la difracción de rayos x y la densidad...
TRANSCRIPT

La difracción de rayos X y ladensidad electrónica
Rafael Moreno EsparzaFacultad de Química
UNAM2007
Interpretación de lasestructuras cristalinas

• La cristalografía de rayos X es una de las másimportantes herramientas del análisis químico en laactualidad
• Su uso e importancia son muy clarosparticularmente para los químicos
• Pero ha creado un problema para quienes la usan sinser especialistas, es decir los químicos
• Especialmente al emplear la gran cantidad deinformación estructural acumulada a la fecha
• Los seres humanos y sin duda los químicos tenemosla tendencia a aceptar ciertas cosas implícitamente
• Una de ellas es la de las estructuras químicasobtenidas por medio de métodos cristalográficos
Introducción

• Como tendencia es comprensible, pero en realidades finalmente un acto de fe verdaderamenteconmovedor
• Pues como los mismos cristalógrafos admiten,muchas estructuras reportadas están plagadas deerrores
• Dado que, hoy por hoy son los químicos losprincipales usuarios de la parafernalia cristalográfica
• Resulta conveniente entonces, que tengan una guíacon las ideas primordiales que le permitan reconocerlas limitaciones y los alcances de las estructurascristalinas publicadas.
• Y a la vez les permita usar esta herramienta sinsufrimientos innecesarios
Introducción

• Considerando que este tema nos ha permitido generarun nuevo grupo que pretende estudiar la densidadelectrónica tanto desde el punto de vista teórico comodesde el punto de vista experimental
• Tomando en cuenta que este grupo cuenta con losexpertos en el lado teórico del problema, como quienestienen la experiencia en el manejo de los datosexperimentales
• Siendo químico he tenido que lidiar con este tipo desituaciones.
• Basado en algo de experiencia en el tema y en algunosartículos y revisiones he pretendido mostrar aquí comoenfrentarse primero al problema de la interpretaciónestructural y después al de la misma densidad
¿Por que estoy aquí?

• El experimento de difracción de monocristal• El valor de R• Desviación estándar estimada (esd)• Grupo espacial incorrecto• Correcciones de absorción• Los elipsoides térmicos• Los problemas con los átomos ligeros• Estructuras acéntricas• Datos insuficientes e interpretación errónea
Temas a tratar

• Este experimento lo llevamos a cabo empleandoun difractómetro
• El cual mide las intensidades (I) difractadas porun cristal y sus desviaciones estándar [σ (I)]
• Estas se reducen matemáticamente,obteniéndose la cantidad (F2) con suscorrespondientes desviaciones estándarasociadas [σ (F2)].
• Los antiguos difractómetros generaban además,empleando un programa de reducción de datos,lo que llamamos factores de estructura F.
El experimento de difracción de monocristal

• Con los datos colectados de ángulo de incidencia(hkl) y factores de estructura al cuadrado F2 consus correspondientes desviaciones estándarσ(F2)
• Haremos un ajuste de mínimos cuadrados nolineal (iterativo) a un modelo estructural de ladensidad ρ
• Obteniéndose como resultado los valores(ajustados/refinados) de las coordenadasatómicas y los factores de movimiento térmico,parámetros térmicos vibracionales o parámetrosde desplazamiento anisotrópico de los átomos.
El experimento de difracción de monocristal

• A pesar de que los datos publicados son cada vezmenos dependientes del error humano, puescada vez es necesario transcribir una menorcantidad de datos numéricos al hacer unapublicación
• El gran esfuerzo hecho por el CambridgeCrystallographic Data Center revisandoprácticamente todas las estructuras publicadas,obligando a los autores a corregirlas, haconvertido este problema en un mal menor.
• Aunque todavía es sorprendente el número depublicaciones que se presentan con este tipo deerrores
Los errores humanos

• Es por ello que es una muy buena práctica, quedespués de terminar de refinar una estructura ycompletar el procedimiento para publicarla,revisar cuidadosamente cada uno de los datosantes de enviarlos.
• Lo más conveniente es emplear los programasde revisión del CIF, CrystallographicInformation File que proporciona laInternational Union of Crystallography en sudirección electrónica:http://www.iucr.ac.uk/
Los errores humanos

• Las determinaciones de estructurascristalinas son cuantitativas.
• Su interpretación, su pertinencia y loscriterios de exactitud deben discutirsecuantitativamente.
• Los parámetros más importantesempleados para esto son:
índices de ajuste de los parámetrosdesviaciones estándar estimadasesquemas de ponderaciónrelación datos/parámetros
Los índices R

• El valor de R, es un criterio estadístico quediscutiremos adelante debido a su importancia.
• Las desviaciones estándar estimadas, son otrocriterio estadístico y que designaremos con sussiglas en inglés esd (estimated standarddeviation)
• Usualmente los parámetros atómicos seexpresan usando la forma: 1.554(6) Å
• Lo que significa que un enlace que mide 1.554 Åtiene una esd de 0.006 Å
Los índices R

• Existen otras dos cantidades Rint y Rsigma queindican la dispersión que presentan en los datosmedidos.
• Estas dos cantidades se expresan así:
• y
• No confundirlos con el índice R que indica labondad del modelo empleado y discutiremosdespués
Otros índices R
Rint=
FO
2 ! FO
2"F
O
2"
Rsigma
=! F
O
2( )"F
O
2"

• Sirve para poder cuantificar la cercanía delmodelo estructural propuesto a los datosexperimentales.
• A falta de un mejor nombre, le llamamos R• Los valores de R intentan expresar el acuerdo
entre los factores de estructura calculados pormedio del ajuste de nuestro modelo FC y losobservados experimentalmente FO.
• El valor convencional de R se expresa así:
El índice de ajuste R
R =F
O! F
C"F
O"

• La expresión anterior explica cual es la razón porla que deseamos un valor pequeño de R.
• Además nos indica cuantitativamente la bondadde un modelo
• A menor R mejor ajustada está la estructura• Desafortunadamente, existen una gran variedad
de trucos cristalográficos para reducirartificialmente el valor de la R
• Para poder ver a través de estos trucos, esnecesario que profundicemos un poco en elexperimento.
El índice de ajuste R

• Todos los cristales muestran patrones de difracción(difractogramas) con algunas reflexiones débiles(especialmente en ángulos de difracción 2θ altos)
• Estas reflexiones tienen un cociente I/σ(I) pequeño• En la práctica al inicio se omiten de los cálculos
aquellas reflexiones con valores de I/σ(I) menores aun cierto umbral, esto es en el intervalo de 1.5 a 3.0
• Con esto, el tiempo de cómputo requerido para elajuste se reduce y de esta manera el valor de R sereduce pues las reflexiones con esd grande seeliminan y el problema de tratar reflexiones conintensidades negativas se evita.
Las reflexiones débiles

• La primera ventaja prácticamente es innecesariadado el estado del cómputo actual, la segunda esmeramente cosmética y la tercera se ha resueltoal emplear las F2 en vez de las F
• Dado que R puede reducirse a un valorarbitrariamente bajo empleando un umbraladecuadamente alto, conviene tener otroscriterios de la exactitud de nuestra estructura queresulten ser más válidos que este parámetro.
• Uno de estos criterios es el del valor de Rponderado (en inglés weighted), cuyo símbolo esR’ o wR.
Las reflexiones débiles

• Los valores ponderados toman en cuenta eltamaño de los valores de σ (F), en el ajustedurante el refinamiento.
• Consideran que las reflexiones con valores de σ (F) pequeños son más exactos que aquellas cuyosvalores de σ (F) son grandes
• Es decir que las reflexiones de σ (F) pequeñapesan más que las de σ (F) grande.
• Así se define un factor (factor de peso o factor deponderación w), que nos permita, al multiplicarsepor la (F), disminuir la contribución de lasreflexiones con σ (F) grande.
Los esquemas de ponderación

• Existen una gran cantidad de esquemas deponderación entre los cuales uno de los másempleados es el siguiente:
• Considerándose que muchos de los erroressistemáticos dependen de manera aproximadamentelineal de la intensidad (I). wR se define así:
• Los valores de wR incorporan más información (laσ), en el ajuste
• Y son menos dependientes del umbral de σ empleado
Los esquemas de ponderación
w
!1 = "2F( ) + g F
2( )
wR =
w1
2 FO! F
C
"#$
%&'(
w1
2 FO(

• Pueden considerarse como más parecidas a larealidad, haciendo que Rw sea un mejor criteriode la exactitud del modelo que R.
• Estos esquemas, conducen a menores valores delas esd de las coordenadas atómicas
• Así por ejemplo SHELXL, refina y ajusta entérminos de la cantidad F2 en vez de F, aúncuando los datos de entrada sean F
• Porque el refinar contra todos los valores de F 2,es superior a refinar contra los valores de Fmayores a un umbral (digamos 4σ (F))
Los esquemas de ponderación

• Esto ocurre porque se incorpora más informaciónexperimental
• Los factores convencionales R están basados en Fde manera que su valor sea igual a cero paravalores negativos de I.
• La expresión del umbral F2 > 2s(F2) se usaúnicamente para calcular los índices tales comoRgt y no es relevante para la elección dereflexiones para el refinamiento.
• Adicionalmente la posibilidad de atorarse en unmínimo local se reduce.
Los esquemas de ponderación

• Cuando hay pseudosimetría, el empleo de lasreflexiones débiles permite discriminar entre dosposibles soluciones alternativas
• Es muy difícil refinar contra todos los valores de Fdebido a la dificultad que hay al estimar el valor de σ (F) a partir de σ (F2) o de un valor de cero o negativode F2 (error experimental del aparato).
• Una desventaja cosmética al refinar con F2 en vez derefinar con F es que los índices R basados en F2 sonestadísticamente dos y hasta tres veces mayores quelos obtenidos al emplear F y un umbral determinado
•• Los valores de este Los valores de este ííndice para todos los datos serndice para todos los datos seráánnaaúún mayores.n mayores.
Los esquemas de ponderación

• El valor del índice wR2 se expresa así:
• Cuyo esquema de ponderación es este:
• Donde:
Los esquemas de ponderación
wR2 =w F
O
2 ! FC
2( )2
"
w FO
2( )2
"
#
$%
&%
'
(%
)%
1 2
w
!1 = "2F
2( ) + aP( )2
+ bP
P =F
C
2+ F
O
2
3

• Es una buena idea utilizar el mismo factor depeso inicial durante todo el proceso de ajuste(WGHT 0.1 en el caso de SHELXL) hasta que elrefinamiento es casi completo y solo entoncesusar el esquema recomendado por el programa.
• Si el esquema se hace variar demasiado pronto, laconvergencia del programa puede verseperjudicada, pues algunas características delsistema (como los átomos faltantes) puedenponderarse negativamente, generando mínimoslocales o de plano espurios.
Los esquemas de ponderación

• Todas las esd (excepto las de los ángulos diedralesentre dos planos de mínimos cuadrados) seestiman usando la matriz de covarianciacompleta
• Las esd de la celda, también se toman enconsideración individualmente en la estimaciónde las esd de las distancias, ángulos y ángulos detorsión
• En el caso de la estimación de las esd de losángulos entre planos de mínimos cuadrados seemplea una aproximación isotrópica
Los esquemas de ponderación

• Otro criterio es el de la bondad de ajuste (eninglés goodness of fit), también basado en F2 elcual se expresa así:
Bondad de ajuste
GooF = S =w F
O
2 ! FC
2( )2
"n ! p( )
#
$
%%
&
'
((
1 2

• Considerando que la información estructural deun experimento de Rayos X está contenida en losparámetros atómicos ajustados y sus esd y no enla R, será necesario juzgar la exactitud de unaestructura cristalina en términos de sus esd y nosolamente en términos de R.
• Esto quiere decir que cuando se omitendemasiadas reflexiones, se tendería al mismotiempo a incrementar las esd de las coordenadasatómicas puesto que están relacionadas demanera inversamente proporcional a la raízcuadrada del número de reflexiones.
La relación datos/parámetros

• Algo relacionado directamente a lo anterior esque las esd serán mayores inevitablemente, si elcociente entre el número de reflexiones medido yel número de parámetros empleados en el ajustees demasiado pequeño .
• Normalmente para el caso de sustanciasorgánicas conviene hacer las mediciones a unángulo de difracción 2θ de al menos 45° para laradiación de Mo-Kα o de 100° para la de Cu-Kα.
• Las estructuras con 2θmax menor que el valorindicado o aquellas cuyo valor no se muestra,seguramente tienen problemas
La relación datos/parámetros

• De esta manera es claro que la relacióndatos/parámetros no debe ser menor a 8 y para teneruna estructura perfectamente determinada sesugiere que sea al menos de 20.
• A pesar de ello, es posible encontrar en la literaturaalgunas estructuras con valores inferiores a 5.
• Se ha argumentado convincentemente acerca de quela omisión de reflexiones débiles conlleva un errorsistemático y tendencioso (sesgado o biased en inglés)que a pesar de ser pequeño se refleja en losparámetros calculados.
• Se ha demostrado de manera concluyente que las esdse incrementan al omitir reflexiones débiles.
La relación datos/parámetros

• Por tanto, es evidente que los refinamientos másexactos se obtienen empleando todas las reflexionesque están por debajo de un cierto umbral (el menorposible).
• Desafortunadamente, el efecto en las esd resulta serrelativamente pequeño, de manera que es posibleconcluir que la práctica de omitir reflexionesprobablemente continuará. Así:La exactitud de una estructura deberá juzgarse por
medio de las esd de los parámetros atómicos obtenidosen vez únicamente emplear la R.
Los umbrales de reflexiones débiles altos, los valoresde 2θmax bajos o las relaciones datos/parámetrospequeñas pueden ser síntomas de estructuras conproblemas de interpretación o pobrementedeterminadas.
La relación datos/parámetros

• En una estructura orgánica determinadacuidadosamente y con exactitud a 25°C,únicamente puede ambicionarse que laslongitudes de enlace tengan una desviaciónestándar esd de 0.002 Å.
• Las esd obtenidas empleando los programasbasados en este método son correctasmatemáticamente, desafortunadamente se basansolamente en la información alimentada a lacomputadora: hkl, F2 y σ(F2) que como tales,tienen error.Es decir, las fuentes de error sistemático se ignoran,
siendo la fuente más seria de inexactitud, la mediciónde las constantes de la celda.
Desviación estimada estándar esd

• Los parámetros derivados de las intensidadesmedidas (I) en el proceso de ajuste de mínimoscuadrados, son las coordenadas fraccionales de losátomos, las cuales se calculan empleando los ejes dela celda unitaria medida
• Para obtener las longitudes de enlace (y lasdimensiones angulares moleculares) en unidadesabsolutas (Å o °), es necesario hacer las operacionesvectoriales apropiadas, considerando las dimensionesde la celda unitaria.
• Por ejemplo, para determinar la distancia (r) entredos átomos cuyas coordenadas fraccionales son (x1 ,y1 , z1) y (x2 , y2 , z2) en una celda definida por los ejesvectoriales
Las coordenadas fraccionales
!a ,!b y!c

• es necesario hacer las siguiente operación:
• donde la distancia de enlace está definida como:
Las coordenadas fraccionales
!r = x
2! x
1( )
!a + y
2! y
1( )
!b + z
2! z
1( )
!c
r =
!r !!r
!r
1
!r
2
!r
0
!a
!b
!c
x
1y
1z
1
x
2y
2z
2

• Por tanto, cualquier error en las dimensiones dela celda da como resultado que aparezcan erroresadicionales en los parámetros moleculares,mayores y por encima de los calculados en elajuste.
• En el caso de los átomos pesados, el errorsistemático en las longitudes de enlace puedellegar a ser mucho mayor que las esd obtenidaspor medio del ajuste de mínimos cuadrados.
Las coordenadas fraccionales

• Convencionalmente para obtener los valores delas constantes de la celda con un difractómetrode cuatro círculos, se requiere de ladeterminación exacta de las posiciones de variasreflexiones fuertes (usualmente 25) en términosde los ángulos de cada círculo (2θ, ω, χ y φ), paracon ellos refinar la celda inicial y así obtener losvalores refinados de las constantes de la celda.
• Como en cualquier ajuste de mínimos cuadrados,las esd se pueden estimar y típicamente están enel intervalo de 0.001 a 0.01 Å para un eje de 10Å, con errores equivalentes para los demás ejes.
Constantes de la celda y el “cero” del aparato

• Para determinar las posiciones exactas de cadauna de las reflexiones fuertes, es necesarioasegurarse que la medición de los ángulos seamuy precisa.
• Para ello, requerimos que el cero del aparato encada círculo sea 0.00, es decir es necesariocalibrar cada círculo y asegurar que el cero estéen posición.
• El problema del error sistemático apareceamenazante de nuevo, pues las longitudes de losejes son inversamente proporcionales al sen(θ)(ley de Bragg), un pequeño error en el cero delcírculo 2θ, causará errores sistemáticos serios enlas constantes de la celda.
Constantes de la celda y el “cero” del aparato

• Una estimación del tamaño de este tipo de error sepuede comprender con el siguiente ejemplo:
• Como se puede ver este error adicional es muchomayor que las esd calculadas.
• Debido a que los ceros en los círculos tienden a variaraún en los difractómetros mejor construidos y surecalibración es extremadamente tediosa, muy amenudo se olvidan (ignoran) estos errores, hasta quese empiezan a hacer incómodos para la propiacolección de datos.
Constantes de la celda y el “cero” del aparato
10.025Åeje calculadoca. 20°2θ de las reflexiones usadas
0.05°error en 2θ10 ÅTamaño del eje
MoKαTipo de radiación:

• En el ejemplo que hemos analizado antes, estoproduce errores sistemáticos adicionales de 0.004 Åen las longitudes de enlace.
• El 4-nitrofenil α-D-glucopiranósido.
• En un 1er reporte se indica queel grupo espacial para este sistemaes P21 en una celda:a = 28.810Å, b = 6.747Å, c = 6.729Å, β = 103.68°.
• En un 2º reporte independiente se obtiene el mismogrupo espacial pero con una celda diferente:a = 28.045Å, b = 6.767Å, c = 6.719Å, β = 90.30°.
Constantes de la celda y el “cero” del aparato

• Para tener valores equiparables es necesariotransformar la primera celda para obtener:a = 27.993Å, b = 6.747Å, c = 6.729Å, b = 90.17°
• Sin embargo, las diferencias entre las medicionesaún son difíciles de explicar (en b hay 10σ deerror).
• De manera, que es claro que las esd reportadaspueden ser extremadamente optimistas.
• Ello lo atestigua la aparición de una cuarta cifradecimal de vez en cuando, nivel en el cual laexpansión térmica a temperatura ambiente essignificativa.
Constantes de la celda y el “cero” del aparato

• Para tener valores equiparables es necesariotransformar la primera celda para obtener:a = 27.993Å, b = 6.747Å, c = 6.729Å, b = 90.17°
• Sin embargo, las diferencias entre las mediciones aúnson difíciles de explicar (en b hay 10σ de error).De manera, que es claro que las esd reportadas pueden
ser extremadamente optimistas.Ello lo atestigua la aparición de una cuarta cifra decimal
de vez en cuando, nivel en el cual la expansión térmica atemperatura ambiente es significativa.
Las esd de las dimensiones moleculares se debenincrementar para poder incluir los errores de lasconstantes de la celda.
Esto ya está instrumentado en algunos programas.
Constantes de la celda y el “cero” del aparato

• Es un hecho triste de la vida, que siendo seres humanos,hasta los cristalógrafos se equivocan de cuando en cuando,por ejemplo, asignando mal el grupo espacial a unaestructura.
• A veces, esto hace que sea imposible resolver la estructura,evitándose con ello que el error llegue a la literatura.
• En otras, la estructura se resuelve, se refina y se publica enun grupo espacial cuya simetría es innecesariamente baja.Es decir que uno o varios elementos de simetría se hanquedado sin reconocer.Puede ocurrir entonces que se hayan refinado dos
moléculas idénticas en vez de una y excepto por el gastode tiempo de computadora, no hay más problema, losparámetros atómicos obtenidos son confiables.
Grupo espacial incorrecto

• Cuando el elemento de simetría omitido es uncentro de simetría, las consecuencias serán muchomás serias, el refinamiento será lento y errático ylos parámetros obtenidos no serán confiables.
• Todo esto es inevitable y está basado en lasmatemáticas del álgebra matricial del propiorefinamiento.
• Para poder detectar este problema, nos convienepresentar algunos problemas típicos y una manerasencilla (aunque no infalible) de atacarlos yresolver el problema. Para ello necesitamos decierta familiaridad (aunque poca) con los gruposespaciales
Los elementos de simetría omitidos

• En general son muy pocos los químicos que seatreven a aventurarse en las recónditasprofundidades de las Tablas de grupos espaciales.
• La razón tiene que ver con el hecho de que losoperadores que relacionan moléculas son pocoimportantes químicamente en comparación con lasdimensiones de una molécula.
• Sin embargo, es muy ilustrativo para los legos, leeralguno de las revisiones acerca de grupos espacialesasignados incorrectamente.
• Aunque sea nomás para darse cuenta de donde sepuede uno equivocar.
Los elementos de simetría omitidos

• El óxido ternario X4YO3, tiene cuatrocaracterísticas que hacen que sospechemos de laasignación.
• Es muy improbable que las cuatro ocurran almismo tiempo.
1. el grupo espacial asignado es P1 y ello es muypoco usual en estos
2. Uno de los ángulos de la celda es 89.99°3. Hay dos unidades-fórmula independientes en la
unidad asimétrica.4. Existen relaciones aritméticas sencillas entre las
coordenadas de pares de átomos, lo cual por símismo sugiere un problema de simetría.
Los elementos de simetría omitidos

• La celda unitaria obtenida experimentalmentepuede transformarse de esta:a = 8.144 Å, b = 6.220 Å, c = 5.758 Å,α = 117.54°, β = 89.99°, γ = 111.24°
• a esta:a’ = 11.030 Å, b’ = 5.758 Å, c’ = 8.144Å,α’ = 90.0°, β’ = 114.10°, γ ’= 90.0°
• usando la siguiente matriz
Los elementos de simetría omitidos
T =
0 !2 !1
0 0 1
!1 0 0
"
#
$$$
%
&
'''

• Esta misma matriz transforma los índices dereflexión.
• Al hacer la transformación, las siguientesausencias sistemáticas aparecen: h0l, l impar y hkl,h+k impar.
• Estas corresponden a los grupos monoclínicos Cc yC2/c.
• Para transformar las coordenadas se emplea lamatriz inversa transpuesta:
Los elementos de simetría omitidos
T!1=
0 !0.5 0
0 !0.5 1
!1 0 0
"
#
$$$
%
&
'''

• Al trasladar a través del origen, los pares de átomos serelacionan por medio del operador (x, -y, 0.5+z).
• Esto demuestra que el grupo de este sistema es en realidadCc, lo cual se confirma cuando al refinar se obtiene unamejor R.
Los elementos de simetría omitidos
Átomo Primera asignación Asignación correcta x y z x’ y’ z’
X ! "#$$ "#%& "'#& X( "! $( "#%) "! &*
"#'! "()) "*&&
X* "'&$ ")(* "*&# X+ "(& ! ")(% "'*% "%)) "! #& "'$&
X% "*') "+#$ "((% X# "))! "+%' "%+$ "' '! "! %) "! ($
X' "#&) "(#) "&+) X) "! &) "(#) "#() ")## "*+$ ")$(
Y ! "$$$ "$$$ "$$$ Y( "%$$ "$$$ "*$( "$$$ ")+& "$$$
O ! ")%& "##& "&)# O( "*# ! "##& "&)# "! #% "$$* "#+$
O* "%+# "& ! ! "% %& O+ "$+' "&$& "#%& "*+$ "+)& "& !)
O% "%)* "*(( "*(# O# "$)( "*($ "*$+ "$+% "% %! "&%*

• El óxido hidratado H3M5O4, que fue publicadosuponiendo el grupo espacial P21/m y la siguientecelda unitaria:a = 5.548, b = 16.50, c = 5.519 y β = 107.0°.
• En este tipo de problemas (monoclínicos con a y ciguales o triclínicos con dos ejes iguales), se puedeconstruir una celda con un ángulo recto adicional(es decir pasar de monoclínico a ortorrómbico o detriclínico a monoclínico ) por medio de unatransformación matricial.
• En este caso la matriz:
Los elementos de simetría omitidos
T =
1 0 1
1 0 !1
0 1 0
"
#
$$$
%
&
'''

• Al aplicarse a los datos originales, genera lacelda siguiente:a = 6.565, b = 8.872 y c = 16.50,con todos los ángulos iguales a 90°.
• El análisis de las reflexiones en este caso nosmuestra que la estructura puede describirse enel grupo ortorrómbico Cmcm.
Los elementos de simetría omitidos

• En ninguno de los casos anteriores se haomitido el centro de simetría y en general serecomienda considerar lo siguiente:
Las dimensiones moleculares reportadas eranconfiables.
La mera existencia de una celda con dos o tresángulos rectos no implica necesariamente quedicha estructura pertenezca a esa clasecristalina.
Cuando es posible construir una celda con lasmétricas indicadas, pero ni las coordenadasatómicas ni los datos de intensidad observadosson coherentes con dicha transformación,entonces no necesariamente pertenecen a talred.
Los elementos de simetría omitidos

• El problema que representa decidir si un centro desimetría está presente o no (es decir, distinguir entrepares de grupos espaciales como P1, P-1; Pnma,Pna21) es un problema mucho más complicado.
• Al describir una estructura centrosimétrica con ungrupo espacial no centrosimétrico, puede conducir aque la R sea buena (hasta menor que cuando elsistema se describe en el grupo correcto), debido a queel número de parámetros en el refinamiento se duplica
• Esto es una consecuencia del viejo principio: “Dame elsuficiente número de parámetros y te ajusto undinosaurio” haciendo que la R no nos sirva comocriterio para definir la bondad de nuestro modelo.
Centro de simetría

• Es entonces menester recurrir a otros métodosque nos permitan solucionar nuestroproblema.
• Entre estos, se encuentran las pruebasestadísticas.
• Estas, si se emplean cuidadosamente, puedenresolver esta ambigüedad.
• Desafortunadamente su empleo descuidadopuede llevarnos a la conclusión equivocadaconduciendo ello a errores muy graves en losdatos publicados de dimensiones moleculares.
Centro de simetría

• La absorción de rayos-X está gobernada por lasiguiente ecuación:
• donde I/Ia representa la fracción de laintensidad del haz incidente, l es la longitud depaso del haz y µ es el coeficiente de absorciónde la sustancia sobre la que incide el haz.
• En los cristales orgánicos si empleamos unafuente de radiación de Mo-Kα, el valor de µ esdel orden de 0.1 mm-1, si además la l es menorque 0.8 mm, es razonable despreciar y nopreocuparse más del efecto de la absorción.
Correcciones de absorción
I
Ia
= e!µ l( )

• Sin embargo, la introducción de átomos pesadosen la sustancia examinada incrementasustancialmente el valor de µ.
• En casos extremos el valor de µl puede llegarhasta 10 (l = 0.1mm y µ = 110 mm-1).
• En estos aparecerán errores sistemáticos severosdebido a las longitudes de paso del cristalestudiado, para las diferentes geometrías dedifracción (ie. diferentes ángulos de reflexión).
• Algo derivado de este problema y que sin embargono se aprecia muy a menudo, es que aunquetuviéramos un cristal esférico perfecto, sepresentarán también errores sistemáticos serios.
Correcciones de absorción

¿Cuándo es imprescindible corregir los efectos deabsorción?
• Para responder a esta pregunta analizaremoslas consecuencias observadas cuando no seaplica la corrección de la absorción.
• Se ha demostrado que la absorción es mássevera cuando los valores de ángulo 2θ sonpequeños; así el decremento general de laintensidad al incrementar 2θ(independientemente de la absorción) secancela en cierta medida.
Correcciones de absorción

• Como este decremento general es causado por losmovimientos térmicos vibracionales de lamolécula, el comportamiento térmico aparente severá afectado artificialmente (decrecerá) debido alos efectos de absorción.
• En los casos más severos esto puede conducir alprograma a ajustar movimientos térmicosnegativos de un átomo
• Estos en jerga cristalográfica se conocen como:non positive definite, los cuales son parámetrosque claro, son físicamente imposibles.
Correcciones de absorción

• Otros efectos relacionados con los problemasde absorción, son:
Movimientos térmicos aparentes de losátomos muy anisotrópicos, cuando la propiaabsorción es anisotrópica, por ejemplo,cuando los cristales difractados tienen formade aguja o de lámina.
Aparición de picos espurios de gran densidadelectrónica cerca de los átomos pesados.
En el caso de tener demasiados picosespurios, la localización de los átomos ligerosse complica.
Sin embargo, las posiciones atómicas no seven demasiado afectadas.
Correcciones de absorción

¿Como corregir estos efectos?• Para contestar específicamente a esta pregunta, se
ha llegado al acuerdo general de que estos efectosempiezan a ser apreciables cuando µl > 0.4.
• Desafortunadamente, la aplicación de estascorrecciones era hasta hace poco un ejercicio muypoco trivial.
• Había de calcularse el tamaño y la forma delcristal difractor y después, se suponía unageometría idealizada manejable, e.gr. cilíndrica.
• Esto permitía calcular de la longitud de paso delhaz respecto a su ángulo de incidencia, para cadareflexión, empleando una serie de cálculos más omenos complicados.
Correcciones de absorción

• Si el cristal no exhibía caras regulares ysimples, el problema se volvía intratable.
• En la actualidad (¡vivan las computadoras y laestadística!), existen métodos más generalesy convenientes que resuelven el problema demanera rutinaria .
• Lamentablemente, se ha desperdiciado muchomás esfuerzo para justificar la ausencia decorrecciones de absorción que el requeridopara hacerlas.
• Estas justificaciones de lo injustificable puedentraducirse libremente así: sabíamos que los datostenían errores sistemáticos serios, pero no nosimportó y no hicimos nada al respecto.
Correcciones de absorción

Las estructuras cuyo producto entre elcoeficiente de extinción del cristal y el tamañomáximo de paso del haz (µl ) es mayor que 0.4,requieren corrección de absorción.
En general es factible determinar el valor de µl,pues tanto el coeficiente de extinción, como eltamaño del cristal se obtienen y publican concada estructura reportada.
La ausencia de la corrección de absorción en loscristales indicados (con átomos pesados ymayores a un cierto tamaño) reduce la exactitudde los parámetros obtenidos y cancela elsignificado de los parámetros térmicos.
Correcciones de absorción

• La representación convencional del movimientotérmico de un átomo, es la de un elipsoide dentrodel cual el átomo tiene una determinadaprobabilidad de encontrarse (generalmente del50%).
• Tales elipsoides están definidos por medio de loque se conoce como factores de temperatura yque se ajustan isotrópica o anisotrópicamente.
• Si se obtienen elipsoides grandes y muyanisotrópicos, es muy factible que representanmovimientos térmicos genuinos, por ejemplo, unacadena de C larga que no está estabilizada porinteracciones secundarias (puentes de hidrógeno,efecto hidrofóbico, etc.)
Los elipsoides térmicos

• Cuando los elipsoides son insólitamente grandesdeberán verse con cautela, ya que losmovimientos térmicos permiten ocultar muy bienlos errores sistemáticos. Hemos discutido una delas maneras en que esto ocurre (e.gr. despreciar laabsorción cuando no se debe).
• Sí el modelo ajustado muestra elipsoides queapuntan en la misma dirección, puede atribuirse aesta causa, existen además otras posibilidades:
Decaimiento del cristal durante el experimento. El cristal es mayor que el haz de Rayos X. El modelo empleado es erróneo. El grupo espacial no es el correcto.
Elipsoides y error sistemático

• Entre las causas mas probables de errores en elmodelo, se encuentra la posibilidad de que enel compuesto difractado existan átomosdesordenados, esto es, uno o más átomos sedeben promediar en varias posiciones,
ya sea porque hay rotación libre (PF6–, ClO4
–,etc.)
o bien porque existe rotación estática (dos o másposiciones posibles para un átomo, ej. gr. unracemato)
• En ambos casos el modelo mostrará un sitiopromedio, donde hay un átomo embarradoanisotrópicamente en una direcciónprivilegiada.
Elipsoides y error sistemático

• Cuando nuestro modelo muestra factoresde temperatura insólitos, debe considerarsesiempre la posibilidad del desorden.
• Ahora bien, la afirmación conversa, es decir,que los factores de temperatura normalesgarantizan la ausencia del desorden, no esnecesariamente cierta.
• En algunas ocasiones (afortunadamentepocas), la existencia de desorden puede pasardesapercibida si la estructura en cuestiónpresenta ciertas características.
Elipsoides y error sistemático
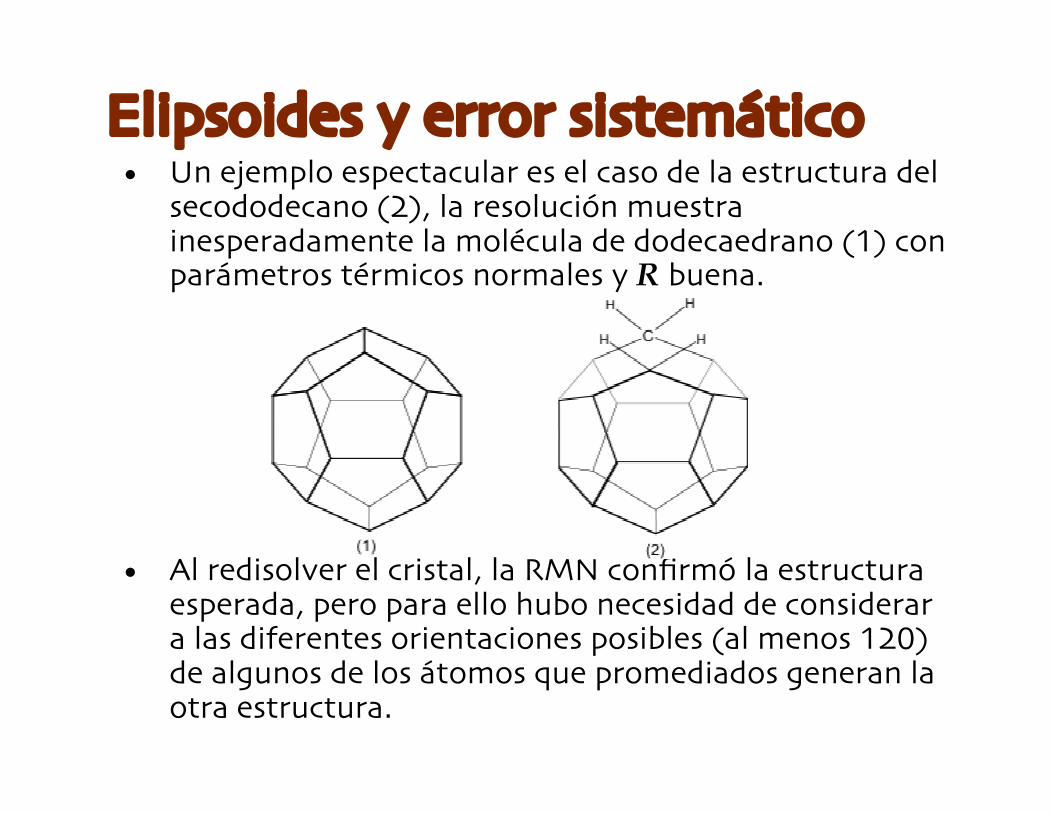
• Un ejemplo espectacular es el caso de la estructura delsecododecano (2), la resolución muestrainesperadamente la molécula de dodecaedrano (1) conparámetros térmicos normales y R buena.
• Al redisolver el cristal, la RMN confirmó la estructuraesperada, pero para ello hubo necesidad de considerara las diferentes orientaciones posibles (al menos 120)de algunos de los átomos que promediados generan laotra estructura.
Elipsoides y error sistemático

• La representación molecular de los movimientostérmicos por medio de elipsoides es una aproximación(como todas las representaciones) y tiene uninconveniente significativo.
• Surge del hecho de que un grupo terminal al moverse,más bien describe una especie de plátano que unelipsoide, el uso de los elipsoides produce unacortamiento sistemático de las longitudes de enlaceaparentes.
Elipsoides y error sistemático

• Este efecto se conoce con el nombre de oscilación (eninglés libration) y se presenta en metilos u otrosgrupos terminales o en los carbonilos unidos ametales.
• Obviamente no se le puede dar un gran significado alos datos donde se presenta este fenómeno si sepretende obtener correlaciones estructurales a partirde diferencias pequeñas (0.01 Å), pues los erroressistemáticos introducidos por la oscilación pueden sersignificativos e inclusive invalidar la discusión.
• Cuando el caso lo amerita, las correcciones a los datosque presentan oscilación, deben aplicarse, haciendo unanálisis detallado de los parámetros térmicosanisotrópicos.
Elipsoides y error sistemático

Los factores térmicos anisotrópicos grandespueden estar reflejando errores sistemáticos enlos datos de intensidad (absorción) o errores enel modelo estructural empleado (en particulardesorden atómico).
Los movimientos térmicos apreciables,especialmente si son perpendiculares a ladirección del enlace, causan un acortamientoaparente de la longitud del enlace determinadacristalográficamente.
De esta manera en el caso de estructuras dondese requiere de la máxima exactitud, la correcciónpor oscilación debe aplicarse, ya que si no sehace, las discusiones basadas en pequeñasdiferencias de las longitudes de enlace puedenser inválidas.
Elipsoides y error sistemático

• Los métodos cristalográficos de difracción derayos X, proveen una imagen de la densidadelectrónica del material estudiado.
• De esta manera, las posiciones de los átomoscon mayor número de electrones, se puedendeterminar con mayor precisión, esto quedasupuesto implícitamente en la primeraafirmación.
• Ahora bien, si la corrección de absorción seaplica adecuadamente, es factible localizar aúnen presencia de átomos pesados a todos losátomos no hidrogenoides sin dificultad.
Problemas con los átomos ligeros

• Sin embargo un problema muy serio es el de lalocalización de los átomos más ligeros de la tablaperiódica es decir, el H y sus congéneres ya quetodos tienen un solo electrón.
• Un electrón es difícil de localizar y además suposición aparente se verá modificada(dependiendo de la electronegatividad del átomoal que esté unido el H) más o menos hacia elátomo X en el enlace H-X.
• El análisis de los datos espectroscópicos y dedifracción de neutrones ha establecido que ladistancia C-H es de ca. 1.08 Å, en tanto que elmismo análisis de los datos de difracción de rayosX, establece que esta distancia es de únicamente0.96 Å.
Problemas con los átomos ligeros

• La diferencia entre las densidades electrónicascalculadas, con el modelo apropiado, del materialestudiado (cuando todos los átomos han sidoencontrados y refinados satisfactoriamente)respecto a las procedentes de nuestros datos(observadas), producen un mapa de densidadelectrónica que al examinarse muestra lascaracterísticas residuales de la densidadelectrónica del material en cuestión.
• Este mapa de densidad al examinarse y siempreque contemos con buenos datos, sentido común,buen juicio y un poco de suerte, nos indicará lasposiciones de la densidad electrónica en laestructura química propuesta.
Localización de H en presencia de átomos pesados

• Si la densidad observada está localizada en lasposiciones químicamente adecuadas, será factibleconsiderarla como átomos de H; aunque su ajuste serámuy impreciso, a menos que se hagan ciertasconsideraciones y se empleen algunas restricciones(fijar la distancia C-H en 0.96 y considerar que ladensidad electrónica del H es pequeña).
• Es muy importante aclarar que también en este casoes imprescindible hacer las correcciones apropiadas deabsorción, para minimizar los valores espurios dedensidad electrónica.
• Un método mucho más confiable que el de difracciónde rayos X para la localización de átomos de H, es el dela difracción de neutrones, donde las contribuciones ala difracción de los átomos de H no se ven eliminadaspor las contribuciones de los átomos pesados.
• Claro que para hacer difracción de neutrones serequiere de cristales grandes y desde luego, del accesoa un reactor atómico.
Localización de H en presencia de átomos pesados

• En vista de que el análisis del mapa de densidadresidual normalmente es tedioso y los datos que serequieren deben ser cuando menos muy buenos, esmuy complicado localizar todos los átomos de H enuna estructura empleando este método.
• Para hacer este problema menor, se han desarrolladométodos indirectos de localización. Donde, los sitiosprobables de H (agujeros en la estructura) se analizanen función de su energía potencial respecto a ladistancia al átomo al que se une.
• Basados en las curvas de interacción entre el protón yotros átomos, los sitios cuyas energías de interacciónson demasiado grandes o demasiado pequeñas seeliminan.
Localización de H en presencia de átomos pesados

• Los sitios restantes se asignan a los H.
• El éxito de esta técnica es impresionante y le ha dadola confianza necesaria para calcular las posiciones de Hen estructuras que no cuentan con datos deneutrones.
Localización de H en presencia de átomos pesados

• Si no hay desorden y los datos son moderadamenteexactos, los átomos de H aparecen con suficienteobviedad en el mapa de densidad electrónica una vezque se han encontrado todos los átomos nohidrogenoides, como para poderse asignar sindificultad.
• Sin embargo, la tendencia general en nuestros días,especialmente debido a que ya de por sí, las posicionesde los átomos de H son tan imprecisas, consiste enincluir en el modelo empleado un método delocalización geométrica, al que se le conoce comomodelo de cabalgata (riding model), que incorporaciertas restricciones (ejemplo, C-H 0.96Å, H-C-H109.5° y factores de temperatura fijos, para C sp3).
Localización de H en estructuras orgánicas

• De esta manera en la mayoría de las ocasiones, esinnecesario identificar los H en el mapa dediferencias de densidad electrónica, ya que susposiciones idealizadas se generan por el programa.
• Las consecuencias de esta actitud son claras, si losátomos de un compuesto (C, N, O, etc.) se hanidentificado erróneamente (quizá basados en unaestructura preconcebida), automáticamente lasposiciones de los átomos de H serán erróneastambién.
• Vale la pena añadir que este error puededetectarse con la aparición de varios síntomas.
Localización de H en estructuras orgánicas

• Entre estos síntomas se encuentran estas: los factores térmicos de los átomos
incorrectamente asignados resultan demasiadograndes o pequeños.
las distancias de enlace son anómalas los contactos de no enlace Y__H….X, son también
anómalos• Desafortunadamente, casi nadie revisa
cuidadosamente tales síntomas, nuevamentegana la idea preconcebida que se tiene de laestructura.
• Es muy factible suponer que una muchasestructuras publicadas presentan este tipo deincorrecciones.
Localización de H en estructuras orgánicas

• Los candidatos más obvios para estas anomalías,son sin duda los productos naturales de estructuradesconocida con ajustes de poca calidad.
Las posiciones de los átomos de H en estructurascon átomos pesados, pueden ser erróneas si nohay datos que las soporten (por ejemplo,cálculos de interacción de energía potencial vsdistancia).
Las posiciones de los átomos de H en lasestructuras orgánicas son muy probablementecorrectas, siempre y cuando se haya hecho unanálisis concienzudo del mapa de diferencias dedensidad electrónica.
El modo de enlace de los residuos con C/N/O esdifícil de determinar empleando únicamente latécnica de difracción de Rayos X.
Localización de H en estructuras orgánicas

• Las estructuras acéntricas (no centrosimétricas)tienen algunas peculiaridades que las hacenespeciales.
• Estas peculiaridades surgen debido a que nocumplen con la ley de Friedel, la cual puederesumirse así:
• La intensidad de una reflexión h, k, l debe ser iguala la de la reflexión h, k , l
• La razón por la cual no se cumple esta ley tiene quever con el fenómeno llamado dispersión anómalade Rayos X.
• Cuando la estructura estudiada únicamente tieneátomos ligeros, las diferencias de intensidad entreh, k, l y h, k , l resultan ser demasiado pequeñaspara poderse medir.
Estructuras acéntricas

• Cuando hay átomos pesados , un análisiscuidadoso de las diferencias entre lasreflexiones complementarias puede distinguirentre un modelo estructural y su imagenespecular.
• Es claro que en los materiales quirales estocorresponde a la determinación de laconfiguración absoluta.
• Claramente resulta ser una ventaja tener lacapacidad de determinar la configuraciónabsoluta de un compuesto empleando elmétodo de difracción de Rayos X.
Estructuras acéntricas

• Ahora bien, la afirmación de que: una estructura acéntrica que tiene átomos pesados
puede distinguirse de su imagen especular gracias ala dispersión anómala de Rayos X
• tiene su conversa: para cualquier estructura acéntrica que tenga
átomos pesados debe considerarse la dispersiónanómala
• que aunque igualmente clara, raramente setoma en cuenta.
Dispersión anómala

• Para considerar el fenómeno de dispersiónanómala usualmente se recurre a dos métodos:
1. Refinar el modelo inicial y un modelo alternativocon un cambio de signo en las coordenadas yaceptar el modelo con menor R. Nótese que lasdiferencias entre Rinicial y Ralternativo seránpequeñas.
2. Refinar un parámetro que multiplica loscomponentes imaginarios de las contribucionesde dispersión anómala de todos los átomos.Un valor cercano a +1 indicará que el modelo escorrecto, en tanto que un valor en la vecindad de-1 indicará que se deben cambiar los signos de lascoordenadas atómicas .
Dispersión anómala

Todas las estructuras acéntricas que tenganun átomo pesado deben de mencionar eltratamiento de la dispersión anómala que sele ha aplicado.
¿Que tan pesado es pesado? pues no mucho,la presencia de un átomo de P en unaestructura orgánica normalmente permitirála determinación de su configuraciónabsoluta.
Si estos efectos no se mencionan es posibleque los parámetros atómicos tenganasociados errores mucho mayores que las esdreportadas por el programa.
Dispersión anómala

• Además del ya mencionado problema del corteinadecuado del ángulo 2θ, una revisión cuidadosade los datos cristalográficos puede revelar que noestán presentes todos los datos independientes delsistema.
• Esto ocurre con algunos grupos de gran simetría ycuando el cristalógrafo no está familiarizado con elintervalo de datos únicos.
• En datos acéntricos es cuestión de gusto o detiempo de difractómetro si se colectan o no lasreflexiones opuestas de Friedel, especialmente en elcaso de estructuras orgánicas con átomos ligerosúnicamente.
Datos insuficientes, interpretación errónea

• Obviamente en este caso, sin estas reflexionesla determinación de la configuración absolutano será confiable o inclusive en ocasiones seráimposible de determinar.
• A pesar de lo ocupado que pueda estar undifractómetro, siempre será mejor colectar losdatos completamente, que suponer que elahorro de tiempo de difractómetro vale losuficiente como para sufrir las consecuencias deeste ahorro al no poder publicar la estructura.
Finis coronat opus
Datos insuficientes, interpretación errónea