la ciencia de materiales y la modelación computacional de...
TRANSCRIPT

LA CIENCIA DE MATERIALES Y LA
MODELACIÓN COMPUTACIONAL DE
ESTRUCTURAS Y PROCESOS
NANOSCÓPICOS
Luis Alberto Montero Cabrera Universidad de La Habana
Cuba, 2013

¿Qué es un “material”? La Wikipedia nos dice que los materiales
son elementos agrupados en un conjunto el cual es, o puede ser, usado con algún fin especifico.
Es obvio que los elementos de tal conjunto pueden tener naturaleza real (ser objetos materiales), naturaleza virtual (ser objetos ideales) o ser de naturaleza mixta.
Nota: nada más parecido a la definición de
sistema

¿Qué es un “material”? La Wikipedia nos dice que los materiales
son elementos agrupados en un conjunto el cual es, o puede ser, usado con algún fin especifico.
Es obvio que los elementos de tal conjunto pueden tener naturaleza real (ser objetos materiales), naturaleza virtual (ser objetos ideales) o ser de naturaleza mixta.
Nota: nada más parecido a la definición de
sistema

¿Qué es un “material”? La Wikipedia nos dice que los materiales
son elementos agrupados en un conjunto el cual es, o puede ser, usado con algún fin especifico.
Es obvio que los elementos de tal conjunto pueden tener naturaleza real (ser objetos materiales), naturaleza virtual (ser objetos ideales) o ser de naturaleza mixta.
Nota: nada más parecido a la definición de
sistema

En ciencias...
En las ciencias, un material es cualquier conglomerado de materia o masa.

En ingeniería...
En ingeniería, un material es una sustancia (elemento o, más comúnmente, compuesto químico o mezcla de ellos) con alguna propiedad útil, sea mecánica, eléctrica, óptica, térmica o magnética.

Ciencia de los materiales
La ciencia de los materiales es un campo interdisciplinario que involucra las propiedades de sustancias de potencial interés industrial y sus aplicaciones a varias áreas de la ciencia y la ingeniería.

8
¿Qué es un modelo?
Un modelo es la representación de cualquier objecto, hecho o creado para un propósito dado.
La Encyclopædia Britannica considera que un modelo es una descripción o analogía usada para ayudar a la visualización de algo (como podría ser un átomo) que no puede ser observado directamente.

9
¿Qué es un modelo?
Un modelo es la representación de cualquier objecto, hecho o creado para un propósito dado.
La Encyclopædia Britannica considera que un modelo es una descripción o analogía usada para ayudar a la visualización de algo (como podría ser un átomo) que no puede ser observado directamente.
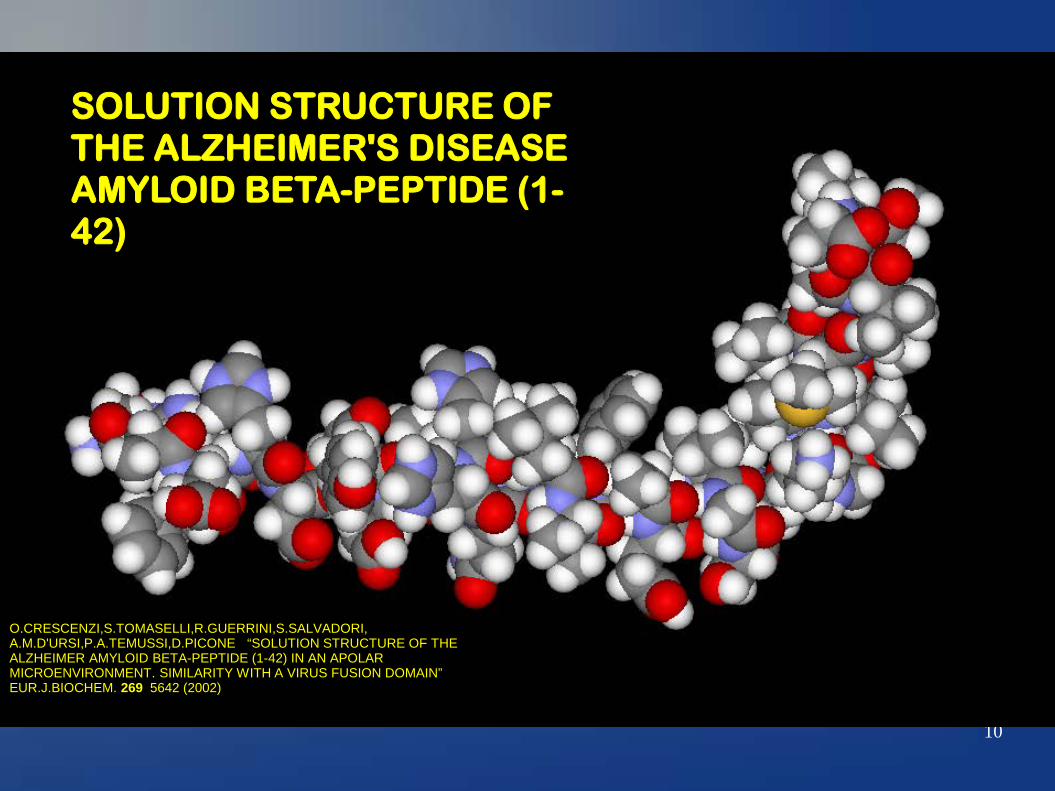
10
SOLUTION STRUCTURE OF THE ALZHEIMER'S DISEASE AMYLOID BETA-PEPTIDE (1-42)
O.CRESCENZI,S.TOMASELLI,R.GUERRINI,S.SALVADORI, A.M.D'URSI,P.A.TEMUSSI,D.PICONE “SOLUTION STRUCTURE OF THE ALZHEIMER AMYLOID BETA-PEPTIDE (1-42) IN AN APOLAR MICROENVIRONMENT. SIMILARITY WITH A VIRUS FUSION DOMAIN” EUR.J.BIOCHEM. 269 5642 (2002)

11
Modelación nanoscópica de materiales
El propósito fundamental de la modelación de materiales es construir modelos virtuales, perceptibles y confiables, de estructuras y procesos que existen esencialmente a escala nanoscópica.
La modelación de materiales trabaja con
herramientas desarrolladas en la química y física computacional.

12
Modelación nanoscópica de materiales
El propósito fundamental de la modelación de materiales es construir modelos virtuales, perceptibles y confiables, de estructuras y procesos que existen esencialmente a escala nanoscópica.
La modelación de materiales trabaja con
herramientas desarrolladas en la química y la física computacional.

13
La segunda ley de Newton y la modelación de materiales
Los modelos confiables de cualquier material a escala nanoscópica se basan en la consideración de que cualquier sistema poliatómico es más estable en las condiciones de mínima energía potencial (o energía interna):
02
2
==
∂∂−=
dtxdm
xVF
i
i

14
El estado físico macroscópico
Los materiales no tienen un estado físico preferente, pero se les suele considerar como sólidos o líquidos.
Es preferible calificarlos como ordenados o amorfos.

15
El estado físico macroscópico
Los materiales no tienen un estado físico preferente, pero se les suele considerar como sólidos o líquidos.
Es preferible calificarlos como ordenados o amorfos.

16
Gas, líquido, sólido
Los materiales gaseosos suelen ser siempre amorfos en ausencia de campos externos.
Los líquidos pueden ser ordenados, aunque predominan los amorfos
Los sólidos pueden ser ordenados, aunque existen sólidos amorfos y también muchos donde hay fases individualmente ordenadas que están distribuidas entre si de forma amorfa.

17
Gas, líquido, sólido
Los materiales gaseosos suelen ser siempre amorfos en ausencia de campos externos.
Los líquidos pueden ser ordenados, aunque predominan los amorfos
Los sólidos pueden ser ordenados, aunque existen sólidos amorfos y también muchos donde hay fases individualmente ordenadas que están distribuidas entre si de forma amorfa.

18
Gas, líquido, sólido
Los materiales gaseosos suelen ser siempre amorfos en ausencia de campos externos.
Los líquidos pueden ser ordenados, aunque predominan los amorfos
Los sólidos pueden ser ordenados, aunque existen sólidos amorfos y también muchos donde hay fases individualmente ordenadas que están distribuidas entre si de forma amorfa.

19
Polimorficos, amorfos y policristalinos
Los polimorficos son materiales predominantemente cristalinos de igual composición química pero diferente estructura nanoscópica, lo que les aporta diferencias en propiedades macroscópicas.
Los amorfos son materiales cuya estructura nanoscópica no es periódica, aunque puedan tener una composición química regular y constante (vidrio).
Los policristalinos son materiales donde coexisten estructuras cristalinas diversas, que pueden tener o no composición química diversa (acero).

20
Polimorficos, amorfos y policristalinos
Los polimorficos son materiales predominantemente cristalinos de igual composición química pero diferente estructura nanoscópica, lo que les aporta diferencias en propiedades macroscópicas.
Los amorfos son materiales cuya estructura nanoscópica no es periódica, aunque puedan tener una composición química regular y constante (vidrio).
Los policristalinos son materiales donde coexisten estructuras cristalinas diversas, que pueden tener o no composición química diversa (acero).

21
Polimorficos, amorfos y policristalinos
Los polimorficos son materiales predominantemente cristalinos de igual composición química pero diferente estructura nanoscópica, lo que les aporta diferencias en propiedades macroscópicas.
Los amorfos son materiales cuya estructura nanoscópica no es periódica, aunque puedan tener una composición química regular y constante (vidrio).
Los policristalinos son materiales donde coexisten estructuras cristalinas diversas, que pueden tener o no composición química diversa (acero).

22
Superficies de energía potencial
Para encontrar las estructuras moleculares más probables es necesaria una función que exprese la energía potencial total, o simplemente, la energía total del sistema, en términos del número y tipo de núcleos (Z) y de sus respectivas coordenadas espaciales, así como las de sus electrones:
Esta función se conoce como la superficie de
energía potencial (PES) del sistema o hipersuperficie.
),,( rRZEE =

23
Superficies de energía potencial
Para encontrar las estructuras moleculares más probables es necesaria una función que exprese la energía potencial total, o simplemente, la energía total del sistema, en términos del número y tipo de núcleos (Z) y de sus respectivas coordenadas espaciales, así como las de sus electrones:
Esta función se conoce como la superficie de
energía potencial (PES) del sistema o hipersuperficie.
),,( rRZEE =

24
El principal problema de los métodos para la modelación molecular es hallar la función
apropiada, analítica o numérica de tales hipersuperficies:
),,( rRZEE =
25
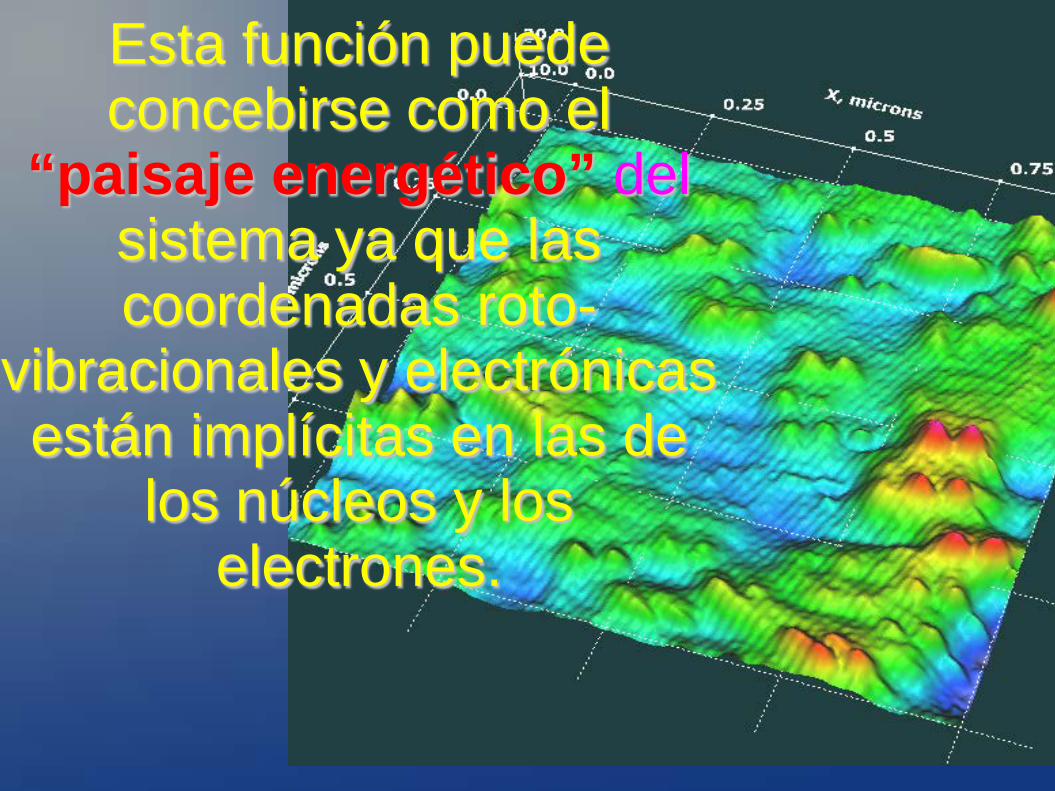
Esta función puede concebirse como el
“paisaje energético” del sistema ya que las coordenadas roto-
vibracionales y electrónicas están implícitas en las de
los núcleos y los electrones.
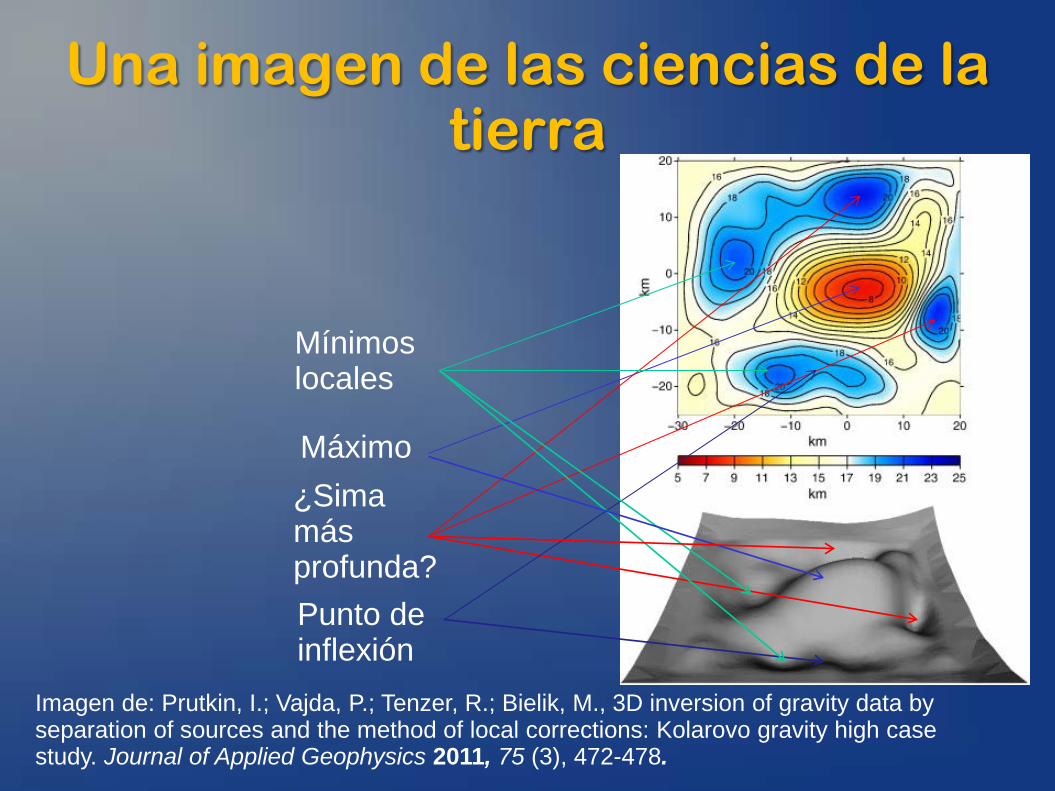
Una imagen de las ciencias de la tierra
Imagen de: Prutkin, I.; Vajda, P.; Tenzer, R.; Bielik, M., 3D inversion of gravity data by separation of sources and the method of local corrections: Kolarovo gravity high case study. Journal of Applied Geophysics 2011, 75 (3), 472-478.
Máximo ¿Sima más profunda? Punto de inflexión
Mínimos locales

27
La Mecánica Cuántica es la teoría física para los sistemas nano y
picoscópicos
La mecánica cuántica es la única teoría conocida hasta ahora que resulta válida a priori para la modelación y la descripción de los fenómenos nanoscópicos y picoscópicos, tal y como es el caso de las interacciones moleculares, los estados electrónicos, las densidades de carga y el enlace químico en los materiales.
Katsnelson, M. I., Graphene: carbon in two dimensions. Materials Today 2007, 10, (1-2), 20-27.

28
Hipersuperficies Cuánticas
Los modelos cuánticos son aquellos donde la hipersuperficie se calcula a partir de las functiones de onda de las partículas involucradas y suelen ser los más confiables si las funciones son bien obtenidas.

29
Hipersuperficies Cuánticas
Los modelos cuánticos son aquellos donde la hipersuperficie se calcula a partir de las functiones de onda de las partículas involucradas y suelen ser los más confiables si las funciones son bien obtenidas.
( )( )( )tr
trHE
tr
,
,ˆ,
Ψ
Ψ=
Ψ=Ψ

30
El reto “Simulations of materials behavior are an important
component of materials science research, partly because measurements are indirect, requiring
theoretical interpretation, and partly because often the ideal experiment simply cannot be performed (due to technological limitations). Empirical physical models used in this context often rely on parameters drawn
from experiments on simpler systems, and so introduce various inaccuracies. In contrast, a quantum mechanical model can potentially offer an independent
source of data more closely attuned to the complexities of the system at hand.” (Carter, E. A.,
Challenges in Modeling Materials Properties Without Experimental Input. Science 2008, 321, 800-803.)
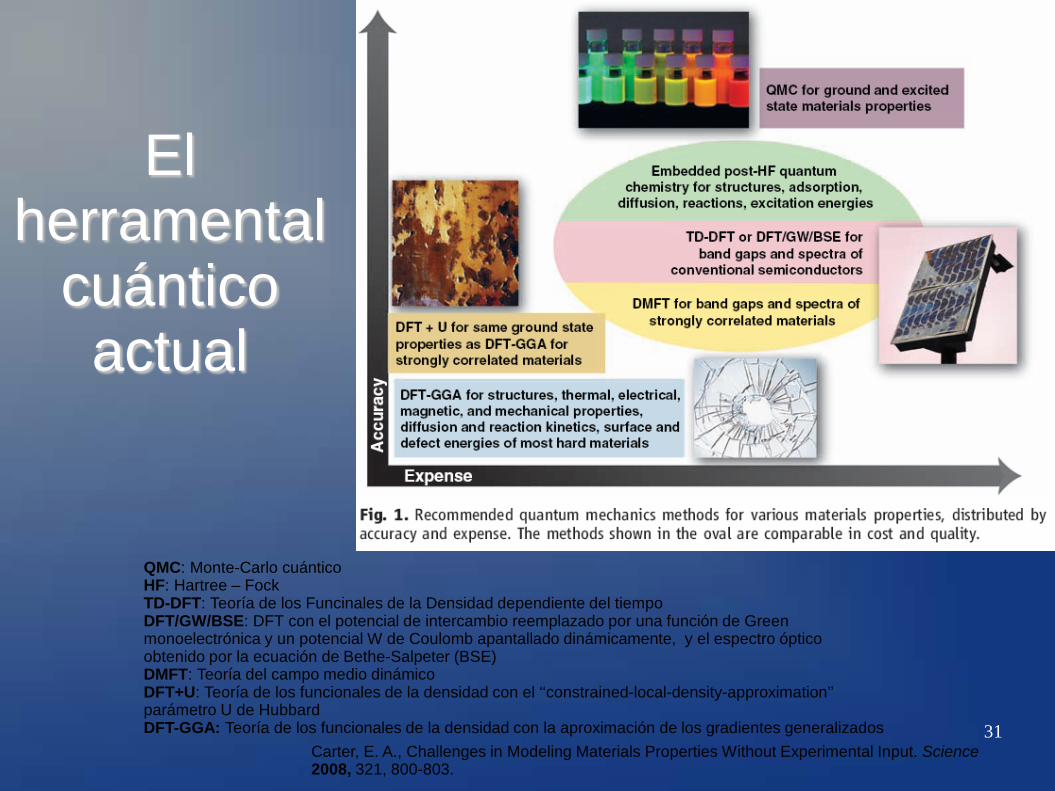
31 Carter, E. A., Challenges in Modeling Materials Properties Without Experimental Input. Science 2008, 321, 800-803.
QMC: Monte-Carlo cuántico HF: Hartree – Fock TD-DFT: Teoría de los Funcinales de la Densidad dependiente del tiempo DFT/GW/BSE: DFT con el potencial de intercambio reemplazado por una función de Green monoelectrónica y un potencial W de Coulomb apantallado dinámicamente, y el espectro óptico obtenido por la ecuación de Bethe-Salpeter (BSE) DMFT: Teoría del campo medio dinámico DFT+U: Teoría de los funcionales de la densidad con el ‘‘constrained-local-density-approximation’’ parámetro U de Hubbard DFT-GGA: Teoría de los funcionales de la densidad con la aproximación de los gradientes generalizados
El herramental
cuántico actual

32
¿Puede haber modelos clásicos?
Los modelos clásicos son aquéllos donde las hipersuperficies se construyen con funciones conocidas de la mecánica clásica que se “ajustan” para reproducir resultados experimentales o cálculos cuánticos previos confiables.

33
¿Puede haber modelos clásicos?
Los modelos clásicos son aquéllos donde las hipersuperficies se construyen con funciones conocidas de la mecánica clásica que se “ajustan” para reproducir resultados experimentales o cálculos cuánticos previos confiables.
( )( )
( )tRftrtrHE
,,,ˆ
≈ΨΨ
=

34
Geometrías moleculares “optimizadas”
El valor del conjunto de coordenadas que arroja la energía mínima E0 para todo el sistema se conoce como la geometría optimizada.
Se trata del “pozo” o “valle” más profundo
del paisaje energético del material.
[ ]eR

35
Geometrías moleculares “optimizadas”
El valor del conjunto de coordenadas que arroja la energía mínima E0 para todo el sistema se conoce como la geometría optimizada.
Se trata del “pozo” o “valle” más profundo
del paisaje energético del material.
[ ]eR

36
Hipersuperficie de la molécula de H2 de acuerdo con un potential de Morse
-1 0 1 2 3 4 5 60
1
2
3
4
5
6
r (10-10 m)
E (e
v)
E=DH2(1-e-1.1(0.74-r))2

37
Las hipersuperficies poliatómicas son mucho más complejas
Katsnelson, M. I., Graphene: carbon in two dimensions. Materials Today 2007, 10, (1-2), 20-27.
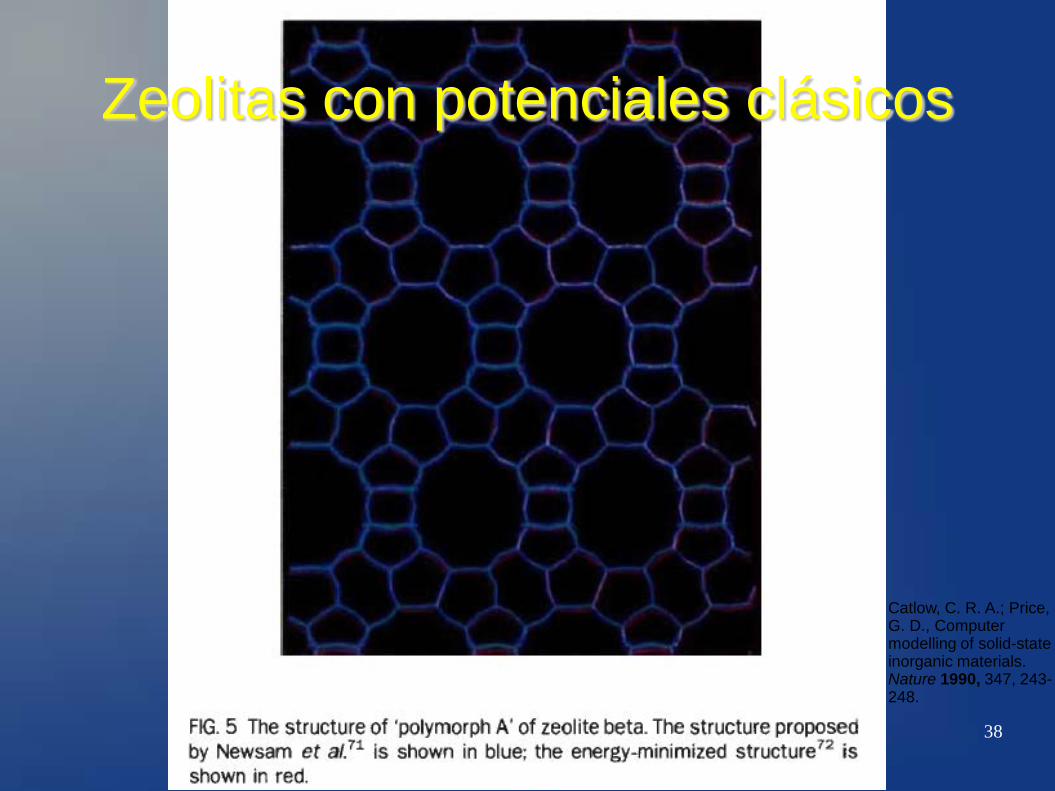
38
Zeolitas con potenciales clásicos
Catlow, C. R. A.; Price, G. D., Computer modelling of solid-state inorganic materials. Nature 1990, 347, 243-248.

39
Múltiples mínimos
Los mínimos globales de cada hipersuperficie
corresponden a la energía optimizada, ya sea cuántica o clásica.
Las hipersuperficies de los sistemas poliatómicos
pueden mostrar también uno o muchos otros minimos locales o secundarios, que representan geometrías alternativas del sistema, que pueden ser algo menos estables, aunque son capaces de ocupar un sitio en el espacio de configuraciones estadístico con poblaciones significativas.

40
Múltiples mínimos
Los mínimos globales de cada hipersuperficie
corresponden a la energía optimizada, ya sea cuántica o clásica.
Las hipersuperficies de los sistemas poliatómicos
pueden mostrar también uno o muchos otros minimos locales o secundarios, que representan geometrías alternativas del sistema, que pueden ser algo menos estables, aunque son capaces de ocupar un sitio en el espacio de configuraciones estadístico con poblaciones significativas.

41
La Entropía en los modelos nanoscópicos
La modelación de un sistema molecular aislado o de un cristal se suele realizar con el mínimo global o absoluto de la hipersuperficie.
En tales casos, la entropía estructural del
sistema macroscópico correspondiente está ausente.

42
La Entropía en los modelos nanoscópicos
La modelación de un sistema molecular aislado o de un cristal se suele realizar con el mínimo global o absoluto de la hipersuperficie.
En tales casos, la entropía estructural del
sistema macroscópico correspondiente está ausente.

43
La Entropía en los modelos nanoscópicos
Sin embargo, la gran mayoría de los sistemas
moleculares o los materiales se comportan macroscópicamente como sistemas reales que presentan múltiples mínimos locales con probabilidades (y poblaciones) significativas cada uno, además del mínimo global.
Esto es equivalente a tomar en consideración
la entropía del sistema cuando se desea proyectar los modelos a la realidad.

44
La Entropía en los modelos nanoscópicos
Sin embargo, la gran mayoría de los sistemas
moleculares o los materiales se comportan macroscópicamente como sistemas reales que presentan múltiples mínimos locales con probabilidades (y poblaciones) significativas cada uno, además del mínimo global.
Esto es equivalente a tomar en consideración
la entropía del sistema cuando se desea proyectar los modelos a la realidad.

45
¿Qué tipos de casos están determinados por la entropía de
asociación?
Existen evidencias teóricas (y lógicas) de que las asociaciones entre un soluto hidrofóbico y el agua están determinadas por la entropía y no por la entalpía de asociación.

46
¿Qué tipos de casos están determinados por la entropía de
asociación? El caso de simulaciones de dinámica clásica de
la disolución de láminas planas de grafenos en agua arrojan lo siguiente:
Choudhury, N.; Pettitt, B. M., Enthalpy-Entropy Contributions to the Potential of Mean Force of Nanoscopic Hydrophobic Solutes. J. Phys. Chem. B 2006, 110, 8459-8463.

47
Una primera proposición
La primera proposición de este trabajo es la de que los materiales en cualquier estado físico requieren de una modelación que explore TODO el paisaje energético si se desea proyectar la modelación nanoscópica al macrouniverso al igual que los sistemas moleculares menos “atados” entre si.

48
Buscando los múltiples mínimos...
La identificación de los múltiples mínimos o valles en una hipersuperficie o paisaje energético solo se puede realizar comenzando desde una serie perfectamente aleatoria de configuraciones espaciales iniciales de las especies canónicas (moléculas libres, cristalitos, monómeros, etc) de cada material.
Estas configuraciones se pueden optimizar posteriormente o tomarse un número elevado de ellas para lograr un conjunto representativo una vez que se evalúen sus energías.

49
Buscando los múltiples mínimos...
La identificación de los múltiples mínimos o valles en una hipersuperficie o paisaje energético solo se puede realizar comenzando desde una serie perfectamente aleatoria de configuraciones espaciales iniciales de las especies canónicas (moléculas libres, cristalitos, monómeros, etc) de cada material.
Estas configuraciones se pueden optimizar posteriormente o tomarse un número elevado de ellas para lograr un conjunto representativo una vez que se evalúen sus energías.

50
Los estados accesibles de un material
El número de estados accesibles o más poblables que caracterizan a cualquier material policristalino o amorfo puede evaluarse clásicamente según la función de partición del espacio de configuraciones explorado:
donde la ∆εi se refiere a las diferencias de
energía de cada configuración con respecto a la de “no interacción” entre las especies canónicas del material.
∑−
=i
RT*i
eqε∆

51
Las funciones macroscópicas del material
Si R es la constante de los gases y T la temperatura absoluta, tendremos que las funciones macroscópicas del material referido a su composición será:
*
*2 '
qqRTEEE assocref ==−
*
** 'ln
qqRTqRSSS assocref +==−
*ln qRTAEA assocref −==−
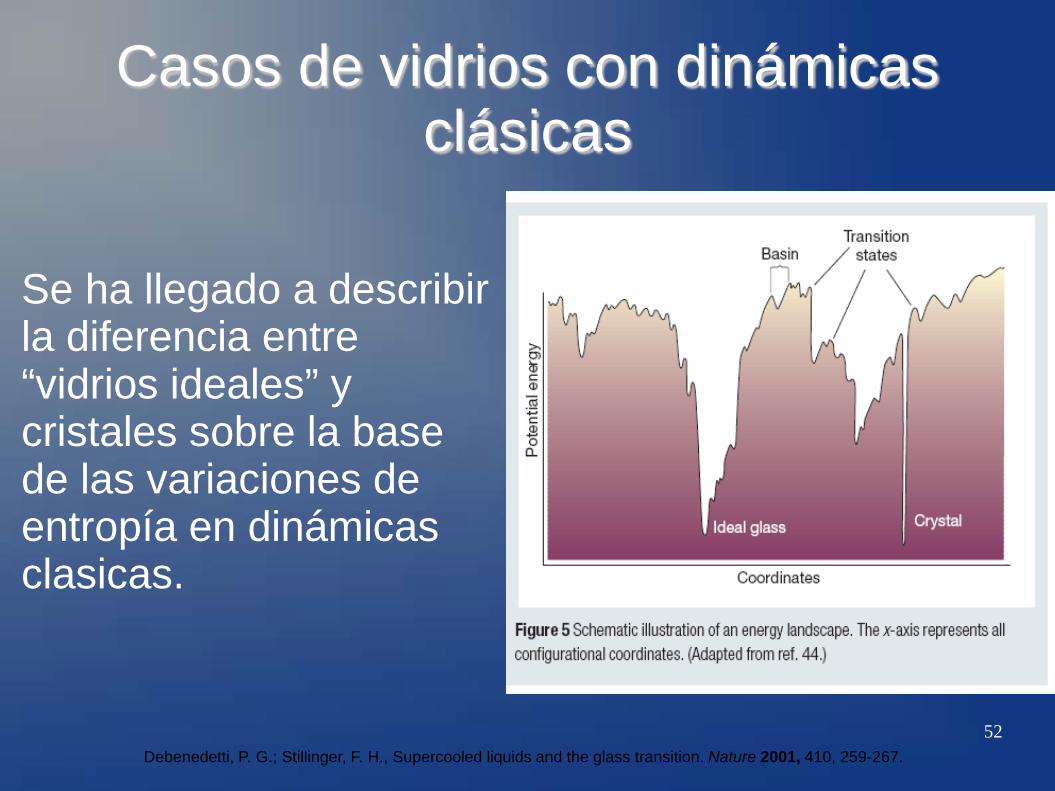
52
Casos de vidrios con dinámicas clásicas
Se ha llegado a describir la diferencia entre “vidrios ideales” y cristales sobre la base de las variaciones de entropía en dinámicas clasicas.
Debenedetti, P. G.; Stillinger, F. H., Supercooled liquids and the glass transition. Nature 2001, 410, 259-267.

53
¿Por qué esta no es una proposición acabada?
La consideración de las especies canónicas de materiales amorfos o policristalinos es motivo de un cuidadoso análisis para cada material. Esto requiere generalización.
La evaluación de la energía de cada especie canónica y de sus interacciones debe ser coherente. No es creible un modelo en el que se usen unos potenciales para una cosa y otros para la otra. Lo ideal sería disponer de un hamiltoniano único y confiable.

54
¿Por qué esta no es una proposición acabada?
La consideración de las especies canónicas de materiales amorfos o policristalinos es motivo de un cuidadoso análisis para cada material. Esto requiere generalización.
La evaluación de la energía de cada especie canónica y de sus interacciones debe ser coherente. No es creible un modelo en el que se usen unos potenciales para una cosa y otros para la otra. Lo ideal sería disponer de un hamiltoniano único y confiable.

55
¿Por qué esta no es una proposición acabada?
“...the formation of quasicrystalline nanoparticle assemblies does not require a unique combination of interparticle interactions, but is a general sphere-packing phenomenon governed by the entropy and simple interparticle potentials.”
Talapin, D. V.; Shevchenko, E. V.; Bodnarchuk, M. I.; Ye, X.; Chen, J.; Murray, C. B., Quasicrystalline order in self-assembled binary nanoparticle superlattices. Nature 2009, 461, 964-967.

56
Pero puede existir una aproximación diferente...
Con una visión similar, pero a partir de moléculas libres como especies canónicas, que son referencias naturales, se puede modelar cómo se hidrata un silicato cíclico y cuales estructuras aportan más del 12 % de la población
Montero-Cabrera, L. A.; Perez-Badell, Y.; Mora-Fonz, M. J., An Approach to Hydration of Model Silica Materials by Exploring Their Multiple Minima Hypersurfaces. The Role of Entropy of Association. J. Phys. Chem. A 2008, 112, (13), 2880-2887.

57
Casos en los que solo los hamiltonianos dan soluciones

58
Sólidos a alta presión
¿Cómo encontrar los valles del paisaje energético en sistemas para los que no se puede optimizar ninguna función clásica?
Tal es el caso de sólidos sometidos a muy altas
presiones.

59
Sólidos a alta presión
¿Cómo encontrar los valles del paisaje energético en sistemas para los que no se puede optimizar ninguna función clásica?
Tal es el caso de sólidos sometidos a muy altas
presiones.

60
Sólidos a alta presión
Feng, J.; Hennig, R. G.; Ashcroft, N. W.; Hoffmann, R., Emergent reduction of electronic state dimensionality in dense ordered Li-Be alloys. Nature 2008, 451, 445-448.
¿Qué nos parece un compuesto de Li y Be?
Un tratamiento cuántico, en este caso con el típico DFT-GGA, puede abordar el problema a presión normal y a muy altas presiones se tuvo que calcular explícitamente a todos los electrones mediante funciones de ondas planas.

61
La catálisis industrial
Las mediciones de eficiencia y el diseño de reactores eran hasta hace poco el patrimonio exclusivo del trabajo ingenieril en plantas piloto.
Los modelos cuánticos se han convertido en
elementos imprescindibles del diseño de procesos en reactores catalíticos

62
La catálisis industrial
Las mediciones de eficiencia y el diseño de reactores eran hasta hace poco el patrimonio exclusivo del trabajo ingenieril en plantas piloto.
Los modelos cuánticos se han convertido en
elementos imprescindibles del diseño de procesos en reactores catalíticos

63
La catálisis industrial Los catalizadores para la síntesis de amoniaco
mediante el proceso Haber pueden optimizarse con un conocimiento previo de modelos de su composición y estructura.
Figure 9 | Linking DFT calculations with industrial reactor design and catalyst selection. a, Schematic illustration of an industrial ammonia synthesis reactor with three adiabatic catalyst beds and two cooling stages. The diagram shows the equilibrium line, the optimal operating line, which can also be called maximum rate line (red), and the operating line for the reactor configuration shown. The closer the operating line approaches the optimal operating line, the lower the catalyst volume required. b, Volcano curves for the turnover frequency, TOF, calculated based on micro-kinetic modelling using parameters calculated by DFT.

64
La catálisis industrial
c, An optimal catalyst curve expresses the reaction conditions under which a transition metal surface with a given nitrogen binding energy is the theoretically optimal catalyst. The optimal catalyst curves make it possible to identify desirable catalysts for relevant reaction conditions. EN* is the metal–nitrogen binding energy (negative values signify exothermic adsorption). d, The interpolation concept illustrating that the binding energy for a CoMo catalyst is intermediate between that of the elemental catalysts Co and Mo (ref. 46). With this concept, or other alloy models, it is possible to identify suitable catalyst leads to be used with the optimal catalyst curves, that is, to design catalysts for specific rector design and process conditions.
Nørskov, J. K.; Bligaard, T.; Rossmeisl, J.; Christensen, C. H., Towards the computational design of solid catalysts. Nature Chemistry 2009, 1, 37-46.
Jacobsen, C. J. H.; Dahl, S.; Boisen, A.; Clausen, B. S.; Topsøe, H.; Logadottir, A.; Nørskov, J. K., Optimal Catalyst Curves: Connecting Density Functional Theory Calculations with Industrial Reactor Design and Catalyst Selection. J. Catal. 2002, 205, (2), 382-387.

65
Un “material” muy especial: las plaquetas del mal de Alzheimer
La acumulación de polipéptidos amiloideos en plaquetas al nivel neuronal se considera uno de los marcadores moleculares de la aparición del mal de Alzheimer, que hace degenerar el tejido nervioso en el humano y muchos animales.

66
Un “material” muy especial: las plaquetas del mal de Alzheimer
La “cristalización” de los polipéptidos en tales plaquetas y su estabilidad demanda la interpretación de este efecto desde el punto de vista del paisaje energético del sistema.
http://www.news.cornell.edu/stories/June07/Relkin.Alzheimers.html

Un “material” muy especial: las plaquetas del mal de Alzheimer
Una modelación cuántica indica que a partir de cálculos sistemáticos de estructuras canónicas seleccionadas cuidadosamente, pero suficientemente grandes, se puede aproximar el paisaje energético del esqueleto de las proteinas.

Se ha demostrado que las estructuras de láminas plegadas β, que son los ladrillos de las fibras amiloideas (DSAS, double stranded anti-parallel β-sheet), constituyen el ordenamiento supramolecular termodinámicamente más estable entre todos los oligomeros y dímeros peptídicos posibles, tanto en el vacío (sin interacciones con moléculas vecinas) como en entornos acuosos, basados en su alta complejidad (T∆S).
Perczel, A.; Hudaky, P.; Palfi, V. K., Dead-End Street of Protein Folding: Thermodynamic Rationale of Amyloid Fibril Formation. J. Am. Chem. Soc. 2007, 129, (48), 14959-14965.

La segunda proposición
La segunda proposición de este trabajo es la de que la modelación de materiales requiere de un hamiltoniano capaz de explorar el paisaje energético de sistemas nanoscópicos y hasta microscópicos en un tiempo de cálculo al alcance de un laboratorio común si se desea tener idea de aspectos cuánticos, como la reactividad química y los efectos de la luz, y así proyectar la modelación al macrouniverso.

El formalismo NDOL
El formalismo NDOL es un procedimiento iterativo y aproximado de Hartree-Fock para el cálculo de la energía y composición de orbitales moleculares de valencia que se se evalua a priori y brinda resultados consistentes:
( )lkAB
BA
lkAB
kB
llAB
lB
pHF
PPHF
γ
γγ
µνµνµν
µµµµ
21−=
++= ∑≠
Montero, L. A.; Alfonso, L.; Alvarez, J. R.; Perez, E., From PPP-MO theory to all-valence electron calculations of ionic and excited states in organic molecules. Int. J. Quantum Chem. 1990, 37, (4), 465-83.
Montero-Cabrera, L. A.; Röhrig, U.; Padron-García, J. A.; Crespo-Otero, R.; Montero-Alejo, A. L.; García de la Vega, J. M.; Chergui, M.; Röthlisberger, U., CNDOL: A fast and reliable method for the calculation of electronic properties of very large systems. Applications to retinal binding pocket in rhodopsin and gas phase porphine. J. Chem. Phys. 2007, 127, (14), 145102.

La calidad de las funciones de onda moleculares NDOL
La correspondencia entre los potenciales de ionización en fase gaseosa de moléculas simples y los valores propios de orbitales llenos más altos es satisfactoria para sistemas simples
4 5 6 7 8 9 10 11 124
5
6
7
8
9
10
11
12
-eH
OM
O (e
v)
Experimental IP (ev)
CNDOL/21 CNDOL/22

Interacción de configuraciones electrónicas monoexcitadas
Las transiciones electrónicas calculadas según: Se optmizan mediante la interacción de
configuraciones monoexcitadas (CIS):
**3
***1 2
ijij
ijijij
JH
KJH
−−=
+−−=
εε
εε
σσ
σσ
( ) 0=−
Φ=Ψ ∑aEH
σσσM
CISM a

Fulerenos
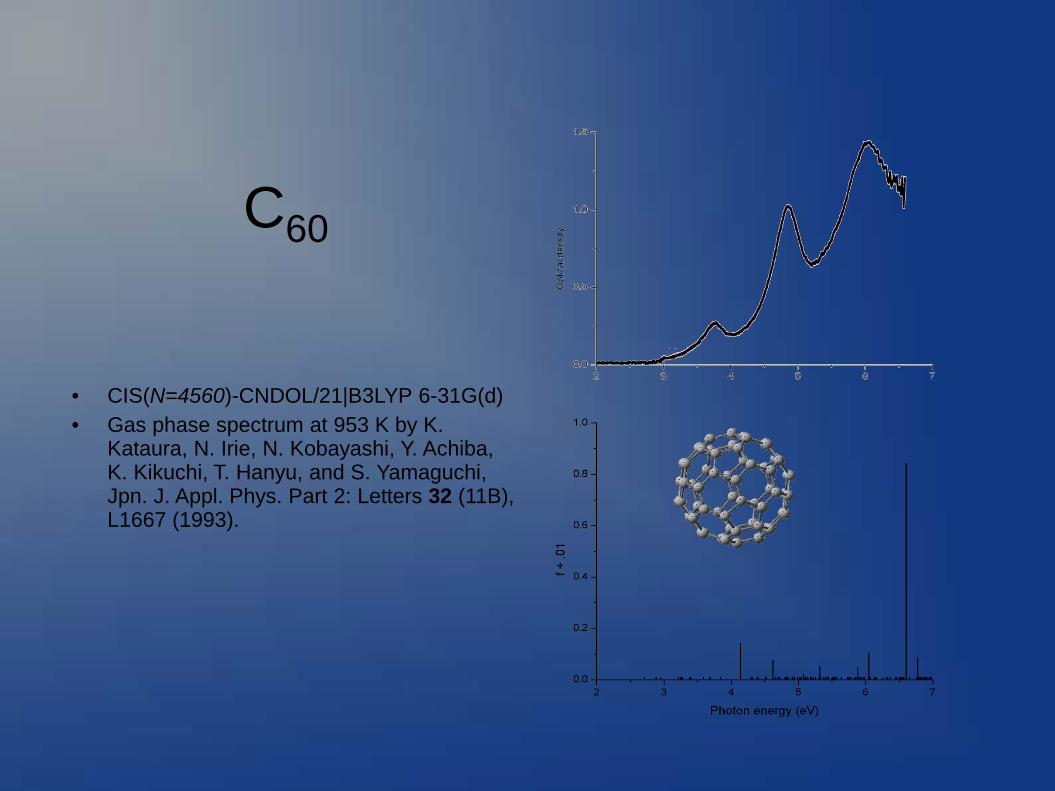
C60
• CIS(N=4560)-CNDOL/21|B3LYP 6-31G(d) • Gas phase spectrum at 953 K by K.
Kataura, N. Irie, N. Kobayashi, Y. Achiba, K. Kikuchi, T. Hanyu, and S. Yamaguchi, Jpn. J. Appl. Phys. Part 2: Letters 32 (11B), L1667 (1993).

C70
• CIS(N=5380)-CNDOL/21|B3LYP 6-31G(d) • Gas phase spectrum at 953 K by K.
Kataura, N. Irie, N. Kobayashi, Y. Achiba, K. Kikuchi, T. Hanyu, and S. Yamaguchi, Jpn. J. Appl. Phys. Part 2: Letters 32 (11B), L1667 (1993).

Nanotubos de carbono

Excitationes electrónicas en nanotubos de carbono de paredes simples (SWCNT)
L Length (nm)
2 0.71
4 1.56
6 2.42
8 3.27
10 4.12
12 4.98
14 5.83
16 6.68
18 7.54
20 8.39
Zig-zag type SWCNT, within (n-m=3M) rule
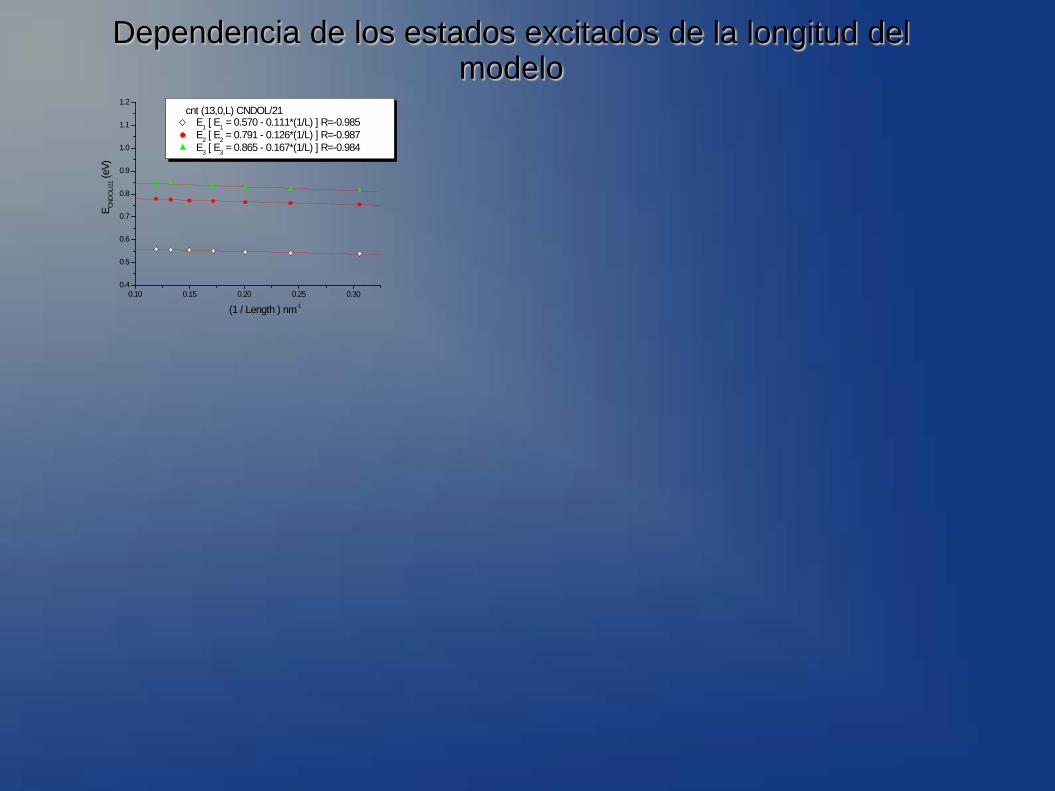
Dependencia de los estados excitados de la longitud del modelo
0.10 0.15 0.20 0.25 0.300.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2E CN
DOL/
21 (e
V)
(1 / Length ) nm-1
cnt (13,0,L) CNDOL/21 E1 [ E1 = 0.570 - 0.111*(1/L) ] R=-0.985 E2 [ E2 = 0.791 - 0.126*(1/L) ] R=-0.987 E3 [ E3 = 0.865 - 0.167*(1/L) ] R=-0.984
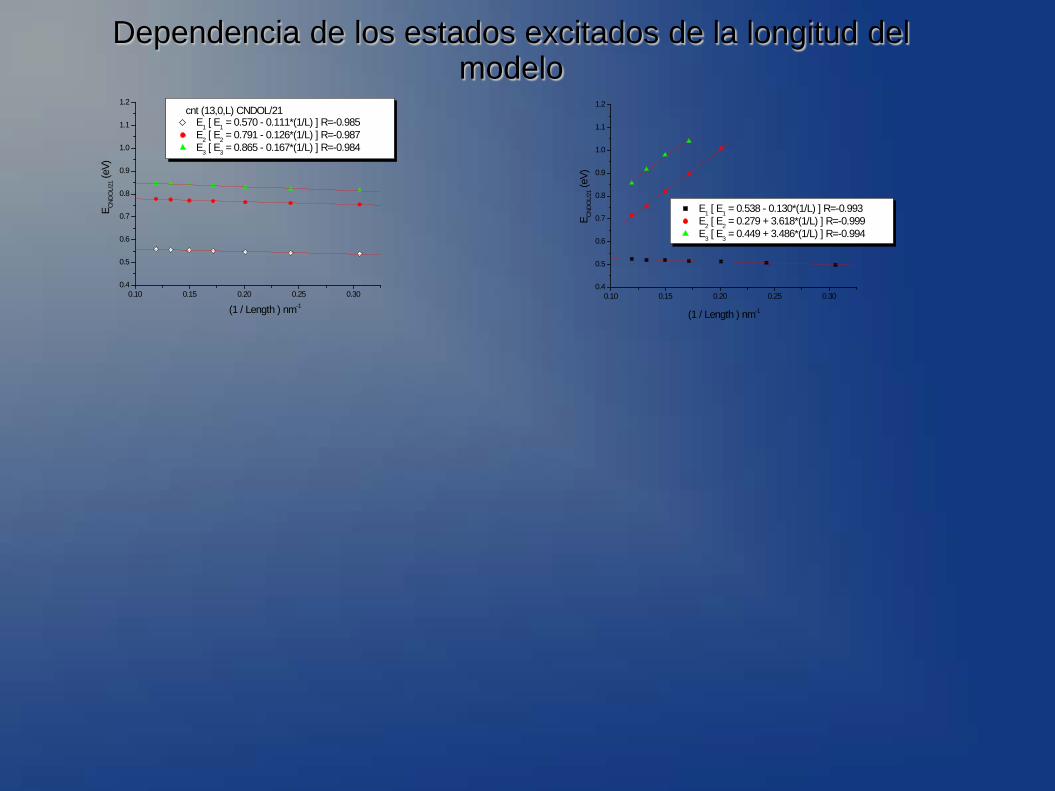
Dependencia de los estados excitados de la longitud del modelo
0.10 0.15 0.20 0.25 0.300.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2E CN
DOL/
21 (e
V)
(1 / Length ) nm-1
cnt (13,0,L) CNDOL/21 E1 [ E1 = 0.570 - 0.111*(1/L) ] R=-0.985 E2 [ E2 = 0.791 - 0.126*(1/L) ] R=-0.987 E3 [ E3 = 0.865 - 0.167*(1/L) ] R=-0.984
0.10 0.15 0.20 0.25 0.300.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2
E CNDO
L/21
(eV)
(1 / Length ) nm-1
E1 [ E1 = 0.538 - 0.130*(1/L) ] R=-0.993 E2 [ E2 = 0.279 + 3.618*(1/L) ] R=-0.999 E3 [ E3 = 0.449 + 3.486*(1/L) ] R=-0.994
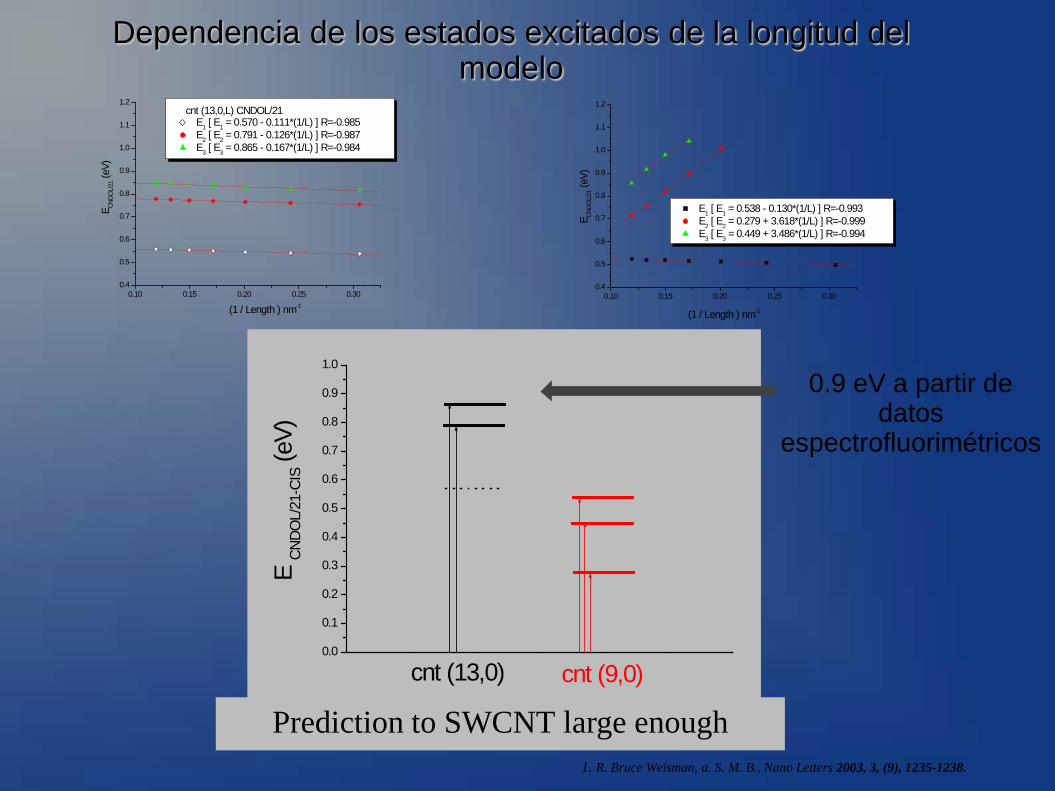
Dependencia de los estados excitados de la longitud del modelo
0.10 0.15 0.20 0.25 0.300.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2E CN
DOL/
21 (e
V)
(1 / Length ) nm-1
cnt (13,0,L) CNDOL/21 E1 [ E1 = 0.570 - 0.111*(1/L) ] R=-0.985 E2 [ E2 = 0.791 - 0.126*(1/L) ] R=-0.987 E3 [ E3 = 0.865 - 0.167*(1/L) ] R=-0.984
0.10 0.15 0.20 0.25 0.300.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2
E CNDO
L/21
(eV)
(1 / Length ) nm-1
E1 [ E1 = 0.538 - 0.130*(1/L) ] R=-0.993 E2 [ E2 = 0.279 + 3.618*(1/L) ] R=-0.999 E3 [ E3 = 0.449 + 3.486*(1/L) ] R=-0.994
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
cnt (13,0) cnt (9,0)
E CN
DOL/
21-C
IS (e
V)
Prediction to SWCNT large enough
0.9 eV a partir de datos
espectrofluorimétricos
1. R. Bruce Weisman, a. S. M. B., Nano Letters 2003, 3, (9), 1235-1238.

Silicatos y aluminosilicatos microporosos

Las zeolitas necesitan simulaciones cuánticas
Una reciente y abarcadora reseña (Smit, B.; Maesen, T. L. M., Molecular Simulations of Zeolites: Adsorption, Diffusion, and Shape Selectivity. Chem. Revs. 2008, 108, 4125–4184) acerca de la modelación molecular de procesos en zeolitas plantea que el principal problema de hacer aplicaciones cuánticas en estos materiales es la importante complejidad estructural y que muchos efectos solo se pueden proyectar en escalas nanoscópicas, no solo picoscópicas, por lo que los cálculos suelen ser prohibitivamente caros.

Las zeolitas necesitan simulaciones cuánticas
Ya en 2002, un hamiltoniano aproximado como el CNDOL/22 pudo explicar por qué los hidrocarburos encerrados en canales de zeolitas se pueden excitar electrónicamente en el visible, lo que facilita su oxidación en el ambiente.
Montero, L. A.; Diaz, L. A.; Castillo, N., UV-Vis spectrum of simple hydrocarbons in a zeolite cavity. A supramolecular charge transfer. Chem. Phys. Lett. 2002, 364, (1,2), 176-179.

Las zeolitas necesitan simulaciones cuánticas
Usando el hamiltoniano CNDOL/21 se logró reproducir el espectro UV de un enigma histórico como es el “azul maya”, donde un colorante orgánico se estabiliza por siglos en un retículo de aluminosilicatos.
Fuentes, M. E.; Peña, B.; Contreras, C.; Montero, A. L.; Chianelli, R.; Alvarado, M.; Olivas, R.; Rodríguez, L. M.; Camacho, H.; Montero-Cabrera, L. A., Quantum Mechanical Model for Maya Blue. Int. J. Quantum Chem. 2008, 108, 1664–1673.

La segunda proposición tiene respuestas aunque requiere
desarrollo La familia de hamiltonianos NDOL requiere trabajo de programación y
prueba para lograr, entre otras, las siguientes prestaciones: Energías del estado base comparables. Capacidades numéricas de exploración de las hipersuperficies (a través
de gradientes o de exploraciones aleatorias del espacio de configuraciones de cualquier sistema).
Cálculos de sistemas de capa abierta. Cálculo con funciones de base extendidas, incluyendo orbitales
atómicos d. Incluir interacción de configuraciones con dobles excitaciones. Obtener funciones de onda confiables de los estados excitados.

Reconocimientos Los autores están muy agradecidos a la Universidad
de La Habana, en La Habana, Cuba, el Alma Mater de muchos, que nos ha acogido siempre y permitido revolucionar muchas veces nuestro propio pensamiento y acción científica. La Agencia Sueca de Cooperación Investigativa (SAREC), en Estocolmo, Suecia, la Deutscher Akademischer Austauschdienst (DAAD) en Bonn, Alemania, y después la Agencia Española de Cooperación Internacional para el Desarrollo (AECID), en Madrid, España, nos han apoyado materialmente de forma decisiva para la realización de este trabajo. El Consejo Superior de Investigaciones Científicas, la Universidad Autónoma de Madrid y el Ministerio de Educación de España han proporcionado también la facilidad esencial de hacer posible una ventana al mundo en todas las etapas desde 1986.

Colaboradores Laboratorio de Química Computacional y Teórica, Universidad de La Habana Ana L. Montero-Alejo Yoana Pérez Badell Rachel Crespo-Otero Susana González-Santana Luis A. Montero-Cabrera Universidad Autónoma de Madrid José Manuel García de la Vega Raúl H. González – Jonte Universidad Nacional Autónoma de México Carlos F. Bunge Universidad Autónoma de Chihuahua María E. Fuentes
y Salvador Dalí y Wilfredo Lam, por sus pinturas