guia transfusión 2010 1
TRANSCRIPT

Guía sobre la transfusión de componentes sanguíneos y derivados plasmáticos4ª edición, 2010
Sociedad Española de Transfusión Sanguínea y Terapia Celular
Guí
a so
bre
la t
rans
fusi
ón
de c
om
pone
ntes
san
guín
eos
y de
riva
dos
plas
mát
ico
s

GUÍA SOBRE LA TRANSFUSIÓN DECOMPONENTES SANGUÍNEOS Y
DERIVADOS PLASMÁTICOS

4ªEdición 2010© SETS 2010
Edita:Sociedad Española de Transfusión Sanguínea y Terapia CelularC/ Mariano Cubí, 408006 Barcelona
Diseño, maquetación e impresión:Texto y Color 65, S.L.Depósito Legal: B.27061-2010
2

PRÓLOGOEntre las principales líneas estratégicas de la Sociedad Española de Transfusión San-guínea y Terapia Celular cabe destacar dos. Por un lado la continuada promoción ymejora de la calidad de la medicina transfusional que se practica en nuestros centrosy hospitales y, por otro, el necesario uso racional de los recursos del sistema, mediantela promoción continua de las buenas prácticas. La cuarta edición de la “Guía sobre latransfusión de componentes sanguíneos y derivados plasmáticos” que ahora me com-place en presentarles tiene que ver con ambos aspectos.La red hemoterápica española, y esto es bien conocido, tiene unos niveles elevados decalidad, que se han conseguido gracias a que se asienta sobre una base muy sólida: lareconocida solvencia científica y calidad humana de sus profesionales que vienen ocu-pando desde hace tiempo lugares destacados en los ámbitos científicos y académicosde nuestra comunidad científica, caracterizada por operar en un entorno muy diná-mico, con grandes cambios estructurales, organizativos y tecnológicos que han te-nido un fuerte impacto en la realidad asistencial y al que han sabido adaptarse desdela promoción continuada de políticas de calidad, tanto en la vertiente técnica comode servicio.Las organizaciones de conocimiento, como son los bancos de sangre, se caracterizanpor el mantenimiento de un equilibrio entre innovación y eficiencia. La innovación escostosa pero imprescindible para mantenerse y aun más para crecer. Por ello hay quebasar muchas decisiones que afectan a la innovación en una gestión basada en re-querimientos como la información fiable de resultados, la práctica habitual de la eva-luación y procesos de decisión para asignar recursos basados en la evidencia.La Sociedad Española de Transfusión Sanguínea y Terapia Celular ha actualizado y publi-cado esta Guía sobre la transfusión de componentes sanguíneos y derivados plasmáticos,con el propósito de hacer accesible a los profesionales de habla hispana la información ne-cesaria para trabajar bien. En esta guía, los profesionales hospitalarios encontrarán, desti-lados a través de mucha experiencia, las indicaciones para el manejo adecuado de lacadena de valor transfusional. La revisión por parte de los profesionales que han partici-pado en ella y a los que en nombre de la SETS quiero agradecer su colaboración, es am-plia, excelente i representa una actualización de los conocimientos contenidos. Esta guía puede constituir un magnífico instrumento para todos aquellos que quieranmejorar la Medicina Transfusional.
Ramon Pau Pla IllaPresidente de la SETSMarzo 2010
3

AGRADECIMIENTOS
La junta directiva de la SETS quiere expresar su agradecimiento a las perso-nas, que han colaborado en la revisión y actualización de la Guía sobre latransfusión de componentes sanguíneos y derivados plasmáticos, SETS edición2006, haciendo, con ello, posible la publicación de esta cuarta edición.
Cristina Arbona CastañoM. Alba Bosch LlobetNelly Carpio MartínezDolors Castellà Cahiz Azucena Castrillo FernándezMercedes Corral AlonsoCarmen Fernández ÁlvarezEsther González GarcíaMaría Luisa Lozano AlmelaPedro Madoz ResamoEduardo Muñiz-DíazManuel Nieto TorviscoPilar Ortiz MurilloJosé Rivera PozoJulia Rodríguez VillanuevaConcha Zamora de Pedro
4

5
ÍNDICE
ASPECTOS GENERALES DE LA DONACIÓN, EL PROCESAMIENTO DE LA SANGRE Y LA TRANSFUSIÓN 7LA DONACIÓN DE SANGRE 7EL PROCESAMIENTO DE LA SANGRE TOTAL 8EL ETIQUETADO DE LOS COMPONENTES SANGUÍNEOS 9EL ALMACENAMIENTO DE LOS COMPONENTES SANGUÍNEOS 10LAS DETERMINACIONES ANALÍTICAS 10LA TRANSFUSIÓN DE LOS COMPONENTES SANGUÍNEOS 11
EFECTOS ADVERSOS DE LA TRANSFUSIÓN 13COMPLICACIONES AGUDAS 13COMPLICACIONES RETARDADAS 25
HEMOVIGILANCIA 31LA HEMOVIGILANCIA EN ESPAÑA 34
TRANSFUSIÓN DE LOS DIFERENTES COMPONENTES SANGUÍNEOSTRANSFUSIÓN EN LOS PACIENTES ADULTOS 43CONCENTRADO DE HEMATÍES 43CONCENTRADO DE PLAQUETAS 54REFRACTARIEDAD A LA TRANSFUSIÓN DE PLAQUETAS 60PLASMA 62CRIOPRECIPITADO 67
TRANSFUSION EN NEONATOS Y NIÑOS 69SANGRE TOTAL 69CONCENTRADO DE HEMATÍES 69CONCENTRADO DE PLAQUETAS 70PLASMA 72TRANSFUSIÓN INTRAUTERINA 73EXANGUINOTRANSFUSIÓN 74ACTUACIÓN EN LOS DÉFICITS DE INMUNOGLOBULINA A 75COMPONENTES SANGUÍNEOS IRRADIADOS 75PARTICULARIDADES DE LA TRANSFUSIÓN EN NIÑOS Y NEONATOS 75

COMPONENTES SANGUÍNEOS IRRADIADOS 81INDICACIONES DE LA IRRADIACIÓN DE COMPONENTES SANGUÍNEOS 81
ALTERNATIVAS A LA TRANSFUSIÓN DE SANGRE ALOGÉNICA 85PRINCIPIOS BÁSICOS DE UN PROGRAMA DE AHORRO DE SANGRE 85USO DE ERITROPOYETINA ALFA Y AGENTES HEMATÍNICOS 86DONACIÓN AUTÓLOGA PREDEPÓSITO 88HEMODILUCIÓN NORMOVOLÉMICA 88RECUPERACIÓN INTRA Y POSTOPERATORIA 89
DERIVADOS PLASMÁTICOS 91ALBÚMINA HUMANA 91INMUNOGLOBULINAS 94FACTOR VIII 102FACTOR VON WILLEBRAND (FVW) 103CONCENTRADOS DE COMPLEJO PROTROMBÍNICO (CCP) Y FIX 104FIBRINÓGENO 105FACTOR VII 105FACTOR XI 106FACTOR XIII 106SISTEMA ADHESIVO DE FIBRINA 106CONCENTRADOS DE ANTITROMBINA 107CONCENTRADOS DE PROTEÍNA C 107
PRÁCTICA TRANSFUSIONAL 111SOLICITUD DE TRANSFUSIÓN 111MUESTRA PRETRANSFUSIONAL 111ACTO TRANSFUSIONAL 112REGISTROS 116
ASPECTOS ÉTICOS Y LEGALES SOBRE LA TRANSFUSIÓN DE COMPONENTES SANGUÍNEOS Y DERIVADOS PLASMÁTICOS 119LA PRESCRIPCIÓN DE COMPONENTES SANGUÍNEOS 119INFORMACIÓN AL PACIENTE. CONSENTIMIENTO INFORMADO 119PRODUCTOS NO SEÑALADOS ESPECIFICAMENTE EN LA NORMATIVA 125MEDICAMENTOS DERIVADOS DEL PLASMA 126HEMOVIGILANCIA 127
6

ASPECTOS GENERALES DE LA DONACIÓN, EL PROCESAMIENTODE LA SANGRE Y LA TRANSFUSIÓN
La Medicina Transfusional (MT) comprende una sucesión de procesos dirigidos ala obtención de componentes sanguíneos (CS) seguros y de buena calidad, paratransfundirlos de forma eficiente y segura. El desarrollo de las acciones involucra-das en este objetivo precisa de unas estructuras que definan y aseguren la calidad,manteniendo la uniformidad de unos requisitos mínimos en cada uno de los pun-tos de la cadena transfusional. La MT se caracteriza por ser multidisciplinaria, conintervención de diferentes profesionales, y se sirve de tecnologías que incorporanlos últimos avances en convivencia con metodologías más clásicas aún vigentes.
LA DONACIÓN DE SANGRE
A pesar de que han transcurrido más de 100 años desde las primeras trans-fusiones terapéuticas, la única fuente disponible de CS para la transfusiónsigue siendo la donación. Un colectivo suficientemente numeroso de perso-nas altruistas y responsables, que donan sangre de forma repetida, consti-tuye el primer elemento para garantizar la seguridad transfusional. Elconcepto de donación no retribuida es el expresado por el Consejo de Eu-ropa: “La donación se considera voluntaria y no remunerada cuando la per-sona dona sangre, plasma o componentes celulares de forma altruista y norecibe compensación económica, ya sea en efectivo o en otra forma que pu-diera ser considerada como sustituto del dinero, incluyendo un tiempo libreen el trabajo superior al razonable para el desplazamiento y la donación”. Ladonación retribuida no es segura, porque puede inducir al donante a ocultardatos de su salud que pueden comprometer al receptor y a sí mismo.
Los donantes aptos para la donación han de cumplir unos criterios de selec-ción contemplados en la normativa estatal que recoge el anexo II del RealDecreto 1088/2005. La encuesta de selección añade criterios suplementariosrelacionados, por ejemplo, con el país de procedencia y otros datos epide-miológicos del potencial donante. Existen donaciones especiales, como algu-nas procedentes de aféresis y las donaciones predepósito para transfusiónautóloga, regidas por condiciones específicas.
7

Garantizar un suministro adecuado de sangre y CS para las necesidades delpaís, hace imprescindible el mantenimiento de una base de donantes activasuficientemente amplia. Esto requiere ejercer unas medidas para conservar, omejor fidelizar, a los donantes ya conocidos y, además, lograr la incorpora-ción continua de nuevos donantes. En esta tarea la promoción de la dona-ción, dirigida a distintos colectivos ejerce un papel fundamental. Diversosaspectos de la promoción de la donación deberían incluirse en materias edu-cativas del periodo académico, desde la escuela a la universidad, para dar a co-nocer el proceso integral de la donación y difundir principios de utilidad,necesidad y solidaridad inherentes a la donación de sangre.
EL PROCESAMIENTO DE LA SANGRE TOTAL
Incluye el fraccionamiento de la misma para conseguir CS y el estudio analí-tico apropiado (pruebas serológicas e inmunohematológicas) de las donacio-nes y donantes.
La capacidad de separar los CS de la sangre total en la década de los 60, marcóun hito en la historia del tratamiento transfusional. Podemos obtener 3 CS apartir de una sola donación, concentrado de hematíes, unidad de plaquetas yunidad de plasma, lo que significa que se pueden beneficiar diferentes pacien-tes. De este modo se optimiza el uso de un bien escaso, los pacientes reciben sóloel CS necesario y se reduce el riesgo de reacción transfusional. En definitiva, laterapia de componentes tiene ventajas de índole logística, económica y ética.
Los CS son las unidades terapéuticas de la sangre, que pueden ser preparadosmediante centrifugación, filtración y congelación, aplicando la metodologíaconvencional de un banco de sangre. En los últimos años se ha incrementadola automatización y se han incorporado nuevos procedimientos en el área deobtención y preparación de componentes. Así, por ejemplo, en los centros es-pañoles se realiza actualmente la leucodepleción por filtración de todos losCS celulares en el momento de su preparación, siendo el contenido residualde leucocitos inferior al millón por unidad.
Para obtener CS idóneos, a veces es preciso realizar en ellos alguna modifi-cación o tratamiento adicional, ya sea para minimizar algún tipo de reaccióno para aumentar la seguridad transfusional. En este marco se sitúa la irra-
8

diación gamma, que produce una inhibición de los linfocitos T presentes enlos CS. Los pacientes con riesgo clínico de desarrollar la enfermedad injertocontra huésped asociada a la transfusión (EICH-AT), deben recibir CS irradia-dos ya que esta técnica resulta eficaz en la profilaxis de dicha complicación.Son pacientes de riesgo los que se hallan en situación de inmunosupresión ylos que reciben CS de un donante con el que comparten antígenos HLA.
Para la transfusión pediátrica, en ocasiones es preciso fraccionar y/o alicuo-tar un CS en varias dosis, esto facilita el uso de varias fracciones procedentesde un solo donante para el mismo receptor.
Otra modificación en los CS es el lavado de hematíes o plaquetas, con el finde retirar algún posible componente (proteína, anticoagulante) causante dealgunas reacciones.
Aunque se apliquen las medidas de seguridad establecidas para todas las do-naciones, persiste un riesgo microbiológico residual mínimo. Para afrontarlo,se han incorporado a la rutina de los centros estrategias de inactivación oreducción de patógenos, las cuales ya se están aplicando al plasma desdehace años y más recientemente a las plaquetas; para hematíes están en unafase avanzada de investigación. La reducción de patógenos tiene por objetoinactivar virus, bacterias y parásitos presentes en los CS, que puedan tenerefectos nocivos en el receptor de la transfusión, sin comprometer su eficaciaterapéutica ni causar efectos adversos.
EL ETIQUETADO DE LOS COMPONENTES SANGUÍNEOS
Finalmente, cada uno de los CS obtenidos subsidiarios de transfusión, porquecumplen los requisitos vigentes de calidad de producto y de idoneidad en re-lación al cribado analítico, deben ser etiquetados. La etiqueta portará infor-mación sobre los siguientes aspectos:
� Denominación oficial del componente, incluyendo cualquier modificaciónrealizada en su composición final.
� Identificación numérica o alfanumérica exclusiva de la donación.� Nombre y dirección del centro procesador.� Grupo ABO y Rh (D). Otros sistemas antigénicos cuando sea necesario.� Resultado del escrutinio de anticuerpos irregulares antieritrocitarios, y su
9

identificación si procede. � Resultados de las pruebas de detección de agentes infecciosos.� Fechas de extracción y caducidad (y hora, cuando sea necesario).� Volumen del CS.� Denominación, composición y volumen del anticoagulante y/o, en su caso,
de la solución aditiva.� Temperatura y condiciones de almacenamiento.� Instrucciones sobre la utilización del CS.
EL ALMACENAMIENTO DE LOS COMPONENTES SANGUÍNEOS
Los diferentes CS deben almacenarse en las condiciones y a la temperaturaque especifica la normativa para cada componente, así los preparados de he-matíes, generalmente en solución aditiva se mantendrán a temperatura entre2 y 6º C, por un periodo máximo de 42 días. Los preparados de plaquetas seconservarán entre 20 y 24º C durante 5 días, este periodo puede ampliarse a7 días si se emplean métodos o sistemas de detección o reducción de la po-tencial contaminación de bacterias. Los componentes de plasma se almace-nan congelados y su tiempo máximo de almacenamiento dependerá de latemperatura de congelación, pudiendo llegar hasta los 36 meses.
LAS DETERMINACIONES ANALÍTICAS
De acuerdo con la normativa legal, a todas las donaciones se les realizarán unabatería de determinaciones analíticas, entre las que se incluyen:
1. Grupo sanguíneo ABO y Rh (D).2. Escrutinio de anticuerpos irregulares antieritrocitarios.3. Pruebas para la detección de agentes infecciosos:
� Sífilis: pruebas serológicas. � Antígeno de superficie del virus de la hepatitis B (VHB).� Anticuerpos contra el virus de la inmunodeficiencia humana I/II (VIH).� Anticuerpos contra el virus de la hepatitis C (VHC).� Detección genómica directa del VHC.� Otras pruebas que se consideren necesarias para detectar portadores de
otros agentes infecciosos, en determinados donantes, por sus circuns-tancias epidemiológicas concretas.
10

A pesar de las pruebas realizadas (serología y detección genómica) existe unriesgo de transmisión de enfermedades infecciosas por la donación. El riesgoestimado de transmisión de VHC es uno por casi dos millones cuatrocientasmil donaciones (1/2381000), el de VIH (1/806000) es casi tres veces superioral de VHC, y es aun más alto para el VHB (1/177000).
Con el fin de establecer las actuaciones a seguir en relación con el resultado delas pruebas de cribado de agentes infecciosos, existen algoritmos destinados aestandarizar la interpretación de tales resultados. En base a ellos, únicamenteserán aceptadas las donaciones con resultados inequívocamente negativos.
LA TRANSFUSIÓN DE LOS COMPONENTES SANGUÍNEOS
La transfusión de CS tiene como objeto el tratamiento de procesos específi-cos en pacientes que requieren esta terapia, cuando no puede ser sustituida porotra alternativa. La indicación de la transfusión de CS obedece a unos objeti-vos básicos como mantener/aumentar el transporte de oxígeno a los tejidos,corregir una hemorragia, normalizar trastornos de la coagulación y, en algunasocasiones, aportar derivados plasmáticos como albúmina o inmunoglobulinas.
La seguridad del acto transfusional se apoya en tres pilares fundamentales: lacorrecta indicación, basada en una valoración minuciosa del índice benefi-cio/riesgo, la elección del CS más idóneo y la elección de una dosis correcta.En definitiva, es necesario hacer un uso óptimo de la sangre. Esto requiere uti-lizar una serie de herramientas entre las que se encuentran guías o recomen-daciones de uso de los CS, con el fin de facilitar una práctica transfusionaluniforme y eficiente. En la misma línea debe actuar el Comité hospitalario detransfusión, estructura organizativa que debe promover la elaboración e im-plantación de medidas para alcanzar el uso óptimo de CS, y que sirve de nexoentre los servicios de transfusión, los clínicos y los centros procesadores.
Aunque la transfusión proporciona beneficios clínicos incuestionables, tam-bién produce algunos efectos adversos. Con el fin de conocer su magnitud ypoder prevenirlos, se ha desarrollado un programa de hemovigilancia (HV)que incluye la detección, registro y análisis de la información relativa a talesefectos. En realidad, el espectro de actuación de la HV abarca todas las fasesde la cadena transfusional.
11

Para trabajar con unos criterios de calidad y requisitos mínimos en cada unode los eslabones de la cadena transfusional, existe un marco legal expresadoen el Real Decreto 1088/2005 (BOE 225 del 20 de septiembre de 2005) ynormas o guías, que regulan todas las actuaciones desde la donación hasta latransfusión al paciente.
Esta guía pretende proporcionar la información necesaria, para contribuir aque el proceso de establecer la indicación de una transfusión y la adminis-tración de componentes, se realicen de la forma más idónea y segura posible.Esta dirigida a médicos implicados en la prescripción de la transfusión de CSy también al personal de enfermería, que interviene en su administración.
REFERENCIASReal Decreto 1088/2005-BOE 225 del 20/09/2005, por el que se establecen los requi-sitos técnicos y condiciones mínimas de la hemodonación y de los centros y serviciosde transfusión.Guide to the preparation, use and quality assurance of blood components, 14th edition.Council of Europe publishing, 2008.Guidelines the blood transfusion services in the United Kingdom, 7th edition, 2005.Boletín de la sociedad española de transfusión sanguínea y terapia celular Vol. 21 (2);2009.Estándares de acreditación en transfusión sanguínea, 3ª edición. Comité de acredita-ción en transfusión. Asociación española de Hematología y Hemoterapia, Sociedad es-pañola de Transfusión sanguínea. 2006.Safe blood components. Key elements. World Health Organization 2005.WHO/EHT/05.0.1Developing a national policy and guidelines on the clinical use of blood. Recommen-dations. World Health Organization. Blood transfusion safety. WHO/BCT/BTS/01.3.Alvarez M. Actualización del riesgo residual de transmisión de enfermedades infeccio-sas en la era de las técnicas NAT. SETS. 20 Congreso Nacional de la Sociedad Españolade Transfusión Sanguínea y Terapia Celular. Tarragona junio de 2009.
12

La transfusión de sangre y sus componentes presenta hoy en día un alto nivel deseguridad, debido a las innovaciones técnicas que se han ido incorporando pro-gresivamente a las distintas fases del proceso de elaboración de componentes,desde la selección/aceptación de donantes, hasta la incorporación, en el labora-torio de seguridad transfusional, de nuevas técnicas con alta capacidad para de-tectar agentes patógenos, potencialmente transmisibles mediante transfusión.
Sin embargo, la transfusión puede conllevar efectos adversos que obligan aconsiderar en cada indicación los riesgos / beneficios de nuestra actuación.
Los efectos adversos relacionados con la transfusión deben comunicarse a lossistemas de Hemovigilancia, mediante los servicios de transfusión hospitalarios.
Quien primero detecta los efectos adversos de la transfusión es el equipoasistencial de los servicios clínicos que, a cabecera del paciente, debe saber in-terpretar i detectar los signos y síntomas de una posible reacción, y conocerel circuito para notificarlos a su correspondiente servicio de transfusión.
Para exponer los efectos adversos y riesgos asociados a la transfusión de com-ponentes se clasifican, como se expone en la Tabla 1, atendiendo a la crono-logía de su aparición:
� COMPLICACIONES AGUDAS: Aparecen durante el acto transfusional, opoco tiempo después (hasta 24 horas).
� COMPLICACIONES RETARDADAS: Tienen lugar más allá de las 24 horasdespués del inicio de la transfusión.
COMPLICACIONES AGUDAS
Entre ellas, hay algunas, que por su gravedad, pueden comprometer la vida delpaciente y además en la fase inicial o temprana puede ser difícil de distinguir,tanto el tipo de reacción, como su gravedad y el alcance clínico que acabaráteniendo en el paciente. Por este motivo, cualquier sintomatología que apa-rezca durante una transfusión debe ser tomada en cuenta, ya que puede tra-tarse del primer signo de una reacción grave.
13
EFECTOS ADVERSOS DE LA TRANSFUSIÓN

Las principales reacciones agudas que revisten gravedad son las resaltadas enrojo en la tabla 1:
� Reacción hemolítica aguda.� Reacción por infusión de un componentes sanguíneo con contaminación
bacteriana.� Lesión pulmonar aguda asociada a transfusión (TRALI).� Sobrecarga circulatoria.� Reacción alérgica grave o reacción anafiláctica.
El algoritmo (Figura 1) muestra cómo reconocer una reacción aguda transfu-sional y la conducta a seguir ante ella.
14
Tabla 1
Principales efectos adversos de la transfusión� Complicaciones agudas
- De origen inmunológico:• Reacción hemolítica aguda• Reacción febril no hemolítica• Reacción alérgica• Lesión pulmonar aguda asociada a transfusión (TRALI)• Aloinmunización con destrucción plaquetar inmediata
- De origen no inmunológico:• Contaminación bacteriana• Sobrecarga circulatoria• Hemólisis no inmune• Reacciones hipotensivas
� Complicaciones retardadas- De origen inmunológico:
• Reacción hemolítica retardada• Aloinmunización frente antígenos eritrocitarios, plaquetarios, leucocitarios o prote-
ínas plasmáticas• Púrpura postransfusional• Enfermedad del injerto contra el huésped postransfusional• Inmunomodulación
- De origen no inmunológico:• Transmisión de agentes infecciosos• Hemosiderosis transfusional

15
Figura 1: Diagrama de flujo. Actuación ante una reacción transfusional aguda.
Reacción transfusional Febril no-hemolítica • Si temperatura aumenta menos de 1,5ºC, las
constantes se mantienen estables y el pa-ciente está bien, dar paracetamol
• Reiniciar la infusión a un ritmo más lento yobservar más frecuentemente
Síntomas / signos de una reacción aguda: Fiebre, escalofríos, taquicardia, hiper- o hipotensión, shock, rash cutáneo,urticaria, dolor a nivel torácico, muscular, óseo o abdominal, nauseas, sensación de malestar general, taquipnea, disneay/o distress respiratorio
Parar la transfusión, Mantener la vía endovenosa y avisar al médico• Medir constantes vitales (temperatura, pulso, tensión arterial, respiración, y saturación arterial de O2• Comprobar la identidad del receptor y los detalles de la unidad y de la compatibilidad de la misma (etiqueta…)
Reacción Alérgica moderada• Administrar Clorfenamina 10 mg
EV lento y reiniciar la transfusión aritmo más lento y observar conmás frecuencia
Reacción Alérgica GraveBroncoespasmo, angioedema, dolorabdominal, hipotensión:• Parar la transfusión, mantener vía
venosa• Retirar la unidad y el equipo de
transfusión • Remitir al servicio de transfusiones,
junto como las otras unidades queya hayan sido infundidas
• Enviar muestra de sangre coagu-lada al laboratorio de transfusiones
• Administrar Clofeniramina 10 mgEV lento
• Iniciar Oxigenoterapia O2• Broncodilatador: Salbutamol en
nebulizador• Si hipotensión severa: admistrar
adrenalina sc o intramuscular (0,5mL de 1/1000 IM) equivalente a0,5mg IM
• Posiblemente, para las próximastransfusiones, lavar con salina loscomponentes sanguíneos
TRALI• Clínica de insuficiencia respiratoria
aguda con fiebre y escalofríos• Parar la transfusión• Dar O2• Tratar como en el Sdre. Distress
respiratorio agudo, ventilaciónasistida si la hipoxia lo indica
Incompatibilidad ABO• Parar la transfusión,• Mantener la via EV e iniciar infusión EV de
suero fisiológico• Remitir al Banco de Sangre la unidad y el
equipo de transfusión• Monitorizar y mantener la diuresis a > 100
mL/h• Furosemida si la diuresis disminuye o existe
anuria• Si desarrolla CID, tratar con los componentes
sanguíneos adecuados• Informar inmediatamente al Departamento o
Servicio de Transfusiones y enviar muestra desangre coagulada
Reacción Hemolítica/Infección bacterianaen la unidad.• Parar la transfusión• Retirar la unidad y el equipo de transfusión y
remitir al servicio de transfusiones, así comolas otras unidades que ya hayan sido infundi-das
• Cursar hemocultivos , y muestras para análisis(hemograma, bioquímica, estudio coagula-ción, urinanálisis) y banco de sangre (recom-probación grupo sanguíneo, pruebascompatibilidad)
• Monitorizar diuresis• Iniciar antibióticoterapia de amplio espectro• Iniciar O2 y fluidoterapia• Consultar UCI y Hematología
Sobrecarga Circulatoria• O2 y furosemida 40-80 mg EV
Fiebremoderada Urticaria
PVCElevada
PVC normal
NO
NO
NO
NO
SI
SI
SI
Reacción con únicamente fiebre moderada o rash
urticariforme
Sospecha de incompatibilidad
ABO
Otras reacciones
hemolíticas o/ contaminación
bacteriana
Reacción alérgica
grave
Dísnea aguda/ hipotensión
Monitorizar GasometriaMedir PVC / presión
capilar pulmonar

DE ORIGEN INMUNOLÓGICO
Reacción transfusional hemolítica aguda
Es el efecto adverso asociado a la transfusión más grave.
Los hematíes transfundidos son destruidos de forma aguda por anticuerpos pre-sentes en el plasma del receptor. La causa más frecuente es la incompatibilidadABO, que ocurre con una frecuencia entre 1/6.000 y 1/20.000 unidades trans-fundidas, debida a errores de identificación en cualquiera de las fases de la ca-dena transfusional (Tabla 2) es la causa más frecuente de muerte evitableasociada a la transfusión, entre 1/100.000 y 1/500.000 unidades transfundidas.
Sintomatología
La sintomatología inicial esfrecuentemente dolor torácicoo lumbar, taquicardia, disnea,escalofríos, fiebre, sangrado, eincluso shock. Esta sintomato-logía puede acompañarse conlas siguientes alteracionesanalíticas: hemoglobinemia,hemoglobinuria, aumento dela bilirrubina sérica, prueba dela antiglobulina humana posi-
16
Tabla 2
Causas más frecuentes de error asociado con reacciones transfusionales� Identificación no correcta del paciente en la solicitud� Identificación errónea de la muestra� Equivocación en la toma de la muestra� Error de trascripción� Error técnico en el Servicio de transfusión� Confusión en la distribución del componente sanguíneo� Confusión en la administración del componente sanguíneo, al no seguir el protocolo de
identificación del receptor
Reacción transfusional hemolítica aguda:
Signos y síntomas
� Dolor torácico� Dolor lumbar� Fiebre� Escalofríos� Hipotensión� Dolor abdominal� Disnea� Nauseas y vómitos� Hemoglobinuria
� Hemoglobinemia� Shock� Anemia� Oliguria o anuria� Sangrado
generalizado� CID� Urticaria � Diarrea

tiva y alteración de las pruebas de coagulación. En pacientes anestesiados losprimeros signos pueden ser la hipotensión y los secundarios a la instauraciónde una coagulación intravascular diseminada (CID). Debe distinguirse de otrashemólisis no inmunes como: infusión de líquidos hipotónicos, ciertos fárma-cos o medicaciones administrados en la misma vía, toxinas bacterianas porcontaminación del componente, temperatura anómala de los hematíes (so-brecalentamiento o congelación).
Ante la sospecha de un episodio hemolítico agudo, la transfusión debe ser in-terrumpida inmediatamente, mantener la vía venosa y notificar al Servicio detransfusión (remitiendo los equipos, documentación y muestras de sangre so-licitadas para realizar análisis), comprobando a la vez que no existen más pa-cientes implicados en un probable error de identificación.
La gravedad de la reacción suele ser proporcional al volumen de producto in-compatible transfundido.
Tratamiento
� Interrumpir la transfusión inmediatamente. � Mantener la vía endovenosa y resucitar con soluciones cristaloides. � Considerar el soporte inotrópico si se prologa la hipotensión. � Cursar hemocultivos del contenido de la unidad transfundida. � Informar al Servicio de transfusión. � Consulta urgente a la Unidad de Cuidados Intensivos (UCI). � Ingreso en UCI si es posible.
El tratamiento debe instaurarse rápidamente y de manera agresiva con flui-doterapia que prevenga la hipotensión para intentar impedir el fracaso renal.La perfusión renal debe ser monitorizada con control de diuresis, que se man-tendrá, mínimo, de 100 ml/hora las primeras 18-24 horas.
Puede utilizarse furosemida e.v. a dosis de 1-2 mg/ kg de peso, (40-80 mg/día EV) que además de efecto diurético, aumenta el flujo al nivel de la cor-teza renal. Si no hay respuesta puede ser preciso la administración de dopa-mina a dosis bajas (5 microgramos / kg/ minuto) para favorecer vasodilatacióny aumento de la perfusión renal. Si en la primera hora no hay respuesta, eva-luada por la diuresis, posiblemente se haya producido necrosis tubular y puedeser necesario la realización de diálisis.
17

Si se desarrolla CID se tratará adecuadamente con plasma u otros derivadosplasmáticos, heparina (aunque su uso es muy controvertido) y si fuera pre-ciso plaquetas.
Reacción transfusional febril no hemolítica
Junto con las reacciones alérgicas leves, constituyen el grupo más frecuentede reacciones transfusionales, afectando aproximadamente al 1-2% de losreceptores.
La causa más frecuente es la presencia de citocinas en el producto transfun-dido, liberadas por los leucocitos o las plaquetas principalmente durante el pe-riodo de almacenamiento. También podría deberse a la presencia deanticuerpos antileucocitarios en el plasma del receptor. La leucorreducciónuniversal ha disminuido notablemente los episodios de reacción febril no he-molítica. La disminución es significativa, cercana al 50 %, en el caso de latransfusión de hematíes y aún más importante, superior al 90 %, en las aso-ciadas a la transfusión de concentrados de plaquetas.
Sintomatología
Se produce un aumento de temperatura corporal superior a 1ºC durante, ohasta 2h después de finalizada la transfusión, suele acompañarse de escalo-fríos y/o tiritonas, no hay hipotensión, ni shock.
Se trata de un diagnóstico de exclusión, y debe tenerse en cuenta que una re-acción febril puede ser el primer síntoma de reacciones muy graves, como lacontaminación bacteriana o la reacción hemolítica aguda.
Tratamiento
La mayoría de las reacciones febriles pueden controlarse parando o enlente-ciendo la velocidad de infusión de la transfusión y con la administración deantipiréticos como el acetaminofeno o los anti-inflamatorios no esteroideos.
Aunque estas reacciones son frecuentes y muy desagradables para el paciente,no comprometen su vida. Sin embargo, es importante recordar que la fiebrey los escalofríos pueden ser también el primer signo de una reacción trans-fusional grave.
18

Reacciones transfusionales alérgicas
Se debe a la existencia de alguna sustancia en el producto transfundido (pro-teínas, fármacos, etc.) a la cual el receptor es alérgico. La sintomatología esmuy variada, desde manifestaciones cutáneas localizadas (habones, eritema,prurito, etc.) a reacciones anafilácticas generalizadas (broncoespasmo, larin-goespasmo, shock).
Según su gravedad, distinguimos dos tipos de reacciones alérgicas:
� Anafilácticas: muy graves y poco frecuentes.� Moderadas y leves: son las más frecuentes, se presentan en aproximada-
mente el 1% de los pacientes transfundidos.
Anafilaxia
Es muy poco frecuente, pero es una de las complicaciones transfusionalesgraves que pueden comprometer la vida del paciente. Sucede generalmentedurante la trasfusión o inmediatamente después.
Los síntomas incluyen disnea, dolor torácico, dolor abdominal y nauseas. Sig-nos: hipotensión, broncoespasmo, edema laríngeo y periorbital, vómitos, eri-tema cutáneo, urticaria y conjuntivitis.
La anafilaxia ocurre cuando un paciente está pre-sensibilizado a un alergenoy ante la re-exposición al antígeno particular, produce anticuerpos IgE.
Los anticuerpos IgG del paciente frente a alergenos infundidos con la trans-fusión pueden causar también reacciones anafilácticas graves.
Un pequeño grupo de pacientes, con déficit de IgA severo, desarrollan anticuer-pos anti IgA y pueden presentar reacciones anafilácticas graves si son expuestosa IgA en la transfusión. En esos casos la clínica comienza tras la transfusión depequeñas cantidades de cualquier componente sanguíneo que contenga plasma.
El déficit de IgA, definido como valores de IgA plasmática inferiores a 50mg/dl, afecta a una de cada 700 personas, y aproximadamente el 30% deellos presentan anticuerpos anti-IgA de clase IgE. A pesar de estas cifras, la re-acción anafiláctica grave post transfusional es muy poco frecuente, no todoslos pacientes con déficit de IgA y portadores de anti IgA presentan reaccio-nes anafilácticas cuando son transfundidos).
19

Si aparece esta complicación, debe notificarse al servicio de transfusión, paraque las transfusiones posteriores se realicen con componentes celulares la-vados con salino, para garantizar la ausencia de proteínas plasmáticas.Cuando se requiera la transfusión de plasma se debe contemplar la posibi-lidad de transfundir plasma de donantes deficitarios en IgA. Si la urgenciano lo permite y se debe transfundir plasma con IgA, se debe instaurar el tra-tamiento preventivo correcto (con hidrocortisona, antihistamínicos, y vigi-lancia constante para tratamiento inmediato con adrenalina si es preciso).
Reacciones alérgicas moderadas y leves
Son frecuentes, se presentan en aproximadamente el 1% de los pacientestransfundidos.
Urticaria, prurito a los pocos minutos de iniciada la transfusión, particular-mente con componentes que contienen mayor volumen de plasma (concen-trados de plaquetas y unidades de plasma fresco congelado).
Tratamiento
En las reacciones leves (la mayoría), responden bien al tratamiento con anti-histamínicos y no se vuelven a producir.
En las reacciones severas y anafilácticas, la transfusión debe interrumpirse in-mediatamente e iniciarse el tratamiento de soporte cardiorrespiratorio apro-piado (que puede incluir: tratamiento vasopresor con adrenalina y corticoides,intubación, etc.).
Lesión pulmonar aguda asociada a transfusión (TRALI)
Se trata de un edema pulmonar no cardiogénico. No existe certeza en relacióncon la patogénesis del TRALI, aunque en todos los supuestos juega un papelpreponderante la infusión pasiva de anticuerpos del donante, que reacciona-rían directamente con los correspondientes antígenos presentes en los leuco-citos del receptor. Una de las hipótesis más aceptadas es la denominada “teoríade los dos eventos”, en la que se postula que el TRALI estaría ocasionado pordos eventos independientes, el primero respondería a circunstancias clínicaspropias del receptor, que provocarían daño endotelial pulmonar y el segundovendría ocasionado por la infusión pasiva de anticuerpos o modificadores dela respuesta biológica, incluyendo lípidos activos, procedentes del donante.
20

La incidencia real de TRALI es desconocida. En Estados Unidos la incidencia deTRALI comunicados es aproximadamente de 1/5.000 transfusiones, en Eu-ropa la incidencia es menor, aproximadamente 1/8.000 transfusiones. Mu-chas opiniones de expertos coinciden en que probablemente el TRALI es unacomplicación transfusional infradiagnosticada.
Sintomatología
Se caracteriza por escalofríos, fiebre, cianosis, hipotensión, insuficiencia respira-toria, después de la transfusión de un volumen de componente sanguíneo quehabitualmente no produce hipervolemia. La expresividad clínica del cuadro puedeser variable: desde una caída en la saturación de oxígeno de la sangre hasta unsíndrome de pulmón blanco bilateral. La causa es un incremento en la permea-bilidad de la microcirculación pulmonar que provoca la salida de líquido a losespacios alveolar e intersticial. Generalmente aparece entre 2 y 4 horas despuésde la transfusión. Suele ser difícil el diagnóstico diferencial entre TRALI y otrosedemas pulmonares no cardiogénicos e incluso de la insuficiencia cardíaca.
Tratamiento
Requiere tratamiento en unidad de cuidados intensivos con soporte respira-torio que puede incluir la intubación endotraqueal. Deben evitarse los diuré-ticos, y los corticoides tienen un dudoso beneficio.
Ante la sospecha de su aparición, debe informarse al Centro de transfusión parael estudio de los donantes implicados y la retirada inmediata de otros productosde los donantes sospechosos para evitar ser transfundidos. El estudio de un epi-sodio de TRALI debería incluir la detección de anticuerpos antigranulocitarios yanti-HLA en el donante y en el receptor y el fenotipo leucocitario del receptor.
Aloinmunización con destrucción plaquetar inmediata
Se produce en pacientes con anticuerpos anti-HLA o anti antígenos plaquetariosespecíficos, por transfusiones o embarazos previos. Estos anticuerpos producen ladestrucción de las plaquetas que contengan el antígeno correspondiente, mani-festándose generalmente en incrementos escasos inmediatamente tras la trans-fusión de plaquetas. Debe diferenciarse de aquellos casos de supervivencia acortadade las plaquetas por razones no inmunológicas (CID, sepsis, esplenomegalia; etc.).La refractariedad plaquetar es una complicación relativamente frecuente en pa-cientes que reciben soporte crónico con concentrados de plaquetas (5 – 15 %).
21

Sintomatología
Puede no presentar ninguna clínica añadida a la propia de la plaquetopeniaque indujo a la transfusión de plaquetas. En ocasiones se observa una reac-ción transfusional de tipo escalofríos e hipertermia cuando se administra latransfusión de plaquetas incompatibles.
Tratamiento
Si aparece fiebre se administrará antipiréticos como el acetaminofeno o losanti-inflamatorios no esteroideos. Detectada la refractariedad para transfu-siones posteriores de plaquetas, estas deberán ser HLA compatibles.
DE ORIGEN NO INMUNOLÓGICO
Contaminación bacteriana
Se trata de una complicación poco frecuente, pero de consecuencias poten-cialmente mortales. Se sospecha que entre el 0.002 y el 0,4 % de los con-centrados de hematíes y el 0,01 y el 1 % de los concentrados de plaquetaspueden estar contaminados con bacterias, mayoritariamente procedente dela flora saprofita cutánea existente en la piel del donante.
La presencia de las bacterias en los componentes sanguíneos suele deberse ala persistencia de los gérmenes en la zona de la punción. En general los gérme-nes Gram negativos se asocian a la contaminación de los concentrados de he-matíes, mientras que los Gram positivos suelen ser los responsables de las sepsisproducidas por los concentrados de plaquetas. Cambios en la coloración de losconcentrados de hematíes o la desaparición en los “remolinos” de los concen-trados de plaquetas nos deben poner sobre aviso de riesgo de contaminación.
Sintomatología
Clínicamente se caracteriza por la presencia de fiebre alta, escalofríos, hipo-tensión y shock durante o inmediatamente después de la transfusión.
Tratamiento
Ante la sospecha de su aparición, debe interrumpirse inmediatamente latransfusión e iniciarse el adecuado tratamiento antibiótico y de soporte car-diovascular. Simultáneamente se iniciará el estudio microbiológico en las
22

muestras de producto transfundido, paciente, equipo de transfusión, etc. Nodebe olvidarse la notificación urgente al Servicio de transfusión y éste al Cen-tro de transfusión, con la finalidad de retirar y/o reclamar aquellos productosprocedentes de la misma unidad.
Actuar como en la reacción hemolítica aguda, y administrar una combina-ción de antibióticos que sean activos contra el espectro de bacterias que pue-den estar implicadas. En ausencia de la valoración de un experto enMicrobiología, en general se considera apropiado seguir el protocolo local deantibioticoterapia para sepsis en pacientes neutropénicos. Si éste no está dis-ponible, una combinación de los siguientes antibióticos se considera activacontra bacterias Gram positivas y Gram negativas:
Sobrecarga circulatoria
Existe el riesgo de provocar una sobrecarga circulatoria con velocidades detransfusión superiores a 2-4 ml/kg /hora, sobre todo en pacientes ancianos,con anemia crónica (con volumen plasmático normal o aumentado) y en pa-cientes con funciones cardiacas o renales comprometidas. El riesgo de sobre-carga circulatoria es extremadamente alto en la administración de solucionesde albúmina al 20%.
Sintomatología
Los signos y síntomas de sobrecarga son los de la insuficiencia ventricular iz-quierda aguda e incluyen: disnea, taquipnea, tos no productiva, elevación de lapresión venosa central, crepitantes bibasales, hipertensión, taquicardia…, En de-finitiva, todo el cortejo de signos de la insuficiencia cardiaca congestiva aguda.
23
Bacterias Gram NegativasPiperacilina/tazobactam 4,5 g /12 horas EV, oCeftriaxona 1g /día EV ( 2 g si infección grave), o Meropenem 1g /12h EV
Bacterias Gram PositivasTeicoplanina 400 mg EV /12h x 2 dosis, y continuar con 1 dosis diaria (no nefrotóxico)Vancomicina 1g/12h EV y seguidamente ajustar según monitorización niveles sanguí-neos. Es igualmente efectivo, pero añade posible nefrotoxicidad.Ceftriaxona i Teicoplanina tienen la ventaja de una única dosis diaria y menor nefrotoxicidad.

Tratamiento
La interrupción inmediata de la transfusión, además tratamiento posturaljunto con la administración de oxígeno y diuréticos. En las siguientes trans-fusiones se realizarán lentamente o mediante alícuotas de una unidad.
Los pacientes con anemia crónica se hallan generalmente en situación de nor-movolemia o hipervolemia, y pueden presentar signos de insuficiencia cardíacapreviamente a la infusión de la transfusión. En estos pacientes es aconsejable:
� Administrar una dosis de diurético (furosemida 20 – 40 mg) después de cadaunidad transfundida y monitorizar con frecuencia sus constantes vitales.
� Restringir el ritmo transfusional a una unidad de hematíes cada 12 horas,suele reducir el riesgo de fallo ventricular izquierdo.
Hemólisis no inmune
Existen diversas situaciones capaces de provocar la hemólisis de hematíes deldonante o del receptor durante el acto transfusional, y cuyo origen no es in-mune: hemólisis mecánica por ciertas válvulas cardiacas o circulación extra-corpórea, la infusión de soluciones hipotónicas o determinadas medicacionesen la vía de transfusión, el calentamiento excesivo de los hematíes, contami-nación bacteriana de la unidad de sangre, etc.
Sintomatología
No hay clínica asociada a esta hemólisis, salvo en el caso de la contaminación bac-teriana, la primera manifestación suele ser la emisión de orinas obscuras, hemo-globinuria, y la presencia de hemoglobinemia, que alertan de la posible hemólisisintravascular. Posteriormente se producirá un aumento de la bilirrubina sérica.
Tratamiento
Parar inmediatamente la transfusión, investigar la causa de la hemólisis. El diag-nóstico diferencial con las reacciones hemolíticas agudas de origen inmunedebe quedar confirmado lo antes posible para instaurar el tratamiento urgente.
Reacciones hipotensivas
Se las ha relacionado con la generación de citocinas (generalmente bradici-nina) durante la filtración de componentes sanguíneos celulares en la cabe-
24

cera del enfermo, especialmente si éste está recibiendo tratamiento con fár-macos inhibidores del enzima convertidor de la angiotensina. Debido a lacorta vida media de la bradicinina, estas reacciones no se observan cuando laleucorreducción es realizada pre-almacenamiento.
Sintomatología
Cuadro de hipotensión sistólica y/o diastólica agudo al poco de inicio de latransfusión. Se suele acompañar de síntomas o signos respiratorios (disneay/o hipoxemia) y un tercio de los casos presentan manifestaciones alérgicas(urticaria, prurito, eritema facial).
Tratamiento
Habitualmente con parar la transfusión es suficiente, revirtiendo rápidamente, de noser así se instaurará tratamiento con fluidoterapia e incluso aminas vasoactivas.
COMPLICACIONES RETARDADAS
DE ORIGEN INMUNOLÓGICO
Reacción hemolítica retardada
La transfusión de hematíes puede inducir la formación de anticuerposcontra antígenos eritrocitarios después de días (respuesta anamnésica auna inmunización previa) o semanas (inmunización primaria) de la trans-fusión. El riesgo de sensibilización por cada unidad transfundida a antíge-nos eritrocitarios (exceptuando el antígeno Rh D) es entre 1-2%. Lareacción de estos anticuerpos con los hematíes recientemente transfun-didos puede producir una reacción hemolítica de carácter extravascular,que rara vez compromete la vida del paciente, o precisa tratamiento desoporte.
Sintomatología
La inmunización primaria pocas veces produce hemólisis de los hematíestransfundidos y por lo tanto no se suele acompañar de ninguna sintoma-tología clínica. En la respuesta anamnésica a una inmunización previa, losdatos clínicos más frecuentes son febrícula, malestar general, ligera icteri-cia a los 3 a 7 días de la transfusión, lo que hace difícil su asociación con
25

la transfusión. La sospecha diagnóstica se produce ante una caída inexpli-cable de la Hb con aparición de una prueba de antiglobulina directa posi-tiva y elevación de LDH o bilirrubina. El escrutinio de anticuerposirregulares suele ser entonces positivo .
Tratamiento
No precisa habitualmen -te otro tratamiento queel sintomático.
Aloinmunización frente a antígenos eritrocitarios, plaquetarios, leu-cocitarios o de proteínas plasmáticas
La inmunización puede evidenciarse tiempo después de la transfusión, y ge-neralmente sin sintomatología clínica. Si posteriormente se administran com-ponentes portadores de los antígenos correspondientes, puede provocarse unacortamiento de la vida media de los mismos acompañado, o no, de clínicageneral.
Sintomatología
No hay sintomatología clínica acompañante en el momento de la aloinmu-nización.
Tratamiento
Solamente los anticuerpos contra antígenos eritrocitarios pueden detectarseen las pruebas de compatibilidad pretransfusional ordinarias. Para posteriorestransfusiones, éstas se deberán realizar con componentes sanguíneos caren-tes del antígeno contra el que va dirigido el /los anticuerpo/s.
Púrpura postransfusional
La púrpura postransfusional (PPT) se manifiesta por un descenso brusco deplaquetas, después de una transfusión, en un paciente con sensibilización pre-
26
Reacción transfusional hemolítica retardada:
Signos y síntomas
Presentación frecuente� Anemia� Fiebre� Ictericia
Presentación excepcional� Hemoglobinura� Hemoglobinemia� Shock� Oliguria o anuria

via, por transfusión o gestación. Casi siempre se trata de mujeres multíparas,en las que se produce una brusca respuesta anamnésica dirigida frente el an-tígeno de alta frecuencia plaquetar HPA-1 (conocido formalmente como PL-A1). Con mucha menos frecuencia se han visto implicados otros antígenos,diferentes del HPA-1 en casos de PPT.
El anticuerpo, paradójicamente, se comporta, como si fuera un autoanticuerpo,destruyendo tanto las plaquetas transfundidas HPA-1-positivo como las del pa-ciente, que son HPA-1-negativo. El mecanismo por el que ocurre es poco cono-cido, una hipótesis es que en el plasma de la unidad transfundida hay antígenoHPA-1, que se adsorbe sobre las plaquetas del receptor. Otros mecanismos su-geridos son la existencia de complejos inmunes que se adhieren a las plaquetasantígeno-negativos, dando lugar a una destrucción acelerada por el sistema re-ticuloendotelial, ó la producción precoz, en la respuesta inmune, de autoanti-cuerpos o aloanticuerpos con reacción cruzada con las plaquetas autólogas.
Sintomatología
La aparición de trombopenia, muchas veces acompañada de púrpura pe-tequial, en los 3-10 días siguientes a la transfusión de concentrado de he-matíes o plaquetas. En un cuadro de PPT los niveles de plaquetas puedenser tan bajos como 10.000-20.000/μL y, pueden persistir durante semanas.
Tratamiento
El sintomático de la plaquetopenia. Como las plaquetas autólogas del re-ceptor, que son HPA-1-negativas son destruidas, lo lógico sería pensar quesi se transfunden plaquetas HPA-1-negativas también serán rápidamentedestruidas, esto es realmente lo que sucede. Sin embargo, la transfusión, enpacientes con trombopenia severa por PPT, de plaquetas HPA-1-negativasjunto a la administración de gammaglobulinas endovenosas se ha mostradoeficaz.
Para evitar nuevas estimulaciones de la respuesta inmune del paciente, seríarecomendable, evitar las transfusiones que contengan plasma en pacientescon PPT. Por ello si se ha de transfundir concentrados de hematíes, se han delavar, para retirar el antígeno plaquetar soluble y fragmentos de membranasde plaquetas residuales.
27

A pesar de que el hecho de desplasmatizar o lavar las unidades de plaquetas,disminuye la efectividad de las plaquetas a transfundir, si el paciente está in-tensamente trombopénico es una alternativa.
Enfermedad de Injerto contra huésped postransfusional (EICH-PT)
Se trata de una complicación, casi siempre fatal originada por la transfusiónde linfocitos T viables a pacientes con una inmunodepresión intensa (recep-tores de progenitores hematopoyéticos, transfusión intrauterina, enfermedadde Hodgkin, etc.) o receptores inmunocompetentes que comparten algún ha-plotipo con el donante (familiares en primer o segundo grado, o pacientestransfundidos con productos HLA compatibles seleccionados). Los linfocitostransfundidos injertan y proliferan, atacando diversos órganos y tejidos del re-ceptor (piel, intestino, hígado, bazo y médula ósea principalmente).
Sintomatología
El cuadro clínico comienza unos días después de la transfusión (entre 10 y 15)con fiebre, diarrea, erupción cutánea, alteraciones de la analítica hepática ypancitopenia.
Tratamiento
Los tratamientos ensayados se han mostrado ineficaces, por lo que la pre-vención de su aparición en pacientes susceptibles es imprescindible. Ésta serealiza mediante la transfusión de componentes celulares sometidos a irra-diación gamma, a dosis no inferiores a 25 Gy, en pacientes inmunocompro-metidos o bien en pacientes que reciben transfusión de familiares de primero segundo grado (debido a que comparten un haplotipo HLA).
Inmunomodulación
La transfusión de componentes sanguíneos puede originar una disregulaciónde la inmunidad celular, y ello está asociado, en parte, con la infusión de leu-cocitos y sus productos (IL-4, IL-10, TGF-1).
Sintomatología
Cuando la transfusión se sigue de un estado de hiporrespuesta o inmunoto-lerancia antigénica puede tener implicaciones en mecanismos que dependen
28

de la respuesta inmune normal como son el crecimiento tumoral y el des-arrollo de infecciones o procesos autoinmunes.
Tratamiento
Será preventivo. En la medida que se eliminen los leucocitos de los diferen-tes componentes sanguíneos se podrá controlar estos eventos inmunes.
DE ORIGEN NO INMUNOLÓGICO
Transmisión de agentes infecciosos
Todas las donaciones son analizadas para la detección de agentes infecciososcomo la hepatitis B, hepatitis C, VIH 1 y 2, y sífilis. A pesar de las pruebas re-alizadas (serología y detección genómica) existe un riesgo de transmisión deenfermedades infecciosas por la donación. El riesgo estimado de transmisiónde VHC es uno por casi dos millones cuatrocientas mil donaciones(1/2381000), el de VIH (1/806000) es casi tres veces superior al de VHC, y esaun más alto para el VHB (1/177000).
Hemosiderosis inducida por transfusión.
En pacientes que requieren transfusiones de concentrados de hematíes demanera continuada y durante largos períodos de tiempo, se produce acumulode hierro y puede desarrollarse una hemosiderosis. Una unidad de concen-trado de hematíes contiene unos 250 mg de hierro y después de múltiplestransfusiones, la sobrecarga de hierro del organismo puede llegar a ser dehasta 100g.
En general, a partir de la transfusión de 20 unidades de hematíes hay riesgode hemosiderosis secundaria. Debe monitorizarse la ferritina sérica y valorarel riesgo/beneficio del tratamiento quelante (subcutáneo u oral)
Sintomatología
El hierro se acumula en el corazón, el hígado y otros órganos, siendo princi-palmente preocupante el desarrollo de una miocardiopatía. La determinaciónperiódica del nivel de ferritina sérica, permite realizar un seguimiento precisodel hierro acumulado.
29

Tratamiento
Requiere un tratamiento especializado. Para su prevención y en el caso dedesarrollo para su tratamiento es útil la administración de quelantes del hie-rro, como la desferrioxamina, con o sin vitamina C, por vía subcutanea, quefavorece la eliminación urinaria del hierro, o los nuevos quelantes orales (des-ferasirox). También se puede utilizar la realización de sangrías terapéuticas,en caso de normalización de la cifra de hemoglobina.
REFERENCIASHandbook of Transfusion Medicine. DBL McClelland. United Kindom Blood Services.4th edition 2007. ISBN-10 011 322677 2. www.tshop.co.ukSerious hazards of transfusion (SHOT) Annual report 2008. http://www.shotuk.orgPathogen Reduction: A Precautionary Principle Paradigm. Harvey J. Alter TransfusionMedicine Reviews, Vol 22, No 2 (April), 2008: pp 97-102.Alvarez M. Actualización del riesgo residual de transmisión de enfermedades infeccio-sas en la era de las técnicas NAT. SETS. 20 Congreso Nacional de la Sociedad Españolade Transfusión Sanguínea y Terapia Celular. Tarragona junio de 2009.
Casamitjana N, Piron M, Bes M, Cerezo B, Gon-zález A, Puig L, Sauleda S. Riesgo residual deVIH-1, VHC y VHB en donaciones en periodoeclipse. SETS. XXI Congreso Nacional de la So-ciedad Española de Transfusión Sanguínea yTerapia Celular. Valladolid, junio de 2009. j
30
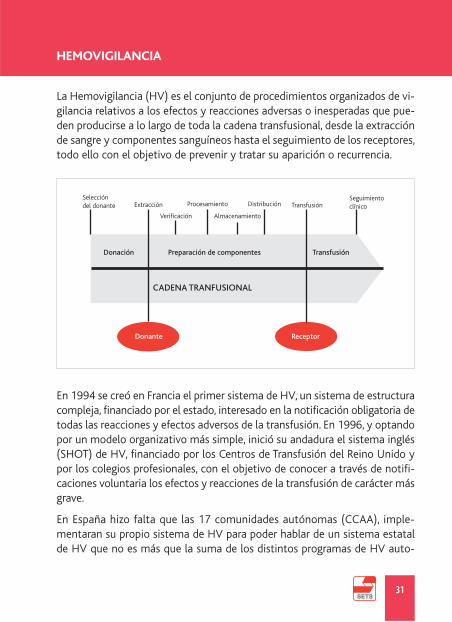
La Hemovigilancia (HV) es el conjunto de procedimientos organizados de vi-gilancia relativos a los efectos y reacciones adversas o inesperadas que pue-den producirse a lo largo de toda la cadena transfusional, desde la extracciónde sangre y componentes sanguíneos hasta el seguimiento de los receptores,todo ello con el objetivo de prevenir y tratar su aparición o recurrencia.
En 1994 se creó en Francia el primer sistema de HV, un sistema de estructuracompleja, financiado por el estado, interesado en la notificación obligatoria detodas las reacciones y efectos adversos de la transfusión. En 1996, y optandopor un modelo organizativo más simple, inició su andadura el sistema inglés(SHOT) de HV, financiado por los Centros de Transfusión del Reino Unido ypor los colegios profesionales, con el objetivo de conocer a través de notifi-caciones voluntaria los efectos y reacciones de la transfusión de carácter másgrave.
En España hizo falta que las 17 comunidades autónomas (CCAA), imple-mentaran su propio sistema de HV para poder hablar de un sistema estatalde HV que no es más que la suma de los distintos programas de HV auto-
31
HEMOVIGILANCIA
Seleccióndel donante Extracción
Donación
Donante Receptor
Preparación de componentes
CADENA TRANFUSIONAL
Transfusión
Verificación
Procesamiento
Almacenamiento
Distribución TransfusiónSeguimientoclínico

nómicos. El Ministerio de Sanidad, como autoridad competente en Europa,ha venido coordinando los distintos programas, elaborando un informe anualy proporcionando a la Comisión Europea la información que regularmente sele solicita. Aunque con un grado de implantación y desarrollo desigual, en elaño 2004 quedó constituido el sistema estatal de HV y, hoy en día, los pro-cedimientos de HV constituyen una herramienta integrada en el conjunto deactividades desarrolladas por los Centros y Servicios de transfusión hospita-larios.
La aparición de la Directiva 2002/98/CE que fija las normas de calidad y se-guridad de la sangre y de los componentes sanguíneos y, muy especialmente,la entrada en vigor de la Directiva 2005/61/CE, también llamada coloquial-mente Directiva de HV, que regula la notificación de las reacciones y efectosadversos de la transfusión, así como la trazabilidad de los componentes san-guíneos, han sido dos elementos clave para impulsar el desarrollo de la HV yacelerar la creación de sistemas de HV en los países miembros de la comu-nidad europea (CE). El cambio más sobresaliente que introducen ambas di-rectivas es la obligatoriedad de que los países de la comunidad dispongan deun sistema de HV y, por ende, el carácter obligatorio de las notificaciones.
32

Gracias a este impulso legal, a finales de 2005, los países de más larga tra-yectoria en la CE ya disponían de un sistema de HV. Y en el momento actual,ya es el conjunto de los 27 países miembros de la comunidad quien disponede su propio sistema de HV. No obstante, la estructura de los diferentes pro-gramas y, sobretodo, el espectro de las complicaciones abarcadas por cadauno de ellos carece de la homogeneidad necesaria para hablar de un autén-tico sistema europeo de HV.
Desde su creación a finales de los años noventa, la Red Europea de HV (EHN,European Haemovigilance Network) ha desarrollado una importante labor afavor del desarrollo de la HV en Europa. Fundada por iniciativa de profesio-nales interesados en esta temática, la EHN ha sido durante años la estructuraaglutinante en Europa de toda la información generada por los distintos sis-temas de HV. Los objetivos perseguidos por esta organización han sido losde promover el desarrollo de la HV, centralizar las notificaciones graves decarácter urgente, conseguir la homogeneización progresiva de los diferentesprogramas, e intercambiar experiencias entre los países miembros. A partir de2010 esta red, europea en origen, se ha convertido en una red de ámbito in-ternacional, pasándose a denominar IHN (International Haemovigilance Net-work).
Una de las aportaciones más relevantes de la EHN ha sido la iniciativa de es-tablecer unas definiciones comunes para las reacciones y efectos adversos dela transfusión, de la donación y del procesamiento de la sangre. Esta tarea se hallevado a cabo con la colaboración y el apoyo de la ISBT (International Societyof Blood Transfusion), a través de su Grupo de trabajo en Hemovigilancia.
Las complicaciones relacionadas con la donación de sangre ya han sido ela-boradas, validadas y aprobadas. El documento original (“Standard for survei-llance of complications related to blood donation”) puede ser consultado enla página web de la IHN y de la ISBT (www.ISBT-web.org / www.EHN-org.net).
Las reacciones y efectos adversos de la transfusión se encuentran pendientesde aprobación en el momento de redactar este capítulo, pero es previsibleque estén disponibles en el curso de 2010.
Finalmente, las definiciones en torno a las incidencias que pueden producirsedurante el procesamiento de la sangre, la preparación y el suministro de los
33

componentes sanguíneos, han sido cedidas para su elaboración a la propiaComisión Europea, ya que este tipo de incidentes tocan de pleno en lo que po-demos considerar la “calidad y la seguridad de los componentes sanguíneos”que es competencia exclusiva de dicha Comisión.
En el contexto de esta Guía cabe hablar de forma preferente de los efectos yreacciones adversas que se producen en el tramo final de la cadena transfu-sional, dentro del ámbito hospitalario y, más concretamente, en la cabeceradel paciente.
LA HEMOVIGILANCIA EN ESPAÑA
El Real Decreto 1088/2005 es la normativa estatal que recoge la transposi-ción de la Directiva Europea 2002/98/CE y, posteriormente, la OrdenSCO/322/2007 recoge la transposición de la Directiva Europea 2005/61/CE.Al igual que en el resto de países europeos esta normativa ha sido decisivapara la implantación y desarrollo del sistema estatal de HV.
El sistema estatal de HV se estructura en tres niveles:
� Nivel 1: Servicios y Centros de Transfusión. La persona responsable deHV en cada Centro y Servicio de transfusión hospitalario es la encargada
34

de efectuar las notificaciones de efectos y reacciones adversas, mediantelos formularios elaborados por el Ministerio de Sanidad, y de remitirlos alcoordinador autonómico de HV.
� Nivel 2: Nivel autonómico. El coordinador autonómico de HV es quien re-cibe las notificaciones y las transmite al Ministerio. Asimismo, es respon-sable de elaborar un informe anual de HV que debe remitir a todos losCentros y Servicios de transfusión hospitalarios. Es la figura que actúa comoenlace entre la comunidad autónoma y el Ministerio.
� Nivel 3: Nivel Estatal. La unidad de HV del Ministerio es la encargada decoordinar la actividad de HV de las diferentes comunidades autónomas yde transmitir a la Comisión Europea cuanta información sea solicitada entorno a esta temática.
DOCUMENTACIÓN
Los documentos elaborados por el grupo de trabajo de HV del Ministerio deSanidad para la notificación de efectos y reacciones adversas son los si-guientes:
35

Formularios de notificación
� Notificación de reacción transfusional. Es el único formulario de carácter“interno” para su uso exclusivo en todos los centros en los que se efectúauna transfusión.
� Notificación de un incidente relacionado con la transfusión.� Notificación de un incidente relacionado con la donación.� Notificación de un incidente relacionado con la preparación de compo-
nentes sanguíneos.
Cuestionarios
Un total de 11, relativos a los principales efectos y reacciones adversas de latransfusión en los que se recogen con detalle las características clínicas de cadacomplicación, los estudios necesarios para efectuar el diagnóstico y una valo-ración del grado de gravedad e imputabilidad de la complicación observada.
� Reacciones hemolíticas agudas y retardadas.� Reacción febril y/o hipotensiva.� Reacción alérgica/anafiláctica.� Edema pulmonar: cardiogénico y no cardiogénico.� Púrpura postransfusional.� Enfermedad del injerto contra el huésped asociada a transfusión. � Contaminación bacteriana.� Infección postransfusional vírica. � Hemosiderosis transfusional.� Errores en la administración de componentes sanguíneos.� Incidentes sin efecto/casi incidentes.
Información mínima necesaria en las notificaciones.
La información incluida en las notificaciones debe de cumplir con los criterios deconfidencialidad establecidos por el sistema de HV y la normativa legal vigente.
El paciente
La identificación del paciente debe incluir al menos la fecha de nacimiento,el sexo, y un número de identificación exclusivo. Deben consignarse los prin-cipales signos y síntomas de los diversos efectos adversos conocidos, las prue-
36

bas de laboratorio realizadas para documentar la reacción y, finalmente, laevolución de paciente.
El componente
Debe aportarse una prescripción detallada del componente implicado que in-cluya el número de unidad y el código relativo a la donación y al donante. Sedebe indicar el tipo de componente (hematíes, plaquetas, plasma, otros), eltipo de donación del que procede el componente (de sangre total, de afére-sis), otras características (irradiado, desplasmatizado), condiciones y duracióndel almacenamiento antes de la transfusión.
Gravedad
Se expresa con arreglo a la siguiente escala de grados de gravedad:
� (0): Ausencia de signos y síntomas. � (1): Signos inmediatos sin riesgo vital para el paciente y resolución total de
la complicación.� (2): Signos inmediatos con riesgo vital.� (3): Morbilidad de larga duración.� (4): Muerte del paciente.� (NC): No constan datos relativos a la gravedad, o no se han podido recabar.
Imputabilidad
Es la probabilidad de que una reacción en el paciente pueda atribuirse a latransfusión de un determinado componente sanguíneo. Debe establecerse elgrado de relación existente entre el efecto adverso observado y el compo-nente transfundido con arreglo a una escala de grados de imputabilidad:
� (0): “Sin relación”. El efecto adverso observado está aparentemente rela-cionado con la transfusión, pero hay evidencia de que el componente noes el responsable.
� (1): “Posible”. El efecto adverso observado está aparentemente relacionadocon la transfusión, pero podría ser, o no, debido a otra causa distinta a latransfusión.
� (2): “Probable”. El efecto adverso observado no parece explicable por otracausa distinta a la transfusión.
37

� (3): “Seguro”. Se ha probado que el efecto adverso observado se debe opuede ser muy probablemente debido a la transfusión.
� (NC): “No consta”. No constan datos relativos a la imputabilidad en la no-tificación, o no se han podido recabar.
� (NE): “No evaluable”. Los datos son insuficientes para evaluar la imputabi-lidad.
Medidas correctoras y preventivas
Además de implementar las medidas correctoras y preventivas que el sistemade HV autonómico, estatal o europeo vaya dictando, cada Centro y cada Ser-vicio de transfusión debe de establecer las suyas propias, de manera que la no-tificación del efecto adverso o de la reacción adversa observada conduzca ala instauración de una medida inmediata de carácter corrector o preventivo.
Se recomienda que las medidas correctoras o preventivas adoptadas quedenregistradas junto al formulario correspondiente y que regularmente se llevea cabo un seguimiento y análisis de su grado de cumplimiento y eficacia.
El comité hospitalario de transfusión puede tener asignada, entre otras muchas, lafunción de implementar medidas correctoras y preventivas, así como el seguimientode su grado de cumplimiento y eficacia. La existencia de un comité hospitalario detransfusión activo fortalece el sistema de HV, y avala el trabajo realizado y los pro-cedimientos implementados por el Servicio de transfusión hospitalario.
Trazabilidad
Se entiende por Trazabilidad, la capacidad para identificar al receptor de cadacomponente sanguíneo y, a la inversa, a todos los donantes que han interve-nido en la transfusión de un determinado paciente. La trazabilidad no quedagarantizada por el hecho de conocer el destinatario teórico de un compo-nente sanguíneo, sino que es necesaria la confirmación en el punto de des-tino conforme el paciente ha sido finalmente transfundido con el componenteprevisto para él. Igualmente, en el caso de que el paciente sufra algún tipo decomplicación, ésta debe ser registrada y notificada de inmediato al Serviciode transfusión.
En definitiva, la HV exige que la información relativa al acto transfusional ya las posibles incidencias que pudieran producirse durante el mismo, se co-
38

muniquen sistemáticamente al Servicio de transfusión. Y, aún más, que el Ser-vicio de transfusión esté informado del destino final de cada componentesanguíneo.
Alerta rápida
Un aspecto de interés adicional de los sistemas de HV es la “alerta rápida”.Se trata del circuito a seguir para la comunicación rápida de aquellos efectoso reacciones indeseables que puedan afectar a más de un donante o recep-tor, a fin de actuar, en cada caso, con la máxima celeridad y eficacia. Un ejem-plo paradigmático de la necesidad de emplear este circuito corresponde a loscasos de receptores que han adquirido una infección supuestamente trans-mitida por un componente sanguíneo que nos obliga a localizar a los restan-tes receptores o a aquellos componentes sanguíneos que todavía no sehubieran transfundido En otro contexto, los problemas, probados o poten-ciales, asociados a los materiales y equipos empleados en el procesamientode los componentes sanguíneos también podrían ser motivo de una alerta rá-pida que permitiera la transmisión inmediata de esta información a los Cen-tros que pudieran estar utilizando los mismos equipos y, así, proceder a laretirada provisional de los mismos.
Información adicional
Se recomienda que cada Servicio de transfusión hospitalario establezca la es-trategia necesaria para conocer con exactitud el número de componentes san-guíneos transfundidos anualmente, de manera que pueda llegar a conocersela prevalencia de incidentes y efectos adversos por unidades transfundidas.
Igualmente se recomienda que cada Servicio de transfusión disponga de una“Guía de uso e indicación de componentes sanguíneos”, de manera que loscriterios empleados para indicar la transfusión de componentes sanguíneosvengan avalados por la Guía de consenso entre los diferentes profesionales in-volucrados en la transfusión de sangre y componentes sanguíneos.
Los riesgos actuales de la transfusión sanguínea y la Hemovigilancia
Desde los primeros informes anuales de HV emitidos por Francia y el ReinoUnido se evidenció que los riesgos actuales de la transfusión sanguínea estánasociados principalmente a las reacciones transfusionales de mecanismo in-
39

mune y a los errores en la administración de los componentes sanguíneos(EAC). En el primer grupo, la lesión pulmonar aguda asociada a la transfusión(LPA-AT), o TRALI (Transfusion related acute lung injury) según la terminolo-gía anglosajona, se ha impuesto como una de las complicaciones más gravesde la transfusión, con tasas de morbilidad y mortalidad muy elevadas en todoslos países. En el grupo de EAC se ha comprobado que la deficiente, insufi-ciente o negligente identificación de los pacientes en el momento de la ex-tracción y/o en el de administración del componente constituye la principalcausa de error. Lejos de la percepción ciudadana del riesgo, todavía ligada alas enfermedades transmisibles por transfusión, año tras año, la HV europeaviene demostrando que las complicaciones más graves y frecuentes de latransfusión sanguínea se producen en el último tramo de la cadena transfu-sional, en el ámbito hospitalario y, más concretamente, en la cabecera delpaciente.
El programa inglés SHOT, en una reciente revisión, refiere algunos de los re-sultados obtenidos tras la implementación de diferentes medidas destinadasa reducir o evitar las principales complicaciones de la transfusión. En el casode los EAC, desde el año 2004 se exige que el personal que transfunde en elReino Unido disponga de un certificado oficial que acredite su competenciatras realizar un curso de formación y entrenamiento de práctica transfusio-nal que incluye las nociones necesarias para efectuar una extracción correctade las muestras y una administración segura de los componentes sanguíneos.Por otra parte, los errores de identificación se han ido reduciendo a medidaque se han ido implementando sistemas de identificación basados funda-mentalmente en el código de barras. Ambas medidas han resultado especial-mente efectivas reduciendo el número de errores de grupo ABO y el de lasreacciones hemolíticas subsiguientes. El uso de plasma para transfusión ex-clusivamente procedente de donantes masculinos no transfundidos tambiénha implicado una notable reducción del número de casos de LPA-AT, con 22casos anuales en 2003, 13 en 2004 y tan sólo 6 en 2005.
La HV nos brinda la oportunidad de conocer cuáles son las complicaciones dela transfusión sanguínea en cada momento, cuál es el nivel de calidad y se-guridad de nuestros componentes, y cuál el grado de seguridad de nuestrosprocedimientos de trabajo. La implementación de medidas correctoras y pre-
40

ventivas, y el posterior análisis de la eficacia de las mismas es una parte fun-damental e ineludible para retroalimentar el sistema de HV y para conven-cer a los diversos profesionales sanitarios que intervienen en la prescripción,preparación y administración de la sangre, de la importancia de su contribu-ción al sistema de HV.
Después de más de quince años de HV en Europa, el balance es positivo, perotodavía no se han alcanzado plenamente algunas de las metas que nos habí-amos propuesto. La trazabilidad total de los componentes sanguíneos no esun tema bien resuelto, ni que todos interpreten por igual y, probablemente,para conseguir una trazabilidad total será necesario un nuevo cambio, el quesupone pasar de una HV pasiva a una HV activa que nos lleve sistemática-mente a la cabecera del paciente para controlar cada transfusión y para co-nocer, de forma inequívoca, el destino final de cada componente sanguíneo.
Finalmente, y a la luz de la información suministrada por todos los sistemasde HV vigentes, a través de sus informes anuales, nos queda por alcanzar elreto más importante, el de conseguir que la transfusión sanguínea adquiera,como mínimo, el grado de calidad y seguridad que ya hemos conseguido connuestros componentes sanguíneos.
REFERENCIASH Andreu G, Morel P, Forestier F et al. Haemovigilance network in France: organisationand analysis of immediate transfusion incident reports from 1994 to 1998. Transfusion2002; 42: 1356-1364.Directive 2002/98/CE setting standards of quality and safety for the collection, tes-ting, processing, storage and distribution of human blood and blood components. Of-ficial J Eur Communities, 8/2/2003.Muñiz-Díaz E. Los riesgos de la transfusión en su punto justo. Boletín de la SETS2003(3):1-2.Love E, Jones H, Williamsom LM. SHOT: A voluntary system for the reporting of serioushazards of transfusion in the UK. TATM 2003; 5:249-255.Faber JC. Work of the European Haemovigilance Network (EHN). Transfusion CliniqueBiologique 2004; 11:2-10.Directive 2005/61/EC as regards traceability requirements and notification of seriuosadverse reactions and events. Official J Eur Communities, 1/10/2005.Martín MªP. Informe anual 2004. Programa Estatal de Hemovigilancia. Boletín de la
41

SETS 2005; Nº 57: 17-24.Hemovigilancia. Estándares de acreditación en transfusión sanguínea. Asociación Es-pañola de Hematología y Hemoterapia. Sociedad Española de Transfusión Sanguínea.Editorial: Acción Médica. Madrid. 3ª Edición 2006: 103-110.Robillard P. Hemovigilance: A review of world data. TATM 2006;8:10-14.Stainsby D, Jones H, Asher D, Atterbury C, Bonicelli A, Brant L et al. Serious hazards oftransfusions: A decade of Hemovigilance in the UK. Transfus Med Rev 2006; 20:273-282.Quaranta JF, Canivet N, Courbil R, Raucoles-Aimé M. Transfusion safety and haemovi-gilance committees. Transfusion Clinique et biologique 2007; 14:107-111.Stainsby D. Haemovigilance, not just a register. The impact of transfusion safety initia-tives in the UK. ISBT Science Series 2007; 2: 189-193.Faber JC. Revue des principaux systems d’hémovigilance dans le monde. Transfus ClinBiol 2009;16:86-92. Council of Europe. Guide to the preparation, use and quality assurance of blood com-ponents. Ed: Council of Europe Publishing. Estrasburgo. 15ª Edición, 2009: 137-48, 299-300.Aynob Y. Hemovigilance in developing countries. Biologicals 2010;38:91-96. �
42

CONCENTRADO DE HEMATÍES
Los concentrados de hematíes (CH) proceden habitualmente de donacionesde sangre total o bien, ocasionalmente, de sangre procesada con un separa-dor celular (donación por aféresis, eritroaféresis).
Actualmente, en la gran mayoría de los Centros de transfusión de nuestro país,se procede a la eliminación de la mayoría de los leucocitos (leucorreducción)presentes en los componentes sanguíneos (CS) celulares, lo que mejora la ca-lidad, reduce el riesgo de inmunización frente a antígenos leucocitarios (HLA),y disminuye la transmisión de virus intracelulares (p. ej. CMV).
Los constituyentes activos de los CH son los hematíes morfológica y funcio-nalmente intactos. Otros constituyentes como el plasma, las plaquetas y leu-cocitos residuales, carecen de efecto terapéutico propio y no influyen en laeficacia terapéutica de los CH.
CONCENTRADO DE HEMATÍES POBRE EN LEUCOCITOS EN SOLU-CIÓN ADITIVA (CH-SAGM).
Es el CH estándar en nuestro país. Se obtiene a partir de una donación de450 ± 45 ml, mediante la eliminación de la mayoría de los leucocitos, pla-quetas y plasma, con posterior resuspensión de los hematíes en unos 100ml de una solución conservadora (SAG-Manitol). La reducción de los leu-cocitos residuales se consigue mediante filtración. El hematocrito resul-tante de este concentrado de hematíes se sitúa sobre un 55%, con uncontenido de Hemoglobina (Hb) superior a los 40g. El volumen aproxi-mado es de 280 ± 50 ml.
CONCENTRADO DE HEMATÍES POBRE EN LEUCOCITOS LAVADO.
Se trata un CH-SAGM del que se han eliminado las proteínas plasmáti-cas mediante repetidos lavados de los hematíes con suero fisiológico es-téril.
43
TRANSFUSIÓN DE LOS DIFERENTES COMPONENTES SANGUÍNEOS
TRANSFUSIÓN EN LOS PACIENTES ADULTOS

CONCENTRADO DE HEMATÍES POBRE EN LEUCOCITOS EN SOLU-CIÓN ADITIVA IRRADIADOS.
Es un CH-SAGM sometido a irradiación a dosis de 25 – 50 Gy, con la finali-dad de inactivar los linfocitos T, responsables de la Enfermedad del InjertoContra el Huésped Postransfusional (EICH-PT). El único efecto desfavorable dela irradiación sobre los CH es la hiperkaliemia debido a la acelerada salida delpotasio intraeritrocitario que se acumula en el sobrenadante. Este efecto esde poca importancia en las transfusiones normales en adultos pero puedecausar graves problemas casos de transfusión masiva, especialmente en niños(TIU y exanguinotransfusión).
CONSERVACIÓN
Los CH- SAGM deben ser almacenados a una temperatura entre 2-6º C ytransportados a una temperatura comprendida entre 1-10º C. La fecha decaducidad es la que consta en la etiqueta del componente.
El almacenamiento de los hematíes fuera del organismo causa complejos cam-bios, en conjunto conocidos como “lesión de almacenamiento”. Entre otros, tallesión incluye cambios morfológicos (p.ej. desarrollo de esferocitos y equinoci-tos), alteraciones funcionales (p.ej. disminución de la concentración del 2,3-di-fosfoglicerato (2,3-DPG) con el consiguiente aumento de la afinidad de la Hb porel oxígeno, pérdida de deformabilidad de los hematíes, etc.). Una parte de estoscambios revierten en las 48-72 horas siguientes a la transfusión. Actualmentesu importancia en términos de oxigenación tisular no es bien conocida.
INDICACIONES
La transfusión de concentrado de hematíes (TCH) está indicada cuando se requierealcanzar un rápido aumento en la capacidad de transporte de oxígeno de la sangrey, como consecuencia, del aporte de oxígeno a los tejidos. El objetivo es evitar, o tra-tar, una anemia hipóxica antes de que se produzcan lesiones irreversibles.
En cualquier paciente con anemia, aguda o crónica, el médico que lo atiendedebe intentar identificar la causa y tratarla si es posible. La TCH solo está jus-tificada si la salud del enfermo puede verse gravemente comprometida porla hipoxia anémica y no existe una terapéutica alternativa.
44

ANEMIA AGUDA
Anemia aguda por hemorragia
La principal estrategia terapéutica en el tratamiento de la hemorragia agudaes prevenir, o corregir, el shock hipovolémico, junto con el rápido control dela hemorragia. Para asegurar la oxigenación tisular es esencial restaurar el vo-lumen circulatorio infundiendo soluciones cristaloides / coloides en cantida-des suficientes para mantener la presión de perfusión arterial. Una vezalcanzada y conservada la normovolemia, los mecanismos fisiológicos decompensación pueden mantener un aporte de oxígeno a los tejidos normalcon concentraciones de Hb tan bajas como 6 g/dl.
En pacientes normovolémicos y con anemia los síntomas y signos clínicos quepueden indicar una oxigenación tisular insuficiente (desencadenantes fisioló-gicos de transfusión) son los que se muestran en la tabla (Tabla 3).
45
Tabla 3
Síntomas y signos clínicos indicativos de oxigenación tisular insuficiente en pacien-tes normovolémicos y anemia� Oxigenación tisular global insuficiente:
– Inestabilidad hemodinámica: Hipotensión relativa (TA media <70-80% de la basal) ytaquicardia relativa (120-130% de la basal).
– Incremento de la fracción de extracción de oxigeno (%) >50%.– Saturación de oxigeno en sangre venosa mezclada (%) (SvO2) <50%– Presión parcial de oxigeno en sangre venosa mezclada (PvO2) (mmHg) < 32 mmHg– Caída en la saturación de oxígeno venoso central <60%– Disminución del consumo de oxígeno (%) > 10% del valor inicial– Aumento del lactato sérico (>2 mmol/L) y acidosis metabólica (déficit de base).
� Oxigenación tisular regional insuficiente: Puede conducir a isquemia miocárdica o ahipotensión y taquicardia, incluso en casos de normovolemia. – La isquemia miocárdica puede ser detectada mediante ECG por:
• Depresión reciente del segmento ST >0,1 mV• Elevación reciente del segmento ST > 0,2 mV durante más de 1 minuto.• Trastornos del ritmo recientes• Anomalías de la contracción miocárdica regional recientes (ECG transesofágica, ECG
transtorácica).

Oxigenación tisular global insuficiente
En pacientes en shock hemorrágico puede estar indicada la TCH desde la faseinicial de la reanimación. El Manual “Advanced Trauma Life Support (ATLS)” delAmerican College of Surgeons, incluye la temprana administración, empírica,de TCH en pacientes con shock hemorrágico, que no han respondido a la re-animación con 2 litros de solución cristaloide.
La TCH inicial puede estar indicada también en pacientes con hemorragiaaguda e inestabilidad hemodinámica, en los que persiste la hemorragia oexiste algún signo de oxigenación insuficiente (p.ej. aumento del lactato enplasma y acidosis).
En casos con hemorragia masiva incontrolada puede ser necesario adminis-trar ya desde el inicio de la reanimación, además de CH, plasma y plaquetas(“paquetes transfusionales”) siguiendo un protocolo estricto, con el objetivode mantener la coagulación sanguínea dentro de límites normales.
Necesidad de transfusión basada en la concentración de hemoglobina(Hb) y/o del hematocrito (Htc)
Estos parámetros no son apropiados para estimar las pérdidas y la necesidad detransfusión en los pacientes con hemorragia aguda, ya que los valores de Hb/Htcpueden ser normales hasta que se haya restablecido la volemia, aunque la masaeritrocitaria esté disminuida. La decisión de transfundir CH debe basarse en el es-tado del volumen intravascular del paciente, las evidencias de shock, la duración eintensidad de la anemia, y los parámetros fisiológicos cardiopulmonares (Tabla 4).
Anemia perioperatoria.
Se aplican criterios similares a los descritos para la hemorragia (Tabla 4).
� Fase preoperatoria. La presencia de anemia preoperatoria es un marcador decomorbilidad y constituye un factor de riesgo independiente de mortalidady de complicaciones postoperatorias graves. El hallazgo de una anemia du-rante la visita preoperatoria debe conducir a la búsqueda de las causas y asu tratamiento específico (hierro, vitamina B12, ácido fólico, Eritropoyetina).
� Fase intraoperatoria. En condiciones de anestesia y de vigilancia continua,se ha demostrado que Hb de 6-7 g/dl (Htc 18-22%) son aceptables en pa-
46

cientes sin comorbilidades graves, y que pacientes de edad avanzada, co-ronarios o con valvulopatías mitrales y aórticas graves toleran bien unaanemia moderada (Hb entre 8,5-10 g/dL; Htc entre 25-30%).
� Fase postoperatoria. En un paciente joven, estable, normovolémico y sin evi-dencias de sangrado es difícil justificar TCH con niveles de Hb superiores a 7-8g/dl. Sin embargo, en pacientes con enfermedad vascular cerebral o coronaria,insuficiencia respiratoria, sepsis, etc., y en pacientes con inestabilidad hemodi-námica es más prudente mantener una Hb entre 9-10 g/dl (Htc entre 27-30%).
Pacientes bajo cuidados intensivos (UCI)
Una estrategia transfusional “restrictiva” (TCH cuando la Hb < 7g/dl, con elobjetivo de mantener la Hb entre 7-9 g/dl) es tan efectiva como una estra-tegia transfusional “liberal” (THC cuando la Hb < 10 g/dl, para mantener laHb del paciente entre 10 y 12 g/dl) en pacientes de UCI hemodinámicamente
47
Tabla 4
Decisión de transfusión de CH en pacientes con hemorragia aguda según el nivel dehemoglobina� < 7 g/dl:
– Está justificada la transfusión. Valores más bajos pueden ser bien tolerados, pero me-recen valoración individual.
� Entre 7 y 8 g/dl:– Con síntomas o signos de anemia hipóxica (Tabla D-1): Deben ser transfundidos. – Hemodinámicamente estables, sin previsión de nueva hemorragia y sin factores de
riesgo asociados (enfermedad arterial coronaria, insuficiencia cardíaca, insuficienciavascular cerebral, etc): NO deben ser transfundidos.
� Entre 8 y 10 g/dL:– Con factores de riesgo asociados (enfermedad arterial coronaria, insuficiencia cardíaca,
insuficiencia vascular cerebral, etc): Deben ser transfundidos. – Con síntomas o signos de anemia hipóxica (Tabla D-1): Deben ser transfundidos.
� > 10 g/dl:– Con infarto de miocardio agudo o angor inestable: Deben ser transfundidos– Con síntomas o signos de anemia hipóxica (Tabla D-1): Deben ser transfundidos– En situación de Hemorragia incontrolada masiva: Pueden ser transfundidos (efecto
beneficioso de hematocritos más elevados sobre la hemostasia primaria).– Resto de situaciones: No deben ser transfundidos.

estables, excepto en pacientes con Infarto agudo de miocardio o angina in-estable en que es aconsejable una Hb más elevada.
En los pacientes que requieren ventilación mecánica y en traumatizadosgraves reanimados considerar la TCH si la Hb < 7 g/dl, Estos pacientes no sebenefician de una estrategia transfusional liberal.
La TCH puede ser beneficiosa en pacientes con síndrome coronario agudoque están anémicos (Hb ″ 8 g/dl) al ingreso en el Hospital.
En caso de sepsis la necesidad de transfusión de cada paciente séptico debevalorada individualmente debido a que no se conoce cuál es el umbral trans-fusional óptimo y no hay pruebas claras de que la transfusión mejore la oxi-genación tisular. Algunos autores recomiendan mantener una Hb entre8-10g/dl.
Los pacientes con alto riesgo de / o con Lesión Pulmonar Aguda y Síndromede Ditress Respiratorio Agudo se recomienda efectuar todos los esfuerzosposibles para evitar la TCH.
En caso de lesiones y enfermedades neurológicas no se ha visto que los pa-cientes con lesión cerebral traumática, moderada o grave, se beneficien deuna estrategia transfusional “liberal”. En pacientes con Hemorragia Sub-
48
Tabla 5
Signos desencadenantes de transfusión fisiológicosParámetro Intraoperatorio - Sala
UCI convencionalHipotensión relativa Si SiTaquicardia relativa Si SiDepresión reciente del segmento ST >0,1mV Si SiElevación reciente del segmento ST > 0,2 mV Si SiAnomalías recientes en la contracción miocárdica Si SiPresión parcial de oxígeno en sangre venosa mezclada (mmHg). <32 No se aplicaFracción de extracción de oxigeno (%) >40 No se aplicaSaturación de oxigeno en sangre venosa mezclada (SvO2) (%) <60 No se aplicaDisminución del consumo de oxígeno (%) >10 No se aplica
aracnoidea no se conoce cuál es el umbral de transfusión óptimo y no haypruebas claras de que la TCH se asocie con mejores resultados.
En términos generales y en el caso de pacientes que mantienen la normo-volemia, se puede considerar la TCH si está presente alguno de los criteriossobre el nivel de Hb, criterios circulatorios, o una de las variables de oxigena-ción. Una Hb por debajo del umbral indicado permite la TCH (tablas 5 y 6).
ANEMIA CRÓNICA
La anemia crónica se instaura a lo largo de varias semanas o meses, dandotiempo a que se desarrollen mecanismos de compensación que aseguran, encondiciones normales, una oxigenación tisular adecuada. La reserva de la ca-pacidad de aporte de oxígeno es tal que el gasto cardíaco, generalmente, noaumenta hasta que la Hb es < 7 g/dl. Por ello, no es raro que estos pacienteslleguen a cifras muy bajas de Hb (5 g/dl) sin apenas sintomatología.
La decisión de administrar o no CH debe estar basada en la etiopatogenia dela anemia, la valoración del cuadro clínico global y la concentración de la Hb.Si existe tratamiento específico, éste será el de elección. La TCH estará indi-cada, únicamente, cuando no se pueda esperar a que el tratamiento específicohaga efecto, bien por la gravedad de la sintomatología anémica que presentael paciente, bien por la urgencia de una intervención quirúrgica pendiente.
En pacientes con anemia crónica sin tratamiento específico que requierentransfusiones periódicas, se deben mantener la cifra de Hb pretransfusional
49
Tabla 6
Decisión de transfusión de CH en pacientes en situación mantenida de normovole-mia según el nivel de hemoglobina (g/dl)
Parámetro Intraoperatorio - SalaUCI convencional
En todos los pacientes 6-7 7-8Pacientes de > 80 años 7-8 8-9Pacientes con enfermedad arterial coronaria 8 8-9Paciente con insuficiencia cardiaca congestiva 8 8-9Paciente con SvO2<90% 8-9 9Fiebre / hipermetabolismo 7-8 8-9

justo por encima del nivel de Hb que no se asocia con síntomas de anemia.Este nivel es muy variable dependiendo de la edad del paciente, actividad ycoexistencia de comorbilidades cardiovasculares o pulmonares.
En pacientes con anemia crónica sin patología cardiovascular, la TCH no estáindicada mientras la Hb no descienda por debajo de 7-8 g/dl y no produzcasíntomas clínicos. Cuando se asocian anomalías en la función cardiovascularo respiratoria, la TCH puede estar indicada con Hb > 8 g/dl.
ß Talasemia mayor
El régimen transfusional debe mantener una Hb pretransfusional de 9,5-10g/dl. Las TCH se administran cada 2-3 semanas. Con ello se suprime la eri-tropoyesis e inhibe la absorción gastrointestinal aumentada de hierro. Todoslos pacientes necesitan tratamiento con quelantes de hierro.
Anemia drepanocítica (anemia falciforme)
En el paciente adulto. El tratamiento transfusional de estos pacientes es com-plejo y exige experiencia. Es recomendable buscar el asesoramiento de un es-pecialista en la enfermedad. Los pacientes con HbSS suelen tener Hb de 7-9g/dl con buena tolerancia. La TCH sólo está indicada en caso de mala tole-rancia (insuficiencia cardíaca, disnea, hipotensión y astenia intensa) y en au-sencia de regeneración. Es fundamental evitar transfusiones innecesariasdebido a los riesgos de aloinmunización, reacciones hemolíticas postransfu-sionales y sobrecarga de hierro, y debe procurarse que la Hb postransfusionalno supere los 10 g/dl para evitar complicaciones por hiperviscosidad. En elpreoperatorio de cirugía con anestesia general de estos pacientes, la transfu-sión simple para elevar la Hb a 10 g/dl, es igual de eficaz que la eritroaféresispara reducir la HbS por debajo del 30%.
Anemia hemolítica autoinmune
Con frecuencia, las pruebas de compatibilidad pretransfusional dan un resul-tado positivo debido a la presencia de autoanticuerpos en el plasma del pa-ciente, pero ello no debe impedir que el paciente se transfunda si lo precisa.La indicación de la TCH se basa en la agudeza del comienzo, la rapidez deprogresión de la anemia, y la gravedad de los síntomas y signos clínicos cau-sados por la anemia. La presencia de hipoxemia grave (en general con Hb ″ 5
50

g/dl pero que puede aparecer con Hb superiores), indica TCH urgente, pero nohay que esperar a que aparezcan estos síntomas para transfundir. Se aconsejatransfundir pequeños volúmenes de hematíes, suficientes para evitar la hi-poxemia, repetidos cada vez que sean necesarios.
INDICACIONES DE LOS CONCENTRADOS DE HEMATÍES LAVADOS
Pacientes que presentan repetidas reacciones alérgicas causadas por anti-cuerpos dirigidos contra las proteínas plasmáticas.
INDICACIONES DE LOS CONCENTRADOS DE HEMATÍES IRRADIADOS
Ver apartado “COMPONENTES SANGUÍNEOS IRRADIADOS”
DOSIS
Una dosis de 4 ml/kg (equivalente a 1 unidad de CH /70 kg peso receptor) ele-vará, como media, la Hb, en unos 0,8 g/dl en un varón de 70 kg. La recupera-ción real puede ser valorada a partir de los 15 minutos de finalizar latransfusión, haciendo una determinación de Hb al receptor.
En muchas ocasiones, un solo concentrado de hematíes es suficiente para ali-viar la sintomatología del paciente y proseguir con el tratamiento etiológico.
ADMINISTRACIÓN
Antes de comenzar la transfusión el médico que la ha prescrito debe recabarel Consentimiento Informado del paciente.
Para la infusión deben usarse equipos de infusión específicos, provistos de unfiltro de 170-200 micrones.
Debe respetarse la compatibilidad de grupo ABO entre los hematíes del do-nante y los anticuerpos circulantes del receptor. Excepto en casos de extremaurgencia, se requiere la realización de pruebas de compatibilidad serológicaentre donante y receptor.
Antes de iniciar la transfusión se verificará la identificación correcta y co-rrespondencia del receptor y la unidad a administrar. ¡Este es uno de los pun-tos clave para asegurar una transfusión segura! Además, se recomienda
51

recomprobar el grupo ABO del paciente mediante una nueva muestra de san-gre extraída en la cabecera del receptor.
La transfusión se comenzará lentamente, a un ritmo de 10 gotas por minuto,vigilando la aparición de posibles efectos adversos durante los primeros 5 a10 minutos. La velocidad de infusión se mantendrá posteriormente a la ve-locidad que tolere la situación cardiovascular del paciente. Habitualmente,para un adulto sin disfunciones cardiovasculares una unidad de concentradode hematíes se transfundirá en 90 a 120 minutos. Dado que para la mayoríade los equipos de transfusión la equivalencia se sitúa aproximadamente en 15gotas por 1 ml, ello supone un ritmo de infusión de 40 a 50 gotas por minuto.
Se ha de evitar el alargamiento del tiempo de transfusión, excepto en pa-cientes que no toleran bien el aumento de volumen (oligoanúricos, cardió-patas, edad avanzada o anemia severa). En estos la transfusión debe ser máslenta (p. ej. 1 ml de CH/kg peso del receptor/hora), con cuidadoso control he-modinámico, sin superar las 4 horas. En algunos pacientes es adecuado ad-ministrar un diurético (p.ej. furosemida 20 a 40 mg por vía oral).
En el curso de una hemorragia masiva puede ser necesario administrar 1 CH, omás, cada 5-10 minutos, con adecuado control clínico y hemodinámico. En estassituaciones es necesario calentar hasta 37ºC todos los líquidos infundidos paraevitar la hipotermia, incluidos los CS, utilizando equipos especialmente diseñadospara ello.
El paciente debe ser adecuadamente controlado durante y después de latransfusión.
REFERENCIASNapolitano LM, Kurek S, Luchette FA, Corwin HL, et al. Clinical practice guideline: Red bloodcell transfusion in adult trauma and critical care. Crit Care Med 2009; 37(12): 3124-3157.So-Osman C, Nelissen R, Te-Slaa R, Coene L, Brand A. A randomized comparison oftransfusion triggers in elective orthopaedic surgery using leukocyte-depleted red bloodcells. Vox Sang 2010;98:56-64Spahn DR, Dettori N, Kocian R, Chassot PC Transfusion in the cardiac patient. Crit. CareClin, 2004; 20:269-279.Vallet B, Adamczyk S, Barreu O, Lebuffe G. Physiologic transfusion triggers. Best PractRes.Clin Anaesthesiol 2007;21:173-181.
52

Madjdpour C, Spahn DR, Weiskopf RB. Anemia and operative red blood cell transfusion:a matter of tolerance. Crit Care Med 2006; 34(5) suppl: S102-S108.Pattel MS, Carson JL. Anemia in the Perioperative Patient. Anaesthesiology Clin 2009;27:751-760.Practice guidelines for preoperative blood transfusion and adjuvant Therapies: an up-dated report by the American Society of Anesthesiologists Task Force on PerioperativeBlood Transfusion and Adjuvant Therapies. Anesthesiology 2006; 105:198-208.Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill:a systematicreview of the literature. Crit Care Med 2008; 36:2667-2674.Pape A, Stein P, Horn O, Habler O. Clinical Evidence of blood transfusion effectiveness.Blood Transfusion 2009; 7:250-258.Wang JK, Klein HG. Red blood cell transfusion in the treatment and management ofanemia: the search for the elusive transfusion trigger. Vox Sang 2010; 98(1):2-11.Rossaint Rolf, Bouillon B, Cerny V, Coats TJ, Duranteau J, Martinez-Mondejar E, Hunt BJ,Komadina R el al. Management of bleeding following major trauma: an updated Euro-pean guidelines. Crit. Care 2010, 14(2) R52. http://ccforum.com/content/14/2/R52Leal-Noval SR, Muñoz-Gomez M,; Murillo- Cabezas F. Optimal hemoglobin concentra-tion in patients with subarachnoid hemorrhage, acute ischemic stroke and traumaticbrain injury. Cuur Opin Crit Care 2008; 14:156-162.Dillinger RP, Levy MM, Carlet JM, Bion J, Parker MM; et al. for the International Survi-ving Sepsis Campaign Guidelines Committee. Surviving Sepsis Campaign: internationalguidelines for management of severe sepsis and septic shock:2008. Crit Care Med2008:38:296-327.Porter J. Blood transfusion: quality and safety issues in thalassemia, basic requirementsand new trends. Hemoglobin 2009; 33(S1):S28-S36.:Foss NB, Kristensen MT, Jensen PS, Palm H, Krasheninnikoff M, Kehlet H. The effectsof liberal versus restrictive transfusion thresholds on ambulation after hip fracture sur-gery. Transfusion 2009; 49: 227-234.Lionnet F, Arlet JB, Bartolucci P, Habibi A, Ribeil JA; Stankovic K, pour le group de re-commendations el d´etude de la drepanocytose de l´adulte. Recommendations prac-tiques practiques de prise en charge de la drepanocytose de l´adulte. Rev Med Interne2009; 30S: S162-S223.Garcia Bueno MJ. Tratamiento transfusional en la enfermedad falciforme. En, Ricard MPy Villegas A Guía de manejo de las enfermedades falciformes.. AEHH 2009 pag 189-198.Schiferer A, Panzer S, Reesink HW, Baulig W, Belisle S, Gerrard C et al. Red cell transfu-sion in elective cardiac surgery patients Vox Sang 2009: 97(2) 172-182.Harris AM, Atterbury CLJ, Chaffe B, Elliot C, Hawkins T et al. British Committee for Standarsin Haematology. British Society for Haematology. “Guidelines on the Administration ofBlood Components”. www.bschguidelines/com/pdf/Admin_blood_components050110.pdf
53

Hebert Pc, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinicaltrial of transfusion requirements in critical care. Transfusion Requirements in CriticalCare Investigators. Canadian Critical Care Trials Group. N Engl J Med 1999; 340: 409-417.Liumbrano G, Bennardello F, Lattanzio A, Piccoli P, Rossetti G. Recommendations forthe transfusion of red blood cells. Blood Transfusion 2009; 7:49-64.Sihler KC, Napolitano LM. Massive transfusion: new insights. Chest 2009; 136(6): 1654-1667.
CONCENTRADO DE PLAQUETAS
Las plaquetas son elementos sanguíneos esenciales para el control de las he-morragias. Se considera normal una cifra de 125 a 300 x109/L.
Las plaquetas disponibles para transfusión se obtienen por dos métodos di-ferentes:
Concentrados de plaquetas obtenidos a partir de donaciones de sangre total
Dependiendo del tipo de fraccionamiento realizado pueden encontrarse enforma:
1. Individual: obtenidas desde el plasma rico en plaquetas o la capa leucopla-quetar de una única donación de sangre total. Contienen entre 0,45-0,80x1011 plaquetas suspendidas en un volumen de plasma que varía entre 50y 70 ml. Para obtener una dosis terapéutica adecuada es necesario mezclar1 concentrado individual por cada 10 kg de peso de receptor.
2. Mezcla: la mayoría de los centros de transfusiones de nuestro país utilizaneste método. Tras una centrifugación fuerte de la unidad de sangre total, seobtiene la capa leucoplaquetaria que contiene la mayoría de las plaquetasy leucocitos, esta se resuspende, bien en plasma o en una solución aditivapara plaquetas (precisan un 30% de plasma), y se vuelve a centrifugar paraeliminar restos de hematíes y leucocitos. Mezclando de 4 a 6 de estos com-ponentes, mediante dispositivos estériles, conseguimos una unidad tera-péutica de plaquetas, con un contenido mínimo de 2,5 x1011 plaquetas yun volumen total de 250-300 ml.
54

Plaquetoaféresis
Son concentrados de plaquetas obtenidos de un único donante medianteprocedimientos de aféresis. Deben contener más de 2,5 x 1011 plaquetas, sus-pendidas en unos 250 ml. de plasma o solución aditiva.
Ambos productos son terapéuticamente equivalentes, tanto en el aumento deplaquetas como en la eficacia hemostática, los efectos secundarios produci-dos por ambas son similares. La plaquetoaféresis tiene la ventaja de una ex-posición a un menor número de donantes.
Actualmente en la mayoría de los Centros de transfusión españoles, se pro-cede a la leucorreducción pre-almacenamiento de todos los componentessanguíneos celulares, resultando un contenido leucocitario inferior al millónde elementos por unidad.
CONSERVACIÓN
Independientemente del método de obtención los concentrados de plaque-tas se almacenan a 22° C (±2° C) en agitación continua, durante un máximode 5 días.
Este periodo puede ampliarse a 7 días si se combina con un sistema de de-tección o reducción de la contaminación bacteriana.
INDICACIONES
Los concentrados de plaquetas se transfunden para prevenir o tratar hemo-rragias en pacientes con defectos cualitativos, cuantitativos, o ambos, de lasplaquetas.
Transfusión profiláctica:
Su indicación se basa en el recuento de plaquetas y en otros datos clínicos delpaciente.
En pacientes estables con trombopenia central es infrecuente que se pro-duzca una hemorragia espontanea grave con plaquetas superiores a 10 x109/l,siendo este el nivel aceptado para la transfusión en estas situaciones. Incluso
55

en pacientes con trombopenia crónicas de larga evolución (síndrome mielo-displásico, anemia aplásica), sin antecedentes hemorrágicos graves (≥ grado2 de la OMS, Tabla 8) y fuera de tratamiento activo, y con el fin de evitar oretrasar la aparición de refractariedad y otros posibles efectos secundarios,se aceptan niveles < 5 x109/l plaquetas.
Es fundamental la monitorización de los pacientes para la detección precozde los síntomas y signos que indican un aumento del riesgo hemorrágico, yen estos casos incrementar el nivel de transfusión a 20 x109/l. Estos factoresde riesgo hemorrágico son: fiebre > 38,5ºC, sepsis, mucositis ≥2, descensobrusco de la cifra de plaquetas (50%) en 24 horas, hipertensión arterial nocontrolada y alteraciones concomitantes de la hemostasia (Coagulación in-travascular diseminada, fibrinólisis, tratamiento anticoagulante, etc).
En la profilaxis para cirugía y otros procedimientos invasivos (biopsia hepá-tica, endoscopia con biopsia, colocación catéter venoso central, punción lum-bar, etc.) la cifra de plaquetas por debajo de la cual se recomienda transfundires 50x109/l, aunque realmente no existen ensayos aleatorizados que permitanestablecer una guía basada en evidencias científicas, y estas están basadas en re-comendaciones de expertos. En estas situaciones no solo hay que tener encuenta la cifra de plaquetas sino también el riesgo global de hemorragia rela-cionada con el tipo y duración de la cirugía, la capacidad para controlar la he-morragia intraoperatoria, las posibles consecuencias de una hemorragia nocontrolada y la presencia de factores que puedan afectar a la función plaquetar.
En caso de intervenciones en territorios en los cuales, incluso pequeñas pér-didas hemáticas pueden tener consecuencias graves, como por ejemplo el
56
Concentrados de plaquetas: Transfusión profiláctica en pacientes adultos
� Contraindicación: En púrpura trombótica trombocitopénica, trombopenia inducida porheparina, en trombopenia inmune (PTI y púrpura postransfusional).
� Indicación: Según cifra de plaquetas y situación clínica. Transfusión en:– <10 x 109/l (<5 x 109/l en trombopenia estable de larga evolución)– <20 x 109/l y factor de riesgo (infección grave, anticoagulación,…)– <50 x 109/l y procedimiento invasivo o hemorragia– <100 x 109/l y cirugía SNC o globo ocular

sistema nervioso central o el globo ocular, se recomienda transfundir si elrecuento plaquetario es inferior a 100 x109/l.
En general la transfusión profiláctica de plaquetas se reserva para los pacientes quepresentan un defecto en la producción medular y muy raramente se necesita enlas trombocitopenias secundarias a un aumento de la destrucción como en lapúrpura trombocitopénica autoinmune. Dichas transfusiones están relativamentecontraindicadas en los pacientes afectos de púrpura trombótica trombocitopénicao de trombocitopenia inducida por la heparina, debido al riesgo potencial de con-tribuir a la aparición de fenómenos trombóticos y deben quedar reservadas paraaquellas situaciones en las que exista una hemorragia con riesgo vital.
Transfusión terapéutica
La transfusión terapéutica de plaquetas se realiza cuando existe una altera-ción cuantitativa, cualitativa o ambas de las plaquetas y el paciente presentauna hemorragia atribuible al defecto plaquetario. En ausencia de otros de-fectos funcionales, se recomienda transfundir plaquetas si existe hemorragiay el recuento de plaquetas es inferior a 50x109/l.
En la transfusión masiva y en pacientes politraumatizados, en los que se es-pera cambios continuos y muy rápidos no solo en la cifra de plaquetas sinotambién en los factores de la coagulación, la recomendación es manteneruna cifra de plaquetas > 75 x109/l. Mientras que en pacientes con Coagula-ción intravascular diseminada la actitud ante la presencia de hemorragia ytrombopenia, tras tratar la causa de la misma y corregir los defectos de la co-agulación, será mantener una cifra > 50x109/l plaquetas.
Una situación especial lo constituye la cirugía cardiopulmonar con bombaextracorpórea, donde se produce una trombopenia dilucional y una altera-ción funcional transitoria de las plaquetas. La transfusión profiláctica se ha de-mostrado ineficaz para disminuir la incidencia y gravedad de los episodioshemorrágicos. Esta debe quedar reservada para aquellos pacientes con cifrasbajas y que tras finalizar la intervención continúen con hemorragia micro-vascular no atribuible a la cirugía ni a alteraciones de la hemostasia.
Hoy día, existen cada vez más datos, de que la transfusión terapéutica tras he-morragia grado 2, independientemente de la cifra de plaquetas, en pacientescon trombopenia esperada de corta duración (por ejemplo, en trasplante au-
57

tólogo de progenitores), estables y en monitorización continua, no incrementala mortalidad ni la aparición de hemorragias grado 3 y 4, comparada con latransfusión profiláctica. Esta opción permite disminuir de forma considerableel uso de plaquetas.
58
Escala de la OMS para definir la gravedad de las hemorragia
Grado 0• ninguna
Grado 1 (Hemorragia menor)• petequias/equimosis• epixtasis/hemorragia orofaringea <1 hora• hemorragia oculta en heces (de trazas a +1)• hemoglobinuria (de trazas a +1)• hemorragia retiniana sin alteración de la visión• hemorragia vaginal mínima
Grado 2 (Hemorragia moderada)• melenas, hematemesis, hemoptisis, hematuria, hematoquecia o hemorragia vaginal que
no requiera transfusión de hematíes• epixtasis/hemorragia orofaringea >1 hora• hemorragia oculta en heces (superior a +2)• hemoglobinuria (superior a +2)
Grado 3 (Hemorragia grave)• melenas, hematemesis, hemoptisis, hematuria, hematoquecia o hemorragia vaginal que
requiere transfusión de hematíes• hemorragia del SNC detectado por TAC, sin consecuencias clínicas• hemorragia en los sitios de punción o inserción de catéteres que requiere transfusión de
hematíes
Grado 4 (Hemorragia incapacitante)• hemorragia retiniana con reducción de la visión• hemorragia del SNC con signos y síntomas neurológicos• hemorragia en órganos vitales (pulmón, intrapericardio)• hemorragia masiva con compromiso hemodinámico• hemorragia fatal independientemente de la localización

DOSIS
Aunque la dosis profiláctica óptima todavía no está bien establecida, para unadulto la dosis habitual oscila entre 0,5-0,8 x 1011 plaquetas por cada 10 kgde peso. Esto corresponde, en la práctica, a una unidad de plaquetoaféresis oa una mezcla de 4 a 6 unidades, siempre con contenidos superiores a 2,5x1011 plaquetas. En el caso de los pacientes pediátricos la dosis recomendadaes de 0,5 x1011 plaquetas por cada 10 Kg peso, o aproximadamente 10 ml/kgde peso.
En condiciones normales, la transfusión a un adulto de una dosis terapéuticade plaquetas obtenida de donaciones de sangre total o bien por plaquetoa-féresis, causa un aumento en el recuento de unas 30 a 50 x109/L plaquetasque puede ser valorado realizando un recuento plaquetario entre los 10 y 60minutos después de finalizar la transfusión.
El rendimiento de la transfusión de plaquetas puede calcularse de forma másexacta mediante el cálculo del incremento corregido del recuento (ICR):
ICR = (Recuento post transfusión – Recuento pre transfusión) (x109/l) x Su-perficie corporal (en m2) / Plaquetas transfundidas (x1011)
Si repetidamente el ICR a la hora de la transfusión de un concentrado de pla-quetas de obtención reciente y ABO compatible es inferior a 7,5 x109/l o a las18 horas es inferior a 4,5 x 109/l, el paciente se considerará refractario a lastransfusiones de plaquetas.
ADMINISTRACIÓN
No se precisan pruebas de compatibilidad eritrocitaria si el contenido de he-matíes del concentrado de plaquetas es inferior a 2 ml. Si las plaquetas estánconservadas en plasma o para la transfusión en neonatos, es conveniente quela transfusión sea ABO compatible entre el plasma del donante y los hema-tíes del receptor.
Para prevenir la aloinmunización Rh, tras la transfusión de plaquetas Rh (D) po-sitivas a mujeres en edad fértil Rh (D) negativas, se recomienda la administra-ción de una dosis de inmunoglobulina anti-D (50 μg), dicha profilaxis permitehasta 5 administraciones de plaquetas Rh incompatibles durante 6 semanas.
59

La transfusión debe realizarse a través de un filtro de 170-200 μm. Se realizarátan rápidamente como sea tolerada por el receptor, por lo general entre 20 y 30minutos. Dado que la mayoría de los equipos de transfusión la equivalencia sesitúa en 15 gotas = 1 ml, ello supone un ritmo de infusión de 125 a 225 gotaspor minuto. En caso de riesgo de sobrecarga hídrica, se enlentecerá el ritmo deinfusión. En cualquier caso el tiempo de transfusión nunca excederá de 4 horaspara reducir el riesgo de contaminación bacteriana del producto.
REFRACTARIEDAD A LA TRANSFUSIÓN DE PLAQUETAS
Se define como refractariedad la ausencia de repuesta a las transfusiones, de-terminada como al menos dos ICR < 7,5 x109/l a la hora. En el 72-88% de loscasos, ésta es debida a factores no inmunes, mientras que en el 25-39% de loscasos es debido a aloinmunización, fundamentalmente por anticuerpos anti-HLA y menos frecuente por anticuerpos antiplaquetarios específicos. La com-patibilidad ABO de la transfusión de plaquetas también es importante, ya que elincremento de títulos anti-A o anti-B se asocia con disminución del rendimiento.
Entre las causas habituales de refractariedad no inmune están la esplenome-galia, infección, hemorragia, CID y la administración de ciertos fármacos entrelos que destacan la anfotericina, vancomicina, ciprofloxacilo y la heparina.
Es fundamental la profilaxis de la misma mediante la leucorreducción de loscomponentes sanguíneos y la transfusión adecuada a la indicación.
El manejo del paciente con refractariedad aloinmune es complicado, y re-quiere un centro especializado. Se proponen varias estrategias:
� Transfusión terapéutica (hemorragia > grado 2 OMS)� Transfusión ABO compatible� Transfusión de productos frescos� Uso de plaquetas compatibles:
– Donantes HLA compatibles: requiere la tipificación HLA de muchos do-nantes para encontrar compatibles.
– Donantes HLA parcialmente compatibles: amplia el número de donantesal agrupar antígenos HLA-A y B que comparten epítopos comunes(CREGs, grupos con reactividad cruzada)
60

– HLA Matchmaker: selecciona según un algoritmo donantes HLA compa-tibles, basado en que solo tres aminoácidos (tripletes) constituyen loscomponentes críticos de los epítopos alo-reactivos del HLA.
� Prueba cruzada para plaquetas y selección de no reactivas.
Los dos últimos métodos se han demostrado igualmente útiles, con respues-tas adecuadas en un 70-80% de los pacientes.
Para aquellos pacientes con HLA infrecuentes o con pobre respuesta se haintentado la utilización de inmunosupresores: inmunoglobulinas intraveno-sas, ciclosporina A, vinblastina entre otras.
En caso de hemorragia grave, se puede utilizar la transfusión de dosis bajas deplaquetas aumentando la frecuencia de administración, asociar inhibidoresde la fibrinólisis e incluso utilizar el factor VII recombinante que se ha de-mostrado eficaz en algunos trabajos, aunque su uso no está aprobado paraesta indicación.
REFERENCIASComité de Acreditación en Transfusión (CAT). Estándares de acreditación. AsociaciónEspañola de Hematología y Hemoterapia. Sociedad Española de Transfusión Sanguí-nea. Ind. Gráf. El Instalador. Madrid. 3ª Edición 2006.Council of Europe. Guide to the preparation, use and quality assurance of blood com-ponents. Ed: Council of Europe Publishing. Estrasburgo. 13ª Edición, 2009. Andreu G, Vasse J,Tardivel R, Semana G. Platelet transfusion: Products, indications, dose,threshold and efficacy. Transfus Clin Biol 2009; 16: 118-133.Cid J, Ramiro L, Escoda L, Llorente A. Efficacy of transfusion of platelet concentratesobtained by manual pooling or by semiautomated pooling of buffy-coats: a retros-pective analysis of count increment, corrected count increment and transfusion inter-val. Vox Sang 2009; 96:29–33.Stroncek DF, Rebulla P: Platelet transfusions. The Lancet 2007; 370: 427-438.Want H, Schaefer-Eckart K, Frank M, Birkmann J, Wilhelm M. A therapeutic platelettransfusion strategy is safe and feasible in patients after autologous peripheral bloodstem cell transplantation. Bone Marrow Transplant 2006;37:387–92.Ronald A, Thomas S, Charles A, Laurence A, Arthur W, Ira A. Management of patient re-fractory to platelet transfusion. Arch. Pathol. Lab. Med. 2003; 127: 406-414.Vidarsson B, Ondundarson PT. Recombinant factor IIa for bleeding in refractory throm-bocytopenia. Throm Haemost 2000;83:634-635.
61

PLASMA
El Plasma Fresco Congelado (PFC) es el componente sanguíneo obtenido biena partir de una unidad de sangre total tras la separación de los hematíes, omediante aféresis, y congelado idealmente en las 8 horas postcolecta.
El volumen del plasma obtenido a partir de la donación de sangre total de 450ml. es de 200-300 ml. El volumen de una donación de plasmaféresis puedeser de 300-600 ml. Contiene todos los factores plasmáticos de la coagulación,incluidos los factores lábiles (Factor V y Factor VIII).
En España es preceptivo, para aumentar la seguridad relativa a transmisión deenfermedades infecciosas, que el PFC sea inactivado para patógenos a travésde métodos de inactivación autorizados, o bien que se mantenga en “cua-rentena” por un periodo 4 meses, hasta que la negatividad del donante paramarcadores de enfermedades infecciosas en una nueva donación, permita li-berar el mismo para transfusión. Puede conservarse hasta 36 meses, depen-diendo del procedimiento de extracción, procesamiento y temperatura deconservación utilizado.
El PFC es la fuente fundamental de derivados plasmáticos: concentrados defactores de la coagulación, albúmina, inmunoglobulinas, etc. La mayoría delplasma obtenido de las donaciones se utiliza con este fin.
CONSIDERACIONES PREVIAS A LA PRESCRIPCIÓN DE PLASMA
No debe prescribirse plasma antes de conocer: hemograma, Razón Normali-zada Internacional (INR), Tiempo Parcial de Tromboplastina (TPT) y fibrinó-geno del paciente.
La prescripción debe incluir el diagnóstico clínico en el que se basa la solici-tud de plasma, y la dosis estimada en función del objetivo a conseguir.
Debe tratarse la causa, si se conoce, de los resultados anómalos de la coagu-lación, incluso aunque tenga que realizarse la transfusión de plasma.
La transfusión de plasma no debe basarse en los resultados anómalos de la-boratorio (INR, TPT), sino en el riesgo y consecuencias de la hemorragia en elpaciente individual.
62

La hemostasia es satisfactoria con niveles de factores de coagulación del 20-30% de lo normal, y con concentración de fibrinógeno ≥ 100 mg/dl.
No hay relación directa entre hemorragia y tests anormales de coagulación,aunque la hemorragia es más probable si el INR y el TPT superan en 1,5 vecesel límite superior del rango de referencia.
Debe controlarse la eficacia postransfusional.
INDICACIONES
Deficiencias de un único factor plasmático de la coagulación
Generalmente en coagulopatias congénitas y específicamente en déficit defactor V (Grado de recomendación C; nivel de evidencia IV).
El uso de PFC en las deficiencias congénitas de otros factores solo se justificasi no se dispone del concentrado específico del factor.
En hemorragias por deficiencias de múltiples factores y/o Coagula-ción Intravascular Diseminada (CID)
El plasma fresco congelado debe usarse solamente cuando además de lasanomalías de la coagulación hay hemorragia activa.
63
Niveles de evidencia sobre la eficacia de tratamientos (U.S. Preventive Services Task Force)
Ia Evidencia obtenida de metanálisis de ensayos controlados aleatorizados.
Ib Evidencia obtenida de al menos un ensayo controlado aleatorizado.
IIa Evidencia obtenida de al menos un estudio controlado bien diseñado, sin aleatorización.
IIb Evidencia obtenida de al menos un estudio “quasi experimental” bien diseñado.
III Evidencia obtenida de estudios descriptivos, no experimentales, bien diseñados, tales
como estudios comparativos, estudios de correlación y estudios de casos.
IV Evidencia obtenida de recomendaciones de Comités de expertos u opiniones y o expe-
riencias clínicas de expertos de reconocido prestigio.

Púrpura Trombótica Trombocitopénica (PTT)
El plasma fresco congelado, preferiblemente cuarentenado, está indicado comosolución de intercambio en el recambio plasmático terapéutico. La elimina-ción del plasma del enfermo implica la eliminación del anticuerpo y de loscomplejos de alto peso molecular del Factor Von Willebrand implicados en lapatogenia de la PTT. El plasma normal infundido aporta la proteasa en la queel paciente es deficiente. Grado de recomendación C, nivel de evidencia IV.
Reversión urgente del efecto de los anticoagulantes orales
La transfusión de plasma no es el tratamiento de elección. Solo está indicadosi el paciente tiene hemorragia grave, o precisa cirugía de emergencia u otroprocedimiento invasivo, y no se dispone de concentrado de complejo de pro-trombina, o este está contraindicado.
Deficiencia de vitamina K
Solo indicado si el paciente tiene hemorragia y/o precisa con urgencia pro-cedimientos invasivos.
Hepatopatía en el contexto de biopsia hepática
Los tests anómalos de coagulación en caso de hepatopatía no son predicto-res del riesgo hemorrágico, probablemente más relacionado con lesiones vas-culares u otros factores.
Hay poca evidencia que apoye el uso de plasma fresco pre-biopsia hepática,dado que además no hay evidencia de que el plasma fresco reduzca el riesgode complicaciones hemorrágicas.
64
Grados de recomendación de las actuaciones clínicos (U.S. Preventive Services Task Force)
A Se requiere al menos un ensayo controlado aleatorizado como parte del conjunto de pu-blicaciones consistentes, de buena calidad, en las que se basa la recomendación especí-fica (niveles de evidencia Ia y Ib).
B Requiere la disponibilidad de estudios clínicos bien diseñados y realizados, peor no ale-atorizados, sobre el tema objeto de recomendación (niveles de evidencia IIa, IIb, III).
C Se basa en la recomendación de Comités de expertos u opiniones y/o experiencias clí-nicas de expertos de prestigio. Esto implica que no existen estudios clínicos de buena ca-lidad directamente aplicables (nivel de evidencia IV).

Puede aceptarse la transfusión de plasma fresco profiláctica basadaen el juicio clínico si: INR y TPT superan en > 1,5 al rango de referen-cia y se asocia trombopenia (< 50000/ml).
Profilaxis de procedimientos invasivos en enfermos con coagulopatía:
� Inserción de catéter venoso central� Toracocentesis / paracentesis� Endoscopia gastrointestinal/ biopsia� Broncoscopia / biopsia pulmonar transbronquial� Biopsia renal� Anestesia epidural, punción lumbar, procedimientos neuroquirúrgicas� Angiografía
No está justificado el uso de plasma fresco congelado si la actividad basal delos factores de coagulación es mayor del 40%.
Si está indicado en el tratamiento de la hemorragia asociada a estos proce-dimientos (grado de recomendación B, nivel de evidencia IIa).
Cirugía de bypass cardiopulmonar
La evidencia actual no apoya el uso rutinario de PFC (grado de recomenda-ción B, nivel de evidencia IIb).
Transfusión masiva
La decisión de transfundir PFC debe basarse en el juicio clínico y los tests decoagulación puntuales.
El uso precoz de PFC (asociado al tratamiento global de la coagulopatía conhematíes, plaquetas, crioprecipitado, Factor VII activado) en el contexto de lahemorragia masiva postraumatismo, puede mejorar la supervivencia.
No hay evidencia de que fórmulas concretas de combinación de componen-tes sanguíneos (por ejemplo: 1Unidad de CH/ 1Unidad PFC/ 1Unidad de CP)reduzcan la morbimortalidad.
Deficiencia del inhibidor plasmático de la C1 esterasa
Indicación de plasma aceptada para el tratamiento de la complicación aso-ciada a la deficiencia: angioedema hereditario que puede poner en peligro lavida del enfermo. Extraordinariamente rara.
65

SITUACIONES EN LAS QUE EL PLASMA NO ESTÁ INDICADO� Hipovolemia: los expansores de volumen tienen menos riesgo y son más
baratos� Para revertir los anticoagulantes orales si el enfermo no sangra: se debe
suspender el anticoagulante, y si es preciso tratamiento con vitamina K� En hipoproteinemia, o como soporte nutricional (hay tratamientos alter-
nativos más eficaces)� Como solución de intercambio en los Recambios Plasmáticos Terapéuticos
(RPT), excepto en la PTT� En Inmunodeficiencias, como fuente de inmunoglobulinas: las preparacio-
nes de inmunoglobulinas para uso endovenoso. son el tratamiento alter-nativo
DOSIS
El volumen de plasma administrado debe ser suficiente para mejorar la he-mostasia. En el adulto la dosis típica es de 10- 15 ml/ Kg, lo que aumentaráen aproximadamente un 20% los factores de coagulación.
Cuando se usa para corregir la deficiencia aislada de un factor de coagulación,la dosis dependerá de la vida media de ese factor específico, de la concen-tración pretransfusional del factor, del nivel postransfusional que queramosconseguir, y del tiempo que queramos mantener los niveles establecidos.
Tras la transfusión tiene que evaluarse tanto la respuesta clínica del enfermo(cese de la hemorragia), como los tests de coagulación.
Si la transfusión no es eficaz puede deberse a:
� Dosis insuficiente� Hemorragia activa/ coagulopatía de consumo� Inhibidores de factores de la coagulación
ADMINISTRACIÓN
El plasma debe ser compatible, respecto al sistema ABO, con el receptor.
No es necesario realizar pruebas de compatibilidad directa.
66

Debe descongelarse a temperatura controlada de 37ºC.
Una vez descongelado debe transfundirse lo antes posible; si fuera necesariomantenerlo almacenado tras la descongelación debe estar a entre 1ºC y 6ºC.
Si se retrasa la transfusión más de 6 horas debe tenerse en cuenta que seproducirá una reducción de la concentración de los factores V y VIII.
La velocidad de infusión debe adecuarse a la situación clínica del enfermo,teniendo muy en cuenta la situación hemodinámica del mismo.
CRIOPRECIPITADO
El crioprecipitado es la fracción plasmática que precipita al descongelar a 4ºCel plasma fresco congelado. Tras la descongelación de una unidad de PFC serealiza la centrifugación a 4ºC, para sedimentar el precipitado; el plasma so-brenadante se elimina, dejando el precipitado en 5 a 20 ml. de plasma, quees nuevamente congelado, pudiendo almacenarse hasta 36 meses, depen-diendo del procedimiento de extracción, procesamiento y temperatura deconservación utilizado.
Las proteínas que precipitan son: fibrinógeno, Factor XIII, Factor VIIIC, FactorVIII von Willebrand y fibronectina. El contenido de Factor VIIIC de la Unidaddebe ser > de 80 UI, y el de fibrinógeno > 150 mg.
INDICACIONES
En hemorragias asociadas a déficits de los factores que contiene, siempre queno estén disponibles concentrados de los factores específicos.
En la práctica clínica la indicación más frecuente es la hipofibrinogenemia entransfusión masiva. Menos frecuente es su uso en disfibrinogenemias congé-nitas o adquiridas.
DOSIS
El tratamiento habitual en hipofibrinogenemia es 1 Unidad por cada 7/10 kgde peso, que debe repetirse hasta lograr un fibrinógeno > 100 mg/dl.
67

ADMINISTRACIÓN
Debe descongelarse a temperatura controlada de 37ºC, y transfundirse deforma inmediata.
El almacenamiento tras la descongelación, que debe evitarse y no superar las6 horas, debe ser a 22ºC, e implica la reducción en la concentración de los fac-tores lábiles.
REFERENCIASAIorio, M Basileo, E Marchesine at el. The good use of plasma. A critical analysisof fiveinternational guidelines. Blood Transfus 2008; 6: 18-24.Cross-sectional Guidelines for Therapy with Blood Components and Plasma Derivati-ves. Executive Committee of the German Medical Association on the recommenda-tion of the Scientific Advisory Board. 4 th revised edition. 2009.Practice Guidelines for Blood Transfusion. A Compilation from Recent Peer-Reviewed Li-terature. American Red Cross. 2ed. 2007.PC Spinella and JB Holcomb. Resuscitation and transfusión principles for traumatic he-morragic shock. Blood Reviews 2009; 23: 231- 240. Thawed plasma components: a framework for preparation, storage and use.Australian & New Zealand Society of Blood Transfusion Ltd. 1st edition, April 2009.MP. Wong, N Droubatchevskaia, KM. Chipperfield, L D. Wadsworth, MB, D J. Ferguson.Guidelines for frozen plasma transfusion. BC Medical Journal. 2007; 49:6, 311- 19. �
68

La transfusión pediátrica es compleja, los niños menores de 4 meses no pue-den ser considerados receptores “pequeños” ya que los requerimientos, laspérdidas por extracciones repetidas para pruebas analíticas, la anemia fisio-lógica postnatal, la limitada respuesta medular, la presencia de hemoglobinaF y A, la inmadurez inmunológica, los hace diferentes.
Los neonatos y especialmente los prematuros son receptores vulnerables a lapotencial inefectividad y toxicidad de la sangre.
Los criterios para la transfusión de un niño de menos de 4 meses son dife-rentes de los de más de 4 meses, no solo por su pequeño volumen sanguíneo,sino por factores fisiológicos como la producción de eritropoyetina endógenaen respuesta a la anemia del prematuro, la anemia fisiológica postnatal y elsistema inmune humoral.
SANGRE TOTAL
Podríamos decir que la única indicación de sangre total es para la realizaciónde exanguinotransfusión, también se utiliza sangre total para el purgado delos diferentes tipos de circuitos de circulación extracorpórea antes de conec-tar el niño a ellos.
CONCENTRADO DE HEMATÍES
INDICACIONES
La tolerancia del grado de anemia es diferente según la edad del niño y de lasituación clínico patológica en la que se encuentre.
Niño prematuro
Está indicado transfundir concentrado de hematíes si:
� Hemoglobina < 7 g/d: estable y ganando peso.� Hemoglobina < 10 g/dl: síntomas moderados de anemia (apnea, bradicar-
dia, taquicardia, taquipnea y escaso aumento de peso).
69
TRANSFUSION EN NEONATOS Y NIÑOS

� Hemoglobina < 12 g/dl: síntomas severos (acidosis, enfermedad cardiaca,requerimientos de oxigeno, hipotensión).
Niños a término < 4 meses
Está indicado transfundir concentrado de hematíes si:
� Hemoglobina < 7 g/dl: manifestación clínica de anemia (apnea, taquicar-dia y escaso aumento de peso).
� Hemoglobina < 10 g/dl: anemia peroperatoria. � Hemoglobina < 10 g/dl: shock clínico o descenso severo de la presión ar-
terial.� Hemoglobina < 13 g/dl: enfermedad cardiaca cianótica congénita, oxige-
nación por membrana extracorpórea (OMEC).� Hemorragia aguda > 10% del volumen que no responde a otros trata-
mientos.
Niños mayores de 4 meses
� Hemoglobina < 7 g/dl: pacientes a tratamiento con quimioterapia o ra-dioterapia o con anemia crónica que no responden a tratamiento médico.
� Hemoglobina < 10 g/dl: postoperatorio con signos de anemia, shock clínicoo descenso severo de la presión sanguínea. Preoperatorio en pacientes conrégimen de transfusiones crónicas.
� Hemoglobina < 12g/dl: enfermedad cardiopulmonar severa.� Hemorragia aguda con una pérdida ≥15% del volumen sanguíneo o hipo-
volemia que no responde a otros tratamientos.� Previo a la obtención de Células Progenitoras Hematopoyéticas.
CONCENTRADO DE PLAQUETAS
El recuento plaquetario en los recién nacidos es similar al de los adultos. Sinembargo, en el período neonatal, y especialmente en los prematuros, se ob-servan alteraciones del funcionalismo plaquetario y de los factores de la co-agulación que hace recomendable mantener recuentos superiores al de losadultos.
La incidencia de hemorragia cerebral clínicamente silente es relativamentealta en prematuros de < 37 semanas con trombopenia.
70

INDICACIONES
Transfusión profiláctica
Para evitar hemorragia se aconsejan las transfusiones profilácticas de pla-quetas para mantener unos recuentos plaquetarios que van a depender de laedad del niño y del proceso a que va a ser sometido. Así:
� Prematuro estable: > 30 x 109/l. � Prematuro enfermo: > 50 x 109/l.� Niño a término < 4 meses: > 20 x 109/l.� Niño a término > 4 meses: > 10 x 109/l.� Previo a punción lumbar y recuento de plaquetas < 10 x 109/l sin hemo-
rragia.� Previo a procedimientos invasivos o cirugía menor mantener el recuento de
plaquetas > 50 x109/l.
Transfusión terapéutica
En caso de hemorragia se aconsejan las transfusiones de plaquetas para man-tener unos recuentos plaquetarios que van a depender del tipo de hemorragiaque se desee tratar. Por lo que la transfusión de plaquetas estará indicada si:
� Pacientes con hemorragia activa y recuentos de plaquetas < 50 x 109/l.� Hemorragia microvascular difusa con bypass cardiopulmonar o oxigena-
ción de membrana extracorpórea (OMEC), cirugía neurológica o cirugíamayor y recuento < 100 x 109/l.
� Hemorragia en paciente con defecto cualitativo de las plaquetas, en es-pera del recuento de plaquetas.
Actuación en casos de Trombopenia Inmune
La trombocitopénia inmune está causada por la presencia de anticuerpos an-tiplaquetas maternos que atraviesan la placenta e interactúan con las pla-quetas fetales y neonatales.
Los neonatos con recuentos de plaquetas superiores a 50 x 109/l no requie-ren transfusión de plaquetas, con cifras inferiores a 20 x 109/l se debe trans-fundir, en los neonatos con cifras entre 20 y 50 x 109/l está indicada latransfusión si hay patología asociada o evidencia de sangrado.
71

Trombopenia alloinmune
El tratamiento de elección consiste en la administración de gammaglobulina en-dovenosa (1g/Kg/día 2 días o 400 mg/Kg/día 5 días, para conseguir recuentosplaquetares superiores a 30 x 109/l, que se debe repetir si en 48 horas el recuentoplaquetario desciende a 20 x 109/l). La transfusión de plaquetas antígeno ne-gativo y/o pruebas cruzadas negativas está en función del recuento plaquetarioy la situación clínica . En caso de que precise tratamiento no es necesario espe-rar por los resultados del laboratorio dado el riesgo de hemorragia severa.
La transfusión de plaquetas HPA 1a negativo y HPA -5b negativo son efecti-vas en el 95% de los casos de trombopenia neonatal alloinmune.
Cuando no se evidencie respuesta a la transfusión de plaquetas HPA (1a y 5bnegativo), se podría valorar la transfusión de plaquetas maternas irradiadas ylavadas, para evitar la presencia de aloanticuerpos maternos que prolongaríanla trombopenia neonatal. En este caso las pruebas reglamentarias para unadonación deben ser realizadas a la madre, pues la donante (madre) y el re-ceptor (niño) son personas distintas.
Trombopenia autoinmune
El tratamiento consiste en la administración de gammaglobulina endovenosacomo en en los casos de trombopenia alloinmunes. La transfusión de pla-quetas no está indicada.
PLASMA
INDICACIONES� Previamente a procedimientos invasivos o cirugía, si el valor de la Razón
Normalizada Internacional (INR) es superior 1,5 a 2 veces el valor normal.� En caso de Hemorragia microvascular difusa, en pacientes con determina-
ciones del Tiempo de Protrombina (TP) o del Tiempo Parcial de Trombo-plastina (TPT) superior en 1,5 veces el valor normal o resultados delaboratorio pendientes.
� En caso de déficit de factores de coagulación, con hemorragia o previo aprocedimientos invasivos o cirugía, si el factor de coagulación especificono está disponible para la transfusión.
72

� En situaciones clínicas con déficit de Vitamina K, con hemorragia o previoa procedimientos invasivos o cirugía.
� En la Purpura Trombótica Trombocitopénica congénita o adquirida.� En caso de déficit de Antitrombina III y Púrpura fulminante del recién na-
cido, secundaria a deficiencia congénita de la Proteína C o Proteína S, si nose dispone de concentrados de estos factores.
� Para la reconstitución del concentrado de hematíes, cuando no se disponede sangre total, para la realización de exanguinotransfusión.
� Para la prevención de hemólisis asociada a activación de antígeno T eritro-citario, activado en el contexto de la enterocolitis necrotizante es una in-dicación controvertida.
CONTRAINDICACIONES
El plasma no debe ser utilizado en las siguientes situaciones:
� Como expansor del volumen sanguíneo.� Para la prevención de hemorragia periventricular en niños prematuros.� Como aporte de inmunoglobulinas.
TRANSFUSIÓN INTRAUTERINA
Los concentrados de hematíes utilizados para realizar la transfusión intrau-terina han de ser de menos de 5 días desde la donación, leucodeplecionadoso de donante con determinación de anticuerpos anti-Citomegalovirus (CMV)negativos, e irradiados.
Los hematíes serán del grupo (ABO) O y Rh(D) negativos, excepto en pre-sencia de anticuerpos maternos incompatibles. Una vez irradiados se trans-fundirán antes de 24 horas postirradiación.
En los casos que se necesite transfundir unidades de plaquetas, éstas, al igualque los concentrados de hematíes, han de ser leucodeplecionadas o de do-nante con determinación de anticuerpos anti-Citomegalovirus (CMV) nega-tivos, e irradiadas. Se deben concentrar para eliminar parte del sobrenadante,se dejarán en reposo durante 1 hora y se transfundirán antes de transcurri-das 4 horas desde inicio del proceso de concentración.
73

EXANGUINOTRANSFUSIÓN
Este procedimiento terapéutico está indicado en neonatos con anemia se-vera al nacimiento o hiperbilirrubinemia, generalmente por Enfermedad He-molítica del Recien Nacido (EHRN).
Es importante tener en cuenta que la exanguinotransfusión tiene una alta inciden-cia de complicaciones severas. La mortalidad es de 2-3 /100 procesos realizados
La indicación por hiperbilirrubinemia depende del peso del niño, del nivel debilirrubina, del ritmo de incremento, de las condiciones clínicas del niño.
COMPONENTE SANGUÍNEO UTILIZADO
La exanguinotransfusión se realiza con sangre total o, en caso de no disposi-ción de ésta, con concentrado de hematíes al que le elimina parcialmente elsobrenadante y se reconstituye con plasma, para obtener un hematocritofinal de 0,45-0,55%.
Los hematíes han de ser del grupo (ABO) O ó compatible con el niño y elplasma de la madre, respecto al grupo Rh(D) han de ser negativo o idénticoque el niño, y carentes del antígeno correspondiente al anticuerpo detectadoen el suero materno (causante de la EHRN). El plasma de grupo AB o delmismo que el niño.
Los hematíes han de ser de menos de 5 días desde la donación, y preferible-mente irradiados, excepto en los casos que no pueda retrasarse la transfu-sión y no haya habido una transfusión intrauterina, una vez irradiados seutilizarán dentro de las 24 horas post-irradiación.
El volumen de sangre total, o concentrado de hematíes reconstituido con plasma,que se utilizará para realizar la exanguinotransfusión, se calcula entre 80-160 ml/kgen niños a término y de 100-200ml/kg en niños prematuros (entre 1 y 2 veces el
74
Edad y peso Exanguinotransfusión< 1.500 g Bilirrubina 13-16
1500-2000 g Bilirrubina 16-182000- 2.500 g Bilirrubina 18-20
A termino Bilirrubina 17-22

volumen sanguíneo). En caso de hiperbilirrubinemia es preciso realizar un recam-bio de dos volúmenes, se debe realizar a un ritmo de 5-20 ml en 3 a 5 minutos. Laduración de un recambio de doble volumen es de aproximadamente 120 minutos.
El niño debe estar debajo de un calentador con temperatura monitorizada ola sangre será transfundida con un calentador mecánico con temperaturacontrolada.
ACTUACIÓN EN LOS DÉFICITS DE INMUNOGLOBULINA A
Plaquetas: Pacientes deficientes de IgA con anti-IgA deben recibir plaquetasde donantes con déficit de IgA.
Hematíes: Pacientes con déficit de IgA pueden recibir hematíes lavados perolos hematíes congelados son los de elección.
Plasma: Pacientes deficientes de IgA con anti-IgA precisan plasma con défi-cit de IgA
COMPONENTES SANGUÍNEOS IRRADIADOS
Ver apartado “COMPONENTES SANGUÍNEOS IRRADIADOS”.
PARTICULARIDADES DE LA TRANSFUSIÓN EN NIÑOS Y NEONATOS
IDENTIFICACIÓN
Se debe garantizar una identificación inequívoca, legible y segura del niño.Los riesgos de una identificación errónea del receptor son mayores que enlos adultos: confusión de las muestras madre y niño por etiquetado previo delos tubos, identificación errónea de la muestra cuando ocurren varios partossimultáneos, dificultad de colocación de pulseras de identificación en pre-maturos, identificación del niño con el nombre de la madre.
PRUEBAS PRETRANSFUSIONALES
Se ha de realizar en la muestra de sangre del niño: La determinación del gruposanguíneo ABO, Rh (D), Coombs Directo y escrutinio de anticuerpos irregula-res eritrocitarios en caso de no disponer de plasma de la madre.
75

En la muestra de la madre: La determinación del grupo sanguíneo ABO, Rh(D)y escrutinio de anticuerpos irregulares eritrocitarios.
Si no se detectan anticuerpos irregulares antieritrocitarios, ni anticuerpos anti-A o anti- B de naturaleza IgG, no es necesario realizar pruebas cruzadas pre-vias a la transfusión de hematíes.
Dada la reducida capacidad inmunológica de los niños menores de 4 mesesla realización de estas pruebas al nacimiento son suficientes y no es precisorepetir en ingresos posteriores hasta los 4 meses de vida.
En caso de que anticuerpos irregulares eritrocitarios estén presentes en elniño y/o presente una prueba de Coombs directo positiva, debe identificarseel anticuerpo y seleccionar unidades carentes del antígeno correspondientea la especificidad del anticuerpo correspondiente, y realizar pruebas cruzadasen técnica de Coombs indirecto.
Los hematíes se seleccionarán de grupo O u otro grupo ABO compatible conel niño y el plasma de la madre, y Rh(D) negativo o idéntico al del niño.
Si el niño es de grupo ABO distinto de O y recibe componentes sanguíneosque contienen al aglutininas anti-A y/o Anti-B, y posteriores unidades selec-cionadas no son de grupo O, se determinará en el plasma del niño la presen-cia de anti-A o anti-B mediante una prueba de Combos indirecto.
Las transfusiones de plaquetas y plasma deben ser ABO compatibles, paraevitar transfusiones incompatibles entre el anticuerpo del plasma del donantey los antígenos de los hematíes del receptor.
A los niños mayores de 4 meses se les realizarán las mismas pruebas y con lamisma frecuencia que a los receptores adultos.
ADMINISTRACIÓN
Todos los componentes han de ser transfundidos mediante el uso de sistemaestéril provisto de cámara con un filtro de 170-200μm. También se puedenusar filtros específicos pediátricos.
La administración mediante bombas mecánicas con jeringa permite una in-fusión constante con control del volumen a transfundir y un sistema de
76

alarma de la presión. Este sistema es adecuado para volúmenes de 10-50 ml.Transfusiones de volumen inferior a 10 ml se realizan con una jeringa a bolosintermitentes, cuyo volumen puede ser aspirado del sistema de filtro de launidad de sangre. Volúmenes superiores a 50 ml se administran con equipode infusión con filtro.
El ritmo de la transfusión depende del componente, del volumen total que va aser transfundido, del acceso venoso y de la tolerancia del niño. Si el tiempo pre-visto de una transfusión de hematíes es superior a 4 horas, el componente san-guíneo será alicatado y su segunda alícuota se conservará a 4º C hasta su uso.
VOLUMEN Y RITMO DEL COMPONENTE SANGUÍNEO
REDUCCIÓN DE VOLUMEN
En caso de que la tolerancia de volumen esté restringida se debe reducir el vo-lumen de las plaquetas mediante centrifugación, aunque no es frecuente por-que la dosis de 10 ml/kg es bien tolerada en general. Una dosis superior a 10ml/kg requiere eliminar el plasma excepto 10-15 ml mediante centrifuga-ción, dejar el concentrado de plaquetas en reposo 1 hora antes de la resus-pensión y transfundir antes de 4 horas.
CALENTAMIENTO
Todos los componentes sanguíneos que se van a transfundir a niños deberíanestar, en la medida de lo posible, a una temperatura similar a la ambiente, y es-pecialmente si la infusión se realiza por una vena central. Los pequeños volú-menes a transfundir no permiten el uso de calentadores mecánicos que seemplean para grandes volúmenes, por lo que se puede calentar pasivamente co-
77
COMPONENTE VOLUMEN RITMO DURACIÓNHEMATÍES 5-15 ml/kg 3-5ml/kg/hora 2-4 horasPLASMA 10-15 ml/kg 10-15 ml/Kg /hora 1 horaPLAQUETAS Niño < de 15 Kg:5-10 ml/kg 5-10 1/2 hora
Niño > de 15 Kg: 1 aféresis o ml/kg/30minutosun Concentrado de plaquetas (aprox. 300ml)

locando el volumen a transfundir a temperatura ambiente 30 minutos previo ala transfusión, pero nunca serán calentados directamente, ni introducidos enagua caliente.
FOTOTERAPIA
Cuando las transfusiones se realizan durante el tratamiento con fototerapia,el tubo se debe colocar de tal forma que se minimice la exposición del com-ponente a la luz de la fototerapia.
VIGILANCIA
La transfusión debe estar controlada en todo momento e interrumpida si elniño presenta:
� Apnea, taquipnea, o dificultad respiratoria.� Taquicardia, bradicardia o arritmia.� Cianosis.� Cambio significativo en la presión arterial sistólica.� Aumento o disminución importante (> 1ºC) de la temperatura.� Hemoglobinuria.
REDUCIR LA EXPOSICIÓN A DONANTES
Para reducir el riesgo de transmisión de enfermedades infecciosas se em-pleará una misma donación para un mismo receptor mediante la disponi-bilidad de unidades alicuotadas, separadas de la unidad principal o unaunidad principal con bolsas satélites múltiples. Esta última opción permiteretirar el volumen preciso que se requiere para cada transfusión en circuitocerrado.
ALTERNATIVA A LA TRANSFUSION DE HEMATÍES
La Eritropoyetina puede reducir las necesidades de transfusión de hematíesen niños, pero no las reduce en neonatos de menos de 2 semanas de vida, pe-riodo de mayor demanda de transfusión por las reiteradas extracciones demuestras. La vía de administración, la dosis, la duración del tratamiento y elsoporte férrico no han sido bien definidos. Recientes estudios han asociado
78

la administración de Eritropoyetina y retinopatía del prematuro. La Eritropo-yetina no está indicada como alternativa la reducción del uso de concentra-dos de hematíes en los neonatos.
REFERENCIASBell EF, StraussRG, Widness JA. Randomized trial of liberal versus restrictive guidelinesfor blood cell transfusion in preterm infants. Pediatrics 2005;115(6):1685-91.Blood Transfusion:Neonatal Transfusion Recommendations at RCH Melbourne.www.rch.org. British Committee for Standardars in Haematology Blood Trasnfusion taskforce (2004)Amendment to the Transfusion guidelines for neonates and older children, 2004.www.bcshguidelines.org . British Committee for Standardars in Haematology Guidelines on the administrationof blood components, 2009. www.bcshguidelines.org.Bulto F. Transfusion guidelines for neonates and older children. British Journal of Hae-matology 2004; 124:433-45.Faucher D, Fetus and Newborn Committee Canada Paediatric Society. Red blood celltransfusions in newborn infants: Revised guidelines. Paedritrics and Child Health 2002;7(8): 553-58.Guidelines for Transfusion Therapy of Infants from Birth to Four Months of Age. Wads-worth Center, NY State Dept of Health, 2004. www.wadsworth.org.Guidelines for Blood Transfusion 2007 (2 thed). American Croos http://www.red-cross.org.Kirpalani H, Whyte RK, Andersen C. The premature infants in need of transfusion (PINT)study: a randomized controlled trial of a restrictive (low) versus liberal (high) transfu-sion threshold for extremely low birth weight infants. J Pediat 2006:149(3):301-307.Luban NLC. Massive transfusion un the neonates. Transfus Med Rev 1995;9:200-14.McClelland, DBL (2007) Handbook of Transfusion Medicine (4 thed) . The Stationery Of-fice, London.New H. Paediatric Transfusion. Vox Sang 2006;90:1-9.Potter C. Anemia of Prematurity: Treatment & Medication 2009. emedicine.meds-cape.com.Roseff SD , Luban NLC, Manno CS. Guidelines for assessing appropriateness of pedia-tric transfusion. Transfusion 2002; 42.1398-13.Rodriguez-Villanueva. La Transfusion Pediatrica. Boletin SETS 2005;56 (2):13-16.Strauss RG. Managing the anemia of prematurity; Red blood cell transfusions versusrecombinant erythropoietin. Transfus. Med Rev 2001;15:;213-23.
79

Strauss RG. Data-driven blood banking practices for neonatal RBC transfusions. Trans-fusion 2000,40:1528-40. Vamvakas EC, Strauss RG. Meta-analysis of controlled clinical trials studying the effi-cacy of rHuEPO in reducing blood transfusions in the anemia of prematurity. Transfu-sion 2001;41:406-15.�

La transfusión de componentes sanguíneos (CS) que contienen linfocitos Tcon capacidad de proliferación comporta el peligro de causar Enfermedad delInjerto contra el Huésped Postransfusional (EICH-PT) en receptores inmuno-deficientes o en ciertas combinaciones donante / receptor. La irradiación delos CS celulares, con una dosis media de 25-50 Gy, es el único método dis-ponible para prevenir esta grave complicación de la transfusión, mientras quela eficacia de estos CS no es afectada.
Hasta ahora, la EICH-PT ha sido sólo observada después de la transfusión deCS celulares (sangre total, concentrados de hematíes, plaquetas y granuloci-tos). No se han descrito casos de EICH-PT después de la transfusión de plasmacongelado y crioprecipitado. Los CS irradiados se pueden transfundir sin nin-guna limitación a receptores normales.
El médico que prescribe la transfusión debe hacer constar en la solicitud trans-fusional la característica de “irradiado” cuando un paciente deba ser trans-fundido con este tipo de CS.
INDICACIONES DE LA IRRADIACIÓN DE COMPONENTES SANGUÍNEOS
Se requieren utilizar CS irradiados para transfusión en las siguientes circunstancias:
Indicaciones de irradiación aceptadas por la mayoría de los autores(pacientes en riesgo confirmado de sufrir EICH postransfusional)
1.- Pacientes que reciben CS provenientes de familiares de primer o se-gundo grado del receptor, incluso si son pacientes inmunocompetentes, de-bido a la elevada probabilidad de compartir haplotipos HLA.
2.- Pacientes transfundidos con CS de donantes HLA idénticos y/o HLA-compatibles, familiares y no familiares.
3.- Transfusiones de concentrados granulocitos, debido a las elevadas can-tidades de linfocitos T (5-10 x 109 linfocitos por unidad) que contienen.
81
COMPONENTES SANGUÍNEOS IRRADIADOS

4.- Síndromes de Inmunodeficiencia Celular Congénita:
� Síndrome de Inmunodeficiencia Combinada Grave.� Síndrome de DiGeorge.� Síndrome de Wiskott-Aldrich.� Ataxia Telangiectasia.� Deficiencia de Purina – Nucleósido – Fosforilasa.
Los estados de inmunodeficiencia humoral no conllevan el mismo riesgo, porlo que no es necesario emplear CS irradiados. No obstante, en recién nacidoscon sospecha de Inmunodeficiencia, deben ser transfundidos con CS irradia-dos hasta que se establezca el diagnóstico definitivo.
5.- Transfusión intraútero (TIU) de hematíes o plaquetas. Los neonatos querecibieron TIU deben seguir siendo transfundidos con CS irradiados duranteel periodo neonatal.
7.- Exanguinotransfusión. En ausencia de otras indicaciones de irradiación(antecedentes de TIU, CS provenientes de familiares) se recomienda irradiarla sangre utilizada para exanguinotransfusión, si ello no supone un excesivoretraso.
8.- En los pacientes con Trasplante de Células Progenitoras Hematopoyé-ticas (CPH), se transfundirán CS irradiados en las siguientes situaciones:
� Pacientes sometidos a autotrasplante de CPH: deben recibir CS irradiadosdesde 14 días antes de la recolección de las CPH hasta finalizada ésta (paraevitar la recogida de linfocitos alogénicos viables), y desde 14 días antesdel trasplante hasta al menos 3 meses después, o hasta que haya eviden-cia de implante del injerto y fiable detección de reconstitución inmunoló-gica (recuperación de células T).
� Pacientes sometidos a alotrasplante de CPH: deben recibir CS irradiadosdesde 14 días antes del trasplante hasta que el paciente ya no reciba profi-laxis de EICH o la cifra de linfocitos supere 1x109/l. Los pacientes con EICHpostrasplante crónica activa deben continuar recibiendo CS irradiados.
� Donantes de CPH para alotrasplante: se deben utilizar CS irradiados si ne-cesitan ser transfundidos en los 14 días anteriores o durante la recolecciónde CPH, o para el cebado del separador celular.
82

9.- Linfoma de Hogdkin: estos pacientes tienen una disfunción inmune in-trínseca que persiste durante periodos desconocidos, por lo que deben reci-bir CS celulares irradiados en todas las fases de la enfermedad y detratamiento.
10.- Pacientes tratados con análogos de la purina (fludarabina, deoxicofor-micina –pentostatin- y chlorodeoxyadenosina -cladribine-). Actualmente nohay datos para establecer un período concreto para el uso de CS irradiados enestos pacientes una vez finalizado el tratamiento. Se aconseja durante almenos 1 año o, mejor, de forma indefinida.
Indicaciones de irradiación en las que la revisión de la literaturamuestra falta de acuerdo (Pacientes sobre los que no hay acuerdosobre el riego)
1.- Recién nacidos prematuros y niños de bajo peso. Aunque no existeacuerdo, se considera que los neonatos, especialmente los extremada-mente prematuros, tienen un elevado riesgo de desarrollar EICH-PT. Mu-chos centros sólo irradian los CS celulares que se van a transfundir aniños prematuros por debajo de un determinado peso (p. ej. ″ 1500 g) ode una determinada edad gestacional (p ej. ″ 30 semanas). Pero otroscentros irradian todos los CS celulares destinados a ser transfundidos aniños de menos de 4 meses (la posibilidad de que un recién nacido afectode inmunodeficiencia de células T congénita reciba transfusiones antesde que la enfermedad haya sido diagnosticada se considera motivo sufi-ciente).
2.- Linfoma no Hogdkin. Se han descrito al menos 17 casos de EICH-PT enpacientes con Linfoma No Hogdkin.
3.- Pacientes con anemia aplásica que reciben tratamiento inmunosupre-sor.
Actualmente no hay datos que apoyen el uso de CS irradiados parapacientes en alguna de las siguientes situaciones:
1.- SIDA2.- Leucemia
83

3.- Tumores sólidos (incluyendo neuroblastoma y rabdomiosarcoma)4.- Trasplante de órgano sólido (corazón, riñón, pulmón, hígado).
Dada la ausencia de efectos nocivos de la irradiación y la gravedad de la EICH-PT cuando se tienen dudas lo más prudente es usar CS irradiados.
REFERENCIASDwyre DM, Holland PV. Transfusion-associated graft-versus-host disease. Vox Sang2008; 95:85-93.Rühl H, Bein G, Sachs JH Transfusion-associated graft-versus-host disease. Transfus MedRev 2009;23(1): 62-71.Gelly KJ, Kerr R, Rawlinson S et al. Transfusion-associated graft-vs -host disease in apatient with high-grade B-cell lymphoma. Should cellular products for patients withnon-Hodgkin`s lymphoma be irradiated? Br J Haematol.2000; 110:228-229. �
84

La transfusión de componentes sanguíneos (CS) para la corrección de los dé-ficits generados por el sangrado que acontece durante o después de una in-tervención quirúrgica es una práctica estandarizada y ampliamente extendida.Si bien los CS utilizados en la actualidad están sometidos a exigentes con-troles de calidad y su utilización puede considerarse como muy segura, exis-ten determinados riesgos que deben ser tenidos en consideración a la hora deprescribir una transfusión:
� Transmisión de enfermedades virales.� Aloinmunización frente a antígenos celulares.� Reacciones transfusionales que incluyen reacciones alérgicas o o lesión pul-
monar aguda asociada a transfusión (TRALI).� Errores en la administración de componentes que se repiten con una de-
terminada frecuencia en las unidades de transfusión de todo el mundo.
La prevención de reacciones adversas a la transfusión pasa siempre, en primerlugar, por una correcta optimización del uso de los distintos CS. Para ello es ne-cesario hacer una correcta indicación terapéutica que permita no sólo un uso res-tringido de los mismos, sino también su administración en el momento adecuado.Aún así, es innegable que los CS son necesarios y que su consumo es especial-mente elevado en determinados procesos quirúrgicos. Cuando estos procesosquirúrgicos son programados y conocemos de antemano la fecha en la que vana tener lugar, el hematólogo puede poner a disposición de los pacientes, ciruja-nos y anestesistas un programa de ahorro de sangre (PAS) destinado a disminuirel riesgo de recibir sangre homóloga sin incrementar el riesgo de aumentar unatransfusión. Estos programas de ahorro de sangre emplean distintas estrategias,solas o en combinación, y deben basarse en una serie de principios básicos.
PRINCIPIOS BÁSICOS DE UN PROGRAMA DE AHORRO DE SANGRE
Los riesgos que queremos evitar mediante la restricción del uso de CS homó-logos tienen una frecuencia muy baja, por lo que las medidas encaminadas asu prevención deben ser sencillas y con una toxicidad muy baja para el paciente.
85
ALTERNATIVAS A LA TRANSFUSIÓN DE SANGRE ALOGÉNICA

Por otro lado, algunos de los efectos adversos de la transfusión no son inheren-tes al hecho de que los CS sean homólogos, como sucede por ejemplo con las so-brecargas circulatorias o la administración de componentes erróneos. Todo aquelprograma de ahorro de sangre que, disminuyendo el uso de sangre homóloga, in-cremente el número total de transfusiones, a expensas de un incremento exce-sivo en uso de CS autólogos, podrá resultar en un riesgo mayor de aparición deestas complicaciones no vinculadas a los hemoderivados homólogos.
Así pues, un PAS debe seguir las siguientes reglas fundamentales:
� Las medidas utilizadas para la reducción de la transfusión de componenteshomólogos deben ser lo más inocuas posibles, de forma que no se sustituyala toxicidad de un procedimiento por otra de magnitud similar.
� Los PAS deben quedar reservados para intervenciones quirúrgicas que su-pongan un riesgo significativo de transfusión debido al sangrado que elproceso quirúrgico implica. Por tanto, cada servicio de transfusión debe de-finir las indicaciones de inclusión en un PAS y evaluar de forma individua-lizada aquellos casos que no entren dentro de las mismas. Un PAS no debebasarse de forma general en la solicitud de inclusión en el mismo por elpropio paciente, sino en criterios exclusivamente médicos.
� El empleo de PAS no debe relajar el uso de indicaciones claras y estrictaspara la transfusión de hemoderivados.
ESTRATEGIAS DISPONIBLES PARA UN PAS
USO DE ERITROPOYETINA ALFA Y AGENTES HEMATÍNICOS
La eritropoyetina alfa (EPO) puede incrementar la producción de sangre au-tóloga tanto en pacientes incluidos en programas de predonación como enpacientes que van a ser sometidos a cirugía y en los que no se puede o no seconsidera apropiado el uso de este tipo de programas. En el contexto de lasestrategias de ahorro de sangre, la eritropoyetina alfa tiene las siguientes in-dicaciones aprobadas:
� Aumentar la producción de sangre autóloga en pacientes incluidos en unprograma de predonación, fundamentalmente si se precisa la recolecciónde un número importante de unidades. Su uso en esta indicación debe sersopesado frente al riesgo conocido de sucesos tromboembólicos.
86

� Disminuir la exposición a las transfusiones de sangre alogénica en pacien-tes adultos sin deficiencia de hierro antes de una cirugía mayor ortopédicaelectiva, en los que se considere que existe un riesgo elevado de complica-ciones transfusionales si no se dispone de un programa de predonación desangre autóloga y en los que se espera una pérdida de sangre moderada(900 a 1800 ml).
En términos generales el tratamiento con EPO se administrará a pacientescon anemia moderada (Hb 10-13 g/dl), sin deficiencia de hierro. La dosishabitual es de 600 UI/kg 2 veces por semana, durante las 3 semanas an-teriores a la cirugía en el caso de predonación y de 600 UI/kg semanal-mente durante tres semanas en el caso de preparación sin predonación(días -21, -14 y -7) antes de la intervención quirúrgica y el día de la ciru-gía. Si por necesidades médicas, el tiempo previsto antes de la cirugía sereduce a menos de tres semanas, se pueden administrar 300 UI/kg de eri-tropoyetina alfa diariamente durante 10 días consecutivos antes de la ci-rugía, el día de la cirugía y los cuatro días inmediatamente posteriores. Sial realizar evaluaciones hematológicas durante el período preoperatorio,el nivel de hemoglobina alcanza 15 g/dl, o superior, se interrumpirá la ad-ministración de erotropoyetina alfa y no se administrarán las dosis poste-riores.
El uso de eritropoyetina en estos casos está contraindicado en pacientes concoronariopatías, artropatías periféricas, enfermedades de las arterias caróti-das o cerebrovasculares de carácter grave, incluidos los pacientes que hayansufrido recientemente infarto de miocardio o accidente cerebrovascular, asícomo en pacientes quirúrgicos que por algún motivo no puedan recibir untratamiento profiláctico antitrombótico adecuado.
Hay que tener precaución con las cifras de tensión arterial que deben sercontroladas. Su uso es controvertido en el caso de hipertensión arterialmal controlada, situación en la que nosotros desaconsejaríamos su uso.Hay que advertir acerca del riesgo tromboembólico al paciente, tratar ladeficiencia de hierro si existiese y acompañar el tratamiento con eritro-poyetina con hierro. En caso de anemia hay que investigar y corregir otrascausas.
87

DONACIÓN AUTÓLOGA PREDEPÓSITO
Consiste en la extracción, almacenamiento y, finalmente, la transfusión de la pro-pia sangre del paciente. La indicación se establece generalmente en pacientesprogramados para diferentes tipos de cirugía. Esta alternativa posibilita la elimi-nación de ciertos riesgos como la transmisión de enfermedades virales, la aloin-munización a antígenos celulares o la aparición de la mayoría de las reaccionestransfusionales. Sin embargo, no evita efectos indeseables como la contaminaciónbacteriana, la sobrecarga de volumen, o los derivados de los errores transfusionales.
Los responsables de cada Centro o Servicio de transfusión, deben establecerlos criterios de selección, extracción y otros elementos clave del procedi-miento. Cada centro debe diseñar los criterios de exclusión. En principio, el cri-terio absoluto de exclusión es la anemia (hemoglobina inferior a 11 g/dl), lapresencia de un proceso infeccioso actual o reciente que puedan asociarse abacteriemia o, según establece la ley, la positividad de alguno de los marca-dores de infección de determinación obligatoria.
Los componentes autólogos que no hayan sido transfundidos no deberán uti-lizarse para transfusión homóloga ni para fraccionamiento.
Los pacientes que participan en un programa deben conocer exactamente lascaracterísticas del procedimiento de selección, extracción, caducidad, efectosadversos, posibilidad de necesitar transfusiones homólogas, etc., y firmar elcorrespondiente consentimiento informado.
El protocolo habitual de donación es de 1 unidad por semana, y la última do-nación debe efectuarse al menos 72 horas antes de la intervención quirúrgica.En determinadas situaciones podría utilizarse eritropoyetina para aumentarel número de unidades extraídas.
HEMODILUCIÓN NORMOVOLÉMICA
Consiste en la extracción de 1 ó más unidades de sangre poco antes de la in-tervención con infusión de coloides o cristaloides, para su transfusión pos-terior. Estas unidades se pueden mantener a temperatura ambiente hasta6h. Para almacenamientos durante más tiempo, la temperatura debe ser de1-6º C.
88

El procedimiento debe asegurar la esterilidad de la extracción, así como eletiquetado y almacenamiento correctos, junto con la realización de las prue-bas de laboratorio adecuadas para garantizar la idoneidad del producto y evi-tar errores de identificación.
RECUPERACIÓN INTRA Y POSTOPERATORIA
Mediante la recolección de sangre recuperada del campo operatorio o de uncircuito extracorpóreo (o de drenajes, cavidades articulares, etc., en el caso dela recuperación postoperatoria).
Existen diferentes métodos de obtención y procesamiento estéril, precisán-dose su filtración antes de la reinfusión. Hasta las 6h puede mantenerse atemperatura ambiente, y a partir de ahí, y hasta 24h, mediante refrigeracióna 1-6º C (sólo en el caso de la recuperación intraoperatoria).
Generalmente el método está contraindicado en caso de padecer el pacienteinfección o neoplasia, así mismo no podrá realizarse si se utilizan solucionesyodadas sobre el campo quirúrgico o si se produce contaminación del mismo.
REFERENCIASHenry D, Carless P, Moxey A, O´Connell D, Ker K, Fergusson D. Pre-operative autologousdonation for minimising perioperative allogeneic blood transfusion. The Cochrane Library: http://mrw.interscience.wiley.com/cochane/clsysrev/articles/CD003602/frame.html �
89


El término de derivados plasmáticos hace referencia a una serie de produc-tos obtenidos a partir de plasma humano. Son preparados en plantas frac-cionadoras industriales a partir de mezclas de plasma provenientes de entre5.000 a 10.000 donantes, siguiendo el método desarrollado por Cohn. Losproductos finales se presentan como productos farmacéuticos, en forma lí-quida o liofilizada tras ser sometidos a procesos físicos y/o químicos de in-activación viral.
ALBÚMINA HUMANA
La albúmina se prepara mediante precipitación con alcohol de plasma hu-mano, siendo posteriormente sometida a pasteurización durante 10 horas a60ºC para la inactivación viral. Es el derivado plasmático más seguro desde elpunto de vista de la transmisión de enfermedades infecciosas conocidas, conla excepción, probablemente, del parvovirus B19. De acuerdo con la Farma-copea Europea, estos preparados deben contener al menos un 95% de albú-mina, siendo el resto globulinas y otras proteínas, con un contenido en sodiode 145 mmol/l y 2 mmol/l de potasio.
Participa en actividades fisiológicas fundamentales como el mantenimientode la presión osmótica intravascular o el transporte de proteínas, minerales,fármacos, etc.
PRESENTACIÓN Y CONSERVACIÓN
La albúmina humana se presenta en concentraciones isooncóticas al 5% ohiperonçoticas, al 20%, y con diferentes volúmenes que varían según el su-ministrador. Existen productos menos purificados (PPL ó PPF) en los que la al-búmina constituye algo más del 80%. El almacenamiento se realizageneralmente a temperatura ambiente, excepto en los casos especificadospor el fabricante.
91
DERIVADOS PLASMÁTICOS

INDICACIONES
Siguen siendo polémicas las situaciones clínicas susceptibles de beneficiarsede la albúmina humana, e incluso un meta-análisis de 1998 sugiere que su usose asocia con una mayor mortalidad en los pacientes críticos en comparacióncon los cristaloides. Otros meta-análisis, revisiones críticas y estudios reali-zados posteriormente han intentado definir las indicaciones adecuadas de laalbúmina, teniendo siempre en cuenta su disponibilidad limitada y alto coste.
Hipovolemia
Solo está indicada la albúmina al 5% en caso de contraindicación para la ad-ministración de cristaloides o coloides sintéticos, o de falta de eficacia deéstos
Hipoalbuminemia
En caso de concentraciones plasmáticas inferiores a 20 g/l estaría indi-cada la administración de soluciones de albúmina al 20%, pero sólo du-rante períodos muy limitados de tiempo. Si existen edemas puede sernecesaria la administración concomitante de diuréticos. Se consideraapropiado su uso en recambios plasmáticos terapéuticos de más de másde 20ml/kg, en la peritonitis bacteriana espontánea y en caso de pare-centesis de más de 5 litros en pacientes con ascitis refractaria. En casosindividualizados puede estar indicada en el periodo post-operatorio en ci-rugía una vez normalizado el volumen circulatorio, en la cirrosis hepáticacon ascitis refractaria, en el síndrome hepatorenal, en el síndrome nefró-tico con hipovolemia y/o edema pulmonar, en el periodo post-operatoriodespués del trasplante hepático para controlar la ascitis y edema perifé-rico, y en quemaduras de más del 30% de superficie corporal después delas primeras 24 horas.
CONTRAINDICACIONES
Antecedentes de alergia a la albúmina o al preparado en concreto. No está in-dicada la administración de albúmina humana en enfermedades que cursancon hipoalbuminemia crónica por síntesis disminuida o problemas de la per-meabilidad capilar. Como la infusión de albúmina provoca aumento del vo-lumen intravascular, las situaciones acompañadas de hipervolemia deben
92

considerarse una contraindicación relativa, y ha de tenerse en cuenta la fun-ción cardiaca del paciente.
EFECTOS ADVERSOS
Pueden producir reacciones alérgicas leves o, más rara vez, reacciones anafilác-ticas. Puede producirse sobrecarga circulatoria y situaciones de hipotensión enpacientes tratados con EACAS. No se ha descrito transmisión de agentes víricos.
DOSIS
En general la dosis debe ajustarse a las necesidades de cada paciente. En te-rapia sustitutiva, la dosis necesaria se determina a partir de los parámetros cir-culatorios usuales.
En adultos la dosis puede determinarse de forma aproximada a partir de la si-guiente fórmula:
Dosis (g) = Concentración deseada de albúmina (g/dl) - concentración actual de albúmina (g/dl) x (kg peso x 0,8)
93
ALBÚMINA HUMANA:Presentación, indicaciones, dosificación y administración
Presentación Al 5% y 20%Principales indicaciones • Hipovolemia: cuando existe contraindicación de coloides y
cristaloides, o por ineficacia de los mismos• Hipoalbuminemia; Si albúmina< 20 g/l, en periodos cortos• Recambios plasmáticos de >20 ml/kg/d• Peritonitis bacteriana espontánea• Paracentesis de > 5 l en ascitis refractarias• Ocasionalmente en otras patologías
Dosificación En terapia sustitutiva: [Concentración deseada de albúmina (g/dl) –concentración ac-tual de albúmina (g/dl)] x (kg x 0,8)
Administración Infusión endovenosaRitmo infusión Velocidad individualizada
Orientativo:Albúmina 20%: 1 - 2 ml/minuto Albúmina 5%: 5 ml/minuto

ADMINISTRACIÓN
En infusión endovenosa. La velocidad se ajustará según las circunstancias in-dividuales, no obstante ésta suele ser de 1-2 ml/min para los preparados al20% y 5 ml/min para los de concentración al 5%.
REFERENCIASLiumbruno G, Bennardello F, Lattanzio A, Piccoli P, Rossetti G. Recomendattions for theuse of albumin and immunoglobulins. Blood Transfus 2009,7:216-34. Boldt J. Use of albumin: an update. Br J Anaesth 2010,104(3):276-84.
INMUNOGLOBULINAS
Las inmunoglobulinas (Ig) se obtienen del plasma mediante el procedi-miento de fraccionamiento de Cohn con alcohol. Los preparados para ad-ministración intramuscular son sometidos posteriormente a unprocedimiento de concentración proteica, mientras que los de administra-ción intravenosa son tratados para eliminar los agregados de Ig de alto pesomolecular, de intensa acción anticomplementaria, haciéndolos así segurospara su infusión intravenosa. Estos preparados contienen fundamental-mente IgG (95%) y muy pequeñas cantidades de IgM e IgA. La Ig tiene unavida media de 18-32 días y, una vez administrada, se distribuye rápidamenteentre el plasma y el espacio extravascular, alcanzándose un equilibrio a los3-5 días.
Las Ig inespecíficas contienen anticuerpos con un amplio espectro de es-pecificidades, que incluye desde los dirigidos contra múltiples agentes in-fecciosos hasta autoanticuerpos y anticuerpos anti-idiotipo. Por ello nosólo actúan aumentando los mecanismos inespecíficos de defensa sinoque también intervienen en la modulación de la respuesta inmune o en elbloqueo temporal de los receptores Fc del sistema mononuclear fagocí-tico. Existen otros preparados que se obtienen a partir de plasmas de do-nantes hiperinmunizados que poseen títulos mayores de anticuerposdirigidos contra determinados agentes infecciosos, y son conocidos comoIg monoespecíficas.
94

EFECTOS ADVERSOS
La administración de Ig por vía intramuscular se ha asociado a la aparición dereacciones anafilácticas, especialmente en aquellos pacientes que las recibende forma repetida. La infusión intravenosa se acompaña hasta en el 10 % delos casos de reacciones adversas, en general leves, como escalofríos, cefalea,mareo, fiebre, erupción cutánea y prurito. Muchos de estos efectos adversosestán relacionados con la velocidad de infusión, por lo que se recomiendaempezar con velocidades de infusión de 0,5 mg/kg/min que se van aumen-tando progresivamente hasta 3 mg/kg/min como máximo, dependiendo dela tolerancia. En ocasiones puede ser necesario premedicar con paracetamolo antihistamínicos. El riesgo de transmisión de enfermedades infecciosas esactualmente muy bajo, gracias a la introducción de técnicas de PCR para elVHC y otros virus, y a los distintos procedimientos de inactivación viral a queson sometidos estos preparados.
INMUNOGLOBULINAS INESPECÍFICAS
Inmunoglobulinas intramusculares
Se utilizan como alternativa a ciertas Ig específicas si no se dispone de ellas(hepatitis A, sarampión, rubéola o parotiditis) (Tabla 8). También se han utili-zado como infusiones subcutáneas en inmunodeficiencias primarias y se-cundarias. Se debe evitar administrar más de 5 ml en un solo sitio deinyección.
Hepatitis A
Profilaxis en individuos que han sufrido exposición al virus de la hepatitis A:0,02 ml/kg (corresponde a 3 mg/kg del preparado disponible comercialmenteal 16,5%, 165 mg/ml) en dosis única, lo antes posible y siempre antes detranscurridas 2 semanas de la exposición.
Para profilaxis en viajes a zonas endémicas, sin vacunación o en las 2 -3 se-manas después de ésta, si la estancia es menor a 3 meses: 0,02 ml/kg en dosisúnica. Si la estancia es superior a 3 meses: 0,06 ml/kg una dosis cada 4-6 se-manas. La inmunización pasivo-activa con Ig y vacunación puede ser útil enlas situaciones en que se precise combinar una protección rápida y duradera.
95
Sarampión
Profilaxis dentro de los 7 días siguientes a la exposición. : 0,25 ml/kg en pa-cientes inmunocompetentes y 0,5 ml/kg en pacientes inmunodeprimidos, conuna dosis máxima de 15 ml en ambos casos.
Inmunodeficiencias primarias y secundarias
Para profilaxis de infecciones. La dosis inicial es de 1-2 ml/kg, y la dosis demantenimiento de 0,7 ml/kg (corresponde a 100 mg/kg de una solución al16,5%) una vez cada 2-4 semanas, debiendo ajustarse para antenern un nivelde aproximadamente 200 mg/dl de IgG circulante. La dosis máxima únicaserá de 30-50 ml en adultos y 20-30 ml en niños.
96
Tabla 8
INMUNOGLOBULINAS INESPECÍFICAS INTRAMUSCULARES:Principales indicaciones y dosis
INDICACIÓN DOSISHepatitis A • Profilaxis en contacto 0,02 ml/kg peso dosis
única antes de 2 semanas• Viaje zona endémica sin vacunación 0,02 ml/kg peso dosis
(o en las 2-3 semanas post) para únicaestancias <3 meses
• Viaje zona endémica sin vacunación 0,06 ml/kg peso cada(o en las 2-3 semanas post) con 4 – 6 semanasestancia>3 meses
Sarampión • Profilaxis en contacto 0,25 ml/kg peso dosis única) antes de los 7 días (dosis máxima: 15 mlPacientes inmunodepri-midos: 0,5 ml/kg peso dosis única antes de los 7 días (dosis máxima 15 ml)
Inmunodeficiencias • Profilaxis de infecciones Dosis inicio: 1 – 2 ml/kgprimarias y Dosis mantenimiento: 0,7 secundarias ml/kg cada 2-4 semanas
Dosis máxima: Adultos: 30 – 50 mlNiños: 20 – 30 ml

Inmunoglobulinas intravenosas (IgIV)
La administración de IgIV está indicada para la profilaxis de infecciones bac-terianas en pacientes con concentraciones séricas disminuidas de Ig o ba-sándose en su efecto inmunomodulador, en el tratamiento de enfermedadesmuy diversas (Tabla 9).
Inmunodeficiencias primarias
Está indicado el tratamiento mantenido con inmunoglobulinas inespecíficas,con ajuste de la dosis en función de las concentraciones plasmáticas de IgGen inmunodeficiencias como la agammaglobulinemia ligada al cromosoma X,la inmunodeficiencia común variable, el síndrome de hiper-IgM, y en inmu-nodeficiencias combinadas graves.
En otras inmunodeficiencias y defectos de subclases de IgG, el tratamientosólo está justificado si se producen infecciones severas recidivantes o es-casa capacidad de inmunización tras las vacunaciones. La dosis inicial ha-bitual es de 0,4-0,8 g/kg, seguida de una dosis de mantenimiento de 0,25g/kg cada 3-4 semanas. La dosis se ajustará individualmente para intentarconseguir concentraciones de unos 500 mg/dl y prevenir las infecciones.Durante las infecciones activas puede ser necesario administrar dosis adi-cionales.
Inmunodeficiencias secundarias
En pacientes con inmunodeficiencias secundarias, como en la Leucemia Lin-fática Crónica o en el mieloma, con niveles plasmáticos bajos de Ig (<3 ó 6mg/dl) y con infecciones bacterianas recurrentes se utilizan a dosis de 0,2-0.4g/kg cada 3-4 semanas.
Infecciones pediátricas por VIH
En niños con CD4>200/ml la administración de IgIV parece ofrecer ciertaprotección frente a las infecciones bacterianas, utilizando dosis de 0,2-0,4g/kg cada 3-4 semanas.
Trasplante ologénico de médula ósea
Como profilaxis de infecciones y EICH, en el periodo pre y post-trasplante.Dosis: 0,5 mg/kg una o dos semanas antes del trasplante, seguida de 0,5
97

mg//semana durante 3 meses. En caso de déficit persistente de formaciónde anticuerpos administrar 0,5 mg/kg cada 4 semanas.
Púrpura trombocitopénica idiopática (PTI)
Indicado en niños y adultos en situaciones en que interese aumentar rápida-mente la cifra de plaquetas (hemorragia o pre-cirugía) Dosis: 0,8-1,0 g / Kg /día durante 1-2 días o 0,4 g / Kg / día durante 3-5 días.
98
Tabla 9
INMUNOGLOBULINAS INESPECÍFICAS INTRAVENOSAS:Principales indicaciones y dosis
INDICACIÓN DOSISInmunodeficiencias • Tratamiento de mantenimiento en: Dosis inicio: primarias Agammaglobulinemia ligada al 0,4 – 0,8 g/Kg
cromosoma X, I nmunodeficiencia cada 3 – 4 semanascombinada severa, Síndrome de hiper-IgM, Inmunodeficiencia Ajustar dosis paracomun variable conseguir nivel
• Si infecciones graves recidivantes en IgG>500 mg/lotras inmunodeficiencias
Inmunodeficiencias • Si >3 infecciones graves/año o sepsis 0,2 – 0,4 g/ kg pesosecundarias cada 2 – 4 semanas Niños con infección • Iniciar antes de que CD 4 sea <200/l 0,2 – 0,4 g/ kg pesopor HIV cada 3 – 4 semaTrasplante alogénico • Profilaxis infecciones y EICH 0,5 mg/kg al iniciomédula ósea • Déficit persistente de anticuerpos 0,5 mg/kg/semana
durante 3 meses0,5 g/kg cada 4 semanas
Púrpura trombo- • En situación de riesgo (hemorragia o 0,8 – 1 g/ kg/ día, 1 ó 2citopénica idiopática pre-cirugía) días ó 0,4 g/ kg/ día,
3 – 5 díasOtras posibles • Síndrome de Guillen- Barré 0,8 – 1 g/ kg/ día, indicaciones • Síndrome de Kawasaki 1 ó 2 días ó
• Refractariedad plaquetaria 0,4 g/ kg/ día,• Esclerosis múltiple recidivante 3 – 5 días• Hemofilia con inhibidor de FVIII• Necrólisis epidérmica• Púrpura postransfusional

Otras posibles indicaciones
En el síndrome de Guillain-Barré y en la enfermedad de Kawasaki su eficaciaestá claramente establecida. Hay otras enfermedades en las que las IgIV pa-recen útiles, pero requieren más estudios de confirmación: esclerosis múlti-ple recidivante, hemofilia con inhibidores de FVIII, necrolisis epidérmica,púrpura postransfusional, enfermedad hemolítica del recién nacido, refracta-riedad plaquetaria, etc
INMUNOGLOBULINAS ESPECÍFICAS
En la actualidad se dispone de los siguientes preparados (Tabla 10):
Inmunoglobulina anti Rh (D)
Su indicación principal es en mujeres Rh (D) negativas, para prevenir la en-fermedad hemolítica del recién nacido por isoinmunización anti-D
� Profilaxis ante parto: Se administrará 300 μg en la semana 28 del emba-razo.
� En caso de aborto, interrupción del embarazo, embarazo ectópico, amnio-centesis, biopsia de corion, mola y placenta previa: se administrará lo antesposible después y no más tarde de las 72 horas siguientes, una dosis de 50μg si se produce en el primer trimestre del embarazo y de 300 μg si es pos-teriormente
� Profilaxis postparto: Si el recién nacido es Rh (D) positivo se administrarálo antes posible después del parto y no más tarde de las 72 horas siguien-tes una dosis de 300 μg.
� Hemorragia trasplacentaria: si e mayor de 30 ml de sangre fetal se debe in-crementar la dosis de Ig.
� Tras transfusión de componentes sanguíneos con hematíes Rh (D) posi-tivo: En el caso de transfusión de plaquetas la dosis será de 250-300 μg,en caso de transfusión de concentrado de hematíes la dosis será de 10-15 μg por mililitro de hematíes transfundidos. Siempre se administrará loantes posible después de la transfusión y no más tarde de las 72 horas si-guientes.
� No es necesario administrar la Inmunoglobulina anti Rh (D) a pacientes Rh(D) negativo que sean varones o mujeres que no se encuentran en edad
99

gestacional, tras la transfusión de componentes plaquetarios obtenidos apartir de donantes Rh (D) positivo.
Inmunoglobulina anti hepatitis B
En situaciones de exposición al virus de la hepatitis B percutánea o mucosaen personas no inmunizadas o cuyo nivel de anticuerpos sea inferior a 10UI/ml se administrará una dosis de 12-20 UI/kg lo antes posible, preferible-mente en el plazo de 48 horas y se iniciará la vacunación. En personas cuyasituación inmune sea desconocida se comprobará el nivel de anticuerpos anti-HBs y se procederá según éste.
En recién nacidos de madres con determinación de Antígeno de Superficie dela Hepatitis B (HBsAg) positivas en el plazo de las 12 horas siguientes al na-cimiento se administrará una dosis de 40 UI/Kg junto con la primera dosis dela vacuna.
Innmunoglobulina anti tetánica
Profilaxis de tétanos en personas que han sufrido heridas recientemente yque no han recibido la vacuna en los 10 últimos años, o cuya pauta de vacu-nación ha sido incompleta o se desconoce, se administrará 250 UI. Si el des-bridamiento de la herida es tardío o incompleto la dosis será de 500 UI.Siempre se iniciará vacunación con toxoide tetánico.
Tratamiento del tétanos clínicamente manifiesto se administrará 3.000-6.000UI/24 horas en inyección única. La duración del tratamiento dependerá delcuadro clínico.
Inmunoglobulina anti rábica
Profilaxis de la rabia después de una mordedura por animal con posibilidad decontagio de rabia se administrará una dosis de 20 UI/kg. Se administrará si-multáneamente la vacuna antirrábica.
Inmunoglobulina anti Citomegalovirus
Profilaxis en trasplante de órganos se administrará 100-150 mg/kg ó 50-100U-PEI/kg cada 2-3 semanas
100

101
Tabla 10
INMUNOGLOBULINAS ESPECÍFICAS: Principales tipos, indicaciones y dosis
INMUNOGLOBULINA ANTI Rh (D)INDICACIÓN DOSISMujeres Rh (D) • Gestación 300 μg en semana 28 gestaciónnegativo • Aborto, embarazo ectópico, Administrar antes de 72 horas:
mola, amniocentesis, biopsia 50 μg si gestación <12 semanasde corion, placenta previa 300 μg si gestación >12 semanas
• Postparto Administrar antes de 72 horas: 300 μg• Tras transfusión de Administrar antes de 72 horas:componentes sanguíneos 250 – 300 μg si transfusión plaquetas.conhematíes Rh(D) 10 – 15 μg / ml de c. hematíespositivo transfundido
INMUNOGLOBULINA ANTI HEPATITIS BINDICACIÓN DOSISExposición al virus percutánea o mucosa en Administrar antes de 48 horaspersonas no inmunizadas 12 – 20 UI / kg Recién nacidos de madres HBsAg positivas Administrar antes de 12 horas
40 UI / kg INMUNOGLOBULINA ANTI TETÁNICAINDICACIÓN DOSISHeridas y no vacunación en 10 años Dosis usual: 250 UI
Si desbridamiento tardío: 500UI(iniciar vacunación)
Tratamiento de tétanos 3.000 – 6.000 UI/24 horasDuración: según evolución
INMUNOGLOBULINA ANTI RÁBICAINDICACIÓN DOSISTras mordedura animal con posible contagio Administrar 20 UI / kg
(iniciar vacunación)INMUNOGLOBULINA ANTI CITOMEGALOVIRUSINDICACIÓN DOSISPrevio a trasplante de órganos Administrar 100 – 150 mg / kg
ó 50 – 100 U-PEI / kgcada 2 – 3 semanas

REFERENCIAS
Liumbruno G, Bennardello F, Lattanzio A, Piccoli P, Rossetti G. Recomendat-tions for the use of albumin and immunoglobulins. Blood Transfus2009,7:216-34.
Protocolo de diagnóstico y prevención de la enfermedad hemolítica del fetoy del recién. SETS. SEGO. Marzo 2008.
FACTORES DE COAGULACIÓN
El tratamiento de las complicaciones hemorrágicas en pacientes con coagu-lopatías congénitas, mediante la reposición en el plasma de la proteína defi-citaria es una modalidad terapéutica ampliamente difundida y cada vez máscompleja. Puesto que el tratamiento con concentrados de factores de la co-agulación derivados del plasma ha causado la transmisión de enfermedadesvirales, como la hepatitis y el VIH, es importante asegurar que los productosque se emplean sean seguros y libres de carga vírica. Desde la década de los80, las agencias reguladoras y los productores de derivados plasmáticos parauso clínico se han hecho eco de esta preocupación, y han desarrollado unaserie de medidas, para reducir –si no eliminar- este riesgo.
Básicamente, estas se basan en:
� Seleccionar adecuadamente a los donantes.� Realizar un cribado riguroso del plasma.� Eliminación de los virus contaminantes durante el proceso de producción.
Además, con el desarrollo de nuevas tecnologías, se ha conseguido obtenerconcentrados de gran pureza. En los últimos años, la aplicación de técnicas deingeniería genética ha conducido al desarrollo de concentrados de factores re-combinantes altamente seguros y eficaces.
FACTOR VIII
Es el factor de coagulación derivado del plasma que presenta una mayor de-manda. De acuerdo con el grado de pureza los concentrados de FVIII se co-nocen como preparados de pureza intermedia, alta, o muy alta.
102

� Los concentrados de pureza intermedia se obtienen con métodos conven-cionales de precipitación y fraccionamiento plasmático.
� Los de pureza alta habitualmente son preparados por purificación croma-tográfica con metodología de intercambio iónico o de afinidad a heparina,mientras que los de muy alta pureza lo son por inmunoafinidad cromato-gráfica, con la utilización de anticuerpos monoclonales.
� Los productos de alta y muy alta pureza están prácticamente desprovistosde contaminantes como inmunoglobulinas, fibrinógeno y fibronectina, ysu purificación implica un “fraccionamiento” de la carga viral, lo que puedeconsiderarse como un complemento a los procedimientos de atenuaciónviral empleados.
INDICACIONES
Los concentrados de pureza intermedia se indican en los casos de enfermedadde von Willebrand (EvW) donde el tratamiento con desmopresina (DDAVP) noestá indicado o es ineficaz. En pacientes con hemofilia A se emplean prioritaria-mente concentrados plasmáticos de alta o muy alta pureza. En la actualidad, eltratamiento de elección de los pacientes recién diagnosticados con hemofiliasevera en los países desarrollados es con factores recombinantes. Aún así, exis-ten razones clínicas que pueden aconsejar, en determinadas ocasiones, la utili-zación de concentrados plasmáticos de FVIII, por ejemplo el deseo personal yparticular del propio paciente; que el paciente tenga una historia anterior de for-mación de inhibidores tras el uso de factores plasmáticos o recombinantes; la ne-cesidad de borrar un inhibidor producido por un factor plasmático (tratamientopor inmunotolerancia) o el desabastecimiento de factores recombinantes.
A nivel mundial, los factores derivados del plasma posiblemente seguirán siendola única opción terapéutica en el 80% de los hemofílicos, pues debido a su mayorcoste tienen acceso nulo o muy limitado a cualquier terapia de reemplazo.
FACTOR VON WILLEBRAND (FVW)
La mayoría de pacientes con enfermedad de von Willebrand (EvW) tipo 1 ex-presan sólo un sangrado moderado y requieren tratamiento en caso de trau-matismos o de cirugía, siendo el DDAVP el tratamiento de elección, al igualque en pacientes con hemofilia A moderada o leve.
103

Los pacientes con EvW con variantes tipo 2, los pacientes con EvW tipo 1 o3 graves (20-30% de pacientes con EvW), los que no responden al DDAVP oque exhiben efectos adversos importantes, son candidatos para una terapiade reemplazo con productos que contengan FvW.
Los derivados plasmáticos de FVIII/FvW purificados y sometidos a inactiva-ción viral, son los más frecuentemente empleados en este contexto. Además,ha sido desarrollado un FvW derivado del plasma, purificado, casi totalmentedeplecionado de FVIII. Hay que tener en cuenta que la hemostasia no puedellevarse a cabo hasta que la actividad del FVIII coagulante ha alcanzado el40%, y que la infusión de FvW sólo no aumenta los niveles endógenos delFVIII durante al menos 6-12 horas, por lo que en situaciones de emergenciahay que administrar FVIII junto a la primera infusión de FvW. En la actuali-dad se están llevando a cabo un estudio aleatorizado y controlado para eva-luar la farmacocinética, seguridad, y tolerancia FvW de origen recombinanteen pacientes con EvW tipo 3.
CONCENTRADOS DE COMPLEJO PROTROMBÍNICO (CCP) Y FIX
Durante mucho tiempo, el único tratamiento existente para la hemofilia Bha sido el CCP. Las proteínas del CCP son separadas del sobrenadante del crio-precipitado y posteriormente purificadas por adsorción a sales inorgánicas opor cromatografía de intercambio iónico. Contienen principalmente factoresII, VII, IX y X y anticoagulantes naturales como la Proteína C, Proteína S y laProteína Z. Para prevenir o reducir la activación de los factores durante el pro-ceso de producción se añade heparina, y en algunos preparados antitrom-bina.
Los efectos adversos fundamentales son el desarrollo de episodios trombóti-cos y de coagulación intravascular diseminada (CID), fundamentalmente enpacientes con hepatopatías graves.
INDICACIONES
Su indicación es en el tratamiento del sangrado o en la profilaxis periopera-toria de pacientes con:
� Deficiencias congénitas de factores de la coagulación dependientes de la vi-
104

tamina K, cuando el producto de factor de coagulación específico purificadono está disponible.
� Deficiencias adquiridas de factores de la coagulación dependientes de la vi-tamina K –p. ej. en tratamientos anticoagulantes- cuando por hemorragiagrave se requiere una reversión rápida del efecto.
� Los concentrados de complejo protrombinasa activados son CCP en losque las proteínas de coagulación han sido activadas. Se emplean para tra-tar hemofílicos con inhibidores. Este producto es efectivo en controlar elsangrado de articulaciones y tejidos blandos en el 80% de estos pacientes
En la actualidad existen concentrados altamente purificados de FIX conse-guidos por diferentes procedimientos de purificación, bien por cromatografíade intercambio iónico o por inmunoafinidad. Está ampliamente demostradasu eficacia terapéutica, así como la ausencia de complicaciones trombóticas.En aquellas situaciones en las que no se dispone de concentrados de FIX re-combinante, es el tratamiento de elección para pacientes con hemofilia B.
FIBRINÓGENO
Los concentrados de fibrinógeno sometidos a inactivación vírica se obtienende la primera fracción precipitada del proceso de Cohn.
Se emplea para el control de las hemorragias en las deficiencias congénitasde fibrinógeno (hipofibrinogenemia / afibrinogenemia). Teniendo en cuenta lalarga vida media del fibrinógeno (2-4 días), se puede llevar a cabo una profi-laxis regular con infusiones semanales del concentrado. Esto se recomiendaen pacientes que sangran frecuentemente y en órganos críticos, como mús-culos, articulaciones, tracto gastrointestinal y sistema nervioso central.
En España, no existe ningún concentrado de fibrinógeno comercializado, de-biéndose obtener como medicamento extranjero.
FACTOR VII
La indicación clínica de este producto es para el tratamiento o profilaxis desangrado en situaciones de riesgo, de pacientes con deficiencias congénitasde factor VII y niveles de FVII:C por debajo del 25%. No obstante, el empleo
105

de forma satisfactoria del FVIIar en este contexto lleva a considerar comoadecuado el empleo de este producto en este tipo de pacientes.
El factor VII derivado de plasma, a diferencia del FVIIar no contiene FVII ac-tivo, por lo que no debe ser administrado en pacientes con hemofilia A o B coninhibidores.
FACTOR XI
Aunque el plasma fresco congelado (PFC) se ha empleado con éxito en pa-cientes con deficiencias de FXI, sin embargo, para alcanzar los niveles tera-péuticos, la administración de grandes cantidades de volumen puedeocasionar sobrecarga de volumen.
Los concentrados de FXI están disponibles, de manera controlada, para pacien-tes con deficiencias de FXI. Puesto que estos factores pueden ser trombogéni-cos, los niveles postinfusión de FXI no deben exceder las 70 U/dl. Además, sedebe evitar el ácido tranexámico en pacientes que reciben concentrados de FXI.
FACTOR XIII
Los concentrados de factor XIII se utilizan como tratamiento sustitutivo parael sangrado de pacientes con déficit congénito de factor XIII, favoreciendotambién la cicatrización de heridas en estos enfermos.
Debido a la larga vida media del FXIII (11-14 días), estos concentrados pue-den ser infundidos a intervalos largos de tiempo (20-30 días).
La seguridad, farmacocinética, e inmunogenicidad de una preparación de FXIIIrecombinante en personas sanas ha sido demostrado en un estudio fase 1,aunque en monos ha producido una coagulopatía generalizada a dosis su-praterapéuticas.
SISTEMA ADHESIVO DE FIBRINA
El término sistema hemostático de fibrina se refiere a un producto que con-tiene factores de coagulación a ser administrados de forma local para producirun coágulo de fibrina.
106

De forma típica, el sistema adhesivo de fibrina consiste en al menos dos compo-nentes, uno de ellos fibrinógeno con/sin FXIII, y el otro trombina, que se mantienenseparados hasta que su mezcla, en presencia de calcio, induce la formación de fibrina.
El sistema adhesivo de fibrina se ha venido empleando en los últimos 30 años,y cada vez su uso se expande, al haberse demostrado una reducción signifi-cativa de pérdida sanguínea en el periodo perioperatorio, en la exposición asangre alogénica, y en la cantidad de sangre transfundida.
En concreto, este producto está indicado en situaciones donde:
� Existe un deficiente sistema coagulativo en el paciente.� No es posible la aplicación de presión mecánica. � Tejidos difíciles de suturar (p.ej. el pulmón o el hígado).� Es crítico una adecuada hemostasia (p.ej. cerebro)
CONCENTRADOS DE ANTITROMBINA
El concentrado de antitrombina es un derivado del plasma humano some-tido a inactivación viral, existiendo también comercialmente disponibles pre-paraciones de antitrombina obtenida por tecnología recombinante.
Los concentrados de antitrombina se administran en pacientes con deficien-cias congénitas de antitrombina con trombosis establecida, o como profila-xis en el periodo peri-parto, o en cirugías. En deficiencias adquiridas deantitrombina su empleo es más controvertido, existiendo pocos estudios clí-nicos realizados en este contexto.
CONCENTRADOS DE PROTEÍNA C
En los últimos años se encuentra aprobado el uso de concentrados de Prote-ína C como terapia de sustitución en pacientes con problemas severos de co-agulación ligados a una deficiencia congénita grave de proteína C, comonecrosis cutáneas al comienzo de tratamiento con antagonistas de la vitaminaK, eventos trombóticos severos en neonatos.
Respecto a los concentrados de proteína C activada de origen recombinante, cuyaindicación es la reducción de la mortalidad por sepsis severa asociada a fallo or-gánico son necesarios estudios adicionales que muestren su utilidad y eficacia.
107
108
Tratamiento de sustitución de deficiencias de proteínas de coagulación
Deficiencia Cirugía mayor Cirugía menor Sangrado espontáneo ComentariosFibrinógeno Concentrado (20-30 mg/kg) PFC (15-20 ml/kg) PFC (15-20 ml/Kg) Profilaxis con
PFC (15-20 ml/kg) Nivel: >50 mg/dl, 2-3d Nivel: >50 mg/dl hasta concentrado semanalCrioprecipitado (1/10kg) cese hemorragia (20-30 mg/kg) si elNivel: >50 mg/dl hasta sangrado espontáneocicatrización es frecuente y severo
Protrombina CCP (20-30 U/kg) PFC (15-20 ml/kg) PFC (15-20 mal/kg) Tras CCP, FVII, FIX, y FXPFC (15-20 ml/kg) Nivel: >30%, 2-3 d Nivel: >30% hasta no deberían excederNivel: >30% hasta cese hemorragia el 150%cicatrización
V PFC (15-20 ml/kg) Como en cirugía mayor Como en cirugía mayor No hay concentradosNivel: >20% hasta Nivel: >20%, 2-3 d Nivel: >20% hasta disponiblescicatrización cese hemorragia
VII Fiar (15-30 mg/kg) a Como en cirugía mayor Como en cirugía mayor Monitorizar conintervalos de 12h) Nivel: >20%, 2-3 d Nivel: >20% hasta ensayos de actividadConcentrado (30-40 U/kg cese hemorragia FVIIa intervalos de 12h)Nivel: >20% hasta cicatrización
VIII Leve: DDAVP (0.3 μg/kg) Leve: DDAVP (0.3 μg/kg) Como en cirugía mayor En pacientes conSevera: Concentrado Severa: Concentrado Nivel: 40-100% inhibidores, administrar(40-50 U/kg) (25 U/kg) dependiendo de FVIIar (90 μg/kg), CCPNivel: Inicial del 60-100%, Nivel: Inicial del 50%, localización de (30-50 U/kg/12h), seguido del 50% 2-3 d hemorragia CCPa (75-100 U/kg)
IX Concentrado (60-80 U/kg) Concentrado (40 U/kg) Como en cirugía mayor En pacientes conNivel: Inicial del 60-80%, Nivel: Inicial del 40%, Nivel: 30-80% inhibidores, administrarseguido del 30% 2-3 d dependiendo de FVIIar (90 μg/kg), CCP
localización de (30-50 U/kg/12h), hemorragia CCPa (75-100 U/kg)
X CCP (20-30 U/kg) PFC (15-20 mL/kg) Como en cirugía mayor Tras CCP, FVII, FIX, yPFC (15-20 mL/kg) Nivel: >30%, 2-3 d Nivel: >20% hasta FX no deberíanNivel: >20% hasta cese hemorragia exceder el 150%cicatrización
XI PFC (15-20 mL/kg) Como en cirugía mayor Como en cirugía mayor Los niveles de FXI gene-Nivel: >20% hasta Nivel: >20%, 2-3 d Nivel: >20% hasta ralmente pueden conse-cicatrización cese hemorragia guirse con infusiones a
días alternos

109
REFERENCIASPeyvandi F, Palla R, Menegatti M, Mannucci PM. Rare Bleeding Disorders: General Aspectsof Clinical Features, Diagnosis, and Management. Seminars of Thrombosis and He-mostasis 2009; 35:349-355.Liras, A. Recombinant proteins in therapeutics: haemophilia treatment as an example.International Archives of Medicine 2008; 1:1-4.Pipe SW. Recombinant clotting factors. Trombosis and Haemostasis 2008; 99: 840–850.Key NS, Negrier C. Coagulation factor concentrates: past, present, and future. Lancet2007; 370: 439-48.Mannucci M, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood2004; 104: 1243-1252. �
Deficiencia Cirugía mayor Cirugía menor Sangrado espontáneo ComentariosXIII Concentrado(10-20 U/kg) Como en cirugía mayor Como en cirugía mayor Profilaxis en todos los
PFC (15-20 mL/kg) Nivel: >5%, 2-3 d Nivel: >5% hasta pacientes. La reposiciónCrioprecipitado (1/10kg) cese hemorragia se puede llevar a caboNivel: >5% hasta cada 20-30dcicatrización
FvW Tipo 1: DDAVP (0,3 µg/kg) Tipo 1: DDAVP (0,3 Tipo 1: DDAVP (0,3 En pacientes conµg/kg) µg/kg) inhibidores, FVIII
Tipos 2 y 3: Concentrados Tipos 2 y 3: Tipos 2 y 3: recombinante o FVIIarde FVIII/FvW (50U/kg) Concentrados de FVIII/ Concentrados de FVIII/Nivel: FVIII >50% hasta FvW (40U/kg) FvW 25U/kgcicatrización Nivel: FVIII >30%, 2-3 d Nivel: FVIII >30%
hasta cese de hemorragiaPC Concentrados (40 UI/kg/ - - Administrar también
12h) hasta reinicio de concentrados al inicio anticoagulación oral - - del tratamiento con
cumarínicosAntitrombina Concentrados (60 U/kg/24h)
Niveles >80-100%
Abreviaturas: PFC: plasma fresco congelado; CCP: concentrado de complejo protrombinasa; CCPa:concentrado de complejo protrombinasa activado; FVIIar: FVII activo recombinante; DDAVP: des-mopresina.


SOLICITUD DE TRANSFUSIÓN
La solicitud de transfusión o instrucción escrita para solicitar una transfusión,es una prescripción médica, que deberá contener todos los elementos nece-sarios para una correcta práctica transfusional.
En toda solicitud ha de especificarse claramente toda la información que per-mita:
� La correcta identificación del receptor: nombre y apellidos, fecha de naci-miento, sexo y localización.
� Indicación de la transfusión: motivo de la transfusión y diagnóstico.� Componente requerido: producto y cantidad (número de unidades o volu-
men en pacientes pediátricos), así como indicaciones específicas como irra-diación, de donante con serología Citomegalovirus (CMV) negativo.
� Identificación clara del médico prescriptor.� Fecha y hora de la solicitud y grado de urgencia (urgente, desesperada, en
el día, reserva operatoria,…).� Antecedentes personales: transfusiones previas, historia obstétrica, anti-
cuerpos conocidos o reacciones adversas anteriores.� Parámetros hematológicos.
MUESTRA PRETRANSFUSIONAL
La identificación del paciente en el momento de la extracción de la mues-tra es clave para evitar el mayor riesgo transfusional. Los errores de identi-ficación del receptor son los responsables de la mayoría de los accidentestransfusionales. La administración equivoca de un concentrado de hematíesAB0 incompatible provoca la mayoría de las reacciones hemolíticas morta-les. Los errores de identificación pueden ocurrir en cualquiera de los trespuntos críticos: en el momento de la extracción de la muestra, durante larealización de las pruebas de compatibilidad o en el momento de la trans-fusión.
111
PRÁCTICA TRANSFUSIONAL

Los Servicios de transfusión deberán tener un procedimiento que permita ga-rantizar la adecuada identificación del paciente, de las muestras pretransfu-sionales y de los componentes administrados
� Debe realizarse siempre la identificación positiva del receptor (nombre yapellidos) así como establecer todos los mecanismos propios de cada cen-tro (brazaletes de identificación, registro con código de barras, identifica-ción con radiofrecuencia (RFID),…) e identificar los tubos siempre en lacabecera del enfermo en el momento de la extracción.
� Cuando la identificación positiva no sea posible (enfermos pediátricos, in-conscientes, o con barreras de lenguaje) la identificación se realizará a tra-vés del familiar, cuidador o responsable hospitalario.
� Las etiquetas predefinidas con los datos del receptor solo pueden utilizarsesi, son solicitadas en el momento. confirmados los datos y colocadas en elmomento de la extracción.
� La muestra que se extraerá será como mínimo en tubo de EDTA (5ml). Enniños de menos de 20 Kg la muestra se adecuará al peso.
� Debe reflejar el estado inmunológico del paciente teniendo una caducidadde 72 horas si, existen antecedentes de embarazo o transfusión en los úl-timos 3 meses. En el resto de casos la muestra tendrá una validez a valo-rar según las características de cada centro.
� Si la muestra se obtiene de una vía central o periférica en uso, será nece-sario desechar los 10 primeros ml.
� La persona responsable de la extracción ha de estar claramente identifi-cada, así como la fecha y hora de la extracción.
� En situaciones de emergencia cada hospital debe tener un mecanismo es-pecífico para la identificación de la muestra y del receptor.
ACTO TRANSFUSIONAL
ACTUACIONES PREVIAS� Revisar las órdenes médicas para confirmar la transfusión y la forma en
que ha de realizarse, componente, cantidad, ritmo y si ha de administrarsealguna premedicación.
112

� Realizar una identificación positiva del receptor (nombre y apellidos y siem-pre que se pueda fecha de nacimiento) así como los mecanismos de segu-ridad propios de cada centro (brazaletes de identificación, registro concódigo de barras, RFID…). Recordar que los errores de identificación del re-ceptor son los responsables de la mayoría de accidentes transfusionales.La administración equivocada de un concentrado de hematíes AB0 incom-patible provoca la mayoría de las reacciones hemolíticas mortales.
� Cuando la identificación positiva no sea posible (enfermos pediátricos, in-conscientes, o con barreras de lenguaje) la identificación se realizará a tra-vés del familiar, cuidador o responsable hospitalario.
� Comprobar el número de identificación de la bolsa (en la cabecera del en-fermo) comprobando que toda la información coincide con la esperada yque la unidad es la destinada al receptor.
� Comprobar que el grupo sanguíneo del paciente se corresponde o, es com-patible con el de la unidad de sangre o componente.
� Comprobar la existencia de algún requerimiento especial de la unida: irra-diada, CMV negativa…
� Realizar inspección visual de la bolsa: poros, roturas…
– Hematíes: la unidad que presente hemólisis, coágulos, distinto color res-pecto a los segmentos u otras unidades no deberán ser transfundidas,retornándose al Servicio de transfusión para descartar una posible con-taminación.
– Plaquetas: se ha de comprobar la presencia de remolinos.– Plasma: comprobar su total descongelación.
� Comprobar la fecha de caducidad de la bolsa.
EQUIPO DE TRANSFUSIÓN
Todos los componentes sanguíneos, hematíes, plaquetas, plasma y criopreci-pitados deben ser administrados a través de un equipo estéril libre de piró-genos y con un filtro capaz de retener coágulos, fibrina y otras partículasdañinas para el receptor de la unidad.
El equipo estándar de transfusión incluye una cámara de goteo con un filtroen línea, con un tamaño de poro de 170-260 μm.
113

Es conveniente no llenar la cámara de goteo más de la mitad y purgar pos-teriormente el resto del equipo.
Aunque algunas instituciones aconsejan el uso de un equipo de infusión di-ferente por cada unidad, puede utilizarse el mismo equipo para 2-4 unidadesen un periodo máximo de 4-6 horas.
Cuando esté indicado el calentamiento de la sangre (transfusión masiva,exanguinotransfusion, título alto de crioaglutininas,…) debe realizarse, conaparatos especialmente diseñados y validados para este uso con control detemperatura y alarma. El calentamiento se realizará durante el paso de la san-gre por estos equipos especiales de transfusión, nunca en baños, ni estufas. Re-cordar que temperaturas superiores a 40ºC pueden producir hemólisis de lasangre.
En situaciones en que se precise la infusión muy rápida existen en el mer-cado equipos diseñados para acelerar el ritmo de infusión. Es imprescindibleseguir en cada caso las instrucciones del fabricante.
En casos de ritmo de infusión lento o en pacientes pediátricos pueden uti-lizarse bombas de infusión con equipos específicos para la administraciónde sangre. En estos casos es también imprescindible seguir las instruccio-nes del fabricante. Solo utilizar bombas de infusión específicamente apro-badas.
VÍA DE INFUSIÓN
Aunque se aconseja que el calibre mínimo de la aguja para transfusión sea de18-20 G, los pacientes pediátricos y los adultos con venas de diámetro muypequeño pueden requerir el uso de agujas de menor calibre. Los calibres de23G han sido utilizados, con éxito en estas circunstancias aunque con flujosmucho más lentos.
Con excepción de solución salina isotónica al 0,9% no debe añadirse ningúnmedicamento o solución a los componentes sanguíneos (por ejemplo la so-lución Ringer Lactato contiene calcio que neutralizará el anticoagulante ydesencadenará la cascada de la coagulación, o la solución de Dextrosa queproducirá hemólisis y aglutinación).
114

Los catéteres centrales venosos de múltiples luces son una excepción, ya quehan sido diseñados para una infusión simultánea de fluidos sin que se mez-clen. Sin embargo, la infusión simultánea de fluidos y/o medicamentos, a tra-vés de catéteres centrales de múltiples luces no debería recomendarse demanera rutinaria. Si el paciente presenta una reacción adversa y se han in-fundido múltiples fluidos simultáneamente, puede ser difícil, y a veces impo-sible determinar que fluido o fármaco ha sido el causante de la reacción.
VELOCIDAD DE INFUSIÓN
Los primeros 15 minutos de la transfusión deben realizarse a velocidad lenta,10 gotas/minuto, y con control estricto del personal de enfermería. Es en esteperíodo cuando se detectan frecuentemente las reacciones hemolíticas agu-das.
Nunca debe pasarse una unidad de concentrado de hematíes en más de 4horas, si es necesario, por las características del paciente (pacientes con ane-mia crónica, con compromiso cardiovascular) fraccionar las unidades y pasarcada fracción en un máximo de 2-3 horas).
Tiempos superiores a 4 horas favorecen el riesgo de contaminación bacte-riana.
115
VOLUMEN CONSERVACIÓN DURACIÓN VELOCIDADTRANSFUSIÓN INFUSIÓN
HEMATIES 200-300ml 2-6ºC 35 a 42 días 60-120 minutos 30-60 gotas/mPLAQUETAS 200-400ml 20-24ºC en agitación 5 días 20-40 minutos 125-225 gotas/mPLASMA 300-400ml Congelado < -25ºC 3años 30-60 minutos 125-175 gotas/mAFERESIS Congelado -18ºC
a -25ºC 3 meses postdescongelación 24h
PLASMA 200-300ml Congelado < -25ºC 3años 20-30 minutos 125-175 gotas/mCongelado -18ºCa -25ºC 3 mesespostdescongelación 24h

Como término medio se transfundirán los componentes sanguíneos a unavelocidad de 5-10 ml/kg/h aunque en pacientes hipovolémicos puede au-mentarse esta velocidad. En pacientes con una reserva cardiorespiratoria baja,se deberá reducir el ritmo de infusión hasta 2,5ml/Kg/h.
MONITORIZACIÓN DEL PACIENTE
Antes de iniciar la transfusión debe registrarse: temperatura, pulso, tensiónarterial, frecuencia respiratoria.
Transcurridos los primeros 15 minutos, si no existe reacción ni modificacionesen las constantes vitales, el paciente debe observarse de forma intermitente,hasta finalizar la transfusión en que volverán a tomarse todas las constantes.
Los controles deben ser más estrictos en pacientes de edad avanzada o concompromiso cardiovascular en los que existirá una adicional monitorización.
Si en cualquier momento durante la transfusión, el paciente presenta signoso síntomas que puedan sugerir una reacción transfusional, la transfusión debeinterrumpirse y evaluar la reacción.
REGISTROS
EN LA HISTORIA CLINICA DEL RECEPTOR
En la historia clínica del paciente debe constar, para su trazabilidad:
� Constancia de la solicitud de transfusión con el nombre del médico prescriptor.� Consentimiento informado.� Personal de enfermería responsable del enfermo en el momento de la
transfusión.� Etiqueta identificativa donde conste el tipo de producto, el número iden-
tificatorio del mismo, el volumen y el grupo sanguíneo.� Fecha y hora de inicio y fin de la transfusión.� Signos vitales pre y postransfusión.� Cualquier efecto adverso relacionado con la transfusión.
116

EN EL SERVICIO DE TRANSFUSIÓN� El Servicio de transfusión tendrá un registro de todas las solicitudes, las
unidades, tipo de producto, pruebas de compatibilidad realizadas y el des-tino final de todos los componentes.
� El sistema de registro de datos, debe garantizar en todo momento la con-tinuidad en la documentación de todos los procesos, desde el donantehasta el receptor, de manera que pueda asegurarse la trazabilidad desde laprimera etapa hasta la última.
REFERENCIASPractical Guide to Transfusion Medicine. Marian Petrides. MD. AAbb press 2001.
Manual Práctico de Medcina transfusional. L.Barbolla; E.Contreras; MM.Pujol. AcciónMédica 2002.
Guidelines for the Administration of Blood and Bloos components. Issued by the Na-tional Users Group. Irish Blood Service 2004.
Australian and New Zeland Society of blood Transfusión Inc. Royal College of Nursing.Guidelines for the Administration of Blood Components. October 2004.
Procedimientos Banc de Sang i Teixits. P-TR-011 Administración de Componentes San-guíneos. Revisión Septiembre 2008.
Comité de acreditación en Transfusión (CAT).Estándares de acreditación. Asociación Es-pañola de Hematologia Hemoterapia. Sociedad Española de Transfusión Sanguínea.23de septiembre 2009.
Guideline on the Administration of Blood Components. British Committee for Stan-darts in Haematology. December 2009.
117


SOBRE LA TRANSFUSIÓN
LA PRESCRIPCIÓN DE COMPONENTES SANGUÍNEOS
La orden de administrar componentes sanguíneos es responsabilidad del mé-dico que toma dicha decisión en un momento clínico determinado del pro-ceso asistencial. A lo largo de este proceso el paciente puede ser atendido demanera sucesiva por un equipo de facultativos, siendo cada uno de ellos elresponsable de la prescripción de los componentes que haya personalmenteordenado.
Como toda orden terapéutica ésta debe ser emitida por escrito en el docu-mento de la historia clínica donde se registren dichas órdenes, y el facultativodebe ser identificable mediante nombre y apellidos, firma registrada, númeroo código. La identificación puede ser manuscrita en papel o mediante aplica-ciones informatizadas.
La prescripción debe de ser clara y precisa en cuanto al tipo de componente,la cantidad y el ritmo de infusión. La orden debe ir acompañada de otras ins-trucciones, si fuera preciso, tales como la administración de una premedica-ción o la necesidad de administrar un componente con característicasespecíficas. En situaciones de urgencia inmediata son admisibles las órdenesverbales que deben ser transcritas en cuanto sea posible.
INFORMACIÓN AL PACIENTE. CONSENTIMIENTO INFORMADO
La información al paciente es un proceso gradual dentro de la relación que seestablece entre el profesional sanitario y el paciente. El profesional debe res-petar las decisiones del paciente de acuerdo a sus valores como clara expre-sión de su autonomía moral siendo aplicables las siguientes disposiciones:
� Convenio de Oviedo para la protección de los derechos humanos y la dig-nidad del ser humano respecto a las aplicaciones de la biología y la medi-cina. Oviedo 1997.
119
ASPECTOS ÉTICOS Y LEGALES SOBRE LA TRANSFUSIÓN DE COMPONENTES SANGUÍNEOS Y DERIVADOS PLASMÁTICOS

� Ley 41/2002 de 14 de Noviembre, básica reguladora de la autonomía delpaciente y de derechos y obligaciones en materia de información y docu-mentación clínica.
El proceso consta de dos fases: información acorde a las necesidades delpaciente y toma de decisión libre y voluntaria. La información es un procesofundamentalmente verbal y progresivo.
La decisión del paciente para algunos procedimientos como la administra-ción de componentes sanguíneos debe constar por escrito en un documentode consentimiento informado.
El profesional responsable del paciente y que ordena la administración de loscomponentes, es el que fundamentalmente debe facilitar la información, sinperjuicio de que todo profesional que interviene en el proceso asistencial estéobligado a cumplir los deberes de información y documentación clínica, es-pecialmente a instancias del paciente.
El Real Decreto 1088/2005, de 16 de Septiembre, por el que se establecen losrequisitos técnicos y condiciones mínimas de la hemodonación y de los centrosy servicios de transfusión, establece literalmente en su capítulo IV que “siem-pre que sea posible el médico que establezca la indicación recabará, después deexplicarle los riesgos y beneficios de este terapéutica, así como sus posibles al-ternativas, la conformidad del paciente según los dispuesto en la Ley 41/2002”.
CARACTERÍSTICAS DE LOS DOCUMENTOS DE CONSENTIMIENTOINFORMADO
La información contenida en el documento debe ser comprensible, veraz, ade-cuada, oportuna y suministrada con la suficiente antelación.
Los documentos de consentimiento deben ser propuestos por grupos multi-disciplinares y aprobados por los Comités de Transfusión Hospitalarios y/o deÉtica Asistencial de cada Hospital. Los documentos de consentimiento nor-malizados garantizan la uniformidad de la información administrada. No obs-tante, de manera verbal el facultativo adaptará esta información a lascircunstancias de cada paciente y resolverá sus posibles dudas. La solicitud deconsentimiento no estará nunca dirigida a buscar una decisión determinadadel paciente y no se convertirá en un mero acto administrativo.
120

El paciente recibirá información en función de sus facultades y grado de com-prensión, incluso si tiene su capacidad limitada, sin perjuicio de que se faci-lite a quien asuma su representación, o si carece de representante legal, de laspersonas vinculadas a él por razones familiares o de hecho.
Los documentos de consentimiento contendrán al menos los siguientes as-pectos: datos identificativos del centro, el servicio y los actores (paciente ymédico); identificación, naturaleza, descripción y finalidad del procedimiento;alternativas al procedimiento si las hubiera, riesgos típicos, atípicos graves ypersonalizados si los hubiera; firmas de los actores, lugar y fecha; posibilidadde revocación del consentimiento y de solicitar aclaraciones o informaciónadicional.
INFORMACIÓN A FAMILIARES O ALLEGADOS
Las personas del entorno del paciente tendrán derecho a la información siem-pre que el paciente lo permita de manera expresa o tácita, pudiendo éste pro-hibir la información a cualquier persona.
RENUNCIA DEL PACIENTE A SER INFORMADO. DELEGACIÓN DE LAINFORMACIÓN
Si el paciente renuncia voluntariamente a ser informado se deberá respetarsu voluntad solicitando señale a la persona en quien delega. La renuncia ex-presa, así como el nombre de la persona que recibirá la información, deberánser recogidos por escrito en la historia del paciente o documento oportuno.
La renuncia voluntaria estará limitada por el interés de la salud del paciente,estando el médico obligado a informar en situaciones en las que se debantomar decisiones inmediatas para salvaguardar la integridad física del paciente.
La persona en quien el paciente delegue no podrá tomar decisiones que, ajuicio del médico, puedan ocasionar daño al paciente.
CONSENTIMIENTO POR REPRESENTACIÓN: MENORES Y PACIEN-TES INCAPACITADOS
Los supuestos en los que la ley prevé la prestación del consentimiento por re-presentación son: pacientes incapacitados legalmente, pacientes que por su
121

estado físico o psíquico no sean competentes, pacientes menores de 16 añosno emancipados.
La ley establece la mayoría de edad sanitaria con carácter general en los 16años. Los pacientes con 16 años cumplidos serán quienes otorguen el con-sentimiento, con independencia de que sus padres o tutores sean informados.Si el paciente es menor de 16 años la representación la ejercen sus padres otutores. En caso de que el paciente tenga un cónyuge mayor de edad la re-presentación se otorga generalmente a éste con preferencia sobre los padres,seguida de la de otros familiares de grado próximo, y dentro del mismo gradolos de mayor edad.
No obstante los menores de 12 a 15 años deberán ser escuchados, solici-tando su consentimiento tácito, especialmente en los casos en los que éstosmuestren capacidad y madurez.
El consentimiento que se preste por sustitución obedecerá siempre al princi-pio general de actuación a favor del paciente y del respeto a su dignidad. Entodos los supuestos se hará constar en la historia las circunstancias que con-curran y en caso de que exista sentencia declarativa de incapacidad, se in-corporará una copia de la misma.
EXCEPCIONES AL CONSENTIMIENTO INFORMADO
Existen dos excepciones: urgencia vital y situación de necesidad terapéu-tica.
Cuando exista una situación de riesgo inmediato y grave para la integridad delpaciente, salvo que el paciente haya expresado de manera reciente y por es-crito su negativa a recibir el tratamiento (instrucciones previas), el médicoestará exento de solicitar el consentimiento informado verbal o escrito, yaque el tiempo requerido para obtener dicho consentimiento puede suponeruna pérdida de oportunidad para salvar la vida del paciente o evitar secuelaspermanentes. Esta situación de riesgo inmediato deberá ser debidamente do-cumentada en la historia del paciente.
Si bien la mayoría de edad sanitaria es de 16 años, para otorgar el documentode instrucciones previas la ley 41/2002, en su artículo 11.1, establece como
122

necesaria la mayoría de edad legal. No obstante en algunas Comunidades Au-tónomas existen regulaciones específicas que confieren capacidad a algunosmenores en las condiciones que aquellas prevén. Por tanto en cuanto a la ca-pacidad de los menores para otorgar instrucciones previas se estará a tenorde lo que dispongan las regulaciones en cada comunidad autónoma.
En el caso de la administración de componentes es excepcional que se pro-duzca lo que se conoce como situación de necesidad terapéutica. Esta se pro-duce cuando a juicio del facultativo el proceso de información yconsentimiento puede dañar gravemente al paciente, en este caso el facul-tativo podría actuar en interés del paciente. De considerarse esta situación de-berá justificarse debidamente en la historia clínica del paciente.
NEGACIÓN DEL CONSENTIMIENTO
El paciente puede negarse tanto a recibir información como a prestar su con-sentimiento. Sin embargo, cuando la negativa a recibir un tratamiento puedaponer en peligro su vida u ocasionar secuelas permanentes, el paciente estaráobligado a escuchar toda la información que el médico considere necesaria ya ratificar por escrito que ha recibido y entendido la información.
El paciente adulto y capacitado, plenamente informado, en ejercicio absolutode su voluntad tiene pleno derecho a rehusar el tratamiento. El médico de-berá respetar su decisión aun cuando ésta vaya en perjuicio de su salud oponga en riesgo su vida. Lo mismo cabría decir para el paciente que hayacumplido los 16 años o haya sido emancipado (mayor de 14 con emancipa-ción legal). En este último caso si el paciente se negara pero no lo hicieran sustutores (o su cónyuge si este fuera mayor de edad) la ley establece que éstosserán informados y su opinión tenida en cuenta para la toma de decisiones,pero no autoriza a que la opinión de estos últimos prevalezca sobre la del pa-ciente. Por tanto legalmente los mayores de edad legal (18 años cumplidos),los mayores de edad sanitaria (16 años cumplidos) y los menores emancipa-dos (14 años cumplidos con emancipación legal) son legalmente competen-tes para rehusar el tratamiento.
Si el paciente tiene una edad comprendida entre 12 y 16 años, la ley estableceque su opinión deberá ser escuchada. Si la renuncia al tratamiento fuera de
123

los padres o de los tutores, pero no del menor y aquella pusiera en riesgo susalud, prevalecerá la opinión del menor sobre la de los padres. Si el menor senegara pero no sus padres, prevalecerá la opinión de los tutores legales enbeneficio de la salud del menor. Si ambos se negaran se solicitará de inme-diato la intervención judicial.
En el caso de menores de 12 años se considera que éstos no tienen madurezsuficiente para manifestar su determinación, y que los padres o tutores nopueden tomar decisiones que vayan en contra de su derecho a la vida. Portanto en los casos en los que los padres niegan el tratamiento y el menorconsiente, o no tiene madurez para expresar su opinión, el médico se consi-dera está autorizado a actuar en beneficio del menor, aunque es recomenda-ble solicitar la intervención judicial.
En los casos en los que el paciente es competente para rehusar el tratamientola ley establece que, si no existiera ningún tipo de tratamiento alternativo elpaciente deberá solicitar el alta voluntaria, si éste se negara la ley estableceigualmente que se puede solicitar la intervención judicial para que dirimasobre la conveniencia del ingreso.
Siempre que se producen estas extremas circunstancias se plantea un dilematanto ético como jurídico, sobre si debe prevalecer el derecho fundamental dela libertad y autonomía del paciente o el deber ético del profesional sanita-rio de salvaguardar la vida. El tribunal constitucional en un caso en que seprodujo un óbito de un menor de 13 años de edad estimó como prevalenteel derecho de libertad religiosa de los tutores legales y del propio menor quese opuso al procedimiento, no condenando a los padres ni a los médicos poromisión del tratamiento.
En aquellos casos en que la autoridad judicial refrende la negativa del pa-ciente, el médico que no ordene la administración de componentes no incu-rre en ningún tipo de responsabilidad civil o penal. Así mismo, el médico queordenara transfundir a un menor de edad de 16 años o mayor de 14 no eman-cipado, en espera de una decisión judicial por un agravamiento o situación deemergencia tampoco incurrirá en dicha responsabilidad.
En todos estos casos se recomienda siempre la consulta urgente con el Co-mité de Ética Asistencial de cada Hospital.
124

Si el facultativo tuviera objeciones de conciencia a la atención de un pacienteque rechace el tratamiento podrá solicitar ser sustituido por otro profesional,sin perjuicio de que, si no fuera posible dicha sustitución, o en situación de ur-gencia, estará obligado a atender al paciente y administrar cualquier otro tra-tamiento, salvo el que el paciente niega.
PRODUCTOS NO SEÑALADOS ESPECIFICAMENTE EN LA NORMATIVA
La legislación vigente establece las normas tanto para la administración decomponentes sanguíneos alogénicos y autólogos. Sin embargo, aunque el RealDecreto 1088/2005 habla de productos autólogos, en su desarrollo se refierea la donación predepósito, y determinados productos no figuran en el catá-logo de componentes, por ejemplo el suero autólogo para uso oftálmico, lasplaquetas o fibrina autóloga para uso quirúrgico. Tampoco en la Ley de In-vestigación Biomédica 14/2007, de 3 de Julio, se hace mención clara a estosproductos y quedan igualmente excluidos en el RD 1301/2006 sobre la ob-tención y uso de células y tejidos humanos.
“Sensu estricto” estos productos no se destinan a la “transfusión”, pero sí seadministran al paciente. En algunos casos su utilización podría recaer en el ám-bito de la investigación biomédica. Ante la ausencia de una normativa especí-fica se considera que estos tratamientos deben ser objeto de procedimientoescrito de consentimiento informado con el mismo rigor que el empleado paralos componentes sanguíneos habituales, siguiendo las recomendaciones delReal Decreto 1088/2005. Si se desconocieran los efectos de dicho tratamientoen el corto o medio plazo, este extremo deberá hacerse constar en el docu-mento. Si el proceso es experimental, en proceso de validación, así como siforma parte de una investigación o ensayo clínico, se informará al paciente. Siel producto debe ser almacenado, conservado bajo ciertas condiciones, mani-pulado o autoadministrado por el propio paciente deberán darse instruccionesescritas al paciente, con copia de las mismas en el historial clínico del paciente.
Si el uso de un componente aunque sea autólogo no se encuesta respaldadopor la existencia de guías de uso elaborados por la administración o socieda-des científicas, se recomienda que el paciente sea explícitamente informadosobre este aspecto.
125

MEDICAMENTOS DERIVADOS DEL PLASMA
La albúmina, los concentrados de factores de la coagulación no recombinan-tes, las inmunoglobulinas, tienen la consideración de “medicamento hemo-derivado” según se recoge en el Real Decreto 710/2002, de 19 de julio, por elque se modifica el Real Decreto 414/1996, de 1 de marzo, por el que se re-gulaban los productos sanitarios, en lo referente a los que incorporen deriva-dos estables de la sangre o plasma humanos.
Así mismo el Real Decreto 1345/2007, de 11 de octubre, regula el procedi-miento de autorización, registro y condiciones de dispensación de los medi-camentos de uso humano fabricados industrialmente. En él se establece quedichos medicamentos se someterán a prescripción restringida:
� A causa de sus características farmacológicas o por su novedad, o por mo-tivos de salud pública, se reserven para tratamientos que sólo puedan utili-zarse o seguirse en medio hospitalario o centros asistenciales autorizados(Medicamentos de Uso Hospitalario).
� Se utilicen en el tratamiento de enfermedades que deban ser diagnostica-das en medio hospitalario, o en establecimientos que dispongan de mediosde diagnóstico adecuados o por determinados médicos especialistas, aun-que la administración y seguimiento pueda realizarse fuera del hospital (Me-dicamentos de Diagnóstico Hospitalario de prescripción por determinadosmédicos especialistas).
� Estén destinados a pacientes ambulatorios, pero cuya utilización pueda pro-ducir reacciones adversas muy graves, lo que requerirá, en su caso, pres-cripción por determinados médicos especialistas y una vigilancia especialdurante el tratamiento (Medicamentos de Especial Control Médico).
No se establece explícitamente en ninguna norma o regulación la obligato-riedad del documento de consentimiento informado escrito para la prescrip-ción de los medicamentos derivados de la sangre, pero parece razonablerecomendar el uso de un documento de consentimiento específico cuandoexistan riesgos significativos con su administración, se administren para indi-caciones no aprobadas en la ficha técnica o en la Guía de uso del Fármaco es-tablecida en la institución sanitaria.
126

En cualquier caso existe siempre la obligación de informar verbalmente al pa-ciente de la naturaleza del medicamento que se le suministra y de sus even-tuales efectos secundarios. La prescripción de estos fármacos deberá estardocumentada en un documento de órdenes terapéuticas con total trazabilidaddel tipo de producto y del facultativo responsable. Los efectos adversos no co-nocidos o muy graves estarán sujetos a su comunicación según lo establecidoen las regulaciones de farmacovigilancia de ámbito estatal o autonómico.
HEMOVIGILANCIA
La Orden SCO/322/2007, de 9 de febrero, establece los requisitos de trazabili-dad y de notificación de reacciones y efectos adversos graves de la sangre y delos componentes sanguíneos. En ella se establece la obligatoriedad de comuni-car dichos efectos a la autoridad sanitaria competente siguiendo los principiosde confidencialidad y protección de datos. En cada comunidad autónoma se dis-pone de regulaciones que desarrollan esta orden y establecen el procedimiento.
REFERENCIASLey 41/2002, de 14 de Noviembre, básica reguladora de la autonomía del paciente yde derechos y obligaciones en materia de información y documentación clínica (BOE274, de 15 de noviembre de 2002).Convenio para la protección de los derechos humanos y la dignidad del ser humano res-pecto a las aplicaciones de la biología y la medicina. Oviedo 4 de abril de 1997. (BOE251, de 20 de octubre de 1997).Real Decreto 1088/2005 de 16 de septiembre por el que se establecen los requisitostécnicos y condiciones mínimas de la hemodonación y de los centros y servicios detransfusión (BOE 225, de 20 de septiembre de 2005).Real Decreto 710/2002, de 19 de julio, por el que se regulan los productos sanitarios(BOE 173, de 20 de julio de 2002).La Orden SCO/322/2007, de 9 de febrero por la que se establecen los requisitos detrazabilidad y de notificación de reacciones y efectos adversos graves de la sangre y delos componentes sanguíneos (BOE 42, de 17 de febrero de 2007).Real Decreto 414/1996, de 1 de marzo, por el que se regulaban los productos sanita-rios (BOE 99, de 24 de abril de 1996). Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento deautorización, registro y condiciones de dispensación de los medicamentos de uso hu-mano fabricados industrialmente (BOE 267, de 7 de noviembre de 2007).LEY 14/2007, de 3 de julio, de Investigación biomédica (BOE 159, de 4 de julio de 2007). �
127


Guía sobre la transfusión de componentes sanguíneos y derivados plasmáticos4ª edición, 2010
Sociedad Española de Transfusión Sanguínea y Terapia Celular
Guí
a so
bre
la t
rans
fusi
ón
de c
om
pone
ntes
san
guín
eos
y de
riva
dos
plas
mát
ico
s