memoria del trabajo de investigación (programa de doctorado de estudios avanzados en ......
TRANSCRIPT

Memoria del trabajo de investigación (Programa de Doctorado de Estudios Avanzados en Química) presentado
por Jorge Rencoret Pazo.
Instituto de Recursos Naturales y Agrobiología de Sevilla (IRNAS-CSIC)


ABREVIATURAS
a Etapa ácida suave (en secuencia de blanqueo)
Ah Alcoholes
Alc Alcanos
ABTS Ácido 2,2’-azino-bis(3-etilbenzotiazolin-6-sulfónico)
AOX Compuestos organoclorados
AQ Antraquinona
BSTFA N,O-bis-(trimetilsilil)-trifluoroacetamida
δC Desplazamiento químico del carbono
δH Desplazamiento químico del protón
DCM Diclorometano
DFRC Derivatización seguida de rotura reductora (derivatization followed by
reductive cleavage)
DHP Polímero deshidrogenativo
DTPA Ácido dietilentriaminopentaacético
2D-NMR Espectroscopia de resonancia magnética nuclear de dos dimensiones
ECF Secuencia de blanqueo libre de cloro elemental (elemental chlorine free)
EPA Agencia de protección medioambiental de EE.UU.
FID Detector de ionización de llama (flame ionization detector)
G Unidad guayacilpropano (o guayacilo)
GC Cromatografía de gases (gas chromatography)
GC/MS Cromatografía de gases/espectrometría de masas (gas
chromatography/mass spectrometry)
H Unidad 4-hidroxifenilpropano (o 4-hidroxifenilo)
HBT 1-Hidroxibenzotriazol
HMBC Espectroscopía 2D de correlaccción de múltiples enlaces
(heteronuclear multiple bond correlation)
HSQC Espectroscopía 2D de correlación heteronuclear de cuanto simple
(heteronuclear single-quantum correlation)
ICP-OES Espectrometría de emisión óptica con plasma acoplado inductivamente
ITD Detector de trampa de iones (ion trap detector)

MW Masa molecular (molecular weight)
L Etapa lacasa-mediador (en secuencia de blanqueo)
L/Q Etapa de quelato incluyendo lacasa-mediador (en secuencia de
blanqueo)
LM Lámina media
MTBE terc-butil metil eter
MWL Lignina de madera molida (milled wood lignin)
NMR Espectroscopía de resonancia magnética nuclear (nuclear
magnetic resonance)
O Etapa de deslignificación con oxígeno (en secuencia de blanqueo)
PoP Doble etapa de blanqueo con peróxido de hidrógeno, la primera
bajo oxígeno presurizado
Py-GC/MS Pirólisis acoplada a cromatografía de gases/espectrometría de masas
(pyrolysis-gas chromatography/mass spectrometry)
Q Etapa de quelato (en secuencia de blanqueo)
rpm Revoluciones por minuto
S Unidad siringilpropano (o siringilo)
SLM Sistema lacasa-mediador
SPE Extracción en fase sólida (solid phase extraction)
Secuencia de blanqueo totalmente libre de cloro (totally chlorine free)
TMP Tipo de pasta de papel obtenida mediante procesos termo-mecánicos
(thermomechanical pulp)
TMSD Trimetilsilildiazometano
TMSi Trimetilsililo
U Unidad de actividad enzimática

ÍNDICE 1. INTRODUCCIÓN……………………………………………………………1
1.1 Estructura y composición de los materiales lignocelulósicos…………….3
1.1.1 Estructura de la lignocelulosa………………………………………3
1.1.2 Composición de la lignocelulosa………………………...................3
1.1.2.1 Celulosa……………………………………………………….5
1.1.2.2 Hemicelulosa.…………………………………………………5
1.1.2.3 Lignina………………………………………………………..6
1.1.2.4 Extractos……………………………………………………...8
1.1.2.5 Compuestos minerales……………………………………….11
1.2 Fabricación de pasta de papel……………………………………………11
1.2.1 Procesos de producción de pasta de papel…………………………11
1.2.1.1 Procesos físicos………………………………………………12
1.2.1.2 Procesos químicos……………………………………………12
1.2.2 Blanqueo de la pasta de papel……………………………………...12
1.2.3 Calidad de las pastas de papel……………………………...............13
2. RESUMEN DE LA LABOR EXPERIMENTAL REALIZADA………….15
2.1 Caracterización química detallada de la madera de cinco especies
de eucalipto: Eucalyptus globulus, E. nitens, E. maidenii,
E. grandis y E. dunnii……………………………………………………...17
2.1.1 Extraíbles lipofílicos.………………………………………………..17
2.1.2 Composición y estructura de la lignina…………………..................17
2.2 Aplicaciones biotecnológicas como posibles alternativas a la proble-
mática del pitch……………………………………………………………18
2.2.1 Utilización del sistema lacasa-mediador para la eliminación de
los lípidos residuales presentes en pastas de distinta naturaleza……18
2.2.2 Inclusión de una etapa enzimática en una secuencia de blanqueo
TCF de pasta de eucalipto…………………………………………..18
3. MATERIALES Y MÉTODOS……………………………………………….21
3.1 Materiales…………………………………………………………………..23
3.1.1 Maderas de las diferentes especies de eucalipto…………………….23
3.1.2 Pastas de eucalipto, picea y lino tratadas con el SLM………………23

3.1.3 Lacasa y HBT………………………………………………………23
3.2 Métodos……………………………………………………………………23
3.2.1 Tratamientos de la pasta con el SLM…………………...………….23
3.2.2 Tratamiento de la pasta de E. globulus con el SLM en una
secuencia de blanqueo TCF………………………………………..24
3.2.3 Extracción de los compuestos extraíbles lipofílicos………………..24
3.2.4 Fraccionamiento de los compuestos extraíbles lipofílicos
mediante extracción en fase sólida (SPE)…………………………...24
3.2.5 Saponificación de ésteres de esteroles……………………………...25
3.2.6 Análisis mediante GC y GC/MS de los extractos lipofílicos
totales y sus fracciones……………………………………………...25
3.2.6.1 GC……………………………………………………………...26
3.2.6.2 GC/MS........................................................................................26
3.2.7 Métodos de derivatización………………………………………….26
3.2.7.1 Silanización…………………………………………………….27
3.2.7.2 Metilación……………………………………………………...27
3.2.8 Determinación de la fracción hidrosoluble………………................27
3.2.9 Determinación de la lignina Klason………………………………..27
3.2.10 Determinación de la composición de polisacáridos……………….27
3.2.11 Análisis de los metales y otros elementos…………………………28
3.2.12 Determinación del contenido en cenizas…………………………..28
3.2.13 Aislamiento de la lignina de la madera. Método Björkman……….28
3.2.14 Determinación de la composición química de la lignina…………..29
3.2.14.1 Pirólisis-cromatografía de gases/espectrometría
de masas ……………………………………………………29
3.2.14.2 Tioacidolisis de la lignina…………………………………….30
3.2.14.3 DFRC…………………………………………………………31
3.2.15 Análisis de la estructura de la lignina mediante resonancia
magnética nuclear bidimensional (2D NMR)………………………32
4. RESULTADOS Y DISCUSIÓN……………………………………………..33
4.1 Composición general de las maderas de las especies de eucalipto
estudiadas…………………………………………………………………..35
4.1.1 Contenido en lignina klason………………………………………...35
4.1.2 Contenido en cenizas y composición de metales…………….……..36

4.1.3 Composición química de los polisacáridos de la madera………….36
4.1.4 Composición lipídica………………………………………………37
4.1.5 Estudio de la composición y estructura de la lignina de las
maderas de las diferentes especies de eucalipto……………………46
4.1.5.1 Análisis de composición de la lignina mediante Py-GC/MS….46
4.1.5.2 Análisis de composición de la lignina mediante DFRC……….50
4.1.5.3 Análisis de composición de la lignina mediante tioacidolisis…51
4.1.5.4 Estudio de la estructura de la lignina de las diferentes
especies de eucalipto…………..………………………………51
4.1.6 Conclusiones………………………………………………………...56
4.2 Utilización del SLM para la eliminación de los lípidos residuales
presentes en pastas de distinta naturaleza…………………………………57
4.2.1 Tratamiento de la pasta kraft de eucalipto con el SLM……………57
4.2.2 Tratamiento de la pasta TMP de picea con el SLM………………..60
4.2.3 Tratamiento de la pasta soda-AQ de lino con el SLM……………..62
4.3 Inclusión de una etapa enzimática en una secuencia TCF………………...65
5. REFERENCIAS……………………………………………………………...71


1. INTRODUCCIÓN
1

2 2

1. Introducción
3
1.1 ESTRUCTURA Y COMPOSICIÓN DE LOS MATERIALES LIGNOCELULÓSICOS
La madera y otros materiales lignocelulósicos representan la mayor fuente de energía y materia orgánica renovables de la biosfera. La madera es el más abundante de los materiales lignocelulósicos y representa la principal materia prima en la industria de pasta de papel. En Europa, en el año 2000, el 99% de la producción de fibra virgen se llevó a cabo a partir de madera (47 millones de toneladas); el resto, se produjo a partir de residuos agrícolas como el bagazo de caña, paja de trigo, o de plantas anuales como bambú, lino, abacá, sisal, etc; si bien estas últimas materias primas se emplean, sobre todo, en países en vías de desarrollo o con escasos recursos forestales. La estructura y composición de la madera (Fengel y Wegener, 1984; Hon y Shiraiski, 1991) condiciona su utilización industrial y la posible aplicación de métodos biotecnológicos. Los materiales lignocelulósicos se pueden clasificar en dos categorías: gimnospermas, árboles de especies coníferas y, angiospermas, que incluyen monocotiledóneas y dicotiledóneas o árboles de especies frondosas (Praham et al.,1984). 1.1.1 Estructura de la lignocelulosa
La pared celular de las plantas vasculares es una estructura compleja y semirrígida constituida por un entramado de microfibrillas cristalinas de celulosa, recubiertas por una matriz de hemicelulosa y lignina (Figura 1). La pared celular vegetal está constituida por una pared primaria y una pared secundaria. Las paredes primarias de dos células adyacentes están unidas entre sí por lignina, denominándose a esta capa lámina media (LM). La pared secundaria está constituida por tres capas que difieren en la orientación de las fibrillas de celulosa. En las capas exterior e interior (S1 y S3) las fibrillas están dispuestas formando un amplio ángulo con el eje de la célula, mientras que la disposición de las fibrillas en la capa intermedia gruesa (S2) es prácticamente paralela al eje (Figura 2). La formación de la pared celular tiene lugar a través de los siguientes procesos: a) Depósito de microfibrillas de celulosa, constituyéndose el marco de la pared celular. b) Adición de las cadenas principales de hemicelulosa alrededor de las microfibrillas y reforzamiento de la pared. c) Adición de las ramificaciones de las hemicelulosas. d) Acumulación de la lignina incrustándose en todos los espacios del entramado.
1.1.2 Composición de la lignocelulosa
La celulosa, junto con la hemicelulosa y la lignina, son los tres polímeros mayoritarios de la lignocelulosa, variando el porcentaje de cada uno de ellos con el tipo de planta, la edad o la parte del tejido vegetal de donde provengan (Fengel y Wegener, 1984). Además de estos tres, están presentes otros compuestos como proteínas, lípidos y minerales. Los constituyentes químicos de la lignocelulosa, por lo tanto, se pueden clasificar en: - Componentes mayoritarios: que incluyen celulosa y hemicelulosas, polisacáridos que representan entre el 60 y el 82% de la madera, según especies, y la lignina, que supone entre el 17 y el 30%. - Componentes minoritarios: que incluyen una amplia variedad de compuestos, que por su naturaleza se clasifican en extractos (representan entre un 1 y un 10% de la madera) y en compuestos minerales (representan hasta un 1%).

1. Introducción
4
Puentes de hidrógeno Celulosa Hemicelulosa Lignina Enlances entre hemicelulosas
Figura 1. Representación esquemática de las relaciones entre los principales constituyentes de la pared secundaria (celulosa, lignina y hemicelulosa) de una angiosperma herbácea. Se muestran las microfibrillas de celulosa inmersas en una matriz de lignina en la que también se incluye la hemicelulosa. Las uniones entre estos polímeros se completan mediante puentes intermoleculares formados por los ácidos p-hidroxicinámicos (Bidlack et al., 1992).
Tangencial
S 3
S 2
S 1
P
L M
Dire
cció
n de
las f
ibra
s
Sección tangencial
F ib ra s d ec e lu lo sa
H e m ic e lu lo saM a triz d e lig n in a -h e m ic e lu lo sa
S ecc ió n R ad ia l
Figura 2. Esquema de la localización de la lignina, celulosa y hemicelulosa en la pared de las células vegetales (P: pared primaria, S1-S3: capas de la pared secundaria y LM: lámina media) adaptado de Kirk y Cullen (1998).

1. Introducción
5
1.1.2.1 Celulosa
La celulosa es el constituyente principal de las células vegetales ya que representa entre un 43 y un 47% en maderas de coníferas y ente un 42 y un 44% en maderas de frondosas (Aitken et al., 1988). La celulosa es un polímero lineal formado por unidades de β-D-glucopiranosa unidas entre sí por enlaces glucosídicos β(1 4) (Figura 3). Dos unidades de glucosa adyacentes se unen eliminando una molécula de agua entre los grupos hidroxilo del C1 y el C4 de la otra unidad para formar la molécula de celobiosa, un disacárido que representa la unidad constitutiva de la celulosa. El grado de polimerización de la celulosa en la madera está comprendido entre 7000 y 10000.
1.1.2.2 Hemicelulosa
Las hemicelulosas, también llamadas poliosas, son polisacáridos ramificados formados por una cadena lineal de monosacáridos unidos principalmente por enlaces β (1→ 4) (en algunos casos β (1→ 3)), a la cual se unen cadenas laterales cortas a menudo de azúcares distintos a los de la cadena principal. Los residuos de monosacáridos (Figura 4) incluyen pentosas (D-xilosa y L-arabinosa), hexosas (D-glucosa, D-galactosa, L-galactosa, D-manosa, L-ramnosa y L-fucosa) y ácidos urónicos (ácido D-glucurónico y ácido D-galacturónico), que pueden encontrarse acetilados o en forma de ésteres metílicos. La fracción hemicelulósica representa entre un 25 y un 30% en especies coníferas y entre un 20 y un 43% en frondosas (Aitken et al., 1988). Las cadenas de hemicelulosas presentan grados de polimerización medios de 150 y actúan junto con la lignina como matriz soporte para las microfibrillas de celulosa en la pared celular. Las hemicelulosas son de menor peso molecular, más fácilmente degradables y más fáciles de disolver que la celulosa. La estructura de la celulosa es la misma en los diferentes tipos de madera y otros materiales lignocelulósicos, sin embargo, en las hemicelulosas existe una considerable variación entre las diferentes maderas. Mientras que en las frondosas (maderas duras) se encuentran principalmente hemicelulosas con unidades de cinco carbonos (pentosanos) como el xilano, en las coníferas (maderas blandas) abundan más las hemicelulosas con unidades de seis carbonos (hexosanos) como los glucomananos que se forman por polimerización de unidades de glucosa y manosa, con una relación de 3 unidades de manosa por una unidad de glucosa. Las plantas herbáceas presentan al igual que las maderas de frondosas un mayor porcentaje de pentosanos.
Figura 3. Estructura de la celulosa.
O
HOOH
O
OH
O
HOOH
O
OH
O
HOOH
O
OH
O
HOOH
O
OH
O

1. Introducción
6
1.1.2.3 Lignina
La lignina es el segundo compuesto orgánico más abundante en la superficie de la tierra sobrepasada sólo por la celulosa. Representa entre un 25 y un 33% en especies coníferas y entre un 18 y un 34% en frondosas (Aitken et al., 1988), siendo una de las moléculas orgánicas más recalcitrantes. La lignina da soporte estructural a los tejidos de las plantas e impermeabilidad a los elementos vasculares, y proporciona resistencia frente al ataque de microorganismos y el estrés mecánico. Se trata de un polímero aromático tridimensional sintetizado a partir de la fenilalanina a través de la ruta de los ácidos cinámicos (Higuchi, 1997; Boerjan et al., 2003). De esta forma se sintetizan los alcoholes p-cumarílico (4-hidroxicinamílico), coniferílico (4-hidroxi-3-metoxicinamílico) y sinapílico (4-hidroxi-3,5-dimetoxicinamílico), que actúan como precursores de la lignina (Figura 5). Su polimerización deshidrogenativa da lugar respectivamente a las unidades 4-hidroxifenilpropano (H), guayacilpropano (G) y siringilpropano (S). Recientemente se ha descrito, utilizando pirólisis analítica, la existencia en plantas herbáceas de unidades acetiladas derivadas de los correspondientes alcoholes p-hidroxicinamílicos acetilados (del Río et al., 2004). En la polimerización de los alcoholes p-hidroxicinamílicos participan peroxidasas y/o fenoloxidasas vegetales que forman radicales libres de tipo fenoxilo, estabilizados por resonancia. Estos radicales polimerizan dando lugar a una variedad de dímeros y posteriormente al polímero de lignina. El tipo de unidades presentes en la lignina, así como los tipos de unión entre las mismas puede variar entre los distintos grupos de plantas (coníferas, frondosas y herbáceas). Incluso en una misma planta la composición de la lignina puede variar en los distintos tejidos y capas de la pared celular. La lignina de coníferas se forma mayoritariamente a partir del alcohol coniferílico (unidad G), mientras que la lignina de frondosas se sintetiza a partir de los alcoholes coniferílico y sinapílico (unidades G y S, respectivamente). La lignina de las plantas herbáceas está compuesta de unidades H procedentes del alcohol p-cumarílico, además de las unidades G y S en distintas proporciones. En la composición de la pared celular de las plantas herbáceas participan los ácidos p-hidroxicinámicos (principalmente p-cumárico y ferúlico). Estos ácidos contribuyen a la unión de la lignina con la hemicelulosa mediante enlaces tanto éster como éter por reacción de sus grupos carboxilo e hidroxilo fenólico (Sun et al., 2002). En estas plantas el ácido ferúlico es el principal responsable de los puentes entre
O
H
HO
H
HO
H
H
OHHOH
OH
O
H
HO
H
HO
OH
H
HHOH
OH
O
H
HO
H
HO
H
H
OHH
H
OH
O
OH
H
H
HO
H
H
OHH
H
OH
A
B
C
D
O
H
HO
H
HO
H
H
OHHOH
OH
O
H
HO
H
HO
OH
H
HHOH
OH
O
H
HO
H
HO
H
H
OHH
H
OH
O
OH
H
H
HO
H
H
OHH
H
OH
A
B
C
D Figura 4. Estructura de algunos de los monosacáridos componentes de las hemicelulosas: D-glucosa (A), D-manosa (B), D-xilosa (C) y L-arabinosa (D).

1. Introducción
7
la lignina y los polisacáridos. De un 40 a un 70% del ácido ferúlico está eterificado con la lignina formando un puente de unión con los polisacáridos a los que se une por enlaces éster. Sin embargo, un 70% del ácido p-cumárico está solamente esterificado con la posición γ de la cadena lateral de las unidades de la lignina. Las uniones entre monómeros dan lugar a una estructura tridimensional (Figura 6).
Los primeros modelos estructurales de lignina de coníferas y frondosas se propusieron en los años 70 (Nimz, 1974; Adler, 1977). En 1951 Freudemberg describió por primera vez un polímero deshidrogenativo (DHP) formado a partir del alcohol coniferílico, cuyas propiedades eran similares a la lignina de las coníferas (Freudenberg and Neish,
165 4 3
2
OH
OCH3
OH
H3CO
αβ
γ
165 4 3
2
OH
OCH3
OH
αβ
γ
165 4 3
2
OH
OH
αβ
γ
Figura 5. Estructura de los alcoholes p-hidroxicinamílicos precursores de la lignina.
Figura 6. Primer modelo estructural de la lignina de angiospermas propuesto por Nimz en 1974.
SS
SS
S
S
S
S
SS
S
H
G
G G
GG
GG
GGG
G
GG
G
Enlaces entre unidades β/α-O-4: 65% β-1: 15% β−β: 8% β-5: 6% α−β: 3% 4-O-5: 2% 5-5’: 2%

1. Introducción
8
1968). No existen modelos detallados de la lignina de las plantas herbáceas excepto algunas aproximaciones (Sun et al., 1997). Aunque la variedad de uniones es amplia, se pueden diferenciar dos tipos: uniones de tipo éter y uniones carbono-carbono. - Uniones éter: se diferencian principalmente dos tipos, uno de ellos basado en la unión de la cadena de propano de un monómero de la lignina a un átomo de carbono del anillo bencénico de otra unidad fenilpropano (Figura 7) la unión más corriente de este tipo es la β-O-4’. La unión α-O-4’ es menos frecuente pero más fácil de romper. El otro tipo lo constituyen las uniones entre dos átomos de carbono de dos anillos bencénicos diferentes, como por ejemplo la unión 4-O-5’.
- Uniones de tipo carbono carbono (Figura 8): más difíciles de romper que las uniones éter, incluyen las uniones de dos cadenas alifáticas (del tipo β-β’), las debidas a la unión de un carbono de un anillo bencénico con el de una cadena alifática de otra unidad (uniones β-5’ o β-1’) y las uniones entre carbonos de anillos bencénicos (unión 5-5’). La lignina también se encuentra unida covalentemente a las hemicelulosas y a la celulosa, formando el llamado complejo lignina-carbohidrato. Existen al menos 3 tipos de estas uniones: uniones éter, éster y fenil glucosídico. 1.1.2.4 Extractos
El término extractos cubre una amplia variedad de compuestos de bajo peso molecular que pueden ser separados de la madera mediante extracciones con agua (extraíbles hidrofílicos) o con disolventes orgánicos (extraíbles lipofílicos) (Hillis, 1962; Fengel y Weneger, 1984; Rowe, 1989; Sjöström, 1993). Los extractos se encuentran en la madera en pequeña cantidad, pero su presencia puede interferir en la deslignificación, pricipalmente los de naturaleza lipofílica. La cantidad y la composición de los extractos varían según la especie considerada. Entre ellos se encuentran los taninos, ácidos grasos y resínicos, alcoholes grasos, esteroles (libres y esterificados), glicéridos y ceras entre otros. Una de las principales funciones de los compuestos extraíbles es la protección de la planta contra patógenos. La baja degradabilidad de muchos de estos compuestos
O
OCH3H3CO
O
HO
HO
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
165
4 32
3'
1'6'5' 4'
2'
α
βγ OCH3
OHO
O OCH3
H3CO
A B
O
OCH3
O
OCH3
OCH3
61 2
45
36'1' 2'
4'5'
3'
CO
OCH3H3CO
O
HO
HO
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
165
4 32
3'
1'6'5' 4'
2'
α
βγ OCH3
OHO
O OCH3
H3CO
A B
O
OCH3
O
OCH3
OCH3
61 2
45
36'1' 2'
4'5'
3'
C
Figura 7. Uniones eter presentes en la molécula de lignina: enlace β-O-4’ (A), enlace α-O-4’ (B) y el enlace 4-O-5’ (C).

1. Introducción
9
contribuye a este fin, pero también origina problemas en ciertos aprovechamientos industriales de la biomasa vegetal que se describen a continuación (Hillis y Sumimoto, 1989).
Los extraíbles polares engloban diferentes compuestos fenólicos libres de bajo peso molecular, lignanos (dilignoles y compuestos relacionados), taninos y flavonoides, entre otros. Los compuestos fenólicos libres incluyen precursores de la lignina (ácidos p-hidroxicinámicos y aldehídos p-hidroxicinamílicos), ácidos bencenocarboxílicos relacionados (como ácido p-hidroxibenzoico, vainíllico y siríngico), aldehídos y cetonas aromáticos (p-hidroxibezaldehído, vanillina, siringaldehído y propioguaiacona). Los taninos son un grupo de compuestos caracterizados más por su acción curtiente sobre las proteínas de las pieles que por su estructura química común. Los taninos hidrolizables son esteres del ácido gálico y sus dímeros (incluyendo el ácido elágico típico del eucalipto) con monosacáridos. Los flavonoides presentan diferentes estructuras derivadas del anillo de flavona (2-fenilbenzopirona). Los taninos no hidrolizables están constituidos por varias unidades de flavonoides condensadas. A pesar de que muchas especies vegetales contienen cantidades muy significativas de extraíbles polares, la fracción lipídica es predominante en algunas de ellas. Además, cuando estas especies vegetales son utilizadas para la fabricación de pasta de papel, estos compuestos lipofílicos deben ser tenidos muy en cuenta ya que pueden deteriorar significativamente la calidad de la pasta, debido a la formación de depósitos de brea, conocidos como depósitos de pitch (Figura 9).
5'
3'2'1' 6'
4'O
OCH3
OCH3O
O654
321
O
OCH3
H3CO
α'
β'
γ'α
β
γ
165
4 32
3'
1'6'5' 4'
2'
α
βγ OCH3
OHO
O OCH3
H3CO
O
OCH3H3CO
HO
HO
165 4 3
2
αβ
γ
4'
2'1'6' 5'
3'
O
OCH3
OCH3
O
OCH3
Oα
β
γ OAr
OHHO
O
H3CO
1'6'5' 4' 3'
2'
α' β'γ'
OCH3
H3CO
1654 3
2
OCH3
HO
OCH3
OH
16543
2 1' 2'3'4'
5'6'
βα
γ
β'α'
γ'OHHO
A B C
D E
5'
3'2'1' 6'
4'O
OCH3
OCH3O
O654
321
O
OCH3
H3CO
α'
β'
γ'α
β
γ
165
4 32
3'
1'6'5' 4'
2'
α
βγ OCH3
OHO
O OCH3
H3CO
O
OCH3H3CO
HO
HO
165 4 3
2
αβ
γ
4'
2'1'6' 5'
3'
O
OCH3
OCH3
O
OCH3
Oα
β
γ OAr
OHHO
O
H3CO
1'6'5' 4' 3'
2'
α' β'γ'
OCH3
H3CO
1654 3
2
OCH3
HO
OCH3
OH
16543
2 1' 2'3'4'
5'6'
βα
γ
β'α'
γ'OHHO
A B C
D E
Figura 8. Uniones tipo C-C presentes en la molécula de lignina: enlace β-β’ (A), enlace β-1’ (B), enlace β-5’/α-O-4’ (C), enlace β-1’/α-O-α’ (D) y enlace 5-5’(E).

1. Introducción
10
Entre estos compuestos se encuentran los ácidos grasos libres, alcoholes grasos libres, hidrocarburos, ceras, grasas, terpenos y esteroides (Figura 10). Las ceras se definen como ésteres de ácidos grasos con alcoholes de cadena larga, mientras que las grasas son ésteres de ácidos grasos con glicerol (mono-, di- y triglicéridos). Los triglicéridos se encuentran entre los principales constituyentes de la fracción lipofílica de los extraíbles de muchas maderas (hasta un 50%). Los principales ácidos grasos esterificados en grasas y ceras son el ácido oleico, linoleico y linolénico, juntos con porcentajes muy inferiores de los ácidos palmítico y esteárico, entre otros. Se han identificado y aislado de la madera, principalmente de coníferas, más de 4000 terpenos diferentes. Estos compuestos se subdividen en varias clases dependiendo del número de unidades de isopreno que los constituyen: monoterpenos (2 unidades), sesquiterpenos (3 unidades), diterpenos (4 unidades), sesterpenos (5 unidades) y triterpenos (6 unidades). Entre los compuestos de tipo diterpenos se encuentran los ácidos resínicos que son muy abundantes en la madera de coníferas (y están ausentes en las frondosas). Los ácidos resínicos se caracterizan por su elevada toxicidad, comparado con los otros constituyentes de la fracción lipofílica de los extraíbles de la madera (Peng y Roberts, 2000). Los principales compuestos tipo triterpenoides son los esteroides que difieren de los triterpenos en su biosíntesis (Sjöström, 1993). Los esteroles son los esteroides más abundantes en la madera pero también existen cetonas e hidrocarburos esteroidales. Los esteroles pueden encontrarse libres o esterificados con ácidos grasos, siendo en ambos casos el sitosterol el principal esterol, tanto en coníferas como en frondosas. También se han descrito formando glicósidos y acilglicósidos en diferentes maderas (Gutiérrez y del Río, 2001). El sitosterol y los esteres de sitosterol se caracterizan por ser compuestos con alto grado de adherencia por lo que a menudo se encuentran en el origen de los depósitos de pitch, que se forman por acumulación de extraíbles lipofílicos durante la fabricación de pasta de papel (Back y Allen, 2000). Una caracterización química adecuada de las materias primas utilizadas para la fabricación de pasta de papel constituirá un requisito fundamental para llegar a diseñar estrategias adecuadas para el control del pitch.
Figura 9. Fotografía de un depósito de pitch en una pasta.

1. Introducción
11
1.1.2.5 Compuestos minerales
La cantidad de materia inorgánica es pequeña (generalmente menos de un 1% del total de la madera), pero indispensable para el crecimiento del árbol. 1.2 FABRICACIÓN DE PASTA DE PAPEL 1.2.1 Procesos de producción de pasta de papel
La fabricación de pasta de papel consiste en la separación de las fibras de celulosa, que se encuentran cementadas por la lámina media compuesta fundamentalmente de lignina utilizando métodos físicos o químicos (Fengel y Wegener, 1984; Sjöström, 1993). El proceso de fabricación de pasta de papel es conocido como pasteado.
Figura 10. Estructuras de algunos extraíbles lipofílicos presentes en materiales lignocelulósicos: hexacosanol (A), ácido palmítico (B), ácido dehidroabiético (C), escualeno (D), β-sitosterol (E), estigmasta-3,5-dien-7-ona (F), α-tocoferol (G), trilinoleina (H) y sitosteril linoleato (I).
OH A
OH
O
B
COOH
C
D
HO
E
O
F
O
HO
G
CO-O-CH2
CO-O-CH
CO-O-CH2
H
O
O I
OH AOH A
OH
O
BOH
O
OH
O
OH
O
OH
O
B
COOH
CCOOHCOOH
C
DD
HO
E
O
FOOO
F
O
HO
GO
HO
O
HO
G
CO-O-CH2
CO-O-CH
CO-O-CH2
HCO-O-CH2
CO-O-CH
CO-O-CH2
CO-O-CH2
CO-O-CHCO-O-CH
CO-O-CH2CO-O-CH2
H
O
O IO
O
O
O
O
O I

1. Introducción
12
1.2.1.1 Procesos físicos
El pasteado físico separa las fibras por fragmentación mecánica utilizando molinos y refinos de discos, lo que supone un considerable gasto energético. Es un proceso que ofrece un gran rendimiento (hasta un 99% del material inicial), obteniéndose pastas que resultan ventajosas para algunos tipos de papel, ya que confieren rigidez, volumen y opacidad. No obstante, el alto contenido de lignina en la pasta va en detrimento de la calidad del papel ya que las fibras son poco flexibles, no están bien unidas entre sí y el papel es poco resistente y tiende a amarillear con el sol (por oxidación de la lignina residual y otros componentes de la pasta). 1.2.1.2 Procesos químicos
El pasteado químico disuelve y extrae una buena parte de la lignina utilizando reactivos químicos a altas temperaturas. Puede realizarse en condiciones alcalinas, utilizando por ejemplo sulfuro sódico e hidróxido sódico (pasteado kraft), o únicamente hidróxido sódico (pasteado a la sosa de fibras no madereras) y en condiciones ácidas, utilizando por ejemplo bisulfito sódico que disuelve la lignina en forma de lignosulfonatos (pasteado al sulfito). En la deslignificación en condiciones alcalinas a menudo se utiliza antraquinona como catalizador. También se emplean solventes orgánicos para el pasteado organosolv de diferentes maderas incluyendo el eucalipto (Gilarranz et al., 1999). El pasteado kraft es el más extendido a nivel mundial (más del 50% del total de pasta) al permitir obtener pastas con una gran resistencia, aunque con menor rendimiento (alrededor del 50% del material inicial), a partir de diferentes tipos de maderas incluyendo el eucalipto (García Hortal y Colom, 1992; Santos et al., 1997). Este proceso libera una lignina degradada, denominada lignina kraft, en la que se ha producido la ruptura de los enlaces éter, entre otras reacciones. La lignina kraft se utiliza como fuente de energía en la fábrica al mismo tiempo que se recupera los reactivos químicos. La lignina residual que queda en la pasta kraft ha sufrido reacciones de oxidación y es responsable del color oscuro de estas pastas. Por ello, para la obtención de papeles de escritura y otros es necesario eliminar la lignina residual mediante un proceso de blanqueo.
1.2.2 Blanqueo de la pasta de papel
Una vez obtenida la pasta, ésta debe ser sometida a un proceso de blanqueo. Durante las cocciones químicas se elimina la mayor parte de la lignina presente en la materia prima utilizada para la fabricación de la pasta de papel, pero la lignina residual que queda en la pasta presenta un color oscuro como consecuencia de las reacciones de oxidación durante la cocción. El blanqueo tiene como objetivo eliminar estos restos de lignina que son responsables del color de las pastas. Mediante esta operación se modifica el color de las pastas y se obtiene un incremento de su blancura consiguiéndose así una materia fibrosa casi completamente blanca, o por lo menos mucho más blanca que el producto inicial, susceptible de ser empleada en la producción de papeles blancos o poco coloreados. Los reactivos comerciales más utilizados para el blanqueo son o han sido el cloro gaseoso, el hipoclorito, el peróxido de hidrógeno y el dióxido de cloro. El proceso de blanqueo ha sido una de las secciones de la industria de la pasta y papel que ha sufrido más cambios durante los últimos años. La decisión de eliminar el cloro y, en algunos casos, el dióxido de cloro, en las secuencias de blanqueo se debe a la necesidad de reducir las emisiones de compuestos organoclorados (AOX) en los efluentes. El

1. Introducción
13
desarrollo de leyes más restrictivas con respecto a los procesos contaminantes (Brooks et al., 1994) y en especial contra los compuestos clorados, catalogados como contaminantes prioritarios por la Agencia de Protección Ambiental de EE.UU. (EPA), por su persistencia en el suelo y el agua, ha llevado a una parte de la industria europea de pasta y papel (especialmente a la de los países nórdicos) a introducir secuencias de blanqueo de la pasta de papel totalmente libre de cloro (secuencias TCF, “Totally Chlorine Free”) basadas principalmente en el blanqueo con peróxido de hidrógeno, oxígeno y ozono (Figura 11). Otra parte de la industria papelera mundial, incluyendo buena parte de la industria norteamericana de este sector, ha eliminado el cloro gaseoso pero continua utilizando dióxido de cloro como agente de blanqueo (secuencia ECF, “Elementary Chlorine Free”). La novedad de la secuencia de blanqueo TCF obliga a solucionar nuevos problemas ya que la eficacia en el blanqueo de estas secuencias es inferior a la de las secuencias que incluían cloro.
1.2.3 Calidad de las pastas
La fabricación de pastas de papel mediante tecnologías más respetuosas con el medio ambiente ha traído nuevos problemas en el blanqueo de la pasta, que no existían cuando se utilizaban reactivos más agresivos (aunque también más contaminantes) y/o en sistemas con un menor grado de cierre de los circuitos. La acción deslignificante del oxígeno se conoce desde hace tiempo pero el desarrollo del blanqueo con oxígeno ha sido bastante lento debido a la degradación de la celulosa y demás polisacáridos de la madera. Como consecuencia, el blanqueo con oxígeno viene acompañado a menudo por perdidas de rendimiento y viscosidad de la pasta. Las ventajas del peróxido de hidrógeno se apoyan en su facilidad de manipulación y aplicación, su versatilidad y la naturaleza relativamente inocua de los productos de reacción. Una parte de los problemas que han surgido con la introducción de estos reactivos está relacionada con la blancura de las pastas, porque de momento ni el oxígeno ni la combinación de oxígeno y peróxido pueden igualar la eficacia de la cloración para la eliminación de los productos derivados de la lignina, responsables del color de las pastas. La modificación de los procesos de cocción, con objeto de obtener pastas con menor contenido en lignina, representa otra alternativa para facilitar la blanqueabilidad de las pastas.
A la máquina de secado
Blowtank
Digestor O Ofiltro filtro filtro filtro filtroStock StockQ Po P
O2 O2H2O2NaOH O2
DTPAH2SO4
98ºCpH 11
98ºCpH 11
105ºCpH 11
98ºCpH 11
Na2SNaOH
Stock
Astillas demadera
Pasta sin blanquear
Pasta blanqueada
A la máquina de secado
Blowtank
Digestor O Ofiltro filtro filtro filtro filtroStock StockQ Po P
O2 O2H2O2NaOH O2
DTPAH2SO4
98ºCpH 11
98ºCpH 11
105ºCpH 11
98ºCpH 11
Na2SNaOH
StockBlowtank
Digestor O Ofiltro filtro filtro filtro filtroStock StockQ Po P
O2O2 O2O2H2O2NaOHH2O2NaOH O2O2
DTPAH2SO4
98ºCpH 11
98ºCpH 11
105ºCpH 11
98ºCpH 11
Na2SNaOH
Stock
Astillas demadera
Pasta sin blanquear
Pasta blanqueada
Figura 11. Diagrama del proceso Kraft seguido de una secuencia de blanqueo TCF.

1. Introducción
14
Otra parte de los problemas está relacionada con los extraíbles de los compuestos lignocelulósicos descritos anteriormente. Entre los problemas causados por este tipo de compuestos ocupa un lugar importante la formación de depósitos de pitch en las máquinas de papel (que obligan a realizar paradas técnicas) y en la misma pasta (Allen, 1980; Hillis y Sumimoto, 1989). Estos últimos reducen drásticamente la calidad del producto final. Generalmente el término pitch se aplica tanto a extraíbles lipofílicos de la madera como a los depósitos que estos lípidos causan durante el proceso de fabricación de la pasta y el papel. Muchos de estos compuestos estaban presentes en concentraciones más bajas cuando se utilizaban volúmenes de agua mayores en el lavado de la pasta y menor grado de cierre de los circuitos. Por otro lado, aunque no se conoce adecuadamente la posible incidencia medioambiental de estos compuestos en los vertidos (Swan, 1989), ya se ha indicado anteriormente que algunos de ellos pueden ser fuertemente tóxicos. La problemática del pitch es muy compleja porque varía con la materia prima así como con el proceso empleado para la fabricación de pasta y papel. La formación de depósitos de pitch durante la fabricación de pastas de papel a partir de madera de coníferas ha sido estudiado durante años, dado el uso mayoritario de este tipo de madera en los principales países productores de pasta (USA, Canadá y países nórdicos de la UE), sin embargo la formación de estos depósitos en procesos que utilizan maderas de frondosas se encuentra menos estudiada.

2. RESUMEN DE LA LABOR EXPERIMENTAL REALIZADA
15

16 16

2. Resumen de la labor experimental realizada
2.1 CARACTERIZACIÓN QUÍMICA DETALLADA DE LA MADERA DE CINCO ESPECIES DE EUCALIPTO: Eucalyptus globulus, E. nitens, E. maidenii, E. grandis y E. dunnii.
La madera de eucalipto es utilizada como materia prima para la fabricación de pasta de papel en el suroeste de Europa, Brasil, Sudáfrica, Japón y otros países. La caracterización química de estas especies se ha centrado fundamentalmente en los compuestos que presentan mayor influencia en el proceso de fabricación de la pasta de papel: la lignina y los extraíbles lipofílicos. Entre las diferentes especies de eucalipto, objetos de nuestro estudio, la madera de E. globulus es considerada la mejor materia prima para la fabricación de pasta kraft, obteniendose de ella el mayor rendimiento de pasta (del Río et al., 2005). La composición lipídica de la madera de E. globulus (Gutiérrez et al., 1998a, 1999, 2001a, b; Gutiérrez y del Río 2001; Freire et al., 2002, 2006), así como la composición de su lignina (Ona et al., 1997; Rodrigues et al., 1999; Evtuguin et al., 2001; del Río et al., 2001a,b, 2002, 2005) es ampliamente conocida, sin embargo, además del E. globulus, las maderas de E. nitens, E. maidenii, E. grandis y E. dunnii también son utilizadas para la fabricación de pasta de papel, aunque su composición química es poco conocida, existiendo una información muy limitada sobre ellas (Freire et al., 2006; Ona et al., 1997; González-Vila et al., 1999; Yokoi et al., 1999, 2001; Capanema et al., 2005). El objetivo final de este estudio, además de realizar una caracterización química detallada de cada una de las especies de eucalipto, es intentar encontrar una explicación de por qué la madera de E. globulus presenta un rendimiento de obtención de pasta de papel superior al resto de las otras especies de eucalipto. 2.1.1 Extraíbles lipofílicos
Los extraíbles lipofílicos son liberados de las fibras durante el pasteo dando lugar a la formación de depósitos pitch, tanto en los distintos puntos del proceso (maquinaria y circuitos) como en el producto final provocando una reducción de la calidad del producto y grandes pérdidas económicas (Hillis y Sumimoto 1989 ; Back y Allen 2000; Gutiérrez et al., 2001c). En la producción de las pastas alcalinas, una gran parte de los lípidos de las fibras utilizadas se eliminan durante la cocción, no obstante algunas especies químicas sobreviven a este proceso (Swan, 1967; Affleck y Ryan, 1969; Leone y Breuil, 1998) y son detectadas como extraíbles de la pasta. Los depósitos pitch representan uno de los mayores problemas en la obtención de pasta de papel a partir de la madera de E. globulus, siendo los esteroles libres y conjugados (esterificados y formando glicósidos) los principales compuestos presentes en los depósitos de pitch (del Río et al., 1998, 2000; Gutiérrez et al., 1998a, 1999; Silvestre et al., 1999; Gutiérrez y del Río 2001). Los extraíbles lipofílicos se aislaron de las maderas y de las pastas, para ser analizados mediante técnicas analíticas que se detallan en el siguiente apartado. 2.1.2 Composición y estructura de la lignina
La composición de la lignina esta directamente relacionada con el rendimiento de la deslignificación (Chang y Sarkanen, 1973; Tsutsumi et al., 1995; González-Vila et al., 1999; del Río et al., 2005). En general, la eficiencia de la cocción está directamente relacionada con la cantidad de unidades siringilo (S) y guayacilo (G) presentes en la molécula de lignina, que se suele expresar mediante la relación S/G. Las unidades guayacilo, a diferencia de las unidades siringilo, presentan en el C5 un posible punto de unión que posibilita la formación de enlaces carbono-carbono, que hace que la
17

2. Resumen de la labor experimental realizada
estructura de la lignina con mayor número de unidades G sea más condensada (presenta mayor número de enlaces C-C), siendo más difícil de degradar. La composición de la lignina de las maderas de las diferentes especies de eucalipto se determinará mediante técnicas y métodos analíticos que se detallan más adelante. Por otro lado, con respecto al estudio de la estructura de la lignina, se ha producido un gran avance debido a la utilización de la resonancia magnética nuclear bidimensional (Nimz et al., 1981; Ralph et al., 1999; Capanema et al., 2001, 2004, 2005; Ralph et al,. 1999, 2004; Liitiä et al., 2003; Balakshin et al,. 2003), ya que hasta el momento solo se habían utilizado técnicas de resonancia magnética nuclear monodimensionles (1H y 13C, las cuales daban lugar a señales solapadas), además de técnicas degradativas (tioacidolis, pirólisis…) que sólo aportan información sobre la composición (contenido de unidades H, G y S). El conocimiento de la estructura de la lignina es fundamental tanto para entender como este polímero se degrada durante las etapas de cocción y blanqueo, así como para el establecimiento de nuevas aplicaciones biotecnológicas dirigidas a eliminar la lignina mediante el uso de hongos y enzimas.
2.2 APLICACIONES BIOTECNOLÓGICAS COMO POSIBLES ALTERNA- TIVAS A LA PROBLAMÁTICA DEL PITCH
2.2.1 Utilización del sistema lacasa-mediador para la eliminación de los lípidos residuales presentes en pastas de distinta naturaleza (Gutiérrez et al., 2006a) Una alternativa para disminuir los depósitos de pitch es el uso de enzimas (Fischer y Messner, 1992; Fischer et al., 1993; Fujita et al., 1992) y microorganismos (Behrendt y Blanchette, 1997; Farrell et al., 1993; Gao et al., 1994; Gutiérrez et al., 1999b, 2001b). Sin embargo, las preparaciones comerciales disponibles no son del todo efectivas, ya que están basadas en enzimas (lipasas) y microorganismos que hidrolizan preferentemente triglicéridos. Además de las lipasas se han propuesto otras enzimas como las estearasas (Calero-Rueda et al., 2002; Kontkanen et al., 2004), no solucionándose el problema del pitch, puesto que los esteroles libres son tan recalcitrantes como los esteres de esteroles. Las lacasas, a diferencia de las lipasas y las estearasas, son enzimas oxidativas que actúan principalmente sobre compuestos fenólicos. Una característica muy importante de estas enzimas es que pueden actuar junto a medidores (sistema lacasa-mediador, SLM) aumentando considerablemente su capacidad en la degradación de lignina y otros compuestos (Bourbonnais y Paice, 1990). Se han realizado muchos estudios sobre la utilización del SLM en la deslignificación y blanqueo de las pasta de papel (Bajpai, 1999; Call y Mücke, 1997), sin embargo, nunca antes se había utilizado el SLM en el tratamiento de los extraíbles lipofílicos. Con la finalidad de evaluar la capacidad de degradación del SLM sobre los extraíbles lipofílicos, se trataron tres pastas de papel (no blanqueadas) representativas de diferentes materias primas lignocelulósicas y de diferentes procesos de pasteo (ver Tabla 1) con el sistema lacasa-HBT (1-Hidroxibenzotriazol). 2.2.2 Inclusión de una etapa enzimática en una secuencia de blanqueo TCF de pasta de eucalipto (Gutiérrez et al., 2006b) El uso de secuencias de blanqueo de la pasta totalmente libres de cloro (procesos TCF) en lugar de procesos libres de cloro elemental (procesos ECF), junto con el cierre de los
18

2. Resumen de la labor experimental realizada
circuitos para disminuir los consumos de agua, está incrementando la severidad de los problemas de pitch debido a la baja reactividad con los lípidos de la pasta. Se propuso llevar a cabo experimentos basados en una secuencia de blanqueo TCF incluyendo una etapa enzimática. La pasta kraft de eucalipto se trató con la lacasa termoestable de elevado potencial redox procedente del hongo P. Cinnabarinus y HBT. Este tratamiento lacasa-mediador se incluyó en una secuencia TCF a escala de laboratorio, realizada en reactores presurizados (secuencia O-O-L-Q-PoP). Esta secuencia reproduce la secuencia industrial TCF usada para el blanqueo de la pasta kraft de E. globulus, con el adicional tratamiento enzimático (etapa L). La secuencia empezó con deslignificaciones con oxígeno (dos etapas) y se completó con dos fases de blanqueo con peróxido (etapa PoP, con un primer paso bajo oxígeno a presión) después de un tratamiento de quelación (etapa Q) para eliminar los metales que destruyen el peróxido. Las pastas y los líquidos (licores) de los tratamientos se tomaron en diferentes etapas de la secuencia que contenía la enzima así como para la secuencia control (industrial). Los extraíbles lipofílicos se aislaron de las pastas y de los líquidos, y se analizaron. Tabla 1. Diferentes tipos de materias primas lignocelulósicas y sus respectivos procesos de pasteo.
Especie Tipo de materia prima Proceso de pasteo
Eucalipto (Eucalyptus globulus) Frondosa Kraft
Picea (Picea abies) Conífera Termomecánico (TMP)
Lino (Linum usitatissimun) No maderera Soda antraquinona (soda-AQ)
19

2. Resumen de la labor experimental realizada 2. Resumen de la labor experimental realizada
20 20

3. MATERIALES Y MÉTODOS
21

22 22

3. Materiales y métodos
3.1 MATERIALES
3.1.1 Maderas de las diferentes especies de eucalipto
Las maderas de eucalipto utilizadas para el estudio de su composición química fueron suministradas por la empresa papelera ENCE (Pontevedra, España). En concreto, las especies analizadas: E. globulus, E. nitens, E. maidenii, E. grandis y E. dunnii, corresponden a árboles adultos (10-12 años). Las especies E. globulus y E. nitens crecieron en el noroeste de España, mientras que las restantes lo hicieron en Uruguay. Las astillas de las maderas fueron molidas tras su recepción y se almacenaron a –20 ºC para preservar al máximo sus fracciones mas alterables. Los resultados del pulpeo de estas cinco especies se encuentran en la Tabla 2. Tabla 2. Evaluación de las diferentes especies de eucalipto realizada en ENCE.
3.1.2 Pastas de eucalipto, picea y lino tratadas con el SLM
- La pasta kraft de eucalipto (Eucalyptus globulus) fue sumistrada por la fábrica de ENCE en Pontevedra (España).
- La pasta termomecánica de picea (Picea abies) fue suministrada por la fábrica de UPM en Valkeakoski (Finlandia).
- La pasta de lino (Linum usitatissimun) sin blanquear y obtenida mediante cocción soda-AQ fue suministrada por la fábrica de CELESA en Tortosa (España).
3.1.3 Lacasa y HBT
La lacasa usada para tratar las pastas fue suministrada por Beldem (Andenne, Bélgica) y se obtuvo de una cepa hiperproductiva del hongo Pycnoporus cinnabarinus proporcionada por el INRA (Marsella, Francia). Una unidad de actividad se definió como la cantidad de enzima que transforma 1 µmol del ácido 2,2’-azino-bis(3-etilbenzotiazolin-6-sulfónico) (ABTS) por min al correspondiente radical catiónico (ε436 29300 M-1 cm-1) en tampón acetato sódico 0,1 M (pH 5). El mediador HBT fue suministrado por Sigma-Aldrich.
3.2 MÉTODOS
3.2.1 Tratamientos de la pasta con el SLM
Los tratamientos de la pasta se llevaron a cabo en reactores de acero inoxidable, de una capacidad de 4 litros, usando 200 g de pasta (peso seco) al 10 % de consistencia bajo una presión de oxígeno de 6 Kg/cm2, excepto para la pasta TMP de picea, la cual se trató bajo condiciones atmosféricas y con burbujeo de oxígeno. Los tratamientos se realizaron utilizando lacasa (20 U/g pasta) y HBT (1,5%, referido al peso de pasta seco)
Especie Densidad Alcalis
activo,% Indice Rto Rechazos Viscosidad
mL/g Alcalis
residual,g/LKappa Bruto,% %
E. globulus 600 13.0 16.1 59.5 8.4 1413 3.6 E. nitens 450 17.5 16.3 50.4 1.7 1177 6.2 E. maidenii 600 18.0 16.5 50.8 1.4 1093 11.3 E. grandis 435 17.0 15.7 49.7 0.2 1148 9.2 E. dunnii 595 20.0 16.1 48.7 1.6 931 15.5
23

3. Materiales y métodos
a pH 4 (para el ajuste del pH se utlizó tampón tartrato), durante 2 h a 50 ºC. Los controles para la evaluación de la acción de la enzima se trataron bajo las mismas condiciones pero sin lacasa ni mediador. Otros controles adicionales como tratamientos solo con enzima (sin mediador) y otros solo con mediador (sin enzima) también se realizaron.
3.2.2 Tratamiento de la pasta de E. globulus con el SLM en una secuencia de blanqueo TCF Las condiciones del tratamiento con el SLM son idénticas a las del apartado anterior pero en este caso el tratamiento se ha introducido como una etapa dentro de una secuencia de blanqueo TCF (O-O-L-Q-PoP), la cual incluye: - Dos etapas alcalinas de oxígeno (O) utilizando una presión de oxígeno de 6 Kg/cm2,
1,5% de NaOH y 0.5% de MgSO4 durante 60 min a 98 ºC. - Una etapa (L) de tratamiento con el SLM descrito en el apartado anterior. - Una etapa de quelato (Q) utilizando ácido dietilentriamino-N,N,N’,N’’,N’’-
pentaacético (DTPA) a una concentración del 0,3% durante 60 min a 85 ºC (pH 5-6). - Una etapa alcalina de peróxido (PoP) utilizando 3% de H2O2, 2% de NaOH, 0.1%
MgSO4 y 0.5% de Na2Si2O3 durante 140 min a 105 ºC bajo 6 Kg/cm2 de O2, seguido de 180 min a 98 ºC sin presión de oxígeno (Ibarra et al., 2006). Todos los porcentajes anteriores referidos al peso de pasta seco.
Las pastas se separaron de los líquidos del tratamiento por filtración y mediante exhaustivos lavados con agua destilada después de cada etapa. Para identificar mejor los efectos producidos por el tratamiento con el SLM, se realizó una secuencia que incluía una etapa control bajo las mismas condiciones que las utilizadas con el SLM pero sin añadir ni lacasa ni mediador (etapa a, debido a que el medio es ligeramente ácido). 3.2.3 Extracción de los compuestos extraíbles lipofílicos
Para el análisis de los compuestos extraíbles lipofílicos es necesario su previo aislamiento. Por ello, los compuestos extraíbles lipofílicos de las maderas de eucalipto (molida), así como los de las pastas utilizadas en los tratamientos con el SLM, se extrajeron con acetona en un extractor de tipo Soxhlet durante ocho horas. Este extractor está constituido por un condensador, un tubo de extracción y un matraz, donde la extracción se lleva a cabo por contacto continuo de un solvente con la muestra sólida (que se encuentra en el interior de un cartucho de celulosa propio para la extracción). El disolvente se evaporó a sequedad en un rotavapor. La cantidad de extracto se determinó por gravimetría. Los análisis de los líquidos, obtenidos de las diferentes etapas de la secuencia de blanqueo TCF aplicada a la pasta de E. globulus, se realizaron mediante una extracción líquido-líquido utilizando terc-butil metil éter (MTBE) como disolvente orgánico. Las extracciones se realizaron en un embudo de decantación a pH=14. Una vez extraídos los extraíbles lipofílicos, los cuales pasan de la fase acuosa (líquidos) a la fase orgánica (MTBE), se secaron bajo una corriente nitrógeno. Los extractos lipofílicos obtenidos de las pastas y los líquidos se redisolvieron en cloroformo para su posterior análisis por cromatografía de gases (GC) y cromatografía de gases/espectrometría de masas (GC/MS). 3.2.4 Fraccionamiento de los compuestos extraíbles lipofílicos mediante extracción en fase sólida (SPE) Para llevar a cabo una caracterización más detallada de algunos compuestos presentes en los extractos, los extraíbles lipofílicos obtenidos de las maderas de eucalipto se
24

3. Materiales y métodos
separaron en distintas fracciones por elución en una columna en orden creciente de polaridad (Gutiérrez et al., 1998b). La columna consiste en un cartucho con fase de aminopropilo (500 mg) Millipore, Milford, MA, USA. Se utilizó una cantidad de extracto lipofilico total entre 5-20 mg que se redisolvió en una cantidad mínima (< 0,5 mL) de hexano-cloroformo (4:1). La columna se acondicionó previamente con hexano (4 mL). La primera fracción se eluyó con 8 mL de hexano (fracción A, contiene los compuestos más apolares como esteres de esteroles, ceras e hidrocarburos), la segunda con 6 mL de una solución de hexano-cloroformo (5:1) (fracción B, rica en triglicéridos y cetonas esteroidales), la tercera con 10 mL de cloroformo (fracción C, contiene principalmente esteroles libres y alcoholes) y la cuarta fracción con 10 mL de una solución de éter etílico-ácido acético (98:2) (fracción D, contiene ácidos grasos). Cada fracción aislada se secó con nitrógeno, se pesó y se procedió a su análisis por GC y GC/MS. 3.2.5 Saponificación de ésteres de esteroles
La identificación completa de la estructura de los ésteres de esteroles requiere una rotura previa del enlace éster, ya que los espectros de masas de estos compuestos se caracterizan por ser similares a los del esteroles libres que lo componen, no apareciendo generalmente ningún fragmento correspondiente al ión molecular (Lusby et al., 1984; Evershed et al., 1989). Para el análisis de los esteres de esteroles, la fracción A, separada por SPE, se hidrolizó a reflujo con una disolución de KOH 0,5 M al 90% en etanol durante 8 h (Gutiérrez et al., 1999a). Una vez finalizada la saponificación, se realizó una extracción líquido-líquido con hexano (3 x 20 ml) extrayéndose la fracción neutra (contiene los esteroles libres), la cual se secó en el rotavapor. Por otro lado, la fase acuosa se acidificó con HCl 6 M y la fracción ácida (contiene los ácidos grasos libres) se extrajo igualmente con hexano (3 x 20ml) y se secó en el rotavapor. Ambas fracciones se analizaron por GC y GC/MS. 3.2.6 Análisis mediante GC y GC/MS de los extractos lipofílicos totales y sus fracciones La cromatografía de gases se utiliza para separar compuestos que se encuentran en mezclas complejas y como método de identificación y determinación cuantitativa de cada uno de esos componentes. En este tipo de cromatografía la fase móvil es un gas inerte y la fase estacionaria es líquida depositada en un soporte sólido apropiado, donde su principio de operación envuelve la volatilización de la muestra, la separación de cada componente en la columna y la detección de los diferentes componentes por el detector. En el análisis de lípidos por GC y GC/MS, es muy importante las características de las columnas cromatográficas, que permitan separar e identificar compuestos de alto peso molecular como ceras, ésteres de esteroles, triglicéridos, etc. Se han realizado estudios sobre procedimientos para el análisis de los extractos lipofílicos de las maderas (Gutiérrez et al., 1998b, 2004) por GC y GC/MS, en donde se usaron diversas columnas de diferente longitud y diferentes programas de temperatura. En estos estudios, las columnas capilares seleccionadas para el análisis de lípidos por GC fueron cortas (de 5 m) ya que proporcionan una conveniente elución y separación de lípidos de alto peso molecular (Wakeham y Frew, 1982; Lusby et al., 1984; Evershed et al., 1989; Sitholé et al., 1992; Örså y Holmbom, 1994), además de que el análisis ocurre en un corto período de tiempo (20 min). Columnas menores de 5 m no son convenientes ya que no proporcionan la resolución necesaria para análisis cuantitativos. En el caso de los análisis por GC/MS, los cromatogramas obtenidos tienen que ser reproducibles con los obtenidos por GC usando columnas capilares de 5 m para identificar los diferentes
25

3. Materiales y métodos
compuestos. No obstante, en el sistema GC/MS no se pueden usar columnas tan cortas, usándose normalmente columnas de 10-15 m. Esta longitud de columna es apropiada para el análisis de lípidos de alto peso molecular por GC/MS proporcionando resultados en un corto período de tiempo (30 min), incrementando para esto la temperatura final a 380 ºC.
3.2.6.1 GC
Los análisis cromatográficos de los extractos se realizaron en un cromatógrafo de gases HP 6890 (Hewlett-Packard, Hoofddorp, Netherlands) equipado con un detector de ionización de llama (FID, flame ionization detector) y una columna capilar corta de alta temperatura de sílice fundida (DB-5HT J&W; 5 m × 0,25 mm I.D., con 0,1 µm de espesor de película) para análisis por GC. Las temperaturas del inyector y del detector se fijaron a 300 ºC y 350 ºC, respectivamente. Se utilizó Helio como gas portador y la inyección se realizó en modo splitless. El programa de temperaturas del horno fue de 100 ºC (1 min) a 350 ºC (3 min) a 15 ºC/min, manteniendo la temperatura final por 5 minutos. Los picos cromatográficos correspondientes a los extraíbles lipofílicos se cuantificaron a partir de sus áreas en los cromatogramas. Se utilizó una mezcla de compuestos patrones suministrados por Sigma-Aldrich (octadecano, hexadecanol, ácido palmítico, sitosterol, 7-oxocolesterol, sitosteril 3β-D-glucopiranósido, colesterol oleato y triheptadecanoina) en la obtención de la recta de calibrado utilizada para la cuantificación de los extraíbles lipofílicos, con un rango de concentración comprendido entre 0.1 y 1 mg/ml. En todos los casos se obtuvo un coeficiente de correlación mayor de 0.99. 3.2.6.2 GC/MS
La combinación de la cromatografía de gases con la espectrometría de masas resulta en una técnica analítica muy útil para el análisis de mezclas de compuestos orgánicos volátiles o semi-volátiles, siendo por lo tanto útil en la identificación de cada uno de los componentes existentes en los extractos lipofílicos obtenidos de los métodos de extracción anteriormente citados. Para la separación e identificación de los compuestos individuales se usó un cromatógrafo de gases Varian Star 3400 acoplado a un detector de trampa de iones (ITD, Varian Saturn 2000) usando una columna capilar de 15 m × 0,25 mm DB-5HT (0,1 µm, J&W Scientific). Se utilizó Helio como gas portador y la inyección se realizó en modo splitless. Las muestras se inyectaron con un inyector automático (Varian 8200). La temperatura del inyector durante la inyección fue de 120 ºC y 0,1 min después de la inyección se programó a 380 ºC (10 min) a una velocidad de 200 ºC/min. El horno se programó a 120 ºC durante 1 min y a 380 ºC durante 5 min a 10 ºC/min. Las temperaturas del ITD y de la línea de transferencia fueron de 200 ºC y 300 ºC respectivamente. La identidad de cada componente se determinó por comparación de sus espectros de masas con los espectros existentes en las librerías (Willey y NIST) y con espectros publicados anteriormente, por sus fragmentaciones y cuando fue posible por sus patrones.
26

3. Materiales y métodos
3.2.7 Métodos de derivatización
Para el análisis por GC/MS es esencial que los compuestos existentes en la muestra sean suficientemente volátiles a presión atmosférica para pasar a través del cromatógrafo de gases, por lo que es necesario recurrir a métodos de derivatización cuando los compuestos a analizar no son volátiles, esto es, métodos de conversión de ciertos compuestos en otros que sean compatibles con el método analítico de GC/MS.
3.2.7.1 Silanización
Compuestos como alcoholes, esteroles y otros con grupos funcionales polares pueden ser derivatizados para conferirles estabilidad térmica y química, además de la volatilidad necesaria. Para esto, se le pueden introducir grupos trimetilsililo (TMS) formando así éteres de trimetilsililo que presentan características de fragmentación más favorables para la identificación de los compuestos por sus espectros de masas. Varios reactivos pueden ser usados para la preparación de los derivados TMS, pero en este caso se ha utilizado el N,O-bis-(trimetilsilil)-trifluoroacetamida (BSTFA) en presencia de piridina. Una vez secada la muestra, se añadió 0,1 ml de BSTFA y 0,05 de piridina. A continuación se calentó a 70 ºC durante 2 h y se evaporó la mezcla con nitrógeno hasta sequedad. Posteriormente, se redisolvió en cloroformo para ser analizada por GC/MS. 3.2.7.2 Metilación
Los grupos carboxilos de los ácidos grasos libres e hidroxiácidos, además de los grupos fenólicos se metilaron con trimetilsilildiazometano (TMSD) en metanol. Para ello, a la muestra seca se le añadió 0,1 ml de metanol y 0,025 ml de una solución 2 M de trimetilsilildiazometano en hexano y se mantuvo durante 20 minutos en el baño de ultrasonido. 3.2.8 Determinación de la fracción hidrosoluble
El porcentaje de los compuestos solubles en agua para las maderas de las diferentes especies de eucalipto se determinó según la norma Tappi T 207 om-88. Para ello, las maderas libres de compuestos extraíbles lipofílicos, se extrajeron con agua a 100 ºC durante 3 horas. El extracto se evaporó y se secó a 100 ºC para su determinación gravimétrica.
3.2.9 Determinación de la lignina Klason
La lignina Klason se determinó según la norma Tappi T222 om-88 con algunas modificaciones. En este método, las maderas molidas de las distintas especies de eucalipto, libres de compuestos extraíbles e hidrosolubles se sometieron a una hidrólisis con H2SO4. Para ello, aproximadamente 300 mg de cada muestra se colocaron en frascos pirex con 3 mL de H2SO4 al 72% y se mantuvieron en un baño de agua a 30 ºC durante 1 hora. Posteriormente se diluyó con 84 mL de agua para obtener la concentración de ácido de 4% y se colocó en una autoclave durante 1 hora a 120 ºC. La lignina Klason se retuvo por filtración en filtros de vidrio poroso (previamente tarados tras 4 horas a 100 ºC). El residuo insoluble (lignina Klason) se lavó con agua destilada hasta pH neutro y seco para cuantificación gravimétrica.
27

3. Materiales y métodos
3.2.10 Determinación de la composición de los polisacáridos
La composición en monosacáridos de las maderas de eucalipto se determinó según la norma Tappi T 249 om-85. Del hidrolizado proveniente de la obtención de la lignina Klason, se retiraron 3 mL, se colocaron en un tubo de centrífuga y se adicionó a cada uno 1 mL de inositol (1 mg/mL), como patrón interno. Las mezclas se neutralizaron con carbonato de bario, se diluyeron y se centrifugaron durante 5 min a 9000 rpm. El sobrenadante de cada tubo se recogió y se secó. A continuación, se añadió borohidruro sódico, para promover la reducción de los monosacáridos. Después de 12 horas las muestras se acidificaron con HCL y se secaron en un rotavapor a 50 ºC. Al residuo de cada tubo se le adicionaron 2 mL de metanol y se secó, repitiendo esta operación 3 veces, para eliminar el borohidruro sódico. Una vez reducidas las muestras, se acetilaron con 0,250 mL de piridina y 0,250 mL de anhídrido acético durante 1 hora a 100 ºC y evaporando de nuevo hasta obtención de un extracto seco. Después de preparadas las muestras, los acetatos de alditol, se analizaron en un cromatógrafo de gases Perkin Elmer Sigma 3, utilizando una columna (2m × 2mm) con 3% SP-2340 sobre 100/120 Supelcoport como fase estacionaria, nitrógeno como gas portador (30 mL/min), una rampa de temperatura de 10 ºC/ min desde 200 ºC (3 min) hasta 230 ºC (8 min) y un detector FID.
3.2.11 Análisis de los metales y otros elementos
Las muestras de maderas de eucalipto, una vez lavadas y secas, se molieron y se realizó la digestión ácida. Para ello, se añadieron 4 mL de ácido nítrico concentrado a 0,5 mg de muestra, dejándolas 15 min en un horno microondas (Jones &Case, 1990). Posteriormente se filtraron con filtro Whatman del número 2 y se recogieron en un matraz que se enrasó hasta 50 mL. La concentración de metales en la disolución obtenida se determinó por espectrometría de emisión por plasma (ICP-OES) en un espectrómetro Termo Jarrel Ash, modelo IRIS Advantages.
3.2.12 Determinación del contenido en cenizas
Se determinó mediante la norma Tappi 211 om-85. Se depositaron 200 mg de cada una de las especies de eucalipto en crisoles de porcelana previamente tarados y se introdujeron en la mufla a 575 ºC durante 6 h. Para tararlos se limpiaron con HCl y se introdujeron en la mufla a 575 ºC durante 1 h.
3.2.13 Aislamiento de la lignina de la madera. Método Björkman
El método Björkman (Björkman, 1956) consiste en la extracción de la lignina directamente de la madera, libre de extraíbles y de hidrosolubles, con ayuda de un molino de bolas durante 170-200h y utilizando tolueno como refrigerante. Después de este tratamiento, la madera molida se trata con una mezcla de dioxano-agua (9:1, utilizando 250 ml por cada 10 g de muestra), agitándose durante 12 h. Este proceso se repite de forma consecutiva 2 veces, centrifugándose la mezcla (25 min, 4 ºC y 11000 rpm) y recogiéndose el sobrenadante (contiene la lignina) en un matraz para ser posteriormente secado en el rotavapor a 40 ºC. A continuación, el residuo seco se disuelve en una mezcla ácido acético: agua (9:1, añadiéndose 20 ml por cada gramo de residuo seco). Esta disolución es añadida lentamente a un vaso con agua destilada (225 ml por cada gramo de lignina) en constante agitación. La lignina precipita y se recoge
28

3. Materiales y métodos
tras centrifugación. El residuo seco se muele en un mortero de ágata para facilitar su disolución en una mezcla 1,2-dicloroetano-etanol (2:1) y de nuevo se vuelve a centrifugar a baja velocidad (5000 rpm durante 5 min) y nos quedamos con el sobrenadante, que contiene la lignina. Éste se dispersa gota a gota, sin agitar en éter dietílico (225 ml es suficiente para 0.5-1 g de lignina), precipitando la lignina, volviéndose a centrifugar (5000 rpm durante 5 min) y resuspendiendo el residuo en éter de petróleo de bajo punto de ebullición (40-60 ºC) en el que se deja durante 12 horas. Finalmente se centrifuga y se seca mediante corriente de nitrógeno y se coserva a 4 ºC, preservándola de la luz y el aire para evitar su oxidación hasta el momento de su análisis. Con este método se obtiene una lignina poco degradada y con una estructura muy similar a la de la lignina nativa de la madera.
3.2.14 Determinación de la composición química de la lignina
La composición química de la lignina se determinó mediante el uso de técnicas degradativas como la pirólisis acoplada a GC/MS, la tioacidolis y la derivatización seguida de rotura reductora (DFRC). 3.2.14.1 Pirólisis-cromatografía de gases/ espectrometría de masas
La pirólisis es un método degradativo que transforma compuestos complejos no volátiles en una mezcla de fragmentos volátiles por descomposición térmica en ausencia de oxígeno, que se lleva a cabo habitualmente a temperaturas de entre 400ºC y 800ºC y en condiciones de atmósfera inerte. En la pirólisis, se producen roturas de los enlaces por acción del calor, pues cuando la energía aplicada a la molécula es mayor que la energía de enlaces específicos ocurre la disociación de éstos de una forma predecible y reproducible, pudiendo obtener cierta información en relación a la molécula original a través del análisis de los productos de degradación formados. Los fragmentos resultantes de la pirólisis pueden ser separados por cromatografía de gases e identificados por espectrometría de masas, utilizándose para esto el método de la pirólisis acoplado a GC/MS, ahora Py-GC/MS. La Py-GC/MS es un poderoso método para el análisis de materiales lignocelulósicos, especialmente de la lignina pues aunque las cadenas laterales de los monómeros de la lignina sean parcialmente destruidas, los substituyentes de las diferentes unidades H, G, S de la lignina no lo son. La lignina es pirolizada para producir una mezcla de compuestos fenólicos (Figura 12) que resultan de la rotura no sólo de enlaces éter, sino también de ciertos enlaces C-C, reteniendo estos fenoles las características de substitución del polímero de lignina y siendo posible por lo tanto identificar los diferentes componentes de ligninas provenientes de unidades H, G y S.
Levoglusano y otros Levoglusano y otros Levoglusano y otros
Celu los aHemicelu losas
Lignina
Glicoaldehídos
Ácido acético y fórmico
Furfura l y productos resinosos de su reacción confenoles en el me dio ácido d e la pirólis is
Carbón
Fenoles
Celu los a
Hemicelu losas
Lignina
Glicoaldehídos
Ácido acético y fórmico
Furfura l y productos resinosos de su reacción confenoles en el me dio ácido d e la pirólis is
Carbón
Fenoles
Celu los a
Hemicelu losas
Lignina
Glicoaldehídos
Ácido acético y fórmico
Furfura l y productos resinosos de su reacción confenoles en el me dio ácido d e la pirólis is
Carbón
Fenoles
Figura 12. Productos de pirólisis de los materiales lignocelulósicos.
29

3. Materiales y métodos
La Py-GC/MS presenta diversas ventajas frente a otros métodos degradativos, pues es una técnica analítica rápida que proporciona resultados en apenas un paso, siendo necesaria poca cantidad de muestra y una simple preparación de la misma (secar y pesar). También presenta ventajas frente a los métodos clásicos de análisis de la lignina, pues no es necesario aislar la lignina de la muestra, permitiendo el análisis de la lignina in situ. Además, es una técnica reproducible permitiendo resultados válidos tanto cualitativamente como cuantitativamente
Para la realización de las pirólisis de las maderas y de las ligninas de eucalipto se utilizó un pirolizador de micro-horno (Frontier Lab), acoplado a un equipo Agilent GC/MS, con una columna de 30 m × 0,25 mm DB-5 (0,25 µm de espesor de película). En una cápsula metálica, se depositaron 0,5-1 mg de muestra finamente dividida y se introdujo en la cámara del microhorno donde se pirolizó a 600 ºC. El cromatógrafo se programó desde 40 ºC (1 min) hasta 300 ºC, con una rampa de 6 ºC/min, manteniéndose 20 min la temperatura final. El inyector y el detector se mantuvieron a 300 ºC. Los compuestos se identificaron por comparación con la literatura (Faix et al., 1990; Ralph y Hatfield, 1991) y con los incluidos en las bibliotecas electrónicas Wiley y NIST. 3.2.14.2 Tioacidolisis de la lignina El principio general de la tioacidolisis consiste en una solvolisis en una mezcla de dioxano y etanotiol en presencia de eterato de trifloruro de boro en medio anhidro (Lapierre et al., 1985; Rolando et al., 1992; Grabber et al., 1996). Las condiciones de la solvolisis son suaves y combinan el efecto de un ácido de Lewis fuerte, el eterato de trifloruro de boro (Et2O-BF3) y el efecto de un nucleófilo suave, el etanotiol (EtSH). La tioacidolisis provoca el corte específico de las uniones alquil-aril-éter (β-O-4 y α-O-4) de las ligninas. Permite por lo tanto, mediante el análisis de los productos de degradación, estimar el porcentaje de monómeros de la lignina unidos por este tipo de enlace. Este análisis se lleva a cabo mediante la separación de los monómeros en una columna de cromatografía de gases. Procedimiento: la preparación del reactivo de tioacidolisis se lleva a cabo mezclando 20 ml de dioxano puro con 2,5 ml de eterato de trifloruro de boro y 10 ml de etanotiol. Esta mezcla se completa hasta 100 ml con dioxano. En cada tubo de tioacidolisis se introducen aproximadamente 5 mg de lignina y se añaden 15 ml del reactivo de tioacidolisis, preparado anteriormente y 200 µl de patrón interno (C22H46). Los tubos se tapan y se mantienen durante 4 horas con agitación a 100ºC en un baño de aceite. Para llevar a cabo la extracción de los productos de tioacidolisis, en cada uno de los vasos donde ésta tendrá lugar se introducen 20 ml de diclorometano y 7,5 ml de bicarbonato de sodio 0,4 M. Pasadas 4 horas de reacción, se trasvasa el contenido de los tubos y se mide el pH del producto, ajustando éste a 2-3 con gotas de HCl 6N. El contenido de los vasos se transfiere a unos embudos de decantación donde la fase orgánica (zona inferior) es recogida en erlenmeyers en los que, previamente, se ha añadido una pequeña cantidad de sulfato de sodio para que adsorba el agua del medio. La fase acuosa superior se extrae dos veces mas con diclorometano, recogiendo de nuevo y combinando todas las fases orgánicas. El contenido de los erlenmeyers se concentra por evaporación en rotavapor a 60ºC.
30

3. Materiales y métodos
Previo al análisis de los productos de tioacidolisis por GC, éstos son silanizados. Los análisis en GC se realizaron en una columna capilar de sílice, siendo la fase estacionaria de polidimetilxilano. Se utilizó Helio como gas portador (presión 1,5 bar) y la temperatura del horno se programó para pasar de 160 a 260ºC en 50 minutos (+ 2ºC/min). El aparato de GC se encontraba equipado con un detector FID. El porcentaje de unidades guayacilo y siringilo se obtuvo utilizando el método de calibrado del patrón externo usando como patrón el hidrocarburo tetracosano. 3.2.14.3 DFRC
La composición (H:G:S) de las ligninas obtenidas de las diferentes maderas de eucalipto también se obtuvo mediante DFRC (Lu y Ralph, 1997a,1997b,1998) que permite el análisis cromatográfico separado de los monómeros unidos con enlaces éteres β-O-4 (los más abundantes en la estructura de la lignina) o α-O-4. El protocolo de la DFRC se divide en cuatro pasos que se detallan a continuación: a) Etapa de formación de βbromo-éteres
Se pesan 10 mg de lignina Björkman y se añaden a un matraz de fondo redondo, junto con una mosca para agitar. Se añaden 2,5 ml de una disolución de bromuro de acetilo: ácido acético (8:92) y se deja la mezcla en agitación durante 2 h a 50 ºC. A continuación, se retira la mosca y se elimina el disolvente en el rotavapor (sin sobrepasar los 50 ºC).
b) Etapa de ruptura reductora
Se añaden, al matraz que contiene el residuo seco, 50 mg de Zn metálico en polvo, 2,5 ml de una disolución dioxano:ácido acético:agua (5:4:1) y una mosca. Se deja en agitación durante 30-40 min a Tª ambiente. Se saca la mosca y se ajusta el pH con una disolución de HCl al 3% hasta que tenga pH < 3. Se añade tetracosano como patrón interno y a continuación se realiza una extracción líquido-líquido en un embudo de extracción, para ello se añaden 10 ml de una disolución saturada de cloruro de amonio y 10 ml de diclorometano (DCM). Sea agita vigorosamente y dejar reposar hasta tener dos fases bien diferenciadas. Se recoge la fase inferior (fase orgánica: DCM) y se procede a dos extracciones sucesivas con 5 ml de DCM en cada una de ellas. La fase orgánica extraída se seca añadiéndole sulfato de sodio anhidro y se filtra, recogiéndose el filtrado en un matraz para la posterior eliminación del disolvente en el rotavapor.
c) Etapa de acetilación
Al residuo que queda tras la evaporación, se le añaden: 1.1 ml de DCM; 0.2 ml de anhídrido acético y 0.2 ml de piridina. La piridina y el anhídrido siempre tienen que estar en la misma proporción (1:1). Se añade una mosca y se deja en agitación 1h. Se elimina la mosca y se añade etanol (se añade de 10 en 10 ml hasta 30 ml en total, para ayudar a eliminar el disolvente).
d) Análisis
El extracto resultante de la etapa de acetilación se resuspende en DCM para poder analizado por GC/MS. Los análisis se realizaron en un equipo Varian Star 3400 acoplado a un detector de trampa de iones (ITD, Varian Saturn 2000) usando una
31

3. Materiales y métodos
columna capilar de 12 m × 0,25 mm DB-5HT (0,1 µm, J&W Scientific). Se utilizó Helio como gas portador y la inyección se realizó en modo splitless. El horno se programó desde 50 ºC (0,2 min) hasta 100 º C con una rampa de 30 ºC/min y desde 100 ºC hasta 300 ºC con una rampa de 5 ºC/min, temperatura a la que permaneció durante 5 min. Las temperaturas del ITD y de la línea de transferencia se establecieron en 200 ºC y 300 ºC respectivamente.
3.2.15 Análisis de la estructura de la lignina mediante 2D NMR.
Los espectros de RMN se registraron en un espectrómetro Bruker AVANCE 500 MHz, equipado con una sonda triple con gradientes en el eje z, a una temperatura de 298ºK. Se disolvieron aproximadamente 40 mg de lignina en 0,75 ml de dimetilsulfóxido deuterado (DMSO-d6).
Los experimentos que se llevaron a cabo se trataron de 2D HSQC (Heteronuclear Single Quantum Correlation) y 2D HMBC (Heteronuclear Multiple Bond Correlation). El análisis HSQC proporciona correlaciones a través de acoplamiento escalar a un enlace entre un protón y el heteronúcleo al que está directamente unido. En cuanto al HMBC. Este experimento 2D sirve para asignar señales del esqueleto de una molécula orgánica. Los picos que se observan en este experimento son correlaciones entre un protón y un heteronúcleo (por ej. 13C, o 15N) situado a una distancia de 2 ó 3 enlaces. En ocasiones se puede llegar a observar correlaciones hasta 4 enlaces. El experimento HMBC permite establecer correlaciones con carbonos cuaternarios que tengan un protón a dos o tres enlaces con mucha mejor sensibilidad que en el espectro monodimensional de 13C. En este experimento se suprimen las correlaciones del isotopómero de 1H directamente unido al heteronúcleo (a un enlace), por lo que este experimento es complementario del HSQC. Las señales de cruce observadas en los espectros bidimensionales se asignaron principalmente por la comparación de éstos con los existen en la literatura (Capanema et al., 2001, 2004, 2005; Ralph et al., 1999, 2004; Liitiä et al., 2003; Balakshin et al., 2003) y mediante la combinación del HSQC y el HMBC.
Los experimentos HSQC se realizaron con 96 acumulaciones, empleando una matriz de datos de 256, 320 o 384 incrementos en la dirección indirecta, y adquiriendo 1024 puntos en la dimensión de adquisición. La anchura espectral fue de 150 o 176 ppm en la dimensión indirecta y de 11,90 ppm en la de adquisición. Los experimentos HMBC se realizaron con 96 o 128 acumulaciones empleando una matriz de datos de 256 o 384 incrementos en la dimensión indirecta y adquiriendo 2048 puntos en la dimensión de adquisición. La anchura espectral fue de 209 o 250 ppm en la dimensión indirecta y de 11,90 ppm en la de adquisición.
32

4. RESULTADOS Y DISCUSIÓN
33

34 34

4. Resultados y discusión
35
4.1 COMPOSICIÓN GENERAL DE LAS MADERAS DE LAS ESPECIES DE EUCALIPTO ESTUDIADAS
La caracterización general de las maderas de las cinco especies de eucalipto: E, globulus, E, nitens, E, maidenii, E, grandis, E, dunnii incluye la cuantificación de las principales fracciones, polisacáridos, lignina, extraíbles totales, material hidrosoluble y cenizas (Figura 13).
Los resultados se muestran en la Tabla 3 y en ella se puede observar que las maderas están constituidas principalmente por los polisacáridos celulosa y hemicelulosa (74,1-78,0%) además de cantidades considerables de lignina (18,7-22,6%), mientras que las fracciones restantes representan bajos porcentajes.
4.1.1 Contenido en lignina klason
La cuantificación de la lignina se realizó mediante el método klason. Este método es el más conocido y utilizado para cuantificar el contenido de este polímero en diferentes sustratos lignocelulósicos (Effland, 1977). Sin embargo, este método no se considera útil para el estudio de la estructura de la lignina debido a que el tratamiento ácido es muy agresivo y da lugar a una lignina muy condensada y alterada. La especie de eucalipto que presenta un menor contenido de lignina klason resultó ser la de E. globulus (18,7%), lo que a priori es una ventaja desde el punto de vista papelero, ya que la cantidad de lignina que hay que eliminar (cocción y blanqueo) es menor. El porcentaje mayor de lignina klason se obtuvo para la especie E. maidenii.
Muestra Extraíbles* Hidrosolubles Lignina Klason Polisacáridos Cenizas
E. globulus 0,3 (1,5) 1,2 18,7 78,0 0,5
E. nitens 0,6 (1,4) 1,2 22,5 74,2 0,4
E. maidenii 0,5 (1,3) 1,3 22,6 74,1 0,3
E. grandis 0,5 (1,3) 1,6 21,1 75,3 0,3
E. dunnii 0,6 (1,2) 1,4 21,6 75,2 0,2
* Los porcentajes entre paréntesis se refieren al extracto de acetona
Tabla 3. Composición general (%) de las madera de las especies de eucalipto.
Figura 13. Diagrama de los principales compuestos constituyentes de los materiales lignocelulósicos.
Material lignocelulósico
Compuestos de bajo peso molecular
Compuestos macromoleculares
Material orgánico Material inorgánico Polisacáridos Lignina
Celulosa HemicelulosaExtraíbles Cenizas
Material lignocelulósico
Compuestos de bajo peso molecular
Compuestos macromoleculares
Material orgánico Material inorgánico Polisacáridos Lignina
Celulosa HemicelulosaExtraíbles Cenizas

4. Resultados y discusión
36
4.1.2 Contenido en cenizas y composición de metales
Los componentes inorgánicos o sustancias minerales dependen de las condiciones del suelo de cultivo y de factores intrínsecos de las plantas (Anglés et al., 1997; Angelova et al., 2004). En la planta varían dependiendo de la especie, de la edad y de la parte estudiada (Anglés et al,. 1997). Los contenidos en cenizas son bajos desde 0,2 a 0,5%, lo que está de acuerdo con la bibliografía. El conocimiento de las cantidades de cationes metálicos presentes en las maderas es de especial interés ya que la presencia de estos en la pasta producen unos efectos negativos en el blanqueo de las pastas que se resumen en la descomposición de los agentes de blanqueo, el incremento de consumo de reactivos, reversión de la blancura, deposiciones en los circuitos y en propiedades fisicomecánicas y ópticas de los papeles menos favorables. En la Tabla 4 se muestran las cantidades de los distintos cationes presentes en la madera de eucalipto. Los elementos más abundantes en las diferentes maderas de las especies de eucalipto resultaron ser K, Ca, Mg, S, P y Mn, siendo la madera de E. globulus la que presenta un contenido global menor.
4.1.3 Composición química de los polisacáridos de la madera
Los azúcares neutros se analizaron, para las diferentes especies, libres de compuestos extraíbles lipofílicos y compuestos hidrosolubles, por cromatografía de gases tras su conversión en acetatos de alditol. Mediante dicha conversión, los isómeros de cada monosacárido se transforman en una sola especie a cuantificar, presentando el cromatograma un único pico para cada tipo de azúcar. Otra ventaja es que mediante este método sólo se analizan aquellas especies moleculares susceptibles de ser convertidas en acetatos de alditol (azúcares), evitando la interferencia con otro tipo de compuestos que pueden estar presentes en la muestra. La Tabla 5 recoge los resultados
Tabla 4. Contenido en metales y otros elementos (mg/Kg) de las especies de eucalipto.
Metal E. globulus E. nitens E. maidenii E. grandis E. dunnii
Al 7,6 3,9 7,5 22,6 83,1
B 2,7 5,5 2,1 3,6 3,4
Ca 260,0 551,0 900,0 930,0 1.900,0
Cd <0,1 0 <0,1 0 <0,1
Cr 1,1 0,2 0,3 0,8 1,1
Cu 0,8 0,7 0,5 1,3 0,7
Fe 14,7 3,9 4,3 24,9 36,7
K 440,0 1.070,0 940,0 370,0 470,0
Mg 120,0 130,0 150,0 90,0 390,0
Mn 77,2 6,1 3,5 50,3 42,5
Na 55,0 290,0 10,0 120,0 20,0
Ni 0,4 0,3 0,4 0,5 0,5
P 50,0 120,0 70,0 60,0 140,0
Pb 1,4 1,5 1,2 0,5 1,0
S 64,0 130,0 83,5 74,0 100,0
Sr 7,2 2,6 5,4 3,4 13,8
Ti 0,0 0,0 0,1 0,3 0,7
Zn 0,6 0,7 0,0 0,4 0,0

4. Resultados y discusión
37
del análisis cuantitativo de los azucares neutros presentes en las distintas maderas de eucalipto estudiadas.
Además de la glucosa, que proviene fundamentalmente de la celulosa, la xilosa es el monosacárido predominante, confirmando la importancia de los xilanos en la estructura de la pared celular (Fengel y Weneger, 1984; Sjöström, 1993; Shimizu, 2001). Manosa, arabinosa y galactosa se encuentran en menores cantidades, especialmente la galactosa no siendo detectada en la madera de E. maidenii.
4.1.4 Composición lipídica
Los extractos lipofílicos, resuspendidos en cloroformo, se analizaron por GC y GC/MS de acuerdo con el método propuesto por Gutiérrez et al., 1998b, los cromatogramas de los extractos lipofílicos totales de cada una de las especies de eucalipto estudiadas se muestran en la Figura 14. Se observa que para una misma cantidad de madera de partida el E. nitens presenta una mayor cantidad de extraíbles lipofílicos. Con la finalidad de obtener una mejor caracterización de las diferentes series homologas y de los compuestos minoritarios, los extractos lipídicos se fraccionaron por SPE (Figura 15) y las fracciones aisladas se analizaron por GC y GC/MS. La composición lipídica detallada de las diferentes especies de eucalipto seleccionadas para este estudio se muestran en la Tabla 6, los compuestos lipofílicos más predominantes en las maderas de eucaliptos estudiadas son los esteroides, incluyendo esteroles, ésteres de esteroles y esterilglucósidos, con cantidades menores de cetonas e hidrocarburos esteroidales (Figura 16) (Rencoret et al., 2007). La madera de E, globulus y E, dunnii son las que presentan un menor contenido de esteroles libres y conjugados (ésteres y glicósidos), los cuales son los principales responsables de la formación de los depósitos de pitch durante la fabricación de la pasta kraft (del Río et al., 1998,2000; Silvestre et al., 1999; Gutiérrez y del Río 2001), mientras que la madera de la especie E, nitens presenta el mayor contenido de estos compuestos causantes de los depósitos de pitch. Otros compuestos lipofílicos presentes en los extractos de las diferentes maderas son series de ácidos grasos, alcoholes grasos, ω-hidroxiácidos, mono- di- y triglicéridos, tocoferoles (libres y esterificados), alquilferulatos y pequeñas cantidades de escualeno (Figura 17).
Especie Arabinosa Xilosa Manosa Galactosa Glucosa E. globulus 2.7 12.1 5.1 1.1 79.0 E. nitens 3.5 16.3 5.1 0.7 74.2 E. maidenii 2.7 11.3 5.5 0.0 80.6 E. grandis 3.6 13.4 5.3 0.7 77.0 E. dunnii 2.4 14.0 4.8 2.1 76.5
Tabla 5. Composición de los monosacáridos de las maderas de eucalipto seleccionadas.

4. Resultados y discusión
38
2.5 5 7.5 10 12.5 15 17.5 20tiempo de retención (min)
E. globulus
E. nitens
E. maidenii
E. grandis
E. dunnii
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
Sitosterol
Estigmasterol+
fucosterol
Cetonas esteroidales
Ésteres de esteroles
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
Sitosterol
Estigmasterol+
fucosterol
Cetonas esteroidales
Ésteres de esteroles
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
SitosterolEstigmasterol
+fucosterol
Cetonas esteroidales
Ésteres de esteroles
Triglicéridos
Triglicéridos
Triglicéridos
Triglicéridos
Triglicéridos
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
Sitosterol
Estigmasterol+
fucosterol
Cetonas esteroidales
Ésteres de esteroles
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
SitosterolEstigmasterol
+fucosterol
Cetonas esteroidales Ésteres de esteroles
2.5 5 7.5 10 12.5 15 17.5 20tiempo de retención (min)
E. globulus
E. nitens
E. maidenii
E. grandis
E. dunnii
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
Sitosterol
Estigmasterol+
fucosterol
Cetonas esteroidales
Ésteres de esteroles
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
Sitosterol
Estigmasterol+
fucosterol
Cetonas esteroidales
Ésteres de esteroles
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
SitosterolEstigmasterol
+fucosterol
Cetonas esteroidales
Ésteres de esteroles
Triglicéridos
Triglicéridos
Triglicéridos
Triglicéridos
Triglicéridos
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
Sitosterol
Estigmasterol+
fucosterol
Cetonas esteroidales
Ésteres de esteroles
C16
C18:1+
C18:2
Ácidos grasos
Hidrocarburos esteroidales
SitosterolEstigmasterol
+fucosterol
Cetonas esteroidales Ésteres de esteroles
Figura 14. Cromatogramas de los extractos de las maderas de eucalipto sin derivatizar.

4. Resultados y discusión
39
Hexano8 mL
A
Hexano/CHCl3(5:1) 6mL
B
CHCl310 mL
Éter/AcOH(98:2) 10 mL
D
Acondicionamiento
Hexano 4 mL
Fase aminopropilo
Extracto lipídico total enHexano /CHCl3 (4:1) 0.5 mL
Extracto total
Ésteres de esteroles
Esteroles
C
Ácidos grasos
D
C
Hidrocarburosesteroidales
AÉsteres de esteroles
Hidrocarburosesteroidales
Cetonas esteroidales BTriglicéridos
Sitosterol
Cetonas esteroidales
Ácidos grasos
2.5 5 7.5 10 12.5 15 17.5 t(min)
2.5 5 7.5 10 12.5 15 17.5 t(min)
Hexano8 mL
A
Hexano/CHCl3(5:1) 6mL
B
CHCl310 mL
Éter/AcOH(98:2) 10 mL
D
Acondicionamiento
Hexano 4 mL
Fase aminopropilo
Extracto lipídico total enHexano /CHCl3 (4:1) 0.5 mL
Extracto total
Ésteres de esteroles
Esteroles
C
Ácidos grasos
D
C
Hidrocarburosesteroidales
AÉsteres de esteroles
Hidrocarburosesteroidales
Cetonas esteroidales BTriglicéridos
Sitosterol
Cetonas esteroidales
Ácidos grasos
2.5 5 7.5 10 12.5 15 17.5 t(min)2.5 5 7.5 10 12.5 15 17.5 t(min)
2.5 5 7.5 10 12.5 15 17.5 t(min)2.5 5 7.5 10 12.5 15 17.5 t(min)
Figura 15. Fraccionamiento SPE aplicado a los extractos totales.

4. Resultados y discusión
40
Tabla 6. Composición lipídica detallada de los extractos lipofílicos de las especies de eucalipto. Compuestos E. globulus E. nitens E. maidenii E. grandis E. dunnii Ácidos n-grasos 306.7 432.2 200.9 307.4 428.9 ácido n-tetradecanoico 1.6 1.2 0.6 1.5 3.2 ácido n-pentadecanoico 1.7 4.7 0.8 2.6 2.7 ácido n-hexadecanoico 88.3 112.7 39.3 75.4 112.3 ácido n-heptadecanoico 2.6 6.8 2.0 3.9 4.9 ácido 9,12-octadecadienoico 48.6 95.9 35.9 45.0 60.4 ácido 9-octadecenoico 39.6 63.9 51.6 41.5 112.2 ácido n-octadecanoico 8.5 14.0 9.3 12.0 23.7 ácido n-nonadecanoico 0.8 2.1 0.7 1.5 1.5 ácido n-eicosanoico 5.6 11.0 4.1 8.3 9.3 ácido n-heneicosanoico 2.8 8.6 2.5 6.2 5.7 ácido n-docosanoico 20.7 27.7 10.9 16.9 14.9 ácido n-tricosanoico 4.1 9.8 3.8 11.0 7.9 ácido n-tetracosanoico 34.2 46.1 17.9 39.9 28.8 ácido n-pentacosanoico 4.7 7.1 3.5 10.6 6.5 ácido n-hexacosanoico 31.9 16.5 14.2 25.1 26.7 ácido n-heptacosanoico 0.7 1.1 0.5 0.9 1.2 ácido n-octacosanoico 7.3 2.2 2.1 4.5 5.5 ácido n-triacontanoico 3.0 0.8 1.2 0.6 1.5 ω-Hidroxiácidos 23.9 17.4 13.9 9.1 10.5 ácido 22-hidroxidocosanoico 15.1 11.1 8.1 5.7 5.9 ácido 24-hidroxitetracosanoico 8.8 6.3 5.8 3.4 4.5 Esteroles 268.3 543.5 336.2 361.5 272.3 campesterol 2,8 6.9 2.7 2.9 2.4 sitosterol 190,3 396.2 263.2 287.0 212.1 estigmastanol 45,7 84.9 53.9 55.6 43.0 fucosterol 4,4 24.2 3.9 7.4 1.8 cicloartenol 8,1 22.8 2.4 0.1 2.7 24-metilencicloartanol 5,7 - - - 1.4 citrostadienol 2,6 1.1 6.0 2.0 5.0 7-oxositosterol 8,7 7.5 4.3 6.5 4.0 Cetonas esteroidales 37.2 72.0 113.0 59.7 95.4 estigmastan-3-ona 4.0 5.8 13.7 4.2 12.4 estigmasta-3,5-dien-7-ona 14.7 1.5 6.2 4.9 5.1 estigmast-4-en-3-ona 13.0 51.2 74.8 39.0 60.4 estigmasta-3,6-diona 5.5 13.5 18.3 11.6 17.5 Hidrocarburos esteroidales 31.3 30.5 22.2 19.4 26.6 estigmastano 3.3 4.3 2.1 2.2 3.2 estigmasta-4,22-dieno 2.6 2.8 1.8 1.6 2.0 estigmasta-3,5-dieno 15.7 17.6 12.6 10.0 13.1 estigmasta-3,5,22-trieno 8.1 4.6 4.1 5.4 6.3 Otros hidrocarburos 1.6 1.2 1.6 0.2 2.0 escualeno 1.6 1.2 1.6 0.2 2.0 Alcoholes grasos 2.4 7.2 3.9 4.1 2.0 n-hexadecanol 0.1 0.4 0.1 tr tr n-octadecanol 0.1 0.5 0.1 0.1 tr n-eicosanol tr 0.1 tr 0.1 tr n-docosanol 0.6 1.9 0.6 1.2 0.7 n-tetracosanol 0.5 2.3 0.6 0.8 0.3 n-hexacosanol 0.3 1.1 0.9 0.3 0.3

4. Resultados y discusión
41
n-octacosanol 0.8 0.9 1.8 1.6 0.7 Tocoferoles 8.5 6.5 11.5 8.3 10.5 α-tocoferol 6.4 4.6 7.4 6.2 7.2 β-tocoferol 2.1 1.9 4.1 2.1 3.3 Monoglicéridos 117.4 121.4 80.3 58.7 61.7 tetradecanoato de 2,3-dihidroxipropilo 0.2 tr 0.1 0.2 0.2 hexadecanoato de 2,3-dihidroxipropilo 3.3 7,0 1.0 2.5 3.0 heptadecanoato de 2,3-dihidroxipropilo 0.2 0,4 tr 0.1 0.1 octadecanoato de 2,3-dihidroxipropilo 2.6 5,6 1.0 2.3 6.0 nonadecanoato de 2,3-dihidroxipropilo tr 0,1 tr tr tr eicosanoato de 2,3-dihidroxipropilo 0.2 0,7 tr 0.1 0.2 heneicosanoato de 2,3-dihidroxipropilo tr 0,7 tr tr tr docosanoato de 2,3-dihidroxipropilo 1.8 8,9 2.9 3.3 3.0 tricosanoato de 2,3-dihidroxipropilo 0.2 1,5 0.3 0.1 0.4 tetracosanoato de 2,3-dihidroxipropilo 24.8 50,0 24.5 17.3 14.5 pentacosanoato de 2,3-dihidroxipropilo 2.9 4,5 4.6 2.5 1.0 hexacosanoato de 2,3-dihidroxipropilo 55.5 35,3 35.6 31.2 26.9 heptacosanoato de 2,3-dihidroxipropilo 1.0 0,8 1.5 1.3 1.4 octacosanoato de 2,3-dihidroxipropilo 20.0 5,8 7.9 4.6 4.2 nonacosanoato de 2,3-dihidroxipropilo 1.7 0,1 0.5 1.1 0.5 triacontanoato de 2,3-dihidroxipropilo 3.0 tr 0.3 1.6 0.3 Monoglicéridos de ω-hidroxiácidos 2.4 5.1 1.3 2.3 1.7 24-hidroxitetracosanoato de 2,3- dihidroxipropilo
0.9 4.1 0.7 1.1 0.7
26-hidroxihexacosanoato de 2,3- dihidroxipropilo
1.2 1.0 0.5 0.9 0.8
28-hidroxioctacosanoato de 2,3- dihidroxipropilo
0.3 tr 0.1 0.3 0.2
Diglicéridos 0.4 0.3 tr 0.2 0.6 dipalmitina tr tr tr tr 0.1 palmitinestearina 0.2 0.2 tr 0.1 0.3 diestearina 0.2 0.1 tr 0.1 0.2 Ferulatos de n-alquilo 5.8 3.4 1.2 1.6 7.9 trans-ferulato de eicosilo tr 0.1 tr tr tr trans-ferulato de docosilo 2.5 1.9 0.5 0.7 1.8 trans-ferulato de tetracosilo 2.0 1.2 0.4 0.5 3.8 trans-ferulato de hexacosilo 1.3 0.2 0.3 0.4 2.3 ω-Carboxialquilferulatos 22.4 1.2 tr 1.1 3.8 ácido trans-feruloiloxihexadecanoico 1.7 tr tr tr 0.3 ácido trans-feruloiloxioctadecanoico 1.5 tr tr tr 0.1 ácido trans-feruloiloxidocosanoico 9.0 0.6 tr 0.2 0.5 ácido trans-feruloiloxitetracosanoico 9.5 0.6 tr 0.9 2.3 ácido trans-feruloiloxihexacosanoico 0.7 tr tr tr 0.6 Glicósidos esteroidales 65.1 206.0 68.1 159.5 89.5 Sitosteril 3β-D-glucopiranósido 65.1 206.0 68.1 159.5 89.5 Ésteres de tocoferoles 5.8 15.9 6.1 6.4 9.7 dodecanoato de α-tocoferilo tr tr tr tr tr tetradecanoato de α-tocoferilo 0.2 0.2 0.2 0.1 0.1 hexadecanoato de α-tocoferilo 0.6 1.3 0.6 0.3 0.4 oleato/linoleato de α-tocoferilo 2.4 8.2 3.3 3.4 4.1 octadecanoato de α-tocoferilo 0.4 0.6 0.5 0.3 0.5 eicosanoato de α-tocoferilo tr tr 0.1 tr tr

4. Resultados y discusión
42
dodecanoato de β-tocoferilo tr tr tr tr tr tetradecanoato de β-tocoferilo 0.2 1.0 0.2 0.1 0.3 hexadecanoato de β-tocoferilo 0.4 1.4 0.3 0.2 0.8 oleato/linoleato de β-tocoferilo 1.4 2.4 0.8 1.8 3.3 octadecanoato de β-tocoferilo 0.2 0.8 0.1 0.2 0.2 eicosanoato de β-tocoferilo tr tr tr tr tr Ésteres de esteroles 250.2 828.5 434.4 214.3 289.6 ésteres de sitosterol 153.1 515.3 297.1 149.2 197.2 ésteres de estigmastanol 52.3 163.3 87.3 41.8 57.6 otros ésteres de esteroles 44.8 149.9 50.0 23.3 34.8 Triglicéridos 11.2 21.0 38.1 15.7 28.0 Lípidos totales identificados 1160.6 2313.3 1332.7 1229.5 1340.7
Los esteroles libres son una de las más abundantes clases de compuestos presentes en los extractos de todas las maderas estudiadas (268-543 mg KgP
-1P), siendo el sitosterol (I)
y el estigamstanol (II) los principales esteroles en todas las maderas analizadas. Otros esteroles, menos abundantes, como campesterol (III), fucosterol (IV), cicloartenol (V), 24-metilencicloartenol (VI), citrostadienol (VII) y 7-oxositosterol (VIII) también se detectaron en todas las especies de eucalipto estudiadas. Los esteroles libres pueden ser analizados tanto en los extractos sin derivatizar como silanizados. Las maderas de las especies de E. globulus y E. dunnii son las que presentan menores cantidades de esteroles libres mientras que la de la especie E. nitens, por el contrario, es la que más esteroles libres contiene. Cantidades significativas (65-206 mg Kg P
-1P) de
esterilglicósidos, principalmente sitosteril 3β-D-glucopiranósido, se encontraron en todos los extractos de las maderas de eucalipto. Estos compuestos se identificaron por primera vez, en la madera de E. globulus por Gutiérrez y del Río (2001), mediante la correspondiente derivatización (solo son detectables cuando se encuentran silanizados) y por comparación con sus patrones. El sitosteril 3β-D-glucopiranósido es especialmente abundante en las maderas de E. nitens y E. grandis, mientras se presentan en menor proporción en las de E. globulus y E. maidenii.

4. Resultados y discusión
43
Los ésteres de esteroles también se encuentran entre los extraíbles lipofílicos más abundantes (214-828 mg Kg P
-1P), siendo la madera del E. nitens la que presenta un mayor
contenido en ésteres de esteroles y las maderas de E. grandis y E. globulus las que menos. Los ésteres de esteroles están compuestos principalmente por series de ésteres de sitosterol y estigmastanol, con menores cantidades de las series de campesterol, fucosterol, y 24-metilencicloartenol. La saponificación de la fracción de ésteres de esteroles, aislada mediante SPE, muestra que el principal éster de esterol presente en los extractos de las maderas de eucalipto es el sitosteril linoleato (X). Otros compuestos esteroidales como las cetonas son detectados en cantidades considerables en los extractos lipofílicos de las maderas de eucalipto seleccionadas para este estudio. Las cetonas esteroidales (37-113 mg Kg P
-1P de madera) están constituidas
principalmente por estigmastan-3-one (XI), estigmasta-3,5-dien-7-ona (XII), estigmast-4-en-3-one (XIII) y estigmasta-3,6-diona (XIV).
HO
IHO
IIHO
IIIHO
IV
HO
V
HO
VI
HO
VII
HO O
VIII
O
XII
O
XIII
OO
XIV XV
O
XI
O
OH
O
OHHO
CH2OH
IX
O
O
X
HO
IHO
IIHO
IIIHO
IVHO
IHOHO
IHO
IIHOHO
IIHO
IIIHOHO
IIIHO
IVHOHO
IV
HO
V
HO
VI
HO
VII
HO O
VIII
HO
V
HOHO
V
HO
VI
HOHO
VI
HO
VII
HOHO
VII
HO O
VIII
HO OHO OO
VIII
O
XII
O
XIII
OO
XIV XV
O
XII
OO
XII
O
XIII
OO
XIII
OO
XIV
OO
OO
XIV XVXV
O
XI
OO
XI
O
OH
O
OHHO
CH2OH
IX
O
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH
IX
O
O
X
O
O
O
O
O
O
X
Figura 16. Estructura de los principales compuestos esteroidales identificados en las maderas de eucalipto a los que se hace referencia en el texto.(I) sitosterol, (II) estigmastanol, (III) campesterol, (IV) fucosterol, (V) cicloartenol, (VI) 24-metilencicloartanol, (VII) citrostadienol, (VIII) 7-oxositosterol, (IX) sitosteril 3β-D-glucopiranósido, (X) sitosteril linoleato, (XI) estigmastan-3-ona, (XII) estigmast-4-en-3-ona, (XIII) estigmasta-3,5-dien-7-ona, (XIV) estigmasta-3,6-diona, (XV) estigmasta-3,5-dieno.

4. Resultados y discusión
44
Con respecto a los hidrocarburos esteroidales, se detectaron con una, dos y hasta con tres instauraciones, aunque en pequeñas cantidades, siendo el estigamasta-3,5-dieno (XV) el más predominante. Los ácidos grasos libres representan una fracción importante de los extraíbles lipofílicos de las maderas estudiadas. Las series de ácidos grasos libres se encontraron en cantidades de 201-432 mg Kg P
-1P de madera, y con un predominio de aquellos que
presentan un número par de átomos de carbono, en el rango de CB14 B a CB30 B. Esta serie puede ser analizada tanto en los extractos sin derivatizar, así como en los metilados y en los silanizados. Los ácidos grasos predominantes resultaron ser el ácido palmítico (XVI), el oleico (XVII) y el linoleico (XVIII). Los ácidos grasos de mayor peso molecular como el ácido docosanoico (CB22B), tetracosanoico (CB24 B) y hexacosanoico (CB26B) se encontraron en elevadas cantidades en todos los extractos de las maderas analizadas. Con respecto a los ácidos grasos hidroxilados, es de destacar que se encontraron ω-hidroxiácidos como el 22-hidroxidocosanoico (XIX) y el 24-hidroxitetracosanoico en todos los extractos. Otros compuestos encontrados, aunque en bajas cantidades, son los tocoferoles, incluyendo α-tocoferoles (XX) y β-tocoferoles (XXI), con un predominio de los primeros. Los tocoferoles, además de encontrarse en su forma libre, también se detectaron esterificados con ácidos grasos de cadena larga en todos los extractos. Las series correspondientes a los α-tocoferol ésteres (XXII) y β-tocoferol ésteres (XXIII) se caracterizaron intactas por GC/MS, mediante sus espectros de masas. Los tocoferoles se encuentran esterificados a ácidos grasos con un número par de átomos de carbono, comprendido en el rango desde CB12B a CB20 B, incluyendo los ácidos grasos insaturados oleico (CB18:1B) y linoleico (CB18:2B) (Rencoret et al., 2006). Se encontraron unas series de compuestos en las cuales el ácido ferúlico está unido mediante un enlace ester con n-alcoholes grasos de cadena larga (XXIV) o con ω-hidroxiácidos (XXV). La caracterización de estos compuestos intactos se realizó mediante sus espectros de masas, tanto de las muestras sin derivatizar, así como con los espectros de las muestras derivatizadas (metiladas y/o silanizadas) publicados por del Río et al., 2004. La serie de n-alquil trans ferulatos se detectó con alcoholes que presentaban entre 20 y 26 átomos de carbono, con predominio de los pares, siendo los más predominantes los CB22 B y CB24B en todas los extractos de las maderas a excepción de la del E. dunnii, donde los predominantes resultaron ser los CB24B y CB26 B. Los feruloil ésteres de ω-hidroxiácidos, los cuales habían sido raramente detectados en algunas plantas (Pereira et al., 2002), se identificaron en el rango desde CB16 B a CB26 B, siendo los más predominantes los CB22B y CB24 B en todos los extractos excepto en el del E. dunnii donde los predominantes fueron los de CB24 B y CB26 B. Pequeñas cantidades de alquilferulatos y ω-carboxialquilferulatos también se encontraron en el extracto de E. globulus (Freire et al., 2002). Los glicéridos, incluyendo mono-, di- y triglicéridos, se detectaron en todos los extractos, con un predominio de monoglicéridos. Los monoglicéridos (XXVI) se encuentran en los extractos en cantidades relativamente grandes (59-121mg KgP
-1P de
madera). El número de carbonos de la molécula de ácido graso se encuentra en el rango de CB14 B a CB30B y de nuevo se observa un predominio de aquellos monoglicéridos constituidos por ácidos grasos con número par de átomos de átomos de carbono, siendo los más predominantes los CB24B, CB26 B y CB28 B. Además, se detectó una serie de monoglicéridos constituidos por ω-hidroxiácidos (Freire et al., 2006) de CB24B, CB26 B y CB28 B en todos los extractos de las diferentes maderas estudiadas, aunque en cantidades muy

4. Resultados y discusión
45
bajas. Las cantidades de triglicéridos encontradas oscilaron entre 11-38 mg KgP
-1P de
madera, constituidos principalmente por tripalmitina (XXVII), mientras los diglicéridos se encuentran en muy baja proporción en todos los extractos de las maderas estudiadas. Finalmente, se encontraron, en todos los extractos, pequeñas cantidades de otros compuestos, como escualeno (XXVII) y una serie de alcoholes de cadena larga (XXIX) desde CB14B a CB28 B, solo detectable en las muestras silanizadas.
OH
O
XVIOH
O
XVII
OH
O
XVIII
HOOH
O
XIX
XXVIII
O
HO
XXO
HO
XXI
O
O
OXXII
O
O
O
XXIII
O
OCH3
OH
O
XXIV
OOH
OO
OCH3
OH
XXV
CO-O-CH2
HO-O-CH
HO-O-CH2
XXVI XXVII
CO-O-CH2
CO-O-CH
CO-O-CH2
OH
XXIX
OH
O
XVIOH
O
OH
O
OH
O
XVIOH
O
XVIIOH
O
OH
O
OHOH
O
XVII
OH
O
XVIIIOH
O
OH
O
OHOH
O
XVIII
HOOH
O
XIX
HOOH
OHO
OH
O
OHOH
O
XIX
XXVIIIXXVIII
O
HO
XXO
HO
O
HO
XXO
HO
XXIO
HO
O
HO
XXI
O
O
OXXII O
O
OO
O
O
O
OXXII
O
O
O
XXIII
O
O
OO
O
OO
O
OO
O
O
O
OO
XXIII
O
OCH3
OH
O
XXIV
O
OCH3
OH
OO
OCH3
OH
O
XXIV
OOH
OO
OCH3
OH
XXV
OOH
OO
OCH3
OH
OOH
OO
OCH3
OH
OOH
OO
OCH3
OH
OCH3
OH
XXV
CO-O-CH2
HO-O-CH
HO-O-CH2
XXVI
CO-O-CH2
HO-O-CH
HO-O-CH2
CO-O-CH2
HO-O-CH
HO-O-CH2
CO-O-CH2
HO-O-CH
HO-O-CH2
XXVI XXVII
CO-O-CH2
CO-O-CH
CO-O-CH2
XXVII
CO-O-CH2
CO-O-CH
CO-O-CH2
CO-O-CH2
CO-O-CH
CO-O-CH2
CO-O-CH2
CO-O-CH
CO-O-CH2
OH
XXIXOHOH
XXIX
Figura 17. Estructuras de los compuestos no esteroidales identificados en las maderas de eucalipto a las que se hace referencia en el texto: (XVI) ácido palmítico, (XVII) ácido oleico, (XVIII) ácido linoleico, (XIX) ácido 22-hidroxidocosanoico, (XX) α-tocoferol, (XXI) β-tocoferol, (XXII) α-tocoferil linoleato, (XXIII) β-tocoferol linoleato, (XXIV) trans-docosanil ferulato, (XXV) trans- ácido feruloiloxidocosanoico, (XXVI) docosanoato de 2,3-dihidroxipropilo, (XXVII) tripalmitina, (XXVIII) escualeno, (XXIX) n-docosanol.

4. Resultados y discusión
46
4.1.5 Estudio de la composición y estructura de la lignina de las maderas de las diferentes especies de eucalipto
El análisis de la composición química de la lignina se llevó a cabo mediante el uso de las siguientes técnicas: Py-GC/MS, DFRC y tioacidolisis. Por otro lado, el estudio de la estructura se realizó mediante 2D-NMR.
4.1.5.1 Análisis de la composición de la lignina mediante Py-GC/MS
Las técnicas pirolíticas presentan diversas ventajas sobre otros métodos degradativos en el estudio de las macromoléculas. Junto con su rapidez, el análisis por Py-GC/MS se caracteriza por la pequeña cantidad de muestra requerida y por permitir el análisis de la lignina “in situ”, frente a los métodos clásicos que requieren su extracción de la madera. Se han publicado numerosos estudios sobre pirólisis de materiales lignocelulósicos (Pouwels et al., 1987; Saiz-Jiménez et al., 1987; Elder, 1991; Yokoi et al., 1999; del Río et al., 2001b, 2002, 2005). Las pirólisis se realizaron tanto a las maderas de las especies de eucalipto seleccionadas para este estudio como a las ligninas Björkman (MWL) obtenidas de éstas. Los pirogramas obtenidos de las diferentes maderas estudiadas se muestran en la Figura 18 y las asignaciones y las abundancias molares relativas de los compuestos obtenidos por pirólisis de las maderas se encuentran en la Tabla 7. Entre los compuestos derivados de los carbohidratos los principales son hidroxiacetaldehído (1), furfural (7), 5(H)-furan-2-ona (10), 2,3-dihidro-5-metilfuran-2-ona (11), 4-hidroxi-5,6-dihidro-2(H)-piran-2-ona (13), y levoglucosano (32). Entre los compuestos derivados de la lignina, los más típicos son los que provienen de las unidades fenólicas del tipo S y G, los más relevantes son: guaiacol (16), 4-metilguaiacol (17), 4-vinilguaicol (20), siringol (21), 4-metilsiringol (26), trans-isoeugenol (27), 4-vinilsiringol (35), siringaldehído (40), homosiringaldehído (43), acetosiringona (44) y trans-sinapaldéhido (52). En todos los pirogramas de las diferentes especies, los derivados fenólicos de tipo S fueron más abundantes que los derivados de tipo G. Las áreas molares relativas de los picos se calcularon para los productos de pirólisis de carbohidratos así como para los derivados de tipo S y G de la lignina. Las relaciones S/G y lignina/carbohidrato (L/CH) se determinaron para cada una de las maderas como el valor medio de dos replicados (Tabla 8). Se debe tener en cuenta que la relación L/CH observada no refleja el contenido real de cada molécula, ya que es sabido que la pirólisis subestima en demasía el contenido en celulosa debido a la gran cantidad de productos no cromatografiables que se producen a partir de ella. Sin embargo, la relación L/CH obtenida por pirólisis de las maderas pueden ser utilizadas para comparar las cantidades relativas de lignina y carbohidratos de las diferentes especies ya que se realizaron en las mismas condiciones. Los resultados obtenidos mostraron que la madera de E. globulus presenta la relación L/CH más baja, además de mostrar que las ligninas procedentes de las diferentes especies de eucalipto estudiadas presentan elevadas relaciones S/G, aunque hay que tener en cuenta que la pirólisis, como otros métodos degradativos, sobreestima las cantidades de unidades S obtenidas (Sarkanen and Hergert, 1971).
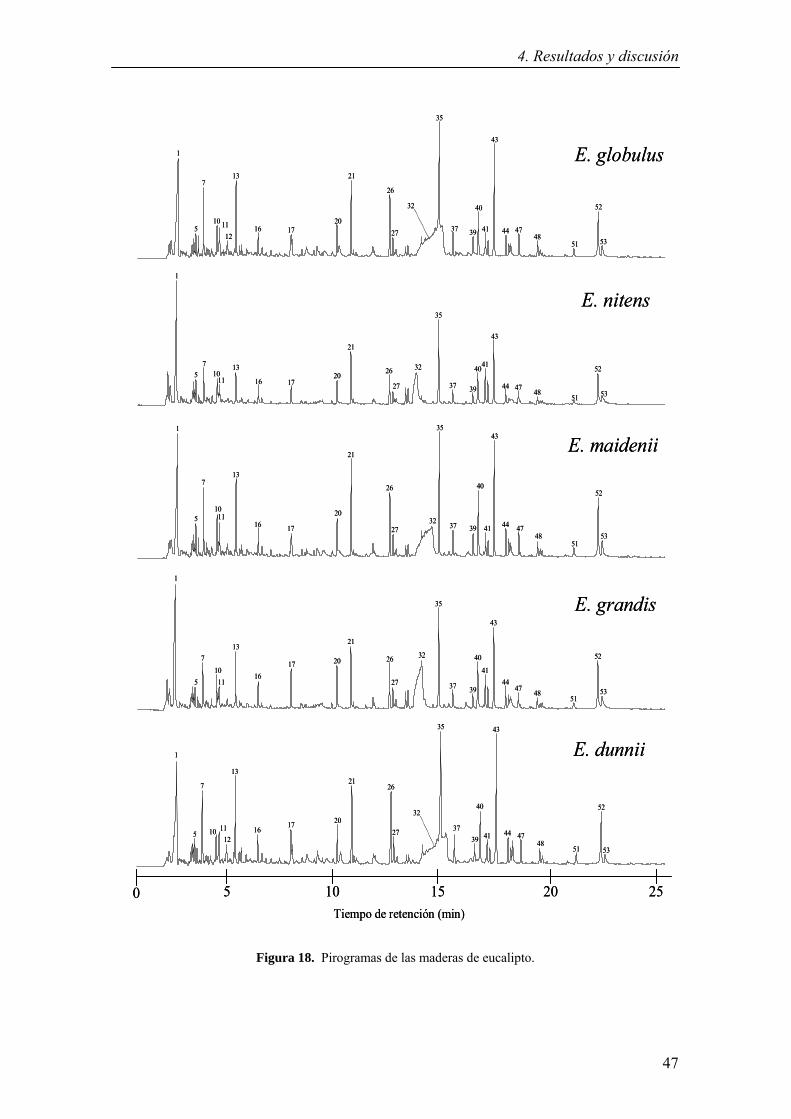
4. Resultados y discusión
47
Figura 18. Pirogramas de las maderas de eucalipto.
5 10 252015Tiempo de retención (min)
E. globulus
E. grandis
E. maidenii
E. nitens
0
1
7
10 11
13
1217
20
21
16
26
27
32
35
37 39
40
41
43
44 4748
51 53
52
5
1
710
11
13
1720
21
1626
27
32
37 39
4041
43
44 47 4851 53
525
35
1
7
1011
13
17
20
21
16
26
2732
35
37 39
40
41
43
44 4748
5153
52
5
1
71011
13
17 20
21
16
26
27
32
35
37 39
40
41
43
4447 48
5153
52
5
35
E. dunnii1
7
10 11
13
12
17 20
21
16
26
27
32
3739
40
41
43
44 4748
51 53
52
5
5 10 252015Tiempo de retención (min)
E. globulus
E. grandis
E. maidenii
E. nitens
0
1
7
10 11
13
1217
20
21
16
26
27
32
35
37 39
40
41
43
44 4748
51 53
52
5
1
710
11
13
1720
21
1626
27
32
37 39
4041
43
44 47 4851 53
525
35
1
7
1011
13
17
20
21
16
26
2732
35
37 39
40
41
43
44 4748
5153
52
5
1
71011
13
17 20
21
16
26
27
32
35
37 39
40
41
43
4447 48
5153
52
5
35
E. dunnii1
7
10 11
13
12
17 20
21
16
26
27
32
3739
40
41
43
44 4748
51 53
52
5

4. Resultados y discusión
48
Tabla 7. Abundancias molares relativas de los compuestos obtenidos por pirólisis de las diferentes maderas de eucalipto.
Nº Compuestos Origen E. globulus E. nitens E. maidenii E. grandis E. dunnii 1 hidroxiacetaldehido CH 24,6 34,9 26,8 26,4 24,0 2 (3H) Furan-2-ona CH 0,6 0,6 0,9 0,7 0,3 3 desconocido CH 1,0 1,2 0,9 0,8 1,0 4 (2H) Furan-3-ona CH 0,8 1,1 0,6 0,9 0,7 5 1,3-hidroxidihidro-2(3H)-furanona CH 1,4 1,7 1,6 1,2 0,9 6 desconocido CH 1,1 0,7 0,9 0,7 0,8 7 furfural CH 3,7 3,5 3,4 3,1 3,2 8 2,3 dihidro-5-metilfuran-2-ona CH 0,3 0,3 0,2 0,3 0,3 9 ciclopent-1-en-3,4-diona CH 0,5 0,6 0,5 0,5 0,0 10 (5H)-furan-2-ona CH 2,5 0,3 2,5 2,4 2,0 11 2,3-dihidro-5-metilfuran-2-ona CH 2,4 1,2 1,5 0,9 1,4 12 aldehido 5-metil-2-furfural CH 0,4 0,2 0,3 0,2 0,4 13 4-hidroxi-5,6-dihidro-(2H)-piran-2-ona CH 3,8 2,6 3,1 2,8 4,9 14 3-hidroxi-2-metil-2-ciclopenten-1-ona CH 0,6 0,3 0,5 0,7 0,9 15 2-hidroxi-3-metil-2-ciclopenten-1-ona CH 0,5 0,4 0,5 0,3 0,4 16 Guaiacol LG 1,2 1,5 1,2 1,4 1,2 17 4-metilguaiacol LG 1,3 1,3 1,3 1,9 1,4 18 3-metoxicatecol LG 0,5 0,2 0,7 0,1 0,8 19 4-etilguaiacol LG 0,2 0,3 0,3 0,2 0,2 20 4-vinilguaicol LG 1,3 1,7 1,6 2,3 1,5 21 siringol LS 3,5 4,0 3,9 3,1 2,7 22 eugenol LG 0,2 0,3 0,2 0,4 0,2 23 propilguaiacol LG 0,1 0,1 0,1 0,1 0,1 24 vainillina LG 0,7 0,5 0,6 0,7 0,6 25 c-isoeugenol LG 0,2 0,3 0,2 0,3 0,2 26 4-metilsiringol LS 2,6 2,1 2,5 2,1 2,5 27 t-isoeugenol LG 0,7 0,7 0,8 0,9 0,9 28 homovainillina LG 0,3 0,4 0,3 0,5 0,5 29 propine-guaiacol LG 0,4 1,2 0,5 0,9 0,3 30 propine-guaiacol LG 0,6 0,9 0,4 0,9 0,6 31 acetoguiacona LG 0,1 0,4 0,4 0,3 0,2 32 levoglucosano CH 18,8 11,3 17,0 18,2 21,5 33 4-etilsiringol LS 0,5 0,4 0,5 1,0 0,4 34 guaiacilacetona LG 0,2 0,2 0,5 0,5 0,4 35 4-vinilsiringol LS 5,5 5,1 4,7 4,4 5,1 36 guaiacilvinilcetona LG 0,2 0,2 0,3 0,4 0,2 37 4-alilsiringol LS 0,9 0,9 0,9 0,9 0,9 38 propilsiringol LS 0,2 0,0 0,1 0,0 0,3 39 c-propenilsiringol LS 0,7 0,6 0,7 0,6 0,5 40 siringaldehido LS 2,0 2,4 2,8 2,4 2,0 41 propine-siringol LS 1,0 2,0 0,8 1,3 1,0 42 propine-siringol LS 0,7 1,3 0,5 0,9 0,7 43 t-propenilsiringol+ homosiringaldehido LS 4,7 3,7 4,1 3,5 4,5 44 acetosiringona LS 0,8 1,0 1,0 1,0 0,8 45 t-coniferilaldehido LG 0,2 0,4 0,8 0,8 0,6 46 alcohol c-coniferílico LG 0,8 0,7 0,9 1,1 1,0 47 siringilacetona LS 0,7 0,7 0,8 0,6 0,7 48 propiosiringona LS 0,3 0,2 0,3 0,2 0,3 49 siringilvinilcetona LS 0,1 0,3 0,2 0,2 0,1 50 alcohol dihidrosinapílico LS 0,1 0,0 0,1 0,1 0,1 51 alcohol c-sinapílico LS 0,3 0,3 0,5 0,4 0,4 52 t-sinapaldehido LS 2,4 2,4 2,9 2,7 2,4 53 alcohol t-sinapílico LS 0,7 0,8 0,9 1,1 1,3
Abreviaturas: c: cis t: trans
La madera de E. globulus presenta la mayor relación S/G (3,0), lo cual hace que sea más fácil de su deslignificación bajo las condiciones de pasteo Kraft debido a la mayor reactividad de las unidades S de la lignina en sistemas alcalinos (Chang y Sarkanen 1973; Tsutsumi et al., 1995).

4. Resultados y discusión
49
Tabla 8. Porcentajes y relaciones obtenidas de las pirólisis de las maderas.
La pirólisis también se realizó a las MWL obtenidas de las diferentes maderas de eucalipto. A diferencia de las pirólisis de las maderas, en estos pirogramas, los picos procedentes de CH no suelen aparecer, en la Figura 19 se muestra el pirograma de la lignina de E. dunni. Las asignaciones y las abundancias molares relativas de los compuestos obtenidos por pirólisis de las MWL se encuentran en la Tabla 9. Las relaciones S/G obtenidas de las MWL presentan unos valores ligeramente más pequeños que aquellos obtenidos de sus maderas, sin embargo, la tendencia en las relaciones S/G se mantienen, siendo la relación S/G mayor para la especie E. globulus (2,6) y la menor para la especie E. grandis (1,9).
Tabla 9. Identificación y abundancia molar relativa de los compuestos obtenidos por Py-GC/MS de las MWL de las maderas de las diferentes especies de eucalipto. Nº Compuestos E. globulus E. nitens E. maidenii E. grandis E. dunnii 1 guaiacol 5,5 5,5 6,3 5,7 5,1 2 4-metilguaiacol 3,8 3,5 4,1 4,6 2,6 3 3-metoxicatecol 1,3 2,4 1,7 0,2 1,3 4 4-etilguaiacol 1,2 1,2 1,4 1,3 0,9 5 4-vinilguaicol 4,8 4,6 5,1 5,9 4,0 6 siringol 13,8 13,5 14,2 11,8 14,5 7 eugenol 0,7 0,9 0,7 1,2 0,9 8 4-propilguaiacol 0,3 0,4 0,3 0,3 0,2 9 vainillina 1,5 2,6 2,8 2,1 2,4 10 c-isoeugenol 0,3 0,1 0,3 0,8 0,4 11 4-methylsyringol 7,9 7,1 7,3 7,4 5,9 12 t-isoeugenol 2,6 3,9 3,4 3,7 3,1
Especie de eucalipto %G %S S/G %L %C L/C E. globulus 24.8 75.2 3.0 37.1 62.9 0.6 E. nitens 27.8 72.2 2.6 39.4 60.6 0.7 E. maidenii 27.2 72.8 2.7 39.0 61.0 0.6 E. grandis 33.3 66.7 2.0 40.1 59.9 0.7 E. dunnii 28.4 71.5 2.5 37.5 62.5 0.6
Figura 19. Pirograma de la MWL obtenida de la madera de E. dunnii.
Tiempo de retención (min)
6.00 8.00 10.00 12.00 14.00 16.00 18.00 20.000
2000000
4000000
6000000
8000000
1e+07
1.2e+07
1.4e+07
1.6e+07
1.8e+07
2e+07
2.2e+07
2.4e+07
2.6e+07
Time-->
AbundanceTIC: DUNII.D
1
2
5
6
11
12
17
19
21 23
24
27+28
2930
31
37
38
- - - - - - - - -
6 8 10 12 14 16 18 20 226.00 8.00 10.00 12.00 14.00 16.00 18.00 20.00
0
2000000
4000000
6000000
8000000
1e+07
1.2e+07
1.4e+07
1.6e+07
1.8e+07
2e+07
2.2e+07
2.4e+07
2.6e+07
Time-->
AbundanceTIC: DUNII.D
1
2
5
6
11
12
17
19
21 23
24
27+28
2930
31
37
38
- - - - - - - - -
6 8 10 12 14 16 18 20 22

4. Resultados y discusión
50
13 homovainillina 1,2 1,0 1,1 0,9 0,8 14 propine-guaiacol 0,6 0,5 0,6 1,0 0,7 15 propine-guaiacol 0,5 0,4 0,5 0,7 0,5 16 acetoguaiacona 0,6 0,9 1,2 1,4 1,0 17 4-etilsiringol 4,2 2,0 1,9 1,5 1,5 18 guaiacilacetona 0,6 0,5 0,5 0,4 0,4 19 4-vinilsiringol 9,9 8,2 7,5 8,1 7,9 20 guaiacilvinilcetona 0,6 0,6 0,7 0,8 0,8 21 4-alilsiringol 1,7 1,9 1,5 1,6 2,0 22 propilsiringol 0,2 0,2 0,2 0,2 0,2 23 c-propenilsiringol 1,4 1,4 1,6 1,8 1,8 24 siringaldehído 6,4 7,0 7,2 7,5 8,4 25 propine-siringol 1,7 1,4 1,2 1,3 1,5 26 propine-siringol 1,1 0,7 0,5 0,8 0,7 27 t-propenilsiringol 5,7 6,8 5,1 5,8 6,0 28 homosiringaldehído 4,9 4,0 3,3 2,6 3,1 29 acetosiringona 2,6 2,7 3,0 3,4 3,4 30 t-coniferaldehído 1,5 1,7 2,2 2,5 2,3 31 alcohol t-coniferílico 0,8 2,1 1,7 1,5 2,5 32 siringilacetona 1,5 1,2 1,1 0,9 1,0 33 propiosiringona 0,9 0,9 1,0 1,0 0,9 34 siringil vinil cetona 0,8 0,6 0,9 1,3 1,2 35 alcohol dihidrosinapílico 0,2 0,4 0,3 0,3 0,4 36 alcohol c-sinapílico 0,3 0,6 0,5 0,3 0,8 37 t-sinapaldehído 5,6 4,6 5,0 6,2 6,2 38 alcohol t-sinapílico 1,0 2,2 2,0 1,2 2,7 S/G ratio 2,6 2,1 2,0 1,9 2,4
4.1.5.2 Análisis de la composición química de la lignina mediante DFRC
La técnica DFRC se aplicó a las MWL aisladas de las maderas de las diferentes especies de eucaliptos. Los cromatogramas obtenidos mediante esta técnica se caracterizan por presentar cuatro picos intensos, correspondientes a los isómeros cis y trans de las unidades S y G. Mediante el cálculo de las áreas molares relativas es posible obtener las relaciones S/G para cada una de las diferentes MWL (Tabla 10).
4.1.5.3 Análisis de la composición química de la lignina mediante tioacidolisis
Al igual que la DFRC, la tioacidolisis se aplicó a las MWL. En este caso los cromatogramas se caracterizan por presentar dos dobletes, cada uno de ellos correspondientes a las formas eritro y treo de cada unidad. Las relaciones S/G al igual que en la DFRC se calculan a partir de las áreas molares relativas. Los valores de relación S/G obtenidos para las diferentes MWL se muestran en la Tabla 10, donde se aprecia que la composición de la MWL, en términos de relación S/G, es bastante similar a la obtenida por Py-GC/MS y muestra la misma tendencia que la obtenida mediante DFRC, siendo la mayor relación S/G para el E. globulus, seguida de E. dunnii, E. nitens, E. maidenii y por último E. grandis.
4. Resultados y discusión
51
4.1.5.4 Estudio de la estructura de la lignina de las diferentes especies de eucalipto
El estudio de la estructura de la lignina se realizó a partir de las MWL, aisladas de las maderas de las diferentes especies de eucalipto, mediante espectroscopia de resonancia magnética nuclear bidimensional. Los experimentos que se realizaron se trataron de 2D HSQC y 2D HMBC. Las señales de cruce observadas en los espectros bidimensionales se asignaron principalmente por la comparación de éstos con los existen en la literatura (Capanema et al., 2001, 2004, 2005; Ralph et al., 1999, 2004; Liitiä et al., 2003; Balakshin et al., 2003) y mediante la combinación del HSQC y el HMBC. El espectro 2D HSQC de la lignina se caracteriza porque presenta tres regiones claramente diferenciadas: la región alifática, la región alifática oxigenada y la región aromática (Figura 20). La región alifática (no oxigenada) presenta señales correspondientes a lípidos, a productos de degradación de la lignina y a otros compuestos no identificados, por lo que es la menos interesante desde el punto de vista del estudio de la estructura de la lignina.
Tabla 10. Relaciones S/G de las MWL de las maderas de eucalipto obtenidas por diferentes técnicas analíticas degradativas.
Técnica E. globulus E. nitens E. maidenii E. grandis E. dunnii
Py-GC/MS 2,6 2,1 2,0 1,9 2,4 DFRC 2,1 1,8 1,6 1,4 2,1 Tioacidolisis 2.5 2.1 1.8 1.6 2.3

4. Resultados y discusión
52
En la Figura 20 se observa que los HSQC de las ligninas aisladas de las maderas de las diferentes especies de eucalipto son muy similares. Para llevar a cabo un análisis de los enlaces entre las unidades es necesario estudiar a fondo la región alifática oxigenada y la región aromática de los espectros HSQC, las cuales se muestran ampliadas para el espectro 2D HSQC de la lignina de E. dunnii en la Figura 21. Las asignaciones de las señales y sus desplazamientos químicos observados en los espectros HSQC de las diferentes ligninas se muestran en la Tabla 11. Las subestructuras elucidadas a partir de los espectros HSQC se pueden observar en la Figura 22.
Figura 20. Espectros HSQC totales de las ligninas de las diferentes especies de eucalipto.
ppm
Región aromática
Región alifática oxigenada
Región alifática
DMSO
E. globulus
1.02.03.04.05.06.07.08.0δH δH
0
δC
1.02.03.04.05.06.07.08.0 ppmppm
100
50
E. nitens
Región aromática
Región alifática oxigenada
Región alifática
ppm1.02.03.04.05.06.07.08.0δH
E. maidenii
Región aromática
Región alifática oxigenada
Región alifática
δC
1.02.03.04.05.06.07.08.0 ppmppm
100
50
δH
E. grandis
Región aromática
Región alifática oxigenada
Región alifática
ppm1.02.03.04.05.06.07.08.0δH
E. dunnii
Región aromática
Región alifática oxigenada
Región alifática
δC
100
50
ppm
Región aromática
Región alifática oxigenada
Región alifática
DMSO
E. globulus
1.02.03.04.05.06.07.08.0δH δH
0
δC
1.02.03.04.05.06.07.08.0 ppmppm
100
50
E. nitens
Región aromática
Región alifática oxigenada
Región alifática
δH
0
δC
1.02.03.04.05.06.07.08.0 ppmppm
100
50
E. nitens
Región aromática
Región alifática oxigenada
Región alifática
ppm1.02.03.04.05.06.07.08.0δH
E. maidenii
Región aromática
Región alifática oxigenada
Región alifática
ppm1.02.03.04.05.06.07.08.0δH
E. maidenii
Región aromática
Región alifática oxigenada
Región alifática
δC
1.02.03.04.05.06.07.08.0 ppmppm
100
50
δH
E. grandis
Región aromática
Región alifática oxigenada
Región alifática
ppm1.02.03.04.05.06.07.08.0δH
E. dunnii
Región aromática
Región alifática oxigenada
Región alifática
δC
100
50
ppm1.02.03.04.05.06.07.08.0δH
E. dunnii
Región aromática
Región alifática oxigenada
Región alifática
ppm1.02.03.04.05.06.07.08.0δH
E. dunnii
Región aromática
Región alifática oxigenada
Región alifática
δC
100
50

4. Resultados y discusión
53
Tabla 11 Asignaciones de las señales de correlación P
13PC- P
1PPPH que aparecen los espectros HSQC de
las MWL de las diferentes especies de eucaliptos.
δBCB/δ BH B (ppm) Asignaciones
9-37/0.8-2.6 diferentes C-H en cadenas alifáticas saturadas
32.0/2.53 CBαBH BαB en alcohol dihidrosinapílico (E)
35.0/1.68 CBβBH BβB en alcohol dihidrosinapílico (E)
53.7/3.46 CBβ B-HBβ B en subestructuras de fenilcumarano (C)
54.1/3.06 CBβ B-HBβ B en subestructuras de resinol (B)
55.4/2.76 CBβ B-HBβ Ben subestructuras β-1’ (H)
56.2/3.73 C-H en metoxilos (MeO)
60.1/3.40 y 3.72 CBγ BHBγ B en subestructuras β-O-4’ (A)
60.3/2.75 CBβ B-HBβ B en subestructuras de espirodienonas (D)
61.9/4.09 CBγ B-HBγ B en alcohol p-hidroxicinamílico (sinapílicol/coniferílico) (I)
71.7/4.74 CBα B-HBα B en subestructuras β-O-4’ unida a unidades tipo G (A)
71.7/3.83 y 4.19 CBγ B-HBγ B en subestructuras de resinol (B)
72.4/4.86 CBα B-HBα B en subestructuras β-O-4’ unida a unidades tipo S (A)
74.8/5.02 CBαBH Bα B en subestructuras β-1’ (H)
79.8/4.11 CBβ’ B-HBβ’ B en subestructuras de espirodienonas (D)
81.7/5.09 CBαBH Bα B en subestructuras de espirodienonas (D)
83.8/5.23 CBβ B-HBβ B en subestructuras β-O-4’ con CBαB=O (F)
84.2/4.29 CBβ B-HBβ B en subestructuras β-O-4’ unida a unidades tipo G (A)
85.5/4.67 CBα B-HBα B en subestructuras de resinol (B)
86.6/4.39 CBα’ BH Bα’ B en subestructuras de espirodienonas (D)
86.4-87.3/4.01-4.11 CBβ B-HBβ B en subestructuras β-O-4’ unida a unidades tipo S (A)
87.4/5.47 CBα B-HBα B en subestructuras de fenilcumarano (C)
104.7/6.69 CB2,6B-HB2,6 B en unidades siringilo (S)
106.8/7.32 CB2,6B-HB2,6 B en unidades siringilo oxidadas (CBα B=O) (S’)
107.0 /7.19 CB2,6B-HB2,6 B en unidades siringilo oxidadas (CBα BOOH) (S’’)
107.4/6.33 CB2BH B2 B en subestructuras de espirodienonas (D)
111.6/6.99 CB2B-HB2B en unidades guaiacilo (G)
112.1/7.53 CB2B-HB2B en unidades guaiacilo oxidadas (G’)
114.1/6.26 CB6BH B6 B en subestructuras de espirodienonas (D)
115.4/6.72 y 6.94 CB5B-HB5B en unidades guaiacilo (G)
119.5/6.77 CB6B-HB6B en unidades guaiacilo (G)
123.8/7.58 CB6B-HB6B en unidades guaiacilo oxidadas (G’)

4. Resultados y discusión
54
Figura 21. Regiones alifática oxigenada y aromática expandida del espectro 2D HSQC de la lignina de E. dunnii.
δH
3.03.54.04.55.05.5 ppm
MeO Cβ Bβ
Aα (S)
Bγ
Aγ
Aβ (G)Aβ (S)
Bα
Cα
Aα (G)
Dβ
A
δC
65
60
70
80
90
55
75
85
ppm
Fβ Dα Dβ’
Dα’
Iγ
S2,6S’2,6
G6
ppm6.06.57.07.58.0δH
B
G2
G5
110
105
115
125
100
120
130
ppm
95
δC
D2
D6
S’’2,6
G’2
δH
3.03.54.04.55.05.5 ppm
MeO Cβ Bβ
Aα (S)
Bγ
Aγ
Aβ (G)Aβ (S)
Bα
Cα
Aα (G)
Dβ
A
δC
65
60
70
80
90
55
75
85
ppm
Fβ Dα Dβ’
Dα’
Iγ
δH
3.03.54.04.55.05.5 ppm
MeO Cβ Bβ
Aα (S)
Bγ
Aγ
Aβ (G)Aβ (S)
Bα
Cα
Aα (G)
Dβ
A
δC
65
60
70
80
90
55
75
85
ppm
Fβ Dα Dβ’
Dα’
Iγ
S2,6S’2,6
G6
ppm6.06.57.07.58.0δH
B
G2
G5
110
105
115
125
100
120
130
ppm
95
δC
D2
D6
S’’2,6
G’2
S2,6S’2,6
G6
ppm6.06.57.07.58.0δH
B
G2
G5
110
105
115
125
100
120
130
ppm
95
δC
D2
D6
S’’2,6
G’2

4. Resultados y discusión
55
El espectro HMBC de la muestra de lignina de E. globulus se muestra en la Figura 23. En los espectros HMBC se pueden observar diferentes señales de correlaciones (figura 23A) entre protones alquílicos (1.8-2.2 ppm) y los carbonos carboxílicos (170-175 ppm) asignadas a ácidos grasos libres o esterificados. Sin embargo, la información más interesante se encuentra en la región expandida δBHB 6-8 ppm (Figura 23B). Esta región muestra las correlaciones entre los protones HB2,6B de las unidades S (6.69 ppm) y los CB1 B, CB2,6 B, CB3,5 B, y CB4 B situados a uno o dos enlaces de distancia.
O
OCH3H3CO
O
HO
HO
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
A
5'
3'2'1' 6'
4'O
OCH3
OCH3O
O654
321
O
OCH3
H3CO
α'
β'
γ'α
β
γ
B
165
4 32
3'
1'6'5' 4'
2'
α
βγ OCH3
OHO
O OCH3
H3CO
C
O
OCH3H3CO
O
HO
O
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
F
165 4 3
2
OH
OCH3
O
H3CO
αβ
γ
EO
OCH3
Oα
β
γ OAr
OHHO
O
H3CO
1'6'5' 4' 3'
2'
α' β'γ'
OCH3
H3CO
1654 3
2
D
HO
OCH3H3CO
HO
HO
165 4 3
2
αβ
γ
4'
2'1'6' 5'
3'
O
OCH3
OCH3
165 4 3
2
OH
OCH3
O
H3CO
αβ
γ
I
O
OCH3
OH
165 4 3
2
α
G
O
OCH3
O
165 4 3
2
α
G’
O
OCH3H3CO
OH
165 4 3
2
α
S
O
OCH3H3CO
O
165 4 3
2
α
S’
O
OCH3H3CO
O
165 4 3
2
αHO
S’’
O
OCH3H3CO
O
HO
HO
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
A
O
OCH3H3CO
O
HO
HO
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
A
5'
3'2'1' 6'
4'O
OCH3
OCH3O
O654
321
O
OCH3
H3CO
α'
β'
γ'α
β
γ
B
5'
3'2'1' 6'
4'O
OCH3
OCH3O
O654
321
O
OCH3
H3CO
α'
β'
γ'α
β
γ
B
165
4 32
3'
1'6'5' 4'
2'
α
βγ OCH3
OHO
O OCH3
H3CO
C
165
4 32
3'
1'6'5' 4'
2'
α
βγ OCH3
OHO
O OCH3
H3CO
C
O
OCH3H3CO
O
HO
O
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
FO
OCH3H3CO
O
HO
O
H3CO
OCH316
5 4 32
2'
6'5'4' 3'
1'
αβ
γ
F
165 4 3
2
OH
OCH3
O
H3CO
αβ
γ
E
165 4 3
2
OH
OCH3
O
H3CO
αβ
γ
EO
OCH3
Oα
β
γ OAr
OHHO
O
H3CO
1'6'5' 4' 3'
2'
α' β'γ'
OCH3
H3CO
1654 3
2
DO
OCH3
Oα
β
γ OAr
OHHO
O
H3CO
1'6'5' 4' 3'
2'
α' β'γ'
OCH3
H3CO
1654 3
2
D
HO
OCH3H3CO
HO
HO
165 4 3
2
αβ
γ
4'
2'1'6' 5'
3'
O
OCH3
OCH3
HO
OCH3H3CO
HO
HO
165 4 3
2
αβ
γ
4'
2'1'6' 5'
3'
O
OCH3
OCH3
165 4 3
2
OH
OCH3
O
H3CO
αβ
γ
I
165 4 3
2
OH
OCH3
O
H3CO
αβ
γ
I
O
OCH3
OH
165 4 3
2
α
G
O
OCH3
OH
165 4 3
2
α
G
O
OCH3
O
165 4 3
2
α
G’
O
OCH3
O
165 4 3
2
α
G’
O
OCH3H3CO
OH
165 4 3
2
α
S
O
OCH3H3CO
OH
165 4 3
2
α
S
O
OCH3H3CO
O
165 4 3
2
α
S’
O
OCH3H3CO
O
165 4 3
2
α
S’
O
OCH3H3CO
O
165 4 3
2
αHO
S’’
O
OCH3H3CO
O
165 4 3
2
αHO
S’’
Figura 22. Subestructuras encontradas en los espectros HSQC a las que se hace referencia en la tabla 11.

4. Resultados y discusión
56
4.1.6 Conclusiones
La caracterización química detallada de las maderas de las diferentes especies de eucalipto ha puesto de manifiesto que:
• Existe una gran similitud entre las composiciones químicas de las diferentes especies, lo cual es lógico ya que todas son maderas de eucalipto.
• En cuanto al contenido de extraíbles lipofílicos, se observa que la especie E. globulus presenta el contenido más bajo, tanto globalmente, como de aquellos compuestos responsables de la formación de los depósitos de pitch. Ésta es una de las ventajas de utilizar la madera de E. globulus en la fabricación de pasta de papel frente a la de las otras especies.
• La madera de las especies de eucalipto estudiadas presentan lignina del tipo G y S, mientras que no se han detectado unidades del tipo H.
• En todas las especies de eucalipto estudiadas existe un mayor porcentaje de lignina S frente a lignina G, lo cual desde el punto de vista papelero es muy positivo, ya que la lignina de tipo S es más fácilmente degradable que la lignina de tipo G (Chang y Sarkanen 1973; Tsutsumi et al., 1995; González-Vila et al., 1999; del Río et al., 2005) lo que, a priori, las convierte en materias primas interesantes para la industría de la pasta y el papel. La especie E. globulus resultó ser la especie con una mayor relación S/G, lo que unido a su menor contenido en lignina, hace que sea la especie que presente un mayor rendimiento en la obtención de pasta de papel.
1.02.03.04.05.06.07.08.0 ppmppm
100
50
δH
δC
150
200
100
δH
δC
150
200
105
7.5 7.0 6.5 ppmppm
C
MeO
PEO-PPO
S2,6S’2,6
C2,6C1C4
C3,5
Cα (β-O-4’)BA
C3,5/HMeO
1.02.03.04.05.06.07.08.0 ppmppm
100
50
δH
δC
150
200
100
δH
δC
150
200
105
7.5 7.0 6.5 ppmppm
C
MeO
PEO-PPO
S2,6S’2,6
C2,6C1C4
C3,5
Cα (β-O-4’)BA
C3,5/HMeO
Figura 23. Espectro HMBC total de la lignina de E. globulus (A), región expandida δH 6-8 ppm del HMBC de la lignina de E. globulus (B), región expandida del HSQC de la lignina de E. globulus (C).

4. Resultados y discusión
57
4.2 UTILIZACIÓN DEL SLM PARA LA ELIMINACIÓN DE LOS LÍPIDOS RESIDUALES PRESENTES EN PASTAS DE DISTINTA NATURALEZA
El tratamiento con el sistema lacasa mediador afectó a todas las principales clases de lípidos encontrados en las diferentes tipos de pastas (Figura 24), como se puede observar por comparación con los correspondientes controles sin enzima. Sin embargo, no se observaron modificaciones durante tratamientos de la pasta con lacasa sola (sin el mediador).
4.2.1 Tratamientos de la pasta kraft de eucalipto con el SLM
Las abundancias de los principales compuestos lipofílicos presentes en la pasta control y la tratada enzimáticamente se muestran en la Tabla 12. Los esteroles en sus formas libres y conjugadas (como glicósidos y ésteres) resultaron ser los compuestos mayoritarios. Algunos hidrocarburos esteroidales y cetonas esteroidales, como la estigmasta-3,5-dien-7-ona y el 7-oxositosterol, se identificaron también junto con una serie de ácidos grasos libres. Los cromatogramas de la pasta control y la pasta tratada con el sistema lacasa-mediador se muestran en la Figura 25. La Tabla 12 muestra que el tratamiento con lacasa-HBT elimina casi en su totalidad los esteroles libres, ésteres de esteroles y el sitosteril-3-β-D-glucopiranósido. El sitosterol que representa más del 80 % de los esteroles libres en la pasta de eucalipto se eliminó en su totalidad, y solo una pequeña cantidad de estigmastanol permaneció en la pasta tratada. Por otro lado, se produjo un aumento en las cantidades de estigmasta-3,5-dien-7-ona, estigamastan-3-ona, y especialmente de 7-oxo-sitosterol, después del tratamiento de las pasta con el sistema lacasa-HBT. El sistema lacasa-mediador resultó ser muy eficiente en la eliminación de esteroles (95-100 % son eliminados), incluyendo sitosterol libre, sus glicósidos y sus ésteres con ácidos grasos, los cuales son los lípidos más característicos de la pasta de eucalipto. Estos compuestos proceden de los extraíbles lipofílicos presentes en la madera de eucalipto, los cuales sobreviven a la cocción y al blanqueo con oxigeno, dando lugar a los problemas de pitch en la pasta. Entre ellos, el estigmastanol (esterol saturado) resultó ser el más resistente a la acción del sistema lacasa-mediador que el sitosterol (esterol con una insaturación). La abundancia de las cetonas esteroidales en la pasta de eucalipto aumentó durante el tratamiento, lo que sugiere que éstas son productos oxidados de los esteroles. Las pequeñas cantidades de 7-oxositosterol que estaban presentes en la pasta de eucalipto inicial se formó problamente durante el pasteo y la deslignificación con oxígeno. El incremento de estos oxo-derivados en la pasta tratada es congruente con la naturaleza oxidativa del tratamiento con el SLM.

4. Resultados y discusión
58
Figura 24. Estructura química de los compuestos representativos de las principales clases de lípidos presentes en las pastas de papel analizadas: ácido palmítico (A), ácido dehidroabiético (B), n-octacosanol (C), campesterol (D), sitosterol (E), estigmastanol (F), 7-oxositosterol (G), estigmasta-3,5-dien-7-ona (H), sitosteril 3-β-D-glucopiranósido (I), 7-oxositosteril 3-β-D-glucopiranósido (J), sitosteril linoleato (K) y trilinoleina (L).
OH
O
A
COOH
B
OH
C
HO
D
HO
E
HO
F
HO OG
OH
O
OH
OOH
HO
CH2OH
I
OO
OH
OOH
HO
CH2OH
J
O
O K
CO-O-CH2
CO-O-CH
CO-O-CH2
L
OH
O
OH
O
OH
O
OH
O
A
COOH
BCOOHCOOH
B
OHOH
C
HOHOHO
D
HOHOHO
E
HOHO
F
HO OHO OG
OOOH
O
OH
OOH
HO
CH2OH
O
OH
OOH
HO
CH2OH
O
OH
OOH
HO
CH2OH
I
OO
OH
OOH
HO
CH2OH
OOO
OH
OOH
HO
CH2OH
O
OH
OOH
HO
CH2OH
J
O
O
O
O
O
O K
CO-O-CH2
CO-O-CH
CO-O-CH2
CO-O-CH2
CO-O-CHCO-O-CH
CO-O-CH2CO-O-CH2
L

4. Resultados y discusión
59
Tiempo de retención (min)
Tiempo de retención (min)2.5 5 7.5 10 12.5 15 17.5 20
Sitosterol
Sitosteril 3-β-D-glucopiranósido
Ésteres deesteroles
7-Oxositosterol
Ácidos grasos Estigmasta-3,5-dien-7-ona
100Re
spue
sta
rela
tiva
C-16C-14
C-18C-22
C-24 C-26C-28
C-20
C-18:1+
C-18:2
Hidrocarburosesteroidales
2.5 5 7.5 10 12.5 15 17.5 20
7-Oxositosterol
Estigmastanol+
Estigmasta-3,5-dien-7-onaEstigmastan-3-ona
7-Oxositosteril 3-β-D-glucopiranósido
100
Ácidos grasos
C-16
C-14 C-18 C-22 C-24C-26
C-20
B
A
Estigmastanol+
Fucosterol
Resp
uest
a re
lativ
a
Tiempo de retención (min)
Tiempo de retención (min)2.5 5 7.5 10 12.5 15 17.5 20
Sitosterol
Sitosteril 3-β-D-glucopiranósido
Ésteres deesteroles
7-Oxositosterol
Ácidos grasos Estigmasta-3,5-dien-7-ona
100Re
spue
sta
rela
tiva
C-16C-14
C-18C-22
C-24 C-26C-28
C-20
C-18:1+
C-18:2
Hidrocarburosesteroidales
2.5 5 7.5 10 12.5 15 17.5 20
7-Oxositosterol
Estigmastanol+
Estigmasta-3,5-dien-7-onaEstigmastan-3-ona
7-Oxositosteril 3-β-D-glucopiranósido
100
Ácidos grasos
C-16
C-14 C-18 C-22 C-24C-26
C-20
B
A
Estigmastanol+
Fucosterol
Resp
uest
a re
lativ
a
Tiempo de retención (min)2.5 5 7.5 10 12.5 15 17.5 20
Sitosterol
Sitosteril 3-β-D-glucopiranósido
Ésteres deesteroles
7-Oxositosterol
Ácidos grasos Estigmasta-3,5-dien-7-ona
100100Re
spue
sta
rela
tiva
C-16C-14
C-18C-22
C-24 C-26C-28
C-20
C-18:1+
C-18:2
Hidrocarburosesteroidales
2.5 5 7.5 10 12.5 15 17.5 20
7-Oxositosterol
Estigmastanol+
Estigmasta-3,5-dien-7-onaEstigmastan-3-ona
7-Oxositosteril 3-β-D-glucopiranósido
100100
Ácidos grasos
C-16
C-14 C-18 C-22 C-24C-26
C-20
B
A
Estigmastanol+
Fucosterol
Resp
uest
a re
lativ
a
Figura 25. Cromatogramas de los extraíbles lipofílicos de la pasta de eucalipto sin lacasa-mediador (A) y de la pasta tratada enzimáticamente (B).

4. Resultados y discusión
60
4.2.2 Tratamiento de la pasta TMP de picea con el SLM
Las abundancias de los principales compuestos lipofílicos presentes en la pasta de picea control y la pasta tratada enzimáticamente se muestran en Tabla 13. En la pasta TMP de picea, la composición de los extraíbles lipofílicos resultó ser diferente a la encontrada en la pasta de eucalipto, con presencia de grandes cantidades de triglicéridos, ésteres de esteroles y ácidos resínicos. Además, presenta pequeñas cantidades de ácidos (ácidos grasos y ácidos resínicos) y esteroles libres. Los cromatogramas de la pasta de picea control y la pasta tratada con el sistema lacasa-mediador se muestran en la Figura 26. Los valores que se observan en la Tabla 13 muestran que el tratamiento enzimático elimina la mayoría de los triglicéridos, ésteres de esteroles y esteril glicósidos. Se produce además una significativa eliminación de ácidos resínicos y de los esteroles libres. La incompleta eliminación de los compuestos anteriores podría ser debida a que el pasteado termomecánico tiene lugar bajo condiciones de oxígeno atmosférico. Finalmente los ácidos grasos y los alcoholes grasos se eliminaron en una menor extensión que los otros compuestos lipofílicos. Los compuestos oxidados 7-oxocampesterol y 7-oxositosterol, además de las respectivas cetonas doblemente insaturadas, la ergosta-3,5-dien-7-ona y la estigmasta-3,5-dien-7-ona aumentaron
Compuestos Pasta de eucalipto control Pasta de eucalipto tratada
enzimáticamente
Ácidos grasos totales 48.5 27.9 ácido mirístico (C 14:0) 2.1 1.7 ácido palmítico (C 16:0) 4.8 4.8 ácido linoleico (C 18:2) 2.9 0.7 acido oleico (C 18:1) 1.4 1.1 ácido esteárico (C 18:0) 2.7 2.1 ácido araquidónico (C 20:0) 2.1 0.7 ácido behénico (C 22:0) 6.9 3.4 ácido lignocérico (C 24:0) 9.4 4.2 ácido cerótico (C 26:0) 9.2 5.3 ácido montánico (C 28:0) 7.0 3.9 Hidrocarburos esteroidales totales 14.2 4.9 Esteroles libres totales 140.3 5.5 sitosterol 111.0 - estigmastanol 25.8 5.5 fucosterol 3.5 - Cetonas esteroidales totales 9.0 97.2 estigmastan-3-ona 0.6 3.6 estigamast-3,5-dien-7-ona 2.9 24.1 7-oxositosterol 5.5 69.5 Esteril glicósidos totales 17.1 6.9 sitosterol-3-β -D-glucopiranósido 17.1 - 7-oxositosteril-3-β-D-glucopiranósido - 6.9
Ésteres de esteroles totales
95.4
-
Tabla 12. Composición de los extraíbles lipofílicos de las pastas kraft de eucalipto, tanto de la utilizada como control como la tratada con el sistema lacasa-HBT (mg/Kg pasta).

4. Resultados y discusión
61
después del tratamiento enzimático, como también ocurre en la pasta de eucalipto tratada enzimáticamente.
Figura 26. Cromatogramas de los extraíbles lipofílicos de la pasta de picea sin lacasa-mediador (A) y de la pasta tratada enzimáticamente (B). Abreviaturas: Ah, alcohol; Alc, alcano.
Sitosterol
Campesterol
Ergosta-3,5-dien-7-ona
Estigmasta-3,5-dien-7-ona
Ah-24
Ácidos grasos y resínicos
TriglicéridosÉsteres deesteroles
B
7-Oxositosterol7-Oxocampesterol
100
Res
pues
ta re
lativ
a
2.5 5 7.5 10 12.5 15 17.5 20Tiempo de retención (min)
Triglicéridos
Ésteres de esteroles
Sitosterol
Campesterol
Alc-24
Ácidos grasos y resínicos
A100
Res
pues
ta re
lativ
a
Sitosteril3-β-D-glucopiranósido
Sitosterol
Campesterol
Ergosta-3,5-dien-7-ona
Estigmasta-3,5-dien-7-ona
Ah-24
Ácidos grasos y resínicos
TriglicéridosÉsteres deesteroles
B
7-Oxositosterol7-Oxocampesterol
100100
Res
pues
ta re
lativ
a
2.5 5 7.5 10 12.5 15 17.5 20Tiempo de retención (min)
2.5 5 7.5 10 12.5 15 17.5 20Tiempo de retención (min)
Triglicéridos
Ésteres de esteroles
Sitosterol
Campesterol
Alc-24
Ácidos grasos y resínicos
A100100
Res
pues
ta re
lativ
a
Sitosteril3-β-D-glucopiranósido

4. Resultados y discusión
62
4.2.3 Tratamiento de la pasta soda-AQ de lino con el SLM
La abundancia de los principales compuestos presentes en la pasta de lino control (pasta soda-AQ) y en la pasta de lino tratada se muestran en la Tabla 14. La composición de la fracción lipídica del lino resultó ser diferente a la de los otros tipos de pasta. Los principales compuestos, que se encontraron, resultaron ser los alcoholes grasos desde el docosanol hasta el dotriacontanol. Además, se encontraron grandes cantidades de esteroles libres y esteril glicósidos, mientras las cetonas esteroidales y las ceras se encontraron en una baja proporción. Los cromatogramas de la pasta de lino control y la pasta tratada con el sistema lacasa-mediador se muestran en la Figura 27. Los valores de la Tabla 14 ponen de manifiesto que el tratamiento con el sistema lacasa-mediador elimina completamente los esteroles libres (sitosterol, campesterol, y estigmasterol) inicialmente presentes en la pasta de lino, mientras algunas cantidades de
Compuestos Pasta de picea control Pasta de picea tratada enzimáticamente
Ácidos grasos totales 116.1 68.3 ácido palmítico (C 16:0) 26.6 1.5 ácido margárico ( 17:0) 21.0 14.1 ácido linoleico (C 18:2) 33.7 6.0 ácido oleico (C 18:1) 23.1 21.4 ácido esteárico (C 18:0) 11.7 5.3 Ácidos resínicos totales 1278.4 400.9 Alcoholes grasos totales 18.6 14.9 n-tetracosanol 17.2 13.7 n-hexacosanol 0.4 0.3 n-octocosanol 1.0 0.9 Esteroles libres totales 120.5 41.4 campesterol 30.4 8.0 sitosterol 90.1 33.4 Cetonas esteroidales totales 7.0 67.0 ergosta-3,5-dien-7-ona 2.0 8.0 estigamasta-3,5-dien-7-ona 3.3 27.0 estigmast-4-en-3-ona 1.7 1.7 7-oxocampesterol - 8.3 7-oxositosterol - 22.0 Esteril glicósidos totales 4.5 tr campesteril-3-β-D-glucopiranósido 0.7 - sitosteril-3-β-D-glucopiranósido 3.8 - 7-oxocampesteril-3-β-D-glucopiranósido - tr 7-oxositosteril-3-β-D-glucopiranósido - tr Ésteres de esteroles totales 1274.6 218.3 Triglicéridos totales 1982.5 151.2
Tabla 13. Composición de los extraíbles lipofílicos de la pasta de picea control y de la pasta tratada con el sistema lacasa mediador (mg/Kg pasta).

4. Resultados y discusión
63
los correspondientes compuestos oxidados (7-oxositosterol, 7-oxocampesterol, y 7-oxoestigamasterol, además de las respectivas cetonas di-insaturadas, ergosta-3,5-dien-7-ona y la estigamasta-3,5-dien-7-ona) aparecen en el tratamiento de la pasta. Asimismo, los tres esteril glicósidos (sitosteril, campesteril y estigmasteril β-D-glucopiranósidos), presentes en la pasta control, también se eliminaron completamente, y aparecieron pequeñas cantidades de los respectivos 7-oxoesteril glicósidos. Alcoholes grasos y n-alcanos se eliminaron en una significativa extensión, aunque éstos resultaron ser, comparativamente, más resistentes a la acción del sistema lacasa-mediador que el resto de los compuestos lipofílicos presentes en esta pasta. Además, una mezcla de picos, que no aparecían bien definidos en los cromatogramas, correspondientes a los antiespumantes usados en los procesos industriales, también se eliminaron durante el tratamiento con el sistema lacasa-mediador.
Tabla 14. Composición de los extraíbles lipofílicos de la pasta de lino control y de la pasta de lino tratada con el sistema lacasa-mediador (mg/Kg pasta).
Compuestos Pasta de lino control Pasta de lino tratada enzimáticamente
Antiespumante 849.5 125.2 Alcoholes grasos totales 121.3 49.7
n-docosanol 3.8 1.8 n-tetracosanol 4.0 1.5 n-hexacosanol 25.6 9.6 n-octocosanol 70.8 29.0 n-triacontanol 15.0 7.8 n-dotriacontanol 2.1 - Alcanos totales 12.8 8.0
n-heptacosano 2.7 1.5 n-nonacosano 8.1 5.5 n-hentriacontano 2.0 1.0 Esteroles libres totales 81.5 - campesterol 11.96 - estigmasterol 3.7 - sitosterol 6.2 - Cetonas esteroidales totales 2.4 19.1 ergosta-3,5-dien-7-ona 0.4 1.4 estigmasta-3,5-dien-7-ona 2.0 6.0 7-oxocampesterol - 2.9 7-oxoestigmasterol - tr 7-oxositosterol - 8.8 Esteril glicósidos totales 14.0 5.4
campesteril 3-β-D-glucopiranósido 2.4 - estigmasteril 3-β-D-glucopiranósido 0.8 - sitosteril 3-β-D-glucopiranósido 10.8 - 7-oxocampesteril 3-β-D-glucopiranósido - 0.9 7-oxoestigmasteril 3-β-D-glucopiranósido - tr 7-oxositosteril 3-β-D-glucopiranósido - 4.5 Ceras totales 2.6 0.6

4. Resultados y discusión
64
2.5 5 7.5 10 12.5 15 17.5 20Tiempo de retención (min)
Antiespumante
Ah-28Sitosterol
Ah-22Ah-24
Ah-26
Ah-30
Campesterol+
Estigmasterol
Ah-32Ceras
Esteril glicósidos
Estigmasta-3,5-dien-7-ona
Res
pues
ta re
lativ
a
100
2.5 5 7.5 10 12.5 15 17.5 20Tiempo de retención (min)
Ceras
7-Oxoesterilglicósidos
7-Oxositosterol
Ah-24Ah-26
Ah-30Ah-22
Ah-28
7-Oxocampesterol+
7-Oxoestigmasterol
Res
pues
ta re
lativ
a
100
Alc-29
B
A
2.5 5 7.5 10 12.5 15 17.5 20Tiempo de retención (min)
Antiespumante
Ah-28Sitosterol
Ah-22Ah-24
Ah-26
Ah-30
Campesterol+
Estigmasterol
Ah-32Ceras
Esteril glicósidos
Estigmasta-3,5-dien-7-ona
Res
pues
ta re
lativ
a
100100
2.5 5 7.5 10 12.5 15 17.5 20Tiempo de retención (min)
Ceras
7-Oxoesterilglicósidos
7-Oxositosterol
Ah-24Ah-26
Ah-30Ah-22
Ah-28
7-Oxocampesterol+
7-Oxoestigmasterol
Res
pues
ta re
lativ
a
100100
Alc-29
B
A
Figura 27. Cromatogramas de los extraíbles lipofílicos de la pasta de lino sin lacasa-mediador (A) y de la pasta tratada enzimáticamente (B). Abreviaturas: Ah, alcohol; Alc, alcano.

4. Resultados y discusión
65
4.2 INCLUSIÓN DE UNA ETAPA ENZIMÁTICA EN UNA SECUENCIA DE BLANQUEO TCF
La composición de los extraíbles lipofílicos se analizó primeramente en las pastas de la secuencia industrial TCF (O-O-a-Q-PoP), la cual incluía una etapa de control bajo las mismas condiciones de la etapa L pero sin enzima (etapa a) (Figuras 28 A-B). Los filtrados correspondientes también se analizaron, y las abundancias de los principales compuestos lipofílicos identificados en las pastas y en los líquidos se muestran en las Tablas 15 y 16 respectivamente. Se puede observar que la mayor parte de los lípidos identificados se encontraban en la pastas, mientras sólo una mínima fracción fue liberada a los líquidos.
Los esteroles libres, esteril glicósidos, y ésteres de esteroles (Figuras 29 A-C) resultaron ser los principales compuestos identificados en la pasta deslignificada con oxígeno y en los correspondientes líquidos, donde representan el 80 % de los lípidos totales (muestras O-O-a en las tablas 15 y 16) junto con cantidades menores de hidrocarburos esteroidales (Figura 29 D). El sitosterol resultó ser el esterol predominante tanto en su forma libre como conjugado. Por otro lado, algunos esteroles oxidados se encontraron en la pasta y en los líquidos. El análisis de las pastas y de los líquidos de filtrado al final del blanqueo industrial TCF (O-O-a-Q-PoP) reveló que el contenido de los extraíbles lipofílicos disminuía con respecto a la pasta de la secuencia O-O-a (con excepción del estigmastanol que permanecía prácticamente invariable), aunque la composición relativa no varió considerablemente tras las 2 etapas adicionales. El descenso en el contenido de extraíbles lipofílicos es debido más a la disolución/dispersión y
Figura 28. Análisis cromatográfico de los extraíbles lipofílicos (silanizados) de la pasta de eucalipto en diferentes etapas de la secuencia TCF con lacasa (derecha) y TCF control (izquierda): (A) pasta después del doble oxígeno y etapa de control (O-O-a), (B) pasta final después de la secuencia control completa (O-O-a-Q-PoP), (C) pasta después del doble oxígeno y la etapa de la lacasa-mediador (O-O-L), (D) pasta final obtenida tras la aplicación de la secuencia completa con lacasa-mediador (O-O-L-Q-PoP). Todos los cromatogramas corresponden a la misma cantidad de pasta.
D
min2.5 5 7.5 10 12.5 15 17.5 20
100 Sitosterol
Sitosteril-3-β-D-glucopiranósido
Ésteres de esteroles
7-OxositosterolHidrocarburos
esteroidales
Ácidos grasos
C-14C-16
C-18C-22
C-26
min2.5 5 7.5 10 12.5 15 17.5 20
100
Sitosterol
Sitosteril-3-β-D-glucopiranósido
Ésteres de esteroles7-OxositosterolHidrocarburos
esteroidales
C-16 C-22C-24
C-24
A100
min2.5 5 7.5 10 12.5 15 17.5 20
100
7-Oxositosterol
Estigmastan-3-onaEstigmasta-3,5-dien-7-ona
C
min2.5 5 7.5 10 12.5 15 17.5 20
7-Oxositosterol
Estigmasta-3,5-dien-7-onaEstigmastan-3-ona 7-Oxositosteril-3-β-D-
glucopiranósido
Estigmasta-3,5-dien-7-ona
C-16C-22 C-26
B D
min2.5 5 7.5 10 12.5 15 17.5 20
100 Sitosterol
Sitosteril-3-β-D-glucopiranósido
Ésteres de esteroles
7-OxositosterolHidrocarburos
esteroidales
Ácidos grasos
C-14C-16
C-18C-22
C-26
min2.5 5 7.5 10 12.5 15 17.5 20
100
Sitosterol
Sitosteril-3-β-D-glucopiranósido
Ésteres de esteroles7-OxositosterolHidrocarburos
esteroidales
C-16 C-22C-24
C-24
A100
min2.5 5 7.5 10 12.5 15 17.5 20
100
7-Oxositosterol
Estigmastan-3-onaEstigmasta-3,5-dien-7-ona
C
min2.5 5 7.5 10 12.5 15 17.5 20
7-Oxositosterol
Estigmasta-3,5-dien-7-onaEstigmastan-3-ona 7-Oxositosteril-3-β-D-
glucopiranósido
Estigmasta-3,5-dien-7-ona
C-16C-22 C-26
C
min2.5 5 7.5 10 12.5 15 17.5 20
7-Oxositosterol
Estigmasta-3,5-dien-7-onaEstigmastan-3-ona 7-Oxositosteril-3-β-D-
glucopiranósido
Estigmasta-3,5-dien-7-ona
C-16C-22 C-26
B

4. Resultados y discusión
66
eliminación de estos compuestos bajo condiciones alcalinas de la etapa PoP que a la propia acción del peróxido de hidrógeno. Este hecho se puso en evidencia tras la realización de un detallado balance de masa de los extraíbles lipofílicos presentes en las pastas, filtrados de blanqueo, y el total de los líquidos de lavado. Este balance reveló que la mayoría de esteroles libres y conjugados no se vieron afectados y sólo una pequeña cantidad de esteroles libres (un máximo del 8 %) se degradó en la etapa PoP.
HO
A
O
OH
O
OHHO
CH2OH B
O
OC
D
HOHO
A
O
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OH B
O
O
O
O
O
OC
D
Figura 29. Estructura de los principales compuestos presentes en las pastas O-O-a: sitosterol (A), Sitosteril 3-β-D-glucopiranósido (B), sitosteril linoleato (C) y estigmasta-3,5-dieno (D).

4. Resultados y discusión
67
Los análisis anteriores revelaron que los principales compuestos lipofílicos presentes en la pasta de eucalipto deslignificada con oxígeno y en los correspondientes líquidos de filtrado, principalmente sitosterol, sitosteril β-D-glucopiranósido y ésteres de sitosterol (Figuras 29 A-C), se eliminaron completamente (98%) por el tratamiento con el sistema lacasa-mediador. Los ácidos grasos presentes en la pasta también se eliminaron en una gran extensión. Solo algunos esteroides oxidados procedentes de la oxidación del sitosterol, principalmente estigmastan-3-ona, estigmasta-3,5-dien-7-ona y 7-oxositosterol (Figuras 30 A-C), fueron identificados en las pastas tratadas enzimáticamente (O-O-L y O-O-L-Q-PoP) junto con pequeñas cantidades de estigmastanol. Los esteroles oxidados en la pasta O-O-a, como 7 α- y 7β- y sitostanotriol (Figuras 30 D-E), fueron también eliminados tras el tratamiento con el sistema lacasa-mediador, y aparecieron pequeñas cantidades de 7-oxositosterol β-D-glucopiranósido (Figura 30 F). Es interesante apuntar que el estigmastanol (esterol saturado), el cual permanecía inalterado tras las etapas de oxígeno y de peróxido, fue degradado (hasta un 75%) por el tratamiento con la lacasa-mediador.
Los resultados obtenidos demuestran como un tratamiento enzimático podría ser integrado en secuencias de blanqueo más respetuosas con el medio ambiente (secuencias TCF), las cuales representan la tendencia actual en las industrias de este sector.
HO OH
HOOH
OH
HO O
B E
OO
OH
O
OHHO
CH2OHF
C
A D
O
O HO OH
HOOH
OH
HO OHHO OH
HOOH
OHHO
OHOH
HO OHO O
B E
OO
OH
O
OHHO
CH2OH
OOO
OH
O
OHHO
CH2OH
O
OH
O
OHHO
CH2OHF
C
A D
OOO
OO
Figura 30. Estructura de los principales compuestos presentes en las pastas tratadas enzimáticamente: estigmastan-3-ona (A),estigmasta-3,5-dien-7-ona (B), 7-oxositosterol (C), 7 α-hidroxisitosterol (D), sitostanotriol (E), y 7-oxositosteril 3-β-D-glucopiranósido (F).

4. Resultados y discusión
68
Compuestos O-O-a O-O-L O-O-a-Q-PoP O-O-L-Q-PoP Esteroles libres totales 140.3 5.5 98.2 3.9 sitosterol 103.1 - 69.3 - estigmastanol 34.4 5.5 28.1 3.9 fucosterol 2.8 - 0.8 Esteroles oxidados totales 11.9 98.5 11.7 49.3 estigmastan-3-ona 0.6 3.6 0.2 3.7 estigmasta-3,5-dien-7-ona 2.9 24.1 1.1 6.6 7α-hidroxisitosterol 0.8 0.2 2.2 0.2 7β-hidroxisitosterol 1.1 0.1 2.2 sitostanetriol 1.0 1.0 0.5 0.5 7-oxositosterol 5.5 69.5 5.5 38.3 Esteril glicósidos totales 17.1 6.9 5.1 0.9 sitosteril-3-β-D-glucopiranósido 17.1 - 5.1 - 7-oxositosteril-3-β-D- - 6.9 - 0.9 Ésteres de esteroles 95.4 - 41.0 - Hidrocarburos esteroidales 14.2 2.6 6.1 1.8 Esteroides totales 278.9 113.5 162.1 55.9 Ácidos grasos totales 44.5 18.1 3.9 0.7 ácido tetradecanoico (C 14:0) 2.1 1.7 - - ácido palmítico (C 16:0) 4.8 4.6 0.4 ácido linoleico (C 18:2) 2.9 0.7 - - ácido oleico (C 18:1) 1.4 1.1 - - ácido esteárico (C 18:0) 2.7 0.5 0.4 ácido eicosanoico (C 20:0) 2.1 0.5 0.2 ácido docosanoico (C 22:0) 6.9 2.5 1.2 0.5 ácido tetracosanoico (C 24:0) 9.4 3.0 1.5 0.2 ácido hexacosanoico (C 26:0) 9.2 2.8 0.2 ácido octacosanoico (C 28:0) 3.0 0.7 Lípidos totales 323.4 131.6 166.0 56.6
Tabla 15. Composición de los extraíbles lipofílicos de la pasta kraft de eucalipto en diferentes etapas de secuencias TCF, con lacasa (O-O-L-Q-PoP) y sin lacasa (control, O-O-a-Q-PoP) expresada en mg/Kg de pasta seca.
Abreviaturas: O, etapa de oxígeno; L, etapa lacasa-mediador; a, etapa control (etapa L sin lacasa ni mediador); Q, etapa de quelato; PoP, doble etapa de peroxido con un primer paso de oxigeno a presión; tr, trazas; y -, no detectado.
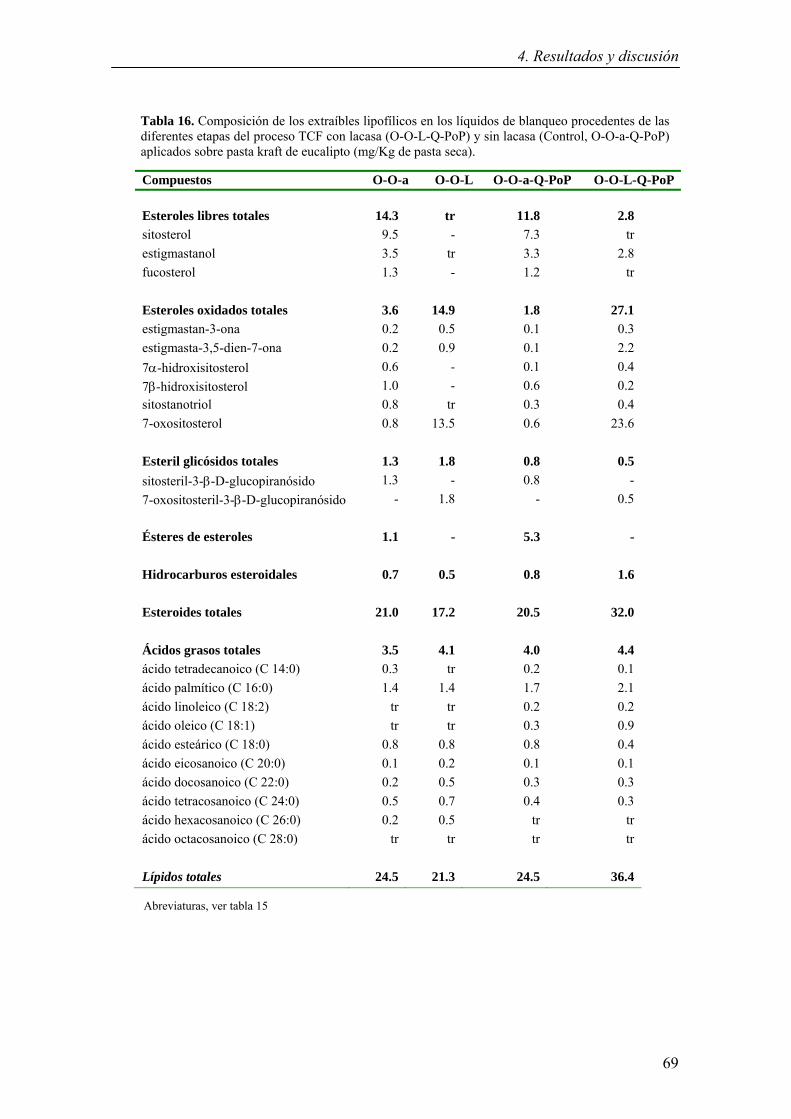
4. Resultados y discusión
69
Compuestos O-O-a O-O-L O-O-a-Q-PoP O-O-L-Q-PoP Esteroles libres totales 14.3 tr 11.8 2.8 sitosterol 9.5 - 7.3 tr estigmastanol 3.5 tr 3.3 2.8 fucosterol 1.3 - 1.2 tr Esteroles oxidados totales 3.6 14.9 1.8 27.1 estigmastan-3-ona 0.2 0.5 0.1 0.3 estigmasta-3,5-dien-7-ona 0.2 0.9 0.1 2.2 7α-hidroxisitosterol 0.6 - 0.1 0.4 7β-hidroxisitosterol 1.0 - 0.6 0.2 sitostanotriol 0.8 tr 0.3 0.4 7-oxositosterol 0.8 13.5 0.6 23.6 Esteril glicósidos totales 1.3 1.8 0.8 0.5 sitosteril-3-β-D-glucopiranósido 1.3 - 0.8 - 7-oxositosteril-3-β-D-glucopiranósido - 1.8 - 0.5 Ésteres de esteroles 1.1 - 5.3 - Hidrocarburos esteroidales 0.7 0.5 0.8 1.6 Esteroides totales 21.0 17.2 20.5 32.0 Ácidos grasos totales 3.5 4.1 4.0 4.4 ácido tetradecanoico (C 14:0) 0.3 tr 0.2 0.1 ácido palmítico (C 16:0) 1.4 1.4 1.7 2.1 ácido linoleico (C 18:2) tr tr 0.2 0.2 ácido oleico (C 18:1) tr tr 0.3 0.9 ácido esteárico (C 18:0) 0.8 0.8 0.8 0.4 ácido eicosanoico (C 20:0) 0.1 0.2 0.1 0.1 ácido docosanoico (C 22:0) 0.2 0.5 0.3 0.3 ácido tetracosanoico (C 24:0) 0.5 0.7 0.4 0.3 ácido hexacosanoico (C 26:0) 0.2 0.5 tr tr ácido octacosanoico (C 28:0) tr tr tr tr Lípidos totales 24.5 21.3 24.5 36.4
Abreviaturas, ver tabla 15
Tabla 16. Composición de los extraíbles lipofílicos en los líquidos de blanqueo procedentes de las diferentes etapas del proceso TCF con lacasa (O-O-L-Q-PoP) y sin lacasa (Control, O-O-a-Q-PoP) aplicados sobre pasta kraft de eucalipto (mg/Kg de pasta seca).

4. Resultados y discusión
70

5. REFERENCIAS
71

72 72

5. Referencias
73
Adler, E. (1977) Lignin chemistry - Past, present and future. Wood Sci. Technol. 11: 169-218. Affleck, R.R. y Ryan, R.G. (1969) Pitch control in a kraft pulp mill. Pulp Paper Mag. Can. 70 (12), T563–T567. Aitken Y., Cadel F. y Voillot C. (1988) «Constituants fibreux des pâtes, papiers et cartons. Pratique de l’analyse ». Editado por: Centre Technique de l’Industrie des Papiers, Cartons et Cellulose y l’Ecole Français de Papeterie et des Industries Graphiques, Grenoble. Allen, L.H. (1980) Mechanisms and control of pitch deposition in newsprint mills. Tappi J. 63(2), 81-87. Angelova, V., Ivanova, R., Delibaltova, V. y Ivanov, K. (2004) Bio-accumalation and distribution of heavy metals in fibre crops (flax, cotton and hemp). Ind. Crops Prod. 19,197-205. Anglés, M.N., Reguant, J., Martínez, J.M., Farriol, X., Montané, D. y Salvadó J. (1997) Influence of the ash fraction on the mass balance during the summative analysis of high-ash content lignocellulosics. Biores. Technol. 59, 185-193. Back, E.L. y Allen, L.H. (2000) Pitch Control, Wood Resin and Deresination. Tappi Press, Atlanta, GA. Bajpai, P. (1999) Application of enzymes in the pulp and paper industry. Biotechnol Progr 15:147–157. Balakshin, M.Y., Capanema, E.A., Chen, C.-L. y Gracz, H.S. (2003) Elucidation of the structures of residual and dissolved pine kraft lignins using an HMQC NMR technique. J. Agric. Food Chem., 51, 6116-6127. Behrendt C.J. y Blanchette R.A. (1997) Biological processing of pine logs for pulp and paper production with Phlebiopsis gigantea. Appl Environ Microbiol 63:1995–2000. Bidlack, J., Malonge, M. y Benson, R. (1992) Molecular structure and component integration of secondary cell walls in plants. Proc. Okla. Acad. Sci. 72: 51-56. Björkman, A. (1956) Studies on finely divided wood. Part I. Extraction of lignin with neutral solvents. SVen. Papperstidn, 13,477-485. Boerjan, W., Ralph, J. y Baucher, M. (2003) Lignin biosynthesis. Annu. Rev. Plant Biol. 54: 519-546. Bourbonnais R. y Paice M.G. (1990) Oxidation of non-phenolic substrates. An expanded role for laccase in lignin biodegradation. FEBS Lett 267:99–102. Brooks, R.T., Edgard, L.L., Nepote, J.C. y Caldwell, M.R. (1994) Bleach plants closeup and conversion to TCF: A case study using mill data and computer simulation. Tappi J. 77, 83-92.

5. Referencias
74
Calero-Rueda O, Plou FJ, Ballesteros A, Martínez AT, Martínez MJ (2002) Production, isolation and characterization of a sterol esterase from Ophiostoma piceae. BBA Proteins Proteom 1599:28–35. Call HP, Mücke I (1997) History, overview and applications of mediated lignolytic systems, especially laccase–mediator-systems (Lignozym(R)-process). J Biotechnol 53:163–202. Capanema, E. A.; Balakshin, M. Y.; Chen, C. L. An improved procedure for isolation of residual lignins from hardwood kraft pulps. Holzforschung 2004, 58, 464-472. Capanema, E. A.; Balakshin, M. Y.; Chen, C.-L.; Gratzl, J. S.; Gracz, H. Structural analysis of residual and technical lignins by 1H-13C correlation 2D NMR-spectroscopy. Holzforschung 2001, 55, 302-308. Capanema, E.A., Balakshin, M.Y., Kadla, J.F. (2005) Quantitative characterization of a hardwood milled wood lignin by nuclear magnetic resonance spectroscopy. J. Agric. Food Chem. 53:9639–9649. Chang, H.-M., Sarkanen, K.V. (1973) Species variation in lignin. Effect of species on the rate of kraft delignification. Tappi 56:132–134. del Río, J.C., Gutiérrez, A. y Martínez, A.T. (2004) Identifying acetylated lignin units in non-wood fibers using pyrolysis-gas chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 18: 1181-1185. del Río, J.C., Gutiérrez, A., González-Vila, F.J., Martín, F. y Romero, J. (1998) Characterization of organic deposits produced in the kraft pulping of Eucalyptus globulus wood. J. Chromatogr. A 823:457–465. del Río, J.C., Gutiérrez, A., Hernando, M., Landín, P., Romero, J. y Martínez, A.T. (2005) Determining the influence of eucalypt lignin composition in paper pulp yield using Py-GC/MS. J. Anal. Appl. Pyrolysis 74:110–115. del Río, J.C., Gutiérrez, A., Martínez, M.J. y Martínez, A.T. (2001b) Py-GC-MS study of Eucalyptus globulus wood treated with different fungi. J. Anal. Appl. Pyrolysis 58/59:441–453. del Río, J.C., Gutiérrez, A., Romero, J., Martínez, M.J. y Martínez, A.T. (2001a) Identification of residual lignin markers in eucalypt kraft pulp by Py-GC-MS. J. Anal. Appl. Pyrolysis 58/ 59:425–439. del Río, J.C., Romero, J. y Gutiérrez, A. (2000) Analysis of pitch deposits produced in kraft pulp mills using a totally chlorine free bleaching sequence. J. Chromatogr. A 874:235–245. del Río, J.C., Speranza, M., Gutiérrez, A., Martínez, M.J. y Martínez, A.T. (2002) Lignin attack during eucalypt wood decay by selected basidiomycetes: a Py-GC/MS study. J. Anal. Appl. Pyrolysis 64:421–431.

5. Referencias
75
Effland, M.J. (1977) Modified procedure to determine acid-insoluble lignin in wood and pulp. Tappi J. 6(10), 143-144. Elder, T. (1991) Pyrolysis of Wood. In wood and Cellulosic Chemistry. Hon, D.N.S., Shiraishi, N., Eds., Marcel Dekker, N.Y. Evershed, R.P., M.C. Prescott, N. Spooner y L.J. Goad. (1989) Negative ion ammonia chemical ionization and electron impact ionization mass spectrometric analysis of steryl fatty acyl esters. Steroids 53, 285–309. Evtuguin, D.V., Pascoal Neto, C., Silva, A.M.S., Domingues, P.M., Amado, F.M.L., Robert, D., Faix, O. (2001) Comprehensive study on the chemical structure of dioxane lignin from plantation Eucalyptus globulus wood. J. Agric. Food Chem. 49: 4252–4261. Faix, O., Meier, D. y Fortmann, I. (1990) Thermal degradation products of wood. A collection of electron of electron-impact (EI) mass spectra of monomeric lignin derived products. Holz Roh Werkst. 48:351–354. Fengel, D. y Wegener, G. (1984) Wood: Chemistry, ultrastructure, reactions. De Gruyter, Berlin. Fischer, K. y Messner, K. (1992) Reducing troublesome pitch in pulp mills by lipolytic enzymes. Tappi J 75:130–135. Fischer, K., Puchinger, L. y Schloffer, K. (1993) Enzymatic pitch control of sulfite pulp on pilot scale. J Biotechnol 27:341–348. Freire, C.S.R., Pinto, P.C.R., Santiago, A.S., Silvestre, A.J.D., Evtuguin, D.V. y Neto C.P. (2006) Comparative study of lipophilic extractives of hardwoods and corresponding ECF bleached kraft pulps. Bioresources 1:3–17. Freire, C.S.R., Silvestre, A.J.D. y Neto, C.P. (2002) Identification of new hydroxy fatty acids and ferulic acid esters in the wood of Eucalyptus globulus. Holzforschung 56:143–149. Freudenberg, K. y Neish, A.C. (1968) Constitution and biosynthesis of lignin. Springer-Verlag, New York. Fujita Y., Awaji H., Taneda H., Matsukura M., Hata K., Shimoto H., Sharyo M., Sakaguchi H. y Gibson K. (1992) Recent advances in enzymic pitch control. Tappi J 75:117–122. Gao, Y., Breuil, C. y Chen, T. (1994) Utilization of triglycerides, fatty acids and resin acids in lodgepole pine wood by a sapstaining fungus Ophiostoma piceae. Mater Org 28:105–118. García Hortal, J.A. y Colom, J.F. (1992) El proceso al sulfato. Vol. I. Universitat Politècnica de Catalunya, Terrassa (Spain).

5. Referencias
76
Gilarranz, M.A., Oliet, M., Rodríguez, F. y Tijero, J. (1999) Methanol-based pulping of Eucalyptus globulus. Can. J. Chem. Engin. 77: 515-521. González-Vila, F.J., Almendros, G., del Río, J.C., Martín, F., Gutiérrez, A. y Romero, J. (1999) Ease of delignification assessment of wood from different Eucalyptus species by pyrolysis (TMAH)-GC/MS and CP/MAS 13C-NMR spectrometry. J. Anal. Appl. Pyrolysis 49:295–305. Grabber, J. H., Quideau, S. y Ralph, J. (1996) p-Coumaroylated syringyl units in maize lignin: Implications for b-ether cleavage by thioacidolysis. Phytochemistry 43, 1189-1194. Gutiérrez, A., del Río, J.C., Martínez, M.J. y Martínez, A.T. (1999b) Fungal degradation of lipophilic extractives in Eucalyptus globulus wood. Appl Environ Microbiol 65:1367–1371. Gutiérrez, A., Romero, J. y del Río, J.C. (2001b) Lipophilic extractives from Eucalyptusglobulus pulp during kraft cooking followed by TCF and ECF bleaching. Holzforschung 55:260–264. Gutiérrez, A., del Río, J.C., González-Vila, F.J. y Romero, J. (1998a) Variation in the composition of wood extractives from Eucalyptus globulus during seasoning. J. Wood Chem. Technol. 18:439–446. Gutiérrez, A. y del Río, J.C. (2001) Gas chromatography/mass spectrometry demonstration of steryl glycosides in eucalypt wood, Kraft pulp and process liquids. Rapid Commun. Mass Spectrom. 15:2515–2520. Gutiérrez, A., del Rio, J.C., González-Vila, F.J. y Martín, F. (1998b) Analysis of lipophilic extractives from wood and pitch deposits by solid-phase extraction and gas chromatography. J. Chromatogr. A 823:449–455. Gutiérrez, A., del Río, J.C., González-Vila, F.J. y Martín, F. (1999) Chemical composition of lipophilic extractives from Eucalyptus globulus Labill. wood. Holzforschung 53:481–486. Gutiérrez, A., del Río, J.C., Ibarra, D., Rencoret, J., Romero, J., Speranza, M., Camarero, S., Martínez, M.J. y Martínez, A.T. (2006b) Enzymatic removal of free and conjugated sterols forming pitch deposits in environmentally sound bleaching of eucalypt paper pulp. Environ. Sci. Technol. 40: 3416-3422. Gutiérrez, A., del Río, J.C. y Martínez, A.T. (2004) Chemical analysis and biological removal of wood lipids forming pitch deposits in paper pulp manufacturing. In Methods in Biotechnology: Environmental Microbiology; Methods and Protocols ;pp 189-202. Gutiérrez, A., del Río, J.C., Martínez, M.J. y Martínez, A.T. (2001c) Biotechnological control of pitch troubles in paper pulp manufacturing. Trends Biotechnol. 19:340–348. Gutiérrez, A., del Río, J.C., Rencoret, J., Ibarra, D. y Martínez, A.T. (2006a) Main lipophilic extractives in different paper pulp types can be removed using the laccase-

5. Referencias
77
mediator system. Appl. Microbiol. Biotechnol., 72: 845-851. Gutiérrez, A., Romero, J. y del Río, J.C. (2001a) Lipophilic extractives in process waters during manufacturing of totally chlorine free Kraft pulp from eucalypt wood. Chemosphere 44:1237–1242. Gutiérrez, A., Romero, J. y del Río, J.C. (2001b) Lipophilic extractives from Eucalyptus globulus pulp during kraft cooking followed by TCF and ECF bleaching. Holzforschung 55: 260–264. Higuchi, T. (1997) Biochemistry and molecular biology of wood. Springer Verlag, London. Hillis, W.E (1962) Wood extractives. Academic press, London. Hillis, W.E. y Sumimoto, M. (1989) Effect of extractives on pulping. In: Natural Products of Woody Plants II; Ed. Rowe, J.W. Springer-Verlag, Berlin. pp. 880–920. Hon, D.N.S. y Shiraiski, N. (1991) Wood and cellulosic chemistry. Marcel Dekker, N.Y. Ibarra, D., Camarero, S., Romero, J., Martínez, M.J. y Martínez, A. (2006) Integrating laccase-mediator treatment into an industrial-type sequence for totally chlorine free bleaching eucalypt kraft pulp. J. Chem. Technol. Biotechnol. 81: 1159-1165. Kirk, T.K. y Cullen, D. (1998) Enzymology and molecular genetics of wood degradation by white-rot fungi. In: Environmentally friendly technologies for the pulp and paper industry. (Eds.: Young, R.A. y Akhtar, M.), TAPPI Press, Atlanta, pp. 273-308. Kontkanen, H., Tenkanen, M., Fagerstrom, R. y Reinikainen, T. (2004) Characterisation of steryl esterase activities in commercial lipase preparations. J Biotechnol 108:51–59. Lapierre, C., Monties, B. y Rolando, C. (1985) Thioacidolysis of Lignins Comparison with Acidolysis. J. Wood Chem. Technol. 5, 277-292. Leone, R. y Breuil, C. (1998). Filamentous fungi can degrade aspen steryl esters and waxes. Int. Biodeter. Biodegr. 41,133–137. Liitiä, T.M., Maunu, S.L., Hortling, B., Toikka, M. y Kilpeläinen, I. (2003) Analysis of technical lignins by two- and three dimensional NMR spectroscopy. J. Agric. Food Chem. 51, 2136-2143. Lu, F.C. y Ralph, J. (1997) Derivatization followed by reductive cleavage (DFRC method), a new method for lignin analysis: Protocol for analysis of DFRC monomers. J. Agric. Food Chem. 45, 2590-2592. Lu, F.C. y Ralph, J. (1997) DFRC method for lignin analysis. 1. New method for beta aryl ether cleavage: Lignin model studies. J. Agric. Food Chem. 45, 4655-4660.

5. Referencias
78
Lu, F.C. y Ralph, J. (1998) The DFRC method for lignin analysis. 2. Monomers from isolated lignins. J. Agric. Food Chem. 46, 547-552. Lusby, W.R., Thompson, M.J. y Kochansky, J. (1984). Analysis of sterol esters by capillary gas chromatography electron impact and chemical ionization-mass spectrometry. Lipids 19 (11), 888–901. Nimz, H. (1974) Beech lignin- Proposal of a constitutional scheme. Angew. Chem. Int. Edit. 13: 313-321. Ona, T., Sonoda, T., Itoh K. y Shibata, M. (1997) Relationship of lignin content, lignin monomeric composition and hemicellulose composition in the same trunk sought by their withintree variations in Eucalyptus camaldulensis and E. globulus. Holzforschung 51:396– 404. Örså, F. y Holmbom, B. (1994) A convenient method for the determination of wood extractives in papermaking process waters and effluents. J. Pulp Paper Sci. 20(12), J361–J366. Peng, G.M. y Robert, J.C. (2000) Solubility and toxicity of resin acids. Water Res. 34, 2779-2785. Pereira, A.S., Sequeira, D.S., Elias, V.O., Simoneit, B.R.T., Cabral, J.A. y Aquino Neto, F.R. (2002) Three series of high molecular weight alkanoates found in Amazonian plants, Phytochemistry 61, 711-719. Pouwels, A.D., Tom, A., Eijkel, G.B. y Boon, J.J. (1987) Characterization of beech wood and its holocellulose and xylan fractions by pyrolysis-gas chromatography-mass spectrometry. J. Anal. Appl. Pyrolysis 11, 417-436. Praham, R.A., Gray, R.L. y Rowell, R.M. (1984) “The chemistry of solid wood”, American Chemical Society, Washington DC. Ralph, J. y Hatfield, R.D. (1991) Pyrolysis-GC/MS characterization of forage materials. J. Agric. Food Chem. 39:1426–1437. Ralph, J., Marita, J.M., Ralph, S.A., Hatfield, R.D., Lu, F., Ede, R.M., Peng, J., Quideau, S., Helm, R.F., Grabber, J.H., Kim, H., Jimenez-Monteon, G., Zhang, Y., Jung, H.-J.G., Landucci, L., MacKay, J., Sederoff, R., Chapple, C. y Boudet, A.M. (1999) Solution-state NMR of lignin. In Advances in Lignocellulosics Characterization; Argyropoulos, D. S., Ed.; Tappi Press: Atlanta, pp 55-108. Ralph, S.A., Ralph, J. y Landucci, L. (2004) NMR Database of Lignin and Cell Wall Model Compounds; U.S. Forest Prod. Lab.: Madison, WI; http://ars.usda.gov/Services/ docs.htm?docid)10491H (accessed: July 2006). Rencoret, J., Gutiérrez, A. y del Río, J.C. (2006) Identification of three different tocopheryl ester series in wood extractives from several species of eucalyptus, In: Proceedings of the 9th European Workshop on Lignocellulosics and Pulp, Vienna, pp, 451–454.

5. Referencias
79
Rencoret, J., Gutiérrez, A. y del Río, J.C (2007) Lipid and lignin composition of woods from different eucalypt species. Holzforschung 61:165–174. Rodrigues, J., Meier, D., Faix, O. y Pereira, H. (1999) Determination of tree to tree variation in syringyl/guaiacyl ratio of Eucalyptus globulus wood lignin by analytical pyrolysis. J. Anal. Appl. Pyrolysis 48:121–128. Rolando, C.B., Monties, B. y Lapierre, C. (1992) Methods in lignin chemistry. In Springer Series in Wood Science; Lin, S. Y., Dence, C. W., Eds.; Springer-Verlag: Berlin, Germany; pp 334-349. Saiz-Jiménez, C., Boon, J.J., Hedges, J.I., Hessels, J.K.C y de Leeuw, J.W. (1987) Chemical characterization of recent and buried woods by analytical pyrolysis. Comparison of pyrolysis data with 13C NMR and wet chemical data. J. Anal. Appl. Pyrolysis 11, 417-436. Sarkanen, K.V. y Hergert, H.L. (1971) Classification and Distribution, In: Lignins – Occurrence, Formation, Structure and Reactions, Eds, Sarkanen, K.V., Ludwig, C.H. Wiley, New York, pp, 43–94. Shimizu, K. (2001) Chemistry of hemicelluloses; in: Wood and Cellulosic Chemistry. 2 nd edition, Eds. D.N.-S. Hon, N. Shiraishi, Marcel Dekker Inc., New York, USA, 177-214. Sigoillot, C., Camarero, S., Vidal, T., Record, E., Asther, M., Pérez-Boada, M., Martínez, M.J., Sigoillot, J.C., Asther, M., Colom, J. y Martínez, A.T. (2005) Comparison of different fungal enzymes for bleaching high-quality paper pulps. J Biotechnol 115:333–343 Silvestre, A.J.D., Pereira, C.C.L., Pascoal Neto, C., Evtuguin, D.V., Duarte, A.C., Cavaleiro, J.A.S. y Furtado, F.P. (1999) Chemical composition of pitch deposits from an ECF Eucalyptus globulus bleached kraft pulp mill: its relationship with wood extractives and additives in process streams. Appita J. 52:375–382. Sitholé, B.B., Sullivan, J.L. and Allen, L.H. (1992) Identification and quantitation of acetone extractives of wood and bark by ion exchange and capillary GC with a spreadsheet program. Holzforschung 46(5), 409–416. Sjöström, E. (1993) Wood chemistry. Fundamentals and applications. Academia Press, San Diego. Sun, R., Lawther, J.M. y Banks, W.B. (1997) A tentative chemical structure of wheat straw lignin. Ind. Crops Prod. 6: 1-8. Sun, R.C., Sun, X.F., Wang, S.Q., Zhu, W. y Wang, X.Y. (2002) Ester and ether linkages between hydroxycinnamic acids and lignins from wheat, rice, rye, and barley straws, maize stems, and fast-growing poplar wood. Ind. Crops Prod. 15: 179-188. Swan, B. (1967). Extractives of unbleached and bleached prehydrolysis-kraft pulp from

5. Referencias
80
Eucalyptus globulus. Svensk Papperstidn.70, 616–619. Swan, E.P. (1989) Health hazards associated with extractives, p. 931-952. In J. W. Rowe (ed.), natural products of woody plants II. Springer-Verlag. Tappi standard (2004) T222 om-88. 2004–2005 Tappi Test Methods.Tappi Press, Atlanta, GA. Tsutsumi, Y., Kondo, R., Sakai, K. y Imamura, H. (1995) The difference of reactivity between syringyl lignin and guaiacyl lignin in alkaline systems. Holzforschung 49:423–428. Wakeham, S.G. y Frew, N.M. (1982) Glass capillary chromatography-electron impact and chemical ionization-mass spectrometry. Lipids 17(11), 831–843. Yokoi, H., Ishida, Y., Ohtani, H., Tsuge, S., Sonoda, T. y Ona, T. (1999) Characterization of within-tree variation of lignin components in Eucalyptus camaldulensis by pyrolysis-gas chromatography. Analyst 124:669–674. Yokoi, H., Nakase, T., Ishida, Y., Ohtani, H., Tsuge, S., Sonoda, T. y Ona, T. (2001) Discriminative analysis of Eucalyptus camaldulensis grown from seeds of various origins based on lignin components measured by pyrolysis-gas chromatography. J. Anal. Appl. Pyrolysis 57:145–152.