instituto politÉcnico nacional unidad profesional … · 2017. 11. 7. · marcadores de peso...
TRANSCRIPT

INSTITUTO POLITÉCNICO NACIONAL
UNIDAD PROFESIONAL INTERDISCIPLINARIA DE
BIOTECNOLOGÍA
ESTANCIA DE TITULACIÓN
Producción de un marcador de peso molecular de ADN mediante
PCR
REPORTE FINAL
En la modalidad de Proyecto de Investigación de la opción curricular
que para obtener el título de
Ingeniero Biotecnólogo
presenta:
Rosalina Gómez Sánchez
Director interno: Dr. Jesús Agustín Badillo Corona
México, D.F Agosto 2010


Agradecimientos
Al Dr. Jesús Agustín Badillo Corona, por haberme permitido trabajar a su lado, por ser
tan paciente y dedicado para la realización de este proyecto, pero sobretodo por su
amistad y confianza.
A mis compañeros de laboratorio Marysol, Diana, Jonás y José, por haberme aligerado
el tiempo de trabajo y por brindarme su amistad.

Dedicatorias
Este trabajo se lo dedico principalmente a mis padres José Eduardo Gómez Aguilar y
Rosalina Sánchez López, por todo el amor, confianza, paciencia, apoyo y tiempo
brindado a mi persona para lograr esta gran meta. Los amo, muchas gracias por todo.
A mi hermano Carlos Eduardo, por siempre ayudarme cuando lo necesité, por
haberme brindado sus conocimientos y por aligerarme mis ratos de estudio, te quiero.
A mi hermana Alejandra, por ser mi mejor amiga, por siempre tener las palabras
adecuadas para hacerme entender, pero sobretodo porque en todo momento estas a
mi lado dispuesta a ayudarme, gracias, te quiero.
A mi hermano César Daniel, por siempre hacerme saber que podía lograr mis metas y
por tu apoyo, te quiero.
A mis amigos Alfonso, Omar, Javier, Pepe, Emmanuel, Jessica y Shantal, por haber
compartido mucho de su tiempo a mi lado, por los ratos de diversión vividos y
simplemente por su amistad y confianza, los quiero a todos.
A mis profesores Leobardo Ordaz, Tania Saynes y Luis Fernández Linares, por siempre
brindarme su apoyo.
Esperando no olvidar a nadie, muchas gracias a todos.

ÍNDICE
1. Introducción…………………………………………………………………………………………………..…..1
1.1 Introducción……………………………………………………………………………………..………………1
1.2 Planteamiento del problema…………………………………………………………..……………….2
1.3 Justificación……………………………………………………………………………………..………………2
1.4 Objetivo general…………………………………………………………………………………..………….2
1.5 Objetivos específicos……………………………………………………………………………..…………2
2. Materiales y Métodos………………………………….……………………………………………………..2
2.1 Materiales……………………………………………………………………………………………………....3
2.1.1 Fuentes de reactivos……………………………………………………………………………..………3
2.1.2 Medios y soluciones……………………………………………………………………………..……….3
2.1.3 Cepas bacterianas………………………………………………………………………………..……….3
2.1.4 Plásmidos y primers……………………………………………………………………………..……….4
2.2 Métodos……………………………………………………………………………………………..……………4
2.2.1 Métodos que involucran células bacterianas…………………………………………..……4
2.2.1.1 Obtención de células electro-competentes de Escherichia coli………….………4
2.2.1.2 Transformación de E. coli DH5α por electroporación………………………….…….4
2.2.2 Métodos de ADN…………………………………………………………………………………….…….5
2.2.2.1 Extracción de ADN plasmídico de E.coli…………………………………………….……..5
2.2.2.2 Precipitación de ADN plasmídico con etanol………………………………………….….5
2.2.2.3 Digestión y ligación de fragmentos de ADN…………………………………………….…6
2.2.2.4 Transformación de E. coli XL10 gold mediante células calcio competentes.6
2.2.3 Diseño de Primers…………………………………………………………………………………………6
2.2.4 Reacción en cadena de la polimerasa (PCR)………………………………………………….7
2.2.5 Electroforesis en gel de agarosa de ADN………………………………………………………7

2.2.6 Determinación de la concentración de ADN mediante
espectrofotometría………………………………………………………………………………………….7
3. Resultados y Discusión de Resultados…………………………………………………………………..8
4. Conclusiones……………………………………………………………………………………………………..…14
5. Perspectivas……………………………………………………………………………………………………..…14
6. Bibliografía……………………………………………………………………………………………………….…15

ÍNDICE DE FIGURAS
Figura 1. Representación esquemática del plásmido Bluescript II KS (+)………..……………………....8
Figura 2. Extracción de DNA de pBsksII en gel de agarosa 0.7%, 90 V, 90 min………………….……10
Figura 3. Productos de PCR de 200, 400, 600, 800, 1000, 1500 y 2000 pb, usando un marcador
comercial, gel de agarosa 0.7%, 90 V, 1h…………………………………………………………………….………….11
Figura 4. Marcador de peso molecular de DNA purificado mediante la precipitación con etanol,
del de agarosa 0.7%, 90V, 1h…………………………………………………………………………………..……………..12
Figura 5. Marcador de peso molecular purificado con glicógeno, gel de agarosa al 0.7%, 90V,
1h………………………………………………………………………………………………………………….……………………….12
Figura 6. Marcador de peso molecular de DNA mediante PCR listo para usarse, (1)
Marcador comercial, (2,3 y 4) marcador listo 1, 3 y 5 uL respectivamente, gel de agarosa
0.7%, 90V, 1h………………………………………………………………………………………….……………………….13
Figura 7. Productos de PCR de 200 (2), 400 (3), 600 (4), 800 (5), 1000 (6), 1500 (7) y 2000 (8) pb
ya estandarizados y producto de ligación (9, 10 y 11), gel de agarosa al 0.7%, 90V, 90 min (1)
Marcador comercial……………………………………………………………………………………………….………………13

ÍNDICE DE CUADROS
Cuadro 1. Primers diseñados y sus características………………………………………………………………... 8
Cuadro 2. Volúmenes estandarizados de las PCR realizadas…………………………………………………10
Cuadro 3. Programa utilizado en el termociclador para amplificar diferentes fragmentos de
DNA………………………………………………………………………………………………………………………………………..11
Cuadro 4. Costo total de producción del marcador de peso molecular de DNA realizado
mediante PCR…………………………………………………………………………………………………………………………14

1
Introducción
1.1 Introducción
La mayoría de los polímeros biológicos poseen carga eléctrica y, por lo tanto, son capaces
de migrar bajo la influencia de un campo eléctrico. El transporte de partículas a través de
un disolvente mediante un campo eléctrico recibe el nombre de electroforesis (Freifelder,
1991).
Los marcadores de peso molecular de ADN son necesarios para determinar el peso
molecular o la cantidad de pares de bases de los aminoácidos, identificar el peso
molecular de una clona después de la digestión con enzimas de restricción, comparar el
tamaño de los insertos de PCR, así como para establecer el tamaño de los productos de
PCR (Chang, Wang, & Lee, 2007) (Jong-Yoon, 2004). En el caso del ADN de doble cadena,
los marcadores estándar son típicamente obtenidos de bacteriófagos o plásmidos. Este
proceso requiere la propagación del virus o del plásmido en un huésped apropiado, la
purificación del ADN viral o plasmídico de los ácidos nucleicos del huésped, la digestión
del ADN purificado con enzimas de restricción y la purificación de los fragmentos
resultantes de la digestión. Este proceso requiere de mucho trabajo, equipo y material.
(Dawson, 1988)
Para caracterizar fragmentos de entre 50 y 3000 pares de bases no es adecuado utilizar
marcadores de peso molecular productos de la digestión de ADN con enzimas de
restricción (Dawson, 1988).
El método para realizar marcadores de peso molecular de ADN mediante PCR involucra la
amplificación de un ADN molde usando dos series de primers que definen el tamaño
absoluto del marcador. Sin embargo, consume mucho tiempo, requiere de muchos
primers para producir todas las fragmentos que se quiere que contenga el marcador.
Además la cantidad de cada fragmento producido no es homogéneo, muchos de los
productos de PCR son más intensos que otros y muchos de ellos son más debiles al ser
observados en una electroforesis en gel de agarosa. (Chang, Wang, & Lee, 2007)
Los productos de PCR generalmente tienen longitudes de entre 50 y 2500 pares de bases.
La electroforesis es generalmente utilizada para determinar si efectivamente el producto
de PCR fue amplificado. Durante el desarrollo de la PCR puede ser necesario aplicar
diferentes concentraciones del gel en el que se correrá el marcador de peso molecular con
el objetivo de determinar la concentración apropiada con la cual se pueden obtener
fragmentos con la adecuada resolución y suficiente intensidad, para permitir la perfecta

2
localización de las fragmentos de pares de bases conocidas en el marcador que se utilizará
(Dawson, 1988).
Marcadores con tamaños estándar para geles de electroforesis de agarosa o
poliacrilamida pueden ser adquiridos de proveedores comerciales o pueden ser
preparados fácilmente en el laboratorio. Es una buena idea tener dos intervalos de
tamaños estándar incluyendo rangos de alto peso molecular de 1kb a 20kb e intervalos de
bajo peso molecular desde 100 pb a 1000pb. Una solución base con los tamaños estándar
puede ser preparada con la dilución de un buffer de corrimiento en gel y después ser
usado de la manera que sea necesario en experimentos de electroforesis individuales
(Sambrook, et al, 2001).
1.2 Planteamiento del problema
Los marcadores de peso molecular de ADN presentan muchas ventajas ya que no son
afectados por el ambiente, son universales, muy abundantes, se requiere poca cantidad
de ADN para los análisis y además el ADN es muy estable y específico. Sin embargo, son
relativamente costosos, necesitan personal entrenado, equipos sofisticados y un estricto
control de la contaminación.
1.3 Justificación
Debido a que los marcadores de peso molecular de ADN comerciales son muy costosos y
que son requeridos con mucha frecuencia en los laboratorios de biología molecular de la
UPIBI, en este proyecto, se propone la metodología para la producción de marcadores de
peso molecular de ADN mediante PCR. Estos marcadores se podrán obtener a un menor
costo, en comparación con los disponibles comercialmente, lo cual resultará en un
beneficio económico y práctico para la institución.
1.4 Objetivo general
Desarrollar y aplicar la metodología para la producción de un marcador de peso
molecular de ADN mediante PCR
1.5 Objetivos específicos
Cuantificar los fragmentos generados por PCR para establecer la formulación del
marcador listo para usar
Preparar el marcador para proporcionarlo a la institución

3
Materiales y Métodos
2.1 Materiales
2.1.1 Fuentes de reactivos
Los reactivos fueron obtenidos de los siguientes proveedores: agar de Dibcon (), Extracto de levadura y triptona de Bioxon (Cuautitlán Izcalli, Estado de México), NaCl de J.T. Baker (Xaloxtoc, Estado de México), Taq polimerasa de Takara (Otsu, Shiga, Japón) MgCl2, Buffer 10x de Invitrogen (Carlsbad, California), el Kit de ZymoResearch (U.S.A), los primers de Bioselect (México, D.F), el etanol y NaOH de High Purity (D.F., México), Acetato de potasio, isopropanol y cloroformo de Fermont (Monterrey, Nuevo León, México), Agarosa OMRESCO (Solon, Ohio).
2.1.2 Medios y soluciones
Medio 2xyt (Triptona 1.6%(w/v), extracto de levadura 1.0% (w/v), cloruro de
sodio 0.5%(w/v)).
El pH fue ajustado a 7.0 con NaOH al 0.5 N. El medio fue esterilizado por 15 min
a 121 ºC, 15 psi en autoclave.
Medio SOC (triptona 2%(w/v), extracto de levadura 0.5% (w/v), NaCl 0.05%
(w/v), KCl 2.5 mM). El pH fue ajustado a 7.0 con NaOH al 0.5 N. El medio fue
esterilizado por 15 min a 121 ºC, 15 psi en autoclave. Cuando el medio se
enfrió a temperatura ambiente, a 1 L del medio se le agregó 20 mL de glucosa
1M esterilizada por filtración (0.22 µm).
2.1.3 Cepas bacterianas
Los cultivos de bacteria fueron crecidos en placas de medio 2xyt (1.5% (w/v) de agar) a
4 ºC para almacenamiento a corto plazo
Escherichia coli DH5α. Genotipo: F- endA1 glnV44 thi-1 recA1 relA1 gyrA96
deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK- mK
+), λ– (Grant,
Jessee, Bloom, & Hanahan, 1990)
Escherichia coli XL10 gold. Genotipo: Tetr D(mcrA)183 D(mcrCB-hsdSMR-
mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac Hte [F¢ proAB
lacIqZDM15 Tn10 (Tetr) Amy Camr]. (Stratagene, 2004)

4
2.1.4 Plásmidos y primers
pBsksII (+)
Es un plásmido con origen de fago, son vectores clonados para simplificar los
procedimientos de clonación y secuenciación. Tienen un extenso sitio de unión
múltiple con 21 sitios únicos de enzimas de restricción. (Technologies, 2008)
Primers
Los primers de 200, 400, 600, 800, 1000, 1500 y 2000 pares de bases fueron diseñados
por el Ing. Erick Miguel Ramos Martínez y los primers de 50, 100, 300, 500, 700, 900,
1100, 1200, 1300, 1400, 1600, 1700, 1800 y 1900 fueron diseñados para la realización
de éste proyecto. Estos primers fueron obtenidos de Bioselect.
2.2 Métodos
2.2.1 Métodos que involucran células bacterianas
2.2.1.1 Obtención de células electro-competentes de Escherichia coli
Una caja petri con medio 2xyt sin ampicilina fue inoculada con E. coli DH5α, la cual se
dejó incubando toda la noche. Se tomó una colonia de E. coli DH5α, la cual se inoculó en
un tubo que contenía 5 mL de medio 2xyt, y se dejó incubar toda la noche. Se
transfirieron 0.5 mL de ese inóculo a un matraz que contenía 50 mL de medio 2xyt pre-
incubado a 37ºC, se incubó de 4-5 horas con agitación vigorosa. Después de este período
el cultivo se mantuvo por 30 min en hielo, después fue transferido a un tubo de 50 mL y se
centrifugó a 4000 x g por 10 min a 4ºC. Se decantó el sobrenadante, el pellet fue re-
suspendido con 25 mL agua desionizada fría. La centrifugación y la re-suspensión se
repitieron 3 veces más, reduciendo el volumen de agua de lavado a 12, 7 y 3 mL,
respectivamente. El pellet que quedó listo al terminar el último lavado se re-suspendió en
1 mL de glicerol frío al 10%. Alícuotas de 40 µL fueron colocadas en tubos estériles de 0.5
mL y congeladas inmediatamente en hielo seco. Estas células competentes fueron
almacenadas a -80 ºC.
2.2.1.2 Transformación de E. coli DH5α por electroporación
Las alícuotas de 40 µL de células electro-competentes fueron descongeladas a
temperatura ambiente y transferidas a un bote con hielos. 2 µL de pBsksII fueron
adicionados a las células competentes y mezclados por pipeteo. La mezcla fue transferida
a la celda de electroporación (Eppendorf 2510). Dos pulsos eléctricos de 2500 V fueron

5
liberados. Después del pulso eléctrico, 1 mL de medio SOC a temperatura ambiente fue
adicionado a la celda de electroporación, se mezcló perfectamente y se recuperó el medio
SOC con el producto de la transformación, todo se adicionó a un tubo estéril de 1.5 mL y
se incubó en el termomixer (Eppendorf) a 37ºC durante 1 hora.
2.2.2 Métodos de ADN
2.2.2.1 Extracción de ADN plasmídico de E.coli
Para la extracción a pequeña escala de ADN bacteriano, se usó el Kit Zyppy TM Plasmid
Midiprep de Zymo Research (U.S.A)
Se tomó una colonia de E. coli DH5α transformada con pBsksII, se inoculó en 5 mL de
medio 2xyt con ampicilina (100 µg/mL), se dejo en agitación vigorosa por 8-12 horas. Se
tomó 1 mL de ese inóculo, se adicionó a un matraz que contenía 50 mL de medio 2xyt con
ampicilina (100 µg/ mL), se dejó incubar toda la noche. Se transfirió el inóculo a un tubo
estéril de 50 mL, el cual se centrifugó a 3400 x g por 10 min, se retiró el sobrenadante, se
agregaron 6 mL de agua desionizada y se re-suspendió completamente la pastilla celular.
Se agregó 1 mL de buffer de lisis 7x, se mezcló completamente invirtiendo el tubo de 4-6
veces y se dejó reposar por 2 minutos. Se adicionaron 3.5 mL de buffer de neutralización
frío y se mezcló hasta que tomó una coloración amarilla completamente. Se colocó un
filtro del kit dentro de otro tubo estéril de 50 mL y se agregó todo el medio neutralizado,
se centrifugó a 500 x g por 6 min. Después de la centrifugación se retiró el filtro de la
columna, se retiró la punta del filtro y se colocó en un tubo cónico incluido en el kit, se
adicionaron 400µL de buffer endo-lavado en pequeñas alícuotas de 150µL hasta
completar los 400µL tomando en cuenta que después de cada volumen agregado se
centrifugó a 11000 x g por 30 seg y después de cada centrifugación se desechó el
sobrenadante. Ya que se completó el volumen de 400µL se adicionaron 800µL de buffer
de lavado en pequeños volúmenes y entre cada adición de buffer se centrifugó a 11000 x
g por 1 min. Se colocó el filtro pequeño en un tubo estéril de 1.5 mL, se agregaron 150µL
de buffer de elución Zippy en el centro de la columna, se incubó 1 min a temperatura
ambiente y se centrifugó a 11000 x g por 1 min para eluir el ADN plasmídico.
2.2.2.2 Precipitación de ADN plasmídico con etanol
Para la precipitación de ADN con etanol, la concentración de iones potasio fue ajustada a
0.3 M con acetato de potasio 3 M. Después de esto, se adicionó el volumen total del
producto de PCR a un tubo estéril de 1.5mL, 1/10 parte del volumen total de PCR de
acetato de potasio 3 M y un volumen de isopropanol. Se dejó incubar 30 min a -20 ºC, se
centrifugó a 14000 rpm a 4 ºC por 20 min, se decantó el sobrenadante y la pastilla celular

6
fue lavada con etanol al 70% frío 2 veces cuidando que no se perdiera la pastilla celular al
decantar el etanol, se re-suspendió la pastilla con un volumen de agua desionizada estéril
equivalente a la mitad del volumen inicial del producto de PCR.
2.2.2.3 Digestión y ligación de fragmentos de ADN
Se amplificó un fragmento de ADN de 2000 pb mediante PCR, se llevó a cabo la digestión y
la ligación de la siguiente manera:
Ligación: (1 µL de pBsks II, 1 µL de enzima EcoRV , 2 µL de buffer 10X, 15 µL de inserto
(producto de PCR de 2000 pb), 1 µL de ligasa
Control (-): (1 µL de pBsksII, 1 µL de enzima EcoRV, 2 µL de buffer 10X, 15 µL de agua
desionizada estéril)
Control (+): (1 µL de ADN plasmídico, 3 µL de buffer 10X, 17 µL de agua desionizada
estéril)
Se dejó incubando la reacción a 4 ºC durante 12 h.
2.2.2.4 Transformación de E. coli XL10 gold mediante células calcio
competentes
Para realizar la transformación se realizaron células calcio-competentes realizadas
siguiendo la metodología de las células electro-competentes con la única diferencia que
los lavados de las pastillas celulares se realizo con CaCl2 al 0.1M y las alícuotas son de 100
µL.
Se toman entre 4-6 µL del producto de ligación los cuales se transfieren a una alícuota de
100 µL de células calcio-competentes, se mezclan 2-3 veces mediante pipeteo, se
transfieren a una placa de medio 2xyt agar (1.5% (w/v)) previamente incubadas a 37 ºC,
se distribuyen los 106 µL con una varilla de vidrio estéril, se incuban las placas entre 12-16
h a 37 ºC. Se toma una colonia ya transformada, se transfiere a un tubo estéril con medio
2xyt, se deja incubando a 37ºC con agitación vigorosa durante 8h y se realiza nuevamente
la extracción del nuevo plásmido ya transformado como se mencionó en el método
2.2.2.1.
2.2.3 Diseño de Primers
De la página de NCBI se obtuvo el genoma del plásmido Bluescript KS II (+), ya teniendo el
genoma, con ayuda del programa PRIMER3 v 0.4.0 (Rozen & Skaletsky, 2000), se
obtuvieron los primers de 50, 100,300, 500, 700, 900, 1100, 1200, 1300, 1400, 1600, 1700,

7
1800, 1900 pb, utilizados para este proyecto, aunque también se contaba con primers
para 200, 400, 600, 800, 100, 1500 y 2000 pb (Diseñados por Erik Miguel Ramos Martínez).
Para cada uno de los primers diseñados en este proyecto, se calculó que en la secuencia
que amplificaban solo tuvieran el 50% ±3, lo cual se obtuvo con el programa OLIGO
CALCULATOR.
2.2.4 Reacción en cadena de la polimerasa (PCR)
Se realizaron reacciones de PCR para obtener fragmentos de 200, 400, 600, 800, 1000,
1500 y 2000 bp en un termociclador (Labnet international.INC).
En todos los casos se usaron las mismas cantidades de reactivos, solo se modificaron
algunas condiciones en el termociclador dependiendo del número de pares de bases,
todas las PCR se llevaron a cabo 40 ciclos desde el segmento 2 al 4.
2.2.5 Electroforesis en gel de agarosa de ADN
Los fragmentos de ADN de doble cadena fueron separados por electroforesis en gel de
agarosa (0.7-1.5% (w/v)) dependiendo del tamaño de los fragmentos que iban a ser
analizados. La agarosa AMRESCO (Solon, Ohio) fue suspendida en buffer TBE 0.5x (54 mM
de Tris base, 27.5 mM de ácido bórico y 2 mM de EDTA, pH 8) y calentada en horno de
microondas. Cuando la agarosa quedo completamente disuelta en el buffer, se agregó 1
µL de colorante rojo Texas, toda la mezcla se vertió a una charola para gel incluida en la
cámara de electroforesis (NESS 5000 HM), se dejó gelificar. Se colocó el gel en la cámara y
se llenó con suficiente buffer TBE hasta que cubriera completamente el gel, se agregaron
los fragmentos de ADN con 2 µL de buffer de carga 6x en los pozos del gel antes de dejarlo
correr. La electroforesis se dejo correr de 1-2 h a un voltaje constante de 90 V. Los
tamaños de los fragmentos fueron estimados mediante la comparación con un marcador
de ADN comercial (ZipRuler Express DNA Ladder 2, Fermentas). Las fragmentos de ADN
fueron visualizadas en un transiluminador de UV (KODAK Gel Logic 440 Imaging System)
2.2.6 Determinación de la concentración de ADN mediante
espectrofotometría
5 µL de la solución de ADN fueron diluidos en 995 µL de agua desionizada. El
espectrofotómetro (Perkinelmer Lambda XLS), fue ajustado a cero con la misma agua
utilizada para diluir el ADN. La contaminación por proteínas fue estimada por la relación
A260/A280. La cuantificación de ADN fue determinada por la medida de absorbancia a
260nm. La concentración de ADN fue calculada considerando que A260=1.0 de ADN de
doble cadena es equivalente a 50 µg/mL (Sambrook & Rusell, 2001)

8
Resultados y Discusión de Resultados
Se usó el DNA del plásmido Bluescript II KS (+) (Figura 1) como molde para realizar todas las
amplificaciones del DNA mediante PCR.
Figura 1. Representación esquemática del plásmido Bluescript II KS (+)
En el diseño de primers, se verificó que los fragmentos que amplificarían cada set de
primers contuvieran el 50 ± 3% de contenido de GC esto con el fin de que al momento en
que los fragmentos amplificados del DNA se sometieran a un campo eléctrico, estas
tuvieran una migración constante, ya que si los productos de PCR fueran algunos con
mayor cantidad de AT y otros con mayor cantidad de GC, al someterlos al campo eléctrico
los de mayor contenido de AT migrarían más rápido que los de mayor contenido de GC
debido a los pesos moleculares de estos pares de bases nitrogenadas (Navarro Blaya &
Navarro García, 2003), y una temperatura de alineación de 60 ±2 ºC para con ello lograr
que los primers diseñados fueran compatibles entre sí, pudiendo llevar a cabo mix de
primers de ser necesario. De los primers diseñados (Cuadro 1), solo se realizaron
amplificaciones del DNA utilizando los primers para 200, 400, 600, 800, 1000,1500 y 2000
pb.
Cuadro 1. Primers diseñados y sus características
Producto de 50 pb %GC 52 Producto de 1000 pb %GC 47
Primers Secuencia Tm %GC Forward TTTGCAAGCAGCAGATTACG 60.2 45
Forward TGCAAGGCGATTAAGTTGG 59.82 47.37 Reverse CCAGAAACGCTGGTGAAAGT 60.3 50
Reverse AACGTCGTGACTGGGAAAAC 60.01 50 Producto de 1100 pb %GC 51
Producto de 100 pb %GC 47 Forward GCATTAATGAATCGGCCAAC 60.3 45
Forward GTGCACCCAACTGATCTTCA 59.68 50 Reverse AGCCCTCCCGTATCGTAGTT 59.98 55
Reverse GTCGCCCTTATTCCCTTTTT 59.44 45 Producto de 1200 pb %GC 47

9
Producto de 200 pb %GC 49 Forward GCAGAGCGAGGTATGTAGGC 60.01 60
Forward GTCCTCCGATCGTTGTCAGA 61.2 55 Reverse TTTGCCTTCCTGTTTTTGCT 59.86 40
Reverse CGCGGTATTATCCCGTATTG 60.2 50 Producto de 1300 pb %GC 52
Producto de 300 pb %GC 52 Forward CGCTCACAATTCCACACAAC 60.16 50
Forward ACCCAGCTTTTGTTCCCTTT 59.98 45 Reverse GATAAATCTGGAGCCGGTGA 60.04 50
Reverse GAGTCAGTGAGCGAGGAAGC 60.29 60 Producto de 1400 pb %GC 52
Producto de 400 pb %GC 53 Forward TATCCGCTCACAATTCCACA 60.07 45
Forward AGCTTGGCGTAATCATGGTC 60.1 50 Reverse CTAGCTTCCCGGCAACAAT 60.22 52.63
Reverse TTTTACGGTTCCTGGCCTTT 60. 8 45 Producto de 1500 pb %GC 51
Producto de 500 pb %GC 51 Forward GTTTTCCCAGTCACGACGTT 60.01 50
Forward ATACGGGAGGGCTTACCATC 60.17 55 Reverse GTAAGCCCTCCCGTATCGTA 59.07 55
Reverse GGTCGCCGCATACACTATTC 60.5 55 Producto de 1600 pb %GC 52
Producto de 600 pb %GC 53.5 Forward ATCCGCTCACAATTCCACAC 60.93 50
Forward ACGAGCATCACAAAAATCGAC 60.1 42.9 Reverse GCGGCCAACTTACTTCTGAC 59.88 55
Reverse CGTCAGACCCCGTAGAAAAG 59.7 55 Producto de 1700 pb %GC 53
Producto de 700 pb %GC 50 Forward AGGAAGGGAAGAAAGCGAAA 60.31 45
Forward CTACGATACGGGAGGGCTTA 59.2 55 Reverse GGGAGTCAGGCAACTATGGA 60.07 55
Reverse CCAGAAACGCTGGTGAAAGT 60.29 50 Producto de 1800 pb %GC 51
Producto de 800 pb %GC 47 Forward CATTAATGAATCGGCCAACG 61.21 45
Forward CAACCCGGTAAGACACGACT 60.03 55 Reverse CCTTCCTGTTTTTGCTCACC 59.71 50
Reverse AAGCCATACCAAACGACGAG 60.13 50 Producto de 1900 pb %GC 51
Producto de 900 pb %GC 50 Forward ACATACGAGCCGGAAGCATA 60.63 50
Forward TCACAAAAATCGACGCTCAA 60.38 40 Reverse CCAGAAACGCTGGTGAAAGT 60.29 50
Reverse GATAAATCTGGAGCCGGTGA 60.04 50 Producto de 2000 pb %GC 50
Forward TTAATTTCGAGCTTGGCGTAA 59.87 38.1
Reverse TTTTGCCTTCCTGTTTTTGC 60.22 40
Al realizar la extracción del DNA del plásmido (Figura 2), se observó que aunque éste
plásmido tiene un peso aproximado de 3.0 Kb, el fragmento se visualizó entre 1500 y 2000
pb, ya que la estructura normal del DNA es enrollada y si se hubiera linealizado antes de
correrla en el gel, hubiera coincidido con el número de bases del plásmido (Stainer, 1996).

10
Figura 2. Extracción de DNA de pBsksII en gel de agarosa 0.7%, 90 V, 90 min
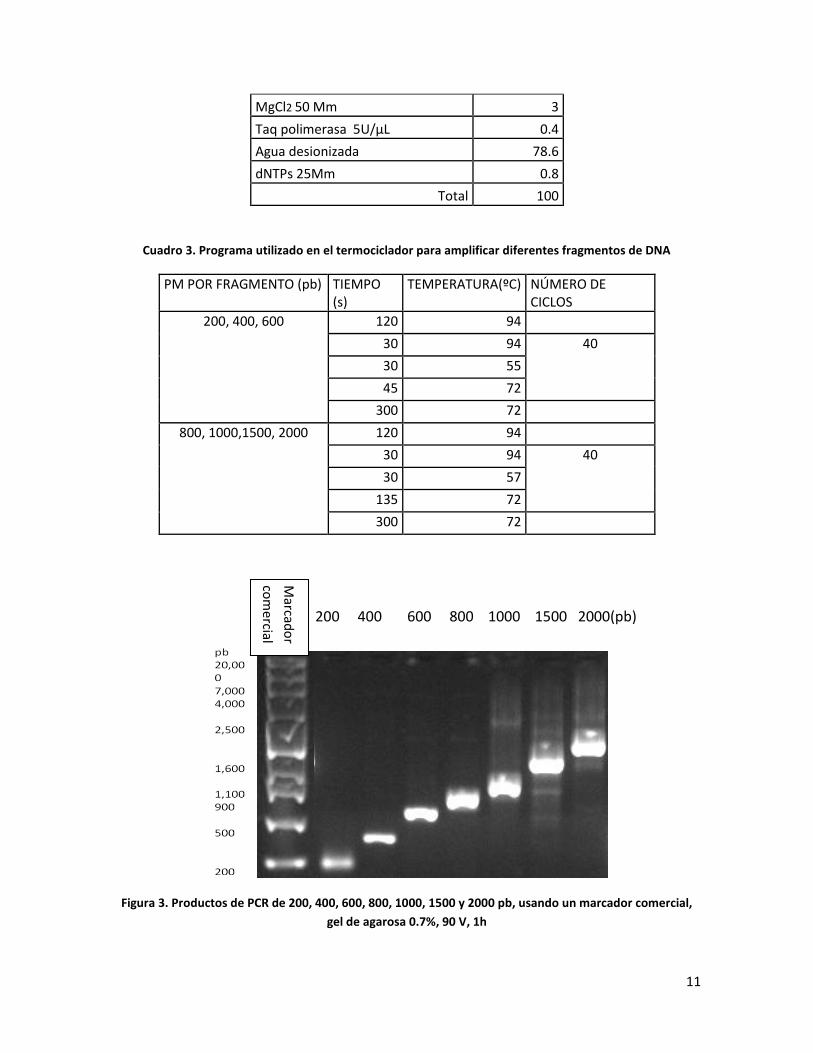
Después de muchas pruebas de PCR, se determinó el programa en el termociclador que
nos daba la mejor amplificación de fragmentos, así como los volúmenes de los reactivos
necesarios para llevar a cabo la reacción en cadena de la polimerasa (Cuadro 2 y Cuadro 3),
con los cuales se llevaron a cabo las amplificaciones de fragmentos de DNA de 200, 400,
600, 800, 1000, 1500 y 2000 pb (Figura 3), los cuales coinciden y se pueden comparar con
los pesos moleculares del marcador comercial, aunque no presentan la misma intensidad
que el marcador comercial, aunque es un problema que se puede arreglar adicionando en
proporciones iguales la cantidad de DNA amplificado al momento de realizar el marcador
de peso molecular. Los barridos que se pueden visualizar en los fragmentos de DNA de
mayor peso molecular se deben a que el DNA se degradó un poco y a restos de reactivos
utilizados en la PCR, por lo tanto se procedió a realizar la purificación.
Cuadro 2. Volúmenes estandarizados de las PCR realizadas
REACTIVO Volumen (µL)
DNA MOLDE pBsksII (dilución 1:20) 2
Primer forward (20pmol/µL) 2.6
Primer reverse (20pmol/µL) 2.6
Buffer 10x 10
pb
20,000
7,000
4,000
2,500
1,500
1,000
700
400
200
11
MgCl2 50 Mm 3
Taq polimerasa 5U/µL 0.4
Agua desionizada 78.6
dNTPs 25Mm 0.8
Total 100
Cuadro 3. Programa utilizado en el termociclador para amplificar diferentes fragmentos de DNA
PM POR FRAGMENTO (pb) TIEMPO (s)
TEMPERATURA(ºC) NÚMERO DE CICLOS
200, 400, 600 120 94
30 94 40
30 55
45 72
300 72
800, 1000,1500, 2000 120 94
30 94 40
30 57
135 72
300 72
200 400 600 800 1000 1500 2000(pb)
Figura 3. Productos de PCR de 200, 400, 600, 800, 1000, 1500 y 2000 pb, usando un marcador comercial,
gel de agarosa 0.7%, 90 V, 1h
Marcad
or
com
ercial

12
En el caso de la purificación, se buscaron alternativas de purificación que brindaran los
mejor resultados, se probó mediante precipitación con etanol (Figura 4), con la cual era
muy difícil recuperar los fragmentos de menor peso molecular como los de 200 y 400 pb,
por ello se eligió utilizar la purificación con glicógeno (Figura 5), el cual es un polisacárido
derivado de las ostras que es muy útil para precipitar DNA, con el cual se pudieron
recuperar todos los fragmentos amplificados.
Figura 4. Marcador de peso molecular de DNA purificado mediante la precipitación con etanol, del de
agarosa 0.7%, 90V, 1h
Figura 5. Marcador de peso molecular purificado con glicógeno, gel de agarosa al 0.7%, 90V, 1h
Para el marcador de peso molecular de 200, 400, 600, 800, 100, 1500 y 2000 pb (Figura 6), se
purificaron los productos de PCR, y se le adiciono EDTA pH 8 a la mezcla de fragmentos de DNA de
esos pesos moleculares así como buffer de carga, para generar un marcador listo para utilizarse.
13
1 2 3 4
Figura 6. Marcador de peso molecular de DNA mediante PCR listo para usarse, (1) Marcador comercial,
(2,3 y 4) marcador listo 1, 3 y 5 uL respectivamente, gel de agarosa 0.7%, 90V, 1h
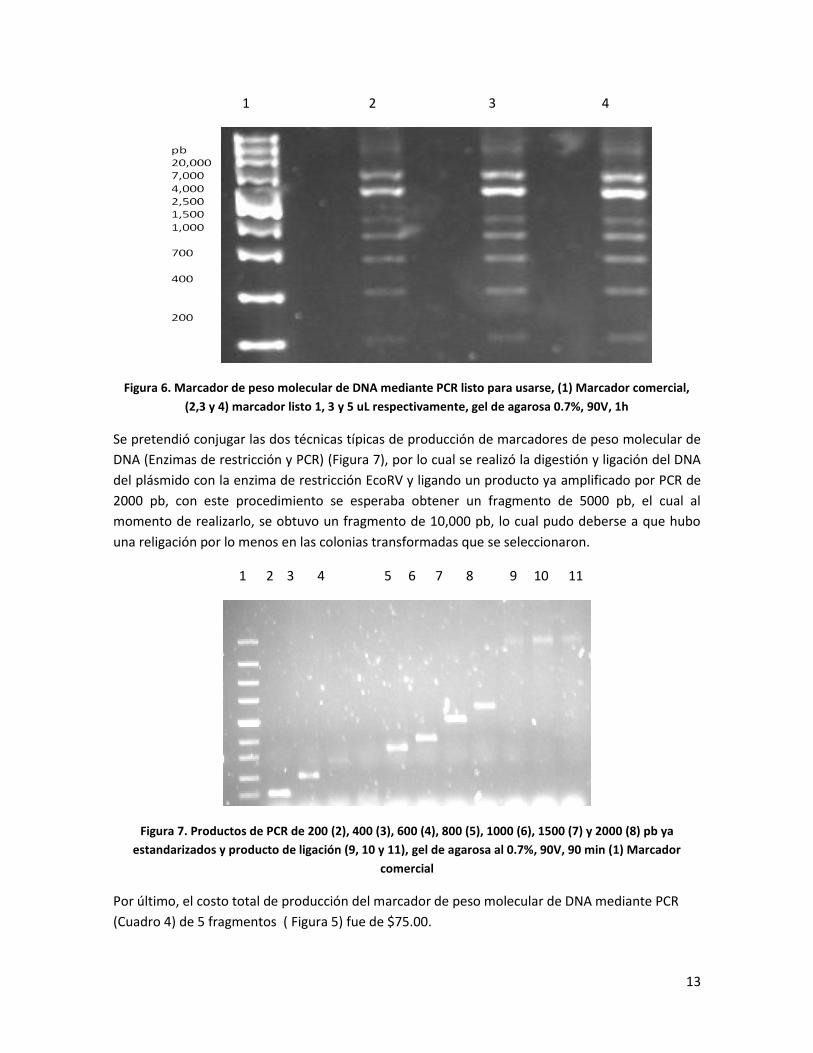
Se pretendió conjugar las dos técnicas típicas de producción de marcadores de peso molecular de
DNA (Enzimas de restricción y PCR) (Figura 7), por lo cual se realizó la digestión y ligación del DNA
del plásmido con la enzima de restricción EcoRV y ligando un producto ya amplificado por PCR de
2000 pb, con este procedimiento se esperaba obtener un fragmento de 5000 pb, el cual al
momento de realizarlo, se obtuvo un fragmento de 10,000 pb, lo cual pudo deberse a que hubo
una religación por lo menos en las colonias transformadas que se seleccionaron.
1 2 3 4 5 6 7 8 9 10 11
Figura 7. Productos de PCR de 200 (2), 400 (3), 600 (4), 800 (5), 1000 (6), 1500 (7) y 2000 (8) pb ya
estandarizados y producto de ligación (9, 10 y 11), gel de agarosa al 0.7%, 90V, 90 min (1) Marcador
comercial
Por último, el costo total de producción del marcador de peso molecular de DNA mediante PCR
(Cuadro 4) de 5 fragmentos ( Figura 5) fue de $75.00.

14
Cuadro 4. Costo total de producción del marcador de peso molecular de DNA realizado mediante PCR
Reactivo Costo por reacción de 100 uL
dNTPs $ 2.10
Taq $ 6.00
Primers F $ 0.29
Primer R $ 0.26
Total $ 8.65
Conclusiones
Se obtuvo un marcador de ADN mediante PCR de 200, 400, 600, 800, 1000, 1500 y
2000 pb listo para ser utilizado
El costo del marcador de 500 µL preparado en laboratorio y purificado con
glicógeno (5 bandas) es de $75.00, 40 veces más barato que el marcador comercial
Perspectivas
Hacer el marcador de peso molecular de ADN con todos los fragmentos de los
primers diseñados.
Mejorar la purificación utilizando una columna de sílice específica para ADN
Realizar digestión y ligación con enzimas de restricción para obtener bandas de
10,000 y 5,000 pb conjuntando los 2 métodos de realización de marcadores de
peso molecular
Realizar marcadores de peso molecular del tamaño que los investigadores de la
UPIBI deseen o necesiten

15
Bibliografía
Amills, M., Francino, O., & Sánchez, A. (1996). Primer-directed synthesis of a molecular weight
marker.
Chang, M., Wang, J.-H., & Lee, H.-J. (2007). Laboratory production of 100 base paor DNA molecular
weight markers.
Dawson, E. P. (1998). Patente nº 5714326. Estados Unidos.
Freifelder, D. (1991). Técnicas de bioquímica y biología molecular. Barcelona: Reverté.
Grant, S. G., Jessee, J., Bloom, F. R., & Hanahan, D. (1990). Differential plasmidrescue from
transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. 87.
Jong-Yoon, C. (2004). Patente nº PCT/KR2004/000046. Corea.
Navarro Blaya, S., & Navarro García, G. (2003). Química agrícola: el suelo y los elementos químicos
esenciales para la vida vegetal. España: Mundi Prensa Libros.
Polyarush, S., Egamberdiev, S., Mansurov, D., & Azimova, S. (2003). Preparation of DNA markers
based on E. coli plasmid DNA. 39 (6).
Rozen, S., & Skaletsky, H. J. (2000). Primer3 on the WWW for general users and for biologist
programmers. Bioinformatics Methods and Protocols: Methods in Molecular Biology . Totowa, New
Jersey, Estados Unidos: Humana Press.
Sambrook, J., & Rusell, D. W. (2001). Molecular Cloning a laboratory manual (Tercera edición ed.,
Vol. 1). New York: Cold Spring Harbor Laboratory Press.
Stainer, R. Y. (1996). Microbiología. España: Reverté.
Stratagene. (2004). XL10 GOLD ULTRACOMPETENT CELLS. La Jolla CA.
Technologies, A. (2008). pBluescript II Phagemids Vectors. La Jolla CA.