instituto politécnico nacional unidad profesional ... didactico... · web viewdisolver en 40 ml de...
TRANSCRIPT

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
INSTITUTO POLITÉCNICO NACIONAL
UNIDAD PROFESIONAL INTERDISCIPLINARIA DE BIOTECNOLOGÍA
DEPARTAMENTO DE BIOPROCESOS
ACADEMIA DE BIOTECNOLOGÍA
MANUAL DEL LABORATORIO DE BIOTECNOLOGÍA MOLECULAR
ELABORADO POR:
M. en C. Paola Berenice Zárate Segura
I. Bt. César A. Jiménez Sierra
México D. F. Abril 2009

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 1. BIOSEGURIDAD
DEFINICIONES OPERACIONALES
1) BIOSEGURIDAD: Debe entenderse como una doctrina de comportamiento encaminada a lograr actitudes y conductas que disminuyan el riesgo del trabajador de la salud de adquirir infecciones en el medio laboral. Compromete también a todas aquellas otras personas que se encuentran en el ambiente asistencial, ambiente éste que debe estar diseñado en el marco de una estrategia de disminución de riesgos.
Los principios de BIOSEGURIDAD se pueden resumir en:
A) Universalidad: Las medidas deben involucrar a todos los pacientes de todos los servicios, independientemente de conocer o no su serología.Todo el personal debe seguir las precauciones estándares rutinariamente para prevenir la exposición de la piel y de las membranas mucosas, en todas las situaciones que puedan dar origen a accidentes, estando o no previsto el contacto con sangre o cualquier otro fluido corporal del paciente.Estas precauciones, deben ser aplicadas para TODAS las personas, independientemente de presentar o no patologías.
B) Uso de barreras: Comprende el concepto de evitar la exposición directa a sangre y otros fluidos orgánicos potencialmente contaminantes, mediante la utilización de materiales adecuados que se interpongan al contacto de los mismos.La utilización de barreras (ej. guantes) no evitan los accidentes de exposición a estos fluidos, pero disminuyen las consecuencias de dicho accidente.
C) Medios de eliminación de material contaminado: Comprende el conjunto de dispositivos y procedimientos adecuados a través de los cuales los materiales utilizados en la atención de pacientes, son depositados y eliminados sin riesgo.
LEY DE BIOSEGURIDAD DE ORGANISMOS GENÉTICAMENTE MODIFICADOS
V. Bioseguridad: Las acciones y medidas de evaluación, monitoreo, control y prevención que se deben asumir en la realización de actividades con organismos genéticamente modificados, con el objeto de prevenir, evitar o reducir los posibles riesgos que dichas actividades pudieran ocasionar a la salud humana o al medio ambiente y la diversidad biológica, incluyendo los aspectos de inocuidad de dichos organismos que se destinen para uso o consumo humano.
VI. Biotecnología moderna: Se entiende la aplicación de técnicas in vitro de ácido nucleico, incluidos el ácido desoxirribonucleico (ADN y ARN) recombinante y la inyección

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
directa de ácido nucleico en células u organelos, o la fusión de células más allá de la familia taxonómica, que supera las barreras fisiológicas naturales de la reproducción o de la recombinación y que no son técnicas utilizadas en la reproducción y selección tradicional, que se aplican para dar origen a organismos genéticamente modificados, que se determinen en las normas oficiales mexicanas que deriven de esta Ley. (LEY DE BIOSEGURIDAD DE ORGANISMOS GENÉTICAMENTE MODIFICADOS Nueva Ley DOF 18-03-2005)
Principios de Bioseguridad
El término “contención” se utiliza para describir métodos seguros para manejar materiales infecciosos en el medio ambiente del laboratorio donde son manipulados o conservados. El objetivo de la contención es reducir o eliminar la exposición de quienes trabajan en laboratorios u otras personas, y del medio ambiente externo a agentespotencialmente peligrosos.La contención primaria, la protección del personal y del medio ambiente inmediatodel laboratorio de la exposición a agentes infecciosos, es provista tanto mediante buenas técnicas microbiológicas como a través del uso de equipos de seguridad adecuados. El uso de vacunas puede brindar un mayor nivel de protección del personal. La contención secundaria, la protección del medio ambiente externo al laboratorio de la exposición a materiales infecciosos, se logra a través de una combinación del diseño de la instalación y prácticas operativas. Por lo tanto, los tres elementos de contención incluyen prácticas y técnicas de laboratorio, equipos de seguridad y el diseño de la instalación. La evaluación del riesgo del trabajo a realizar con un agente específico determinará la combinación apropiada de estos elementos.
Equipos de Seguridad (Barreras Primarias). Los equipos de seguridad incluyen gabinetes de seguridad biológica (BSCs), recipientes cerrados, y otros controles de ingeniería destinados a eliminar o minimizar las exposiciones a materiales biológicospeligrososUn ejemplo de otra barrera primaria es la cubeta centrífuga de seguridad, unrecipiente cerrado destinado a prevenir la liberación de aerosoles durante el centrifugado.Diseño y Construcción de Instalaciones (Barreras Secundarias)El diseño y la construcción de la instalación contribuyen a la protección de quienes trabajan en el laboratorio, proporcionan una barrera para proteger a las personas que se encuentran fuera del laboratorio, y protegen a las personas o animales de la comunidad de agentes infecciosos que pueden ser liberados accidentalmente del laboratorio.
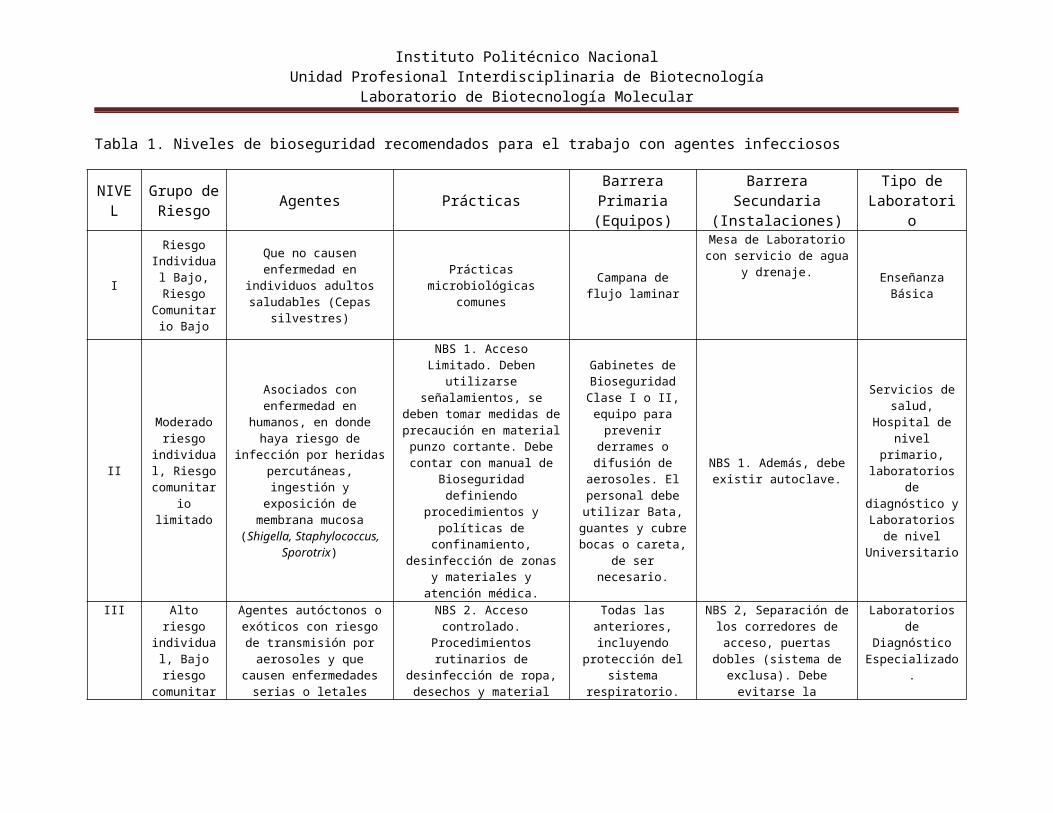
Niveles de Bioseguridad. Existen cuatro niveles de bioseguridad (BSLs), que constan de combinaciones de prácticas y técnicas de laboratorio, equipos de seguridad e instalaciones de laboratorio. Cada combinación es específicamente apropiada para las operaciones llevadas a cabo, las vías de transmisión documentadas o sospechadas de los agentes infecciosos, y la función o la actividad del laboratorio (tabla 1).

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología Laboratorio de Biotecnología Molecular
Tabla 1. Niveles de bioseguridad recomendados para el trabajo con agentes infecciosos
NIVEL
Grupo deRiesgo Agentes Prácticas Barrera Primaria
(Equipos)Barrera Secundaria
(Instalaciones)Tipo de
Laboratorio
I
Riesgo Individual
Bajo, Riesgo Comunitario
Bajo
Que no causen enfermedad en individuos adultos saludables (Cepas
silvestres)
Prácticas microbiológicas comunes
Campana de flujo laminar
Mesa de Laboratorio con servicio de agua y drenaje. Enseñanza
Básica
II
Moderado riesgo
individual, Riesgo
comunitario limitado
Asociados con enfermedad en humanos, en donde haya
riesgo de infección por heridas percutáneas,
ingestión y exposición de membrana mucosa
(Shigella, Staphylococcus, Sporotrix)
NBS 1. Acceso Limitado. Deben utilizarse
señalamientos, se deben tomar medidas de precaución en material punzo cortante. Debe contar con manual de
Bioseguridad definiendo procedimientos y políticas de confinamiento, desinfección
de zonas y materiales y atención médica.
Gabinetes de Bioseguridad Clase I
o II, equipo para prevenir derrames o
difusión de aerosoles. El
personal debe utilizar Bata, guantes
y cubre bocas o careta, de ser
necesario.
NBS 1. Además, debe existir autoclave.
Servicios de salud, Hospital
de nivel primario, laboratorios de diagnóstico y
Laboratorios de nivel
Universitario
III
Alto riesgo individual,
Bajo riesgo comunitario
Agentes autóctonos o exóticos con riesgo de
transmisión por aerosoles y que causen enfermedades serias o letales (Brucella,
Hystoplasma)
NBS 2. Acceso controlado. Procedimientos rutinarios de
desinfección de ropa, desechos y material previo al
lavado.
Todas las anteriores, incluyendo
protección del sistema respiratorio.
NBS 2, Separación de los corredores de acceso,
puertas dobles (sistema de exclusa). Debe evitarse la
recirculación del aire. Presión de aire negativa en
el área de trabajo
Laboratorios de Diagnóstico
Especializado.
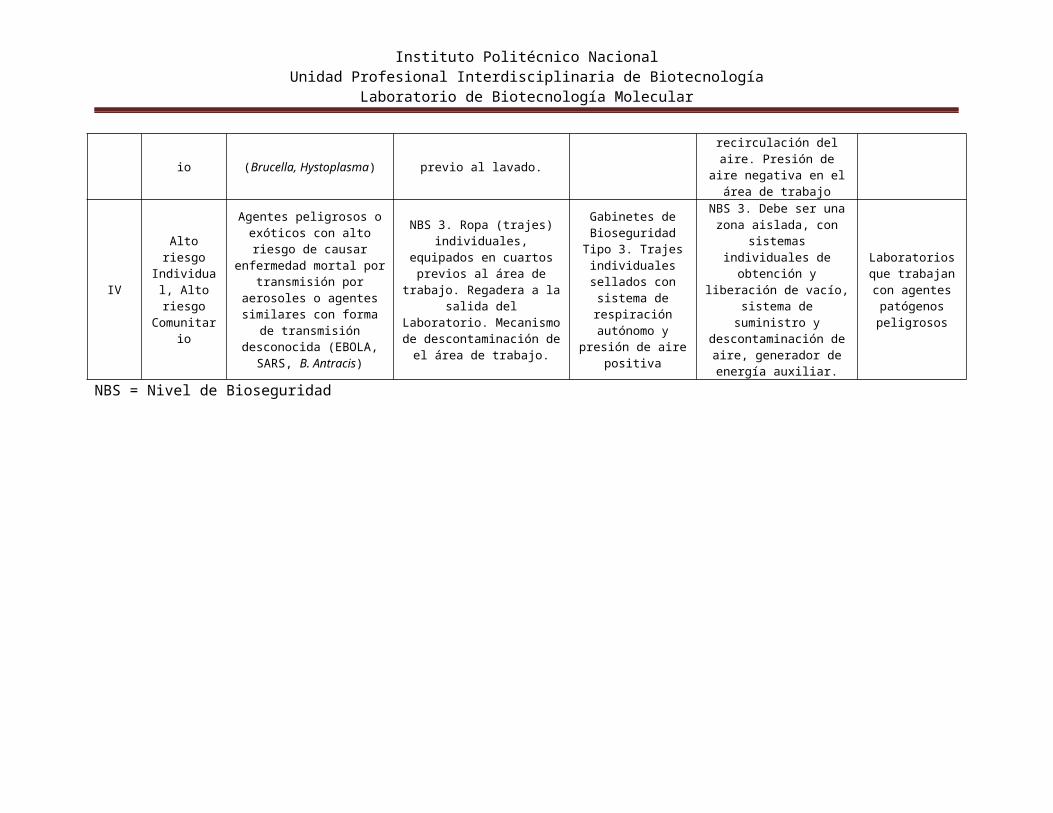
IV
Alto riesgo Individual, Alto riesgo
Comunitario
Agentes peligrosos o exóticos con alto riesgo de causar enfermedad mortal
por transmisión por aerosoles o agentes
similares con forma de transmisión desconocida
(EBOLA, SARS, B. Antracis)
NBS 3. Ropa (trajes) individuales, equipados en cuartos previos al área de
trabajo. Regadera a la salida del Laboratorio. Mecanismo de descontaminación de el
área de trabajo.
Gabinetes de Bioseguridad Tipo 3. Trajes individuales
sellados con sistema de respiración
autónomo y presión de aire positiva
NBS 3. Debe ser una zona aislada, con sistemas
individuales de obtención y liberación de vacío,
sistema de suministro y descontaminación de aire,
generador de energía auxiliar.
Laboratorios que trabajan con
agentes patógenos peligrosos
NBS = Nivel de Bioseguridad

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Objetivo:
Informar y motivar a los alumnos sobre principios y normas de bioseguridad para su aplicación activa en el trabajo de laboratorio
Actividades:
Actividad dinámica por los alumnos
Identificación y descripción de equipo en el laboratorio
Referencias
http://www.cdc.gov/od/ohs/pdffiles/bmbl4_spanish.pdf
LEY DE BIOSEGURIDAD DE ORGANISMOS GENÉTICAMENTE MODIFICADOS Nueva Ley DOF 18-03-2005
Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 2. CUANTIFICACIÓN DE ADN
La cuantificación del ADN se puede estimar por visualización en geles de agarosa en el que el ADN es teñido por algún colorante com Bromuro de Etidio, Syber Green o Gel Red y mediante espectrofotómetro
La estimación de ADN por método espectrofotométrico a 260 mn y 280 nm en una celda de cuarzo, para que la lectura sea adecuada se requiere la habilidad de medir de manera exacta y reproducible, el transferir volúmenes pequeños de líquidos son críticos para obtener resultados útiles. Para los volúmenes menores de 1 mL, el método más común para medir líquidos requiere el uso de un dispositivo conocido como micropipeta.
Para el uso adecuado de la micropipeta asegúrese que está usando la micropipeta correcta para el volumen que necesita. También, asegúrese que la micropipeta está realmente lista para el volumen que necesita revisando la “ventana de volumen”, y, si es necesario, cambiando el volumen mediante el “dispositivo” del control o mando del volumen hasta que la micropipeta corrija el volumen (la micropipeta no lee su mente; varias personas usarán las pipetas, no siempre se encontrará como usted la necesita).
No Trate de colocar la micropipeta para volúmenes mayores que el máximo, o para volúmenes menores del cero, esto descalibrará y dañará la micropipeta.
OBJETIVOS
Objetivo General
El alumno determinará la cantidad de ADN por método espectrofotométrico,
Objetivos específicos
Conocerá las partes y el funcionamiento de las micropipetas,
Adquirirá destreza en la utilización de pipetas para la medición de volúmenes entre 1 y 1000 μL.
Determinará algunos límites de confianza para el uso de micropipetas
Cuantificará el ADN por espectrofotometría
MATERIALES
1 juego de micropipetas P1000, P200 y P10 por equipo Agua desionizada Balanzas granataria que detectan 0,0001 g Tapas de cajas de petri de plástico
Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Puntas de diferentes capacidades (10, 200 y 1000 µL)ADN de concentración desconocida
METODOLOGÍA EXPERIMENTAL
MANEJO DE LA MICROPIPETA
1. Partes de micropipeta
2. Llenado de la pipeta
Colocar una punta según correspondaColocar el pulgar sobre el mando o émbolo de pipeteado. Oprimir el émbolo hasta el primer un punto de resistencia “alto” o "tope". Si continúa presionando encontrará un punto donde el émbolo ya no se mueve hacia abajo, este corresponde al segundo punto de resistencia “alto” o "tope". Oprimir el émbolo hasta el primer tope y colocar la punta dentro del líquido hasta una profundidad menor a 1 mm. De una manera lenta y controlada, disminuya la presión del émbolo para permitir que se desplace hacia arriba. No suelte el émbolo abruptamente, al permitirlo causará que el líquido pueda salpicar dentro de la punta produciendo volúmenes inexactos y generando contaminación de la pipeta. Una vez el émbolo se haya desplazado hasta arriba mantenga la micropipeta en el líquido durante un segundo, esto evita que se aspire aire en la parte final.
3. Expulsión de la muestra
Llevar la micropipeta al tubo de polipropileno de 1.5 mL. Se apoya la punta en la pared inferior del tubo. Oprima el émbolo hasta el primer tope y luego hasta el segundo tope. Haga este procedimiento a una velocidad moderada, hacerlo muy rápido hará que queden

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
gotas de muestra en la punta. Si observa cuidadosamente, entonces notará que al oprimir hasta el segundo alto se expele todo el líquido de la punta. Lo anterior es cierto para la mayoría de soluciones acuosas, excepto para las soluciones de alta viscosidad como el Glicerol. Si es usada inadecuadamente, la micropipeta transferirá volúmenes inexactos.
4. Calibración de Micropipeta
La micropipeta puede perder su calibración. Comprobar la calibración de la pipeta es un procedimiento simple que puede ahorrar tiempo considerable, energía, y reactivos. En esta práctica aprenderá a usar la micropipeta de tamaños diversos medir su exactitud, precisión y calibración. Para cada micropipeta revisar el porcentaje de exactitud (E%) y el coeficiente de variación (CV%) en todo el rango usando al menos dos volúmenes diferentes; por ejemplo: Para la micropipeta P1000 revisar los volúmenes de 300 μL y 1000 μL. Para P200 revisar los volúmenes de 60 y 200 μL. Para P10 revisar 3 y 10 μL.
1. Seleccionar la micropipeta y las puntas adecuadas. 2. Colocar la tapa de la caja de petri en la balanza y tarar a cero. 3. Tomar el volumen de agua destilada con la micropipeta y dispénselo en la tapa de la caja de petri. Registre el peso del agua añadido. La densidad del agua es 1 g/mL a 25 º C, por lo tanto un volumen de 1 mL corresponde aproximadamente a una masa de 1 g, 300 μL a 0.3 g, 200 μL a 0.2 g, 60 μL a 0.06 g, 10 μL a 0.01 g y 3 μL a 0.003 g. 4. Repita el procedimiento cinco veces para cada volumen. 5. Repetir todo el procedimiento para glicerol. La densidad del glicerol es de 1,2656 g/mL a 25 ° C. Calcular la masa esperada para cada volumen.
5. CALCULO DE LA EXACTITUD (E%) Y DEL COEFICIENTE DE VARIACIÓN (CV%)
Valor medioX = Σ Xi /n donde Xi= pesadas, n= numero de pesadas
Volumen medioV=X.Z donde X= valor medio y Z= factor z (1/densidad)
Valore del control a 21,5 ° C (Z = 1,0032)
Exactitud
E(%)= ((V-Vnominal)/Vnominal) *100
Desviación estándarS = Z . Σ (Xi – X)2 n – 1
Coeficiente de Variación
CV= (S*100)/V

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Límites de tolerancia para micropipetas
Volumen (μL) E% nominal CV% nominal
5-10 1 0.820-50 0.7 0.4100-1000 0.5 0.2
Una vez determinadas la exactitud y el coeficiente de variación de las micropipetas proporcionadas, realice la cuantificación de las muestras proporcionadas.
CUANTIFICACIÓN DE ADN
6. Preparación de las muestras.
- Realice diluciones 1:10, 1:100 y 1:1000 de las muestras proporcionadas por triplicado con agua Milli Q estéril, en tubos de polipropileno estériles.
- Encienda el espectrofotómetro al menos 15 minutos antes de iniciar las lecturas.
-Mantenga las muestras en hielo o en refrigeración hasta ser cuantificadas
-Cuantifique las muestras a 260 y 280mn
Muestra Dilución Lectura 260 Lectura 280
Relación 260/280
ADN (ng/µL)
ADN (fg/µL)
ADN (mg/µL)
ADN 1
ADN 2
-Realice los cálculos correspondientes
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.
Molecular cloning

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 3. EXTRACCIÓN DE ADN
OBJETIVOS
Objetivo General
El alumno extraerá ADN de diferentes muestras por métodos físico-químicos
Objetivos específicos
Conocerá la metodología básica para extracción de ADN,
Adquirirá destreza la manipulación de diversas muestras.
Determinará la integridad del ADN por electroforesis en Agarosa
MATERIALES
Reactivos y MaterialesAcetato de Sodio (3 M)Etanol absolutoEtanol 70%FenolCloroformoAgua desionizada TE (pH 8.0)1 juego de micropipetas P1000, P200 y P10 por equipo GradillasMedio LB líquidoTermobloquesTubos de polipropileno 1.5 mLCentrifugaVortex
Muestras:-E. coli-Suelo

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
METODOLOGÍA EXPERIMENTAL
Condiciones de trabajo:Uso de bata obligatoria, zapatos cerrados, cubreboca, guantes de látex o vinilo,
Extracción de ADN de E. coli
1. Crecer E. coli en medio LB (3 mL) durante toda la noche.2. Colocar 1 mL del cultivo en un tubo de polipropileno de 1.5 mL.3. Centrifugar el tubo a 7000 G durante 2 min.4. Decantar el sobrenadante.5. Agregar 250 L de agua desionizada estéril al paquete celular.6. Agitar vigorosamente utilizando un vortex hasta lograr la resuspensión de las células
(aproximadamente 30 s).7. Agregar 250 L de fenol (equilibrado con Tris 1M pH 8.0).8. Agitar vigorosamente durante 30 s utilizando un vortex.9. Agregar 250 L de cloroformo.10. Agitar vigorosamente utilizando un vortex durante 30 s. 11. Centrifugar a 19000 G durante 5 min.12. Recuperar la fase acuosa (fase superior, aprox. 225 L) y colocarla en otro tubo de
1.5 mL. En muchas ocasiones en la interfase se presenta un precipitado blanco, este precipitado no debe de ser recuperado, ya que son proteínas que pueden contaminar la muestra e inhibir reacciones posteriores.
13. Agregar 225 L de cloroformo.14. Agitar vigorosamente durante 30 s utilizando un vortex.15. Centrifugar a 19000 G durante 5 min.16. Recuperar la fase acuosa (fase superior, aprox. 200 L) y colocarla en otro tubo de
1.5 mL, teniendo las mismas precauciones que en el paso 12.17. Agregar 20 L de Acetato de Sodio (3M) a la fase acuosa.18. Agitar en vortex 20 s.19. Agregar 440 L de etanol Absoluto frío a la fase acuosa. 20. Centrifugar a 19000 G durante 30 min. Es importante colocar los tubos un la misma
orientación siempre, para saber en qué lado del tubo está el ADN precipitado. Mientras se está llevando a cabo la centrifugación, se debe hacer el gel de agarosa.
21. Decantar el sobrenadante a un tubo nuevo y mantenerlo en hielo durante el resto del experimento.
22. Agregar 500 L de etanol al 70% al tubo que ha sido vaciado, evitando resuspender la pastilla.
23. Centrifugar a 19000 G durante 5 min.24. Decantar el sobrenadante a un tubo nuevo y mantenerlo en hielo durante el resto del
experimento.25. Colocar el tubo que ha sido vaciado encima de una servilleta de papel y boca abajo,
hasta que todo el etanol haya sido evaporado.26. Resuspender el ADN en 50 L de TE pH 7.8 o agua desionizada.
GUARDAR A -20°C DEPENDIENDO DEL AVANCE CONTINUAR27. Realizar una electroforesis en gel de agarosa para visualizar el ADN (práctica 4).28. Cuantificar el ADN utilizando un espectrofotómetro a una longitud de onda de 260

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
nm.
Extracción de ADN de Suelo
29. Pesar 0.1 mg de suelo en un tubo de polipropileno de 1.5 mL.30. Agregar 250 L de agua desionizada estéril.31. Agitar vigorosamente utilizando un vortex hasta lograr la resuspensión de las células
(aproximadamente 30 s).32. Agregar 250 L de fenol (equilibrado con Tris 1M pH 8.0).33. Agitar vigorosamente durante 30 s utilizando un vortex.34. Agregar 250 L de cloroformo.35. Agitar vigorosamente utilizando un vortex durante 30 s. 36. Centrifugar a 19000 G durante 5 min.37. Recuperar la fase acuosa (fase superior, aprox. 225 L) y colocarla en otro tubo de
1.5 mL. En muchas ocasiones en la interfase se presenta un precipitado blanco, este precipitado no debe de ser recuperado, ya que son proteínas que pueden contaminar la muestra e inhibir reacciones posteriores.
38. Agregar 225 L de cloroformo.39. Agitar vigorosamente durante 30 s utilizando un vortex.40. Centrifugar a 19000 G durante 5 min.41. Recuperar la fase acuosa (fase superior, aprox. 200 L) y colocarla en otro tubo de
1.5 mL, teniendo las mismas precauciones que en el paso 12.42. Agregar 20 L de Acetato de Sodio (3M) a la fase acuosa.43. Agitar en vortex 20 s.44. Agregar 440 L de etanol Absoluto frío a la fase acuosa. 45. Centrifugar a 19000 G durante 30 min. Es importante colocar los tubos un la misma
orientación siempre, para saber en qué lado del tubo está el ADN precipitado. Mientras se está llevando a cabo la centrifugación, se debe hacer el gel de agarosa.
46. Decantar el sobrenadante a un tubo nuevo y mantenerlo en hielo durante el resto del experimento.
47. Agregar 500 L de etanol al 70% al tubo que ha sido vaciado.48. Centrifugar a 19000 G durante 5 min.49. Decantar el sobrenadante a un tubo nuevo y mantenerlo en hielo durante el resto del
experimento.50. Colocar el tubo que ha sido vaciado encima de una servilleta de papel y boca abajo,
hasta que todo el etanol haya sido evaporado.51. Resuspender el ADN en 50 L de TE pH 7.8 o agua desionizada.
GUARDAR A -20°C DEPENDIENDO DEL AVANCE CONTINUAR52. Realizar una electroforesis en gel de agarosa para visualizar el ADN (práctica 4).53. Cuantificar el ADN utilizando un espectrofotómetro a una longitud de onda de 260
nm.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
CUANTIFICACIÓN DE ADN
Considerando 1Abs 260nm= 50 µg/µL
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.
Molecular cloning

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 4. CUANTIFICACIÓN DE ADN EN GEL DE ELECTROFORESIS
Una vez que se ha realizado la extracción de cualquier tipo de ácido nucleico es necesario revisar la integridad y la cantidad del mismo, esto se efectúa mediante el corrimiento electroforético en geles de agarosa o de poliacrilamida. La agarosa es más comúnmente utilizada puesto que no es tóxica. Los geles son teñidos con diferentes colorantes para visualizar el ADN. Los más comunes son: el bromuro de etidio, el nitrato de plata , y recientemente el SYBR FAFE, Gel Red, Syber green, Syber Gold, etc. Después de ser teñidos se visualizan utilizando un transiluminador o una lámpara UV.
OBJETIVOS
Objetivo General
El alumno determinará la cantidad de ADN por densitometría
Objetivos específicos
Adquirirá destreza el corrimiento de muestras en geles de agarosa
Cuantificará el ADN por densitometría
MATERIALES
Materiales y Reactivos Solución de Agarosa 0.5% w/v Solución para teñir ADN Regulador para electroforesis TBE 1X Regulador de carga 6x 1 juego de micropipetas P1000, P200 y P10 por equipo Agua desionizada Puntas de diferentes capacidades (10, 200 y 1000 µL) Muestras de ADN Cámara de Electroforesis Fuente de Poder Marcador de Peso Molecular (100pb)

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
METODOLOGÍA EXPERIMENTAL
1. Colocar 20 mL de TBE en un vaso de precipitado de 100 mL (para un volumen de gel de 20 mL, si es mayor hacer los cálculos conservando la proporción de la concentración del gel)
2. Agregar 100 mg de agarosa3. Calentar en parrilla de calentamiento hasta fundir completamente4. Adicionar 0.5L de reactivo para teñir ADN (Rojo Texas, Cyber green, etc.)5. Mezclar6. Verter la solución en una canastilla para electroforesis7. Colocar el formador de pozos (peine) en la canastilla8. Dejar gelificar (cubrir el gel con papel aluminio)9. Colocar la canastilla adentro de la cámara de electroforesis10. Agregar buffer de corrimiento (TBE)11. Preparar las muestras a analizar (mezclar con regulador de carga, no más de 10 L
en total)12. Colocar las muestras adentro de los pozos13. Llevar a cabo la electroforesis a 100 V durante 30 a 45 min14. Visualizar el gel en el transiluminador y cuantificar según instrucciones.
REFERENCIAS
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.
Molecular cloning
ANEXOS
6x Gel-loading Buffer (Amortiguador de carga)Azul de bomofenol 0.25% (w/v) Glicerina en agua 30% (v/v)
TBEPrepare una solución madre 5x en 1 litro deH2OTris base 54 g Ácido Bórico 27.5 g EDTA 0.5 M (pH 8.0) 20 ml

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 5. EXTRACCIÓN DE ARN
En una técnica sencilla de extracción de ARN, las células son homogenizadas en tiocianato de guanidina y posteriormente purificado por fenol-cloroformo a pH reducido. El rendimiento del ARN total extraído depende del tipo de muestra y generalmente esta en un rango de 4-7 μg/mL iniciando de 5-10 μg de tejido o 106 células.
OBJETIVOS
Objetivo General
El alumno extraerá ADN de diferentes muestras por métodos físico-químicos
Objetivos específicos
Conocerá la metodología básica para extracción de ARN,
Adquirirá destreza en la manipulación de diversas muestras.
Determinará la integridad del ARN por electroforesis en gel de Agarosa
MATERIALES
Cloroformo : alcohol isoamílico (49:1, v/v)EtanolIsopropanolNitrógeno liquidoFenolAcetato de sodio (2 M, pH 4.0)Solución D (solución desnaturalizante)Agua desionizada 1 juego de micropipetas P1000, P200 y P10 por equipo GradillasTermobloquesTubos de polipropileno 1.5 mLCentrifugaVortex
Muestras:-E. coli

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
METODOLOGÍA EXPERIMENTAL
Condiciones de trabajo:Uso de bata obligatoria, zapatos cerrados, cubre boca, guantes de látex o vinilo,
Extracción de ARN de E. coli
Muestra
1. Crecer E. coli en medio LB (3 mL) durante toda la noche.2. Colocar 1 mL del cultivo en un tubo de polipropileno de 1.5 mL.3. Centrifugar el tubo a 12 000 rpm durante 2 min.4. Decantar el sobrenadante.
Tratamiento de la muestra
1. Preparar las células o tejido 2. Colocar el tejido en un mortero estéril libre de RNAsas y adicionar Nitrógeno
líquido 3. Usando el pistilo macerar el tejido hasta obtener polvo fino4. Transferir el macerado a un tubo nuevo estéril y adicionar 3 mL de solución D5. Homogenizar 15-30 segundos a temperatura ambiente en un homogenizador (en
caso de no tener pasar el macerado en una jeringa de 1mL 3 veces)6. Transferir el homogenizado a un tubo Nuevo y adicionar 0.1 mL de Acetato de
sodio 2M, 1mL de fenol y 0.2 mL de cloroformo-alcohol isoamílico por cada mililitro de solución D.
7. Mezclar en el Vortex vigorosamente por 10 s e incubar en hielo por 15 minutos8. Centrifugar a 9000 rpm por 20 minutos a 4°C9. Transferir la fase acuosa a un tubo nuevo10. Adicionar un volumen de isopropanol e invertir el tubo 5 veces para mezclar e
incubar a -20°C mínimo 1 hr11. Centrifugar a 10,000g (9000 rpm) por 30 minutos a 4°C12. Cuidadosamente decantar y disolver el ARN en 0.3 mL de la solución D.13. Adicionar un volumen de isopropanol e incubar 1 h a -20°C14. Centrifugar 10 minutos a 4°C15. Decantar y adicionar un volumen de etanol al 70%16. Centrifugar a máxima velocidad durante 3 minutos.17. Adicionar 50-100 μl de H2O. Almacenar la solución de ARN a -70°C.18. Estimar la concentración de ARN por lectura a 260nm y correr 5µL en un gel de
agarosa al 1% (práctica 4)

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Anexos
Solución D
4 M Tiocianato de guanidina25 mM citrato de sodio0.5% (w/v) Lauril sarcosianato de sodio0.1 M -mercaptoetanol
Disolver 250 g de tiocianato de guanidina en 293 ml de H2O, 17.6 ml de 0.75 M de citrato de sodio(pH 7.0), y 26.4 ml de 10% (w/v) lauril sarcocianato de sodio. En una parrilla a 65°C mezclar los ingredientes con ayuda de agitación magnética, guardar a temperatura ambiente y adicionar 0.36 ml de 14.4 M mercaptoetanol por cada 50 mL de la solución D justo antes de usar.
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 6. Extracción de ADN plasmídico por lisis alcalina
OBJETIVOS
Objetivo General
El alumno extraerá ADN plasmídico.
Objetivos específicos
Conocerá la metodología básica para extracción de ADN plasmídico. Determinará la integridad del ADN plasmídico por electroforesis en gel de Agarosa.
MATERIALES
Soluciones
Solución de lisis alcalina ISolución de lisis alcalina IISolución de lisis alcalina IIIAmpicilinaEtanol Fenol:cloroformoTE (pH 8.0)
Medios
Cajas con LB ampicilinaCajas con medio LB

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
METODOLOGÍA EXPERIMENTAL
Condiciones de trabajo:
Uso de bata obligatoria, zapatos cerrados, cubreboca, guantes de látex o vinilo,
METODOLOGÍA
1. Inocular 2 mL de medio rico LB ampicilina con una colonia con plásmido. Incubar toda la noche a 37°C con agitación.2. Centrifugar a velocidad máxima por 30 segundos los 2 mL del cultivo a temperatura ambiente. 3. Remover el sobrenadante dejando la pastilla libre del medio.4. Resuspender la pastilla en 100 μL de solución Alcalina I fría, mezclar vigorosamente con ayuda del vortex.5.Adicionar 200 μL de solución de lisis II recién preparada, cerrar el tubo y mezclar invirtiendo el tubo cinco veces, “No usar vortex”. Poner el tubo en hielo6.Adicionar 150 μL de solución alcalina III fría, cerrar el tubo e invertir el tubo cinco veces para mezclar. Guardar el tubo en hielo 3-5 minutos (NO CONGELAR).7. Centrifugar a máxima velocidad por cinco minutos a 4°C. Transferir el sobrenadante a un tubo limpio.8. Adicionar un volumen de fenol : cloroformo (1:1 v/v), mezclar en vortex y centrifugar a velocidad máxima por 2 min a 4°C. Transferir el sobrenadante a un tubo nuevo.9. Precipitar el ADN plasmídico con adición de 2 volúmenes de etanol absoluto, incubar a -20ºC 20 minutos. 10. Centrifugar a máxima velocidad 5 minutos a 4°C.11. Remover el sobrenadante.12. Adicionar 1 mL de etanol al 70% (cuidar no resuspender la pastilla), centrifugar a máxima velocidad por 2 minutos a 4°C.13. Remover el sobrenadante e invertir el tubo sobre una superficie limpia para permitir que la pastilla quede lo más seca posible.14. Resuspender la pastilla en 50 μL de agua desionizada estéril, guardar a -20°C.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Anexos
Solución de lisis alcalina I
50 mM glucosa25 mM Tris-Cl (pH 8.0)10 mM EDTA (pH 8.0)Guardar a 4°C
Solución de lisis alcalina II
0.2 N NaOH 1% (w/v) SDSPreparar la solución al momento.
Solución de lisis alcalina III
5 M acetato de potasio, 60.0 mLÁcido acético glacial, 11.5 mLH2O, 28.5 mlGuardar a 4°C
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 7. Inserción de material genético en un vector plasmídico.
La ligación de fragmentes de interés en plásmidos ha sido una metodología presente en casi todos los estudios que tienen que ver con técnicas de Biología Molecular. Se han utilizado este tipo de sistemas como el pUC 19 linearizado con enzimas de restricción específicas, una vez hecho esto se realiza una reacción de ligación plásmido-inserto con la enzima ligasa.
Objetivo General
El alumno realizará la inserción de material genético dentro de un vector plasmídico.
Objetivos específicos
Conocerá la metodología básica para cortar un vector por medio de enzimas de restricción.
Conocerá la metodología básica para insertar material genético de interés dentro de un plásmido previamente tratado con enzimas de restricción.
MATERIALES
- Plásmido- Enzimas de restricción- Material genético de interés (máximo xxx pdb).- T4 ADN Ligasa- Buffer de restricción.- Buffer de Ligación.- Termo-Bloque
MÉTODO
A. Restricción1. En un tubo estéril, adicionar 2 l de buffer de restricción.2. Adicionar n ng (2 l) de plásmido.3. Colocar 1 l de la enzima de restricción proporcionada por el profesor.4. Aforar con agua desionizada estéril a un volumen total de reacción de 20 l.5. Incubar a la temperatura indicada para la enzima durante 30 min en el termobloque.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
B. Ligaciòn6. Colocar 2 L de buffer de ligación en un tubo estéril.7. Adicionar 1 l de T4 ADN ligasa.8. Añadir (x ng) 1 l de plásmido previamente tratado con enzima de restricción.9. Agregar (x ng) 2 l del inserto deseado.10. Aforar a 20 l de volumen de reacción con agua desionizada estéril.11. Incubar a 22ºC durante 2 horas en el termobloque.12. Almacenar a -20ºC hasta el momento de su uso.
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 8. Reacción en cadena de la Polimerasa (PCR).
La Reacción en Cadena de la polimerasa (PCR) permite obtener más de 10 millones de copias de una cadena de ADN de interés a partir de pocas moléculas de ésta. La sensibilidad de ésta técnica requiere de cuidados para que la muestra no esté contaminada previamente con otras cadenas de ADN que pudieran generar amplificados.
Existen numerosas técnicas que emplean la PCR (RAPD, RFLP, RTPCR) con diferentes aplicaciones en campos de diagnóstico, criminología, e investigación.
Objetivo General
Obtener un producto amplificado por la técnica de Reacción en Cadena de la Polimerasa (PCR).
Objetivos específicos
Conocerá la metodología básica para amplificar una cadena de interés por medio de la reacción en cadena de la polimerasa.
Identificará la correcta o incorrecta inserción de una cadena de interés en un vector en base al tamaño del amplificado por medio de PCR.
MATERIALES
-Termociclador.-ADN molde-Tubos de polipropileno de 200 µL.-Oligonucleótidos iniciadores (primers): sentido y anti sentido-Taq DNA polimerasa-Micro pipetas de diferentes capacidades (1000-100, 200-20 y 10-0.5 µL-10X PCR buffer-MgCl2-dNTPs: 1 mM dATP, 1 mM dGTP, 1 mM dCTP, 0.65 mM dTTP.
MÉTODO

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
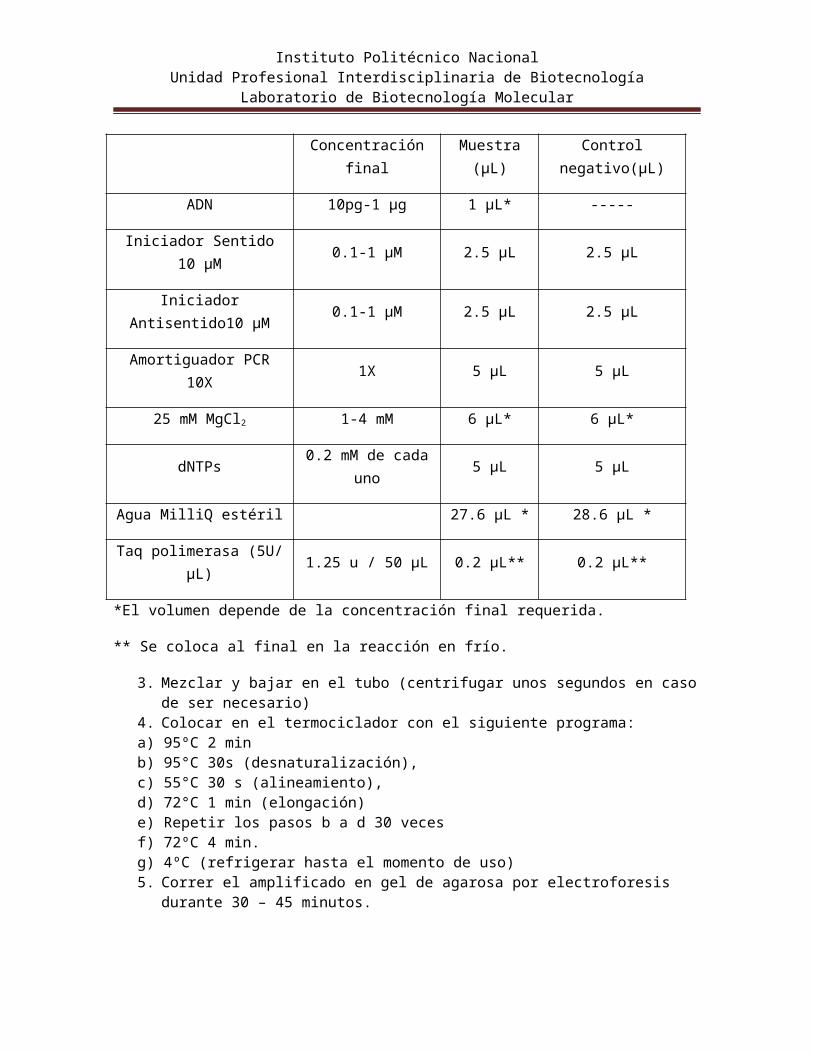
1. En condiciones de esterilidad colocar dos tubos de polipropileno de 200µL de capacidad en hielo.
2. Adicionar lo que se indica en la tabla 1.Tabla 1. Reacción en cadena de la Polimerasa
Concentración finalMuestra
(µL)Control negativo(µL)
ADN 10pg-1 µg 1 µL* -----
Iniciador Sentido 10 µM 0.1-1 µM 2.5 µL 2.5 µL
Iniciador Antisentido10 µM 0.1-1 µM 2.5 µL 2.5 µL
Amortiguador PCR 10X 1X 5 µL 5 µL
25 mM MgCl2 1-4 mM 6 µL* 6 µL*
dNTPs 0.2 mM de cada uno 5 µL 5 µL
Agua MilliQ estéril 27.6 µL * 28.6 µL *
Taq polimerasa (5U/ µL) 1.25 u / 50 µL 0.2 µL** 0.2 µL**
*El volumen depende de la concentración final requerida.
** Se coloca al final en la reacción en frío.
3. Mezclar y bajar en el tubo (centrifugar unos segundos en caso de ser necesario)4. Colocar en el termociclador con el siguiente programa:a) 95ºC 2 minb) 95°C 30s (desnaturalización), c) 55°C 30 s (alineamiento), d) 72°C 1 min (elongación)e) Repetir los pasos b a d 30 vecesf) 72ºC 4 min.g) 4ºC (refrigerar hasta el momento de uso)5. Correr el amplificado en gel de agarosa por electroforesis durante 30 – 45 minutos.
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 9. Método para hacer células competentes de Escherichia coli con CaCl2
Uno de los pasos cruciales en cualquier metodología de Biología Molecular es la obtención de células competentes, listas para poder sobre producir el gen de interés insertado en un plásmido. El método para hacer células competentes por cloruro de calcio es uno de los más rutinarios y eficientes, además de ser extremadamente barato.
OBJETIVOS
Objetivo General
El alumno aprenderá a hacer células competentes de Escherichia coli con CaCl2.
Objetivos específicos
Conocerá la metodología básica para de hacer células competentes de Escherichia coli con CaCl2.
Determinará la vialbilidad de hacer células competentes de Escherichia coli con CaCl2.
MATERIALES
- CaCl2 (0.1 M)- Espectrofotómetro- Gradillas para tubos- Hielo- Medio LB líquido- Micropipetas de 200 μL y 1000 μL- Puntas para micropipetas de 200 μL y 1000 μL- Termo-bloques- Tubos de polipropileno 1.5 mL estériles- Ultracentrífuga
MÉTODO
1. Incubar E. coli a 37°C por 16 horas en placas de agar LB2. Inocular 100 mL de medio LB e incubar a 37°C con agitación, hasta que la densidad
óptica a 590 nm sea de 0.3 a 0.4 (aproximadamente 3 horas)

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
3. Transferir 45 mL de células a un tubo de propileno frío y estéril y mantenerlo por 10 min a 4°C.
4. Centrifugar a 6000 rpm por 4 min5. Decantar el sobrenadante6. Resuspender el paquete celular en 5 mL de CaCl2 estéril (0.1 M, frío) 7. Colocar el tubo en hielo durante 10 min a 4°C.8. Centrifugar a 6000 rpm por 4 min a 4°C.9. Decantar el sobrenadante10. Resuspender el paquete celular en 1 mL de CaCl2 estéril (0.1 M, frío). En este
momento se pueden guardar la células competentes a -70ºC o se puede proseguir con el protocolo de transformación.
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 10. Transformación de células competentes
La ligación de fragmentes de interés en plásmidos ha sido una metodología presente en casi todos los estudios que tienen que ver con técnicas de Biología Molecular. Se han utilizado este tipo de sistemas como el pUC 19 linearizado con enzimas de restricción específicas, una vez hecho esto se realiza una reacción de ligación plásmido-inserto con la enzima ligasa.
Objetivo General
El alumno realizará la transformación de de células competentes con vector plasmídico.
Objetivos específicos
Conocerá la metodología básica para transformar células competentes con un vector plasmídico
Determinará la transformación de células competentes de Escherichia coli con CaCl2.
MATERIALES
- Células competentes- Incubadora- Placas de agar LB-Ampicilina - Reacción de PCR - Termo-Bloque - plásmido
MÉTODO
C. Muestra
13. Adicionar 4 L de reacción de PCR a un tubo de polipropileno.14. Adicionar 1 L del buffer de ligación. 15. Adicionar 1 L del Plásmido.16. Mezclar la reacción suavemente.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
17. Incubar por 16 h a 4°C.18. Adicionar todo el contenido del tubo a un tubo que contiene células competentes.19. Mezclar el tubo suavemente.20. Incubar a -20ºC durante 20 min.21. Colocar el tubo en un baño o Termo-bloque a 42ºC durante 1 min.22. Colocar el tubo a -20ºC durante 2 min.23. Adicionar 800 L de medio LB.24. Colocar el tubo en un baño o un Termo-bloque a 37ºC durante 1 hora.25. Agregar 200 L en una placa con medio LB Ampicilina26. Espátular las células.27. Incubar la caja durante 16 horas a 37ºC
D. Control de viabilidad y control negativo
28. Colocar 200 L de células competentes.29. Mezclar el tubo suavemente.30. Incubar a -20ºC durante 20 min.31. Colocar el tubo en un baño o Termo-bloque a 42ºC durante 1 min.32. Colocar el tubo a -20ºC durante 2 min.33. Adicionar 800 L de medio LB.34. Colocar el tubo en un baño o un Termo-bloque a 37ºC durante 1 hora.35. Agregar 200 L en una placa con medio LB Ampicilina y 200 L a una caja LB36. Espátular las células.37. Incubar la caja durante 16 horas a 37ºC
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 11. Western Blot.
El western blot es una técnica inmuno enzimática utilizada en biología molecular para la detección de proteínas en muestras crudas o extractos celulares. Esta técnica tiene múltiples aplicaciones, tanto en el área de investigación como en la de diagnóstico clínico, en donde es utilizada ya sea independiente o conjuntamente con ELISA.
La técnica consiste en realizar la separación de las proteínas de la muestra por electroforesis en del de poli acrilamida (PAGE) y posteriormente transferirlas a una membrana, ya sea por gravedad, difusión o un campo eléctrico.
Una vez en la membrana, la proteína es identificada mediante un inmunoensayo, en donde se liga a la proteína de interés un anticuerpo específico contra ella ligado a una enzima, la cual permite la detección.
Objetivo General
El alumno conocerá y realizará la técnica de westtern blott para detección de antigeno o anticuerpo.
Objetivos específicos
Conocerá la metodología básica para correr un SDS-PAGEConocerá la metodología básica para realizar una electrotransferencia.Conocerá la metodología básica para detectar proteínas por medio de un
inmunoensayo.
MATERIALES
- Plásmido
MÉTODO
E. Tratamiento de la muestra.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
38. A una muestra de concentración conocida de proteína (ya sea antígeno o anticuerpo) se le adiciona amortiguador de muestra hasta un volumen final de 85 l.
39. Añadir 10 l de solución de azul de bromofenol y 5 l de -mercaptoetanol.40. Incubar en baño maría a ebullición durante 5 minutos.
B. Preparación del Gel.41. Se limpia con etanol al 70% las placas de vidrio para remover contaminantes.42. Colocar las placas de vidrio y los separadores en la base para preparar el gel.43. Preparar el gel separador de acuerdo a lo indicado en la tabla 1 del anexo,
adicionando el persulfato e amonio y el temed al final.44. Vaciar el gel separador en los vidrios evitando la formación de burbujas, y
alcanzando un nivel 3cm por debajo del borde superior. Dejar polimerizar por 20 a 30 minutos.
45. Preparar el gel concentrador de acuerdo a la tabla 2 del anexo.46. Vaciar en la parte superior y colocar el peine, evitando la formación de burbujas.
Dejar polimerizar por 20 a 30 minutos.
C. Montaje de la cámara de electroforesis y corrimiento del gel.47. Montar las placas de vidrio conteniendo el gel en su base, y esta en la cámara.48. Preparar amortiguador decorrida 1x.49. Llenar con amortiguador de corrida la cámara interna hasta el límite y la externa
hasta cubrir los electrodos.50. Retirar con cuidado el peine. Cargar aproximadamente 20 l de muestra y marcador
de peso molecular en los pozos.51. Colocar la tapa de la cámara y conectar a la fuente de poder.52. Correr a 50 V mientras las muestras se encuentren en el gel concentrador.53. Correr a 100 V una vez alcanzado el gel separador y hasta que el frente de corrida
alcance el final del gel.54. Detener la fuente de poder, recuperar el amortiguador de corrida y desmontar la
cámara.55. Cuidadosamente recuperar el gel de los vidrios y colocar en amortiguador de
transferencia.NOTA: En caso de existir un duplicado del gel, este puede ser teñido para observar el corrimiento del gel antes de proseguir con la siguiente etapa.
D. Montaje de la cámara de electro transferencia y electro transferencia.56. Incubar fibras scotch y papel whatmann (0.16 mm de espesor) en amortiguador de
transferencia 30 min. antes de iniciar el montaje de los cartuchos.57. Cortar la membrana de nitrocelulosa a un tamaño ligeramente mayor al de los geles.
Este procedimiento debe ser llevado a cabo con guantes. Colocar en un recipiente con amortiguador de transferencia.
58. Colocar el gel sobre la membrana de nitrocelulosa, y cuidadosamente, marcar sobre la membrana, con lápiz, el frente decorrida y el número de carril del gel.
59. Para el armado de los cartuchos se colocan dos fibras scotch, dos papeles filtro, el gel, la membrana de nitrocelulosa, dos papeles filtro más y dos fibras scotch más.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Hay que eliminar cualquier burbuja que se forme al ir adicionando las diferentes capas.
60. Colocar el cartucho en la cámara de electrotransferencia de manera que el gel quede hacia el ánodo (-) y la membrana hacia el cátodo (+).
61. Llenar la cámara con amortiguador de transferencia hasta el nivel indicado.62. Cerrar la cámara, conectar a la fuente de poder y transferir a 100 V por una hora,
cuidando que la temperatura del amortiguador no sobrepase los 60ºC.63. Al terminar la transferencia, recuperar, con guantes, la membrana del cartucho.
NOTA: En caso de tener duplicado de la membrana, teñir una de ellas para observar la transferencia de las proteínas antes de proseguir con el siguiente paso.
E. Reacción inmunoenzimática64. Colocar la membrana de nitrocelulosa en un recipiente, y agregar l solución de
bloqueo, de manera que cubra completamente la membrana. Incubar 2 horas a temperatura ambiente con agitación continua.
65. Retirar la solución de bloqueo y realizar 3 lavados con PBS-Twen de 5 min. cada uno, con agitación continua.
66. Incubar la membrana con la solución de proteína complementaria a aquella en la membrana (si se tiene antígeno en la membrana, incubar con solución e anticuerpo, y viceversa) durante 2 horas.
67. Retirar la solución de proteínas y repetir el paso 28.68. Se adiciona a la membrana la solución de avidita – HRP (peroxidada de rábano) y se
incuba por 2 horas con agitación continua.69. Retirar la solución de conjugado y repetir el paso 28.70. Se incuba la membrana en la solución de cromógeno / sustrato en agitación continua
hasta que aparezcan las bandas.71. Se enjuaga la membrana con agua desionizada y se deja secar.
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
ANEXO I
Tabla 1. Gel separador (para un volumen de 15 mL)
Sol. stock (mL) / Conc. gel (%) 5 6 7 8 9 10 12 13 15
Sol. Acrilamida – bisacrilamida
2.5 3.0 3.5 4.0 4.5 5.0 6.0 6.5 7.5
Amortiguador pH 8.8 3.75 3.75 3.75
3.75 3.75 3.75
3.75 3.75 3.75
Agua desionizada 8.75 8.25 7.75
7.25 6.75 6.25
5.25 4.75 3.75
Persulfato de amonio 0.05 0.05 0.05
0.05 0.05 0.05
0.05 0.05 0.05
TEMED 0.01 0.01 0.01
0.01 0.01 0.01
0.01 0.01 0.01
Tabla 2. Gel concentrador (para un volumen de 2 mL)
Sol. Acrilamida – bisacrilamida 0.26 mL
Amortiguador pH 6.8 0.5 mL
Agua desionizada 1.22 mL
Persulfato de amonio 0.01 mL
TEMED 0.02
Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PREPARACIÓN DE SOLUCIONES
Acrilamida 30% - Bis-acrilamida 0.8%
Pesar 29.2 g de acrilamida (99.9% pureza) más 0.8 g de bis-acrilamida. Disolver en agua bidestilada y aforar a 100 mL. Filtrar con papel Whatman 1. Guardar en frasco ámbar a 4° C. Usar guantes al pesar ya que la acrilamida es un compuesto neurotóxico.
Amortiguador del gel separador pH 8.8
Pesar 9.081 g de Tris-base, y 0.2 g de SDS, disolver en poca agua desionizada y ajustar el pH con HCl (aprox. 1 mL). Aforar a 50 mL y almacenar a temperatura ambiente.
Amortiguador del gel separador pH 6.8
Pesar 3.027 g de Tris-base y 0.2 g de SDS, disolver en poca agua desionizada y ajustar el pH con HCl (aprox. 1.5 mL). Aforar a 50 mL y almacenar a temperatura ambiente.
Amortiguador de corrida 5X / SDS
Pesar 7.55 g de Tris-base, 36 g de glicina y 2.5 g de SDS, disolver con agua desionizada y aforar a 500 mL. Al usar ajustar a 1X.
Amortiguador de muestra pH 6.8
Pesar 1.52 g de Tris-base y 2.0 g de SDS. Disolver en 40 mL de agua desionizada y ajustar el pH a 6.8 con HCl (aprox. 2.0 mL). Adicionar 20 mL de glicerol y ajustar el volumen a 100 mL.
Persulfato de amonio al 10%
Se prepara al momento de usarlo. Preparar el volumen mínimo necesario.
Solución de azul de bromofenol 10 mg/mL
Pesar 100 mg de azul de bromofenol y disolverlo en 10 mL de agua desionizada. Guardar en refrigeración.
Solución teñidora
Para preparar 100 mL se usan 50 mL de Metanol, 10 mL de ácido acético y 40 mL de agua desionizada y 0.05 g de azul de Coomassie. Disolver el azul de Coomassie en el metanol y adicionar el ácido acético. Aforar con el agua desionizada.
Solución desteñidora
Para preparar 500 mL se usan 25 mL de ácido acético, 82.5 mL de metanol y 392.5 mL de agua desionizada.
Amortiguador de transferencia
(Tris 0.025M, glicina 0.192M, pH 8.3, metanol 20% v/v). Medir 125 mL de Trizma-base 2 M y agregar 14.49 g de glicina, añadir 200 mL de metanol, aforar a 1000 mL con agua bidestilada. Guardar a 4 ºC. Nota: No ajustar pH oscila entre 8.1 y 8.4 dependiendo de la

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
calidad del tris, el metanol debe ser grado analítico, este amortiguador se puede usar hasta 3 veces, después de cada uso filtrar con papel Whatman nº 1.
Solución salina de fosfatos 0.01 M, NaCl 0.15 M, pH 7.2 (PBS)
Medir 800 mL de agua desionizada y agregar 100 mL de PB 10X y 8.75 g de NaCl. Disolver las sales y ajustar el pH. Aforar a 1 L con agua desionizada. La solución de PB se prepara con 2.62 g de fosfato de sodio monobásico monohidratado y 11.5 g de fosfato de sodio dibásico anhidro. Disolver en 200 mL de agua desionizada y aforar a 1 L con agua desionizada.
Amortiguador de lavado (PBS-Tween 20, 0.05%)
A un litro de PBS pH 7.2 añadir 500 µL de Tween 20.
Solución de bloqueo.
Pesar 5 g de leche descremada en polvo y disolverlo en 100 mL de PBS-Tween 20. Guardar a –20 °C.
Rojo de Ponceau 0.2% en ácido tricloroacético 3%
Pesar 200 mg de rojo de Ponceau y añadir 3 g de ácido tricloroacético. Aforar a 100 ml con agua desionizada. Mantener a 4° C en frasco color ámbar.
Solución cromógeno / sustrato
Pesar 25 mg de 4-cloro-1-naftol, añadir 5 mL de metanol absoluto y 25 mL de PBS, agregar 25 l de peróxido de hidrógeno al 3%. Nota: esta solución se prepara inmediatamente antes de usarla.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
ANEXO II
PURIFICACIÓN DE GAMMA GLOBULINA POR PRECIPITACIÓN CON SULFATO DE AMONIO.
INTRODUCCIÓN.
Tiselius y Kabat demostraron que la actividad de anticuerpos está principalmente presente en la fracción gamma de las proteínas del suero. Desde entonces se han diseñado diferentes métodos para la obtención de esta fracción sérica. Los anticuerpos pueden purificarse por métodos específicos e inespecíficos. Los métodos específicos se basan en la propiedad que tienen los anticuerpos para reaccionar con sus antígenos. Así, los anticuerpos pueden ser precipitados por la adición del antígeno en estado soluble, o ser adsorbidos al antígeno previamente insolubilizado. En ambos casos, el complejo resultante puede disociarse por modificación del pH a valores entre 2.5 y 4.0 ó 9.5 y 11.0, o bien por aumento en la concentración de sales o la concentración del antígeno soluble. Después, los anticuerpos se recuperan por alguno de los métodos de separación inespecífica. Las separaciones inespecíficas están basadas en las propiedades fisicoquímicas de los anticuerpos. Entre estos métodos, los más utilizados son la precipitación con etanol, con sulfato de amonio o sulfato de sodio, electroforesis en diversos soportes (papel, acetato de celulosa, gel de almidón, etc.), cromatografía de intercambio iónico y filtración en tamices moleculares. Uno de los métodos inespecíficos más empleados de purificación de gamma globulina es la precipitación con sulfato de amonio a una concentración final de 33%. Generalmente, se considera que con tres precipitaciones se obtiene una gamma globulina con un alto grado de pureza. La precipitación de la gamma globulina por este método se debe al fenómeno conocido como “salting out”, el cual consiste en que las moléculas de agua que solvatan a la

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
molécula de anticuerpo son desplazadas por los iones sulfato y amonio, que además neutralizan los grupos cargados de la proteína, con lo que se insolubiliza y precipita.
OBJETIVO. Purificar gamma globulinas de conejo y de humano por precipitación con sulfato de amonio a una saturación final del 33% y detección por medio de un gel de SDS-PAGE.
MATERIALES Y MÉTODO.
Material por grupo:
1 centrífuga clínica. 6 agitadores magnéticos 5.0 mL de BaCI2 al 10%. 1 parrilla con agitación.
Material por equipo:
10 mL de suero de conejo 10 mL de suero de humano. 20 mL de solución sobresaturada de sulfato de amonio. 5.0 mL de NaOH 2N. 50 mL de solución de salina al 0.85% (SS), pH 7.8. 20 mL de salina-regulador de boratos (SSRB) 0.1 M, pH 7.8. Papel indicador de pH. Tubo para diálisis. 10 cm de hilo grueso. 2 tubos falcón de 15 mL, graduados. 2 vasos de p.p. de 100 mL 2 pipetas serológicas de 5.0 mL. 1 pipeta serológicas de 1.0 mL. 2 pipetas Pasteur y bulbo de hule. 1 matraz Erlenmeyer de 25 mL.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Método: 1. Ajustar a 7.8 el pH de la solución saturada de sulfato de amonio, con NaOH 2N. 2. Adicionar a un volumen de suero, contenido en un vaso de p.p., medio volumen de la solución sobresaturada de sulfato de amonio, LENTAMENTE Y CON AGITACIÓN CONSTANTE. De esta forma, el sulfato de amonio queda a una saturación final del 33%. 3. Continuar con la agitación durante 10-15 min después de terminar la adición del sulfato de amonio. 4. Traspasar el contenido del vaso de p.p. a un tubo falcón y centrifugar los tubos a temperatura ambiente a 3500 rpm durante 15 min. 5. Decantar y disolver el sedimento en SS pH 7.8, hasta alcanzar el volumen original del suero. 6. Repetir los pasos 2, 3, 4 y 5. 7. Repetir nuevamente los pasos 2, 3 y 4. 8. Disolver en SSRB el sedimento después de la última centrifugación hasta alcanzar la mitad o un tercio del volumen original del suero. 9. Dializar contra SSRB en frío (4° C) y con agitación, durante 2 días, realizando 2 cambios diarios de solución de diálisis hasta eliminar completamente al sulfato de amonio. Para saber si efectivamente se eliminó el sulfato de amonio, a 5.0 mL de la solución de diálisis se le agregan unas gotas de cloruro de bario al 10%. La diálisis se considera completa si ya no se observa la formación de un precipitado blanco de sulfato de bario. 10. Transferir la gamma globulina dializada a un tubo y centrifugar en frío a 2500 rpm durante 10 min para eliminar el precipitado que se hubiera formado. 11. Guardar las gamma globulinas para la separación de isotipos. 12. Determinar la concentración de proteínas por el método de Bradford. 13. Correr un gel de SDS-PAGE para observar las gamma globulinas.
REFERENCIAS BIBLIOGRÁFICAS.
Campbell, D.H.; Garvey, LS.; Cremer, N.E.; Sussdorf, D.H. 1970. Methods in Immunology. 2. ed. W.A. Benjamin. Editor. Kabat, E.A.; Mayer, M.M. 1968. Inmunoquímica Experimental. 1a. ed. Editorial La Prensa
Médica Mexicana.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 12. Inmuno ensayo Ligado a enzimas (ELISA)
El inmunoensayo enzimático, conocido como ELISA por sus siglas en inglés (enzyme linked immunosorbant assay) fue descrito en 1971 por Engvall y Perlman.En este método se utilizan anticuerpos conjugados a una enzima. El anticuerpo conserva su capacidad de unión específica al antígeno, mientras la enzima es capaz de catalizar una reacción de oxido-reducción, en la cual el sustrato se transforma en un producto colorido. En este sistema el antígeno o el anticuerpo se adsorbe a una fase sólida insoluble (micro pozos, tubos o perlas de polivinilo o estireno)Existen diversas variantes del método de ELISA, como son los métodos directo, indirecto y en “sándwich”, y el método de ELISA competitivo. Estos permiten la determinación de antígenos en fluidos biológicos, a excepción del método indirecto con que se detectan anticuerpos.En los métodos directos, el anticuerpo dirigido específicamente contra el antígeno es el que lleva unida la enzima. En cambio en los métodos indirectos, el conjugado enzima-anticuerpo reacciona con el primer anticuerpo, el cual ya reaccionó con el antígeno. En el método de sándwich, el conjugado antígeno anticuerpo reacciona con el antígeno que se ha unido antes a un primer anticuerpo, adsorbido a la fase sólida. El o los componentes que no reaccionaron se eliminaron por lavados; por último se adiciona el sustrato de la enzima y se mide la intensidad del color desarrollado, el cual es proporcional a la magnitud de la reacción antígeno-anticuerpo.El método de ELISA tiene aplicaciones muy variadas, lo cual lo hace muy versátil: diagnóstico de enfermedades infecciosas, virales o parasitarias, cuantificación de hormonas, cuantificación de haptenos, titulación de anticuerpos en bajas concentraciones, determinación de anticuerpos no precipitantes.
Objetivo General
El alumno conocerá y realizará la técnica de ELISA para detección de antígeno o anticuerpo.
Objetivos específicos
Conocerá la metodología básica para adsorber antígenos en micro placas de ELISA.
Ligará antígenos o anti anticuerpos conjugados a antígenos adsorbidos en placas de ELISA.
Determinará la presencia del antígeno deseado por la reacción de una enzima conjugada a anticuerpo.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
MATERIALES
Estufa regulada a 37 °C.Placas de ELISA de poliestireno. (fase sólida)Antígeno “H” u “O”, solución al 5% en solución salina 0.85%.Pipetas automáticas de 20-200 mlAnticuerpo anti-IgG purificado y conjugado con HRPSuero humano control positivo y suero humano control negativoSustrato: 4 mg de orto-fenilen diamina, 4 ml de H2O2 al 30% (v/v) en 10 ml de regulador de citratos 0. XM pH 5.0Regulador de bloqueo: 0.25 % de gelatina ó 2% de albúmina sérica bovina (BSA), en regulador de fosfato borato sal (PBS) 0.1M pH 9.6Regulador de lavado: regulador de salina-fosfatos 0.01M pH 7.2 adicionado de 0.05% Tween 20.Diluyente del suero (PBSG): regulador de salina-fosfatos 0.01M, pH 7.2 adicionado de 0.25% de gelatina.
MÉTODO
Método de ELISA indirecto.
1. Adicionar 0.2 ml de antígeno en los pozos de poliestireno. Incubar la placa a 4°C durante 24 horas para permitir la adsorción del antígeno a la fase sólida.
2. Eliminar las soluciones por inversión de la tira de pozos, y desechar el líquido remanente sacudiendo fuertemente la tira sobre papel absorbente.
3. Lavar los pozos con 0.1 ml de solución de lavado. Dejar los pozos llenos con esta solución durante 3 minutos. Al cabo de este tiempo, eliminar la solución de la manera descrita en el paso 2. Repetir dos veces más con el fin de eliminar todo el antígeno no adsorbido.
4. Bloquear con 0.1 ml de una solución de 0.25% de BSA en PBS e incubar los pozos a 37°C durante 30 min para permitir el bloqueo de los sitios a los que no se adsorbió el antígeno.
5. Repetir el paso 2.6. Adicionar 0.1ml de los sueros control negativo, positivo y problema uno en cada pozo. Si lo
requiere puede hacer diluciones del suero. Incubar a 37°C durante 30 minutos para permitir la reacción antígeno-anticuerpo.
7. Repetir los pasos 2 y 3 para eliminar el exceso de suero que no reaccionó.8. Adicionar 0.1ml de conjugado diluido en PBSG, a todos los pozos. Incubar a 37°C durante 30
min. Para permitir que el conjugado reaccione con los anticuerpos humanos que se hayan unido al antígeno en la fase sólida.
9. Repetir los pasos 2 y 3 para eliminar el exceso de conjugado que no haya reaccionado.10. Adicionar 0.1 ml de una solución recién preparada del sustrato de la enzima: H2O2 al 0.04% en
regulador de salina-fosfatos pH 5.0 y adicionado de ortofenilendiamina al 0.04% (como cromógeno). Incubar en oscuridad para permitir la reacción enzimática y el consecuente desarrollo de color (Nota: el cromógeno es fotosensible)
11. Detener la reacción enzimática adicionando 12 l de H2SO4 8N en cada pozo.12. Determinar la presencia o ausencia del antígeno de interés contra los controles de manera
visual, o de contarse, con un lector de placas de ELISA.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Referencias
1. Avrameas S. (1971) Immunochemistry 6:432. Kemeny D. M. (1991) A practical Guide to ELISA. Pergamon Press. Oxford. Pp 21-215.3. Voller A., Bidwell D.E. y Bartlett A. (1979) The enzyme linked immunosorbet assay (ELISA). A
guide with abstracts of microplate application. Dynatech Laboratories, Inc.4. Profesores del Departamento de Inmunología (1992) Manual de Prácticas de Inmunología.
ENCB, IPN pp.47-51.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
PRÁCTICA 13. Transformación de plántulas de tabaco (Nicotiana tabacum) mediada por Agrobacterium tumefaciens
La transformación de plantas es una técnica que permite la inclusión de ADN de interés dentro el genoma de una planta. Esto puede realizarse por diversas técnicas, como son la bobalística, electroporación, microinyección o por Agrobacterium tumefaciens.
El empleo de plantas sobre bacterias para la producción de proteínas de interés presenta ciertas ventajas. Una de las principales es la capacidad de las plantas para hacer modificaciones post traduccionales en las proteínas de interés, ya que al ser organismos eucariontes, poseen la maquinaria celular necesaria para estos procesos.
Sin embargo, también presentan desventajas. Entre ellas el hecho de poseer diferentes tejidos. Debido a esto, si la transformación no está bien dirigida, el producto de interés puede ser sintetizado en órganos u organelos no deseados, dificultando de esta manera la recuperación y purificación del producto.
Agrobacterium es un simbionte natural de las plantas. Se vale de las heridas en las plantas, para infestarlas, y de esta manera aprovechar la maquinaria celular de la planta para sintetizar los metabolitos que requiere. Agrobacterium cuanta con el plásmido Ti, que introduce en la planta para desencadenar ciertos procesos que permiten la invasión de la planta por parte de la bacteria.
Objetivo General
El alumno obtendrá plántulas te tabaco (Nicotiana tabacum) transformadas por Agrobacterium tumefaciens.
Objetivos específicos
Conocerá la metodología básica para obtener explantes de plántulas de tabaco.Conocerá la metodología para transformar explantes por medio de A. tumefaciens.Conocerá la metodología básica para seleccionar explantes transformados.Conocerá la metodología básica para regenerar plántulas a partir de explantes.

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
MATERIALES
- Plásmido- Enzimas de restricción
MÉTODO
F. Preparación de materiales (Día 1)1. En una placa de agar LB antibióticos sembrar por estría simple la cepa de
Agrobacterium tumefaciens e incubar a 28ºC por 24 horas.2. Preparar 5 placas petri y esterilizarlas.3. Preparar 200 ml dem medio MS estéril.4. Preparar 10 cajas de petri con papel filtro whatmann en su interior.5. Preparar 20 cajas de medio NBM estéril.6. Preparar 20 cajas de medio NBM antibióticos estéril.
G. Propagación de Agrobacterium (Día 2).
7. Tomar una colonia de A. tumefaciens e inocular 5 ml de LB antibióticos, incubar en agitación a 28ºC por 24 horas.
C. Propagación de Agrobacterium (Día 3).
8. Tomar 200 l del cultivo de A. tumefaciens e inocular 50 ml de LB antibióticos. Incubar en agitación a 28ºC por 24 horas.
D. Recuperación de la cepa (Día 4).
9. Centrifugar el cultivo de A. tumefaciens a 7500 rpm por 10 min.10. Decantar y resuspender la pastilla en 5 ml de MS0.
E. Cosecha de explantes.
11. Disectar las hojas de plántulas axénicas de tabaco.12. Colocar las hojas en las cajas estériles conteniendo papel filtro.13. Obtener explantes de 0.5 cm x 0.5 cm aproximadamente con hojas de bisturí estériles.14. Recuperar los explantes e incubar en la suspensión de A. tumefaciens por 30 minutos.
F. Siembra y Recuperación de explantes.
15. Recuperar los explantes, secar en papel filtro estéril y colocar en las cajas con medio NBM con la epidermis inferior hacia arriba (haz en contacto con le medio).

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
16. Incubar los explantes a 25ºC con un fotoperiodo de 16h luz durante 5 -7 días.17. Transferir los explantes a medio NBM con kanamicina (100 g/ml) y carbenicilina (500 g/ml). Incubar a las condiciones antes mencionadas por 3 días.18. Transferir los explantes resistentes a kanamicina (aqueyos que no hayan muerto o se encuentren oxidados) a medio MSB5 con kanamicina (100 g/ml) y carbenicilina (200 g/ml) para enraizar.
ANEXO
Medio Luria Bertani (LB) Antibióticos.Extracto de LevaduraTriptona de CaseínaCloruro de SodioEstreptomicina 300 g/mlKanamicina 25 g/ml
Medio Base Murashigue y Skoog (MS)
MS I ( Macronutrientes ) mg/LNH4NO3 1650
KNO3 1900CaCl2.2H2O 440
MgSO4.7H2O 370KH2PO4 170
MS II ( Micronutrientes )IK 0.83
H3BO3 6.2MnSO4.4H2O 22.3ZnSO4.7H2O 8.6
NaMoO4.2H2O 0.25CuSO4..5H2O 0.025CoCl2.6H2O 0.025
MS III ( Fe- EDTA )EDTA 37.3
FeSO4.7H2O 27.8
MS0

Instituto Politécnico Nacional Unidad Profesional Interdisciplinaria de Biotecnología
Laboratorio de Biotecnología Molecular
Medio Base MSSacarosa 30 g/LpH 5.5
MSB5
Medio MS0Inositol 1 mg/LPiridoxina 0.1 mg/LTiamina 2 mg/LÁcido Nicotínico 0.1 mg/LpH 6
NBM
Medio MSB5Ácido Naftalen Acético (NAA) 0.1 mg/LBenzil Aminopurina (BAP) 1 mg/LpH 5.9
Referencias
Sambrook, J. y Russell, D. W. Molecular Cloning: A Laboratory Manual. 3ª ed. Ed Cold Spring Harbor Laboratories. Nueva York. USA 2001. 2344 págs.