Índice de contenidos - centogene.com · la enfermedad de m. fabry presenta un desarrollo...
TRANSCRIPT


1
Índice de contenidos
Novedades – Breve descripción de las características de la enfermedad 3
Recomendaciones más importantes 3
Introducción 5
Objetivos y campo de aplicación 5
1 Clasificación y epidemiología 5
2 Fisiopatología 5
3 Historia natural de la enfermedad 6
3.1 Dolores 7
3.2 Piel 7
3.2.1 Angioqueratomas 7
3.2.2 Dishidrosis 7
3.2.3 Linfedemas 8
3.3 Tracto gastrointestinal 8
3.4 Riñónes 9
3.5 Corazón 10
3.6 Sistema nervioso central 11
3.7 Órganos sensoriales 12
3.7.1 Ojos 12
3.7.2 Oídos 13
3.8 Calidad de vida 13
3.9 Otras manifestaciones de M. Fabry 13
4 Diagnóstico en caso de sospecha de M. Fabry 13
4.1 Determinación de la actividad de AGLA 14
4.1.1 Determinación de la actividad de AGLA en hombres 14
4.1.2 Determinación de la actividad de AGLA en mujeres 14
4.2 Análisis genético-molecular 14
4.2.1 Mutaciones patógenas 14
4.2.2 Análisis de mutaciones para confirmar el diagnóstico en hombres 15
4.2.3 Análisis de mutaciones para confirmar el diagnóstico en mujeres 15
4.3 Análisis familiar y asesoramiento genético 16
4.4 Determinación de Gb3 y liso-Gb3 16
4.5 Diagnóstico diferencial 16

2
5 Diagnóstico con M. Fabry confirmado 16
5.1 Riñones 17
5.1.1 Abordaje inicial 17
5.1.2 Seguimiento 17
5.2 Corazón 18
5.2.1 Abordaje inicial 18
5.2.2 Seguimiento 18
5.3 Sistema nervioso 18
5.3.1 Abordaje inicial 18
5.3.2 Seguimiento 18
5.4 Tracto gastrointestinal 19
5.5 Oídos 19
5.5.1 Abordaje inicial 19
5.5.2 Seguimiento 19
6 Depresión y calidad de vida 19
7 Terapia y asistencia 20
7.1 Terapia de sustitución enzimática 20
7.1.1 Datos disponibles sobre la eficacia 20
7.1.2 Indicación e inicio de la terapia 22
7.1.3 Tolerancia 23
7.1.4 Particularidades de la terapia en mujeres 23
7.1.5 Particularidades de la terapia en la infancia 23
7.1.6 Calidad de vida 24
7.1.7 Terapia de infusión en casa 24
7.2 Terapias concomitantes 25
7.2.1 Terapia nefrológica concomitante 25
7.2.2 Terapia cardiológica concomitante 25
7.2.3 Terapia neurológica concomitante 26
8 Organización de autoayuda 26
Bibliografía 27
Índice de abreviaturas 34
Grupo de expertos 34
Procedimiento de consenso 35
Conflicto de intereses 35
Vigencia 35

3
Novedades – Breve descripción de las características de la enfermedad
El cuadro clásico de la enfermedad por depósito lisosomal M. Fabry, hereditaria y ligada al cromosoma X, se observa en general entre los varones de las familias. Las mujeres enferman más tarde que los hombres y presentan un desarrollo de la enfermedad más leve.
La enfermedad de M. Fabry presenta un desarrollo progresivo. En comparación con la población normal, la calidad de vida disminuye significativamente en los pacientes con M. Fabry.
Los primeros síntomas (acroparestesias, dolores crónicos en las articulaciones, trastornos gastrointestinales, sensibilidad al calor, tinnitus) se manifiestan en general durante la infancia o la adolescencia.
Otras manifestaciones son angioqueratomas y telangiectasias no específicas, alteraciones en la sudoración, pérdida de la audición neurosensorial y daños en el órgano vestibular. Otros efectos posibles son también trastornos obstructivos de las vías respiratorias, osteopenia y anemia. Son frecuentes los diagnósticos erróneos. A menudo la enfermedad de M. Fabry se diagnostica hasta muchos años después de la aparición de los primeros síntomas.
Una característica oftalmológica frecuente es la córnea verticillata: en general los cambios en la córnea pueden ser detectados fácilmente en una revisión con una lámpara de hendidura.
Hasta un 18% de los niños(as) con M. Fabry (menores de 18 años) pueden tener proteinuria al momento del diagnóstico. La nefropatía no es pronóstica, independientemente del sexo del paciente.
Entre las manifestaciones cardiacas se encuentra la miocardiopatía (hipertrofia del ventrículo izquierdo) y/o trastornos del ritmo cardiaco. Los(as) niños(as) pueden presentar un mayor índice de masa ventricular izquierda y una reducida variabilidad de la frecuencia cardiaca al momento del diagnóstico. La hipertrofia ventricular y la fibrosis intramiocárdica son indicadores de trastornos malignos del ritmo cardiaco.
Entre las manifestaciones en el SNC se encuentran los ataques isquémicos transitorios
(TIA por sus siglas en inglés) (también detectados en la infancia) y accidentes cerebrovasculares. Las alteraciones en las señales de la TRM en la zona del núcleo pulvinar del tálamo son indicadores de una implicación del SNC.
Raras veces se diagnostican clínicamente los procesos con pocos síntomas, en muchas ocasiones con una sola manifestación orgánica; estos casos son descritos, sobre todo, en personas afectadas que fueron diagnosticadas en el marco de un programa de tamizaje.
En todos los pacientes con M. Fabry, el pronóstico depende de forma decisiva de un diagnóstico y tratamiento temprano.
En los casos de enfermedad sin tratamiento, las posibles complicaciones en riñones, corazón y SNC disminuyen con frecuencia y claramente la esperanza de vida de los afectados. Las principales causas de muerte en pacientes con M. Fabry son insuficiencia renal, paro cardiaco súbito por trastornos del ritmo cardiaco e infarto cerebral.
La terapia de sustitución enzimática (TSE) en los casos de M. Fabry puede detener la progresión de la enfermedad. En el perfil de compatibilidad de la TSE no se han observado diferencias entre niños y adultos.
Desde el año 2001 existen dos registros internacionales de pacientes fomentados respectivamente por uno de los dos fabricantes del preparado de sustitución enzimática: Fabry Outcome Survey (FOS; Shire HGT) y Fabry Registry (Genzyme). La Canadian Fabry Disease Initiative (CFDI) ha establecido en el año 2007 un registro nacional para pacientes con M. Fabry independiente de la industria.
Recomendaciones más importantes
En los varones, el método a elegir para el diagnóstico es determinar la actividad de la α-galactosidasa A (AGLA) en los leucocitos de la sangre. Para confirmar el diagnóstico se recomienda realizar un análisis molecular del gen de la α-galactosidasa A (gen GLA) para comprobar si existe una mutación causante de la enfermedad. Según el estado actual de la ciencia, si la actividad de AGLA se sitúa en rangos normales, se puede excluir el diagnóstico de M. Fabry en los varones.
En mujeres, la medición de la actividad de

4
AGLA ofrece un valor informativo muy limitado y se requiere del análisis molecular para el diagnóstico.
Si la actividad enzimática presenta un valor patológicamente reducido, o se detecta una mutación genética, el paciente debería recibir asesoramiento genético.
En los pacientes con proteinuria o función renal limitada de origen idiopático, así como en los pacientes jóvenes (< 55 años, en particular con ectasia basilar) es necesario descartar la existencia de un M. Fabry.
El tratamiento de M. Fabry exige la estrecha cooperación de diferentes especialidades médicas.
Tras el diagnóstico de M. Fabry se recomienda una evaluación clínica básica de los órganos y sistemas a los que afecta de forma típica. Estos son piel, ojos, riñones, corazón, cerebro, sistema nervioso periférico (incluido dolor), oídos, tracto gastrointestinal y calidad de vida
El abordaje médico inicial y de seguimiento son idénticos para hombres y mujeres.
Las manifestaciones orgánicas que disminuyen la calidad de vida de los afectados como la nefropatía, las enfermedades cardiacas y cerebrovasculares, los trastornos del oído y la polineuropatía de pequeña fibra deben ser controladas en el curso de las consultas médicas de seguimiento.
Las personas asintomáticas con una mutación patogénica en el gen GLA deberían ser controladas cada 6 meses.
Los parámetros para la exploración del riñón son: creatinina, excreción de creatinina, tasa de filtración glomerular o GFR (por sus siglas en inglés), precipitación de proteínas en orina, ultrasonidos, medición de la presión sanguínea 24 horas; se recomienda realizar reconocimientos médicos de control al menos una vez al año.
Si existe proteinuria y/o hipertonía se requiere una terapia adicional, como es habitual aplicar también en otras afecciones renales crónicas para retrasar la progresión de la insuficiencia renal. Como terapias concomitantes se dispone de los medicamentos inhibidores ECA o sartanos.
Durante la diálisis y tras un trasplante de riñón debe continuarse la TSE sin cambios.
Los parámetros para la exploración del corazón son: ECG, ECG de larga duración, ecocardiografía, RM cardiaca; se recomienda realizar pruebas de seguimiento cada 12 meses cuando existan síntomas cardiológicos y cada 24 meses en ausencia de síntomas cardiológicos o con resultados patológicos negativos.
Los parámetros para la exploración del SNC y sistema nervioso periféricos son: sonografía doppler y dúplex de las arterias cerebrales (en particular la arteria basilar), RM cerebral, neurografía, evaluación de la intensidad del dolor y de la calidad de vida; se recomienda realizar pruebas de seguimiento cada 12 meses en caso de síntomas neurológicos, particularmente vasculares, y cada 24 meses incluida una RM cerebral si no existen síntomas neurológicos.
El tratamiento de M. Fabry comprende la TSE y el tratamiento paralelo de las afecciones orgánicas y los síntomas.
En la actualidad, la TSE es la única terapia causal de la enfermedad de M. Fabry. Es recomendable iniciar la TSE lo antes posible, tanto en hombres como en mujeres, tras confirmarse el diagnóstico de M. Fabry y en caso de existir síntomas clínicos. La eficacia de la TSE ha sido probada también en niños(as).
Los objetivos de la terapia son: reducir las molestias (sobre todo mitigar el dolor), mejorar la calidad de vida, evitar un malfuncionamiento orgánico (sobre todo de riñones, corazón y SNC) y aumentar la esperanza de vida.
No existe ninguna indicación para interrumpir la TSE durante el embarazo.
La terapia de infusión en casa: cuando el paciente haya recibido unos 6 tratamientos de TSE en la clínica o en la consulta médica sin producirse reacciones adversas, se puede ofrecer la posibilidad de realizar la terapia en el domicilio del paciente.

5
Introducción M. (Anderson) Fabry es una enfermedad de depósito lisosomal hereditaria ligada al cromosoma X. La afección se caracteriza por una deficiencia de la enzima α-galactosidasa A (AGLA) que, en un caso típico, causa la acumulación del esfingolípido globotriaosilceramida (Gb3) en numerosos órganos del cuerpo. Los pacientes se presentan tanto con síntomas aislados (p. ej. solo dolores o solo trastornos cerebrovasculares) como con múltiples síntomas, en general con diferentes grados de intensidad. Entre estos se encuentran sobre todo dolores (acroparestesias), alteraciones en la sudoración, trastornos del ritmo cardiaco, disnea, además de molestias gastrointestinales y pérdida de audición. Diagnósticos importantes son nefropatía, miocardiopatía, infarto cerebral y angioqueratomas. Sin tratamiento, M. Fabry no solo merma la calidad de vida de los pacientes, también reduce en gran medida su esperanza de vida. Las principales causas de morbilidad y mortalidad son la concurrencia de afecciones renales, cardiacas y cerebrovasculares que provocan infartos cerebrales y fallos cardiacos y renales prematuros. M. Fabry presenta una elevada viriabilidad clínica. El diagnóstico se establece con frecuencia solo tras una búsqueda específica y en cooperación con diferentes médicos especialistas. Es frecuente, sobre todo en la infancia, que no se identifiquen los indicios de M. Fabry como tales o se atribuyan a otros trastornos. Objetivos y campo de aplicación La presente guía comprende la diversidad clínica de M. Fabry según el estado actual del conocimiento y ofrece pautas para llegar a un diagnóstico y una terapia de la enfermedad mediante la cooperación interdisciplinaria. El objetivo central es poner a disposición la información y las directivas de actuación siguientes:
guía para un diagnóstico rápido y cuidadoso con el paciente y los recursos
líneas de decisión racionales para decidir la terapia
Esta guía es multidisciplinaria. Está dirigida en primera línea a médicos que ejercen en clínicas y organizaciones de pacientes.
1 Clasificación y epidemiología M. Fabry es una enfermedad de depósito lisosomal (EDL) hereditaria ligada al cromosoma X. Estudios sobre la frecuencia muestran prevalencias de 1:40.000 a 1:117.000 nacidos vivos (Meikle et al., 1999; Desnick et al., 2001), con considerables variaciones entre los diferentes países. Sin embargo, existen indicios de que la enfermedad es más frecuente de lo que se pensaba hasta ahora: el tamizaje de aproximadamente 37.000 recién nacidos vivos varones mostró una incidencia de falta de α-galactosidasa A, la base de M. Fabry, cercana a 1:3.100 (Spada et al., 2006). M. Fabry afecta sobre todo a hombres. Las mujeres también pueden padecerla, aunque su desarrollo puede ser más lento y el pronóstico mejor. La edad media de manifestación se sitúa en los pacientes varones entre los 3 y los 10 años, en las mujeres afectadas entre los 6 y los 15 años (MacDermot et al., 2001 [b]; Ramaswami et al., 2006; Ries et al., 2003). En los enfermos sin tratamiento, la insuficiencia renal, las afecciones cerebrovasculares y la miocardiopatía redujeron la esperanza de vida de las personas afectadas en aproximadamente 20 años entre los hombres y 10 años entre las mujeres (Germain, 2010; MacDermot et al., 2001 [a]; MacDermot et al., 2001 [b]). 2 Fisiopatología La causa subyacente de M. Fabry es una mutación del gen GLA que codifica la encima lisosomal α-galactosidasa A (AGLA). Hasta la fecha se han identificado varios cientos de mutaciones en el gen GLA (Gal et al., 2010). Por la deficiencia o la falta de AGLA no se metabolizan suficientemente los glicoesfingolípidos – sobre todo la globotriaosilceramida (Gb3) – y se acumulan en los lisosomas. Las células dañadas por los depósitos de glicoesfinglolípidos se encuentran entre otros en:
riñones (células mesangiales, endoteliales y epiteliales de los túbulos, podocitos)
corazón (células miocardiales, endoteliales y fibrocitos)
sistema nervioso periférico (neuronas de los ganglios de la raíz dorsal y del sistema nervioso autónomo) vasos sanguíneos (células endoteliales, periteliales y células musculares lisas).

6
La acumulación progresiva de glicoesfingolípidos afecta a numerosos órganos y tejidos, por lo que los pacientes se presentan a menudo con toda una serie de características y síntomas de diferente severidad. (Peters et al., 2001).
3 Historia natural de la enfermedad Los síntomas de M. Fabry varían según la edad (ver tabla 1) y, no raras veces, en primer plano se encuentran manifestaciones orgánicas aisladas. En general, el número de sistemas orgánicos implicados y la gravedad de los síntomas aumentan con la edad de los pacientes ((Mehta et al., 2004). La variabilidad de las manifestaciones clínicas es mayor en las mujeres que en los hombres (Deegan et al., 2006; Wang et al., 2007; Whybra et al., 2001; Wilcox et al., 2008).
Tabla 1. Características y síntomas típicos de la enfermedad de Fabry en función de la edad (adaptada de Hughes et al., 2005; Mehta et al., 2010).
Edad típica Características y síntomas Infancia y adolescencia (< 16 años) Dolores neuropáticos (p. ej. acroparestesias)
Dolores crónicos en las articulaciones Anomalías oftalmológicas (córnea verticillata, tortuositas vasorum) Pérdida de audición, tinnitus Mareos Dishidrosis (hipohidrosis, menos frecuente hiperhidrosis) Hipersensibilidad al calor y el frío Trastornos gastrointestinales, dolores abdominales Letargo y fatiga Angioqueratomas Primeras anomalías renales y cardiacas (p. ej. microalbuminuria, proteinuria, variabilidad anómala de la frecuencia cardiaca)
Edad adulta temprana (17- 30 años) Además de los síntomas anteriores: Otros angioqueratomas
Linfedemas en las extremidades inferiores Riñones: proteinuria e insuficiencia renal avanzada Corazón: miocardiopatía hipertrófica e
HVI, angina de pecho, arritmias SNC: TIA, infarto cerebral Depresión
Edad adulta avanzada (> 30 años) Empeoramiento de los síntomas anteriores y además: Insuficiencia cardiaca, trastornos del ritmo cardiaco Recidiva de TIA e infarto cerebral
Insuficiencia renal, necesidad de diálisis HVI = hipertrofia ventricular izquierda; SNC = sistema nervioso central; TIA = ataque isquémico transitorio

7
3.1 Dolores Los dolores neuropáticos son uno de los síntomas más frecuentes de la enfermedad de M. Fabry (grado de evidencia I b). Comentario: los datos sobre la prevalencia de los dolores oscilan en los hombres entre un 33% (Üceyler et al., 2011) y un 80% (Hoffmann et al., 2005; MacDermot et al., 2001). También en las mujeres los datos sobre la prevalencia de los dolores comprenden desde un 25% (Üceyler et al., 2011) hasta un 70% (Hoffmann et al., 2005; MacDermot et al., 2001 [b]). Las molestias se presentan de forma típica como acroparestesias con escozor, hormigueo o entumecimiento de las extremidades. Pero también se conocen casos de accesos de dolor punzante o lancinante en forma de las denominadas crisis de dolor (Hughes et al., 2005). En principio puede afectar a cualquier región del cuerpo (p. ej. columna cervical y cabeza; Hoffmann et al., 2007 [a]). Son desencadenantes frecuentes de las crisis de dolor la actividad corporal, cambios de temperatura, estrés psíquico y/o afecciones intercurrentes (Burlina et al., 2011). Los dolores neuropáticos pueden causar padecimientos considerables ya en edades tempranas (grado de evidencia II a). Comentario: en concordancia con los datos de prevalencia en adultos, la frecuencia de ataques de dolor en la infancia se sitúa en más del 60% para ambos sexos. Sin embargo, solo 1/3 de los(as) niños(as) han declarado padecer dolores crónicos (Ramaswami et al., 2006). Un análisis retrospectivo ha mostrado que los dolores agudos se presentan entre los pacientes varones a partir de los 14 años de edad, y en las mujeres a partir de los 19 años (Hoffmann et al., 2007 [a]). Datos del registro de Fabry evidencían un inicio de los síntomas a edades aún más tempranas (9 años en los niños y 10 en las niñas) (Eng et al., 2007). La causa de los dolores es un deterioro de los nervios periféricos debido a los depósitos de Gb3 (grado de evidencia IIb). Comentario: análisis histopatológicos han podido demostrar una implicación de los ganglios en las raíces dorsales de la médula espinal y una degeneración axonal de pequeñas fibras nerviosas desmielinizadas, responsables de la transmisión del
dolor (Kahn, 1973; Dütsch et al., 2002). Posiblemente los neurilemas protegen a las fibras nerviosas contra los depósitos de Gb3 dañinos (Dütsch et al., 2002). Existe una relación entre la gravedad en el deterioro de los nervios por un lado y el funcionamiento renal por otro (Schiffmann et al., 2006; Üceyler et al., 2011). 3.2 Piel 3.2.1. Angioqueratomas Los angioqueratomas constituyen un indicio de M. Fabry, pero no son patognomónicos. Se trata de pequeños ensanchamientos de los vasos próximos al tronco en general agrupados. Su expansión está en correlación con la gravedad general de la enfermedad (grado de evidencia IV). Comentario: los angioqueratomas son característicos de la enfermedad de M. Fabry. El nombre designa pequeños engrosamientos rojizo marrones de los vasos, en parte hiperqueratósicos, de manifestación agrupada, principalmente en las zonas glúteas, periumbical, escrotal y en los muslos. Otros puntos de manifestación para las eflorescencias son las manos, los pies y las mucosas (p. ej. la mucosa bucal), si bien pueden presentarse en cualquier región cutánea. Los primeros angioqueratomas se ponen de manifiesto en la infancia y aumentan en número a lo largo de la vida. Las mujeres muestran en general menos angioqueratomas. En un análisis de las manifestaciones cutáneas en 714 pacientes se encontraron angioqueratomas en un 66% de los varones y en un 36% de las mujeres (Orteu et al,. 2007). Además de los angioqueratomas se ha observado también teleangiectasias no específicas. 3.2.2 Dishidrosis Muchos pacientes con M. Fabry sufren una disminución en la secreción de sudor, lo que en entornos cálidos y en casos de esfuerzo físico puede provocar fiebre con síntomas vegetativos como mareos y vómitos (grado de evidencia IV). Comentario: la hipohidrosis, o en algunos casos anhidrosis, se explica en parte por una disfunción del sistema nervioso vegetativo y por otra se discute la acumulación de Gb3 directamente en las glándulas sudoríparas como causa patogénica. En algunos casos no se desarrolla hipohidrosis ni anhidrosis, sino el efecto contrario, una

8
hiperhidrosis, también desagradable (Lidove et al., 2006). 3.2.3 Linfedemas Otro síntoma constatado en algunos pacientes son los linfedemas, que se presentan sobre todo en las extremidades inferiores. Este síntoma ha sido observado en casi un 25% de los varones y un 17% de las mujeres (Orteu et al., 2007). 3.3 Tracto gastrointestinal Aproximadamente la mitad de todos los pacientes con M. Fabry sufren en el curso de la enfermedad al menos un síntoma gastrointestinal (grado de evidencia II b). Comentario: numerosos informes de casos describen trastornos gastrointestinales en pacientes con M. Fabry (Anderson, 1898; Fabry, 1898; van Wayjen, 1958; Flynn et al., 1972; Rowe et al., 1974; Bryan et al., 1977; Sheth et al., 1981; O´Brien et al., 1982; Cable et al., 1982; Friedman et al., 1984; Nelis und Jacobs, 1989; Argoff et al., 1998; Hoffmann et al., 2004). MacDermot et al. reportaron primera vez la sintomatología de la mayor cohorte de hombres con M. Fabry. En esta cohorte se encontró una prevalencia total de trastornos gastrointestinales del 69% (MacDermot et al., 2001). La mayoría de los pacientes sufrían sensación de plenitud, trastornos en la digestión y contracciones intestinales. El 40% de los pacientes informó de un cuadro alternante entre diarrea y estreñimiento, siendo también posibles los vómitos. No se citan datos sobre la frecuencia de cada uno de los síntomas. La evaluación de los datos sobre los trastornos gastrointestinales de 342 pacientes del Fabry Outcome Survey (FOS) indicó una prevalencia del 52% (Hoffmann et al., 2007). Independientemente de la edad y el sexo, los dolores gastrointestinales fueron las molestias más frecuentes (32,5%), seguidas de diarrea (20,5%), estreñimiento (13,5%), náuseas (12,3%) y vómito (6,7%). Las combinaciones de tales molestias pueden, en determinadas circunstancias, imitar el denominado síndrome de intestino irritable. Ocasionalmente los pacientes han informado intolerancias a diversos alimentos (Hoffmann et al., 2004; Banikazemi et al., 2005). En algunas ocasiones, las molestias gastrointestinales son la única manifestación de M. Fabry, o la que afecta con más intensidad a los
pacientes (Flynn et al., 1972; Rowe et al., 1972; Hoffmann et al., 2004). Todos los datos disponibles hasta ahora en relación a los trastornos gastrointestinales en pacientes con M. Fabry, más allá de casos aislados o de series de casos, tienen un valor informativo limitado por haber sido tomados con carácter retrospectivo. Los datos se basan, en parte, en las indicaciones de los pacientes afectados, sin confirmación por análisis de laboratorio, rayos X o endoscopia. Los informes de casos aislados y series de casos pueden completar esta información en el sentido de que pueden describir también manifestaciones más raras o aquellas eventualmente relacionadas con M. Fabry en el curso de la enfermedad. Las mujeres pueden sufrir trastornos gastrointestinales en parte con mayor frecuencia que los hombres (grado de evidencia IIb) Comentario: aunque en la bibliografía no existe ningún informe de casos que describa los trastornos gastrointestinales como síntoma principal en una mujer, MacDermot et al. encontraron una prevalencia total del 60% para los síntomas gastrointestinales en mujeres con M. Fabry (MacDermot et al., 2001). Las molestias gastrointestinales con M. Fabry pueden comenzar en la infancia temprana y, en determinadas ocasiones, ser el primero y el único síntoma de la enfermedad (grado de evidencia II b). Comentario: varios autores exponen que los(as) niños(as) pueden padecer molestias gastrointestinales considerables en fases tempranas de la enfermedad de M. Fabry (Sheth et al., 1981; Ries et al., 2003; Ries et al., 2005). De hecho, análisis retrospectivos de cohortes mayores han mostrado una prevalencia de los síntomas gastrointestinales del 60,8% en la infancia (adultos: 48,9%). Se ha señalado el inicio de las molestias con aprox. 12 años (Hoffmann et al., 2007). A pesar de que la mayoría de los pacientes con M. Fabry padece molestias gastrointestinales, en parte con un cuadro de trastornos considerables, los pacientes con M. Fabry no presentan en general indicios de carencias nutritivas (grado de evidencia II b). Comentario: el BMI de los niños se situaba en la cohorte del Fabry Outcome Survey entre los

9
percentiles 50 y 75 (Hoffmann et al., 2007), y no había diferencias del BMI entre los niños con molestias gastrointestinales o sin ellas. El BMI medio de los pacientes adultos con trastornos gastrointestinales se situó incluso ligeramente sobre el de los adultos sin tales síntomas (24,7 vs. 23,9 kg/m²) (Hoffmann et al., 2007). 3.4 Riñones Hasta la fecha no se ha analizado suficientemente la importancia de los resultados de la biopsia renal en relación a las consecuencias terapéuticas. En principio se puede solicitar la biopsia renal para asegurar el diagnóstico; pero no suministra ningún dato clínico adicional relevante cuando el diagnóstico de M. Fabry ya ha sido confirmado mediante análisis de laboratorio químicos, moleculares y/o clínicamente en base a otras manifestaciones típicas. El diagnóstico de una nefropatía de Fabry se realiza en general mediante un microscopio óptico, de forma ideal con un colorante azul de toluidina. Un examen inmunohistológico o por microscopio electrónico muestra los depósitos de ceramida con su forma en general de bulbo. En los casos de deterioro avanzado del parénquima renal, con el empeoramiento funcional que eso conlleva, se produce fibrosis intersticial y esclerosis focal de los glomérulos con diferentes grados de intensidad. En base a las biopsias renales de 59 pacientes con Fabry de 11 centros se ha desarrollado un algoritmo estandarizado, tanto de modificaciones específicas de Fabry como también generales, que permite estimar el grado de gravedad de la nefropatía causada por Fabry. (Fogo et al., 2010). No obstante, no permite deducir una estimación pronóstica, la cual queda reservada a otras pruebas y análisis. Los pacientes con microalbuminuria o proteinuria de origen idiopático deberían ser informados de la existencia de un M. Fabry (grado de evidencia III). Comentario: la enfermedad crónica del riñón con una pérdida progresiva de la capacidad renal, combinada con frecuencia con una proteinuria, es un síntoma cardinal de M. Fabry (Ortiz et al., 2008). La nefropatía de Fabry es tanto un término morfológico como también clínico funcional. Puede ser caracterizada, desde un punto de vista de diagnóstico y evolución, mediante biopsia y análisis químicos de laboratorio sobre los parámetros de funcionamiento del riñón. El metabolismo deficiente de Gb3 provoca la acumulación en diferentes células del tejido renal. De ello puede
derivarse una esclerosis del glomérulo y fibrosis intersticial con proteinuria e insuficiencia renal. Del total de pacientes nefrológicos entre los pacientes de diálisis en los EE.UU se ha constatado M. Fabry en 42 de 250.352 pacientes, es decir en un 0,16 ‰, (Thadhani et al., 2002). En comparación con los estudios de tamizaje, realizados entre pacientes de diálisis, ese dato de prevalencia es posiblemente demasiado bajo (Linthorst et al., 2010). Los pacientes con funcionamiento renal limitado de origen idiopático (GFR < 60 ml/min/1,73 m2) deberían ser informados de la existencia de la enfermedad de M. Fabry (grado de evidencia III). Comentario: una recopilación de datos de 105 afectados muestra que en los hombres la nefropatía comienza en general con una proteinuria y, tras manifestarse una hipertonía y una insuficiencia renal crónica, se requiere iniciar la diálisis (Branton et al., 2002). Los primeros indicios clínicos de la nefropatía han sido observados a una edad media de 20 años y la insuficiencia renal terminal ocurre en promedio a los 38 años. (Eng et al., 2007; Ortiz et al., 2010). La variabilidad por edades es alta y los primeros indicios de nefropatía por Fabry han sido descritos en la infancia y la adolescencia (Tondel et al., 2008). Un estudio de Wanner y sus colaboradores ha observado una disminución anual de eGFR superior a 1 ml/min*1,73m2 en un 71% de los hombres y un 39% de las mujeres con M. Fabry (Wanner et al., 2010). En un análisis retrospectivo, la edad media de los hombres con enfermedad renal crónica (GFR< 60 ml/min*1,73m2) se situó en los 42 años, los pacientes varones con un GFR superior a 60 ml/min*1,73m2 tenían una edad media de 27 años. La pérdida anual de GFR en el primer grupo fue el doble que en el grupo con un funcionamiento renal normal inicial (-3,0 y -6,8 ml/min*año) (Schiffmann, 2009). Entre las mujeres la afección de los riñones comienza, por regla general, más tarde y evoluciona con menos intensidad (Ortiz et al., 2008). No obstante el riesgo de una nefropatía por Fabry es también considerable entre las mujeres (Schiffmann, 2009). El primer indicio de una afección renal clínica relevante con M. Fabry es la proteinuria, que ha sido constatada en el 10% de todos los(as) niños(as) con M. Fabry menores de 18 años (grado de evidencia II a).

10
Comentario: en algunos casos aislados puede producirse a la edad de 2 años (Tondel et al., 2008). Entre las causas de la insuficiencia renal por enfermedad de M. Fabry se encuentra el deterioro del glomérulo (Tondel et al., 2008; Branton et al., 2002). Las biopsias renales en niños han mostrado depósitos de glicoesfinglolípidos en los podocitos, así como cambios glomerulares, tubulointersticiales y/o vasculares, que al parecer se producen en etapas tempranas y antes del deterioro funcional renal (Tondel et al., 2008). También entre los adultos se pueden observar cambios histológicos en un estadio temprano de enfermedad renal. La gravedad de la insuficiencia renal está en correlación con la dimensión de la
esclerosis arteriolar y glomerular (Fogo et al., 2010).
3.5 Corazón Poco más de la mitad de todos los pacientes con M. Fabry desarrolla una miocardiopatía típica a lo largo de su enfermedad (grado de evidencia II a). Comentario: en principio, la consecuencia metabólica de la deficiencia de α-galactosidasa puede afectar a todas las estructuras cardiacas, incluidos miocardio, sistema de conducción y válvulas del corazón (Frustaci et al., 2007; Linhart et al., 2007; Mehta A et al. 2004; Weidemann et al., 2009). En un estudio realizado por Linhart y sus colaboradores, más del 50% de todos los pacientes con M. Fabry mostraron una afección cardiaca con una edad media de 36 años (Linhart et al., 2007). Manifestaciones cardiacas típicas de la enfermedad de M. Fabry son:
hipertrofia ventricular izquierda (HVI, en su mayoría en forma concentrada) (Kampmann et al., 2008; Linhart et al., 2007)
fibrosis intramiocárdica (Weidemann et al., 2005; Moon et al., 2003; Weidemann et al., 2009)
prominencia del músculo papilar (Niemann et al., 2010)
cambios en el ECG, con onda P e intervalo PR cortos, aumento de la amplitud QRS y alteraciones de la reporalización (Namdar et al., 2011)
alteración del ritmo cardiaco, cuya frecuencia aumenta con la edad (aleteo auricular paroxístico o sostenido, taquicardias ventriculares sostenidas o no sostenidas) (Shah et al., 2005)
disfunción valvular (válvula mitral, válvula aórtica) (Weidemann et al., 2009)
También en la infancia y la juventud pueden manifestarse síntomas cardiacos (grado de evidencia II a). Comentario: en un estudio realizado en 20 pacientes con enfermedad de Fabry pediátricos, Kampmann y sus colaboradores encontraron en todos los afectados un elevado índice de masa ventrivular izquierda (masa ventricular izquierda superior percentil 75) y reducción variable de la frecuencia cardiaca (Kampmann et al., 2008). En un estudio sobre la manifestación cardiaca, realizado en 8 niños y 12 niñas, todos mostraron una masa ventricular izquierda superior al percentil 75 en las ecocardiografías. 35% de los(as) niños(as) con M. Fabry presentaron una hipertrofia ventricular izquierda clásica (Kampmann et al., 2008; Hopkin et al., 2008; Linhart et al., 2007). El aumento del tamaño del corazón no se debía a una disfunción sistólica o diastólica. En el ECG de larga duración se observó en jóvenes una limitada variabilidad de la frecuencia cardiaca como posible primer indicio de un trastorno cardiaco funcional. La hipertrofia ventricular izquierda es el diagnóstico guía en el marco de la miocardiopatía debida a Fabry (grado de evidencia II a). Comentario: en más del 50% de los hombres y en cerca del 33% de las mujeres con M. Fabry se observa una hipertrofia ventricular izquierda (HVI) (Linhart et al., 2007, Weidemann et al., 2005). Los hombres desarrollan una HVI antes que las mujeres (Niemann et al., 2011). En la hipertrofia ventricular izquierda existe una correlación positiva entre la arritmia y la afección de las válvulas mitral y aórtica. La hipertrofia ventricular izquierda evoluciona de forma progresiva si no es tratada. En el marco de la hipertrofia se ha observado muy raras veces una obstrucción de la vía de salida del ventrículo izquierdo. En la mayoría de los pacientes se trata de una hipertrofia ventricular izquierda concéntrica (Linhart et al., 2007, Weidemann et al., 2005). Solo en casos de miocardiopatías avanzadas se puede producir una hipertrofia asimétrica acentuada del septo debido a la dilatación de la pared posterolateral (Weidemann et al., 2005; Moon et al., 2003; Weidemann et al., 2009; Weidemann et al., 2011, Niemann et al., 2011).

11
Típico de la miocardiopatía avanzada es una fibrosis intramiocardiaca (grado de evidencia II a). Comentario: la fibrosis miocardiaca se presenta en el estadio avanzado de la miocardiopatía, típicamente en los segmentos posterolaterales basales (Weidemann et al., 2005; Moon et al., 2003; Weidemann et al., 2009; Weidemann et al., 2011, Niemann et al., 2011). Esta puede ser constatada mediante la técnica "late enhancement imaging" durante una tomografía por resonancia magnética nuclear. En el estadio final se muestra también una dilatación de esa sección de la pared con alteraciones regionales de movimiento de la pared que pueden ser confundidos fácilmente con un infarto de miocardio antiguo. Esta fibrosis denominada de reemplazo tiene un efecto negativo sobre el pronóstico (Weidemann et al., 2009). Las complicaciones en el marco de las manifestaciones cardiacas se cuentan entre las causas principales de muerte en la enfermedad de M. Fabry (grado de evidencia II a). Comentario: Patel y sus colaboradores analizaron en 2.869 pacientes con Fabry el desarrollo natural de la enfermedad desde el punto de vista de los accidentes cardiovasculares (infarto de miocardio, fallo cardiaco o muerte debida a causas cardiacas). Un total de 5,8% de los hombres y 3,7% de las mujeres habían sufrido un accidente cardiovascular a una media de edad de 45 y 54 años. El accidente documentado con mayor frecuencia es el fallo cardiaco (3,5% de los hombres y 2,3% de las mujeres). Se ha constatado que la hipertrofia ventricular es el predictor más seguro de accidentes cardiovasculares (valores umbral: diástole final septo >10 en mujeres, diástole final septo >11 mm en hombres) (Patel et al., 2011). Los trastornos del ritmo cardiaco parecen ser los responsables de la muerte cardiaca súbita en muchos pacientes Fabry (Shah et al., 2005). 3.6 Sistema nervioso central Los ataques isquémicos transitorios (TIA) o el infarto cerebral son un suceso frecuente en pacientes con M. Fabry (grado de evidencia II a). Comentario: en una evaluación de los datos de 366 pacientes Fabry, registrados en Fabry Outcome Survey (FOS), Mehta y sus colaboradores (2004) informaron de una mayor prevalencia de infarto cerebral en mujeres que en hombres (27% frente a
12%). En una revisión de 2001 se subraya la importancia del infarto cerebral para la morbilidad y la mortalidad de los pacientes con M. Fabry (Feldt-Rasmussen, 2011). En un análisis de Buechner y sus colaboradores, casi el 25% de los pacientes sufrieron un evento cerebrovascular en el curso de su enfermedad de Fabry. La edad media en el momento de producirse el TIA o el infarto cerebral es de 34 años para los hombres y de 54 años para las mujeres (Buechner et al., 2008; Sims et al., 2009). Otros estudios confirman que, por término medio los hombres lo sufren antes que las mujeres y se ha informado también de TIA en la infancia (Mehta et al., 2004; Ramaswami et al., 2006; Pintos-Morell und Beck, 2009). También los pacientes sin manifestaciones previas de TIA o infarto cerebral pueden presentar la denominada "small vessel disease" (enfermedad de vasos sanguíneos pequeños) en los glanglios basales, el tálamo y el tronco del encéfalo, así como en la región periventricular, como pudieron mostrar Reisin y sus colaboradores en 16 de 36 pacientes adultos sintomáticos con M. Fabry. (Reisin et al., 2011). La recidiva de infarto cerebral o TIA puede ser en casos aislados la primera manifestación de la enfermedad (grado de evidencia II a). Comentario: el trastorno cerebrovascular puede presentarse conjuntamente con manifestaciones renales y cardiacas (Mehta et al., 2005; Schiffmann et al., 2009). Un estudio de Sims y sus colaboradores mostró, no obstante, que antes del primer infarto cerebral no se había diagnosticado ninguna afección renal o cardiaca en casi el 71% de los pacientes Fabry varones y el 77% de las mujeres. 50% de los pacientes varones y 38% de las mujeres sufrieron su primer infarto cerebral antes de diagnosticarse M. Fabry (Sims et al., 2009). En total, la recidiva de infartos cerebrales y TIA es frecuente y se asocia a un peor pronóstico de la enfermedad (Mitsias et al., 1996; Grewal., 1994; Burlina et al., 2011). En pacientes jóvenes que sufran un infarto cerebral de origen idiopático se debería descartar siempre que la existencia de un cuadro de M. Fabry, sobre todo si existen otros síntomas adicionales como ectasia de la arteria basilar (grado de evidencia II a). Comentario: la prevalencia de M. Fabry es mayor en determinadas poblaciones, así como en pacientes jóvenes con infarto cerebral (18 a 55 años de edad) y debe ser considerada como causa de infarto

12
cerebral en al menos 1% de esos pacientes (Brouns et al., 2010). Fellgiebel y sus colaboradores (2011) han presentado la ectasia de la arteria basilar como un instrumento potencial de tamizaje para M. Fabry. Por lo tanto, en los pacientes jóvenes de ambos sexos con infarto cerebral (18 a 55 años de edad) se debería excluir la posibilidad de padecer M. Fabry mediante el análisis de la arteria basilar (v. abajo). Rolfs y sus colaboradores analizaron el gen GLA en 721 adultos con infarto cerebral criptogénico (18 a 55 años de edad). En total, 4,9% de los hombres y 2,4% de las mujeres con infarto cerebral presentaron una mutación en el gen GLA. Estos habían desarrollado sobre todo infartos cerebrales isquémicos en la región cerebrobasilar (Rolfs et al., 2005). En los reconocimientos por TRM se observaron los siguientes cambios en el SNC de pacientes con M. Fabry:
lesiones de la sustancia blanca cerebral (Crutchfield et al., 1998; Fellgiebel et al., 2006; Ginsberg et al., 2005; Viana-Baptista, 2011) – estas fueron constatadas en casos aislados también en niños/as (grado de evidencia II a).
alteración en el riego sanguíneo del cerebro (Moore et al., 2001) (grado de evidencia II a).
modificaciones en los vasos vertebrobasilares (Ginsberg et al., 2005; Fellgiebel et al., 2011) (grado de evidencia II a).
vasos sanguíneos dilatados y tortuosos (Fellgiebel et al., 2006; Ginsberg et al., 2005). (grado de evidencia II b). una indicación típica, pero solo constatada en cerca del 30% de todos los casos, de la afectación del SNC por M. Fabry son los cambios de las señales del TRM-T1 en el núcleo pulvinar thalami (Mehta et al., 2010). (grado de evidencia II a).
3.7 Órganos sensoriales 3.7.1 Ojos La mayoría de los pacientes con M. Fabry presentan modificaciones oculares que no pueden ser diagnosticadas como no invasivas (grado de evidencia II b). Comentario: los cambios oftalmológicos con M. Fabry afectan a la córnea, el cristalino y los vasos de la conjuntiva y la retina. En cerca del 60-70% de los(as) niños(as) con M. Fabry se presentan hallazgos oftalmológicos relevantes (Ramaswami et al., 2006; Pintos-Morell und Beck, 2009). Por regla general esos cambios no conllevan una limitación de la capacidad visual (Nguyen et al., 2005).
La característica ocular específica más frecuente es la córnea verticillata (CV, enturbiamiento superficial de la córnea localizado por debajo del centro de la córnea de forma espiral), observada en el 40-90% de los pacientes (Nguyen et al., 2005; Sodi et al., 2007, Samiy, 2008). Esas modificaciones de la córnea pueden ser identificadas en general mediante exploración con lámpara de hendidura en los reconocimientos oftalmológicos de rutina. Mediante estudio histopatológico se ha podido probar cambios en el forma de la córnea verticillata en un feto de 22 semanas de gestación (Tsutsumi et al., 1984); por otra parte, las modificaciones oculares pueden manifestarse después de las primeras décadas (Samiy, 2008).
Además de las terapias con amiodarona, en menor número de casos con cloroquina y otros fármacos anfífilicos, la enfermedad de M. Fabry es la causa más frecuente de este enturbiamiento de la córnea. Mientras la CV inducida por fármacos es reversible al suspender la terapia respectiva, en los pacientes con M. Fabry persiste por regla general incluso bajo la TSE (Falke et al., 2008; Wasielica-Poslednik et al., 2011). Está menos caracterizado otro enturbiamiento de la córnea conocido como "haze", un enturbiamiento difuso del estroma corneal. Esa modificación parece ser claramente menos frecuente que la CV, siendo omitida también por su discreta acusación. Otras posibles manifestaciones son vasos tortuosos (Tortuositas vasorum) de la retina y la conjuntiva. Si bien esas alteraciones de los vasos no son patognomónicas; en la región de la conjuntiva se observan además aneurismas en los vasos. La existencia de múltiples vasos tortuosos ha sido descrita en un estudio como posible indicador de una evolución desfavorable de la afección cardiaca y renal (Sodi et al., 2007). Al cuadro de la enfermedad se asocian dos opacidades diferentes del cristalino: la denominada catarata de Fabry subcapsular anterior o posterior (Sher et al., 1979). Esta última se considera específica de la enfermedad, pero también ha sido observada en otras afecciones por depósito lisosomal como la manosidosis. Las dos formas de catarata pueden ser evaluadas adecuadamente solo mediante dilatación de pupilar con medicamentos (por eso los datos de prevalencia de la bibliografía seguramente son demasiado bajos). Mientras la CV es solo ligeramente más frecuente en hombres que en mujeres, los otros trastornos oculares se presentan al menos el doble de veces en hombres que en mujeres (Sodi et al., 2007; Samiy, 2008).

13
En la literatura más antigua se encuentra una serie de informes de casos sobre complicaciones vasculares y de la retina (ver resumen en Samiy, 2008); es interesante constatar la ausencia de tales informes en las publicaciones actuales, aunque se podrían esperar más informes sobre tales complicaciones por la existencia actual de mayores bases de datos. Aproximadamente el 30% de los pacientes con M. Fabry sufren una neuropatía óptica subclínica con discretas pérdidas del campo visual, subjetivas e imperceptibles. El diagnótico de la modificación ocular más frecuente, la córnea verticillata, depende menos de la exploración oftalmológica con lámpara de hendidura que la tortuositas vasorum o la "catarata por Fabry". 3.7.2 Oídos En pacientes con M. Fabry se produce con frecuencia una pérdida auditiva neurosensorial, sobre todos de las frecuencias elevadas, y
tinnitus (grado de evidencia I b).
Comentario: análisis de cohortes pequeñas (Germain et al., 2002) y grandes han arrojado como resultado que la pérdida de audición es, por regla general, un proceso progresivo (Hegemann et al., 2006; Ries et al., 2007). En la mayoría de los casos se trata de una pérdida de audición principalmente coclear (Ries et al, 2007). En el 5 al 30% de los pacientes se informa además una pérdida de audición aguda con un desarrollo de pocas horas hasta días (Hegemann et al., 2006; Ries et al., 2007). El tinnitus puede presentarse desde la infancia y parece estar en correlación con la gravedad de la enfermedad (Hegemann et al., 2006; Keilmann et al., 2009). Independientemente de la pérdida de audición y el tinnitus, muchos pacientes con M. Fabry presentan también un daño del órgano vestibular (Palla et al., 2007; Sergi et al., 2010). 3.8 Calidad de vida La calidad de vida de pacientes con M. Fabry que no reciben tratamiento enzimatico es significativamente menor que la calidad de vida de la población normal. Esto es así tanto para hombres (grado de evidencia II b) como para las mujeres (grado de evidencia I b).
Comentario: existen estudios sobre la calidad de vida en pacientes que desconocen su enfermedad (es decir sin TSE) (Miners et al., 2002) realizados con hombres de Gran Bretaña y EE.UU. (Gold et al., 2002). Un estudio de la fase III-B ha demostrado que también las mujeres con M. Fabry que no reciben tratamiento declaran tener una calidad de vida claramente menor que la población normal (Baehner et al., 2003). En un análisis retrospectivo, efectuado por el Fabry Outcome Survey con 120 hombres y mujeres con M. Fabry, la calidad de vida frente a personas de igual sexo y edad de un grupo de personas normales de Gran Bretaña era también significativamente menor (Hoffmann et al., 2005). Varios autores han demostrado que ese menoscabo puede ser importante desde la infancia (Ries et al., 2005; Ramaswami et al., 2006; Hopkin et al., 2008). 3.9 Otras manifestaciones de M. Fabry Magage y sus colaboradores (2007) encontraron alteraciones obstructivas de las vías respiratorias en el 61% de los hombres y el 26% de las mujeres con M. Fabry. En pacientes con M. Fabry se ha observado a menudo una osteopenia en la región lumbar de la columna vertebral y en el cuello femoral (Germain et al., 2005). Kleinert y sus colaboradores analizaron la prevalencia de una anemia en 345 pacientes con M. Fabry: 47% de los hombres y 20% de las mujeres presentaron un nivel de hemoglobina <13 g/dl (hombres) y <12 g/dl (mujeres). La existencia de una anemia estaba asociada, en la mayoría de los casos, a un funcionamiento renal disminuido, una insuficiencia renal y/o indicios de procesos inflamatorios (Kleinert et al., 2005). 4 Diagnóstico en caso de sospecha de M. Fabry El diagnóstico en caso de sospecha de la enfermedad de M. Fabry se realiza en base a un análisis enzimático y/o molecular. Los pacientes con un M. Fabry confirmado deberían ser trasladados a un centro que posea experiencia en el diagnóstico y la terapia de las enfermedades por depósito lisosomal.
Las pruebas básicas en hombres consisten en determinar la actividad enzimática de la α-galactosidasa A (AGLA) en los leucocitos; y en las mujeres el análisis de la mutación del gen GLA.

14
Las mutaciones consideradas patogénicas van asociadas en los hombres – pero no obligatoriamente en las mujeres – a una reducción patológica de la actividad enzimática.
En los hombres que presenten una reducción patológica de la actividad de AGLA se puede presuponer la existencia de un M. Fabry.
El 20-30% de las mujeres con mutaciones causantes de enfermedad presentan una actividad de AGLA en sangre normal. Por esa razón, una actividad enzimática normal en una mujer no excluye con seguridad la existencia de un M. Fabry. En mujeres es necesario realizar siempre el análisis molecular del gen GLA para el diagnóstico.
4.1 Determinación de la actividad de
AGLA (α-galactosidasa A) Costo aproximado 60 euros, disponibilidad de los resultados en un plazo de 1-2 semanas. 4.1.1 Determinación de la actividad de
AGLA en hombres En los hombres el método a elegir para confirmar el diagnóstico es la determinación de la actividad de la α-galactosidasa A (AGLA) en los leucocitos de la sangre (grado de evidencia IV). Comentario: material apto para las pruebas: muestras de sangre con Liquemin o EDTA. Transporte a temperatura ambiente como máximo 2 días. La determinación de la actividad enzimática también puede realizarse con sangre seca sobre papel filtro. En caso de baja actividad se recomienda realizar un segundo análisis con otro material (p. ej. muestra sangre EDTA). Una reducción patológica de la actividad de AGLA indica, por regla general, la existencia de un M. Fabry. La actividad enzimática se sitúa claramente por debajo del nivel normal (0 - 24%) en todos los hombres con un M. Fabry clínico aparente, pero también si es leve ("oligosintomático") o (aún) asintomático. Los hombres con una actividad enzimática de 11-24% del valor normal presentan con frecuencia un fenotipo leve. Según el estado actual de la ciencia, si el valor de AGLA en los hombres se sitúa en rangos normales se puede excluir un M. Fabry (grado de evidencia IV b).
4.1.2 Determinación de la actividad de AGLA en mujeres
La determinación de la actividad de AGLA en las mujeres carece de valor informativo (grado de evidencia IV b). Comentario: las portadoras de M. Fabry tienen un alelo de GLA patógeno (mutado) y uno intacto. Eso explica que una parte de las portadoras presente niveles normales de actividad enzimática. La actividad enzimática medida está influida por diferentes factores genéticos y no genéticos y el valor de AGLA se sitúa en niveles patógenos en solo un 10% de las portadoras (≤ 24% del valor normal). En las mujeres no se puede observar una relación entre la actividad enzimática y la sintomatología clínica. Aparte de los casos de portadoras confirmados genéticamente, para realizar el diagnóstico en mujeres es indispensable demostrar una mutación muy probablemente patógena del gen GLA. 4.2 Análisis genético molecular Las mutaciones en el gen GLA son en general mutaciones "privadas" específicas de la familia (grado de evidencia IV). Comentario: M. Fabry está causado por una mutación del gen GLA, codificado para la α-galactosidasa A (AGLA) y localizado en el brazo largo del cromosoma X, en la banda q22. La proteína precursora de AGLA comprende 429 aminoácidos, incluido el péptido señal de 31 bases en la región N-terminal. La parte codificadora de las proteína (1290 pares de bases) del gen está clasificada en 7 exones, separados a su vez por 6 intrones. Se conoce un gran número de diferentes mutaciones en el gen GLA (ver sinopsis en Gal, 2010). La mayoría de los pacientes/familias (~90%) son portadores de la misma mutación. Esta puede ser demostrada – según la transmisión hereditaria - en parientes del caso indexado. Un M. Fabry también puede estar condicionado por una nueva mutación. 4.2.1 Mutaciones patogénicas La importancia patogenética de algunas mutaciones en el gen GLA puede ser juzgada en base a tres categorías (Gal et al., 2011). La siguiente clasificación refleja los resultados de los análisis de más de 500 diferentes mutaciones en el gen GLA

15
en varios miles de pacientes con M. Fabry.
Categoría 1: mutaciones muy probablemente patógenas
(A) Del grupo de mutaciones puntuales
Mutaciones sin sentido: el intercambio de un nucleótico en uno de los 7 exónes de GLA forma un codón de paro prematuro. Aún cuando se pudiera sintetizar una proteína estable a partir de ese alelo de GLA, se produce una proteína truncada y no apta para el funcionamiento debido a la interrupción prematura de la síntesis de la proteína AGLA.
Mutaciones en las zonas de splicing: estas transforman una de las dos bases nucleotídicas, las primeras (GT) o las últimas (AG), en uno de los 6 intrones de GLA. Los dinucleotidos GT y AG de las denominadas secuencias consenso para splicing representan secuencias de señales que son imprescindibles para el splicing normal del ARNm del gen GLA.
Mutaciones de sentido equivocado: la actividad enzimática de AGLA puede verse afectada considerablemente cuando una mutación genética cambia un aminoácido, que desempeña un papel crítico en la formación y/o mantenimiento de la estructura biológica activa o participa directamente en el proceso catabólico. Las variantes de sentido equivocado en todos los genes son por principio problemáticas desde el punto de vista de su asignación como patogénicas, porque también pueden ser polimorfismos. En la actualidad se emplean los denominados análisis in silico, es decir, programas de predicción para llegar a una interpretación. Este grupo de mutaciones puntuales muy probablemente patógenas supone aproximadamente un 25% de todas las mutaciones de GLA.
(B) Recombinaciones Este grupo de mutaciones comprende cambios que varían el número de bases nucleotídicas o que afectan a dos o más bases nucleotídicas consecutivas. Entre ellas se encuentran las delecciones (pérdida de nucleótidos), duplicaciones e inserciones (exceso de nucleótidos), inversiones (secuencia de nucleótidos invertida) y las combinaciones de las anomalías anteriores. Aún cuando de los alelos del gne GLA aquí citados se sintetizase una proteína estable, el resultado es un AGLA no funcional. Este grupo supone un 29% de
todas las mutaciones del gen GLA.
Categoría 2: mutaciones posiblemente patogénicas
Todas las demás mutaciones de sentido equivocado diferentes de las citadas en la categoría 1.
Todas las demás mutaciones de las zonas de splicing diferentes de las citadas en la categoría 1.
Categoría 3: mutaciones muy probablemente no patogénicas
Las mutaciones del gen GLA en hombres con una actividad de AGLA normal o solo ligeramente reducida son consideradas como no relevantes para la enfermedad. Es necesario, sin embargo, señalar que los hombres con un M. Fabry confirmado enzimáticamente raras veces pueden portar dos cambios en la secuencia del gen GLA, uno patogénico y otro no patogénico. Hay que citar aquí en particular la variante no patogénica p.Asp313Tyr, que origina valores bajos en la determinación de la actividad de AGLA en el suero (pero no en los leucocitos) por una pseudodeficiencia, similares al M. Fabry clásico. 4.2.2 Análisis de mutaciones para confirmar
el diagnóstico en hombres En los hombres se realiza el análisis genético cuando la actividad enzimática de AGLA sea patológica (grado de evidencia IV). Comentario: en los hombres con una actividad patológica de AGLA se recomienda efectuar una comprobación de mutaciones patogénicas en el gen GLA como medio de confirmación de los resultados enzimáticos y para la tipificación posterior de familiares interesados. Con un diagnóstico enzimático confirmado, aún cuando no se detecten mutaciones en el gen GLA relevantes para la enfermedad en el diagnóstico genético molecular rutinario, debe mantenerse el diagnóstico de un M. Fabry. 4.2.3 Análisis de mutaciones para confirmar
el diagnóstico en mujeres En las mujeres es obligatorio realizar siempre un análisis molecular del gen GLA para diagnosticar la enfermedad (grado de evidencia III).

16
Comentario: aunque la enfermedad afecta a las mujeres de múltiples formas, no es raro encontrar una actividad normal de α-galactosidasa A. Por lo tanto, para establecer el diagnóstico es necesario realizar siempre un análisis molecular. Si se conocen mutaciones en la familia resulta más sencillo el diagnóstico mediante un análisis de mutaciones dirigido. Si la anamnesis familiar es negativa, hay que llevar a cabo un análisis molecular del gen GLA para establecer el diagnóstico. 4.3 Análisis familiar y asesoramiento
genético Los pacientes deben recibir una explicación del diagnóstico del ADN en el marco de un asesoramiento genético (grado de evidencia IV). Comentario: para el análisis molecular es necesaria la explicación correspondiente y el consentimiento informado por escrito del paciente. Conforme a la ley de diagnóstico genético, los pacientes, independientemente de su sexo, deben recibir asesoramiento genético en caso de diagnóstico enzimático positivo o de resultados moleculares positivos. Para las personas asintomáticas en riesgo es obligatorio ofrecer asesoramiento genético por parte de un médico cualificado para ello, al menos antes del análisis genético y tras el mismo para la notificación de los resultados; así como permitir un tiempo de reflexión adecuado entre el primer asesoramiento genético y la extracción de sangre. El objetivo es informar a los pacientes y a sus familiares sobre todas las cuestiones relacionadas (p. ej. herencia del cromosoma X, evaluación de los resultados del ADN, diagnóstico prenatal). Cuando se utilicen los resultados de familiares es muy importante cumplir las disposiciones respectivas sobre la protección de datos. Se recomienda siempre un análisis del árbol genealógico. Una anamnesis familiar detallada debe formar parte del diagnóstico cuando exista sospecha de M. Fabry (Gal et al., 2012). 4.4 Determinación de Gb3 y liso-Gb3 En principio, la determinación de Gb3 en la orina o en las biopsias de tejidos puede suministrar indicaciones sobre la existencia de M. Fabry. Sin embargo, hasta ahora no se dispone de evidencia suficiente para un diagnóstico seguro en base a Gb3. Un nuevo biomarcador, liso-Gb3, puede contribuir a mejorar el diagnóstico y, sobre todo, el seguimiento (Aerts et al., 2008).
4.5 Diagnóstico diferencial La gama de diagnósticos diferenciales posibles para M. Fabry es muy amplia y debe ser contemplada en relación con los síntomas clínicos principales (grado de evidencia II a). Comentario: son diagnósticos erróneos frecuentes por ejemplo dolores de crecimiento, síndrome de intestino irritable, M. Menière, M. Osler, afecciones de las formas reumatoides, esclerosis múltiple, infarto cerebral criptogénico o trastornos neuropsicológicos (Mehta A et al., 2004).
5. Diagnóstico con M. Fabry confirmado
Tras el diagnóstico de un M. Fabry debería llevarse a cabo una exploración inicial de los órganos y sistemas orgánicos típicamente afectados. Entre ellos se encuentran obligatoriamente piel, tracto gastrointestinal, ojos, oídos, cerebro, sistema nervioso (incluidos dolores y calidad de vida), corazón y riñones (ver figura 1). Comentario:
Las manifestaciones como nefropatía, afecciones cardiacas y cerebrovasculares pueden acortar la vida y deberían ser controladas en el marco de las consultas médicas de seguimiento cada 12 meses.
El primer abordaje y las pruebas periódicas de seguimiento son idénticos para hombres y mujeres.

17
Figura 1 Algoritmo para el diagnóstico y la asistencia de pacientes con M. Fabry (adaptado de Mehta A et al., 2010). 5.1 Riñone
5.1 Riñones 5.1.1 Abordaje incial Para diagnosticar oportunamente el M. Fabry y para detectar a tiempo la nefropatía de pronóstico desfavorable se debería efectuar un diagnóstico detallado de cada paciente. Comentario: los(as) niños(as) y adultos con M. Fabry deberían ser sometidos a una exploración inicial de las manifestaciones renales (reducida tasa de filtración del glomérulo, nefropatía de Fabry). Ésta debería comprender los análisis siguientes:
creatinina, excreción de creatinina, estimación de la tasa de filtrado glomerular (GFR por sus siglas en inglés) con la fórmula MDRD o la fórmula de Schwartz (estimación de la GFR en niños/as, Schwartz et al., 2009)
precipitación de proteínas en orina - tiras reactivas (microalbuminuria o
proteinuria) - cociente total de proteínas y
albúmina/creatinina en orina espontánea - proteínas en orina de 24 horas
ultrasonidos (lesiones vasculares)
medición de la presión sanguínea de 24h En caso de M. Fabry confirmado y de sospecha de una segunda enfermedad puede ser práctico o
necesario realizar una biopsia renal como diagnóstico diferencial. No se recomienda realizar la biopsia renal fuera de estudios controlados (voto de ética) para confirmar el diagnóstico ni para controlar la evolución. 5.1.2 Seguimiento Independientemente de la existencia de síntomas nefrológicos se debería efectuar análisis de seguimiento cada 12 meses. Comentario: análisis que deberían efectuarse en niños(as) y adultos:
creatinina, evacuación de creatinina, GFR (fórmula MDRD) o fórmula de Schwartz (estimación de GFR en niños/as)
precipitación de proteínas en la orina
- tiras reactivas (microalbuminuria o proteinuria)
- cociente total de proteínas/creatinina y albúmina/creatinina en orina espontánea
- dado el caso, proteínas en orina de 24 horas
medición de la presión sanguínea Los ultrasonidos solo son necesarios en caso de requerimiento (no ofrece resultados específicos de Fabry). Una biopsia renal (histología) solo debería ser considerada en caso de progresión de la
Familiares con M. Fabry
Síntomas indicadores de M. Fabry
p. ej. acroparestesias, angioqueratomas, hipersensibilidad al calor
y el frío, trastornos gastrointestinales, etc.)
Hombres: análisis bioquímico y genético actividad de α-galactosidasa A en leucocitos de la sangre
confirmación: análisis molecular del gen GLA
Mujeres: análisis genético análisis molecular del gen GLA
Dolores /
calidad de vida
p. ej. BPI,
WHO5,
MDI10,
SF-36
Sistema
neurológico sonografía
doppler y
dúplex
TRM cerebral
neurografía
Ojos / oídos
exploración
con lámpara
de hendidura
audiograma
Función renal
albuminuria
creatinina,
GFR
ultrasonidos
presión
sanguínea
Función
cardiaca
ECG
ecocardio-
grafía
ECG 24
horas
TRM
cardiaca, si
procede
Tracto
gastro-
intestinal
diarrea
estreñi-
miento
dolores en
el abdomen
Piel
angioquera-
tomas
dishidrosis
teleangiec-
tasias

18
enfermedad a pesar de la TSE (por razones de pronóstico). 5.2 Corazón 5.2.1 Abordaje inicial Todos los pacientes con la enfermedad de Fabry diagnosticada deberían ser sometidos a una exploración cardiaca completa. Comentario: en la primera consulta se debería llevar a cabo en todos los pacientes con M. Fabry una exploración de la afectación cardiaca por un lado, y por otro, la evaluación de una potencial miocardiopatía. A este respecto se debería solicitar las siguientes pruebas de diagnóstico:
ECG
ECG de 24 horas
ecocardiografía
tomografía por resonancia magnética nuclear del corazón (con Late Enhancement Imaging)
Estas recomendaciones son válidas para niños(as) y adultos. En el marco de los ECG se debe buscar en particular trastornos de repolarización, indicios de hipertrofia y acortamiento del intervalo PQ (ECG de descanso) y trastornos malignos del ritmo cardiaco (ECG 24 horas). En el marco de la ecocardiografía debería dejarse constancia de un conjunto de datos estándar y cuantificar en particular el grosor de la pared posterior y del septo en la diástole final, así como el funcionamiento sistólico y diastólico. Con ayuda de la resonancia magnética nuclear se debería buscar la existencia de una posible fibrosis. Una angiografía coronaria y una biopsia miocárdica solo deberían efectuarse en caso de indicaciones muy especiales. La biopsia del miocardio permite esclarecer, a nivel de diagnóstico, si existe afección cardiaca. Pero, según los conocimientos actuales, no está indicada si se ha comprobado la existencia de mutación o, en los hombres, una clara actividad enzimática reducida. 5.2.2 Seguimiento
Cuando existan diagnósticos o síntomas cardiológicos se debería llevar a cabo un seguimiento cada 12 meses. En caso contrario, si no existen síntomas ni diagnósticos cardiológicos, son suficientes pruebas de evolución cada 24 meses. Básicamente deben realizarse los mismos análisis y pruebas que en el abordaje inicial.
5.3 Sistema nervioso La evaluación de las manifestaciones neurológicas específicas de la enfermedad de M. Fabry en órganos se efectúa básicamente en el contexto de las manifestaciones generales y según el grado de concurrencia de otros órganos (principalmente el corazón y los riñones). 5.3.1 Abordaje inicial Para evaluar los síntomas neurológicos de la enfermedad de M. Fabry se debería efectuar un reconocimiento clínico centrado en los síntomas vasculares con inclusión del sistema nervioso autónomo. Comentario: los análisis siguientes deberían efectuarse en niños(as) y adultos:
sonografía doppler y dúplex: en la enfermedad de M. Fabry se encuentran vasos macroangiopáticos solo en algunos casos; por otra parte es típico el engrosamiento íntima-media (homogéneo, por regla general sin ulceraciones en íntima). Un indicio importante es la ectasia de la arteria basilar. TRM cerebral
evaluación de la intensidad del dolor y la calidad de vida: p. ej. BPI (cuestionario sobre dolores), WHO5 (cuestionario sobre depresión), MDI10 (permite una clasificación según ICD10) o SF-36 (desventaja: muy voluminoso)
5.3.2 Seguimiento Si existen síntomas neurológicos (sobre todo vasculares) se debería efectuar seguimiento cada 12 meses. En caso contrario, sin síntomas neurológicos, es conveniente realizar el seguimiento cada 24 meses. Comentario: análisis y pruebas recomendables para niños(as) y adultos:
sonografía doppler y dúplex
SF-36, BPI o cuestionario alternativo
TRM cerebral En caso de progresión clínica o de nueva aparición de síntomas a pesar de la TSE:
sonografía doppler y dúplex
TRM cerebral
SF-36, BPI o cuestionario alternativo

19
5.4 Tracto gastrointestinal El diagnóstico gastrointestinal sigue básicamente el mismo procedimiento que en los casos de trastornos abdominales y normalmente comprende un examen de ultrasonido transabdominal, gastroscopia (EGD con biopsia) y una colonoscopia (con biopsia). Dado el caso se puede recurrir también a una prueba de aliento de H2 y una cápsula endoscópica. Comentario: debe tenerse en cuenta que, a pesar de un cuadro de trastornos gastrointestinales, las pruebas mediante aparatos y los análisis químicos de laboratorio pueden no arrojar ningún resultado (MacDermot et al., 2001). 5.5 Oídos 5.5.1 Abordaje inicial En todos los pacientes con Fabry debería efectuarse una consulta otorrinolaringológica durante el abordaje inicial, sobre todo cuando ya existan pérdida de audición, tinnitus o trastornos en el equilibrio. Comentario: análisis y pruebas recomendables para niños(as) y adultos:
evaluación por parte de un médico otorrinolaringólogo con diagnóstico de audición y, dado el caso, tinnitus
examen del aparato vestibular
5.5.2 Seguimiento Si ya existen pérdida de audición, tinnitus o trastornos del equilibrio se deberían efectuar seguimiento cada 12 meses, en caso contrario es conveniente un seguimiento cada 24 meses. Comentario: análisis y pruebas recomendables para niños(as) y adultos:
pruebas audiométricas tonales
diagnóstico de tinnitus y equilibrio, si procede
6. Depresión y calidad de vida La calidad de vida relacionada con la salud debería ser analizada periódicamente en niños
y adultos como instrumento de seguimiento complementario. Comentario: con una enfermedad multisistémica, caracterizada por molestias crónicas (sobre todo dolores), una larga duración del diagnóstico y una menor esperanza de vida se puede presuponer un elevado riesgo de trastornos afectivos. En un estudio de Cole y sus colaboradores, casi la mitad de los pacientes con Fabry consultados sufrían depresión clínica manifiesta (Cole et al., 2007). Los estudios sobre la calidad de vida relacionada con la salud (bienestar corporal, psíquico y social, así como capacidad funcional) muestran asimismo limitaciones correspondientes. Una encuesta realizada en Gran Bretaña entre 38 hombres con M. Fabry (cuestionarios SF-36 y EQ-5D) dio como resultado una merma significativa de la calidad de vida en todas las dimensiones cuestionadas en comparación con la población normal (Miners et al., 2002). Un estudio similar entre hombres estadounidenses con M. Fabry confirmó esos resultados (Gold et al., 2002). Un estudio en la fase III-B realizado en Alemania para mujeres con M. Fabry mostró una reducida calidad de vida en todas las dimensiones del SF-36 en comparación con la población normal (Baehner et al., 2003). Otros estudios con mujeres heterocigotas han demostrado también restricciones esenciales en la calidad de vida (Street et al., 2006; Wang et al., 2007). Otro estudio de Hoffmann y sus colaboradores confirmó la significativa merma en la calidad de vida (cuestionario EQ-5D) entre 120 hombres y mujeres (Hoffmann et al., 2005). También entre los niños con M. Fabry se ha constatado ya una menor calidad de vida en algunas dimensiones del HRQOL en comparación con otros niños de la misma edad (Hopkin et al., 2008; Ries et al., 2005).

20
7. Terapia y asistencia La terapia de M. Fabry exige la estrecha cooperación de diferentes especialidades médicas. Entre estas se encuentran fundamentalmente médicos de familia, internistas, pediatras, nefrólogos, cardiólogos, neurólogos, gastroenterólogos, dermatólogos, oftalmólogos, otorrinolaringólogos o audiólogos pediátricos y expertos en genética humana. Será necesaria una asistencia psiquiátrica o psicológica en función del estado individual de la enfermedad. Tras confirmarse el diagnóstico, el paciente debería ser enviado a un centro con experiencia en el diagnóstico y las terapias de esta enfermedad, para someterse a un detallado abordaje inicial. Comentario: la terapia persigue los siguientes objetivos:
reducir las molestias (sobre todo mitigación del dolor)
impedir la progresión de manifestaciones orgánicas (sobre todo en riñones, corazón y SNC)
mejorar la calidad de vida y la manifestación orgánica
normalizar la esperanza de vida El tratamiento de M. Fabry disponible en la actualidad comprende la TSE y terapias paralelas de las afecciones orgánicas y síntomas. Los pacientes con M. Fabry necesitan en general una asistencia especialmente intensa, costosa y multimodal. 7.1 Terapia de sustitución enzimática
La TSE, disponible desde 2001, es una opción de tratamiento causal para compensar la deficiecia o la pérdida funcional de α-galactosidasa A (AGLA). La enzima AGLA fabricada por procedimiento tecnológico genético para la TSE es administrada cada 14 días por vía intravenosa. La terapia se mantiene durante toda la vida. Hay dos preparados de sustitución enzimática aprobados:
agalsidasa alfa (Replagal®) se fabrica en una línea celular humana y se administra en una dosis de 0,2 mg/kg de peso corporal.
agalsidasa beta (Fabrazyme®) se fabrica recombinado en células CHO (CHO = Chinese Hamster Ovary) y se administra con una dosis de 1,0 mg/kg de peso corporal.
El objetivo de TSE es mantener el funcionamiento
orgánico normal y, en un caso ideal, provocar un retroceso de las manifestaciones orgánicas existentes. La TSE es, por el momento, la única posibilidad de ralentizar el avance de la enfermedad y prevenir sus consecuencias (Mehta et al., 2010). 7.1.1 Datos disponibles sobre la eficacia
La terapia de sustitución enzimática tiene efecto clínico, sobre todo en la mejoría de la calidad de vida, los dolores y el funcionamiento renal y cardiaco (grado de evidencia I b). Comentario: para la TSE con agalsidasa alfa o agalsidasa beta solo se dispone de un número limitado de estudios clínicos controlados y aleatorios (Eng et al., 2001, Schiffmann et al., 2001; Hajioff et al., 2003; Banikazemi et al., 2007; Hughes et al., 2008). Tras la aprobación de los dos preparados enzimáticos en el año 2001, en general se ha administrado la TSE a todos los pacientes diagnosticados. Los datos disponibles sobre el tratamiento a largo plazo con agalsidasa alfa o agalsidasa beta han sido generados, por regla general, a partir de estudios prolongados abiertos de los análisis de la fase III o bien a partir de los dos registros de pacientes "Fabry Outcome Survey (FOS)" y "Fabry Registry" (ver capítulo 8 "Registros internacionales de pacientes"). Se han publicados datos de larga duración con periodos de observación de hasta 5 años. Los estudios sobre las terapias con agalsidasa alfa y agalsidasa beta han constatado una reducción de Gb3 en la orina y el plasma, así como una reducción de los depósitos de Gb3 en el endotelio microvascular de riñones, corazón y piel (Eng et al., 2001; Schiffmann et al., 2001; Baehner et al., 2003; Wilcox et al., 2004; Eto et al., 2005; Ries et al., 2006; Germain et al., 2007; Hughes et al., 2008; Wraith et al., 2008; Whybra et al., 2009). El descenso del nivel de Gb3 en el plasma pudo constatarse ya a los tres meses de terapia (van Bremen et al., 2011).
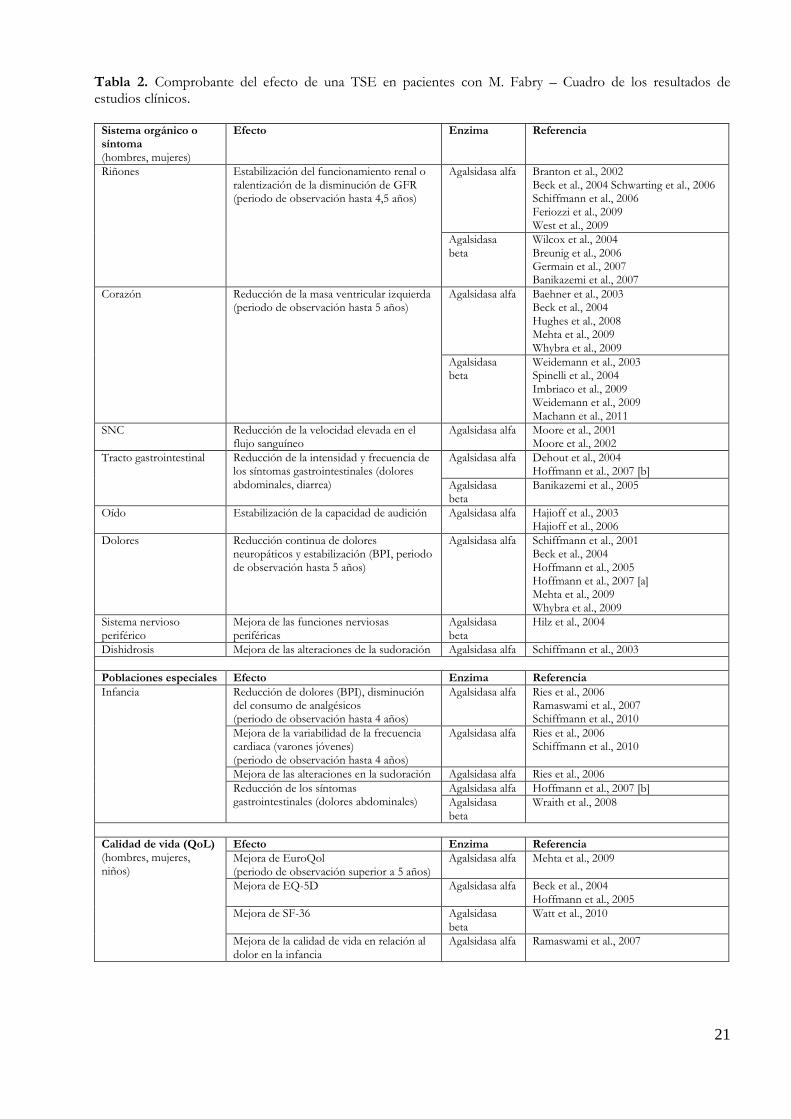
21
Tabla 2. Comprobante del efecto de una TSE en pacientes con M. Fabry – Cuadro de los resultados de estudios clínicos.
Sistema orgánico o síntoma (hombres, mujeres)
Efecto Enzima Referencia
Riñones Estabilización del funcionamiento renal o ralentización de la disminución de GFR (periodo de observación hasta 4,5 años)
Agalsidasa alfa Branton et al., 2002 Beck et al., 2004 Schwarting et al., 2006 Schiffmann et al., 2006 Feriozzi et al., 2009 West et al., 2009
Agalsidasa beta
Wilcox et al., 2004 Breunig et al., 2006 Germain et al., 2007 Banikazemi et al., 2007
Corazón Reducción de la masa ventricular izquierda (periodo de observación hasta 5 años)
Agalsidasa alfa Baehner et al., 2003 Beck et al., 2004 Hughes et al., 2008 Mehta et al., 2009 Whybra et al., 2009
Agalsidasa beta
Weidemann et al., 2003 Spinelli et al., 2004 Imbriaco et al., 2009 Weidemann et al., 2009 Machann et al., 2011
SNC Reducción de la velocidad elevada en el flujo sanguíneo
Agalsidasa alfa Moore et al., 2001 Moore et al., 2002
Tracto gastrointestinal Reducción de la intensidad y frecuencia de los síntomas gastrointestinales (dolores abdominales, diarrea)
Agalsidasa alfa Dehout et al., 2004 Hoffmann et al., 2007 [b]
Agalsidasa beta
Banikazemi et al., 2005
Oído Estabilización de la capacidad de audición Agalsidasa alfa Hajioff et al., 2003 Hajioff et al., 2006
Dolores Reducción continua de dolores neuropáticos y estabilización (BPI, periodo de observación hasta 5 años)
Agalsidasa alfa Schiffmann et al., 2001 Beck et al., 2004 Hoffmann et al., 2005 Hoffmann et al., 2007 [a] Mehta et al., 2009 Whybra et al., 2009
Sistema nervioso periférico
Mejora de las funciones nerviosas periféricas
Agalsidasa beta
Hilz et al., 2004
Dishidrosis Mejora de las alteraciones de la sudoración Agalsidasa alfa Schiffmann et al., 2003
Poblaciones especiales Efecto Enzima Referencia Infancia Reducción de dolores (BPI), disminución
del consumo de analgésicos (periodo de observación hasta 4 años)
Agalsidasa alfa Ries et al., 2006 Ramaswami et al., 2007 Schiffmann et al., 2010
Mejora de la variabilidad de la frecuencia cardiaca (varones jóvenes) (periodo de observación hasta 4 años)
Agalsidasa alfa Ries et al., 2006 Schiffmann et al., 2010
Mejora de las alteraciones en la sudoración Agalsidasa alfa Ries et al., 2006 Reducción de los síntomas gastrointestinales (dolores abdominales)
Agalsidasa alfa Hoffmann et al., 2007 [b] Agalsidasa beta
Wraith et al., 2008
Calidad de vida (QoL) (hombres, mujeres, niños)
Efecto Enzima Referencia Mejora de EuroQol (periodo de observación superior a 5 años)
Agalsidasa alfa Mehta et al., 2009
Mejora de EQ-5D Agalsidasa alfa Beck et al., 2004 Hoffmann et al., 2005
Mejora de SF-36 Agalsidasa beta
Watt et al., 2010
Mejora de la calidad de vida en relación al dolor en la infancia
Agalsidasa alfa Ramaswami et al., 2007

22
En un estudio realizado por Banikazemi y sus colaboradores entre pacientes con una disminución de ligera a moderada del funcionamiento renal, una TSE retrasó el tiempo hasta el primer evento clínico (renal, cardiaco, cerebrovascular o muerte) en comparación con el grupo placebo. El periodo medio de observación comprendió en ese estudio 18,5 meses (Banikazemi et al., 2007). Otros estudios sobre la TSE han confirmado la eficacia clínica, principalmente en relación al dolor, la calidad de vida y las manifestaciones orgánicas renales y cardiacas limitadoras de la vida (ver tabla 2). La eficacia de la TSE ha sido constatada también en niños(as) (ver tabla 2). Las dos sustancias disponibles presentan variaciones en el patrón de glicosilación (Lee et al., 2003). De los datos disponibles hasta la fecha no se deduce ningún indicio de diferencias clínicas relevantes en cuanto a la eficacia. Un estudio independiente de Canadá analiza en la actualidad una comparación directa de agalsidasa alfa 0,2 mg/kg y agalsidasa beta 1,0 mg/kg – se esperan más resultados de ese estudio a corto plazo (Sirrs et al., 2010). La TSE puede mitigar los dolores neuropáticos (grado de evidencia I b). Comentario: en un estudio de la fase I/II, la infusión de 5 dosis de agalsidasa beta (0,3-3,0 mg/kg cada dos semanas) ha producido una mejoría en la sensación de dolor y en la intensidad actual del dolor frente al estado antes de iniciarse el estudio (Eng et al., 2001). También en el estudio doble ciego controlado y aleatorio sobre la agalsidasa alfa los pacientes informaron de una significativa mitigación de los dolores con la TSE, lo que no ocurrió en el grupo de placebo (Schiffmann et al., 2001). La observación de esos pacientes a lo largo de 3 años ha confirmado estos resultados (Schiffmann et al., 2003). El estudio doble ciego controlado y aleatorizado para agalsidasa beta también ha mostrado una significativa mejoría en la sensación de dolor frente al estado previo al inicio del estudio ya al cabo de 20 semanas, sin embargo ocurrió lo mismo en el grupo de placebo (Eng et al., 2001). Tampoco la evaluación retrospectiva a largo plazo de grupos de pacientes mayores permite valoraciones unificadas sobre la efectividad de la TSE en relación al dolor. Un análisis retrospectivo de 752 pacientes con M. Fabry (393 mujeres y 353 hombres) de la base de datos de FOS ha mostrado
una mejora de los dolores al cabo de 24 y 36 meses (Hoffmann et al., 2007 [b]). Otra evaluación de la base de datos de FOS, aplicada solo a mujeres, no pudo confirmar estos resultados (Hughes et al., 2011). Los autores concluyeron que la mejoría en los dolores posiblemente se consiga sobre todo en un estadio temprano de la terapia y a largo plazo sea menos acusada. Esto se contrapone a Mehta et al., que informaron de una significativa mejoría de los dolores en 181 adultos (de ellos 126 hombres) tras cinco años de TSE (Mehta et al., 2010). Las experiencias de la TSE en niños(as) con M. Fabry son también heterogéneas en relación a la mejoría de los dolores. Los resultados de un estudio prospectivo abierto sobre la seguridad y la eficacia de la TSE en niños indican una mejoría significativa constante en la intensidad del dolor en comparación con el momento anterior al inicio del estudio (Schiffman et al., 2010). Ramaswami et al., por el contrario, no pudieron constatar ninguna reducción significativa de la prevalencia del dolor tras periodos de hasta dos años en un análisis retrospectivo de los datos de FOS de 98 niños (Ramaswami et al., 2011). No obstante, los pacientes que al comienzo del estudio padecían ya ataques de dolor o dolores crónicos, informaron de una mejoría de esas molestias al cabo de dos años. Los distintos enfoques de los estudios y la selección de instrumentos, en parte muy diferentes, para registrar los dolores tienen que ser tomados en cuenta al evaluar estos resultados. En general parece que la TSE puede producir la regeneración de las fibras nerviosas en regiones proximales de la piel en pacientes con M. Fabry (Üceyler et al., 2011). El hecho de que no todos los pacientes acusen una mejoría de los dolores puede estar relacionado con un deterioro ya irreversible de las fibras nerviosas, indicando la necesidad de comenzar la TSE para M. Fabry lo más tempranamente posible (Hilz et al., 2004). Cuando no sea posible dominar el dolor con los analgésicos habituales, ni el paciente responda a una TSE, puede ser conveniente administrar gabapentina (Ries et al., 2003) y carbamacepina (Filling-Katz et al., 1989). 7.1.2 Indicación e inicio de la terapia La única posibilidad de prevenir la progresión y consecuencias de M. Fabry es la TSE (grado de evidencia I b).

23
Comentario: para evitar los depósitos primarios de Gb3 y las manifestaciones clínicas asociadas a ellos, una vez confirmado el diagnóstico de un M. Fabry, y tras documentar la gravedad clínica o la progresión de los síntomas, se debería iniciar la TSE lo antes posible. Recomendación para pacientes varones y mujeres:
la indicación de la TSE es independiente de la edad, siempre que existan indicios de manifestaciones orgánicas relevantes (corazón, riñones, cerebro, dolores) El tratamiento es necesario durante toda la vida.
7.1.3 Tolerancia La tolerancia a la TSE es buena en general, sin diferencias entre hombres y mujeres o niños y adultos (grado de evidencia II b). Comentario: el suceso no deseado más frecuente en la TSE puede ser la reacción adversa al medicamento. Esta puede comprender sobre todo dolores de cabeza, parestesias, enrojecimiento, sofocos, fiebre, escalofríos, sensación de frío, náuseas, vómitos y fatiga (información especializada de Fabrazyme® 2010, información especializada de Replagal® 2010). La mayoría de las reacciones adversas fueron ligeras o moderadas. Por regla general se presentan durante los tres primeros meses tras el inicio de la terapia y vuelven a disminuir en frecuencia a lo largo de la misma. La experiencia ha demostrado que, tras un primer tratamiento de las molestias (reducción de la tasa de infusión, administración de antiflogísticos, antihistamínicos y/o glucocorticoides no esteroides) se puede continuar la infusión tras algunas semanas/meses sin las medidas concomitantes. Formación de anticuerpos Cuando disminuya la eficacia de la TSE se debería realizar una determinación de los anticuerpos. Si el resultado es positivo se puede considerar un cambio de preparado (grado de evidencia IV). Comentario: una posible causa de las reacciones adversas al medicamento es la conversión del suero con formación de anticuerpos (Ac) contra las proteínas humanas. En estudios clínicos, la mayoría de los pacientes tratados con agalsidasa beta (>80%) desarrollaron anticuerpos IgG (información especializada de Fabrazyme® 2010),
en los tres primeros meses de tratamiento y con agalsidasa alfa fueron cerca del 24% en el grupo de pacientes varones, no constatándose la formación de anticuerpos entre las mujeres (información especializada de Replagal® 2010). Hasta ahora no está claro el efecto de la formación de anticuerpos sobre la eficacia del tratamiento. Tras la infusión de agalsidasa beta se han constatado anticuerpos IgE en un número limitado de pacientes (información especializada de Fabrazyme® 2010). 7.1.4 Particularidades de la terapia en
mujeres No existe ninguna indicación para interrumpir la TSE durante el embarazo (grado de evidencia III). Comentario: la indicación de la TSE para mujeres heterocigotas debería establecerse en base a los mismos criterios que para los hombres hemicigotos (Baehner et al., 2003, Mehta et al., 2004). La TSE en mujeres está indicada en particular cuando se presenten los síntomas o manifestaciones orgánicas siguientes: proteinuria, insuficiencia renal, miocardiopatía o/y acroparestesias que no son transitorias y que no responden, o no en la medida suficiente, a los analgésicos (Weidemann et al., 2011). La TSE debe continuar también en el embarazo, pues en varios estudios de casos no se ha observado hasta ahora ningún efecto negativo en la madre ni en el bebé debidos al tratamiento enzimático (Wendt et al., 2005; Germain et al., 2010; Politei et al., 2010), pero, por otro lado, sí se ha constatado una progresión de la enfermedad sin la TSE. 7.1.5 Particularidades de la terapia en la
infancia Varios estudios han demostrado la eficacia de la TSE también en la infancia (grado de evidencia II a). Comentario: bajo el tratamiento se observó un retroceso de las acroparestesias (Ries et al., 2006; Ramaswami et al., 2007; Schiffmann et al., 2010) y de los trastornos abdominales (Hoffmann et al., 2007 [b]; Wraith et al., 2008). Igualmente se ha observado una normalización de la variabilidad en la frecuencia cardiaca (Ries et al., 2006; Schiffmann et al., 2010) y una mejoría en las alteraciones de la sudoración (Ries et al., 2006). En todos los estudios se ha comprobado un retroceso de las concentraciones de Gb3 en el suero y la orina.

24
7.1.6 Calidad de vida Para una gran parte de los pacientes hombres y mujeres con M. Fabry bajo la TSE se puede conseguir una mejora de la calidad de vida, independientemente del preparado empleado (grado de evidencia II a). Comentario: de los estudios dobles ciegos aleatorizados sobre la TSE para M. Fabry, tanto con agalsidasa alfa como también con agalsidasa beta, no se ha podido deducir con seguridad una primera mejoría significativa de la calidad de vida. Schiffmann et al. han registrado la calidad de vida asociada a dolores antes y durante la TSE en hombres con M. Fabry, pero estos parámetros no tratan de una dimensión de un instrumento de calidad de vida, sino de una dimensión del cuestionario de dolor Brief-Pain-Inventor (Schiffmann et al., 2001). Con todo, los hombres tratados con agalsidasa alfa mostraron a las 24 semanas una mejoría significativa de la calidad de vida relacionada al dolor en comparación con el grupo placebo (evidencia I b). En el estudio doble ciego controlado y aleatorio de Eng et al. se ha podido observar una mejoría en dos dimensiones del cuestionario SF-36 (funciones corporales y emocionales) bajo la terapia con agalsidasa alfa; pero también los hombres con M. Fabry tratados con placebo declararon una mejoría de las funciones y dolores corporales tras 6 meses (Eng et al., 2001). Entretanto, numerosos trabajos han mostrado claramente una mejoría en la calidad de vida de pacientes con M. Fabry bajo la terapia, tanto con agalsidasa alfa como con agalsidasa beta y tanto en hombres como en mujeres (Baehner et al., 2003; Hoffmann et al., 2005; Eto et al., 2005; Watt et al., 2010). Una evaluación de cuatro años del Fabry-Outcome-Surveys aportó únicamente una tendencia hacia la mejora de la calidad de vida en hombres y mujeres bajo la TSE (Hughes et al., 2011). Hay que tener en cuenta aquí que el tamaño de la población seguramente era demasiado pequeño para mostrar con seguridad un efecto. En un análisis de cinco años de la misma base de datos se pudo demostrar una mejoría significativa de la calidad de vida (Mehta et al., 2009). También entre niños(as) se ha observado una mejoría significativa de la calidad de vida después de iniciada la TSE –al cabo de un periodo de intervención de 12 a 23 semanas – (Ramaswami et al., 2007).
La recopilación periódica de datos sobre la calidad de vida en pacientes con M. Fabry bajo la TSE sería deseable y conveniente (grado de evidencia I b). Comentario: el uso de un cuestionario estandarizado no sustituye la evaluación médica del bienestar del paciente ni una supervisión meticulosa de la terapia. No obstante, el uso del un cuestionario estandarizado, validado y adaptado a la edad para recoger datos de la calidad de vida puede contribuir a objetivizar cambios ocurridos bajo la terapia. 7.1.7 Terapia de infusión en casa Cuando el paciente haya recibido unos 6 tratamientos de TSE en la clínica o en la consulta médica y no se hayan presentado reacciones adversas al medicamento, se puede ofrecer la posibilidad de trasladar el tratamiento a la casa del paciente (grado de evidencia II b). Comentario: el traslado del tratamiento al entorno habitual facilita la relación con la enfermedad y mejora la observancia en la mayoría de los casos. Ya existen experiencias con la terapia de infusión en casa en diferentes países (Milligan et al., 2006; Linthorst et al., 2006; Schiffmann et al., 2006; Cousins et al., 2008; Guest et al.; 2010). Las siguientes condiciones son obligatorias para establecer el tratamiento en el hogar:
formación de personas (p. ej. enfermeras o cuidores), que se hagan cargo del tratamiento en casa (conocimientos sobre M. Fabry y la TSE, rutina práctica en la colocación de un acceso venoso) tienen que conocerse sobre todo las medidas a adoptar en caso de efectos secundarios antes de comenzar la terapia en casa es obligatorio aclarar las cuestiones legales (p. ej. responsabilidad del médico asistente)
7.2 Terapias concomitantes Además de la TSE pueden ser necesarios otros tratamientos según los síntomas y manifestaciones orgánicas (ver tabla 3).

25
Tabla 3. Opciones terapéuticas complementarias para los síntomas o efectos de M. Fabry (adaptado de Hughes et al., 2005; Eng et al., 2006; Mehta et al., 2010; Weidemann et al., 2011).
Dolores Evitar mecanismos activadores (esfuerzo corporal, calor, fuertes oscilaciones de temperatura, estrés, sobrefatiga); anticonvulsivos (p. ej. fenitoina, carbamacepina, gabapentina, topiramat), antidepresivos tricíclicos
Trastorno del riñón, proteinuria Inhibidores ECA, sartanos, control de anemia Fallo renal Dialisis o trasplante Hipertonía Antidepresivos como inhibidores ECA (sin bloqueadores beta
en casos de bradicardia sinusal) Taquicardia Antiarrítmicos, implante de desfibrilador (ICD) Bradicardia Implante de marcapasos Insuficiencia cardiaca Diuréticos, inhibidores ECA (eventl. sartanos en caso de
intolerancia a los inhibidores ECA), marcapasos o terapia ICD, trasplante de corazón
Estenosis de la arteria coronaria PTCA, CABG Afección cerebrovascular Profilaxis de infarto cerebral (p. ej. aspirina u otros inhibidores
de la agregación de trombocitos) Dislipidemia Estaninas Obstrucción de las vías respiratorias Prescindir de nicotina, intento de terapia con dilatores
bronquiales Alteraciones en la sudoración Eventual inyección de la toxina botulínica para inhibir la
secreción de sudor en casos de hiperdrosis Retraso en el vaciamiento gástrico, dispepsia Comidas menos copiosas y frecuentes, metoclopramida,
bloqueador H2 Pérdida severa de audición Audífonos, implante coclear Depresión Asistencia psiquiátrica o psicológica, inhibidor de la
readsorción de serotonina 7.2.1 Terapia nefrológica concomitante Si existe proteinuria y/o hipertonía debe aplicarse una terapia adicional, como es habitual también para otras afecciones renales crónicas con objeto de retrasar la progresión (grado de evidencia III). Comentario: la consecuente utilización de inhibidores ECA o bloqueadores AT-2 (sartanos) en combinación con la TSE ha sido analizada hasta ahora para la enfermedad de M. Fabry en series de casos no controladas (Tahir et al., 2007). En relación con ello puede ser problemática una presión sanguínea relativamente baja que afecte a la observancia en la toma de medicamentos. Ahora bien, el objetivo primario de la terapia concomitante citada para M. Fabry no es bajar la presión sanguínea hasta alcanzar un valor predeterminado. Se aplica en primera línea para estabilizar el funcionamiento renal y para reducir la proteinuria al mínimo.
Una insuficiencia renal terminal debe ser tratada igual que en los pacientes sin M. Fabry (grado de evidencia III). Comentario: en los pacientes con insuficiencia renal terminal está indicada la aplicación de hemodiálisis o diálisis peritoneal. No existen contraindicaciones para un trasplante de riñón. Tras el trasplante de riñón no se ha descrito recidiva en el trasplante debido al funcionamiento normal de la α-galactosidasa en el órgano trasplantado. Al persistir la situación patológica del metabolismo se debería continuar la TSE sin modificación para evitar daños extrarrenales. Ya se han publicado datos sobre la tolerancia y la eficacia de la TSE en pacientes con trasplante de riñón (Ojo et al., 2000; Pastores et al., 2007; Mignani et al., 2008; Cybulla et al., 2009). 7.2.2 Terapia cardiológica concomitante Todos los pacientes con M. Fabry y miocardiopatía deberían recibir una terapia cardiológica adicional (grado de evidencia III)

26
Comentario: la terapia concomitante es especialmente necesaria en los casos de miocardiopatía avanzada. En relación con esto desempeñan un papel central los inhibidores ECA, que pueden tener efectos positivos en la regresión de la hipertrofia. Hay que tener en cuenta, no obstante, que los pacientes con M. Fabry tienden a la hipotensión. Por eso, tras el inicio de una terapia de inhibición ECA se requiere un seguimiento exhaustivo de la presión sanguínea (Weidemann et al., 2010). Por otra parte, todos los pacientes con alteraciones del ritmo (taquicardias) deberían recibir un ß-bloqueador. Este es eficaz al mismo tiempo contra los trastornos del ritmo ventricular. Pero como muchos pacientes con Fabry también sufren de bradicardias (Shah et al., 2005), se debería excluir previamente ésta mediante un ECG de 24 horas, sobre todo en los casos con síntomas típicos como mareos o síncopes. En el marco de la terapia antiarrítmica debe observarse que en pacientes con M. Fabry se ha descrito una interacción entre amiodarona y la enzima a sustituir (Whitley et al., 1983). Por lo tanto se debería iniciar una terapia de amiodarona solo tras una valoración exhaustiva. Los pacientes con miocardiopatía avanzada sufren con frecuencia bradicardias sintomáticas. Por eso, para esos pacientes está indicada una terapia con marcapasos. Antes del implante es importante excluir los trastornos de ritmo ventricular malignos, típicos en las etapas finales de la cardiomiopatía. En ese caso se debería implantar un desfibrilador (ICD) "de acuerdo con las directivas generales para el implante de ICD en caso de alteración maligna del ritmo (ACC/AHA/ESC 2006 Guidelines for management of patients with ventricular arrhythmias, Weidemann et al., 2010). Los pacientes con una implicación cardiaca padecen con frecuencia molestias anginosas, no atribuibles en general a una afección cardiaca coronaria primaria, sino a una enfermdad de vasos pequeños. Aquí se puede considerar la administración de preparados de nitrógeno o antagonistas de calcio en dosis bajas tras haber excluido una afección cardiaca coronaria. 7.2.3 Terapia neurológica concomitante Aproximadamente un 25% de todos los pacientes con Fabry sufren TIA o infartos cerebrales recurrentes (grado de evidencia II a). Comentario: los pacientes con M. Fabry presentan un elevado riesgo de sufrir un TIA o un infarto cerebral (Feldt-Rasmussen, 2011). En estos
pacientes los TIA e infartos cerebrales se presentan a menudo antes que en otros pacientes y el riesgo de recidiva es mayor (Mehta et al., 2010; Viana-Baptista, 2011). Por lo tanto son necesarias terapias profilácticas contra el infarto cerebral (inhibidores de la agregación plaquetaria con ASS 75-100 mg o clopidogrel 75 mg, así como el tratamiento de la hipertonía arterial) y medidas preventivas secundarias (abstinencia de nicotina, actividad corporal, tratamiento con estaninas, ajuste del metabolismo en los casos de Diabetes mellitus) de forma análoga a la guía AWMF "Infarto cerebral" (número de registro 053-011) .
8. Organización de autoayuda M. Fabry Selbsthilfegruppe in Deutschland: MFSH – M. Fabry Selbsthilfegruppe e.V. Guilleaumestr. 13 51065 Köln Correo electrónico: [email protected] Internet: http://www.fabry-selbsthilfegruppe.de

27
Bibliografía
1. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias J Am Coll Cardiol. 2006 Sep 5;48(5):e247-346.
2. Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008; 105(8): 2812-7.
3. Anderson W. A case of angiokeratoma. Br J Dermatol. 1898; 10: 113-7.
4. Argoff CE, Barton NW, Brady RO, Ziessman HA. Gastrointestinal symptoms and delayed gastric emptying in Fabry's disease: response to metoclopramide. NuclMedCommun. 1998;19(9): 887-91.
5. Baehner F, Kampmann C, Whybra C, Miebach E, Wiethoff CM, Beck M. Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J Inherit Metab Dis. 2003; 26: 617-627.
6. Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, Finkel R, Packman S, Bichet DG, Warnock DG, Desnick RJ. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007; 146: 77-86.
7. Banikazemi M, Ullman T, Desnick RJ. Gastrointestinal manifestations of Fabry disease: clinical response to enzyme replacement therapy. Mol Genet Metab. 2005; 85: 255-259.
8. Beck M, Ricci R, Widmer U, Dehout F, de Lorenzo AG, Kampmann C, Linhart A, Sunder-Plassmann G, Houge G, Ramaswami U, Gal A, Mehta A. Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest. 2004; 34: 838-844.
9. Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin Iii HA, Kopp JB. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002; 81: 122-138.
10. Breunig F, Weidemann F, Strotmann J, Knoll A, Wanner C. Clinical benefit of enzyme replacement therapy in Fabry disease. Kidney Int. 2006; 69(7): 1216-21.
11. Brouns R, Thijs V, Eyskens F, Van den Broeck M, Belachew S, Van Broeckhoven C, Redondo P, Hemelsoet D, Fumal A, Jeangette S, Verslegers W, Baker R, Hughes D, De Deyn PP; BeFaS Investigators. Belgian Fabry study: prevalence of Fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke. 2010; 41(5): 863-8.
12. Bryan A, Knauft RF, Burns WA. Small bowel perforation in Fabry's disease. AnnInternMed. 1977; 86(3): 315-6.
13. Buechner S, Moretti M, Burlina AP, Cei G, Manara R, Ricci R, Mignani R, Parini R, Di Vito R, Giordano GP, Simonelli P, Siciliano G, Borsini W. Central nervous system involvement in anderson-fabry diease: A clinical and MRI retrospective study. J Neurol Neurosurg Psychiatry. 2008; 79: 1249-1254.
14. Burlina AP, Sims KB, Politei JM, Bennett GJ, Baron R, Sommer C, Møller AT, Hilz MJ. Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. BMC Neurol. 2011; 11: 61.
15. Cable WJ, Kolodny EH, Adams RD. Fabry disease: impaired autonomic function. Neurology. 1982; 32(5): 498-502.
16. Cole AL, Lee PJ, Hughes DA, Deegan PB, Waldek S, Lachmann RH. Depression in adults with Fabry disease: a common and under-diagnosed probleM. J Inherit Metab Dis. 2007; 30: 943-951.
17. Cousins A, Lee P, Rorman D, Raas-Rothschild A, Banikazemi M, Waldek S, Thompson L. Home-based infusion therapy for patients with Fabry disease. Br J Nurs. 2008; 17: 653-657.
18. Crutchfield KE, Patronas NJ, Dambrosia JM, Frei KP, Banerjee TK, Barton NW, Schiffmann R. Quantitative analysis of cerebral vasculopathy in patients with Fabry disease. Neurology. 1998; 50: 1746-1749.
19. Cybulla M, Walter KN, Schwarting A, Divito R, Feriozzi S, Sunder-Plassmann G. Kidney transplantation in patients with Fabry disease. Transpl Int. 2009; 22: 475-481.
20. Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006; 43: 347-352.
21. Dehout F, Roland D, Treille de Granseigne S, Guillaume B, Van Maldergem L. Relief of gastrointestinal symptoms under enzyme replacement therapy [corrected] in patients with Fabry disease. J Inherit Metab Dis. 2004; 27: 499-505.
22. Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease. 2001; pp. 37–174.
23. Dütsch M, Marthol H, Stemper B, Brys M, Haendl T, Hilz MJ. Small fiber dysfunction predominates in Fabry neuropathy. J Clin Neurophysiol. 2002; 19(6): 575-86.
24. Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, Breunig F, Charrow J, Germain DP, Nicholls K, Banikazemi M. Fabry disease: baseline

28
medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007; 30(2): 184-92.
25. Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, Bultas J, Lee P, Sims K, Brodie SE, Pastores GM, Strotmann JM, Wilcox WR. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006; 8: 539-548.
26. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ. Safety and efficacy of recombinant human alpha-galactosidase A-replacement therapy in Fabry's disease. N Engl J Med. 2001; 345: 9-16.
27. Eto Y, Ohashi T, Utsunomiya Y, Fujiwara M, Mizuno A, Inui K, Sakai N, Kitagawa T, Suzuki Y, Mochizuki S, Kawakami M, Hosoya T, Owada M, Sakuraba H, Saito H. Enzyme replacement therapy in Japanese Fabry disease patients: the results of a phase 2 bridging study. J Inherit Metab Dis. 2005; 28: 575-583.
28. Fabry J. Ein Beitrag zur Kenntnis der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Arch Derm Syph. 1898; 43: 187-200.
29. Fachinformation Fabrazyme®, Stand der Information: 01/2010
30. Fachinformation Replagal®, Stand der Information: 12/2010
31. Falke K, Büttner A, Schittkowski M, Stachs O, Kraak R, Zhivov A, Rolfs A, Guthoff R. The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratopathy: a confocal laser-scanning microscopy study. Graefes Arch Clin Exp Ophthalmol. 2009; 247(4): 523-34.
32. Feldt-Rasmussen U. Fabry disease and early stroke. Stroke Res Treat. 2011; 2011: 615218.
33. Fellgiebel A, Keller I, Martus P, Ropele S, Yakushev I, Böttcher T, Fazekas F, Rolfs A. Basilar artery diameter is a potential screening tool for fabry disease in young stroke patients. Cerebrovasc Dis. 2011; 31: 294-299.
34. Fellgiebel A, Müller MJ, Ginsberg L. CNS manifestations of Fabry’s disease. Lancet Neurology. 2006; 5: 791-795.
35. Feriozzi S, Schwarting A, Sunder-Plassmann G, West M, Cybulla M. Agalsidase alfa slows the decline in renal function in patients with Fabry disease. Am J Nephrol. 2009; 29: 353-361.
36. Filling-Katz MR, Merrick HF, Fink JK, Miles RB, Sokol J, Barton NW. Carbamazepine in Fabry's disease: effective analgesia with dose-dependent exacerbation of autonomic dysfunction. Neurology. 1989; 39(4): 598-600.
37. Flynn DM, Lake BD, Boothby CB, Young EP. Gut lesions in Fabry's disease without a rash. ArchDisChild. 1972; 47(251): 26-33.
38. Fogo AB, Bostad L, Svarstad E, Cook WJ, Moll S, Barbey F, Geldenhuys L, West M, Ferluga D, Vujkovac B, Howie AJ, Burns A, Reeve R, Waldek S, Noël LH, Grünfeld JP, Valbuena C, Oliveira JP, Müller J, Breunig F, Zhang X, Warnock DG; all members of the International Study Group of Fabry Nephropathy (ISGFN). Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol Dial Transplant. 2010; 25(7): 2168-77.
39. Friedman LS, Kirkham SE, Thistlethwaite JR, Platika D, Kolodny EH, Schuffler MD. Jejunal diverticulosis with perforation as a complication of Fabry's disease. Gastroenterology. 1984; 86(3): 558-63.
40. Frustaci A, Chimenti C. Images in cardiovascular medicine. Cryptogenic ventricular arrhythmias and sudden death by Fabry disease: prominent infiltration of cardiac conduction tissue. Circulation. 2007; 116: 350-351.
41. Gal A. Molecular genetics of Fabry disease and genotype-phenotype correlation; in Elstein D, Altarescu G, Beck M (eds). Fabry Disease. 2010; pp 34–50.
42. Gal A, Beck M, Winchester B. Clinical utility gene card for: Fabry disease. Eur J Hum Genet. 2012; 20(2).
43. Gal A, Hughes DA, Winchester B. Toward a consensus in the laboratory diagnostics of Fabry disease - recommendations of a European expert group. J Inherit Metab Dis. 2011; 34(2): 509-14.
44. Gal A, Schäfer E, Rohard I. The genetic basis of Fabry disease. In: Mehta A, et al (eds). In: Fabry disease: perspectives from five years of FOS. 2006; pp. 323-330.
45. Germain DP, Avan P, Chassaing A, Bonfils P. Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty-two hemizygous male patients. BMC Med Genet. 2002; 3: 10.
46. Germain DP, Benistan K, Boutouyrie P, Mutschler C. Osteopenia and osteoporosis: previously unrecognized manifestations of Fabry disease. Clin Genet. 2005; 68: 93-95.
47. Germain DP, Bruneval P, Tran TC, Balouet P, Richalet B, Benistan K. Uneventful pregnancy outcome after enzyme replacement therapy with agalsidase beta in a heterozygous female with Fabry disease: A case report. Eur J Med Genet. 2010; 53: 111-112.
48. Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, Lee P, Loew T, Vedder AC, Abichandani R, Wilcox WR, Guffon N. Sustained, long-term renal stabilization after 54 months

29
of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007; 18: 1547-1557.
49. Ginsberg L, Valentine A, Mehta A. Fabry disease. Practical Neurology. 2005; 5: 110-113.
50. Gold KF, Pastores GM, Botteman MF, Yeh JM, Sweeney S, Aliski W, Pashos CL. Quality of life of patients with Fabry disease. Qual Life Res. 2002; 11: 317-327.
51. Grewal RP. Stroke in Fabry’s disease. J Neurol. 1994; 241: 153-156.
52. Guest JF, Jenssen T, Houge G, Aaseboe W, Tøndel C, Svarstad E. Modelling the resource implications of managing adults with Fabry disease in Norway favours home infusion. Eur J Clin Invest. 2010; 40: 1104-1112.
53. Hajioff D, Enever Y, Quiney R, Zuckerman J, Mackermot K, Mehta A. Hearing loss in Fabry disease: the effect of agalsidase alfa replacement therapy. J Inherit Metab Dis. 2003; 26: 787-794.
54. Hajioff D, Hegemann S, Hegemannn S, Conti G, Beck M, Sunder-Plassmann G, Widmer U, Mehta A, Keilmann A. Agalsidase alpha and hearing in Fabry disease: data from the Fabry Outcome Survey. Eur J Clin Invest. 2006; 36: 663-667.
55. Hegemann S, Hajioff D, Conti G, Beck M, Sunder-Plassmann G, Widmer U, Mehta A, Keilmann A. Hearing loss in Fabry disease: data from the Fabry Outcome Survey. Eur J Clin Invest. 2006; 36: 654-662.
56. Hilz MJ, Brys M, Marthol H, Stemper B, Dütsch M. Enzyme replacement therapy improves function of C-, Adelta-, and Abeta-nerve fibers in Fabry neuropathy. Neurology. 2004; 62: 1066-1072.
57. Hoffmann B, Beck M, Sunder-Plassmann G, Borsini W, Ricci R, Mehta A [a]. Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy—a retrospective analysis from the Fabry Outcome Survey. Clin J Pain. 2007; 23: 535-542.
58. Hoffmann B, Garcia de Lorenzo A, Mehta A, Beck M, Widmer U, Ricci R. Effects of enzyme replacement therapy on pain and health related quality of life in patients with Fabry disease: data from FOS (Fabry Outcome Survey). J Med Genet. 2005; 42: 247-252.
59. Hoffmann B, Reinhardt D, Koletzko B. Effect of enzyme-replacement therapy on gastrointestinal symptoms in Fabry disease. Eur J Gastroenterol Hepatol. 2004; 16(10): 1067-9.
60. Hoffmann B, Schwarz M, Mehta A, Keshav S [b]: Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007; 5: 1447–1453.
61. Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, Lemay R, Tylki-Szymanska A,
Wilcox WR. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008; 64: 550-555.
62. Hughes DA, Barba Romero MA, Hollak CE, Giugliani R, Deegan PB. Response of women with Fabry disease to enzyme replacement therapy: comparison with men, using data from FOS--the Fabry Outcome Survey. Mol Genet Metab. 2011; 103(3): 207-14.
63. Hughes DA, Elliott PM, Shah J, Zuckerman J, Coghlan G, Brookes J, Mehta AB. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008; 94: 153-158.
64. Hughes DA, Ramaswam U, Elliott P, Deegan P, Lee P, Waldek S, Apperley G, Cox T and Mehta AB for the National Specialist Commissioning Advisory Group (NSCAG). Guidelines for the diagnosis and management of Anderson-Fabry Disease. UK Department of Health 2005; http://www.dh.gov.uk/en/ Publicationsand statistics/Publications/ PublicationsPolicy And Guidance /DH_4118404.
65. Imbriaco M, Pisani A, Spinelli L, Cuocolo A, Messalli G, Capuano E, Marmo M, Liuzzi R, Visciano B, Cianciaruso B, Salvatore M. Effects of enzyme-replacement therapy in patients with Anderson-Fabry disease: a prospective long-term cardiac magnetic resonance imaging study. Heart. 2009; 95: 1103-1107.
66. Inagaki M, Ohno K, Ohta S, Sakuraba H, Takeshita K. Relief of chronic burning pain in Fabry disease with neurotropin. Pediatr Neurol. 1990; 6(3): 211-3.
67. Kahn P. Anderson-Fabry disease: a histopathological study of three cases with observations on the mechanism of production of pain. J Neurol Neurosurg Psychiatry. 1973; 36(6): 1053-62.
68. Kampmann C, Wiethoff CM, Whybra C, Baehner FA, Mengel E, Beck M. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 2008; 97: 463-469.
69. Keilmann A, Hajioff D, Ramaswami U. Ear symptoms in children with Fabry disease: data from the Fabry Outcome Survey. J Inherit Metab Dis. 2009; 32: 739-744.
70. Kleinert J, Dehout F, Schwarting A, de Lorenzo AG, Ricci R, Kampmann C, Beck M, Ramaswami U, Linhart A, Gal A, Houge G, Widmer U, Mehta A, Sunder-Plassmann G. Anemia is a new complication in Fabry disease: data from the Fabry Outcome Survey. Kidney Int. 2005; 67: 1955-1960.
71. Lee K, Jin X, Zhang K et al. Lee K, Jin X, Zhang K, Copertino L, Andrews L, Baker-Malcolm J, Geagan L, Qiu H, Seiger K, Barngrover D, McPherson JM, Edmunds T. A biochemical and pharmacological comparison of enzyme replacement therapies for the

30
glycolipid storage disorder Fabry disease. Glycobiology. 2003; 13: 305-313.
72. Lidove O, Ramaswami U, Jaussaud R, Barbey F, Maisonobe T, Caillaud C, Beck M, Sunder-Plassmann G, Linhart A, Mehta A. Hyperhidrosis: a new and often early symptom in Fabry disease. International experience and data from the Fabry Outcome Survey. Int J Clin Pract. 2006; 60: 1053-1059.
73. Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, Elliott PM. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007; 28: 1228-1235.
74. Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet. 2010; 47(4): 217-22.
75. Linthorst GE, Vedder AC, Ormel EE, Aerts JM, Hollak CE. Home treatment for Fabry disease: practice guidelines based on 3 years experience in The Netherlands. Nephrol Dial Transplant. 2006; 21: 355-360.
76. MacDermot KD, Holmes A, Miners AH [a]. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001; 38: 750-760.
77. MacDermot KD, Holmes A, Miners AH [b]. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001; 38: 769-775.
78. Machann W, Breunig F, Weidemann F, Sandstede J, Hahn D, Köstler H, Neubauer S, Wanner C, Beer M. Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A. Eur J Heart Fail. 2011; 13: 278-283.
79. Magage S, Lubanda JC, Susa Z, Bultas J, Karetová D, Dobrovolný R, Hrebícek M, Germain DP, Linhart A. Natural history of the respiratory involvement in Anderson-Fabry disease. J Inherit Metab Dis. 2007; 30: 790-709.
80. Mehta A, Beck M, Elliott P, Giugliani R, Linhart A, Sunder-Plassmann G, Schiffmann R, Barbey F, Ries M, Clarke JT. Enzyme replacement therapy with agalsidase alfa in patients with Fabry's disease: an analysis of registry data. Lancet. 2009; 374: 1986-1996.
81. Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U, Rolfs A, Rivera A, Waldek S, Germain DP. Fabry disease: a review of current management strategies. QJM. 2010; 103: 641-659.
82. Mehta A, Ginsberg L. Natural history of the cerebrovascular complications of Fabry disease. Acta Paediatr Suppl. 2005; 94: 24-27.
83. Mehta A, Ricci R, Widmer U, Dehout F, Garcia De Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004; 34: 236-242.
84. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999; 281: 249–254.
85. Mignani R, Feriozzi S, Pisani A, Cioni A, Comotti C, Cossu M, Foschi A, Giudicissi A, Gotti E, Lozupone VA, Marchini F, Martinelli F, Bianco F, Panichi V, Procaccini DA, Ragazzoni E, Serra A, Soliani F, Spinelli L, Torti G, Veroux M, Cianciaruso B, Cagnoli L. Agalsidase therapy in patients with Fabry disease on renal replacement therapy: a nationwide study in Italy. Nephrol Dial Transplant. 2008; 23: 1628-1635.
86. Milligan A, Hughes D, Goodwin S, Richfield L, Mehta A. Intravenous enzyme replacement therapy: better in home or hospital? Br J Nurs. 2006; 15: 330-333.
87. Miners AH, Holmes A, Sherr L, Jenkinson C, MacDermot KD. Assessment of health-related quality-of-life in males with Anderson Fabry Disease before therapeutic intervention. Qual Life Res. 2002; 11: 127-133.
88. Mitsias P, Levine SR. Cerebrovascular complications of Fabry’s disease. Ann Neurol. 1996; 40: 8-17.
89. Moon JC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennell DJ, Leed PJ, Elliott PM. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitiuM. Eur Heart J. 2003; 24(23): 2151-5.
90. Moore DF, Altarescu G, Ling GS, Jeffries N, Frei KP, Weibel T, Charria-Ortiz G, Ferri R, Arai AE, Brady RO, Schiffmann R. Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. Stroke. 2002; 33: 525-531.
91. Moore DF, Scott LTC, Gladwin MT, Altarescu G, Kaneski C, Suzuki K, Pease-Fye M, Ferri R, Brady RO, Herscovitch P, Schiffmann R. Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease. Circulation. 2001; 104: 1506-1512.
92. Namdar M, Steffel J, Vidovic M, Brunckhorst CB, Holzmeister J, Lüscher TF, Jenni R, Duru F. Electrocardiographic changes in early recognition of Fabry disease. Heart. 2011; 97(6): 485-90.
93. Nelis GF, Jacobs GJ. Anorexia, weight loss, and diarrhea as presenting symptoms of angiokeratoma corporis diffusum (Fabry-Anderson´s disease). Dig Dis Sci. 1989; 34(11): 1798-800.
94. Nguyen TT, Gin T, Nicholls K, Low M, Galanos J, Crawford A: Ophthalmological

31
manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin Experiment Ophthalmol. 2005; 33: 164–168.
95. Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, Machann W, Voelker W, Ertl G, Wanner C, Weidemann F. Differences in Fabry cardiomyopathy between female and male patients: consequences for diagnostic assessment. JACC Cardiovasc Imaging. 2011; 4(6): 592-601.
96. Niemann M, Liu D, Hu K, Herrmann S, Breunig F, Strotmann J, Störk S, Voelker W, Ertl G, Wanner C, Weidemann F. Prominent papillary muscles in Fabry disease: a diagnostic marker? Ultrasound Med Biol. 2011; 37(1): 37-43.
97. O'Brien BD, Shnitka TK, McDougall R, Walker K, Costopoulos L, Lentle B, Anholt L, Freeman H, Thomson AB. Pathophysiologic and ultrastructural basis for intestinal symptoms in Fabry's disease. Gastroenterology 1982; 82: 957-962.
98. Ojo A, Meier-Kriesche HU, Friedman G, Hanson J, Cibrik D, Leichtman A, Kaplan B. Excellent outcome of renal transplantation in patients with Fabry's disease. Transplantation. 2000; 69(11): 2337-9.
99. Orssaud C, Dufier JL, Germain DP. Ocular manifestations in Fabry disease: A survey of 32 hemizygous male patients. Ophthalmic Genet. 2003; 24: 129-139.
100. Orteu CH, Jansen T, Lidove O, Jaussaud R, Hughes DA, Pintos-Morell G, Ramaswami U, Parini R, Sunder-Plassman G, Beck M, Mehta AB. Fabry disease and the skin: data from FOS, the Fabry outcome survey. Br J Dermatol. 2007; 157: 331-337.
101. Ortiz A, Oliveira JP, Waldek S, Warnock DG, Cianciaruso B, Wanner C. Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant. 2008; 23: 1600-1607.
102. Palla A, Hegemann S, Widmer U, Straumann D. Vestibular and auditory deficits in Fabry disease and their response to enzyme replacement therapy. J Neurol. 2007; 254: 1433-1442.
103. Pastores GM, Boyd E, Crandall K, Whelan A, Piersall L, Barnett N. Safety and pharmacokinetics of agalsidase alfa in patients with Fabry disease and end-stage renal disease. Nephrol Dial Transplant. 2007; 22: 1920-1925.
104. Patel MR, Cecchi F, Cizmarik M, Kantola I, Linhart A, Nicholls K, Strotmann J, Tallaj J, Tran TC, West ML, Beitner-Johnson D, Abiose A. Cardiovascular events in patients with fabry disease natural history data from the fabry registry. J Am Coll Cardiol. 2011; 57: 1093-1099.
105. Peters FPJ, Vermeulen A, Kho TL. Anderson-Fabry’s disease: α-galactosidase deficiency. Lancet. 2001; 357: 138-140.
106. Pintos-Morell G, Beck M. Fabry disease in children and the effects of enzyme replacement treatment. Eur J Pediatr. 2009; 168: 1355-1363.
107. Pitz S Grube-Einwald K, Renieri G, Reinke J. Subclinical optic neuropathy in Fabry disease. Ophthalmic Genet. 2009; 30: 165-171.
108. Politei JM. Treatment with agalsidase beta during pregnancy in Fabry disease. J Obstet Gynaecol Res. 2010; 36: 428-429.
109. Ramaswami U, Parini R, Pintos-Morell G, Kalkum G, Kampmann C, Beck M. Fabry disease in children and response to enzyme replacement therapy: results from the Fabry Outcome Survey. Clin Genet. 2012; 81(5): 485-90.
110. Ramaswami U, Wendt S, Pintos-Morell G, Parini R, Whybra C, Leon Leal JA, Santus F, Beck M. Enzyme replacement therapy with agalsidase alfa in children with Fabry disease. Acta Paediatr. 2007; 96: 122-127.
111. Ramaswami U, Whybra C, Parini R, Pintos-Morell G, Mehta A, Sunder-Plassmann G, Widmer U, Beck M. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 2006; 95: 86-92.
112. Reisin RC, Romero C, Marchesoni C, Nápoli G, Kisinovsky I, Cáceres G, Sevlever G. Brain MRI findings in patients with Fabry disease. J Neurol Sci. 2011; 305(1-2): 41-4.
113. Ries M, Clarke JT, Whybra C, Timmons M, Robinson C, Schlaggar BL, Pastores G, Lien YH, Kampmann C, Brady RO, Beck M, Schiffmann R. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics. 2006; 118: 924-932.
114. Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ, Rosing DR, Robinson C, Schaefer E, Gal A, Dambrosia JM, Garman SC, Brady RO, Schiffmann R. Pediatric Fabry disease. Pediatrics. 2005; 115: e344-355.
115. Ries M, Kim HJ, Zalewski CK, Mastroianni MA, Moore DF, Brady RO, Dambrosia JM, Schiffmann R, Brewer CC. Neuropathic and cerebrovascular correlates of hearing loss in Fabry disease. Brain. 2007; 130: 143–50.
116. Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, Gal A, Beck M. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003; 162: 767-772.
117. Rolfs A, Böttcher T, Zschiesche M, Morris P, Winchester B, Bauer P, Walter U, Mix E, Löhr M, Harzer K, Strauss U, Pahnke J, Grossmann A, Benecke R. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005; 366: 1794-1796.

32
118. Rozenfeld P, Neumann PM. Treatment of fabry disease: current and emerging strategies. Curr Pharm Biotechnol. 2011; 12(6): 916-22.
119. Rowe JW, Gilliam JI, Warthin TA. Intestinal manifestations of Fabry's disease. AnnInternMed. 1974; 81(5): 628-31.
120. Samiy N. Ocular Features of Fabry disease: Diagnosis of a treatable life-threatening disorder. Surv Ophthalmol. 2008; 53: 416-423.
121. Schiffmann R, Floeter MK, Dambrosia JM, Gupta S, Moore DF, Sharabi Y, Khurana RK, Brady RO. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve. 2003; 28: 703-710.
122. Schiffmann R, Kopp JB, Austin HA, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001; 285: 2743-2749.
123. Schiffmann R, Martin RA, Reimschisel T, Johnson K, Castaneda V, Lien YH, Pastores GM, Kampmann C, Ries M, Clarke JT. Four-year prospective clinical trial of agalsidase alfa in children with Fabry disease. J Pediatr. 2010; 156: 832-837.
124. Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO. Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant. 2006; 21: 345-354.
125. Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, Sorensen SA, Wilcox WR, Desnick RJ. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009; 24(7): 2102-11.
126. Schwarting A, Dehout F, Feriozzi S, Beck M, Mehta A, Sunder-Plassmann G. Enzyme replacement therapy and renal function in 201 patients with Fabry disease. Clin Nephrol. 2006; 66: 77-84.
127. Sergi B, Conti G, Paludetti G, Interdisciplinary Study Group On Fabry Disease. Inner ear involvement in Anderson-Fabry disease: long-term follow-up during enzyme replacement therapy. Acta Otorhinolaryngol Ital. 2010; 30: 87-93.
128. Shah JS, Hughes DA, Sachdev B, Tome M, Ward D, Lee P, Mehta AB, Elliott PM. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol. 2005; 96: 842-846.
129. Sher NA, Letson Rd, Desnick RJ. The ocular manifestations in Fabry’s disease. Arch Ophthalmol. 1979; 97: 671-676.
130. Sheth KJ, Werlin SL, Freeman ME, Hodach AE. Gastrointestinal structure and function in Fabry's disease. Am J Gastroenterol. 1981; 76(3): 246-51.
131. Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009; 40: 788-794.
132. Sirrs S, Clarke JT, Bichet DG, Casey R, Lemoine K, Flowerdew G, Sinasac DS, West ML. Baseline characteristics of patients enrolled in the Canadian Fabry Disease Initiative. Mol Genet Metab. 2010; 99: 367-373.
133. Sodi A, Ioannidis AS, Mehta A, Davey C, Beck M, Pitz S: Ocular manifestations of Fabry's disease: data from the Fabry Outcome Survey. Br J Ophthalmol. 2007; 91: 210-214.
134. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006; 79: 31–40.
135. Spinelli L, Pisani A, Sabbatini M, Petretta M, Andreucci MV, Procaccini D, Lo Surdo N, Federico S, Cianciaruso B. Enzyme replacement therapy with agalsidase beta improves cardiac involvement in Fabry's disease. Clin Genet. 2004; 66: 158-165.
136. Street NJ, Yi MS, Bailey LA, Hopkin RJ. Comparison of health-related quality of life between heterozygous women with Fabry disease, a healthy control population, and patients with other chronic disease. Genet Med. 2006; 8: 346–353.
137. Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol 2007; 18: 2609-2617.
138. Thadhani R, Wolf M, West ML, Tonelli M, Ruthazer R, Pastores GM, Obrador GT. Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002; 61: 249-255.
139. Tondel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008; 51: 767-776.
140. Üçeyler N, He L, Schönfeld D, Kahn AK, Reiners K, Hilz MJ, Breunig F, Sommer C. Small fibers in Fabry disease: baseline and follow-up data under enzyme replacement therapy. J Peripher Nerv Syst. 2011; 16(4): 304-14.
141. van Bremen MJ, Rombach SM, Dekker N, Poorthuis BJ, Linthorst GE, Zwinderman AH, Breunig F, Wanner C, Aerts JM, Hollak CE. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta. 2011; 1812: 70-76.
142. Van Wayjen RG. [Enterocolitis as a symptom of angiokeratoma corporis diffusum (Ruiter-Pompen-Wyers-Kuhnau thesaurismosis).]. NedTijdschrGeneeskd. 1958; 102(39):1941-3.

33
143. Viana-Baptista M. Stroke and Fabry disease. J Neurol. 2012; 259(6): 1019-28.
144. Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007; 9: 34-45.
145. Wanner C, Oliveira JP, Ortiz A, Mauer M, Germain DP, Linthorst GE, Serra AL, Maródi L, Mignani R, Cianciaruso B, Vujkovac B, Lemay R, Beitner-Johnson D, Waldek S, Warnock DG. Prognostic indicators of renal disease progression in adults with Fabry disease: natural history data from the Fabry Registry. Clin J Am Soc Nephrol. 2010; 5: 2220-2228.
146. Wasielica-Poslednik J, Pfeiffer N, Reinke J, Pitz S. Confocal laser-scanning microscopy allows differentiation between Fabry disease and amiodarone-induced keratopathy. Graefes Arch Clin Exp Opthalmol. 2011; 249: 1689-1696.
147. Watt T, Burlina AP, Cazzorla C, Schönfeld D, Banikazemi M, Hopkin RJ, Martins AM, Sims K, Beitner-Johnson D, O'Brien F, Feldt-Rasmussen U. Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: findings from the Fabry Registry. Genet Med. 2010; 12: 703-712.
148. Weidemann F, Breunig F, Beer M, Sandstede J, Störk S, Voelker W, Ertl G, Knoll A, Wanner C, Strotmann JM. The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. Eur Heart J. 2005; 26(12): 1221-7.
149. Weidemann F, Breunig F, Beer M, Sandstede J, Turschner O, Voelker W, Ertl G, Knoll A, Wanner C, Strotmann JM. Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation. 2003; 108: 1299-1301.
150. Weidemann F, Linhart A, Monserrat L, Strotmann J. Cardiac challenges in patients with Fabry disease. Int J Cardiol. 2010; 141(1): 3-10.
151. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Störk S, Voelker, W, Ertl G, Wanner C, Strotmann J Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation 2009; 119: 524-529.
152. Weidemann F, Niemann M, Warnock DG, Ertl G, Wanner C. The Fabry cardiomyopathy: models for the cardiologist. Annu Rev Med. 2011; 62: 59-67.
153. Weidemann F, Strotmann JM, Niemann M, Herrmann S, Wilke M, Beer M, Voelker W, Ertl G, Emmert A, Wanner C, Breunig F. Heart valve involvement in Fabry cardiomyopathy. Ultrasound Med Biol. 2009; 35(5): 730-5.
154. Wendt S, Whybra C, Kampmann C, Teichmann E, Beck M. Successful pregnancy outcome in a patient with Fabry disease receiving enzyme replacement therapy with agalsidase alfa. J Inherit Metab Dis. 2005; 28: 787-788.
155. West M, Nicholls K, Mehta A, Clarke JT, Steiner R, Beck M, Barshop BA, Rhead W, Mensah R, Ries M, Schiffmann R. Agalsidase alfa and kidney dysfunction in Fabry disease. J Am Soc Nephrol. 2009; 20: 1132-1139.
156. Whitley CB, Tsai MY, Heger JJ, Prystowsky EN, Zipes DP. Amiodarone phenocopy of Fabry's keratopathy. JAMA. 1983; 249(16): 2177-8.
157. Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J, Bruhl K, Gal A, Bunge S, Beck M. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001; 24: 715-724.
158. Whybra C, Miebach E, Mengel E, Gal A, Baron K, Beck M, Kampmann C. A 4-year study of the efficacy and tolerability of enzyme replacement therapy with agalsidase alfa in 36 women with Fabry disease. Genet Med. 2009; 11: 441-449.
159. Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004; 75: 65-74.
160. Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008; 93: 112-128.
161. Wraith JE, Tylki-Szymanska A, Guffon N, Lien YH, Tsimaratos M, Vellodi A, Germain DP. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr. 2008; 152: 563-570.

34
Índice de abreviaturas BPI Brief Pain Inventory, cuestionario sobre dolores CV córnea verticillata ECG electrocardiograma TSE terapia de sustitución enzimática FOS Fabry Outcome Survey GFR tasa de filtración gomerular, (por sus siglas en inglés) Sistema GI sistema gastrointestinal HRQRL calidad de vida en función de la salud (Health related quality of life) ECC enfermedad cardiaca coronaria HVI hipertrofia ventricular izquierda MDI-0 Major Depression Inventory TRM tomografía por resonancia magnética QOL Quality of Life SF-36 Short Form 36; cuestionario de salud para analizar la calidad de vida TIA ataque isquémico transitorio (por sus siglas en inglés) WHO5 cuestionario rápido SNC sistema nervioso central Grupo de expertos (por orden alfabético)
Prof. Dr. med. Michael Beck, Zentrum für Kinder- und Jugendmedizin, Universitätsmedizin Mainz
Dr. med. Frank Breunig, KfH Nierenzentrum Ochsenfurt
Prof. Dr. med. Andreas Gal, Institut für Humangenetik, Universitätsklinikum Hamburg
Dr. med. Björn Hoffmann, Facharzt für Kinder- und Jugendmedizin, Hückeswagen
Prof. Dr. med. Christoph Kampmann, Zentrum für Kinder- und Jugendmedizin, Universitätsmedizin Mainz
Prof. Dr. med. Annerose Keilmann, Universitätsklinik für HNO und Kommunikationsstörungen, Universitätsmedizin Mainz
Prof. Dr. med. Matthias Löhr, Abteilung Gastroenterologie, Karolinska Institut Stockholm
Prof. Dr. Dr. med. h.c. Hartmut Neumann, Abteilung Innere Medizin, Universitätsklinikum Freiburg
Prof. Dr. med. Susanne Pitz, Funktionsbereich Kinder- und Neuroophthalmologie, Strabologie, Augenklinik, Universitätsmedizin Mainz
Prof. Dr. med. Arndt Rolfs, Albrecht-Kossel-Institut für Neuroregeneration, Universitätsmedizin Rostock
Prof. Dr. med. Frank Weidemann, Med. Klinik und Poliklinik I, Universitätsklinik Würzburg

35
Procedimiento de consenso La presente guía ha obtenido el consenso por la técnica de grupo nominal (TGN). Véase también el informe de la guía. http://www.awmf.org/leitlinien/detail/ll/030-134.html Conflictos de intereses Todos los miembros del grupo de expertos han expuesto sus conflictos de intereses ante AWMF. Vigencia
Esta guía tiene una vigencia de 3 años hasta su revisión. Responsable de la redacción de esta guía Prof. Dr. med. Arndt Rolfs Albrecht-Kossel-Institut für Neuroregeneration Zentrum für Nervenheilkunde Universität Rostock Gehlsheimer Str. 20 18147 Rostock Correo electrónico: [email protected]

