introducción grupo heterogéneo manifestaciones debido a la infiltración mastocitaria en...
TRANSCRIPT

Seminario Biblioráfico
Melisa Paula Celoria . HPC
Click icon to add picture

Introducción Grupo heterogéneo manifestaciones debido a la infiltración mastocitaria
en diferentes órganos
Los mastocitos presentan predilección por la piel
Mayoría esporádica aunque existen formas familiares
cutáneas (MC), sistémicas (MS)
prevalecen en población blanca de origen europeo, en ambos sexos por igual,
2/3 de los casos se presentan en niños como formas cutáneas aisladas
Adultos jóvenes con compromiso cutáneo, en el 60-80% de los casos tb presentan afectación sistémica. Evolución crónica e indolente
Mayores de 60 años: en su mayoría presentan trastorno hematológico clonal

Manifestaciones clínicas
Liberación de mediadores mastocitarios
Infiltración cutánea de mastocitos
Infiltracion mastocitaria en otros órganos

Manifestaciones clínicasMediadores mastocitarios
(Histamina, PGD2)
flush, cefalea
prurito, urticaria
mareos,palpitaciones
disnea, precordialgia
nauseas, vomitos, diarrea
30% MC y 50% MS
Factores que favorecen la degranulación mastocitaria
Los síntomas sistémicos (enrojecimiento, malestar, dolor abdominal ...) no son necesariamente sinónimos de afectación sistémica.


Manifestaciones clínicas
Infiltración mastocitaria cutánea
Clasificación según la OMS:
o Urticaria pigmentosa
o Pápulo-Nodular (mastocitoma cutáneo, Xantelasmoide, Multinodular)
o Cutánea difusa
o Telangiectasia macular eruptiva persistente (TMEP)

Urticaria pigmentosa (UP)
Más frecuente, a cualquier edad, principalmente luego de los 6 meses de vida
Erupción pruriginosa maculo-papular cuyas lesiones van desde 1 mm a más de 1 cm, de número variable y de color rojo-purpúrico o marrón-amarillento.
Ante la fricción se tornan turgentes y eritematosas: Signo de Darier. Patognomónico.
La pigmentación de las lesiones en la dermatoscopía puede impresionar una red debido a la hipermelanogénesis
Generalmente presentan distribución simétrica en tronco y miembros inferiores, raramente en cara y cuero cabelludo. Palmas, plantas y mucosas en casos excepcionales.

UP en niños Lesiones de mayor tamaño,
escasas, ovalada o alargada en los pliegues, de tono marrón claro, ligeramente sobreelevadas, de consistencia elástica, dando un aspecto atigrado o de "piel de leopardo".
Puede presenterse formas ampollares hasta en un 60% de los casos. Éstas resuelven sin dejar cicatríz aunque pueden quedar áreas hipopigmentadas.
En los casos ampollares no se recomiendo la búsqueda del signo de Darier ya que exacerbar la reacción.
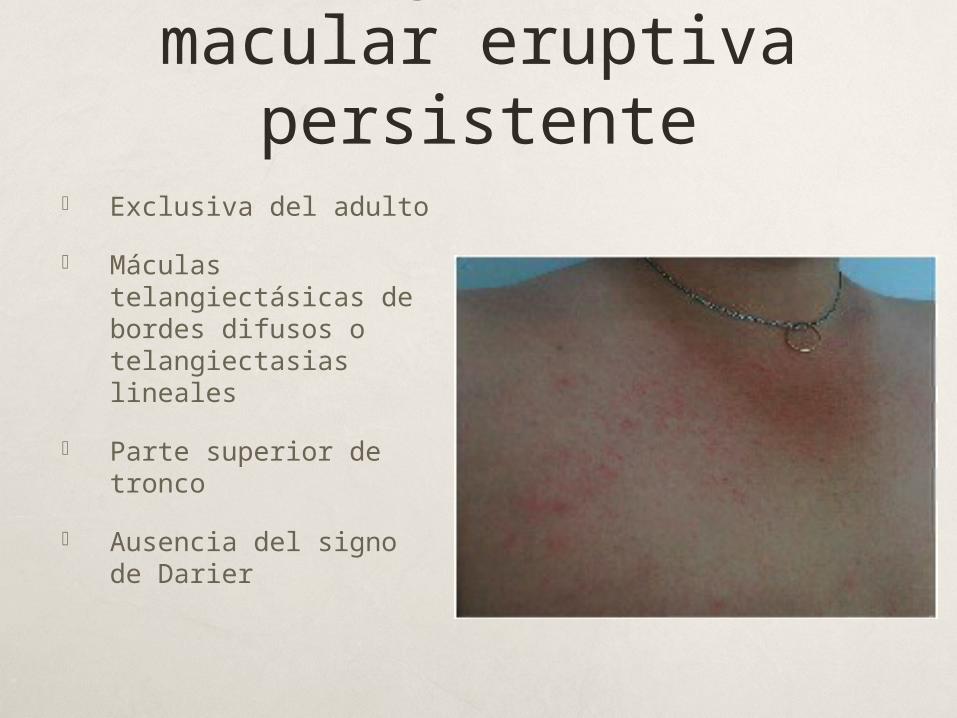
Telangiectasia macular eruptiva persistente
Exclusiva del adulto
Máculas telangiectásicas de bordes difusos o telangiectasias lineales
Parte superior de tronco
Ausencia del signo de Darier

Mastocitosis pápulo-nodular
Tres variedades
oMastocitoma
oXantelasmoide
oMultinodular
Principalmente en infantes

Mastocitoma
Mastocitosis
xantelasmoide
Suele presenterse desde el nacimiento
Elementos ovales xantomatosos
El signo de Darier es inconstante pero cuando está presente puede desencadenar la formación de bullas y síntomas sistémicos
Esta variedad tiende a persistir hacia la edad adulta.
40% presente al nacimiento. Principal manifestación cutánea de mastocitosis antes de los tres meses
de edad
Común dentro de los tres primeros años de vida
Nódulo único, turgente, de superficie lisa, amarillento, rosado o
marrón generalmente en extremidades. Signo de Darier positivo (su búsqueda excesiva puede desencadenari síntomas
sistémicos)
D/D: xantogranuloma juvenil, nevus de Spitz, melanoma nodular

Mastocitosis multinodular Elementos nodulares hemiesféricos de superficie
lisa, de color rosado pálido al amarillo perlado y hasta a veces blanco.
Con el tiempo, los nódulos pierden volumen evolucionando a una etapa macular.
Tiende a persistir

Mastocitosis cutánea difusa
Muy rara (menos de 50 casos)
Niños menores de 3 años, rara vez en los adultos
Piel amarillenta, gruesa, pastosa, con lesiones micropápulares que se acentúan en los pliegues.
El prurito es muy intenso y puede dar lugar a la formación de ampollas y erosiones.
Forma eritrodérmica es la de mayor gravedad
Adultos: la piel adopta una apariencia de piel de naranja, engrosada, liquenificada y con aspecto poiquilodérmico.

Manifestaciones clínicas Compromiso óseo
Generalmente asintomático
Osteoporosis
Fracturas de huesos largos o cuerpos vertebrales más frecuentemente
Debe realizarse desitometría a todo adulto con diagnóstico de mastocitosis
Compromiso hepático y digestivo
Diarrea, dolor abdominal y dispepsia
Hepatitis colestácica
Hepatomegalia

Cardiovasculares
Shock anafiláctico
(forma de presentación frecuente en el adulto)
Hematológicas
Hepatoesplenomegalia
Poliadenopatías
Anemia, leucocitosis con eosinofilia, trombocitopenia
Síndromes mieloproliferativos(leucemia mielomonocítica crónica), síndromes mielodisplásicos, más raramente linfoma no hodgking, neutropenia crónica, mieloma.

Fisiopatología Los mastocitos se originan en la MO como células agranulares
indiferenciadas CD34+, CD117+ (KIT). Al migrar a los tejidos adoptan la morfología granular.
KIT es un receptor tirosina cinasa producto del prooncogen c-kit en el cromosoma 4q12. Éste, en condiciones normales, requiere del ligando factor de células madre (SCF) para ser activado permitiendo asi la supervivencia de los mastocitos maduros.
Las mutaciones detectadas (D816V la más frecuente) provocan actividad tirosina quinasa en ausencia del ligando SCF.
Esta activación permite la diferenciación de progenitores hematopoyéticos (CD34 +), la migración, proliferación, supervivencia y activación de los mastocitos en los tejidos, incluído la piel.
Tanto en los adultos como en los niños, se ha confirmado la teoría clonal, pero el 5-30% de los pacientes no tiene una mutación KIT, lo que lleva a pensar en otros mecanismos

Histología Anatomía patológica es el método de confirmación tanto de la
afectación cutánea como sistémica
En condiciones normales los mastocitos se encuentran alrededor del folículo piloso, los vasos y los nervios. Su número varía normalmente según el áreas del cuerpo y su aumento puede estar relacionado a patologías inflamatorias.
Si el infiltrado es denso, los mastocitos pueden evidenciarse con tinciones de hematoxilina-eosina. De lo contrario, se utilizan tinciones como el azul de toloudina o Giemsa.
La inmunohistoquímica puede ser de gran ayuda poniendo de manifiesto el marcador CD117. El mastocito es la única célula que lo expresa a nivel dérmico (el melanocito también lo expresa pero a nivel epidérmico).

En las MS, la MO se haya comprometida en el 90% de los casos, por lo tanto, la PAMO en el método diagnóstico de elección
Visualizaremos células cebadas de aspecto fusiformes, agrupadas en acúmulos paratrabeculares
CD117 y CD25 positivas (CD2 negativas)

HISTOLOGÍA

Diagnóstico Fundamentalmente clínico e histopatológico.
Confirmada una MC en niños no se justifica continuar su estudio excepto que se trate de una mastocitosis cutánea difusa.
En el paciente adulto debe siempre realizarse un hemograma, función hepática, dosaje de triptasa sérica así como radiografías y DMO.
La existencia de anormalidades hematológicas, radiológicos y densitométricas, debe conducir a practicar una biopsia de médula ósea.
La triptasa es actualmente el marcador plasmático de la degranulación mastocitaria y su cuantificación se correlaciona con el tamaño del infiltrado y con el valor predictivo de afectación sistémica:
25-75 ng /ml: 50%
superior a 75 ng /ml: casi 100%

Se ha demostrado que la MS se acompaña de un aumento de los niveles séricos de CD117 soluble y CD25 (receptor de IL-2)
Dosaje de histamina, ácido acético metil-4-imidazol y metabolitos urinarios de PGD2 e histaminuria. Uso limitado debido a los posibles falsos positivos (alergia)
Mutación del proto-oncogén KIT D816V en piel o en MO podría tener valor terapéutico y pronóstico.

Criterios diagnósticos de MS
Mayores:
infiltrado multifocal de mastocitos en órganos extracutáneos
Menores:
mastocitos fusiformes o con morfología atípica en más del 25% del extendido de MO o biopsias tisulares
Mutación D816V del C-KIT en sangre, MO o tejidos lesionados
Expresión de CD25 o CD2 en mastocitos de MO, sangre u otro tejido lesionado
Concentración basal de triptasa mayor a 20 ng/ml
Un criterio mayor + un criterio menor: diagnóstico
Tres criterios menores: diagnóstico

Tratamiento Objetivos
Disminuír los síntomas relacionados con la liberación de mediadores mastocitarios
Reducir la infiltración mastocitaria (depende de la presentación clínica)
Medidas generales: evitar alimentos histaminoliberadores, los cambios bruscos de temperatura, el ejercicio vigoroso,los contrastes yodados, los fármacos histaminoliberadores y tener precaución en las intervenciones quirúrgicas. Kit de Epinefrina autoinyectable.

Tratamiento sintomático Fármacos de primera línea:
o Antihistamínicos anti H1 (desloratadina, levocetirizina, fexofenadina…)
o Antihistamínicos anti H2 (ranitidina, cimetidina)
o Su asociación controla el flush, el prurito y los síntomas gastrointestinales
Cromoglicado disodico: estabilizador de la membrana. Mejora las manifestaciones digestivas y el prurito.

Antileucotrienos (Montelukast): controla el prurito y el flush
AAS: puede ser útil si los anteriores han fracasado
Adrenalina: shock anafiláctico
Corticoides sistémicos: en formas agresivas y asociadas a sindrome de malaabsorción o ascitis. Dosis: 1 mg/kg/día con reducción progresiva.
Bifosfonatos: EV fracturas patológicas. VO en osteoporosis. VD y Ca++ en osteopenia.

Tratamiento dermatológico
Adultos: PUVA tto clásico en la UP. Duración 5 a 8 meses. Riesgo de cancerización. Laser vascular o luz pulsada en TEMP.
Niños: regresan espontáneamente el 50% de los casos. En el mastocitoma aislado pueden utilizarse CTC potentes oclusivos. Cirugía si no hay resultado con lo anterior. En formas cutáneas difusas PUVA (pocos casos)

Terapia inmunomoduladora y
citorreductora

Terapia inmunomoduladora y citorreductora
Interferón alfa: en MSA con o sin enfermedad hematológica asociada. Eficaz en reducir la liberación de mediadores mastocitarios y la infiltración. Sin embargo su efecto antiproliferativo es moderado y presenta recaídas así como una baja tolerancia por sus efectos adversos (depresión, síntomas sistémicos)
2 Clorodesoxiadenosina (2-CdA), análogo de las purinas, disminuye síntomas asociados a la liberación de mediadores así como la infiltración específica en MO. Especialmente en pacientes de edad avanzada. Riesgo de mielosupresión.
Quimioterapia con ciclofosfamida, vincristina y prednisona: mastocitosis asociada a trastornos hematológicos
Mesilato de Imatinib (inhibibor de la tirosinquinasa) ha demostrado eficacia en pacientes con mutación de KIT en la posición F522C.
Disatinib inhibiría in vitro a aquellos mastocitos que presentan la mutación D816V. Sin buena respuesta en disminuír síntomas o signos de la enfermedad en pacientes.
Talidomida, acido trans retinoico, bexaroteno, anticuerpos monoclonales anti CD25 (daclizumab) y anti CD2 y CD30 (brentixumab)

Pronóstico En los niños raramente amenaza la vida excepto las
formas cutaneas difusas
Factores independientes de mal pronóstico:
o edad mayor de 65 años
o pérdida de peso
o anemia
o trombocitopenia
o hipoalbuminemia
o blastos en MO mayor al 5%