intolerancia a la galactosa
TRANSCRIPT

Protocolo de diagnóstico y tratamiento delos errores congénitos del metabolismo de
la galactosa
Baldellou A1; Briones P2; Ruiz M3
1Hospital Universitario Miguel Servet, Zaragoza.
2Institut de Bioquímica Clínica. Hospital Clinic y CSIC, Barcelona.
3Hospital Nuestra Señora de la Candelaria. Santa Cruz de Tenerife.
Palabras clave: galactosemia, dieta exenta de galactosa.
Correspondencia:Dr. Antonio Baldellou.Unidad de Enfermedades Metabólicas.Hospital Miguel Servet. Zaragoza.e-mail: [email protected]
153
Galactosemia7
P7 17/5/07 17:57 Página 153

Introducción
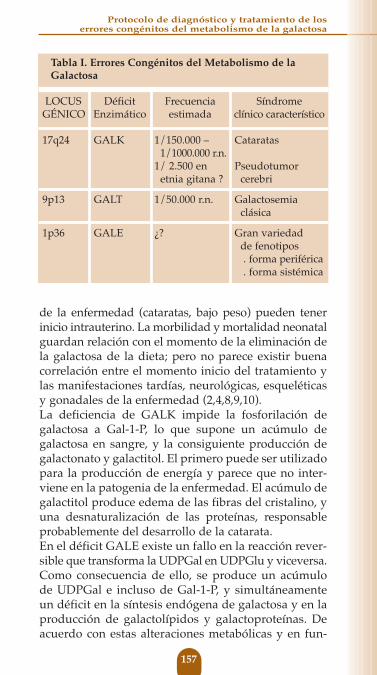
La galactosa es un monosacárido de seis átomos decarbono que forma parte importante de la dieta de laespecie humana desde el nacimiento. En forma delactosa se halla presente en la leche de los mamíferos;en forma de galactosa soluble, o ligada con enlacestipo β en ciertas legumbres, verduras y frutas; enforma de galactósidos ligada con enlaces tipo α enpolisacáridos de origen vegetal; y en forma de galac-tocerebrósidos y gangliósidos en algunas vísceras deanimales.La fuente más importante de galactosa es la lactosa dela leche, la cual es hidrolizada en el intestino por unaβ-galactosidasa (lactasa) localizada en el “borde encepillo” de la mucosa intestinal. Aunque algunos ali-mentos de uso habitual poseen galactosa ligadamediante enlaces α o enlaces β y el organismo posee β-galactosidasas en el intestino, y α-galactosidasas enmuchos tejidos, no está claro el grado de disponibilidadde estas galactosas, y su importancia en la dieta humanadesde el punto de vista cuantitativo. Una vez liberada en el intestino la galactosa esabsorbida a través del enterocito, mediante un trans-portador activo Na-dependiente que es común para laglucosa. Trasladada por el torrente circulatorio estransformada en glucosa, principalmente por el hígado,aunque también el cerebro y los hematíes poseen losenzimas necesarios para su utilización. El 80% de la galactosa ingerida se utiliza como fuente ener-gética en la vía de la glucólisis, y el 20% para la sín-tesis de glicoproteínas y glicolípidos que resultanfundamentales para la acción de muchas proteínas yhormonas y para la estabilidad de las membranascelulares (1).En circunstancias normales la conversión de galacto-sa en glucosa tiene lugar a través de la “vía Leloir”.La α-D- galactosa es fosforilada mediante el enzimagalactokinasa (GALK) a Galatosa-1-fosfato, la cual porla acción del enzima galactosa-1-fosfato-uridiltransferasa
154
Galactosemia
P7 17/5/07 17:57 Página 154

(GALT) y utilizando la UDPGlucosa como sustrato, estransformada en Glucosa-1-fosfato y UDPGalactosa,que a su vez, mediante la acción del enzima UDP-galactosa 4-epimerasa (GALE) se mantiene en equilibriocon la UDPGlucosa. De este modo, por cada moléculade galactosa que entra en esta vía metabólica, se pro-duce una de Glu-1-P. Existen además, tres vías accesorias en el metabolismode la galactosa. La “vía de la pirofosforilasa” es capazmediante unas pirofosforilasas (UDPGal pirofosforilasa yUDPGlc pirofosforilasa) de incorporar la galactosa de laGalactosa-1-fosfato a la UDPGalactosa, que a su vezes convertida en UDPGlucosa que produce Glucosa-1-fosfato, con lo que este mecanismo sería capaz deoxidar cerca del 1% de la galactosa en la deficienciade GALT. Pero además esta vía, permite sintetizarUDPGalactosa (a partir de la Glucosa-1-fosfato queno proceda del metabolismo normal de la galactosa)que puede ser convertida en Galactosa-1-fosfato yésta, en galactosa, mediante la acción de una fosfatasa.De este modo se asegura una síntesis endógena degalactosa, que puede ser utilizada en la síntesis deglucoconjugados, de alrededor de 10-20 mg/k/díaen los niños y de 4-5 mg/k/día en los adultos. Lasotras dos vías alternativas sólo adquieren importan-cia cuando se produce un acúmulo de galactosa acausa de un déficit enzimático en la vía clásica.Mediante una aldosa reductasa puede ser reducida agalactitol que en parte es excretado en el riñón y en parte se acumula en algunos tejidos donde juegaun papel importante en la patogenia de la enferme-dad; o puede ser oxidada mediante la galactosa-deshi-drogenasa a galactonato capaz de ser utilizado para la producción de energía a través de la vía de las pento-sas. Fig. 1No parece existir regulación hormonal del metabolismode la galactosa, y sus niveles sanguíneos dependenfundamentalmente de la ingesta, de la saturación delos enzimas hepáticos y de la capacidad de excreciónrenal (1-5).
155
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
P7 17/5/07 17:57 Página 155

Etiología
En la especie humana se conocen tres errores congénitosdel metabolismo de la galactosa, cada uno de ellosdebido a la mutación de un gen de carácter autosómicorecesivo. Para los tres han sido identificadas diversasmutaciones alélicas responsables de un fenotipo bio-químico característico, que condiciona a su vez unasmanifestaciones clínicas determinadas (6,7). Tabla I.Su incidencia global está alrededor de 1/50.000 reciénnacidos vivos, y la trascendencia de su diagnósticoprecoz radica en que el adecuado tratamiento dietéticopuede salvar la vida y asegurar la casi total integridadfuncional de los pacientes (2).
Patogenia
La vulnerabilidad de los tejidos a la disfunción homeos-tática de la galactosemia parece ser mayor en los pri-meros momentos de la vida y algunas manifestaciones
156
Galactosemia
Fig. 1. Resumen del metabolismo de la galactosa.1 Galcatokinasa (GALK), 2 Galactosa-1-fosfato uridiltransferasa(GALT), 3 UDP-Galactosa 4-epimerasa (GALE), 4 UDP-Clc pirofosfo-rilasa y UDP-Gal pirofosforilasa (considerado el mismo), 5 Fosfatasa,6 Aldosa reductasas, 7 Galactosa dehidrogenasa.
P7 17/5/07 17:57 Página 156
de la enfermedad (cataratas, bajo peso) pueden tenerinicio intrauterino. La morbilidad y mortalidad neonatalguardan relación con el momento de la eliminación dela galactosa de la dieta; pero no parece existir buenacorrelación entre el momento inicio del tratamiento ylas manifestaciones tardías, neurológicas, esqueléticasy gonadales de la enfermedad (2,4,8,9,10).La deficiencia de GALK impide la fosforilación degalactosa a Gal-1-P, lo que supone un acúmulo degalactosa en sangre, y la consiguiente producción degalactonato y galactitol. El primero puede ser utilizadopara la producción de energía y parece que no inter-viene en la patogenia de la enfermedad. El acúmulo degalactitol produce edema de las fibras del cristalino, yuna desnaturalización de las proteínas, responsableprobablemente del desarrollo de la catarata.En el déficit GALE existe un fallo en la reacción rever-sible que transforma la UDPGal en UDPGlu y viceversa.Como consecuencia de ello, se produce un acúmulode UDPGal e incluso de Gal-1-P, y simultáneamenteun déficit en la síntesis endógena de galactosa y en laproducción de galactolípidos y galactoproteínas. Deacuerdo con estas alteraciones metabólicas y en fun-
157
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
Tabla I. Errores Congénitos del Metabolismo de laGalactosa
LOCUS Déficit Frecuencia SíndromeGÉNICO Enzimático estimada clínico característico
17q24 GALK 1/150.000 – Cataratas1/1000.000 r.n.
1/ 2.500 en Pseudotumor etnia gitana ? cerebri
9p13 GALT 1/50.000 r.n. Galactosemia clásica
1p36 GALE ¿? Gran variedad de fenotipos. forma periférica. forma sistémica
P7 17/5/07 17:57 Página 157

ción de los tejidos en los que se expresa el déficitenzimático se produce la gran variabilidad en las mani-festaciones clínicas de la enfermedad. El déficit GALT impide la adecuada oxidación de lagalactosa previa transformación en glucosa ya que blo-quea la transformación de Gal-1-P en Glu-1-P; y la sínte-sis de UDPGal. Como consecuencia de ello, se produceun aumento de Gal-1-P eritrocitaria y de la galactosaplasmática con la consiguiente galactosuria. La galactosaen exceso en el plasma es convertida en galactonato, yen galactitol. El primero puede ser en parte oxidado y enparte eliminado por la orina. El galactitol no puede sermetabolizado, y en parte es eliminado por la orina, peroel resto tiene seguramente efectos tóxicos importantespara el organismo. En esta deficiencia enzimática semantiene a través de la vía de la pirofosforilasa, una pro-ducción endógena significativa de galactosa (que tienegran trascendencia en los mecanismos patogénicosde la enfermedad) y de UDPGal necesaria para la sín-tesis de macromoléculas glicosiladas. Seguramente laconcurrencia simultánea de niveles elevados de galacto-sa-1-fosfato y de galactitol, el déficit de UDPGal y laconsiguiente alteración de la síntesis de compuestosgalactosilados, junto a trastornos de la apoptosis celular,son los principales responsables del daño neurológico,hepático, renal, esquelético y gonadal de los pacientes.
Diagnóstico
I. Diagnóstico de Sospecha
En ausencia de un examen sistemático neonatal, eldiagnóstico de un error congénito del metabolismo dela galactosa debe sospecharse en todo paciente quepresenta manifestaciones clínicas y bioquímicas com-patibles con la existencia de la enfermedad (6,7). Sunúmero e intensidad dependerá del enzima implicado,de la mutación génica responsable, y de la actividadresidual de cada paciente; pero en todos los casos daránlugar a un síndrome metabólico de “intoxicación” del
158
Galactosemia
P7 17/5/07 17:57 Página 158

organismo por acúmulo de los productos no metaboli-zados y que resultan especialmente tóxicos para elcristalino, el hígado y el riñón (2,3,4,5,11).En la deficiencia de galactoquinasa la única manifesta-ción de enfermedad en los individuos homozigotos esla catarata bilateral a causa de la desnaturalización delas proteínas y de la tumefacción osmótica que produceen sus fibras. Existe un aumento de galactosa y galac-titol en plasma y la galactosuria concomitante.Por el contrario, en la deficiencia de UDP galactosa 4-epimerasa de leucocitos y eritrocitos, los pacientesson asintomáticos con ingesta dietética normal; y losafectos del déficit enzimático sistémico presentan clínicamuy parecida a la galactosemia clásica sin una buenarespuesta al tratamiento dietético. En las dos formasclínicas existe un aumento de galactosa-1-fosfato, y deUDPGalactosa en sangre (3,8).Los individuos afectos de una deficiencia de GALTpresentan la clínica de “galactosemia clásica”. Mani-festaciones tóxicas generales, en forma de rechazo delalimento vómitos, falta de medro y depresión neuro-lógica; que aparecen tras un intervalo libre después delnacimiento, y sólo después de la ingesta de galactosaprocedente de la leche de la madre o de una fórmulacon lactosa. Catarata nuclear “en gota de aceite” quepuede tener un inicio intrauterino, en casos excepcio-nales. Fracaso hepático grave, con ictericia, hepatoes-plenomegalia, ascitis y diátesis hemorrágica. Tubulo-patía proximal con acidosis, glucosuria, aminoaciduriay albuminuria. Déficit inmunitario con tendencia a sepsispor E. Coli.Uno de los aspectos menos conocidos de la galactosemiaes el motivo por el cual manifestaciones tardías de laenfermedad pueden presentarse a pesar del tratamientodietético adecuado. Afectación del Sistema NerviosoCentral en forma de declive progresivo del IQ, dispraxiaverbal, temblores cerebelosos y movimientos extrapira-midales. Disfunción ovárica con hipogonadismo hiper-gonadotrópico. Retraso del crecimiento prepuberal.Osteoporosis (12-19).
159
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
P7 17/5/07 17:57 Página 159

160
Galactosemia
Tab
la I
I. D
iagn
ósti
co d
e S
osp
ech
a d
e la
Gal
acto
sem
ia
I. C
lín
ica
de
Sos
pec
ha
A. S
ínto
mas
Tóx
icos
Vóm
itos
Rec
hazo
del
alim
ento
Falt
a d
e m
edro
Dep
resi
ón n
euro
lógi
caB
. Afe
ctac
ión
ofta
lmol
ógic
aC
atar
atas
C. F
raca
so h
epát
ico
grav
eIc
teri
cia
Hep
ato-
espl
enom
egal
iaA
scit
isD
iáte
sis
hem
orrá
gica
D. T
ubul
opat
ía p
roxi
mal
E. D
éfic
it i
nmun
itar
ioSe
psis
E. C
oli
II. B
ioq
uím
ica
Ines
pec
ífic
aA
. Dis
func
ión
hepá
tica
Hip
erbi
lirru
bine
mia
Hip
oalb
umin
emia
Déf
icit
com
plej
o pr
otro
mbi
naA
umen
to G
OT,
GPT
, GG
T, L
DH
Aum
ento
de
ácid
os b
iliar
es p
lasm
átic
osH
ipog
luce
mia
B. T
ubul
opat
ía p
roxi
mal
ren
alA
cid
osis
hip
ercl
orém
ica
Glu
cosu
ria
Am
inoa
cid
uria
Alb
umin
uria
III.
Bio
qu
ímic
a E
spec
ífic
aA
. Gal
ctos
uria
B. A
umen
to d
e ga
lact
osa
en p
lasm
aC
. Aum
ento
Gal
-1-P
erit
roci
tos
D. G
alac
tito
l en
pla
sma
y or
ina
P7 17/5/07 17:57 Página 160

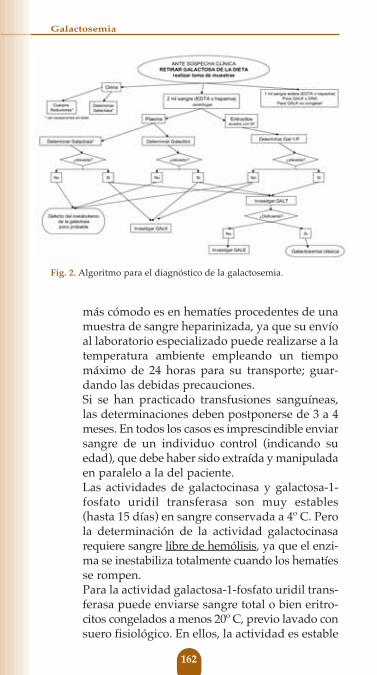
La identificación de galactosa en orina; junto a nivelesaumentados de galactosa en plasma, de galactitol ensangre y orina, y de Galactosa-1-P eritrocitaria, sonespecíficos de un bloqueo en cualquiera de las víasmetabólicas del catabolismo de la galactosa, que mientrasno se demuestre lo contrario está producido por undéficit enzimático congénito.La galactosuria debe identificarse con la reacción deClinitest® para cuerpos reductores; y es importanterecordar que su presencia exige la ingesta de galactosaen las horas previas a su determinación, ya que una dietasin galactosa de sólo 8 - 12 horas de duración puedehacerla desaparecer de la orina.Sin embargo, hay que tener presente que pequeñas can-tidades de galactosa en orina pueden presentarse enpacientes con enfermedades hepáticas, y que puedeexistir una glucosuria inespecífica como consecuenciadel daño tubular en las galactosemias.
II. Diagnóstico de certeza
A. EnzimáticoLa comprobación de la deficiencia enzimáticapuede practicarse en hematíes, fibroblastos culti-vados o en biopsia hepática. En la práctica los
161
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
Tabla III. Galactosemia por déficit GALT.Manifestaciones clínicas tardía
ALTERACIÓN DEL SISTEMA NERVIOSO CENTRALDisminución del DQ/IQ.Dispraxia verbal.Ataxia cerebelosa, temblor, movimientos extrapiramidales.
DEFICIENCIA OVÁRICAHipogonadismo hipergonadotrópico.
RETRASO DEL CRECIMIENTO
OSTEOPOROSIS
P7 17/5/07 17:57 Página 161
más cómodo es en hematíes procedentes de unamuestra de sangre heparinizada, ya que su envíoal laboratorio especializado puede realizarse a latemperatura ambiente empleando un tiempomáximo de 24 horas para su transporte; guar-dando las debidas precauciones.Si se han practicado transfusiones sanguíneas,las determinaciones deben postponerse de 3 a 4meses. En todos los casos es imprescindible enviarsangre de un individuo control (indicando suedad), que debe haber sido extraída y manipuladaen paralelo a la del paciente.Las actividades de galactocinasa y galactosa-1-fosfato uridil transferasa son muy estables(hasta 15 días) en sangre conservada a 4º C. Perola determinación de la actividad galactocinasarequiere sangre libre de hemólisis, ya que el enzi-ma se inestabiliza totalmente cuando los hematíesse rompen.Para la actividad galactosa-1-fosfato uridil trans-ferasa puede enviarse sangre total o bien eritro-citos congelados a menos 20º C, previo lavado consuero fisiológico. En ellos, la actividad es estable
162
Galactosemia
Fig. 2. Algoritmo para el diagnóstico de la galactosemia.
P7 17/5/07 17:58 Página 162

durante varios meses. El plasma separado y con-gelado inmediatamente es de utilidad para lavaloración de galactosa y de galactitol.La actividad de UDP-galactosa 4-epimerasa esmuy inestable incluso en eritrocitos congelados.Pueden enviarse sangre total o eritrocitos conge-lados, pero la actividad debe analizarse en los 3 – 4 días siguientes a la extracción.Para los tres enzimas es posible el diagnóstico deportadores, ya que los individuos heterozigotostienen una actividad enzimática intermedia.
B. MolecularPara los estudios mutacionales se precisan 2 - 5 mlde sangre con EDTA, pero la búsqueda selectivade una mutación concreta (por ejemplo la fre-cuente Q188R para el gen GALT) puede realizarsea partir de sangre seca sin heparina impregnadaen papel de filtro.
III. Diagnóstico prenatal
El diagnóstico prenatal de homozigotos es posiblemediante cuantificación de galactitol en líquido ami-niótico; valorando la actividad enzimática en vellosi-dades coriónicas o amniocitos cultivados; o por análisismutacional en aquellos casos en los que se conoce elgenotipo del caso índice.
IV. Diagnóstico sistemático del recién nacido
La investigación de la presencia de un error congénitodel metabolismo de la galactosa no ha sido universal-mente incluida en los programas de “screening” neo-natal de muchos países debido fundamentalmente alos problemas técnicos, y al hecho de que siempre quese instaure el tratamiento antes del segundo mes devida, no parece existir correlación entre el inicio deltratamiento y la aparición de complicaciones tardíasde la enfermedad (4). Sin embargo, puesto que el diag-nóstico y el tratamiento precoz están en relación directa
163
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
P7 17/5/07 17:58 Página 163

con la prevención de la morbilidad y mortalidad de laenfermedad durante la época neonatal, cada vez estámás extendida la investigación sistemática neonatalde la galactosemia y la implantación progresiva del“screening ampliado” mediante espectrometría de masasen tandem, que permite la medición, de galactosa total(galactosa y galactosa-1-fosfato) y de la de la actividadGALT seguramente facilitará la implantación universalde este diagnóstico (20).
Tratamiento
A. Deficiciencia de GALT
La galactosa de la dieta debe ser eliminada ante lamenor sospecha, incluso antes de confirmar el diag-nóstico; y el tratamiento debe mantenerse de porvida. Probablemente la ingesta diaria no deberíacontener nunca más de 125 mg de galactosa (frentea los 6.500 mg que por término medio tiene unadieta de un adulto normal); y una dieta estricta con-tiene aproximadamente unos 40 mg; pero puesto queno se conoce exactamente la cantidad de galactosaque puede ingerirse sin que resulte tóxica para elhígado, el riñón o el sistema nervioso central, debeprocurarse que los pacientes de riesgo reciban entodo momento la menor cantidad posible de galac-tosa (3,4,5,21). La principal fuente de galactosa de la dieta es la lac-tosa procedente casi exclusivamente de la leche demamíferos y de los derivados lácteos, pero tambiéndebe considerarse la presencia de lactosa en losexcipientes medicamentosos, en los alimentos manu-facturados y en una gran variedad de productoscomerciales. En el recién nacido y lactante debe suprimirse la lac-tancia materna y utilizar una leche exenta de lactosa.Lo ideal, es administrar una fórmula sin lactosacuyas proteínas procedan de la soja, ya que las fór-mulas sin lactosa, pero con hidrolizados de proteínas
164
Galactosemia
P7 17/5/07 17:58 Página 164

vacunas pueden seguir conteniendo lactosa en lacaseína y en la seroalbúmina. A partir del año de vidase puede pasar de una fórmula infantil de soja a unpreparado líquido de soja, algunos de los cuales vie-nen enriquecidos en calcio. Sin embargo, no son unabuena fuente de vitaminas y minerales, por lo que siel niño no tiene un buen apetito es mejor continuarcon una fórmula de soja.Con la introducción de la alimentación complemen-taria es algo más complicado mantener una dietaabsolutamente libre de galactosa, debido a las difi-cultades existentes para conocer el contenido realde galactosa libre o ligada de los alimentos, y al des-conocimiento que se posee acerca de la capacidadde utilización por parte del organismo de la galac-tosa ligada en enlaces α o β (22,-25).Existe galactosa ligada con enlaces tipo β en ciertaslegumbres, verduras y frutas; ligada con enlacestipo α (en forma de galactósidos) en algunos polisa-cáridos de origen vegetal y en forma de galactocere-brósidos y gangliósidos la galactosa es componentede algunas vísceras de animales (cerebro, riñón,hígado, páncreas o bazo).
165
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
Tabla IV. Contenido aproximado en galactosa solublede algunos alimentos (mg/100 gr de alimento)
Menos de 5 mgAlbaricoque, aguacate, melón, uva, pomelo, naranja,
fresas, mango, olivas verdes.Espárrago, remolacha de azúcar, repollo, coliflor, apio,
pepino, berenjena, col rizada, patata, rábano, espinaca,calabacín, lechuga y maíz.
Entre 5 y 10 mgManzana, melocotón, plátano, pera.Judías, brócoli, zanahoria, cebolla, nabo, patata.
Entre 10 y 20 mgKiwi, sandía, dátil, piña.Col de Bruselas, calabaza, sandía, batata.
P7 17/5/07 17:58 Página 165

El organismo humano tiene α-galactosidasas enmuchos tejidos, capaces de degradar los enlaces α,pero no tiene en el intestino delgado, y por ello estetipo de enlaces parece que sólo pueden ser escindidosa partir de la α-galactosidasa presente en bacteriasdel intestino grueso cuando colonizan el delgado acausa de una diarrea, por ejemplo. Por el contrario,β-galactosidasas, capaces de hidrolizar enlaces β, sehan encontrado en muchos alimentos (manzanas,guisantes, pimienta, tomate, etc..), y en el intestinohumano dónde digieren la lactosa y en diferentemedida los productos con galactosa ligada en enlacesβ-1,4.De cualquier manera, y desde un punto de vistapráctico estos alimentos no lácteos son en todos loscasos una fuente insignificante de galactosa compa-rados con la producción endógena de galactosa, porlo que en este momento no existe evidencia científicaque apoye su exclusión.Debe recordarse que lactosa se utiliza en la indus-tria como ingrediente y vehículo de transporte desaborizantes y en medicamentos. En España, lareglamentación vigente sobre “excipientes de decla-
166
Galactosemia
Tabla IV. Contenido aproximado en galactosa solublede algunos alimentos (mg/100 gr de alimento)
Entre 20 y 30 mgPapaya, caqui, arándano.Tomate.
Entre 30 y 50 mgSoja, alubia pinta.
Más de 100 mgLentejas, judías blancas, guisantes.
Más de 400 mgHigos secos, pasas, avellanas.Garbanzos.
Continuación de la tabla IV.
P7 17/5/07 17:58 Página 166

ración obligatoria” incluye a la lactosa siempreque la dosis máxima diaria exceda de 5 gramos. Encambio, la galactosa se debe declarar siempre a cual-quier dosis.Es prácticamente imposible por el contrario, conocerla cantidad de galactosa libre o ligada de la mayoríade los productos comestibles manufacturados de unmodo artesanal, y debe desconfiarse de cualquierreceta no preparada en casa y con productos básicosbien conocidos. Los manufacturados de un modoindustrial tampoco son fiables debido a que las nor-mas legales que regulan el etiquetado permiten quepuedan pasar desapercibidas cantidades indetermi-nadas de galactosa (lactosa), especialmente en formade saborizantes o edulcorantes artificiales. Muchosderivados lácteos se añaden a productos manufac-turados como galletas, postres, pasta y algunospanes. Por otro lado, puede existir confusión conpalabras que pueden hacer pensar a los padres quela leche es un ingrediente de la dieta cuando real-mente no lo es (Tabla V).
Confección de la dieta diariaCon el fin de añadir la menor cantidad posible degalactosa a la síntesis endógena propia, es precisovalorar todas las fuentes de galactosa exógena queel paciente recibe en la confección práctica de ladieta diaria. Los protocolos de soporte nutricional para la galac-tosemia (24,25) han venido dividiendo los alimentos
167
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
Tabla V. Derivados no lácteos
Ácido láctico E270, lactato sódico E325, lactato patásicoE325, lactato cálcico E327.
Lactitiol, lactoalbúmina, lactoglobulina, licasina, lactilatos,lactosa-glucona-delta, glutamato monosódico,mantequilla de coco, crema no-láctea
P7 17/5/07 17:58 Página 167

en tres grupos: los de libre utilización porque prác-ticamente no poseen galactosa en su composición(≤ 5 mg/100 gr); los que deben ser usados con pru-dencia y bajo controles analíticos seriados, porquecontienen por término medio entre 5-20 mg/100 grde galactosa; y los prohibidos porque son muy ricosen galactosa (≥ 20 mg/dl) (Tabla VI). En la actualidad existe acuerdo prácticamente uná-nime para eliminar la leche y derivados de la dietade por vida, porque observaciones aisladas queparecen evidenciar que después de la infancia laintroducción de la galactosa en la dieta no causaproblemas clínicos en algunos pacientes necesitanser confirmadas con seguridad. No existe el mismo consenso acerca del uso de pro-ductos naturales (legumbres, frutas y vísceras ) en ladieta de la galactosemia. Parece que el aporte efec-tivo de galactosa procedente de estos alimentos esmenor que la producción endógena diaria de galac-tosa del propio paciente, no parece que su inclusióno eliminación de la dieta modifique parámetros bio-químicos o clínicos de los pacientes y además no hayuna relación clara entre la rigurosidad de la dieta yla aparición de manifestaciones tardías de la enfer-medad. Probablemente evitar el abuso de estos ali-mentos, sin excluirlo por completo de la dieta es unaconducta prudente por el momento (4).Mención especial merece el uso de quesos(Emmentaler, Gruyère, Tilster) confeccionadosmediante un proceso de fermentación natural que“consume la lactosa” y les hace seguros, en teoría,para la dieta de los individuos afectos. Cuando nose tiene constancia absoluta de este procesado natural(y es posible que los quesos hayan sido sometidos auna producción industrial masiva), no deben serutilizados porque no compensa correr un riesgoinnecesario con estos alimentos. Sólo deben ser uti-lizados con las debidas garantías de origen.
168
Galactosemia
P7 17/5/07 17:58 Página 168

169
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
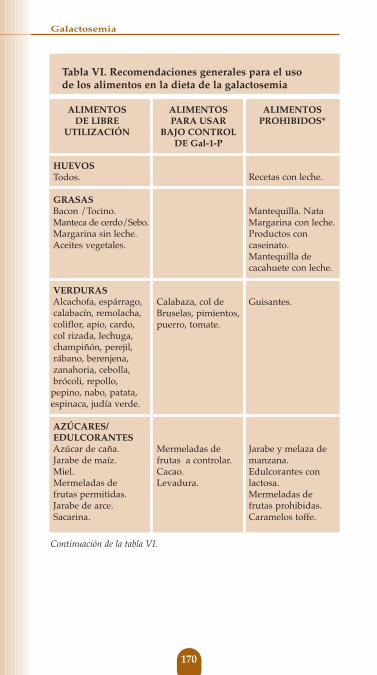
Tabla VI. Recomendaciones generales para el uso de los alimentos en la dieta de la galactosemia
ALIMENTOS ALIMENTOS ALIMENTOSDE LIBRE PARA USAR PROHIBIDOS*
UTILIZACIÓN BAJO CONTROLDE Gal-1-P
BEBIDASCafé, vino, cerveza, té.Bebidas carbónicas.
LECHE Y DERIVADOSFórmulas de soja.“Leche de soja” enriquecida con Ca.Queso de soja, yogur de soja, helado de soja, postres de soja.
CEREALESTrigo, cebada, avena, centeno, maíz, avena, arroz.
Todas las pastas manufacturadas sin leche: fideos, macarrones, espaguetis,tortitas, palomitas de maíz sin mantequilla, etc..
REPOSTERÍACabello de ángel, gelatina.Todos los manufacturados con productos sin leche.
Cualquier bebida con leche, lactosa, caseína, caseinato cálcico, caseinato sódico, hidrolizado de caseína.
Leche y derivados (Flanes, cremas, yogur, quesos, helados, nata, mantequilla, grasa animal, ghee, crema artificial, seroalbúmina, margarinas que contengan seroalbúmina, seroalbúmina vegetariana).Bebidas con leche.
Todos los manufacturados con leche.
Todos los manufacturados con elementos prohibidos.
Fórmulas de soja con harina de soja.
Harina de soja.Harina de girasol.
P7 17/5/07 17:58 Página 169
170
Galactosemia
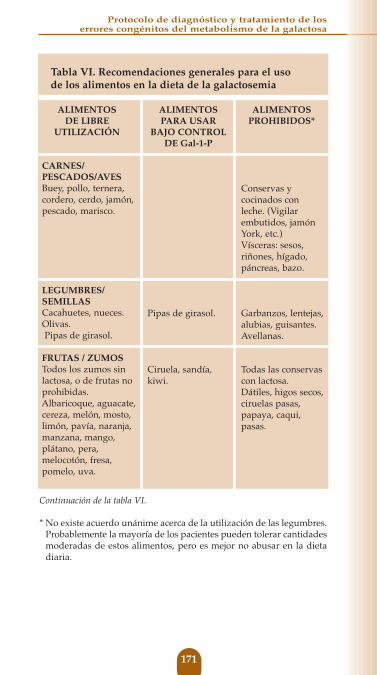
Tabla VI. Recomendaciones generales para el uso de los alimentos en la dieta de la galactosemia
ALIMENTOS ALIMENTOS ALIMENTOSDE LIBRE PARA USAR PROHIBIDOS*
UTILIZACIÓN BAJO CONTROLDE Gal-1-P
HUEVOSTodos.
GRASASBacon /Tocino.Manteca de cerdo/Sebo.Margarina sin leche.Aceites vegetales.
VERDURASAlcachofa, espárrago, calabacín, remolacha, coliflor, apio, cardo, col rizada, lechuga, champiñón, perejil, rábano, berenjena, zanahoria, cebolla, brócoli, repollo,
pepino, nabo, patata, espinaca, judía verde.
AZÚCARES/EDULCORANTESAzúcar de caña.Jarabe de maíz.Miel.Mermeladas de frutas permitidas.Jarabe de arce.Sacarina.
Recetas con leche.
Mantequilla. NataMargarina con leche.Productos con caseinato.Mantequilla de cacahuete con leche.
Guisantes.
Jarabe y melaza de manzana.Edulcorantes con lactosa.Mermeladas de frutas prohibidas.Caramelos toffe.
Calabaza, col de Bruselas, pimientos, puerro, tomate.
Mermeladas de frutas a controlar.Cacao.Levadura.
Continuación de la tabla VI.
P7 17/5/07 17:58 Página 170
171
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
Tabla VI. Recomendaciones generales para el uso de los alimentos en la dieta de la galactosemia
ALIMENTOS ALIMENTOS ALIMENTOSDE LIBRE PARA USAR PROHIBIDOS*
UTILIZACIÓN BAJO CONTROLDE Gal-1-P
CARNES/PESCADOS/AVESBuey, pollo, ternera, cordero, cerdo, jamón, pescado, marisco.
LEGUMBRES/SEMILLASCacahuetes, nueces.Olivas.Pipas de girasol.
FRUTAS / ZUMOSTodos los zumos sin lactosa, o de frutas no prohibidas.Albaricoque, aguacate, cereza, melón, mosto, limón, pavía, naranja, manzana, mango, plátano, pera, melocotón, fresa, pomelo, uva.
Conservas y cocinados conleche. (Vigilarembutidos, jamónYork, etc.)Vísceras: sesos, riñones, hígado, páncreas, bazo.
Garbanzos, lentejas,alubias, guisantes.Avellanas.
Todas las conservascon lactosa.Dátiles, higos secos,ciruelas pasas, papaya, caqui,pasas.
Pipas de girasol.
Ciruela, sandía, kiwi.
Continuación de la tabla VI.
* No existe acuerdo unánime acerca de la utilización de las legumbres.Probablemente la mayoría de los pacientes pueden tolerar cantidadesmoderadas de estos alimentos, pero es mejor no abusar en la dietadiaria.
P7 17/5/07 17:58 Página 171

Suplemento de CalcioEs habitual que, después de la época de la lactancia,una dieta para niños galactosémicos que no estéexpresamente suplementada, no asegure la ingestade los aportes diarios adecuados de calcio; y porello, a partir de esa edad es probable que sea nece-sario utilizar un suplemento de Ca por vía oral (26).La dosis depende de la ingesta dietética en cada caso,y a ser posible se usará Carbonato cálcico (1 gramoproporciona 400 mg de calcio elemental) o Pidolatocálcico (250 ml proporcionan 25 mg de Ca) por sumenor efecto quelante. No debe utilizarse lactobio-nato de calcio porque es una fuente significativa degalactosa. Mientras dure el tratamiento es precisomonitorizar el Ca en orina con el fin de detectarhipercalciuria por sobredosificación. Tabla VII.
Tratamiento hormonalEn las mujeres con déficit GALT e hipergonadismohipergonadotropo es necesario iniciar la terapiacon etinilestradiol a partir de los 13 años de vida, silas gonadotropinas son altas y el estriol bajo. Poste-riormente, a los tres años de evolución se instauratratamiento de contracepción oral con etinilestradioly progesterona (4-21).
172
Galactosemia
Tabla VII. Ingestas dietéticas recomendadas de calcio(National Academy of Sciences, 1997)
EDAD mg/día
0 – 6 meses 210
6 – 12 meses 270
1 – 3 años 500
4 – 8 años 800
9 – 18 años 1.300
P7 17/5/07 17:58 Página 172

Otros tratamientosLos intentos de tratamiento del déficit GALT coninhibidores de la galactokinasa, con el fin de con-vertir los pacientes GALT en pacientes GALK, o conuridina, no han resultado efectivos por el momento.
Control del tratamientoLos marcadores utilizados para el seguimiento deltratamiento dietético son los niveles de Galactosa-1-Peritrocitarios, y galactitol plasmático. Al iniciar ladieta Gal-1-P, y galactitol descienden rápidamentehasta los 2-3 meses de vida. A partir de ese momentolo hacen de un modo expotencial hasta los 8-10 años;pero sólo llegan a normalizarse por completo en“variantes” con mucha actividad residual (27-28).Valores de 4 mg % de Gal-1-P, y de 25-30 μmol/L degalactitol plasmático (mg/dl : 38 × μmol/L), es pro-bable que sean los máximos que puedan aceptarsecomo buenos, bajo dieta exenta de galactosa; perode cualquier modo, debido a la gran variabilidadbiológica individual y a la falta de correlación biendefinida con la ingesta de galactosa, su máximo sig-nificado lo adquieren cuando se usan de un modoseriado a lo largo del tiempo para ver la evoluciónde cada individuo.Para su determinación se requieren un mínimo de2-3 ml de sangre heparinizada. Lo idóneo es separarel plasma inmediatamente por centrifugación ycongelarlo. Los eritrocitos deben lavarse dos vecescon suero fisiológico, y desechando cuidadosamenteel total del suero sobrenadante, congelar el sedimen-to de hematíes y enviarlo al laboratorio, junto con elplasma, en nieve carbónica.
Tratamiento dietético de la mujer embarazadaEl embarazo puede ocurrir en una madre galactosé-mica (madre homozigota con feto heterozigoto) oen una madre que ha tenido previamente un niñocon galactosemia (madre heterozigota con posiblefeto homozigoto). En el primer caso debe mantenerse
173
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
P7 17/5/07 17:58 Página 173

la dieta exenta en lactosa, ya que a pesar de que lagalactosa-1-fosfato no atraviesa la placenta, si lohacen la galactosa y el galactitol, y este último podríadañar el cristalino del feto. En el segundo casoaunque la madre siga una dieta sin lactosa, se acu-mula galactosa–1-fosfato y galactitol en el líquidoamniótico provenientes de la síntesis endógena querealiza el feto, pero es prudente evitar la lactosadurante el embarazo con el fin de limitar en lo posiblelas fuentes de galactosa del feto (4,21).
Tratamiento de la deficiencia parcial de GALTNo existe por el momento un consenso universalacerca del tratamiento de los pacientes asintomáticos,identificados por “screening” neonatal o familiar, ycon una actividad GALT del 10-50%. La mayoría deautores se inclina por tratar a todos los recién nacidoshasta los 4 meses de vida; y a partir de ese momentocomprobar de un modo progresivo, y bajo controlesde los niveles de galactosa-1-fosfato, el efecto de laintroducción de la galactosa en la dieta (21).
B. Deficiencia de GALK
Debe procederse a la eliminación de la leche de ladieta; pero parecen tolerarse otras fuentes menoresde galactosa como derivados de la leche, legumbres,etc. Si el tratamiento es precoz, las cataratas puedenresolverse. Si el diagnóstico es tardío es habitual quelas cataratas deben ser operadas y debido al peligrode recidiva, la dieta sin leche debe mantenerse depor vida (21).
C. Deficiencia de Epimerasa
Las formas “periféricas” no precisan tratamiento, perodeben ser controladas cuidadosamente. Las formasgraves deben ser tratadas de por vida con dieta res-trictiva en galactosa; pero puesto que estos pacientesson galactosa-dependientes para sintetizar los com-
174
Galactosemia
P7 17/5/07 17:58 Página 174

puestos glicosilados necesarios para el S.N.C., esdifícil conseguir el equilibrio adecuado entre aportey necesidades de galactosa de la dieta (21).
Control general de la evolución de los pacientesLa frecuencia de los controles y los exámenes a rea-lizar en cada uno de ellos, se establecerá en funciónde las necesidades individuales; pero un protocolosistemático favorece la calidad del tratamiento.(27,28).
1. Déficit GALT Frecuencia teórica de los controles< 1 año: cada 3 meses.1-4 años: cada 4 meses.> 4 años: cada 6 meses.> 18 años: anual.Examen clínicoAntropometría: Peso, talla, perímetro craneal entodos los controles.Examen neurológico. En todos los controles.Valoración del CD/IQ y del lenguaje. A los 3, 6, 12meses y una vez al año hasta los 6 años. Despuésuna vez cada dos años.Encuesta dietética y del tratamientoEn todos los controles.Exámenes ComplementariosI. Bioquímicos
- Generales: Hemograma, iones, proteínas totales,función hepática y renal, calcio, fósforo, fosfa-tasas alcalinas, y cociente calcio/creatinina enorina. Al diagnóstico y semestralmente hastalos 18 años de vida. Posteriormente cada dosaños, y en función de las necesidades de cadapaciente.
- Específicos: Galactosa-1-fosfato eritrocitaria,y/o galactitol. Al diagnóstico, y trimestral-mente durante el primer año de vida. Cada 6meses entre los 2 y los 18 años. Posteriormenteuna vez al año.
175
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
P7 17/5/07 17:58 Página 175

II. Examen oftalmológicoExamen con lámpara de hendidura una vezal año hasta los 18 años. Posteriormente unavez cada 2 años.
III. Densitometría óseaA los 2, 4, 8, 12 y 16 años.No se recomienda el uso de radiografías paravaloración de masa ósea. Durante la infancia es buena alternativa a laDEXA, la densitometría ósea por ultrasonidos.
IV. EEGEn función de la evolución del paciente.
V. Resonancia magnética nuclearEn función de la evolución del paciente.
VI. Examen de la función gonadal.Mujeres:
Test de LH-RH lo más precoz posible (si esposible antes de los 6 meses de vida, y siem-pre antes de los 3 años). Repetir a los 10 añosy a los 12 años. Si es normal repetir siempreque exista sospecha de alteración gonadal.ECO pélvica. Simultáneamente con el testLH-RH.
2. Déficit de GALKControl oftalmológico y presencia de galactosaen orina una vez al año, hasta los 18 años y unavez cada dos años, posteriormente.
3. Déficit de GALEControl clínico, examen oftalmológico, hemogra-ma, función hepática y renal, examen neurológico yvaloración cognitiva. Cada tres meses el primeraño de vida, cada 4 meses hasta los 4 años, semes-tralmente hasta los 18 años y una vez al añoposteriormente.
Referencias
1. Robert JJ. Métabolisme des hydrates de carbone. En RicourC, Ghisolfi J, Putet G, Goulet O (eds). Traité de NutritionPédiatrique. Paris. Maloine, 1993; pag. 28-32.
176
Galactosemia
P7 17/5/07 17:58 Página 176

2. Holton JB, Walter JH, Tyfield LA. Galactosemia. EnScriver Ch R, Beaudet A L, Sly W S, Valle D (eds). Themetabolic and molecular bases of inherited disease. NewYork, McGraw Hill, 2001; pag. 1553-1588.
3. Gitzelmann R. Disorders of galactose metabolism. EnFernandes J, Saudubray JM, van den Berghe G, Walter JH(eds). Inborn metabolic diseases. Diagnosis and treatment.,Springer Medizin Verlag, Heidelberg 2006; pag. 121-130.
4. Bosch A. Classical galactosaemia revisted. J Inher MetabDis, 2006: 29: 516-525.
5. Baldellou A. Errores congénitos del metabolismo de lagalactosa, en Sanjurjo P, Baldellou A, ed. Diagnóstico yTratamiento de las enfermedades metabólicas hereditarias.Ergon, SA. Madrid, 2006, pag. 283-292.
6. Boleda M, Girós ML, Briones P, Sanchis A, Álvarez L,Balaguer S, Holton JB. Severe neonatal galactose-depen-dent disease with low-normal epimerase activity. J InherMetab Dis 1995; 18: 88-89.
7. Gort L, Boleda MD, Tyfield L, Vilarinho L, Rivera I, CardosoML, Santos –Leite M, Girós M, Briones P. Mutationalspectrum of classical galactosaemia in Spain and Portugal.J Inher Metab Dis 2006; Oct 13 (Epub ahead of print).
8. Holton J. Galactosemia: pathogenesis and treatment. J InherMetab Dis 1996; 19: 3-7.
9. Berry GT, Nissim I, Gibson JB, Mazur AT, Lin Z, Elsas LJ,Singh RH, Klein PD, Segal S. Quantitative assessment ofwhole body galactose metabolism in galactosemic patients.Eur J Pediatr 1997; 156 (supp 1); 43-49.
10. Segal S. Galactosemia today. The enigma and the challenge.J Inher Metab Dis 1998; 21: 455-471.
11. Martínez Pardo M. Trastornos del metabolismo de loshidratos de carbono. Actualización en su diagnóstico ytratamiento. Act Nutr 1998; 24: 35-42.
12. Waggoner DD, Buist NRM, Donell GN. Long-term prog-nosis in galactosaemia. Results of a survey of 350 cases. JInher Metab Dis 1990: 13; 802-818.
13. Nelson CD, Waggoner DD, Donell GN, Tuerk JM, Buist NR.Verbal dispraxia in treated galactosaemia. Pediatrics 1991;88: 346-350.
14. Honeyman MM, Green A, Holton JB, Leonard JV.Galactosaemia: results of the British Paediatric SurveillanceUnit study, 1988-90. Arch Dis Child 1993; 69: 339-341.
15. Schweitzer Y, Shin C, Jacobs I, Brodehl J. Long-termoutcome in 134 patients with galactosaemia. Eur J Pediatr1993; 152: 36-43.
177
Protocolo de diagnóstico y tratamiento de los errores congénitos del metabolismo de la galactosa
P7 17/5/07 17:58 Página 177

16. Kaufman FR, Loro ML, Azen C, Wenz E, Gilsanz V. Effect ofhypogonadism and deficient calcium intake on bone densi-ty in patients with galactosemia. J Pediatr 1993; 123: 365-70.
17. Widhalm B, Mirande da Cruz B, Koch M. Diet does notensure normal development in galactosemia. J Am CollNutr 1997; 16(3): 204-208.
18. Prestoz LLC, Couto AS, Shin YS, Petry KG. Altered folliclestimulating hormone isoforms in female galactosaemiapatients. Eur J Pediatr 1997; 156: 116-120.
19. Ridel KR, Leslie ND, Gilbert DL. An updated review of thelong-term neurological effects of galactosemia. PediatrNeurol 2005; 33:153-161.
20. Schweitzer-Krantz S. Early diagnosos of inherited meta-bolic disorders towards improving outcome: the contro-versial issue of galactosaemia. Eur J Pediatr 2003; 162(supp 1): S50-53.
21. Raque JP, Visser G, Smit PA. Disorders of carbohydrateand glycogen metabolism, in Blau N, Hoffmann GF,Leonard J, Clarke J TR ed. Physicians guide to the treatmentand follow-up of metabolic diseases. Springer Verlag,Berlin-Heilderberg, 2006: pag. 161-180.
22. Gross KC, Acosta PB. Fruits and vegetables are a sourceof galactose: Implications in planning the diets of patientswith galactosemia. J Inher Metab Dis 1991; 14: 253-258.
23. White F. Overview of diet classical galactosaemia. InbornErrors Review Series, number 7, SHS, 1997.
24. Acosta PB, Yanicelli S. The Ross Metabolic Formula System.Nutrition Support Protocols. 3 rd ed. Columbus OH: RossProduct Division, 1997: 277-296.
25. Walter JH, Collins JE, Leonard JV, on behalf of the UKGalactosaemia Steering Group. Recommendations forthe management of galactosaemia. Arch Dis Child 1999;80: 93-96.
26. Dietary Reference Intakes (DRIs) for calcium, Phosphorus,Magnesium, Vitamin D and Fluoride. Food and NutritionBoard. Institute of Medicine, National Academy of Sciences.National Academy Press. Washington DC, 1997.
27. Poll-The BT, Schweitzer SM. Monitoring of the treatmentin classical galactosemia. 27th Conference of the EuropeanMetabolic Group. Milupa, 1994.
28. Hutchesson ACJ, Murdoch-Davis C, Green A, Preece MA,Allen J, Holton JB, Rylance G. Biochemical monitoringof treatment for galactosemia: Biological variability inmetabolite concentrations. J Inher Metab Dis 1999; 22:139-148.
178
Galactosemia
P7 17/5/07 17:58 Página 178