guia serv 4 - docenciaoftalmologia.org · objetivo principal estandarizar el manejo de las...
TRANSCRIPT

Guías de práctica clínica de la SERV
4 | Guía clínica para el diagnósticodiferencial y el manejo de lasenfermedades hereditarias dela retina y la coroides

Guías de práctica clínica de la SERV
4 | Guía clínica para el diagnósticodiferencial y el manejo de lasenfermedades hereditarias dela retina y la coroides

Coordinadora:
Rosa M.ª Coco Martín
Unidad de Mácula y Degeneraciones RetinianasIOBA, Universidad de Valladolid
Equipo de trabajo:
Rosa M.ª Coco Martín
Unidad de Mácula y Degeneraciones RetinianasIOBA, Universidad de Valladolid
Rafael Navarro Alemany
Especialista en Retina del IMOBarcelona
Escenarios clínicos a los que se refiere la GPC
y condiciones de aplicación
Definición de situaciones en las que no es
aplicable la GPC
Esta guía clínica no es aplicable enenfermedades hereditarias diferentes de lasseñaladas.
La validación de esta Guíaha sido realizada por lossiguientes revisores:
M.ª José Blanco
Hospital Provincial de ConxoComplejo Hospitalario Universitario de SantiagoSantiago de Compostela
David Salom
Servicio de OftalmologíaHospital General Universitario de Valencia
Robert H. Rosa, Jr., M.D.
Professor and Vice Chair for ResearchDepartment of OphthalmologyScott & White Eye InstituteTexas A&M Health Science Center2401 South 31st StreetTemple, Texas 76508
Patrocinado por:
Publicación de la Guía: marzo de 2009Próxima revisión: diciembre de 2012
Este documento debe ser citado como «Guiaclínica para el diagnóstico diferencial y el manejode las enfermedades hereditarias de la retina y lacoroides». Guías de Práctica Clínica de la SERV».Disponible en www.serv.es
Copyright © 2009, Sociedad Españolade Retina y Vítreo.
Autores
Realización y Producción: MACLINE, S.L.
D.L.: M-10562-2009
ISBN: 978-84-936943-1-9

3
La elaboración de las GUÍAS DE PRÁCTICA CLÍNICA de la Sociedad Española de Retina
y Vítreo (en adelante GPCdeSERV), ha sido un objetivo establecido por la Junta Directiva dela Sociedad en su reunión administrativa celebrada durante su XII Congreso anual.
Entendemos por GPCdeSERV el conjunto de instrucciones, directrices, afirmaciones o reco-mendaciones, desarrolladas de forma sistemática con el objetivo de ayudar a los miembrosde nuestra Sociedad y a los oftalmólogos en general, en su práctica clínica del cuidado de lasenfermedades oftalmológicas que afectan a la retina y a la toma de decisiones, sobre la mo-dalidad de asistencia sanitaria apropiada para unas circunstancias clínicas específicas. Por lotanto, diferentes guías se irán publicando progresivamente con este objetivo a lo largo de lospróximos años.
Para la elaboración de cada guía se nombra una comisión de expertos o grupo de trabajo en-cargada específicamente por la SERV a tal efecto, y unos revisores externos a la comisión.Las guías se basan en los mejores datos científicos publicados, interpretados y discutidos porel grupo de trabajo, obtenidos de la revisión de otros protocolos clínicos ya existentes, datosde ensayos clínicos, y de la evidencia científica disponible.
Las GPCdeSERV sugieren líneas de actuación en la mayoría de los pacientes, pero no pre-tenden establecer criterios de obligado cumplimiento, ni eximir al oftalmólogo de su respon-sabilidad de reflexionar ante un caso concreto y actuar según su buen criterio profesional.Además, en modo alguno, limitan o vinculan la libertad del oftalmólogo en su toma de deci-siones para el tratamiento de un paciente determinado, que puede así optar por otra pautadistinta, dentro de las técnicas normales requeridas, si entiende que, según su experiencia,el resultado buscado exige otro tipo de terapia. El que dicha opción no esté contemplada enlas GPCdeSERV como pauta de actuación recomendada, no puede considerarse en modo al-guno como una mala praxis profesional o una vulneración de la «lex artis ad hoc».
Seguir las recomendaciones de las GPCdeSERV no asegura el éxito en cada paciente y enocasiones puede ser necesario otros abordajes diagnósticos o terapéuticos que no están es-pecíficamente recomendados en estas guías. Es el oftalmólogo/retinólogo el que tiene la res-ponsabilidad de tomar la mejor decisión en el adecuado manejo clínico-terapéutico de un pa-ciente concreto teniendo en cuenta todas las circunstancias que le rodean y, en relación conla respuesta obtenida, continuar en una u otra línea. La adaptación de las GPCdeSERV a losdistintos sistemas sanitarios puede suponer la variación de algunos de los aspectos de lasmismas.
Ya que la innovación en la práctica médica es algo consustancial a la misma, la SERV consi-dera que, dada la evolución y los avances médicos que se producen continuamente, las Guíastiene una validez que está fijada en cada una de ellas debiendo ser revisadas en el plazo se-ñalado.
La SERV declina específicamente toda responsabilidad por lesiones, daños de cualquier cla-se, negligencia u otras situaciones, o por cualquier reclamación que pueda surgir por el usode cualquier información o recomendación contenida en las GPCdeSERV. Incluso las deriva-das de referencias a determinadas, técnicas, instrumentos o fármacos que no están especí-ficamente aprobados y que se realizan con fines ilustrativos, siendo el propio médico el quetiene la responsabilidad de actuar de acuerdo con las leyes locales vigentes.

4
Declaración de conflicto de intereses de los participantes ____________ 5
Resumen estructurado ________________________________________________________________________ 5
Lista de abreviaturas ___________________________________________________________________________ 6
Definición de objetivos ________________________________________________________________________ 7
Diagnóstico electrofisiológico ____________________________________________________________ 8
Diagnóstico diferencial de las enfermedades extensasde fotorreceptores ________________________________________________________________________________ 24
Distrofias maculares ____________________________________________________________________________ 29
Bibliografía ____________________________________________________________________________________________ 39
Índice de contenidos

5
Resumen estructurado
Declaración de conflictode intereses de los participantes
La Dra. Coco no tiene intereses comerciales en este tema.
El Dr. Navarro no tiene intereses comerciales en este tema.
A. Propósito de la GPC
El objetivo de la Guía de Práctica Clínica es estandarizar el estudio diagnóstico y elmanejo clínico de las enfermedades hereditarias de la retina y la coroides.
B. Metodología
Para ello, se dan unas recomendaciones básicas de cómo realizar los test electrofi-siológicos, de qué pruebas diagnósticas realizar en cada caso y de los tratamientosrecomendados en distintas circunstancias.

ISCEV Sociedad Internacional de Electrofisiología Clínica y Visión
EOG Electrooculograma
ERG Electrorretinograma
PERG ERG en Patrón
PVE Potenciales Visuales Evocados
MERG ERG Multifocal
CV Campo Visual
AV Agudeza Visual
OCT Tomografía de Coherencia Óptica
CAR Retinopatía asociada al carcinoma
MAR Retinopatía asociada al Melanoma
CRAO Oclusión arteria central de la retina
CSNB Ceguera Nocturna Congénita Estacionaria
RP Retinitis pigmentosa
DB Distrofia de Bastones
DC Distrofia de conos
DCB Distrofia de conos y bastones
LCA Amaurosis congénita de Leber
MCLX Monocromatismo de conos azules ligado a X
ADRP Retinitis pigmentosa autosómica dominante
ARRP Retinitis pigmentosa autosómica recesiva
XLRP Retinitis pigmentosa ligada a X
EPR Epitelio pigmentario de la retina
DHA Ácido docosahexaenoico
OAT Ornitina aminotransferasa
FF Fundus Flavimaculatus
STDG Enfermedad de Stargardt
DMAE Degeneración Macular Asociada a la Edad
ABCR o bien ABCA4 ATP-binding Cassette transporter gene, subfamilia A, miembro 4
A-2E N-retinilidene-N-retiniletanolamina
CACD Distrofia Coroidea Areolar Central
periferina/RDS Gen periferina/Retinal Degeneration Slow
AFG Angiografía fluoresceínica
FO Fondo de ojo
6
Lista de abreviaturas

Objetivo principal
■ Estandarizar el manejo de las enfermedades hereditariasde la retina más frecuentes: Retinitis pigmentosa y Dis-trofias maculares
Objetivos secundarios
■ Unificar los criterios para indicar pruebas diagnósticas encasos de enfermedades hereditarias de la retina.
■ Unificar los criterios de tratamiento o de manejo de lasenfermedades hereditarias de la retina
■ Promover y extender el conocimiento respecto al modode realización y la interpretación de la electrofisiologíaen las enfermedades hereditarias de la retina.
■ Promover y extender el conocimiento básico de la gené-tica de las enfermedades monogénicas.
■ Identificar aquellas familias que necesitan ser vistas yorientadas para la realización de consejo genético y/odiagnóstico genético molecular.
7
Definición de objetivos

Se recomienda seguir los «Standards» dela Sociedad Internacional de Electrofisiolo-gía Clínica y Visión (ISCEV) para la realiza-ción de todas y cada una de las pruebaselectrofisiológicas, pues de otro modo losresultados no son interpretables por otrapersona, ni reproducibles, ni evidente-mente comparables con otros que seanrealizados por otro equipo profesional:http://www.iscev.org/1,2. A continuaciónse hace un breve resumen de ello.
Electrooculograma (EOG)
En condiciones normales existe una dife-rencia de potencial entre la córnea y elpolo posterior del ojo que es conocidocomo el potencial de reposo3. En la prác-tica su evaluación clínica más fiable seobtiene haciendo una ratio entre el me-
nor valor del potencial de reposo en os-curidad y el máximo en la luz, lo que seconoce como Índice de Arden [ArdenIndex = (mm con luz/mm en oscuridad) x100] cuyo valor de normalidad es mayoro igual a 170-185% (en miopes ≥150%)4.Es básicamente útil en la enfermedad deBest, en la que de forma casi patogno-mónica aparece un EOG alterado conelectrorretinograma (ERG) normal tantoen enfermos como en portadores. En lasenfermedades retinianas extensas siem-pre está alterado junto con el ERG.
Electrorretinograma
El ERG mide el potencial de «acción»de la retina5,6. Puede registrarse po-niendo un electrodo en la córnea y otroen un área de piel cercana al globo ocu-
8
Diagnósticoelectrofisiológico
Figura 1. EOG normal a la derecha y de un Best a la izquierda.

lar7. Para que el ERG nos de informa-ción fiable es vital establecer una estan-darización del estímulo y la luz de fondoque deberán estar perfectamente cali-bradas8. El ERG estándar de la ISCEVincluye las siguientes estimulaciones(luminosidad del flash en cd·s·m2):
1) Escotópico 0,01 ERG («respuestapura de bastones»).
2) Escotópico 3,0 ERG («respuesta má-xima o estándar mixta de bastones yconos»).
3) Escotópico 3,0 dos estímulos segui-dos separados 15» («potenciales os-cilatorios»).
4) Fotópico 3,0 ERG («respuesta de co-nos»).
5) Fotópico 3,0 flicker («respuesta purade conos»).
Se recomienda además añadir un esco-tópico 10,0 ó 30,0.
Interpretación del ERG
■ La onda a grande negativa se originaen los segmentos internos de los foto-receptores y por lo tanto en la retinaexterna.
■ La onda b positiva nace en la retina in-terna y concretamente en células bi-polares y células de Müller.
■ Los potenciales oscilatorios (OP’s)
aparecen superpuestos al principio dela onda b. Estos van a tener su origenen retina interna.
Electrorretinograma enpatrón (PERG)
Ante la sospecha de enfermedad macu-lar se aconseja realizar un ERG en Pa-trón (PERG), en el que el estímulo es undamero de ajedrez iluminado en el quelos cuadros blancos pasan a negro con-tinuamente9,10. Las ondas registradasson N35, P50 y N95.■ La onda P50 se origina en las células
ganglionares que recogen la respues-ta de los conos maculares.
■ La onda N95 tiene su origen en axo-nes de células ganglionares que reci-ben estímulo desde fotorreceptoresmaculares.
Por esa razón, el PERG junto con los Po-tenciales Visuales Evocados (PVE) esútil para realizar el diagnóstico diferen-cial entre enfermedad desmielinizantedel nervio óptico y patología macular. SiP50 es normal y N95 está abolida lomás probable es que estemos ante unaneuropatía óptica que ha producido una
9
Diagnóstico electrofisiológico
Figura 2. ERG normal.

pérdida selectiva de células gangliona-res. Sin embargo, si el PERG es planocon pérdida también de la P50 es másprobable que se trate de un problemamacular.
ERG multifocal (MERG)
El MERG proporciona una medida obje-tiva topográfica de la función retinianacentral en un tiempo aceptable11. Es útilen comorbilidad comparando con cam-
po visual (CV) pues puede permitir se-parar el componente retiniano. Usadopor neuroftalmólogos (junto con PVE yPERG)12. Permite diferenciar defecto or-gánico de funcional. También es útil enel diagnóstico de la patología macularhereditaria o adquirida en que ERG es-tándar suele ser normal (junto conPERG) y en su monitorización.
Indicaciones de la electrofisiología
■ Confirmar un diagnóstico que se sos-pechaba.
■ Descartar un diagnóstico.■ Realizar un diagnóstico que no se sos-
pechaba.■ Evaluar discrepancias entre signos y
síntomas (identificar simuladores).■ Identificar portadores (ej: de Retinitis
pigmentosa ligada a X o Best’s ca-rriers).
■ Pronóstico sobre la conservación de lavisión central usando el PERG
■ En el diagnóstico diferencial tener encuenta las enfermedades autoinmu-nes como la retinopatía asociada alcarcinoma (CAR) o la retinopatía aso-ciada al melanoma (MAR)13,14.
10
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 3. PERG normal a la izquierda y en medioPERG con pérdida selectiva de la onda N95 enatrofia óptica dominante (útil en el diagnóstico di-ferencial con una distrofia macular incipiente en elniño).
Figura 4. Arriba ERG normal (izquierda respuestas sectoriales, en medio representación en 2 dimensio-nes, y derecha en 3 dimensiones). Abajo a la izquierda respuestas y 2D en Retinosis pigmentaria. Abajoa la derecha respuestas sectoriales y 3D en un Stargardt’s).

■ Diagnóstico diferencial entre enferme-dad desmielinizante y problema macular.
■ Caracterizar un fenotipo para un geno-tipo conocido (investigación)15.
Causas de ERG negativo(ausencia de la onda b)
Existe ERG negativo si se observa ERGde bastones plano, y en el ERG mixtode bastones y conos se observa unaonda a grande sin onda b visible. LosOP’s están ausentes. Todo esto indicauna afectación de la retina interna, conrespuesta conservada de la capa de fo-torreceptores.16, 17
En este caso el diagnóstico definitivohay que buscarlo en la siguiente listarestringida de enfermedades:■ Melanoma associated retinopathy
(MAR).■ Oclusión arteria central de la retina
(CRAO).
■ Ceguera nocturna congénita estacio-naria (CSNB) ligada al X/Oguchi’s.
■ Retinosquisis ligada a X.■ Síndrome de Goldmann Favre.■ Toxicidad por metanol.■ Toxicidad por quinina.■ Siderosis.■ Enfermedades por acúmulo de lipo-
pigmentos como la Lipofucsinosis o laenfermedad de Batten.
Test electrodiagnósticos mínimospor enfermedad
■ Retinitis pigmentosa y en general en-fermedades extensas de la retina:EOG/ERG.
■ Distrofias maculares: EOG/PERG (siestá disponible)/ERG.
Retinitis pigmentosa(= distrofia de bastones= distrofia de bastones yconos)
Definición de la enfermedad
Consiste en una distrofia de la retina ex-terna producida por la muerte progresi-va de fotorreceptores por apoptosis,que afecta fundamental e inicialmente abastones, aunque cuando la enferme-dad avanza acaba por afectar también alos conos18-21.El término de retinitis pigmentosa seutiliza para englobar a un grupo ampliode enfermedades retinianas heredita-rias genéticamente heterogéneo perofenotípicamente muy similar, lo que sig-nifica que se origina por múltiples alte-raciones genéticas distintas en un nú-mero elevado de genes diferentes, peroque conducen al mismo cuadro clínicoque conocemos como RP. Estas dege-neraciones retinianas progresivas com-parten unas características clínicas co-munes que incluyen electrorretinogra-ma de bastones (ERG) disminuido o
11
Diagnóstico electrofisiológico
Ejemplo de ERG negativo.
ERG escotópico normal arriba de OD y ERG nega-tivo sólo en OI abajo.

ausente, ceguera nocturna, reducciónconcéntrica del campo visual22, migra-ción intrarretiniana de pigmento desdela retina externa, atrofia progresiva de laretina externa, palidez del nervio ópticopor gliosis, atenuación de los vasos reti-nianos y, eventualmente, pérdida de vi-sión central23,24.
Es la más frecuente de las enfermeda-des hereditarias de la retina, afectandoalrededor de 1 de cada 4.000 personasde la población general. Es una enfer-medad con una enorme heterogenei-dad genética25.
Modo de herencia
Su modo de herencia puede ser autosó-mico dominante (ADRP que generalmen-te suelen ser las menos severas), autosó-
mico recesivo (ARRP) o recesivo ligado alcromosoma X (XLRP). Las portadoras deXLRP suelen tener reflejo tapetorretinia-no anómalo y tiempo implícito anormalde la onda b en el ERG lo que ayuda aidentificar a las portadoras y por lo tantoa las familias con este tipo de herencia.
No siempre es posible hacer el diag-nóstico genético molecular, es decirencontrar la mutación exacta causantede la enfermedad en cada caso. La difi-cultad se deriva de que no se conocentodos los genes implicados y de queademás habría que mirar un númeroelevadísimo de genes lo que hace invia-ble estudiar casos esporádicos, porejemplo. De entre las enfermedades he-reditarias de la retina, se conocen 191genes y 142 de ellos ya han sido clona-dos: http://www.sph.uth.tmc.edu/Retnet/
12
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Tabla 1. CLASIFICACIÓN DE LAS ENFERMEDADES HEREDITARIAS EXTENSAS
DE LA RETINA Y LA COROIDES MÁS IMPORTANTES
1. PROGRESIVAS
• Distrofias de bastones y conos: Retinitis pigmentosa (RP); Amaurosis congénita de Leber(LCA) y RP sindrómicas (Usher y Bardet Biedl)
• Coroideremia• Atrofia girata• Distrofias de conos• Distrofias de conos y bastones
2. ESTACIONARIAS
a) Ceguera nocturna congénita estacionaria (CNCE) (ceguera nocturna como en la RP perono progresiva)• CNCE con FO normal• CNCE con FO anormal (Fundus Albipunctatus, Oguchi´s y Retina parcheada de Kandori)
b) Disgenesias no progresivas de conos (electrofisiología similar a la distrofia de conos, perono progresivas)• Anomalías pigmento conos (Tricromatismo): Deuteranomalía/Protanomalía/Tritanomalía• Pérdida un sistema conos (Dicromatismo): Deuteranopia/Protanopia/Tritanopia• Pérdida dos sistemas conos (Monocromatismos):
– Monocromatismo de conos rojos o verdes. Ambos tienen buena visión con cegue-ra completa a colores.
– Monocromatismo de conos azules ligado a X (mala visión pues sólo hay conos azu-les y éstos no están en la fóvea).
– Monocromatismo de bastones = Acromatopsia congénita (hay pocos conos y losque hay son anómalos). Es autosómico recesivo y ello permite diferenciarlo del mo-nocromatismo de conos azules con quién comparte la clínica.
Se estima que el 16% de las RP sonsindrómicas siendo las más frecuentesel síndrome de Usher26-30 (con RP+Sor-dera, supone un 20% de las sindrómi-cas y responsable de un 6-10% de to-das las sorderas neurosensoriales con-génitas) y el Bardet-Biedl31-35 (9% de lassindrómicas con RP+obesidad+polidac-tilia+hipogonadismo), ambas de heren-cia autosómica recesiva.
Exploración
Si hay dudas sobre si la queja sobre lamala visión nocturna es real y en el fon-do no se ve nada determinante, hacerinicialmente una curva de adaptación ala oscuridad para confirmar o descartarla ceguera nocturna.36 Si se confirma laceguera nocturna proceder a hacer unERG. Si no se dispone de pruebas deadaptación a la oscuridad hacer directa-mente un ERG.
Según los resultados de los trabajosrealizados por Berson37, podría descar-tarse el diagnóstico de retinosis pig-mentaria en todo aquel individuo que,aún bajo sospecha clínica, presenteERGs normales después de los seisaños de edad. Este autor ha descritotambién que la afectación electrorreti-nográfica en las familias con RP sigueuna distribución mendeliana, encontran-do relación entre el grado de afectaciónelectrofisiológica y la forma de heren-cia. Estimó que la pérdida media anual
en el ERG de los pacientes afectos deRP oscila entre el 16 y el 18,5%.
Citar también la importancia del ERGpara la detección de portadoras en lasformas de RP ligadas al sexo38,39. Losregistros del ERG de las portadoras obli-gadas están reducidos en su amplitud ytienen latencias aumentadas. La anor-malidad en los registros del ERG de lasportadoras obligadas de las formas liga-das al sexo contrastan con los registrosnormales de los portadores de las for-mas recesivas.
Respecto al fondo de ojo bajo midriasis,la apariencia de una RP avanzada clásicaincluye atenuación de los vasos retinia-nos, palidez del nervio óptico, epiteliopigmentario de la retina (EPR) moteadoy granulado, migración de pigmento ha-cia la retina interna (pigmentación en es-pículas) y eventualmente atrofia del EPRy la coriocapilar. En general, hay una si-metría importante entre los dos ojos yes un axioma con pocas excepcionesque las enfermedades hereditarias sonbilaterales y simétricas, teniendo quedudar de un diagnóstico de hereditariaen caso de enfermedad monocular omuy asimétrica. En los casos en que seobserva todo lo dicho previamente eldiagnóstico clínico ofrece pocas dudas.
En los estadios iniciales de la RP la reti-na puede aparecer normal o casi nor-mal, incluso cuando el CV muestra sóloun defecto relativo. Los pacientes quetienen RP sin cambios pigmentarios amenudo son diagnosticados como reti-
13
Diagnóstico electrofisiológico
Figura 5. Genes identificados en enfermedadesretinianas.
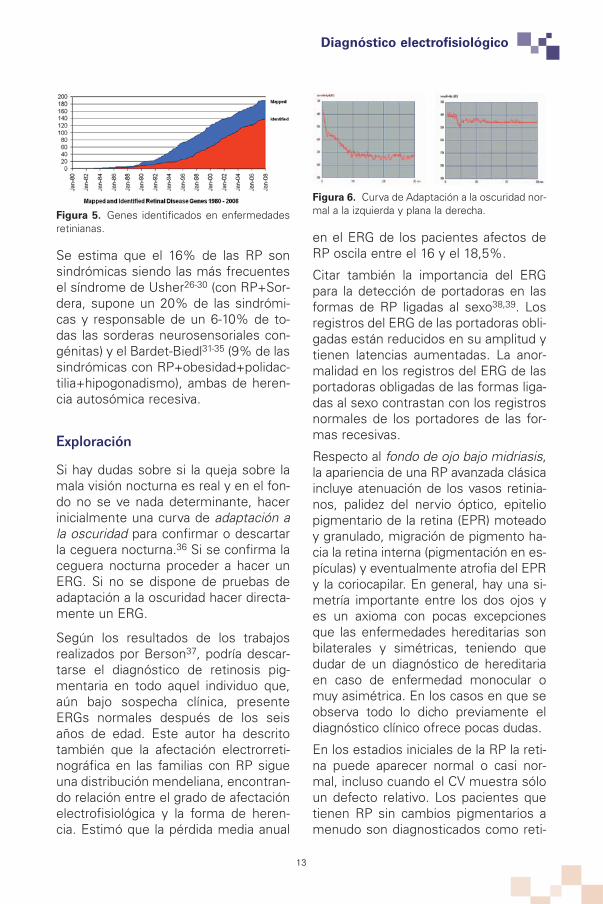
Figura 6. Curva de Adaptación a la oscuridad nor-mal a la izquierda y plana la derecha.

nitis pigmentosa sine pigmento. Estosuele durar poco tiempo, pero en otrasocasiones puede durar muchos años.40
También existe un fenotipo especial deRP llamado RP12 en el que aparecenvasos y nervio óptico prácticamentenormales, una maculopatía leve y unapigmentación que parece más numularque tipo espículas.
Si el paciente presenta ya una RP clási-ca y es la primera vez que se ve al pa-ciente, pedir un campo visual41. Los pa-cientes no notan siempre la pérdida decampo, sobre todo, si mantienen biensu visión central. Como muchos pacien-tes no son conscientes de su pérdidade CV, es importante explorarlo de vezen cuando, sobre todo, si el pacienteconduce (recordemos que según nues-tra legislación no se puede conducir siexiste un defecto en los 120° centralesen visión binocular en la línea horizontal;ésto se puede explorar con el CV de Es-terman del Humphrey en binocular quees más rápido o bien hacer un CV decampo completo de cada ojo siguiendo
nuestra legislación). Recordemos quese produce una pérdida media del 4-5%anual del CV y por lo tanto no deberíande realizarse con una frecuencia mayorque la de un CV cada año.
Tomar la agudeza visual en cada visita.Recordar que la visión central puedeafectarse seriamente por otras compli-caciones asociadas. También puede serútil revisar la sensibilidad al contraste,ya que cuanto más baja sea ésta másse beneficiarán los pacientes de uncambio de polaridad en los textos cuan-do se apliquen técnicas de rehabilita-ción visual42,43.
Como causas de disminución de visiónpor complicaciones asociadas, puedenaparecer catarata, edema macular cis-toide o rezume macular difuso, mem-branas epirretinianas y telangiectasiasperiféricas. Se descartarán estas even-tualidades cuando la disminución de vi-sión se produzca de forma más acen-tuada de lo que es habitual en esta en-fermedad. Para ello, se utilizarán laangiofluoresceíngrafía y/o el OCT40.
El OCT es casi obligado en la RP debidoa la alta prevalencia de EMC en estospacientes que puede ascender a un38%44. El OCT de alta resolución tam-
14
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 7. RP avanzada (FO y AF).
Figura 8. Aspecto del FO en RP12, generalmen-te debido a mutaciones en CRB1. Figura 9. Edema macular en RP (AFG y OCT).

bién puede ayudar en el pronóstico dela RP al ser capaz de evaluar la presen-cia de fotorreceptores o su degenera-ción mediante el «foveal outer seg-ment/pigment epithelium thickness»(FOSPET)45.
Si se dispone de Autofluorescencia,puede ser interesante realizar estaprueba, ya que, en la RP se observa unanillo parafoveal de aumento de la auto-fluorescencia. El radio de este anillo secorrelaciona de forma inversa con laspruebas electrofisiológicas como elPERG y el MERG (las áreas dentro delanillo muestran áreas de función fotópi-ca preservada). Ello demuestra que lasanomalías de la autofluorescencia tie-nen un significado funcional y puedenayudar a identificar pacientes y áreas re-tinianas en los que pueda merecer lapena realizar alguna intervención tera-péutica46-48.
Se aconseja revisar los dedos de manosy pies, pues el Bardet-Biedl se asocia apolidactilia, pero el Refsum (otra RP sin-drómica) también se asocia con anoma-lías de los dedos de manos y pies.
La electrofisiología hay que realizarlasiempre en los diagnósticos de novo.Una vez que el ERG es plano, ya nomerece la pena repetirlo, pues siem-pre será plano. Los potenciales oscila-torios están conservados si la AV esbuena.49 Para monitorizar el resto defunción central también se puede usarel MERG50,51. El PERG también es deayuda a la hora de determinar el pro-nóstico a corto-medio plazo de la visióncentral. Un PERG plano nos indica quehay elevadas posibilidades de pérdidade la AV central a lo largo de los si-guientes 5 años.
El ERG puede ser de ayuda en el diag-nostico del Usher tipo I y será necesariorealizarlo siempre en el contexto de una
sordera profunda congénita, con el finde identificar aquellos individuos en losque pueda estar indicado realizar un im-plante coclear. Para distinguir el Ushertipo I del tipo II se puede usar el test delcalor, ya que el calor provoca nistagmusen el tipo I pero no en el II. En los pa-cientes con Usher’s se recomienda queel Otorrinolaringólogo además de explo-rar la audición, compruebe si el pacien-te tiene también afectación olfatoria52.
15
Diagnóstico electrofisiológico
Figura 10. Autofluorescencia en RP.
Figura 11. Ejemplo de ERG en RP (a la izquierda)comparado con ERG normal (a la derecha). En las3 figuras superiores se observa afectación de to-das las pruebas en oscuridad, con conservaciónde los resultados en pruebas fotópicas.

Manejo
El manejo de la RP incluye un diagnós-tico acertado, el consejo social y gené-tico, y el manejo medico de las compli-caciones oculares asociadas. El conse-jo genético puede ser muy difícil si nohay historia familiar positiva y tambiénen algunos casos de ADRP con expre-sividad variable. Se debe obtener tantainformación como sea posible y haceranálisis mutacional si se puede53,54. Enfamilias con XLRP se debe ofrecerconsejo genético prenatal (esto lo ha-rán los genetistas clínicos)55. Se reco-mienda realizar consejo genético por loque se hará siempre un árbol genealó-gico para realizar una orientación inicialsi es posible dado que las implicacio-nes pronósticas son diferentes en cadatipo de herencia56. Además, este pasoes obligado para poder orientar el diag-nóstico genético molecular si fuera ne-cesario y posible57-60. Recomendamosrealizar personalmente la exploraciónde los familiares si ello fuera posible,pues ello muchas veces nos aclara eldiagnóstico permitiéndonos observarhallazgos sugestivos de una patologíaconcreta en un miembro determinadode la familia. Además, podremos estarseguros de qué miembros son afectosy sanos con total seguridad, lo cualpuede ser crucial si se necesita realizardiagnóstico genético molecular o si se
va a estudiar a la familia con fines deinvestigación.
Actualmente, no existe un tratamientoefectivo en la modelación del procesoprimario. Algunos han propuesto, haceaños, el uso de dosis altas de vitaminaA (15.000 UI) puesto que se ha visto unefecto beneficioso manteniendo los re-gistros del ERG, aunque no se ha en-contrado efecto beneficioso sobre elCV61. Según estos mismos ensayos pa-rece que la vitamina E tendría un efec-to perjudicial, por lo que no se puedenusar suplementos vitamínicos de losque se usan habitualmente para la de-generación macular asociada a laedad62-65. Más reciente es la propuestade añadir suplementos de 1.200 mg/dde ácido docosahexaenoico (DHA) e ini-ciarlos junto con la vitamina A66-69. De lamisma forma, la toma de suplementosde luteína (10 mg/d durante 12 semanasseguidos de 30 mg/d) parece producircierto beneficio en el CV de los pacien-tes con RP70. Habrá que tener cuidadode que los pacientes a quienes se lesrecomiende terapias con vitamina A yderivados tengan RP típicas y, por lotanto, que no sean susceptibles de por-tar una mutación en el gen ABCA4, esdecir, que no sean pacientes con Star-gardt´s que acaben desarrollando sig-nos y síntomas tardíos de RP, y que nosean pacientes con distrofia de conos ybastones. En general parece que los an-tioxidantes son beneficiosos y esteefecto se ha encontrado sin usar vitami-na A con una combinación de luteína,zeaxanthina, ácido alpha lipoico y l-glu-tathion reducido71-73.
Los problemas oculares asociados de-ben ser manejados apropiadamente.Debe ofrecerse la cirugía de cataratacuando esté indicada en pacientes conRP. Sin embargo se debe discutir con elpaciente las posibilidades reales de me-
16
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 12. Catarata SCP típica de RP.

joría, dado que la incidencia de opacifi-cación capsular y de contracción capsu-lar anterior es alta, aunque la incidenciadel edema de mácula secundario es pe-queña74.
El edema macular cistoide secundariopuede responder a terapia con inhibido-res de la anhidrasa carbónica en un por-centaje importante de pacientes, aun-que no todos lo toleran a largo plazo yademás se puede producir un efecto re-bote en tratamientos prolongados75-77.Se ha utilizado la dorzolamida en gotaspudiendo ser eficaz en algunos pacien-tes sin muchos riesgos. También se haintentado la cirugía78 el bebacizumab79
y la triamcinolona intravítrea80.
Para el manejo de los síndromes pseu-do Coats se puede necesitar realizar fo-tocoagulación y/o crioterapia, sobretodo si están produciendo una exuda-ción que pueda comprometer el globoocular induciendo desprendimientosexudativos de la retina81.
A veces las ayudas de baja visión puedenser útiles, sobre todo la utilización de fil-tros de absorción selectiva para el con-trol del deslumbramiento, lo que ademásprotegerá la retinas82,83. Las ayudas queproducen magnificación no suelen fun-cionar en este tipo de pacientes. Ade-más habrá que ponerles la mejor refrac-ción posible y aconsejarles sobre la me-jor iluminación para la lectura y lamobilidad. También se les puede ense-ñar técnicas de escaneo del campo, sepueden probar prismas para tratar de ex-pandir el campo o a caminar con un pe-rro si fuera necesario84-86. Incluso se hanprobado gafas de vision nocturna quehan funcionado en algunos casos87,88.
En otras RP sindrómicas como el sín-drome de Refsum el oftalmólogo puedeser el primero en hacer el diagnóstico,por lo que hay que hacer las pruebas de
laboratorio adecuadas y referir al neuró-logo para el tratamiento dietético ade-cuado (dieta baja en phytol-ácido fitáni-co), ya que el tratamiento puede dete-ner la retinopatía. Otras RP sindrómicasque además son tratables son la abeta-lipoproteinemia o enfermedad de Bas-sen-Kornzweig, que será tratada con vi-tamina A y vitamina E y, en algunos ca-sos, con vitamina K también. Tampocoolvidemos la terapia con vitamina E parala ataxia Friedreich-like asociada con RPque parece eficaz a corto plazo89. En ni-ños con sordera en que se sospecheUsher’s es importante establecer lo an-tes posible el diagnóstico de RP, puesen estos casos está muy claro que elimplante coclear tiene un éxito muy su-perior si se coloca lo antes posible yesto tiene implicaciones a la hora de ad-quirir la habilidad de hablar de forma in-teligible e incluso de tener una vida aca-démica lo más normalizada posible90.
Por último, es importante aconsejar alos pacientes acudir a la ONCE y al IM-SERSO para registrarse como ciegos sicumplen los criterios de ceguera legal(AV< 0,1 y/o CV<10°)91. A veces es ne-cesario tratamiento psicológico para en-cajar la pérdida visual y también paraencajar que se está transmitiendo unaenfermedad hereditaria92.
Normalmente estos pacientes pregun-tan sobre posibilidades de tratamientosque aún se encuentran en fase experi-mental. Por eso puede ser bueno infor-marles de ello pues esto suele mejorarsu estado de depresión, siempre que
17
Diagnóstico electrofisiológico
Figura 13. Telangiectasias periféricas en RP.

se disponga del tiempo necesario. En elfuturo se buscan nuevos fármacos93 ytambién tratamientos que controlen laapoptosis94. Para algunas enfermeda-des será posible la realización de terapiagénica95-100. También se trabaja en tera-pia celular101 mediante algunas formasde trasplantes de EPR102 del complejofotoreceptor-EPR o de fotorrecepto-res103, o de células madre104-106. Tam-bién hay ensayos clínicos en fase III confactores de crecimiento que impiden lamuerte celular107 y aparatos electróni-cos para visión artificial108-110. Es impor-tante que el paciente comprenda que laidentificación de los genes causantes yel entendimiento de su función debenayudar en la puesta en marcha de estasnuevas terapias.
Casos en que resultaobligada la consulta conel genetista clínico
■ Casos infantiles, pues es importantepara el pronóstico del paciente, y tam-bién porque se trata de familias enque los padres se encuentran en edadde tener más hijos y, por lo tanto, sehace necesario dar consejo genético ala familia completa.
■ Casos no esporádicos porque tambiénhay que dar consejo genético a la fa-milia e identificar pacientes de riesgoy portadores.
■ Casos sindrómicos. En estos el diag-nóstico ha de ser correcto por las po-sibles complicaciones en otros órga-nos que han de ser siempre descarta-das o confirmadas.
■ Para afinar el diagnóstico en casospoco claros debido a que exista varia-bilidad familiar en la expresión clínicade la enfermedad.
■ Enfermedad de Stargardt, ya que enestos las probabilidades de encontrar
las mutaciones concretas son muyelevadas, permitiendo el diagnóstico yel consejo genético exacto en cadacaso.
■ Sospecha de mutaciones en el genCERKL, que se pueden sospechar porel aspecto clínico algo diferenciado(con una típica atrofia macular muyprecoz) debido al mal pronóstico ma-cular que implica si se confirma estamutación en el estudio genético mole-cular111.
Detección de mujeresportadoras de RP recesivaligada a X
La identificación de mujeres portadorasde RP ligada a X es importante para elconsejo genético. En algunos casos, laidentificación de estas portadoras esmuy fácil porque pertenecen a familiasya filiadas o porque son portadoras obli-gadas (han tenido o tienen un padre o unhijo afectos). Sin embargo, la identifica-ción del estado portador a veces resultamás difícil en el caso de mujeres que tie-nen una historia familiar menos docu-mentada, o en las que la segregación dela enfermedad se sitúa en el lado mater-no (la enfermedad se transmite a travésde mujeres portadoras sin que existanvarones afectados conocidos en la histo-ria familiar), o en el caso de aquellas mu-jeres que aún teniendo un hijo con RPsevera, carecen de historia familiar de laenfermedad. El diagnóstico del estadoportador heterozigoto en estas mujeresse basa, en la práctica clínica, en los ha-llazgos oftalmoscópicos y en las prue-bas electrofisiológicas.
La presencia de anormalidades fundus-cópicas (parches de movilización pig-mentaria con morfología de osteoclas-tos en media periferia retiniana, reflejotapetal en polo posterior, palidez de dis-
18
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...

co óptico o atenuación del árbol vascu-lar retiniano) observables en estas pa-cientes es variable.
En general, existe acuerdo en que loshallazgos oftalmoscópicos característi-cos del estado portador de RP ligada aX incluyen los siguientes fenotipos: re-flejo tapetal, cambios pigmentarios pe-riféricos con o sin morfología espiculaday ausencia de anormalidades detecta-bles oftalmoscópicamente. Aunque elreflejo tapetal es un signo de diagnósti-co positivo del estado portador, su inci-dencia varía en las diferentes series ysu ausencia no excluye el estado porta-dor. Esta variabilidad aparece tambiénentre pacientes portadoras obligadas.La mayoría de las portadoras no presen-tan este signo patognomónico del esta-do portador.
La presencia de cambios pigmentariosperiféricos es muy sugestiva del estadoportador, sin embargo, su valor diagnós-tico es menor que el del reflejo tapetal,sobre todo si su aspecto no se aseme-ja al de la RP clásica y especialmente enpresencia de cambios degenerativosperiféricos miópicos. En mujeres jóve-nes portadoras de RP ligada a X estehallazgo es menos frecuente que el delreflejo tapetal.
Es necesario ser especialmente cuida-doso en la evaluación de mujeres mio-pes que además presentan riesgo deser portadoras de RP ligada a X y, sobretodo, en ausencia de hallazgos oftalmos-cópicos típicos del estado portador. Losporcentajes de anormalidades electro-
rretinográficas detectables en mujeresportadoras varían desde el 50 al 96% delos casos según los diferentes autores,pero la identificación de alteracioneselectrofisiológicas puede verse dificul-tada por la alta prevalencia de miopía enestas pacientes.
Formas atípicas de RP
– RP inversa o lo que es lo mismo
distrofia de conos y bastones112. Lasdistrofias de conos son un subgrupogenéticamente heterogéneo de distro-fias coriorretinianas hereditarias carac-terizadas por degeneración progresivade todos los conos de la retina con pre-servación de la función de bastones encasos de distrofias puras de conos(DC), y con afectación más tardía delsistema de bastones en el caso de lasdistrofia de conos y bastones (DCB)113-
115. Es posible encontrarnos con todoslos tipos de herencia: autosómica do-minante116-119, autosómica recesiva yrecesiva liga a X120.
Los individuos afectos de distrofia deconos y bastones sufren inicialmentede fotofobia, disminución de visión, ce-guera a colores y escotomas centralesen el CV. En algunos existe palidez deldisco y apariencia granular del EPR ma-cular, mientras que en otros aparece lamaculopatía en ojo de buey típica. Enestadíos avanzados se observa atrofiacentral, pero lo que es más importantecomienzan a aparecer signos y sínto-mas de RP con ceguera nocturna y pig-
19
Diagnóstico electrofisiológico
Figura 14. Distrofia de conos y bastones, fotos de fondo y AF.

mentación en espículas en la periferiade la retina.
En distrofias de conos en estadíos ini-ciales, el ERG es obligado pues nos per-mitirá diferenciar una distrofia de conosde una distrofia macular, de forma queen la DC las pruebas fotópicas estaránafectadas de forma marcada, mientrasque en las distrofias maculares (comoel Stargardt´s o la distrofia coroidea are-olar central) las pruebas fotópicas pue-den ser normales o estar sólo levemen-te afectadas. Además, el ERG nos per-mitirá distinguir entre DC y DCB, ya queen la última observaremos, además deuna afectación importante de las prue-bas fotópicas, una disminución más omenos marcada de la onda b tambiénen el ERG escotópico.
Los pacientes con distrofia de conostienen problemas con la fotofobia y sue-len mejorar bastante llevando gafas olentes de contacto oscuras121,122. Tam-bién suelen responder bien a las ayudasvisuales que producen magnificación (alcontrario que los pacientes con RP) in-cluyendo ayudas ópticas tipo lupas oelectrónicas del tipo del circuito cerradode televisión.
– Amaurosis congénita de Leber
(LCA)123. Se trata de un grupo de distro-fias retinianas que podrían ser conside-radas como las RP de comienzo másprecoz causando una ceguera congéni-ta124,125. Su modo de herencia es auto-sómico recesivo126. Estos enfermossuelen tener fotofobia y se frotan conti-nuamente los ojos (signo de Frances-chetti o signo oculodigital). Los pacien-tes suelen tener inicialmente movi-mientos oculares irregulares debido asu imposibilidad de realizar la fijación,pero posteriormente desarrollan nistag-mus. Las reacciones pupilares son le-ves y ocasionalmente se puede encon-trar una respuesta paradógica. La mayorparte de los pacientes suelen ser hiper-métropes (al contrario que el resto delas RP en que los pacientes suelen sermiopes)127,128. También se conoce suasociación con keratocono, además depoder asociarse a otras anomalías sisté-micas.
Inicialmente el fondo de ojo es normal,pero con el tiempo aparecerán puntea-dos amarillentos129 y después la típicapigmentación en espículas, la atenua-ción de los vasos y la palidez del nervioóptico produciendo un aspecto indistin-guible de cualquier otra RP. Los cambiosmaculares se encuentran frecuente-mente, en forma de alteraciones de pig-mento inicialmente, y más tarde con laaparición de atrofia macular que puedenllegar a ser cambios colobomatosos130-
132. El ERG está abolido sobre los 3 me-ses de edad133.
20
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 15. Distrofia de conos.
Figura 16. A la izquierda ERG escotópico mixto normal, y al lado en DCB. La 3.ª figura muestra ERG fo-tópico normal, y a la derecha en DCB.

Actualmente se encuentra en fase deensayo clínico en humanos, la terapiagénica para LCA, pero sólo para las cau-sadas por RPE65. El gen RPE65 es res-ponsable de un gran porcentaje de ca-sos de LCA en Europa Central y del Nor-te, sin embargo en España son másfrecuentes las mutaciones CRB1, nohabiéndose encontrado en nuestro paísningún caso de LCA debida a mutaciónRPE65 hasta este momento.
– Distrofia tapetoretiniana cristalina
de Bietti Distrofia con depósitos amari-llentos birrefringentes en el fondo deojo134,135.
– Retinitis punctata albecens en queen lugar de pigmentación en espículasaparecen depósitos blanquecinos136
(producidas a mutaciones en el genRLBP1 que codifica la proteína CRALBPque participa en el metabolismo de la vi-
tamina A y que por ello mejoran espe-cialmente con suplementos con vitami-na A)137,138.
– RP sine pigmento (son en realidadformas iniciales de RP que suelen poderdemostrarse por alteración clara delERG). Antes de hacer este diagnósticopensar en otras posibilidades, y tenerclaro que si el ERG es normal se descar-ta este diagnóstico139.
21
Diagnóstico electrofisiológico
Figura 17. LCA.
Figura 18. Bietti’s.
Figura 19. Retinitis punctata albecens.

Formas localizadaso segmentarias de RP
Todas estas, a priori, tienen mucho me-jor pronóstico que las formas más ex-tensas de RP.■ RP en sector (AD): simétrica, bilateral,
inferonasal y con reducción leve de laamplitud de la onda b del ERG escotó-pico140.
■ RP anular o pericentral o circinada o
peripapilar, suele ser autosómica do-minante141.
■ RP perivenosa o paravenosa, en quelas zonas degeneradas están cercanasa las venas142.
■ RP con retina para-arterial preserva-
da, en que la pigmentación en espícu-las sólo aparece cuando nos alejamosde las arteriolas retinianas143.
■ RP unilateral (A priori dudar de estediagnóstico, ya que es raro, podríantratarse de portadoras de XLRP con
afectación en un ojo por inactivacióndel X «bueno» en el mismo)144,145.
Diagnóstico diferencialde los nistagmuscongénitos146
1. Amaurosis Congénita de Leber (ERGplano desde los 3 meses de edad).
22
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 20. RP en sector.
Figura 21. RP en anillo y su AF.
Figura 23. OI normal, en OD pseudo RP ocasionada por Diffuse Unilateral Subacute Neuroretinitis(DUSN), ver la larva que señala la flecha. La foto de la izquierda muestra la retina periférica inferior del OD.
Figura 22. RP perivenosa.

2. Acromatopsia congénita o mono-cromatismo de bastones (en el ERGsólo se alteran las pruebas fotópicaslo que ayuda al diagnóstico diferen-cial con LCA). ¡Ojo! no confundircon distrofias de conos, estas últi-mas son progresivas tanto en loscambios que se producen en el FOcomo en el deterioro de la funciónvisual.
3. Monocromatismo de conos azulesligado a X (MCLX) (en el ERG sólose alteran las pruebas fotópicas loque ayuda al diagnóstico diferencialcon LCA). Clínicamente indistingui-ble de la acromatopsia congénitapero, el MCLX es mucho menos fre-cuentes y ambas se diferencian porel modo de herencia, el ERG tam-bién puede ayudar (ERG con luz azulplano en el MCLX) e incluso se pue-de pedir diagnóstico genético mole-cular.
4. Distrofia conos (8% tienen nistag-mus, generalmente vertical).
5. Hipoplasia nervio óptico (ERG nor-mal con PVE plano).
6. En el albinismo147 realizar PVE de almenos 3 canales que muestra«missrouting» típico de esta enfer-medad en el 80% de los pacientespor el que se observa asimetría enla respuesta registrada en ambos ló-bulos occipitales y de amplitud ma-yor en el lóbulo contralateral al ojoestimulado. Se diferencia de la co-roideremia porque en el albinismo
existe aplasia macular (los vasos nosiguen dirección centrípeta hacia lafóvea) y porque además existe tran-siluminación del iris.
7. Alström (distrofia retiniana de co-mienzo precoz, sordera neurosenso-rial grave con obesidad, hipogona-dismo, fallo renal, diabetes mellitusinsulín resistente, y cardiomiopatíadilatada).
8. Sdr. Joubert: apraxia oculomotora(movimientos anormales de ojos ycabeza) asociada a distrofia retinia-na de comienzo precoz con hipopla-sia del vermis cerebeloso, y proble-mas respiratorios neonatales
9. Maple syrup urine disease (leuco-distrofia: error congénito del meta-bolismo de los aminoácidos quecursa con encefalopatía).
10. Síndrome de Senior-Loken (distrofiaretiniana y nefronoptisis).
11. Nistagmus idiopático, para hacereste diagnóstico sería necesario ha-cer ERG y descartar las otras causas.
Las enfermedades sindrómicas se des-cartan siempre en estrecha colabora-ción con el pediatra del niño.
23
Diagnóstico electrofisiológico
Figura 24. Albinismo.
1. Coroideremia
Esta es una enfermedad rara de heren-cia recesiva ligada a X. Generalmenteafecta a varones y las mujeres son por-tadoras mostrando cambios típicos en elfondo de ojo. Los síntomas típicos inclu-yen ceguera nocturna y constricciónconcéntrica del campo visual por lo quepuede ser confundida con RP. La visióncentral generalmente se conserva hastala década de los 50, lo que puede ayu-darnos a distinguirla de la XLRP en quese suele afectar mucho más pronto148.
La coroideremia es una de las enferme-dades hereditarias para la que el diag-nóstico prenatal está disponible ya quese conocen el locus y el gen causantes.
En los varones afectos la ceguera noc-turna y la pérdida de campo se inicianen la primera o segunda décadas de lavida. Las características fundoscópicastípicas cuando la enfermedad progresa,
incluyen pérdida total de pigmento tan-to a nivel del EPR como de la coriocapi-lar, acompañada de una pérdida extensade la capa coriocapilar. La retina internay el nervio óptico permanecen norma-les. Generalmente se trata de pacientesmiopes149,150.
Las mujeres portadoras suelen tener unfenotipo muy típico que puede ayudar aldiagnóstico del estatus de portadoracuando el diagnóstico genético molecu-lar no está disponible151. Las portadoraspueden estar afectas en distintos gra-dos de severidad. Puede haber sólocambios pigmentarios leves en mediaperiferia, o presentar degeneración delEPR y la coroides en parches más ex-tensos de la retina. También puede ha-ber cambios peripapilares degenerati-vos y depósitos drusenoides blanqueci-nos en la media periferia. Se puedenencontrar numerosos depósitos blan-cos puntiformes repartidos por toda laperiferia media, o alternativamente la
24
Diagnósticodiferencial de lasenfermedades extensasde fotorreceptores
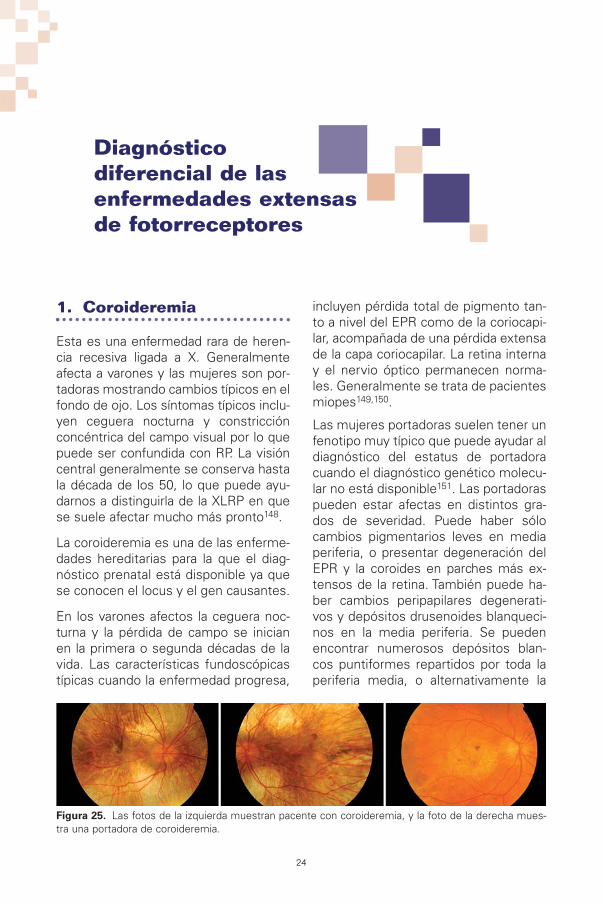
Figura 25. Las fotos de la izquierda muestran pacente con coroideremia, y la foto de la derecha mues-tra una portadora de coroideremia.

aparencia puede ser muy similar a la delos varones afectos. Unas pocas muje-res portadoras pueden progresar hastauna coroideremia franca a pesar de por-tar la mutación en heterocigosis.
La electrofisiología en pacientes afectoses similar a la de la RP. Sin embargo, laelectrofisiología en las portadoras decoroideremia suele ser completamentenormal, al contrario de lo que sucedecon las portadoras del XLRP en las quesuele encontrarse alguna anomalía, ayu-dando a diferenciar ambas enfermeda-des.
Es importante hacer el diagnostico dife-rencial con la XLRP de mucho peor pro-nóstico y con otras atrofias coroideasdifusas o con la atrofia girata152,153. Paraello se pueden usar los análisis de liga-miento, el análisis mutacional, y los testde anticuerpos anti-gen de la coroidere-mia. Los test de anticuerpos no puedenusarse para testar el estado de mujerportadora. También es posible que ha-cer diagnóstico prenatal si fuese nece-sario154.
2. Atrofia girata
La atrofia girata es una rara enfermedadde herencia autosómica recesiva que sedebe a la deficiente actividad de una en-zima de la matriz mitocondrial, la orniti-na aminotransferasa (OAT) que intervie-ne en el metabolismo de la arginina. Siexiste déficit de la OAT se produce unaumento de la ornitina en suero. La ele-vación de ornitina en el suero por si mis-ma no parece causar anomalías retinia-nas, ya que otros errores del metabolis-mo que cursan con hiperornitiemia noproducen afectación ocular.
Suele comenzar con nictalopia en la pri-mera década de la vida seguida de pér-dida de campo periférico y también pér-
dida de agudeza visual, por lo tanto lasintomatología es indistinguible de laRP. Es frecuente encontrar miopía ele-vada, astigmatismo y catarata subcap-sular. En el fondo de ojo se observanparches circulares de atrofia coroideaconfluentes en la periferia que se ex-tienden desde la retina anterior a la pos-terior. Algunos de los vasos coroideosgrandes pueden estar conservados155.Las lesiones permanecen separadasentre sí por bordes finos de pigmento yla región peripapilar se atrofia. La mácu-la se hace anormal en estadíos muy tar-díos de forma que la pérdida de agude-za visual suele suceder en la quinta osexta décadas de la vida, y presentan al-gunas alteraciones del ERG y el EOG156.
Encontrar una elevación de ornitina enel plasma o la orina confirma el diagnós-tico157. La actividad de la ornitina amino-transferasa está muy reducida y se pue-de medir en varios tejidos incluídos losfibroblastos de la piel. Los heterocigo-tos pueden tener reducida su actividadal 50% de lo normal.
Se han intentado tres aproximaciones ala terapia de la atrofia girata. En una pe-queña proporción de casos hay actividadresidual de ornitina aminotransferasaque puede ser estimulada dando suple-mentos de piridoxina (vitamina B6) conla subsecuente reducción de los nivelesde ornitina plasmáticos. Se ha demos-trado mejoría al menos en un pacientetratado de esta forma. Una segundaaproximación consiste en reducir el
25
Diagnóstico diferencial de las enfermedades extensas de fotorreceptores
Figura 26. Atrofia Girata.

aporte diario en la dieta de arginina por-que se han documentado casos de me-joría en la función visual con el uso dedietas restrictivas estrictas. Por último,se ha propuesto aumentar la eliminaciónrenal de ornitina para lo que se han usa-do la lisina y el ácido alpha-aminoiso-butyrico que interferirían con el transpor-te del aminoácido en el riñón158,159.
3. Ceguera nocturnacongénita estacionaria
Introducción
La ceguera nocturna congénita no pro-gresiva, o estacionaria, (CSNB) se ca-racteriza por el comienzo de nictalopiaen la infancia que no progresa y conbuena AV. La adaptación a la oscuridadpuede estar ausente o se encuentramuy prolongada y suelen ser mio-pes160,161. El modo de herencia más co-mún es el recesivo ligado a X aunqueexisten familias con herencia autosómi-ca recesiva y dominante, y parece quese deben a un fallo en la transmisión dela información desde los fotorrecepto-res a la retina interna162-164. Se puedensubdividir en dos grupos, los que tienenFO normal y los que tienen anormalida-des claras de fondo. Las que cursan confondo anormal son el fundus albipuncta-tus y la enfermedad de Oguchi.
Los pacientes con CSNB recesiva ligadaa X (los más frecuentes) son miopes y
típicamente tienen mala AV. La mayorparte de las portadoras son asintomáti-cas, aunque suelen cambios tener evi-dentes en el ERG. Como este grupo tie-ne mala AV con fondo de ojo normal yERG alterado, se puede confundir eldiagnóstico con la LCA en la infancia, yaque el ERG puede llegar a tardar en ex-tinguirse aproximadamente un año enel Leber. Para distinguirlas hay que fijar-se en que el ERG mejora dramática-mente en condiciones fotópicas en laCSNB.
Con excepción de las familias con he-rencia recesiva liga a X los pacientescon CSNB tienen buena visión de díapero una importante ceguera nocturna.Los errores de refracción son frecuen-tes, sobre todo, la miopía. Puede aso-ciarse con una pobre agudeza visual ynistagmus desde el primer momentode la vida en la CSNB tipo II.
El ERG puede ser útil para clasificar laCSNB en dos grupos: el tipo I (tipoRiggs/tipo Nougaret) no se observaonda a en pruebas escotópicas (ERGescotópico positivo con una onda b exa-gerada) lo que sugiere un defecto inhe-rente de los bastones165. En el tipo II(tipo Shubert-Bornshein) el ERG escotó-pico es negativo (la onda a está conser-vada comparada con la onda b) y se pre-sume que esto indicaría una anomalíapostransduccional, es decir la respues-ta se genera en la retina externa perono se transmite bien a la retina interna.
26
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 27. A la izquierda CSNB con FO normal (como se ve puede ser difícil sospecharla, pues se pue-de atribuir la ceguera nocturna a la propia miopía del paciente, pues son fondos miópicos). En el centroFO de un Oguchi’s y su coloración normalizada después de 3 horas en la oscuridad. A la derecha un fun-dus albipunctatus (el diagnóstico diferencial entre el fundus albipunctatus y la retinitis punctata albecenslo da el ERG, que es igual al de la RP en ésta última).

Un correcto estudio electrofisiológicoobservando los detalles descritos arribaes importante en el diagnóstico de estaenfermedad estacionaria y así poderdescartar degeneraciones retinianasprogresivas de peor pronóstico166.
4. Monocromatismocongénito de bastones =acromatopsia congénita
El término monocromatismo de basto-nes se usa para describir una enferme-dad en la que hay pocos conos y anó-malos, que también se llama acroma-topsia congénita. Es una enfermedadno progresiva que afecta a 1 de cada100.000 personas. Es la enfermedad deconos más frecuentes, pero a menudose falla en su diagnóstico. Los pacien-tes no tienen respuestas mediadas porconos, pero tienen función normal debastones. Su modo de herencia es au-tosómico recesivo.
Se caracteriza por una ceguera comple-ta de la visión de colores (acromatopsiacongénita completa) o una visión de co-lores gravemente limitada (acromatop-sia incompleta) y una gran aversión a laluz o fotofobia. Además, tienen baja AVdesde el nacimiento y nistagmus. Losque tienen una forma incompleta sue-len tener mejor AV que los que tienen laforma completa y se cree que es debi-do a la existencia de algunos conos delongitud de onda media (verdes) o larga(rojos). El ERG de conos está ausente omuy reducido. El FO habitualmente es
normal, aunque con el tiempo puedenobservarse cambios granulares en lamácula, se puede observar ausencia delreflejo foveal y en algunos casos apare-ce maculopatía en ojo de buey. La histo-patología muestra una reducción en elnúmero de conos extrafoveales y unaestructura anormal de los conos fovea-les.
En el ERG se observa una respuesta debastones en adaptación a la oscuridadnormal, con ausencia de respuesta deconos debido a la reducción en el nú-mero de éstos. Como el fondo es nor-mal, son los síntomas y el ERG los queconducen al diagnóstico. Además lacurva de adaptación a la oscuridad esmonofásica con ausencia de la adapta-ción inicial de los conos. La sensibilidadal contraste con colores puede revelarumbrales muy elevados o ausentes de-pendiendo del tipo de acromatopsia.
Estos pacientes mejoran con gafas olentes de contacto que permitan unatransmitancia entre el 10% y el 2% porlo que puede sugerirles su utiliza-ción167,168.
5. Monocromatismo deconos azules ligado a X
El MCLX es una rarísima enfermedad enque sólo los bastones y los conos S tie-nen una sensibilidad y un funcionamien-to normales. Los pacientes afectos sonvarones con mala AV, nistagmus, mio-pía, cambios mínimos en el FO desde elnacimiento, por lo tanto, con un cuadro
27
Diagnóstico diferencial de las enfermedades extensas de fotorreceptores
Figura 28. Acromatopsia completa y su AF.
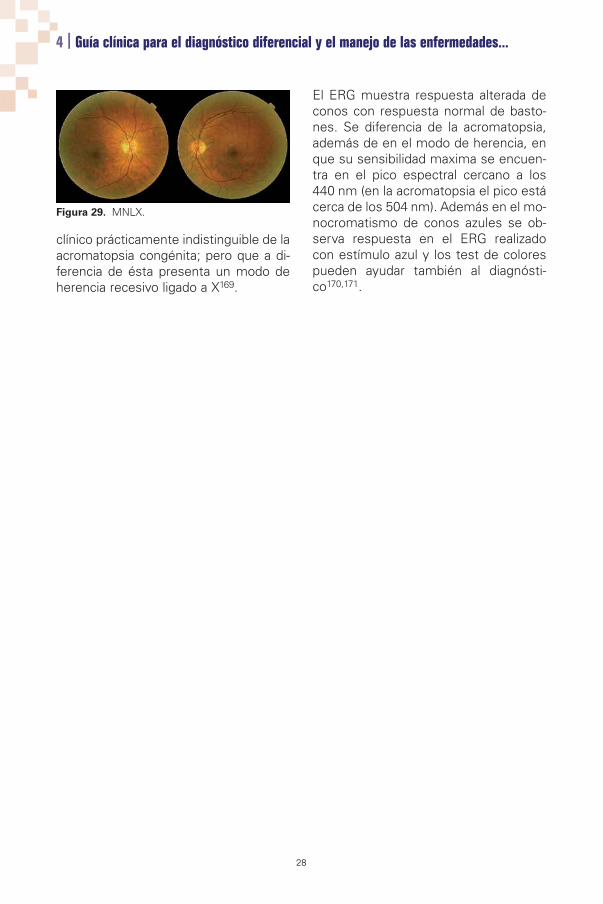
clínico prácticamente indistinguible de laacromatopsia congénita; pero que a di-ferencia de ésta presenta un modo deherencia recesivo ligado a X169.
El ERG muestra respuesta alterada deconos con respuesta normal de basto-nes. Se diferencia de la acromatopsia,además de en el modo de herencia, enque su sensibilidad maxima se encuen-tra en el pico espectral cercano a los440 nm (en la acromatopsia el pico estácerca de los 504 nm). Además en el mo-nocromatismo de conos azules se ob-serva respuesta en el ERG realizadocon estímulo azul y los test de colorespueden ayudar también al diagnósti-co170,171.
28
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 29. MNLX.

Las distrofias maculares hereditariasse caracterizan por una disminuciónde la agudeza visual de forma bilateraly por el hallazgo de unas alteracionesen el área macular de forma general-mente simétrica. La disminución en laagudeza visual puede manifestarsedesde el nacimiento o en cualquiermomento posterior, pero generalmen-te esta presente en las primeras dosdécadas de la vida. Estas distrofiaspueden afectar únicamente la macula,o pueden debutar en la macula y se-guir en la retina periférica. Estas enfer-medades pueden ser heredadas poraberraciones cromosómicas o por he-rencia autosómica dominante, autosó-mica recesiva o ligada al cromosomaX. Otros patrones de herencia más in-frecuentes son el digénico y el mito-condrial.
Fundus flavimaculatus/Enfermedad de Stargardt
Actualmente fundus flavimaculatus (FF)y enfermedad de Stargardt (STDG) sontérminos que se pueden usar para nom-brar un grupo heterogéneo de trastor-nos hereditarios de la retina ocasiona-dos por mutaciones en un mismo gen,ABCA4, cuyo modo de herencia es au-tosómico recesivo172. Hasta el 6% de la
población tiene mutaciones en este genen al menos uno de sus dos cromoso-mas 1, lo que hace que sea la segundaenfermedad hereditaria de la retina másfrecuente tras la retinitis pigmentosa yla distrofia macular más frecuente queexiste173.
Patofisiología
En 1997 Allikmets identificó el gen res-ponsable de esta enfermedad, el genABCR, o más concretamente el ATP-binding Cassette transporter gene,subfamilia A, miembro 4 (ABCA4)174.Este gen contiene 50 exones que codi-fican una proteína del borde de lamembrana del disco de los segmentosexternos de los fotorreceptores. Paraidentificar la función de esta proteínase creó un ratón transgénico deficien-te en ABCA4 observándose un aumen-to de all-trans retinal en los segmentosexternos tras el fotoblanqueamientoque indicaba un déficit parcial de suaclaramiento175. Estos ratones tam-bién mostraron un retraso en la adap-tación a la oscuridad que ya había sidodescrito en sujetos con STDG y queprobablemente se debe a una acumu-lación transitoria de un complejo no co-valente de opsina y all-trans retinal enlos discos176-178.
29
Distrofias maculares

ABCA4 pertenece a una familia de trans-portadores ABC (proteínas transmembra-na dependientes de energía) que partici-pan en transporte de substratos a travésde membranas. Cuando hay un defectoen este transportador el complejo N-reti-nilideno-PE se acumulará en el interiordel disco y pasará a las células del EPRmediante fagocitosis. La acumulación deeste complejo en las células del EPR lle-va a la formación de un fluoróforo de lipo-fucsina, el N-retinilidene-N-retiniletanola-mina (A-2E) cuya acumulación es tóxicapara esta célula. Secundariamente al fallode ésta célula se produciría la muerte delos fotorreceptores179,180.
Los últimos hallazgos sugieren la hipó-tesis de que mutaciones severas enABCR se asocian con enfermedad decomienzo precoz con pérdida primariade fotorreceptores (RP y DCB). En pa-cientes con mutaciones moderadas, losfotorreceptores funcionan al principio,sin embargo la falta de transporte deproductos de desecho conduce a unaacumulación del mismo en el EPR ypérdida secuencial de fotorreceptores
(FF/STDG). Finalmente la asociación deABCR a DMAE puede ser debida a laacumulación gradual de estos mismosproductos de desecho con eventual pér-dida de fotorreceptores181.
Clínica
Evolución de la enfermedad en el tiem-po: Aunque la mayoría de estos pacien-tes experimentan pérdida visual en lasdos primeras décadas de la vida, éstees un grupo heterogéneo de enferme-dades, y en algunos pacientes la enfer-medad se hace sintomática en periodosmedios de la vida o incluso más tarde.La pérdida de la visión central puede es-tar acompañada de síntomas así comode evidencias electrofisiológicas de unadisfunción de conos. Igualmente, algu-nos pacientes, particularmente aquellosque desarrollan flecos amarillentos ex-tendiéndose en la periferia pueden desa-rrollar evidencia de disfunción de conosy de bastones o presentar cuadros deretinitis pigmentaria franca con afecta-ción añadida del ERG de bastones182.
Tabla 2. ACTIVIDAD DE ABCR
Actividad de ABCR GENOTIPO FENOTIPO
NULA nulo/nulo RP+ hipo/nulo DCB
++ hipo/nulo o hipo/hipo Stargardt’s /FF+++ hipo/wild type DMAE+++ wild type/wild type Normal
30
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Disco Segm Ext Fr
ABCA4
FlipasaNretinilideno-PE all-trans retinal
OpsinaRodopsinaN-retinilideno-fosfatidletanolamina
A-2E tóxicopara EPR
Nretinilideno-N-retinletanolamina(A-2E)
Figura 30. Patofisiología en patología de retina debida a mutaciones en el gen ABCA4.
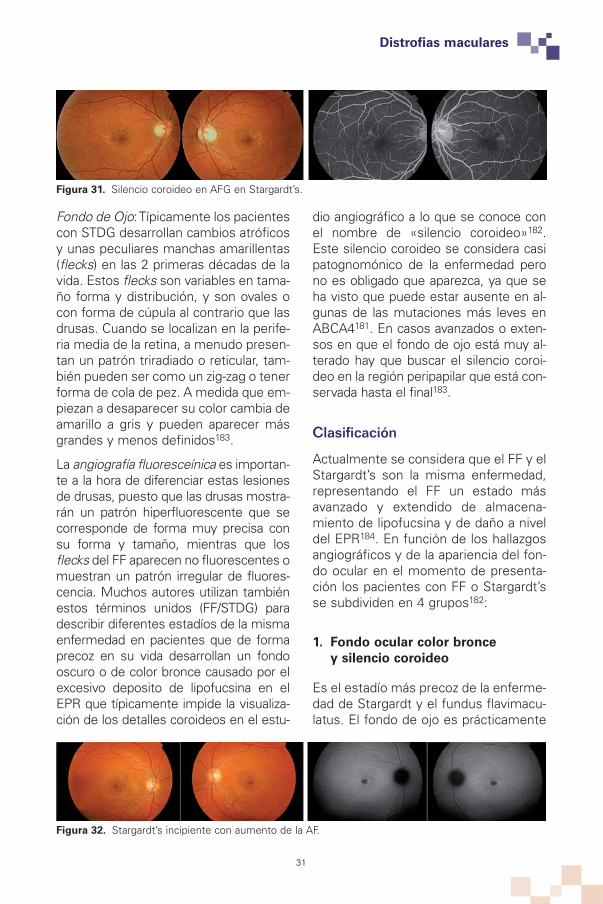
Fondo de Ojo: Típicamente los pacientescon STDG desarrollan cambios atróficosy unas peculiares manchas amarillentas(flecks) en las 2 primeras décadas de lavida. Estos flecks son variables en tama-ño forma y distribución, y son ovales ocon forma de cúpula al contrario que lasdrusas. Cuando se localizan en la perife-ria media de la retina, a menudo presen-tan un patrón triradiado o reticular, tam-bién pueden ser como un zig-zag o tenerforma de cola de pez. A medida que em-piezan a desaparecer su color cambia deamarillo a gris y pueden aparecer másgrandes y menos definidos183.
La angiografía fluoresceínica es importan-te a la hora de diferenciar estas lesionesde drusas, puesto que las drusas mostra-rán un patrón hiperfluorescente que secorresponde de forma muy precisa consu forma y tamaño, mientras que losflecks del FF aparecen no fluorescentes omuestran un patrón irregular de fluores-cencia. Muchos autores utilizan tambiénestos términos unidos (FF/STDG) paradescribir diferentes estadíos de la mismaenfermedad en pacientes que de formaprecoz en su vida desarrollan un fondooscuro o de color bronce causado por elexcesivo deposito de lipofucsina en elEPR que típicamente impide la visualiza-ción de los detalles coroideos en el estu-
dio angiográfico a lo que se conoce conel nombre de «silencio coroideo»182.Este silencio coroideo se considera casipatognomónico de la enfermedad perono es obligado que aparezca, ya que seha visto que puede estar ausente en al-gunas de las mutaciones más leves enABCA4181. En casos avanzados o exten-sos en que el fondo de ojo está muy al-terado hay que buscar el silencio coroi-deo en la región peripapilar que está con-servada hasta el final183.
Clasificación
Actualmente se considera que el FF y elStargardt’s son la misma enfermedad,representando el FF un estado másavanzado y extendido de almacena-miento de lipofucsina y de daño a niveldel EPR184. En función de los hallazgosangiográficos y de la apariencia del fon-do ocular en el momento de presenta-ción los pacientes con FF o Stargardt’sse subdividen en 4 grupos182:
1. Fondo ocular color bronce
y silencio coroideo
Es el estadío más precoz de la enferme-dad de Stargardt y el fundus flavimacu-latus. El fondo de ojo es prácticamente
31
Distrofias maculares
Figura 31. Silencio coroideo en AFG en Stargardt’s.
Figura 32. Stargardt’s incipiente con aumento de la AF.

normal excepto por el excesivo pigmen-to en el EPR que produce el color bron-ce y que oscurece los detalles de fondoen la coroides.
2. Maculopatía atrófica con o sin
flecks amarillentos
Es lo que clásicamente se ha llamado en-fermedad de Stargardt. Inicialmente lapérdida de EPR puede ser tan mínimaque en algunos pacientes sólo se pone demanifiesto al hacer la angiografía. Tambiénla aparición de fondo color bronce y silen-cio coroideo puede no ser evidente en laprimera década de la vida. Sin embargo,más tarde aparecen las lesiones amari-llentas y el almacenamiento de lipofucsi-na se hace evidente. Es muy común queaparezca un patrón en ojo de buey y enestos casos los pacientes se suelen que-jar de un escotoma anular, que en algunoscasos les permite conservar buenas agu-dezas visuales hasta la edad de 40 años.Estos pacientes pueden desarrollar enocasiones cambios excéntricos a nivel delEPR que incluyen hipertrofia, hiperplasia,metaplasia fibrosa y atrofia. Ocasional-mente también pueden desarrollar mem-branas neovasculares subretinianas y le-siones disciformes en la mácula.El test de visión de colores normalmentemuestra una discromatopsia rojo-verde
leve. Muchos pacientes muestran unalargamiento de la adaptación a la oscuri-dad185. Los hallazgos electrorretinográfi-cos generalmente son normales o discre-tamente alterados. El EOG puede ser dis-cretamente anormal en algunos casos186.
3. Maculopatía atrófica con signos
y síntomas tardíos de RP
Estos son similares a los del grupo dospero en edades más tardías de la vidaaparecen signos y síntomas de retinitispigmentosa incluyendo nictalopia, pér-dida difusa de pigmento del EPR, estre-chamiento de los vasos retinianos yanormalidades del ERG fotópico y esco-tópico186. Actualmente se consideraque muchos de estos pacientes pade-cen realmente una DCB.
4. Fundus flavimaculatus
Es el cuadro clínico descrito por Fran-ceschetti. Estos pacientes pueden te-ner lesiones amarillentas centrales y pa-racentrales asociados con mínima evi-dencia angiográfica o fundoscópica deatrofia del EPR entre estas lesiones.Suele haber silencio coroideo. La agu-deza visual puede ser normal si el cen-tro de la fóvea no está afectado por unade estas lesiones, aunque muchos pa-
32
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 33. Stargardt’s típico y su AF.
Figura 34. FF y su AF.

cientes tienen un gran fleck en la foveo-la que produce disminución de la visión.
Exploración para dar un pronóstico
Los hallazgos fundoscópicos y la pérdidavisual generalmente son simétricos enambos ojos, aunque ocasionalmenteuno de los dos ojos puede estar másafectado. También se sabe que general-mente los miembros de una misma fa-milia suelen evolucionar igual en cuantoal comienzo desarrollo y gravedad de lapérdida visual, aunque existen excepcio-nes, ya que puede haber heterogenei-dad genética en una misma familia187.
En cuanto al pronóstico visual, en un es-tudio observacional hecho en 95 pacien-tes con FF o Stargardt´s se ha visto quea los 19 años las probabilidades de man-tener una AV > 20/40 (0,5) en al menosun ojo son del 52%, del 32% a los 29años y del 22% a los 39 años. Una vezque la AV ha alcanzado este nivel de20/40, tiende a disminuir rápidamente yse estabilizará en 20/200 (0,1)188.
El OCT de alta resolución también per-mite evaluar el grado de pérdida de lacapa de fotorreceptores, lo que puedeayudar en el pronóstico de estos pa-cientes189.
La autofluorescencia puede ayudar a deli-near áreas de atrofia y éstas se puedencorrelacionar con la microperimetría me-jor que las fotos de fondo190. Sin embar-go, las áreas de disminución de autofluo-rescencia sólo se corresponden con lasáreas de escotoma profundo en la mitadde los pacientes. Por lo que se hace nece-sario medir la sensibilidad retiniana conotras técnicas y no fijarse sólo en la AF191.
La presencia de flecks en la media peri-feria, especialmente si aparece en eda-des tempranas de la vida, implica unpeor pronóstico par los pacientes afec-
tos de Stargardt en comparación conlos que sólo tienen afectación macu-lar192. Las posibilidades de encontrarcambios en el ERG y el EOG son mayo-res también en pacientes con FF y sig-nos y síntomas de distrofia de conos ybastones. Por ello, los estudios electro-fisiológicos van a tener también un im-portante valor pronóstico, sobre todo,en pacientes jóvenes en los que las le-siones amarillentas son todavía míni-mas186. Sin embargo, no se ha encon-trado una correlación entre los pacien-tes que muestran anomalías en el ERGcon uno o varios genotipos dados193.
Otras consideraciones
Se aconseja valorar la posibilidad de hacerdiagnóstico genético mediante un chipautomatizado que analiza hasta 501 muta-ciones diferentes de ABCA4 enviando elDNA a Asper Biotech (http://www.aspe-rophthalmics.com) y que en este momen-to es capaz de diagnosticar de formaexacta hasta un 98% de las variantes ge-néticas existentes de esta enfermedad.
La realización de técnicas de rehabilita-ción visual y el uso de ayudas visuales eneste grupo de pacientes suelen ser unéxito, por lo que hay que recomendarloencarecidamente.194, 195 Además, hay quetener en cuenta que afecta a pacientesmuy jóvenes que pueden necesitar ayu-das en su escolarización. Es especialmen-te importante recomendar la utilización defiltros de absorción selectiva tipo Corning,ya que además de mejorar la adaptación ala oscuridad propia de esta enfermedad,podrían disminuir la acumulación de mate-rial anómalo a nivel del EPR, retrasando eldaño que éste ocasiona83,196.
Estos pacientes presentan también deforma más frecuente lesiones secunda-rias a traumatismos oculares, y así se handescrito la aparición de edema de Berlin
33
Distrofias maculares

sostenido o de cambios degenerativosextensos del EPR con fibrosis subretinia-na182, por lo que hay que recomendarlesque eviten actividades en que puedanproducirse contusiones oculares.
Además, la toma de b-carotenos (Provita-mina A) y Retinol (Vitamina A) está FOR-MALMENTE CONTRAINDICADA ya queel acúmulo de A-2E podría aumentar siaumenta la cantidad de sus sustratos197.
Paradójicamente, hay algunos trabajosque sugieren un posible efecto benefi-cioso de un derivado de la Vitamina A (laisotretinoína, un medicamento usado enel tratamiento del acné grave cuyo nom-bre comercial es Roacután®) al bloquearla formación de A-2E198. Parece queeste fármaco disminuiría la acumulaciónde lipofucsina por microscopía electróni-ca en ratones transgénicos ABCR-/-.Este fármaco no debe tomarse en nin-gún caso si no es bajo control facultati-vo. Cuando se usa en el tratamiento delAcné nunca se prescribe de forma inde-finida, y cuando se utiliza en tratamien-tos prolongados los dermatólogos ha-cen controles hepáticos y de los lípidosséricos (fundamentalmente los triglicéri-dos) cada 3 meses. Por ese motivo, serecomienda esperar a tener mayoresevidencias sobre la eficacia y seguridadde este fármaco en esta enfermedad.
Se está ensayando ya la terapia génicaen animales de experimentación paraesta enfermedad, parece que esta tera-pia sí consigue reducir los niveles deA2E en el ratón, por lo que los resulta-dos han sido esperanzadores199.
Distrofia macularautosómica dominanteStargardt-like
La distrofia macular Stargardt-like auto-sómica dominante es una de las distro-
fias maculares de comienzo precoz enque se ha identificado el gen causante(ELOVL4) en 6p, una región con numero-sos genes implicados en otras distrofiasretinianas. El gen codifica para una prote-ína implicada en la elongación de los áci-dos grasos de cadena larga. Zhang y co-laboradores han hipotetizado que la si-militud con el fenotipo de Stargardt’srecesivo puede ser consecuencia de unaalteración de la actividad de ABCA4 enun ambiente anormal de los lípidos de lamembrana plasmática, lo que conduciríaa alteraciones bioquímicas similares enambas enfermedades200,201.
El Stargardt’s dominante se caracterizapor una pérdida precoz de la visión, cam-bios atróficos en la mácula con o sinflecks, relativamente buena función vi-sual, mínimos defectos en la visión de co-lores y ausencia de cambios significativosen el ERG. En ellos se hace especialmen-te importante hacer el diagnóstico dife-rencial con las distrofias en patrón y lasdistrofias de conos dominantes (tabla 3).
Central areolar choroidaldystrophy
La distrofia coroidea areolar central(CACD) fue descrita por primera vez en1884 por Nettleship y redescrita en1953 por Sorsby y Crick con 5 pedigre-es de los que sólo 1-2 eran autosómi-cos dominantes (varios tenían drusas oflecks por lo que podrían ser STDG oDMAE)202. CACD es el término más uti-lizado, aunque otros términos descripti-vos se han usado también para esta en-fermedad incluyendo «coroiditis centralsenil», «angio-esclerosis coroidea»,«esclerosis coroidea areolar central», y«atrofia coroidea areolar central»202-204.Es una enfermedad caracterizada por lapérdida de la coriocapilar en un área cir-cunscrita que afecta a la región foveal y
34
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...

parafoveal, y que a priori no se asocia adrusas cuticulares o flecks, aunque ve-remos que existen excepciones205. Esimportante conocer esta enfermedad,por su frecuencia (se trata de una en-fermedad dominante) y porque ennuestro país parece que es más fre-cuente que en otras poblaciones en-contrarnos con distrofias macularesproducidas por mutaciones en periferi-na/RDS entre las que se encuentraesta enfermedad206.
CACD se hereda normalmente de for-ma autosómica dominante, aunque se
han publicado pedigrees recesivos207.Se ha encontrado ligamiento de casosde CACD autosómica dominante al cro-mosoma 17p13, región en que se en-cuentran varios genes cuyas mutacio-
35
Distrofias maculares
Tabla 3. DIAGNÓSTICO DIFERENCIAL
Stargardt’s
DominanteDistrofia en Patrón
Distrofia de Conos
Dominante
Modo de herencia AutosómicoDominante
AutosómicoDominante
AutosómicoDominante
Gen ELOVL4 Periferin/RDS Variable
Presentación ↓ AV inexplicableMácula atrófica
moteada
Variable Hemeralopia +fotofobia +
mácula granular
Síntoma principal Disminuciónde visión
Disminuciónde visión
Disminuciónde visiónFotofobia
HemeralopiaDefecto visión colores
Visión 50% 0,5 a los20 años
Progresivo
Variable 20% < 0,5 a los20 años
Mácula Lesión atróficaFlecks en el FO
Acúmulo de pigmentoRaro flecks
Mácula granularAtrofia mácula en
ptes > Raro flecks
Nervio óptico Palidez temporal Normal NormalSilencio coroideo No No NoCampo visual periférico Normal Generalmente normal Generalmente normal
Visión colores Normal Normal Discromatopsia coneje claro
ERG Amplitud flickerpuede estar ↓ enpacientes mayores
con t implícitonormal
Variable ↓ Respuestasfotópicas
Figura 35. DCAC debida a mutación en periferinaen paciente de 58 años.

nes causan enfermedades hereditariasde retina208. También en otra región convarios genes que causan hereditariasen la retina está el gen peripherina/RDS(CR 6p), y se han descrito algunos pe-digríes en que la CACD es producidapor mutaciones en este gen, que curio-samente sí pueden tener drusas aso-ciadas209.
Clínica
Deutman en una serie larga de pacien-tes describió 4 estadios210:■ Estadío I: depigmentación moteada
fina y leve parafoveal, de comienzo enla edad adulta y asintomática.
■ Estadío II: moteado rodeando la fó-vea.
■ Estadío III: aparecen gradualmenteáreas redondas u ovales de atrofia geo-gráfica (ausencia de coriocapilar) simé-tricas, bien delimitadas, inferiores a 3-4 diámetros papilares y sin afectaciónfoveal.
■ Estadío IV: ya hay afectación central.
El diagnóstico puede ser difícil en ca-sos iniciales, porque los cambios ma-culares son inespecíficos y puede exis-tir solamente un moteado sin otros sig-
nos de enfermedad. El inicio de la en-fermedad se produce en la 4ª.-5.ª déca-das de la vida y se conservan AV de 0,1hasta edades avanzadas de la vida. Porello, muchos de ellos están mal diag-nosticados de DMAE seca. Con eltiempo aparecerá un reflejo tapetorre-tiniano y finalmente un área circunscri-ta de atrofia del EPR. En muchos ca-sos, hay un borde marcado que separala retina sana de la enferma, y es muytípico que persista una pequeña islacentral de 1-2° rodeada por una zonade atrofia que produce un escotomaen anillo de los 20° centrales. Los gran-des vasos coroideos subyacentes sehacen claramente visibles en estadíosavanzados, momento en que se hadescrito disfunción generalizada de FRcomprobada en AV, CV, fondo de ojo,AFG y ERG.
La AFG es útil en detección de cambiospigmentarios precoces (pueden tenerconfiguración en ojo de buey). Es raroque se compliquen con membranasneovasculares coroideas. La AFG en es-tadíos precoces muestra pequeñas zo-nas de efecto ventana del EPR en la re-gión parafoveal con dispersión de pig-mento. En casos más avanzados se veclaramente la ausencia de coriocapilaren la AFG. Finalmente lo único que seobservan son los grandes vasos coroi-deos en esa zona.
ERG y adaptación oscuridad suelen sernormales en esta enfermedad. El PERGy el PVE en patrón reverso están altera-dos (como en toda distrofia macular)desde fases precoces y en miembros
36
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 36. Paciente con DCAC en estadío I. Eshermano del anterior de 49 años (diagnósticopoco claro si no fuese por el hermano).
Figura 37. AF en DCAC

jóvenes. El EOG puede ser subnormal.El MERG suele mostrar disfunción másallá de las áreas de atrofia observadasoftalmoscópicamente. La exploracióncon autofluorescencia también puedeser útil.
Diagnóstico diferencialde enfermedades asociadasa atrofia central areolar ogeográfica del EPRy la coroides no asociadacon «flecks»205
■ Distrofias maculares familiares concoloboma macular:– Distrofia coroidea areolar central.– Distrofia anular concéntrica benig-
na.■ Atrofia central en distrofia de conos o
en distrofia de bastones y conos.■ Distrofia macular autosómica recesiva
asociada a FF-STDG.■ Distrofia pseudoinflamatoria de Sorsby
(raro, es más frecuente que desarro-llen CNV).
■ Drusas maculares dominantes queevolucionan a atrofia.
■ Drusas laminares basales (o lo que eslo mismo «drusas cuticulares») queevolucionan a atrofia.
■ DMAE atrófica.■ Atrofia 2.ª a miopía degenerativa.■ Atrofia 2.ª a desprendimiento seroso o
hemorrágico del EPR de causas varias(Ej.: coriorretinopatía serosa central ocoroiditis).
Distrofia viteliformemacular o enfermedadde Best
La enfermedad de Best, descrita en1905 por Friedrich Best, presenta carac-terísticamente, en estadios iníciales, eldepósito de un material amarillento enel área macular en forma de yema dehuevo211,212. Esta lesión puede ser úni-ca o múltiple. En la evolución, este de-pósito se descompone y evoluciona ha-cia una atrofia o fibrosis. Generalmente,el pronostico visual es bueno, mante-niéndose la capacidad de lectura hastala 5.ª década o incluso de por vida. Enocasiones, puede aparecer un neovas-cularización coroidea en el área macular,que suele notar el paciente como perdi-da de agudeza visual o incremento en lametamorfopsia.
La enfermedad de Best sigue un patrónde herencia autosómica dominante. Elgen responsable de la enfermedad hasido localizado en cromosoma 11q, ydenominado VMD2, el cual codifica unaproteína denominada bestrofina, la cualha sido localizada en la membrana plas-mática basolateral del epitelio pigmen-tario de la retina213-216. A pesar de tratar-se de una enfermedad de herencia do-minante, la expresividad de este gen esvariable (baja penetrancia), y las altera-ciones maculares a veces nunca se de-sarrollan en portadores obligados.
El EOG demuestra una marcada reduc-ción del ratio luz-oscuridad con un índi-ce de Arden patológico217, 218. El ERG esnormal. El EOG puede ser útil para de-
37
Distrofias maculares
Figura 38. Enfermedad de Best y su AF en estadío viteliforme en la primera década de la vida.

tectar aquellos pacientes que presen-tan la mutación sin lesiones en el áreamacular, y que pueden transmitir la en-fermedad a la siguiente generación.
La angiografía fluoresceínica no es útilen esta enfermedad (a menos que sesospeche neovascularización coroidea).La tomografía de coherencia óptica de-muestra el depósito del material hipe-rreflectante en el espacio subretiniano.Las imágenes por autofluorescencia de-muestran la marcada hiperautofluores-cencia del material viteliforme, y en fa-ses avanzadas, cuando predomina laatrofia o fibrosis, es característica la hi-perautofluorescencia en los márgenes
de la lesión con hipoautofluorescenciaen el área de atrofia del EPR.Recordemos que el estadio viteliforme(lesiones amarillas simétricas subfovea-les) aparece en las primeras décadas dela vida. Una lesión de tipo viteliforme enun paciente adulto no debe ser diagnos-ticada de Best a menos que se confir-me un EOG anómalo, pues puede de-berse a otras causas219.El diagnostico diferencial de las lesionesviteliformes maculares incluye: adult-on-set foveomacular dystrophy, patterndystrophy of the RPE, pseudo-vitelliformmacular detachment associated with ba-sal laminar or cuticular drusen220-222.
38
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...
Figura 40. Best avanzado con su autofluorescencia. Atrofia en OD y cicatriz de CNV en OI.
Figura 39. Best en estadío pseudohipopión, con su AFG y el OCT en la tercera década de la vida.

1. Fishman GA, Birch DG, Holder GA, BrigeMG. Electrophysiologic Testing in Disordersof the Retina, Optic nerve, and VisualPathway. In: Ophthalmology Monographs 2of the American Academy ofOphthalmology. 2nd Edition. Singapore.LEO. 2001.
2. Heckenlively JR, Arden GB, eds. Principlesand Practice of Clinical Electrophysiology ofVision. St. Louis. Mosby Year Book, Inc. 1991.
3. Brown M, Marmor MF, Vaegan, Zrenner E,Brigell M, Bach M. ISCEV Standard forClinical Electro-oculography (EOG) 2006,205-212.
4. Marmor MF, Zrenner E. Standard for clinicalelectro-oculography. Doc Ophthalmol 1993;85: 115-124.
5. Marmor MF, Holder GE, Seeliger NW,Yamamoto S: Standard for clinicalelectroretinography (2003 update). DocOphthalmol 2004; 108: 107-114.
6. Marmor MF, Fulton A, Holder GE, Miyake Y(Chair), Brigell M, Bach M. ISCEV Standardfor clinical electroretinography (2008update).
7. Brown M, Marmor MF, Vaegan, Zrenner E,Brigell MMarmor MF, Zrenner E. Standardfor clinical electroretinography. DocOphthalmol 1998; 97: 143-156.
8. Brigell M, Bach M, Barber C, Moskowitz A,Robson J: Guidelines for calibration ofstimulus and recording parameters used inclinical electrophysiology of vision (Revised2002). Doc Ophthalmol 2003; 107: 185-193.
9. Bach M, Hawlina M, Holder GE, Marmor M,Meigen T, Vaegan, Miyake Y. Standard forPattern Electroretinography. DocOphthalmol 2000; 101: 11-18.
10. Holder GE, Brigell MG, Hawlina M, MeigenT, Vaegan, Bach M. ISCEV standard forclinical pattern electroretinography - 2007update. Doc Ophthalmol 2007, 114: 111-116.
11. Hood DC, Bach M, Brigell M, Keating D,Kondo M, Lyons JS, Palmowski-Wolfe AM.ISCEV guidelines for clinical multifocalelectroretinography (2007 edition). DocOphthalmol 2008; 116: 1-11.
12. Odom JV, Bach M, Barber C, Brigell M,Holder G, Marmor MF, Tormene AP, Vaegan:Visual evoked potentials standard. DocOphthalmol 2004; 108: 115-123.
13. Marmor MF. The dilemma of the late-onset«dystrophy». Doc Ophthalmol 2007; 114:107-109.
14. Seles S, Lang GE. CAR syndrome(carcinoma-associated retinopathysyndrome) associated with anadenocarcinoma of the cervix. KlinMonatsbl Augenheilkd 2005; 222: 736-739.
15. Goodwin P. Hereditary retinal disease. CurrOpin Ophthalmol 2008; 19: 255-262.
16. Koh AH, Hogg CR, Holder GE. The incidenceof negative ERG in clinical practice. DocOphthalmol 2001; 102: 19-30.
17. Renner AB, Kellner U, Cropp E, FoersterMH. Dysfunction of transmission in theinner retina: incidence and clinical causes ofnegative electroretinogram. Graefes ArchClin Exp Ophthalmol 2006; 244: 1467-73.Epub 2006 Apr 13.
18. Bird AC. Retinal photoreceptor dystrophies;Edward Jackson Memorial Lecture. Am JOphthalmol 1995; 119: 543-562.
19. Hamel C. Retinitis pigmentosa. Orphanet JRare Dis. 2006; 11: 1: 40.
39
Bibliografía

20. Fain GL. Why photoreceptors die (and whythey don’t). Bioessays 2006; 28: 344-354.
21. Ben-Arie-Weintrob Y, Berson EL, Dryja TP.Histopathologic-genotypic correlations inretinitis pigmentosa and allied diseases.Ophthalmic Genet 2005; 26: 91-100.
22. Somani S, Brent MH, Markowitz SN. Visualfield expansion in patients with retinitispigmentosa. Can J Ophthalmol 2006; 41:27-33.
23. Tsujikawa M, Wada Y, Sukegawa M, SawaM, Gomi F, Nishida K, Tano Y. Age at onsetcurves of retinitis pigmentosa. ArchOphthalmol 2008; 126: 337-340.
24. van Soest S, Westerveld A, de Jong PT,Bleeker-Wagemakers EM, Bergen AA.Retinitis pigmentosa: defined from amolecular point of view. Surv Ophthalmol1999; 43: 321-334.
25. Daiger SP, Bowne SJ, Sullivan LS.Perspective on genes and mutationscausing retinitis pigmentosa. ArchOphthalmol 2007; 125: 151-158.
26. Skeik N, Jabr FI. Usher’s syndrome. InternMed. 2008; 47: 807-808. Epub 2008 Apr 16.
27. Kimberling WJ, Moller C. Clinical andmolecular genetics of Usher syndrome. JAm Acad Audiol 1995; 6: 63-72.
28. Smith RJH, Berlin CI, Hejtmancik JF, et al.Clinical diagnosis of the Usher syndromes.Am J Med Genet. 1994; 50: 32-38.
29. Usher CH. Clinical and geneticheterogeneity of Usher syndrome. R. LondOphthalmol Hosp Rep 1914; 19: 130-236.
30. Jacobson SG, Cideciyan AV, Aleman TS,Sumaroka A, Roman AJ, Gardner LM,Prosser HM, Mishra M, Bech-Hansen NT,Herrera W, Schwartz SB, Liu XZ, KimberlingWJ, Steel KP, Williams DS. Ushersyndromes due to MYO7A, PCDH15,USH2A or GPR98 mutations share retinaldisease mechanism. Hum Mol Genet. 2008;17: 2405-2415. Epub 2008 May 7.
31. Bruford A, Riise R, Teague PW, et al.Linkage mapping in 29 Bardet-Biedlsyndrome families confirms loci inchromosomal regions 11q13, 15q22.3-q23,and 16q21. Genomics 1997; 41: 93-99.
32. Campo RV, Aaberg TM. Ocular and systemicmanifestations of the Bardet-Biedlsyndrome. Am J Ophthalmol1982; 94: 750-756.
33. Green JS, Parey PS, Hamett JD, et al. Thecardinal manifestations of Bardet-Biedlsyndrome a form of Laurence-Moon-Bardet-Biedl syndrome. N Engl J Med 1989; 321:1002-1009.
34. Keith CG. Bardet-Biedl syndrome. Aust JOphthalmol 1984; 12: 143-148.
35. Rizzo JF, Berson EL Lessell S. Retinal andneurological findings in the Laurence-Moon-Bardet-Biedl phenotype. Ophthalmology1986; 93: 1452-1456.
36. Hansen RM, Eklund SE, Benador IY, MockoJA, Akula JD, Liu Y, Martinez-Perez ME,Fulton AB. Retinal degeneration in children:dark adapted visual threshold and arteriolardiameter. Vision Res 2008; 48: 325-31. Epub2007 Aug 31.
37. Berson EL. Electroretinographic findings inretinitis pigmentosa. Jpn J Ophthalmol1987: 31: 327-348.
38. Berson EL. Retinitis Pigmentosa. TheFriedenwald Lecture. Invest Ophthalmol VisSci 1993; 34: 1659-1675.
39. Stavrou P, Good PA, Broadhurst EJ, BundeyS, Fielder AR, Crews SJ. ERG and EOGabnormalities in carriers of X-linked retinitispigmentosa. Eye 1996; 10: 581-589.
40. Hartong DT, Berson EL, Dryja TP. Retinitispigmentosa. Lancet 2006; 368: 1795-1809.
41. Gerth C, Wright T, Héon E, Westall CA.Assessment of central retinal function inpatients with advanced retinitis pigmentosa.Invest Ophthalmol Vis Sci 2007; 48: 1312-1318.
42. Sandberg MA, Gaudio AR. Reading speed ofpatients with advanced retinitis pigmentosaor choroideremia. Retina 2006; 26: 80-88.
43. Kiser AK, Mladenovich D, Eshraghi F,Bourdeau D, Dagnelie G. Reliability andconsistency of visual acuity and contrastsensitivity measures in advanced eyedisease. Optom Vis Sci. 2005; 82: 946-954.
44. Hajali M, Fishman GA, Anderson RJ. Theprevalence of cystoid macular oedema inretinitis pigmentosa patients determined byoptical coherence tomography. Br JOphthalmol 2008; 92: 1065-1068.
45. Witkin AJ, Ko TH, Fujimoto JG, Chan A,Drexler W, Schuman JS, Reichel E, DukerJS. Ultra-high resolution optical coherencetomography assessment of photoreceptorsin retinitis pigmentosa and related diseases.Am J Ophthalmol 2006; 142: 945-952. Epub2006 Sep 1.
40
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...

46. Robson AG, Michaelides M, Saihan Z, BirdAC, Webster AR, Moore AT, Fitzke FW,Holder GE. Functional characteristics ofpatients with retinal dystrophy that manifestabnormal parafoveal annuli of high densityfundus autofluorescence; a review andupdate. Doc Ophthalmol 2008; 116: 79-89.Epub 2007 Nov 6.
47. Meyerle CB, Fisher YL, Spaide RF.Autofluorescence and visual field loss insector retinitis pigmentosa. Retina 2006; 26:248-250.
48. Kellner U, Kellner S, Weber BH, Fiebig B,Weinitz S, Ruether K. Lipofuscin- andmelanin-related fundus autofluorescencevisualize different retinal pigment epithelialalterations in patients with retinitispigmentosa. Eye 2008 Sep 12. [Epub aheadof print].
49. Ikenoya K, Kondo M, Piao CH, Kachi S,Miyake Y, Terasaki H. Preservation ofmacular oscillatory potentials in eyes ofpatients with retinitis pigmentosa andnormal visual acuity. Invest Ophthalmol VisSci 2007; 48: 3312-3317.
50. Wolsley CJ, Silvestri G, O’Neill J, SaundersKJ, Anderson RS. The association betweenmultifocal electroretinograms and OCTretinal thickness in retinitis pigmentosapatients with good visual acuity. Eye 2008Oct 31. [Epub ahead of print].
51. Nagy D, Schönfisch B, Zrenner E, Jägle H.Long-term follow-up of retinitis pigmentosapatients with multifocal electroretinography.Invest Ophthalmol Vis Sci 2008; 49: 4664-4671. Epub 2008 Jun 19.
52. Seeliger M, Pfister M, Gendo K, Paasch S,Apfelstedt-Sylla E, Plinkert P, Zenner HP,Zrenner E. Comparative study of visual,auditory, and olfactory function in Ushersyndrome. Graefes Arch Clin ExpOphthalmol 1999; 237: 301-307.
53. Daiger SP, Sullivan LS, Gire AI, Birch DG,Heckenlively JR, Bowne SJ. Mutations inknown genes account for 58% ofautosomal dominant retinitis pigmentosa(adRP). Adv Exp Med Biol 2008; 613: 203-209.
54. Koenekoop RK, Lopez I, den Hollander AI,Allikmets R, Cremers FP. Genetic testing forretinal dystrophies and dysfunctions:benefits, dilemmas and solutions. ClinExperiment Ophthalmol 2007; 35: 473-485.
55. Chong NHV, Bird AC. Management of inheritedouter retinal dystrophies: present and future.Br J Ophthalmol 1999; 83: 120-122.
56. Hamosh A, Scott AF, Amberger J, Bocchini C,Valle D, Mckusick VA. Online MendelianInheritance in Man (OMIM), aknowledgebase of human genes and geneticdisorders. Nucl. Acids Res 2002; 30: 52-55.
57. Dryja TP, Hahn LB, Cowley GS, McGee TL,Berson EL. Mutation spectrum of therhodopsin gene among patients withautosomal dominant retinitis pigmentosa.Proc Natl Acad Sci USA. 1991; 88: 9370-9374.
58. Heckenlively JR Autosomal dominantretinitis pigmentosa. In Heckenlively JR.(ed.) Retinitis Pigmentosa. Philadelphia.Lippincott. 1988: pp. 125-149.
59. Inglehearn CF. Molecular genetics of humanretinal dystrophies. Eye 1998; 12: 571-579.
60. Kajiwara K, Hahn LG, Mukai S, Travis GH,Berson EL,Dryja TP. Mutations in the humandegeneration slow gene in autosomaldominant retinitis pigmentosa. Nature 1991;354: 480-483.
61. [No authors listed]. NEI statement: Updateon vitamin A as a treatment for retinitispigmentosa. Optom Vis Sci 2008; 85: E790-E791.
62. Berson EL, Rosner B, Sandberg MA, et al. Arandomized trial of vitamina a and vitaminaE supplementation for retinitis pigmentosa.Arch Opthalmol 1993; 111: 761-772.
63. Marmor MF. A randomized trial of vitaminaA and vitamina E suplementation forretinitis pigmentosa. Arch Ophthalmol 1993;111: 1460-1461.
64. Massof RW, Finkelsein D. Supplementalvitamina A retards loss of ERG amplitude inretinitis pigmentosa. Arch Ophthalmol 1993;111: 751-754.
65. Norton EWD. A randomized trial of vitaminA and vitamin E supplementation forretinitis pigmentosa. Arch Ophthalmol 1993;111: 1460.
66. Hodge WG, Barnes D, Schachter HM, PanYI, Lowcock EC, Zhang L, Sampson M,Morrison A, Tran K, Miguelez M, Lewin G.The evidence for efficacy of omega-3 fattyacids in preventing or slowing theprogression of retinitis pigmentosa: asystematic review. Can J Ophthalmol 2006;41: 481-490.
41
Bibliografía

67. Berson EL, Rosner B, Sandberg MA,Weigel-DiFranco C, Moser A, Brockhurst RJ,Hayes KC, Johnson CA, Anderson EJ,Gaudio AR, Willett WC, Schaefer EJ. Clinicaltrial of docosahexaenoic acid in patientswith retinitis pigmentosa receiving vitaminA treatment. Arch Ophthalmol 2004; 122:1297-1305.
68. Berson EL, Rosner B, Sandberg MA,Weigel-DiFranco C, Moser A, Brockhurst RJ,Hayes KC, Johnson CA, Anderson EJ,Gaudio AR, Willett WC, Schaefer EJ. Furtherevaluation of docosahexaenoic acid inpatients with retinitis pigmentosa receivingvitamin A treatment: subgroup analyses.Arch Ophthalmol 2004; 122: 1306-1314.
69. Bazan NG. Cell survival matters:docosahexaenoic acid signaling,neuroprotection and photoreceptors. TrendsNeurosci 2006; 29: 263-271. Epub 2006 Apr 3.
70. Bahrami H, Melia M, Dagnelie G. Luteinsupplementation in retinitis pigmentosa: PC-based vision assessment in a randomizeddouble-masked placebo-controlled clinicaltrial [NCT00029289]. BMC Ophthalmol2006; 6: 23.
71. Sanz MM, Johnson LE, Ahuja S, Ekström PA,Romero J, van Veen T. Significantphotoreceptor rescue by treatment with acombination of antioxidants in an animalmodel for retinal degeneration. Neuroscience2007; 145: 1120-1129. Epub 2007 Feb 9.
72. Berson EL. Nutrition and retinaldegenerations. Int Ophthalmol Clin 2000;40: 93-111.
73. Komeima K, Rogers BS, Lu L, CampochiaroPA. Antioxidants reduce cone cell death in amodel of retinitis pigmentosa. Proc NatlAcad Sci U S A 2006; 103: 11300-11305.Epub 2006 Jul 18.
74. Jackson H, Garway-Heath D, Rosen P, BirdAC, Tuft SJ. Outcome of cataract surgery inpatients with retinitis pigmentosa. Br JOphthalmol. 2001 Aug; 85(8): 936-8.
75. Wolfensberger TJ. The role of carbonicanhydrase inhibitors in the management ofmacular edema. Doc Ophthalmol 1999; 97:387-397.
76. Fishman GA, Apushkin MA. Continued useof dorzolamide for the treatment of cystoidmacular oedema in patients with retinitispigmentosa. Br J Ophthalmol 2007; 91: 743-745. Epub 2007 Jan 10.
77. Grover S, Apushkin MA, Fishman GA.Topical dorzolamide for the treatment ofcystoid macular edema in patients withretinitis pigmentosa. Am J Ophthalmol2006; 141: 850-858. Epub 2006 Mar 20.Comment in: Am J Ophthalmol 2006; 142:707; author reply 707-708.
78. García-Arumí J, Martinez V, Sararols L,Corcostegui B. Vitreoretinal surgery forcystoid macular edema associated withretinitis pigmentosa. Ophthalmology 2003;110: 1164-1169.
79. Melo GB, Farah ME, Aggio FB. Intravitrealinjection of bevacizumab for cystoid macularedema in retinitis pigmentosa. ActaOphthalmol Scand 2007; 85: 461-463. Epub2007 Feb 20.
80. Scorolli L, Morara M, Meduri A, Reggiani LB,Ferreri G, Scalinci SZ, Meduri RA. Treatmentof cystoid macular edema in retinitispigmentosa with intravitreal triamcinolone.Arch Ophthalmol 2007; 125: 759-764.
81. Gass JDM, Sterescopic atlas of maculardiseases: diagnosis and treatment, 4th ed.St Louis, MO: CV Mosby; 1997: pp. 357.
82. Adrian W, Everson RW, Schmidt I.Protection against photic damage in retinitispigmentosa. Adv Exp Med Biol 1977; 77:233-247.
83. Paskowitz DM, LaVail MM, Duncan JL.Light and inherited retinal degeneration. BrJ Ophthalmol 2006; 90: 1060-1066. Epub2006 May 17.
84. Weiss NJ. Low vision management ofretinitis pigmentosa. J Am Optom Assoc1991; 62: 42-52.
85. Croughs P. Filtering lenses in retinopathy. BullSoc Belge Ophtalmol 1997; 264: 119-124.
86. Somani S, Brent MH, Markowitz SN. Visualfield expansion in patients with retinitispigmentosa. Can J Ophthalmol 2006; 41:27-33.
87. Hartong DT, Kooijman AC. Night-visiongoggles for night-blind subjects: subjectiveevaluation after 2 years of use. OphthalmicPhysiol Opt 2006; 26: 490-496.
88. Mancil RM, Mancil GL, King E, Legault C,Munday J, Alfieri S, Nowakowski R, BlaschBB. Improving nighttime mobility in personswith night blindness caused by retinitispigmentosa: A comparison of two low-visionmobility devices. J Rehabil Res Dev 2005;42: 471-486.
42
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...

89. Grant CA, Berson EL. Treatable forms ofretinitis pigmentosa associated withsystemic neurological disorders. IntOphthalmol Clin 2001; 41: 103-110.
90. Blanchet C, Roux AF, Hamel C, Ben Salah S,Artières F, Faugère V, Uziel A, Mondain M.Usher type I syndrome in children:genotype/phenotype correlation andcochlear implant benefits. Rev Laryngol OtolRhinol (Bord) 2007; 128: 137-143.
91. Jangra D, Ganesh A, Thackray R, Austin L,Ulster A, Sutherland J, Levin AV.Psychosocial adjustment to visual loss inpatients with retinitis pigmentosa.Ophthalmic Genet 2007; 28: 25-30.
92. Mezer E, Babul-Hirji R, Wise R, Chipman M,DaSilva L, Rowell M, Thackray R, ShumanCT, Levin AV. Attitudes regarding predictivetesting for retinitis pigmentosa. OphthalmicGenet 2007; 28: 9-15.
93. Ohguro H. New drug therapy for retinaldegeneration. Nippon Ganka Gakkai Zasshi2008; 112: 7-21.
94. Marigo V. Programmed cell death in retinaldegeneration: targeting apoptosis inphotoreceptors as potential therapy forretinal degeneration. Cell Cycle 2007; 6:652-655. Epub 2007 Mar 17.
95. Buch PK, MacLaren RE, Ali RR.Neuroprotective gene therapy for thetreatment of inherited retinal degeneration.Curr Gene Ther 2007; 7: 434-445.
96. Gorbatyuk MS, Hauswirth WW, Lewin AS.Gene therapy for mouse models of ADRP.Adv Exp Med Biol 2008; 613: 107-112.
97. Stieger K, Mendes-Madeira A, Meur GL,Weber M, Deschamps JY, Nivard D, ProvostN, Moullier P, Rolling F. Oral administrationof doxycycline allows tight control oftransgene expression: a key step towardsgene therapy of retinal diseases. Gene Ther2007; 14: 1668-1673. Epub 2007 Oct 4.
98. Tomita H, Sugano E, Yawo H, Ishizuka T,Isago H, Narikawa S, Kügler S, Tamai M.Restoration of visual response in ageddystrophic RCS rats using AAV-mediatedchannelopsin-2 gene transfer. InvestOphthalmol Vis Sci 2007; 48: 3821-3826.
99. Shinohara T, Mulhern ML, Madson CJ.Silencing gene therapy for mutantmembrane, secretory, and lipid proteins inretinitis pigmentosa (RP). Med Hypotheses2008; 70: 378-380. Epub 2007 Jun 27.
100. O’Reilly M, Palfi A, Chadderton N,Millington-Ward S, Ader M, Cronin T, Tuohy T,Auricchio A, Hildinger M, Tivnan A, McNallyN, Humphries MM, Kiang AS, Humphries P,Kenna PF, Farrar GJ. RNA interference-mediated suppression and replacement ofhuman rhodopsin in vivo. Am J Hum Genet2007; 81: 127-135. Epub 2007 May 23.
101. Lund R. Cell-based therapies to limitphotoreceptor degeneration. Arch Soc EspOftalmol 2008; 83: 457-464.
102. Pinilla I, Cuenca N, Sauvé Y, Wang S, LundRD. Preservation of outer retina and itssynaptic connectivity following subretinalinjections of human RPE cells in the RoyalCollege of Surgeons rat. Exp Eye Res 2007;85: 381-392. Epub 2007 Jun 14.
103. Vecino E. Gene therapy against retinosispigmentary. Arch Soc Esp Oftalmol 2008;83: 213-214.
104. Mooney I, LaMotte J. A review of thepotential to restore vision with stem cells.Clin Exp Optom 2008; 91: 78-84.
105. Arnhold S, Absenger Y, Klein H, Addicks K,Schraermeyer U. Transplantation of bonemarrow-derived mesenchymal stem cellsrescue photoreceptor cells in the dystrophicretina of the rhodopsin knockout mouse.Graefes Arch Clin Exp Ophthalmol 2007;245: 414-422. Epub 2006 Aug 4.
106. Haruta M. Embryonic stem cells: potentialsource for ocular repair. Semin Ophthalmol2005; 20: 17-23.
107. Sieving PA, Caruso RC, Tao W, Coleman HR,Thompson DJ, Fullmer KR, Bush RA. Ciliaryneurotrophic factor (CNTF) for human retinaldegeneration: phase I trial of CNTFdelivered by encapsulated cell intraocularimplants. Proc Natl Acad Sci U S A 2006;103: 3896-3901. Epub 2006 Feb 27.
108. Besch D, Sachs H, Szurman P, Gülicher D,Wilke R, Reinert S, Zrenner E, Bartz-SchmidtKU, Gekeler F. Extraocular surgery forimplantation of an active subretinal visualprosthesis with external connections:feasibility and outcome in seven patients. BrJ Ophthalmol 2008; 92: 1361-1368. Epub2008 Jul 28.
109. Thanos S, Heiduschka P, Stupp T.Implantable visual prostheses. ActaNeurochir Suppl 2007; 97: 465-472.
43
Bibliografía

110. Gekeler F, Szurman P, Grisanti S, Weiler U,Claus R, Greiner TO, Völker M, Kohler K,Zrenner E, Bartz-Schmidt KU. Compoundsubretinal prostheses with extra-ocular partsdesigned for human trials: successful long-term implantation in pigs. Graefes Arch ClinExp Ophthalmol 2007; 245: 230-241.
111. Avila-Fernandez A, Riveiro-Alvarez R,Vallespin E, Wilke R, Tapias I, CantalapiedraD, Aguirre-Lamban J, Gimenez A, Trujillo-Tiebas MJ, Ayuso C. CERKL mutations andassociated phenotypes in seven Spanishfamilies with autosomal recessive retinitispigmentosa. Invest Ophthalmol Vis Sci2008; 49: 2709-2713.
112. Michaelides M, Hardcastle AJ, Hunt DM,Moore AT. Progressive cone and cone-roddystrophies: phenotypes and underlyingmolecular genetic basis. Surv Ophthalmol2006; 51: 232-258.
113. Simunovic MP, Moore AT. The conedystrophies. Eye 1998; 12: 553-565.
114. Hamel CP. Cone rod dystrophies. OrphanetJ Rare Dis 2007; 2: 7.
115. Weleber RG, Eisner A. Cone Degeneration(Bull’s eye dystrophies) and colour visiondefects in retinal dystrophies anddegenerations. DA Newsome. New York.Raven Press. 1988; pp. 233-56.
116. Kelsell RE, Evans K, Gregory C, Moore AT,Bird AC, Hunt DM. Localisation of a genefor autosomal dominant cone-rod dystrophyCORD 6 to chromosome 17p. Hum MolGenet 1997; 6: 587-600.
117. Nakazawa M, Kikawa E, Chida Y, Wada Y,Shiono T, And Tamai M. Autosomal dominantcone-rod dystrophy associated withmutations in codon 244 (Asn211His) andcodon 184 (Tyr184Ser) of theperipherin/RDS gene. Arch Ophthalmol1996; 114: 72-78.
118. Payne AM, Downes SM, Bessant DAR,Taylor R, Holder GE, Warren MJ, Bird AC,Bhattacharya SS. A mutation in guanylatecyclase activator 1A (GUCA1A) in anautosomal dominant cone dystrophypedigree mapping to a new locus onchromosome 6p21.1. Hum Mol Genet 1998;2: 273-277.
119. Small KW, Syrquin M, Mullen L, Gehrs KMapping of autosomal dominant conedegeneration to chromosome 17p. Am. J.Ophthalmol 1996; 121: 13-18.
120. Jacobsen DM, Thompson HS, Bartley JA. X-linked progressive cone dystrophy. Clinicalcharacteristics in affected males and femalecarriers. Ophthalmology 1989; 96: 885.
121. Young RS, Krefman RA, Fishman GA. Visualimprovements with red-tinted glasses in apatient with cone dystrophy. ArchOphthalmol 1982; 100: 268-271.
122. Jonsson AC, Burstedt MS, Golovleva I,Sandgren O. Tinted contact lenses inBothnia dystrophy. Acta Ophthalmol Scand2007; 85: 534-539. Epub 2007 Mar 22.
123. Yzer S, Leroy BP, De Baere E, de Ravel TJ,Zonneveld MN, Voesenek K, Kellner U,Ciriano JP, de Faber JT, Rohrschneider K,Roepman R, den Hollander AI, CruysbergJR, Meire F, Casteels I, van Moll-RamirezNG, Allikmets R, van den Born LI, CremersFP. Microarray-based mutation detection andphenotypic characterization of patients withLeber congenital amaurosis. InvestOphthalmol Vis Sci 2006; 47: 1167-1176.
124. Alström CH, Olson OA. Heredo-retinopathiacongenitalis monohybrida recessivaautosomalis. Hereditas 1957; 43: 1-178.
125. Edwards WC, Price WD, Macdonald R Jr.Congenital amaurosis of retinal origin (Leber).Am J Ophthalmol 1971; 72: 724-728.
126. den Hollander AI, Roepman R, KoenekoopRK, Cremers FP. Leber congenitalamaurosis: genes, proteins and diseasemechanisms. Prog Retin Eye Res 2008; 27:391-419. Epub 2008 Jun 1.
127. Dagi LR, Leys MJ, Hansen RM, Fulton AB.Hyperopia in complicated Leber´s congenialamaurosis. Arch Ophthalmol 1990; 108:709-712.
128. Wagner Rs, Caputo AR, Nelson LB, ZanoniD. High hyperopia in Leber’s congenitalamaurosis. Arch Opthalmol 1985; 103: 1507-1509.
129. Chew E, Deutman A, Pinckers A, Aan deKerk A. Yellowish flecks in Leber´scongenital Amaurosis. Br J Ophthalmol1984; 68: 727-731.
130. Heher Kl, Traboulsi EI; maumenee IH. Thenatural history of Leber’s congenitalamaurosis: age related findings in 35patients. Ophthalmology 1992; 99: 241-245.
131. Lambert SR, Kriss A, Taylor d, et al. Follow-up and diagnostic reappraisal of 75 patientswith Leber´s congenital amaurosis. Am JOphthalmol 1989; 107: 624-631.
44
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...

132. Smith D, Oestreicher J, Musarella MA.Clinical spectrum of Leber´s congenitalamaurosis in the second to fourth decadesof life. Ophthalmology 1990; 97: 1156-1161.
133. Franceschetti A, Dieterle P. Importancediagnostique et pronostique del´électroretinogramme (ERG) dans lesdégénérescences tapéto-rétiniennes avecrétrécissement du champ visuel ethéméralopie. Confin Neurol 1954; 14: 184-186.
134. Chaker N, Mghaieth F, Baccouri R, MerdassiA, Turki F, El Matri L. Clinical andangiographic characteristics of Bietti’scorneoretinal dystrophy: a case study of an8-year-old girl. J Fr Ophtalmol 2007; 30: 39-43.
135. Giuffre G. Progression of Bietti’s crystallinedystrophy. J Fr Ophtalmol 1991; 14: 249-254.
136. Humbert G, Delettre C, Sénéchal A,Bazalgette C, Barakat A, Bazalgette C,Arnaud B, Lenaers G, Hamel CP.Homozygous deletion related to Alu repeatsin RLBP1 causes retinitis punctataalbescens. Invest Ophthalmol Vis Sci 2006;47: 4719-4724.
137. Ellis DS, Heckenlively JR. Retinitis punctataalbecens; fundus appearance and functionalabnormalities. Retina 1983; 3: 27-31.
138. Marmor MF. Defining fundus albipunctatus.Doc Ophthalmol Proc Ser 1977; 13: 227-234.
139. Pearlman JT, Flood TP, Siff SR. Retinitispigmentosa sine pigmento. Am JOphthalmol 1976; 81: 417-419.
140. Van Woerkom C, Ferrucci S. Sector retinitispigmentosa. Optometry 2005; 76: 309-317.
141. de Crecchio G, Alfieri MC, Cennamo G,D’Esposito F, Forte R. Pericentralpigmentary retinopathy: long-term follow-up.Eye 2006; 20: 1408-1410. Epub 2006 Feb 3.
142. Choi JY, Sandberg MA, Berson EL. Naturalcourse of ocular function in pigmentedparavenous retinochoroidal atrophy. Am JOphthalmol 2006; 141: 763-765.
143. Porta A, Pierrottet C, Aschero M, Orzalesi N.Preserved para-arteriolar retinal pigmentoepithelium retinitis pigmentosa. Am JOphthalmol 1992; 113: 161-164.
144. Henkes HE. Does Unilateral retinitispigmentosa really exist? An ERG and EOGstudy of the fellow eye. In Burian HM,Jacobson JH, editors: Clinicalelectroretinography: proceedings of theThird International Symposium held inOctober, 1964. Oxford, New York. PergamonPress. 1966, pp. 327-350.
145. Carr RE, Siegel IM. Unilateral retinitispigmentosa. Arch Ophthalmol 1973; 90: 21-26.
146. Foxman SG, Heckenlively JR, Bateman JB,Wirtschafter JD. Classification of congenitaland early onset retinitis pigmentosa. ArchOphthalmol 1985; 103: 1502-1506.
147. Spekreijse H, Apkarian P. The use of asystem analysis approach toelectrodiagnostic (ERG and VEP)assessment. Vision Res 1986; 26: 195-219.
148. Rubin ML, Fishman RS, McKay RA.Choroideremia; study of a family andliterature review. Arch Opthalmol 1966: 76:563-574.
149. McCulloch C, McCulloch RJP. A hereditaryand clinical study of choroideremia. TransAm Acad Ophthalmol Otolaryntol 52ndAnnual Mtg. 1947, publ 1948, pp. 160-190.
150. Renner AB, Kellner U, Cropp E, PreisingMN, MacDonald IM, van den Hurk JA,Cremers FP, Foerster MH. Choroideremia:variability of clinical and electrophysiologicalcharacteristics and first report of a negativeelectroretinogram. Ophthalmology 2006;113: 2066.e1-10. Epub 2006 Aug 28.
151. Seabra MC, Brown MS, Goldstein JL.Retinal degeneration I choroideremia:deficiency of Rab geranylgeranyltransferase. Science 1993; 259: 377.
152. Hayasaka S, Shoji K, Kanno C-I et al.Differential diagnosis of diffuse choroidalatrophies, diffuse choriocapillaris atrophy,choroideremia, and gyrate atrophy of thechoroid and retina. Retina 1985; 5: 30-37.
153. Kurstjens JH. Choroideremia and Gyrateatrophy of the choroid and retina. DocOphthalmol 1965; 19: 1.
154. Van der HurkJA, Van Zandvoort PM,Brunsmann F, et al. Prenatal exclusion ofchoroideremia. Am J Med Genet 1992; 44:822-823.
155. Wilson DJ, Weleber RG, Green WR. Ocularclinicopathologic study of gyrate atrophy.Am J Ophthalmol. 1991; 111: 24-33.
45
Bibliografía

156. Kaiser-Kupfer MI. et al. Systemicmanifestations of gyrate atrophy of the choroidand retina. Ophthalmology 1981; 88: 302.
157. Takki K Gyrate atrophy of the choroid andretina with hyperornithinemia Br JOphthalmol 1974; 58: 53.
158. Kaiser-Kupfer MI et al. Gyrate atrophy of thecohoroid and retina: improved visualfunction following reduction of plasmaornithine by diet. Science 1980; 210: 1128.
159. Kaiser-Kupfer MI, Caruso RC, Valle D. Gyrateatrophy of the choroid and retina. Long-termreduction of ornithine slows retinaldegeneration. Arch Ophthalmol 1991; 109:1539-1548.
160. Carr RE. Congenital stationary nightblindness. Trans Am Ophthalmol Soc 1974;72: 448-487.
161. Watanabe I, Taniguchi Y, Morioka K, Kato M.Congenital stationary night blindness withmyopia: a clinicopathologic study. DocOphthalmol 1986; 63: 55-62.
162. Strom TM, Nyakatura G, Apfelstedt-Sylla E, etal. An L-type calcium-channel gene mutated inincomplete X-linked congenital stationarynight blindness. Nat Genet 1998; 19: 260-263.
163. Rao VR, Cohen GB, Oprian DD. Rhodopsinmutation G90D and a molecular mechanismfor congenital night blindness. Nature 1994;367: 639-642.
164. Ripps H. Night blindness revisted from manto molecules. Invest Ophthalmol Vis Sci1982; 23: 588-609.
165. Dryja TP, Hahn LB, Reboul T, Arnaud B.Missense mutation in the gene encodingthe alpha subunit of rod transducin in theNougaret form of congenital stationary nightblindness. Nat Genet 1996; 13: 358-360.
166. Miyake Y, Yagasaki K, Horiguchi M, KawaseY, Kanda T. Congenital stationary nightblindness with negative electroretinogram: anew classification. Arch Ophthalmol 1986;104: 1013-1020.
167. Young RS, Krefman RA, Anderson RJ,Fishman GA. Two additional benefits of darkglasses on rod vision in patients withcongenital achromatopsia. Am J OptomPhysiol Opt. 1983; 60: 56-60.
168. Rajak SN, Currie AD, Dubois VJ, Morris M,Vickers S. Tinted contact lenses as analternative management for photophobia instationary cone dystrophies in children. JAAPOS 2006; 10: 336-339.
169. Nathans J, Thomas D, Hogness DS.Molecular genetics of human colour vision:the genes encoding blue green and redpigments. Science 1986; 232: 193-202.
170. Reichel E, Bruce AM, Sandberg MA, BersonEL An electroretinographic and moleculargenetic study of x-linked cone degeneration.Am J Ophthalmol 1989; 108: 540-547.
171. Neitz J, Neitz M. Color vision defects. InMolecular Genetics of Inherited EyeDisorders. Jay B. and Wright AF eds.Harwood academic publishers. New York,New York. 1994: Vol 2: Chap 9; pp. 217-57.
172. Van Driel MA, Maugeri A, Klevering BJ, etal. ABCR unites what ophthalmologistdivide. Ophthalmic Genet 1998; 19: 117-122.
173. Riveiro-Alvarez R, Aguirre-Lamban J, Lopez-Martinez MA, Trujillo-Tiebas MJ,Cantalapiedra D, Vallespin E, Avila-FernandezA, Ramos C, Ayuso C. Frequency of ABCA4mutations in 278 Spanish controls: an insightinto the prevalence of autosomal recessiveStargardt disease (arSTGD). Br J Ophthalmol2008 Oct 31. [Epub ahead of print].
174. Allikmets R, Singh N, Shroyen SH, et al. Aphotoreceptor cell-specific ATP-bindingtransporter gene (ABCR) is mutated inrecessive Stargardt macular Dystrophy. NatGenet 1997; 15: 236-246.
175. Mandal NA, Ambasudhan R, Wong PW, etal. Characterization of mouse orthologue ofELOVL4: genomic organization and spatialand temporal expression. Genomics 2004;83: 626-635.
176. Cideciyan AV, Aleman TS, Swider M, et al.Mutations in ABCA4 results in accumulationof lipofuscin before slowing of the retinoidcycle: a reappraisal of the human diseaseséquense. Hum Mol Genet 2004; 13: 525-534.
177. De Laey JJ, Veroughstraete C.Hyperlipofuscinosis and subretinal fibrosis inStargardt’s disease. Retina 1995; 15: 399-406.
178. Shroyen NF, Lewis RA, Allikmets R, et all.The rod photoreceptor ATP-binding cassettetransporter gene, ABCR and retinal disease:from monogenic to multifactorial. Vis Res1999; 39: 2537-2544.
179. Mata NL, Weng J, Travis GH. Biosynthesisof a major lipofuscin fluorophore in miceand human retinal and maculardegeneration. Proc Natl Acad Sci 2000; 13:7154-7159.
46
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...

180. Pacione LR, Szego MJ, Ikeda S, et al.Progress toward understanding the geneticand biochemical mechanisms of inheritedphotoreceptor degenerations. Annu RevNeurosci 2003; 26: 657-700.
181. Paloma E, Coco R, Martínez-Mir A, VilageliuL, Balcells S, González-Duarte R. Análisis ofABCA4 in mixed Spanish Familiassegregating different retinal dystrophies.Hum Mutat 2002; 20: 476-483.
182. Gass JDM, Sterescopic atlas of maculardiseases: diagnosis and treatment, 4th ed.St Louis, MO: CV Mosby; 1997: pp. 326-333.
183. Cideciyan AV, Swider M, Aleman TS,Sumaroka A, Schwartz SB, Roman MI,Milam AH, Bennett J, Stone EM, JacobsonSG. ABCA4-associated retinal degenerationsspare structure and function of the humanparapapillary retina. Invest Ophthalmol VisSci 200; 46: 4739-4746.
184. Fishman GA, Stone EM, Grover S, et al.Variation of clinical expression in patientswith Stargardt dystrophy and sequencevariations in the ABCR gene. ArchOphthalmol 1999; 117: 504-510.
185. Kang DerwentJJ, Derlacki DJ, Hetling JR, etal. Dark Adaptation of rod photoreceptors innormal subjects, and in patients withStargardt disease and an ABCA4 mutations.Invest Ophthalmol Vis Sci 2004; 45: 2447-2456.
186. Lachapelle P, Little JM, Roy MS. Theelectroretinogram in Stargardt´s disease andfundus flavimaculatus. Doc Ophthalmol1989; 73: 395-404.
187. Shastry BS. Evaluation of the commonvariants of the ABCA4 gene in families withStargardt disease and autosomal recessiveretinitis pigmentosa. Int J Mol Med 2008;21: 715-720.
188. Lois N, Holder GE, Fitzke FW, et al.Intrafamilial variation of phenotype inStargardt Macular Dystrophy-FundusFlavimaculatus. Invest Ophthalmol Vis Sci1999; 40: 2668-2675.
189. Ergun E, Hermann B, Wirtitsch M,Unterhuber A, Ko TH, Sattmann H, ScholdaC, Fujimoto JG, Stur M, Drexler W.Assessment of central visual function inStargardt’s disease/fundus flavimaculatuswith ultrahigh-resolution optical coherencetomography. Invest Ophthalmol Vis Sci2005; 46: 310-316.
190. Sunness JS, Ziegler MD, Applegate CA.Issues in quantifying atrophic maculardisease using retinal autofluorescence.Retina 2006; 26: 666-672.
191. Sunness JS, Steiner JN. Retinal functionand loss of autofluorescence in stargardtdisease. Retina 2008; 28: 794-800.
192. Oh KT, Weleber RG, Oh DM, Billingslea AM,Rosenow J, Stone EM. Clinical phenotypeas a prognostic factor in Stargardt disease.Retina 2004; 24: 254-262.
193. Oh KT, Weleber RG, Stone EM, Oh DM,Rosenow J, Billingslea AM.Electroretinographic findings in patientswith Stargardt disease and fundusflavimaculatus. Retina 2004; 24: 920-928.
194. Schuchard RA. Adaptation to macularscotomas in persons with low vision. Am JOccup Ther 1995; 49: 870-876.
195. Altpeter E, Mackeben M, Trauzettel-KlosinskiS. The importance of sustained attention forpatients with maculopathies. Vision Res2000; 40: 1539-1547.
196. Radu RA, Mata NL, Bagla A, et al. Lightexposure stimulates formation of A2Eoxiranes in a mouse model of Stargardt’smacular degeneration. Proc Natl Acad Sci2004; 101: 5928-5923.
197. Radu RA, Yuan Q, Hu J, Peng JH, Lloyd M,Nusinowitz S, Bok D, Travis GH. Acceleratedaccumulation of lipofuscin pigments in theRPE of a mouse model for ABCA4-mediatedretinal dystrophies following Vitamin Asupplementation. Invest Ophthalmol Vis Sci2008; 49: 3821-3829. Epub 2008 May 30.
198. Radu RA, Mata NL, Nusinowitz S, et al.Treatment with isotretinoin inhibitslipofuscin accumulation in a mouse modelof recessive Stargardt’s maculardegeneration. Proc Natl Acad Sci 2003; 100:4742-4747.
199. Kong J, Kim SR, Binley K, Pata I, Doi K,Mannik J, Zernant-Rajang J, Kan O, Iqball S,Naylor S, Sparrow JR, Gouras P, Allikmets R.Correction of the disease phenotype in themouse model of Stargardt disease bylentiviral gene therapy. Gene Ther 2008; 15:1311-1320. Epub 2008 May 8.
200. Zhang K, Kniazeva M, Hutchinson A, et al.The ABCR gene in recessive and dominantStargardt diseases: a genetic pathway inmacular degeneration. Genomics1999; 60:234-237.
47
Bibliografía

201. Donoso LA, Edwards AO, Frost A, et al.Autosomal Dominant Stargardt-like maculardystrophy. Surv Ophthalmol 2001, 46: 149-163.
202. Sorsby A, Crick RP. Central areolar choroidalsclerosis. Br J Ophthalmol 1953; 37: 129-139.
203. Carr RE Central areolar choroidal dystrophy.Arch Ophthalmol 1965; 73: 32-35.
204. Noble KG: Central areolar choroidal dystrophy.Am J Ophthalmol 1977; 84: 310-318.
205. Gass JDM, Sterescopic atlas of maculardiseases: diagnosis and treatment, 4th ed.St Louis, MO: CV Mosby; 1997: pp. 334-336.
206. Gamundi MJ, Hernan I, Muntanyola M,Trujillo MJ, García-Sandoval B, Ayuso C,Baiget M, Carballo M. High prevalence ofmutations in peripherin/RDS in autosomaldominant macular dystrophies in a Spanishpopulation. Mol Vis 2007; 13: 1031-1037.
207. Sorsby A: Choroidal angiosclerosis withspecial reference to its hereditary character.Br J Ophthalmol 1939; 23: 433-444.
208. Wells J, Wroblewski J, Keen J, et al:Mutations in the human retinaldegeneration slow (rds) gene can causeeither retinitis pigmentosa or maculardystrophy. Nat Genet 1992; 3: 213-217.
209. Klevering BJ, van Driel M, van HogerwouAJ, van De Pol DJ, Deutman AF, PinckersAJ, Cremers FP, Hoyng CB. Central areolarchoroidal dystrophy associated withdominantly inherited drusen. Br JOphthalmol 2002; 86: 91-96.
210. Hoyng CB, Deutman AF. The developmentof central areolar choroidal dystrophy.Graefes Arch Clin Exp Ophthalmol 1996;234: 87-93.
211. Best F. Über eine hereditaremaculaaffektion: Bietrage zurvererbungslehre. Z Augenheilkd 1905; 13:199-212.
212. Blodi C, Stone E. Best’s vitelliformdystrophy. Ophthalmic Paediatr Genet 1990;11: 49-59.
213. Stone E, Nichols B, Streb L, Kimura A,Sheffield V. Genetic linkage of vitelliformmacular degeneration (Best disease) tochromosome 11q13. Nat Genet 1992; 1:246-250.
214. Forsman K, Graff C, Nordstrom S, et al. Thegene for Best’s macular dystrophy is locatedat 11q13 in a Swedish family. Clin Genet1992; 42: 156-159.
215. Marquardt A, Stohr H, Passmore L, Kramer F,Rivera A, Weber B. Mutations in a novelgene, VMD2, encoding a protein of unknownproperties cause juvenile onset vitelliformmacular dystrophy (Best’s disease). Hum MolGenet 1998; 7: 1517-1525.
216. Petrukhin K, Koisti M, Bakall B, et al.Identification of the gene responsible forBest macular dystrophy. Nat Genet 1998;19: 241-247.
217. Franc¸ois J, De Rouck A, Fernandez-SassoD. Electro-oculography in vitelliformdegeneration of the macula. ArchOphthalmol 1967; 77: 726-733.
218. Deutman AF. Electro-oculography in familieswith vitelliform dystrophy of the fovea. ArchOphthalmol 1969; 81: 305-316.
219. Marmor MF. Vitelliform lesions in adults.Ann Ophthalmol 1979; 11: 1705-1712.
220. Gass JD, Jallow S, Davis B.Adult vitelliformmacular detachment occurring in patientswith basal laminar drusen. Am J Ophthalmol1985; 99: 445-459.
221. Meunier I, Cohen SY, Debibie C, Quentel G.Five-year evolution of basal laminar drusencombined with vitelliform maculardetachment. Arch Ophthalmol 2004; 122:1566-1567.
222. Barbazetto IA, Yannuzzi NA, Klais CM,Merriam JE, Zernant J, Peiretti E, YannuzziLA, Allikmets R. Pseudo-vitelliform maculardetachment and cuticular drusen: exclusionof 6 candidate genes. Ophthalmic Genet2007; 28: 192-197.
48
4 | Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades...

Guías de práctica clínica de la SERV
4 | Guía clínica para el diagnósticodiferencial y el manejo de lasenfermedades hereditarias dela retina y la coroides