas
potenciales, también las puede activar.

Modelos de Calidad de Aguas 116
Hidrólisis química.
La hidrólisis química es el proceso mediante el cual un compuesto tóxico reacciona con el
agua. En concreto, un agente nucleófilo (hidróxido, agua o hidrogeniones), N, desplaza a un grupo
donante X. La hidrólisis no incluye reacciones ácido-base, de hidratación, de adición o
eliminación. Consiste en la destrucción de un enlace molecular y la formación de uno nuevo con
los componentes de la molécula de agua (H+, OH-). La reacción está fuertemente afectada por el
pH.
Los tipos de compuestos que son susceptibles de hidrolizarse son:
- Haluros de alquilo
- Amidas
- Aminas
- Carbamatos
- Ácidos de ésteres carboxílicos
- Epóxidos
- Nitrilos
- Ácidos de ésteres fosfónicos
- Ácidos de ésteres fosfóricos
- Ácidos de ésteres sulfónicos
- Ácidos de ésteres sulfúricos
Por ejemplo, un haluro de alquilo es degradado a su alcohol correspondiente mediante este
mecanismo:
H + Br + CH CHOH CH CH CH OH + CH CHBr CH CH CH +-322323223 _
La expresión cinética para la hidrólisis Skh es:
C] OH[ k - C k - C] H[ k - = S -HOHHN
+HHkh Ec.( 142)
donde:
kHH : constante de reacción de hidrólisis catalizada por ácidos (L/mol [H+] día) (0-107).
kHN : constante de reacción de hidrólisis neutra (día-1) (0-100).
kHOH : constante de reacción de hidrólisis catalizada por bases (L/mol [OH-] día) (0-107).
Está reconocida como la mayor vía de degradación de numerosos compuestos orgánicos.

Balances de Materia en Sistemas Naturales 117
Oxidación Química.
Las reacciones de oxidación en aguas naturales tienen lugar cuando los oxidantes (radicales
libres a menudo originados mediante reacciones fotoquímicas) se encuentran presentes en
concentraciones lo suficientemente elevadas como para favorecer dicha reacción. El cloro y el
ozono son los principales agentes oxidantes.
La descomposición de los compuestos tóxicos sigue una cinética de segundo orden, según:
C[oxid] k - = S oxkox Ec.( 143)
donde:
kox : contante de reacción de segundo orden (L/mol día).
[oxid] : concentración del agente oxidante (mol/L).
En aguas naturales, el oxidante es generalmente un radical libre presente en bajas
concentraciones. Si la velocidad de formación de estos radicales libres en aguas naturales es relati-
vamente constante, entonces la oxidación del compuesto tóxico puede ser considerada como una
reacción de primer orden:
C k - = S ,oxkox Ec.( 144)
En otros casos, si el agua está muy turbia o el cauce o lago es muy profundo, los radicales
libres tenderán a generarse únicamente en la superficie, cerca de la interfase aire-agua, con lo que
el proceso de oxidación perderá importancia. La oxidación por ozono está fuertemente influida por
el pH.

Modelos de Calidad de Aguas 118
Fototransformaciones.
Se entiende por fotólisis las reacciones de degradación iniciadas por la luz. Es una función
de la energía que incide sobre la molécula y el rendimiento cuántico del compuesto químico. Dicho
rendimiento cuántico se define como el número de moles de compuesto reaccionados por número
de einsteins absorbidos (un einstein es la unidad de luz en una base molar así como el cuanto o
fotón lo es en una base molecular).
El proceso interno sería como sigue: cuando la luz alcanza una molécula del contaminante,
el contenido energético de la molécula se incrementa, alcanzando un estado de excitación elec-
trónica. Dicho estado es inestable y la molécula alcanza un nivel de energía normal (más bajo) por
dos caminos:
- Emitiendo la energía que le sobra: procesos de fluorescencia y fosforescencia.
- Convirtiéndose en una molécula diferente mediante una nueva distribución electrónica,
distinta a la que presentaba en el estado excitado.
La fotólisis puede ser directa o indirecta. La fotólisis indirecta se produce cuando una
molécula intermedia absorbe inicialmente la energía y posteriormente la transmite al compuesto
tóxico, produciéndose su transformación. La expresión cinética Skfi sería como la siguiente:
C k - = C X k - = S ,fifikfi Ec.( 145)
en donde:
X : concentración de la especie intermedia (mol/L).
Para la fotólisis directa, la ecuación básica de degradación Skf es de la forma:
C k - = S fkf φ Ec.( 146)
en donde:
kf : constante de velocidad de absorción de luz por parte del compuesto tóxico.
φ : rendimiento cuántico de la reacción.
El rendimiento cuántico podría ser considerado como una medida de la eficiencia de la
fotoreacción. El valor de kf el producto de la intensidad luminosa I (a una profundidad z

Balances de Materia en Sistemas Naturales 119
determinada o un valor medio para un espesor de columna de agua dado)por la absorción de luz por
parte del compuesto químico. El valor de I para una profundidad z viene dado por la expresión:
e I = I z K - oz e Ec.( 147)
en donde Io es la intensidad de la luz en superficie y Ke es el coeficiente de extinción lumínica. Este
coeficiente es una función de las impurezas que lleve el agua y la longitud de onda de la luz
incidente. Se puede determinar mediante la siguiente expresión:
[SS] c +[COD] b +[Cla] a + K = K ewe Ec.( 148)
en donde a, b y c son coeficientes específicos de extinción luminosa, Kew es el coeficiente de
extinción para el agua pura y [Cl a], [COD] y [SS] son las concentraciones de fitoplancton, materia
orgánica y sólidos, respectivamente.
Volatilización.
La volatilización es un proceso de transferencia de un compuesto químico entre el aire y el
agua o viceversa. Su resultado no es la ruptura y desaparición del compuesto en cuestión, sino
únicamente su desplazamiento de un medio a otro. El proceso es similar a la reaireación en aguas
naturales y la velocidad a la que tiene lugar se puede relacionar con las velocidades de
transferencia de oxígeno conocidas, como se verá más adelante. La velocidad de volatilización
depende fundamentalmente del tamaño de la molécula (peso molecular).
Los modelos de trasferencia de un gas entre una fase líquida y una gaseosa están basados en
la teoría de la doble capa. Según esta teoría, la volatilización es una función de la constante de
Henry, la resistencia de la película gaseosa y la de la película líquida. La constante de Henry es una
relación entre la presión de vapor del compuesto químico y su solubilidad, como ya se comentó en
el apartado referente al oxígeno disuelto. El equilibrio se establece cuando la concentración
disuelta iguala a la presión parcial dividida por la constante de Henry. Sin entrar en los detalles, se
puede expresar la variación cinética Skv que sufre el compuesto tóxico de la siguiente manera:
C) - C( Hk = S s
Lkv Ec.( 149)
en donde:
Cs : concentración de saturación o solubilidad del compuesto tóxico (mol/L).
H : profundidad media del río (m).
C : concentración del compuesto en el agua (mol/L).

Modelos de Calidad de Aguas 120
kL : velocidad de transferencia global (m/día).
El coeficiente kL reflejaría la velocidad a la que el compuesto químico penetra en la
película estacionaria. Depende de la turbulencia del agua y de la atmósfera. Se puede relacionar
con el coeficiente de reaireación puesto que se ha comprobado que la relación entre kL y el
coeficiente de reaireación es constante independientemente de las condiciones de turbulencia del
sistema. Igual que éste, se puede estimar a partir de velocidades y profundidades del río o en
función de velocidades del viento en el caso de lagos o embalses.
La volatilización suele tener menor importancia en lagos y embalses que en ríos y
corrientes.
Adsorción.
La teoría de los procesos de sorción ha sido suficientemente desarrollada en el apartado
2.3.1.1.1. En este punto se va a mostrar su aplicación en aguas superficiales.
La adsorción de compuestos tóxicos a sólidos suspendidos y sedimentos es otro mecanismo
de transferencia bastante significativo. La partición de un compuesto entre la materia particulada
presente y la fase disuelta no es una vía de transformación, sino que únicamente relaciona los
estados disueltos y adsorbidos del compuesto.
El coeficiente de partición octanol/agua Kow es un parámetro básico en la evaluación de
este mecanismo y representa la medida de la solubilidad de un compuesto químico en agua. Cuanto
menos soluble es un compuesto en el agua, mayor será su tendencia a adsorberse en superficies de
sedimentos o microorganismos. La forma de determinar Kow es la siguiente:
- Se añade el compuesto químico a la mezcla de octanol puro (un disolvente no polar) y
agua pura (un disolvente polar). La relación de volúmenes entre el octanol y el agua se se-
lecciona para el valor estimado de Kow.
- La mezcla es agitada hasta que se alcanza el equilibrio.
- Se centrifuga con el fin de separar las dos fases. En cada una de ellas se determina la
concentración de compuesto químico.

Balances de Materia en Sistemas Naturales 121
- Kow es la relación entre la concentración de compuesto en la fase octanol y la
concentración en la fase agua. Es un valor adimensional.
La expresión cinética para el proceso de adsorción-desorción es la siguiente:
C k +] C - C[ C k - = S p2ppc1kad Ec.( 150)
en donde:
C : concentración disuelta de compuesto tóxico.
Cp : concentración particulada de compuesto tóxico.
Cpc : concentración máxima de contaminante que puede ser adsorbida por los sólidos.
k1 : constante de velocidad de adsorción.
k2 : constante de velocidad de desorción.
Para determinar la cantidad de compuesto adsorbido o disuelto se suele emplear la conocida
Isoterma de Langmuir. En estado estacionario es independiente de la concentración de sólidos y se
cumple que la cantidad adsorbida es lineal para bajas concentraciones de compuesto tóxico, pero
progresivamente, conforme aumenta la concentración del compuesto en el medio se alcanza el
valor de saturación.
C + kk
r C = r
1
2c Ec.( 151)
en donde:
r : cantidad de compuesto tóxico adsorbido por unidad de masa.
rc : capacidad máxima de adsorción por parte de los sólidos.
En general, la capacidad de adsorción de los sedimentos es inversamente proporcional al
tamaño de las partículas: arcilla>sedimentos inorgánicos>arena. La adsorción de compuestos
orgánicos también es función del contenido de materia orgánica del sedimento.
La separación entre formas disueltas y particuladas se realiza empleando las fracciones
disuelta y particulada fd y fp. La concentración total de contaminante puede expresarse como:

Modelos de Calidad de Aguas 122
m) k + (1m k = f
m) k + (1
1 = f
C f + C f = C + C = C
d
dp
dd
tpTdpT
Ec.( 152)
Las reacciones de adsorción alcanzan rápidamente el equilibrio y, por consiguiente, se
puede suponer que las relaciones cinéticas se desarrollan en estado estacionario. Esta situación se
suele denominar como de "equilibrio local" y refleja el hecho de que las reacciones de
adsorción/desorción son relativamente más rápidas que las demás cinéticas o procesos de
transporte que suceden en el sistema de estudio.
Bioconcentración.
La bioconcentración de compuestos tóxicos puede ser definida como la toma directa de
dichos compuestos a través de las agallas o el tejido epitelial de los organismos acuáticos. El
interés de este mecanismo es que puede ayudarnos a predecir el riesgo de exposición del ser
humano a los compuestos tóxicos por vía de la alimentación, especialmente del pescado.
Este proceso engloba otros dos procesos de sumo interés:
- Bioacumulación. Hace referencia al hecho de la toma de compuestos tóxicos por parte de
los peces desde un variado número de fuentes: presas, sedimentos, la misma bioconcen-
tración desde el agua.
- Biomagnificación. Es el proceso por el cual la bioacumulación aumenta en cada escalón
de la cadena trófica, desde los productores primarios hasta el máximo depredador.
La forma más habitual de medir este efecto en los seres vivos es mediante el índice BCF,
Factor de Bioconcentración. Es la relación entre la concentración en el organismo y la concen-
tración en el agua cuando se ha alcanzado el equilibrio después de varios días de exposición del ser
vivo al medio contaminado:
F k - BC k e =
tdF d
21 Ec.( 153)
en donde:

Balances de Materia en Sistemas Naturales 123
F : residuo del compuesto tóxico en el organismo, µg/kg.
e : eficiencia de la adsorción de compuestos tóxicos en las agallas.
k1 : constante de velocidad de bioacumulación, día-1.
k2 : constante de velocidad de depuración (excreción, etc), día-1 .
C : concentración del compuesto tóxico disuelta µg/L.
B : Biomasa del organismo, kg/L.
En estado estacionario:
C BCF = B kC k e = F
2
1 Ec.( 154)
El mecanismo intrínseco del proceso es similar al de la adsorción. En este caso los
compuestos orgánicos tóxicos tienden a una partición en los tejidos grasos de los peces u otros
organismos acuáticos. El significado del índice BCF es análogo al coeficiente de partición
sedimento-agua, Kp.
La mayor parte de los compuestos químicos de interés son de tipo hidrofóbicos (lipofílicos),
por lo que muestran una gran tendencia a la bioconcentración en los tejidos grasos de los seres
vivos. La concentración de sustancias lipofílicas en los tejidos de los seres vivos es esperable
puesto que la concentración de lípidos en las células es mucho mayor que en el agua.
Por último señalar que el hecho de que un compuesto químico se bioacumule es indicación
de su resistencia a la biodegradación y su persistencia en el medio.
2.3.2.4.2 Compuestos químicos inorgánicos: metales pesados.
Por el término "metales pesados" se entienden habitualmente aquellos metales situados
entre el número atómico 21 (escandio) y el 84 (polonio), que aparecen en las aguas naturales desde
fuentes antropogénicas o naturales. También se incluyen tradicionalmente los no-metales Arsénico
y Selenio y el Aluminio. En ocasiones, dependiendo de su concentración y especiación química
(forma como se presenta en un momento dado en el medio natural) pueden ser tóxicos para los
organismos acuáticos.

Modelos de Calidad de Aguas 124
Las principales características de estos contaminantes, que los diferencian de los anteriores
son las siguientes:
- Su origen tiene una importante componente natural: disolución de estratos geológicos o
actividad volcánica.
- En la naturaleza, la cantidad total de metal se mantiene constante: únicamente cambia su
especiación química, es decir, la forma como aparecen en cada momento.
Los metales pueden formar complejos solubles con ligandos orgánicos o inorgánicos.
El principal mecanismo por el cual un metal es "secuestrado" del medio es por adsorción en
superficies sólidas. Este proceso presenta algunas diferencias con respecto a la adsorción de com-
puestos orgánicos descritos en el apartado anterior. En ellos se cumplía la regla de "lo semejante
disuelve a lo semejante" y los compuestos orgánicos se adsorbían en la matriz orgánica de los
sólidos suspendidos o los sedimentos. En el caso de metales existen tres vías para realizarse este
fenómeno:
• Adsorción física en superficies sólidas.
• Adsorción química o ligadura por ligandos en la interfase agua-sólido.
• Intercambio iónico con otro ión en la interfase sólido-agua.
Además, si el metal pesado es acomplejado en disolución por un ligando de tipo orgánico
puede ser adsorbido tal y como si fuera un compuesto orgánico.
La formulación matemática que describe la partición de un metal entre una fase sólida y una
fase acuosa es llamada "coeficiente de distribución" para metales pesados o coeficiente de
partición.
M CC = k p
D Ec.( 155)
en donde:
kD : coeficiente de distribución (L/mg).
Cp : concentración de metal en la fase adsorbida (mg/L).
C : concentración de metal en la fase disuelta (mg/L).
M : concentración de sólidos (mg/L).

Balances de Materia en Sistemas Naturales 125
El coeficiente de distribución o coeficiente de partición es función de carácter del
adsorbente: propiedades mineralógicas. estructura química, composición, propiedades eléctricas,
etc.
Para un completo conocimiento de la situación del medio natural se requiere, además de la
distribución del metal entre la fase líquida y la sólida: su especiación. Un metal puede tener distinto
nivel de toxicidad en función de la forma como esté en la naturaleza. Esto puede conducir a la
definición de estándares de calidad del agua específicos para cada zona considerada. Por ejemplo:
una zona que tenga una gran cantidad de materia orgánica disuelta de origen natural puede no
necesitar unos estándares de calidad de agua muy severos puesto que los compuestos orgánicos
pueden acomplejar a los metales y hacerlos no tóxicos para los seres vivos.
A continuación se va a realizar una introducción al proceso de equilibrio y reacciones
cinéticas para una serie de importante de metales pesados: Cd, As, Hg, Se, Pb, Ba, Zn, Cu. Estos
elementos son frecuentemente citados en la literatura por su carácter tóxico y carcinogénico sobre
los seres vivos. Su origen más común es:
• Aguas residuales industriales: Cd, Zn, Cu, Pb.
• Combustión de carbón y demás combustibles fósiles: Hg, Pb, Cd, Zn, con el transporte
intermedio desde la atmósfera hasta el agua.
• Filtraciones desde minas o retornos de riego: As, Se, Ba.
• Fuentes naturales: As, Se, Ba, Cu..
Cadmio.
El cadmio divalente Cd2+ es la especie predominante de Cadmio encontrada en las aguas
superficiales. Habitualmente entre el 60-90% está en esta forma aunque la presencia de ligandos
acomplejantes sea elevada. Es importante conocer este hecho puesto que el cadmio libre juega un
papel muy importante en la toxicidad de las aguas naturales.
La adsorción física o química sobre la superficie de diversos sólidos es el principal
mecanismo de eliminación del cadmio de las aguas naturales. La adsorción en fangos de río es
rápida y el equilibrio se suele alcanzar en muy pocos minutos. Los factores principales que afectan
a este mecanismo son:
- Tipo de sólido.

Modelos de Calidad de Aguas 126
- Estado de subdivisión del sólido.
- Tiempo de contacto.
- Concentración de agentes formadores de complejos químicos.
La presencia de materiales orgánicos tales como ácidos húmicos aumenta el fenómeno de la
adsorción.
En la tabla siguiente se muestran algunos valores para el coeficiente de partición del cadmio
en función de los sólidos suspendidos existentes en el cauce natural:
SS (mg/L) kD (L/kg) % Disuelto % Adsorbido
1 4 106 20 80
10 3 105 25 75
100 2 104 30 70
1000 2 103 40 60
Arsénico.
El arsénico puede aparecer en cuatro estados de oxidación estables en el medio (+5, +3, 0, -
3). Tiene por lo tanto una inusual y compleja química. El estado de -3 es el más raro de todos
debido al extremadamente bajo potencial red-ox que requiere para su presencia.
El arsénico es eliminado de la disolución mediante adsorción en arcillas o coprecipitación
con precipitados de otros iones metálicos (hidróxido de hierro, óxidos de manganeso, etc.).
Un proceso peligroso que puede sufrir el arsénico es el de su metilación a dimetil arsina
(CH3)2AsH, altamente tóxica.
En la tabla siguiente se muestran algunos valores para el coeficiente de partición del
arsénico en función de los sólidos suspendidos existentes en el cauce natural:
SS (mg/L) kD (L/kg) % Disuelto % Adsorbido

Balances de Materia en Sistemas Naturales 127
1 5 105 70 30
10 9 104 50 50
100 2 104 30 70
1000 3 103 24 76
Mercurio.
El mercurio aparece en el medio natural en tres estados de oxidación (0, +1, +2). La
presencia de una especie u otra depende de:
- Potencial redox.
- pH del sistema natural.
- Existencia de aniones u otros ligandos que podrían formar complejos con el mercurio.
El mercurio es expulsado a la atmósfera desde diversas fuentes:
- Transpiración y descomposición de la vegetación.
- Procesos de combustión y volatilización.
En el aire es fijado mediante adsorción sobre las partículas suspendidas existentes. Es
eliminado de la atmósfera mediante deposición seca o húmeda. Gran parte del mercurio forma
complejos con material húmico acumulado en zonas aluviales, por lo que sólo una pequeña parte
soluble podrá ser asimilada por los seres vivos. Debido a la baja velocidad de sedimentación de las
arcillas, éstas se distribuyen ampliamente a través de los océanos. Los organismos pelágicos
aglomeran el mercurio transportado por las arcillas promoviendo la sedimentación y afectando el
destino del mismo en la cadena medio-oceánica. Otro proceso que sufre es la toma de mercurio
disuelto por parte del fitoplancton y algas.
En aguas naturales, con pH comprendidos entre 6 y 8 y potenciales Eh en torno a 0.5v, el
mercurio metálico Hg0 y el HgS son las especies que participan en mayor medida en el equilibrio
con otras especies en disolución. Las especies más abundantes en aguas naturales son el Hg(OH)2 y
HgCl2. En sedimentos reductores, con bajos potenciales redox, el mercurio es inmovilizado por

Modelos de Calidad de Aguas 128
iones sulfuro. Si el potencial redox es extremadamente bajo y el pH es mayor que 9, la solubilidad
del mercurio aumenta mucho debido a la formación de iones HgS22-.
Un proceso importante que sufre el mercurio en los sedimentos es el de metilación,
mediante el cual el mercurio metálico es transformado en un compuesto organometálico, altamente
tóxico. El mercurio metálico es tóxico, pero también es un metal inerte, no reactivo. El vertido de
numerosas toneladas de mercurio en ríos, lagos o estuarios provoca que, al ser un metal tóxico,
numerosas bacterias se protejan a sí mismas transformándolo en iones metil mercúricos (CH3Hg+)
y en dimetil mercurio gaseoso (CH3)2Hg. Estos compuestos orgánicos, o ligeras modificaciones de
los mismos, han pasado a la cadena alimenticia a través de peces hasta llegar al hombre donde los
iones metil mercurio actúan como un veneno nervioso mortal. Entre 1953 y 1961, 116 personas se
envenenaron en Minamata (Japón) al comer pescado que contenía compuestos de metil mercurio.
Los mecanismos de metilación pueden ser microbiano, catalizado por enzimas, o químico,
no catalizado por metilcobalamina.
Diversas características que presenta este proceso:
- La velocidad de la metilación aumenta con la temperatura.
- La velocidad es mayor si hay sólidos suspendidos y en la parte superior del sedimento
progresa más que en las profundas.
- A bajas concentraciones de oxígeno, la velocidad también es mayor.
- La formación de dimetilmercurio se ve inhibida en medios ácidos. El pH óptimo para la
formación de metil mercurio es 6, mientras que para la formación de dimetil mercurio es 9.
En la tabla siguiente se muestran algunos valores para el coeficiente de partición del
mercurio en función de los sólidos suspendidos existentes en el cauce natural:
SS (mg/L) kD (L/kg) % Disuelto % Adsorbido
1 3 106 25 75
10 2 105 30 70
100 2 104 30 70

Balances de Materia en Sistemas Naturales 129
1000 1 103 45 55
Selenio.
El selenio es uno de los minerales más ampliamente distribuidos en la corteza terrestre,
habitualmente asociado a minerales de azufre. El selenio entra en los medios acuáticos por pro-
cesos de meteorización natural, combustión de combustibles fósiles, extracción, fundición y
refinado de metales y otras actividades industriales. Los dos estados de oxidación más comunes son
+4 y +6. Las concentraciones promedio en los ríos son de aproximadamente 0.2 µg/L. En el agua
del mar tiene un valor medio de 0.1 µg/L.
No se conoce mucho sobre la especiación del selenio en aguas naturales. Las
concentraciones de iones selenito y selenato son mayores a pH 7 que a pH 4.5, por lo que parece
ser que son más estables en medios básicos o ligeramente ácidos.
Bario.
El bario suele aparecer en la naturaleza en forma de carbonato de bario o sulfato. Ambas
sales son altamente insolubles e inocuas, aunque la mayor parte de las sales de bario son solubles y
tóxicas. Estas últimas pueden afectar a los sistemas musculares y nerviosos. También parece ser
que existe una correlación positiva entre las enfermedades cardiovasculares y la concentración de
bario en aguas de bebida.
El comportamiento de los iones de bario en el terreno es similar al del resto de los metales
estudiados. Puede formar complejos orgánicos con ácidos grasos, lo cual aumenta su movilidad en
el terreno. Parte de él es adsorbido o precipitado como sulfato, quedando fijado en arenas o arcillas.
La falta de conocimiento sobre las interacciones del bario en el medio natural es bastante grande.
Su similitud en muchos aspectos con el calcio ha abierto la posibilidad de asemejar el
comportamiento del bario en los procesos de intercambio iónico, formación de complejos, y
adsorción, con el fin de conseguir un modelo de comportamiento general para el bario.

Modelos de Calidad de Aguas 130
Plomo.
El plomo en el suelo puede ser originado desde fuentes naturales o antropogénicas. Entre
las fuentes naturales destacan la meteorización de rocas o depósitos minerales y los procesos de
vulcanismo.
Entre las fuentes antropogénicas, relativamente recientes (menos de 100 años), destaca su
empleo como antidetonante en gasolinas. Efectivamente, desde 1923 se vienen empleando el te-
traetilo de plomo y otros compuestos de alquil plomo para este fin. Se estima que en el hemisferio
norte hay una media de 10 mg de plomo por metro cuadrado procedentes de la combustión de
gasolinas. La cantidad de plomo que alcanza los cauces naturales y el mar por escorrentía es muy
importante.
El movimiento del plomo a largas distancias está relacionado con el transporte de sus
formas particuladas o adsorbidas. En las cercanías de depósitos minerales el plomo puede ser
rápidamente movilizado debido a la producción de compuestos ácidos como consecuencia del
lavado de sulfuros. Sin embargo, este plomo puede ser fijado mediante su adsorción sobre otras
superficies formado sulfatos o carbonatos de plomo. En general, se puede afirmar que este metal
tiene una baja movilidad geoquímica.
En disolución acuosa el plomo existe casi únicamente en forma de Pb(II), mientras que el
Pb(IV) sólo existe bajo condiciones oxidantes muy marcadas. El plomo forma complejos con li-
gandos orgánicos como aminoácidos, proteínas, polisacáridos y ácidos fúlvicos y húmicos.
Los compuestos de alquil plomo son sumamente tóxicos y su posible formación en los
sedimentos de los sistemas naturales ha supuesto un nuevo motivo de alarma (además de su origen
antropogénico). La metilación del plomo se produce a partir de la reacción de Pb(II) con agentes
donantes de CH3+.
Otro proceso importante que sufre el plomo en el medio natural es de su adsorción sobre
superficies sólidas, principalmente sobre materiales arcillosos. Gracias a ello, los sedimentos de
lagos o embalses, o en general, de aquellos sistemas cuyo tiempo de residencia sea elevado, actúan
como sumideros de este metal pesado.

Balances de Materia en Sistemas Naturales 131
En la tabla siguiente se muestran algunos valores para el coeficiente de partición del plomo
en función de los sólidos suspendidos existentes en el cauce natural:
SS (mg/L) kD (L/kg) % Disuelto % Adsorbido
1 3 105 75 25
10 2 105 30 70
100 1 105 10 90
1000 9 104 1 99
Cinc.
La toxicidad del cinc no está directamente relacionada con la concentración total de metal,
sino con la presencia de iones Zn libres.
Los complejos de cinc más habituales se dan con iones hidróxido, carbonato, bicarbonato y
sulfato. También con iones cloruro en agua de mar.
La adsorción de cinc en arcillas es el principal proceso que afecta a la desaparición de este
elemento en sistemas naturales. Dicho proceso origina un enriquecimiento en este metal de los
sedimentos. Las principales variables que afectan a la movilidad del cinc son:
- Concentración y composición de los sólidos suspendidos y sedimentos.
- Concentraciones disueltas y particuladas de hierro y manganeso.
- pH.
- Salinidad.
- Concentración de ligandos acomplejantes.
- Concentración de Zn.
En la tabla siguiente se muestran algunos valores para el coeficiente de partición del cinc en
función de los sólidos suspendidos existentes en el cauce natural:
SS (mg/L) kD (L/kg) % Disuelto % Adsorbido

Modelos de Calidad de Aguas 132
1 1 106 40 60
10 2 105 30 70
100 5 104 17 83
1000 1 104 10 90
Cobre.
El cobre puede estar presente en el medio natural en tres formas:
- Particulado.- Precipitados de óxidos, sulfuros y carbonatos. Complejos inorgánicos
insolubles. Cobre adsorbido en arcillas u otros materiales sólidos.
- Disuelto.- Ion cúprico libre y complejos solubles.
- Coloidal.- Material polipeptídico y arcillas finas y precipitados hidróxidos metálicos.
Las reacciones de precipitación y de formación de complejos son los procesos más
característicos que puede sufrir el cobre en aguas naturales. La precipitación puede dar lugar a
óxidos de cobre (CuO) o malaquita (Cu2(OH)2CO3) en función del pH y la alcalinidad del medio.
En presencia de precipitados de cobre, el Cu(II) libre puede acomplejarse con iones bicarbonato
para formar especies solubles de CuCO3. Esto explicaría por qué la fracción de Cu(II) soluble es
pequeña con respecto al cobre soluble total.
En la tabla siguiente se muestran algunos valores para el coeficiente de partición del cobre
en función de los sólidos suspendidos existentes en el cauce natural:
SS (mg/L) kD (L/kg) % Disuelto % Adsorbido
1 1 106 50 50
10 2 105 30 70
100 3 104 25 75
1000 6 103 14 86

Balances de Materia en Sistemas Naturales 133
2.3.2.5 Contaminación radiactiva.
El origen de la contaminación radiactiva está en los procesos de generación de energía
nuclear, pruebas nucleares y numerosas aplicaciones industriales. Las principales características de
los contaminantes radiactivos son las siguientes:
- Se adsorben sobre material particulado, por lo que pueden acumularse en los
sedimentos.
- No se volatilizan.
- Se descomponen siguiendo una cinética simple de 1er orden.
C k - = S radrad k
Ec.( 156)
El término habitualmente empleado para expresar la persistencia de un contaminante
radiactivo en el entorno es el de periodo de semidesintegración, t50. Este periodo es el tiempo para
el cual se descompone el 50% del producto inicial. La relación entre t50 y krad es:
t0.693 = k
50rad Ec.( 157)
Aunque un análisis superficial de problema parecería mostrar que la contaminación
radiactiva es un fenómeno de tipo atmosférico única y exclusivamente, lo cierto es que no es así.
La atmósfera es el vehículo de transporte de estos contaminantes, que poco a poco se van
depositando en terrenos y zonas húmedas, incrementando los niveles de radiactividad en ellos.

Modelos de Calidad de Aguas 134
2.3.2.6 Interacciones con los sedimentos.
En los sistemas naturales, los sedimentos juegan un papel sumamente importante como
fuente/sumidero de las sustancias que están siendo vertidas o lo fueron en el pasado. Constituyen,
pues, un registro histórico de lo sucedido en el sistema natural y cuyas consecuencias pueden
detectarse en la actualidad debido al almacenamiento producido en los sedimentos.
Los depósitos bénticos son el resultado de diversos procesos, algunos de los cuales afectan
a material particulado y otros a sustancias disueltas. Los más importantes son, en un principio, los
que afectan al material particulado, puesto que éste es el constituyente originario del sedimento.
Los procesos principales son la sedimentación de material particulado (materia orgánica, sólidos
inorgánicos, etc.), la resuspensión de dicho material bajo ciertas circunstancias y la movilidad de
las capas superficiales, menos cohesionadas, del mismo. Las sustancias disueltas se intercambian
entre la columna de agua y el agua intersticial del sedimento en función de la diferencia de
concentraciones entre ambas zonas.
Todo el material particulado: sólidos inorgánicos, metales adsorbidos en ellos, materia
orgánica, compuestos químicos adsorbidos en la materia orgánica, fitoplancton... es transportado
aguas abajo a una velocidad aproximadamente igual a la velocidad media de la corriente. Por otra
parte, también es transportado en la dirección vertical con una velocidad de caída que se puede
estimar a partir de la ley de Stokes, como ya se vió para el caso del fitoplancton:
νρρ ) - ( R g
92 = V wp
2
s Ec.( 158)
donde:
Vs : velocidad de caída de la partícula (m/día).
g : aceleración de la gravedad (m/día2).
R : radio de la partícula (m).
ρp : densidad de la partícula (g/m3).
ρw : densidad del agua (g/m3).
ν : viscosidad cinemática (m2/día).
Una vez la partícula alcanza la superficie del sedimento existe cierta probabilidad de que
sea resuspendida y no contribuya a la sedimentación neta. En sistemas naturales, donde la

Balances de Materia en Sistemas Naturales 135
sedimentación sea bastante superior a la resuspensión, es bastante común emplear una velocidad
de sedimentación neta:
HV = k s
s Ec.( 159)
en donde H es la profundidad de la columna de agua (m).
En un sistema natural, los sedimentos de granulometría más fina tienden a depositarse en
zonas de baja energía. En ríos, por ejemplo, se depositan en aquellas zonas donde la velocidad baja
notablemente. En estuarios se suelen depositar preferentemente en la "zona muerta" donde se
anulan las energías del río y las mareas. En lagos, la turbulencia debida a las corrientes provoca
que el material más grueso sedimente en las zonas someras mientras que el fino lo hace en las
profundas, hacia el centro del lago. En embalses se suele formar una zona de sedimentación o delta
en la entrada del mismo y una acumulación de finos en la base de la presa.
El transporte de sedimentos que se produce por el desplazamiento de la capa superior del
sedimento suele contribuir muy poco al balance global de la sedimentación. Únicamente en
estuarios el transporte de arcillas finas puede ser importante en el movimiento de contaminantes,
aunque no se han desarrollado expresiones que puedan dar cuenta de este mecanismo.
Para una capa de sedimento de espesor hj, las ecuaciones que describen las variaciones de
concentración con el tiempo Sk para las fracciones particuladas y disueltas de una sustancia son las
siguientes (Ambrose, 1988):
C k - )C - C( h
E = t
C = S
C k - C hv - C
hv - C
hv =
tC
= S
wjwwjwi2j
DIFwjk
pjppjj
Cpj
j
Rpij
Dpjk
w
p
∂
∂
∂
∂
Ec.( 160)
en donde:
j, i : índices correspondientes a la capa de sedimento y la columna de agua.
p, w : índices correspondientes al material particulado y disuelto.
C : concentraciones (mg/L).
vD : velocidad de deposición del material particulado (m/día).
vR : velocidad de resuspensión del material particulado (m/día).

Modelos de Calidad de Aguas 136
vc : velocidad de compactación del material particulado (m/día).
EDIF : coeficiente de difusión de las sustancias disueltas entre la columna de agua y el sedimento
(m2/día).
kp, kw : constantes de velocidad de reacción asociadas a las fracciones particulada y disuelta de la
sustancia (m/día).
hj : espesor de la capa del sedimento (m).
El comportamiento de los sólidos en el sedimento viene reflejado por la relación entre la
velocidad de resuspensión y la de compactación, ya que ambos son mecanismos que "eliminan"
sólidos del sedimento. La relación entre ambas velocidades viene determinada por el coeficiente Fr:
v + vv = F
cr
rr Ec.( 161)
Si la velocidad de compactación es mucho mayor que la de resuspensión, el valor de Fr
tiende a cero, lo cual significa que todo el material suspendido queda fijado en el sedimento y no
vuelve a la columna de agua. En este caso el sedimento actúa como un sumidero más de materia. Si
la velocidad de resuspensión es mucho mayor que la de compactación, Fr tiende a 1, por lo que la
concentración de sólidos en el agua a largo plazo será igual a la de la entrada, puesto que no se
produce sedimentación.
El intercambio de nutrientes entre la columna de agua y los sedimentos es particularmente
importante en los problemas de eutrofización y, sobre todo, con sustancias como el fósforo
inorgánico.

Balances de Energía 137
3. BALANCES DE ENERGÍA.
3.1 Introducción.
La atmósfera absorbe un 10% de la energía que emite el sol. El resto llega a la superficie
terrestre en forma de luz y calor. Parte de esa energía es reflejada en forma de radiación calorífica
(radiación electromagnética de longitud de onda más larga que la luminosa). La atmósfera es casi
transparente a la radiación de onda corta pero muy absorbente de las radiaciones de onda larga
(efecto invernadero). La atmósfera, en vez de calentarse de una forma regular por efecto de la
radiación solar, lo hace de una forma local en función de la temperatura de la superficie terrestre.
La transferencia de energía entre la atmósfera y un lago profundo y en el interior del mismo
puede dar lugar a la generación de una estratificación térmica en el seno del mismo. La
aparición/desaparición estacional de esta estratificación puede tener efectos importantes en la
calidad de sus aguas. Esta es una de las razones por las cuales la modelización de la temperatura y
de la transferencia de calor es un aspecto importante de los modelos de calidad de aguas.
Otro motivo de plantear balances de energía en sistemas naturales es la contaminación
térmica de las aguas, que puede llegar a ser tan grave como cualquier otro tipo de contaminación
más conocido. El origen de esta contaminación está en el vertido de aguas procedentes de los
intercambiadores de calor de los circuitos de refrigeración de factorías industriales, centrales
térmicas y nucleares. A modo de ejemplo cabe señalar que en una central clásica pasan al menos 2
kcal de calor al agua de refrigeración por cada kcal de energía eléctrica producida. En una planta
nuclear la relación puede llegar a ser de 3 a 1.
El efecto inmediato del vertido es la elevación de la temperatura del agua del sistema
natural. Las principales consecuencias medioambientales de este aumento de la temperatura son:
- Producción de un cambio de las especies que pueblan la zona, provocando el
desplazamiento de especies que predominaban por otras. El salmón y la trucha, por
ejemplo, no pueden vivir en aguas a más de 25ºC.
- Disminución de la solubilidad del oxígeno en el agua.

Modelos de Calidad de Aguas 138
- Aumento del metabolismo microbiano y de la demanda biológica de oxígeno, en
general.
Por otra parte, la entrada de agua con distinta densidad puede modificar el régimen de flujo
de nuestro sistema. Esto podría suceder, por ejemplo, en lagos poco profundos donde se vertiera
agua caliente a través de emisarios sumergidos.
3.2 Ecuación general del balance de energía.
A efectos de balances de energía, se puede considerar el calor como una sustancia más cuya
ecuación de conservación es la misma que la que se vió en el apartado anterior. Se puede suponer
que el calor tiene una concentración determinada (cal/m3) y que se relaciona con la temperatura por
medio de la siguiente expresión:
) T - T ( C = C 0pρ Ec.( 162)
en donde:
ρ: es la densidad del agua (g/m3).
Cp: es el calor específico del agua (1 cal/g ºC).
T: es la temperatura absoluta (K).
To: es una temperatura base (K).
Las principales aproximaciones que se hacen son:
- Considerar ρ y Cp constantes.
- Despreciar la generación de calor mediante mecanismos internos (disipación viscosa de
energía).
La ecuación general del Balance de Energía tendría una forma similar a la Ecuación
General del Balance de Materia, y se expresa como:
Vs
c1 + )
xT
E(x
+ T) u(x
- = tT
xx ρ∂∂
∂∂
∂∂
∂∂ Ec.( 163)

Balances de Energía 139
donde s/V es el término de fuente/sumidero, representa todas las transferencias de calor a través del
sistema aire-agua o sedimento-agua. Este último término suele ser despreciable, por lo que
únicamente tienen importancia las transferencias a través de la interfase aire-agua.
Introduciendo el concepto de flujo de energía HN (cal/m2 día), la relación entre s/V y HN
viene dado por:
dH =
Vs N Ec.( 164)
en donde d es la profundidad de la corriente. La ecuación del balance quedaría:
d c
H + )xT
E(x
+ T) u(x
- = tT N
xx ρ∂∂
∂∂
∂∂
∂∂ Ec.( 165)
Cuando exista un vertido cuya temperatura pueda modificar ostensiblemente la temperatura
del agua de nuestro sistema, es necesario plantear un balance de energía. Las entradas y salidas de
calor son las siguientes:
- Caudales de entrada: ríos, afluentes, descargas puntuales, aguas marinas, etc.
- Caudales de salida.
- Calor generado por reacciones químicas y biológicas.
- Calor intercambiado con el sedimento.
- Calor intercambiado con la atmósfera a través de la superficie del agua.
Sin lugar a dudas, el proceso dominante es el intercambio de calor entre el aire y la
atmósfera, pero el proceso problemático es la entrada externa de calor con los caudales de entrada.
El intercambio de calor a través de la interfaz aire-agua puede realizarse por medio de los
siguientes procesos:
- Radiación.- Emisión de energía calorífica que emite todo cuerpo a una temperatura. No
requiere medio material para transmitirse.
- Evaporación.- Pérdida de calor asociada a la evaporación del agua.
- Conducción: las moléculas más calientes ceden parte de su energía cinética a la más frías
que están junto a ellas. No se produce un desplazamiento de las moléculas.
- Convección.- Transmisión de calor por desplazamiento de las masas de fluido debido a las
diferencias de densidad entre distintas zonas de éste.

Modelos de Calidad de Aguas 140
El flujo neto de calor H es la suma algebraica de los diversos flujos de energía:
Qc Qe -Qbr -Qar - Qa Qsr - Qs H ±+= Ec.( 166)
donde:
H : flujo de calor neto a través de la superficie (kcal/m2/hr).
Qs : Radiación de onda corta incidente sobre la superficie del agua (30 a 300 kcal/m2/hr)
Qsr : Radiación de onda corta reflejada desde la superficie del agua (5 a 25 kcal/m2/hr)
Qa : Radiación de onda larga que entra desde la atmósfera (225 a 336 kcal/m2/hr)
Qar : Radiación de onda larga reflejada (5 a 15 kcal/m2/hr)
Qbr : Radiación emitida por el volumen de agua (220 a 345 kcal/m2/hr)
Qe : Energía empleada por la evaporación del agua (25 a 900 kcal/m2/hr)
Qc : Energía transmitida por convección desde o hacia el volumen de agua ( -35 a 50 kcal/m2/hr en
la superficie).
Cada uno de estos componentes puede ser estimado a partir de relaciones semiteóricas,
ecuaciones empíricas y datos meteorológicos básicos. En función de las expresiones elegidas, se
requeriría la siguiente información:
- Presión atmosférica.
- Nubosidad.
- Velocidad y dirección del viento.
- Humedad relativa.
- Radiación solar de onda corta.
- Temperatura del agua.
- Latitud.
- Longitud.
- Punto de rocío.
3.2.1 Radiación neta solar de onda corta Qsn.
La radiación neta solar de onda corta es la diferencia entre la radiación solar incidente Qs y
la reflejada Qsr. La técnicas para determinarla se basan en el empleo de datos meteorológicos. La

Balances de Energía 141 interacción de la radiación con gases, vapor de agua, polvo, nubes, etc. dificulta el empleo de
expresiones semiempíricas que obligan al empleo de gran número de datos experimentales.
Una de las ecuaciones más comúnmente usadas para la estimación de este término es:
) C 0.65 - 1 ( Q 0.94 = Q 2scsn Ec.( 167)
donde:
Qsc: es la radiación solar incidente kcal/m2/hr.
C: es la fracción de cielo cubierto por nubes.
El valor de Qsc se determina en función de la localización geográfica, época del año y hora
del día.
Un tema importante a la hora de determinar Qsn es el efecto que producen las sombras de
los árboles, terraplenes, etc en el río o lago, puesto que dichas sombras reducen la radiación solar
incidente y las temperaturas que alcanzará el agua serán menores que en zonas sin sombras.
3.2.2 Radiación atmosférica neta Qan.
La radiación atmosférica neta está caracterizada por valores de longitudes de onda mucho
mayores que la radiación solar puesto que sus principales emisores son vapor de agua, dióxido de
carbono y ozono.
Una de las fórmulas más empleadas es:
)e 0.031 + A ( ) 273 + T ( )R - 1 ( = Q - Q = Q air4
aLaraan σ Ec.( 168)
en donde:
Qan : radiación atmosférica neta de onda larga (cal/m2/día).
A : coeficiente (0.5-0.7).
eair : presión de vapor del aire a 2 metros sobre la superficie del agua (mm Hg).
Ta : temperatura del aire a 2 metros sobre la superficie del agua, ºF.
RL : coeficiente de reflexión (0.03).
σ : Constante de Stefan-Boltzman = 11.7 10-8 (cal/cm2/d/K4).

Modelos de Calidad de Aguas 142 3.2.3 Radiación emitida por el agua Qbr.
Todos los objetos cuya temperatura absoluta es superior a cero emiten radiación. La
radiación emitida por el agua es, con mucho, el mayor de todos los flujos que intervienen en el
balance de calor de un sistema natural. Su valor se puede determinar con bastante precisión a partir
de la temperatura de la superficie del agua, tal y como se expresa mediante la Ley de Stefan-
Boltzmann:
T 0.97 = Q 4sbr σ Ec.( 169)
en donde:
Ts : temperatura de la superficie del agua (K).
σ: Constante de Stefan-Boltzman = 1.357 10-8 (cal/m2/s/K4).
0.97 : factor de emisividad.
Como una aproximación aplicable en numerosos casos, entre 0 y 30ºC, se puede emplear la
siguiente expresión en función de la temperatura del agua:
T 1.17 + 73.6 = Qbr Ec.( 170)
la cual presenta una desviación menor del 2.1% con respecto a la ecuación 158.
3.2.4 Flujo de calor por Evaporación, Qe.
Las pérdidas de calor por evaporación ocurren como resultado del cambio de estado del
agua de líquido a vapor. El valor de Qe puede ser calculado como:
) e - e ( ) W 0.95 + 19.0 ( =Q airs2
e Ec.( 171)
en donde:
W : velocidad del viento a una determinada altura sobre el nivel del agua (m/s).
es : presión de vapor de saturación a la temperatura de la superficie del agua (mm Hg). Función no
lineal de la temperatura de la superficie del agua.
eair = presión de vapor de la atmósfera (mm Hg).

Balances de Energía 143 3.2.5 Flujo de calor convectivo Qc.
El calor convectivo es transferido entre el aire y el agua por conducción y transportado lejos
desde (o hacia) la interfase aire-agua por convección asociada con las masas de aire en
movimiento.
) T - T ( ) W 0.95 + 19.0 ( c =Q airs2
1c Ec.( 172)
en donde:
c1 : 0.47 mm Hg/ºC.
W : velocidad del viento a una determinada altura sobre el nivel del agua (m/s).
Ts : temperatura de la superficie del agua (ºC).
Tair : temperatura del aire (ºC).
3.2.6 Linealización de la transferencia de calor.
Las ecuaciones anteriores representan flujos de calor mediante ecuaciones no lineales en
función de Ts, fundamentalmente. Estas aproximaciones son adecuadas para simulaciones de cortos
periodos de tiempo, donde la necesidad de predecir las variaciones de la temperatura del agua se
produce cada pocas horas. Para simulaciones con intervalos de tiempo mayores o en estado
estacionario es más económico emplear aproximaciones lineales de la transferencia de calor. La
expresión que se suele emplear es la siguiente:
) T - T ( K = H es Ec.( 173)
en donde:
Ts : temperatura de la superficie del agua (ºC).
Te : temperatura de equilibrio (ºC).
K : coeficiente de intercambio de calor a través de la superficie (cal /m2 d ºC).
La temperatura de equilibrio es aquella a la que tendería un sistema natural dadas unas
determinadas condiciones meteorológicas y para la cual el valor del flujo de energía, H, sería cero.
El valor de K depende de la temperatura del agua y de la velocidad del viento y se define
para proporcionar incrementos del flujo de calor en función del incremento de la temperatura del
agua.

Modelos de Calidad de Aguas 144
3.2.7 Calor intercambiado con el sedimento.
En el caso de sistemas naturales profundos, el flujo de calor hacia el sedimento suele jugar
un papel poco importante en el balance global de calor. Sin embargo, puede ser un término
importante para evaluar la variación diurna de la temperatura del agua en sistemas naturales poco
profundos (menos de tres metros).
Se han hecho algunos intentos para determinar el calor intercambiado en base a propiedades
del lecho, tales como su difusividad térmica y la capacidad de almacenamiento de calor.

Modelación Matemática de la Calidad de las Aguas 145
4. MODELACIÓN MATEMÁTICA DE LA CALIDAD DE LAS AGUAS.
Un modelo matemático es una formulación idealizada que representa la respuesta de un
sistema físico ante un estímulo externo. En el campo de estudio que nos ocupa, se trataría de
determinar el valor de la concentración de cualquier sustancia en el agua en función de un vertido.
Expresado esto mediante una sencilla formulación matemática:
Wa1 = c Ec.( 174)
en donde:
c : concentración de la sustancia vertida (g/m3).
W : caudal másico vertido (g/s).
a : factor de asimilación (m3/s).
El factor de asimilación engloba todos aquellos factores físicos, químicos y biológicos que
actúan sobre una sustancia en las aguas receptoras del vertido. A partir de esta formulación, puede
establecerse que los modelos se pueden emplear de tres maneras:
- Como herramientas para la simulación: prever la concentración a que daría lugar un
determinado vertido. Sería la aplicación directa de la expresión anterior.
- Como herramienta para el diseño: una vez conocido o fijado el objetivo de calidad del
agua de la zona de estudio calcular el vertido permisible:
c a =W Ec.( 175)
- Como herramienta para llevar a cabo una modificación medioambiental:
cW = a Ec.( 176)
Los modelos de calidad de aguas se dividen en dos grandes grupos:
- Modelos inductivos o empíricos. Basados en el establecimiento de relaciones entre
variables a partir de un gran número de datos de campo.
- Modelos deductivos o mecanicistas. Aplican la ley de conservación de la materia para
llegar a la ecuación general de transporte. Establecen una serie de relaciones teóricas entre

Modelos de Calidad de Aguas 146
las variables que intervienen en el proceso, por lo que necesitan los valores de una serie de
parámetros químicos, físicos y/o biológicos para poder ser resueltos.
4.1 Introducción histórica.
El desarrollo histórico de los modelos de calidad de aguas ha estado íntimamente ligado con
los tipos de sustancias contaminantes cuyo conocimiento permitía su estudio y al tipo de sistema
que se pretendía modelizar. Los tipos de contaminantes que, a lo largo de los años han ido
introduciéndose en diversos modelos, por el orden en que, aproximadamente, fueron incluidos son:
- Oxígeno disuelto - Materia orgánica.
- Fósforo total - Fitoplancton.
- Nutrientes - Fitoplancton.
- Microorganismos patógenos.
- Compuestos orgánicos tóxicos - Metales pesados.
- Sólidos sedimentables - Fangos.
- Lluvia ácida.
- Bioacumulación.
Las dimensiones del sistema natural objeto de estudio han determinado dos tipos de
soluciones:
- Soluciones analíticas en sistemas sencillos. Ríos y estuarios.
- Resolución numérica en 1-2-3 dimensiones. Ríos, lagos, estuarios y zonas costeras.
A continuación se realiza una breve descripción cronológica del desarrollo de los modelos
de calidad de aguas superficiales.
1925. Streeter y Phelps. Obtienen una expresión analítica para determinar el contenido de oxígeno
a lo largo de un río sometido a una descarga de contínua de materia orgánica biodegradable.
Hipótesis de partida:
- Caudal y composición del vertido constantes con el tiempo.
- Velocidad del río constante.
- La concentración sólo varía con la distancia a lo largo del río.
- La degradación de la materia orgánica sigue una cinética química de 1er. orden.

Modelación Matemática de la Calidad de las Aguas 147
- Se produce transferencia de oxígeno por reaireación superficial con la atmósfera.
- Estado estacionario.
1969. Vollenweider. Solución analítica para el cálculo de la concentración de fósforo total en el
agua de un lago.
Hipótesis de partida:
- Caudal y composición del vertido constantes con el tiempo.
- Lago completamente mezclado.
- No hay variación en el nivel del lago.
- Desaparición del fósforo por sedimentación.
- Estado estacionario.
1973. Aparición de QUALII (USEPA). Modelo matemático por ordenador para ríos.
Esquema de trabajo:
- División del río en celdas interconectadas.
- Resolución numérica de la ecuación diferencial en una dimensión.
- Problemas de eutrofización e intercambio de calor.
- Simulaciones dinámicas.
1983. Aparición de WASP (USEPA). Modelo matemático por ordenador para ríos, estuarios, lagos
y zonas costeras.
Esquema de trabajo:
- División del sistema natural en celdas interconectadas.
- Resolución numérica de la ecuación diferencial en una dimensión.
- Problemas de eutrofización y toxicidad de compuestos orgánicos y metales pesados.
- Simulación dinámica.
- Introducción detallada de intercambios con el sedimento.
-
Años 80-90. Amplio desarrollo de modelos bidimensionales y pseudobidimensionales.
- Desarrollo de técnicas numéricas de resolución de la ecuación general.

Modelos de Calidad de Aguas 148
- Desarrollo de ordenadores más potentes.
- Avances en la investigación sobre el comportamiento de las sustancias (obtención de
parámetros característicos).
- Inicio de comercialización de algunos paquetes informáticos para PC's y Estaciones de
Trabajo.
Tendencias actuales.
- Desarrollo de pantallas gráficas de comunicación con el usuario (entorno Windows, por
ejemplo).
- Desarrollo de modelos biológicos cuyos parámetros varían en función de la evolución
del tipo de microorganismos presente.
- En modelos de eutrofización: introducción de varias especies de fitoplancton,
introducción de más elementos de la red trófica: zooplancton, peces planctívoros, peces
piscívoros.
- Empleo de modelos basados en redes neuronales.
4.2 Modelos empíricos.
Los modelos empíricos establecen una serie de relaciones, habitualmente mediante técnicas
de regresión, entre las distintas variables que intervienen en un proceso a partir de un gran número
de datos experimentales. En el estudio de eutrofización en lagos, por ejemplo, estas
aproximaciones han tenido un notable desarrollo y una aplicación válida.
El desarrollo de estos modelos se inició a partir de los trabajos de Vollenweider (1968,
1975). A partir de una recolección de datos de vertidos de fósforo a lagos, profundidad, tiempos de
residencia y estado trófico de los mismos, estableció una correlación entre estas variables, de forma
que se podía determinar el grado de eutrofización que podía llegar a tener un lago sometido a cierto
vertido. Se podía determinar, en suma, su capacidad de asimilación. Esta aproximación partió de la
evidencia real de que los lagos profundos son menos susceptibles a la eutrofización que los
someros y de la suposición de que el fósforo actúa como nutriente limitante en estos sistemas.
El trabajo de Vollenweider ha tenido consecuencias muy importantes, incluso en el
desarrollo de modelos de tipo determinista. Sin salir del campo empírico, se han establecido
modelos que permiten calcular variables relacionadas con el estado trófico de un lago:

Modelación Matemática de la Calidad de las Aguas 149
concentración de clorofila a, profundidad del disco de Secchi, demanda de oxígeno desde zonas
profundas de los lagos a partir de datos de fósforo total en el agua del lago.
Algunos ejemplos de estas correlaciones son los siguientes:
0.259 - [PT] log 0.76 = [Cla] log Ec.( 177)
0.803 + a] [Cl log 0.473 - = [SD] log Ec.( 178)
][PT 0.086 = AHOD 0.478 Ec.( 179)
donde:
[Cl a] : concentración de clorofila a (mg/m3).
[PT] : concentración de fósforo total (mg/m3).
SD : profundidad de lectura del disco de Secchi (m).
AHOD : demanda de oxígeno por unidad de superficie en las zonas profundas del lago (g O /m2 d).
La limitación fundamental de estos modelos estriba en su carácter excesivamente local, que
impide su extrapolación a otros lugares. No obstante, constituyen una buena herramienta para
conocer algunas relaciones entre distintas variables que luego pueda ser introducidas en un modelo
de calidad de aguas más complejo.
Un caso singular y sumamente interesante en el campo de la modelización matemática del
transporte de contaminantes en el agua lo constituyen los llamados modelos de campo cercano,
modelos empíricos obtenidos para el estudio de descarga de aguas residuales a través de emisarios
submarinos. Mediante estos modelos se calcula el valor de la dilución inicial, esto es, el grado de
mezcla del agua residual que sale a través de las boquillas difusoras del emisario submarino
durante el proceso de ascensión hacia la superficie.
Estos modelos, obtenidos a partir de experiencias en laboratorio, relacionan variables
ambientales como la velocidad ambiente y el grado de estratificación vertical con variables de
diseño como el caudal de vertido y la profundidad del emisario con el fin de obtener la dilución
inicial S y otros resultados. En la actual legislación española referente al diseño de conducciones de
vertido (Orden de 13 de Julio de 1993 por la que se aprueba la Instrucción para el proyecto de
conducciones de vertido desde tierra al mar, BOE 27 de Julio de 1993) se proporcionan varios

Modelos de Calidad de Aguas 150
modelos de este tipo aplicables bajo distintas condiciones. En el capítulo 5 se tratará con mayor
profundidad este tema.
4.3 Soluciones aproximadas.
La resolución analítica de la ecuación general del balance de materia es posible únicamente
en el caso de que podamos simplificar mucho el funcionamiento de nuestro sistema natural. Estas
simplificaciones son cinéticas de tipo lineal, geometrías sencillas y caudales y niveles constantes.
A continuación se muestran dos simplificaciones de dicha ecuación general aplicables a ríos y
lagos en las condiciones que se describen. Posteriormente se explicará la forma de abordar
problemas más complejos.
4.3.1 Lago completamente mezclado.
En este caso se va a considerar a un lago como un sistema de dimensión cero. Se supone
que está perfectamente mezclado, por lo que no hay gradientes de concentraciones en superficie ni
en profundidad. Su comportamiento se asemeja a lo que en terminología de Ingeniería Química se
denomina Reactor Contínuo de Tanque Agitado.
Al lago afluye un río con un caudal QR y una concentración del contaminante a estudiar de
CR. El caudal del único vertido que se realiza es QV y su concentración es CV.
La descomposición natural del contaminante se ajusta a una cinética de primer orden con
una constante de velocidad de desaparición k.
C V
QV, CV
QR+QV, C QR, CR
Figura 11. Representación esquemática de un lago somero totalmente mezclado.
El planteamiento del balance de materia en este caso es:

Modelación Matemática de la Calidad de las Aguas 151
CVkC)Q(QCQCQdtdCV VRVVRR −+−+= Ec.( 180)
La concentración del contaminante en el lago al cabo de un tiempo t de haberse iniciado el
vertido vendrá dada por:
e C + )e - 1 ( V BW = C t B -
o t B - Ec.( 181)
siendo:
C Q + C Q = W ) Q + Q (
V = t VVRRVR
o Ec.( 182)
k + t1 = Bo
Ec.( 183)
La solución en estado estacionario es la siguiente:
V B W = Ce Ec.( 184)
El parámetro B incluye los efectos de la morfología del lago, las entradas externas de
caudal y la desaparición propia de la sustancia, si tiene lugar. La respuesta de un lago a una entrada
externa puede observarse mediante este parámetro aunque no es útil a efectos prácticos. Para ello
se introduce el término tiempo de respuesta, que es el tiempo que se emplearía para que se
completara un porcentaje fijo de recuperación si el vertido cesara:
φφ - 100100 ln
B1 = t
B100 ln = t
B2 ln = t
99
50
Ec.(185)
Si el contaminante fuera conservativo para las escalas de tiempo que se estuvieran
manejando (k=0), entonces
( ) ( )e C + )e - 1 (
QQW = C
Vt QQ
-o
V
t QQ -
VR
VRVR ++
+
Ec.(186)
Y la concentración de equilibrio sería:

Modelos de Calidad de Aguas 152
W = CVR
e + Ec.(187)
Las ecuaciones anteriores serían válidas para un contaminante tal como la materia orgánica
carbonosa, L, que se degradara siguiendo una cinética de 1er. orden. En ese mismo caso, para la
concentración de oxígeno disuelto en el lago, el balance de materia es como sigue:
[ ] [ ] [ ] [ ] [ ] [ ]( 22'2
'12VRV2VR2R
2 OO VkLVkO)Q(QOQOQdtOdV −+−+−+= sat ) Ec.(188)
La integración de esta ecuación diferencial permite calcular la evolución temporal de la
concentración de oxígeno disuelto en un lago:
( )
2i
Bt02
Btsat22'1O
2
k'VQB
e][Oe1B
][Ok'B
LkBV
W(t)][O 2
+=
+−
+−=
∑
−−
Ec.(189)
A partir de la ecuación anterior, la concentración de oxígeno disuelto en estado estacionario
queda:
+−=
B][Ok'
BLk
BV
W][O sat22e
'1O
22
e Ec.(190)
Por último, si se trata de una sustancia que estuviera sometida a varios procesos, unos de
transformación y otros de generación, representados matemáticamente mediante cinéticas de
primer orden, el término B los englobaría a todos ellos. Por ejemplo, si estudiamos el nitrógeno
amoniacal, originado a partir del nitrógeno orgánico y transformado en nitritos (ver apartado
2.3.2.2.2.), el término B que correspondería a este caso sería:
'1
'min nitri
okk +
t1 = B − Ec.(191)

Modelación Matemática de la Calidad de las Aguas 153
4.3.2 Ríos unidimensionales.
La forma más sencilla de representar a un río es mediante un sistema unidimensional. Se
considera que la concentración del contaminante en la sección transversal del río es uniforme y no
existe mezcla (transporte dispersivo) a lo largo del eje del río (esto supone que el transporte
asociado a la velocidad del agua es bastante superior al difusivo/dispersivo en esa dirección). Este
sistema es lo que se conoce como Reactor de Flujo de Pistón.
Como en el caso anterior, existe un único vertido, de caudal QV y de concentración de
contaminante CV. El caudal del río es QR y su concentración de contaminante problema es CR. La
velocidad del río permanece constante. La descomposición natural del contaminante se ajusta a una
ley de primer orden con una constante de velocidad cinética k.
dxx
CC
∂
∂+ Q , Q, C
QR, CR
QV, CV
dx Figura 12.- Representación esquemática de un río sometido a un vertido puntual.
El balance de materia al elemento dV en este caso es:
CVkxxCCQCQ=
tCV ∆−∆
∂∂
+−∂∂
∆ )( Ec.(192)
CVkxxCQ=
dtCdV ∆−∆
∂∂
−∆ Ec.(193)
sustituyendo ∆V = A ∆x:
CkxC
AQ=
tC
−∂∂
−∂∂ Ec.(194)
si ∆x tiende a cero, el valor de C tenderá a C, por lo que:
CkxCu=
tC
x −∂∂
−∂∂ Ec.(195)

Modelos de Calidad de Aguas 154
En estado estacionario, la variación de la concentración con respecto al tiempo es cero, por lo que:
CkxCu
CkxCu=
tC
x
x
−=∂∂
=−∂∂
−∂∂ 0
Ec.(196)
La integración de la ecuación anterior para las condiciones iniciales t = 0, C = C0 , permite
calcular la concentración del contaminante a lo largo del eje del río:
e C = xC ) ux ( k -
o)( Ec.(197)
siendo:
x : distancia aguas abajo del punto de vertido.
u : velocidad de la corriente.
Co : concentración inicial en el punto de vertido.
Q + QC Q + C Q = C
PR
PPRRo Ec.(198)
4.3.2.1 Oxígeno Disuelto y Materia Orgánica Carbonosa.
Mediante la aplicación del balance de oxígeno disuelto a un río y considerando que:
- La fuente de oxígeno principal es la reaireación.
- El consumo de oxígeno principal es debido a la demanda biológica carbonosa.
Streeter y Phelps (1925) obtuvieron una expresión que permite determinar el déficit de oxígeno, D
(diferencia entre la concentración de saturación y la existente en cada momento) en cualquier punto
de un río afectado por un vertido constante de agua residual.
e D + )e - e( J - J
L J =x D x J -0x J -x J -
12
01 221)( Ec.(199)
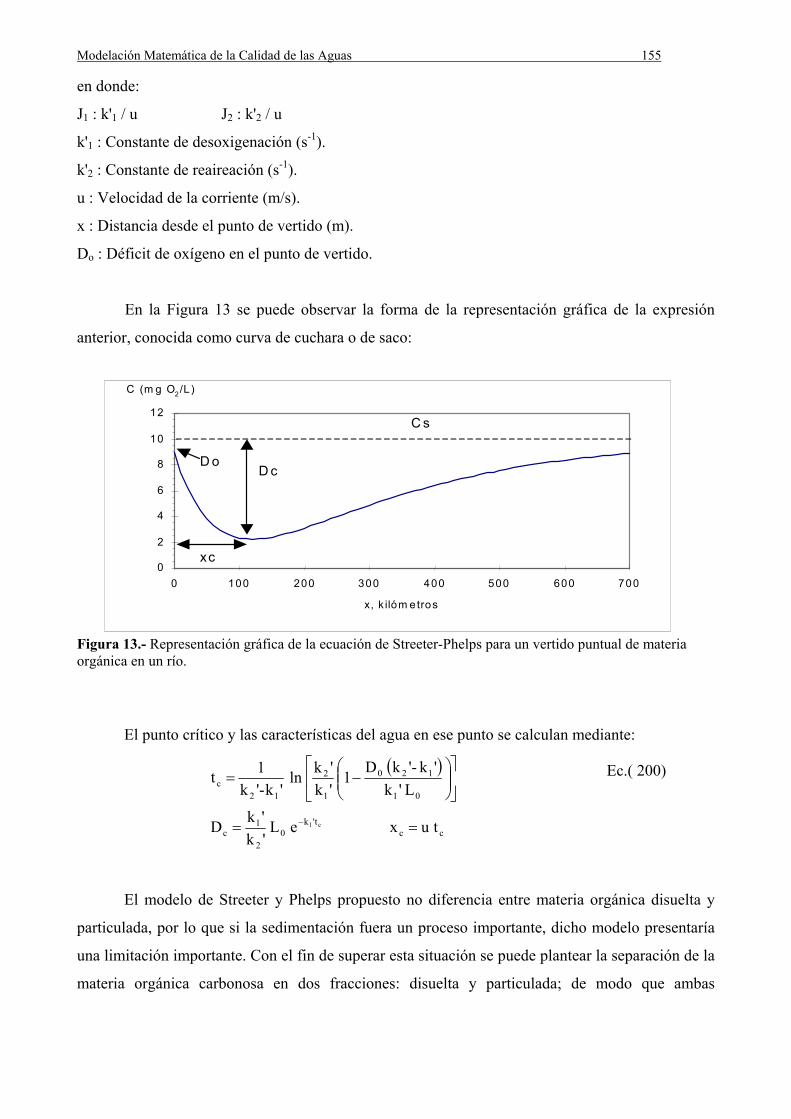
Modelación Matemática de la Calidad de las Aguas 155
en donde:
J1 : k'1 / u J2 : k'2 / u
k'1 : Constante de desoxigenación (s-1).
k'2 : Constante de reaireación (s-1).
u : Velocidad de la corriente (m/s).
x : Distancia desde el punto de vertido (m).
Do : Déficit de oxígeno en el punto de vertido.
En la Figura 13 se puede observar la forma de la representación gráfica de la expresión
anterior, conocida como curva de cuchara o de saco:
0 2 4 6 8
10 12
0 100 200 300 400 500 600 700 x, k ilóm etros
C (m g O 2 /L )
C s
D c D o
xc
Figura 13.- Representación gráfica de la ecuación de Streeter-Phelps para un vertido puntual de materia orgánica en un río.
El punto crítico y las características del agua en ese punto se calculan mediante:
( )
cct'k
02
1c
01
120
1
2
12c
tuxeL'k'kD
L'k'k'-kD1
'k'kln
'k'-k1t
c1 ==
−=
−
Ec.( 200)
El modelo de Streeter y Phelps propuesto no diferencia entre materia orgánica disuelta y
particulada, por lo que si la sedimentación fuera un proceso importante, dicho modelo presentaría
una limitación importante. Con el fin de superar esta situación se puede plantear la separación de la
materia orgánica carbonosa en dos fracciones: disuelta y particulada; de modo que ambas

Modelos de Calidad de Aguas 156
fracciones sufrieran procesos de biodegradación, pero únicamente la fracción particulada
sedimentara con una constante de velocidad de sedimentación ks.
La aplicación de la Ecuación General del Balance de Materia a esta situación da lugar al
siguiente conjunto de ecuaciones diferenciales en derivadas parciales:
0 L'kD'kxDu
tD
0 Lk-L'kx
Lut
L
0 L'kx
Lut
L
12
PSP1PP
D1DD
=+−∂∂
−=∂∂
=−∂
∂−=
∂∂
=−∂
∂−=
∂∂
Ec.(201)
cuya solución en estado estacionario permite obtener las concentraciones a lo largo del río de
materia orgánica disuelta, particulada y oxígeno disuelto:
( )( )
0xDD
ee'k'k
L'k
eek - 'k'k
L'keDD(x)
0xLLeL (x)L
0xLLeL (x)L
00
x/u'kx/u'k
12
D01
x/u'k)x/uk'k(
S12
P01x/uk'0
0P0Px/u)'k'k(
P0P
0D0Dx/u'k
D0D
21
2S12
S1
1
==
−−
+
+−−
+=
===
===
−−
−+−−
+−
−
Ec.(202)
En el caso de que el vertido de materia orgánica sea tan elevado que provoque una situación
de falta de oxígeno en algún punto de río, la ecuación de Streeter-Phelps ya no puede aplicarse. La
condición que se cumple en ese punto es que D = [O2]sat. A partir de ese momento, el consumo de
oxígeno ya no sucede a la velocidad determinada por la ecuación 78, sino que está limitado a otros
procesos. Una aproximación al problema consistiría en considerar que la velocidad de degradación
de materia orgánica carbonosa es proporcional al oxígeno que se transfiere por reaireación
superficial. La cinética de primer orden que seguía el proceso normal se ha transformado en una
cinética de orden cero:
[ ]
[ ] ) t- t( O k - L = L
O k - = tdL d
ifsat2'2i
sat2'2
Ec.(203)
en donde:
ti : tiempo para el cual se produce el inicio del proceso de anoxia (d).

Modelación Matemática de la Calidad de las Aguas 157
Li : valor de la materia orgánica en ese punto(mg O2/L).
Se puede demostrar que, en estas condiciones, el tiempo para el cual finaliza el periodo de
anoxia es:
[ ][ ]sat22
sat22i1
1if O k
O k - L k k1 + t = t
′
′′
′
Ec.(204)
Esta aproximación no considera la actividad de degradación de materia orgánica carbonosa
por parte de microorganismos anaerobios que, aunque lenta, puede tener cierta importancia. Por
otro lado, parte de la materia orgánica carbonosa puede ser empleada por bacterias desnitrificantes
que llevan a cabo la transformación de nitratos en nitrógeno gas, por lo que su DBO sobre el medio
disminuirá.
4.3.2.2 Oxígeno disuelto y nitrificación.
La presencia de nitrógeno orgánico y/o amoniacal en un vertido a un río puede provocar la
diminución del oxígeno disuelto en el mismo, como vimos que puede suceder de forma genérica.
En este caso, el balance de materia para cada una de las especies de nitrógeno y para el Déficit de
Oxígeno Disuelto en estado estacionario es:
Ec.(2053)
0141433
0
0
0
0
22412
3233
224122
41min44
min
=++−∂∂
−=∂∂
=+∂
∂−=
∂∂
=−+∂
∂−=
∂∂
=−+∂
∂−=
∂∂
=−∂
∂−=
∂∂
−+
−−−
−+−−
+++
][NOk.][NHk.Dk'xDu
tD
][NOkx
][NOu
t][NO
][NOk][NHkx
][NOut
][NO
][NHk[NO]kx
][NHut
][NH
[NO]kx
[NO]utd
[NO]
'nitri
'nitri
'nitri
'nitri
'nitri
'nitri
'
'
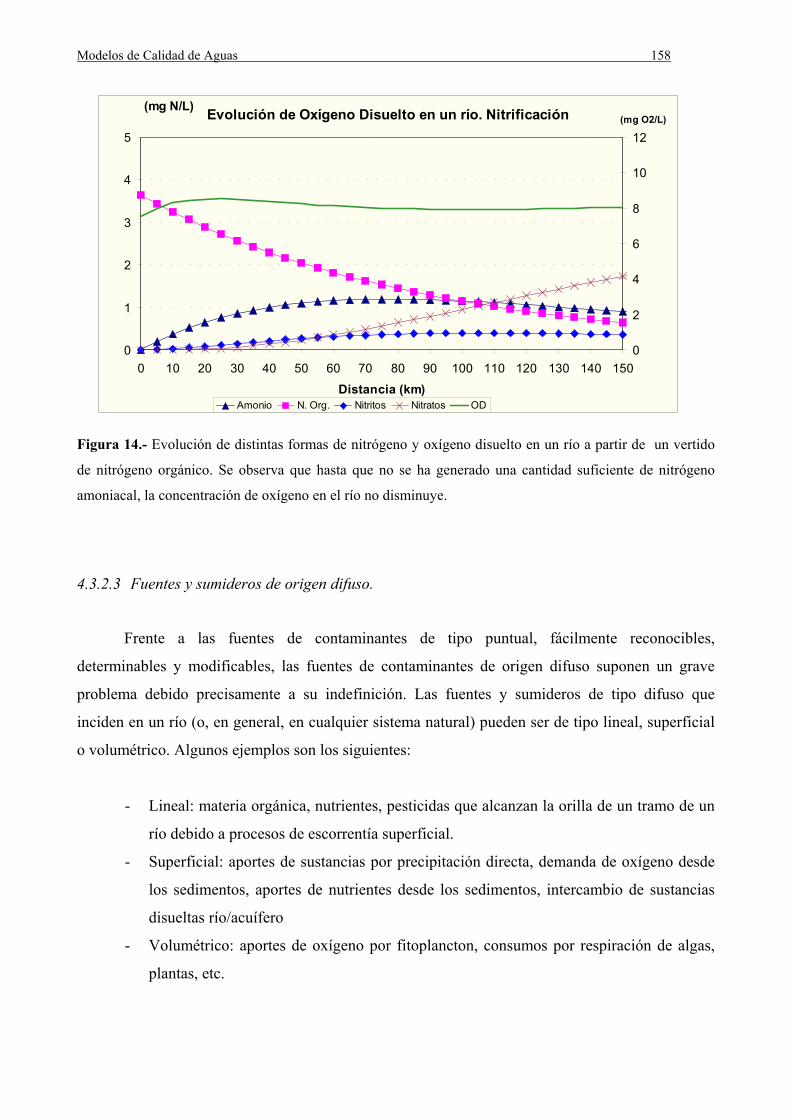
Modelos de Calidad de Aguas 158
Evolución de Oxígeno Disuelto en un río. Nitrificación
0
1
2
3
4
5
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150Distancia (km)
(mg N/L)
0
2
4
6
8
10
12(mg O2/L)
Amonio N. Org. Nitritos Nitratos OD
Figura 14.- Evolución de distintas formas de nitrógeno y oxígeno disuelto en un río a partir de un vertido
de nitrógeno orgánico. Se observa que hasta que no se ha generado una cantidad suficiente de nitrógeno
amoniacal, la concentración de oxígeno en el río no disminuye.
4.3.2.3 Fuentes y sumideros de origen difuso.
Frente a las fuentes de contaminantes de tipo puntual, fácilmente reconocibles,
determinables y modificables, las fuentes de contaminantes de origen difuso suponen un grave
problema debido precisamente a su indefinición. Las fuentes y sumideros de tipo difuso que
inciden en un río (o, en general, en cualquier sistema natural) pueden ser de tipo lineal, superficial
o volumétrico. Algunos ejemplos son los siguientes:
- Lineal: materia orgánica, nutrientes, pesticidas que alcanzan la orilla de un tramo de un
río debido a procesos de escorrentía superficial.
- Superficial: aportes de sustancias por precipitación directa, demanda de oxígeno desde
los sedimentos, aportes de nutrientes desde los sedimentos, intercambio de sustancias
disueltas río/acuífero
- Volumétrico: aportes de oxígeno por fitoplancton, consumos por respiración de algas,
plantas, etc.

Modelación Matemática de la Calidad de las Aguas 159
Un caso típico es el correspondiente a la demanda de oxígeno procedente de la
descomposición aerobia de la materia orgánica en el mismo. Si se define SB’ como la demanda de
oxígeno en g O2/m2 d, la ecuación del déficit en un sistema lineal con dispersión turbulenta
despreciable y sin aportes de materia orgánica es:
0 H
S DkxDu
tD '
B'2 =+−
∂∂
−=∂∂
Ec.(206)
Cuya integración permite obtener la variación del déficit de oxígeno a lo largo del río en estado
estacionario:
01
0x0e1k
/HSD(x)
002
0
0x/uk
'2
'B
22
'2
==
−+=
==
−=
−−
−
xDDsiek
/HSeDD(x)
Dsi
x/uk'
'Bx/uk ''
Ec.(207)
Se puede observar que el valor del déficit tiende a un valor constante dado por:
'
'Bk
/HSD2
=
El asumir un valor de SB
’ constante no es realista en simulaciones a largo plazo, en donde
las condiciones del sedimento pueden ir cambiando. En esos casos, lo correcto es simular las
interacciones en el sedimento empleando expresiones similares a las planteadas en el tema anterior.
4.3.3 Sistemas unidimensionales con dispersión longitudinal.
En aquellos sistemas naturales unidimensionales en los cuales el transporte convectivo sea
pequeño, el transporte difusivo/dispersivo aumenta su importancia relativa. Algunas zonas de ríos y
los estuarios pueden, en muchos casos, asimilarse a sistemas lineales en una dimensión, en los
cuales, además de transporte convectivo, es apreciable el transporte dispersivo turbulento.
Q, C dxx
C
∂
∂+ Q ,, C
QV, CV

Modelos de Calidad de Aguas 160
QR, CR
dx Figura 15. Representación esquemática de un estuario sometido a un vertido puntual.
El balance de materia al elemento dV en este caso es:
CVkxxC
xxCAE
xCAEx
xCCQCQ=
tCV xx ∆−
∆
∂∂
∂∂
+∂∂
+∂∂
−∆∂∂
+−∂∂
∆ )( Ec.(208)
CVkxxCAEx
xCQ=
tCV x ∆−∆
∂
∂+∆
∂∂
−∂∂
∆ 2
2 Ec.(209)
sustituyendo ∆V = A ∆x:
CkxCE
xC
AQ=
tC
x −∂
∂+
∂∂
−∂∂
2
2 Ec.(210)
si ∆x tiende a cero, el valor de C tenderá a C, por lo que:
CkxCE
xCu=
tC
xx −∂
∂+
∂∂
−∂∂
2
2 Ec.(211)
En estado estacionario, la variación de la concentración con respecto al tiempo es cero, por lo que:
02
2=−
∂
∂+
∂∂
−∂∂ Ck
xCE
xCu=
tC
xx Ec.(212)
La integración de la ecuación anterior, permite calcular la concentración del contaminante a lo
largo del eje del río en estado estacionario da lugar a la solución general:
eG + e F = c xλ xλ 21 Ec.(213)
Siendo:

Modelación Matemática de la Calidad de las Aguas 161
u
E k =
) 4 + 1 - 1 ( E 2u =
) 4 + 1 + 1 ( kE 2
u =
2x
x2
x1
η
ηλ
ηλ
Ec.(214)
y F y G constantes de integración calculables a partir de las condiciones de frontera.
Hay que tener en cuenta la transferencia de materia que se produce debida a la dispersión.
Parte de esta transferencia se produce en sentido contrario a la corriente dominante puesto que
viene determinada por el gradiente de concentraciones (ley de Fick).
Si el vertido de una masa M de contaminante conservativo, se realiza en un breve instante
de tiempo (función pulso) comparado con el tiempo de transporte, la solución de la ecuación de
transporte es la siguiente:
( )
−−=
tEtux
tEMtxc
xx 4)(exp
4),(
2
2/1π Ec.(215)
En donde M es la masa por unidad de superficie transversal que entra en el río.
Si el contaminante no es conservativo, sino que se degrada siguiendo una cinética de 1er.
orden:
( )
−
−−= tk
tEtux
tEMtxc
xx 4)(exp
4),(
2
2/1π Ec.( 216)
En el caso concreto del estudio de materia orgánica, L, y oxígeno disuelto, la aplicación de
un balance de materia en un sistema con dispersión da lugar a las siguientes ecuaciones:
L'kD'kxDE
xDu
tD
L'kxLE
xLu
tL
122
2x
12
2x
+−∂
∂+
∂∂
−=∂∂
−∂
∂+
∂∂
−=∂∂
Ec.( 217)
cuya solución en estado estacionario es la siguiente:
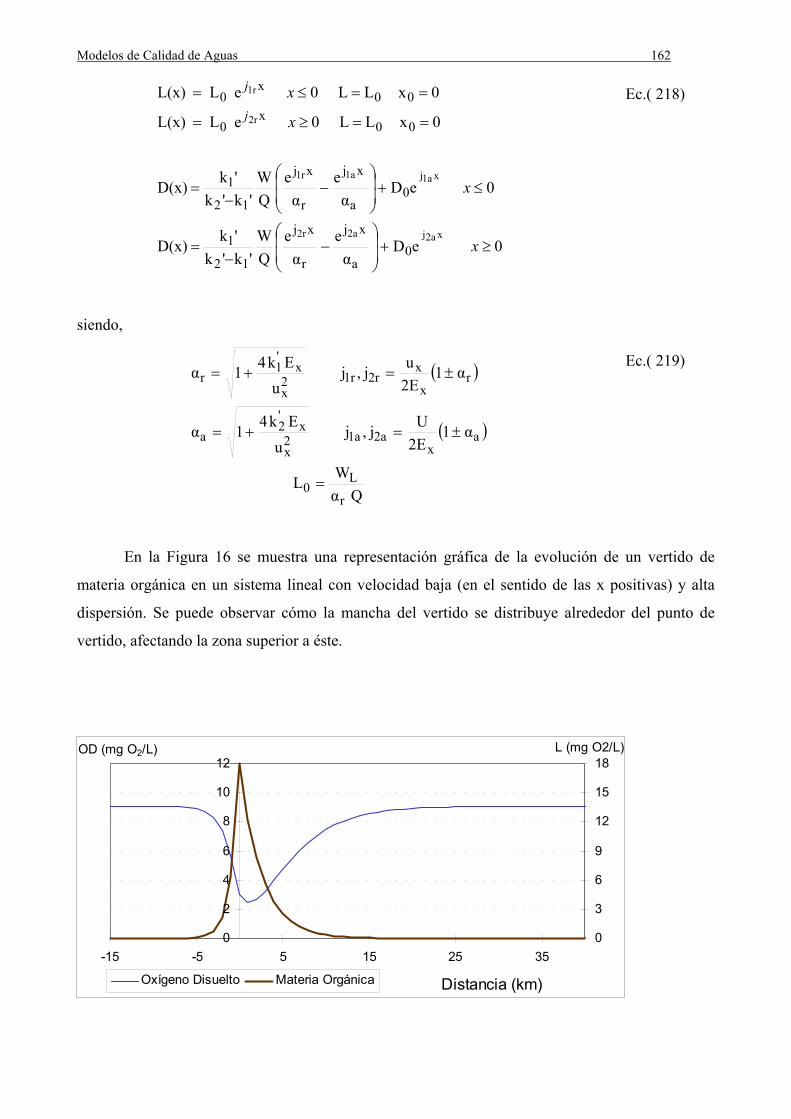
Modelos de Calidad de Aguas 162
0eDα
eα
eQW
'k'k'kD(x)
0eDα
eα
eQW
'k'k'kD(x)
0xLL0eL L(x)
0xLL0eL L(x)
x2aj2a2r
x1aj1a1r
2r
1r
0a
xj
r
xj
12
1
0a
xj
r
xj
12
1
00x
0
00x
0
≥+
−
−=
≤+
−
−=
==≥=
==≤=
x
x
x
xj
j
Ec.( 218)
siendo,
( )
( )
QαWL
α12EUj,j
uEk41α
α12Euj,j
uEk41α
r
L0
ax
2a1a2x
x'2
a
rx
x2r1r2
x
x'1
r
=
±=+=
±=+=
Ec.( 219)
En la Figura 16 se muestra una representación gráfica de la evolución de un vertido de
materia orgánica en un sistema lineal con velocidad baja (en el sentido de las x positivas) y alta
dispersión. Se puede observar cómo la mancha del vertido se distribuye alrededor del punto de
vertido, afectando la zona superior a éste.
0
2
4
6
8
10
12
-15 -5 5 15 25 35
Distancia (km)
OD (mg O2/L)
0
3
6
9
12
15
18L (mg O2/L)
Oxígeno Disuelto Materia Orgánica

Modelación Matemática de la Calidad de las Aguas 163
Figura 16.- Evolución de la concentración de materia orgánica y oxígeno disuelto aguas arriba y aguas
abajo de un punto de vertido. La velocidad del río es de 0.01 m/s y el coeficiente de dispersión turbulenta es
de 105 cm2/s
4.3.4 Sistemas de lagos conectados.
Un problema que suele ser de interés en la naturaleza es la conexión entre lagos o embalses
situados de manera consecutiva, de manera que los vertidos que se produzcan en algunos de ellos
repercutan en los que hay aguas abajo. Una forma muy sencilla de abordar este problema es
considerar que se trata de lagos completamente mezclados y calcular las concentraciones de cada
uno de ellos en estado estacionario.
W1
QR QR, C1 V1 C1
W2
QR, C2 V2 C2
W3
QR,C3 V3 C3
Figura 17.- Sistema de lagos conectados en serie.
Se supone que los vertidos Wn son del mismo contaminante y que se degrada siguiendo una
cinética de 1er. orden, con una constante k. y que el vertido no modifica ostensiblemente el caudal
circulante.
Para el primer lago, la concentración en estado estacionario es:
1
1
11
11 V kQ
WV B
W = C
R += Ec.( 220)
Para el segundo, la concentración en estado estacionario es:

Modelos de Calidad de Aguas 164
2
1
12
2
1
12
22
122
/1V kQ
QV k W
W
V kQ V kQ
WQW
V B CQW
= CR
R
R
R
R
R+
++
=+
++
=+
Ec.( 221)
En general:
nR
Rn
nn
n V kQ QV k
WW = C
++
+−
−/1 1
1
Ec.( 222)
Estas expresiones pueden ser útiles también para simular un sistema lineal, mediante su
descomposición en secciones, constituyendo cada una de ellas un pequeño lago completamente
mezclado.

Modelación Matemática de la Calidad de las Aguas 165
4.3.5 Aguas subterráneas: transporte en zona no saturada y zona saturada.
4.3.5.1 Zona no saturada.
La zona no saturada de un sistema acuífero es aquella capa del suelo que separa la
superficie del terreno del nivel piezométrico del acuífero. El transporte de contaminantes en esta
zona se caracteriza por estar ligado a la velocidad de percolación del fluido desde la superficie. El
transporte es, por lo tanto, unidimensional en la dirección descendente.
Los factores principales que afectan a este transporte son los siguientes:
- Tipos de materiales distintos que conforman el terreno.
- Características de estos materiales:
- Porosidad.
- Contenido residual de agua.
- Permeabilidad (conductividad hidráulica).
En este caso, la ecuación general de conservación de la materia se expresa como sigue:
C R k - zC V -
zC D =
tC R vvv2
2
vv ∂∂
∂∂
∂∂ Ec.( 223)
donde:
C : concentración de la fase disuelta del contaminante en la zona no saturada (mg/L).
Dv : coeficiente de dispersión en la zona no saturada (m2/año).
kv : constante de velocidad de reacción de primer orden en el interior de la zona no saturada (año-1).
Rv : Factor de retardo de la zona no saturada (adim).
Vv : velocidad de filtración a través de la zona no saturada (m/año).
t : tiempo (año).
z : coordenada vertical, positiva en la dirección descendente (m).
En ella se tiene en cuenta la dispersión en la dirección del flujo (vertical), procesos de
adsorción y de descomposición mediante una cinética de primer orden del contaminante.
Esta ecuación presenta soluciones analíticas si es simplificada convenientemente. Por
ejemplo, en el caso de que se considere el estado estacionario:

Modelos de Calidad de Aguas 166
C R k - zC V -
zC D = 0 vvv2
2
v ∂∂
∂∂ Ec.( 224)
cuya solución analítica si Dv <<< es la siguiente:
V
R L k - exp = CC
v
vv
0 Ec.( 225)
en donde:
C0 : concentración de contaminante en la parte superior de la zona no saturada (mg/L)
L : el espesor de la zona no saturada (m).
4.3.5.2 Zona saturada.
La zona saturada es aquella en la que el flujo de agua a través de la matriz sólida del terreno
es permanente y está bien establecido. En este caso, la ecuación general del balance de materia se
representa como:
θ∂∂
∂∂
∂∂
∂∂
∂∂
BC q R - C R k - z
C V - zC D +
yC D +
xC D =
tC R ssss2
2
z2
2
y2
2
xs Ec.( 226)
donde:
x, y, z : coordenadas espaciales en las direcciones longitudinal, lateral y vertical (m).
C : concentración de disuelta del contaminante (mg/L).
Dx, Dy, Dz : coeficientes de dispersión en las direcciones x, y, z (m2/año).
Vs : velocidad uniforme en la dirección x (m/año).
Rs : factor de retardo en la zona saturada.
t : tiempo transcurrido (año).
ks : coeficiente global de degradación de primer orden (año-1).
q : recarga directa fuera de la zona de vertido directa que diluye la pluma contaminante (m/año).
B : espesor de la zona saturada.
θ : porosidad efectiva de la zona saturada.
La resolución analítica de la ecuación anterior es sumamente complicada, por lo que se
recurre a métodos numéricos, tal y como se explicó en el tema de aguas superficiales. En este caso
también es necesaria la creación de una malla de cálculo en cada una de cuyas celdas se calcula en

Modelación Matemática de la Calidad de las Aguas 167
cada instante la concentración de contaminante. Aún así se hace necesario realizar una serie de
simplificaciones, las más comunes de las cuales son las siguientes:
- Acuíferos sencillos de espesor uniforme.
- Las propiedades del medio poroso son isotrópicas y homogéneas.
- La velocidad del agua en el acuífero es estacionaria y uniforme.
- La recarga debida al vertido contaminante es despreciable frente al flujo global.
- Las transformaciones cinéticas de los contaminantes se reducen a reacciones de primer
orden.
- La adsorción de contaminantes sigue una isoterma de adsorción lineal. Equilibrio local.
4.4 Métodos numéricos.
Cuando la geometría del sistema natural es tan compleja que no se pueden realizar las
simplificaciones anteriores para reducir el sistema natural a dimensiones 1 ó 0, cuando los
parámetros (caudal, volumen, etc.) son variables, cuando las cinéticas de reacción no son lineales o
cuando los vertidos no responden a un tipo ideal, se hace necesario recurrir a métodos matemáticos
para resolver la ecuación diferencial en derivadas parciales que representa el balance de materia.
Para el cálculo de la concentración del contaminante en el espacio es necesario construir un
sistema reticular en cada una de cuyas celdas se calculará la concentración de dicha sustancia para
un tiempo determinado. Cuanto mayor sea el número de celdas mayor también será la
aproximación al valor correcto, pero a costa de un mayor tiempo de cálculo.
4.4.1 Método de Diferencias Finitas.
El método de diferencias finitas es hasta el momento el más empleado para la resolución de
este tipo de ecuaciones. En estas técnicas se introduce una malla de tipo rectangular en la cual los
valores de C son conocidos para un tiempo t y en los puntos centrales de la red uni, bi o
tridimensional. Dichos valores conocidos se representan mediante la notación Cj-1,n, Cj,n, Cj+1,n en la
cual el primer subíndice representa la celda y el segundo el tiempo. Los valores desconocidos son
representados como Cj-1,n+1, Cj,n+1, Cj+1,n+1.

Modelos de Calidad de Aguas 168
1 2 3 4 5 6 7
Cj,n Cj+1,n Cj-1,n
Mediante un esquema de cálculo explícito se representan las derivadas a partir de valores
conocidos mediante alguna de las siguientes aproximaciones:
x 2)C - C(
= xC
x)C - C(
= xC
x)C - C(
= xC
n1,-jn1,+j
nj,n1,+j
n1,-jnj,
∆∂∂
∆∂∂
∆∂∂
Ec.( 227)
Un esquema de cálculo implícito es aquel en el que la derivada se calcula a partir de valores
conocidos y desconocidos de C:
∆∆∂
∂x 2
)C - C( +
x 2)C - C(
21 =
xC n1,-jn1,j+1+n1,-j1+n1,j+ Ec.( 228)
La técnica explícita es más sencilla de programar, mientras que en el esquema implícito un
conjunto de ecuaciones simultáneas debe ser resuelto para obtener las concentraciones en el
siguiente intervalo de tiempo. La ventaja es que permite el uso de intervalos de tiempo mayores y
es más estable.
La aplicación más sencilla de la técnica de diferencias finitas da lugar al denominado
método de Euler, que aplicado a un lago completamente mezclado, sería como sigue:
c B - V
W(t) = t dc d Ec.( 229)
)t - t(] c B - V(t) W [ + c = c i1+iii1 + i Ec.( 230)
c B - V(t) W =
t - tc - c
ii1+i
i1+i Ec.( 231)

Modelación Matemática de la Calidad de las Aguas 169
h )c ,t( f + c = c iii1+i Ec.( 232)
Es decir, este método permite calcular la concentración en un instante determinado a partir
del valor del tiempo anterior, de la pendiente a la curva c = f(t) en el punto ti, ci y del paso de
tiempo. Hay que hacer notar que esta solución aproximada se ajustará más a la solución analítica
dada por la ecuación (182) cuanto menor sea el valor del paso de tiempo h.
Una mejora del método de Euler, que limita el error producido en la aproximación por
aplicar el valor de la derivada en el inicio del intervalo de tiempo (ti) a todo el intervalo (ti+1), es el
método de Heun o predictor-corrector. Este método evalúa la derivada en dos puntos: ti y ti+1 y
calcula el valor de ci+1 a partir del valor medio de ambas:
)c,t( f = tdc d
iii Ec.( 233)
2)c ,tf( + )c ,tf(
= dtdc o
1+i1+iii Ec.( 234)
h )c ,tf( + c = c iiio
1+i Ec.( 235)
h2
)c ,tf( + )c ,tf( c=c
o1+i1+iii
jj ++1 Ec.( 236)
)c,tf( = dtcd o
1+i1+i1+i Ec.( 237)
El método predictor-corrector es una aproximación de segundo orden. Esto significa que si
la solución de la ecuación diferencial fuera un polinomio cuadrático proporcionaría los resultados
perfectos. Es una aproximación superior al método de Euler pero el hecho de que haya que efectuar
dos evaluaciones de la derivada para cada paso de tiempo significa que es necesario pagar cierto
precio computacional para ganar en precisión.

Modelos de Calidad de Aguas 170
Un método ampliamente extendido en el campo de la modelización de la calidad del agua
es el método de Runge-Kutta. Este método tiene la forma general:
h + c = c i1+i φ Ec.( 238)
en donde φ es la denominada función incremento. Al igual que en el método de Heun, la función
incremento representa un valor promedio de la derivada en el intervalo temporal. Si la función
incremento es de cuarto orden se obtiene una clásica expresión para el cálculo de ci+1:
h)] k + k 2 + k 2 + k( 61 [ + c = c 4321i1+i Ec.( 239)
donde:
)kh + c h, + tf( = k
)kh 21 + c h,
21 + tf( = k
)kh 21 + c h,
21 + tf( = k
)c ,tf( = k
3ii4
2ii3
1ii2
ii1
Ec.( 240)
La aparición de la inestabilidad numérica puede evitarse conociendo el intervalo de tiempo
máximo que se puede emplear, determinable en función de las entradas y salidas de materia a cada
celda. Para mantener la estabilidad del sistema, la masa que es transportada por convección,
dispersión y transformada en cada celda debe ser menor que la masa residente. Esta condición se
traduce en una expresión del siguiente tipo:
)C/V S( + R + Q
V = t
jjjkk
ijk
iji
j
∆∑∑∑
Minmax Ec.( 241)
en donde :
Qij : es el caudal que entra o sale de la celda i a la j.

Modelación Matemática de la Calidad de las Aguas 171
Rij : es el flujo dispersivo entre las celdas i y j.
Sjk Vj/Cj : es el término de reacción del componente k en la celda j.
Otro problema asociado al empleo de este método numérico es la aparición de una mezcla
artificial. Esta dispersión numérica puede determinarse mediante el cálculo de un coeficiente de
dispersión:
t)u - (L 2u = Enum ∆ Ec.( 242)
en donde:
u : es la velocidad del agua (m/s)
L : es la longitud de la celda (m)
t : intervalo de tiempo (s)
Esta expresión refleja los dos efectos incluidos en la dispersión numérica: la discretización
espacial y la discretización temporal. Dicha dispersión será menor cuanto menor sea la velocidad
de la corriente en nuestro sistema natural, menor sea la longitud de la celda y mayor sea el intervalo
de tiempo empleado. Esta última condición está delimitada por el intervalo de tiempo máximo que
se puede emplear según el criterio de estabilidad mencionado anteriormente.
4.5 Factores para la elección del modelo.
La elección de una herramienta de trabajo como son los modelos matemáticos de calidad de
aguas no es un hecho arbitrario, sino que depende de una serie de factores de entre los cuales
habría que destacar los siguientes:
Sistema Natural. En función del tipo de sistema natural: lago, río, estuario, zona costera, etc.
se podrán realizar distintas simplificaciones que determinarán el tipo de
modelo (en una, dos o tres dimensiones) que es conveniente emplear.
Definición del Problema. El objetivo del estudio es la evaluación del impacto de cierto
contaminante en el medio natural. En función de este contaminante

Modelos de Calidad de Aguas 172
se elegirán las formulaciones más adecuadas y se definirán las
escalas de tiempo y espacio que empleará el modelo.
Obtención de datos. La obtención de datos de campo mediante una campaña de muestreo
en la zona permitirá un conocimiento adecuado de la situación actual.
Cuanto más numerosos sean éstos más detallado podrá ser el modelo
que se emplee y un mayor número de aspectos del problema podrán
ser abordados.
Capacidad computacional. La disponibilidad de las herramientas de cómputo es otro
factor limitante del empleo de los modelos. La resolución de
la ecuación de transporte en tres dimensiones requiere
equipos altamente avanzados, mientras que numerosos
problemas en una o dos dimensiones pueden ser resueltos con
sencillos ordenadores personales.
Las escalas temporal y espacial en las que se desenvuelve el problema a estudiar son
determinantes a la hora de definir el modelo a emplear. En la siguiente tabla se muestra, por
ejemplo, los diferentes procesos implicados en el vertido de aguas residuales al mar a través de
emisarios submarinos:
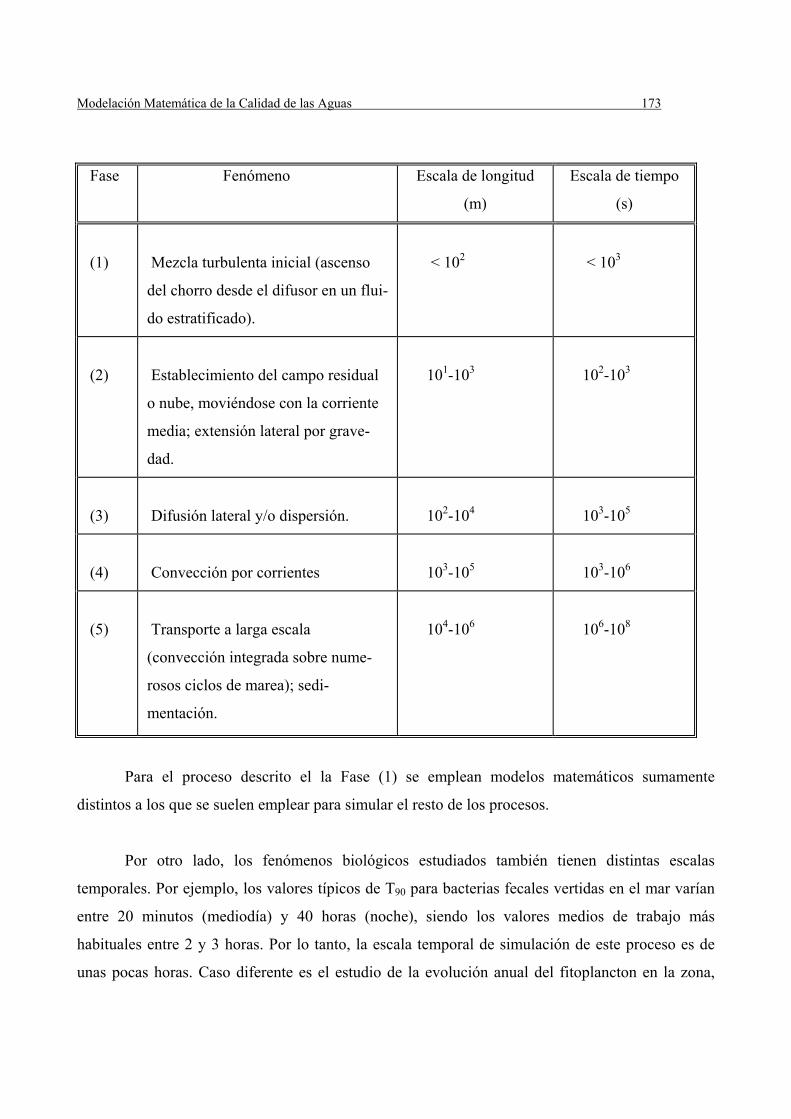
Modelación Matemática de la Calidad de las Aguas 173
Fase Fenómeno Escala de longitud
(m)
Escala de tiempo
(s)
(1)
Mezcla turbulenta inicial (ascenso
del chorro desde el difusor en un flui-
do estratificado).
< 102
< 103
(2)
Establecimiento del campo residual
o nube, moviéndose con la corriente
media; extensión lateral por grave-
dad.
101-103
102-103
(3)
Difusión lateral y/o dispersión.
102-104
103-105
(4)
Convección por corrientes
103-105
103-106
(5)
Transporte a larga escala
(convección integrada sobre nume-
rosos ciclos de marea); sedi-
mentación.
104-106
106-108
Para el proceso descrito el la Fase (1) se emplean modelos matemáticos sumamente
distintos a los que se suelen emplear para simular el resto de los procesos.
Por otro lado, los fenómenos biológicos estudiados también tienen distintas escalas
temporales. Por ejemplo, los valores típicos de T90 para bacterias fecales vertidas en el mar varían
entre 20 minutos (mediodía) y 40 horas (noche), siendo los valores medios de trabajo más
habituales entre 2 y 3 horas. Por lo tanto, la escala temporal de simulación de este proceso es de
unas pocas horas. Caso diferente es el estudio de la evolución anual del fitoplancton en la zona,

Modelos de Calidad de Aguas 174
para la cual se requiere tiempos de simulación elevados. En el primer ejemplo, el intervalo de
tiempo que empleará el modelo de calidad de aguas será sensiblemente menor que en el segundo,
puesto que la biodegradación de las bacterias fecales transcurre a más velocidad. También el
tamaño de las celdas de la malla computacional será menor en el primer caso que en el segundo.
Otro factor que influye de una forma importante en la elección del modelo es el coste
computacional, entendido como capacidad de almacenamiento y capacidad de cálculo. Los criterios
de estabilidad numérica impuestos por las técnicas de resolución de ecuaciones diferenciales
implican que, para problemas de tipo bidimensional por ejemplo, si se divide por dos la red de
cálculo, aumenta por 16 el coste de cálculo. Esto hace que, en numerosas aplicaciones prácticas, se
desarrollen y empleen modelos de tipo unidimensional.
4.6 Calibración-verificación de los modelos.
El empleo de modelos matemáticos de simulación requiere un trabajo previo de
conocimiento del entorno en donde van a ser aplicados. Dicho conocimiento permitirá ajustar las
constantes cinéticas que intervienen en los procesos químicos y biológicos descritos en las páginas
anteriores a la realidad del sistema natural. A este proceso se le denomina calibración del modelo.
Para ello se hace necesaria la realización de una campaña de toma de muestras y análisis de los
parámetros más significativos. Dicha campaña debe tener una duración adecuada a las condiciones
de trabajo.
Si, por ejemplo, el problema consiste en analizar la descarga de agua residual en un río, la
campaña de muestreo se deberá realizar (si no es posible durante el ciclo anual completo) en las
condiciones más desfavorables. En este caso sería en aquella época del año durante la cual el
caudal fuera más bajo, pero nunca, por ejemplo, en época de avenidas. Si el estudio que se está
llevando a cabo versa sobre la problemática de la eutrofización, la campaña de muestreo debería
cubrir un periodo representativo de la evolución del fitoplancton: al menos un año.
Durante la fase de calibración de un modelo de simulación de cualquier proceso natural hay
que ajustar los resultados de la simulación efectuada por el modelo durante un período
determinado, con los datos de campo obtenidos durante ese mismo período. Para ello es necesario

Modelación Matemática de la Calidad de las Aguas 175
establecer las funciones matemáticas que representan adecuadamente los procesos que tienen lugar
en el sistema natural estudiado, así como los valores de todos los parámetros que intervienen en el
modelo. Esta labor se lleva a cabo mediante las siguientes etapas:
- Selección de las funciones matemáticas que mejor representen la evolución de las
variables del modelo para ese sistema natural y obtención de los parámetros
correspondientes a dichas funciones.
- Realización del análisis de sensibilidad. Estudio de la influencia de todos los parámetros
que intervienen en el modelo de calidad en el resultado final de la simulación.
- Determinación de aquellos parámetros que puedan ser obtenidos a partir de datos de
campo.
- Asignación de valores a aquellos parámetros que, tras el análisis de sensibilidad, se
hayan mostrado poco significativos. Estos valores se asignan a partir de la información
recogida de la bibliografía.
- Diseño del proceso de calibración: realización de series de simulaciones agrupadas por
parámetros significativos que afectan a una o a pocas variables. En cada serie de
simulaciones se modifica solamente el valor de uno de los parámetros. El proceso se
inicia a partir de datos de la bibliografía.
Un tema de sumo interés en la fase de calibración de los modelos numéricos es la
sensibilidad que presenta una variable (concentraciones de fitoplancton, de oxígeno disuelto, etc.) a
las variaciones de los valores de los distintos parámetros que son objeto de la calibración. Cuando
se maneja un número considerable de parámetros es conveniente conocer, antes de iniciar la
calibración, aquellos que modificarán de forma más apreciable las variables. El objetivo es doble:
- Determinar los parámetros más significativos cuyo valor sea característico del sistema
natural estudiado y no pueda ser conocido más que realizando el proceso de calibración. A
aquellos poco significativos se les asigna valores característicos obtenidos de la
bibliografía, lo que permite reducir el tiempo de trabajo en la calibración.

Modelos de Calidad de Aguas 176
- Observar si un parámetro tiene incidencia significativa sobre todas las variables de estado
o únicamente sobre una o un grupo determinado de ellas. De esta forma la calibración se
puede iniciar por los parámetros más generales y acabar por los más particulares.
Existen diversas maneras de llevar a cabo el análisis de sensibilidad de un modelo de
calidad de aguas. A modo de ejemplo, Larsen (1992) en su modelo de oxígeno disuelto en el río
Usk, calcula los incrementos de oxígeno que se producen en distintos puntos del río cuando el
parámetro considerado se reduce en un 50%.
Otra forma de realizar el análisis de sensibilidad es mediante el cálculo de la sensibilidad
relativa, Sx (Hopkins, 1983):
paramparam
xx
= S x ∆
∆
Ec.( 243)
en donde Sx es la sensibilidad relativa, x es la variable de estado y param es el parámetro
considerado.
La sensibilidad relativa depende de las expresiones matemáticas empleadas en la
descripción de los modelos físicos, químicos y biológicos, por lo que es una característica propia
del modelo de calidad de aguas desarrollado. Este análisis es independiente del sistema natural
sobre el cual se vaya a aplicar el modelo. Únicamente si la formulación del modelo cambiara,
habría que volver a realizarlo.
Además de averiguar qué parámetros afectan en mayor medida a los valores de las
variables, es importante conocer el grado de incertidumbre que introducen en los resultados finales.
Esta incertidumbre es debida a que no se conoce exactamente el valor del parámetro, sino su
intervalo de variación. El resultado de este análisis permitirá conocer sobre qué parámetros es
necesario realizar un esfuerzo mayor para su estimación.

Vertidos al Mar _____________________________ 177
5. VERTIDOS AL MAR MEDIANTE EMISARIOS SUBMARINOS. UN CASO PRÁCTICO.
5
t
r
a
s
b
e
s
n
r
a
v
l
m
q
El diseño y la operación de emisarios submarinos son ejemplos típicos de uso del medio receptor como auxiliar en la depuración de sustancias contaminantes. Es por ello también un caso de estudio para la aplicación de modelos de calidad de aguas superficiales de varios tipos. Finalmente, otro aspecto de interés se resume en el hecho de que la legislación actual obliga al empleo de una serie de modelos de cálculo, cuyo origen uso y limitaciones debe ser familiar para el estudiante.
.1 - Introducción.
Un emisario submarino es una conducción que transporta aguas residuales parcialmente
ratadas desde la costa hasta un punto mar adentro situado a cierta profundidad. Dado que el agua
esidual vertida tiene una menor salinidad y mayor temperatura que la del agua del mar, tiende a
scender. Durante este ascenso se mezcla con agua limpia del mar provocando la dilución de las
ustancias contaminantes. La inyección del chorro en el mar se suele efectuar mediante múltiples
oquillas situadas en el denominado tramo difusor, tramo final del emisario. La disposición de
ste tramo se estudiará más detalladamente en un siguiente apartado.
El objetivo del emisario submarino es lograr que los efectos de los contaminantes vertidos
obre el medio marino sean mínimamente perjudiciales, facilitando su asimilación en el medio
atural. Ha de quedar claro que un emisario submarino no es un sistema de tratamiento de aguas
esiduales, sino un sistema de evacuación que basa su efectividad en una elevada capacidad de
utodepuración del mar. Si ésta es muy baja, no será adecuado emplear un emisario submarino para
erter las aguas residuales.
Esta capacidad de autodepuración, como ya se vió en temas anteriores, está relacionada con
os mecanismos de transformación de los diversos contaminantes que puedan ser vertidos. Estos
ecanismos de transformación se ven complementados por los procesos de transporte y mezcla
ue sufre el efluente.

Modelos de Calidad de Aguas 178
Corrientes
Viento
COSTA
EDAR
Red de Saneamiento
Emisario Submarino
Tramo difusor
Figura 19. Emisario submarino.
A grandes rasgos, los procesos implicados en la disminución de la concentración de
contaminantes vertidos a través de un emisario submarino son tres:
- Dilución inicial. Cuando el chorro de agua residual sale a través de las boquillas difusoras
del tramo final del emisario submarino, se produce una primera fase de mezcla rápida con
agua del mar, que da lugar a una dilución elevada. Esta mezcla es debida a la interacción
del agua que sale a través del emisario con el agua ambiente y fundamentalmente está
condicionada por la cantidad de movimiento del agua residual y la diferencia de densidades
(denominado efecto de flotabilidad) entre el agua dulce y la salada. Esta fase dura unos
pocos minutos y se desarrolla en unas pocas decenas de metros. Finaliza cuando la mancha
de agua residual se ha estabilizado en superficie o en una zona intermedia de la columna de
agua, como más adelante se explicará.

Vertidos al Mar _____________________________ 179
- Dilución secundaria. Tras la etapa de ascenso inicial, la mancha de agua residual es
transportada por las corrientes y la mezcla se produce por mecanismos de dispersión
turbulenta en las tres direcciones del espacio. La velocidad de mezcla es más lenta en este
caso.
- Autodepuración. El término autodepuración corresponde a las transformaciones de tipo
físico, químico y/o biológico que pueden sufrir las sustancias contaminantes. Se ha
empleado este término debido a que su uso es muy frecuente en problemas relacionados con
el diseño y la operación de emisarios submarinos, aunque sólo es estrictamente aplicable a
aquellas sustancias cuya concentración disminuya debido a su transformación. No es
necesario recordar que los procesos de transformación pueden dar lugar a nuevas sustancias
contaminantes, en cuyo caso no se puede hablar de autodepuración para estas últimas.
Dadas las diferentes escalas de tiempo y espacio a las que operan estos mecanismos se
asume que, por ejemplo, en el proceso de mezcla inicial no se dan mecanismos de transformación
de sustancias, mientras que los procesos de dispersión turbulenta y autodepuración sí que se dan
simultáneamente. En la Tabla siguiente se muestra esquemáticamente esta situación.
Fase
Fenómeno
Escala de longitud (m)
Escala de tiempo (s)
(1)
Dilución Inicial (ascenso de un chorro en el seno de un fluido).
< 102
< 103
(2)
Difusión lateral y/o dispersión. Advección por corrientes.
102-105
103-105
(3)
Procesos físicos, químicos, biológicos. Mortalidad bacteriana.
103-105
3 6 10 -10

Modelos de Calidad de Aguas 180
5.2 - Aspectos ambientales del vertido por medio de emisarios submarinos.
En este apartado se va a realizar un análisis pormenorizado de los procesos que afectan al
vertido de aguas residuales a través de emisarios submarinos, se introducirán los factores más
importantes y se aportarán los modelos matemáticos más habituales para el cálculo de dichos
procesos.
5.2.1 .- Dilución Inicial.
La Dilución Inicial representa el grado de mezcla que alcanza un chorro de agua residual
desde que sale a través de la boquilla de un emisario submarino hasta que llega a la superficie (o se
estabiliza en una zona intermedia de la columna de agua, como posteriormente se verá). Este
término es de suma importancia, puesto que la legislación establece su valor mínimo en 100:1
(aunque se admite una condición de excepcionalidad que la sitúa en 80:1).
El chorro que sale a través de las boquillas difusoras puede tener dos comportamientos
extremos en función de sus características de salida: si su densidad no difiere de la densidad
ambiente y posee cierta velocidad inicial, se considera como un chorro propiamente dicho; su
comportamiento (trayectoria, dilución) viene determinado por la variación de su cantidad de
movimiento inicial y no por la variación de densidad (flotabilidad). Si la velocidad inicial es nula,
pero existe una diferencia entre las densidades de ambos fluidos, el chorro actúa como una pluma;
asciende por efecto de la diferencia de densidades entre el agua salada y agua dulce hasta que la
flotabilidad se anula. En realidad, el comportamiento de cualquier efluente es una combinación de
ambos y el resultado final será consecuencia tanto de las condiciones de salida de éste, como del
estado del ambiente externo (variación de densidad del agua del mar en función de la profundidad
y velocidad de ésta, principalmente). En adelante, se empleará la denominación de "chorro" para
los vertidos que se comporten de esta forma, reservándose las denominaciones de "chorro puro", y
"pluma" para los casos límite ya comentados.
Se comprueba también que, a la larga, todos los chorros acaban convirtiéndose en plumas,
puesto que el efecto asociado a la cantidad de movimiento desaparece rápidamente conforme el
efluente se aleja del difusor, mientras que el efecto de la flotabilidad suele permanecer más tiempo.

Vertidos al Mar _____________________________ 181
Chorros y plumas pueden tener un comportamiento laminar o turbulento y, al igual que el
flujo en conducciones, es posible definir un número de Reynolds, y asumir que por encima de
cierto valor, el flujo es completamente turbulento. En los casos habituales de trabajo se supone que
el flujo turbulento está plenamente establecido.
El comportamiento de un chorro turbulento depende de tres clases de parámetros:
• Parámetros propios del chorro. Este grupo incluye la velocidad inicial del chorro, el caudal,
la cantidad de movimiento del fluido, y el caudal másico de cualquier elemento trazador tal
como salinidad, agua caliente, o sustancias contaminantes.
• Parámetros ambientales. Este segundo grupo comprende los factores del medio receptor,
tales como la velocidad ambiente, la estratificación debida a diferencias de densidad del
agua del mar, etc.
• Factores geométricos. El tercer grupo hace referencia al diámetro de los orificios de salida,
la separación entre ellos, y su orientación con respecto a la corriente marina, entre otras
características.
A continuación se va a realizar un análisis de cada uno de los factores mencionados con el fin
de evaluar su importancia en el proceso de dilución inicial.
5.2.1.1 .- Velocidad de salida/orientación de la salida.
La velocidad de salida, u0, del chorro a través de las boquillas de salida afecta de manera
importante al tiempo que tarda en llegar a la superficie y, por tanto, al tiempo que dispone para
mezclarse con agua salada. Sin embargo, este factor está muy relacionado con la orientación de la
boquilla de salida con respecto al fondo marino, pudiéndose dar dos situaciones extremas:
• Que la salida del chorro sea vertical (perpendicular al fondo marino). En este caso, una
velocidad elevada dará lugar a un rápido ascenso del chorro y a una menor dilución inicial.
En esta situación el ascenso por flotabilidad se suma al ascenso por cantidad de
movimiento.
• Que la salida del chorro sea horizontal (paralela al fondo marino). A diferencia del caso
anterior, la fuerza ascensional sólo es debida a la diferencia de flotabilidad, por lo que el

Modelos de Calidad de Aguas 182
chorro describirá una trayectoria curvilínea hacia la superficie. La dilución inicial sería
mucho mayor que en el caso anterior. En una situación extrema es posible incluso que el
chorro se diluya antes de llegar a la superficie (caso muy poco probable).
5.2.1.2 .- Densidad del agua residual/densidad del agua del mar.
La relación entre la densidad del agua del efluente y la densidad del agua del mar en el punto de vertido determinan el efecto de ascensión por flotabilidad. Cuanto más marcada sea esta diferencia mayor será el grado de turbulencia en la mezcla y, por consiguiente, mayor será la dilución inicial (aunque también el chorro tienda a ascender más rápidamente). Con el fin de determinar este efecto en el diseño de los emisarios submarino se ha de calcular la aceleración reducida por flotabilidad, definida como:
0
0'ρ
= aggρρ −
en donde: g : aceleración gravitacional (9.81 m/s2). ρa : densidad del agua del mar (kg/m3). ρ0 : densidad del agua efluente (kg/m3). Un caso particular de vertidos de aguas residuales al mar consiste en el vertido de salmueras
procedentes de instalaciones de desalación de aguas. En estos casos, podría ser que la densidad de
la salmuera fuera mayor que la del mar, y por lo tanto la flotabilidad sería negativa, por lo que el
chorro, si ascendiera hacia la superficie, sólo lo haría debido a su cantidad de movimiento. Sería
conveniente, por lo tanto, que la velocidad de salida fuera elevada y en dirección vertical (nunca
horizontal).
5.2.1.3 .- Corrientes marinas.
Es bastante habitual que el medio en donde descarga el emisario esté en movimiento con
una velocidad media Ua; y también es de esperar que esta circunstancia modifique el compor-

Vertidos al Mar _____________________________ 183
tamiento del chorro, variando su trayectoria y aumentando la dilución del vertido. Los modelos que
se emplean para el cálculo de la dilución inicial suelen considerar valores de Ua despreciables, por
lo que sus resultados quedan dentro de un margen de seguridad elevado.
La consideración de Ua y, más aún, de su distribución vertical en función de la profundidad,
z, da lugar a mejores aproximaciones en el cálculo de la dilución inicial y del punto de salida del
chorro a la superficie.
5.2.1.4 .- Estratificación marina.

Model 184os de Calidad de Aguas
En las zonas costeras es fre-
cuente encontrar zonas
estratificadas en profundidad
debido a las diferencias de tem-
peratura y salinidad de las masas
de agua. La presencia de este
fenómeno puede alterar no-
tablemente el comportamiento
del chorro estudiado hasta ahora.
La densidad del agua del mar
aumenta con la profundidad,
mientras que la de la pluma as-
cendente aumenta con la altura,
conforme se va mezclando con
agua exterior. Puede ocurrir que,
a cierta profundidad, sus densi-
dades se igualen, y la pluma deje
de ascender. Varios casos típicos
son ilustrados en la Figura
contigua. En ella aparece
representada la termoclina, una
zona de pocos metros de espesor en la que existe un gradiente muy fuerte de temperaturas, y por
consiguiente, de densidades. El valor de H1 indica la altura de la termoclina, mientras que H
representa la profundidad total. En el caso extremo (A), el efluente es atrapado entre dos capas y se
extiende horizontalmente. Esta es una situación bastante frecuente, y puede ser observada bajo con-
diciones ambientales particularmente favorables, en emisarios profundos, durante el verano. En el
caso de existir una estratificación débil (B), el efluente rebasa el nivel de la termoclina y alcanza la
superficie. Si la estratificación es moderada (C), el vertido se distribuye entre el nivel de la
termoclina y la superficie.
Si la estratificación del agua del mar es lineal en profundidad, es necesario calcular el paráme-
tro de estratificación, Γ, definido como:
dzd
g - = a
a
ρ
ρ0
Γ

Vertidos al Mar _____________________________ 185
en donde:
ρa: densidad del mar (kg/m3)
ρa 0: densidad del mar en z = 0 (kg/m3)
5.2.1.5 .- Diámetro de los orificios de salida.
Para un mismo caudal de salida a través de cada boquilla, una disminución en su diámetro
provoca un aumento de la velocidad, por lo que las consecuencias derivadas de ello se expusieron
en el apartado 5.2.1.1. La elección del diámetro no es arbitraria: si es pequeño, la velocidad de
salida puede ser muy grande y las pérdida de carga elevadas, además de que las incrustaciones
marinas pueden llegar a obturar el orificio. Si es muy grande se puede producir la intrusión del
agua marina al interior de la conducción. Valores habituales de los diámetros se encuentran entre
60 y 120 mm.
5.2.1.6 .- Separación entre las boquillas de salida.
El mecanismo de funcionamiento del tramo difusor puede ser de dos tipos: si las boquillas
difusoras de salida están muy cerca entre sí, el chorro que salga por ellas se unirá rápidamente con
los contiguos, de modo que no se puede hablar de chorros individuales sino de una línea-fuente de
longitud igual a la longitud total del tramo difusor. Si, por el contrario, los difusores se sitúan lo
suficientemente separados de modo que cada chorro mantenga su individualidad, se está hablando
de chorros circulares turbulentos.
Se suele considerar que si la separación es de, al menos, el 20% de la profundidad, los chorros
no se mezclan hasta que no llegan a la superficie, mientras que si están separados menos del 3% de
la profundidad, sí lo hacen.
Los modelos matemáticos para el cálculo de la dilución inicial son, en ambos casos diferentes.
5.2.2 .- Dilución Secundaria.
Una vez establecida la “nube” de agua residual en superficie, ésta es transportada por las
corrientes y sometida a procesos de dispersión turbulenta. Estos procesos no modifican la cantidad

Modelos de Calidad de Aguas 186
total de contaminantes, sino que los distribuyen en el espacio (el mismo efecto que se consigue en
la dilución inicial), por lo que es de suma importancia conocer esta distibución.
A continuación se van a analizar los procesos de transporte convectivo y
difusivo/dispersivo.
5.2.2.1 .- Transporte convectivo.
Con el fin de evaluar correctamente el transporte convectivo de contaminantes es
fundamental contar con un campo de velocidades típico de la zona de estudio. Este campo de
velocidades puede obtenerse mediante el empleo de:
• Currentímetros. Son aparatos que miden velocidad y orientación de la corriente en puntos
fijos. Se pueden instalar en varios puntos de la zona de estudio y a varias profundidades
(con el fin de obtener el perfil vertical de la velocidad).
• Flotadores de vela sumergida. Consisten en una boya móvil de la que pende una aspa
sumergida a cierta profundidad. La boya se mueve a la velocidad y con la orientación de la
vela sumergida.
La velocidad y la orientación de las corrientes determinará el tiempo de recorrido de la mancha
de agua residual hasta la zona a proteger. Este aspecto es crucial puesto que cuanto mayor sea ese
tiempo, mayor será la degradación natural de los contaminantes.
5.2.2.2 .- Transporte dispersivo.
El cálculo experimental de los coeficientes de dispersión turbulenta Ex, Ey y Ez en el mar es
sumamente complejo y no suele ser objeto de estudio en la mayoría de los trabajos relacionados
con emisarios submarinos.
Las expresiones más habituales para el cálculo de la dispersión lateral están basadas en la
“ley de los 4/3”. En expresiones tienen la forma general:
3/4LaEH =

Vertidos al Mar _____________________________ 187
en donde L es una longitud característica del problema que se esté estudiando. En el caso de
emisarios submarinos, se suele tomar como L el valor de la anchura de la mancha en superficie,
denominada B.
La Instrucción española propone la expresión:
si B viene expresada en metros. 3/452 103)( BmEH−=
Aunque Fischer (1979) indica que los valores de α caen en el rango 4.64 10.4- 9.28 10-5
(m2/s) por lo que sería más correcto emplear la siguiente:
3/452 102831.9)( BmEH−=
Las expresiones para el cálculo de la dispersión horizontal parten, por consiguiente, de un
valor de B. Esto significa que, para un mismo emisario submarino, la orientación del tramo difusor
con respecto a la corriente marina puede dar lugar a un transporte dispersivo distinto.
Aunque parezca contradictorio, se ha comprobado que un mayor valor de B produce una
menor dilución en el centro de la trayectoria debido a la dispersión lateral, aunque los coeficientes
de dispersión sean mayores.
La dilución que se obtiene mediante los procesos de dispersión turbulenta suele ser muy
pequeña: del orden de 10:1 a 50:1 como mucho. Por ello es el proceso menos relevante de todos y
al que, en caso de necesidad, se le podría dedicar menos esfuerzos.
5.2.3 .- Autodepuración.
Cuando la mancha de agua residual es transportada y dispersada por las corrientes marinas
se producen simultáneamente diversos procesos de transformación, característicos de cada
sustancia contaminante. En temas anteriores se han estudiado los términos fuente/sumidero para
diversos contaminantes. En este caso, los contaminantes que se van a analizar son los
microorganismos patógenos, contaminantes típicos en los vertidos de aguas residuales urbanas.

Modelos de Calidad de Aguas 188
La contaminación del agua debido a la presencia de microorganismos patógenos ha sido
desde muy antiguo un serio problema de sanidad pública. En países subdesarrollados, en donde los
sistemas de potabilización y abastecimiento todavía no están totalmente implantados, se dan
todavía graves epidemias que afectan a amplios sectores de la población. En los países
desarrollados, en donde el riesgo de contraer enfermedades infecciosas por el consumo de agua
potable es muy reducido, la problemática principal se centra en los problemas sanitarios en zonas
de baño y áreas de cultivos marinos, fundamentalmente debidos a vertidos de aguas residuales
urbanas parcialmente tratadas.
El origen de virus, bacterias y protozoos causantes de enfermedades es
fundamentalmente animal y proceden en su mayor parte del aparato digestivo del ser humano. Otra
fuente de bacterias es el arrastre por agua de lluvia del lavado de calles y plantas.
Desde finales del siglo XIX se intentó buscar un indicador fiable de la presencia de
microorganismos patógenos, como Salmonella (fiebres tifoideas), Pseudomonas, Staphylococus,
Aeromonas, puesto que la técnicas para su detección son complejas. El indicador más empleado
son las bacterias del grupo Coliforme y, dentro de ellas, las de origen Fecal. El grupo Coliforme se
suele dividir en dos grupos:
- Fecales. Géneros Escherichia y Klebsiella.
- No fecales. Géneros Enterobacteria y Citrobacteria (asociados a suelos y plantas).
Por otro lado, se está imponiendo cada vez más el empleo de otro tipo de bacterias fecales,
los Estreptococos Fecales y, dentro de ellas, los Enterococos, como un indicador más seguro del
riesgo de contraer alguna enfermedad. Ello es debido a que se ha comprobado una mayor
persistencia en las aguas que los CT y CF, siendo aproximadamente su velocidad de desaparición
la mitad (su persistencia en el medio es el doble).
La relación entre Coliformes Fecales y Estreptococos Fecales es un indicador del origen de
la contaminación. Si el valor es ligeramente mayor de 1, la contaminación suele ser de origen fecal
humano, mientras que si es inferior a 1, es de origen fecal animal.
5.2.3.1 .- Modelos matemáticos de Bacterias Fecales.

Vertidos al Mar _____________________________ 189
El objetivo de la simulación mediante modelos matemáticos de los niveles de bacterias
fecales vertidas en el medio natural es el establecimiento del nivel de contaminación potencial
debida a agentes patógenos. Es conocido que dichos niveles disminuyen en el medio más
rápidamente de lo que lo harían si únicamente sufrieran procesos de transporte y dispersión
turbulenta. Existe cierta capacidad de autodepuración natural por parte del medio. Por
consiguiente, la concentración de bacterias al cabo de un tiempo de haber sido vertidas dependerá
de dos factores principales:
- La descarga inicial.
- La tasa de desaparición o coeficiente de autodepuración.
Hay que tener en cuenta que algunos microorganismos de tipo no-fecal pueden aumentar su
número si las condiciones ambientales son adecuadas, pero esto raramente sucede.
Son muy diversos los factores que afectan a la velocidad de desaparición de bacterias del
agua. Se pueden agrupar en tres grandes grupos: Físicos, Físico-químicos y Biológicos-
bioquímicos, que se desarrollan a continuación.
Factores Físicos:
- Fotooxidación. Efecto de la luz solar. Es el factor más importante de todos. Aumenta diez
veces la velocidad de desaparición existente bajo condiciones de oscuridad.
- Adsorción. Los microorganismos pueden quedar fijados en sólidos suspendidos,
principalmente si son de tipo arcilloso.
- Coagulación.
- Floculación. Formación de agregados junto a sustancias coloidales coaguladas.
- Sedimentación. Deposición de los agregados de bacterias formados. Hace disminuir su
presencia en la columna de agua pero aumentan en los sedimentos. Puede ser importante en
función del grado de tratamiento del agua residual: cuanto menor sea el nivel de tratamiento
mayor será la influencia de este proceso.
- Temperatura. Es un factor general que afecta a todos los demás. En la oscuridad y en
aguas dulces, es el factor más importante.
- Físico-químicos:

Modelos de Calidad de Aguas 190
- Efectos osmóticos: la supervivencia de E. Coli es inversamente proporcional a la
salinidad. El vertido de bacterias habituadas a agua dulce en aguas con elevado contenido
en sales provoca, por efecto osmótico, el paso del agua intracelular de las bacterias a través
de su membrana al medio.
- pH.
- Toxicidad debida a metales pesados.
- Potencial Redox. Afecta a la solubilidad de las sales de los metales pesados, los cuales, al
ser movilizados aumentan el efecto tóxico.
- Variaciones bruscas de oxígeno disuelto. El paso de un medio pobre en oxígeno como el
agua residual a otro rico en oxígeno como el agua del mar puede promover una mayor
desaparición bacteriana.
- Biológicos y bioquímicos:
- Nutrientes. La presencia de sales nutrientes puede hacer disminuir la toxicidad del medio
debido a la formación de complejos con iones metálicos tóxicos. Su presencia o ausencia
también afectaría a las poblaciones de depredadores de los coliformes.
- Depredadores. Otras bacterias, amebas. Su influencia global depende de la densidad de
población.
- Bacteriófagos. Poca importancia. Se inactivan rápidamente.
- Algas. Generan sustancias antibacterianas que son excretadas al entorno. Otro efecto
indirecto es aquel que se produce cuando aparece una proliferación rápida ( bloom) de
algas; los organismos que actúan como depredadores de algas y bacterias aumentan, por lo
que aumenta la tasa de desaparición bacteriana.
La formulación habitual para dar cuenta de la desaparición de bacterias fecales del sistema
natural ha sido mediante una expresión cinética de primer orden, como la mayoría de las empleadas
hasta el momento:
e C = C
C k - = dtdC
t k -ot
Ec.( 244)
en donde:

Vertidos al Mar _____________________________ 191
C : concentración de coliformes (NMP2/100 ml).
Co : concentración inicial.
Ct : concentración a tiempo t.
k : constante de desaparición (día-1, ó hr-1).
t : tiempo transcurrido (días u horas).
Es muy habitual emplear otra expresión en la que el valor de la constante k es sustituido por
otra constante denominada T90. Este valor de T90 representa el tiempo para el cual ha desaparecido
el 90% de las bacterias que fueron vertidas en el medio.
10 N = NT 90
t -
ot Ec.( 245)
en donde:
Nt : número de bacterias presentes a tiempo t (NMP/100 ml).
No : número de bacterias inicial (NMP/100 ml).
t : tiempo transcurrido (días u horas).
La relación entre los valores de k y de T90 es la siguiente:
T90 = ln 10 / k
En el caso de que se consideren varios tipos de bacterias, se representaría cada una de ellas
mediante un valor particular de k o de T90.
Dado que los coliformes y otros indicadores también desaparecen en la oscuridad (aunque a
velocidades mucho menores que con luz), algunos autores han propuesto expresiones para la
determinación de k que tienen en cuenta los componentes dependientes de la luz y los que no
dependen de ella:
L k + 071. 24
(%SW)) 0.006 + (0.8 = k L20) - (T Ec.( 246)
en donde:
%SW: Porcentaje de agua de mar.
T : Temperatura (ºC).
L : Intensidad luminosa media en la capa de agua (cal/cm2/hr).
2 NMP son las siglas de Número Más Probable. Se trata de una unidad de medición habitual del número de colonias de bacterias que aparecen en un cultivo a partir de aguas contaminadas. Dado que se emplean métodos estadísticos para el recuento de estas colonias, el resultado se expresa de esa manera, como una estimación estadística de la concentración.

Modelos de Calidad de Aguas 192
kL :Constante de proporcionalidad que depende del organismo considerado (cm2/cal).
En la Instrucción para el proyecto de conducciones de vertido desde tierra al mar se aporta
la siguiente expresión, aplicable a bacterias del grupo coliforme, para el cálculo del término T90:
135/)20(2
90 1002.0800
1)65.01(60
−−
+
−−= aTSSCT α
Ec.( 247)
en donde:
α : ángulo que forma el sol con respecto al horizonte en grados sexagesimales (valor mínimo 0).
C : fracción del cielo cubierto por nubes (entre 0 y 1).
SS: concentración de sólidos en suspensión (mg/L, valor máximo 800 mg/L).
Ta : temperatura del agua (ºC).
Como se puede deducir de lo anterior, los valores de k o T90 varían mucho a lo largo del día,
por lo que una de las tareas más complejas es la determinación experimental de estos valores.
Dicha determinación puede realizarse mediante ensayos de laboratorio o experiencias in situ. Esta
última opción es preferible a la primera.
Si el régimen del flujo no es bien conocido, como suele ocurrir en los vertidos de aguas
residuales al mar a través de emisarios submarinos, un procedimiento para la determinación del
valor de k es el siguiente:
- Se añade un trazador al caudal estacionario vertido. Dicho trazador puede ser un
contaminante que se considere conservativo durante el tiempo que dura el proceso, por
ejemplo los nitratos. Pueden emplearse colorantes, radioisótopos, etc.
- Se van tomando muestras a distintos tiempos y siguiendo la trayectoria del contaminante
(para lo cual hay que emplear boyas señalizadoras). La dilución física se determina con el
trazador. La concentración de bacterias de cada muestra, corregida a partir de los valores de
la dilución física permitirá calcular los valores de k.
Esta determinación tiene un grave inconveniente y es que puede dar resultados erróneos si
la dilución del trazador no se realiza con agua "limpia" sino con agua contaminada por la propia
descarga.

Vertidos al Mar _____________________________ 193
Si el régimen del flujo es conocido, por ejemplo en el caso de vertidos a ríos o canales, el
método es bastante más sencillo y seguro:
- En primer lugar se sitúa un punto fijo aguas abajo del vertido en donde la mezcla en la
dirección transversal del flujo sea completa.
- Se toman muestras en dicho punto y en varios más aguas abajo. Se determina en cada uno
de ellos la concentración de bacterias. Conocida la velocidad del río se determina el tiempo
transcurrido entre cada estación y suponiendo que el vertido se mantiene constante se puede
determinar el valor de k.
En este caso, el inconveniente principal es que el río sea excesivamente ancho y no se
pueda asumir la hipótesis de mezcla completa en la dirección transversal. También se ha supuesto
que la velocidad de la corriente se mantenía constante y es uniforme. En el caso de que el vertido
no sea estacionario, habría que seguir ese volumen de agua contaminada a lo largo del río o
calcular el tiempo para el cual habría que tomar la muestra en función de la localización de la
estación de muestreo.
Los valores de T90 pueden oscilar entre 15 minutos para las horas centrales del día y
decenas de horas para la noche. La legislación española permite, en el caso de que el vertido sea
efectuado por una población con menos de 10000 h-e, emplear un valor fijo de T90 igual o superior
a 2 horas para el mar Mediterráneo y de 3 horas para el Atlántico.
5.3 Diseño de emisarios submarinos.
En este apartado se desarrolla la aplicación de los métodos de cálculo propuestos por la
legislación española de referencia sobre el diseño de conducciones de vertido: Orden de 13 de

Modelos de Calidad de Aguas 194
Julio de 1993 por la que se aprueba la Instrucción para el proyecto de conducciones de vertido
desde tierra al mar (BOE 27 de Julio de 1993). La misma Instrucción indica que, si el emisario
submarino es de gran entidad pueden emplearse modelos más complejos siempre que se asegure su
fiabilidad.
El diseño de emisarios submarinos consiste en el cálculo del tramo difusor en una
localización que permita alcanzar los objetivos de Dilución Inicial mayor que 100 y que cumpla
con los objetivos de calidad en las zonas costeras que se deseen proteger. Estos objetivos de
calidad dependen del uso al cual vaya destinada la zona a considerar: recreativo, cultivos marinos,
etc. El último aspecto del diseño es el diseño hidráulico del sistema de boquillas difusoras. En la
figura siguiente se muestra el diagrama de flujo del proceso de diseño.
5.3.1 .- Cálculo del tramo difusor.
Los datos de partida, conocidos, son:
• Caudal vertido, Q.
• Pendiente de la línea de costa.
• Pendiente de la zona del tramo difusor. PTE.
• Concentración de contaminantes.
• Valor de T90.
• Velocidad de la corriente, Ua.
Los datos seleccionados por el diseñador son:
• Profundidad de vertido, Y.
• Forma y diámetro de las boquillas difusoras, d.
• Separación entre las boquillas.
• Colocación y diámetro, D, de la conducción.
• Orientación del tramo difusor.
Una vez obtenidos los datos anteriores hay que plantear el siguiente conjunto de ecuaciones:

Vertidos al Mar _____________________________ 195
Ecuaciones para el diseño del tramo difusor cuando la separación entre boquillas es, al menos, del
20% de la profundidad H:
(1) ( )
54.0' 3/2
0
220 =
+
HgBu
HUB au Condición de estabilidad de la capa de mezcla.
Esta ecuación se puede escribir como ( )
BHUHgBu
u a23/2
00
54.0' −=
(2) 02
4udQb
π=
(3)
+= 1
bd Q
Qdeenteraparten
(4) 2
0
4
/
d
nQu d
π=
(5) PTEn
nHLd
dT )1(2.01
)1(2.0−−
−=
(6) 1
3 '−
=
Ta L
QgUF
(7) [ ]3/193.0, −= FLsenLmáxB TT θ
en donde:
u0 : velocidad de salida del agua a través de cada boquilla (m/s).
Ua : velocidad de la corriente (m/s).
B : anchura de la mancha en superficie (m).
H: profundidad efectiva (m).
g’ : aceleración reducida por flotabilidad (m/s2).
d : diámetro de las boquillas del difusor (m).
Q : caudal total (m3/s).
Qb : caudal a través de cada boquilla (m3/s).
LT : longitud del tramo difusor (m).
nd : número de boquillas difusoras.

Modelos de Calidad de Aguas 196
PTE: pendiente en la zona del tramo difusor.
Todas estas ecuaciones, salvo la ecuación (5), vienen dadas en la Instrucción. Su resolución
simultánea proporcionará los valores finales de las características del tramo difusor. El valor de H
se calcula como la profundidad del punto de vertido Y menos la elevación sobre el fondo de la
salida de la boquilla difusora.
5.3.2 .- Cálculo de la Dilución Inicial.
Una vez calculado el tramo difusor se emplean las ecuaciones propuestas en la Instrucción
para el cálculo de la dilución inicial, S, y el espesor de la mancha en superficie. En el caso en el
que la separación entre boquillas sea, al menos, del 20% de la profundidad H, las ecuaciones son:
Ec.(248) ( )
a
b
UBQSe
QeHg'.S
=
−=−
32
35
31
0890
Si la dilución calculada es mayor que 100, el proceso ha finalizado de momento, a falta de comprobar el cumplimiento de los objetivos de calidad en la zona a proteger.
5.3.3 .- Cálculo de los objetivos de calidad.
La ecuación propuesta en la Instrucción para el cálculo de la concentración de
microorganismos patógenos en un punto alejado de la zona de vertido es:

Vertidos al Mar _____________________________ 197
eUKBK
tKBσ
σπB
He
SC
C(t)
azy
/
yy
Tt
yh
334
5
212
0
104103
216
102
90
−−
−
==
+=
=
Ec.( 249)
en donde:
C0 : concentración inicial de coliformes (NMP/100 ml).
e : espesor inicial de la mancha en superficie (m).
Hh: espesor de la mancha máximo (cuando llega a la costa), (m).
B: anchura de la mancha en superficie (m).
t : tiempo de recorrido hasta la zona a proteger (hr.).
T90 : constante de degradación del 90% de C0 (hr.)
Ky : coeficiciente de dispersión turbulenta lateral (m2/s).
Kz : coeficiciente de dispersión turbulenta vertical (m2/s).
El término Hh se calcula, para una localización muy alejada del punto de surgencia, a partir
de las expresiones para el cálculo de la dilución por dispersión vertical (F3(Y,t)) en zonas próximas
al punto de surgencia:
tK
HezeerfzeerftzF
zz
hzz
2
2221),(3
=
=
−+
+=
σ
σσ
Ec.( 250)
Para resolver esta ecuación y calcular Hh se asume que, al estar suficientemente lejos del
punto de surgencia, el valorde F3 ya no depende de la coordenada vertical, z, por lo que se puede
hacer z igual a 0 en la expresión anterior, de modo que:

Modelos de Calidad de Aguas 198
tK
HeeerftF
zz
hz
2
2)(3
=
=
=
σ
σ Ec.( 251)
La función error es una función estadística fuertemente relacionada con el área bajo la curva de distribución
normal de la concentración de una sustancia vertida a un sistema lineal (río) con dispersión. Se define como:
∫ −= z dzerf 0 )2(exp2
)( ξξπ
en donde ξ representa la coordenada espacial en donde se realiza el vertido.
Su cálculo se puede realizar de varias maneras. Una aproximación sencilla es la siguiente:
( )z
Msiendo
zeMMMzerf
4705.01
1
237478.0209588.0348.01)(
+=
−+−−=
Otra forma de calcularla es mediante la expresión:
( ) ( ) ( )∑∞
= −−
−−−=
1 112
1211
2)(
m mm
mzmzerfπ
5.3.4 .- Análisis de la Instrucción española.
A continuación se realiza un breve análisis sobre algunos aspectos de la Instrucción,
fundamentalmente de algunas de las expresiones utilizadas.
5.3.4.1 .- Coeficientes de dispersión turbulenta.
A partir de las expresiones propuestas por Okubo y Osmidov (1970 para coeficientes
de dispersión turbulenta en el mar (ec. 20, pág 27) sería más adecuado emplear la siguiente
expresión para Ky:
3/45102831.9)2( BmHE −=

Vertidos al Mar _____________________________ 199
Esto daría lugar a mayores coeficientes de dispersión. La Instrucción emplea valores
menores, luego proporciona mayor margen de seguridad.
Un problema más grave aparece en el término
yσπB
2
de la ecuación para el cálculo de C(t), puesto que su valor debe ser siempre menor o igual a uno (es
un factor de dilución). Se puede comprobar que en muchos casos puede alcanzar valores superiores
a 1, lo cual carece de sentido. Por ejemplo, si la velocidad de la corriente Ua es de 0.2 m/s y la
distancia a la costa es de 2000 m, un valor de B superior a 15 m. ya da valores mayores que 1. Y no
es difícil que B sea superior a 15 m.
Se recomienda el uso de la ecuación propuesta por Brooks (1960) para evaluar la dilución
en el centro de la trayectoria debido a la dispersión horizontal:
2/1
322 1)/81(2/3
−+=
BtKerfF
y
5.3.4.2 .- Orientación de las boquillas difusoras.
La Instrucción no indica en ningún momento si las boquillas difusoras tienen una
orientación vertical u horizontal. Las ecuaciones propuestas en los apartados B.2.2.1., B.2.3.1. y
B.2.3.2. se encuentran en el libro Mixing in Inland and Coastal Waters (Fischer et al., 1979, cap.
10) y no queda claro si se trata de chorro vertical u horizontal.
5.4 .- Diseño hidráulico de Emisarios Submarinos.
Un aspecto muy importante que se debe cumplir en el tramo difusor de un emisario
submarino es que a través de todas las boquillas salga el mismo caudal. De esa manera nos

Modelos de Calidad de Aguas 200
aseguraremos de que la dilución inicial se cumple para todas las boquillas. En el punto anterior se
ha calculado la dilución inicial y los objetivos de calidad suponiendo un diámetro de la primera
boquilla difusora d. En este apartado se van a calcular los diámetros teóricos primero, y reales,
después, de todas las boquillas difusoras. También se determinarán los diámetros de la conducción
principal, sometidos a la condición de que la velocidad en su interior ha de ser superior a 0.8 m/s.
Para los cálculos hidráulicos se va a suponer que las boquillas difusoras tienen forma que se
puede observar en la Figura 20.
d
d
d
2 d
d
D
Figura 20. Esquema típico de la sección del tramo difusor y una boquilla-
4 1
1
2
2
z4z
6
z1
Vertidos al Mar _____________________________ 201
Figura 21. Sección longitudinal del tramo difusor. Las boquillas están orientadas de forma alternada.
El proceso consiste en aplicar balances de energía entre los puntos situados en el interior de
la conducción principal y las boquillas de salida. En cada caso hay que observar las pérdidas de
carga que se producen en cada etapa. El esquema de trabajo es el siguiente:
3 5
z3
1) Balance de energía entre los puntos 1 y 2. Mediante la aplicación de la ecuación de
Bernouilli entre los puntos 1 y 2 se determina el valor de la energía en el punto 1, justo
debajo de la primera boquilla difusora.
2) Comprobación de la velocidad en la conducción principal tras el punto 1. Tras haber sacado
el caudal Qb a través de la 1ª boquilla, hay que comprobar que la velocidad en la
conducción principal es superior a 0.8 m/s. Si no lo es hay que reducir el diámetro de la
conducción principal.
3) Balance de energía entre los puntos 2 y 3. Se determinan las pérdidas de carga entre esos
dos puntos con el fin de obtener la energía en el punto 3.
4) Balance de energía entre 3 y 4. Igual al realizado en el apartado 1, pero en este caso la
incógnita es el diámetro de la boquilla difusora.
5) Se repiten los pasos 2-3-4 hasta finalizar con todas las boquillas.
Una vez finalizada esta etapa se habrán obtenido los valores de los diferentes diámetros
necesarios en cada boquilla. Estos diámetros son diámetros teóricos que hay que transformar en
diámetros comerciales, por lo que hay que asignar a cada boquilla el diámetro comercial más
próximo al teórico calculado. Cuando se hace esto va a cambiar la energía con la que ha de
llegar el agua a través del emisario hasta el punto 1. Por ello, hay que rehacer los cálculos
mediante los siguientes pasos:
1) Se supone un valor a la energía en el punto 1. Este valor será próximo al calculado en
el proceso anterior.
2) Se plantea el balances de energía entre los puntos 1 y 2. Como el diámetro está fijado
y la energía se supone, se calcula el caudal evacuado.

Modelos de Calidad de Aguas 202
3) Se calcula el caudal en cada boquilla panteando los balances correspondientes. El
caudal en la última boquilla debe ser similar al de las boquillas anteriores. Si es muy
distinto hay que volver al principio suponiendo otro valor para la energía en 1.
El proceso de cálculo esquematizado anteriormente se refleja a continuación.
1) Balance de Energía entre el punto 1 y 2: Cálculo de 1γ1p
Se conoce el diámetro de la boquilla = 100 mm
3/mfkg10262γ
3/mfkg10001γ
=
=
pérdidas2g
22v
1γ2p
2z2g
21v
1γ1p
1z +++=++
+−=+=+
1
212γ
γγ2z
1γ2z
2z1γ2p
2z
pérdidas = bifurcación + codo + rozamiento + estrechamiento bifurcación = ver tablas correspondientes codo = 0.29 rozamiento = ec. de colebrook estrechamiento = ver tablas correspondientes
2) Balance de Energía entre el punto 1 y 3: Cálculo de 1γ3p

Vertidos al Mar _____________________________ 203
Sólo existen pérdidas por la bifurcación y en la conducción principal 3) Balance de Energía entre el punto 3 y 4: Cálculo del diámetro de la boquilla. Igual que el punto apartado 1), pero la incógnita es “d”, diámetro de la boquilla de salida.
Una regla que se ha demostrado muy conveniente, a la hora de diseñar un emisario
submarino, es que la suma del área de las boquillas difusoras aguas abajo de un punto, debe der
menor que el área de la sección principal en dicho punto. La experiencia indica que la mejor
relación tuberíaárea
boquillastotalárea está entre 1/3 y 2/3. El diámetro de las boquillas debe ser lo
suficientemente pequeño como para distribuir el caudal adecuadamente, pero no tanto como para
aumentar la pérdida de carga.

Modelos de Calidad de Aguas 204
6. BIBLIOGRAFÍA.
Ambrose, R.B. et al. (1988). WASP4, A Hydrodynamic and Water Quality Model. Model Theory,
User's Manual and Programmer's Guide. EPA/600/3-87/039.
Barnwell, T. O. et al. (1985). Rates, Constants, and Kinetics Formulations in Surface Water
Quality Modeling (2nd. edition). EPA/600/3-85/040.
Barnwell, T. O., Ambrose, R.B. et al. (1987), Processes, Coefficients, and Models for Simulating
Toxic Organics and Heavy Metals in Surface Waters. EPA/600/3-87/015.
Benet, J.M., Ferrer, J. (1993). Ingenieria Sanitaria y Ambiental. Primera Parte. DIHMA-UPV.
SPUPV-93.168.
Biswas, A. K. Ed. (1981). Models for Water Quality Management. McGraw-Hill Series in Water
Resources and Environmental Engineering. McGraw-Hill Book Company. New York.
Chapra, S.C. (1997). Surface Water Quality Modelling. Mc-Graw Hill. New York.
Clark, Mark M. (1996). Transport Modeling for Environmental Engineers and Scientist. John
Wiley & Sons, Inc. New York, USA.
Falconer, R.A. (Ed.) (1992). Water Quality Modelling.Ashgate Publishing Ltd.
Fischer, H.B. et al. (1979). Mixing in Inland and Coastal Waters. Academic Press, Inc.
Gardiner, R.D., M.T. Aver y R.P. Canale (1984). Sediment Oxygen Demand in Green Bay, Lake
Michigan. In: Pirbazari and DeVinney (eds.), Environmental Engineering. Proceedings of the 1984
Specialty Conference. ASCE. NY, pp. 514-519.

Bibliografía _____________________________ 205
Hopkins, T. (ed.) (1983). Quantitative Analysis and Simulation of Mediterranean Coastal
Ecosystem: the Gulf of Naples, a case study. Unesco Reports in Marine Science.
Melli, P., Zannetti P. (Ed.) (1992). Environmental Modelling. Computational Mechanics
Publications. Southampton, UK.
Metcalf-Eddy, Inc. (1995). Ingeniería Sanitaria: Tratamiento, evacuación y reutilización de aguas
residuales. Ed. Labor.
Mills, W.B., D.B. Porcella, M.J. Ungs, S.A. Gherini, K.V. Summers, Lingfung Mok, G.L. Rupp,
G.L. Bowie y D. A. Haith. (1985). Water Quality Assessment: A Screening Procedure for Toxic
and Conventional Pollutants, Parts 1 and 2. U.S. Environmental Protection Agency, Athens, GA.
EPA-600/6-85-002 a y b.
Nemerow, N.L. (1974). Scientific Stream Pollution Analysis. McGraw-Hill Series in Water
Resources and Environmental Engineering. McGraw-Hill Book Company. New York.
Stumm W., Morgan J.J. (1981). Aquatic Chemistry. John Wiley&Sons, Inc.

ÍNDICE
1. INTRODUCCIÓN. ...................................................................................................... 1
1.1 Contaminación de las aguas. ................................................................................... 1 1.1.1 Principales problemas en aguas subterráneas.................................................... 1 1.1.2 Principales problemas en aguas superficiales. ................................................... 4
1.2 Gestión de la calidad de las aguas........................................................................... 7
2. BALANCES DE MATERIA EN SISTEMAS NATURALES. .............................. 11
2.1 Enunciado general.................................................................................................. 11
2.2 Transporte convectivo y dispersivo en sistemas naturales. ................................ 15 2.2.1 Aguas subterráneas. .......................................................................................... 15
2.2.1.1 Definición y tipos de acuíferos............................................................................................... 16 2.2.1.2 Flujo del agua en un medio poroso. ....................................................................................... 18 2.2.1.3 Dispersión en acuíferos. ......................................................................................................... 19 2.2.1.4 Transporte de contaminantes en medios porosos. .................................................................. 20
2.2.2 Aguas superficiales............................................................................................ 21 2.2.2.1 Ríos. ....................................................................................................................................... 21 2.2.2.2 Lagos. ..................................................................................................................................... 25 2.2.2.3 Embalses................................................................................................................................. 27 2.2.2.4 Estuarios. ................................................................................................................................ 28 2.2.2.5 Sistemas naturales tridimensionales. ...................................................................................... 31
2.3 Contaminantes no conservativos........................................................................... 31 2.3.1 Aguas subterráneas. .......................................................................................... 32
2.3.1.1 Modelación de las interacciones físico-químicas. .................................................................. 33 2.3.1.1.1 Equilibrio químico........................................................................................................... 34 2.3.1.1.2 Procesos controlados por la cinética................................................................................ 47
2.3.2 Aguas superficiales............................................................................................ 54 2.3.2.1 Alcalinidad y pH. ................................................................................................................... 54 2.3.2.2 Oxígeno disuelto y materia orgánica. ..................................................................................... 56
2.3.2.2.1 Materia orgánica carbonosa............................................................................................. 60 2.3.2.2.2 Nitrificación..................................................................................................................... 63 2.3.2.2.3 Fracciones disueltas y particuladas.................................................................................. 66 2.3.2.2.4 Demanda de oxígeno desde los sedimentos..................................................................... 67 2.3.2.2.5 Fotosíntesis y respiración. ............................................................................................... 69 2.3.2.2.6 Reaireación superficial. ................................................................................................... 70 2.3.2.2.7 Resumen .......................................................................................................................... 72
2.3.2.3 Interacciones fitoplancton-nutrientes. .................................................................................... 73 2.3.2.3.1 Fitoplancton..................................................................................................................... 75 2.3.2.3.2 Nutrientes. ....................................................................................................................... 96
2.3.2.4 Sustancias tóxicas................................................................................................................. 109 2.3.2.4.1 Compuestos químicos orgánicos. .................................................................................. 109 2.3.2.4.2 Compuestos químicos inorgánicos: metales pesados. ................................................... 123
2.3.2.5 Contaminación radiactiva..................................................................................................... 133 2.3.2.6 Interacciones con los sedimentos. ........................................................................................ 134
3. BALANCES DE ENERGÍA. .................................................................................. 137
3.1 Introducción.......................................................................................................... 137

3.2 Ecuación general del balance de energía............................................................ 138 3.2.1 Radiación neta solar de onda corta Qsn. ......................................................... 140 3.2.2 Radiación atmosférica neta Qan. ..................................................................... 141 3.2.3 Radiación emitida por el agua Qbr. ................................................................. 142 3.2.4 Flujo de calor por Evaporación, Qe. ............................................................... 142 3.2.5 Flujo de calor convectivo Qc. .......................................................................... 143 3.2.6 Linealización de la transferencia de calor. ..................................................... 143 3.2.7 Calor intercambiado con el sedimento............................................................ 144
4. MODELACIÓN MATEMÁTICA DE LA CALIDAD DE LAS AGUAS. ......... 145
4.1 Introducción histórica.......................................................................................... 146
4.2 Modelos empíricos................................................................................................ 148
4.3 Soluciones aproximadas....................................................................................... 150 4.3.1 Lago completamente mezclado........................................................................ 150 4.3.2 Ríos unidimensionales. .................................................................................... 153
4.3.2.1 Oxígeno Disuelto y Materia Orgánica Carbonosa................................................................ 154 4.3.2.2 Oxígeno disuelto y nitrificación. .......................................................................................... 157 4.3.2.3 Fuentes y sumideros de origen difuso. ................................................................................. 158
4.3.3 Sistemas unidimensionales con dispersión longitudinal. ................................ 159 4.3.4 Sistemas de lagos conectados.......................................................................... 163 4.3.5 Aguas subterráneas: transporte en zona no saturada y zona saturada. ......... 165
4.3.5.1 Zona no saturada. ................................................................................................................. 165 4.3.5.2 Zona saturada. ...................................................................................................................... 166
4.4 Métodos numéricos. ............................................................................................. 167 4.4.1 Método de Diferencias Finitas. ....................................................................... 167
4.5 Factores para la elección del modelo. ................................................................. 171
4.6 Calibración-verificación de los modelos............................................................. 174
5. VERTIDOS AL MAR MEDIANTE EMISARIOS SUBMARINOS. UN CASO PRÁCTICO........................................................................................................................ 177
5.1 - Introducción. ...................................................................................................... 177
5.2 - Aspectos ambientales del vertido por medio de emisarios submarinos........ 180 5.2.1 .- Dilución Inicial. ........................................................................................... 180
5.2.1.1 .- Velocidad de salida/orientación de la salida. .................................................................... 181 5.2.1.2 .- Densidad del agua residual/densidad del agua del mar..................................................... 182 5.2.1.3 .- Corrientes marinas. ........................................................................................................... 182 5.2.1.4 .- Estratificación marina. ...................................................................................................... 183 5.2.1.5 .- Diámetro de los orificios de salida.................................................................................... 185 5.2.1.6 .- Separación entre las boquillas de salida............................................................................ 185
5.2.2 .- Dilución Secundaria..................................................................................... 185 5.2.2.1 .- Transporte convectivo....................................................................................................... 186 5.2.2.2 .- Transporte dispersivo........................................................................................................ 186
5.2.3 .- Autodepuración. ........................................................................................... 187 5.2.3.1 .- Modelos matemáticos de Bacterias Fecales. ..................................................................... 188
5.3 Diseño de emisarios submarinos. ........................................................................ 193 5.3.1 .- Cálculo del tramo difusor. ........................................................................... 194

5.3.2 .- Cálculo de la Dilución Inicial...................................................................... 196 5.3.3 .- Cálculo de los objetivos de calidad. ............................................................ 196 5.3.4 .- Análisis de la Instrucción española. ............................................................ 198
5.3.4.1 .- Coeficientes de dispersión turbulenta. .............................................................................. 198 5.3.4.2 .- Orientación de las boquillas difusoras. ............................................................................. 199
5.4 .- Diseño hidráulico de Emisarios Submarinos.................................................. 199
6. BIBLIOGRAFÍA. .................................................................................................... 204